Ингибиторы гиразы и их применение для лечения бактериальной инфекции
Номер патента: 5680
Опубликовано: 28.04.2005
Авторы: Чарифсон Пол, Стамос Дин, Трюдо Мартэн, Ронкин Стивен, Бадиа Майкл, Грийо Анн-Лор





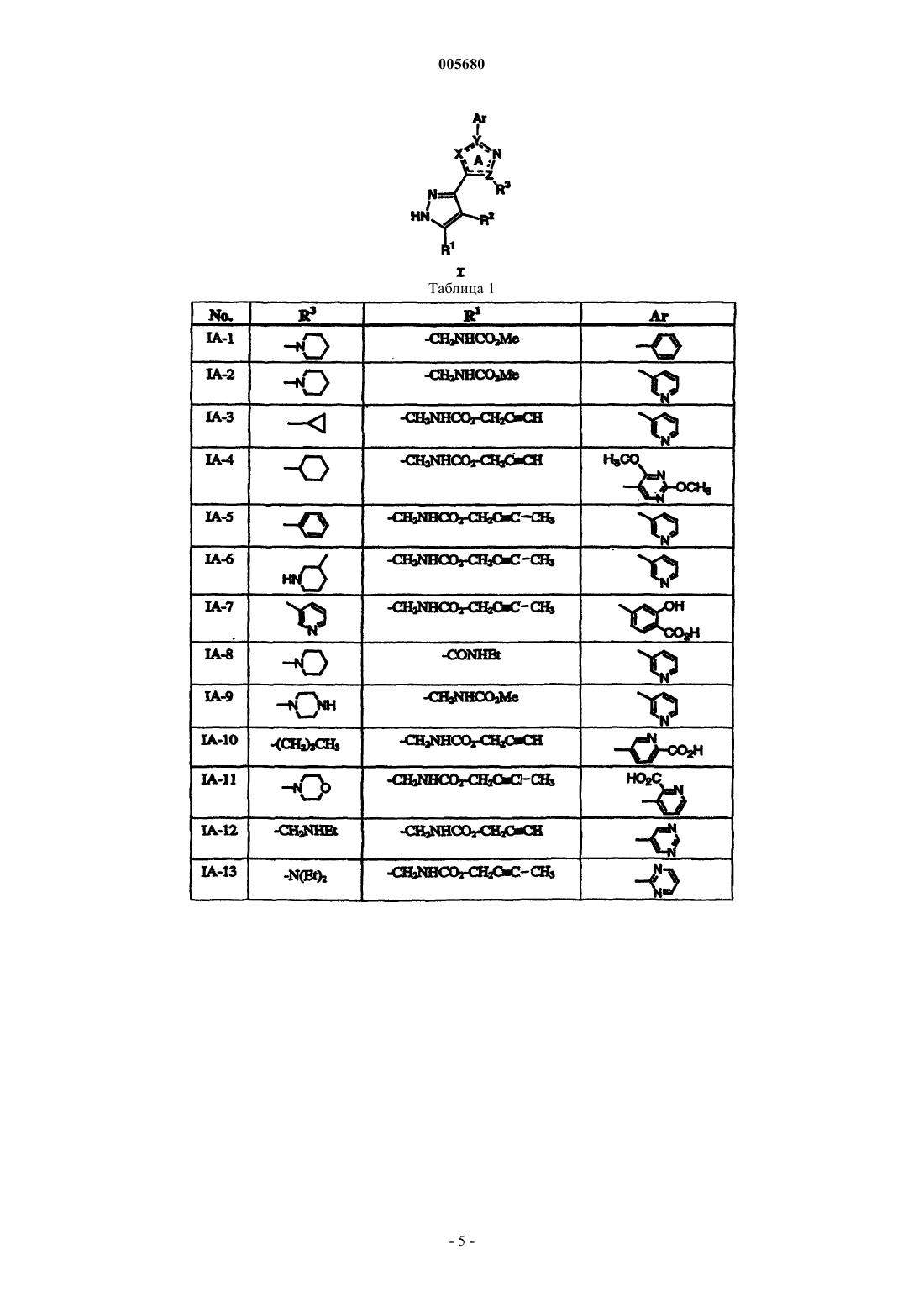
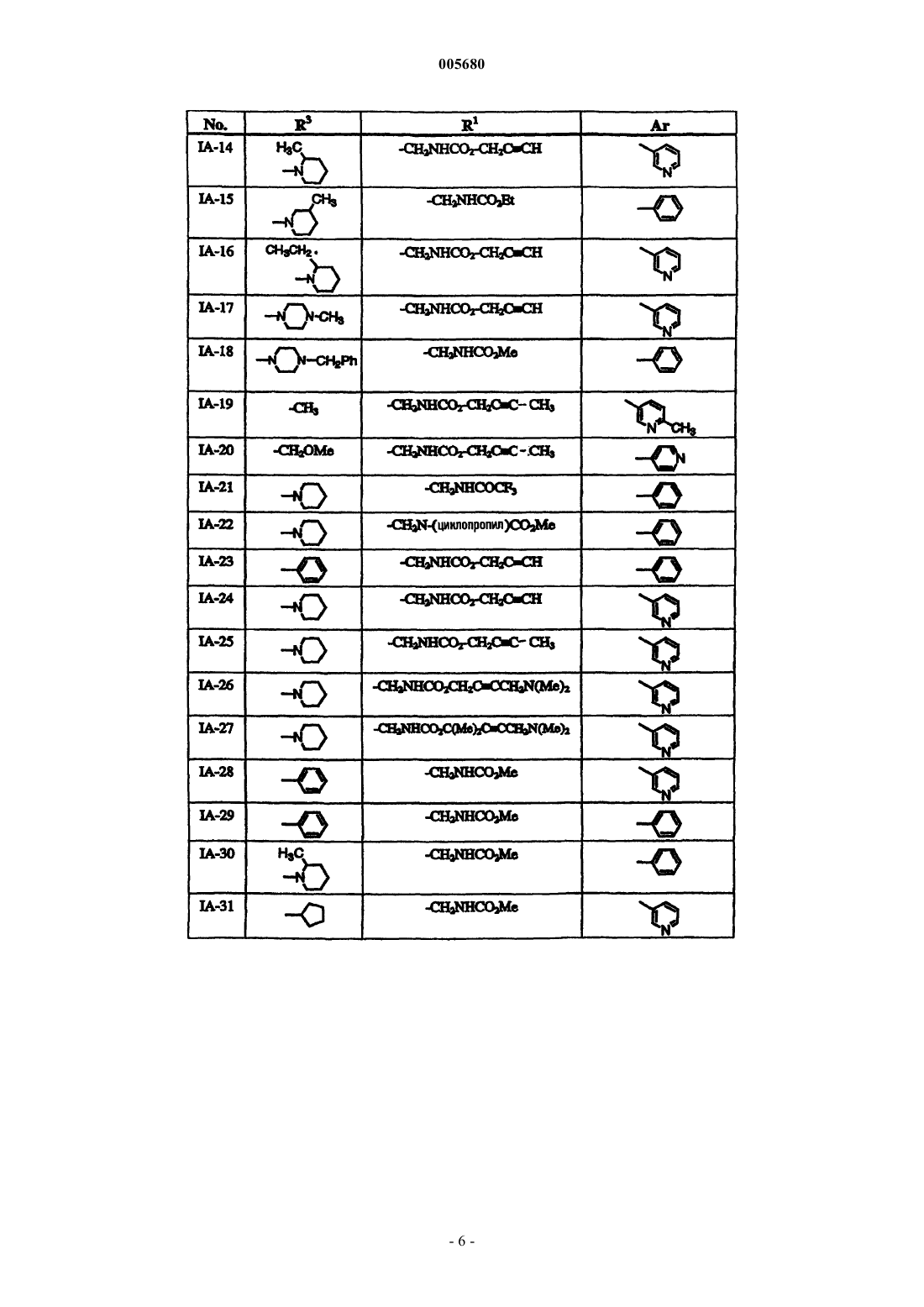
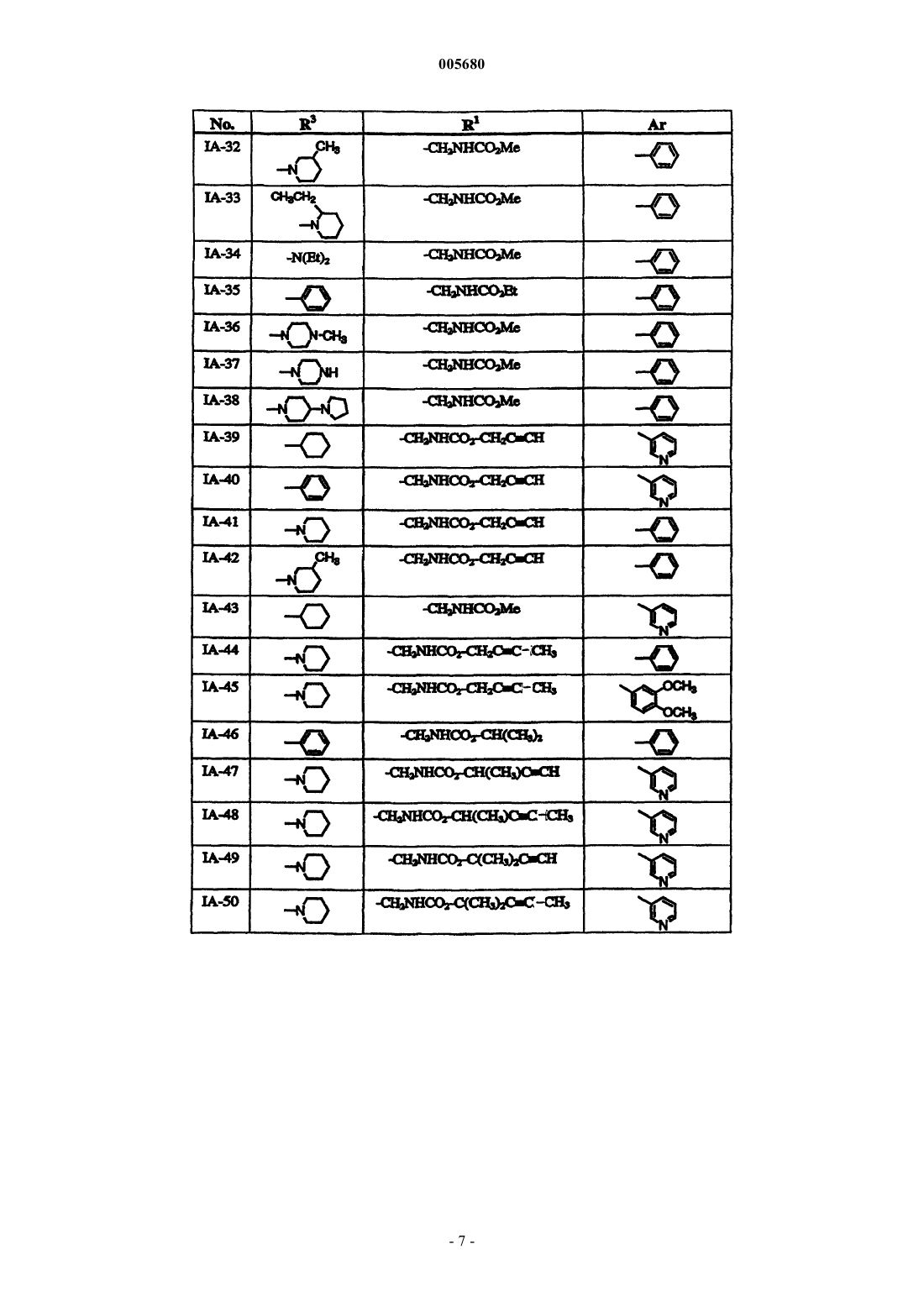
Формула / Реферат
1. Способ лечения бактериальной инфекции у млекопитающего, в случае необходимости такого лечения, включающий стадию введения указанному млекопитающему терапевтически эффективного количества соединения формулы

или его фармацевтически приемлемой соли, где
R1 обозначает необязательно замещеннную группу, выбранную из C1-6 алифатической группы, -C (R4)2(CH2)nNRCOR, -C(R4)2(CH2)nNRCO2(C1-6 алифатической группы), -CO2(C1-6 алифатической группы), -CON(R)2, -C(R4)2(CH2)nCON(R)2, -C(R4)2(CH2)nSO2N(R)2 или -SO2N(R)2;
n обозначает ноль или один;
каждый R независимо выбран из водорода или необязательно замещенной C1-6 алифатической группы;
R2 выбран из водорода, галогена или C1-4 алифатической группы;
кольцо A вместе с X, Y, Z и атомом азота в них обозначает гетероарильное кольцо, выбранное из 1,3-тиазола, 1,3-оксазола, имидазола или 1,2- пиразола;
Z обозначает C-R3 или N-R3;
X обозначает S, O или NH;
Y обозначает C или N;
R3 обозначает -(CH2) PN (R5)2 или необязательно замещенную группу, выбранную из C1-6 алкила, C2-6 алкенила или алкинила, циклопропила, циклопентила, циклогексила, пирролидинил-[C1-6 алкила или C2-6 алкенила или алкинила], пиперидинил-[C1-6 алкила или C2-6 алкенила или алкинила], пиперазинил-[C1-6алкила или C2-6 алкенила или алкинила], морфолинил-[C1-6 алкила или C2-6 алкенила или алкинила], пирролидинила, пиперидинила, пиперазинила, морфолинила, фенила или пиридила;
каждый R4 независимо выбран из водорода, необязательно замещенной C1-6 алифатической группы, или два R4, взятые вместе с углеродом, к которому они присоединены, образуют трех-шестичленное алифатическое кольцо;
каждый R5 независимо выбран из водорода, необязательно замещенной C1-4 алифатической группы, или два R5, взятые вместе с азотом, к которому они присоединены, образуют пяти- или шестичленное гетероциклическое кольцо, причем каждое кольцо содержит в общем до 2 гетероатомов, выбранных из N, O или S;
p обозначает целое число от нуля до четырех, когда Z обозначает C-R3, или целое число от одного до четырех, когда Z обозначает N-R3; и
Ar обозначает необязательно замещенные фенил, пиридил или тиофен.
2. Способ по п.1, где соединение имеет формулу IA

и где R1, R2, R3 и Ar определены в п.1.
3. Способ по п.2, где соединение имеет один или более следующих признаков:
(a) R1 выбран из -C(R4)2NHCOR, -C(R4)2NHCO2R, -CO2R и -CONHR, где R обозначает необязательно замещенную C1-4 алифатическую группу и каждый R4 независимо выбран из водорода, C1-3 алкильной группы, или два R4, взятые вместе с углеродом, к которому они присоединены, образуют трех- или четырехчленное алифатическое кольцо; и/или
(b) R3 обозначает C1-6 алкил, C2-6 алкенил или алкинил, циклопропил, циклопентил, циклогексил, необязательно замещенные алкокси, алкиламино или диалкиламино, необязательно замещенные морфолинил, пиперазинил, пиперидинил, пиридил или фенил; и/или
(c) Ar обозначает необязательно замещенное кольцо, выбранное из фенила или пиридила.
4. Способ по п.3, где соединение имеет следующие признаки:
(a) R1 выбран из -C(R4)2NHCOR, -C(R4)2NHCO2R, -CO2R и -CONHR, где R обозначает необязательно замещенную C1-4 алифатическую группу и каждый R4 независимо выбран из водорода, C1-3 алкильной группы, или два R4, взятые вместе с углеродом, к которому они присоединены, образуют трех- или четырехчленное алифатическое кольцо;
(b) R3 обозначает C1-4 алифатическую группу, необязательно замещенную алкокси, диалкиламино, необязательно замещенные морфолинил, пиперазинил, пиперидинил, пиридил или фенил; и
(c) Ar обозначает необязательно замещенное кольцо, выбранное из фенила или пиридила.
5. Способ по п.4, где R выбран из -C1-6 алкила, -C1-4 галогеналкила, -аллила, -CH2Cу CR6 и -CH(C1-3 алкил) Cу CR6, а R6 выбран из водорода, -C1-4 алифатической группы, -CH2N(Me)2 или -CH2O(C1-3 алкила).
6. Способ по п.1, где соединение формулы I выбрано из группы, включающей











и где R2, X, Y и Z определены в п.1.
7. Способ по любому из пп.1-6, где бактериальная инфекция, подлежащая лечению, характеризуется присутствием одного или более следующих микроорганизмов: Streptococcus pneumoniae, Streptococcus pyogenes, Enterococcus fecalis, Enterococcus faecium, Klebsiella pneumoniae, Enterobacter sps., Proteus sps., Pseudomonas aeruginosa, E. coli, Serratia marcesens, S. aureus, Coag. Neg. Staph., Acinetobacter sps., Salmonella sps., Shigella sps., Helicobacter pylori, Mycobacterium tuberculosis, Mycobacterium avium, Mycobacterium intracellulare, Mycobacterium fortuitum, Mycobacterium chelonae, Mycobacterium kansasii, Haemophilus influenzae, Stenotrophomonas maltophilia и Streptococcus agalactiae.
8. Способ по любому одному из пп.1-6, где бактериальная инфекция, подлежащая лечению, выбрана из одного или более следующих заболеваний: инфекций мочевыводящих путей, пневмонии, послеоперационных раневых инфекций и инфекционных заболеваний крови, простатита, инфекционных заболеваний кожи и мягких тканей, инфекционных заболеваний костей и суставов, внутрибрюшинных инфекций, менингита, абсцесса мозга, инфекционной диареи, желудочно-кишечных инфекций, хирургической профилактики и лечения лихорадочной нейтропении у пациентов.
9. Соединение формулы IA

или его фармацевтически приемлемая соль, где
R1 обозначает необязательно замещенную группу, выбранную из C1-6 алифатической группы, -C(R4)2(CH2)nNRCOR, -C(R4)2(CH2)nNRCO2(C1-6 алифатической группы), -CO2(C1-6 алифатической группы), -CON(R)2, -C(R4)2(CH2)nCON(R)2, -C(R4)2(CH2)nSO2N(R)2 или -SO2N(R)2;
n обозначает ноль или один;
каждый R независимо выбран из водорода или необязательно замещенной C1-6 алифатической группы;
R2 выбран из водорода, галогена или C1-4 алифатической группы;
R3 обозначает -(CH2)PN (R5)2 или необязательно замещенную группу, выбранную из C1-6 алкила, C2-6 алкенила или алкинила, циклопропила, циклопентила, циклогексила, пирролидинил-[C1-6 алкила или C2-6 алкенила или алкинила], пиперидинил-[C1-6 алкила или C2-6 алкенила или алкинила], пиперазинил-[C1-6 алкила или C2-6 алкенила или алкинила], морфолинил-[C1-6 алкила или C2-6 алкенила или алкинила], пирролидинила, пиперидинила, пиперазинила, морфолинила, фенила или пиридила;
каждый R4 независимо выбран из водорода, необязательно замещенной C1-6 алифатической группы, или два R4, взятые вместе с углеродом, к которому они присоединены, образуют трех-шестичленное алифатическое кольцо;
каждый R5 независимо выбран из водорода, необязательно замещенной C1-4 алифатической группы, или два R5, взятые вместе с азотом, к которому они присоединены, образуют пяти- или шестичленное гетероциклическое кольцо, причем каждое кольцо включает в общем до 2 гетероатомов, выбранных из N, O или S;
p обозначает целое число от нуля до четырех, когда Z обозначает C-R3, или целое число от одного до четырех, когда Z обозначает N-R3; и
Ar обозначает необязательно замещенные фенил, пиридил или тиофен.
10. Соединение по п.9, где указанное соединение имеет один или более следующих признаков:
(a) R1 выбран из -C(R4) 2NHCOR, -C(R4) 2NHCO2R, -CO2R и -CONHR, где R обозначает необязательно замещенную C1-4 алифатическую группу и каждый R4 независимо выбран из водорода, C1-3 алкильной группы, или два R4, взятые вместе с углеродом, к которому они присоединены, образуют трех- или четырехчленное алифатическое кольцо; и/или
(b) R3 обозначает C1-6 алкил, C2-6алкенил или алкинил, циклопропил, циклопентил, циклогексил, необязательно замещенные алкокси, алкиламино или диалкиламино, необязательно замещенные морфолинил, пиперазинил, пиперидинил, пиридил или фенил; и/или
(c) Ar обозначает необязательно замещенное кольцо, выбранное из фенила или пиридила.
11. Соединение по п.10, где указанное соединение имеет следующие признаки:
(a) R1 выбран из -C(R4) 2NHCOR, -C(R4) 2NHCO2R, -CO2R и -CONHR, где R обозначает необязательно замещенную C1-4 алифатическую группу и каждый R4 независимо выбран из водорода, C1-3 алкильной группы, или два R4, взятые вместе с углеродом, к которому они присоединены, образуют трех- или четырехчленное алифатическое кольцо; и
(b) R3 обозначает C1-4 алифатическую группу, необязательно замещенную алкокси, диалкиламино, необязательно замещенные морфолинил, пиперазинил, пиперидинил, пиридил или фенил; и
(c) Ar обозначает необязательно замещенное кольцо, выбранное из фенила или пиридила.
12. Соединение по п.11, где R выбран из -C1-4 алкила, -C1-4 галогеналкила, -аллила, -CH2Cу CR6, -CH(C1-3алкил)Cу CR6 и -C(Me)2Cу CR6, а R6 выбран из водорода, C1-4 алифатической группы, -CH2N(Me)2 или -CH2O(C1-3 алкила).
13. Соединение по п.9, где соединение формулы IA выбрано из группы, включающей




и где R2 обозначает водород.
14. Фармацевтическая композиция, включающая соединение по п.9 и фармацевтически приемлемый носитель.
Текст
005680 По данной заявке испрошен приоритет по предварительной заявке на патент США, серийный номер 60/17 6671, поданной 18 января 2000 г., и предварительной заявке на патент США, серийный номер 60/254331, поданной 8 декабря 2000 г. Область техники Данное изобретение относится к области медицинской химии и направлено на создание новых соединений и их фармацевтических композиций, которые ингибируют ДНК-гиразы. Изобретение также относится к способам применения соединений и фармацевтических композиций данного изобретения для лечения бактериальных инфекций, включая внутрибольничные инфекции, которые являются чувствительными к ингибированию гираз. Предпосылки к созданию изобретения Устойчивость бактерий к антибиотикам давно установлена и в настоящее время рассматривается как серьезная всемирная проблема здравоохранения. В результате резистентности некоторые бактериальные инфекции или вызывают затруднения при лечении антибиотиками, или даже не поддаются лечению. Названная проблема становится особенно серьезной в связи с недавним обнаружением множественной лекарственной устойчивости у определенных штаммов бактерий, таких как Streptococcus pneumoniae (SP), Mycobacterium tuberculosis и Enterococcus. Появление устойчивого к ванкомицину энтерококка вызвало особую тревогу, так как прежде ванкомицин был единственным эффективным антибиотиком для лечения этой инфекции и считался в отношении многих инфекций лекарством, представляющим собой "последнее средство". В то время как другие устойчивые к лекарствам бактерии, такие как энтерококки, не вызывают опасного для жизни заболевания, существуют опасения, что гены, которые индуцируют резистентность, могут передаваться другим более смертоносным микроорганизмам, таким какStaphylococcus aureus, среди которых устойчивость к метициллину уже является широко распространенной (De Clerq, et al., Current Opinion in Anti-infective Investigational Drugs, 1999, 1, 1; Levy, "The Challengeof Antibiotic Resistance", Scientific American, March, 1998). Другой причиной для беспокойства является проблема, как быстро может распространяться устойчивость к антибиотикам. Например, до 1960 г. SP был универсально чувствителен к пенициллину, а в 1987 г. в США резистентными оказались только 0,02% штаммов SP. Однако к 1995 г. было установлено,что устойчивость SP к пенициллину составляла приблизительно 7% и до 30% в некоторых районах США(Lewis, FDA Consumer magazine (September, 1995); Gershman in The Medical Reporter, 1997). В частности, больницы оказываются центрами появления и передачи устойчивых к лекарствам микроорганизмов. Встречающиеся в больницах инфекции, известные как внутрибольничные инфекции,все больше становятся проблемой, вызывающей опасения. У двух миллионов американцев, инфицируемых в больницах каждый год, более половины внутрибольничных инфекций оказались устойчивыми по крайней мере к одному антибиотику. Центр по контролю заболеваемости сообщил, что в 1992 г. свыше 13000 пациентов больниц умерло от бактериальных инфекций, которые оказались резистентными к терапии антибиотиками (Lewis, "The Rise of Antibiotic-Resistant Infections", FDA Consumer magazine, Sept,1995). Как результат необходимости бороться с устойчивыми к лекарствам бактериями и возрастающей несостоятельностью доступных лекарственных средств, возрождается интерес к открытию новых антибиотиков. Одной привлекательной стратегией разработки новых антибиотиков является ингибирование ДНК-гиразы, бактериального фермента, необходимого для репликации ДНК, и поэтому необходимого для роста и деления бактериальных клеток. Активность гиразы также ассоциирована с событиями в транскрипции ДНК, репарацией и рекомбинацией ДНК. Гираза является одной из топоизомераз, группы ферментов, которые катализируют взаимное превращение топологических изомеров ДНК (смотри в общем, Kornberg and Baker, DNA Replication, 2d Ed.,Chapter 12, 1992, W.H. Freeman and Co.; Drlica, Molecular Microbiology, 1992, 6, 425; Drlica and Zhao, Microbiology and Molecular Biology Reviews, 1997, 61, 377). Сама гираза регулирует сверхспиральность ДНК и облегчает топологический стресс, который имеет место, когда цепи родительской двухцепочечной ДНК раскручиваются в процессе репликации. Гираза также катализирует превращение релаксированной,закрытой кольцевой двойной ДНК в отрицательную сверхспиральную форму, которая является более благоприятной для рекомбинации. Механизм реакции сверхспирализации включает закручивание гиразы вокруг участка ДНК, разрыв двойной цепи в этом участке, прохождение второго участка ДНК через разрыв и соединение разорванных цепей. Такой механизм разрыва является характерным для топоизомераз типа II. Реакция сверхспирализации осуществляется посредством связывания АТФ с гиразой. Затем во время реакции АТФ гидролизуется. Такое связывание АТФ и последующий гидролиз вызывают конформационные изменения в ДНК-связанной гиразе, что необходимо для ее активности. Также было установлено, что уровень сверхспирализованности ДНК (или релаксации) зависит от соотношения АТФ/АДФ. В отсутствие АТФ гираза способна только релаксировать сверхспиральную ДНК. Бактериальная ДНК-гираза является белковым тетрамером 400 килодальтон, состоящим из двух A(gyrA) и двух В (gyrB) субъединиц. Связывание и разрыв ДНК связаны с gyrA, тогда как белок gyrB связывает и гидролизует АТФ. GyrB состоит из аминоконцевого домена, который проявляет АТФ-азную активность, и карбоксиконцевого домена, который взаимодействует с gyrA и ДНК. В отличие от этого-1 005680 эукариотические топоизомеразы типа II являются гомодимерами, которые могут релаксировать отрицательные и положительные сверхспирали, но не могут вставлять отрицательные сверхспирали. Идеально,чтобы антибиотики, действие которых основано на ингибировании бактериальной ДНК-гиразы, были бы селективными в отношении этого фермента и были бы относительно неактивными в отношении эукариотических топоизомераз типа II. Широко используемые хинолоновые антибиотики ингибируют бактериальную ДНК-гиразу. Примеры хинолонов включают известные соединения, такие как налидиксовая кислота и оксолинивая кислота,а также последующие, более сильнодействующие фторхинолоны, такие как норфлоксацин, ципрофлоксацин и гатифлоксацин. Эти соединения связываются с gyrA и стабилизируют расщепленный комплекс,таким образом ингибируя полную функцию гиразы, что приводит к гибели клетки. Однако лекарственная резистентность также осознана как проблема этого класса соединений (доклад Всемирной организации здравоохранения, "Use of Quinolones in Food Animals and Potential Impact on Human Health", 1998). Что касается хинолонов, а также других классов антибиотиков, бактерии, подвергнутые действию соединений, предшествующего уровня, часто быстро вырабатывают перекрестную устойчивость к более сильнодействующим соединениям того же класса. Имеются менее известные ингибиторы, которые связываются с gyrB. Примеры включают кумарины, новобиоцин и кумермицин А 1, циклотиалидин, циподин и клероцидин. Показано, что кумарины очень прочно связываются с gyrB. Например, новобиоцин создает сеть водородных связей с белком и несколько гидрофобных контактов. Оказалось, что в то время как новобиоцин и АТФ связываются в пределах АТФ-связывающего сайта, имеется минимальное, частичное перекрывание в ориентации связи двух соединений. Перекрывающиеся участки являются фрагментом сахара новобиоцина и аденина АТФ(Maxwell, Trends in Microbiology, 1997, 5, 102). Для устойчивых к кумарину бактерий наиболее преобладающей точковой мутацией является поверхностный остаток аргинина, который связывается с карбонилом кольца кумарина (Argl36 в gyrB Е.coli). В то время как ферменты с описанной мутацией демонстрируют более низкую сверхспирализованность и АТФ-азную активность, они также оказываются менее чувствительными к ингибированию кумариновыми лекарственными средствами (Maxwell, Mol. Microbiol., 1993, 9, 681). Несмотря на существование сильных ингибиторов гиразной сверхспирализации, кумарины широко не использовались как антибиотики. Они не подходят в основном вследствие их низкого проникновения в бактерии, эукариотической токсичности и плохой растворимости в воде (Maxwell, Trends in Microbiology, 1997, 5, 102). Желательно иметь новые эффективные ингибиторы gyrB, которые преодолевают описанные недостатки. Такие ингибиторы были бы привлекательными кандидатами в антибиотики без проблем резистентности, которые присущи другим классам антибиотиков. Поскольку устойчивость бактерий к антибиотикам становится важной проблемой общественного здравоохранения, имеется постоянная потребность в разработке более новых и сильнодействующих антибиотиков. Точнее, существует потребность в антибиотиках, которые представляют новый класс соединений, ранее не использованных для лечения бактериальной инфекции. Такие соединения оказались бы особенно полезными для лечения внутрибольничных инфекций в больницах, где появление и распространение резистентных бактерий становится все более преобладающим явлением. Описание изобретения Установлено, что соединения данного изобретения и их фармацевтические композиции полезны для лечения бактериальных инфекций. Эти соединения имеют общую формулу I или его фармацевтически приемлемой соли, гдеR1 обозначает необязательно замещеннную группу, выбранную из C1-6 алифатической группы, -С 4n обозначает ноль или один; каждый R независимо выбран из водорода или необязательно замещенной C1-6 алифатической группы;R2 выбран из водорода, галогена или C1-4 алифатической группы; кольцо А вместе с X, Y, Z и атомом азота в них обозначает гетероарильное кольцо, выбранное из 1,3-тиазола, 1,3-оксазола, имидазола или 1,2- пиразола;R3 обозначает -(CH2)PN(R5)2 или необязательно замещенную группу, выбранную из C1-6 алкила, С 2-6 алкенила или алкинила, циклопропила, циклопентила, циклогексила, пирролидинил-[C1-6 алкила или С 2-6 алкенила или алкинила], пиперидинил-[C1-6 алкила или С 2-6 алкенила или алкинила], пиперазинил-[C1-6 алкила или С 2-6 алкенила или алкинила], морфолинил-[C1-6 алкила или С 2-6 алкенила или алкинила], пирролидинила, пиперидинила, пиперазинила, морфолинила, фенила или пиридила; каждый R4 независимо выбран из водорода, необязательно замещенной C1-6 алифатической группы,или два R4, взятые вместе с углеродом, к которому они присоединены, образуют трех-шестичленное алифатическое кольцо; каждый R5 независимо выбран из водорода, необязательно замещенной C1-4 алифатической группы,или два R5, взятые вместе с азотом, к которому они присоединены, образуют пяти- или шестичленное гетероциклическое кольцо, причем каждое кольцо содержит в общем до 2 гетероатомов, выбранных изN, О или S; р обозначает целое число от нуля до четырех, когда Z обозначает C-R3, или целое число от одного до четырех, когда Z обозначает N-R3; и Аr обозначает необязательно замещенные фенил, пиридил или тиофен. Использованные в описании следующие определения следует применять, если не указано особо. Термин "алифатический" означает C1-C12 углеводороды с прямой, разветвленной цепью или циклические, которые являются полностью насыщенными или которые содержат один или более участков ненасыщенности. Например, подходящие алифатические группы включают замещенные или незамещенные линейные, разветвленные или циклические алкильные, алкенильные, алкинильные группы и их производные, такие как (циклоалкил)алкил, (циклоалкенил)алкил или (циклоалкил)алкенил. Термины "алкил" и "алкокси", используемые самостоятельно или как часть более крупной группы, относятся как к прямым, так и разветвленным цепям, содержащим от одного до двенадцати атомов углерода. Термины "алкенил" и "алкинил", используемые самостоятельно или как часть более крупной группы, должны включать как прямые, так и разветвленные цепи, содержащие от двух до двенадцати атомов углерода. Термины "галогеналкил", "галогеналкенил" и "галогеналкокси" означают алкил, алкенил или алкокси, в зависимости от обстоятельств замещенные одним или более атомами галогена. Термин "галоген" означает F,Cl, Вr или I. Термин "гетероатом" подразумевает N, О или S. Азотсодержащие соединения данного изобретения также включают соответствующие N-оксиды соединений, а также соединения, имеющие кватернизованную форму любого основного азота. Кольца, содержащие от одного до четырех гетероатомов, выбранных из N, О или S, включают гетероциклические ароматические (или гетероарильные) кольца и неароматические гетероциклические кольца. Примеры ароматических гетероциклических колец включают 2-фуранил, 3-фуранил, N-имидазолил,2-имидазолил, 4-имидазолил, 5-имидазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2 оксадиазолил, 5-оксадиазолил, 2-оксазолил, 4-оксазолил, 5-оксазолил, 2-пирролил, 3-пирролил, 2 пиридил, 3-пиридил, 4-пиридил, 2-пиримидил, 4-пиримидил, 5-пиримидил, 3-пиридазинил, 2-тиазолил,4-тиазолил, 5-тиазолил, 5-тетразолил, 2-триазолил, 5-триазолил, 2-тиенил или 3-тиенил. Примеры неароматических гетероциклических колец включают 2-тетрагидрофуранил, 3-тетрагидрофуранил, 2 тетрагидротиофенил,3-тетрагидротиофенил,2-морфолино,3-морфолино,4-морфолино,2 тиоморфолино, 3-тиоморфолино, 4-тиоморфолино, 1-пирролидинил, 2-пирролидинил, 3-пирролидинил,1-пиперазинил, 2-пиперазинил, 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-пиперидинил, 4 тиазолидинил, диазолонил, N-замещенный диазолонил, 1-фталимидинил, бензоксан, бензотриазол-1-ил,бензопирролидин, бензопиперидин, бензоксолан, бензотиолан, тетрагидроизохинолин, декагидроизохинолин и бензотиан. Арильная группа (карбоциклическая и гетероциклическая) или аралкильная группа, такая как бензил или фенетил, может содержать один или более заместителей. Примеры подходящих заместителей на ненасыщенном атоме углерода арильной группы включают галоген, -R, -OR, -ОН, -SH, -SR, защищенный ОН (такой как ацилокси), фенил (Ph), замещенный Ph, -OPh, замещенный -OPh, замещенное или незамещенное пяти-шестичленное кольцо, содержащее от одного до четырех гетероатомов, -NO2, -CN, -NH2, NHR, -N(R)2, -NHCOR, -NHCONHR, -NHCON(R)2, -NRCOR, NHCO2R, -CO2R, -CO2H, -COR, -CONHR, CON(R)2, -S(O)2R, -SONH2, -S(O)R, -SO2NHR или -NHS(O)2R, в которых R соответствует алифатической группе или замещенной алифатической группе. Алифатическая группа или неароматическое гетероциклическое кольцо могут содержать один или более заместителей. Примеры подходящих заместителей у насыщенного углерода алифатической группы или неароматического гетероциклического кольца включают заместители, перечисленные выше для ненасыщенного углерода, также, как и следующие: =O, =S, =NNHR, =NNR2, =N-OR, =NNHCOR,=NNHCO2R, =NNHSO2R или =NR. Цепь алкилидена представляет собой углеводородную цепь, которая может быть насыщенной или ненасыщенной, такой как -(CH2)n-, -(CH=CH)m(CH2)n- или -(СС)m(СН 2)n-, в-3 005680 которых m и n соответствуют целым числам от нуля до шести. Цепь алкилидена может быть замещена таким же образом, как и алифатическая группа. Замещаемый азот в ароматическом или неароматическом гетероциклическом кольце необязательно может быть замещен. Приемлемые заместители азота включают R, COR, S(O)2R и CO2R, где R соответствует алифатической группе или замещенной алифатической группе. Специалистам в данной области будет ясно, что некоторые соединения данного изобретения могут существовать в таутометрических формах, все такие формы соединений включены в объем изобретения. Если не указано особо, также подразумевается, что описанные в заявке структуры включают все стереохимические формы структуры, то есть R и S конфигурации для каждого асимметричного центра. Поэтому индивидуальные стереохимические изомеры, а также смеси энантиомеров и диастереомеров данных соединений включены в объем изобретения. Данное изобретение также относится к способу лечения бактериальной инфекции у млекопитающего, включающему стадию введения указанному млекопитающему терапевтически эффективного количества соединения формулы I, при необходимости такого лечения. Подгруппы соединений данного изобретения включают I-A, I-B, I-C, I-D и I-Е, представленные ниже в которых R1, R2, R3 и Аr являются такими, как описано выше, a R7 означает водород или C1-6 алифатическую группу. Соединения формулы I-A являются новыми соединениями. Предпочтительные группы R1 включают -С(R4) 2NHCOR, -С(R4)2NHCO2R, -CO2R и -CONHR, в которых R представляет собой необязательно замещенную C1-4 алифатическую группу, а каждый R4 независимо выбран из водорода, C1-3 алкильной группы или два R4, взятые вместе с углеродом, к которому они присоединены, образуют трех- или четырехчленное алифатическое кольцо. Примеры предпочтительного R включают -C1-4 алкил, -C1-4 галогеналкил, -аллил, -CH2C=CR6, -СН (C1-3 алкил) OCR6 и -С (Me)2CCR6, в которых R6 означает водород, -C1-4 алифатическую группу, -CH2N(Me)2 и -CH2O(C1-3 алкил). Предпочтительной группой R2 является водород. Когда R1 означает -CONH(C1-3 алкил) или -СO2(C1-3 алкил), другими предпочтительными R2 являются галоген и -C1-4 алкильные группы. Предпочтительные группы R3 включают C1-6 алкильную, С 2-6 алкенильную или алкинильную группу, необязательно замещенную алкокси, алкиламино или диалкиламино, необязательно замещенные морфолинил, пиперазинил, пиперидинил, пиридил или фенил. Предпочтительные группы Аr представляют собой необязательно замещенные фенил, пиридил или тиофен. Примеры необязательных заместителей, присоединенных к Аr, включают один или более из следующих: алкил, алкокси, гидрокси, карбокси, галоген, SO2R, SO2NHR, амино, алкиламино, диалкиламино и пиридил. Конкретные соединения формулы I представлены в табл. 1 (R2 означает водород). Нумерация представленных примеров произведена на основании вышеописанных подгрупп: IA относится к кольцевым тиазолам А (X представляет серу), IB относится к оксазолам (X означает кислород), IС относится к имидазолам (X означает NH), ID относится к пиразолам (Y означает азот) и IE относится к пиразолам (Z означает азот). Соединения данного изобретения, в общем, могут быть получены способами, известными специалистам в данной области для аналогичных соединений, и по схемам синтеза, представленным ниже. Основной ссылкой является Katritzky and Rees, Comprehensive Heterocyclic Chemistry, vol.5, 1984, PergamonPress. В показанном ниже способе, группа Аr формулы I может быть представлена фенильным кольцом. Специалистам в данной области должно быть понятно, что описанные способы, в общем, применимы к соединениям, содержащим арильные группы, отличные от фенила. Реагенты и условия: (a) (EtO2C)2 СНВr, пиридин, толуол, нагревание; (b) ангидрид трифторметансульфокислоты, 2,6-лутидин, СН 2 Сl2, 0 С; (с) Ме 2 АlСl, MeNHOMeHCl, CH2Cl2, 0 С; (d) пиперидин, толуол, нагревание; (е) LiCCCH2N (Li) СО 2 трет-Вu, THF, 0 СRT (комнатная температура); (f) H2NNH2,EtOH, комнатная температура; (g) трифторуксусная кислота, CH2Cl2, (h) метиловый эфир имидазол-1 карбоновой кислоты, ацетонитрил, нагревание. Выше на схеме I представлен путь получения соединений тиазола данного изобретения, в соответствии с которым, как проиллюстрировано в описании, в положении 4 (R3) кольцо тиазола замещается аминогруппой, где Аr означает фенил, a R3 означает пиперидин. Специалистам в данной области должно быть ясно, что пиперидиновый реагент на стадии (d) может быть заменен другими аминами для получения других 4-(замещенная аминогруппа)тиазолов. Схема II Реагенты и условия: (a) EtO2CCH(Cl)С(=O)R3, EtOH, нагревание; (b) Ме 2 АlСl, MeNHOMeHCl,CH2Cl2, 0C; (c) LiCCCH2N(Li)СО 2 трет-Bu, THF, 0Cкомнатная температура; (d) H2NNH2, EtOH,комнатная температура; (е) трифторуксусная кислота, СН 2 Сl2; (f) метиловый эфир имидазол-1 карбоновой кислоты, ацетонитрил, нагревание. Выше на схеме II представлен обычный путь для тиазольных соединений формулы IA, в которой R3 является алкилом или арилом. Реагенты и условия: (a) EtO2CCH (Cl) COCH2OCH3, EtOH, нагревание; (b) Ме 2 АlСl, MeNHOMeHCl,СН 2 Сl2, 0 С; (с) MeMgBr, THF, 0C; (d) КО-трет-Bu, диэтилоксалат, THF, комнатная температура; (е) H2NNH2,уксусная кислота, EtOH; (f) BBr3, CH2Cl2; (g) (R4)2NH, THF; (h) LiAlH4, THF; (i) SOCl2, CH2Cl2, 0C; (j) NH3, диоксан; (k) метиловый эфир имидазол-1-карбоновой кислоты, ацетонитрил, нагревание; (1) EtNH2, MeOH, нагревание. Выше на схеме III представлен основной путь получения соединений формулы IA, в которой R3 означает (CH2)pN(R4)2, a p равно 1. Схема IV Реагенты и условия: (a) EtO2CCHS+(Me)2Br-, 60% NaH, THF; (b) декалин, 195 С; (с) ангидрид трифторметансульфокислоты, 2,6-лутидин, СН 2 Сl2, 0 С; (d) Ме 2 АlСl, MeNHOMeHCl, CH2Cl2, 0 Скомнатная температура; (е) пиперидин, толуол, 90 С; (f) СНССН 2NНСО 2-трет-Вu, н-BuLi, -15 С 10 С; (g) H2NNH2H2O, EtOH,комнатная температура; (h) (4:1) CH2Cl2-TFA; (i) ClCO2Me, EtOAc, 1,0 н NaHCO3. Выше на схеме IV представлен путь получения соединений оксазола IB данного изобретения, в соответствии с которым, как проиллюстрировано в описании, в положении 4 (R3) кольцо оксазола замещается аминогруппой, где Аr означает фенил, a R3 означает пиперидин. Образование кольца оксазолона в- 16005680 соответствии со стадиями (а) и (b) осуществлено по способу, описанному в Tetrahedron, Vol.29, 19831990 (1973). Схема V(e) H2NNH2H2O, EtOH, комнатная температура; (f) (4:1) CH2Cl2-TFA; (i) ClCO2Me, EtOAc, 1,0 н NaHCO3. Выше на схеме V представлен путь получения оксаэолов IB, в соответствии с которым кольцо оксазола в положении 4 (R3) замещается различными группами, например алифатической группой. Согласно стадии (а) образование кольца оксазола осуществлено по способу, описанному в J. Chem. Soc, Сhem. Соmmun., 29-30 (1995). Схема VIClCO2Me, EtOAc, 1,0 н NaHCO3. Выше на схеме VI представлен способ получения соединений IB, при котором кольцо оксазола в положении 4 замещается группой арила, что продемонстрировано в заявке, используя фенильную группу. Реагенты и условия: (a) PhNHNH2, Et2O, комнатная температура; (b) водная NaOH, MeOH; (с) карбонилдиимидазол, THF; (d) MeNHOMeHCl, диизопропилэтиламин, DFM, 80 С; (е) LiСССН 2N(Li)СО 2 трет-Вu, THF, 0 Скомнатная температура; (f) H2NNH2, EtOH, комнатная температура; (g) CH2Cl2,TFA; (h) метиловый эфир 1-имидазолкарбоновой кислоты, ацетонитрил, нагревание. Выше на схеме VII представлен общий способ получения пиразолов формулы ID. Представленный способ особенно подходит для соединений, в которых заместитель R3 означает алифатическую группу или арил. Схема VIII(g) H2NNH2, EtOH, комнатная температура; (h) CH2Cl2, TFA; (i) метиловый эфир имидазолкарбоновой кислоты, ацетонитрил, нагревание. Выше на схеме VIII представлен общий способ получения пиразолов формулы IE. Фармацевтические композиции и способы данного изобретения полезны в основном для борьбы с бактериальными инфекциями in vivo. Примеры бактериальных организмов, с которыми можно бороться с помощью композиций и способов данного изобретения, включают следующие микроорганизмы:agalactiae, не ограничиваясь ими. Поэтому композиции и способы полезны для борьбы, лечения или снижения распространения, тяжести или действия внутрибольничных или невнутрибольничных инфекций. Не ограничиваясь, примеры применений при внутрибольничных инфекциях включают применение при инфекциях мочевыводящих путей, пневмонии, операционных раневых инфекциях, костных и сус- 18005680 тавных инфекционных заболеваниях и инфекционных заболеваниях крови. Примеры применений при невнутрибольничных инфекциях, не ограничиваясь ими, включают применение при инфекциях мочевыводящих путей, пневмонии, простатите, инфекционных заболеваниях кожи и мягких тканей, инфекционных заболеваниях костей и суставов, внутрибрюшинных инфекциях, менингите, абсцессе мозга, инфекционной диареи и желудочно-кишечных инфекционных заболеваниях, хирургической профилактике и для лечения пациентов с лихорадочной нейтропенией."Невнутрибольничные инфекции" также называются приобретенными при контакте инфекциями. Фармацевтические композиции данного изобретения включают соединение формулы I или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. Такие композиции необязательно могут включать дополнительные терапевтические агенты. Не ограничиваясь ими, такие агенты включают антибиотик, противовоспалительное средство, ингибитор матриксных металлопротеиназ, ингибитор липоксигеназы, антагонист цитокина, иммунодепрессант, противораковое средство, противовирусное средство, цитокин, фактор роста, иммуномодулятор, простагландин или соединение против сосудистой гиперпролиферации. Термин "фармацевтически приемлемый носитель" относится к нетоксичному носителю, который может быть введен пациенту вместе с соединением данного изобретения и который не оказывает вредного влияния на его фармакологическую активность. Фармацевтически приемлемые носители, которые могут быть использованы в фармацевтических композициях данного изобретения, включают, но не ограничены ими, ионообменники, оксид алюминия,стеарат алюминия, лецитин, сывороточные белки, такие как сывороточный альбумин человека, буферные вещества, такие как фосфаты, глицин, сорбиновую кислоту, сорбат калия, смеси частичных глицеридов насыщенных растительных жирных кислот, воду, соли или электролиты, такие как протамин сульфат, вторичный кислый фосфат натрия, кислый фосфат калия, хлорид натрия, соли цинка, коллоидный кремнезем, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, натрийкарбоксиметилцеллюлоза, полиакрилаты, воски, полиэтиленполиоксипропиленблоксополимеры, ланолин и системы доставки самоэмульгирующихся лекарственных средств (SEDDS),таких как -токоферол, сукцинат полиэтиленгликоля 1000 или другие полимерные матрицы систем доставки. В случае фармацевтической композиции, содержащей только соединение формулы I в качестве активного компонента, способы введения названных композиций могут дополнительно включать стадию введения субъекту дополнительного вещества. Такие дополнительные вещества включают, но не ограничены ими, антибиотик, противовоспалительное средство, ингибитор матриксных металлопротеиназ,ингибитор липоксигеназы, антагонист цитокина, иммунодепрессант, противораковое средство, противовирусное вещество, цитокин, фактор роста, иммуномодулятор, простагландин или соединение, препятствующее сосудистой гиперпролиферации. Термин "фармацевтически эффективное количество" относится к количеству, эффективному для лечения или снижения интенсивности симптомов бактериальной инфекции у пациента. Термин "фармацевтически эффективное количество" относится к количеству, эффективному в предупреждении или, по существу, уменьшении бактериальной инфекции у пациента. Соединения данного изобретения можно применять обычным способом для контроля уровней бактериальных инфекций in vivo и для лечения заболеваний или снижения распространенности или тяжести эффектов, которые опосредованы бактериями. Такие способы лечения, схемы дозирования и требования могут быть определены специалистом в данной области из доступных способов и методик. Например, для введения пациенту, страдающему бактериальной инфекцией или заболеванием, соединение данного изобретения можно комбинировать с фармацевтически приемлемым адъювантом фармацевтически подходящим способом и в количестве, эффективном для уменьшения тяжести такой инфекции или заболевания. Альтернативно, соединения данного изобретения можно использовать в композициях и способах для лечения или защиты индивидуумов от бактериальных инфекций или заболеваний в течение продолжительных периодов времени. Соединения можно использовать в таких композициях, или отдельно, или вместе с другими соединениями данного изобретения по способу, соответствующему общепринятым способам применения ингибиторов ферментов в фармацевтических композициях. Например, соединение данного изобретения можно комбинировать с фармацевтически приемлемым адъювантом, обычно употребляемым в вакцинах, и вводить в профилактически эффективных количествах для защиты индивидуумов от бактериальных инфекций или заболеваний в течение продолжительного периода времени. Соединения формулы I также могут быть введены одновременно с другими антибиотиками, чтобы усилить терапевтическое или профилактическое действие против различных бактериальных инфекций. Если соединенияданного изобретения вводят в комбинации в другими агентами, их можно вводить пациенту последовательно или одновременно. Альтернативно, фармацевтические или профилактические композиции согласно данному изобретению содержат соединение формулы I и другое терапевтическое или профилактическое средство.- 19005680 Фармацевтические композиции данного изобретения можно вводить перорально, парентерально, с помощью ингаляционного спрея, местно, посредством глазного раствора или мази, ректально, назально,трансбуккально, вагинально или через имплантированный резервуар. Фармацевтические композиции данного изобретения могут содержать любые обычные нетоксичные фармацевтически подходящие носители, адъюванты или наполнители. В некоторых случаях рН композиции можно регулировать фармацевтически приемлемыми кислотами, основаниями или буферами, чтобы повысить стабильность приготовленного соединения или формы его доставки. Используемый в описании термин "парентеральный способ" включает подкожный, внутрикожный, внутривенный, внутримышечный, внутрисуставной, внутрисиновиальный, интрастернальный, внутриоболочечный, внутрираневой и внутричерепной инъекционные или инфузионные способы. Фармацевтические композиции могут быть в виде стерильного препарата для инъекций, например,в виде стерильной водной или маслянистой суспензии для инъекций. Такая суспензия может быть приготовлена в соответствии со способами, известными в данной области, используя подходящие диспергирующие или увлажняющие вещества (например, такие как Твин 80) и суспендирующие агенты. Стерильный препарат для инъекций также может представлять собой стерильный раствор или суспензию для инъекций в нетоксичном разбавителе или растворителе, подходящими для парентерального введения,например, в виде раствора в 1,3-бутандиоле. В число приемлемых наполнителей и растворителей, которые могут использоваться, входят маннит, вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные, нелетучие масла обычно используют как среду для растворения или суспендирования. Для этой цели можно использовать любые успокаивающие нелетучие масла, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота и ее глицеридные производные, используют в препаратах для инъекций, которые являются натуральными фармацевтически приемлемыми маслами, такими как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных производных. Указанные масляные растворы или суспензии также могут содержать растворитель-спирт с длинной цепью или диспергатор, такие как вещества, описанные в Фармакопеи Швейцарии, или подобный спирт. Фармацевтические композиции данного изобретения можно вводить перорально в любой дозированной форме, подходящей для перорального введения, включая капсулы, таблетки и водные суспензии и растворы, не ограничиваясь ими. В случае таблеток для перорального применения обычно используемые носители включают лактозу и кукурузный крахмал. Также обычно добавляют смазывающие вещества, такие как стеарат магния. Для перорального введения в капсульной форме полезные разбавители включают лактозу и высушенный кукурузный крахмал. Когда перорально вводят водные суспензии и растворы и пропиленгликоль, активный ингредиент комбинируется с эмульгирующими и суспендирующими веществами. Если необходимо, можно добавлять определенные подсластители, и/или вкусовые добавки, и/или красители. Фармацевтические композиции данного изобретения также можно вводить в виде суппозиториев для ректального введения. Указанные композиции могут быть приготовлены смешиванием соединения данного изобретения с подходящим, не вызывающим раздражение наполнителем, который представляет собой твердое вещество при комнатной температуре, но жидкость при ректальной температуре и поэтому будет плавиться в прямой кишке с высвобождением активных соединений. Такие материалы включают масло какао, пчелиный воск и полиэтиленгликоли, не ограничиваясь ими. Местное применение фармацевтических композиций данного изобретения оказывается особенно полезным, если требуемое лечение вовлекает области или органы, легко доступные для местной аппликации. Для местного применения на кожу фармацевтическую композицию следует готовить с подходящей мазью, содержащей активные компоненты, суспендированные или растворенные в носителе. Не ограничиваясь, носители для местного применения соединений данного изобретения включают минеральное масло, вазелиновое масло, белый вазелин, пропиленгликоль, соединение полиоксиэтиленполиоксипропилена, эмульгирующийся воск и воду. Альтернативно, фармацевтическая композиция может быть приготовлена в виде подходящего лосьона или крема, содержащего активное соединение, суспендированное илирастворенное в носителе. Подходящие носители включают минеральное масло, моностеарат сорбитана, полисорбат 60, сложные цетиловые эфиры, воск, цетеариловый спирт, 2 октилдодеканол, бензиловый спирт и воду, не ограничиваясь ими. Фармацевтические композиции данного изобретения также можно применять местно в нижнем отделе кишечника с помощью ректальных суппозиториев или подходящей композиции для клизм. Применяемые местно пластыри также предусмотрены в данном изобретении. Фармацевтические композиции данного изобретения можно вводить посредством назального аэрозоля или ингаляции. Такие композиции готовят в соответствии с технологиями, хорошо известными в области технологии приготовления лекарственных средств, и получают в виде растворов в физиологическом растворе, употребляя бензиловый спирт или другие подходящие консерванты, усилители абсорбции для увеличения биодоступности, фторуглероды и/или другие солюбилизаторы или диспергаторы, известные в данной области.- 20005680 Дозы приблизительно от 0,01 до 100 мг/кг массы тела в день, предпочтительно от 0,5 до 75 мг/кг массы тела в день и наиболее предпочтительно приблизительно от 1 до 50 мг/кг массы тела в день, соединения активного ингредиента оказываются эффективными при монотерапии для предупреждения и лечения бактериальных инфекционных заболеваний, вызванных бактериями, такими как Streptococcuspneumoniae, Streptococcus pyogenes, Enterococcus fecalis, Enterococcus faecium, Klebsiella pneumoniae, Enterobacter sps., Proteus sps., Pseudomonas aeruginosa, E. coli, Serratia marcesens, S. aureus и Coag. Neg. Staph. Вообще, фармацевтические композиции данного изобретения следует вводить приблизительно от 1 до 5 раз в день или, альтернативно, в виде длительной инфузии. Такое введение можно использовать как хроническую или экстренную терапию. Количество активного ингредиента, которое можно комбинировать с носителями, чтобы получить разовую дозированную форму, изменяется в зависимости от субъекта, которого лечат, и определенного способа введения. Обычный препарат должен содержать приблизительно от 5 до 95% активного соединения (мас./мас.). Предпочтительно, такие препараты содержат приблизительно от 20 до 80% активного соединения. Если композиции данного изобретения содержат комбинацию соединения формулы I и одного или более дополнительных терапевтических или профилактических веществ, дозы как соединения, так и дополнительного вещества должны находится в диапазоне доз приблизительно от 10 до 80% дозы, обычно вводимой при режиме монотерапии. При улучшении состояния пациента, если необходимо, можно применять поддерживающую дозу соединения, композиции или комбинации данного изобретения. Впоследствии может потребоваться модификация дозы, дозированной формы или частоты введения, или обеих. В некоторых случаях, однако,пациентам может требоваться прерывистое лечение на долговременной основе в зависимости от появления любых рецидивов или симптомов заболевания. Квалифицированным специалистам будет понятно, что могут требоваться более низкие или более высокие дозы, чем указанные выше. Специальные схемы дозирования и лечения для любого отдельного пациента будут зависеть от различных факторов, включающих активность определенного применяемого соединения, возраста, массы тела, общего состояния здоровья, пола, диеты, времени введения, скорости экскреции, комбинации лекарств, тяжести и течения заболевания и предрасположенности пациента к заболеванию и заключения лечащего врача. Одним из аспектов данного изобретения является способ лечения или предупреждения бактериальной инфекции или заболевания у субъекта, включающий введение субъекту любого соединения, фармацевтической композиции или комбинации, описанных в заявке, и фармацевтически приемлемого носителя. Соединения данного изобретения также используются как коммерческие реагенты, которые эффективно связываются с ферментом гиразой В. В качестве коммерческих реагентов соединения данного изобретения и их производные можно использовать для блокирования активности гиразы В в биохимических и клеточных исследованиях бактериальной гиразы В или ее гомологов, или могут быть преобразованы для связывания со стабильной смолой в качестве связанного субстрата для целей аффинной хроматографии. Описанные и другие применения коммерческих ингибиоров гиразы В будут очевидны специалистам в данной области. Для того чтобы данное изобретение было более понятным, приведены следующие примеры. Указанные примеры представлены только с целью иллюстрации и никоим образом не предназначены для ограничения объема изобретения. Примеры синтеза Пример 1. Этиловый эфир 2-фенил-4-трифторметансульфонилокситиазол-5-карбоновой кислоты Исходное вещество, этиловый эфир 4-гидрокси-2-фенилтиазол-5-карбоновой кислоты, получали согласно способу, описанному в Kedersky et al., J. Med. Chem., 34, 2158 (1991). К раствору исходного вещества (2,3 ммоль) в СН 2 Сl2 (10 мл) при 0 С последовательно добавляли 2,6-лутидин (2,53 ммоль) и ангидрид трифторметансульфоновой кислоты (2,53 ммоль). Реакционную смесь перемешивали при температуре от 0 С до комнатной температуры в течение двух часов. Реакционную смесь разбавляли СН 2 Сl2 и последовательно промывали 5% NAHSO4, водой, NаНСО 3 и насыщенным солевым раствором, затем сушили над MgSO4 и концентрировали в вакууме. Хроматография на силикагеле неочищенного вещества давала 82% требуемого указанного в заголовке соединения в виде белого кристаллического твердого вещества, совместимого с 1H ЯМР (CDCl3):1,4 (т, 3 Н), 4,4 (кв, 2 Н), 7,4-7,6 (м, 3 Н), 7,95 (м, 2 Н). К раствору полученного выше этилового эфира 2-фенил-4-трифторметансульфонилокситиазол-5 карбоновой кислоты (0,75 ммоль) в толуоле (5 мл) добавляли пиперидин (4,5 ммоль). Реакционную смесь нагревали до 80 С в течение 2 ч. Затем смесь разбавляли этилацетатом, последовательно промывали водой и насыщенным солевым раствором и сушили над MgSO4. Храматография на силикагеле неочищенной смеси давала указанное в заголовке соединение (96%) в виде желтоватого масла. Пример 3. Метоксиметиламид 2-фенил-4-пиперидин-1-илтиазол-5-карбоновой кислоты Раствор гидрохлорида N,О-диметилгидроксиламина (3,62 ммоль) в сухом CH2Cl2 (5 мл) при 0 С по каплям обрабатывали чистым хлоридом диметилалюминия (3,62 ммоль) и полученную смесь перемешивали при 0 С в течение 0,5 ч. Затем смесь оставляли для нагревания до комнатной температуры перед добавлением по каплям полученного выше этилового эфира 2-фенил-4-пиперидин-1-илтиазол-5-карбоновой кислоты (0,724 ммоль) в СН 2 Сl2 (2 мл). Затем желтую смесь перемешивали при комнатной температуре в токе азота в течение одного часа и повторно охлаждали до 0 С. Смесь медленно гасили добавлением по каплям 2,0 нNaOH, нагревали до комнатной температуры и экстрагировали двумя порциями СН 2 Сl2. Органическую фазу последовательно промывали 1,0 н NaOH и насыщенным солевым раствором, сушили над MgSO4 и концентрировали в вакууме, чтобы получить желтое масло. Хроматография на силикагеле давала 3 в виде желтого воскового кристаллического твердого вещества (выход 98%). 1 Н ЯМР (CDCl3):3,35 (с, 3 Н) ,1,6-1,8 (м, 6 Н), 3,3 (с, 3 Н), 3,5 (м, 4 Н), 3,7 (с, 3 Н), 7,3-7,4 (м, 3 Н), 7,95 (м, 2 Н). Пример 4. 1-(2-Фенил-4-пиперидин-1-илтиазол-5-ил)этанон К раствору полученного выше метоксиметиламида 2-фенил-4-пиперидин-1-илтиазол-5-карбоновой кислоты (0,754 ммоль) в THF (5 мл) добавляли MeLiLiBr (0,83 ммоль) при 0 С. Реакционную смесь перемешивали до тех пор, пока реакция не завершалась, затем гасили добавлением насыщенного хлорида аммония и экстрагировали этилацетатом. Органическую фазу промывали насыщенным солевым раствором, сушили над MgSO4 и концентрировали в вакууме, чтобы получить коричневое масло. Хроматография на силикагеле давала указанное в заголовке соединение (72%) в виде желтоватого масла. 1H ЯМР Раствор полученного выше 1-(2-фенил-4-пиперидин-1-илтиазол-5-ил)этанона (0,545 ммоль) в сухомTHF (5 мл) по каплям обрабатывали 1,0 М трет-бутоксидом калия в растворе THF (0,654 ммоль). Суспензию перемешивали при комнатной температуре в токе азота в течение 15 мин. Добавляли диэтилоксалат(0,600 ммоль), а коричневую суспензию разбавляли дополнительным количеством THF (6 мл) и оставляли для перемешивания при комнатной температуре в течение 30 мин. Реакционную смесь гасили добавлением ледяной уксусной кислоты (0,710 ммоль) и этанола (5 мл). Растворитель удаляли в вакууме, отделяя оставшееся масло, которое растворяли в абсолютном EtOH (5 мл). Этанольный раствор обрабатывали моногидратом гидразина (0,655 ммоль) и смесь нагревали при 80 С в течение 1 ч. Полученную желтую суспензию концентрировали в вакууме, отделяя оставшееся масло, которое растворяли в этилацета- 22005680 те. Полученную органическую фазу промывали водой, насыщенным NаНСО 3 и насыщенным солевым раствором, затем сушили над MgSO4 и выпаривали в вакууме, чтобы получить желтое маслянистое твердое вещество. Хроматография на силикагеле давала указанное в заголовке соединение (59%) в виде желтого твердого вещества. 1 Н ЯМР (CDCl3):1,4 (т, 3 Н), 1,6-1,9 (6 Н), 3,1 (м, 4 Н), 4,4 (кв, 2 Н), 6,9 (шс, 1 Н),7,4-7,5 (м, 3 Н), 7,9 (м, 2 Н). Пример 6. Метиловый эфир 4-метоксиметил-2-фенилтиазол-5-карбоновой кислоты Раствор СН 3 О 2 ССН(Сl)СОСН 2 ОСН 3 (68 ммоль, 1,2 эквивал.), полученного согласно De Kimpe et al.,Synthesis, 188 (1986), в абсолютном EtOH (75 мл) обрабатывали тиобензамидом (7,8 г, 56,7 ммоль, 1,0 эквивал.) и полученную коричневую смесь кипятили с обратным холодильником в токе азота в течение 8 ч. Смесь распределяли между этилацетатом и насыщенным NаНСО 3. Органический слой дважды промывали водой и насыщенным солевым раствором, затем сушили над безводным сульфатом натрия и концентрировали в вакууме, чтобы получить коричневое масло. Хроматография на силикагеле с элюированием смесью (9:1) гексан-этилацетат давала 6,98 г (47%) указанного в заголовке соединения в виде желтого кристаллического твердого вещества. 1 Н ЯМР (CDCl3):3,6 (с, 3 Н), 3,9 (с, 3 Н) , 4,95 (с, 2 Н), 7,4-7,5 (м, 3 Н), 8,0 (м, 2 Н). Пример 7. Метоксиметиламид 4-метоксиметил-2-фенилтиазол-5-карбоновой кислотыCH2Cl2 (250 мл) при 0 С по каплям обрабатывали чистым хлоридом диметилалюминия (12,7 мл, 136,3 ммоль, 6,0 эквивал.) и полученную смесь перемешивали при 0 С в течение 2 ч, затем оставляли для нагревания до комнатной температуры. К полученной смеси по каплям добавляли раствор полученного выше метилового эфира 4-метоксиметил-2-фенилтиазол-5-карбоновой кислоты (5,98 г, 22,71 ммоль, 1,0 эквивал.) в CH2Cl2 (20 мл). Затем желтую смесь перемешивали при комнатной температуре в токе азота в течение одного часа и повторно охлаждали до 0 С. Смесь медленно гасили добавлением по каплям 2,0 нNaOH, нагревали до комнатной температуры и экстрагировали двумя порциями CH2Cl2. Органическую фазу последовательно промывали 1,0 н NaOH и насыщенным солевым раствором, сушили над MgSO4 и концентрировали в вакууме, чтобы получить желтое масло. В результате хроматографии на силикагеле,элюируя смесью (4:1) гексан-этилацетат, получали 6,5 г (97%) указанного в заголовке соединения в виде желтого воскового кристаллического твердого вещества. 1 Н ЯМР (CDCl3):3,35 (с, 3 Н), 3,5 (с, 3 Н), 3,7 К раствору полученного выше метоксиметиламида 4-метоксиметил-2-фенилтиазол-5-карбоновой кислоты (6,706 г, 22,9 ммоль, 1,0 эквивал.) в сухом THF (25 мл) при 0 С по каплям добавляли раствор 1,4 М бромида метилмагния в смеси (3:1) толуол-THF (32,7 мл, 45,8 ммоль, 2,0 эквивал.). Полученную желтовато-коричневую суспензию перемешивали в токе азота при комнатной температуре в течение 30 мин,затем гасили добавлением насыщенного хлорида аммония и экстрагировали этилацетатом. Органическую фазу промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме, чтобы получить коричневое масло. Хроматография на силикагеле с элюрованием градиента смеси гексан-этилацетат (9:1) до (4:1) давала указанное в заголовке соединение (6,033 г, 81%) в виде желтого кристаллического твердого вещества. 1 Н ЯМР (CDCl3):2,7 (с, 3 Н), 3,5 (с, 3 Н),4,9 (с, 2 Н), 7,4-7,5 (м, 3 Н), 8,0 (м, 2 Н). К раствору полученного выше 1-(4-метоксиметил-2-фенилтиазол-5-ил)этанона (5,22 г, 21,12 ммоль,1,0 эквивал.) в сухом THF (100 мл) при -15 С по каплям добавляли раствор 1,0 М трет-бутоксида калия вTHF (31,7 мл, 31,7 ммоль, 1,5 эквивал.) и суспензию перемешивали при комнатной температуре в токе азота в течение одного часа. Добавляли диэтилоксалат (4,4 мл, 31,7 ммоль, 1,5 эквивал.), коричевую суспензию разбавляли дополнительным количеством THF (60 мл) и оставляли для перемешивания при комнатной температуре в течение 30 мин. Смесь гасили добавлением ледяной уксусной кислоты (3,2 мл, 2,6 эквивал.). THF удаляли в вакууме, а оставшееся масло растворяли в абсолютном этаноле (175 мл) и обрабатывали моногидратом гидразина (1,4 мл, 30 ммоль, 1,4 эквивал.). Полученную смесь нагревали при 70 С в течение 3 ч. Полученную желтую суспензию концентрировали в вакууме, отделяя оставшееся масло, которое растворяли в этилацетате. Органическую фазу промывали водой, насыщенным NаНСО 3 и насыщенным солевым раствором, затем сушили над безводным сульфатом натрия и концентрировали в вакууме, чтобы получить желтое маслянистое твердое вещество. Хроматография на силикагеле с элюированием градиента смеси гексан-этилацетат (9:1) - (4:1) давала желтое твердое вещество, которое растирали с гексаном, фильтровали и сушили в вакууме для получения 3,69 г (51%) указанного в заголовке соединения в виде не совсем белого твердого вещества. 1H ЯМР (CDCl3):1,4 (т, 3 Н), 3,5 (с, 3 Н), 4,4 (кв,2 Н), 4,8 (с, 2 Н), 7,0 (с, 1 Н), 7,4 (м, 3 Н), 7,9-8,0 (м, 2 Н). Пример 10. Этиловый эфир 5-(4-бромметил-2-фенилтиазол-5-ил)-2 Н-пиразол-3-карбоновой кислоты Раствор, -78 С, полученного выше этилового эфира 5-(4-метоксиметил-2-фенилтиазол-5-ил)-2 Нпиразол-3-карбоновой кислоты (1,5 г, 4,37 ммоль, 1,0 эквивал.) в сухом CH2Cl2 (20 мл) обрабатывали раствором 1,0 М ВВr3 в СН 2 Сl2 (5,24 мл, 5,24 ммоль, 1,2 эквивал.) и смесь перемешивали при -78 С в течение 45 мин, затем оставляли для нагревания до комнатной температуры и перемешивали в течение одного часа. Реакционную смесь гасили добавлением насыщенного NаНСО 3, перемешивали в течение 30 мин, потом дважды экстрагировали СН 2 Сl2. Органическую фазу промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме для получения желтого твердого вещества. Хроматография на силикагеле с элюированием градиента смеси (3:2) - (1:1) гексан-этилацетат давала 610 мг (36%) указанного в заголовке соединения в виде не совсем белого твердого вещества. 1 Н ЯМР (CDCl3):1,4 (т, 3 Н),4,4 (кв, 2 Н), 4,9 (с, 2 Н), 7,2 (с, 1 Н), 7,4 (м, 3 Н), 7,95 (м, 2 Н), 11,1 (шс, 1 Н). Пример 11. Этиловый эфир 5-(4-морфолин-4-илметил-2-фенилтиазол-5-ил)-2 Н-пиразол-3-карбоновой кислоты Раствор полученного выше этилового эфира 5-(4-бромметил-2-фенилтиазол-5-ил)-2 Н-пиразол-3 карбоновой кислоты (20 мг) в сухом THF (1,0 мл) обрабатывали морфолином (2 капли) и Et3N (1 капля) и смесь перемешивали при комнатной температуре в токе азота в течение 2,5 ч. Реакционную смесь распределяли между этилацетатом и водой. Органический слой промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме, чтобы получить маслянистое твердое вещество. Хроматография на силикагеле с элюрованием градиента смеси гексан-ацетон(9:1) - (4:1) давала 18 мг (89%) указанного в заголовке соединения в виде белого твердого вещества. 1 Н ЯМР (CDCl3):1,45 (т, 3 Н), 2,7 (шм, 4 Н), 3,8 (шм, 4 Н), 3,9 (с, 2 Н), 4,45 (кв, 2 Н), 6,95 (с, 1 Н), 7,45 (м, 3 Н),7,9 (м, 2 Н). Смесь 10,0 г (72,9 ммоль) тиобензамида и 17,4 г (146 ммоль) диметилформамиддиметилацетали перемешивали при комнатной температуре в течение 2 ч. Летучие компоненты выпаривали при пониженном давлении. Остаток растворяли в этаноле (40 мл). К полученному раствору добавляли 1,0 г (109 ммоль) хлорацетона и смесь перемешивали при комнатной температуре в течение 3,5 ч. Реакционную смесь разбавляли этилацетатом и промывали дважды водным бикарбонатом натрия, один раз водой, один раз насыщенным солевым раствором и сушили над сульфатом натрия. Растворитель выпаривали при пониженном давлении, а остаток очищали с помощью хроматографии на силикагеле, используя смесь ацетонггексан 3:97 в качестве элюента, чтобы получить 3,5 г указанного в заголовке соединения (25%). 1 Н ЯМР (500 МГц, CDCl3)8,36 (с, 1 Н), 8,01 (д, 2 Н), 7,49 (м, 3 Н), 2,61 (с, 1 Н). Пример 13. Этиловый эфир 5-(2-фенилтиазол-5-ил)-2 Н-пиразол-3-карбоновой кислоты(0,98 ммоль) 1 М трет-бутоксида калия в тетрагидрофуране. Раствор оставляли для перемешивания в течение 0,5 ч. Добавляли 0,15 г (0,98 ммоль) диэтилоксалата и раствор оставляли для перемешивания в течение 2 ч. Реакцию останавливали водным хлоридом аммония и смесь разделяли этилацетатом. Органическую фазу дважды промывали водным хлоридом аммония, один раз водой, один раз насыщенным солевым раствором и сушили над сульфатом натрия. Растворитель выпаривали при пониженном давлении и остаток растворяли в этаноле (10 мл). К этанольному раствору добавляли 0,04 г (0,64 ммоль) ледяной уксусной кислоты с последующим добавлением 0,03 г (0,64 ммоль) моногидрата гидразина. Раствор оставляли для перемешивания в течение 3 ч при комнатной температуре. Растворитель выпаривали при пониженном давлении, а остаток очищали хроматографией на силикагеле, используя смесь гексан:этилацетат 9:1 в качестве элюента, чтобы получить 75 мг указанного в заголовке соединения (51%). 1 К смеси 0,03 г (0,10 ммоль) полученного выше этилового эфира 5-(2-фенилтиазол-5-ил)-2 Нпиразол-3-карбоновой кислоты в ацетонитриле (2 мл) и диметилформамиде (1,5 мл) добавляли 0,02 г(0,10 ммоль) N-бромсукцинамида. Реакционную смесь оставляли для перемешивания в течение 2 ч и разбавляли этилацетатом. Раствор 3 раза промывали водным бикарбонатом натрия, один раз насыщенным солевым раствором и сушили над сульфатом натрия. Растворитель выпаривали при пониженном давлении, а остаток очищали хроматографией на силикагеле, используя смесь гексан:этилацетат 9:1 в качестве элюента, чтобы получить 28 мг указанного в заголовке соединения (74%). 1 Н ЯМР (500 МГц,CDC13)8,53 (с, 1 Н), 8,01 (д, 2 Н), 7,48 (м, 3 Н), 4,47 (кв, 2 Н), 1,44 (т, 3 Н). Пример 15. Этиловый эфир 4-хлор-5-(2-фенилтиазол-5-ил)-2 Н-пиразол-3-карбоновой кислоты К раствору 25 мг (0,084 ммоль) полученного выше этилового эфира 5-(2-фенилтиазол-5-ил)-2 Нпиразол-3-карбоновой кислоты в дихлорметане добавляли 23 мг (0,168 ммоль) сульфурилхлорида и оставляли для перемешивания на всю ночь при комнатной температуре. Раствор разбавляли этилацетатом,один раз промывали водным бикарбонатом натрия, один раз водой, один раз насыщенным солевым раствором и сушили над сульфатом натрия. Растворитель выпаривали при пониженном давлении, а остаток очищали хроматографией на силикагеле, используя смесь этилацетат:гексан 7:93 в качестве элюента,чтобы получить 23 мг указанного в заголовке соединения (82%). 1 Н ЯМР (500 МГц, CDCl3)8,39 (с, 1 Н),7,94 (д, 2 Н), 7,40 (м, 2 Н), 4,40 (кв, 2 Н), 1,38 (т, 3 Н). К 15 мг (0,045 ммоль) полученного выше этилового эфира 4-хлор-5-(2-фенилтиазол-5-ил)-2 Нпиразол-3-карбоновой кислоты добавляли 45 мг (1,0 ммоль) 2 М этиламина в тетрагидрофуране с последующим добавлением 2 капель воды. Смесь нагревали до 60 С в герметично закрытой пробирке и оставляли для перемешивания на всю ночь. Растворитель выпаривали при пониженном давлении, а остаток очищали хроматографией на силикагеле, используя смесь этилацетат:гексан 2:5 в качестве элюента, чтобы получить 5 мг указанного в заголовке соединения (33%). 1 Н ЯМР (500 МГц, CDCl3) 88,39 (с, 1 Н), 8,00 К раствору 3,72 г (93 ммоль) гидроокиси натрия в воде (20 мл) при 0 С добавляли по каплям 3,72 г(23,2 ммоль) брома. Реакционную смесь оставляли для нагревания до комнатной температуры и перемешивали в течение 15 мин. Раствор добавляли к 1,05 г (5,17 ммоль) полученного выше 1-(2-фенилтиазол 5-ил)этанона в диоксане (50 мл) и оставляли для перемешивания в течение 3 ч. Раствор вливали в лед,подкисленный 1 н хлористо-водородной кислотой, и дважды экстрагировали этилацетатом. Объединенные органические части сушили над сульфатом магния, а растворитель выпаривали при пониженном давлении, чтобы получить 1,01 г (4,9 ммоль) карбоновой кислоты. К кислоте в THF (10 мл) добавляли 1,04 г (6,4 ммоль) 1,1-карбонилдиимидазола. Раствор нагревали до 50 С и оставляли для перемешивания в течение 1 ч. Раствор охлаждали до комнатной температуры. Добавляли 0,79 г (7,9 ммоль) триэтиламина и 0,672 г (6,9 ммоль) гидрохлорида N,О-диметилгидроксиламина и оставляли для перемешивания на всю ночь. Раствор разбавляли этилацетатом и промывали один раз водным бисульфатом калия, один раз водой, один раз насыщенным солевым раствором и сушили над сульфатом натрия. Растворитель выпаривали при пониженном давлении, а остаток очищали хроматографией на силикагеле, используя смесь этилацета:гексан 7:93 в качестве элюента, чтобы получить 0,66 г указанного в заголовке соединения (54%). 1 Н ЯМР (500 МГц, CDCl3)8,58 (с, 1 Н), 8,00 (м, 2 Н), 7,46 (м, 3 Н), 3,82 (с, 3 Н), 3,40 (с, 3 Н). Пример 18. 1-(2-Фенилтиазол-5-ил)пропан-1-он К раствору 0,32 г (1,3 ммоль) полученного выше метоксиметиламида 2-фенилтиазол-5-карбоновой кислоты в тетрагидрофуране при комнатной температуре добавляли 0,34 г (2,6 ммоль) 1 М бромида этилмагния в тетрагидрофуране. Реакционную смесь оставляли для перемешивания в течение одного часа. Реакцию останавливали водным хлоридом аммония и разделяли этилацетатом. Органическую фазу промывали один раз водным хлоридом аммония, один раз водой, один раз насыщенным солевым раствором и сушили над сульфатом натрия. Растворитель выпаривали при пониженном давлении, а остаток очищали хроматографией на силикагеле, используя смесь этилацетат:гексан 1:19, чтобы получить 0,26 г указанного в заголовке соединения (93%). 1 Н ЯМР (500 МГц, CDCl3)8,36 (с, 1 Н), 8,00 (м, 2 Н), 7,49 (м,3 Н), 2,98 (кв, 2 Н), 1,27 (т, 3 Н). Пример 19. Этиловый эфир 2-гидрокси-3-метил-4-оксо-4-(2-фенилтиазол-5-ил)масляной кислоты К раствору 0,26 г (1,2 ммоль) полученного выше 1-(2-фенилтиазол-5-ил)пропан-1-она, -78 С, добавляли 0,24 г (1,4 ммоль) 1 М бис(триметилсилил)амида лития в тетрагидрофуране. Смесь оставляли для перемешивания в течение 0,5 ч, а затем добавляли 0,38 г (1,5 ммоль) 1 М триизопропоксида хлортитана в гексане. Реакционную смесь оставляли для нагревания до -20 С и перемешивали в течение 15 мин. Реакционную смесь повторно охлаждали до -78 С и добавляли 0,25 г (0,24 ммоль) этилглиоксалата в толуоле (50%). Раствор нагревали до комнатной температуры и оставляли перемешиваться в течение 0,5 ч. Реакцию останавливали водным тетрагидратом тартрата калия-натрия и распределяли этилацетатом. Органическую фазу дважды промывали водным тетрагидратом тартрата калия-натрия, один раз водой, один раз насыщенным солевым раствором и сушили над сульфатом натрия. Растворитель выпаривали при пониженном давлении, а остаток очищали хроматографией на силикагеле, используя смесь этилаце- 26005680 тат:гексан 1:9, чтобы получить 0,15 г указанного в заголовке соединения (40%). 1 Н ЯМР (500 МГц,CDCl3)8,42 (с, 1 Н), 8,01 (д, 2 Н), 7,48 (м, 3 Н), 5,8 (м, 1 Н), 4,27 (кв, 2 Н), 3,75 (м, 1 Н), 3,28 (м, 1 Н), 1,37 (д,3 Н), 1,26 (т, 3 Н). Пример 20. Этиловый эфир 4-метил-5-(2-фенилтиазол-5-ил)-2 Н-пиразол-3-карбоновой кислоты Смесь 0,39 г (0,91 ммоль) периодинана Десс-Мартина (Dess-Martin) и 0,07 г (0,91 ммоль) третбутилового спирта в дихлорметане (2 мл) оставляли для перемешивания в течение 20 мин при комнатной температуре. Раствор охлаждали до 0 С и к нему добавляли 0,15 г (0,45 ммоль) полученного выше этилового эфира 2-гидрокси-3-метил-4-оксо-4-(2-фенилтиазол-5-ил)масляной кислоты в дихлорметане (2 мл). Реакционную смесь перемешивали при 0 С в течение 3 ч и гасили бисульфитом натрия в 50% водном бикарбонате натрия. Смесь разбавляли дихлорметаном и оставляли для перемешивания в течение 20 мин при комнатной температуре. Органическую фазу дважды промывали водным бикарбонатом натрия,один раз водой, один раз насыщенным солевым раствором и сушили над сульфатом натрия. Растворитель выпаривали при пониженном давлении и остаток растворяли в этиловом спирте (5 мл). Добавляли 41 мг (0,68 ммоль) ледяной уксусной кислоты с последующим добавлением 34 мг (0,68 ммоль) моногидрата гидразина. Раствор оставляли для перемешивания в течение 4 ч при комнатной температуре. Растворитель выпаривали при пониженном давлении и остаток очищали хроматографией на силикагеле,используя смесь этиловый спирт:дихлорметан 1:99 в качестве элюента, чтобы получить 0,035 г указанного в заголовке соединения (25%). 1 Н ЯМР (500 МГц, CDCl3)8,08 (с, 1 Н), 8,00 (д, 2 Н), 7,47 (м, 3 Н), 4,45 Суспензию подходящего коммерческого N-бензоил-DL-аланина (3,0 г, 15,5 ммоль, 1,0 эквивал.) в сухом бензоле (62 мл) и сухом дихлорметане (23 мл) по каплям обрабатывали чистым оксалилхлоридом(13,5 мл, 155 ммоль, 10 эквивал.) и белую суспензию перемешивали в течение ночи в токе азота при комнатной температуре. Затем полученную гомогенную желтую смесь выпаривали в вакууме до масла, дважды получали азеотроп с бензолом и выпаривали, чтобы получить неочищенный хлорангидрид кислоты в виде желтого масла. Продукт использовали непосредственно на следующей стадии без дальнейшей обработки. Смотри Crooks et al., J. Chem. Soc, Chem. Comm., 2335 (1995). Раствор 0 С, полученного выше неочищенного хлорангидрида кислоты (15,5 ммоль, 1,0 эквивал.) в сухом THF (50 мл) обрабатывали гидрохлоридом N,O-диметилгидроксиламина (2,27 г, 23,3 ммоль, 1,5 эксивал.) с последующей обработкой Et3N (6,5 мл, 46,5 ммоль, 3,0 эквивал.) и темно-коричневую суспензию перемешивали в токе азота в течение ночи. Смесь распределяли между этилацетатом и водой. Органическую фазу последовательно промывали 5% раствором KHSO4, водой и насыщенным солевым раствором, затем сушили над безводным сульфатом натрия. Концентрирование в вакууме давало неочищенное, коричневое масло. Неочищенное масло хроматографировали на силикагеле, используя градиентное элюирование смесью гексан-простой эфир (4:1) - (7:3), чтобы получить 1,82 г (48%) указанного в заголовке соединения в виде желтого кристаллического твердого вещества. 1 Н ЯМР: (CDCl3)2,5 (с, 3 Н),3,35 (с, 3 Н), 3,9 (с, 3 Н), 7,4-7,5 (м, 3 Н), 8,05 (м, 2 Н). Пример 22. Сложный трет-бутиловый эфир [4-(4-метил-2-фенилоксазол-5-ил)-4-оксобут-2 инил]карбаминовой кислоты Раствор -15 С, N-BOC-пропаргиламина (651 мг, 4,2 ммоль, 3,5 эквивал.) в сухом THF (12 мл) по каплям обрабатывали 1,6 М н-BuLi в растворе гексана (5,25 мл, 8,4 ммоль, 7,0 эквивал.) и бледно-желтый раствор дианиона перемешивали при -15 С в течение 30 мин в токе азота. Раствор полученного выше метоксиметиламида 4-метил-2-фенилоксазол-5-карбоновой кислоты (296 мг, 1,2 ммоль, 1,0 эквивал.) в сухом THF (3 мл) по каплям добавляли к раствору дианиона при -15 С и смесь перемешивали при 0 С в течение 2 ч в токе азота. Смесь гасили добавлением раствора 2 М NaH2PO4 (5 мл), нагревали до комнатной температуры, а затем экстрагировали этилацетатом. Органическую фазу промывали водой и насыщенным солевым раствором, затем сушили над безводным сульфатом натрия и концентрировали в вакууме, чтобы получить указанное в заголовке соединение в виде неочищенного, коричневого масла. Неочищенное масло сразу использовали на следующей стадии без дальнейшей очистки. Раствор полученного выше сложного трет-бутилового эфира [4-(4-метил-2-фенилоксазол-5-ил)-4 оксобут-2-инил]карбаминовой кислоты (1,2 ммоль) в абсолютном этаноле (7 мл) обрабатывали избытком моногидрата гидразина (6 капель) и коричневую смесь перемешивали при комнатной температуре в течение 30 мин. Смесь выпаривали в вакууме до масла и хроматографировали на силикагеле, элюируя градиентом смеси гексан-этилацетат (4:1). Получено 258 мг (61%) 3 в виде бледно-желтого твердого вещества с хорошим 1 Н ЯМР (CDCl3):1,55 (с, 9 Н), 2,5 (с, 3 Н), 4,35 (д, 2 Н), 5,2 (шт, 1 Н), 6,45 (с, 1 Н), 7,47,5 (м, 3 Н), 8,05 (м, 2 Н). Пример 24. С-[5-(4-метил-2-фенилоксазол-5-ил)-2 Н-пиразол-3-ил]метиламин Раствор полученного выше сложного трет-бутилового эфира [5-(4-метил-2-фенилоксазол-5-ил)-2 Нпиразол-3-илметил]карбаминовой кислоты (258 мг, 0,728 ммоль, 1,0 эквивал.) в сухом СН 2 Сl2 (4 мл) обрабатывали трифторуксусной кислотой (1 мл, избыток) и коричневую гомогенную смесь перемешивали в токе азота при комнатной температуре в течение одного часа. Смесь распределяли между СН 2 Сl2 и 1,0 нNaOH, органическую фазу промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме, чтобы получить 177 мг (96%) указанного в заголовке соединения в виде не совсем белого твердого вещества. Неочищенное твердое вещество использовали без дальнейшей очистки. Пример 25. Этиловый эфир [5-(4-метил-2-фенилоксазол-5-ил)-2 Н-пиразол-3-илметил]карбаминовой кислоты Гетерогенную смесь полученного выше С-[5-(4-метил-2-фенилоксазол-5-ил)-2 Н-пиразол-3-ил]метиламина(31 мг, 0,122 ммоль, 1,0 эквивал.) в этилацетате (0,5 мл) и 1,0 н NаНСО 3 (0,5 мл) обрабатывали избытком метилового эфира хлормуравьиной кислоты (5 капель) и смесь перемешивали при комнатной температуре в течение 30 мин. Смесь распределяли между этилацетатом и насыщенным NaHCO3. Органическую фазу промывали водой и насыщенным солевым раствором, затем сушили над безводным сульфатом натрия и выпаривали в вакууме, чтобы получить желтое твердое вещество. Хроматография на силикагеле с элюированием смесью гексанацетон (4:1) давала 28 мг (74%) указанного в заголовке соединения в виде белого твердого вещества. 1 Н ЯМР Исходный сложный кетоэфир PhCOCH(Cl)CO2Et получали согласно De Kimpe, et al., Synthesis, 188(1986). Исходный сложный кетоэфир (27 ммоль, 1,08 эквивал.) и бензамид (3,0 г, 25,0 ммоль, 1 эквивал.) нагревали неразбавленными при 150 С в течение 4 ч. Затем смесь распределяли между CH2Cl2 и насыщенным NаНСО 3. Органическую фазу промывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме. Оставшийся бензамид осаждали простым эфиром. Фильтрат концентрировали, а затем хроматографировали на силикагеле, элюируя смесью гексан-простой эфир (95:5), чтобы получить 500 мг указанного в заголовке соединения в виде белого твердого вещества. 1H ЯМР (CDCl3):1,4 (т, 3 Н), 4,4 (кв, 2 Н), 7,4-7,6 (м, 3 Н), 8,1 (дд, 1 Н), 8,25 (дд, 1 Н). Пример 27. 2,4-Дифенилоксазол-5-карбоновая кислота Раствор полученного выше этилового эфира 2,4-дифенидоксазол-5-карбоновой кислоты (500 мг,1,70 ммоль, 1,0 эквивал.) в диоксане (6 мл) обрабатывали 2 н NaOH (1,7 мл, 3,4 ммоль, 2,0 эквивал.) и- 28005680 смесь перемешивали в токе азота при комнатной температуре в течение ночи. Затем смесь распределяли между этилацетатом и 2,0 н НСl. Органическую фазу промывали 0,5 н НСl и насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме, чтобы получить 426 мг указанного в заголовке соединения в виде неочищенного желтого твердого вещества. Продукт немедленно использовали на следующей стадии без очистки. Пример 28. Метоксиметиламид 2,4-дифенилоксазол-5-карбоновой кислоты Раствор полученной выше 2,4-дифенилоксазол-5-карбоновой кислоты (427 мг, 1,61 ммоль, 1,0 эквивал.) в сухом THF обрабатывали карбонилдиимидазолом (340 мг, 2,09 ммоль, 1,3 эквивал.) и смесь нагревали при 50 С в течение 3 ч. Добавляли триэтиламин (360 мл, 2,58 ммоль, 1,6 эквивал.) и N,Oдиметилгидроксиламин-НСl (236 мг, 2,42 ммоль, 1,5 эквивал.) и смесь нагревали при 50 С в течение 3 ч. Смесь распределяли между этилацетатом и водой. Органическую фазу промывали 5% KHSO4 и насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме до коричневого масла. Неочищенное масло хроматографировали на силикагеле, элюируя смесью гексанпростой эфир (7:3), чтобы получить 371 мг (75%) указанного в заголовке соединения в виде коричневого кристаллического твердого вещества. 1H ЯМР (CDCl3):3,35 (с, 3 Н), 3,8 (с, 3 Н), 7,3-7,6 (м, 6 Н), 7,95 (дд,2 Н), 8,15 (дд, 2 Н). Пример 29. Сложный трет-бутиловый эфир [4-(2,4-дифенилоксазол-5-ил)-4-оксобут-2 инил]карбаминовой кислоты Раствор -15 С, N-BOC-пропаргиламина (641 мг, 4,13 ммоль, 3,5 эквивал.) в сухом THF (12 мл) обрабатывали по каплям 1,6 М н-BuLi в растворе гексана (5,16 мл, 8,3 ммоль, 7,0 эквивал.) и полученный бледно-желтый раствор дианиона перемешивали при -15 С в течение 30 мин в токе азота. Раствор полученного выше метоксиметиламида 2,4-дифенилоксазол-5-карбоновой кислоты (365 мг, 1,18 ммоль, 1,0 эквивал.) в сухом THF (3 мл) по каплям добавляли к раствору дианиона при -15 С и смесь перемешивали при 0 С в течение 2 ч в токе азота. Смесь гасили добавлением раствора 2 М NaH2PO4 (5 мл), затем нагревали до комнатной температуры и экстрагировали этилацетатом. Органическую фазу промывали водой и насыщенным солевым раствором, затем сушили над безводным сульфатом натрия и концентрировали в вакууме, чтобы получить указанное в заголовке соединение в виде неочищенного коричневого масла. Неочищенное масло, без очистки, использовали непосредственно на следующей стадии. Пример 30. Сложный трет-бутиловый эфир [5-(2,4-дифенилоксазол-5-ил)-2 Н-пиразол-3 илметил]карбаминовой кислоты Раствор полученного выше сложного трет-бутилового эфира [4-(2,4-дифенилоксазол-5-ил)-4 оксобут-2-инил]карбаминовой кислоты (1,2 ммоль) в абсолютном этаноле (7 мл) обрабатывали избытком моногидрата гидразина (6 капель) и коричневую смесь перемешивали при комнатной теспературе в течение ночи. Смесь концентрировали в вакууме до масла и хроматографировали на силикагеле, элюируя смесью гексан-этилацетат (4:1). Указанное в заголовке соединение (251 мг) получали в виде бледножелтого твердого вещества. 1 Н ЯМР (CDCl3):1,50 (с, 9 Н), 2,5 (с, 3 Н), 4,3 (м, 2 Н), 5,2 (шт, 1 Н), 6,5 (с,1 Н), 7,3-7,5 (м, 6 Н), 7,9 (м, 2 Н), 8,15 (м, 2 Н). Пример 31. Метиловый эфир [5-(2,4-дифенилоксазол-5-ил) 2 Н-пираэол-3-илметил]карбаминовой кислоты Раствор полученного выше сложного трет-бутилового эфира [5-(2,4-дифенилоксазол-5-ил)-2 Нпиразол-3-илметил]карбаминовой кислоты (251 мг, 1,0 эквивал.) в сухом CH2Cl2 (8 мл) обрабатывали трифторуксусной кислотой (2 мл, избыток) и коричневую гомогенную смесь перемешивали в токе азота при комнатной температуре в течение 1,5 ч. Смесь распределяли между СН 2 Сl2 и 1,0 н NaOH. Органиче- 29005680 скую фазу промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали в вакууме, чтобы получить 181 мг неочищенного бензиламина в виде светлокоричневого твердого вещества. Неочищенный бензиламин использовали без дальнейшей очистки. Гетерогенную смесь бензиламина (32 мг, 0,101 ммоль, 1,0 эквивал.) в этилацетате (1,5 мл) и 1,0 н NaHCO3(1,5 мл) обрабатывали избытком метилового эфира хлормуравьиной кислоты (5 капель) и смесь перемешивали при комнатной температуре в течение 30 мин. Смесь распределяли между этилацетатом и насыщенным NаНСО 3. Органическую фазу промывали водой и насыщенным солевым раствором, затем сушили над безводным сульфатом натрия и концентрировали в вакууме, чтобы получить желтое масло. Хроматография на силикагеле с элюированием градиентом смеси гексан-ацетон (85:15) - (4:1) давала 24 мг указанного в заголовке соединения в виде белого твердого вещества. 1 Н ЯМР (ДМСО-d6):3,6 (с,3 Н), 4,3 (3, 2 Н), 6,5 (с, 1 Н), 7,35-7,6 (м, 6 Н), 7,7 (шм, 1 Н), 8,1 (м, 2 Н), 8,2 (м, 2 Н). Пример 32. Этиловый эфир 5-фенил-2 Н-пиразол-3-карбоновой кислоты К смеси, комнатной температуры, ацетофенона (1,0 мл, 8,57 ммоль) и диэтилоксалата (1,75 мл,12,86 ммоль) в THF (15 мл) добавляли трет-бутоксид калия (8,57 мл 1,0 М раствора в трет-BuOH) в атмосфере азота. Полученную темную смесь перемешивали при комнатной температуре в течение двух часов. Затем неочищенную реакционную смесь разбавляли этилацетатом, гасили 6 н НСl, а потом разбавляли насыщенным солевым раствором и достаточным количеством воды, чтобы растворить все твердые вещества. Фазы разделяли и органическую фазу сушили над Na2SO4, фильтровали и концентрировали в вакууме. Неочищенный дикетоэфир разбавляли EtOH (10 мл), затем последовательно обрабатывали уксусной кислотой (2 мл) и гидразином (1 мл) и перемешивали при комнатной температуре в течение 1 ч. Неочищенную реакционную смесь концентрировали в вакууме до густого масла, разбавляли этилацетатом,последовательно промывали водой и насыщенным солевым раствором, сушили над Na2SO4, фильтровали, концентрировали в вакууме и проводили флэш-хроматографию (силикагель, градиент смеси гексан/этилацетат), чтобы получить указанное в заголовке соединение (1,76 г, выход 95%) в виде желтого твердого вещества. 1 Н ЯМР (CDCl3, 400 МГц):7,83 (д, 2 Н); 7,25 (дд, 2 Н); 7,28 (дд, 1 Н); 7,09 (с, 1 Н); 4,59 (кв, 2 Н); 1,39 (т, 3 Н). Пример 33. Этиловый эфир 2-этил-5-фенил-2 Н-пиразол-3-карбоновой кислоты К смеси 0 С полученного выше этилового эфира 5-фенил-2 Н-пиразол-3-карбоновой кислоты (350 мг, 1,62 ммоль) и иодэтана (260 мкл, 3,23 моль) в DMF (3 мл) добавляли чистый LiH (на кончике шпателя, избыток) в атмосфере азота. Полученную смесь нагревали вплоть до комнатной температуры и перемешивали в течение ночи. Неочищенную реакционную смесь охлаждали до 0 С, гасили водным NH4Cl,разбавляли этилацетатом и достаточным количеством воды, чтобы растворить все твердые вещества. Фазы разделяли и органическую фазу последовательно промывали водой и насыщенным солевым раствором, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Пространственные изомеры разделяли и очищали флэш-хроматографией (силикагель, градиент смеси гексан/этилацет), чтобы получить указанное в заголовке соединение (167 мг, выход 42%, Rf выше в смеси гексан/этилацетат) и нежелательный пространственный изомер (175 мг, выход 44%) в виде белых твердых веществ. 1 Н ЯМР К раствору комнатной температуры полученного выше этилового эфира 2-этил-5-фенил-2 Нпиразол-3-карбоновой кислоты (165 мг, 67 5 мкмоль) в МеОН (2 мл) добавляли водную NaOH (215 мкл 10 н раствора, 215 мкмоль) в атмосфере азота. Полученную смесь оставляли для перемешивания при комнатной температуре в течение ночи. Реакционную смесь подкисляли 6 н НСl, разбавляли этилацетатом и насыщенным солевым раствором и разделяли фазы. Органическую фазу сушили над Na2SO4,фильтровали и концентрировали в вакууме. Неочищенную кислоту суспендировали в THF (2 мл) и до- 30
МПК / Метки
МПК: A61K 31/4155, C07D 417/04, A61P 31/04
Метки: лечения, применение, бактериальной, гиразы, ингибиторы, инфекции
Код ссылки
<a href="https://eas.patents.su/30-5680-ingibitory-girazy-i-ih-primenenie-dlya-lecheniya-bakterialnojj-infekcii.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы гиразы и их применение для лечения бактериальной инфекции</a>
Способ лечения бактериальной инфекции и фармацевтическая композиция для его осуществления

Номер патента: 3699
Опубликовано: 28.08.2003
Авторы: Форд Чарльз В., Ваттс Джеффри Л.
МПК: A61K 31/422, A61P 31/04
Метки: бактериальной, лечения, способ, композиция, фармацевтическая, инфекции, осуществления
Формула / Реферат:
1. Способ лечения инфекции, выбранной из группы, состоящей из ушных инфекций, инфекций мягких тканей и акне и расположенной не на поверхности кожи полезного теплокровного млекопитающего, нуждающегося в таком лечении, который включает в себя нанесение на такой участок кожи, который не является пораженным участком, фармацевтической композиции, содержащей трансдермально эффективное количество оксазолидинона, выбранного из группы, состоящей из...
Способ лечения или предотвращения бактериальной респираторной или кишечной инфекции домашнего скота

Номер патента: 1530
Опубликовано: 23.04.2001
Авторы: Вилкенинг Роберт Д., Кропп Хельмут, Рэтклифф Рональд В., Фэррингтон Дэниэл О., Кларк Джеффри Н.
МПК: A61K 31/395, A61P 1/00
Метки: инфекции, скота, респираторной, кишечной, предотвращения, домашнего, лечения, способ, бактериальной
Формула / Реферат:
1. Способ лечения или предотвращения бактериальной респираторной или кишечной инфекции домашнего скота, который включает введение нуждающемуся в таком лечении или предотвращении животному терапевтически или профилактически эффективного количества 8а-азалида, причём организмом, вызывающим респираторную или кишечную инфекцию, является Раsteurellа spp., Actinibacillus spp., Haemophilus somnus, Mycoplasma spp., Treponema hyodysenterae или Salmonella...
Применение бактериальной комбинированной композиции для получения средства для профилактики и/или лечения гипероксалурии у людей и животных и способ профилактики и/или лечения указанного заболевания

Номер патента: 4257
Опубликовано: 26.02.2004
Автор: Де Симоне Клаудио
МПК: A61K 39/02, A23L 1/03, A61P 13/04...
Метки: способ, людей, заболевания, средства, бактериальной, получения, комбинированной, применение, животных, указанного, композиции, гипероксалурии, лечения, профилактики
Формула / Реферат:
1. Применение бактериальной комбинированной композиции, включающей в качестве штаммов бактерий (a) Streptococcus thermophilus в смеси с (b) по меньшей мере одним штаммом бактерий, выбранным из группы, состоящей из Lactobacillus brevis, Lactobacillus acidophilus, Lactobacillus plantarum, Bifidobacterium infantis, Bifidobacterium longum и Bifidobacterium breve или их смесей, для получения диетического, фармацевтического или ветеринарного средства...
Ингибиторы металлопротеаз матрикса, способы их получения, фармкомпозиция, применение и способ лечения

Номер патента: 2971
Опубликовано: 26.12.2002
Авторы: Фрэй Майкл Джонатан, Дикинсон Роджер Питер, Дэк Кевин Нейл
МПК: C07C 231/12, A61K 31/165, A61P 43/00...
Метки: способ, применение, способы, фармкомпозиция, получения, лечения, матрикса, ингибиторы, металлопротеаз
Формула / Реферат:
1. Соединение формулы (I) его фармацевтически приемлемая соль или сольват, где R1 представляет Н, ОН, С1-4алкил, С1-4алкокси или С2-4алкенил; R2 представляет C1-6алкил, необязательно замещенный фтором, индолилом, имидазолилом, группой SO2(С1-4алкил), С5-7циклоалкилом либо необязательно защищенной группой ОН, SH, CONH2, CO2H, NH2 или NHC(=NH)NH2, С5-7циклоалкил, необязательно замещенный C1-6алкилом, или бензил, необязательно замещенный...
Пептиды млекопитающих для лечения микробной инфекции

Номер патента: 2242
Опубликовано: 28.02.2002
Авторы: Дубник Бернард, Хоффман Брайан Ф.
МПК: A61P 31/04, A61K 31/74
Метки: микробной, пептиды, инфекции, лечения, млекопитающих
Формула / Реферат:
1. Способ уничтожения бактерий или грибков, включающий контактирование бактерий или грибков с эффективной антимикробной дозой белка гемоглобина млекопитающих, фрагмента белка гемоглобина или их полипептидных фрагментов, выбранных из группы, включающей a-цепь гемоглобина, не содержащую гема, b -цепь гемоглобина, не содержащую гема, фрагмент гемоглобина a-3, фрагмент гемоглобина b -2, последовательность аминокислот SEQ ID NO: 3, последовательность...
Предыдущий патент: Бициклические гетероциклы, содержащие эти соединения лекарственные средства, их применение и способ их получения
Следующий патент: Замещенные 1,2,3,4,5,6-гексагидро-2,6-метано-3-бензазоцины и их применение в качестве лекарственных средств
Случайный патент: Рекомбинантный ген m вируса гриппа b, кодирующий белок m1 с мутацией m86v