Амидные соединения в качестве активаторов противовирусных препаратов
Номер патента: 19616
Опубликовано: 30.05.2014
Авторы: Аше Жервен Ивонн Поль, Халленбергер Беате Забине, Схепенс Вим Берт Грит, Ван`т Клостер Гербен Альберт Элетериус, Йонкерс Тим Хьюго Мария, Баумайстер Юдит Ева, Сасаки Дженнифер Тийоми
























Формула / Реферат
1. Соединение формулы

его фармацевтически приемлемые соли и стереоизомерные формы, где R представляет собой Н, фенил, пиридил, C1-6алкил или
где А и В независимо представляют собой Н, C1-6алкил, необязательно замещенный этинильной, имидазолильной группой или гетероатомом, представляющим собой азот, который необязательно замещен C1-6алкильной группой, или где А и В вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членное насыщенное, частично или полностью ненасыщенное гетероциклическое кольцо, содержащее от 1 до 2 гетероатомов, каждый из которых независимо выбран из азота или кислорода;
R1 выбран из группы, включающей

R2 представляет собой C1-6алкильную группу, необязательно замещенную ОН-группой, аминоС1-6алкильной, пирролидинильной, морфолинильной, этинильной или С3-7циклоалкильной группой, необязательно замещенной атомом галогена;
R3 представляет собой ОН;
R4 представляет собой Н;
R5 представляет собой пиридильную или фенильную группу, необязательно замещенную атомом галогена;
X представляет собой О, S или N, необязательно замещенный C1-6алкильной группой.
2. Соединение по п.1, имеющее формулу

3. Соединение по п.1, имеющее формулу

4. Соединение по п.1, имеющее формулу

5. Применение соединения по любому из пп.1-4 в качестве лекарственного средства для лечения или предотвращения ВИЧ-инфекций.
6. Применение соединения по любому из пп.1-4 для производства лекарственного средства для активации противовирусных ВИЧ-препаратов.
7. Комбинация для лечения или предотвращения ВИЧ-инфекций, включающая
a) соединение по любому из пп.1-4 или его фармацевтически приемлемую соль и
b) ингибитор ВИЧ.
8. Комбинация по п.1, где ингибитор ВИЧ представляет собой дарунавир или гексагидрофуро[2,3-b]фуран-3-иловый эфир (1-бензил-3-{[2-(1-циклопентилпиперидин-4-иламино)бензотиазол-6-сульфонил]изобутиламино}-2-гидроксипропил)карбаминовой кислоты.
9. Комбинация по п.8, где соединение по пп.1-4 представляет собой тиазол-5-илметил (2S,3R)-4-(2-(этил(метил)амино)-N-изобутилбензо[d]оксазол-6-карбоксамидо)-3-гидрокси-1-фенилбутан-2-илкарбамат и ингибитор ВИЧ представляет собой дарунавир или гексагидрофуро[2,3-b]фуран-3-иловый эфир (1-бензил-3-{[2-(1-циклопентилпиперидин-4-иламино)бензотиазол-6-сульфонил]изобутиламино}-2-гидроксипропил)карбаминовой кислоты.
10. Комбинация по п.9, где соединение по пп.1-4 представляет собой тиазол-5-илметил (2S,3R)-4-(2-(этил(метил)амино)-N-изобутилбензо[d]оксазол-6-карбоксамидо)-3-гидрокси-1-фенилбутан-2-илкарбамат и ингибитор ВИЧ представляет собой гексагидрофуро[2,3-b]фуран-3-иловый эфир (1-бензил-3-{[2-(1-циклопентилпиперидин-4-иламино)бензотиазол-6-сульфонил]изобутиламино}-2-гидроксипропил)карбаминовой кислоты.
11. Применение комбинации по любому из пп.7-10 в качестве лекарственного средства для лечения или предотвращения ВИЧ-инфекций.
12. Применение комбинации по любому из пп.7-10 для производства лекарственного средства для лечения или предотвращения ВИЧ-инфекций.
13. Фармацевтическая композиция, содержащая комбинацию по любому из пп.7-10 и фармацевтически приемлемый эксципиент.
Текст
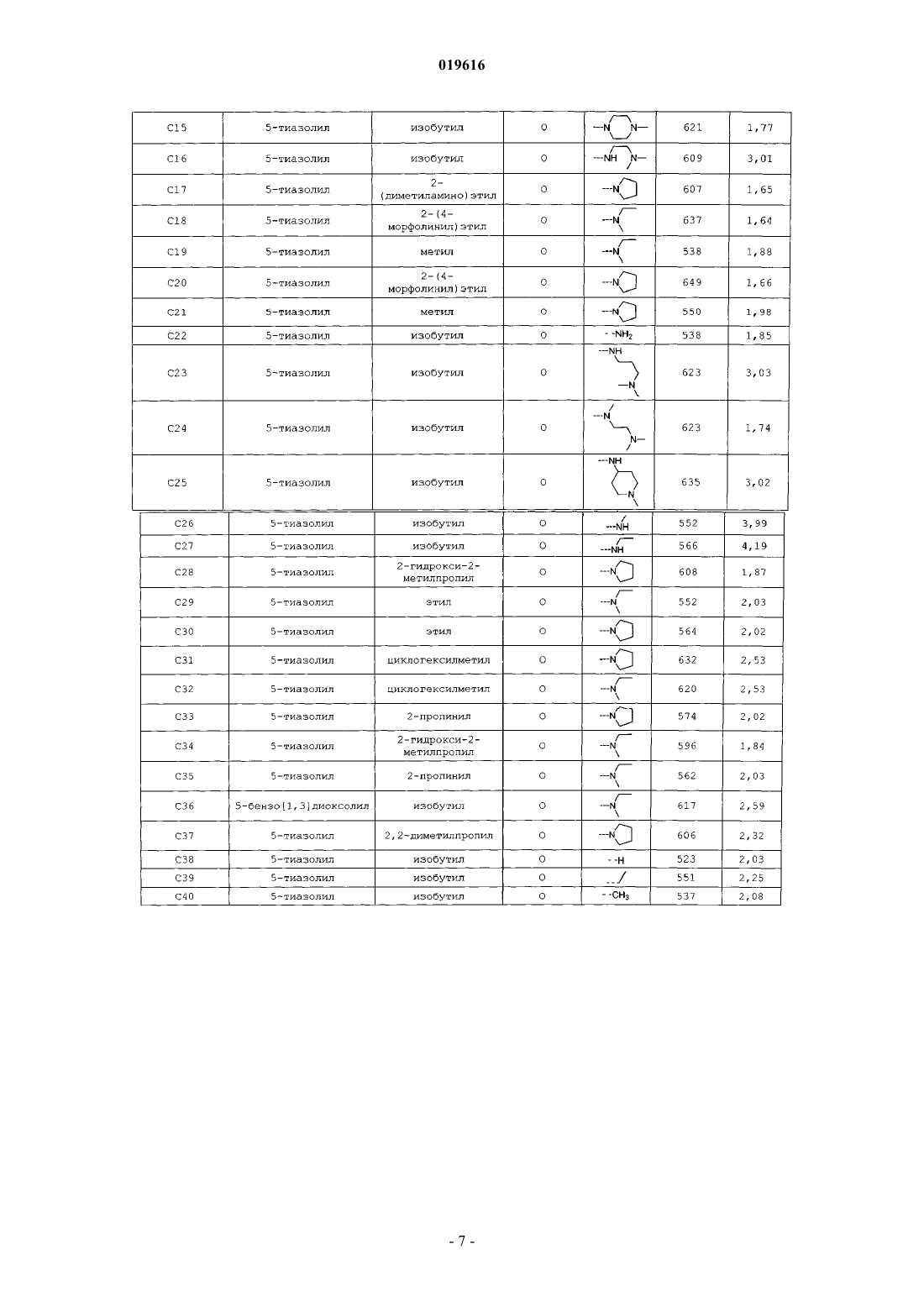
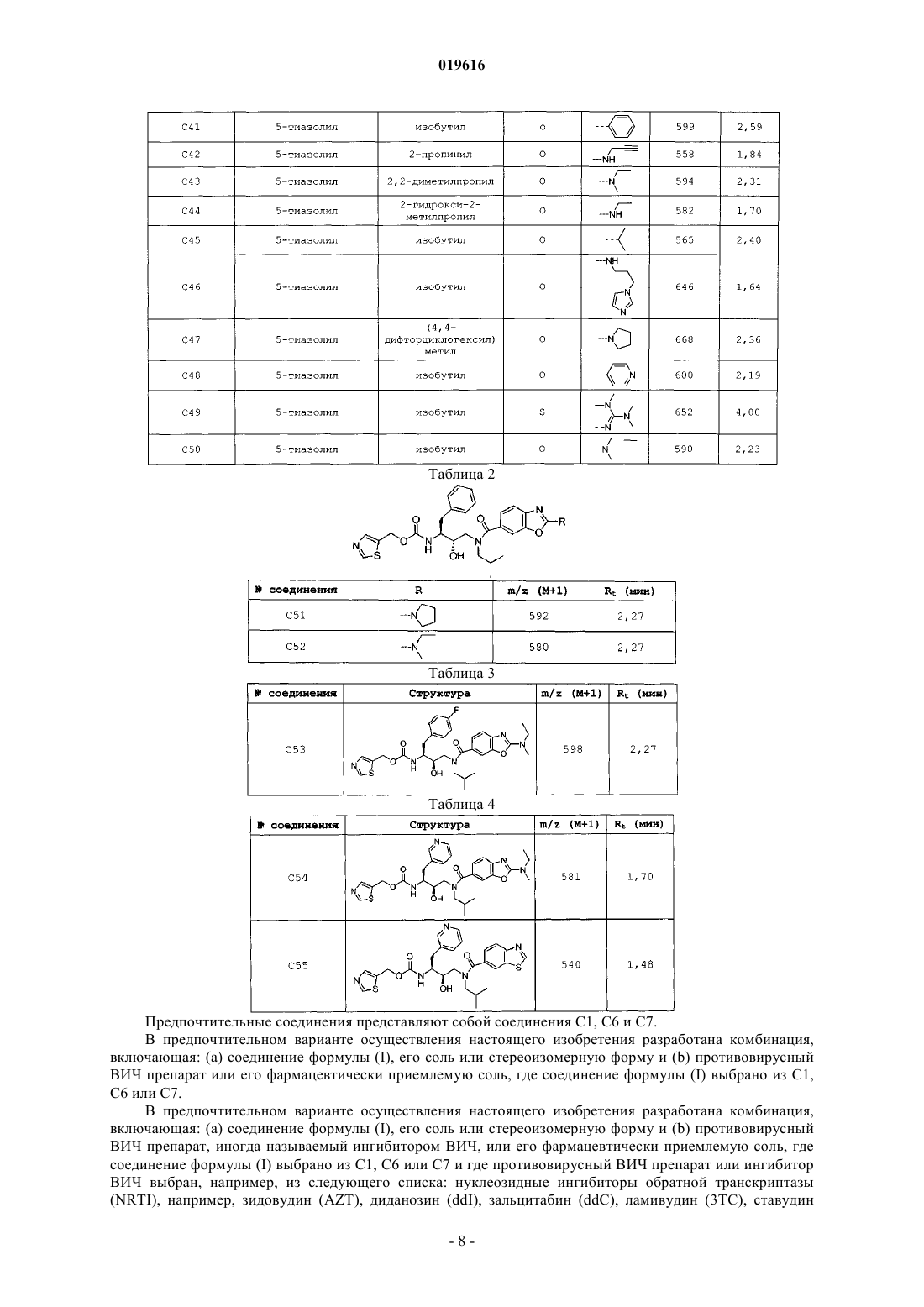
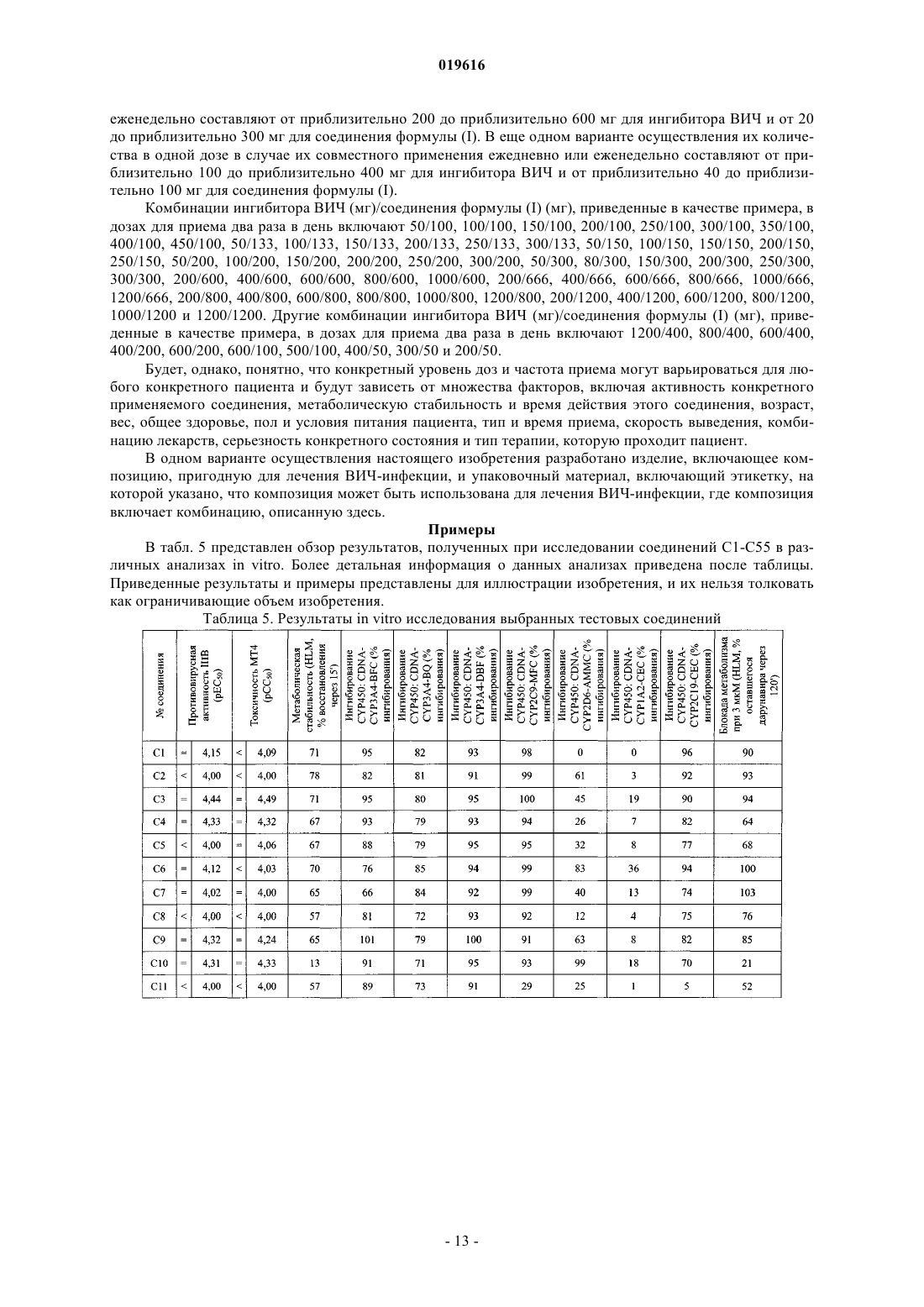
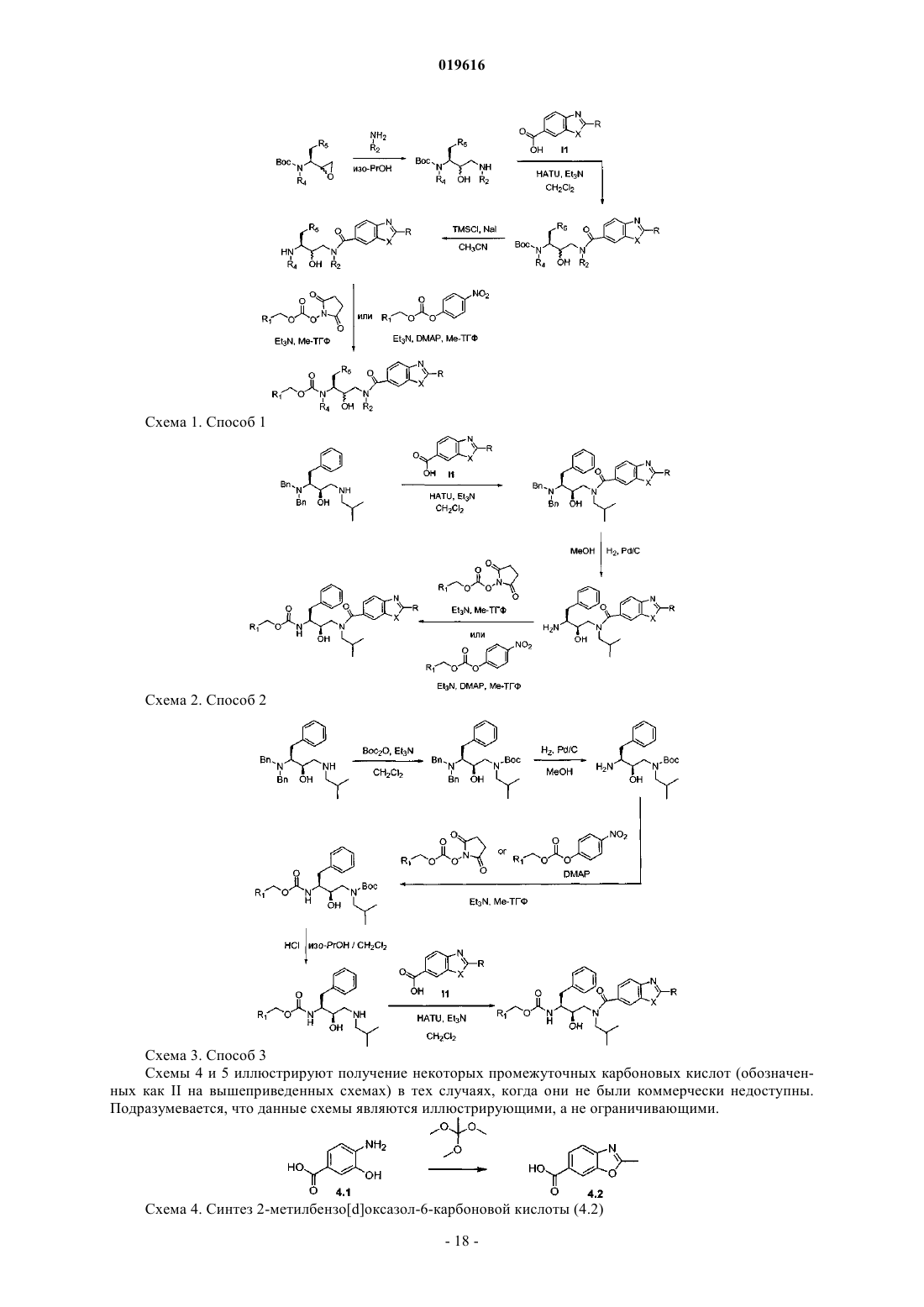
АМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АКТИВАТОРОВ ПРОТИВОВИРУСНЫХ ПРЕПАРАТОВ Настоящее изобретение относится к соединениям, которые обладают ингибирующими свойствами по отношению к CYP450 и, следовательно, полезны в качестве активаторов некоторых лекарств, т.е. они способны увеличивать по меньшей мере одну из фармакокинетических переменных некоторых лекарств при совместном применении. Изобретение также предусматривает применение указанных соединений для улучшения биодоступности некоторых лекарств. Также разработаны способы получения соединений по изобретению и фармацевтических композиций. Йонкерс Тим Хьюго Мария, Схепенс Вим Берт Грит, Аше Жервен Ивонн Поль, Халленбергер Беате Забине, Сасаки Дженнифер Тийоми,Баумайстер Юдит Ева, Ван'Т Клостер Гербен Альберт Элетериус (BE) Медведев В.Н. (RU) Настоящее изобретение относится к соединениям, которые обладают ингибирующими свойствами по отношению к CYP450 и, следовательно, полезны в качестве активаторов некоторых лекарств, т.е. они способны увеличивать по меньшей мере одну из фармакокинетических переменных некоторых лекарств при совместном применении. Изобретение также предусматривает применение указанных соединений в качестве веществ, повышающих биодоступность некоторых лекарств. Также разработаны способы получения соединений по изобретению и фармацевтических композиций, содержащих данные соединения. Многие лекарства, включая некоторые ингибиторы протеазы ВИЧ (PI) и ненуклеозидные ингибиторы обратной транскриптазы (NNRTI), метаболизируются цитохромной системой Р 450. Цитохромная система Р 450 представляет собой группу ферментов, найденных в печени и кишечнике, которые выполняют множество функций в организме человека. Активность цитохрома Р 450 различна для разных индивидуумов и популяций. Небольшие генетические отличия могут оказывать влияние на то, сколько экспрессировано конкретных ферментов и, таким образом, как быстро лекарство подвергается метаболизму. Ферменты цитохрома Р 450, которые являются производными гена, называют изоформами. На основании сходства их химического состава изоформы разделяют на семейства и подсемейства. Варианты ферментов описывают при помощи нумерации и буквенного обозначения, которые отражают их химическую и генетическую структуру. Цитохром Р 450, подсемейство IIIA (нифедипиноксидаза), полипептид 4, также называемыйCYP3A4, относится к одному конкретному пути метаболизма, используемому для разложения и выведения лекарств и других веществ. Метаболическое превращение некоторых лекарств при помощи цитохромной системы Р 450 часто приводит к тому, что указанные лекарства обладают неблагоприятными фармакокинетическими свойствами, и появляется необходимость принимать их более часто и в более высоких дозах, чем желательно. Введение таких лекарств вместе с агентом, который ингибирует метаболизм при помощи цитохромной системы Р 450, может улучшить фармакокинетические свойства лекарства. Исходя из этого, были опубликованы способы улучшения фармакокинетических свойств некоторых лекарств, см., например, USP 6037157, D.E. Kempf с соавт., Antimicrob. Agents Chemother., 41, стр. 654-660 (1997). В заявке WO03/049746 раскрыт способ улучшения фармакокинетических свойств гексагидрофуро[2,3-b]фуранил-содержащих ингибиторов протеазы ВИЧ, который включает введение человеку, который в этом нуждается, комбинации терапевтически эффективного количества гексагидрофуро[2,3b]фуранил-содержащего ингибитора протеазы ВИЧ и терапевтически эффективного количества ингибитора цитохрома Р 450. Большинство ингибиторов протеазы ВИЧ при клинической терапии в настоящий момент сочетают с ритонавиром для улучшения воздействия и, следовательно, повышения клинической эффективности. Данный тип применяемого взаимодействия лекарство-лекарство называют "активацией". Активация также позволяет использовать упрощенные режимы лечения при использовании существующих PI за счет уменьшения количества принимаемых лекарств и ежедневной частоты приема. К сожалению, улучшение режимов лечения, основанных на PI, под действием ритонавира даже при низких дозах не проходит без риска. Ритонавир сам представляет собой ингибитор протеазы ВИЧ. Резистентность к ритонавиру связана с отбором одной или более из нескольких резистентных мутаций. Резистентные мутации, связанные с использованием ритонавира, часто вызывают или способствуют развитию резистентности к другим ингибиторам протеазы. Различные мутации связаны с перекрестной резистентностью к различным лекарствам. Например, M46I связан с перекрестной резистентностью к индинавиру, нелфинавиру и фосампренавиру (но не к саквинавиру); V82A, F,T,S сам по себе связан с перекрестной резистентностью к индинавиру, но в комбинации с другими мутациями также вызывает резистентность к нелфинавиру, фосампренавиру и саквинавиру; и I84V способствует развитию резистентности ко всем доступным ингибиторам протеазы. Хотя ни одна из данных мутаций не связана с полной резистентностью к лопинавиру, каждая частично способствует развитию резистентности, и одновременное наличие нескольких мутаций может вызывать резистентность. Восприимчивость к индинавиру маловероятна при наличии резистентности к ритонавиру. Таким образом, для медицинских нужд необходимы альтернативы ритонавиру в качестве активатора для эффективной и безопасной анти-ВИЧ терапии. Для медицинских нужд также необходимы альтернативы ритонавиру в качестве активатора для эффективной и безопасной анти-ВИЧ терапии, в которой исключена возможность развития резистентности под действием активатора. В соответствии с настоящим изобретением сейчас было обнаружено, что следующие соединения формулы (I) обладают ингибирующими свойствами по отношению к CYP450 и могут использоваться в качестве активаторов. Данные соединения представлены формулой их фармацевтически приемлемые соли и стереоизомерные формы, где R представляет собой Н, фенил, пиридил, C1-6 алкил или где А и В независимо представляют собой Н, C1-6 алкил, необязательно замещенный этинильной,имидазолильной группой или гетероатомом, представляющим собой азот, который необязательно замещен С 1-6 алкильной группой, или где А и В вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членное насыщенное, частично или полностью ненасыщенное гетероциклическое кольцо,содержащее от 1 до 2 гетероатомов, каждый из которых независимо выбран из азота или кислорода;R2 представляет собой C1-6 алкильную группу, необязательно замещенную ОН-группой, аминоС 1-6 алкильной, пирролидинильной, морфолинильной, этинильной или С 3-7 циклоалкильной группой, необязательно замещенной атомом галогена;R5 представляет собой пиридильную или фенильную группу, необязательно замещенную атомом галогена;X представляет собой О, S или N, необязательно замещенный C1-6 алкильной группой. Предпочтительные соединения представляют собой следующие соединения формулы (II), (III) и(IV) соответственно, в качестве альтернативы описанные здесь как С 1, С 6 и С 7 соответственно. В следующем аспекте настоящее изобретение относится к применению соединений формул (I)-(IV) в качестве лекарственного средства для лечения или предотвращения ВИЧ-инфекций. Настоящее изобретение относится также к применению соединений формул (I)-(IV) для производства лекарственного средства для активации противовирусных ВИЧ препаратов. В еще одном аспекте настоящее изобретение относится к комбинации для лечения или предотвращения ВИЧ-инфекций, включающей:a) соединение формулы (I), (II), (III) или (IV) или его фармацевтически приемлемую соль иb) ингибитор ВИЧ. Предпочтительно ингибитор ВИЧ представляет собой дарунавир или гексагидрофуро[2,3-b]фуран 3-иловый эфир (1-бензил-3-[2-(1-циклопентилпиперидин-4-иламино)бензотиазол-6-сульфонил]изобутиламино-2-гидроксипропил)карбаминовой кислоты. Предпочтительно в комбинации соединение формулы (I) представляет собой тиазол-5-илметил(2S,3R)-4-(2-(этил(метил)амино)-N-изобутилбензо[d]оксазол-6-карбоксамидо)-3-гидрокси-1-фенилбутан 2-илкарбамат и ингибитор ВИЧ представляет собой дарунавир или гексагидрофуро[2,3-b]фуран-3 иловый эфир(1-бензил-3-[2-(1-циклопентилпиперидин-4-иламино)бензотиазол-6-сульфонил]изобутиламино-2-гидроксипропил)карбаминовой кислоты. Еще более предпочтительно, когда в комбинации соединение формулы (I) представляет собой тиазол-5-илметил (2S,3R)-4-(2-(этил(метил)амино)-N-изобутилбензо[d]оксазол-6-карбоксамидо)-3-гидрокси 1-фенилбутан-2-илкарбамат и ингибитор ВИЧ представляет собой гексагидрофуро[2,3-b]фуран-3-иловый эфир (1-бензил-3-[2-(1-циклопентилпиперидин-4-иламино)бензотиазол-6-сульфонил]изобутиламино-2 гидроксипропил)карбаминовой кислоты. Настоящее изобретение также направлено на применение указанной комбинации в качестве лекарственного средства для лечения или предотвращения ВИЧ-инфекций. Кроме того, настоящее изобретение касается применения указанной комбинации для производства лекарственного средства для лечения или предотвращения ВИЧ-инфекций. В следующем аспекте настоящее изобретние относится к фармацевтической композиции, содержащей указанную комбинацию и фармацевтически приемлемый эксципиент. Было обнаружено, что соединения формулы (I), (II), (III) и (IV) не вызывают появления резистентности по отношению к ВИЧ или вызывают ее на минимальном уровне, и, следовательно, их можно использовать как альтернативу ритонавиру (RTV) в качестве активаторов ингибиторов ВИЧ. Также было обнаружено, что соединения формулы (I) полезны в качестве активаторов других вирусных ингибиторов, таких как, например, ингибиторы вируса гепатита С (HCV) и/или респираторносинцитиального вируса (RSV). Комбинация соединений формулы (I) и других лекарств, таких как ингибиторы ВИЧ, вируса гепатита С и/или респираторно-синцитиального вируса, дает преимущество в том,что это позволяет разработать терапию для инфицированных пациентов, которая безопасна, эффективна и позволяет использовать более низкие терапевтически эффективные дозы противовирусных препаратов по сравнению с тем случаем, когда такие противовирусные препараты используются сами по себе. Более низкая доза всегда желательна с точки зрения токсичности и количества принимаемых лекарств, позволяя таким образом уменьшить появление побочных эффектов и увеличить восприимчивость к лечению соответственно. Комбинация соединений формулы (I) и ингибиторов ВИЧ или других вирусных ингибиторов приводит к синергическому действию данных противовирусных препаратов при введении указанной комбинации пациенту, нуждающегося в этом. Здесь и далее, как и в предшествующей части, используются следующие определения за исключением тех случаев, где указано обратное. Где бы ни использовался термин "замещенный" в определении соединений по данному изобретению, подразумевается, что он указывает на то, что один или более атомов водорода на атомах, указанных или входящих в выражение, использующее термин "замещенный", заменены на группу из указанного списка, при условии, что не превышена нормальная валентность указанных атомов и что замещение приводит к образованию химически стабильного соединения, т.е. соединения, которое сохраняет свою структурную и молекулярную индивидуальность при используемой степени чистоты и в течение удобного периода времени. Удобный период времени будет зависеть от области применения. Термин "галоген" относится к фтору, хлору, брому и йоду. Используемый здесь термин "C1-4 алкил" в качестве группы или части группы обозначает насыщенные углеводородные радикалы с неразветвленной или разветвленной цепью, содержащие от 1 до 4 атомов углерода, такие как, например, метил, этил, 1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-1-пропил; термин "C1-6 алкил" включает C1-4 алкильные радикалы и их высшие гомологи, содержащие 5 или 6 атомов углерода, такие как, например, 1-пентил, 2-пентил, 3-пентил, 1-гексил, 2-гексил, 2-метил-1-бутил,2-метил-1-пентил, 2-этил-1-бутил, 3-метил-2-пентил и подобные. Интерес среди C1-6 алкилов представляют C1-4 алкилы, в особенности изобутил."С 3-7 циклоалкил" относится к циклопропилу, циклобутилу, циклопентилу, циклогексилу и циклогептилу. Термин "гетероарил" хорошо известен в данной области техники и относится к моноциклической или бициклической системе, содержащей одно или два (конденсированых) ароматических кольца, причем циклическая система содержит по меньшей мере один гетероатом, выбранный из азота, кислорода или серы, и гетероатомы необязательно замещены C1-6 алкилом. Стоит отметить, что радикалы в любом молекулярном фрагменте, используемые в определениях,могут располагаться в любом месте данного фрагмента при условии, что он химически стабилен. Радикалы, используемые в определениях переменных, включают все возможные изомеры, если не указано обратное. Например, пиридил включает 2-пиридил, 3-пиридил и 4-пиридил. При любом использовании здесь и далее подразумевается, что выражения "соединения формулы (I),например", "настоящие соединения", "соединения по изобретению" или аналогичные термины включают соединения формулы (I) и их любую подгруппу, соединения, представленные в таблицах и примерах,приведенных ниже, стереохимически изомерные формы, рацемические смеси, аддитивные соли любого из вышеприведенных соединений. При любом использовании здесь и далее термин "противовирусный(е) препарат(ы) для лечения ВИЧ" и "ингибитор(ы) ВИЧ" взаимозаменяемы и в контексте текущего описания имеют одно и то же значение. Соединения формулы (I) могут содержать в своих заместителях хиральные центры и, следовательно, существовать в виде стереохимически изомерных форм. Термины "стереохимически изомерные формы", "стереоизомерные формы" и их эквиваленты, используемые здесь, определяют все возможные соединения, образованные одними и теми же атомами, связанными в одной и той же последовательности,но обладающие различными трехмерными структурами, которые не являются взаимозаменяемыми, ко-3 019616 торыми могут обладать соединения формулы (I). В тех случаях, когда для обозначения абсолютной конфигурации хирального атома с заместителем используются (R) или (S) или, в качестве альтернативы, символ , в обозначении учитывается соединение целиком, а не его отдельные заместители. Если не указано или не отмечено обратное, химическое обозначение соединения включает смесь любых возможных стереохимически изомерных форм, которыми может обладать данное соединение. Указанная смесь может содержать все диастереомеры и/или энантиомеры основной молекулярной структуры указанного соединения. Подразумевается, что все стереохимически изомерные формы соединений по настоящему изобретению как в чистой форме, так и в смеси друг с другом, включены в объем настоящего изобретения. Чистые стереоизомерные формы соединений и промежуточных соединений, как отмечено здесь,определяются как изомеры, в значительной степени свободные от других энантиомерных или диастереомерных форм той же самой исходной молекулярной структуры, как и у указанных соединений или промежуточных соединений. В частности, термин "стереоизомерно чистый" относится к соединениям или промежуточным соединениям со стереизомерным избытком, составляющим по меньшей мере 80% (т.е. минимум 90% одного изомера и максимум 10% других возможных изомеров), вплоть до стереоизомерного избытка в 100% (т.е. 100% одного изомера и отсутствие другого), более конкретно, к соединениям или промежуточным соединениям со стереизомерным избытком, составляющим от 90 вплоть до 100%,даже более конкретно, к соединениям или промежуточным соединениям со стереизомерным избытком,составляющим от 94 вплоть до 100%, и, наиболее конкретно, к соединениям или промежуточным соединениям со стереизомерным избытком, составляющим от 97 вплоть до 100%. Термины "энантиомерно чистый" и "диастереомерно чистый" необходимо понимать аналогичным образом, но в данном случае подразумевается соответственно энантиомерный избыток и диастереомерный избыток в рассматриваемой смеси. Чистые стереоизомерные формы соединений и промежуточных соединений по данному изобретению можно получить при использовании способов, известных в данной области техники. Например,энантиомеры можно отделить друг от друга при помощи селективной кристаллизации их диастереомерных солей с оптически активными кислотами или основаниями. Их примеры представляют собой винную кислоту, дибензоилвинную кислоту, дитолуоилвинную кислоту и камфорсульфокислоту. В качестве альтернативы, энантиомеры можно разделить при помощи хроматографии с использованием хиральных стационарных фаз. Указанные чистые стереохимически изомерные формы можно также получить из соответствующих чистых стереохимически изомерных форм подходящих исходных материалов при условии, что реакция протекает стереоспецифическим образом. Предпочтительно, если желательно получить конкретный стереоизомер, указанное соединение будет синтезировано при помощи стереоспецифических способов получения. Данные способы будут выгодно использовать энантиомерно чистые исходные материалы. Диастереомерные рацематы соединений по изобретению можно получить отдельно при помощи стандартных способов. Подходящие физические способы разделения, которые можно выгодно использовать, представляют собой, например, селективную кристаллизацию и хроматографию, например колоночную хроматографию. Для некоторых соединений по изобретению абсолютную стереохимическую конфигурацию нельзя определить экспериментально. Специалист в данной области техники способен определить абсолютную конфигурацию таких соединений при помощи способов, известных в данной области техники, таких как,например, дифракция рентгеновских лучей. В настоящих соединениях могут встречаться изотопы атомов. Изотопы включают те атомы, которые имеют одинаковый атомный номер, но различные массовые числа. В качестве общего примера и не ограничиваясь им, изотопы водорода включают тритий и дейтерий. Изотопы углерода включают С-13 и С-14. Термин "пролекарство", используемый в данном тексте, означает фармакологически приемлемые производные, такие как сложные эфиры, амиды и фосфаты, так, что в результате биопревращения производного in vivo получается активное лекарство, как определено в соединениях формулы (I). Работа Гудмана (Goodman) и Гилмана (Gilman) (The Pharmacological Basis of Therapeutics, 8 ред., McGraw-Hill, Int.Ed. 1992, "Biotransformation of Drugs", стр. 13-15), описывающая пролекарства в общем, включена здесь посредством ссылки. Пролекарства предпочтительно обладают отличной растворимостью в воде, повышенной биодоступностью и легко подвергаются метаболизму с образованием активных ингибиторов invivo. Пролекарства на основе соединения по настоящему изобретению можно получить модификацией функциональных групп, присутствующих в соединении, таким образом, что введенные фрагменты отщепляются, либо при помощи стандартных манипуляций, либо in vivo с образованием исходного соединения. Предпочтительны фармацевтически приемлемые сложноэфирные пролекарства, которые гидролизуются in vivo и которые можно получить из тех соединений формулы (I), которые содержат гидроксильную или карбоксильную группу. In vivo гидролизуемый сложный эфир представляет собой сложный эфир, который гидролизуется в теле человека или животного с образованием исходной кислоты или спирта. Подходящие фармацевтически приемлемые сложные эфиры для карбоксильной группы включают C1-6 алкоксиметиловые сложные эфиры, например метоксиметиловый сложный эфир, C1-6 алканоилоксиметиловые сложные эфиры, например пивалоилоксиметиловый, фталидиловые сложные эфиры,С 3-8 циклоалкоксикарбонилокси-C1-6 алкиловые сложные эфиры,например 1-циклогексилкарбонилоксиэтиловый сложный эфир, 1,3-диоксолен-2-онилметиловые сложные эфиры, например 5 метил-1,3-диоксолен-2-онилметиловый сложный эфир, и C1-6 алкоксикарбонилоксилэтиловые сложные эфиры, например, 1-метоксикарбонилоксиэтиловый сложный эфир, которые могут образовываться с участием любой карбоксильной группы в соединениях по данному изобретению. Гидролизуемый in vivo сложный эфир соединения формулы (I), содержащий гидроксильную группу, включает неорганические сложные эфиры, такие как сложные эфиры фосфорной кислоты, ацилоксиалкиловые эфиры и родственные соединения, которые в результате гидролиза сложного эфираin vivo разрушаются с образованием исходной гидроксильной группы. Примеры -ацилоксиалкиловых эфиров включают ацетоксиметоксильный и 2,2-диметилпропионилоксиметоксильный эфиры. Набор групп, образующих с гидроксильной группой гидролизуемые in vivo сложные эфиры, включают алканоил, бензоил, фенилацетил, замещенные бензоил и фенилацетил, алкоксикарбонил (для получения алкиловых эфиров угольной кислоты), диалкилкарбамоил и N-(диалкиламиноэтил)-N-алкилкарбамоил (для получения карбаматов), диалкиламиноацетил и карбоксиацетил. Примеры заместителей на бензоильной группе включают морфолиновую и пиперазиновую группу, присоединенные к 3 или 4 положению бензоильного кольца через атом азота кольца при помощи метиленовой группы. Для использования в терапии соли соединений формулы (I) представляют собой соли, в которых противоион является фармацевтически приемлемым. Однако соли кислот и оснований, которые не являются фармацевтически приемлемыми, также могут найти свое применение, например, в получении или очистке фармацевтически приемлемого соединения. Все соли, независимо от того, являются ли они фармацевтически приемлемыми или нет, включены в объем настоящего изобретения. Подразумевается, что фармацевтически приемлемые аддитивные соли кислот и оснований, указанные выше, включают терапевтически активные аддитивные нетоксичные соли кислот и оснований, которые соединения формулы (I) способны образовывать. Фармацевтически приемлемые кислотноаддитивные соли можно получить удобным образом при обработке основной формы соответствующей кислотой. Соответствующие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например хлористо-водородную или бромисто-водородную кислоты, серную,азотную, фосфорную и подобные кислоты, или органические кислоты, такие как, например, уксусную,пропионовую, гидроксиуксусную, молочную, пировиноградную, щавелевую (т.е. этандионовую кислоту), малоновую, янтарную (т.е. бутандионовую кислоту), малеиновую, фумаровую, яблочную (т.е. гидроксибутандионовую кислоту), винную, лимонную, метансульфоновую, этансульфоновую, бензолсульфоновую, п-толуолсульфоновую, цикламовую, салициловую, п-аминосалициловую, пальмитиновую и подобные кислоты. С другой стороны, указанные соли при обработке соответствующим основанием можно превратить в свободные основания. Соединения формулы (I), содержащие кислый протон, также можно превратить в нетоксичные соли металлов или аддитивные соли аминов при обработке соответствующими органическими или неорганическими основаниями. Соответствующие соли оснований включают, например, аммониевые соли, соли щелочных и щелочно-земельных металлов, например, литиевые, натриевые, калиевые, магниевые, кальциевые соли и подобные, соли с органическими основаниями, например бензатиновые, N-метил-Dглюкаминовые, гидробаминовые соли и соли с аминокислотами, такими как, например, аргинин, лизин и подобные. Термин "сольват" используется здесь для описания молекулярного комплекса, включающего i) соединения по изобретению, а также их соли и ii) одну или более молекул фармацевтически приемлемого растворителя, например, этанола, изопропанола, 1-метокси-2-пропанола, метанола, ацетона, дихлорметана, этилацетата, анизола, тетрагидрофурана или мезилата. Термин "гидрат" используется в том случае,когда указанный растворитель представляет собой воду. Термин "четвертичный амин", используемый здесь и ранее, определяет четвертичные аммонийные соли, которые соединения формулы (I) способны образовывать при взаимодействии между основным атомом азота соединения формулы (I) и соответствующим кватернизующим агентом, таким как, например, необязательно замещенный алкилгалогенид, арилгалогенид или арилалкилгалогенид, например метилиодид или бензилиодид. Также можно использовать другие реагенты с хорошими уходящими группами, такие как алкилтрифторметансульфонаты, алкилметансульфонаты и алкил-п-толуолсульфонаты. Четвертичный амин содержит положительно заряженный азот. Фармацевтически приемлемые противоионы включают хлорид, бромид, иодид, трифторацетат и ацетат. Выбранный противоион можно вводить при помощи ионообменных смол. Подразумевается, что N-оксиды настоящих соединений включают соединения формулы (I), в кото-5 019616 рых один или несколько атомов азота окислены до образования так называемого N-оксида. Будет понятно, что соединения формулы (I) могут обладать металлосвязывающими, хелатирующими, комплексообразующими свойствами и, следовательно, могут существовать в виде комплексов металлов или хелатов металлов. Некоторые соединения формулы (I) могут также существовать в таутомерной форме. Соединения формулы (I) содержат два асимметрических центра, показанных ниже знаком Предпочтительно соединения формулы (I) характеризуются стереохимией, показанной ниже в структурной формуле (I-а) Соединения формулы (I) в соответствии с настоящим изобретением могут быть выбраны из любого из следующих соединений в табл. 1, 2, 3 или 4. Помимо заместителей (обозначенных как R, R1, R2 и X),также приведены данные ЖХ-МС (m/z (M+1) и время удерживания (Rt. Представленные результаты и примеры приведены для иллюстрации изобретения, и их нельзя толковать как ограничивающие объем изобретения. Таблица 1 Предпочтительные соединения представляют собой соединения С 1, С 6 и С 7. В предпочтительном варианте осуществления настоящего изобретения разработана комбинация,включающая: (а) соединение формулы (I), его соль или стереоизомерную форму и (b) противовирусный ВИЧ препарат или его фармацевтически приемлемую соль, где соединение формулы (I) выбрано из С 1,C6 или C7. В предпочтительном варианте осуществления настоящего изобретения разработана комбинация,включающая: (а) соединение формулы (I), его соль или стереоизомерную форму и (b) противовирусный ВИЧ препарат, иногда называемый ингибитором ВИЧ, или его фармацевтически приемлемую соль, где соединение формулы (I) выбрано из С 1, С 6 или С 7 и где противовирусный ВИЧ препарат или ингибитор ВИЧ выбран, например, из следующего списка: нуклеозидные ингибиторы обратной транскриптазы(d4T), эмтрицитабин (FTC), абакавир (ABC), априцитабин (AVX-754), элвуцитабин (АСН-126443), фосфазид, КР-1461, MIV-210, рацивир (PSI-5004) и подобные, или ненуклеозидные ингибиторы обратной транскриптазы (NNRTI), такие как делавирдин (DLV), эфавиренз (EFV), невирапин (NVP), каправирин(CPV), каланолид А, дапивирин (ТМС 120), этравирин (ТМС 125), рилпивирин (ТМС 278), аловудин (MIV310), UC-781 и подобные, или нуклеотидные ингибиторы обратной транскриптазы (NtRTI), например тенофовир и тенофовир дизопроксил фумарат (TDF) и подобные, или ингибиторы трансактивирующих белков, такие как ингибиторы Tat, например, RO-5-3335, BI-201, ингибиторы вируса ретикулоэндотелиоза (REV) или ингибиторы протеазы, например, ритонавир (RTV), саквинавир (SQV), лопинавир (АВТ 378 или LPV), индинавир (IDV), ампренавир (VX-478), ТМС-126, нелфинавир (AG-1343), атазанавир(BMS-232632), дарунавир (ТМС-114), в настоящее время известный под торговой маркой Prezista, SPI256, фосампренавир (GW433908 или VX-175), Р-1946, МК-8122 (PPL-100), типранавир (PNU-140690),или ингибитор протеазы с химическим названием гексагидрофуро[2,3-b]фуран-3-иловый эфир (1-бензил 3-[2-(1-циклопентилпиперидин-4-иламино)бензотиазол-6-сульфонил]изобутиламино-2-гидроксипропил)карбаминовой кислоты и подобные, или ингибиторы вирусной интегразы, например, ралтегравир(МК-518), элвитегравир (GS-9137; JTK-303), BMS-538158 и подобные, или ингибиторы внедрения, которые включают ингибиторы слияния (например, Т-20 или энфувиртид, Т-1249), ингибиторы связывания и ингибиторы сорецепторов, последние включают антагонисты CCR5 и антагонисты CXR4 (например,AMD-3100), примеры ингибиторов внедрения представляют собой PRO-140, PRO-542, TBR-220 (ТАК 220), TBR-652 (ТАК-652), викривирок (SCH-417690), TNX-355, маравирок (UK-427857), BMS-488043,BMS-806, ингибитор развития, например, бевиримат (РА-457), рибонуклеотидные ингибиторы редуктазы(клеточные ингибиторы), например, гидроксимочевина и подобные, или любые сочетания из вышеприведенного. Наиболее предпочтительный вариант осуществления представляет собой комбинацию, включающую: (а) соединение, выбранное из С 1, С 6 или С 7, его соль или стереоизомерную форму и (b) противовирусный ВИЧ-препарат, выбранный из дарунавира или соединения с химическим названием гексагидрофуро[2,3-b]фуран-3-иловый эфир (1-бензил-3-[2-(1-циклопентилпиперидин-4-иламино)бензотиазол-6 сульфонил]изобутиламино-2-гидроксипропил)карбаминовой кислоты. Наиболее предпочтительна комбинация, в которой соединение представляет собой тиазол-5 илметил(2S,3R)-4-(2-(этил(метил)амино)-N-изобутилбензо[d]оксазол-6-карбоксамидо)-3-гидрокси-1 фенилбутан-2-илкарбамат и ингибитор ВИЧ представляет собой гексагидрофуро[2,3-b]фуран-3-иловый эфир (1-бензил-3-[2-(1-циклопентилпиперидин-4-иламино)бензотиазол-6-сульфонил]изобутиламино-2 гидроксипропил)карбаминовой кислоты. В одном варианте осуществления настоящего изобретения разработан способ получения описанной выше комбинации, включающий стадию смешивания соединения формулы (I) или его фармацевтически приемлемой соли и противовирусного ВИЧ-препарата или его фармацевтически приемлемой соли. Альтернативный вариант осуществления данного изобретения предусматривает способ, где комбинация включает один или более дополнительных агентов, как описано здесь. Комбинации по настоящему изобретению, а также сами соединения по настоящему изобретению можно использовать в качестве лекарственных средств. Указанное применение в качестве лекарства или лекарственного средства включает системное введение ВИЧ-инфицированным пациентам количества,эффективного для борьбы с состояниями, связанными с ВИЧ. Соединения по настоящему изобретению,более конкретно - соединения формулы (II)-(IV), особенно пригодны в качестве агентов для активации других химических веществ, таких как противовирусные химические соединения, у млекопитающих. Следовательно, комбинации по настоящему изобретению можно использовать в производстве лекарственного средства, пригодного для лечения, предотвращения или борьбы с инфекцией или заболеванием, связанным с ВИЧ-инфекцией у млекопитающего. В одном варианте осуществления настоящего изобретения разработана фармацевтическая композиция, включающая комбинацию в соответствии с любым из вариантов осуществления, описанных здесь, и фармацевтически приемлемый эксципиент. В частности, настоящее изобретению предусматривает фармацевтическую композицию, включающую: (а) терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли, (b) терапевтически эффективное количество противовирусного ВИЧ-препарата или его фармацевтически приемлемой соли и (с) фармацевтически приемлемый эксципиент. Фармацевтическая композиция также необязательно включает дополнительный агент, выбранный из противовирусных ВИЧ-препаратов. Подразумевается, что использованный здесь термин "композиция" включает продукт, включающий конкретные ингредиенты, а также любой продукт, который прямым или косвенным образом получается из комбинации конкретных ингредиентов. Использованный здесь термин "терапевтически эффективное количество" обозначает, что количество активного соединения или компонента или фармацевтического агента, который вызывает биологическую или лекарственную ответную реакцию в ткани, системе, животном или человеке, которой добивался в контексте настоящего изобретения исследователь, ветеринар, врач или другой специалист, и которая включает облегчение симптомов заболевания, которое лечат. Поскольку настоящее изобретение относится к комбинациям, включающим два или более агентов, "терапевтически эффективное количество" - это такое количество агентов, принятых вместе, что совокупный эффект вызывает желаемую биологическую или лекарственную ответную реакцию. Например, терапевтически эффективным количеством композиции, включающей: (а) соединение формулы (I) и (b) противовирусный ВИЧ-препарат, будет такое количество соединения формулы (I) и такое количество противовирусного ВИЧ-препарата, что вместе они производят совокупный эффект, который является терапевтически эффективным. Фармацевтическую композицию можно получить способом, непосредственно известным специалисту в данной области техники. С данной целью по меньшей мере одно соединение формулы (I) или любой ее подгруппы и противовирусный ВИЧ-препарат вместе с одним или более твердыми или жидкими фармацевтическими эксципиентами и, по желанию, в комбинации с другими фармацевтически активными соединениями переводят в подходящую форму для введения или дозированную форму, которую затем можно использовать в качестве фармацевтического препарата в медицине или ветеринарии. В одном варианте осуществления комбинации по настоящему изобретению составляют в виде комбинированных препаратов для одновременного, раздельного или последовательного применения в терапии ВИЧ, когда это возможно. В таком случае соединение общей формулы (I) или любой ее подгруппы составляют в фармацевтическую композицию, содержащую другие фармацевтически приемлемые эксципиенты, а соответствующий противовирусный ВИЧ-препарат составляют отдельно в фармацевтические композиции, содержащие другие фармацевтически приемлемые эксципиенты. Удобно, что две данные отдельные фармацевтические композиции могут быть частью набора для одновременного, раздельного или последовательного применения. Таким образом, индивидуальные компоненты комбинации по настоящему изобретению можно вводить по отдельности в разное время в ходе терапии или одновременно как в раздельных комбинациях или в одной комбинации. Следовательно, настоящее изобретение необходимо понимать как охватывающее все такие режимы одновременного или чередующегося лечения, и термин "введение" следует интерпретировать аналогичным образом. В предпочтительном варианте осуществления отдельные дозированные формы вводят практически одновременно. Композиции или продукты, включающие комбинацию по настоящему изобретению, независимо от того, включены ли они в один препарат или в препараты для одновременного, раздельного или последовательного применения, можно вводить перорально (включая суспензии, капсулы, таблетки, порошки,растворы, суспензии, эмульсии), сублингвально, парентерально (включая подкожные инъекции, внутривенную, внутримышечную и внутрикожную инъекцию или инфузионные способы), местно, ректально(включая суппозитории), вагинально, при помощи имплантированного резервуара, в единичных дозированных формах, содержащих стандартные нетоксичные фармацевтически приемлемые носители, адъюванты и среды. Предпочтительная композиция или продукт включает соединение любой из формул (I)-(IV) и ингибитор ВИЧ, где ингибитор представляет собой дарунавир или соединение с химическим названием гексагидрофуро[2,3-b]фуран-3-иловый эфир (1-бензил-3-[2-(1-циклопентилпиперидин-4-иламино)бензотиазол-6-сульфонил]изобутиламино-2-гидроксипропил)карбаминовой кислоты. В соответствии с изобретением наиболее предпочтительны композиция или продукт, включающие тиазол-5-илметил (2S,3R)-4-(2-(этил(метил)амино)-N-изобутилбензо[d]оксазол-6-карбоксамидо)-3-гидрокси-1-фенилбутан-2-илкарбамат, и где ингибитор ВИЧ представляет собой гексагидрофуро[2,3-b]фуран 3-иловый эфир (1-бензил-3-[2-(1-циклопентилпиперидин-4-иламино)бензотиазол-6-сульфонил]изобутиламино-2-гидроксипропил)карбаминовой кислоты. В случае формы для перорального введения композиции по настоящему изобретению можно смешивать с подходящими добавками, такими как эксципиенты, стабилизаторы или инертные разбавители,и переводить при помощи стандартных способов в подходящие формы для введения, такие как таблетки,покрытые таблетки, твердые капсулы, водные, спиртовые или масляные растворы. Примеры подходящих инертных носителей представляют собой гуммиарабик, оксид магния, карбонат магния, фосфат калия,лактозу, глюкозу или крахмал, в частности кукурузный крахмал. В данном случае приготовление может проводиться с использованием как сухих, так и влажных гранул. Подходящие масляные эксципиенты или растворители представляют собой растительные или животные масла, такие как подсолнечное масло или масло печени трески. Подходящие растворители для водных или спиртовых растворов представляют собой воду, этанол, растворы сахара или их смеси. В качестве дополнительных вспомогательных веществ для других форм для введения можно также использовать полиэтиленгликоли и полипропиленгликоли. В качестве таблеток немедленного высвобождения данные композиции могут содержать микрокристаллическую целлюлозу, дикальцийфосфат, крахмал, стеарат магния и лактозу и/или другие эксципиенты, связывающие агенты, модифицирующие агенты, дезинтегранты, разбавители и лубриканты, известные в данной области техники. Пероральное введение комбинации по настоящему изобретению осуществляют путем однородного и тщательного смешивания подходящего количества каждого компонента в виде порошка, необязательно также включающего мелкодисперсный твердый носитель, и помещения смеси, например, в твердую желатиновую капсулу. Твердый носитель может включать одно или более веществ, которые выступают в роли связывающих агентов, лубрикантов, дезинтегрантов, красителей и подобных. Подходящие твердые носители включают, например, фосфат кальция, стеарат магния, тальк, сахара, лактозу, декстрин, крахмал, желатин, целлюлозу, поливинилпирролидин, низкоплавкие воски и ионообменные смолы. Пероральное введение комбинации по настоящему изобретению можно также осуществлять путем получения капсул или таблеток, содержащих желаемое количество только соединения формулы (I), необязательно смешанного с твердым носителем, как описано выше, и капсул, содержащих желаемое количество только противовирусного ВИЧ-препарата. Прессованные таблетки, содержащие соединение формулы (I), можно получить при помощи однородного и тщательного смешивания активного ингредиента с твердым носителем, таким как описанный выше, для получения смеси, обладающей необходимыми компрессионными свойствами, и затем придания смеси желаемых формы и размера в подходящем устройстве. Отлитые таблетки могут быть изготовлены при помощи плавления в подходящем устройстве смеси порошкообразного соединения формулы (I) с инертными жидкими разбавителями. Пероральное введение можно также осуществлять путем получения прессованных или плавленых таблеток, содержащих соединение формулы (I), как было только что описано, таблеток подходящего размера для помещения в стандартные капсулы (например, твердые желатиновые капсулы) и затем помещение таблеток в капсулы, содержащие подходящее количество порошкообразного противовирусного ВИЧ-препарата. Для подкожного или внутривенного введения активные компоненты композиций, при желании вместе со стандартными веществами, такими как солюбилизаторы, эмульгаторы или другие вспомогательные вещества, переводят в раствор, суспензию или эмульсию. Компоненты композиций можно также подвергнуть лиофилизации, и полученные лиофилизаты использовать, например, для получения препаратов для инъекций или вливаний. Подходящие растворители представляют собой, например, воду,физиологический солевой раствор или спирты, например этанол, пропанол, глицерин, а также растворы Сахаров, такие как растворы глюкозы или маннита, или, в качестве альтернативы, смеси различных указанных растворителей. Растворы или суспензии для инъекций можно приготовить в соответствии со способами, известными в данной области техники, при использовании подходящих нетоксичных парентерально-приемлемых разбавителей или растворителей, таких как маннит, 1,3-бутандиол, вода, раствор Рингера или изотонический раствор хлорида натрия, или подходящих диспергирующих или увлажняющих и суспендирующих агентов, таких как стерильные смягчающие жирные масла, включая синтетические моно- или диглицериды, и жирные кислоты, включая олеиновую кислоту. Фармацевтические композиции по данному изобретению можно также вводить местно, особенно в тех случаях, когда объект лечения включает области или органы, легкодоступные при помощи местного применения, включая заболевания глаза, кожи или нижнего кишечника. Подходящие препараты для местного применения можно легко приготовить для каждой из данных областей или органов. Местное применение в случае нижнего кишечника может проводиться при помощи состава в виде ректальных суппозиториев (см. ниже) или при помощи подходящего состава для клизмы. Также можно использовать трансдермальные пластыри для местного применения. В случае местного применения фармацевтические композиции можно приготовить в виде подходящей мази, содержащей активные компоненты, суспендированные или растворенные в одном или более носителях. Носители для местного применения соединений по данному изобретению включают минеральное масло, жидкий вазелин, белый вазелин, пропиленгликоль, соединение на основе полиоксиэтилена и полиоксипропилена, эмульгирующий воск и воду, но не ограничиваясь ими. В качестве альтернативы, фармацевтические композиции можно приготовить в виде подходящего лосьона или крема, содержащего активные компоненты, суспендированные или растворенные в одном или более фармацевтически приемлемых носителях. Подходящие носители включают минеральное масло, сорбитмоностеарат,полисорбат 60, воск на основе цетиловых сложных эфиров, цетиариловый спирт, 2-октилдодеканол, бензиловый спирт и воду, но не ограничиваясь ими. При ректальном введении в виде суппозиториев данные препараты можно приготовить путем смешивания индивидуальных компонентов композиции по изобретению с подходящим нераздражающим эксципиентом, таким как какао-масло, синтетические глицеридные сложные эфиры или полиэтиленгликоли, которые при обычных температурах находятся в твердом состоянии, но переходят в жидкое состояние и/или растворяются в ректальной полости с высвобождением лекарства. В другом варианте осуществления способа по изобретению введение можно осуществлять с едой(например, богатой жиром пищей) или отдельно от еды. Термин "с едой" подразумевает прием пищи либо во время, либо не более чем за один час до или после введения одного или обоих компонентов комбинации по изобретению. В одном варианте осуществления комбинации по настоящему изобретению содержат такое количество соединения формулы (I) или его фармацевтически приемлемой соли, которое достаточно для клинического улучшения биодоступности ингибитора ВИЧ или противовирусного ВИЧ-препарата относительно биодоступности в случае, когда вводится только указанный ингибитор ВИЧ или противовирусный ВИЧ-препарат. В другом варианте осуществления комбинации по настоящему изобретению содержат такое количество соединения формулы (I) или его фармацевтически приемлемой соли, которое достаточно для по- 11019616 вышения по меньшей мере одной из фармакокинетических переменных ингибиторов ВИЧ, выбранных изt1/2, Cmin, Cmax, Css, площади под фармакокинетической кривой (AUC) за 12 ч или площади под фармакокинетической кривой (AUC) за 24 ч, относительно по меньшей мере одной указанной фармакокинетической переменной в том случае, когда вводится только указанный ингибитор ВИЧ. Еще один вариант осуществления относится к способу улучшения биодоступности ингибитора ВИЧ, включающему введение индивидууму, нуждающемуся в таком улучшении, описанной здесь комбинации, включающей терапевтически эффективное количество каждого компонента указанной комбинации. В еще одном варианте осуществления изобретение относится к применению соединения формулы(I) или его фармацевтически приемлемой соли для улучшения по меньшей мере одной из фармакокинетических переменных ингибитора ВИЧ, выбранных из t1/2, Cmin, Cmax, Css, площади под фармакокинетической кривой (AUC) за 12 ч или площади под фармакокинетической кривой (AUC) за 24 ч, при условии,что подобное применение не осуществляется в теле человека или животного. Термин "индивидуум", используемый здесь, относится к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, который выступает в качестве объекта лечения, наблюдения или эксперимента. Биодоступность определяют как долю введенной дозы, достигшую большого круга кровообращения, t1/2 представляет собой время полужизни, или время, за которое концентрация в плазме уменьшается до половины исходного значения. Css представляет собой равновесную концентрацию, т.е. концентрацию, при которой скорость поступления лекарства равна скорости его удаления. Cmin определяют как самую низкую (минимальную) концентрацию, измеренную в интервале между приемами лекарственного средства. Cmax представляет собой самую высокую (максимальную) концентрацию, измеренную в интервале между приемами лекарственного средства. AUC определяют как площадь под кривой "концентрация в плазме - время" для определенного периода времени, например 12 или 24 ч. Комбинации по данному изобретению можно вводить людям в диапазонах доз, определенных для каждого компонента, содержащегося в указанных комбинациях. Компоненты, содержащиеся в указанных комбинациях, можно вводить вместе или по-отдельности. Уровни доз ингибиторов ВИЧ и соединения формулы (I) или его фармацевтически приемлемой соли или сложного эфира могут составлять порядка 0,02-5,0 г в день. Когда ингибитор ВИЧ или противовирусный ВИЧ препарат и соединение формулы (I) вводятся в комбинации, весовое отношение ингибитора ВИЧ к соединению формулы (I) может лежать в диапазоне от приблизительно 40:1 до приблизительно 1:15, или от приблизительно 30:1 до приблизительно 1:15,или от приблизительно 15:1 до приблизительно 1:15, обычно от приблизительно 10:1 до приблизительно 1:10, и в основном от приблизительно 8:1 до приблизительно 1:8. Также подходящими являются весовые отношения ингибитора ВИЧ к соединению формулы (I), лежащие в диапазоне от приблизительно 6:1 до приблизительно 1:6, или от приблизительно 4:1 до приблизительно 1:4, или от приблизительно 3:1 до приблизительно 1:3, или от приблизительно 2:1 до приблизительно 1:2, или от приблизительно 1,5:1 до приблизительно 1:1,5. В одном аспекте вес ингибитора ВИЧ равен весу соединения формулы (I) или превышает его, где весовое отношение ингибитора ВИЧ к соединению формулы (I) может лежать в диапазоне от приблизительно 1:1 до приблизительно 15:1, обычно от приблизительно 1:1 до приблизительно 10:1, и в основном от приблизительно 1:1 до приблизительно 8:1. Также используются весовые отношения ингибитора ВИЧ к соединению формулы (I), лежащие в диапазоне от приблизительно 1:1 до приблизительно 6:1, или от приблизительно 1:1 до приблизительно 5:1, или от приблизительно 1:1 до приблизительно 4:1, или от приблизительно 3:2 до приблизительно 3:1, или от приблизительно 1:1 до приблизительно 2:1, или от приблизительно 1:1 до приблизительно 1,5:1. В соответствии с одним вариантом осуществления ингибитор ВИЧ и соединение формулы (I) можно вводить совместно один раз или дважды в день, один раз, два раза, три раза, четыре раза, пять раз или шесть раз в день, предпочтительно перорально, где количество ингибитора ВИЧ в одной дозе составляет от приблизительно 10 до приблизительно 2500 мг, и количество соединения формулы (I) в одной дозе составляет от 10 до приблизительно 2500 мг. В другом варианте осуществления их количества в одной дозе в случае их совместного введения один раз или дважды в день составляет от приблизительно 50 до приблизительно 1500 мг для ингибитора ВИЧ и от приблизительно 50 до приблизительно 1500 мг для соединения формулы (I). В еще одном варианте осуществления их количества в одной дозе в случае их совместного применения ежедневно или еженедельно составляют от приблизительно 100 до приблизительно 1000 мг для ингибитора ВИЧ и от приблизительно 100 до приблизительно 800 мг для соединения формулы (I). В еще одном варианте осуществления их количества в одной дозе в случае их совместного применения ежедневно или еженедельно составляют от приблизительно 150 до приблизительно 8 00 мг для ингибитора ВИЧ и от приблизительно 100 до приблизительно 600 мг для соединения формулы (I). В еще одном варианте осуществления их количества в одной дозе в случае их совместного применения ежедневно или еженедельно составляют от приблизительно 200 до приблизительно 600 мг для ингибитора ВИЧ и от приблизительно 100 до приблизительно 400 мг для соединения формулы (I). В еще одном варианте осуществления их количества в одной дозе в случае их совместного применения ежедневно или еженедельно составляют от приблизительно 200 до приблизительно 600 мг для ингибитора ВИЧ и от 20 до приблизительно 300 мг для соединения формулы (I). В еще одном варианте осуществления их количества в одной дозе в случае их совместного применения ежедневно или еженедельно составляют от приблизительно 100 до приблизительно 400 мг для ингибитора ВИЧ и от приблизительно 40 до приблизительно 100 мг для соединения формулы (I). Комбинации ингибитора ВИЧ (мг)/соединения формулы (I) (мг), приведенные в качестве примера, в дозах для приема два раза в день включают 50/100, 100/100, 150/100, 200/100, 250/100, 300/100, 350/100,400/100, 450/100, 50/133, 100/133, 150/133, 200/133, 250/133, 300/133, 50/150, 100/150, 150/150, 200/150,250/150, 50/200, 100/200, 150/200, 200/200, 250/200, 300/200, 50/300, 80/300, 150/300, 200/300, 250/300,300/300, 200/600, 400/600, 600/600, 800/600, 1000/600, 200/666, 400/666, 600/666, 800/666, 1000/666,1200/666, 200/800, 400/800, 600/800, 800/800, 1000/800, 1200/800, 200/1200, 400/1200, 600/1200, 800/1200,1000/1200 и 1200/1200. Другие комбинации ингибитора ВИЧ (мг)/соединения формулы (I) (мг), приведенные в качестве примера, в дозах для приема два раза в день включают 1200/400, 800/400, 600/400,400/200, 600/200, 600/100, 500/100, 400/50, 300/50 и 200/50. Будет, однако, понятно, что конкретный уровень доз и частота приема могут варьироваться для любого конкретного пациента и будут зависеть от множества факторов, включая активность конкретного применяемого соединения, метаболическую стабильность и время действия этого соединения, возраст,вес, общее здоровье, пол и условия питания пациента, тип и время приема, скорость выведения, комбинацию лекарств, серьезность конкретного состояния и тип терапии, которую проходит пациент. В одном варианте осуществления настоящего изобретения разработано изделие, включающее композицию, пригодную для лечения ВИЧ-инфекции, и упаковочный материал, включающий этикетку, на которой указано, что композиция может быть использована для лечения ВИЧ-инфекции, где композиция включает комбинацию, описанную здесь. Примеры В табл. 5 представлен обзор результатов, полученных при исследовании соединений С 1-С 55 в различных анализах in vitro. Более детальная информация о данных анализах приведена после таблицы. Приведенные результаты и примеры представлены для иллюстрации изобретения, и их нельзя толковать как ограничивающие объем изобретения. Таблица 5. Результаты in vitro исследования выбранных тестовых соединенийnd: не определено Анализы 1 и 2. Противовирусная активность/токсичность Соединения по настоящему изобретению были протестированы на противовирусную активность в ходе клеточного анализа, который проводили в соответствии со следующей процедурой. В линию Т-лимфоцитов человека МТ 4 были встроены гены зеленого флуоресцентного белка (GFP) и ВИЧ-специфичного промотора - ВИЧ-1 длинного концевого повтора (LTR). Данная линия клеток обозначается как МТ 4 LTR-EGFP, и она может быть использована для оценки анти-ВИЧ активности исследуемых соединений in vitro. В ВИЧ-1-инфицированных клетках вырабатывается Tat-белок, который активирует LTR-промотор и в конечном счете приводит к стимуляции выработки репортерского GFP, что позволяет измерять протекание ВИЧ-инфекции флуорометрическим способом. Аналогично, в клетки МТ 4 были встроены GFP и конститутивный промотор цитомегаловируса(CMV). Данная линия клеток обозначается как МТ 4 CMV-EGFP, и она может быть использована для оценки цитотоксичности исследуемых соединений in vitro. В данной линии клеток уровни GFP сравнимы с таковыми для инфицированных клеток МТ 4 LTR-EGFP. Цитотоксичные исследуемые соединения понижают уровни GFP у неинфицированных клеток МТ 4 CMV-EGFP. Можно определить значения эффективной концентрации, такие как 50% эффективная концентрация (ЕС 50); их обычно выражают в мкМ. Значение ЕС 50 определяют как концентрацию тестового соединения, которая уменьшает флуоресценцию ВИЧ-инфицированных клеток на 50%. 50% цитотоксичную концентрацию (СС 50 в мкМ) определяют как концентрацию тестового соединения, которая уменьшает флуоресценцию неинфицированных клеток на 50%. Отношение СС 50 к ЕС 50 определяют как индекс селективности (SI), который показывает селективность анти-ВИЧ активности ингибитора. Окончательный контроль за ВИЧ-1-инфекцией и цитотоксичностью осуществляют при помощи сканирующего микроскопа. Анализ изображений позволяет обнаружить с высокой чувствительностью вирусную инфекцию. Измерение проводят до наступления некроза клеток, который обычно происходит через приблизительно пять дней после инфицирования, в частности, измерения проводят в течение трех дней после инфицирования. В табл. 5 приведены значения pEC50 по сравнению со штаммом ВИЧ-1 IIIB дикого типа, а также значения рСС 50 для выбранного числа соединений по изобретению. Значение pEC50 соответствует -log10(ЕС 50). Значение рСС 50 соответствует log10 (СС 50). Приведены соединения, для которых значения pEC50 лежат в диапазоне от значения, меньшего чем 4,00, до максимального значения в 4,88. Для дарунавира, коммерчески доступного ингибитора протеазы ВИЧ, значение рЕС 50 составляет 8,17. Диапазон значений pEC50 от значения 4 до 4,88 по сравнению с 8,17 соответствует значительно меньшей противовирусной активности, демонстрируя таким образом, что соединения по настоящему изобретению не вызывают развития резистентности к ВИЧ или вызывают ее на минимальном уровне. Аналогично, значения токсичности, приведенные для соединений по настоящему изобретению, лежащие в диапазоне от значения, меньшего чем 4,00, до максимального значения в 4,86, демонстрируют низкую или минимальную токсичность данных соединений. Анализ 3. Метаболическая стабильность тестовых соединений (HLM 15') Субклеточные образцы тканей были приготовлены в соответствии со способом Горрода (Gorrod) с соавт. (Xenobiotica, 5, стр. 453-462 (1975 при помощи разделения в центрифуге после механической гомогенизации ткани. Ткань печени человека промывали в ледяном 0,1 М Трис-HCl буфере (рН 7,4) для вымывания избытка крови. Ткань затем высушивали промоканием, взвешивали и крупно нарезали при помощи хирургических ножниц. Кусочки ткани гомогенизировали в 3 объемах ледяного 0,1 М фосфатного буфера (рН 7,4) либо при помощи гомогенизатора Potter-S (Braun, Италия), оснащенного тефлоновым пестиком, или при помощи гомогенизатора Sorvall Omni-Mix в течение 710 с. В обоих случаях во время процесса гомогенизации сосуд держали на/во льду. Гомогенаты тканей центрифугировали при 9000g в течение 20 мин при 4 С с использованием центрифуги Sorvall или ультрацентрифуги Beckman. Полученный супернатант можно хранить при -80 С, и его обозначают как "S9". Фракцию S9 центрифугировали при 100000g в течение 60 мин при 4 С с использованием ультрацентрифуги Beckman. Из полученного супернатанта тщательно удаляли воздух, отбирали аликвоты и обозначали как "цитозоль". Гранулу повторно суспендировали в 0,1 М фосфатном буфере (рН 7,4) в конечном объеме в 1 мл на 0,5 г веса исходной ткани и обозначали как "микросомы". Из всех субклеточных фракций отбирали аликвоты,немедленно замораживали в жидком азоте и хранили при -80 С вплоть до использования. Тестовые соединения и НАДФН-генерирующую систему добавляли к микросомам печени человека(рН=7,4), до получения конечной реакционной смеси с концентрациями в 5 мкМ тестового соединения,0,8 мМ D-глюкозо-6-фосфата, 0,8 мМ MgCl2 и 0,8 ед/мл глюкозо-6-фосфатдегидрогеназы. Термоинактивируемые (10 мин при 95 С) "S9" или микросомы использовали для контрольных экспериментов. Реакционные смеси выдерживали при 37 С в течение 5 мин, после чего реакцию запускали путем добавления 0,8 мМ -НАДФ. Реакцию проводили в течение 0 или 15 мин. Затем реакцию останавливали путем добавления 2 объемов ДМСО (или ацетонитрила). Образцы центрифугировали (10 минут, 900g) и анализировали при помощи ЖХ-МС. Анализ 4. Ингибирование CYP450 Ингибирование метаболизма тестовых соединений различными изоферментами CYP P450 определяли при помощи белков, экспрессированных в Е. coli (3 А 4, 2 С 9, 2D6, 1 А 2 и 2 С 19), которые превращают свои специфичные субстраты во флуоресцентную молекулу (табл. 6). Данную флуоресцентную молекулу наблюдали с использованием флуоресцентного сканирующего спектрофотометра для прочтения планшетов (Victor2 (Wallac) или Fluoroskan (Labsystems. Соединения, ингибирующие ферментативную реакцию, будут приводить к понижению сигнала флуоресценции. Ферменты CYP Р 450 получали самостоятельно, или использовали коммерческие препараты, и их хранили при -80 С. Таблица 6. Превращения, опосредуемые соответствующими изоферментами CYP Р 450,экспрессированными в Е. coli Анализ проводили в черных 96-луночных планшетах Costar. Тестовые соединения прибавляли к раствору ферментов CYP P450 в присутствии НАДФН-генерирующей системы. После 5 мин предварительного выдерживания при 37 С, прибавляли свежеприготовленный раствор субстрата в фосфатном буфере (рН 7,4). Известные ингибиторы CYP P450 использовали для положительных контрольных экспериментов, отрицательные контрольные эксперименты проводили в отсутствие фермента CYP Р 450. В табл. 7 приведены конечные концентрации в реакционной смеси. Реакционные смеси выдерживали при 37 С в течение 30 мин (CYP3A4-BFC), 30 мин (CYP3A4-BQ), 10 мин (CYP3A4-DBF), 15 мин (CYP1A2CEC), 30 мин (CYP2C9-MFC, CYP2C19-CEC) или 45 мин (CYP2D6-AMMC) соответственно. Затем реакцию останавливали путем добавления 200 мкл ацетонитрила и регистрировали сигнал флуоресценции. Таблица 7. Конечные концентрации в реакционной смеси для анализа ингибирования изоферментами Расчет ингибирования изоферментами CYP P450 (% ингибирования):% активности=(100/(средний положительный контроль средний отрицательный контроль(средний образец - средний отрицательный контроль)% ингибирования=100-% активности Анализ 5: % блокады метаболизма: ингибирование метаболизма ТМС 114 Дарунавир (ТМС 114, в настоящее время известный под торговой маркой Prezista) и тестовые соединения - активаторы прибавляли к микросомам печени человека (фракция "микросомы", концентрация белка 1 мг/мл), суспендированным в калий-фосфатном буфере (рН 7,4), до получения конечной реакционной смеси с концентрациями в 3 мкМ дарунавира и 3 мкМ тестового соединения. В случае неактивированных параллельных реакций тестовое соединение не добавляли. Прокипяченные микросомы печени человека использовали для контрольных экспериментов. После прибавления (в объемном соотношении 1:3) НАДФН-генерирующей смеси, состоящей из никотинамид-аденин-динуклеотидфосфата (-НАДФ, 0,5 мг/мл, 653,2 мкМ), D-глюкозо-6-фосфата (2 мг/мл, 7,1 мМ), глюкозо-6-фосфатдегидрогеназы (1,5 ед/мл) в 2% NaHCO3 реакционную смесь выдерживали при 37 С в течение 30 или 120 мин, после чего реакцию останавливали путем повышения температуры до 95 С. Концентрации дарунавира определяли при помощи ВЭЖХ-МС. Контрольное значение,полученное при определении процентного содержания оставшегося ТМС 114 в неактивированной реакции (отсутствие тестового соединения), составляет 12% (среднее значение для 10 экспериментов). Для анализа "активирующей способности" (способность соединений улучшать фармакокинетику дарунавира) in vivo показательные примеры С 1, С 6 и С 7 вводили перорально (в подходящей среде, такой как, например, PEG400- или PEG400/30% физиологический раствор или HpCD) группе (n=3) сытых кобелей породы бигль в дозе 5 мг/кг массы тела за 15 мин до введения дарунавира в дозе 5 мг/кг массы тела. Пероральное введение осуществляли принудительно. В течение всего времени собаки имели свободный и непрерывный доступ к воде. Образцы крови отбирали из яремной вены после 0 (до введения),0,5, 1, 2, 4, 7 и 24 ч после введения дозы. Образцы центрифугировали при 1900g в течение 10 мин при 5 С для отделения плазмы. Отделенную плазму хранили в морозильной камере в течение 2 ч после отбора пробы крови. В течение всего времени образцы крови и плазмы находились на льду и были защищены от света. Индивидуальные образцы плазмы анализировали на содержание дарунавира и соединенияактиватора при помощи ЖХ-МС/МС. Фармакокинетические параметры для дарунавира рассчитывали при помощи анализа без использования камерной модели, программного обеспечения WinNonLin версии 5.0, Pharsight; они приведены в табл. 8. Приведенные значения представляют собой среднее значение для 3 собак. Кратное изменение (FC) значений показывает различие по сравнению с контрольными экспериментами, в которых вводили только 5 мг/кг дарунавира. Таблица 8. Активирующее влияние на ключевые фармакокинетические параметры дарунавира Эксперимент Реагенты приобретали из коммерческих источников и использовали в том виде, в котором они были получены. Тонкослойную хроматографию проводили на пластинах силикагеля 60 F254 (Merck). ЖХ-МС анализ проводили с использованием одного из следующих способов. Данные для соединений C1-C55 приведены в табл. 1 (см. выше). ЖХМС-способ 1 ВЭЖХ-система: Waters Alliance 2695 (насос плюс система автоматической подачи образца), Waters 996 (фотодиодный матричный детектор) Колонка: Waters XTerra MS C18 2,5 мкм 504,6 мм Температура: 55 С Подвижная фаза: А: 10 мМ HCOONH4+0,1% НСООН в Н 2 О В: CH3CN Градиент: 0 мин: 15% В, 3 мин: 95% В, 4,2 мин: 95% В Время установления равновесия: 1,2 мин Поток: 2 мл/мин Вводимый объем: 5 мкл 0,5 мг/мл раствора МС-детектор: Waters ZQ Ионизация: электрораспыление в положительном и отрицательном режиме ЖХМС-способ 2 ВЭЖХ-система: Waters Alliance 2790 (насос плюс система автоматической подачи образца), Waters 996 (фотодиодный матричный детектор) Колонка: Waters SunFire C18 3,5 мкм 1004,6 мм Температура: 55 С Подвижная фаза: А: 10 мМ NH4OOCH+0,1% НСООН в H2O В: ацетонитрил Градиент: 0 мин: 5% В, 5,4 мин: 95% В, 7,2 мин: 95% В Время установления равновесия: 1,8 мин Поток: 1,5 мл/мин Вводимый объем: 5 мкл 0,5 мг/мл раствора МС-детектор: Waters LCT Ионизация: электрораспыление в положительном и отрицательном режиме Спектры ЯМР регистрировали на спектрометре Bruker Avance 400, работающем при 400 МГц. Химические сдвиги приведены в м.д., а значения J в Гц. Мультиплетность указана с использованием следующих сокращений: д - для дублета, т - для триплета, м - для мультиплета, и т.д. Названия соединений создавали с использованием Chemdraw Ultra, версии 9.0.7 (CambridgeSoft). Химия Соединения формулы (I) получали в соответствии с общими способами 1, 2 и 3, приведенными на схемах 1, 2 и 3. Более детально процедуры описаны ниже. Схема 3. Способ 3 Схемы 4 и 5 иллюстрируют получение некоторых промежуточных карбоновых кислот (обозначенных как II на вышеприведенных схемах) в тех случаях, когда они не были коммерчески недоступны. Подразумевается, что данные схемы являются иллюстрирующими, а не ограничивающими. Раствор соединения 4.1 (1,10 г) и 1,1,1-триметоксиэтана (5 мл) нагревали до 100 С в течение 5 мин в микроволновой печи. Реакционную смесь концентрировали при пониженном давлении до получения 1,26 г соединения 4.2 (95% чистого) в виде коричневого порошка. 1 Схема 5. Синтез 2-(этил(метил)амино)бензо[d]оксазол-6-карбоновой кислоты (5.6) К перемешиваемому раствору безводного МеОН (4 л) на ледяной бане по каплям добавлялиCH3COCl (490 мл) при температуре ниже 10 С. Затем прибавляли соединение 4.1 (486 г, 3,17 моль). Смесь перемешивали при комнатной температуре в течение 2 дней. ТСХ (EtOAc/петролейный эфир равно 1:1) указывала на завершение реакции. МеОН удаляли в вакууме. Воду (4 л) прибавляли к смеси.K2CO3 медленно прибавляли до тех пор, пока значение рН не достигало 10. Органическую фазу отделяли, высушивали над Na2SO4 и концентрировали до получения соединения 5.2 (460 г, 80%) в виде твердого вещества коричневого цвета. Суспензию соединения 5.2 (393 г, 2,35 моль) и о-этилксантата калия (393 г, 2,45 моль) в пиридине(3 л) нагревали при кипении с обратным холодильником в течение 12 ч. ТСХ (EtOAc/петролейный эфир равно 1:2) указывала на завершение реакции. Смесь охлаждали до комнатной температуры и переливали в четыре 5-л колбы, содержащие 1600 мл концентрированной HCl и 10 л ледяной воды. Смесь перемешивали в течение 5 мин и твердый осадок отделяли фильтрованием. Твердое вещество промывали 1 л воды и высушивали в течение 30 мин. Твердое вещество растворяли в 10 л этилацетата, промывали 2 л 1 н. HCl и 1 л солевого раствора, высушивали над Na2SO4 и концентрировали до получения соединения 5.3(390 г, 79,3%) в виде твердого вещества розового цвета. К суспензии соединения 5.3 (320 г, 1,53 моль) и K2CO3 (276 г, 2 моль) в EtOAc (3 л) прибавляли MeI(238,9 г, 1,68 моль) при температуре ниже 20 С. Смесь перемешивали при комнатной температуре в течение 12 ч. ТСХ (EtOAc/петролейный эфир равно 1:3) указывала на завершение реакции. Воду (2 л) прибавляли к смеси. Органическую фазу отделяли, высушивали над Na2SO4 и концентрировали до получения соединения 5.4 (320 г, 93,8%) в виде твердого вещества розового цвета. Соединение 5.4 (5 г) растворяли в 75 мл ТГФ. Прибавляли N-метил-N-этиламин (13,2 г, 10 экв.), и смесь перемешивали при 70 С до тех пор, пока ЖХ-МС не указывала на завершение реакции. Смесь выпаривали до высушивания и соединение 5.5 (5,25 г, 99%) использовали как таковое в следующей реакции. Соединение 5.5 (5,25 г) растворяли в смеси ТГФ-вода 1:1 (100 мл). Прибавляли LiOH (5,36 г, 10 экв.). Смесь перемешивали при 70 С в течение ночи. Концентрированную HCl прибавляли до тех пор,пока не наблюдалось выпадение осадка. Смесь экстрагировали 3 раза 100 мл 2-Me-ТГФ. Объединенные органические слои высушивали над MgSO4. Фильтрование с последующим выпариванием растворителя привело к получению соединения 5.6 в виде твердого вещества желтоватого цвета. Соединение высушивали в вакууме в течение ночи. Было получено 4,33 г (88%). 1 Н-ЯМР (400 МГц, ДМСО-d6)м.д. 1,20 (т, J=7,2 Гц, 3 Н), 3,15 (с, 3 Н), 3,58 (кв, J=7,1 Гц, 2 Н), 7,29(д, J=8,4 Гц, 1 Н), 7,80 (дд, J=8,3, 1,5 Гц, 1 Н), 7,84 (д, J=1,5 Гц, 1 Н), 12,65 (с(шир), 1 Н). Показательные примеры для конечных соединений, полученных при помощи способа 1.(10,0 экв.) в изопропаноле (150 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали при пониженном давлении до количественного получения соединения 6.2. Неочищенный продукт высушивали под высоким вакуумом и использовали как таковой на следующей стадии. Трет-бутил (2S,3R)-3-гидрокси-4-(изобутиламино)-1-фенилбутан-2-илкарбамат (6.2) (31,0 г) в дихлорметане прибавляли к раствору бензотиазол-6-карбоновой кислоты (1,05 экв.), триэтиламина (1,5 экв.) и HATU (2-(7-аза-1 Н-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат, 1,05 экв.) в дихлорметане (500 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Прибавляли воду и разделяли фазы. Органическую фазу три раза промывали насыщенным водным раствором Na2CO3, солевым раствором, высушивали над MgSO4 и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии (элюент: дихлорметандихлорметан/метанол 95:5) до количественного получения соединения 6.3. Хлортриметилсилан (3 экв.) прибавляли к раствору трет-бутил (2S, 3R)-3-гидрокси-4-(Nизобутилбензо[d]тиазол-6-карбоксамидо)-1-фенилбутан-2-илкарбамата (6.3) (45 г) и иодида натрия (4 экв.) в ацетонитриле (300 мл). Реакционную смесь перемешивали при комнатной температуре в течение двух часов. Прибавляли водный 2 M раствор NaOH, и перемешивание продолжали в течение дополнительных 30 мин. Солевой раствор и дихлорметан прибавляли к реакционной смеси. Органическую фазу отделяли, промывали солевым раствором, высушивали над MgSO4 и концентрировали при пониженном давлении. Соединение очищали при помощи колоночной хроматографии (элюент: дихлорметандихлорметан/метанол 9:1), получали 26,6 г (74%) соединения 6.4 в виде твердого вещества белого цвета. Раствор N-2R,3S)-3-амино-2-гидрокси-4-фенилбутил)-N-изобутилбензо[d]тиазол-6-карбоксамида(6.4) (20,9 г), 2,5-диоксопирролидин-1-илтиазол-5-илметилкарбоната (1,0 экв.) и триэтиламина (1,2 экв.) в 2-метилтетрагидрофуране (300 мл) перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь промывали насыщенным водным раствором Na2CO3, высушивали над MgSO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали при помощи колоночной хроматографии (элюент: дихлорметандихлорметан/метанол 95:5) до получения 24,0 г (85%) чистого соединения С 7. 1(кв, J=7,l Гц, 2 Н), 3,87-3,94 (м, 3 Н), 5,00 (с (шир), 1 Н), 5,16-5,30 (м, 3 Н), 7,18-7,21 (м, 2 Н), 7,31 (с, 1 Н),7,32 (д, J=8,0 Гц, 1 Н), 7,58-7,60 (с, 1 Н), 7,82 (с, 1 Н), 8,47 (с(шир), 2 Н), 8,78 (с, 1 Н). Показательные примеры для соединений, полученных при помощи способа 2.(1,05 экв.), триэтиламина (2,0 экв.) и ВОР (бензотриазолил-N-окситрисдиметиламинофосфонийгексафторфосфат, 1,05 экв.) в дихлорметане (500 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Прибавляли воду, и фазы разделяли. Органическую фазу 3 раза промывали насыщенным водным раствором Na2CO3, высушивали над MgSO4 и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии (элюент: гептан/этилацетат 6:4 этилацетат) до получения 38 г (87%) соединения 7.2. Палладий/углерод (5 г, 10% Pd/C по массе) прибавляли к раствору N-2R,3S)-3-(дибензиламино)-2 гидрокси-4-фенилбутил)-2-(этил(метил)амино)-N-изобутилбензо[d]оксазол-6-карбоксамида (7.2) (18 г) в метаноле (400 мл). Реакционную смесь перемешивали при комнатной температуре в атмосфере Н 2 до тех пор, пока ЖХ-МС не указывала на завершение превращения. Смесь фильтровали через целит. После упаривания фильтрата получали 10,6 г (83%) соединения 7.3. Раствор(1,05 экв.), DMAP (0,2 экв.) и триэтиламина (1,0 экв.) в ДМФА (10 мл) перемешивали при комнатной температуре в течение ночи. Прибавляли воду, и фазы разделяли. Органическую фазу 3 раза промывали насыщенным водным раствором Na2CO3, высушивали над MgSO4 и концентрировали при пониженном давлении. Реакционную смесь промывали насыщенным водным раствором Na2CO3, высушивали надMgSO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали при помощи колоночной хроматографии (элюент: дихлорметан/метанол 99:197:3) до получения 263 мг (45%) чистого соединения (C36).H-ЯМР (400 МГц, хлороформ-d)м.д. 0,61-0,78 (м, 6 Н), 1,70-1,85 (м, 1 Н), 2,90-3,02 (м, 1 Н), 3,023,28 (м, 3 Н), 3,39-3,53 (м, 1 Н), 3,86 (с, 3 Н), 3,76-4,07 (м, 3 Н), 5,00-5,30 (м, 4 Н), 7,17-7,32 (м, 6 Н), 7,51 (с,1 Н), 7,76-7,84 (м, 2 Н), 7,94 (с, 1 Н), 8,78 (с, 1 Н). Показательные примеры для соединений, полученных при помощи способа 3. Схема 8. Синтез тиазол-5-илметил (2S,3R)-4-(2-(этиламино)-N-изобутилбензо[d]оксазол-6 карбоксамидо)-3-гидрокси-1-фенилбутан-2-илкарбамата (С 27) Раствор соли щавелевой кислоты и (2R,3S)-3-(дибензиламино)-1-(изобутиламино)-4-фенилбутан-2 ола (8.1) (60,0 г), ди-трет-бутилдикарбоната (0,99 экв.) и триэтиламина (3,3 экв.) в дихлорметане (1000 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь промывали насыщенным водным раствором NaHCO3, высушивали над MgSO4 и концентрировали при пониженном давлении. Неочищенный продукт очищали при помощи колоночной хроматографии (элюент: дихлорметан) до получения 60,3 г (99%) чистого соединения 8.2. Палладий/углерод (12,4 г, 10% Pd/C по массе) прибавляли к раствору трет-бутил (2R,3S)-3(дибензиламино)-2-гидрокси-4-фенилбутил(изобутил)карбамата (8.2) (60,3 г) в метаноле (400 мл). Реакционную смесь перемешивали при комнатной температуре в атмосфере Н 2 до тех пор, пока ЖХ-МС не указывала на завершение превращения. Смесь фильтровали через целит и концентрировали при пониженном давлении. Неочищенный продукт очищали при помощи колоночной хроматографии (элюент: дихлорметандихлорметан/метанол (NH3) 93:7) до получения 32,6 г (83%) чистого соединения 8.3. Раствор трет-бутил (2R,3S)-3-амино-2-гидрокси-4-фенилбутил(изобутил)карбамата (8.3), 2,5 диоксолирролидин-1-илтиазол-5-илметилкарбоната (1,0 экв.) и триэтиламина (1,01 экв.) в 2 метилтетрагидрофуране (600 мл) перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь промывали насыщенным водным раствором NaHCO3, высушивали над MgSO4 и концентрировали при пониженном давлении. Неочищенный продукт (8.4) использовали как таковой на следующей стадии. К раствору трет-бутилтиазол-5-илметил (2R,3S)-2-гидрокси-4-фенилбутан-1,3-диилдикарбамата(8.4) (46,3 г) в дихлорметане (400 мл) прибавляли HCl (5-6 н.) в изопропаноле (100 мл). Реакционную смесь энергично перемешивали при комнатной температуре до тех пор, пока ЖХ-МС не указывала на завершение превращения. Реакционную смесь концентрировали при пониженном давлении. Остаток промывали насыщенным водным раствором NaHCO3, высушивали над MgSO4 и концентрировали при пониженном давлении до получения 33,9 г (92%) чистого соединения 8.5. Тиазол-5-илметил (2S,3R)-3-гидрокси-4-(изобутиламино)-1-фенилбутан-2-илкарбамат (8.5) (1125 мг) прибавляли к смеси 2-(этиламино)бензо[d]оксазол-6-карбоновой кислоты (1,0 экв.), триэтиламина(3,0 экв.) и ВОР (бензотриазолил-N-окситрисдиметиламинофосфонийгексафторфосфат, 1,0 экв.) в дихлорметане (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Прибавляли воду и фазы разделяли. Органическую фазу 3 раза промывали насыщенным водным раствором NaHCO3, высушивали над MgSO4 и концентрировали при пониженном давлении. Остаток очищали при помощи колоночной хроматографии (элюент: дихлорметандихлорметан/метанол (NH3) 98:2) до получения 1165 мг (66%) чистого соединения С 27. 1 его фармацевтически приемлемые соли и стереоизомерные формы, где R представляет собой Н, фенил, пиридил, C1-6 алкил или где А и В независимо представляют собой Н, C1-6 алкил, необязательно замещенный этинильной,имидазолильной группой или гетероатомом, представляющим собой азот, который необязательно замещен C1-6 алкильной группой, или где А и В вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членное насыщенное, частично или полностью ненасыщенное гетероциклическое кольцо,содержащее от 1 до 2 гетероатомов, каждый из которых независимо выбран из азота или кислорода; тельно замещенной атомом галогена;R5 представляет собой пиридильную или фенильную группу, необязательно замещенную атомом галогена; 5. Применение соединения по любому из пп.1-4 в качестве лекарственного средства для лечения или предотвращения ВИЧ-инфекций. 6. Применение соединения по любому из пп.1-4 для производства лекарственного средства для активации противовирусных ВИЧ-препаратов. 7. Комбинация для лечения или предотвращения ВИЧ-инфекций, включающаяa) соединение по любому из пп.1-4 или его фармацевтически приемлемую соль иb) ингибитор ВИЧ. 8. Комбинация по п.1, где ингибитор ВИЧ представляет собой дарунавир или гексагидрофуро[2,3b]фуран-3-иловый эфир(1-бензил-3-[2-(1-циклопентилпиперидин-4-иламино)бензотиазол-6 сульфонил]изобутиламино-2-гидроксипропил)карбаминовой кислоты. 9. Комбинация по п.8, где соединение по пп.1-4 представляет собой тиазол-5-илметил (2S,3R)-4-(2(этил(метил)амино)-N-изобутилбензо[d]оксазол-6-карбоксамидо)-3-гидрокси-1-фенилбутан-2 илкарбамат и ингибитор ВИЧ представляет собой дарунавир или гексагидрофуро[2,3-b]фуран-3-иловый эфир (1-бензил-3-[2-(1-циклопентилпиперидин-4-иламино)бензотиазол-6-сульфонил]изобутиламино-2 гидроксипропил)карбаминовой кислоты. 10. Комбинация по п.9, где соединение по пп.1-4 представляет собой тиазол-5-илметил (2S,3R)-4-(2(этил(метил)амино)-N-изобутилбензо[d]оксазол-6-карбоксамидо)-3-гидрокси-1-фенилбутан-2 илкарбамат и ингибитор ВИЧ представляет собой гексагидрофуро[2,3-b]фуран-3-иловый эфир (1-бензил 3-[2-(1-циклопентилпиперидин-4-иламино)бензотиазол-6-сульфонил]изобутиламино-2 гидроксипропил)карбаминовой кислоты. 11. Применение комбинации по любому из пп.7-10 в качестве лекарственного средства для лечения или предотвращения ВИЧ-инфекций. 12. Применение комбинации по любому из пп.7-10 для производства лекарственного средства для лечения или предотвращения ВИЧ-инфекций. 13. Фармацевтическая композиция, содержащая комбинацию по любому из пп.7-10 и фармацевтически приемлемый эксципиент.
МПК / Метки
МПК: C07D 417/12, C07D 417/14, C07D 277/82, A61P 31/18, A61K 31/427
Метки: амидные, противовирусных, качестве, соединения, препаратов, активаторов
Код ссылки
<a href="https://eas.patents.su/28-19616-amidnye-soedineniya-v-kachestve-aktivatorov-protivovirusnyh-preparatov.html" rel="bookmark" title="База патентов Евразийского Союза">Амидные соединения в качестве активаторов противовирусных препаратов</a>
Фосфонатные и фосфинатные соединения в качестве активаторов глюкокиназы

Номер патента: 15228
Опубликовано: 30.06.2011
Авторы: Рионо Деннис Е., Тино Джозеф А., Мэн Вэй, Чэнь Шон С., Чжан Хао, Ши Ян, Чэн Питер Т.В., Салски Ричард Б., Болтон Скотт А.
МПК: A61K 31/675, C07F 9/32, A61P 3/00...
Метки: активаторов, соединения, фосфонатные, фосфинатные, глюкокиназы, качестве
Формула / Реферат:
1. Соединение формулыгде R1представляет собой гетероарил, замещенный R4и необязательно замещенный одним или двумя заместителями R5 и R6, где R1 представляет собойR4 выбран из группы, состоящей из-(CH2)n-Z-(CH2)m-PO-(OR7)(OR8);-(CH2)n-Z-(CH2)m-PO-(OR7)R9;-(CH2)n-Z-(CH2)m-O-PO-(OR7)R9;-(CH2)n-Z-(CH2)m-O-PO-(R9)R10 и-(CH2)n-Z-(CH2)m-PO-(R9)R10;R7 и R8являются одинаковыми или различными и независимо выбраны из С1-8алкила илиR7 и R8могут быть...
Новые амидные соединения, обладающие антагонистическим в отношении мсн действием, и содержащие эти соединения лекарственные средства

Номер патента: 9040
Опубликовано: 26.10.2007
Авторы: Рудольф Клаус, Лентер Мартин, Леманн-Линтц Торстен, Мюллер Штефан Георг, Штенкамп Дирк, Лотц Ральф P.Х., Арндт Кирстен, Рот Геральд Юрген, Лустенбергер Филиппо, Виланд Хайке-Андреа
МПК: C07C 237/04, C07C 233/29, C07C 235/24...
Метки: средства, лекарственные, соединения, антагонистическим, мсн, обладающие, эти, содержащие, отношении, амидные, действием, новые
Формула / Реферат:
1. Амидные соединения общей формулы I в которой R1, R2 независимо друг от друга обозначают Н, C1-C6алкил, C3-С7циклоалкил, С3-С7циклоалкил-C1-C3алкил, w-гидрокси-C2-C3алкил, w-(С1-С4алкокси)-С2-C3алкил, С1-С4алкоксикарбонил-С1-С4алкил, карбоксил-С1-С4алкил, амино-С2-С4алкил, С1-С4алкиламино-С2-С4алкил, ди-(С1-С4алкил)амино-С2-С4алкил, цикло-C3-C6алкиленимино-С2-С4алкил, пирролидин-3-ил, N-(С1-С4алкил)пирролидинил-C1-C3алкил, пиперидинил,...
Хинолинкарбоксамиды в качестве противовирусных агентов

Номер патента: 3945
Опубликовано: 30.10.2003
Авторы: Тернер Стивен Рональд, Таисривонгс Сувит, Вэйлланкорт Валери А., Шнуте Марк Э., Стробач Джозеф Волтер, Такер Джон Алан
МПК: A61P 31/12, C07D 215/16, A61K 31/47...
Метки: агентов, качестве, хинолинкарбоксамиды, противовирусных
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая соль, где X представляет a) O или b) S; W представляет a) R2, b) NR7R8, c) OR9 или d) SOiR9; R1 представляет a) Cl, b) F, c) Br, d) CN или e) NO2; R2 представляет a) (CH2CH2O)mR10, b) het, где указанный het связан через атом углерода, c) C1-7алкил, который может быть частично ненасыщенным и необязательно замещенным одним или несколькими заместителями, выбранными из группы,...
Три(цикло)замещённые амидные соединения

Номер патента: 12204
Опубликовано: 28.08.2009
Автор: Файф Мэттью
МПК: C07D 277/46, A61P 3/10, A61K 31/427...
Метки: амидные, соединения, три(цикло)замещённые
Формула / Реферат:
1. Соединение формулы (I) или его фармацевтически приемлемая соль, где группа, образованная -НС< и >V, представляет собой 3-оксоциклопентил, 4-оксоциклогексил или 3-гидроксициклопентил; R1 представляет собой SOR3 или SO2R3; R2 представляет собой водород, хлор, фтор или трифторметил; R3 представляет собой С1-4алкильную группу, С3-7циклоалкильную группу или арильную группу; m равно 0 или 1; и пунктирная линия вместе со сплошной линией...
Дикетопиперазиновые и пиперидиновые производные в качестве противовирусных агентов

Номер патента: 14957
Опубликовано: 29.04.2011
Авторы: Ванг Тао, Хан Йинг, Свидорски Джейкоб, Карини Дэвид Дж., Кэдоу Джон Ф., Минвелл Николас А., Йин Живей, Жанг Жонгксинг, Регуэйро-Рен Алисия
МПК: A61K 31/4436, A61K 31/4427, A61K 31/437...
Метки: дикетопиперазиновые, противовирусных, пиперидиновые, агентов, производные, качестве
Формула / Реферат:
1. Соединение, выбранное из группы, состоящей из2. Фармацевтическая композиция, содержащая противовирусное эффективное количество соединения по п.1 или его фармацевтически приемлемую соль и один или большее количество фармацевтически приемлемых носителей, наполнителей или разбавителей.3. Композиция по п.2, дополнительно содержащая второе соединение, обладающее противо-ВИЧ активностью.4. Фармацевтическая композиция по п.3, в которой соединение,...
Предыдущий патент: Система транспортировки убираемой культуры для кормоуборочного комбайна с ускорителем выгрузки и воздуходувкой для создания воздушного потока
Следующий патент: Устройство мобильной станции, устройство базовой станции, способ связи и система связи
Случайный патент: Бициклические аминопроизводные и содержащие их антагонисты pgd2.