Циклические пептидные антигрибковые агенты
Номер патента: 464
Опубликовано: 26.08.1999
Авторы: Родригез Майкл Дж., Борромео Питер С., Джемисон Джеймс Э., Васудеван Венкатрагхаван, Тернер Уильям В.Мл.







Формула / Реферат
1. Соединение формулы I:

где R' обозначает водород, метил или NH2C(O)CH2-;
R" и R'" обозначают, независимо, метил или водород;
Rx1, Rx2, Ry1, Ry2, Ry3, Ry4 обозначают, независимо, гидрокси или водород;
R0 обозначает группу формулы:

R1 обозначает С1-С6 алкил, C1-C6 алкокси, фенил, п-галогенфенил, п-нитрофенил, фенокси, бензил, п-галогенбензил или п-нитробензил;
I) R2 обозначает группу формулы

где
A) R3 обозначает C1-C12 алкил, C1-C6 алкокси или хинолил;
B) R3 обозначает -O-(СН2)m-[O-(СН2)n]р-O(С1-С12алкил);
m и n, независимо, равны 2, 3 или 4;
р равно 0 или 1;
C) R3 обозначает -Y-(C1-C12 алкил);
Y обозначает -СуС- или -СН=СН-; или
D) R3 обозначает - O-(CH2)q-G -;
q равно 2, 3 или 4;
G обозначает C7-C10 бициклоалкил или С7-С14 трициклоалкил;
или
II) R2 является группой формулы

где Z обозначает -О-, -Су С-, -СН=СН-, -СН2-СН2-, -СН2- или связь;
A) R4 обозначает водород, C1-C12 алкил, замещенный C1-C12 алкил, С2-C12 алкенил, замещенный C2-C12 алкенил, C2-C12 алкинил, замещенный С2-C12 алкинил, C1-C12 алкокси, С3-С12 циклоалкил, С7-С10 бициклоалкил, C7-C14 трициклоалкил, С3-С12 циклоалкокси, нафтил, пиридил, тиенил, бензотиенил, хинолил или фенил; или
B) R4 обозначает фенил, замещенный амино, C1-C12 алкилтио, галогеном, C1-C12 алкилом, C2-C12 алкенилом, C2-C12 алкинилом, замещенным C1-C12 алкилом, замещенным C2-C12 алкенилом, замещенным C2-C12 алкинилом, C1-C12 алкокси, трифторметилом, фенилом, замещенным фенилом или фенилом, замещенным группой формулы -O-(CH2)m-[O-(CH2)n]p-O-(С1-С12 алкил), где m, n и р определены выше; или
C) R4 обозначает C1-C12 алкокси, замещенный галогеном, С3-С12 циклоалкилом, С7-С10 бициклоалкилом, C7-C14 трициклоалкилом, C1-C6 алкокси, C2-C12 алкинилом, амино, С1-С4 алкиламино, ди(С1-С4 алкил)амино, формамидо, C2-C12 алканоиламино или фенилом, замещенным группой формулы -O-(CH2)m-[O-(CH2)n]p-O-(C1-C12 алкил), где m, n и р определены выше; или
D) R4 обозначает -O-(CH2)r-W-R5;
r равно 2, 3 или 4;
W обозначает пирролидино, пиперидино или пиперазино;
R5 обозначает водород, C1-C12 алкил, С3-С12 циклоалкил, бензил или С3-С12циклоалкилметил; или
Е) R4 обозначает -Y1-R6;
Y1 обозначает -Су С- или -СН=СН-;
R6 обозначает С3-С12 циклоалкил, С7-С10 бициклоалкил, C7-C14 трициклоалкил, С3-С12 циклоалкенил, нафтил, бензотиазолил, тиенил, инданил, флуоренил или фенил, замещенный C1-C12 алкилтио, C2-C12 алкенилом, C2-C12 алкинилом, галоген(С1-С6 алкокси) или группой формулы -O-(CH2)r-W-R5, где r, W и R5 определены выше; или R6 обозначает фенил, замещенный группой формулы
-O-(CH2)m-[O-(CH2)n]p-O-(C1-C12 алкил), где m, n и р определены выше; или
F) R4 обозначает C1-C12 алкокси, замещенный группой формулы -NHC(O)R7;
R7 обозначает C1-C6 алкокси или фенил(С1-С6 алкокси); или
III) R2 обозначает группу формулы

где R8 обозначает C1-C12 алкокси или группу формулы -O-(СН2)m-[O-(СН2)n]р-O-(С1-С12 алкил), где m, n и р определены выше; или
IV) R2 обозначает группу формулы

где Y и R6 определены выше;
R9 обозначает фенил, С1-С12алкил или C1-C12 алкокси; или
V) R2 обозначает нафтоил, замещенный R4, где R4 определен выше;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, где
R', R" и R'" обозначают, каждый, метил;
Ry1, Ry2, Ry3 и Ry4 обозначают, каждый, гидрокси;
Rx1, Rx2 обозначают водород;
или его фармацевтически приемлемая соль.
3. Соединение по п.2, где R2 обозначает группу формулы

где Z обозначает -Су С- или связь;
A) R4 обозначает водород, C2-C12 алкинил, замещенный C2-C12 алкинил, C1-C12 алкокси, С3-С12 циклоалкокси или фенил; или
B) R4 обозначает фенил, замещенный C1-C12 алкокси или группу формулы -O-(СН2)2-O-(С1-С6алкил), или
C) R4 обозначает C1-C12 алкокси, замещенный С3-С12 циклоалкилом, С7-С14 трициклоалкилом, C7-C14 трициклоалкокси, C1-C6 алкокси, амино, ди(С1-С4 алкил)амино, формамидо или фенил, замещенный группой формулы -O-(СН2)2-O-(С1-С6алкил); или
D) R4 обозначает -O-(CH2)r-W-R5;
r равно 2 или 3;
W обозначает пиперидино;
R5 обозначает водород, С1-С12алкил, C3-C12 циклоалкил, бензил или С3-С12 циклоалкилметил; или
Е) R4 обозначает -Y1-R6;
Y1 обозначает -Су С-;
R6 обозначает фенил, замещенный C1-C12 алкилтио, C2-C12 алкенил, C2-C12 алкинил, галоген(С1-С6алкокси); или
R6 обозначает фенил, замещенный группой формулы -O-(CH2)r-W-R5, где r, W и R5 те же, чтю определены выше;
или R6 обозначает фенил, замещенный группой формулы -O-(СН2)2-O-(С1-С6алкил); или
F) R4 обозначает C1-C12 алкокси, замещенный группой формулы -NHC(O)R7;
R7 обозначает C1-C6 алкокси или фенил(С1-С6 алкокси), или его фармацевтически приемлемая соль.
4. Соединение по п.3, где R1 обозначает C1-C4 алкил, C1-C4 алкокси, фенил, п-хлорфенил, п-бромфенил или нитрофенил, бензил, п-хлорбензил, п-бромбензил или нитробензил; или его фармацевтически приемлемая соль.
5. Соединение по п.4, где R1 обозначает метил, этил, метокси, этокси, фенил, бензил;
или его фармацевтически приемлемая соль.
6. Соединение по п.5, где Z обозначает связь; R4 обозначает фенил, замещенный н-пентокси; и R1 обозначает метил; или его фармацевтически приемлемая соль.
7. Фармацевтический препарат, содержащий соединение формулы I или его фармацевтически приемлемую соль, указанные в любом из пп.1-6, объединенное с одним или несколькими фармацевтически приемлемыми носителями, растворителями или наполнителями.
8. Применение соединения формулы I или его фармацевтически приемлемой соли, указанных в любом из пп.1-6 в виде фармацевтического препарата.
9. Способ лечения грибковых инфекций у млекопитающих, который включает введение таким млекопитающим соединения формулы I или его фармацевтически приемлемой соли, указанных в любом из пп.1-6.
10. Способ получения соединения формулы I или его фармацевтически приемлемой соли, указанных в любом из пп.1-6, заключающийся во взаимодействии соединения формулы IC

где R обозначает водород,
R', R", R'", Rx1, Rx2, Ry1, Ry2, Ry3 и Ry4 и R2 указаны в п.1, с замещенной C1-C6 алкилфосфорной кислотой, фенилфосфорной кислотой, замещенным C1-C6 алкилфосфатом или фенилфосфатом; с последующим, при необходимости, выделением целевого продукта в виде фармацевтически приемлемой соли.
Текст
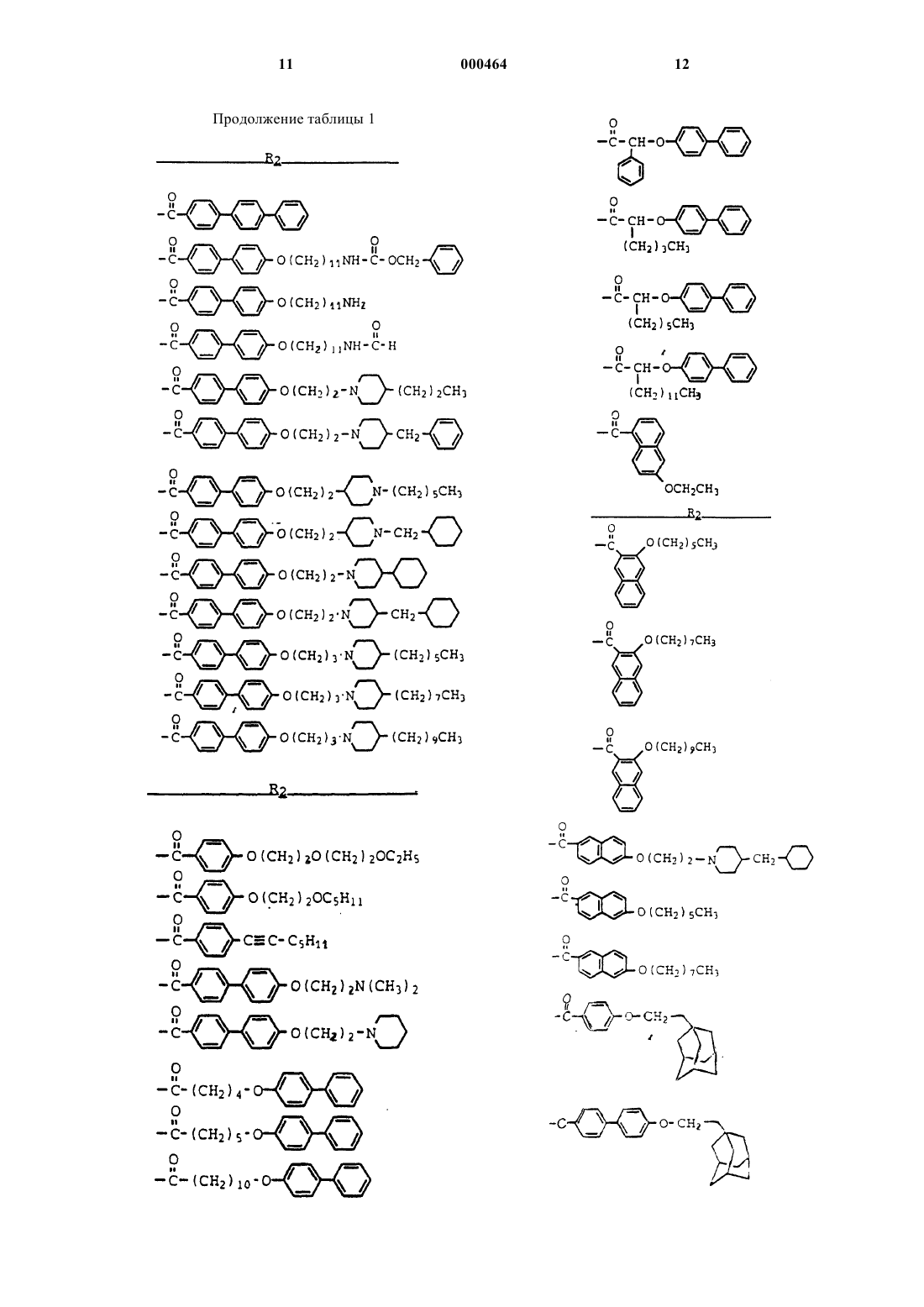
1 Настоящее изобретение относится к полусинтетическим циклическим пептидным соединениям, которые могут использоваться как антигрибковые и антипаразитарные средства и которые обладают улучшенной стабильностью и водорастворимостью. В частности, настоящее изобретение относится к производным эхинокандинового класса циклических пептидов, к способам лечения грибковых и паразитарных инфекций и к лекарственным формам, используемым при этих способах. Соединения охватываемые настоящим изобретением, являются полусинтетическими соединениями, полученными из циклических пептидов, которые продуцируются при культивировании различных микроорганизмов. К настоящему времени известен ряд циклических пептидов, включая эхинокандин В (А 30912 А),акулеацин, мулундокандин, спориофунгин, L671329 и S31794/F1. Обычно циклические пептиды могут быть структурно охарактеризованы как циклическое гексапептидное ядро с ацилированной аминогруппой на одной из аминокислот ядра. Как правило, эта аминогруппа ацилирована жирной кислотой, образуя боковую цепь вне ядра. Например, эхинокандин В имеет линолеоильную боковую цепь, в то время как акулеацин содержит пальмитоильную боковую цепь. Жирнокислотные боковые цепи могут быть удалены из циклического пептидного ядра с получением аминового ядра (например, соединение формулы I, ниже, где R2 является водородом). Аминогруппа может быть затем повторно ацилирована с получением полусинтетических соединений, таких как соединения, заявленные в настоящей заявке. Ядро эхинокандин В было повторно ацилировано некоторыми, не встречающимися в природе группами боковых цепей с получением ряда антигрибковых агентов (см. Debono, патент США 4293489). Одним из таких антигрибковых агентов является цилофунгин, который представлен соединением формулы I, где R', R", R'" обозначают метил, Rx1 и Rx2 обозначают гидрокси, Ry1, Ry2, Ry3 и Ry4 обозначают гидрокси, R0 обозначает гидрокси и R2 обозначает п(октилокси)бензоил. Настоящее изобретение относится к соединению общей формулы I:B) R4 обозначает фенил, замещенный амино, C1-C12 алкилтио, галогеном, C1-C12 алкилом,C2-C12 алкенилом, C2-C12 алкинилом, замещенным C1-C12 алкилом, замещенным C2-C12 алкенилом, замещенным C2-C12 алкинилом, C1-C12 алкокси, трифторметилом, фенилом, замещенным фенилом или фенилом, замещенным группой формулыC) R4 обозначает C1-C12 алкокси, замещенный галогеном, С 3-С 12 циклоалкилом, С 7-С 10 бициклоалкилом, C7-C14 трициклоалкилом, C1C6 алкокси, C2-C12 алкинилом, амино, C1-C4 алкиламино, ди(С 1-С 4 алкил)амино, формамидо,C2-C12 алканоиламино или фенилом, замещен 3 ным группой формулы -O-(CH2)m-[O-(CH2)n]p-O(C1-C12 алкил), где m, n и р определены выше; илиR6 обозначает фенил, замещенный группой общей формулы -O-(CH2)m-[O-(CH2)n]p-O-(C1C12 алкил), где m, n и р определены выше; илиF) R4 обозначает C1-C12 алкокси, замещенный группой формулы -NHC(O)R7; где R8 обозначает C1-C12 алкокси или группу формулы -O-(СН 2)m-[O-(СН 2)n]р-O-(С 1-С 12 алкил), где m, n и р определены выше; илиV) R2 обозначает нафтоил, замещенный R4,где R4 определен выше; или его фармацевтически приемлемая соль. Также предложены фармацевтические препараты, способ лечения грибковых и паразитарных инфекционных заболеваний, в которых используются соединения по изобретению и способ получения соединений общей формулыI. Как здесь используется, термин "C1-C12 алкил" относится к линейной или разветвленной алкильной цепи, имеющей от одного до двенадцати атомов углерода. Типичные C1-C12 алкилильные группы включают метил, этил, пропил,изопропил, бутил, втор-бутил, трет-бутил, пентил, 5-метилпентил, гексил, гептил, 3,3 диметилгептил, октил, 2-метилоктил, нонил,децил, ундецил, додецил и тому подобное. Термин "C1-C12 алкил" включает в себя определения терминов "C1-C6 алкил" и "C1-С 4 алкил". Термин "галоген" относится к хлору, фтору, брому и йоду. Термин "C2-C12 алкенил" относится к линейной или разветвленной алкенильной цепи,имеющей от 2 до 12 атомов углерода. ТипичныеC2-C12 алкенильные группы включают этенил, 1 пропен-2-ил, 3-бутен-1-ил, 1-бутен-2-ил, 1 бутен-1-ил, 1-пентен-3-ил, 2-гексен-3-ил, 1 децен-2-ил, 2-децен-5-ил и тому подобное. Термин "C2-C12 алкинил" относится к линейной или разветвленной алкинильной цепи,имеющей от двух до двенадцати атомов углерода. Типичные C2-C12 алкинильные группы включают этинил, 1-пропин-1-ил, 1-пропин-2 ил, 1-бутин-1-ил, 1-бутин-3-ил, 1-пентен-3-ил,4-пентен-2-ил, 1-гексин-3-ил, 3-гексин-1-ил, 5 метил-3-гексин-1-ил, 5-октин-1-ил, 7-октин-1 ил, 4-децин-1-ил, 6-децин-1-ил и тому подобное. Термин "C1-C12 алкилтио" относится к линейной или разветвленной алкильной цепи,имеющей от двух до двенадцати атомов углерода, присоединенную к атому серы. ТипичныеC1-C12 алкилтиогруппы включают метилтио,этилтио, пропилтио, изопропилтио, бутилтио, 3 метилгептилтио, октилтио, 5,5-диметилгексилтио и тому подобное. Термин "C1-C12 алкокси" относится к линейной или разветвленной алкильной цепи,имеющей от двух до двенадцати атомов углерода, присоединенную к атому кислорода. Типичные C1-C12 алкилокси группы включают метокси, этилокси, пропокси, бутокси, втор-бутокси,пентокси, 5-метилгексокси, гептокси, октилокси, децилокси, додецилокси и тому подобное. Термин "C1-C12 алкил" включает в себя определение терминов "C1-С 6 алкилокси" и "C1-C4 алкокси". Термин "замещенный C1-C12 алкил", "замещенный C2-C12 алкенил" и "замещенный C2C12 алкинил" относятся к конкретным радикалам, замещенным 1 или 2 заместителями, неза 5 висимо выбранным из галогена, гидрокси, защищенного гидрокси, амино, защищенного амино, C1-C7 ацилокси, нитро, карбокси, защищенного карбокси, карбамоила, карбамоилокси,циано, метилсульфониламино, фенила, замещенного фенила или C1-C12 алкокси. Термин "замещенный фенил" относится к фенильной группе, замещенной 1, 2 или 3 заместителями, независимо выбранными из галогена, гидрокси, защищенного гидрокси, циано,нитро, C1-C12 алкила, C1-C12 алкокси, карбокси,защищенного карбокси, карбоксиметила, гидроксиметила, амино, аминометила, трифторметила или N-метилсульфониламино. Термин "С 3-С 12 циклоалкил" относится к насыщенной углеводородной кольцевой структуре, имеющей от трех до двенадцати атомов углерода. Типичные С 3-С 12 циклоалкильные группы включают циклопропил, циклобутил,циклопентил, циклогексил и циклогептил, циклооктил и тому подобное. Термин "С 3-С 12 циклоалкокси" означает циклоалкильную группу, присоединенную к атому кислорода. Типичные С 3-С 12 циклоалкокси группы включают циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси и циклогептилокси и тому подобное. Термин "С 3-С 12 циклоалкенил" означает углеводородную кольцевую структуру, имеющую от трех до двенадцати атомов углерода, по меньшей мере, с одной двойной связью. Типичные С 3-С 12 циклоалкенильные группы включают циклопропенил, циклобутенил, циклопентенил и тому подобное. Термин "метил (С 3-С 12 циклоалкил)" относится к С 3-С 12 циклоалкильной группе, которая замещена метильной группой. Типичные метил(С 3-С 12 циклоалкильные) группы включают 2 метилциклопропил, 2-метилциклобутил, 3 метилциклопентил, 4-метилциклогексил и тому подобное. Термин "C1-C4 алкиламино" означает линейную или разветвленную алкиламиновую цепь, имеющую от одного до четырех атомов углерода, присоединенную к атому азота. Типичные C1-C4 алкиламиногруппы включают метиламино, этиламино, пропиламино, изопропиламино, бутиламино, втор-бутиламино и тому подобное. Термин "ди(С 1-С 4 алкил)амино" означает ди(С 1-С 4 алкил)аминовую цепь, имеющую две линейные или разветвленные алкильные цепочки из от одного до четырех атомов углерода,присоединенных к общему атому азота. Типичные ди(С 1-С 4 алкил)аминогруппы включают диметиламино диэтиламино, этилметиламино,метилизопропиламино, дипропиламино, дибутиламино,метилбутиламино,третбутилизопропиламино, ди-трет-бутиламино и тому подобное. Термин "C2-C12 алканоил" означает линейную или разветвленную алкильную цепь, 000464 6 имеющую от одного до одиннадцати атомов углерода, присоединенную к карбонильному радикалу. Типичные C2-C12 алканоильные группы включают этаноил, пропаноил, изопропаноил, бутаноил, изобутаноил, втор-бутаноил, третбутаноил, пентаноил и тому подобное. Термин "C2-C12 алканоиламино" означает линейную или разветвленную алкильную группу, присоединенную к карбониламино группе. Типичные C2-C12 алканоиламино группы включают этаноиламино, пропаноиламино, изопропаноиламино, бутаноиламино, изобутаноиламино, втор-бутаноиламино, трет-бутаноиламино,пентаноиламино и тому подобное. Термин "С 7-С 10 бициклоалкил" означает два конденсированных циклоалкильных кольца,имеющих всего от семи до десяти атомов углерода, и термин "C7-C14 трициклоалкил" означает три конденсированных циклоалкильных кольца,имеющих всего от семи до четырнадцати атомов углерода. Типичные "С 7-С 10 бициклоалкил" и "C7-C14 трициклоалкил" группы включают бицикло[2,2,1]гепт-2-ил, бицикло[2,2,1]гепт-4 ен-2-ил,бицикло-[3,1,1]нон-3-ил,бицикло[3,3,1]нон-2-ил, бицикло[3,2,1]окт-2-ил, бицикло-[2,2,2]окт-2-ил, бицикло[2,2,2]окт-5-ен-2-ил,адамантил и тому подобное. Термин "гидрокси защитная группа" означает заместитель гидрокси группы, который обычно используют для блокировки или защиты функциональной способности гидроксигруппы,в то время как реакция протекает по другим функциональным группам соединения. Примеры таких гидрокси защитных групп включают тетрагидропиранил, 2-метоксипроп-2-ил, 1 этоксиэт-1-ил, метоксиметил, -метоксиэтоксиметил, метилтиометил, трет-бутил, трет-амил,тритил, 4-метокситритил, 4,4'-диметокситритил,4,4',4"-триметокситритил, бензил, аллил, триметилсилил, (трет-бутил)диметилсилил и 2,2,2 трихлорэтоксикарбонил и тому подобное. Типы защитных групп не являются существенными при условии, что производное гидроксигруппы остается стабильным в условиях проведения последующей реакции (или реакций) и может быть удален в нужный момент без разрушения оставшейся части молекулы. Предпочтительной гидроксизащитной группой является триметилсилил. Другие примеры гидроксизащитных групп описаны в Greene T.W.,"Protective Groups"защищенный гидрокси" относится к гидрокси группе, связанной с одной из перечисленных выше гидроксизащитных групп. Термин "дидеокси" означает соединения формулы I, где Rx1 и Rx2, каждый, означает водород. Термин "ингибирование", т.е. метод ингибирования паразитарной или грибковой активности, включает остановку, замедление либо торможение, или предупреждение в целях про 7 филактики роста, или любых сопутствующих характеристик и результатов жизнедеятельности паразита и грибка. Термин "контактирование", то есть контактирование соединения изобретения с паразитом или грибком, включает объединение или частичное слияние, или видимое касание или взаимное прикосновение соединения по изобретению с паразитом или грибком. Однако, этот термин не накладывает никаких дальнейших ограничений на процесс, такой как механизм ингибирования и способы устанавливают с целью осуществления идеи изобретения, которая заключается в ингибировании паразитарной и грибковой активности под действием соединений и присущих им противопаразитарных и противогрибковых свойства или, иными словами, соединения, использованные в заявленных методах, являются причиной такого ингибирования. Термин "фармацевтически приемлемая соль", как он здесь используется, относится к солям соединений изобретения вышеуказанной формулы, которые по существу не токсичны для живых организмов. Типичные фармацевтически приемлемые соли включают такие соли, которые получают путем взаимодействия соединений по настоящему изобретению с неорганической или органической кислотой или неорганическим основанием. Такие соли известны как соли добавления кислот и соли добавления оснований. Кислотами, обычно применяемыми для получения солей добавления кислот, являются неорганические кислоты, такие как хлороводородная кислота, бромоводородная кислота, йодоводородная кислота, серная кислота, фосфорная кислота и тому подобное, и органические кислоты, такие как п-толуолсульфоновая кислота, метансульфоновая кислота, щавелевая кислота, п-бромфенилсульфоновая кислота, карбоновая кислота, янтарная кислота, лимонная кислота, бензойная кислота, уксусная кислота и тому подобное. Примерами таких фармацевтически приемлемых солей являются сульфат,пиросульфат, бисульфат, сульфит, бисульфит,фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирофосфат, хлорид, бромид, йодид,ацетат, пропионат, деканоат, каприлат, акрилат,формиат, изобутират, капроат, гептаноат, пропиолат, оксалат, малонат, сукцинат, суберат,себацат, фумарат, малеат, бутин-1,4-диоат, гексин-1,5-диоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фталат, сульфонат, ксиленсульфонат,фенилацетат, фенилпропионат, фенилбутират,цитрат, лактат, -гидроксибутират, гликолят,тартрат, метансульфонат, пропансульфонат,нафталин-1-сульфонат, нафталин-2-сульфонат,манделат и тому подобное. Предпочтительными фармацевтически приемлемыми солями добавления кислот являются такие, которые образо 000464 8 ваны с неорганическими кислотами, такими как хлороводородная кислота и бромоводородная кислота, и с органическими кислотами, такими как малеиновая кислота и метансульфоновая кислота. Соли добавления основания включают полученные из неорганических оснований, таких как гидроксиды, карбонаты и бикарбонаты аммония или щелочных или щелочноземельных металлов и тому подобное. Такие основания,которые могут использоваться при получении солей по данному изобретению включают гидроксид натрия, гидроксид калия, гидроксид аммония, карбонат калия, карбонат натрия, бикарбонат натрия, бикарбонат калия, гидроксид кальция, карбонат кальция и тому подобное. Особенно предпочтительны формы соли калия и натрия. Следует отметить, что природа противоиона, являющегося частью любой соли соединения по изобретению, не имеет определяющего значения, при условии, что соль, как целое является фармацевтически приемлемой и при условии, что противоион не привносит нежелательных качеств в соль, как целое. Типичные примеры ацильных групп у R2 в формуле I включают бензоил, замещенный полиоксаалкильной группой, такой как 2 метоксиэтокси (р равно 0, m равно 1), 2 этоксиэтокси (р равно 0, m равно 2), 2-(2 этоксиэтокси)этокси (р равно 1, m равно 2, n равно 2), 3-(2-этокси-этокси)пропокси, 4-(2 метоксиэтокси)бутокси и тому подобное, или бензоил, замещенный алкильными группами(-CC-(C1-C12 алкил, такими как пропинил,бутинил, гексинил, ундецинил и тому подобное,или цис или транс алкенильными группами являются дифениловые эфиры (Z обозначает(Z обозначает связь). Примеры дифенилэфирных групп включают 4-(4-бутоксифенокси)-бензоил, 4-(4-гексоксифенокси)бензоил, 4-(4-этоксифенокси) бензоил, 4-(4-фенилоксифенокси)бензоил, 4-[4-(4 додецилоксифенокси)]бензоил, 4-[4-(3-диметиламинопропокси)фенокси]бензоил и тому подобное. Примеры дифенилацетиленовых и стильбеновых групп включают 4-стирилбензоил, 4-(4 метоксистирил)бензоил,4-(4-бутоксистирил) бензоил, 4-(4-фенилэтинил)бензоил, 4-(4-это 9 ксифенилэтинил)бензоил, 4-(4-циклогексилоксифенилэтинил)бензоил и тому подобное. Примеры бифенильных групп включают 4[4-бутокси)фенил]бензоил, 4-[4-циклобутилметокси)фенил]бензоил,4-[4-циклопентилметокси)фенил]-бензоил,4-[4-циклогексилэтокси) фенил]бензоил, 4-[4-гексокси)фенил]-бензоил,4-фенилбензоил,4-[4-(11-аминоундецилокси) фенил]бензоил,4-[4-(11-формамидоундецилокси)фенил]бензоил, 4-[4-изопентокси)фенил]бензоил и тому подобное. Примеры бифенильных групп, где R4 обозначает -O-(CH2)r-W-R5 включают 4-[4-[2-(Nциклoгeкcилпипepидинo-4-ил)этoкcи]фeнил] бeнзoил,4-[4-[2-(N-гексилпиперидино-4-ил) этокси]фенил]бензоил, 4-[4-[2-(4-бензилпиперидино)этокси]фенил]бензоил, 4-[4-[2-(4-циклогексилпиперидино)этокси]-фенил]бензоил и тому подобное. Примеры бифенильных и дифенилэфирных групп, где R4 обозначает -Y1-R6 включают 4-[4-фенилэтинил)фенил]бензоил, 4-[4-фенилэтинил)-фенокси]бензоил, 4-[4-(гексинил) фенил]бензоил, 4-[4-(стирил)фенокси]-бензоил, 4[4-[4-(метилпиперидино)этокси]фенилэтинил] фенил]бензоил и тому подобное. Ацильные группы, где R4 обозначает-O-(CH2)r-W-R5, могут образовывать соли добавления кислот с помощью основных аминогрупп пиперидиновых и пиперазиновых гетероциклических групп с органическими или минеральными кислотами, такими как хлороводородная кислота, бромоводородная кислота, серная кислота и фосфорная кислота, и с органическими кислотами, такими как сульфоновая кислота, бензолсульфоновая кислота, толуолсульфоновая кислота, метансульфоновая кислота,уксусная кислота, хлоруксусная кислота, трифторуксуная кислота, бензойная кислота, изофталевая кислота, салициловая кислота, лимонная кислота, яблочная кислота, янтарная кислота,малоновая кислота и тому подобное. В таблице I, приведенной ниже, представлены дальнейшие примеры ацильных групп, R2,входящих в состав циклических пептидов общей формулы I.C1-C12 алкилтио, C2-C12 алкенил, C2-C12 алкинил,галоген(С 1-С 6 алкокси) или группой формулы O-(CH2)r-W-R5, где r, W и R5 определены выше; илиR6 обозначает фенил, замещенный группой формулы -O-(CH2)m-[O-(CH2)n]p-O-(C1-C12 алкил), где m, n и р указаны выше; илиF) R4 обозначает C1-C12 алкокси, замещенный группой формулы -NHC(O)R7; R7 обозначает C1-C6 алкокси или фенил(С 1-С 6 алкокси); или фармацевтически приемлемую соль. Более предпочтительными являются ацильные группы, R2 формулы: где Z обозначает -СС- или связь; или их фармацевтически приемлемые соли. В таблице 2, приведенной ниже, представлен перечень предпочтительных ацильных групп, R2, входящих в состав циклических пептидов общей формулы IC2-C12 алкинил, C1-C12 алкокси, трифторметил,фенил, замещенный фенил или группу формулыC) R4 обозначает C1-C2 алкокси, замещенный галогеном, С 3-С 12 циклоалкил, С 7-С 10 бициклоалкил, C7-C14 трициклоалкил, C1-C6 алкокси, C2-C12 алкинил, амино, C1-C4 алкиламино,ди(С 1-С 4 алкил)амино, формамидо, C2-C12 алканоиламино или фенил, замещенный группой формулы -O-(CH2)m-[O-(CH2)n]p-O-(C1-C12 алкил), где m, n и р определены выше; или 15 Более предпочтительными из этих соединений являются соединения формулы I, где:Ry1, Ry2, Ry3, Ry4, каждый, обозначает гидрокси; или их фармацевтически приемлемые соли. Из этих соединений более предпочтительными являются также соединения формулы, где Реакционная схема IRx1 и Rx2 обозначают водород; или их фармацевтически приемлемые соли. Соединения формулы I могут быть получены в соответствии со схемой реакции I, как описано далее. 17 где Rnat является боковой цепью природного циклического пептида;Rв обозначает гидроксизащитную группу иR', R", R'", Rx1, Rx2, Ry1, Ry2, Ry3, Ry4, R, R0 и R2 определены выше. Представленная выше схема реакции I заканчивается проведением реакций А-С (или АС'") в этом порядке. Когда реакция завершена,промежуточное соединение может быть выделено при помощи общеизвестных в настоящее время методов, например, соединение может быть кристаллизовано и затем собрано фильтрованием. Или реакционный растворитель может быть удален экстракцией, выпариванием или декантацией. При желании, промежуточное соединение может быть очищено обычными методами, такими как кристаллизация или хроматография на твердом носителе, таком как силикагель, окись алюминия и тому подобное,перед проведением следующей стадии по схеме реакции. В реакции IА природный циклический пептид формулы (IА) деацилируют известным способом с получением аминоядра формулы(IВ). Эту реакцию обычно проводят с использованием ферментативного ацилирования, подвергая природный циклический пептид действию фермента деацилазы. Фермент деацилазу можно получить из микроорганизма Actinoplanes utahensis и использовать, по существу, так, как описано в патенте США 4293482 и 4304716,включенные здесь в качестве ссылки. Получить фермент деацилазу также можно из микроорганизмов Pseudomonas. Деацилирование может быть осуществлено с использованием цельных клеток Actinoplanes utahensis или Pseudomonas,или их неочищенных или очищенных ферментов, или с использованием иммобилизованных ферментов. Смотри European Patent Application0460882 (11 декабря 1991). Примерами природных циклических пептидов, которые могут быть использованы в качестве исходных продуктов включают акулеацин (пальмитоильная боковая цепь), тетрагидроэхинокандин В (стеароильная боковая цепь), мулундокандин (разветвленная C15 боковая цепь), L-671329 (разветвленная C16 боковая цепь), S-31794/F1 (тетрадеканоильная боковая цепь), спориофунгин(пальмитоильная боковая цепь) и тому подобное. Предпочтительным природным циклическим пептидом является эхинокандин В (соединение формулы IА), где R', R" и R'", каждый,обозначает метил, Rx1 и Rx2, каждый, обозначает гидрокси, Ry1, Ry2, Ry3 и Ry4, каждый, обозначает гидрокси, R обозначает гидрокси и R2 обозначает линолеоил. В реакции IB полученное аминоядро затем реацилируют по известным методам для получения соединения формулы I, где R2 является ацильной группой, описанной выше. 18 Например аминоядро может быть ацилировано взаимодействием с подходящим замещенным ацилгалогенидом, предпочтительно в присутствии акцептора кислоты, такого как третичный амин, например, триэтиламин. Реакцию обычно проводят при температуре приблизительно от -20 до 25 С. Типичные растворители для этой реакции включают полярные апротонные растворители, такие как диоксан и диметилформамид. Выбор растворителя не является определяющим при условии, что используемый растворитель инертен по отношению к протекающей реакции, и вещества, участвующие в реакции, достаточно растворимы для осуществления желаемой реакции. Аминоядро может быть ацилировано путем взаимодействия с подходящей замещенной карбоновой кислотой в присутствии связывающего агента. Типичные связывающие агенты включают дициклогексил-карбодиимид (ДЦК),N,N'-карбонилдиимидазол, бис(2-оксо-3-оксазолидинил)фосфинхлорид(EEDQ),бензотриазол-1-илокситрипиррамидинфосфоний гексафторфосфат (РуВОР) и тому подобное. Кроме того, аминоядро может быть ацилировано активированным эфиром карбоновой кислоты (RCOOH), таким как эфир карбоновой кислоты формулы R2-COOH и п-нитрофенил,2,4,5-трихлорфенил, гидроксибензотриазол гидрат (НОВТ Н 2O), пентафторфенол, Nгидроксисукцинимид и тому подобное. Предпочтительными ацилирующими группами являются активные эфиры карбоновой кислотыR2-COOH, такие как 2,4,5-трихлорфенилэфир и НОВТ эфир. Обычно реакцию проводят в течение от одного до шестидесяти пяти часов при температуре приблизительно от 0 до 30 С в апротонном растворителе. Реакция обычно завершается приблизительно за время от двадцати четырех до сорока восьми часов при температуре приблизительно от 15 до 30 С. Типичными растворителями для этой реакции являются тетрагидрофуран и диметилформамид или смесь таких растворителей. Обычно аминоядро используют в эквимолярном количестве по отношению к активированному эфиру, или с небольшим избытком аминоядра. В реакции IС соединение формулы (IС) фосфорилируют путем взаимодействия с соответственно замещенным алкил- или фенилфосфатом с получением соединения формулы I, гдеC1-С 6 алкокси или фенокси, или путем взаимодействия с соответственно замещенной алкилили фенилфосфоновой кислотой с получением соединения формулы I, где R0 обозначает-P(O)2OH-R1, где R1 является C1-C6 алкилом,соответственно замещенным фенильным или бензильным остатком. 19 Реакцию проводят в присутствии основания, такого как триметилсиланоат лития(LHMDS), пиридин и тому подобное. Обычно,реакцию проводят в течение одного часа при температуре приблизительно от около -30 С до около 0 С в апротонном растворителе, таком как тетрагидрофуран и диметилформамид. Реакцию обычно завершают в течение около пятидесяти минут в этих условиях. Фосфат или фосфатный реагент обычно используют в от эквимольного соотношения до одного мольного избытка аминоядра в присутствии эквимольного или слегка избыточного количества основания. Фосфорилирование аминоядра с незащищенными аминальными гидроксигруппами (Rx1 и Rx2) обычно проводят при температуре от около -30 до около -15 С. Альтернативно, по реакции IC' ацилированное ядро формулы (IC), где Rx1 и/или Rx2 обозначают гидрокси, может быть необязательно защищено с помощью гидроксизащитной группы, используя способы известные в данной области. Например, взаимодействие обычно проводят путем объединения соединения формулы (IC) с подходящей гидроксизащитной группой в присутствии катализатора при температуре в области от около 0 до около 40 С в течение от около одного до пяти часов в растворителе, инертном по отношению ко всем компонентам реакции. Гидроксизащитная группа обычно используется в количестве, изменяющемся от около эквимольного до около 100 мольного избытка по отношению к соединению формулы (IC), предпочтительно, в большом молярном избытке. Подходящие катализаторы включают сильные кислоты, такие как птолуолсульфокислота, камфорсульфоновая кислота (КСК), хлороводородная кислота, серная кислота, трифторуксусная кислота и тому подобное. Типичные растворители, подходящие для использования в данной реакции, включают любые органические растворители, такие как диоксан. Выбор растворителя не является определяющим при условии, что используемый растворитель инертен по отношению к проходящей реакции и реагенты достаточно растворимы для осуществления желаемой реакции. Реакцию,предпочтительно, проводят при температуре в интервале от около 20 до около 30 С в течение от около 2 до 4 ч. Нет необходимости защищать дидеокси соединения формулы (IC), то есть такие соединения формулы (IC), где Rx1 и Rx2 обозначают водород. Реакцию IC" проводят как описано выше для реакции IC, см. выше. В реакции IC'" гидроксизащитные группы, используемые для защиты аминальных гидроксигрупп в реакции IC', описанной выше, удаляют в соответствии с известными способами в данной области с получением целевого соединения формулы I. Например, защитные группы могут быть удалены путем 20 взаимодействия с кислотой Льюиса в обоюдно инертном органическом растворителе, таком как метиленхлорид. Примеры кислот Льюиса включают триметилсилилбромид, эфират трифторбора и тому подобное. Обычно реакцию проводят при температуре от около 0 до около 40 С,предпочтительно, при температуре от около 20 до около 30 С. Предпочтительной кислотой Льюиса является эфират трифторбора. Циклические пептиды, используемые для получения соединений по настоящему изобретению, могут быть получены ферментацией известных микроорганизмов. Например, циклический пептид формулы (IВ), где R', R" и R'" являются метилом, Rx1 и Rx2 являются гидрокси,Ry1, Ry2, Ry3 и Ry4 являются гидрокси и R является гидрокси (циклическое ядро соответствует А 30912 А), может быть получен способом, подробно описанным Abbott et al., патент США,сер.4293482, который включен здесь в качестве ссылки. Циклический пептид формулы(IB), где R', R" и R'" обозначают метил, Rx1 обозначает гидрокси, Rx2 обозначает водород, Ry1,Ry2, Ry3 и Ry4 обозначают гидрокси и R обозначает гидрокси (циклическое ядро соответствует А-30912 В), может быть получен способом, подробно описанным Abbott et al., патент США сер.4299763, который включен здесь в качестве ссылки. Циклический пептид формулы (IB), гдеR', R" и R'" обозначают метил, Rx1 и Rx2 обозначают водород, Ry1, Ry3 и Ry4 обозначают гидрокси, Ry2 обозначает водород и R обозначает гидрокси (циклическое ядро соответствует А 30912D), может быть получен способом, подробно описанным Abbott et al., патент США сер.4299762, который включен здесь в качестве ссылки. Акулеацин может быть получен способом, подробно описанным Mizuno et al., патент США сер.3978210, который включен здесь в качестве ссылки. Циклический пептид формулы(IB), где R' обозначает -CH2C(O)NH2, R" обозначает метил, R"' обозначает водород, Rx1, Rx2,Ry1, Ry2, Ry3 и Ry4 обозначают гидрокси, R обозначает гидрокси, может быть получен деацилированием циклического пептида, полученного способом, подробно описанным Shieh-Shung etal., патент США сер.5198421, который включен здесь в качестве ссылки. Дидеоксисоединения формулы I получены удалением бензильных и аминальных гидрокси групп (Rx1 и Rx2, соответственно). Гидрокси группы могут быть удалены путем обработки нон-дидеокси соединения формулы I (где R2 обозначает водород или ацил) сильной кислотой и восстановителем при температуре между -5 С и 70 С в подходящем растворителе. Типичные сильные кислоты включают трихлоруксусную кислоту или эфират трифторбора. Предпочтительной сильной кислотой является трифторуксусная кислота. Типичные восстановители включают цианоборгидрид натрия или триэтилсилан. Предпочтительным восстановителем яв 21 ляется триэтилсилан. Подходящие растворители включают метиленхлорид, хлороформ или уксусную кислоту, предпочтительно метиленхлорид. Сильная кислота должна присутствовать в количестве от 2 до 60 молей на моль субстрата,и восстановитель должен присутствовать в количестве от 2 до 60 молей на моль субстрата. Этим способом осуществляется селективное удаление аминальных и бензильных гидроксигрупп.R2-COOH кислотные предшественники получают гидролизом нитрила формулы R-CN эфире или формулы R2-COO(C1-C4 алкил). Нитрильные и сложноэфирные промежуточные соединения могут быть получены с использованием хорошо известных способов в данной области. Например, нитрильные и сложноэфирные промежуточные соединения, где R2 обозначает алкоксиарильный радикал, могут быть получены с использованием способа А или способа В,описанных ниже. Способ А. Один эквивалент алкилбромида, йодида или п-толуолсульфоната добавляют к смеси,содержащей один эквивалент основания, такого как трет-бутоксид калия или карбонат калия, и один эквивалент гидрокси арильного соединения в 200-300 мл ацетонитрила. Полученную реакционную смесь кипятят с обратным холодильником в течение приблизительно 6 ч и затем концентрируют в вакууме до получения остатка. Этот остаток растворяют в смеси диэтилового эфира и 2 н. раствора гидроксида натрия. Полученные слои разделяют и органический слой высушивают над сульфатом магния,фильтруют и высушивают с получением желаемого алкоксиарильного продукта. Способ В. Один эквивалент диэтилазодикарбоксилата в течение более десяти минут добавляют по каплям при комнатной температуре к смеси, содержащей один эквивалент гидроксиарильного соединения, один эквивалент алкилового спирта и один эквивалент трифенилфосфина в 200-300 мл тетрагидрофурана. По истечении приблизительно семнадцати часов раствор удаляют в вакууме до получения остатка. Этот остаток растворяют в диэтиловом эфире и полученную смесь промывают 2 н. раствором гидроксида натрия, высушивают над сульфатом магния,фильтруют и концентрируют с получением продукта, который затем кристаллизуют из смеси диэтиловый эфир/пентан, или, если продукт содержит третичный амин, получают его гидрохлоридную соль и кристаллизуют из смеси метанол/этилацетат. Нитрильные и сложноэфирные промежуточные соединения, где R2 обозначает алкинильный или алкенильный арильный остаток,могут быть получены по способу С, описанному ниже. 22 Способ С. Смесь, содержащую два эквивалента триэтиламина, 0,05 эквивалента дихлорида палладия, 0,1 эквивалента трифенилфосфина, 0,025 эквивалента йодида меди (I) и один эквивалент алкина или два эквивалента алкена, добавляют к одному эквиваленту арилбромида, -йодида или трифторметансульфоната в ацетонитриле (600 мл/0,1 моль арильного реагента) в атмосфере азота. Полученную смесь кипятят с обратным холодильником в течение приблизительно семнадцати часов и затем выпаривают растворитель в вакууме до получения остатка. Этот остаток переводят во взвесь в 300 мл диэтилового эфира и затем смесь фильтруют для удаления образовавшегося твердого продукта. Фильтрат промывают 1 н. раствором соляной кислоты, высушивают над сульфатом магния, фильтруют и затем сушат с получением целевого продукта. Промежуточные сложноэфирные соединения, где R2 обозначает терфенильную группу,могут быть получены по способу D, описанному ниже. Способ D. 1. Приготовление бороновой кислоты. Бутиллитий (1,2 эквивалента) добавляют к одному эквиваленту охлажденного (-78 С) арилгалогенида в тетрагидрофуране. После приблизительно пятнадцати минут добавляют два эквивалента триизопропилбората. После приблизительно десяти минут реакционную смесь нагревают до комнатной температуры и затем гасят реакцию путем добавления воды, после чего добавляют раствор 1 н. соляной кислоты. Образовавшиеся соли разделяют и органический слой концентрируют в вакууме с получением твердого остатка. Этот твердый остаток собирают фильтрованием и затем промывают гексаном с получением чистого продукта бороновой кислоты. 2. Приготовление терфенилового эфира Тетракис(трифенилфосфин)палладий (0,03 эквивалента) добавляют к смеси, содержащей один эквивалент арилборной кислоты, 1,5 эквивалента карбоната калия и один эквивалент метил 4-йодбензоата (или трихлорфенилового эфира йодбензоата) в толуоле, продутым азотом. Полученную реакционную смесь кипятят с обратным холодильником в течение приблизительно семи часов, а затем декантируют для удаления карбоната калия и высушивают в вакууме с получением осадка. Этот осадок растирают в ацетонитриле и затем фильтруют с получением желаемого твердого продукта. Арилнитрилы и сложные эфиры, описанные выше, могут быть преобразованы в соответствующие карбоновые кислоты гидролизом по способу Е или по способу F, описанному ниже. Способ Е. Арилнитрил растворяют в этаноле и избытке 50%-ного гидроксида натрия и кипятят с обратным холодильником в течение приблизительно двух часов. К полученной реакционной смеси добавляют воду до выпадения твердого осадка. Этот твердый продукт собирают фильтрацией, добавляют его к смеси диоксан/6 н. соляная кислота и полученную смесь кипятят с обратным холодильником в течение приблизительно семнадцати часов. Когда реакция, в основном, закончена,продукт карбоновой кислоты кристаллизуют путем добавления воды, фильтруют и высушивают в вакууме. Способ F. Избыток 2 н. раствора гидроксида натрия добавляют к ариловому эфиру в метаноле, полученный раствор кипятят с обратным холодильником в течение приблизительно пяти часов и затем подкисляют добавлением избытка соляной кислоты. К полученной смеси добавляют воду до выпадения твердого продукта(карбоновая кислота). Карбоновую кислоту собирают фильтрацией и высушивают в вакууме. Карбоновые кислоты могут быть преобразованы в соответствующие 2,4,5-трихлорфениловые эфиры по способу G, описанному ниже. Такие активированные эфиры затем используют для ацилирования аминоядра, как описано выше в схеме реакции IС. Способ G. Смесь, содержащую один эквивалент арилкарбоновой кислоты, один эквивалент 2,4,5-трихлорфенола и один эквивалент N,Nдициклогексилкарбодиимида (ДЦК) в метиленхлориде перемешивают в течение приблизительно семнадцати часов и затем фильтруют. Фильтрат концентрируют с получением остатка. Этот остаток растворяют в диэтиловом эфире,фильтруют и добавляют пентан до начала кри 24 сталлизации. Кристаллический продукт собирают фильтрацией и высушивают в вакууме. В последующих приготовлениях и примерах описано, как синтезируют соединения по настоящему изобретению. Термины точка плавления, спектры протонного ядерного магнитного резонанса, масс-спектры, инфракрасные спектры, ультрафиолетовые спектры, элементный анализ, высоко эффективная жидкостная хроматография и тонкослойная хроматография обозначены аббревиатурами "т.п.", "ЯМР","МС", "ИК", "УФ", "Анализ", "ВЭЖХ" и "ТСХ",соответственно. Кроме того, в ИК спектрах указаны лишь представляющие интерес, а не все наблюдаемые максимумы поглощения. При обсуждении ЯМР спектров используются следующие аббревиатуры: "с" обозначает синглет, "д" обозначает дублет, "дд" означает дублет дублетов, "т" обозначает триплет, "кв" обозначает квадруплет, "м" обозначает мультиплет, "дм" обозначает дублет мультиплетов, и(Гц). ЯМР данные относятся к свободному основанию целевого соединения, если это не оговорено особо. Спектры ядерного магнитного резонанса были получены на приборе General Electric QE300 300 МГц. Величины химических сдвигов выражены в значениях дельта(миллионные доли относительно тетраметилсилана). Приготовление 1 Следующие нитрильные и эфирные промежуточные соединения, где R2 обозначает алкоксиарильную группу, получают, по существу,в соответствии со способом А, подробно описанным выше. Приготовление 2 Следующие нитрильные и эфирные промежуточные соединения, где R2 обозначает алкоксиарильный остаток, получают в полном соответствии со способом В, подробно описанным выше. Таблица В Приготовление 3 Следующие эфирные промежуточные соединения, где R2 обозначает алкинильный или алкенильный арильный остаток, получают в соответствии со способом С, подробно описанным выше. Таблица С Приготовление 4 Следующие эфирные промежуточные соединения, где R2 обозначает терфенильный радикал, были приготовлены, главным образом, в соответствии со способом D, подробно описанным выше. Таблица D.I Приготовление 5 Следующие активированные эфиры получают в соответствии со способом G, подробно описанным выше. Таблица G Пример 1. N-ацилирование циклических пептидных ядер. Производные N-ацильных циклических пептидов, перечисленных в таблице 3 ниже,получают растворением ядра эхинокандина В(А-30912 А) (соединение формулы IB, где R',R" и R'" обозначают, каждый, метил, Rx1 и Rx2 обозначают, каждый, гидрокси, Ry1, Ry2, Ry3 иRy4 обозначают, каждый, гидрокси и R обозначает гидрокси) и активированных сложноэфирных (эфир 2,4,5-трихлорфенола) промежуточных соединений, описанных в приготовлении 6,в 25-50 мл диметилформамида. Полученную реакционную смесь перемешивают приблизительно 17-65 ч при комнатной температуре и затем удаляют растворитель в вакууме с получением осадка. Этот осадок суспендируют в эфире, собирают фильтрацией, промывают метиленхлоридом и затем растворяют в метаноле в смеси 1:1 (об/об) ацетонитрил/вода. Полученный раствор подвергают обращенно-фазовой ВЭЖХ (С 18, элюент 20-40% водный ацетонитрил, содержащий 0,5% одноосновного фосфата аммония (в/об); 20 мл/мин, 230 нм). После удаления непрореагировавшего ядра А 30912 А целевой продукт элюируют из колонки, используя в качестве элюента водный ацетонитрил. Фракции, содержащие целевой продукт, объединяют и затем концентрируют в вакууме или лиофилизуют с получением целевого ацилированного ядра. Продукт можно проанализировать с использованием обращенно-фазовой ВЭЖХ (С 18,40% водный ацетонитрил, содержащий 0,5% одноосновного фосфата аммония (в/об); 2 мл/мин, 230 нм) или с использованием МС(60,2 ммоль) ядра А 30912 А 26,0 г (48,2 ммоль) 2,4,5-трихлорфенольного эфира (4-пентилокси]-1,1',4',1"-терфенил]-4-карбоновой кислоты в 8,5 л диметилформамида. Полученной реакционной смеси дают взаимодействовать около сорока восьми часов и затем концентрируют в вакууме и очищают с использованием ВЭЖХ; в результате получают 18 г соединения 3II. МС (FAB): 1140, 5103 (М+1). Соединения А-РР (представленное а таблице 3, ниже) получают по существу так, как описано выше. Пример 2. Дидеоксицилофунгин. Раствор 96 мл (602 ммоль) триэтилсилана в 50 мл метиленхлорида добавляют к суспензии 10,00 г (9,71 ммоль) цилофунгина в 100 мл метиленхлорида. Затем медленно в течение более 15 мин, добавляют раствор 46,4 мл (602 ммоль) трифторуксусной кислоты в 50 мл метиленхлорида. Полученный раствор перемешивают при комнатной температуре в течение приблизительно двух часов и затем концентрируют в вакууме с получением осадка. Этот осадок растирают в диэтиловом эфире и затем очищают с использованием обращенно-фазовой ВЭЖХ(С 18; градиентный элюент 10-20% ацетонитрил в воде (об/об), 500 psi). Фракции, содержащие целевое соединение, объединяют, концентри руют в вакууме и затем лиофилизируют из пдиоксана с получением указанного в заголовке целевого соединения. Выход: 6,66 г (67,8%). МС (FAB) для C49P72N7O15. Расч. 998,5086. Найден. 998,512. УФ: 39 К смеси 5 г (4,4 ммоль) соединения из таблицы 3II и 17 мл трифторуксуной кислоты в 250 мл метиленхлорида добавляют 35 мл триэтилсилана. Когда реакция, в основном, заканчивается, по данным ВЭЖХ (С 18, элюент 55% ацетонитрил; 2 мл/мин, 280 нм; RT (исходный продукт) = 4,19 мин; RT (продукт) = 6,40 мин), реакционную смесь концентрируют в вакууме с получением твердого продукта. Этот твердый продукт переводят во взвесь в 100 мл 50% водного ацетона и затем растворяют, доводя рН смеси до около 7. Полученный раствор выливают в большой объем воды (приблизительно 1 л),что приводит к высаживанию белого осадка. Этот твердый продукт выделяют фильтрацией через стеклянный фильтр, промывают диэтиловым эфиром и затем высушивают в вакууме при 55 С с получением 3,718 г указанного в заголовке соединения. Чтобы собрать оставшийся на фильтре осадок, фильтр промывают метанолом, который затем собирают и высушивают в вакууме; при этом получают дополнительно 0,154 г указанного в заголовке соединения. Выход: 3,872 г (79%). МС (FAB) m/e 1108,7(M). ВЭЖХ: (элюент 55% ацетонитрил; 2 мл/мин, 280 нм).RT = 6,43 мин. Пример 4. Получение дидеокси циклических гексапептидов. Следующие дидеоксисоединения были получены в соответствии со способом, подробно описанным в примере 3, используя расчетные количества соединения формулы IC, где R', R" и 40 А. Защита. К раствору 2,00 г (1,75 ммоль) соединения таблицы 3II в 50 мл диоксана при комнатной температуре добавляют 25 мл (175 ммоль) 2(триметилсилил)этанола и п-толуолсульфоновую кислоту (15 мольн. процентов). Получают реакционную смесь и оставляют при комнатной температуре приблизительно на три часа. Когда реакция, в основном, завершается, как показывают данные ВЭЖХ, реакцию гасят добавлением твердого бикарбоната натрия и фильтруют, указанное в заголовке желаемое соединение выделяют из фильтра, используя обращенно-фазовую ВЭЖХ (50% ацетонитрил/50% вода; 50 мл/мин; 280 нм). Выход: 807 мг. В. Получение этилфосфонатного производного. К охлажденному (0 С) раствору 234,1 мг(0,191 ммоль), указанного в заголовке примера 5 А соединения, в 10 мл тетрагидрофурана добавляют по каплям 0,21 мл (0,210 ммоль) 1,0 М раствора бис(триметилсилил)амида лития(LHMDS) в гексане. Полученную смесь перемешивают в течение приблизительно двадцати минут, после чего добавляют к ней по каплям 24,5 мкл (0,223 ммоль) дихлорангидрида этилфосфоновой кислоты. Реакционную смесь перемешивают в течение приблизительно тридцати минут, гасят реакцию добавлением 1 мл воды и затем концентрируют в вакууме с получением твердого продукта. Выход: 42 мг. ВЭЖХ (50% ацетонитрил/50% вода; 50 мл/мин; 280 нм).RT= 1,37. С. Снятие защиты. К смеси 40,0 мг (0,028 ммоль), указанного в заголовке примера 5 В соединения в 20 мл метиленхлорида, добавляют по каплям 35 мкл(0,28 ммоль) эфирата трифторида бора. Полученную реакционную смесь оставляют взаимодействовать приблизительно на тридцать минут и затем гасят реакцию добавлением 1,0 мл воды с получением белого осадка. Реакционную смесь растирают в диэтиловом эфире и затем фильтруют с получением светло-желтого твердого вещества. Выход: 12 мг, MC(FAB): 1238,6 (M+Li). Пример 6. Получение соединения, где R',R" и R"' обозначают, каждый, метил; Rx1 и Rx2 обозначают, каждый, гидрокси; Ry1,Ry2, Ry3, Ry4 обозначают, каждый, гидрокси; R0 обозначает метилфосфонат и R2 обозначает ацильную группу, представленную в таблице 3II. А. Получение производного метилфосфоната. Указанное в подзаголовке соединение получают в соответствии со способом, подробно описанным выше, в примере 5 В, с использованием 271,1 мг (0,221 ммоль) соединения примера 5 А, 0,24 мл (0,24 ммоль) 1,0 М раствораLHMDS в гексане и 35,3 мл (0,266 ммоль) дихлорангидрида метилфосфоновой кислоты в 10 мл тетрагидрофурана с получением 40 мг сырого продукта, который затем используют без дополнительной очистки. В. Снятие защиты. Указанное в подзаголовке соединение получают в соответствии со способом, подробно описанным в примере 5 С, с использованием соединения, выделенного в примере 6 А и 35 мкл(0,28 ммоль) эфирата трифторида бора с получением твердого продукта бело-серого цвета. МС (FAB): 1200,5(М-Н 2O). Пример 7. Получение соединения, где R',R" и R'", каждый, обозначают метил, Rx1 и Rx2 обозначают, каждый, гидрокси, Ry1, Ry2, Ry3, Ry4 обозначают, каждый, гидрокси, R0 обозначает фенилфосфонат и R2 обозначает ацильную группу, представленную в таблице 3II. А. Получение производного фенилфосфоната. Указанное в подзаголовке соединение получают в соответствии со способом, подробно описанным выше, в примере 5 В, с использованием 359,6 мг (0,294 ммоль) указанного в заголовке примера 5 А соединения, 0,333 мл (0,323 ммоль) 1,0 М раствора LHMDS в гексане и 50 мкл дихлорангидрида фосфоновой кислоты с получением 52 мг сырого продукта, используемого без дополнительной очистки. В. Снятие защиты. Указанное в подзаголовке соединение получают в соответствии со способом, подробно описанным в примере 5 С, с использованием соединения, выделенного в примере 6 А и 361 мкл эфирата трифторида бора с получением твердого продукта желтоватого цвета. Выход:32 мг. МС (FAB): 1262,4(М-Н 2O). Пример 8. Получение соединения, где R',R" и R'" обозначают, каждый, метил; Rx1 и Rx2 обозначают, каждый, водород; Ry1, Ry2, Ry3, Ry4 обозначают, каждый, гидрокси; R0 обозначает изопропилфосфат и R2 обозначает ацильную группу, представленную в таблице 3II. А. Изопропилдихлорфосфат. К раствору 1,55 мл (16,6 ммоль) оксихлорида фосфора в 5 мл тетрахлорида углерода добавляют в атмосфере азота 1,28 мл (16,6 ммоль) изопропанола, что приводит к повышению температуры реакционной смеси. Для поддержания температуры в интервале от 20 до 35 С необходимо использовать баню со льдом. Затем реакционную смесь оставляют при комнатной температуре приблизительно на семь часов в атмосфере азота. Полученную смесь конденсируют в вакууме с получением прозрачного масла. Выход: 1,9 г (65 %). В. Получение изопропилфосфатного производного. К охлажденному (0 С) раствору, содержащему 0,5 г (0,45 ммоль) вещества, указанного в 42 заголовке примера 3, в 10 мл тетрагидрофурана и 54 мкл (0,54 ммоль) триметилсиланолата лития (LiOTMS), добавляют 88 мг (0,5 ммоль) вещества, указанного в подзаголовке примера 8 А. Полученную реакционную смесь перемешивают в течение приблизительно десяти минут. К реакционной смеси добавляют дополнительныйLiOTMS до тех пор, пока рН смеси не становится основным. Когда реакция, в основном, заканчивается, на что указывают данные ВЭЖХ, реакцию гасят добавлением воды, реакционную смесь перемешивают в течение приблизительно одного часа и затем концентрируют в вакууме с получением твердого желтого продукта. Этот твердый продукт очищают с использованием ВЭЖХ (элюент 45% ацетонитрил/45% вода/10% трифторуксусная кислота (1% водный раствор с получением белого твердого продукта. Выход: 105 мг. МС (FAB): 1230,4 (М+). Пример 9. Получение соединения, где R',R" и R"' обозначают, каждый, метил; Rx1 и Rx2 обозначают, каждый, водород; Ry1, Ry2, Ry3, Ry4 обозначают, каждый, гидрокси; R0 обозначает бутилфосфат и R2 обозначает ацильную группу,представленную в таблице 3II. А. Бутилдихлорфосфат. Указанное в подзаголовке целевое соединение получают в соответствии со способом,подробно описанным в примере 8 А, используя 1,25 мл (13,5 ммоль) оксихлорида фосфора и 1 г(13,5 ммоль) бутанола в 5 мл тетрахлорида с получением бесцветного масла. Выход: 2,3 г (89%). В. Получение бутилфосфатного производного. Указанное в подзаголовке целевое соединение получают в соответствии со способом,подробно описанным в примере 8 В, при этом используют 0,5 г (0,45 ммоль) указанного в заголовке примера 3 соединения, LiOTMS и 95 мг(0,50 ммоль) соединения, указанного в подзаголовке примера 9 А, с получением твердого желтого продукта. Этот твердый продукт очищают с помощью обращенно-фазовой ВЭЖХ (градиентный элюент 45% ацетонитрила/45% воды/10% (1% водная) трифторуксусной кислоты 50% ацетонитрила/40% воды/10% (1% водная) трифторуксусной кислоты)с получением 126 мг целевого соединения. МС (FAB): 1244,4 (M+). Пример 10. Получение соединения, где R',R" и R'" обозначают, каждый, метил; RX1 и Rx2 обозначают, каждый, водород; Ry1, Ry2, Ry3, Ry4 обозначают, каждый, гидрокcи; R0 обозначает метилфосфат и R2 обозначает ацильную группу,представленную в таблице 3II. К смеси 500 мг (0,45 ммоль) соединения,указанного в заголовке примера 3 и 0,5 мл (0,5 ммоль) LiOTMS в 5 мл тетрагидрофурана добавляют 0,075 мл (0,75 ммоль) метилдихлорфосфата, что приводит к растворению твердого 43 продукта. Реакцию постоянно контролируют с помощью ВЭЖХ (элюент 70% ацетонитрил; 2 мл/мин; 280 нм), после чего в реакционную смесь дополнительно добавляют 0,7 мл LiOTMS и 0,02 мл метилдихлофосфата. Когда реакция, в основном, завершится по данным ВЭЖХ (элюент 50% ацетонитрил, содержащий 0,1% трифторуксусной кислоты, 2 мл/мин; 280 нм), целевое вещество выделяют с помощью ВЭЖХ(81% чистоты по данным ВЭЖХ). Полученное соединение очищают с использованием ВЭЖХ(ступенчатый градиентный элюент 30%35%40% ацетонитрила, содержащего 0,1% трифторуксусной кислоты; 90 мл/мин; 280 нм) с получением 109 мг указанного в заголовке соединения (94% чистоты). МС (FAB): m/e 1202,6 (M+). Пример 11. Получение соединения, где R',R" и R'" обозначают, каждый, метил; Rx1 и Rx2 обозначают, каждый, водород; Ry1, Ry2, Ry3, Ry4 обозначают, каждый, гидрокcи; R0 обозначает гексилфосфонат и R2 обозначает ацильную группу, представленную в таблице 3II. К охлажденной смеси (0 С) 1 г (0,902 ммоль) указанного в заголовке примера 3 соединения, в 5 мл тетрагидрофурана добавляют по каплям 1,35 мл 1 М раствора LHMDS в тетрагидрофуране (1,35 ммоль). После перемешивания полученной смеси в течение приблизительно тридцати минут к ней добавляют 309 мкл(1,804 ммоль) гексилдихлорфосфата и реакционную смесь оставляют нагреваться до комнатной температуры, после чего добавляют воду. Полученную реакционную смесь высушивают в вакууме с получением указанного в заголовке целевого соединения. Выход: 102 мг. МС (FAB): Расч. 1262,5978 (M+Li). Найдено: 1262,5979 (M+Li). Пример 12. Получение соединения, где R',R" и R"' обозначают, каждый, метил; Rx1 и Rx2 обозначают, каждый, водород; Ry1, Ry2, Ry3, Ry4 обозначают, каждый, гидрокси; R0 обозначает метилфосфонат и R2 обозначает ацильную группу, представленную в таблице 3II. Указанное в заголовке соединение получают в соответствии со способом, подробно описанным в примере 11, с использованием 221,9 мг (0,200 ммоль) указанного в заголовке соединения примера 3, 0,240 мл 1 М раствораLHMDS в гексане (0,240 ммоль) и 35 мг (0,26 ммоль) дихлорангидрида метилфосфоновой кислоты в тетрагидрофуране. Выход: 44 мг. МС (FAB): 1192,2 (M+Li). 44 Пример 13. Получение соединения, где R',R" и R'" обозначают, каждый, метил; Rx1 и Rx2 обозначают, каждый, гидрокси; Ry1, Ry2, Ry3, Ry4 обозначают, каждый, гидрокси; R0 обозначает метилфосфат и R2 обозначает ацильную группу,представленную в таблице 3II. А. Получение метилфосфатного производного. К охлажденной смеси (0 С) 400 мг (0,32 ммоль), указанного в подзаголовке примера 5 А соединения, и 0,36 мл (0,36 ммоль) LiOTMS (1M раствор в метиленхлориде) в 5 мл тетрагидрофурана в атмосфере азота добавляют 0,04 мл(0,4 ммоль) метилдихлорфосфата. Когда реакция в основном завершится, на что указывают данные ВЭЖХ (элюент 80% ацетонитрил; 2 мл/мин; 280 нм), к смеси добавляют несколькими порциями гидроксид лития. Целевое соединение выделяют с использованием ВЭЖХ (элюент 60% ацетонитрил, содержащий 0,1% трифторуксусной кислоты: 90 мл/мин; 280 нм). Фракции, содержащие целевое соединение, объединяют и концентрируют в вакууме с получением 129,8 мг указанного в подзаголовке соединения. Выход: 30% ВЭЖХ (элюент 65% ацетонитрил, содержащий 0,1% трифторуксусной кислоты; 2 мл/мин; 280 нм): RT = 4,28 мин. В. Снятие защиты. К охлажденной смеси (0 С) 118 мг (0,09 ммоль), указанного в подзаголовке примера 13 А соединения, в 3 мл метиленхлорида добавляют 35 мкл (0,28 ммоль) эфирата трифторида бора. Полученную реакционную смесь оставляют взаимодействовать приблизительно десять минут и затем гасят реакцию несколькими каплями воды, что приводит к образованию белого осадка. Полученную смесь концентрируют в вакууме с получением остатка. Этот остаток переводят во взвесь в диэтиловом эфире и затем фильтруют с получением твердого осадка, который сушат в вакууме. Чистота полученного продукта по данным ВЭЖХ (элюент 50% ацетонитрил, содержащий 0,1% трифторуксусной кислоты; 2 мл/мин; 280 нм): RT = 3,92 мин) составляет 92%. Выход: 88 мг (80%).MC(FAB): 1216,4(M-H2O), 1256,3(M+Na). Пример 14. Получение соединения, где R',R" и R"' обозначают, каждый, метил; Rx1 и Rx2 обозначают, каждый, гидрокси; Ry1, Ry2, Ry3, Ry4 обозначают, каждый, гидрокси; R0 обозначает этилфосфат и R2 обозначает ацильную группу,представленную в таблице 3II. А. Получение этилфосфатного производного. К охлажденной смеси (0 С) 400 мг (0,32 ммоль), указанного в подзаголовке примера 5 А соединения и 0,36 мл (0,36 ммоль) LiOTMS (1M раствор в метиленхлориде) в 5 мл тетрагидрофурана в атмосфере азота добавляют 0,47 мл(0,4 ммоль) этилдихлорфосфата. Когда реакция, 45 в основном, завершится согласно данным ВЭЖХ (элюент 80% ацетонитрил; 2 мл/мин; 280 нм), к смеси по каплям добавляют приблизительно 0,5 мл воды. Целевое соединение выделяют с помощью ВЭЖХ (элюент 60% ацетонитрил, содержащий 0,1% трифторуксусной кислоты; 90 мл/мин; 280 нм). Фракции, содержащие целевое соединение, объединяют и концентрируют в вакууме с получением остатка. Этот остаток растирают в диэтиловом эфире и затем отфильтровывают с получением 67,8 мг твердого продукта. Чистота, полученного продукта,была определена как - 71% по данным ВЭЖХRT= 5,99 мин). В. Снятие защиты. Указанное в заголовке целевое соединение,получают в соответствии со способом, подробно описанным в примере 13 В, при этом используют 67,8 мг соединения, указанного в подзаголовке примера 14 А и 0,1 мл (0,81 ммоль) эфирата трифторида бора в метиленхлориде. Полученный продукт имеет чистоту 89% по данным ВЭЖХ (элюент 50% ацетонитрил, содержащий 0,1% трифторуксусной кислоты; 2 мл/мин; 280 нм; RT = 5,85 мин ). Выход: 51 мг. МС (FAB): 1230,3(М-Н 2O). Соединения формулы I имеют улучшенные свойства по сравнению с ранее известнымиN-ацилциклическими пептидными противогрибковыми соединениями. Например, настоящие соединения обладают повышенной пероральной биодоступностью, важным свойством для системного противогрибкового соединения. Кроме того, настоящие соединения имеют повышенную противогрибковую активность и улучшенную растворимость в воде по сравнению с ранее известными соединениями. Соединения формулы I проявляют противогрибковую активность и противопаразитарную активность. Например, соединения формулы I ингибируют рост различных инфекционных грибков, включаяspp., Pseudallescheria boydii, Coccidioides immitis,Sporothrix schenckii и тому подобное. Противогрибковую активность исследуемого соединения определяют in vivo, получая минимальную ингибирующую концентрацию(МIС) соединения, для чего используют метод стандартного агарового разведения или дисковый диффузионный метод. Затем соединения тестируют на мышах in vivo для определения эффективной дозы исследуемого соединения, 000464 46 позволяющей контролировать системную грибковую инфекцию. Таким образом, была исследована противогрибковая активность по отношению к С. albicans следующих соединений. Таблица 5. Минимальная ингибирующая концентрация против С. albicans ПримерМIС (мкг/мл) 5 С 0,312 6 В 1,25 7 В 2,5 8 В 80 9 В 80 10 0,312 11 1,25 12 0,039 13 В 0,625 14 В 0,625 Кроме того, in vivo (мыши) была исследована эффективная доза следующих соединений,подавляющих системную грибковую инфекциюED50 (мг/кг) 5 С 1,25 6 В 1,58 7 В 2,5 8 В 1,02 9 В 0,39 10 0,47 11 0,312 12 0,79 13 В 1,86 14 В 1,38 Соединения по изобретению также ингибируют рост определенных организмов, в первую очередь ответственных за вредные инфекции у лиц со сниженным иммунным статусом. Например, соединения по изобретению ингибируют рост Pneumocystis carinii организма,вызывающего пневмоциститную пневмонию(РСР) у больных СПИДом и других пациентов с ослабленным иммунитетом. Другие протазоны ингибируемые соединениями формулы I включают Plasmodium spp., Leishmania spp., Trypanosoma spp., Cryptosporidium spp., Isospora spp.,Cyclospora spp., Trichomonas spp., Microsporidiosis spp., и тому подобное. Соединения формулы I активны in vitro иin vivo и могут использоваться при борьбе как с системными грибковыми инфекциями, так и с грибковыми кожными инфекциями. Соответственно, настоящее изобретение относится к способу ингибирования грибковой активности, заключающемуся в контактировании соединения формулы I или его фармацевтически приемлемой соли с грибком. Предпочтительный способ включает ингибирование активности Candidaalbicans или Aspergillus fumigatis. Настоящее изобретение относится также к способу лечения грибковых инфекций, который включает введение эффективного количества соединения формулы I или его фармацевтически приемлемой соли хозяину, при необходимости такого лечения. Предпочтительный способ включает лечение инфекции Candida albicans или Aspergillusfumigatis По отношению к противогрибковой активности термин "эффективное количество" означает количество соединения по настоящему изобретению, способное ингибировать грибковую активность. Вводимая доза будет меняться в зависимости от таких факторов, как природа и тяжесть инфекции, возраст и общее состояние здоровья носителя инфекции, а также толерантности его организма к противогрибковому агенту. Конкретная схема введения также будет изменяться в зависимости от этих факторов, а дневную дозу можно вводить однократно или распределять ее на несколько приемов в течение дня. Курс может длиться от приблизительно 2-3 дней до приблизительно 2-3 недель и дольше. Обычная дневная доза (введенная однократно или разделенная на несколько доз) будет содержать от 0,01 до 100 мг активного соединения изобретения на один килограмм веса тела пациента. Предпочтительные дневные дозы будут обычно составлять от около 0,1 до около 60 мг/кг и идеально от около 2,5 до около 40 мг/кг. В настоящем изобретении предлагаются также фармацевтические препараты, которые могут использоваться для введения противогрибковых соединений по изобретению. Таким образом, настоящее изобретение относится также к фармацевтическому препарату, включающему в себя один или несколько фармацевтически приемлемых носителей, растворителей или наполнителей, и соединение по п.1 формулы изобретения. Такие препараты содержат от 0,1 до 99,9% (по массе) активного ингредиента, в большинстве случаев от приблизительно 10% до приблизительно 30% (по массе). Под "фармацевтически пригодным" подразумевается такой носитель, растворитель или наполнитель, который совместим с другими ингредиентами препарата и не вреден для пациента. Соединение формулы I может быть введено парентерально, например, путем внутримышечных, подкожных или внутрибрюшинных инъекций, или с помощью оральных или назальных средств. В дополнение к перечисленным способам введения соединение формулы I может быть применено, как лекарственное вещество местного действия для кожных инъекций. Препараты для парентерального введения включают в себя соединение формулы I и физиологически приемлемый растворитель, такой как деионизированная вода, физиологический солевой раствор, 5% декстроза и другие широко 48 известные растворители. Препарат может содержать солюбилизатор, такой как полиэтиленгликоль или пропиленгликоль, или другие известные солюбилизирующие агенты. Такие препараты могут быть составлены в стерильных пузырьках, содержащих противогрибковое соединение и наполнитель в форме сухого порошка или лиофилизованного порошка. Физиологически приемлемый растворитель добавляют непосредственно перед использованием, и раствор отбирают шприцом для введения пациенту. Настоящие фармацевтические препараты приготовлены по известным методикам с использованием известных и легко доступных компонентов. При изготовлении композиции по настоящему изобретению активный ингредиент обычно смешивают с носителем или растворяют в носителе, или помещают внутрь носителя,который может быть в форме капсулы, саше,бумажного или другого контейнера. Когда носитель служит также и растворителем, он может быть твердым, полутвердым или жидким материалом, который выступает в качестве растворителя, наполнителя или среды для активного ингредиента. Таким образом, композиции могут иметь вид таблеток, пилюль, порошков, лепешек, саше, облаток, эликсиров, суспензий,эмульсий, растворов, сиропов, аэрозолей (как,например, твердых или в жидкой среде), мазей,содержащих, например, вплоть до 10% (по массе) активного соединения, мягких или жестких желатиновых капсул, суппозиториев, стерильных инъекционных растворов, стерильно упакованных порошков и тому подобное. Для перорального способа введения противогрибковым соединением заполняют желатиновые капсулы или заформовывают его в таблетки. Таблетки могут также содержать связывающий агент, диспергирующий агент или другой природный наполнитель, подходящий для приготовления таблеток надлежащего размера для тщательной дозировки противогрибкового соединения формулы I. Для педиатрического или гериатрического использования противогрибковое соединение применяют в виде его жидких ароматизированных суспензии, раствора или эмульсии. Предпочтительным пероральным препаратом является линолевая кислота,кремофор RH-60 и вода, предпочтительно, в количестве (по объему) 8% линолевой кислоты,5% кремофора RH-60, 87% стерильной воды и соединение формулы I в количестве от приблизительно 2,5 до приблизительно 40 мг/мл. Для местного применения противогрибковому соединению может быть придана форма сухого порошка для прикладывания к поверхности кожи или оно может иметь форму жидкого препарата, имеющего в своем составе солюбилизирующую водную или неводную жидкость,например, спирт или гликоль. Следующие примеры препаратов являются только иллюстративными и никоим образом не 49 предполагают ограничение области изобретения. Термин "активный ингредиент" означает соединение формулы I или его фармацевтически приемлемую соль. Препарат 1. Жесткие желатиновые капсулы готовят с использованием следующих компонентов: Количество (мг/капсула) Активный ингредиент 250 Крахмал, высушенный 200 Стеарат магния 10 Всего 460 мг Препарат 2. Таблетка приготовлена с использованием ингредиентов, приведенных ниже: Количество (мг/капсула) Активный ингредиент 250 Целлюлоза, микрокристаллическая 400 Двуокись кремния, тонкоизмельченная 10 Стеариновая кислота 5 Всего 665 мг Компоненты смешивают и прессуют в таблетки, вес каждой таблетки 665 мг. Препарат 3. Аэрозольный раствор готовят с содержанием следующих компонентов: Вес Активный ингредиент 0,25 Метанол 25,75 Пропеллент 22 (хлордифторметан) 74,00 Всего 100,00 Активное соединение смешивают с этанолом и смесь добавляют к порции пропеллента 22, охлажденного до -30 С. Затем требуемое количество смеси помещают в стальной коррозионно-стойкий контейнер и разбавляют остатком пропеллента. Затем на контейнер устанавливают клапан. Препарат 4. Таблетки, каждая из которых содержит 60 мг активного ингредиента, готовят следующим образом: Активный ингредиент 60 мг Крахмал 45 мг Микрокристаллическая целлюлоза 35 мг Поливинилпирролидон(в виде 10% раствора в воде) 4 мг Натрий карбоксиметилкрахмал 4,5 мг Стеарат магния 0,5 мг Тальк 1 мг Всего 150 мг Активный ингредиент, крахмал и целлюлозу просеивают через сито с 45 меш (США) и тщательно перемешивают. Водный раствор,содержащий поливинилпирролидон, смешивают с полученным порошком и затем смесь пропускают через сито 14 меш (США). Полученные таким образом гранулы высушивают при 50 С и просеивают через сито 18 меш (США). Натрий карбоксиметилкрахмал, стеарат магния и тальк, предварительно просеянные через сито 60 (США), добавляют к гранулам, которые, 000464 50 после смешивания, прессуют на машине для таблетирования, получая таблетки весом 150 мг каждая. Препарат 5. Капсулы, каждая из которых содержит 80 мг активного ингредиента, изготавливают следующим образом: Активный ингредиент 80 мг Крахмал 59 мг Микрокристаллическая целлюлоза 59 мг Стеарат магния 2 мг Всего 200 мг Активный ингредиент, целлюлозу, крахмал и стеарат магния перемешивают, пропускают через сито с ячейками 45 (США) и помещают в твердые желатиновые капсулы в количестве по 200 мг. Препарат 6. Суппозитории, каждая из которых содержит 225 активного ингредиента, изготавливают следующим образом: Активный ингредиент 225 мг Глицерид насыщенной жирной кислоты 2000 мг Всего 2225 мг Активный ингредиент пропускают через сито с ячейками 60 меш (США) и суспендируют в глицеридах насыщенных жирных кислот, предварительно расплавленных при минимально необходимом нагревании. Затем смесь заливают в формы для производства суппозиториев номинальной вместимости 2 г и дают им охладиться. Препарат 7. Суспензии, каждая из которых содержит 50 мг активного ингредиента на 5 мл дозы, изготавливают следующим образом: Активный ингредиент 50 мг Натрийкарбоксиметилцеллюлоза 50 мг Сироп 1,25 мг Раствор бензойной кислоты 0,10 мг Ароматизаторq.v. Очищенная вода до общего объема 5 мл Активный ингредиент просеивают через сито 45 меш (США) и перемешивают с натрийкарбоксиметилцеллюлозой до образования однородной пасты. Раствор бензойной кислоты,ароматизатор и краситель разбавляют частью воды и добавляют, перемешивая. Большую часть воды добавляют затем до требуемого объема. Препарат 8. Внутривенный препарат можно приготовить следующим образом: Активный ингредиент 100 мг Изотонический солевой раствор 1000 мл Раствор приведенных выше ингредиентов,как правило, вводят внутривенно субъекту со скоростью 1 мл в минуту. Далее в настоящем изобретении предлагается метод лечения или профилактики пневмо 51 нии, который включает в себя введение эффективного количества соединения формулы I или его фармацевтически приемлемой соли, носителю, нуждающемуся в подобном лечении. Соединения формулы I могут быть использованы профилактически для профилактики заражения инфекцией, вызванной организмом Pneumocystiscarinii или, наоборот, могут быть использованы для лечения носителя, уже инфицированногоPneumocystis carinii. Соединение формулы I может быть введено парентерально, например,используя внутримышечную, внутривенную или внутрибрюшинную инъекцию, перорально или путем ингаляции прямо в воздушные пути легких. Предпочтительным способом введения является ингаляция с помощью аэрозольного спрея соединения формулы I. Что касается противопаразитарной активности термин "эффективное количество" означает количество соединения по настоящему изобретению, способное ингибировать паразитарную активность. Эффективное количество соединения формулы I составляет от приблизительно 3 мг/кг веса тела пациента до приблизительно 100 мг/кг. Дневную дозу соединения изобретения можно вводить однократно или в несколько приемов, например, за два, три или четыре раза в течение дня, на протяжении всего курса лечения. Величина индивидуальных доз,способ доставки, частота дозировки, длительность терапии будут меняться в зависимости от таких факторов, как интенсивность и степень инфекции, возраст и общее состояние здоровья пациента, реакция пациента на терапию и толерантность пациента по отношению к лекарственному веществу. Известно, что пневмоциститные пневмонийные инфекции у пациентов,страдающих СПИД, являются высоко рефракторными (с трудом поддаются лечению), благодаря природе самой инфекции. Например, в случае тяжелых, развившихся инфекций, люминальная поверхность дыхательных путей забивается инфекционным материалом и в тканях легких происходит экстенсивное развитие паразита. Пациентам с развившейся инфекцией будут требоваться, соответственно, более высокие дозы в течение более длительного периода времени. В противоположность этому, иммунодефицитных пациентов, не сильно инфицированных и чувствительных по отношению к пневмоциститной пневмонии, можно лечить более низкими и менее частыми профилактическими дозами.B) R4 обозначает фенил, замещенный амино, C1-C12 алкилтио, галогеном, C1-C12 алкилом,C2-C12 алкенилом, C2-C12 алкинилом, замещенным C1-C12 алкилом, замещенным C2-C12 алкенилом, замещенным C2-C12 алкинилом, C1-C12 алкокси, трифторметилом, фенилом, замещенным фенилом или фенилом, замещенным группой формулы -O-(CH2)m-[O-(CH2)n]p-O-(С 1-С 12 алкил), где m, n и р определены выше; илиC) R4 обозначает C1-C12 алкокси, замещенный галогеном, С 3-С 12 циклоалкилом, С 7-С 10 бициклоалкилом, C7-C14 трициклоалкилом, C1C6 алкокси, C2-C12 алкинилом, амино, С 1-С 4 алкиламино, ди(С 1-С 4 алкил)амино, формамидо,C2-C12 алканоиламино или фенилом, замещенным группой формулы -O-(CH2)m-[O-(CH2)n]p-O(C1-C12 алкил), где m, n и р определены выше; илиC1-C12 алкилтио, C2-C12 алкенилом, C2-C12 алкинилом, галоген(С 1-С 6 алкокси) или группой формулы -O-(CH2)r-W-R5, где r, W и R5 определены выше; или R6 обозначает фенил, замещенный группой формулыF) R4 обозначает C1-C12 алкокси, замещенный группой формулы -NHC(O)R7; где R8 обозначает C1-C12 алкокси или группу формулы -O-(СН 2)m-[O-(СН 2)n]р-O-(С 1-С 12 алкил), где m, n и р определены выше; илиV) R2 обозначает нафтоил, замещенный R4,где R4 определен выше; или его фармацевтически приемлемая соль. 2. Соединение по п.1, гдеRx1, Rx2 обозначают водород; или его фармацевтически приемлемая соль. 3. Соединение по п.2, гдеB) R4 обозначает фенил, замещенный C1C12 алкокси или группу формулы -O-(СН 2)2-O(С 1-С 6 алкил), илиR6 обозначает фенил, замещенный группой формулы -O-(CH2)r-W-R5, где r, W и R5 те же,что определены выше; или R6 обозначает фенил, замещенный группой формулы -O-(СН 2)2-O-(С 1-С 6 алкил); илиF) R4 обозначает C1-C12 алкокси, замещенный группой формулы -NHC(O)R7;R7 обозначает C1-C6 алкокси или фенил(С 1 С 6 алкокси), или его фармацевтически приемлемая соль. 4. Соединение по п.3, где R1 обозначает C1C4 алкил, C1-C4 алкокси, фенил, п-хлорфенил, пбромфенил или нитрофенил, бензил, пхлорбензил, п-бромбензил или нитробензил; или его фармацевтически приемлемая соль. 5. Соединение по п.4, где R1 обозначает метил, этил, метокси, этокси, фенил, бензил; или его фармацевтически приемлемая соль. 6. Соединение по п.5, где Z обозначает связь; R4 обозначает фенил, замещенный нпентокси; и R1 обозначает метил; или его фармацевтически приемлемая соль. 7. Фармацевтический препарат, содержащий соединение формулы I или его фармацевтически приемлемую соль, указанные в любом из пп.1-6, объединенное с одним или несколькими фармацевтически приемлемыми носителями, растворителями или наполнителями. 56 8. Применение соединения формулы I или его фармацевтически приемлемой соли, указанных в любом из пп.1-6 в виде фармацевтического препарата. 9. Способ лечения грибковых инфекций у млекопитающих, который включает введение таким млекопитающим соединения формулы I или его фармацевтически приемлемой соли,указанных в любом из пп.1-6. 10. Способ получения соединения формулы I или его фармацевтически приемлемой соли, указанных в любом из пп.1-6, заключающийся во взаимодействии соединения формулы где R обозначает водород,R', R", R'", Rx1, Rx2, Ry1, Ry2, Ry3 и Ry4 и R2 указаны в п.1, с замещенной C1-C6 алкилфосфорной кислотой, фенилфосфорной кислотой,замещенным C1-C6 алкилфосфатом или фенилфосфатом; с последующим, при необходимости,выделением целевого продукта в виде фармацевтически приемлемой соли.
МПК / Метки
МПК: C07K 7/56, A61K 38/12
Метки: агенты, антигрибковые, циклические, пептидные
Код ссылки
<a href="https://eas.patents.su/29-464-ciklicheskie-peptidnye-antigribkovye-agenty.html" rel="bookmark" title="База патентов Евразийского Союза">Циклические пептидные антигрибковые агенты</a>
Предыдущий патент: Соединительный узел для расширяющейся трубной колонны, снабженной прорезями
Следующий патент: Способ получения капролактама
Случайный патент: Клапанный узел контейнера для напитка, контейнер для напитка и способ заполнения и опорожнения контейнера