Ненуклеозидные ингибиторы обратной транскриптазы
Номер патента: 16386
Опубликовано: 30.04.2012
Авторы: Ву Джеффри, Суини Захари Кевин, Кеннеди-Смит Джошуа



























Формула / Реферат
1. Соединение формулы I

в которой А обозначает:
(a) CR3=CR2,
(b) CH2R3CH2R2 или
(c) CHR2;
или (i) R1 обозначает (II) и R2 обозначает водород, C1-C3-алкил, C1-C3-галогеналкил, C2-C6-алкенил или CH2ORc; и, в дополнение, если А обозначает или (a), или (b), то R2 также может обозначать галоген, NRaRb, CN или ORc;
или (ii) R1 обозначает C1-C6-алкил и R2 обозначает (II);
X1 обозначает О или S;
X2 обозначает О или, если А обозначает (а), то X2 обозначает О или NRd;
R3 обозначает водород или C1-C3-алкил;
R4 обозначает галоген, C1-C6-алкил, C3-C6-циклоалкил, C1-C6-галогеналкил или C1-C6-алкоксигруппу;
Ra и Rb независимо обозначают водород, C1-C3-алкил или C1-C6-ацил;
Rc и Rd независимо обозначают водород или C1-C3-алкил;
Ar обозначает фенил, замещенный 1-3 группами, независимо выбранными из группы, включающей галоген, цианогруппу, C1-C6-галогеналкил, C1-C6-алкил, C1-C6-алкоксигруппу и C3-C6-циклоалкил;
n является целым числом, равным от 1 до 3;
или его фармацевтически приемлемые соли.
2. Соединение по п.1, в котором А обозначает (a); R1 обозначает (II); X1 и X2 обозначают О и n равно 1.
3. Соединение по п.2, в котором R2 обозначает водород, галоген, C1-C6-алкил или C1-C6-галогеналкил; R3 обозначает водород; R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил, необязательно замещенный 1-3 группами, независимо выбранными из группы, включающей цианогруппу, галоген и C1-C6-галогеналкил.
4. Соединение по п.3, в котором R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5-дифторметилфенил.
5. Соединение по п.1, в котором А обозначает (а); R1 обозначает (II); X1 обозначает S; X2 обозначает О; R2 обозначает водород, галоген, C1-C6-алкил, C1-C6-галогеналкил; R3 обозначает водород; R4 обозначает галоген или C1-C6-алкил; Ar обозначает фенил, необязательно замещенный 1-3 группами, независимо выбранными из группы, включающей цианогруппу, галоген и C1-C6-галогеналкил, и n равно 1.
6. Соединение по п.1, в котором А обозначает (a); R1 обозначает (II); X1 обозначает О; X2 обозначает NRd; Rd обозначает Н и n равно 1.
7. Соединение по п.6, в котором R2 обозначает водород, галоген, C1-C6-алкил, C1-C6-галогеналкил; R3 обозначает водород; R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил, необязательно замещенный 2 группами, независимо выбранными из группы, включающей цианогруппу, галоген и C1-C6-галогеналкил.
8. Соединение по п.7, в котором R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5-дифторметилфенил.
9. Соединение по п.1, в котором А обозначает (b); R1 обозначает (II); X1 и X2 обозначают О и n равно 1.
10. Соединение по п.9, в котором R2 обозначает водород, галоген, C1-C6-алкил, C1-C6-галогеналкил; R3 обозначает водород; R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил, необязательно замещенный 2 группами, независимо выбранными из группы, включающей цианогруппу, галоген и C1-C6-галогеналкил.
11. Соединение по п.10, в котором R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5-дифторметилфенил.
12. Соединение по п.1, в котором А обозначает (с); R1 обозначает (II); X1 и X2 обозначают О и n равно 1.
13. Соединение по п.12, в котором R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил, необязательно замещенный 2 группами, независимо выбранными из группы, включающей цианогруппу, галоген и C1-C6-галогеналкил.
14. Соединение по п.13, в котором R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5-дифторметилфенил.
15. Соединение по п.1, в котором А обозначает (a); R2 обозначает (II); X1 и X2 обозначают О и R3 обозначает водород.
16. Соединение по п.15, в котором R1 обозначает метил или водород; R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил, необязательно замещенный 2 группами, независимо выбранными из группы, включающей цианогруппу, галоген и C1-C6-галогеналкил.
17. Соединение по п.16, в котором R1 обозначает метил или водород; R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5-дифторметилфенил.
18. Соединение по п.1, в котором А обозначает (с); R2 обозначает (II) и X1 и X2 обозначают О.
19. Соединение по п.18, в котором R1 обозначает метил или водород; R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил, необязательно замещенный 2 группами, независимо выбранными из группы, включающей цианогруппу, галоген и C1-C6-галогеналкил.
20. Соединение по п.19, в котором R1 обозначает метил или водород; R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5-дифторметилфенил.
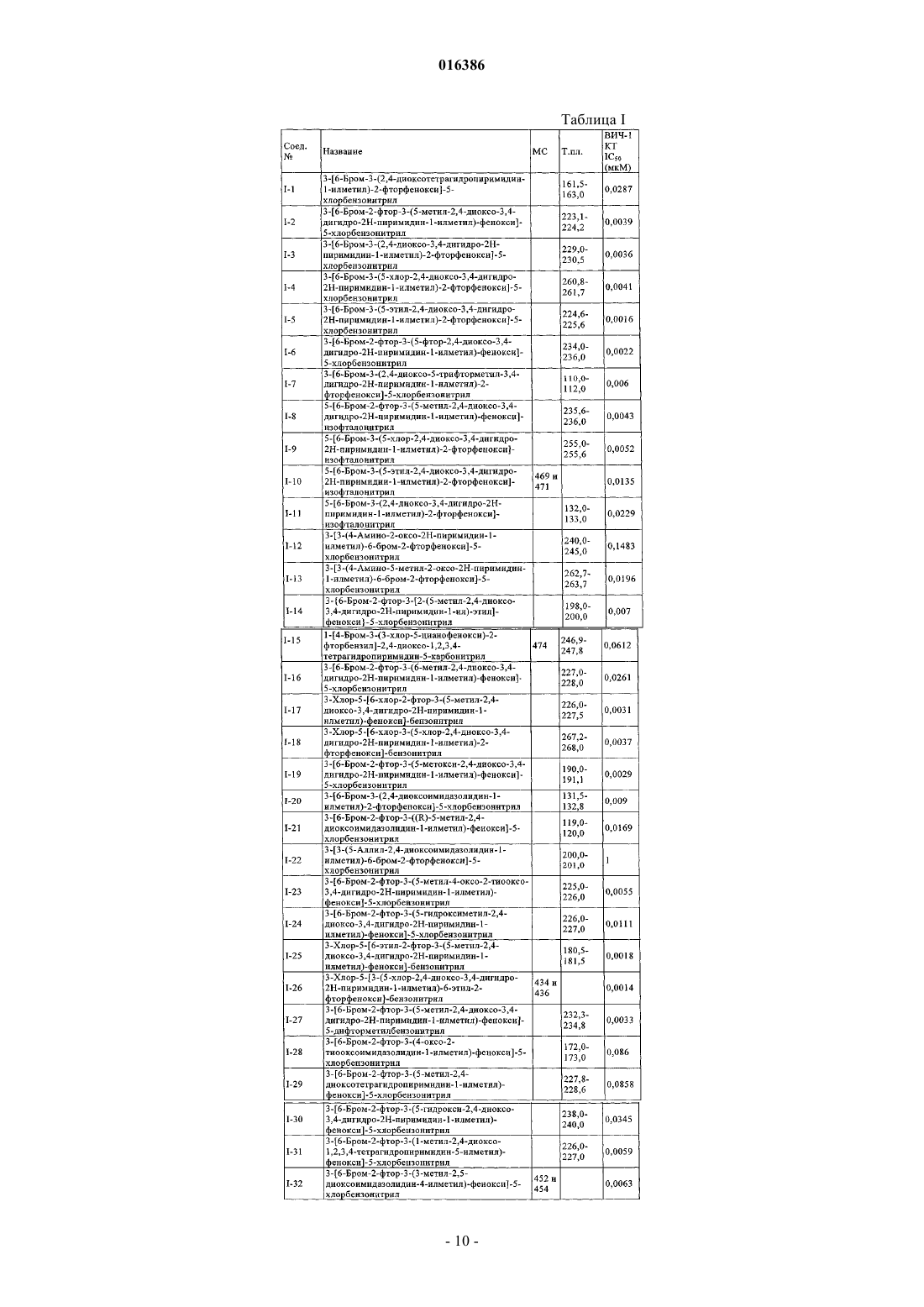
21. Соединение по п.1, выбранное из группы, включающей
3-[6-бром-3-(2,4-диоксотетрагидропиримидин-1-илметил)-2-фторфенокси]-5-хлорбензонитрил;
3-[6-бром-2-фтор-3-(5-метил-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)фенокси]-5-хлорбензонитрил;
3-[6-бром-3-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)-2-фторфенокси]-5-хлорбензонитрил;
3-[6-бром-3-(5-хлор-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)-2-фторфенокси]-5-хлорбензонитрил;
3-[6-бром-3-(5-этил-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)-2-фторфенокси]-5-хлорбензонитрил;
3-[6-бром-2-фтор-3-(5-фтор-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)фенокси]-5-хлорбензонитрил;
3-[6-бром-3-(2,4-диоксо-5-трифторметил-3,4-дигидро-2Н-пиримидин-1-илметил)-2-фторфенокси]-5-хлорбензонитрил;
5-[6-бром-2-фтор-3-(5-метил-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)фенокси]изофталонитрил;
5-[6-бром-3-(5-хлор-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)-2-фторфенокси]изофталонитрил;
5-[6-бром-3-(5-этил-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)-2-фторфенокси]изофталонитрил;
5-[6-бром-3-(2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)-2-фторфенокси]изофталонитрил;
3-[3-(4-амино-2-оксо-2Н-пиримидин-1-илметил)-6-бром-2-фторфенокси]-5-хлорбензонитрил;
3-[3-(4-амино-5-метил-2-оксо-2Н-пиримидин-1-илметил)-6-бром-2-фторфенокси]-5-хлорбензонитрил;
3-{6-бром-2-фтор-3-[2-(5-метил-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-ил)этил]фенокси}-5-хлорбензонитрил;
1-[4-бром-3-(3-хлор-5-цианофенокси)-2-фторбензил]-2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-карбонитрил;
3-[6-бром-2-фтор-3-(6-метил-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)фенокси]-5-хлорбензонитрил;
3-хлор-5-[6-хлор-2-фтор-3-(5-метил-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)фенокси]бензонитрил;
3-хлор-5-[6-хлор-3-(5-хлор-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)-2-фторфенокси]бензонитрил;
3-[6-бром-2-фтор-3-(5-метокси-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)фенокси]-5-хлорбензонитрил;
3-[6-бром-3-(2,4-диоксоимидазолидин-1-илметил)-2-фторфенокси]-5-хлорбензонитрил;
3-[6-бром-2-фтор-3-((R)-5-метил-2,4-диоксоимидазолидин-1-илметил)фенокси]-5-хлорбензонитрил;
3-[3-(5-аллил-2,4-диоксоимидазолидин-1-илметил)-6-бром-2-фторфенокси]-5-хлорбензонитрил;
3-[6-бром-2-фтор-3-(5-метил-4-оксо-2-тиооксо-3,4-дигидро-2Н-пиримидин-1-илметил)фенокси]-5-хлорбензонитрил;
3-[6-бром-2-фтор-3-(5-гидроксиметил-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)фенокси]-5-хлорбензонитрил;
3-хлор-5-[6-этил-2-фтор-3-(5-метил-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)фенокси]бензонитрил;
3-хлор-5-[3-(5-хлор-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)-6-этил-2-фторфенокси]бензонитрил;
3-[6-бром-2-фтор-3-(5-метил-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)фенокси]-5-дифторметилбензонитрил;
3-[6-бром-2-фтор-3-(4-оксо-2-тиооксоимидазолидин-1-илметил)фенокси]-5-хлорбензонитрил;
3-[6-бром-2-фтор-3-(5-метил-2,4-диоксотетрагидропиримидин-1-илметил)фенокси]-5-хлорбензонитрил;
3-[6-бром-2-фтор-3-(5-гидрокси-2,4-диоксо-3,4-дигидро-2Н-пиримидин-1-илметил)фенокси]-5-хлорбензонитрил;
3-[6-бром-2-фтор-3-(1-метил-2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-илметил)фенокси]-5-хлорбензонитрил и
3-[6-бром-2-фтор-3-(3-метил-2,5-диоксоимидазолидин-4-илметил)фенокси]-5-хлорбензонитрил.
22. Применение соединения по любому из пп.1-21 для приготовления лекарственного средства, предназначенного для лечения инфицирования посредством ВИЧ-1 или предупреждения инфицирования посредством ВИЧ-1 или лечения СПИД или ПСП.
23. Фармацевтическая композиция, включающая терапевтически эффективное количество соединения по любому из пп.1-21 и по меньшей мере один носитель, инертный наполнитель или разбавитель.
Текст
НЕНУКЛЕОЗИДНЫЕ ИНГИБИТОРЫ ОБРАТНОЙ ТРАНСКРИПТАЗЫ Изобретение относится к соединениям формулы I, в которых R1, X1, X2 и А являются такими, как определено в настоящем изобретении, или их фармацевтически приемлемым солям,ингибирующим обратную транскриптазу ВИЧ-1, и способу предупреждения и лечения инфицирования посредством ВИЧ-1 и лечения СПИД и/или ПСП. Кроме того, изобретение относится к композициям, содержащим соединения формулы I, применимые для предупреждения и лечения инфицирования посредством ВИЧ-1 и лечения СПИД и/или ПСП. 016386 Настоящее изобретение относится к области противовирусной терапии и, в частности, к ненуклеозидным ингибиторам обратной транскриптазы, предназначенным для лечения заболеваний, опосредуемых вирусом иммунодефицита человека (ВИЧ). Настоящее изобретение относится к новым 1 Н-пиримидин-2,4-дионам, дигидропиримидин-2,4-дионам имидазолидин-2,4-дионам формулы I, предназначенным для лечения или профилактики заболеваний, опосредуемых ВИЧ, СПИД или ПСП, с использованием указанных соединений при монотерапии или комбинированной терапии. Настоящее изобретение относится к области противовирусной терапии и, в частности, к ненуклеозидным соединениям, которые ингибируют обратную транскриптазу ВИЧ и применимы для лечения заболеваний, опосредуемых вирусом иммунодефицита человека (ВИЧ). Настоящее изобретение относится к новым гетероциклическим соединениям формулы I, предназначенным для лечения или профилактики заболеваний, опосредуемых ВИЧ, СПИД или ПСП, с использованием указанных соединений при монотерапии или комбинированной терапии. Вирус иммунодефицита человека (ВИЧ) является возбудителем синдрома приобретенного иммунодефицита (СПИД), т.е. заболевания, характеризующегося поражением иммунной системы, в частностиCD4+ Т-клеток, при сопутствующей восприимчивости к инфекциям, вызываемым условно-патогенными организмами. Инфекция ВИЧ также связана с предшественником, СПИД-ассоциированным комплексом(ПСП), т.е. синдромом, характеризующимся такими симптомами, как хроническая генерализованная лимфаденопатия, лихорадка и потеря массы. Как и другие ретровирусы, геном ВИЧ кодирует белки-предшественники, известные под обозначениями gag и gag-pol, которые подвергаются процессингу вирусной протеазой с образованием протеазы,обратной транскриптазы (ОТ), эндонуклеазы/интегразы и зрелых структурных белков ядра вируса. Прерывание этого процессинга предотвращает продуцирование обычного инфекционного вируса. Были предприняты значительные усилия для борьбы с ВИЧ путем ингибирования кодируемых вирусом ферментов. Для химиотерапии ВИЧ-1 были тщательно исследованы два фермента: протеаза ВИЧ и обратная транскриптаза ВИЧ (J.S.G. Montaner et al., Antiretroviral therapy, 'the state of the art', BiomedE. De Clercq, New Developments in Anti-HIVChemotherap. Curr. Med. Chem. 2001, 8:1543-1572). Выявлены два общих класса ингибиторов ОТ: нуклеозидные ингибиторы обратной транскриптазы (НИОТ) и ненуклеозидные ингибиторы обратной транскриптазы (ННИОТ). В настоящее время корецептор CCR5 стал потенциальной мишенью для химиотерапии ВИЧ (D. Chantry, Expert Opin. Emerg. Drugs. 2004, 9(1):1-7; С.G. Barber, Curr. Opin. Invest. Drugs. 2004, 5(8):851-861; D. Schols, Curr. Topics Med. Chem. 2004,4(9):883-893; N.A. Meanwell and J.F. Kadow, Curr. Opin. Drug Discov. Dev. 2003, 6(4):451-461). На рынок поступили лекарственные средства, воздействующие на новые ферменты-мишени, включая ингибиторы интегразы, примерами которых являются ралтегравир (Merck), утвержденные к применению FDA(Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США) и элвитегравир (Gilead Sciences и Japan Tobacco), находящийся в фазе II клинических исследований. АнтагонистCCR5 маравирок (SELZENTRY, Pfizer) также утвержден FDA к применению в качестве средства лечения ВИЧ-1. НИОТ обычно представляют собой аналоги 2',3'-дидезоксинуклеозида (ddN), которые до взаимодействия с вирусной ОТ должны быть фосфорилированы. Соответствующие трифосфаты выступают в качестве конкурентных ингибиторов или альтернативных субстратов для вирусной ОТ. После включения в нуклеиновые кислоты нуклеозидные аналоги обрывают процесс удлинения цепи. Обратная транскриптаза ВИЧ обладает способностью менять последовательность ДНК, что позволяет резистентным штаммам преодолевать блокаду путем расщепления нуклеозидного аналога и продолжения процесса удлинения цепи. Применяющиеся в настоящее время в клинической практике НИОТ включают зидовудин(AZT), диданозин (ddI), залцитабин (ddC), ставудин (d4T), ламивудин (3 ТС) и тенофовир (РМРА). ННИОТ впервые были открыты в 1989 г. ННИОТ являются аллостерическими ингибиторами, которые обратимо связываются с несубстратным центром связывания обратной транскриптазы ВИЧ и тем самым меняют форму активного центра или активность блокирующей полимеразы (R.W. Buckheit, Jr.,Non-nucleoside reverse transcriptase inhibitors: perspectives for novel therapeutic compounds and strategies forAntiviral Therapy, Drugs. 2001, 61(1):19-26). Хотя в лаборатории идентифицированы более 30 структурных классов ННИОТ, для лечения ВИЧ утверждены к применению только 3 соединения: эфавиренц, невирапин и делавирдин.-1 016386 Хотя ННИОТ сначала рассматривались как перспективный класс соединений, проведенные in vitro и in vivo исследования быстро показали, что ННИОТ создают низкий барьер для появления резистентных по отношению к лекарственным препаратам штаммов ВИЧ и обладают низкой классоспецифической токсичностью. Резистентность по отношению к лекарственным препаратам часто развивается уже после одной точечной мутации в ОТ. Хотя комбинированная терапия с применением различных НИОТ,ИП (ингибитор протеазы) и ННИОТ во многих случаях чрезвычайно значительно снижала вирусную нагрузку и замедляла прогрессирование заболевания, сохранились значительные терапевтические проблемы (R.М. Gulick, Eur. Soc. Clin. Microbiol. and Inf. Dis. 2003, 9(3):186-193). Смеси не являлись эффективными для всех пациентов, часто проявлялись потенциально тяжелые побочные эффекты и было показано, что быстро размножающийся вирус ВИЧ легко образует резистентные по отношению к лекарственным препаратам мутантные варианты протеазы и обратной транскриптазы дикого типа. Сохраняется необходимость в более безопасных лекарственных препаратах, обладающих активностью по отношению к штаммам дикого типа и широко распространенным резистентным штаммам ВИЧ. Пиридазиноновые ненуклеозидные ингибиторы обратной транскриптазы описали J.P. Dunn et al. в патенте US7189718, выданном 13 марта 2007 г., и J.P. Dunn et al. в публикации патентаUS2005021554, поданного 22 марта 2005 г. 5-Арилалкил-2,4-дигидро-[1,2,4]триазол-3-он,5-арилалкил-3 Н-[1,3,4]оксадиазол-2-он и 5-арилалкил-3 Н-[1,3,4]тиадиазол-2-он, как ненуклеозидные ингибиторы обратной транскриптазы, раскрыли J.P. Dunn et al. в патенте US7208059, выданном 24 ареля 2007 г., в публикации патента US20060225874, опубликованной 5 октября 2006 г., и в публикации патента US20060025462, поданного 27 июня 2005 г. Родственные соединения раскрылиY.D. Saito et al. в публикации патента US20070078128, опубликованной 5 апреля 2007 г. Фенилацетамидные ненуклеозидные ингибиторы обратной транскриптазы раскрыли J.P. Dunn et al. в патентеUS7166738, выданном 23 января 2007 г., и способы борьбы с ретровирусными инфекциями с помощью фенилацетамидов раскрыли J.P. Dunn et al. в публикации патента US20050239880, опубликованной 27 октября 2005 г.; Т. Mirzadegan and Т. Silva в публикации патента US20070088053, опубликованной 19 апреля 2007 г., и Z.K. Sweeney and T. Silva в публикации патента US20070088015, опубликованной 19 апреля 2007 г. Эти заявки во всей своей полноте включены в настоящее изобретение в качестве ссылки. В WO 2006/067587, опубликованной 26 июня 2006 г., L.H. Jones et al. раскрыли производные феноксиацетамида и содержащие их композиции, которые связывают обратную транскриптазу ВИЧ-1 и являются модуляторами, в частности ингибиторами. K.R. Romines et al. (J. Med. Chem. 2006, 49(2):727739) и P. Bonneau et al. (в публикации патента US20060069261, опубликованной 30 марта 2006 г.) описали феноксиацетамиды, которые ингибируют обратную транскриптазу ВИЧ-1. В публикации патента US2007/0021442, опубликованной 25 января 2007 г., S.A. Saggar et al. раскрыли дифениловые эфиры, как ингибиторы обратной транскриптазы ВИЧ-1. Настоящее изобретение относится к соединению формулы IR2 также может обозначать галоген, NRaRb, CN или ORc; илиR3 обозначает водород или C1-C3-алкил;Rc и Rd независимо обозначают водород или C1-C3-алкил;Ar обозначает фенил, замещенный 1-3 группами, независимо выбранными из группы, включающей галоген, цианогруппу, C1-C6-галогеналкил, C1-C6-алкил, C1-C6-алкоксигруппу и C3-C6-циклоалкил;n является целым числом, равным от 1 до 3; или к его фармацевтически приемлемым солям.-2 016386 Соединения формулы I ингибируют обратную транскриптазу ВИЧ-1 и приводят к способу предупреждения и лечения инфицирования посредством ВИЧ-1 и лечения СПИД и/или ПСП. Генетический код ВИЧ-1 легко мутирует, что приводит к штаммам, обладающим пониженной восприимчивостью к лечению современными терапевтическими средствами. Настоящее изобретение также относится к композициям, содержащим соединения формулы I, применимым для предупреждения и лечения инфицирования посредством ВИЧ-1 и лечения СПИД и/или ПСП. Настоящее изобретение также относится к соединениям формулы I, которые применимы для монотерапии или комбинированной терапии другими противовирусными средствами. При использовании в настоящем изобретении объект в форме единственного числа относится к одному или большему количеству объектов; например, "соединение" означает одно или большее количество соединений или не менее одного соединения. Форма единственного числа и выражения "один или большее количество" и "не менее чем один" в настоящем изобретении можно использовать, как взаимозаменяемые. Выражение "определенная выше в настоящем изобретении" относится к самому широкому определению каждой группы, приведенному в кратком изложении сущности изобретения. Для всех остальных вариантов осуществления, приведенных ниже, заместители, которые могут использоваться в каждом варианте осуществления и которые не определены явно, соответствуют самому широкому определению,приведенному в кратком изложении сущности изобретения. Если не приведено другое определение, то все технические и научные термины, использованные в настоящем изобретении, обладают такими же значениями, которые обычно подразумевает специалист в области техники, к которой относится настоящее изобретение. В настоящем изобретении приведены ссылки на различные методологии, известные специалистам в данной области техники. Стандартные справочники, в которых изложены общие положения фармакологии, включают публикацию Goodman(2001). При осуществлении настоящего изобретения можно использовать любые подходящие материалы и/или методики, известные специалистам в данной области техники. Однако описаны предпочтительные материалы и методики. Материалы, реагенты и т.п., на которые приведены ссылки в последующем описании и примерах, можно получить из коммерческих источников, если не указано иное. При использовании в настоящем описании, в промежуточной фразе или в содержании пункта формулы изобретения термины "включает" и "включающий" следует интерпретировать, как допускающее изменения значения. Таким образом, термины следует интерпретировать, как синонимичные с выражениями "содержащий не менее" или "включающий не менее". При использовании в контексте способа термин "включающий" означает, что способ включает, по меньшей мере, указанные стадии, но может включать дополнительные стадии. При использовании в контексте соединения или композиции термин"включающий" означает, что соединение или композиция включает, по меньшей мере, указанные характеристики или компоненты, но также может включать дополнительные характеристики или компоненты. Термин "примерно" при использовании в настоящем изобретении означает приблизительно, в области, ориентировочно или около. Если термин "примерно" используется в связи с числовым диапазоном, то он изменяет этот диапазон путем расширения границ за пределы верхней и нижней границы указанного числового диапазона. Обычно термин "примерно" при использовании в настоящем изобретении изменяет числовое значение выше и ниже указанного значения с отклонением на 20%. Термин "необязательно" или "необязательный" при использовании в настоящем изобретении означает, что последующее описанное событие или обстоятельство может, но не должно осуществиться и что описание включает случаи, когда событие или обстоятельство осуществляется, и случаи, когда оно не осуществляется. Например, "необязательно замещенный" означает, что необязательно замещенный фрагмент может включать водород или заместитель. Выражение "необязательная связь" означает, что связь может содержаться или может не содержаться и что описание включает ординарные, двойные или тройные связи. Если заместитель обозначен, как"связь" или как "отсутствующий", то атомы, связанные с заместителями, непосредственно соединены друг с другом. Если любая переменная (например, R1, R4a, Ar, X1 или Het) содержится более одного раза в любом фрагменте или формуле, изображающей и описывающей соединения, использованные или заявленные в настоящем изобретении, ее определение в каждом случае не зависит от ее определения в каждом другом случае. Кроме того, комбинации заместителей и/или переменных допустимы, только если такие соединения приводят к стабильным соединениям."Стабильное" соединение является соединением, которое можно получить и выделить и структура и свойства которого остаются в основном неизменными в течение периода времени, достаточного для того, чтобы обеспечить применение соединения для задач, описанных в настоящем изобретении (например,для терапевтического или профилактического введения субъекту). Если явно не указано иное, то все диапазоны, указанные в настоящем изобретении, включают границы. Например, описание гетероциклического кольца, как содержащего "от 1 до 4 гетероатомов", означает, что кольцо может содержать 1, 2, 3 или 4 гетероатома. Также следует понимать, что любой диапа-3 016386 зон, указанный в настоящем изобретении, включает все поддиапазоны, входящие в этот диапазон. Так,например, арил или гетероарил, описанный как необязательно содержащий "от 1 до 5 заместителей",включает в качестве вариантов любой арил, необязательно содержащий от 1 до 4 заместителей, от 1 до 3 заместителей, от 1 до 2 заместителей, от 2 до 5 заместителей, от 2 до 4 заместителей, от 2 до 3 заместителей, от 3 до 5 заместителей, от 3 до 4 заместителей, от 4 до 5 заместителей, 1, 2, 3, 4 и 5 заместителей. Значки в конце связи или "", проведенный через связь, означают положение присоединения функциональной группы или другого химического фрагмента к остальной части молекулы, частью которой она является. Так, например: Подразумевается, что термины, определенные в настоящем изобретении, можно объединять с образованием химически возможных комбинаций, например "гетероалкиларил", "галогеналкилгетероарил","арилалкилгетероциклил", "алкилкарбонил", "алкоксиалкил" и т.п. Если термин алкил" используется в качестве суффикса после другого термина, как в терминах "фенилалкил" или "гидроксиалкил", это означает, что алкильная группа, определенная выше, замещена одним или двумя заместителями, выбранными из числа других конкретных названных групп. Так, например, "фенилалкил" означает алкильную группу,содержащую 1 или 2 фенильных заместителя, и таким образом включает бензил, фенилэтил и бифенил."Гидроксиалкил" включает 2-гидроксиэтил, 2-гидроксипропил, 1-(гидроксиметил)-2-метилпропил,2-гидроксибутил, 2,3-дигидроксибутил, 2-(гидроксиметил), 3-гидроксипропил и т.п. Соответственно, при использовании в настоящем изобретении термин "гидроксиалкил" используют для определения подмножества гетероалкильных групп, определенных ниже. Термин "-(арил)алкил" означает незамещенный алкил или арилалкильную группу. Термин "(гетеро)арил или (гет)арил" означает арильную или гетероарильную группу. Термин "алкил" при использовании в настоящем изобретении означает обладающий разветвленной или неразветвленной цепью насыщенный одновалентный углеводородный остаток, содержащий от 1 до 10 атомов углерода. Термин "низш. алкил" означает обладающий разветвленной или неразветвленной цепью углеводородный остаток, содержащий от 1 до 6 атомов углерода. "C6-C10-Алкил" при использовании в настоящем изобретении означает алкил, содержащий от 1 до 10 атомов углерода. Примеры алкильных групп включают, но не ограничиваются только ими, низшие алкильные группы, включая метил,этил, пропил, изопропил, н-бутил, изобутил, трет-бутил или пентил, изопентил, неопентил, гексил, гептил или октил. Термин "алкилен" при использовании в настоящем изобретении означает двухвалентный насыщенный линейный углеводородный радикал, содержащий от 1 до 10 атомов углерода (например, (СН 2)n), или разветвленный насыщенный двухвалентный углеводородный радикал, содержащий от 2 до 10 атомов углерода (например, -CHMe- или -СН 2 СН(i-Pr)CH2-), если не указано иное. Свободные валентности алкиленовой группы не выходят из одного и того же атома. Примеры алкиленовых радикалов включают,но не ограничиваются только ими, метилен, этилен, пропилен, 2-метилпропилен, 1,1-диметилэтилен,бутилен, 2-этилбутилен. Термин "алкенил" при использовании в настоящем изобретении означает незамещенный углеводородный цепочечный радикал, содержащий от 2 до 10 атомов углерода и содержащий двойные связи."C2-C10-Алкенил" при использовании в настоящем изобретении означает алкенил, содержащий от 2 до 10 атомов углерода. Примерами являются винил, 1-пропенил, 2-пропенил (аллил) или 2-бутенил (кротил). Термин "циклоалкил" при использовании в настоящем изобретении означает насыщенное карбоциклическое кольцо, содержащее от 3 до 8 атомов углерода, т.е. циклопропил, циклобутил, циклопентил,циклогексил, циклогептил или циклооктил. "C3-C7-Циклоалкил" при использовании в настоящем изобретении означает циклоалкил, содержащий в карбоциклическом кольце от 3 до 7 атомов углерода. Термин "алкоксигруппа" при использовании в настоящем изобретении означает группу -О-алкил, в которой алкил является таким, как определено выше, такую как метокси-, этокси, н-пропилокси-, изопропилокси-, н-бутилокси-, изобутилокси, трет-бутилокси-, пентилокси-, гексилоксигруппу, включая их изомеры. "Низш. алкоксигруппа" при использовании в настоящем изобретении означает алкоксигруппу,содержащую "низш. алкильную" группу, определенную выше. "C6-C10-Алкоксигруппа" при использовании в настоящем изобретении означает -О-алкил, в котором алкил представляет собой C1-C10. Термин "галогеналкил" при использовании в настоящем изобретении обозначает обладающую разветвленной или неразветвленной цепью алкильную группу, определенную выше, в которой 1, 2, 3 или большее количество атомов водорода замещены на галоген. Примерами являются 1-фторметил,1-хлорметил, 1-бромметил, 1-йодметил, дифторметил, трифторметил, трихлорметил, трибромметил,трийодметил, 1-фторэтил, 1-хлорэтил, 1-бромэтил, 1-йодэтил, 2-фторэтил, 2-хлорэтил, 2-бромэтил,2-йодэтил, 2,2-дихлорэтил, 3-бромпропил или 2,2,2-трифторэтил. Термин "галоген" при использовании в настоящем изобретении означает фтор, хлор, бром или йод.-4 016386 Термин "ацил" при использовании в настоящем изобретении означает группу формулы -C(=O)R, в которой R обозначает водород или низш. алкил, определенный в настоящем изобретении. Термин "алкилкарбонил" при использовании в настоящем изобретении означает группу формулы C(=O)R, в которой-C(=O)R, содержащую от 1 до 6 атомов углерода. Термины урацил или 1 Н-пиримидин-2,4-дион, дигидропиримидин-2,4-дион и имидазолидин-2,4 дион или гидантоин означают фрагмент формулы (i), (ii) и (iii) соответственно Соединения формулы I обладают таутомерией. Таутомерные соединения могут существовать в виде двух взаимопревращающихся форм. Прототропные таутомеры образуются вследствие переноса ковалентно связанного атома водорода между двумя атомами. Таутомеры обычно находятся в виде равновесной смеси, и попытки выделить индивидуальные таутомеры обычно приводят к смеси,химические и физические характеристики которой соответствуют характеристиками смеси соединений. Положение равновесия зависит от химического строения молекулы. Например, для многих алифатических альдегидов и кетонов, таких как ацетальдегид, преобладает кетоформа, тогда как для фенолов преобладает енольная форма. Обычно прототропные таутомеры включают таутомеры кетон/енол(-C(=NR)-NH--C(-NHR)=N-). Два последних являются особенно распространенными для гетероарильных и гетероциклических колец, и в объем настоящего изобретения входят все таутомерные формы соединений. Таким образом, производное цитозина эквивалентно можно представить как енаминовый (i) или иминовый (ii) таутомер В одном варианте осуществления настоящее изобретение относится к соединению формулы I, в которой A, X1, X2, R1, R2, R3, R4, Ra, Rb, Rc, Rd, Ar и n являются такими, как определено выше в настоящем изобретении. Во всех вариантах осуществления настоящего изобретения, если "А" обозначает этенилен(a) или этилен (b), то атом углерода, к которому присоединен R3, связан с атомом азота и атом углерода,к которому присоединен R2, связан с карбонилом или его эквивалентом. Во втором варианте осуществления настоящее изобретение относится к соединению формулы I, в которой А обозначает (a); R1 обозначает (II); X1 и X2 обозначают О и n равно 1. В третьем варианте осуществления настоящее изобретение относится к соединению формулы I, в которой А обозначает (a); R1 обозначает (II); X1 и X2 обозначают О; n равно 1; R2 обозначает водород,галоген, C1-C6-алкил, C1-C6-галогеналкил; R3 обозначает водород; R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил, необязательно замещенный 1-3 группами, независимо выбранными из группы,включающей цианогруппу, галоген и C1-C6-галогеналкил. В четвертом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой А обозначает (a); R1 обозначает (II); X1 и X2 обозначают О; n равно 1; R2 обозначает водород,галоген, C1-C6-алкил, C1-C6-галогеналкил; R3 обозначает водород; R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5-дифторметилфенил. В пятом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой А обозначает (a); R1 обозначает (II); X1 обозначает S; X2 обозначает О; n равно 1; R2 обозначает водород, галоген, C1-C6-алкил, C1-C6-галогеналкил; R3 обозначает водород; R4 обозначает галоген илиC1-C6-алкил и Ar обозначает фенил, необязательно замещенный 1-3 группами, независимо выбранными из группы, включающей цианогруппу, галоген и C1-C6-галогеналкил. В шестом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой А обозначает (a); R1 обозначает (II); X1 обозначает О; X2 обозначает NHRd; Rd обозначает Н и n равно 1. В седьмом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой А обозначает (a); R1 обозначает (II); X1 обозначает О; X2 обозначает NHRd; Rd обозначает Н; n равно 1; R2 обозначает водород, галоген, C1-C6-алкил, C1-C6-галогеналкил; R3 обозначает водород; R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил, необязательно замещенный 1-3 группами,независимо выбранными из группы, включающей цианогруппу, галоген и C1-C6-галогеналкил.-5 016386 В восьмом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой А обозначает (a); R1 обозначает (II); X1 обозначает О; X2 обозначает NHRd; Rd обозначает Н; n равно 1; R2 обозначает водород, галоген, C1-C6-алкил, C1-C6-галогеналкил; R3 обозначает водород; R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5 дифторметилфенил. В девятом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой А обозначает (b); R1 обозначает (II); X1 и X2 обозначают О и n равно 1. В десятом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой А обозначает (b); R1 обозначает (II); X1 и X2 обозначают О; n равно 1; R2 обозначает водород,галоген, C1-C6-алкил, C1-C6-галогеналкил; R3 обозначает водород; R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил, необязательно замещенный 1-3 группами, независимо выбранными из группы,включающей цианогруппу, галоген и C1-C6-галогеналкил. В одиннадцатом варианте осуществления настоящее изобретение относится к соединению формулыI, в которой А обозначает (b); R1 обозначает (II); X1 и X2 обозначают О; n равно 1; R2 обозначает водород,галоген, C1-C6-алкил, C1-C6-галогеналкил; R3 обозначает водород; R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5-дифторметилфенил. В двенадцатом варианте осуществления настоящее изобретение относится к соединению формулыI, в которой А обозначает (с); R1 обозначает (II); X1 и X2 обозначают О и n равно 1. В тринадцатом варианте осуществления настоящее изобретение относится к соединению формулыI, в которой А обозначает (с); R1 обозначает (II); X1 и X2 обозначают О; n равно 1; R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил, необязательно замещенный 1-3 группами, независимо выбранными из группы, включающей цианогруппу, галоген и C1-C6-галогеналкил. В четырнадцатом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой А обозначает (с); R1 обозначает (II); X1 и X2 обозначают О; n равно 1; R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5-дифторметилфенил. В пятнадцатом варианте осуществления настоящее изобретение относится к соединению формулыI, в которой А обозначает (a); R2 обозначает (II); X1 и X2 обозначают О; R3 обозначает водород. В шестнадцатом варианте осуществления настоящее изобретение относится к соединению формулыI, в которой А обозначает (a); R1 обозначает водород или метил; R2 обозначает (II); X1 и X2 обозначают О; R3 обозначает водород; R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил, необязательно замещенный 2 группами, независимо выбранными из группы, включающей цианогруппу, галоген иC1-C6-галогеналкил. В семнадцатом варианте осуществления настоящее изобретение относится к соединению формулыI, в которой А обозначает (a); R1 обозначает водород или метил; R2 обозначает (II); X1 и X2 обозначают О; R3 обозначает водород; R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил,3,5-дицианофенил или 3-циано-5-дифторметилфенил. В восемнадцатом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой А обозначает (с); R2 обозначает (II); X1 и X2 обозначают О. В девятнадцатом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой А обозначает (с); R1 обозначает водород или метил; R2 обозначает (II); X1 и X2 обозначают О; R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил, необязательно замещенный 2 группами, независимо выбранными из группы, включающей цианогруппу, галоген и C1-C6-галогеналкил 1. В двадцатом варианте осуществления настоящее изобретение относится к соединению формулы I, в которой А обозначает (с); R1 обозначает водород или метил; R2 обозначает (II); X1 и X2 обозначают О; R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5 дифторметилфенил. В двадцать первом варианте осуществления настоящее изобретение относится к соединению, выбранному из числа соединений I-1-I-32, приведенных в табл. 1. В двадцать втором варианте осуществления настоящее изобретение относится к способу лечения инфицирования посредством ВИЧ-1 или предупреждения инфицирования посредством ВИЧ-1 или лечения СПИД или ПСП, включающему введение нуждающемуся в нем реципиенту терапевтически эффективного количества соединения формулы I, в которой А, X1, X2, R1, R2, R3, R4, Ra, Rb, Rc, Rd, Ar и n являются такими, как определено выше в настоящем изобретении. В двадцать третьем варианте осуществления настоящее изобретение относится к способу лечения инфицирования посредством ВИЧ-1 или предупреждения инфицирования посредством ВИЧ-1 или лечения СПИД или ПСП, включающему совместное введение нуждающемуся в нем реципиенту терапевтически эффективного количества соединения формулы I, в которой А, X1, X2, R1, R2, R3, R4, Ra, Rb, Rc, Rd,Ar и n являются такими, как определено выше в настоящем изобретении и по меньшей мере одного соединения, выбранного из группы, включающей ингибиторы ВИЧ-протеазы, нуклеозидные ингибиторы обратной транскриптазы, ненуклеозидные ингибиторы обратной транскриптазы, ингибиторы интегразы,антагонисты CCR5 и ингибиторы слияния вирусов.-6 016386 В двадцать четвертом варианте осуществления настоящее изобретение относится к способу лечения инфицирования посредством ВИЧ-1 или предупреждения инфицирования посредством ВИЧ-1 или лечения СПИД или ПСП, включающему совместное введение нуждающемуся в нем реципиенту терапевтически эффективного количества соединения формулы I, в которой А, X1, X2, R1, R2, R3, R4, Ra, Rb, Rc, Rd,Ar и n являются такими, как определено в настоящем изобретении, и по меньшей мере одного соединения, выбранного из группы, включающей зидовудин, ламивудин, диданозин, залцитабин, ставудин, рескриптор, сустива, вирамун, эфавиренц, невирапин или делавирдин, саквинавир, ритонавир, нелфинавир,индинавир, ампренавир, лопинавир, ралтегравир калия, энфувиртид и маравирок. В двадцать пятом варианте осуществления настоящее изобретение относится к способу ингибирования обратной транскриптазы ВИЧ-1 у реципиента, инфицированного посредством ВИЧ-1, включающему введение нуждающемуся в нем реципиенту терапевтически эффективного количества соединения формулы I, в которой A, X1, X2, R1, R2, R3, R4, Ra, Rb, Rc, Rd, Ar и n являются такими, как определено выше в настоящем изобретении. В двадцать шестом варианте осуществления настоящее изобретение относится к способу ингибирования обратной транскриптазы ВИЧ-1 у реципиента, инфицированного посредством ВИЧ-1, экспрессирующего обратную транскриптазу, содержащего по меньшей мере одну мутацию по сравнению с ВИЧ-1 дикого типа, включающему введение нуждающемуся в нем реципиенту терапевтически эффективного количества соединения формулы I, в которой А, X1, X2, R1, R2, R3, R4, Ra, Rb, Rc, Rd, Ar и n являются такими, как определено выше в настоящем изобретении. В двадцать седьмом варианте осуществления настоящее изобретение относится к способу ингибирования обратной транскриптазы ВИЧ-1 у реципиента, инфицированного посредством ВИЧ-1, экспрессирующего обратную транскриптазу, обладающего сниженной восприимчивостью к эфавиренцу, невирапину или делавирдину, включающему введение нуждающемуся в нем реципиенту терапевтически эффективного количества соединения формулы I, в которой A, X1, X2, R1, R2, R3, R4, Ra, Rb, Rc, Rd, Ar и n являются такими, как определено выше в настоящем изобретении. В двадцать восьмом варианте осуществления настоящее изобретение относится к фармацевтической композиции, включающей терапевтически эффективное количество соединения формулы I, в которой А, X1, X2, R1, R2, R3, R4, Ra, Rb, Rc, Rd, Ar и n являются такими, как определено выше в настоящем изобретении, и по меньшей мере один носитель, инертный наполнитель или разбавитель. А-М. Vandamme et al. (Antiviral ChemistryChemotherapy, 1998, 9:187-203) раскрыли современные средства клинического лечения ВАРВТ инфицирования посредством ВИЧ-1 у человека, включающие по меньшей мере тройные комбинации лекарственных средств. Высокоактивная антиретровирусная терапия(ВАРВТ) традиционно включает комбинированную терапию нуклеозидными ингибиторами обратной транскриптазы (НИОТ), ненуклеозидными ингибиторами обратной транскриптазы (ННИОТ) и ингибиторами протеазы (ИП). Эти соединения подавляют биохимические процессы, необходимые для репликации вирусов. Хотя ВАРВТ привела к резкому изменению прогноза для лиц, инфицированных посредством ВИЧ, в современной терапии сохранились многие недостатки, включая очень сложные режимы дозировки и побочные эффекты, которые могут быть весьма тяжелыми (A. Carr and D.A. Cooper, Lancet. 2000, 356(9239):1423-1430). Кроме того, эти многолекарственные режимы лечения не уничтожают ВИЧ 1 и длительное лечение обычно приводит к множественной лекарственной устойчивости и тем самым ограничивает их применимость при длительном лечении. Остается приоритетной разработка новых терапевтических средств, которые можно использовать в комбинации с НИОТ, ННИОТ, ИП и ингибиторами слияния вирусов с получением лучших средств лечения инфицирования посредством ВИЧ-1. Типичные подходящие НИОТ включают зидовудин (AZT; РЕТРОВИР); диданозин (ddl; ВИДЕКС); залцитабин (ddC; ГИВИД); ставудин (d4T; ЗЕРИТ); ламивудин (3 ТС; ЭПИВИР); абакавир (ЗИАГЕН); адефовир дипивоксил [бис(РОМ)-РМЕА; ПРЕВОН]; лобукавир (BMS-180194),нуклеозидный ингибитор обратной транскриптазы, раскрытый в ЕР-0358154 и ЕР-0736533; ВСН-10652,ингибитор обратной транскриптазы (в виде рацемической смеси ВСН-10618 и ВСН-10619), разрабатывающийся фирмой Biochem Pharma; эмитрицитабин [(-)-FTC], разрабатывающийся фирмой-L-2',3'-дидезокси-5-фторцитидин), лицензированный Vion Pharmaceuticals; DAPD, пуриновый нуклеозид, (-)D-2,6-диаминопуриндиоксолан, раскрытый в ЕР-0656778, лицензия на который выдана фирмеTriangle Pharmaceuticals; и лоденосин (FddA), 9-(2,3-дидезокси-2-фтор-b-D-треопентофуранозил)аденин,стабильный по отношению к воздействию кислот ингибитор обратной транскриптазы, разрабатывающийся фирмой U.S. Bioscience Inc. В США к применению утверждены четыре ННИОТ: невирапин (BI-RG-587; ВИРАМУН), выпускающийся фирмой Boehringer Ingelheim (BI); делавирадин (ВНАР, U-90152; РЕСКРИПТОР), выпускающийся фирмой Pfizer; эфавиренц (DMP-266, СУСТИВА) бензоксазин-2-он, выпускающийся фирмой BMS; и этравирин (ТМС-125, ИНТЕЛЕНЦ), выпускающийся фирмой Tibotec.-7 016386 Другие исследующиеся в настоящее время ННИОТ включают UK-453061 (L.H. Jones et al.,WO 2005085860), AR806 (J.-L. Girardet et al., WO 2006026356), IDX899 (R. Storer et al. (US 2006074054),TMC-278 (J.E.G. Guillemont et al., WO 03/16306), PNU-142721, фуропиридинтиопиримид, разрабатывающийся фирмой Pfizer; каправирин (S-1153 или AG-1549; 5-(3,5-дихлорфенил)-тио-4-изопропил-1-(4 пиридил)метил-1 Н-имидазол-2-илметилкарбонат), выпускающийся фирмами Shionogi и Pfizer; эмивирин[MKC-442; (1-(этоксиметил)-5-(1-метилэтил)-6-(фенилметил)-(2,4(1 Н,3 Н)-пиримидиндион)], выпускающийся фирмами Mitsubishi Chemical Со. и Triangle Pharmaceuticals; (+)-каланолид A (NSC-675451) и В,производные кумарина, раскрытые в выданном Национальному институту здравоохранения патентеUS5489697, лицензия на который выдана Sarawak/Advanced Life Sciences; DAPY (TMC120; 4-4-[4E)-2-циановинил)-2,6-диметилфениламино]пиримидин-2-иламинобензонитрил),выпускающийся фирмами Tibotec-Virco и JohnsonJohnson; BILR-355 BS (12-этил-8-[2-(1-гидроксихинолин-4 илокси)этил]-5-метил-11,12-дигидро-5 Н-1,5,10,12-тетраазадибензо[а,е]циклооктен-6-он), выпускающийся фирмой Boehringer-Ingleheim, и PHI-236 (7-бром-3-[2-(2,5-димегоксифенил)этил]-3,4-дигидро-1 Нпиридо[1,2-а][1,3,5]триазин-2-тион). Типичные подходящие ИП включают саквинавир (Ro 31-8959; ИНВИРАЗА; ФОРТОВАЗА); ритонавир (АВТ-538; НОРВИР); индинавир (MK-639; КРИКСИВАН); нелфнавир (AG-1343; ВИРАЦЕПТ); ампренавир (141W94; АГЕНЕРАЗА); ТМС 114 (дарунавир, ПРЕЗИСТА); ласинавирBMS-2322623, азапептид, разрабатывающийся фирмой Bristol-Myers Squibb в качестве ИП ВИЧ-1 2-го поколения; АВТ-378, разрабатывающийся фирмой Abbott; и AG-1549 имидазолкарбамат, разрабатывающийся фирмой Agouron Pharmaceuticals, Inc. Пентафузид (ФУЗЕОН) содержит 36 аминокислот синтетического пептида, который ингибирует слияние ВИЧ-1 с мембранами-мишенями. Пентафузид (3-100 мг/день) вводят в виде непрерывного подкожного вливания или инъекции вместе с эфавиренцем и 2 ИП инфицированным посредством ВИЧ-1 пациентам, устойчивым по отношению к тройной комбинированной терапии; применение 100 мг/сутки является предпочтительным. ФУЗЕОН связывается с GP41 на вирусной оболочке и предупреждает образование входной поры для капсиды вируса, выходящей из клетки. ВИЧ-1 инфицирует клетки специфических линий моноцитов и макрофагов и лимфоциты-хелперы Т-клеток путем использования высокоаффинного взаимодействия гликопротеина вирусной оболочки(Env) с антигеном CD-4. Установлено, что наличие антигена CD-4 необходимо, но не достаточно для входа в клетку и для инфицирования клеток необходим по меньшей мере один другой поверхностный белок(Е.A. Berger et al., Ann. Rev. Immunol. 1999, 17:657-700). Позднее было установлено, что два хемокиновых рецептора, рецептор CCR5 или CXCR4, являются корецепторами, наряду с которыми CD4 необходим для инфицирования клеток вирусом иммунодефицита человека (ВИЧ). Для предупреждения слияния вирусов проводили поиск антагонистов связывания CCR5. Маравирок (Pfizer) является антагонистомCCR5 и недавно утвержден к применению FDA. Викривирок (Schering), выпускающийся фирмой Pfizer,находится на последней стадии разработки. У многих других фирм существуют исследовательские программы, находящиеся на различных стадиях исследований и разработок (см., например, A. Palani andJ.R. Tagat, J. Med. Chem. 2006, 49(10):2851-2857, P. Biswas et al. Expert. Opin. Investig. Drugs. 2006,15(5):451-464; W. Kazmierski et al. Biorg. Med. Chem. 2003, 11:2663-76). Антагонисты CCR5, которые попадут на рынок, вероятно, будут применимы в комбинации с ННИОТ, НИОТ и ИП. Другие противовирусные средства включают гидроксимочевину, рибаривин, IL-2, IL-12, пентафузид. Гидроксимочевина (Droxia) представляет собой ингибитор рибонуклеозидтрифосфатредуктазы, и показано, что она оказывает синергетическое воздействие на активность диданозина, и она исследована со ставудином. IL-2 (алдеслейкин; ПРОЛЕЙКИН) раскрыли Ajinomoto в ЕР-0142268, Takeda в ЕР-0176299 и Chiron в патентах USRE 33653, 4530787, 4569790, 4604377, 4748234, 4752585 и 4949314. Рибаривин представляет собой 1D-рибофуранозил-1 Н-1,2,4-триазол-3-карбоксамид. Общепринятые обозначения и сокращения включают ацетил (Ас), атмосфера (атм), трет-бутоксикарбонил (Boc), ди-трет-бутилпирокарбонат или boc-ангидрид (BOC2O), бензил (Bn), бутил (Bu), регистрационныйв журнале Chemical Abstracts (CASRN), бензилоксикарбонил (CBZ или Z), 1,5-диазабицикло[4,3,0]нон-5-ен (ДБН), 1,8-диазабицикло[5,4,0]ундец-7-ен (ДБУ), N,N'-дициклогексилкарбодиимид(EDCI), этил (Et), этилацетат (EtOAc), этанол (EtOH), этиловый эфир 2-этокси-2 Н-хинолин-1-карбоновой кислоты (EEDQ), диэтиловый эфир (Et2O), О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуронийгексафторфосфат (HATU), уксусная кислота (HOAc), 1-N-гидроксибензотриазол (HOBt), высокоэффективная жидкостная хроматография (ВЭЖХ), изопропанол (ИПС), метанол (МеОН), температура плавления (т.пл.), MeSO2- (мезил или Ms), метил (Me), ацетонитрил (MeCN), метахлорбензойная кислота(МХПБК), масс-спектр (МС), метил-трет-бутиловый эфир (МТБЭ), N-метилморфолин (NMM),N-метилпирролидон (NMP), фенил (Ph), пропил (Pr), изопропил (i-Pr), пиридин (pyr), комнатная температура (КТ), трет-бутилдиметилсилил или t-BuMe2Si (ТБДМС), триэтиламин (ТЭА или Et3N), трифлат или CF3SO2- (Tf), трифторуксусная кислота (ТФК), О-бензотриазол-1-ил-N,N,N',N'-тетраметилуронийтетрафторборат (TBTU), тонкослойная хроматография (ТСХ), тетрагидрофуран (ТГФ), триметилсилил или Me3Si (ТМС), моногидрат паратолуолсульфоновой кислоты (TsOH или pTsOH), 4-Ме-C6H4SO2- или тозил (Ts), N-уретан-N-карбоксиангидрид (UNCA). При использовании для алкильного фрагмента общепринятая номенклатура, включая приставки нормальный (н.), изо, вторичный (втор-), третичный (трет-) и нео, обладает обычными значениями(J. Rigaudy and D.P. Klesney, Nomenclature in Organic Chemistry, IUPAC 1979 Pergamon Press, Oxford.). Примеры репрезентативных соединений, входящих в объем настоящего изобретения, приведены в представленной ниже таблице. Эти примеры и методики синтеза приведены для того, чтобы специалисты в данной области техники смогли лучше понять и осуществить настоящее изобретение. Их следует рассматривать не в качестве ограничивающих объем настоящего изобретения, а только в качестве его иллюстративных и репрезентативных примеров. Обычно номенклатура, использованная в настоящем описании, основана на AUTONOM v.4.0,компьютерной системе института Beilstein для генерации систематической номенклатуры ИЮПАК (Международный союз теоретической и прикладной химии). Если имеется расхождение между изображенной структурой и названием, присвоенным этой структуре, более точной является изображенная структура. Кроме того, если стереохимическая конфигурация структуры или части структуры не указана, например, жирными или штриховыми линиями, то структуру или часть структуры следует интерпретировать, как включающую все ее стереоизомеры.- 10016386 Соединения, предлагаемые в настоящем изобретении, можно получить с помощью множества способов, представленных на иллюстративных схемах реакций синтеза и описанных ниже. Исходные вещества и реагенты, использующиеся при получении этих соединений, обычно или продаются поставщиками, такими как Aldrich Chemical Co., или получаются по методикам, известным специалистам в данной области техники по процедурам, описанным в литературе, такой как Fieser and Fieser's Reagents forand C.W. Rees (Eds) Pergamon, Oxford 1996, vol. 1-11 и Organic Reactions, WileySons: New York, 1991,Volumes 1-40. Приведенные ниже схемы реакций синтеза являются лишь иллюстрациями некоторых способов, с помощью которых можно синтезировать соединения, предлагаемые в настоящем изобретении, и специалистом в данной области техники с учетом раскрытия, приведенного в настоящем описании, могут быть выполнены и ему должны быть понятны различные модификации этих схем реакций синтеза. Исходные вещества и промежуточные продукты для этих схем реакций синтеза при необходимости можно выделить и очистить с помощью обычных способов, включая, но не ограничиваясь только ими,фильтрование, перегонку, кристаллизацию, хроматографию и т.п. Такие вещества можно охарактеризовать с помощью обычных средств, включая физические характеристики и спектральные данные. Если не указано иное, то описанные в настоящем изобретении реакции предпочтительно проводить в инертной атмосфере при атмосферном давлении и в диапазоне температур от примерно -78 до примерно 150 С, более предпочтительно от примерно 0 до примерно 125 С и наиболее предпочтительно и удобно при температуре, близкой к комнатной (или окружающей среды), например, приблизительно при 20 С. На приведенных ниже схемах некоторые соединения изображены с заместителями, указанными в общем виде; однако специалист в данной области техники должен сразу же понять, что характер и количество групп R может меняться и приводить к различным соединениям, входящим в объем настоящего изобретения. Общие формулы на схемах предназначены для иллюстрации, а не для внесения ограничений в объем настоящего изобретения, который определяется прилагаемой формулой изобретения. Кроме того, условия проведения реакций являются типичными и хорошо известны альтернативные условия. Последовательности реакций в приведенных ниже примерах не предназначены для ограничения объема настоящего изобретения, который определяется прилагаемой формулой изобретения. Схема А Соединения, предлагаемые в настоящем изобретении, получают алкилированием подходящего замещенного бензилбромида A-2d урацилом или цитозином или его замещенным производным (схема А). Требующийся бензилбромид получают формилированием соль Гриньяра, полученной из А-2 а, который,в свою очередь, получают нуклеофильным замещением одного из фторидных заместителей в А-1(CASRN 1566882-52-9). Получение диариловых простых эфиров описано в обзоре (J.S. Sawyer, RecentAdvances in Diaryl Ether Synthesis, Tetrahedron. 2000, 56:5045-5065). Введение простого арилоксиэфира часто можно провести с помощью прямой реакции SNAr замещения по ароматическому кольцу, содержащему отщепляющуюся группу и электроотрицательные заместители. Известно, что фторароматические соединения с электроотрицательными заместителями чувствительны к нуклеофильной атаке слабыми нуклеофильными реагентами. Фторидные заместители обычно значительно более лабильны, чем другие галогенидные заместители. В то время как сильные нуклеофильные реагенты, такие как вода и гидроксид, не замещают фтор, слабые нуклеофильные реагенты, такие как фенолы, имидазолы, амины,тиолы и некоторые амиды, легко замещают фтор даже при комнатной температуре (D. Boger et al., Biorg.Group VCH Publishers, New York, NY. 1991). Специалист в данной области техники должен понимать,что последовательность реакций, приведенная на схеме А, относится к 3-хлор-5-гидроксибензонитрилу,другие фенолы можно использовать аналогичным образом.- 11016386 Монометаллирование А-2 а с помощью изо-PrMgCl/LiCl/ТТФ и формилирование образовавшейся соли магния с помощью ДМФ дает А-2b. Восстановление полученного альдегида в А-2 с можно провести с помощью хорошо известных реагентов, которые обеспечивают селективное восстановление альдегида. Известно, что борогидрид натрия селективно восстанавливает альдегиды и кетоны в присутствии цианидного заместителя. Восстановление борогидридом натрия обычно проводят в спиртовых или водных средах. Превращение спирта (А-2 с) в галогенид (A-2d) является хорошо известной методикой, которую можно провести с использованием различных реагентов. Обычно использующиеся реагенты включаютSOBr2, PBr3, POBr3 и галогенирующие реагенты на основе фосфора, такие как (RO)3PRX и R3PX2, являются примерами обычно использующихся реагентов. В данном случае тетрабромид углерода и трифенилфосфин или PBr3 образуют эффективный бромирующий реагент (A.R. Katritzky et al. Chem Scr. 1987,27:477). Обработка урацила гексаметилдисилазаном дает 2,4-бис-(триметилсилилокси)пиримидин, который взаимодействует с алкилгалогенидом только в положении N-1 (H. Singh et al., Synthesis. 1990, 520). Соответствующие соединения с этиленовым мостиком (формула II, n=2) можно получить из соответствующей 3-арилоксифенилуксусной кислоты или соответствующих ее эфиров. 3-Арилокси-2-фтор-4 замещенные фенилуксусные кислоты обычно можно получить путем последовательной конденсации 2,3,4-трифтор-1-нитробензола с подходящим замещенным фенолом и диэфиром малоновой кислоты. Эфир гидролизуют и декарбоксилируют и получают фенилуксусную кислоту. Применение третбутилэтил- (или метил-) малоната позволяет провести катализируемый кислотой селективный гидролиз трет-бутилового эфира и декарбоксилирование и непосредственно получить этиловый (или метиловый) эфир. Нитрогруппу можно восстановить и соответствующую аминогруппу можно заместить на галоген с помощью реакции Зандмайера, которая хорошо известна в данной области техники, и получить 4-хлорили 4-бромпроизводные. Фенолы, использующиеся в начальной конденсации, легкодоступны, однако в указанном примере 1 приведены пути, которые представлены в качестве примера и не ограничивают объем настоящего изобретения. Эта методология описана в публикациях J.P. Dunn et al. в патентеUS7166730, опубликованном 23 января 2007 г., и D. Kertesz et al. в публикации US2005/0234236,опубликованной 29 сентября 2005 г. Восстановление арилоксифенилуксусной кислоты в соответствующий спирт, превращение спирта в соответствующий галогенид и замещение галогена цитозином или урацилом можно провести, как описано выше. Соединения, предлагаемые в настоящем изобретении, в которых R4 обозначает C1-C6-алкил, можно получить из 4-бромированного промежуточного продукта, например А-2b, по реакции сочетания Негиши. Сочетание по Негиши цинкорганических галогенидов или диалкилцинка с галогенаренами и арилтрифлатами является эффективной методикой присоединения алкильной группы к арену (E.-I. Negishi,Acc. Chem. Res. 1982, 15:340-348). Реакцию катализируют с помощью Pd(0) и палладий предпочтительно связывают с бидентатным лигандом, например Pd(dppf)Cl2 и Pd(dppe)Cl2 (J.M. Herbert Tetrahedron Lett. 2004, 45:817-819). Обычно реакцию проводят в инертном апротонном растворителе, и подходящими являются обычные простые эфирные растворители, включая диоксан, ДМЭ (диметиловый эфир) и ТГФ. Обычно реакцию проводят при повышенной температуре. Варианты осуществления соединений, предлагаемых в настоящем изобретении, в которых R4 обозначает циклопропил, получают путем введения винильного заместителя с использованием три-нбутилвинилолова по реакции Стилле с последующим катализируемым посредством Pd(II) циклопропанированием полученного стирола диазометаном. Схема В Варианты осуществления соединений, предлагаемых в настоящем изобретении, в которых R4 обозначает хлор и n=1, получают из 3-хлор-5-(6-хлор-2-фтор-3-метилфенокси)бензонитрила (В-3a), который получают конденсацией 3-хлор-5-фторбензонитрила (В-1, CASRN 327056-73-05) и 6-хлор-2-фтор-3 метилфенола (В-2). Свободнорадикальное бромирование метильного заместителя с помощью NBS иAIBN дает В-3b, который превращают в соединения, предлагаемые в настоящем изобретении, как это описано выше (схема В).- 12016386 Варианты осуществления соединений, предлагаемых в настоящем изобретении, содержащие гидантоиновое кольцо, получают с помощью катализируемой основанием циклизации N-карбамоил-Nзамещенной альфа-аминокислоты, которую, в свою очередь, можно получить восстановительным алкилированием. Схема СA-2b и эфир альфа-аминокислоты приведены на схеме С. Специалист в данной области техники должен понимать, что доступность альфа-аминокислот делает возможным введение различных заместителей в положение 5 гидантоинового кольца. Схема D В соединения, предлагаемые в настоящем изобретении, в которых атом углерода С-5 урацилового или гидантоинового кольца связан с 3-арилоксибензильным фрагментом, необходимо введение связи С-С, а не связи С-N, необходимой, когда 3-арилоксибензильный фрагмент присоединен к атому азота(схема D). R4 и Ar на схеме D являются такими, как описано в п.1 формулы изобретения. Гидантоины получают алкилированием А-2b анионом, полученным из амина, защищенного по атому N сложноэфирной группой, который вводит два атома углерода и один атома азота гидантоинового кольца. В примере,приведенном на схеме D, используется N-Boc-метиловый эфир саркозина. После удаления уретановой защитной группы D-1c обрабатывают триметилсилилизоцианатом, что приводит к ацилированию амина и внутримолекулярной циклизации введенного атома азота и сложного эфира. Присоединенные через атом С урацилы получают путем гомологизации А-2b посредством конденсации с малоновым эфиром и декарбоксилирования с получением 3-арилпропионата D-1e. Ацилирование этого эфира этилформиатом дает -формиловый эфир, который циклизуют с тиомочевиной с получением D-3a, который можно гидролизовать с получением мочевины D-3b, который алкилируют путем силилирования более кислого имидного атома азота и алкилирования второго атома азота алкилирующим реагентом. Соединения, предлагаемые в настоящем изобретении, можно приготовить в виде самых различных дозированных форм, предназначенных для перорального введения, и с использованием различных носителей. Пероральное введение можно осуществлять с помощью таблеток, таблеток с покрытием, драже,капсул из мягкого и твердого желатина, растворов, эмульсий, сиропов или суспензий. Соединения, предлагаемые в настоящем изобретении, эффективны при введении другими путями, включая, наряду с другими путями введения, непрерывное (внутривенное капельное), местное, парентеральное, внутримышечное, внутривенное, подкожное, чрескожное (которое может включать агент, увеличивающий проницаемость) трансбуккальное, назальное, ингаляционное и проводимое с помощью суппозиториев введение. Предпочтительным путем введения обычно является пероральный с использованием обычного суточного дозировочного режима, который можно подобрать в соответствии с тяжестью заболевания и реакцией пациента на активный ингредиент. Соединение или соединения, предлагаемые в настоящем изобретении, а также их фармацевтически применимые соли совместно с одним или большим количеством обычных инертных наполнителей, носителей или разбавителей можно приготовить в виде фармацевтических композиций и разовых дозированных форм. Фармацевтические композиции и разовые дозированные формы могут включать обычные ингредиенты в обычных количествах с прибавлением или без прибавления дополнительных активных соединений или действующих веществ, и разовые дозированные формы могут содержать любое подходящее эффективное количество активного ингредиента, соответствующего применяемому суточному дозировочному диапазону. Фармацевтические композиции можно использовать в виде твердых веществ,- 13016386 таких как таблетки или заполненные капсулы, полужидких веществ, порошков, композиций пролонгированного действия, или в виде жидкостей, таких как растворы, суспензии, эмульсии, эликсиры или заполненные капсулы для перорального введения; или в виде суппозиториев для ректального или вагинального введения; или в виде стерильных растворов для инъекций, предназначенных для парентерального введения. Типичный препарат будет содержать от примерно 5 до примерно 95% (мас./мас.) активного соединения или соединений. Подразумевается, что термин "препарат" или "дозированная форма" включает и твердые, и жидкие композиции активного соединения, и специалист в данной области техники должен понимать, что активный ингредиент может находиться в разных препаратах в зависимости от органамишени или ткани-мишени, от требуемой дозы и фармакокинетических параметров. Термин "инертный наполнитель" при использовании в настоящем изобретении означает соединение, которое является пригодным для приготовления фармацевтической композиции, обычно безопасное, нетоксичное и биологически и в других отношениях не являющееся нежелательным, и включает инертные наполнители, которые приемлемы для ветеринарии, а также для фармацевтического применения для людей. Соединения, предлагаемые в настоящем изобретении, можно вводить по отдельности, но обычно их вводят в смеси с одним или большим количеством фармацевтических инертных носителей,разбавителей или носителей, выбранных в соответствии с предполагаемым режимом введения и стандартной фармацевтической практикой."Фармацевтически приемлемое" означает применимое для приготовления фармацевтической композиции, безопасное, нетоксичное и биологически и в других отношениях не являющееся нежелательным, и включает то, что является приемлемым для приготовления фармацевтических средств, предназначенных для людей."Фармацевтически приемлемой соли" активного ингредиента может быть изначально придана желательная фармакокинетическая характеристика активного ингредиента, которая отсутствует у несолевой формы, и она даже может благоприятно влиять на фармакодинамику активного ингредиента с точки зрения его терапевтической активности в организме. Выражение "фармацевтически приемлемая соль" соединения означает соль, которая является фармацевтически приемлемой и которая обладает необходимой фармакологической активностью исходного соединения. Такие соли включают:(2) соли, образованные с кислым протоном, содержащимся в исходном соединении, замещенным ионом металла, например ионом щелочного металла, ионом щелочно-земельного металла или ионом алюминия; или координацией с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п. Твердые формы препаратов включают порошки, таблетки, пилюли, капсулы, облатки, суппозитории и диспергирующиеся гранулы. Твердым носителем может быть одно или большее количество веществ, которые также могут выступать в качестве разбавителей, вкусовых веществ, солюбилизаторов,смазывающих веществ, суспендирующих веществ, связующих, консервантов, веществ, обеспечивающих распадаемость таблеток, или капсулирующего материала. В порошках носитель обычно представляет собой тонкоизмельченное твердое вещество, которое образует смесь с тонкоизмельченным активным компонентом. В таблетках активный компонент обычно в подходящих соотношениях смешан с носителем, обладающим необходимой связывающей способностью, и спрессован с приданием необходимой формы и размера. Подходящие носители включают, но не ограничиваются только ими, карбонат магния,стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, трагакантовую камедь, метилцеллюлозу, натриевую соль карбоксиметилцеллюлозы, легкоплавкий воск, масло какао и т.п. Твердые формы препаратов в дополнение к активному компоненту могут содержать красители, ароматизаторы, стабилизаторы, буферные вещества, искусственные и натуральные подсластители, диспергирующие вещества, загустители, солюбилизирующие вещества и т.п. Жидкие композиции, также пригодные для перорального введения, включают жидкую композицию, включая эмульсии, сиропы, эликсиры, водные растворы, водные суспензии. Они включают твердые формы препаратов, которые предназначены для проводимого незадолго до введения превращения в жидкие формы препаратов. Эмульсии можно приготовить в растворах, например в водных растворах пропиленгликоля, или они могут содержать эмульгирующие вещества, например, такие как лецитин, сорбитанмоноолеат или камедь акации. Водные растворы можно приготовить путем растворения активного- 14016386 компонента в воде и прибавления подходящих красителей, ароматизаторов, стабилизаторов и загущающих веществ. Водные суспензии можно приготовить путем диспергирования тонкоизмельченного активного компонента в воде, содержащей вязкое вещество, такое как натуральные или синтетические камеди,смолы, метилцеллюлоза, натриевая соль карбоксиметилцеллюлозы и другие хорошо известные суспендирующие вещества. Соединения, предлагаемые в настоящем изобретении, можно приготовить для парентерального введения (например, путем инъекции, например инъекции ударной дозы вещества или непрерывного вливания) и можно приготовить в виде разовой дозированной формы в ампулах, предварительно заполненных шприцах, предназначенных для вливания контейнерах небольшого объема или содержащих множество доз контейнерах с прибавленным консервантом. Композиции могут находиться в таких формах, как суспензии, растворы или эмульсии в масляных или водных разбавителях, например в виде растворов в водном растворе полиэтиленгликоля. Примеры масляных и неводных носителей, разбавителей, растворителей и наполнителей включают пропиленгликоль, полиэтиленгликоль, растительные масла (например, оливковое масло) и пригодные для инъекции органические сложные эфиры (например, этилолеат), и они могут содержать образующие композиции вещества, такие как консервирующие, смачивающие, эмульгирующие или суспендирующие, стабилизирующие и/или диспергирующие вещества. Альтернативно, активный ингредиент может находиться в порошкообразном виде, полученном асептическим выделением стерильного твердого вещества или лиофилизацией из раствора для проводимого перед применением восстановления с помощью подходящего разбавителя, например стерильной апирогенной воды. Соединения, предлагаемые в настоящем изобретении, можно приготовить для введения в виде суппозиториев. Сначала расплавляют низкоплавкий воск, такой как смесь глицеридов жирных кислот, или масло какао, и активный компонент равномерно диспергируют, например, путем перемешивания. Затем расплавленную однородную смесь выливают в формы соответствующего размера, дают ей охладиться и затвердеть. Соединения, предлагаемые в настоящем изобретении, можно приготовить для вагинального введения. В данной области техники известно, что подходящими являются пессарии, тампоны, кремы, гели,пасты, пены и аэрозольные препараты, содержащие такие носители в дополнение к активному ингредиенту. При необходимости композиции можно приготовить с энтеросолюбильным покрытием, приспособленным для пролонгированного или регулируемого введения активного ингредиента. Например, соединения, предлагаемые в настоящем изобретении, можно поместить в устройства для чрескожной или подкожной доставки лекарственных препаратов. Такие устройства доставки являются предпочтительными,когда необходимо пролонгированное действие соединения и когда принципиально важным является соблюдение пациентом режима лечения. Системы чрескожной доставки, содержащие соединения, часто прикрепляют к приклеивающейся к коже твердой подложке. Необходимое соединение также можно объединять со средством, увеличивающим проницаемость, например азоном (1-додецилазациклогептан-2 оном). Системы доставки пролонгированного действия вводят подкожно в субдермальный слой оперативным путем или путем инъекции. Подкожный имплантат содержит соединение, капсулированное в растворимой в липидах мембране, например силиконовом каучуке, или в биологически разрушающемся полимере, например полимолочной кислоте. Подходящие композиции, а также фармацевтические носители, разбавители и инертные наполнители описаны в публикации Remington: The Science and Practice of Pharmacy. 1995, edited by E.W. Martin,Mack Publishing Company, 19th edition, Easton, Pennsylvania. После изучения настоящего изобретения специалист по составлению композиций может внести в композиции изменения и получить многочисленные композиции, предназначенные для конкретного пути введения, не делая композиции, предлагаемые в настоящем изобретении, нестабильными и не ухудшая их терапевтическую активность. Модификацию соединений, предлагаемых в настоящем изобретении, например, для того, чтобы сделать их лучше растворимыми в воде или другом разбавителе, можно без труда осуществить путем внесения небольших изменений (состава соли, этерификации и т.п.), которые входят в компетенцию специалиста с общей подготовкой в данной области техники. В компетенцию специалиста с общей подготовкой в данной области техники также входит изменение пути введения и дозировочного режима введения конкретного соединения, так чтобы управлять фармакокинетикой соединений, предлагаемых в настоящем изобретении, для обеспечения максимального благоприятного воздействия на пациента. Выражение "терапевтически эффективное количество" при использовании в настоящем изобретении означает количество, необходимое для ослабления симптомов заболевания у индивидуума. Дозу следует подбирать в каждом конкретном случае в соответствии с индивидуальными потребностями. Эта дозировка может меняться в широких пределах в зависимости от множества факторов, таких как тяжесть подвергающегося лечению заболевания, возраст и общее состояние здоровья пациента, другие лекарственные средства, которыми лечится пациент, путь и форма введения и предпочтения и опыт практикующего врача. При пероральном введении суточная доза, составляющая от примерно 0,01 до примерно 1000 мг/(кг массы тела) в сутки, должна являться подходящей для монотерапии и/или комбинированной- 15016386 терапии. Предпочтительная суточная доза составляет от примерно 0,1 до примерно 500 мг/(кг массы тела), более предпочтительная от 0,1 до примерно 100 мг/(кг массы тела) и наиболее предпочтительная от 1,0 до примерно 10 мг/(кг массы тела) в сутки. Таким образом, для введения лицу массой 70 кг дозировочный диапазон должен составлять от примерно 7 мг до 0,7 г в сутки. Суточную дозу можно вводить в виде одной дозы или разделенных доз, обычно от 1 до 5 доз в сутки. Обычно лечение начинают с меньших доз, которые меньше оптимальной дозы соединения. Затем дозу увеличивают небольшими шагами,пока не будет достигнут оптимальный для данного конкретного пациента эффект. Специалист с общей подготовкой в данной области техники при лечении заболеваний, описанных в настоящем изобретении,без чрезмерного экспериментирования и на основании личной подготовки, опыта и раскрытия настоящего изобретения должен быть способен определить терапевтически эффективное количество соединений,предлагаемых в настоящем изобретении, необходимое для лечения данного заболевания и пациента. В вариантах осуществления настоящего изобретения активное соединение или соль можно вводить в комбинации с другим противовирусным агентом, таким как нуклеозидный ингибитор обратной транскриптазы, другой ненуклеозидный ингибитор обратной транскриптазы или ингибитор протеазы ВИЧ. Если активное соединение или его производное или соль вводится в комбинации с другим противовирусным агентом, то активность по сравнению с исходным соединением может увеличиться. Если методикой лечения является комбинированная терапия, такое введение может быть совместным или последовательным по отношению к нуклеозидным производным. Таким образом, "совместное введение" при использовании в настоящем изобретении включает введение агентов в одно и то же время или в разные моменты времени. Введение двух или большего количества агентов в одно и то же время можно осуществить с помощью одной композиции, содержащей два или большее количество активных ингредиентов,или путем в основном совместного введения двух или большего количества дозированных форм, содержащих по одному активному агенту. Следует понимать, что в настоящем изобретении указания на лечение распространяются на профилактику, а также на лечение имеющихся патологических состояний и что лечение животных включает лечение людей, а также других животных. Кроме того, борьба с инфекцией ВИЧ-1 при использовании в настоящем изобретении также включает лечение или профилактику заболевания или патологического состояния, сопутствующего инфекции ВИЧ-1 или опосредуемого ею, или его клинических симптомов. Предпочтительно, чтобы фармацевтические препараты представляли собой разовые дозированные формы. В такой форме препарат разделяется на разовые дозы, содержащие соответствующие количества активного компонента. Разовая дозированная форма может представлять собой упакованный препарат, и упаковка содержит отдельные количества препарата, такие как упакованные таблетки, капсулы и порошки во флаконах или ампулах. Разовая дозированная форма также может представлять собой капсулу,таблетку, облатку или пастилку или она может представлять собой соответствующее количество любой из них в упакованном виде. Представленные ниже примеры иллюстрируют получение и биологическую оценку соединений,входящих в объем настоящего изобретения. Эти примеры и методики получения, приведенные ниже,предоставлены для того, чтобы специалист в данной области техники мог яснее понять и осуществить настоящее изобретение. Их следует считать не ограничивающими объем настоящего изобретения, а просто иллюстративными и типичными. Пример 1. 3-[6-Бром-3-(2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)-2-фторфенокси]-5-хлорбензонитрил (I-3). Получение 3-хлор-5-гидроксибензонитрила. Стадия 1. В круглодонную колбу объемом 100 мл в токе аргона помещали 3,5-дихлорбензонитрил(7,0 г, 40,69 ммоль) и безводный ДМФ (75 мл). К раствору добавляли метоксид натрия (2,26 г,44,76 ммоль) и затем полученный раствор перемешивали при КТ в течение 24 ч. После завершения реакции в реакционный сосуд по каплям добавляли 10% водный раствор HCl. Неочищенную смесь экстрагировали с помощью EtOAc и последовательно промывали водным раствором кислоты, водой и рассолом. Экстракты, содержащие EtOAc, сушили (Na2SO4), фильтровали и растворитель удаляли в вакууме и получали неочищенное твердое вещество, которое перекристаллизовывали из смеси гексан/ацетон и получали 5,9 г (86%) 5-хлор-3-метоксибензонитрила. Стадия 2. В колбу объемом 250 мл помещали 5-хлор-3-метоксибензонитрил (7,0 г, 41,766 ммоль) и 2,4,6-коллидин (100 мл). Смесь нагревали до 170 С и добавляли LiI (16,76 г, 125,298 ммоль) и реакционную смесь нагревали в течение 4 ч. После израсходования метилового эфира реакционную смесь охлаждали до КТ и реакцию останавливали 10% водным раствором HCl. Полученную смесь экстрагировали с помощью EtOAc и промывали водой и рассолом. Экстракт, содержащий EtOAc, сушили над (Na2SO4) и фильтровали. Растворитель удаляли в вакууме и получали желтое масло, которое очищали с помощью хроматографии на силикагеле, элюируя смесью EtOAc/гексан (10:90), и получали 6,0 г (94%) 3-хлор-5-гидроксибензонитрила.(Схема А) Стадия 1. К раствору 3-хлор-5-гидроксибензонитрила (153 мг, 1 ммоль) и ДМА (1 мл) добавляли NaH (42 мг, 1,05 экв., 60% дисперсия в минеральном масле) и полученную смесь перемешивали при 50 С в течение 30 мин. К раствору добавляли соединение А-1 (2,7 г, 10 ммоль) и полученную смесь нагревали при 125 С в течение 2 ч. Раствор охлаждали и разбавляли с помощью EtOAc и полученный раствор промывали таким же объемом 10% раствора H2SO4. Органический экстракт сушили(MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2, элюируя смесью 10% EtOAc/гексан, и получали 331 мг (82%) соединения А-2 а. Стадия 2. К раствору соединения А-2 а (2,00 г, 4,93 мл) в PhMe (40 мл), выдерживаемому в атмосфере Ar и охлажденному до -78 С, добавляли раствор i-PrMgCl (2 M раствор в ТГФ, 3,08 мл, 6,16 ммоль). Раствор перемешивали в течение 1 ч, затем добавляли раствор CuCN2LiCl (1 M раствор в ТГФ, 0,1 мл). Полученный раствор перемешивали при -50 С в течение 2 ч и затем реакционную смесь с помощью канюли переносили в колбу, содержащую ДМФ (0,57 мл, 7,4 ммоль) и PhMe (10 мл), охлажденные до-78 С. Смесь нагревали до КТ и реакцию останавливали путем добавления насыщенного водного раствора NH4Cl. Органическую фазу отделяли, промывали рассолом, сушили (MgSO4) и выпаривали досуха в вакууме и получали 1,50 г (86%) соединения А-2b в виде почти белого твердого вещества. Стадия 3. К раствору соединения А-2b в ТГФ (5 мл) и МеОН (5 мл) при перемешивании при КТ порциями добавляли борогидрид натрия. После перемешивания в течение 24 ч реакцию останавливали путем добавления насыщенного водного раствора NH4Cl. Органические вещества экстрагировали с помощью EtOAc, промывали рассолом, сушили (MgSO4) и выпаривали досуха в вакууме. Продукт очищали с помощью хроматографии на SiO2, элюируя смесью EtOAc/гексан в градиентном режиме (от 10 до 50%EtOAc), и получали 0,25 г (31%) соединения А-2 с. Стадия 4. К раствору соединения А-2 с (3,00 г, 8,41 ммоль) в ДХМ (100 мл) при перемешивании добавляли раствор PBr3 (1 М раствор в ДХМ, 9,3 мл). После перемешивания в атмосфере N2 при КТ в течение 24 ч реакцию останавливали путем добавления насыщенного водного раствора NaHCO3. Органическую фазу отделяли, промывали рассолом, сушили (MgSO4) и выпаривали в вакууме. Продукт очищали с помощью хроматографии на SiO2, элюируя смесью EtOAc/гексан в градиентном режиме (от 20 до 50%(0,3 мл), ДХЭ (2 мл), йод (каталитическое количество, 1 кусочек) и урацил (0,16 г, 5 экв.) объединяли в атмосфере аргона и перемешивали при 80 С в течение 18 ч. Реакционную смесь охлаждали, органические вещества выпаривали и остаток растворяли в ДХМ и промывали с помощью H2O. Органический раствор сушили (MgSO4), фильтровали и выпаривали. Неочищенный продукт очищали с помощью хроматографии на SiO2, элюируя смесью МеОН/ДХМ в градиентном режиме (1-7% МеОН), и получали 0,07 г (55%) соединения I-3. 3-[6-Бром-2-фтор-3-(5-метил-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)фенокси]-5 хлорбензонитрил получали аналогичным образом, но на стадии 5 вместо урацила использовали тимин. Соединение очищали с помощью хроматографии на SiO2, элюируя смесью МеОН/ДХМ в градиентном режиме (1-6% МеОН), и получали соединение I-2. 3-[6-Бром-3-(5-хлор-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)-2-фторфенокси]-5 хлорбензонитрил получали аналогичным образом, но на стадии 5 вместо урацила использовали 5-хлор 1 Н-пиримидин-2,4-дион. Соединение очищали с помощью хроматографии на SiO2, элюируя смесью МеОН/ДХМ в градиентном режиме (1-7% МеОН), и получали соединение I-4. 3-[6-Бром-3-(5-этил-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)-2-фторфенокси]-5 хлорбензонитрил получали аналогичным образом, но на стадии 5 вместо урацила использовали 5-этил 1 Н-пиримидин-2,4-дион. Соединение очищали с помощью хроматографии на SiO2, элюируя смесью МеОН/ДХМ в градиентном режиме (1-7% МеОН), и получали соединение I-5. 3-[6-Бром-2-фтор-3-(5-фтор-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)фенокси]-5 хлорбензонитрил получали аналогичным образом, но на стадии 5 вместо урацила использовали 5-фтор 1H-пиримидин-2,4-дион, и получали соединение I-6. 3-[6-Бром-3-(2,4-диоксо-5-трифторметил-3,4-дигидро-2 Н-пиримидин-1-илметил)-2-фторфенокси]5-хлорбензонитрил получали аналогичным образом, но на стадии 5 вместо урацила использовали 5-трифторметил-1 Н-пиримидин-2,4-дион (CASRN 54-20-6), и получали соединение I-7. 5-[6-Бром-2-фтор-3-(5-метил-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)фенокси]изофталонитрил,5-[6-бром-3-(5-хлор-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)-2-фторфенокси]изофталонитрил и 5-[6-бром-3-(5-этил-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)-2-фторфенокси]изофталонитрил получали аналогичным образом, но на стадии 1 вместо 3-хлор-5-гидроксибензонитрил использовали 5-гидроксиизофталонитрил и тимин, на стадии 5 вместо урацила использовали 5-хлорурацил и 5-этилурацил соответственно.- 17016386 Пример 2. 3-[3-(4-Амино-2-оксо-2 Н-пиримидин-1-илметил)-6-бром-2-фторфенокси]-5-хлорбензонитрил (I-12). Смесь цитозина (0,04 г, 3 экв.), хлортриметилсилана (1 капля) и гексаметилдисилазана (0,15 мл) нагревали при 120 С в течение 1 ч. Реакционную смесь охлаждали до 80 С и затем добавляли соединениеA-2d (0,05 г, 0,119 ммоль) и йод (1 кусочек). Реакционную смесь нагревали при 80 С в течение 18 ч. Затем реакционную смесь охлаждали, органические вещества выпаривали и полученное твердое вещество растворяли в ДХМ и промывали с помощью H2O и полученную органическую фазу сушили (MgSO4),фильтровали и выпаривали. Неочищенный продукт очищали с помощью хроматографии на SiO2, элюируя смесью МеОН/ДХМ в градиентном режиме (1-7% МеОН), и получали 0,036 г (67%) соединения I-12. 3-[3-(4-Амино-5-метил-2-оксо-2 Н-пиримидин-1-илметил)-6-бром-2-фторфенокси]-5-хлорбензонитрил(I-13) получали аналогичным образом, но вместо цитозина использовали 5-метилцитозин. Пример 3. 3-6-Бром-2-фтор-3-[2-(5-метил-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-ил)этил]фенокси-5 хлорбензонитрил (I-14) Стадия 1. Триметилсилилдиазометан (9,75 мл, 1,5 экв.) при КТ добавляли к раствору соединения 10 а (5,0 г, 13 ммоль) в смеси МеОН (30 мл) и ДХМ (30 мл). Летучие вещества удаляли и получали метиловый эфир, который растворяли в ТГФ (116 мл) и МеОН (12 мл), и медленно добавляли NaBH4 (3,87 г,8 экв.). Смесь нагревали при 80 С в течение 3 ч, охлаждали до 0C и подкисляли, по каплям добавляя 4 М раствор HCl. Смесь экстрагировали эфиром, промывали водой и сушили (MgSO4), фильтровали и выпаривали. Неочищенный продукт очищали с помощью хроматографии на SiO2, элюируя смесьюEtOAc/гексан в градиентном режиме (от 0 до 30% EtOAc), и получали 1,8 г (38%) соединения 10b. Стадия 2. Трифенилфосфин (1,54 г, 1,5 экв.) при 0 С порциями добавляли к раствору соединения 10b (1,45 г, 3,9 ммоль) и CBr4 (1,62 г, 1,25 экв.) в ДХМ (15 мл). Смесь перемешивали в течение 1 ч, концентрировали в вакууме и добавляли Et2O. Полученные твердые вещества удаляли фильтрованием и фильтрат концентрировали в вакууме. Остаток очищали с помощью хроматографии на SiO2, элюируя смесью EtOAc/гексан в градиентном режиме (от 0 до 30% EtOAc), и получали 1,62 г (95%) соединения 10 с. Стадия 3. Тимин (0,09 г, 3 экв.), ГМДС (1,1,1,3,3,3-гексаметилдисилазан) (0,3 мл) и хлортриметилсилан (2 капли) нагревали при 120 С в течение 2 ч. Реакционную смесь охлаждали и добавляли ДХЭ(1,0 мл), соединение 10 с (100 мг, 0,23 ммоль) и йод (1 кусочек). Реакционную смесь перемешивали при 78 С в течение 16 ч. Реакционную смесь охлаждали до температуры окружающей среды и затем органические растворители выпаривали. Полученное неочищенное вещество растворяли в ДХМ и органический слой последовательно промывали водой и рассолом, сушили (MgSO4), фильтровали и выпаривали. Продукт очищали с помощью хроматографии на SiO2, элюируя смесью МеОН/ДХМ в градиентном режиме(8,9 г, 64,28 ммоль). Смесь нагревали в атмосфере аргона при 120 С (масляная баня) в течение 5,5 ч. Реакционную смесь охлаждали до КТ и добавляли воду (150 мл). Смесь экстрагировали с помощью EtOAc(150 мл) и водную фазу подвергали обратной экстракции с помощью EtOAc (2100 мл). Фазу, содержащую EtOAc, сушили (MgSO4), фильтровали и концентрировали в вакууме и получали 11,1 г (чистота 75%) соединения В-3a в виде белого кристаллического твердого вещества. Стадия 2. К раствору соединения В-3a (11,1 г, чистота 75%, 28 ммоль) в CCl4 (100 мл) добавлялиNBS (5,4 г, 30 ммоль), затем AIBN (450 мг, 2,74 ммоль). Смесь нагревали при температуре немного ниже температуры кипения в течение 5 ч. Повторно добавляли NBS (2,7 г) и AIBN (200 мг) и нагревание продолжали в течение еще 5 ч. Вещество охлаждали до температуры окружающей среды и фильтровали для удаления выпавшего в осадок сукцинимида. Фильтрат концентрировали и остаток переносили в EtOAc(100 мл) и встряхивали с рассолом (100 мл). Фазу, содержащую EtOAc, собирали и водную фазу подвергали обратной экстракции с помощью EtOAc (280 мл). Объединенные органические экстракты сушилиSiO2, элюируя смесью EtOAc/гексан в градиентном режиме (1,5-8% EtOAc), и получали 6,8 г (65%) соединения В-3b в виде белого кристаллического твердого вещества. Стадия 3. К суспензии тимина (353 мг) в гексаметилдисилазане (1,2 мл) добавляли триметилсилилхлорид (4 капли) и смесь нагревали при 120 С в течение 1 ч. К этому раствору добавляли соединение В 3b (300 мг, 0,8 ммоль), затем ДХЭ (5 мл) и затем йод (1 кусочек, каталитическое количество) и смесь нагревали при 80 С в течение ночи. Смесь охлаждали до КТ и затем концентрировали в вакууме. Остаток подвергали распределению между ДХМ (40 мл) и водой (40 мл). Фазы разделяли и водную фазу подвергали обратной экстракции с помощью ДХМ (40 мл). Объединенные органические экстракты сушили(MgSO4), фильтровали и выпаривали. Продукт очищали с помощью хроматографии на SiO2, элюируя смесью EtOAc/гексан в градиентном режиме (от 50 до 85% EtOAc), и получали 0,090 г (27%) соединенияI-17 в виде белого твердого вещества. Пример 5. 3-[6-Бром-3-(2,4-диоксоимидазолидин-1-илметил)-2-фторфенокси]-5-хлорбензонитрил (I-20, схема С). Стадия 1. Бензиловый эфир глицина (925 мг, 1,0 экв.) растворяли в ДХЭ (25 мл) и последовательно добавляли соединение А-2b (2 г, 5,6 ммоль) и NaBH(ОАс)3 (1,66 г, 1,4 экв.). После перемешивания смеси в течение ночи реакцию останавливали насыщенным раствором Na2CO3 и смесь экстрагировали с помощью Et2O. Затем органические слои промывали рассолом, сушили над (MgSO4) и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2, элюируя смесьюEtOAc/гексан в градиентном режиме (от 20 до 30% EtOAc), и получали 1,60 г (57%) соединения С-1 а. Стадия 2. Триметилсилилизоционат (336 мкл, 2,5 экв.) и ДМАП (12 мг, 0,10 экв.) добавляли к раствору соединения С-1 а (510 мг, 1,01 ммоль) и ТГФ (5 мл). Полученный раствор нагревали при 50 С в течение 20 ч, охлаждали до КТ и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2, элюируя смесью EtOAc/гексан в градиентном режиме (от 33 до 66%EtOAc), и получали 0,280 г (51%) соединения С-1b. Стадия 3. NaH (20 мг, 1,1 экв., 60% в минеральном масле) при 0 С добавляли к раствору соединения С-1b (250 мг, 0,48 ммоль) в ДМФ (2,5 мл). Реакционную смесь перемешивали при КТ в течение 2 ч, затем реакционную смесь выливали в насыщенный раствор NH4Cl (20 мл) и экстрагировали с помощью EtOAc. Затем органические слои промывали рассолом, сушили (MgSO4) и концентрировали в вакууме и получали 0,190 г (95%) соединения I-20. 3-[6-Бром-2-фтор-3-R)-5-метил-2,4-диоксоимидазолидин-1-илметил)фенокси]-5-хлорбензонитрил(I-21) получали аналогичным образом, но на стадии 1 вместо бензилового эфира глицина использовали бензиловый эфир (L)-аланина. Пример 6. 5-[6-Бром-3-(2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)-2-фторфенокси]изофталонитрил (I-11) Крезолцианат. В реакторе под слой H2O (350 мл) помещали бром (100 мл; 1,06 экв.) и через кожух пропускали хладагент. Для охлаждения использовали баню из воды со льдом. В отдельном сосуде готовили раствор NaCN (100 г, 1,11 экв.) в H2O (350 мл) и этот раствор добавляли к смеси бром/вода с такой скоростью, чтобы температура поддерживалась 30 С. Полученную взвесь бромциана добавляли к раствору ортокрезола (209 г, 1,00 экв.) в толуоле (900 мл). Двухфазную смесь интенсивно перемешивали и охлаждали до температуры ниже 10 С. Добавляли ТЭА (270 мл, 0,98 экв.), поддерживая температуру 10 С. Перемешивание прекращали и водную фазу удаляли и заменяли гептаном (540 мл). Органическую фазу последовательно промывали разбавленным раствором NaOH (1,20 экв.), водой, 2 М растворомHCl (0,4 экв.), водой, насыщенным раствором NaHCO3 и водой, поддерживая температуру 15 С. Раствор, содержащий гептан, сушили путем непродолжительной перегонки в вакууме (температура 35 С) и анализировали по Карлу Фишеру. Раствор, содержащий органическую фазу, хранили до дальнейшего- 19016386 использования. Стадия 1. В дегазированный реактор помещали раствор изо-PrMgCl (1,14 экв., 2 М раствор в ТГФ) в ТГФ и в реактор закачивали соединение 11 а (495,2 г, 1,352 моль; CASRN 136386-79-3), поддерживая температуру ниже 65 С с помощью водяной бани. После уменьшения выделения тепла реакционную смесь перемешивали при КТ до завершения металлирования (аликвоты отбирали, нейтрализовывали разбавленной H2SO4 и анализировали с помощью газовой хроматографии). Полученный раствор, содержащий арильный реагент Гриньяра, добавляли к раствору крезолцианата (см. выше; CASRN 1123-89-3) в гептане, поддерживая температуру реакционной смеси ниже 10 С. За ходом реакции следили путем анализа соотношения крезол/цианат в отобранных аликвотах, нейтрализовыванных разбавленной H2SO4. После израсходования цианата реакционную смесь добавляли к разбавленному раствору H2SO4 (86,5 г H2SO4 и 2,15 л H2O). Водный слой отделяли и оставшуюся органическую фазу разбавляли гептаном и последовательно промывали охлажденным льдом водным раствором NaOH (320 г 50% раствора NaOH и 1 кг льда),водой, насыщенным раствором NH4Cl и водой. Раствор сушили путем азеотропной перегонки и продукт очищали с помощью перегонки в вакууме и получали 395,7 г (93,7%) соединения 11b с примесью 3-6% 3-(трет-бутилдиметилсилокси)бромбензола. Стадии 2-4. В реактор помещали раствор соединения 11b (36 кг) и смесь толуол/гептан (65 кг) и раствор охлаждали до температуры ниже -50 С путем непосредственного впрыскивания жидкого N2 под поверхность раствора. Добавляли раствор изопропилмагнийхлорида (70 кг, 2,0 М раствор в ТГФ) с такой скоростью, чтобы температура реакционной смеси оставалась ниже -20 С (добавляли количество жидкого N2, необходимое для поддержания требующейся температуры). Для добавления требовалось примерно 50 мин. Через кожух реактора пропускали охлаждающий (-20 С) раствор и полученную смесь перемешивали при -20 С в течение по меньшей мере 1 ч. За ходом металлирования следили путем анализа отобранных и нейтрализованных разбавленной H2SO4 аликвот с помощью ВЭЖХ. ДМФ (примерно 30 кг) охлаждали до -10 С и переносили в реакционную смесь с такой скоростью, чтобы температура реакционной смеси в ходе переноса оставалась ниже 0 С. Реакционную смесь медленно нагревали 20 С, отбирали аликвоты, нейтрализовывали и анализировали с помощью ВЭЖХ. Реакционную смесь повторно охлаждали до 0 С и добавляли раствор 8,2 кг H2SO4 и 90 л H2O, поддерживая температуру реакционной смеси ниже 10 С. В реакционный сосуд помещали МТБЭ (50 кг) и сосуд встряхивали в течение по меньшей мере 15 мин. Фазы разделяли и водную фазу удаляли из реакционного сосуда. Оставшийся органический раствор повторно промывали с помощью H2O (110 л) и водную фазу отбрасывали. К реакционному сосуду присоединяли холодильник, охлажденный до 5 С, и ловушку ДинаШтарка, которую можно было переключать от режима кипячения с обратным холодильником в режим отгонки. Сосуд продували с помощью N2 и последовательно добавляли p-TsOH (0,5 кг), этиленгликоль(22 кг) и этиленгликольдиацетат (22 кг). ТГФ и МТБЭ удаляли путем отгонки (температура кожуха составляла от 80 до 95 С). После завершения отгонки ловушку Дина-Штарка переключали в режим кипячения с обратным холодильником и температуру кожуха повышали примерно до 100 С и этиленгликоль и воду удаляли путем азеотропной перегонки. При необходимости дополнительно добавляли толуол. Азеотропную отгонку воды продолжали до тех пор, пока ВЭЖХ не показывала, что содержание альдегида составляло менее 1%. Реакционную смесь охлаждали до 25 С и добавляли насыщенный растворNaHCO3 (25 кг) и воду (75 л), раствор перемешивали, фазы разделяли и водный раствор удаляли. Оставшуюся органическую фазу промывали с помощью H2O (100 л). Реакционный сосуд оснащали всем необходимым для перегонки и растворители удаляли сначала при атмосферном давлении, затем в вакууме при температуре кожуха 60 С. После того как в смеси оставались только толуол и соединение R-3a, реакционную смесь охлаждали до 25C и добавляли ДМЭ (диметиловый эфир) (70 кг). Раствор охлаждали до температуры, равной от(поддерживая температуру реакционной смеси менее -10 С). Из реакционной смеси отбирали аликвотные пробы и после завершения десилилирования реакционную смесь разбавляли с помощью H2O (80 л),охлаждали до менее 0 С и значение рН реакционной смеси доводили до 6-7 холодным 6,0 М растворомH2SO4 (13,2 кг концентрированной H2SO4 и 22 л H2O). Смесь подвергали распределению с МТБЭ(130 кг). Водный слой отделяли и подвергали обратной экстракции с помощью МТБЭ. Объединенные органические экстракты промывали с помощью H2O, водный слой отделяли и летучие растворители отгоняли до получения объема реакционной смеси, равного примерно 50-70 л. Оставшуюся органическую фазу разбавляли гептаном (20 кг) и полученный выпавший в осадок фенол отфильтровывали и сушили на нутч-фильтре и получали соединение 15b. Стадия 5. Раствор соединения 15b (6,0 г, 31,38 ммоль), K2CO3 (4,76 г, 34,52 ммоль) и ДМА (48 мл) перемешивали в течение 5 мин. К раствору добавляли 1,4-дибром-2,3-дифторбензол (85,33 г,0,3138 моль) и раствор нагревали при 125 С в течение 55 мин. Анализ с помощью ВЭЖХ показывал полное израсходование исходных веществ. Реакционную смесь разбавляли с помощью H2O (73 мл), как следует перемешивали и нижний органический слой отделяли. Органическую фазу разбавляли с помощью H2O (900 мл) и затем избыток дибромдифторбензола удаляли путем перегонки с паром. Оставшийся- 20016386 раствор экстрагировали с помощью ДХМ (50 мл) и органическую фазу отделяли и разбавляли с помощью МеОН (115 мл). Колбу оснащали всем необходимым для перегонки и растворители отгоняли до тех пор, пока температура не сохранялась равной 65 С в течение 10 мин. Реакционную смесь медленно охлаждали до 6 С и полученное твердое вещество отфильтровывали и дважды промывали с помощью МеОН. Белое твердое вещество сушили в вакууме и получали 9,7 г соединения 12 а. Стадия 6. Изо-PrMgCl (15,6 мл, 1,4 экв.) по каплям добавляли к раствору соединения R-4 (10 г,22,6 ммоль) и толуола (140 мл), охлажденному до -78 С. Реакционную смесь перемешивали при -78 С в течение 4 ч, быстро нагревали до -20 С и затем повторно охлаждали до -78 С. К реакционной смеси добавляли ДМФ (3,4 мл) и реакционную смесь нагревали до КТ, реакцию останавливали с помощью NH4Cl и смесь экстрагировали с помощью EtOAc. Неочищенный продукт очищали с помощью хроматографии на SiO2, элюируя смесью 25% EtOAc/гексаны, и получали 5,93 г (68%) соединения 12b. Стадия 7. NaBH4 (1,14 г, 2 экв.) добавляли к раствору соединения 12b (5,93 г, 15,1 ммоль) в смеси ТГФ (25 мл) и EtOH (25 мл). Реакционную смесь перемешивали при КТ в течение 2 ч, затем выдерживали при 0 С в течение ночи. Реакцию останавливали с помощью H2O, смесь экстрагировали с помощьюEtOAc, сушили (MgSO4) и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2 (45% EtOAc/гексаны) и получали 5,4 г (91%) соединения 12 с в виде прозрачного маслянистого вспененного твердого вещества. Стадия 8. Водный раствор TsOH (0,14 г в 6 мл H2O, 0,06 экв.) добавляли к раствору соединения 12 с(5,4 г, 13,7 ммоль) в MeCN (20 мл) и H2O (20 мл). Смесь нагревали при 70 С в течение 2 ч, затем перемешивали при КТ в течение ночи. Смесь экстрагировали с помощью EtOAc и объединенные органические экстракты промывали раствором NaHCO3, рассолом, сушили (MgSO4) и концентрировали в вакууме и получали 4,1 г (87%) соединения 14 а. Стадия 9. Гидроксиламингидрохлорид (2,1 г, 1,05 экв.) тремя порциями добавляли к растворуNaHCO3 (2,55 г, 1,05 экв.) в H2O (168 мл). Добавляли раствор соединения 14 а (10,12 г, 28,9 ммоль) в ТГФ(168 мл) и реакционную смесь перемешивали при КТ. После завершения реакции (примерно 3 ч) фазы разделяли, водный слой промывали раствором NH4Cl, разбавленной HCl и экстрагировали с помощьюEtOAc. Объединенные органические слои сушили (MgSO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали с помощью хроматографии на SiO2, элюируя смесью EtOAc/гексаны, и получали 8,62 г (82%) соединения 14b в виде масла, которое медленно затвердевало. Стадия 10. АТФК (ангидрид трифторуксусной кислоты) (6,5 мл, 2 экв.) добавляли к раствору соединения 14b (8,62 г, 24 ммоль) в смеси пиридина (11,5 мл, 6 экв.) и диоксана (57 мл), охлажденному до 0 С. Реакционную смесь нагревали при 65 С в течение нескольких часов, затем охлаждали до КТ и перемешивали в течение ночи. Темно-желтую смесь разбавляли с помощью ДХМ и промывали водой и разбавленной HCl. Органический слой сушили (MgSO4), фильтровали и концентрировали в вакууме и получали желтое масло, которое очищали с помощью хроматографии на SiO2, элюируя смесью 40%EtOAc/гексаны, и получали смесь спирта и соответствующего трифторацетата (5,91 г). Эту смесь растворяли в ТГФ и при 0 С по каплям добавляли раствор LiOH (840 мг, примерно 1,5 экв.) в H2O. Смесь перемешивали при 0 С в течение 1 ч, реакцию останавливали 1 н. раствором HCl и смесь экстрагировали с помощью EtOAc. Объединенные органические слои сушили (MgSO4), фильтровали и концентрировали и получали 4,9 г (59%) соединения 14 с в виде белого твердого вещества, немного загрязненного исходным эфиром. Стадия 11. Раствор PBr3 (15 мл 1,0 М раствора в ДХМ, 1,1 экв.) добавляли к раствору соединения 14 с (4,81 г, 13,9 ммоль) в ДХМ (23 мл). Раствор перемешивали при КТ в течение 2 ч. Реакцию останавливали с помощью NaHCO3, смесь экстрагировали с помощью ДХМ, сушили (MgSO4), фильтровали и концентрировали и получали желтое масло. Продукт очищали с помощью хроматографии на SiO2, элюируя смесью 20% EtOAc/гексаны, и получали 1,9 г соединения 16 в виде белого твердого вещества. 5-[6-Бром-3-(2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)-2-фторфенокси]изофталонитрил получали из соединения 16 и урацила по методике, описанной на стадии 5 примера 1. Пример 7. 3-[6-Бром-2-фтор-3-(3-метил-2,5-диоксоимидазолидин-4-илметил)фенокси]-5-хлорбензонитрил (I-32). Метиловый эфир Boc-N-саркозина (18) получали путем обработки раствора Boc-Sar-OH (1,0 г,5,3 ммоль) в смеси 1:1 ДХМ/МеОН (25 мл), охлажденного до 0 С, порциями диазометана (2 М раствор,избыток) до образования светло-желтой реакционной смеси. Реакционную смесь выдерживали при 0 С в течение 30 мин и затем добавляли НОАс (2 капли). Органические растворители удаляли и получали 1,0 г(90%) соединения 18. Стадия 1. Раствор диизопропиламина (0,14 мл, 1,2 экв.) в ТГФ охлаждали до -78 С и затем добавляли н-бутиллитий (1 экв.). Реакционную смесь нагревали до 0 С и затем перемешивали в течение 15 мин. К полученному раствору добавляли соединение 18 (0,2 г, 1,2 экв.) и полученный раствор перемешивали в течение 30 мин. Раствор енолята охлаждали до -78 С и добавляли раствор соединения D-1a (Ar=3-хлор 5-цианофенил, R4=Br, 0,35 г, 0,833 ммоль) в ТГФ. Реакционную смесь нагревали до КТ и перемешивали в течение 4 ч. Реакционную смесь выливали в водный раствор NH4Cl и экстрагировали с помощью EtOAc. Органический слой промывали с помощью H2O, затем сушили (Na2SO4) и очищали с помощью хромато- 21016386 графии на SiO2 и получали 0,2 г (44%) соединения D-1b (Ar=3-хлор-5-цианофенил, R4=Br). Стадия 2. Раствор соединения D-1b, ТФК (1 мл) и ДХМ перемешивали при 0 С в течение 5 ч. Органические растворители удаляли в вакууме и затем остаток повторно растворяли в ДХМ. Органический слой промывали насыщенным раствором Na2CO3 и затем рассолом. Органический слой сушили (Na2SO4) и растворители выпаривали и получали 0,15 г (92%) соединения D-1c. Стадия 3. К раствору соединения D-1c (0,1 г, 0,226 ммоль) и ДМАП (4 мг, 0,15 экв.) в ТГФ (1 мл) добавляли TMSNCO (примерно 0,9 мл, 2,5 экв.) и реакционную смесь нагревали при 50 С в течение 18 ч. Летучие вещества удаляли в вакууме и остаток растворяли в ДХМ и МеОН. Органический слой промывали насыщенным раствором Na2CO3 и затем рассолом. Органический слой сушили (Na2SO4) и органические растворители выпаривали. Неочищенный продукт очищали с помощью препаративной ТСХ и получали 0,030 г (30%) соединения I-32 (D-2; Ar=3-хлор-5-цианофенил, R4=Br). Пример 8. 3-[6-Бром-2-фтор-3-(1-метил-2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-илметил)фенокси]-5 хлорбензонитрил (I-31, схема D). Стадия 4. К суспензии NaH (0,14 г, 3 экв.) и ДМФ (3 мл), охлажденной до 0 С, добавляли третбутилэтилмалонат. Реакционную смесь нагревали до КТ и перемешивали в течение 30 мин. К полученному раствору, охлажденному до 0 С, добавляли раствор соединения D-1a (Ar=3-хлор-5-цианофенил,R4=Br, 0,5 г, 1,19 ммоль) в ДМФ (2 мл). Реакционную смесь перемешивали при КТ в течение 1,5 ч, затем разбавляли этилацетатом и последовательно промывали насыщенным раствором NH4Cl, водой и рассолом. Органический слой сушили (Na2SO4), фильтровали и выпаривали и получали 0,62 г (99%) соединения D-1d. Стадия 5. К раствору соединения D-1d в ДХМ (2,5 мл), охлажденному до 0 С, добавляли ТФК(2 мл) и реакционную смесь перемешивали при КТ в течение 6 ч. Летучие вещества удаляли в вакууме и остаток разбавляли с помощью ДХМ и органический слой последовательно промывали водным раствором NaHCO3, водой и рассолом. Водный промывочный раствор подкисляли с помощью HCl и повторно экстрагировали с помощью ДХМ. Объединенные органические слои промывали водой, сушили (Na2SO4),фильтровали и выпаривали. Неочищенный остаток растворяли в ДМФ (2 мл) и H2O (0,07 мл) и нагревали в микроволновой печи при 160 С в течение 20 мин. Реакционную смесь разбавляли с помощью EtOAc и последовательно промывали водой и рассолом. Органический слой сушили (Na2SO4), фильтровали и выпаривали. Неочищенный продукт очищали с помощью хроматографии на SiO2, элюируя смесью(1 мл) по каплям добавляли к раствору трет-бутоксида калия в Et2O (2,5 мл). Реакционную смесь нагревали до КТ и перемешивали в течение 18 ч. Летучие вещества удаляли в вакууме и остаток растворяли в ИПС (5 мл). Добавляли тиомочевину и реакционную смесь нагревали при 80 С в течение 4 ч. Летучие вещества удаляли в вакууме и остаток промывали эфиром. Остаток растворяли в H2O и раствор подкисляли с помощью НОАс. Полученный осадок отфильтровывали и промывали водой. Остаток растворяли вEt2O и органический слой промывали водой и рассолом. Органические растворители выпаривали и получали 0,1 г (50%) соединения D-3a. Стадия 7. К раствору соединения D-3a (0,1 г, 0,21 ммоль) и НОАс добавляли 20% водный раствор хлоруксусной кислоты (1 мл). Реакционную смесь нагревали при 100 С в течение 6 ч. Реакционную смесь охлаждали до 0 С и добавляли H2O (2 мл) и полученную смесь перемешивали. Полученный осадок отфильтровывали и промывали водой и с помощью Et2O и получали 0,030 г (31%) соединения D-3b. Стадия 8. N,О-бис-(Триметилсилил)ацетамид (0,1 мл, 6 экв.) при КТ медленно добавляли к соединению D-3b (30 мг, 0,067 ммоль) в ДХМ (0,9 мл) и перемешивали в течение 2 ч. Добавляли метилйодид(0,145 мл, 35 экв.) и реакционную смесь нагревали при 28 С в течение 18 ч. Летучие вещества удаляли в вакууме и органический остаток растворяли в EtOAc. Органический слой промывали водой и затем рассолом, сушили (Na2SO4), фильтровали и выпаривали. Неочищенный продукт очищали с помощью хроматографии на SiO2, элюируя смесью МеОН/ДХМ в градиентном режиме (0-4% МеОН), и получали 0,015 г- 22016386 Стадия 1. Уксусный ангидрид (0,93 г, 1,5 экв.) добавляли к раствору соединения 12 с (2,4 г,6,1 ммоль) и ТЭА (0,93 г, 1,5 экв.) в MeCN (10 мл). Раствор перемешивали при КТ в течение 1 ч, разбавляли с помощью EtOAc, промывали раствором NaHCO3, сушили (Na2SO4), фильтровали и концентрировали и получали 2,2 г (82%) соединения 18 в виде прозрачного масла. Стадия 2. Раствор p-TsOH (60 мг, 0,6 экв.) в H2O (6 мл) добавляли к раствору соединения 18 (2,2 г,5,0 ммоль) в MeCN (8 мл). Полученный раствор нагревали при 70 С в течение 5 ч. Затем раствор охлаждали до КТ, разбавляли с помощью EtOAc и промывали насыщенным раствором NaHCO3 и рассолом. Органический слой сушили (Na2SO4) и концентрировали. Неочищенный продукт очищали с помощью хроматографии на SiO2, элюируя смесью EtOAc/гексан, и получали 1,3 г (62%) соединения 20 в виде прозрачного масла. Стадия 3. К раствору соединения 20 (0,12 г, 0,3 ммоль) в ДХМ (1 мл), охлажденному до 0 С, добавляли каплю EtOH, затем ДАТС (диалкиламинотрифторид серы) (0,92 г, 1,2 экв.). Раствор нагревали до КТ и выдерживали при этой же температуре в течение ночи. Затем смесь осторожно выливали на лед. Добавляли насыщенный раствор NaHCO3 и смесь экстрагировали с помощью ДХМ, сушили (Na2SO4),фильтровали и концентрировали. Этот продукт растворяли в ТГФ (10 мл) и добавляли 2 М раствор LiOH(1,75 мл) в H2O и смесь перемешивали в течение 3 ч. Реакцию останавливали 1 н. раствором HCl, смесь экстрагировали с помощью EtOAc, сушили (Na2SO4), фильтровали и концентрировали. Очистка остатка с помощью хроматографии на SiO2 давала 0,074 г (67%) соединения 22 а. Стадия 4. Бензиловый спирт 22 а можно превратить в соответствующий бензилбромид 22b так, как это описано на стадии 4 примера 1. Стадия 5. Соединение I-27 получали из соединения 22b так, как это описано на стадии 5 примера 1,но используя тимин вместо урацила. Пример 10. Анализ обратной транскриптазы ВИЧ-1. Активность РНК-зависимой ДНК полимеразы определяли с использованием биотинилированного праймерного олигонуклеотида и тритированного dNTP субстрата. Свежесинтезированную ДНК количественно исследовали путем захвата биотинилированных праймерных молекул на покрытых стрептавидином гранулах для сцинтилляционно-проксимального анализа (СПА) (Amersham). Последовательности для субстрата для исследования полимеразы были следующими: содержащий 18 нуклеотидов ДНК праймер, 5'-биотин/GTC CCT GTT CGG GCG ССА-3'; содержащая 47 нуклеотидов РНК матрица,5'-GGG UCU CUC UGG UUA GAC CAC UCU AGC AGU GGC GCC CGA АСА GGG AC-3'. Биотинилированный ДНК праймер получали у фирмы Integrated DNA Technologies Inc. и матрицу РНК синтезировала фирма Dharmacon. Смесь для исследования ДНК полимеразы (конечный объем 50 мкл) содержала 32 нМ биотинилированного ДНК праймера, 64 нМ РНК субстрата, dGTP, dCTP, dTTP (всех по 5 мкМ),103 нМ [3H]-dATP (удельная активность = 29 мКи/ммоль) в 45 мМ Tris-HCl [Tris = трис(гидроксиметиламинометан)], рН 8,0, 45 мМ NaCl, 2,7 мМ Mg(CH3COO)2, 0,045% Triton X-100 мас./об.,0,9 мМ ЭДТК (этилендиаминтетрауксусная кислота). Реакционные смеси содержали 5 мкл серийных разведений соединения в 100% ДМСО для определения IC50, и конечные концентрации ДМСО составляли 10%. Реакции инициировали путем прибавления 30 мкл фермента ВИЧ-КТ (конечные концентрации 1-3 нМ). Концентрации белка подбирали так, чтобы обеспечить равномерное образование продукта в течение не менее 30 мин инкубации. После инкубации при 30 С в течение 30 мин реакцию останавливали путем прибавления 50 мкл 200 мМ ЭДТК (рН 8,0) и 2 гранул мг/мл SA-PVT СПАAmersham, RPNQ0009, восстановлены в 20 мМ Tris-HCl, рН 8,0, 100 мМ ЭДТК и 1% бычьего сывороточного альбумина. Затем гранулам давали осаждаться в течение ночи и сигналы СПА определяли с помощью верхнего счетчика для 96-луночных планшетов NXT (Packard). Значения IC50 получали с помощью сигмоидального регрессионного анализа с использованием программного обеспечения GraphPad, и они приведены в табл. I. Пример 11. Методика исследования противовирусной активности. Активность по отношению к ВИЧ исследовали с помощью модификации методики, предложеннойPauwels et al. (Pauwels et al., 1988, J. Virol. Methods. 20:309-321). Методика основана на способности соединений защищать инфицированные посредством ВИЧ Т-лимфобластные клетки (клетки МТ 4) от гибели, опосредуемой инфекцией. Конечной точкой исследования считали концентрацию соединения, при которой жизнеспособность клеток культуры сохранялась равной 50% (50% ингибирующая концентрация, IC50). Жизнеспособность клеток культуры определяли по потреблению растворимого желтого 3-[4,5-диметилтиазол-2-ил]-2,5-дифенилтетразолийбромида (МТТ) и его восстановлению в пурпурную нерастворимую формазановую соль. После солюбилизации для определения количества формазанового продукта использовали спектрофотометрические методики. Клетки МТ 4 готовили в логарифмической фазе роста и всего получали 210 клеток, инфицированных штаммом НХВ 2 ВИЧ при множественности заражения, равной 0,0001 инфицирующих единиц вируса на клетку, при полном объеме, равном 200-500 мкл. Перед удалением вируса клетки инкубировали с вирусом в течение 1 ч при 37 С. Затем клетки промывали 0,01 М забуференным фосфатом физиологиче- 23016386 ским раствором при рН 7,2 и после этого повторно суспендировали в культуральной среде для инкубации в культуре с серийными разведениями исследуемых соединений. Использовали культуральную среду RPMI 1640 без прибавления фенолового красного, с прибавлением пенициллина, стрептомицина,L-глутамина и 10% фетальной телячьей сыворотки (GM10). Исследуемые соединения готовили в виде 2 мМ растворов в диметилсульфоксиде (ДМСО). Затем готовили по 4 реплики серийных 2-кратных разведения в GM10 и по 50 мкл помещали в 96-луночные планшеты при конечных концентрациях в диапазоне 625-1,22 нМ. Затем в каждую лунку прибавляли 50 мкл GM10 и 3,5104 инфицированных клеток. Также готовили контрольные культуры, не содержащие клеток (холостая проба), содержащие неинфицированные клетки (100% жизнеспособность; 4 реплики) и содержащие инфицированные клетки без соединения (полная опосредуемая вирусом гибель клеток; 4 реплики). Затем культуры в течение 5 дней инкубировали при 37 С в увлажненной атмосфере, содержащей 5% CO2 в воздухе. Свежий раствор, содержащий 5 мг/мл МТТ, готовили в 0,01 М забуференном фосфатом физиологическом растворе, рН 7,2 и 20 мкл прибавляли к каждой культуре. Культуры дополнительно инкубировали, как описано выше, в течение 2 ч. Затем их перемешивали путем втягивания пипеткой и выпускания из нее с 170 мкл Triton Х-100 в подкисленном изопропаноле (10% об./об. Triton Х-100 в смеси состава 1:250 концентрированной HCl с изопропанолом). После полной солюбилизации формазанового осадка путем дополнительного перемешивания поглощение (ОП - оптическая плотность) измеряли при длинах волн, равных 540 и 690 нм (значения при 690 нм использовали в качестве холостых значений для расхождений между данными для лунок). Затем выраженную в процентах степень защиты для каждой обработанной культуры рассчитывали по уравнению Значения IC50 можно было определить по зависимостям выраженной в процентах степени защиты от log10 концентрации лекарственного средства. По данным обоих исследований соединения формул I обладают активностью в диапазоне значенийIC50 от примерно 0,5 до примерно 10000 нМ или от 0,5 до примерно 5000 нМ, а предпочтительные соединения обладают активностью в диапазоне от примерно 0,5 до примерно 750 нМ, более предпочтительные примерно от 0,5 до 300 нМ и наиболее предпочтительные примерно от 0,5 до 50 нМ. Таблица II Пример 12. Фармацевтические композиции из соединений, предлагаемых в настоящем изобретении, предназначенные для доставки различными путями, готовили так, как это описано в этом примере. Композиция для перорального введения (А). Ингредиенты смешивают и дозируют в капсулы, содержащие примерно по 100 мг каждая; одна капсула содержит примерно полную суточную дозу. Композиция для перорального введения (В). Ингредиенты объединяют и гранулируют с использованием растворителя, такого как метанол. Затем композицию сушат и формуют в таблетки (содержащие примерно 20 мг активного соединения) с помощью соответствующей таблетирующей машины.- 24016386 Композиция для перорального введения (С). Ингредиенты смешивают с получением суспензии для перорального введения. Отличительные признаки, раскрытые в приведенном выше описании или в приведенной ниже формуле изобретения, выраженные в конкретных формах или с помощью средств для осуществления раскрытого назначения, или методика или способ для достижения раскрытого результата, если это целесообразно, могут по отдельности или в виде любой комбинации таких отличительных признаков быть использованы для осуществления настоящего изобретения в его различных формах. Приведенное выше изобретение описано довольно подробно для иллюстрации и примера с целью обеспечения ясности и понимания. Для специалиста в данной области техники должно быть очевидно,что в объеме прилагаемой формулы изобретения возможны изменения и модификации. Поэтому следует понимать, что приведенное выше описание является иллюстративным, а не ограничивающим. Поэтому объем настоящего изобретения следует определять не с учетом приведенного выше описания, а по прилагаемой формуле изобретения вместе со всем объемом эквивалентов, на которые распространяется такая формула изобретения. Патенты, опубликованные заявки и научная литература, цитированные в настоящем изобретении,входят в объем знаний специалистов в данной области техники и включены в него в качестве ссылки во всей своей полноте в такой степени, как если бы специально и по отдельности для каждого из них было указано о включении в качестве ссылки. Любое расхождение между любой ссылкой, цитированной в настоящем изобретении, и конкретными положениями, приведенными в настоящем описаниями, следует разрешать в пользу последнего. Аналогичным образом, любое расхождение между принятым в данной области техники определением термина или фразы и определением термина или фразы, специально указанным в настоящем описании, следует разрешать в пользу последнего. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы I(c) CHR2; или (i) R1 обозначает (II) и R2 обозначает водород, C1-C3-алкил, C1-C3-галогеналкил, C2-C6-алкенил или CH2ORc; и, в дополнение, если А обозначает или (a), или (b), то R2 также может обозначать галоген,NRaRb, CN или ORc; или (ii) R1 обозначает C1-C6-алкил и R2 обозначает (II);R3 обозначает водород или C1-C3-алкил;Rc и Rd независимо обозначают водород или C1-C3-алкил;Ar обозначает фенил, замещенный 1-3 группами, независимо выбранными из группы, включающей галоген, цианогруппу, C1-C6-галогеналкил, C1-C6-алкил, C1-C6-алкоксигруппу и C3-C6-циклоалкил;n является целым числом, равным от 1 до 3; или его фармацевтически приемлемые соли. 2. Соединение по п.1, в котором А обозначает (a); R1 обозначает (II); X1 и X2 обозначают О и n рав- 25016386 но 1.C1-C6-галогеналкил; R3 обозначает водород; R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил, необязательно замещенный 1-3 группами, независимо выбранными из группы, включающей цианогруппу, галоген и C1-C6-галогеналкил. 4. Соединение по п.3, в котором R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил,3,5-дицианофенил или 3-циано-5-дифторметилфенил. 5. Соединение по п.1, в котором А обозначает (а); R1 обозначает (II); X1 обозначает S; X2 обозначает 2 О; R обозначает водород, галоген, C1-C6-алкил, C1-C6-галогеналкил; R3 обозначает водород; R4 обозначает галоген или C1-C6-алкил; Ar обозначает фенил, необязательно замещенный 1-3 группами, независимо выбранными из группы, включающей цианогруппу, галоген и C1-C6-галогеналкил, и n равно 1. 6. Соединение по п.1, в котором А обозначает (a); R1 обозначает (II); X1 обозначает О; X2 обозначает NRd; Rd обозначает Н и n равно 1. 7. Соединение по п.6, в котором R2 обозначает водород, галоген, C1-C6-алкил, C1-C6-галогеналкил; 3R обозначает водород; R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил, необязательно замещенный 2 группами, независимо выбранными из группы, включающей цианогруппу, галоген иR обозначает водород; R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил, необязательно замещенный 2 группами, независимо выбранными из группы, включающей цианогруппу, галоген иC1-C6-галогеналкил. 11. Соединение по п.10, в котором R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил,3,5-дицианофенил или 3-циано-5-дифторметилфенил. 12. Соединение по п.1, в котором А обозначает (с); R1 обозначает (II); X1 и X2 обозначают О и n равно 1. 13. Соединение по п.12, в котором R4 обозначает галоген или C1-C6-алкил и Ar обозначает фенил,необязательно замещенный 2 группами, независимо выбранными из группы, включающей цианогруппу,галоген и C1-C6-галогеналкил. 14. Соединение по п.13, в котором R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил,3,5-дицианофенил или 3-циано-5-дифторметилфенил. 15. Соединение по п.1, в котором А обозначает (a); R2 обозначает (II); X1 и X2 обозначают О и R3 обозначает водород. 16. Соединение по п.15, в котором R1 обозначает метил или водород; R4 обозначает галоген илиC1-C6-алкил и Ar обозначает фенил, необязательно замещенный 2 группами, независимо выбранными из группы, включающей цианогруппу, галоген и C1-C6-галогеналкил. 17. Соединение по п.16, в котором R1 обозначает метил или водород; R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5-дифторметилфенил. 18. Соединение по п.1, в котором А обозначает (с); R2 обозначает (II) и X1 и X2 обозначают О. 19. Соединение по п.18, в котором R1 обозначает метил или водород; R4 обозначает галоген илиC1-C6-алкил и Ar обозначает фенил, необязательно замещенный 2 группами, независимо выбранными из группы, включающей цианогруппу, галоген и C1-C6-галогеналкил. 20. Соединение по п.19, в котором R1 обозначает метил или водород; R4 обозначает Br или Cl и Ar обозначает 3-хлор-5-цианофенил, 3,5-дицианофенил или 3-циано-5-дифторметилфенил. 21. Соединение по п.1, выбранное из группы, включающей 3-[6-бром-3-(2,4-диоксотетрагидропиримидин-1-илметил)-2-фторфенокси]-5-хлорбензонитрил; 3-[6-бром-2-фтор-3-(5-метил-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)фенокси]-5 хлорбензонитрил; 3-[6-бром-3-(2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)-2-фторфенокси]-5 хлорбензонитрил; 3-[6-бром-3-(5-хлор-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)-2-фторфенокси]-5 хлорбензонитрил; 3-[6-бром-3-(5-этил-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)-2-фторфенокси]-5 хлорбензонитрил; 3-[6-бром-2-фтор-3-(5-фтор-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)фенокси]-5 хлорбензонитрил; 3-[6-бром-3-(2,4-диоксо-5-трифторметил-3,4-дигидро-2 Н-пиримидин-1-илметил)-2-фторфенокси]-5 хлорбензонитрил; 5-[6-бром-2-фтор-3-(5-метил-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1 илметил)фенокси]изофталонитрил; 5-[6-бром-3-(5-хлор-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)-2- 26016386 фторфенокси]изофталонитрил; 5-[6-бром-3-(5-этил-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)-2 фторфенокси]изофталонитрил; 5-[6-бром-3-(2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)-2-фторфенокси]изофталонитрил; 3-[3-(4-амино-2-оксо-2 Н-пиримидин-1-илметил)-6-бром-2-фторфенокси]-5-хлорбензонитрил; 3-[3-(4-амино-5-метил-2-оксо-2 Н-пиримидин-1-илметил)-6-бром-2-фторфенокси]-5 хлорбензонитрил; 3-6-бром-2-фтор-3-[2-(5-метил-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-ил)этил]фенокси-5 хлорбензонитрил; 1-[4-бром-3-(3-хлор-5-цианофенокси)-2-фторбензил]-2,4-диоксо-1,2,3,4-тетрагидропиримидин-5 карбонитрил; 3-[6-бром-2-фтор-3-(6-метил-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)фенокси]-5 хлорбензонитрил; 3-хлор-5-[6-хлор-2-фтор-3-(5-метил-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1 илметил)фенокси]бензонитрил; 3-хлор-5-[6-хлор-3-(5-хлор-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)-2 фторфенокси]бензонитрил; 3-[6-бром-2-фтор-3-(5-метокси-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)фенокси]-5 хлорбензонитрил; 3-[6-бром-3-(2,4-диоксоимидазолидин-1-илметил)-2-фторфенокси]-5-хлорбензонитрил; 3-[6-бром-2-фтор-3-R)-5-метил-2,4-диоксоимидазолидин-1-илметил)фенокси]-5-хлорбензонитрил; 3-[3-(5-аллил-2,4-диоксоимидазолидин-1-илметил)-6-бром-2-фторфенокси]-5-хлорбензонитрил; 3-[6-бром-2-фтор-3-(5-метил-4-оксо-2-тиооксо-3,4-дигидро-2 Н-пиримидин-1-илметил)фенокси]-5 хлорбензонитрил; 3-[6-бром-2-фтор-3-(5-гидроксиметил-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)фенокси]5-хлорбензонитрил; 3-хлор-5-[6-этил-2-фтор-3-(5-метил-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1 илметил)фенокси]бензонитрил; 3-хлор-5-[3-(5-хлор-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)-6-этил-2 фторфенокси]бензонитрил; 3-[6-бром-2-фтор-3-(5-метил-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)фенокси]-5 дифторметилбензонитрил; 3-[6-бром-2-фтор-3-(4-оксо-2-тиооксоимидазолидин-1-илметил)фенокси]-5-хлорбензонитрил; 3-[6-бром-2-фтор-3-(5-метил-2,4-диоксотетрагидропиримидин-1-илметил)фенокси]-5 хлорбензонитрил; 3-[6-бром-2-фтор-3-(5-гидрокси-2,4-диоксо-3,4-дигидро-2 Н-пиримидин-1-илметил)фенокси]-5 хлорбензонитрил; 3-[6-бром-2-фтор-3-(1-метил-2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-илметил)фенокси]-5 хлорбензонитрил и 3-[6-бром-2-фтор-3-(3-метил-2,5-диоксоимидазолидин-4-илметил)фенокси]-5-хлорбензонитрил. 22. Применение соединения по любому из пп.1-21 для приготовления лекарственного средства,предназначенного для лечения инфицирования посредством ВИЧ-1 или предупреждения инфицирования посредством ВИЧ-1 или лечения СПИД или ПСП. 23. Фармацевтическая композиция, включающая терапевтически эффективное количество соединения по любому из пп.1-21 и по меньшей мере один носитель, инертный наполнитель или разбавитель.
МПК / Метки
МПК: A61K 31/4174, C07D 233/72, C07D 239/553, C07D 239/545, C07D 233/78, C07D 239/54, C07D 239/22, A61K 31/513, A61P 31/18
Метки: ингибиторы, транскриптазы, обратной, ненуклеозидные
Код ссылки
<a href="https://eas.patents.su/28-16386-nenukleozidnye-ingibitory-obratnojj-transkriptazy.html" rel="bookmark" title="База патентов Евразийского Союза">Ненуклеозидные ингибиторы обратной транскриптазы</a>
Ненуклеозидные ингибиторы обратной транскриптазы

Номер патента: 5598
Опубликовано: 28.04.2005
Автор: Симоно Брюно
МПК: A61K 31/55, C07D 471/14
Метки: ненуклеозидные, транскриптазы, ингибиторы, обратной
Формула / Реферат:
1. Соединение общей формулы I где R2 выбирают из группы, включающей H, F, Cl, C1-C4алкил, C3-C4циклоалкил и CF3; R4 обозначает H или Me; R5 обозначает H, Me или Et при условии, что R4 и R5 оба не обозначают Me, и если R4 обозначает Me, то R5 не обозначает Et; R11 обозначает Me, Et, циклопропил, пропил, изопропил или циклобутил и Q выбирают из группы, включающей или его фармацевтически приемлемая соль. 2. Соединение по п.1, где R11 обозначает...
Ненуклеозидные ингибиторы обратной транскриптазы

Номер патента: 9700
Опубликовано: 28.02.2008
Авторы: Сунд Кристьян, Роуэ Натали, Антонов Дмитрий, Сальберг Кристер, Янссон Катарина, Линдстрем Стефан
МПК: A61K 31/44, A61K 31/425, A61K 31/426...
Метки: ингибиторы, ненуклеозидные, транскриптазы, обратной
Формула / Реферат:
1. Соединение формулы Z где А представляет собой СН или N; R1 представляет собой заместитель у атома углерода в кольце, содержащем А, выбранном из -S(=O)pRa, где Ra представляет собой -C1-С4-алкил, -ORx, -NRxRx, -NHNRxRx, -NHNHC(=O)ORx, -NRxOH; -C(=O)-Rb, где Rb представляет собой -C1-С4-алкил, ORx, -NRxRx, -NHNRxRx, -NHC1-С3-алкил-C(=O)ORx; -NRxRc, где Rc представляет собой Н, C1-С4-алкил, -NRxRx; -C(=O)Rd, -CN, S(=O)pRx, где Rd представляет...
Ненуклеозидные ингибиторы обратной транскриптазы

Номер патента: 9478
Опубликовано: 28.02.2008
Авторы: Кох Йунг-Хио, Яо Наньхуа, Де Ла Роса Марта А., Нилар Шахул, Шоу Стефания, Чжан Чжицзюн, Гунич Эсмир, Хун Чжи, Ким Хонгву, Хэматейк Роберт, Жирарде Жан-Люк
МПК: A61K 31/4709, A61K 31/4196, C07D 233/84...
Метки: обратной, транскриптазы, ингибиторы, ненуклеозидные
Формула / Реферат:
1. Способ лечения пациента, инфицированного ВИЧ, включающий введение пациенту фармацевтической композиции, содержащей соединение в дозе, эффективной, чтобы снизить вирусную репродукцию, причем соединение имеет структуру в соответствии с формулой (А) или формулой (В) в которых R1 представляет необязательно замещенный C1-C10алкил, галоген или CF3, R2 представляет необязательно замещенный циклоалкил, необязательно замещенный арил, необязательно...
Ненуклеозидные ингибиторы обратной транскриптазы как антагонисты пролиферации и индукторы дифференцировки клеток

Номер патента: 7004
Опубликовано: 30.06.2006
Авторы: Паломбини Гильельмо, Нерви Клара, Спадафора Коррадо, Лавия Патриция, Лорендзини Родольфо Нелло, Маттеи Элизабетта
МПК: A61K 31/55, A61K 31/535, A61K 31/495...
Метки: пролиферации, ненуклеозидные, индукторы, обратной, антагонисты, транскриптазы, дифференцировки, клеток, ингибиторы
Формула / Реферат:
1. Применение по меньшей мере одного соединения-ингибитора обратной транскриптазы для получения фармацевтической композиции для противодействия утрате дифференцировки клеток и уменьшения скорости пролиферации клеток при раковых и не раковых патологиях, где вышеуказанное соединение выбрано из класса соединений 5,11-дигидро-6Н-дипиридо[3,2-b:2',3'-е][1,4]диазепина. 2. Применение по п.1, где соединение способно стимулировать дифференцировку клеток....
Ненуклезидные ингибиторы обратной транскриптазы

Номер патента: 9178
Опубликовано: 28.12.2007
Авторы: Маленфан Эрик, Ландри Серж, Йоаким Кристиан, Нод Жюли, Тхавонекхам Боункхам, Симоно Брюно, О'мира Джеффри
МПК: A61K 31/55, A61P 31/18, C07D 221/00...
Метки: ингибиторы, транскриптазы, ненуклезидные, обратной
Формула / Реферат:
1. Соединение, представленное формулой I в котором R2 выбирают из ряда, включающего Н, галоген; R4 обозначает Н или CH3; R5 обозначает Н или СН3; R11 обозначает Н, С1-С4алкил или С3-С4циклоалкил; А обозначает соединяющую цепь C1-С3алкила; В обозначает О; n обозначает 0 или 1; причем, когда n обозначает 0, кольцо С обозначает фенил, замещенный (C1-С6)алкилом; и Е выбирают из ряда, включающего: (I) CONR12R13, где R12 и R13, каждый независимо друг...
Предыдущий патент: Высокоэффективный нагреватель питательной воды
Следующий патент: 2-(замещенные амино)бензотиазолсульфонамидные ингибиторы вич-протеазы
Случайный патент: Система и способ для подбора персонала на временные медицинские вакансии