Соединения с серотонинергической активностью, способ их получения и содержащие их фармацевтические композиции
Номер патента: 19300
Опубликовано: 28.02.2014
Авторы: Поленцани Лоренцо, Ди Санто Роберто, Фурлотти Гвидо, Каццолла Никола, Гульельмотти Анджело, Кости Роберта, Ализи Мария Алессандра







Формула / Реферат
1. Соединения формулы (I)
где R1 означает атом водорода, линейную или разветвленную алкильную группу, содержащую 1-6 атомов углерода, или алкоксиалкилгруппу, содержащую 1-6 атомов углерода;
R2 означает атом водорода, атом галогена или линейную или разветвленную алкильную группу, содержащую 1-3 атома углерода;
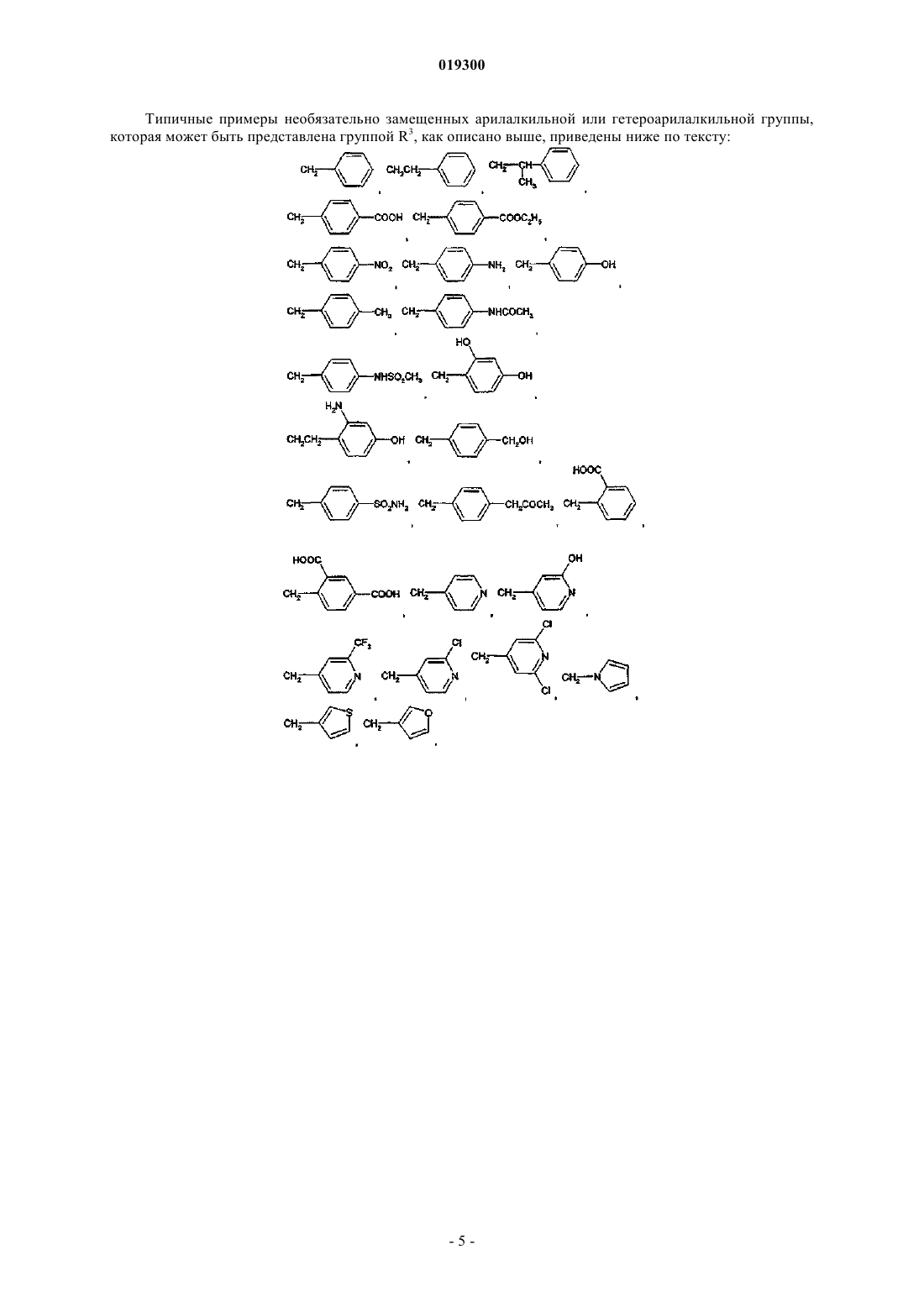
R3 представляет собой (i) атом водорода; (ii) линейную или разветвленную алкильную группу, содержащую 1-6 атомов углерода; (iii) алкоксиалкильную группу, содержащую 1-6 атомов углерода; (iv) арилалкильную группу, в которой алкильная группа содержит 1-3 атома углерода, а арильная группа представляет собой остаток бензола и может быть замещена одним или двумя заместителями, которые могут быть одинаковыми или различными, выбранными из атома галогена, алкильной группы, содержащей 1-3 атома углерода, алкоксигруппы, содержащей 1-3 атома углерода, гидроксиалкильной группы, содержащей 1-3 атома углерода, -ОН, -NR'R", -NO2, -CF3, -CO2R', R'CON(R")-, R'SO2N(R")- и R'R"NSO2-, где R' и R", которые могут быть одинаковыми или различными, представляют собой атом водорода или алкильную группу, содержащую 1-3 атома углерода; (v) RivRvNCO(CH2)n-, где n означает целое число от 0 до 2 и Riv и Rv, которые могут быть одинаковыми или различными, являются атомами водорода, алкильными группами, содержащими 1-3 атома углерода, арильными группами, выбранными из остатков бензола, необязательно замещенными одним или двумя заместителями, которые могут быть одинаковыми или различными, выбранными из атома галогена, алкильной группы, содержащей 1 атома углерода, -ОН, -NH2; и (vi) Cγ-(CH2)m-, где m означает целое число от 0 до 2 и Cγ является алициклической группой, содержащей от 3 до 7 атомов углерода, или насыщенной 5- или 6-членной гетероциклической группой, содержащей по меньшей мере один гетероатом, выбранный из О и N, необязательно замещенный по атому N алкильной группой, содержащей 1 атом углерода,
их кислотно-аддитивные соли с фармацевтически приемлемыми органическими или неорганическими кислотами, а также их основно-аддитивные соли с фармацевтически приемлемыми органическими или неорганическими основаниями.
2. Соединение по п.1, в котором R1 представляет собой атом водорода или линейную или разветвленную алкильную группу, содержащую 1-6 атомов углерода; R2 означает атом водорода или атом галогена и R3 представляет собой (i) атом водорода; (ii) линейную или разветвленную алкильную группу, содержащую 1-6 атомов углерода; (iii) арилалкильную группу, в которой алкильная группа содержит 1-3 атома углерода, а арильная группа представляет собой остаток бензола и может быть замещена одним или двумя заместителями, которые могут быть одинаковыми или различными, выбранными из атома галогена, алкильной группы, содержащей 1-3 атома углерода, алкоксигруппы, содержащей 1-3 атома углерода, гидроксиалкильной группы, содержащей 1-3 атома углерода, -ОН, -NR'R", -NO2, -CO2R', R'CON(R")-, R'SO2N(R")- и R'R"NSO2-, где R' и R", которые могут быть одинаковыми или различными, представляют собой атом водорода или алкильную группу, содержащую 1-3 атома углерода; (iv) RivRvNCO(CH2)n-, где n означает целое число от 0 до 2 и Riv и Rv, которые могут быть одинаковыми или различными, являются атомами водорода, арильными группами, выбранными из остатков бензола; и (v) Cγ-(CH2)m-, где m означает целое число от 0 до 2 и Сγ является насыщенным гетероциклическим фрагментом, выбранным из группы, включающей морфолин, пиперидин, N-метилпиперазин и пирролидин.
3. Соединение по п.1, в котором R1 является линейной или разветвленной алкильной группой, содержащей 1-6 атомов углерода; R2 означает атом водорода и R3 представляет собой (i) линейную или разветвленную алкильную группу, содержащую 1-6 атомов углерода; (ii) арилалкильную группу, в которой алкильная группа содержит 1-3 атома углерода, а арильная группа выбрана из остатков бензола и может быть замещена заместителем, выбранным из алкоксигруппы, содержащей 1-3 атома углерода, гидроксиалкильной группы, содержащей 1-3 атома углерода, -NR'R", -CO2R', R'CON(R")-, R'SO2N(R")- и R'R"NSO2-, где R' и R", которые могут быть одинаковыми или различными, представляют собой атомы водорода или алкильные группы, содержащие 1-3 атома углерода; (iii) RivRvNCO(CH2)n-, где n означает целое число от 0 до 2 и Riv и Rv, которые могут быть одинаковыми или различными, являются атомами водорода или арильными группами, выбранными из остатков бензола; и (iv) Cγ-(CH2)m-, где m означает целое число от 0 до 2 и Cγ представляет собой остаток морфолина или пиперидина.
4. Соединения по п.1, в котором R1, R2 и R3 представлены в следующей таблице:
5. Фармацевтическая композиция, включающая эффективное количество соединения формулы (I) или его кислотно-аддитивной или основно-аддитивной соли по любому из предшествующих пунктов, а также по меньшей мере один фармацевтически приемлемый наполнитель.
6. Фармацевтическая композиция по п.5, которая включает такое количество соединения формулы (I) или его кислотно-аддитивной или основно-аддитивной соли, чтобы обеспечить введение от 0,001 до 100 мг/кг/день соединения формулы (I) в пересчете на соединение в свободной форме.
7. Способ получения соединения формулы (I)
или его фармацевтически приемлемой кислотно-аддитивной или основно-аддитивной соли,
где R1, R2 и R3 соответствуют определениям, данным в пп.1-4,
отличающийся тем, что он включает взаимодействие между соединением формулы (II)
и соединением формулы (III)
где R1, R2 и R3 имеют указанные выше значения и X означает атом галогена.
8. Способ получения по п.7, в котором указанное взаимодействие проводят в органическом растворителе в присутствии органического или неорганического основания.
9. Способ получения по любому из пп.7 и 8, где указанную реакцию проводят в присутствии активирующего агента, выбранного из группы, включающей йодид калия, йодид натрия, йодид цезия, йодид тетрабутиламмония и йодид триметилфениламмония.
10. Способ получения соединения формулы (I)
или его фармацевтически приемлемой кислотно-аддитивной или основно-аддитивной соли,
где R1, R2 и R3 соответствуют определениям, данным в любом из пп.1-4,
отличающийся тем, что он включает взаимодействие между соединением формулы (IV)
и соединением формулы (V)
где R1, R2 и R3 имеют указанные выше значения.
11. Способ получения по п.10, где указанную реакцию проводят в полярном апротонном органическом растворителе в присутствии основания.
12. Способ получения соединения формулы (II)
где R1 и R2 имеют значения, определенные в любом из пп.1-4,
отличающийся тем, что он включает:
(1) реакцию между соединением формулы (IV)
и соединением формулы (VI)
где R1 и R2 имеют указанные выше значения,
с получением соединения формулы (VII)
(2) реакцию дебензилирования пиперидинового атома азота соединения формулы (VII) с получением соединения формулы (II), где указанную реакцию осуществляют путем гидрирования.
13. Способ получения по п.12, где указанную реакцию (1) проводят в полярном апротонном органическом растворителе, выбранном из группы, включающей кетоны, тетрагидрофуран, диметилформамид, диметилсульфоксид, диоксан и ацетонитрил в присутствии основания, выбранного из группы, включающей гидроксид натрия и гидрид натрия, и указанную реакцию (2) проводят в спиртовом растворителе в атмосфере водорода в присутствии палладия на угле в качестве катализатора.
14. Соединение формулы (II)
где R1 является атомом водорода, линейной или разветвленной алкильной группой, содержащей 1-6 атомов углерода, или алкоксиалкильной группой, содержащей 1-6 атомов углерода; и
R2 является атомом водорода, атомом галогена или линейной или разветвленной алкильной группой, содержащей 1-3 атома углерода.
15. Соединение формулы (I)
или его фармацевтически приемлемая кислотно-аддитивная или основно-аддитивная соль,
где R1, R2 и R3 соответствуют определениям, данным в любом из пп.1-4,
для фармацевтического применения.
16. Применение соединения формулы (I)
или его фармацевтически приемлемой кислотно-аддитивной или основно-аддитивной соли,
где R1, R2 и R3 соответствуют определениям, данным в любом из пп.1-4,
для получения фармацевтической композиции, предназначенной для лечения расстройств перистальтики кишечника, расстройств центральной нервной системы, недержания мочи и сердечной аритмии.
17. Применение соединения формулы (I)
или его фармацевтически приемлемой кислотно-аддитивной или основно-аддитивной соли,
где R1, R2 и R3 соответствуют определениям, данным в любом из пп.1-4,
для получения фармацевтической композиции, предназначенной для лечения хронической боли.
18. Применение соединения по п.17 для получения фармацевтической композиции для лечения невропатической боли, вызванной диабетом, раком, иммунодефицитом, травмами, ишемией, рассеянным склерозом, ишиалгией, тригеминальной невралгией и постгерпетическим синдромом.
Текст
СОЕДИНЕНИЯ С СЕРОТОНИНЕРГИЧЕСКОЙ АКТИВНОСТЬЮ, СПОСОБ ИХ ПОЛУЧЕНИЯ И СОДЕРЖАЩИЕ ИХ ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ где R1, R2 и R3 определены в описании изобретения, а также их фармацевтически приемлемым кислотно-аддитивным и основно-аддитивным солям. Кроме того, изобретение относится к способу и промежуточному соединению для получения указанных соединений и к фармацевтической композиции, включающей указанные соединения. Помимо этого, изобретение относится к применению новых 2 Н-пирроло[3,4-с]хинолинов для получения фармацевтической композиции,которая эффективна при лечении нарушений серотонинергической системы.(71)(73) Заявитель и патентовладелец: АЦЬЕНДЕ КИМИКЕ РЬЮНИТЕ АНДЖЕЛИНИ ФРАНЧЕСКО Область техники, к которой относится изобретение Настоящее изобретение относится к новым 2 Н-пирроло[3,4-с]хинолинам, способу их получения и к содержащим их фармацевтическим композициям. Кроме того, изобретение относится к применению новых 2 Н-пирроло[3,4-с]хинолинов для получения фармацевтической композиции, которая эффективна при лечении нарушений серотонинергической системы. Известный уровень техники Среди многих известных классов рецепторов серотонина рецепторы 5-HT4 были обнаружены в мочевом пузыре, в желудочно-кишечной системе, в гладкой мускулатуре и сердечной мышце, а также в отдельных областях центральной нервной системы (Eglen R.M. et.al., Central 5-HT4 receptors. TrendsPharmacol. Sci. 1995; 16(11):391-8; Hedge S.S. and Eglen R.M., Peripheral 5-HT4 receptors. FASEB J. 1996; 10(12):1398-407). Соединения, которые обладают действием агонистов, частичных агонистов и антагонистов рецепторов 5-HT4, являются потенциально эффективными при фармакологическом лечении расстройств перистальтики кишечника, расстройств центральной нервной системы, недержания мочи и сердечной аритмии (Bockaert J. et al., 5-HT4 receptors. Curr. Drug Targets - CNs Neurol. Disord. 2004, 3(1):3851; Gershon M.D. Review article: serotonin receptors and transporters - roles in normal and abnormal gastrointestinal motility. Aliment Pharmacol. Ther. 2004; 20 (Suppl. 7): 3-14). Кроме того, известно, что некоторые лекарственные средства, которые действуют на серотонинергическую систему, способны контролировать хроническую боль, в частности невропатическую боль. Хроническая боль является признаком ряда патологий, от которых в среднем страдает примерно 10-20% взрослого населения. Хроническая боль, как правило, связана с клиническими состояниями, характеризуемыми хроническими и/или дегенеративными поражениями. Хроническая боль отличается от острой боли, главным образом, продолжительностью. Острая боль продолжается в течение нескольких дней или недель, соответственно времени излечения причины боли(травмы, ожога, чрезмерных усилий, хирургического или стоматологического вмешательства и т.п.). С другой стороны, хроническая боль сохраняется в течение нескольких месяцев и даже лет, вызывая мышечное напряжение, ограниченную подвижность, усталость, потерю аппетита и апатию. Хроническая боль может проявляться также в рецидивирующей форме с промежутками, равными неделям, месяцам и даже годам, или она может быть связана с хроническими патологиями. Типичными примерами патологий, характеризующихся хронической болью, являются ревматоидный артрит, остеоартрит, фибромиалгия, невропатии и т.д. [Ashburn M.A., Staats P.S. Management of chronic pain. Lancet. 1999; 353:1865-69]. Хроническая боль, в частности невропатическая боль, часто лишает сил и является причиной потери работоспособности и плохого качества жизни. За этим также следуют экономические и социальные потери. Обезболивающие препараты, применяемые в настоящее время для лечения невропатической боли,включают нестероидные противовоспалительные средства (NSAID), антидепрессанты, опиоидные анальгетики и противосудорожные средства [Woolf C.J., Mannion R.J., Neuropathic pain: aetiology, symptoms,mechanism and management. Lancet. 1999; 353: 1959-1964]. Однако хроническую боль и, в частности, невропатическую боль, к сожалению, трудно лечить с помощью лекарственных средств, доступных в настоящее время. Поэтому разработка новых лекарственных средств всегда была одной из основных целей фармацевтической промышленности. Кроме того, несмотря на многочисленные исследовательские усилия, направленные на выявление подходящих анальгетических соединений, имеется значительное количество пациентов для боли которых не существует подходящего лечения [Scholz J., Woolf C.J. Can we conquer pain Nat Neusci. 2002; 5:1062-76]. Заявки на патент WO 2004/101548 и WO 2005/013989 относятся к применению соединений индазола для получения фармацевтических композиций, который эффективны при лечении невропатической боли. Считалось, что некоторые соединения, которые воздействуют на серотонинергическую систему, в частности антагонисты рецептора 5-HT4, могут иметь обезболивающее действие. Соединения, которые оказывают влияние на серотонинергическую систему, имеют ряд нежелательных и побочных действий из-за их низкой селективности в отношении различных рецепторов серотонина. Конкретно, известно, что вся группа серотонинергических рецепторов подразделяется на различные подтипы, например 5-HT1(A-F), 5-HT2(A-C), 5-HT3, 5-HT4, 5-HT6 или 5-HT7, которые по-разному расположены в различных системах организма, например в центральной и/или периферической нервной системе, в пищеварительной системе и в сердечно-сосудистой системе. Результаты взаимодействия активных соединений с серотонинергическими рецепторами различаются в зависимости от места нахождения этих рецепторов. Указанная низкая селективность часто вынуждает прерывать лечение или избегать его для определенных типов пациентов, у которых помимо наличия хронической боли, присутствуют сопутствующие патологии конкретных систем организма, например сердечно-сосудистой системы. Конкретно, взаимодействие с серотонинергической системой и, еще более конкретно, с определенными субрецепторами, например 5-HT2A, может привести к нежелательным последствиям для сердечно-1 019300 сосудистой системы, включая воздействие на сердечный ритм. Пирролохинолины являются широкоизвестными соединениями. Химическая структура этих соединений включает пиррол, конденсированный с хинолином, с образованием трициклической системы. В зависимости от положения участка соединения обоих циклических компонентов, пирролохинолины можно разделить на различные классы. 2 Н-Пирроло[3,4-b]хинолины были описаны в заявке на патент JP 2005/306774 как фармацевтические соединения с антибактериальной активностью. 2 Н-Пирроло[3,4-b]хинолины были описаны в патенте США 6335346 как фармацевтические вещества с седативным и снотворным действием. Пирроло[3,2-c]хинолины были описаны в международной патентной заявке WO 98/05660 как вещества, способные действовать в качестве ингибиторов кинуренин-3-гидроксилазы (KYN-OH), которые потенциально применимы для профилактики и/или лечения нейродегенеративных нарушений, например мозговой ишемии и/или гипоксии, болезни Паркинсона, эпилепсии, болезни Хантингтона, болезни Альцгеймера и т.п. Пирроло[3,2-c]хинолины были описаны также в международной заявке на патент WO 99/09029 как вещества, способные ингибировать секрецию кислоты в желудке, которые потенциально применимы для лечения язвы желудка. Гексагидро-2 Н-пирроло[3,4-c]хинолины были описаны в патентах США 4268513, 4350814 и 4440768 как фармацевтические соединения с антипсихотическим, анальгетическим и антидепрессантным действием. Пирроло[3,4-c]хинолин-1-оны были описаны в патентах США 6323216 и 6413978 как фармацевтические вещества с антагонистической активностью в отношении серотонинергического рецептора 5-HT3,которые потенциально применимы в качестве средств против рвоты и кашля, а также при различных патологиях центральной нервной системы, например тревоги, депрессии, шизофрении, психозов, деменции Альцгеймера и старческой деменции. 5 Н-Пирроло[3,4-c]хинолины были описаны в патенте США 5908932 как фармацевтические соединения с агонистической, антагонистической и обратной агонистической активностью в отношении рецепторов GABA (-аминомасляной кислоты) мозга, и они потенциально применимы в лечении расстройств сна, тревоги и конвульсий, в лечении передозировки бензодиазепиновых соединений и для улучшения внимания. Описание изобретения Авторы настоящего изобретения неожиданно обнаружили, что новые 2 Н-пирроло[3,4-с]хинолины способны взаимодействовать с серотонинергической системой, а именно с высоким сродством связываться с серотонинергическим рецептором 5-HT4. Кроме того, авторы неожиданно обнаружили, что эти новые соединения имеют незначительное сродство или вообще не имеют сродства к рецептору 5-HT2A и вследствие этого оказывают минимальное нежелательное влияние на сердечно-сосудистую систему. Таким образом, настоящее изобретение относится к соединениям формулы (I) где R1 означает атом водорода, линейную или разветвленную алкильную группу, предпочтительно содержащую 1-6 атомов углерода, необязательно замещенную 1-3 гидроксильными группами, или алкилалкоксигруппу, предпочтительно содержащую 1-6 атомов углерода;R2 означает атом водорода, атом галогена или линейную или разветвленную алкильную группу,предпочтительно содержащую 1-3 атома углерода, -CF3, -OSO2CF3, -SO2CH3, -SO2NHCH3 илиR3 представляет собой (i) атом водорода; (ii) линейную или разветвленную алкильную группу,предпочтительно содержащую 1-6 атомов углерода; (iii) алкилалкоксигруппу, предпочтительно содержащую 1-6 атомов углерода; (iv) арилалкильную или гетероарилалкильную группу, алкильная группа которой предпочтительно содержит 1-3 атома углерода и арильная или гетероарильная группа может быть замещена одним или двумя заместителями, которые могут быть одинаковыми или различными,выбранными из атома галогена, алкильной группы, содержащей 1-3 атома углерода, алкоксигруппы, содержащей 1-3 атома углерода, гидроксиалкильной группы, содержащей 1-3 атома углерода, -ОН, -NR'R",-NO2, -CF3, -CO2R', R'CON(R")-, R'SO2N(R")- и R'R"NSO2-, где R' и R", которые могут быть одинаковыми или различными, представляют собой атом водорода или алкильную группу, содержащую 1-3 атома углерода; (v) RivRvNCO(СН 2)n-, где n означает целое число от 0 до 2, и Riv и Rv, которые могут быть одинаковыми или различными, являются атомами водорода, алкильными группами, содержащими 1-3 атома углерода, арильными группами или гетероарильными группами, необязательно замещенными одним или двумя заместителями, которые могут быть одинаковыми или различными, выбранными из атома галогена, алкильной группы, содержащей 1-3 атома углерода, алкоксигруппы, содержащей 1-3 атома углерода,гидроксиалкильной группы, содержащей 1-3 атома углерода, -ОН, -NO2, -NH2, -CF3, -СО 2 Н,-СО 2-С 1-3 алкил, -SO2NH2 и -NHSO2-C1-3 алкил; и (vi) C-(CH2)m-, где m означает целое число от 0 до 2 и С является алициклической группой, содержащей от 3 до 7 атомов углерода, или насыщенной 5- или 6-членной гетероциклической группой, содержащей по меньшей мере один гетероатом, выбранный из О и N, необязательно замещенный по атому N алкильной группой, содержащей 1-3 атома углерода,их кислотно-аддитивным солям с фармацевтически приемлемыми органическими или неорганическими кислотами, а также их основно-аддитивным солям с фармацевтически приемлемыми органическими или неорганическими основаниями. Способность взаимодействовать с серотонинергической системой и, в том числе, с серотонинергическим рецептором 5-HT4 делает эти соединения особенно применимыми при лечении патологий, в которых принимает участие данный рецептор, и потенциально эффективными при фармакологическом лечении расстройств кишечной перистальтики, расстройств центральной нервной системы, недержания мочи и сердечной аритмии. В частности, авторы установили, что соединения по настоящему изобретению особенно применимы при лечении хронической боли и, в частности, невропатической боли. Высокая селективность по отношению к другим серотонинергическим рецепторам, в частности рецепторам 5-HT2A, снижает нежелательные воздействия на сердечно-сосудистую систему, которые встречаются в случае других лекарственных средств, оказывающих действие на серотонинергическую систему. Таким образом, соединения по настоящему изобретению применимы в качестве фармацевтических действующих начал, в частности для лечения расстройств перистальтики кишечника, расстройств центральной нервной системы, недержания мочи и сердечной аритмии, предпочтительно хронической боли и более предпочтительно невропатической боли. Поэтому во втором аспекте настоящее изобретение относится к фармацевтической композиции,включающей эффективное количество соединения формулы (I) где R1, R2 и R3 соответствуют данному выше определению,его кислотно-аддитивной соли с фармацевтически приемлемой органической или неорганической кислотой и его основно-аддитивной соли с фармацевтически приемлемым органическим или неорганическим основанием, а также по меньшей мере один фармацевтически приемлемый наполнитель. Согласно другому аспекту настоящее изобретение относится к описанному выше соединению формулы (I) и его фармацевтически приемлемым кислотно-аддитивным или основно-аддитивным солям для фармацевтического применения. Согласно следующему аспекту настоящее изобретение относится к описанному выше соединению формулы (I) и его фармацевтически приемлемым кислотно-аддитивным или основно-аддитивным солям для получения фармацевтической композиции, которая является эффективной при лечении расстройств перистальтики кишечника, расстройств центральной нервной системы, недержания мочи и сердечной аритмии. В другом предпочтительном аспекте настоящее изобретение относится к применению описанного выше соединения формулы (I) и его фармацевтически приемлемых кислотно-аддитивных и основноаддитивных солей для получения фармацевтической композиции, которая эффективна при лечении хронической боли и, в частности, невропатической боли. Типичными примерами патологий, характеризуемых невропатической болью, являются диабет, рак,иммунодефицит, травмы, ишемия, рассеянный склероз, ишиас, тригерминальная невралгия и постгерпетический синдром. Преимущественно R1 представляет собой атом водорода или линейную или разветвленную алкильную группу, предпочтительно содержащую 1-6 атомов углерода. Более предпочтительно R1 является линейной или разветвленной алкильной группой, предпочтительно содержащей 1-6 атомов углерода. Преимущественно R1 является линейной или разветвленной алкильной группой, включающей от 1 до 3 групп ОН. Предпочтительно R2 представляет собой атом водорода, атом галогена, -CF3, -OSO2CF3, -SO2CH3,-SO2NHCH3 или -NHSO2CH3. Более предпочтительно группа R2 представляет собой атом водорода, -CF3,-OSO2CF3, -SO2CH3, -SO2NHCH3 или -NHSO2CH3. Преимущественно R3 выбирают из группы, включающей (i) атом водорода; (ii) линейную или раз-3 019300 ветвленную алкильную группу, предпочтительно содержащую 1-6 атомов углерода; (iii) арилалкильную группу или гетероарилалкильную группу, в которой арильная или гетероарильная группа может быть замещена одним или двумя заместителями, которые могут быть одинаковыми или различными, выбранными из атома галогена, алкильной группы, содержащей 1-3 атома углерода, алкоксигруппы, содержащей 1-3 атома углерода, гидроксиалкильной группы, содержащей 1-3 атома углерода, -ОН, -NR'R", -NO2,-CO2R', R'CON(R")-, R'SO2N(R")- и R'R"NSO2-, где R' и R", которые могут быть одинаковыми или различными, представляют собой атом водорода или алкильную группу, содержащую 1-3 атома углерода;(iv) RivRvNCO(CH2)n-, где n означает целое число от 0 до 2, и Riv и Rv, которые могут быть одинаковыми или различными, являются атомами водорода, арильными группами или гетероарильными группами; и(v) C-(СН 2)m-, где m означает целое число от 0 до 2 и C является насыщенным гетероциклическим фрагментом, выбранным из группы, включающей морфолин, пиперидин, N-метилпиперазин и пирролидин. Еще более предпочтительно R3 выбран из группы, включающей (i) линейную или разветвленную алкильную группу, содержащую 1-6 атомов углерода; (ii) арилалкильную группу, в которой арильная группа может быть замещена заместителем, выбранным из алкоксигруппы, содержащей 1-3 атома углерода, гидроксиалкильной группы, содержащей 1-3 атома углерода, -NR'R", -CO2R', R'CON(R")-,R'SO2N(R")- и R'R"NSO2-, где R' и R", которые могут быть одинаковыми или различными, представляют собой атомы водорода или алкильные группы, содержащие 1-3 атома углерода; (iii) RivRvNCO(CH2)n-, гдеn означает целое число от 0 до 2, и Riv и Rv, которые могут быть одинаковыми или различными, являются атомами водорода или арильными группами; и (iv) C-(CH2)m-, где m означает целое число от 0 до 2 и С представляет собой остаток морфолина или пиперидина. Арильная или гетероарильная группа, образующая часть арилалкильной или гетероарилалкильной группы, которая может быть представлена группой R3, как описано выше, может представлять собой группу, являющуюся остатком бензола, нафталина, пиридина, хинолина, изохинолина, пиразина, хиноксалина, пиримидина, хиназолина, пиридазина, циннолина, фурана, бензофурана, изобензофурана, пиррола, индола, изоиндола, тиофена, бензотиофена, изобензотиофена, имидазола, бензимидазола, пиразола,индазола, оксазола, бензоксазола, изоксазола, бензизоксазола, тиазола и бензотиазола; предпочтительно бензола, пиридина, фурана, бензофурана и пиррола. Типичные примеры необязательно замещенных арилалкильной или гетероарилалкильной группы,которая может быть представлена группой R3, как описано выше, приведены ниже по тексту: Типичные примеры соединений, представленных указанной формулой (I), приведены в табл. 1. Таблица 1 Типичными примерами фармацевтически приемлемых неорганических кислот являются хлористоводородная кислота, бромисто-водородная кислота, фосфорная кислота, серная кислота и азотная кислота. Типичными примерами фармацевтически приемлемых органических кислот являются уксусная кислота, аскорбиновая кислота, щавелевая кислота, янтарная кислота, малеиновая кислота, фумаровая кислота, винная кислота, метансульфоновая кислота, паратолуолсульфоновая кислота, лимонная кислота,молочная кислота, дубильная кислота и бензойная кислота. Кроме того, в качестве органических кислот могут использоваться аминокислоты, такие как аспарагиновая кислота и глутаминовая кислота. Типичными примерами фармацевтически приемлемых органических и неорганических оснований являются: моно-, ди- и триалкиламины, например метиламин, диметиламин, триметиламин, этиламин,диэтиламин, триэтиламин, пропиламин, дипропиламин, трипропиламин, этилендиамин, моно-, ди- и триалканоламины, например моноэтаноламин, диэтаноламин и триэтаноламин; гуанидин, морфолин, пиперидин, пирролидин, пиперазин, 1-бутилпиперидин, 1-этил-2-метилпиперидин, N-метилпиперазин,1,4-диметилпиперазин, N-бензилфенилэтиламин, N-метилглюкозамин, трис-(гидроксиметил)аминометан, аммиак, гидроксид натрия, гидроксид кальция, гидроксид калия, гидроксид алюминия, гидроксид железа, гидроксид магния и гидроксид цинка. Кроме того, в качестве органических оснований могут применяться аминокислоты, такие как аргинин и лизин. Описанные выше соединения формулы (I) можно получать согласно приведенной ниже схеме А,исходя из новых промежуточных соединений формулы (II) при их взаимодействии с подходящими производными галогенов формулы (III):-6 019300 где R1, R2 и R3 имеют указанные выше значения и X представляет собой атом галогена, предпочтительно хлор или бром. Реакцию по схеме А предпочтительно проводят в присутствии органического или неорганического основания в органическом растворителе. Примерами подходящих органических оснований являются алифатические или ароматические амины, например моно-, ди- или триалкиламины, моно-, ди- или триалканоламины, бензиламин,N-метилбензиламин и т.п. Примерами подходящих неорганических оснований являются сильные основания, такие как NaOH или KOH, или слабые основания, такие как NH4OH, Na2CO3, K2CO3, NaHCO3 и т.п. Примерами подходящих органических растворителей являются полярные протонные и апротонные органические растворители. Типичными примерами полярных протонных органических растворителей являются метанол, этанол, пропанол или бутанол. Типичными примерами полярных апротонных органических растворителей являются кетоны (например, ацетон или метилэтилкетон), тетрагидрофуран, диметилформамид, диметилсульфоксид, диоксан, ацетонитрил и т.п. Реакцию преимущественно проводят при нагревании, предпочтительно при температуре кипения используемого растворителя. Реакцию преимущественно осуществляют в присутствии активирующего агента, например йодида калия, йодида натрия, йодида цезия, йодида тетрабутиламмония или йодида триметилфениламмония. Новые промежуточные соединения формулы (II) являются еще одним аспектом настоящего изобретения. Новые промежуточные соединения формулы (II) можно получать согласно схеме А 1, представленной ниже: Получение новых промежуточных соединений формулы (II) на первой стадии включает взаимодействие между 4-хлор-2 Н-пирроло[3,4-с]хинолином (IV) и (1-бензилпиперид-4-ил)метанолом (IV) с последующим удалением бензильного заместителя у пиперидинового атома азота с помощью гидрирования. Предпочтительно 4-хлор-2 Н-пирроло[3,4-с]хинолин (IV) вводят в реакцию с натриевой солью(1-бензилпиперид-4-ил)метанола (VI), которую получают взаимодействием соединения (VI) с сильным основанием, например гидридом натрия. Реакцию предпочтительно осуществляют в полярном апротонном растворителе, выбранном из указанных ранее растворителей, предпочтительно в диметилформамиде. Реакцию проводят при нагревании, предпочтительно при кипячении с обратным холодильником. Реакцию дебензилирования предпочтительно проводят с помощью каталитического гидрирования в атмосфере азота, в спиртовом растворителе, предпочтительно используя в качестве катализатора палладий на угле. Типичным примером спиртового растворителя является метанол. Реакцию преимущественно осуществляют при комнатной температуре. В качестве альтернативы, описанные выше соединения формулы (I) можно получать по приведенной ниже схеме В: Реакцию по схеме В предпочтительно осуществляют в присутствии органического растворителя,используя натриевую соль соединения (V), которую получают взаимодействием соединения (VI) с сильным основанием, например гидридом натрия. Примерами подходящих органических растворителей являются полярные апротонные органические растворители. Типичными примерами полярных апротонных органических растворителей являются указанные ранее растворители и предпочтительно N,N-диметилформамид и метилэтилкетон. Промежуточные соединения формулы (IV) известны, и их получение описано в ARKIVOC (2004),number V, 181-195. Предпочтительно фармацевтические композиции по настоящему изобретению получают в виде подходящих лекарственных форм, включающих эффективную дозу по меньшей мере одного соединения формулы (I) либо его кислотно-аддитивной соли с фармацевтически приемлемой органической или неорганической кислотой или основно-аддитивной соли с фармацевтически приемлемым органическим или неорганическим основанием, а также по меньшей мере один фармацевтически приемлемый наполнитель. Предполагается, что термин "фармацевтически приемлемый наполнитель" подразумевает, без каких-либо конкретных ограничений, вещество, которое подходит для получения фармацевтической композиции, которую предполагается вводить живому существу. Такими веществами, известными в технике, являются, например, антиадгезионные средства, связующие средства, дезинтегрирующие средства, наполнители, разбавители, средства для образования суспензий, средства для улучшения скольжения, смазывающие средства, консервирующие средства, стабилизаторы, смачивающие средства, абсорбенты, ПАВ, буферы, соли для регулирования осмотического давления, эмульгаторы, вкусоароматические добавки и подсластители. Примерами подходящих фармацевтически приемлемых наполнителей являются сахара, например лактоза, глюкоза или сахароза, крахмалы, например кукурузный крахмал и картофельный крахмал, целлюлоза и ее производные, например натрий карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы, камедь трагаканта, солод, желатин, тальк, масло какао, воски, масла, например масло арахиса, масло хлопчатника, сафлоровое масло, масло кунжута, оливковое масло, кукурузное масло и соевое масло, гликоли, например пропиленгликоль, полиолы, например глицерин, сорбит, маннит и полиэтиленгликоль,сложные эфиры, например этилолеат и этиллаурат, агар-агар, буферные вещества, например гидроксид магния и гидроксид алюминия, альгиновая кислота, вода, изотонические растворы, этанол, буферные растворы, полиэфиры, поликарбонаты, полиангидриды и т.п. Примерами подходящих лекарственных форм являются таблетки, капсулы, таблетки с покрытием,гранулы, растворы и сиропы для перорального приема; антисептические пластыри, растворы, пасты,кремы и мази для трансдермального введения; суппозитории для ректального введения и стерильные растворы для инъекций или аэрозольного введения. Другими подходящими лекарственными формами являются формы с продолжительным высвобождением или формы на основе липосом, предназначенные для перорального или инъекционного пути введения. При необходимости конкретных видов лечения фармацевтическая композиция по настоящему изобретению может содержать другие фармакологически активные ингредиенты, одновременное введение которых является полезным. Количество соединения формулы (I) или его кислотно-аддитивной или основно-аддитивной соли в фармацевтической композиции по настоящему изобретению может меняться в широком диапазоне в зависимости от известных факторов, например типа патологии, с которой связана подвергаемая лечению невропатическая боль, тяжести заболевания, массы пациента, лекарственной формы, выбранного пути введения, количества приемов в течение дня и эффективности выбранного соединения формулы (I). Однако оптимальное количество может быть легко определено специалистом в данной области по стандартным методикам. Как правило, количество соединения формулы (I) или его кислотно-аддитивной соли или основноаддитивной соли в фармацевтической композиции по настоящему изобретению будет таким, что оно обеспечит количество вводимого соединения формулы (I) от 0,001 до 100 мг/кг/день в пересчете на свободную не солевую форму соединения. Предпочтительно количество вводимого соединения будет находиться в пределах от 0,05 до 50 мг/кг/день и еще более предпочтительно от 0,1 до 10 мг/кг/день. Лекарственные формы фармацевтических композиций по настоящему изобретению можно получать по методикам, хорошо известным специалистам в области фармацевтической химии, включающим смешивание, гранулирование, прессование, растворение, стерилизацию и т.п. Эффективность соединений формулы (I) при лечении хронической боли была продемонстрирована с помощью экспериментальной модели у крыс, представляющей собой аллодинию, вызванную перевязыванием седалищного нерва. Как известно специалистам в данной области, указанная экспериментальная модель может считаться имеющей предсказательную силу с точки зрения эффективности действия на человека. Эта экспериментальная модель перевязывания седалищного нерва у крыс представляет собой невропатию, которая воспроизводит ряд реакций, аналогичных реакциям, наблюдаемым у людей при различных травматических и патологических состояниях, связанных с невропатической болью. Причина этого заключается в том, что перевязывание седалищного нерва способно вызывать синдром, который связан с активацией специфических циклов, предназначенных для управления ощущением боли и характеризуемых появлением аллодинии, гипералгезии и спонтанной боли. Хорошо известно, что эта модель является подходящим инструментом для исследования лекарственных средств, предназначенных для лечения невропатической боли у людей и, в частности, для управления такими состояниями, как аллодиния и гипералгезия. Типичными примерами человеческих патологий, характеризуемых нарушениями, отражаемыми в указанной экспериментальной модели, и характеризуемых наличием невропатической боли, являются диабет, рак, иммунодефицит, травмы, ишемия, рассеянный склероз, ишиас, тригеминальная невралгия и постгерпетический синдром. Подтверждение связывания соединений с серотонинергическими рецепторами проводили с помощью биохимического теста на очищенных мембранах рекомбинантных человеческих клеток, которые устойчиво экспрессируют эти конкретные рецепторы, или на отдельных тканях животных, как описано, в частности, у Grossman C.J. et al. (1993), Br. J. Pharmacol. 109:618-624, Bonhaus D.W. et al. (1995), Br. J.Pharmacol. 115(4):622-628 и Saucier С. et al. (1997), J. Neurochem. 68(5):1998-2011. Как известно специалистам в данной области техники, этот тест является моделью молекулярного взаимодействия и селективности в отношении выбранных рецепторов, имеющей предсказательную силу. Тесты 1. Аллодиния, вызванная перевязыванием седалищного нерва у крыс. В тесте использовали самцов крыс CD, масса которых в момент поступления составляла 200-250 г. Аллодинию вызывали перевязыванием седалищного нерва левой задней лапы под анестезиейdisorders of pain sensation like those seen in man. Pain. 1988; 33:87-107]. По меньшей мере через две недели после перевязывания седалищного нерва отбирали крыс, которые демонстрировали снижение порога чувствительности, зафиксированного перед хирургическим вмешательством, не менее чем на 50%. Болевой порог измеряли на устройстве von Frey, которое с целью регистрации болевой реакции давало возможность постепенно увеличивать выраженное в граммах давление на ступню левой задней лапы крысы,соответствующее моменту, когда животное отдергивает лапу. Через 30 мин, 1, 2 и 4 ч после введения тестируемых соединений, болевой порог, измеренный у контрольных животных, сравнивали с болевым порогом животных, которым вводили тестируемое соединение (соединение 12 а табл. 1). Контрольным животным вводили тот же носитель (метилцеллюлозу), который применяли для введения тестируемого соединения. Результаты показаны на фигуре. 2. Связывание с серотонинергическими рецепторами. Подтверждение связывания с серотонинергическими рецепторами проводили с использованием очищенных мембран отдельных тканей животных или рекомбинантных человеческих клеток, которые устойчиво экспрессируют данные конкретные рецепторы. Связывание с рецептором серотонина 5-HT4 проводили с использованием стандартной методики,описанной в Grossman C.J., Kilpatric G.J. and Bunce K.T. (1993): "Development of a radioligand binding assay for 5-HT4 receptors in guinea-pig and rat brain". Br. J. Pharmacol. 109:618-624. Исходными материалами,использованными в исследовании, были гомогенат полосатого тела морской свинки и RS-23597190 в качестве образца сравнения. Тестируемые соединения исследовали при шести концентрациях для получения значений pKi индивидуальных соединений. Связывание с человеческим рецептором серотонина 5-HT2A проводили по стандартной методике,описанной в Bonhaus, D.W., Bach С., De Souza A., Salazar F.H., Matsuoka B.D., Zuppan P., Chan H.W.,Eglen R.M. (1995): "The pharmacology and distribution of human 5-hydroxytryptamine 2B (5-HT2B) receptoragonist-induced down-regulation involves decreases in receptor RNA and number". J. Neurochem. 68(5):19982011. Тестируемые соединения исследовали при шести концентрациях для получения значений pKi индивидуальных соединений. Значения сродства к рецепторам 5-HT4 и 5-HT2A ряда соединений формулы (I) по настоящему изобретению приведены соответственно в табл. 2 и 3 и выражены в виде pKi, значения которых пропорционально увеличиваются с ростом сродства соединений к рецептору. Данные табл. 2 и 3 демонстрируют, что приведенные в таблицах соединения формулы (I) обладают высоким сродством к рецептору 5-HT4, но небольшим или отсутствующим сродством к рецептору 5-HT2A. Синтетические примеры. 1 а) Этил 3-(2-нитрофенил)пропаноат. Раствор 2-нитробензальдегида (132 ммоль; 20,0 г) в абсолютном этаноле (220 мл) добавляли к смеси ацетата триэтилфосфония (159 ммоль; 35,7 г; 31,9 мл) и безводного карбоната калия (397 ммоль; 54,9 г). Эту смесь кипятили с обратным холодильником при энергичном перемешивании в течение 1 ч 30 мин. После охлаждения удаляли этанол при пониженном давлении, добавляли воду (500 мл) и полученный раствор экстрагировали этилацетатом (3300 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (3300 мл), сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный неочищенный продукт очищали на колонке с оксидом алюминия (элюируя хлороформом) и получали 27,6 г (94%) конечного продукта: желтое масло. 1 Н ЯМР (CDCl3,м.д.): 1,37 (т, 3H); 4,31 (кв, 2 Н); 6,38 (д, 1 Н); 7,55-7,59 (м, 1 Н); 7,68 (м, 2 Н); 8,048,14 (м, 2 Н). 1b) Этил 4-(2-нитрофенил)-1H-пиррол-3-карбоксилат. Раствор этил 3-(2-нитрофенил)пропаноата (120 ммоль; 26,6 г) и 4-толуолсульфонилметил изоцианата (TosMIC) (130 ммоль; 25,4 г) в смеси диметилсульфоксида и безводного этилового эфира(150 и 300 мл) по каплям в токе аргона добавляли к 60% суспензии гидрида натрия в парафине(260 ммоль; 10,4 г) в безводном этиловом эфире (300 мл). По окончании добавления смесь перемешивали при комнатной температуре в течение 25 мин, затем добавляли воду (500 мл) и полученный раствор экстрагировали этилацетатом (3600 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (3300 мл), сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный неочищенный продукт очищали на колонке с оксидом алюминия (элюируя смесью 1/1 хлороформ/этилацетат) и получали 12,8 г (41%) конечного продукта, имеющего следующие характеристики: твердое вещество желтого цвета, т.пл. 159-161C (растворитель кристаллизации: этанол),1 Н ЯМР (CDCl3,м.д.): 2,51 (м, 3H); 4,01 (кв, 2 Н); 6,51 (м, 1 Н); 7,31-7,33 (м, 2 Н); 7,39-7,41 (м, 2 Н); 7,49-7,53 (м, 1 Н); 7,92-7,94 (м, 1 Н); 12,0 (с, 1 Н). 1 с) 2 Н-Пирроло[3,4-с]хинолин-4(5H)-он. К раствору соединения, полученного в примере 1b (7,7 ммоль; 2,0 г) в ледяной уксусной кислоте(100 мл), поддерживая постоянную температуру 85C, в течение 15 мин добавляли порошок железа(120 ммоль; 6,7 г). Смесь оставляли перемешиваться при этой температуре в течение 45 мин. После охлаждения железо удаляли фильтрованием, несколько раз промывали тетрагидрофураном и фильтрат упаривали при пониженном давлении. Неочищенный продукт подвергали хроматографической очистке на колонке с оксидом алюминия (элюируя этилацетатом) и получали 1,05 г (74%) конечного продукта,имеющего следующие характеристики: твердое вещество красного цвета с температурой сублимации 280C (этанол),Н ЯМР (ДМСО-d6,м.д.): 7,05-7,28 (м, 3H); 7,57-7,63 (м, 2 Н); 7,84-7,88 (м, 1 Н); 10,7 (с, 1 Н); 12,1N,N-диметилформамиде (10 мл) добавляли безводный карбонат калия (11,0 ммоль; 1,5 г) и метилйодид(11,0 ммоль; 1,54 г; 0,68 мл). Поддерживали постоянную температуру смеси на уровне 90C и перемешивали в течение ночи. После охлаждения реакционную смесь обрабатывали водой и фильтровали. Твердое вещество сушили под инфракрасной лампой и очищали хроматографией на колонке с оксидом кремния(элюируя смесью 10/1 хлороформ/метанол), получая 0,96 г (45%) чистого продукта со следующими характеристиками: твердое вещество красного цвета с температурой сублимации 225C (толуол),1 Н ЯМР (ДМСО-d6,м.д.): 3,91 (с, 3H); 7,09-7,25 (м, 3H); 7,58-7,81 (м, 3H); 10,73 (с, 1 Н). 1 е) 4-Хлор-2-метил-2 Н-пирроло[3,4-с]хинолин (соединение формулы IV: R1=CH3; R2=H). Смесь продукта, полученного в примере 1d (5,0 ммоль; 1,0 г), оксихлорида фосфора (16,2 мл) и триэтиламина (1,2 мл) выдерживали при 120C в течение 6 ч. После охлаждения реакционную смесь осторожно выливали на лед и экстрагировали этилацетатом (3100 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (150 мл), насыщенным раствором бикарбоната натрия(350 мл) и затем опять насыщенным раствором NaCl (350 мл). Органический раствор сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный неочищенный продукт очищали на колонке с оксидом кремния (элюируя смесью 1/1 н-гексан/этилацетат) и получали 1,0 г(92%) конечного продукта со следующими характеристиками: твердое вещество желтого цвета, т.пл. 123-124C (бензол),1 Н ЯМР (ДМСО-d6,м.д.): 4,09 (с, 3H); 7,51-7,58 (м, 2 Н); 7,76 (м, 1 Н); 7,82-7,85 (м, 1 Н); 7,97 (м,1 Н); 8,15-8,17 (м, 1 Н). 1f) 2-Изопропил-2 Н-пирроло[3,4-с]хинолин-4(5H)-он. К кипящему раствору продукта, полученного в примере 1 с (16,3 ммоль; 3,0 г), в диоксане (150 мл) добавляли металлический калий (14,8 ммоль; 580 мг) и смесь перемешивали при кипячении с обратным холодильником до полного исчезновения металла (примерно 2 ч). После охлаждения добавляли 2-йодпропанол (16,3 ммоль; 2,77 г) и 18-краун-6 эфир (14,8 ммоль; 3,92 г) и полученную смесь кипятили с обратным холодильником в течение 5 ч 30 мин. Затем добавляли дополнительную порцию 2-йодпропанола (8,1 ммоль; 1,38 г) и реакционную смесь кипятили с обратным холодильником при перемешивании в течение еще 15 ч. После охлаждения удаляли диоксан при пониженном давлении остаток смешивали с этилацетатом (100 мл) и промывали насыщенным раствором NaCl (350 мл). Полученный органический раствор сушили над безводным сульфатом натрия и удаляли растворитель при пониженном давлении, получая неочищенный продукт, который очищали хроматографией на колонке с оксидом алюминия (элюируя этилацетатом). Получали 0,9 г (24%) продукта со следующими характеристиками: твердое вещество белого цвета, т.пл. 189-190C (толуол),1 Н ЯМР (ДМСО-d6,м.д.): 1,48 (д, 6 Н); 4,52 (м, 1 Н); 7,02-7,20 (м, 3H); 7,63-7,70 (м, 2 Н); 7,77 (м,1 Н); 10,62 (с, 1 Н). 1g) 4-Хлор-2-изопропил-2 Н-пирроло[3,4-с]хинолин (соединение формулы IV: R1=CH(CH3)2; R2=H). Смесь продукта, полученного в примере 1f (4,4 ммоль; 1,0 г), оксихлорида фосфора (14,3 мл) и триэтиламина (1,1 мл) выдерживали при 120C в течение 25 мин. После охлаждения реакционную смесь осторожно выливали на лед и экстрагировали этилацетатом (3100 мл). Органические фазы объединяли и промывали насыщенным раствором NaCl (150 мл), насыщенным раствором бикарбоната натрия(350 мл) и затем опять насыщенным раствором NaCl (350 мл). После этого органический раствор сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный неочищенный продукт очищали на колонке с оксидом кремния (элюируя хлороформом) и получали 0,9 г (91%) конечного продукта со следующими характеристиками: твердое вещество желтого цвета, т.пл. 70-72C (циклогексан),1 Н ЯМР (CDCl3,м.д.): 1,64 (д, 6 Н); 4,59 (м, 1 Н); 7,46-7,49 (м, 3H); 7,55 (м, 1 Н); 7,93-7,98 (м, 2 Н). 2 а) 1-Бутил-1-пиперидинметанол (соединение формулы V: R3=CH2CH2CH3). Смесь этил изонипекотата (31,8 ммоль, 5,0 г), безводного карбоната калия (63,6 ммоль; 8,8 г) и 1-бромбутана (31,8 ммоль; 4,36 г) в абсолютном этаноле (63 мл) кипятили с обратным холодильником при перемешивании в течение 3 ч. После охлаждения карбонат отделяли фильтрованием и фильтрат упаривали при пониженном давлении. 7,68 г этил 4-бутил изонипекотата получали в форме масла, которое использовали в следующей стадии без дополнительной очистки. Полученный продукт растворяли в безводном этиловом эфире (32 мл) и по каплям добавляли к суспензии литийалюминийгидрида (41,3 ммоль; 1,57 г) в том же растворителе (20 мл), охлажденной до 0C. Полученную реакционную смесь перемешивали при комнатной температуре в течение 15 ч. После охлаждения до 0C осторожно добавляли лед, и 1 полученные при этом гидроксиды отделяли фильтрованием. После разделения двух фаз эфирную фазу сушили над безводным сульфатом натрия и упаривали при пониженном давлении, получая 5,1 г (94%) целевого соединения в виде прозрачного масла. 1 Н ЯМР (CDCl3,м.д.): 0,89 (т, 3H); 1,26-1,32 (м, 4 Н); 1,44-1,51 (м, 3H); 1,72 (д, 2 Н); 1,89-1,95 (м,2 Н); 2,29-2,33 (м, 3H); 2,94-2,97 (м, 2 Н); 3,46 (д, 2 Н). 2b) Этил 4-(2-фенилэтил)изонипекотат. Смесь этил изонипекотата (64 ммоль; 10,0 г) безводного карбоната калия (192 ммоль; 26,5 г) и фенилэтилбромида (77 ммоль; 14,25 г) в безводном N,N-диметилформамиде (100 мл) перемешивали при 70C в течение 5 ч 30 мин. После охлаждения реакционную смесь разбавляли водой (300 мл) и экстрагировали этилацетатом (3200 мл). Объединенные органические фазы промывали насыщенным растворомNaCl (3100 мл), сушили над сульфатом натрия и упаривали при пониженном давлении. Получали неочищенный продукт, который очищали на колонке с оксидом алюминия (элюируя смесью 2/1 н-гексан/этилацетат), получая 14,0 г (84%) чистого продукта в виде желтого масла. 1 Н ЯМР (CDCL3,м.д.): 1,22-1,27 (т, 3H); 1,75-1,85 (м, 2 Н); 1,92-1,96 (м, 1 Н); 2,09-2,14 (т, 2 Н); 2,262,33 (м, 1 Н); 2,57-2,61 (м, 2 Н); 2,79-2,83 (м, 2 Н); 2,95-2,98 (м, 2 Н); 4,10-4,16 (кв, 2 Н). 2 с) 1-(2-Фенилэтил)-4-пиперидинметанол (соединение формулы V: R3=CH2Ph). К суспензии литийалюминийгидрида (70 ммоль; 2,66 г) в безводном ТГФ (40 мл) при 0C по каплям добавляли раствор этил 4-(2-фенилэтил)изонипекотата (54 ммоль; 14,0 г) в том же растворителе (130 мл). Реакционную смесь перемешивали при комнатной температуре в течение 10 мин. После охлаждения до 0C осторожно добавляли лед и образовавшиеся при этом гидроксиды отделяли фильтрованием. После удаления ТГФ при пониженном давлении водную фазу экстрагировали этилацетатом (3100 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (3100 мл), сушили над сульфатом натрия и упаривали при пониженном давлении. Получали неочищенный продукт, который очищали на колонке с оксидом алюминия (элюируя этилацетатом), что позволяло получить 7,9 г (67%) чистого продукта со следующими характеристиками: твердое вещество желтого цвета, т.пл. 89-90C (циклогексан),1H ЯМР (CDCl3,м.д.): 1,25-1,43 (м, 2 Н); 1,50-1,58 (м, 2 Н); 1,76-1,79 (м, 2 Н); 2,03-2,09 (м, 2 Н); 2,592,64 (м, 2 Н); 2,82-2,87 (м, 2 Н); 3,05-3,08 (м, 2 Н); 3,52 (м, 2 Н); 7,18-7,31 (м, 5 Н). 2d) 4-Пиперидинметанол. К суспензии литийалюминийгидрида (8,3 ммоль; 310 мг) в безводном ТГФ (5 мл) при 0C по каплям добавляли раствор этилизонипекотата (6,4 ммоль; 1,0 г) в безводном ТГФ (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 35 мин. После охлаждения до 0C осторожно добавляли водный этанол и отделяли фильтрованием образовавшиеся при этом гидроксиды. Органическую фазу сушили над безводным сульфатом натрия и упаривали при пониженном давлении, получая 1,34 г(100%) чистого продукта в виде масла. 1 Н ЯМР (ДМСО-d6,м.д.): 1,09-1,18 (м, 2 Н); 1,50 (м, 1 Н); 1,66-1,69 (м, 2 Н); 2,53-2,61 (м, 3H); 3,053,08 (м, 2 Н); 3,25 (д, 2 Н); 4,60 (с, 1 Н). 2 е) N-Фенил-2-(4-гидроксиметилпиперид-1-ил)ацетамид (соединение формулы V: R3=CONHPh). К раствору 4-пиперидинметанола (9,65 ммоль; 1,15 г) в N,N-диметилформамиде (15 мл) добавляли 2-хлор-N-фенилацетамид (11,6 ммоль; 1,97 г) и безводный карбонат калия (29 ммоль; 4,0 г). Смесь перемешивали при 70C в течение 2 ч 30 мин. После охлаждения реакционную смесь разбавляли водой(15 мл) и экстрагировали этилацетатом (350 мл). Объединенные органические фазы промывали насыщенным раствором NaCl, сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали хроматографией на колонке с оксидом кремния (элюируя смесью 1/1 хлороформ/этилацетат) и получали 420 мг (27%) чистого продукта со следующими характеристиками: твердое вещество белого цвета, т.пл. 93-95C (бензол),1 Н ЯМР (CDCl3,м.д.): 1,34-1,38 (м, 2 Н); 1,58 (м, 1 Н); 1,81 (м, 2 Н); 2,10 (м, 1 Н); 2,24-2,28 (м, 2 Н); 2,93-2,96 (м, 2 Н); 3,12 (с, 2 Н); 3,54 (д, 2 Н). 2f) Этил 4-бензилизонипекотат. Смесь этил изонипекотата (65 ммоль; 10,2 г), безводного карбоната калия (195 ммоль; 26,9 г) и бензилбромида (78 ммоль; 13,36 г) в безводном N,N-диметилформамиде (100 мл) перемешивали при 70C в течение 18 ч. После охлаждения реакционную смесь разбавляли водой (300 мл) и экстрагировали этилацетатом (3200 мл). Объединенные органические фазы промывали насыщенным раствором NaCl(3100 мл), сушили над сульфатом натрия и упаривали при пониженном давлении. Получали неочищенный продукт, который очищали на колонке с оксидом алюминия (элюируя хлороформом) и получали 13,84 г (86%) чистого продукта в виде желтого масла. 1 Н ЯМР (CDCl3,м.д.): 1,28-1,32 (т, 3H); 1,81 (м, 4 Н); 2,07-2,12 (м, 2 Н); 2,34 (м, 1 Н); 2,91 (м, 2 Н); 3,55 (м, 2 Н); 4,15-4,21 (м, 2 Н); 7,31-7,38 (м, 5 Н). 2g) (1-Бензил-4-пиперидил)метанол (соединение VI). К раствору литийалюминийгидрида (72 ммоль; 2,7 г) в безводном ТГФ (40 мл) при 0C по каплям добавляли раствор этил 4-бензилизонипекотата (55 ммоль; 13,7 г) в том же растворителе (130 мл). Реакционную смесь перемешивали при комнатной температуре в течение 20 мин. После охлаждения до 0C осторожно добавляли лед и образовавшиеся при этом гидроксиды отделяли фильтрованием. После удаления тетрагидрофурана при пониженном давлении водную фазу экстрагировали этилацетатом(3100 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (3100 мл), сушили над сульфатом натрия и упаривали при пониженном давлении. Получали неочищенный продукт,который очищали на колонке с оксидом алюминия (элюируя этилацетатом), получая 10,25 г (90,5%) чистого продукта в виде масла. 1 Н ЯМР (CDCl3,м.д.): 1,29-1,39 (м, 2 Н); 1,53-1,56 (м, 1 Н); 1,74-1,78 (м, 2 Н); 1,86 (с, 1 Н); 1,99-2,05(I): R1=CH3; R2=H; R3=CH2CH2CH3). К 60% суспензии NaH в парафине (6,72 ммоль; 270 мг) в безводном N,N-диметилформамиде (14 мл) по каплям добавляли раствор (1-бутил-4-пиперидил)метанола (6,72 ммоль; 1,14 г) в безводномN,N-диметилформамиде (14 мл). Реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Полученный при этом алкоксид по каплям добавляли к раствору продукта, полученного в примере 1 е (1,92 ммоль; 470 мг), в безводном N,N-диметилформамиде (14 мл), предварительно нагретому до 146C. Полученную реакционную смесь перемешивали при 146C в течение 1 ч 10 мин. После охлаждения смесь выливали на лед и экстрагировали этилацетатом (350 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (320 мл), сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали хроматографией на колонке с оксидом алюминия (используя хлороформ в качестве элюента), получая 520 мг (71%) чистого продукта со следующими характеристиками: твердое вещество, т.пл. 83-85C (бензол/циклогексан),1 Н ЯМР (CDCl3,м.д.): 0,97 (т, 3H); 1,35-1,59 (м, 6 Н); 1,90-2,07 (м, 5 Н); 2,37 (м, 2 Н); 3,03 (м, 2 Н); 3,99 (с, 3H); 4,49 (д, 2 Н); 7,28-7,49 (м, 4 Н); 7,75 (м, 1 Н); 7,88 (м, 1 Н). 4) 4-[1-(2-Фенил)этилпиперидин-4-ил]метокси-2-метил-2 Н-пирроло[3,4-с]хинолин (соединение формулы (I): R1=CH3; R2=H; R3=CH2Ph). К суспензии 60% NaH в парафине (8,05 ммоль; 320 мг) в безводном N,N-диметилформамиде (16 мл) по каплям добавляли раствор 1-(2-фенилэтил)-4-пиперидинметанола (8,05 ммоль; 1,77 г) в безводномN,N-диметилформамиде (16 мл). Реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Полученный при этом алкоксид по каплям добавляли к раствору продукта, полученного в примере 1 е (2,3 ммоль; 500 мг) в безводном N,N-диметилформамиде (16 мл), предварительно нагретому до 146C. Полученную реакционную смесь перемешивали при 146C в течение 7 ч 30 мин. После охлаждения смесь выливали на лед и экстрагировали этилацетатом (350 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (320 мл), сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали хроматографией на колонке с оксидом алюминия (элюируя смесью 4/1 н-гексан/этилацетат), получая 620 мг (68%) чистого продукта со следующими характеристиками: твердое вещество, т.пл. 90-92C (бензол/циклогексан),1 Н ЯМР (CDCl3,м.д.): 1,67 (м, 2 Н); 1,94-1,97 (м, 3H); 2,17 (м, 2 Н); 2,69 (м, 2 Н); 2,90 (м, 2 Н); 3,15(м, 2 Н); 3,98 (с, 3H); 4,49 (д, 2 Н); 7,20-7,42 (м, 9 Н); 7,74 (м, 1 Н); 7,88 (м, 1 Н). 5) 4-[1-(2-Фенил)этилпиперид-4-ил]метокси-2-изопропил-2 Н-пирроло[3,4-с]хинолин (соединение формулы (I): R1=CH(CH3)2; R2=H; R3=CH2Ph). К суспензии 60% NaH в парафине (7,7 ммоль; 310 мг) в безводном N,N-диметилформамиде (15 мл) по каплям добавляли раствор 1-(2-фенилэтил)-4-пиперидинметанола (7,7 ммоль; 1,69 г) в безводномN,N-диметилформамиде (15 мл). Реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Полученный при этом алкоксид по каплям добавляли к раствору продукта, полученного в примере 1g (2,2 ммоль; 500 мг), в безводном N,N-диметилформамиде (15 мл), предварительно нагретому до 146C. Полученную реакционную смесь перемешивали при 146C в течение 1 ч. После охлаждения смесь выливали на лед и экстрагировали этилацетатом (350 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (320 мл), сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали хроматографией на колонке с оксидом алюминия(элюируя смесью 4/1 н-гексан/этилацетат), получая 680 мг (73%) чистого продукта со следующими характеристиками: твердое вещество, т.пл. 90-93C (н-гексан),1 5 а) 4-[1-(2-Фенил)этилпиперид-4-ил]метокси-2-изопропил-2 Н-пирроло[3,4-с]хинолина гидрохлорид. Готовили раствор хлористого водорода в метаноле, добавляя по каплям ацетилхлорид (2,67 ммоль; 200 мг) к 10 мл метанола, охлажденного на ледяной бане. Этот раствор несильно перемешивали в течение нескольких минут и затем по каплям добавляли к раствору продукта, полученного в примере 5(2,34 ммоль; 1,0 г) в метаноле (5,0 мл). По окончании добавления смесь перемешивали при 0C в течение 45 мин, после чего добавляли безводный диэтиловый эфир (примерно 200 мл), пока не наблюдалось осаждение соли. Полученную соль отделяли фильтрованием, промывали безводным этиловым эфиром(32 мл) и сушили в вакууме при 45C в течение 6 ч. Получали 630 мг (58%) чистого продукта со следующими характеристиками: твердое вещество, т.пл. 138-140C (изопропиловый эфир/изопропанол),1H ЯМР (ДМСО-d6,м.д.): 1,57 (д, J=6,59 Гц, 6 Н); 1,79-2,29 (м, 5 Н); 2,86-3,47 (м, 6 Н); 3,63 (д,J=11,71 Гц, 2 Н); 4,55 (д, J=4,03 Гц, 2 Н); 4,68 (септет, J=6,59 Гц, 1 Н); 7,20-7,48 (м, 7 Н); 7,80 (шир.с, 1 Н); 7,95-8,16 (м, 3H); 10,93 (шир.с, 1 Н). б) N-Фенил-2-4-[2-метил-2 Н-пирроло[3,4-с]хинолин-4-илоксиметил]пиперид-1-илацетамид (соединение формулы (I): R1=CH3; R2=H; R3=CONHPh). К суспензии 60% NaH в парафине (30,8 ммоль; 1,23 г) в безводном N,N-диметилформамиде (60 мл) по каплям добавляли раствор N-фенил-2-(4-гидроксиметилпиперид-1-ил)ацетамида (30,8 ммоль; 7,65 г) в безводном N,N-диметилформамиде (60 мл). Реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Полученный при этом алкоксид по каплям добавляли к раствору продукта, полученного в примере 1 е (8,8 ммоль; 1,9 г), в безводном N,N-диметилформамиде (60 мл), предварительно нагретому до 146C. Полученную реакционную смесь перемешивали при 146C в течение 3 ч 30 мин. После охлаждения смесь выливали на лед и экстрагировали этилацетатом (3150 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (3100 мл), сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали хроматографией на колонке с оксидом алюминия (элюируя смесью 2/1 н-гексан/этилацетат), получая 2,9 г (77%) чистого продукта в виде масла. 1N-Фенил-2-4-[2-метил-2H-пирроло[3,4-с]хинолин-4-илоксиметил]пиперид-1-илацетамида гидрохлорид. Готовили раствор хлористого водорода в метаноле, добавляя по каплям ацетилхлорид (1,32 ммоль; 100 мг) к 5 мл метанола, охлажденного на ледяной бане. Реакционную смесь несильно перемешивали в течение нескольких минут и затем по каплям добавляли к раствору амина, полученного в примере 6(1,2 ммоль; 500 мг) в метаноле (3,5 мл). По окончании добавления смесь перемешивали при 0C в течение 45 мин, после чего добавляли безводный диэтиловый эфир (примерно 100 мл), пока не наблюдалось осаждение соли. После растирания при комнатной температуре в течение ночи полученную соль отделяли фильтрованием, промывали безводным этиловым эфиром (32 мл) и сушили в вакууме при 45C в течение 2 дней. Получали 420 мг (75%) чистого продукта со следующими характеристиками: твердое вещество, т.пл. 163-165C (метанол),1 Н ЯМР (ДМСО-d6,м.д.): 1,66-2,41 (м, 5 Н); 3,10-3,32 (м, 2 Н); 3,63 (д, J=11,39 Гц; 2 Н); 4,01 (с, 3H); 4,19 (д, J=3,80 Гц; 2 Н); 4,53 (д, J=5,61 Гц; 2 Н); 7,12 (т, J=7,35 Гц; 1 Н); 7,28-7,50 (м, 4 Н); 7,59-7,93 (м, 5 Н); 8,03 (дд, J=7,43; 1,65 Гц; 1 Н); 10,14 (шир.с, 1 Н); 11,02 (с, 1 Н). 7) 4-[(1-Бензилпиперид-4-ил)метилокси]-2-метил-2H-пирроло[3,4-с]хинолин (соединение формулыVII: R1=CH3; R2=H), (соединение формулы (I): R1=CH3; R2=H; R3=Ph). К суспензии 60% NaH в парафине (16,1 ммоль; 650 мг) в безводном N,N-диметилформамиде (32 мл) по каплям добавляли раствор (1-бензил-4-пиперидил)метанола (16,1 ммоль; 3,32 г) в безводномN,N-диметилформамиде (32 мл). Реакционную смесь перемешивали при комнатной температуре в течение 10 мин. Полученный при этом алкоксид по каплям добавляли к раствору продукта, полученного в примере 1 е (4,6 ммоль; 1,0 г) в безводном N,N-диметилформамиде (32 мл), нагретому до 146C. Полученную реакционную смесь перемешивали при этой температуре в течение 1 ч 30 мин. После охлаждения смесь выливали на лед и экстрагировали этилацетатом (3100 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (350 мл), сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали хроматографией на колонке с оксидом алюминия К раствору продукта, полученного в примере 7 (0,35 ммоль; 1,34 г), в метаноле (150 мл) добавляли 10% палладий на угле (200 мг). Смесь перемешивали в атмосфере Н 2 при комнатной температуре и атмосферном давлении в течение 4 дней, пропуская через реакционную смесь поток Н 2 каждые 3 ч и добавляя 10% палладий на угле (200 мг) каждые 24 ч. После отделения палладия при помощи фильтровальной бумаги удаляли растворитель при пониженном давлении, получая остаток, который подвергали хроматографической очистке на колонке с оксидом алюминия (элюируя смесью 2/1 хлороформ/метанол). Описанным способом получали 410 мг (40%) чистого продукта в виде масла. 1(57 мл) добавляли концентрированную серную кислоту (3,0 мл). Эту смесь кипятили с обратным холодильником при перемешивании в течение 2 ч 30 мин. После охлаждения удаляли этанол при пониженном давлении и полученный неочищенный продукт растворяли в хлороформе (200 мл). Органическую фазу промывали, во-первых, 1 н. NaOH (100 мл) и затем насыщенным раствором NaCl (3100 мл). После высушивания над безводным сульфатом натрия и удаления растворителя при пониженном давлении получали 12,0 г (100%) чистого продукта в виде бледно-желтого масла. 1 Н ЯМР (CDCl3,м.д.): 1,42 (т, 3H); 2,74 (т, 2 Н); 3,34 (т, 2 Н); 4,39 (кв, 2 Н); 7,55 (м, 2 Н); 8,04 (м,2 Н). 8b) 4-(2-Бромэтил)бензоламин. К раствору 4-нитро-1-(2-бромэтил)бензола (13 ммоль; 3,0 г) в этилацетате (200 мл) добавляли 10% палладий на угле (500 мг). Полученную суспензию помещали в атмосферу Н 2 в автоклав для гидрирования Пара при давлении 70 фунтов/кв.дюйм при комнатной температуре на 4 ч. В конце этого периода суспензию фильтровали через фильтровальную бумагу и выпаривали растворитель при пониженном давлении, получая 2,81 г амина (46,8%) в виде масла, которое использовали в следующей стадии без дополнительной очистки. 8 с) N-[4-(2-Бромэтил)фенил]ацетамид. К суспензии 4-(2-бромэтил)бензоламина, полученного в примере 8b (1,0 г; 5 ммоль), в пиридине(1,2 мл) и уксусном ангидриде (1,27 г; 12,4 ммоль), помещенной на ледяную баню на 30 мин, добавляли 1 н. HCl. Полученную смесь экстрагировали этилацетатом (3200 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (3300 мл), сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Полученный таким образом неочищенный продукт очищали на колонке с оксидом кремния (элюируя этилацетатом), получая 620 мг (51%) продукта: твердое вещество, т.пл. 135-136C (бензол),1 Н ЯМР (CDCl3,м.д.): 2,25 (с, 3H); 3,19 (т, 2 Н); 3,59 (т, 2 Н); 7,22 (д, 2 Н); 7,50 (д, 2 Н). 8d) 4'-(2-Бромэтил)метансульфонанилид. К раствору 4-аминофенилэтил бромида (5 ммоль; 1,0 г) в безводном ТГФ (25 мл) добавляли триэтиламин (6,6 ммоль; 0,744 г; 0,81 мл). К этой смеси, помещенной на ледяную баню, добавляли метансульфонилхлорид (6 ммоль, 0,687 г; 0,46 мл). Смесь перемешивали при комнатной температуре в течение 45 мин и затем разбавляли водой, подкисленной 1 н. HCl, до кислой реакции и экстрагировали этилацетатом. Объединенные органические фазы промывали насыщенным раствором NaCl и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении, получая 1,18 г неочищенного продукта в виде желтого масла. Неочищенный продукт очищали хроматографией на колонке с оксидом алюминия (элюируя смесью 1/1 этилацетат/хлороформ), получая 580 мг (41%) продукта со следующими характеристиками: твердое вещество, т.пл. 115-116C (толуол/гексан),1 Н ЯМР (CDCl3,м.д.): 3,07 (с, 3H); 3,21 (т, 2 Н); 3,61 (т, 2 Н); 6,40 (с, 1 Н); 7,25-7,29 (м,4 Н). 8 е) 4-(2-Бромэтил)бензиловый спирт. К суспензии LiAlH4 (4,5 ммоль; 0,172 г) в безводном ТГФ (5,0 мл), охлажденной до 0C, по каплям добавляли раствор метил 4-(2-бромэтил)бензолкарбоксилата (1,0 г; 4,1 ммоль) в безводном ТГФ(10,0 мл). Реакционную смесь перемешивали в течение 5 мин при комнатной температуре и после добавления льда фильтровали через фильтр Бюхнера. Растворитель удаляли при пониженном давлении и остаток экстрагировали этилацетатом (3100 мл). Объединенные органические фазы промывали насыщенным раствором NaCl, сушили над сульфатом натрия и упаривали при пониженном давлении. Таким способом получали 800 мг (82,6%) чистого продукта в виде масла. 1H ЯМР (CDCl3,м.д.): 3,22 (т, 2 Н); 3,62 (т, 2 Н); 4,72 (с, 2 Н); 7,25-7,39 (м,4 Н). 8f) 4-(Метоксиметил)фенилэтил бромид. К суспензии 60% NaH в парафине (13,9 ммоль; 0,56 г) в ТГФ (20 мл), охлажденной до 0C, вопервых, по каплям на ледяной бане добавляли раствор 4-(2-бромэтил)бензилового спирта (9,25 ммоль; 1,99 г) в ТГФ (20 мл) и затем йодметан (13,9 ммоль; 1,972 г; 0,86 мл). Смесь перемешивали при комнатной температуре в течение 1 ч 30 мин, после чего выливали в воду и экстрагировали этилацетатом. Объ- 15019300 единенные органические фазы промывали насыщенным раствором NaCl (320 мл), сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Остаток очищали хроматографией на колонке с оксидом алюминия, элюируя этилацетатом, и получали 1,38 г продукта в виде масла (65%). 1 Н ЯМР (CDCl3,м.д.): 3,24 (т, 2 Н); 3,44 (с, 2 Н); 3,64 (т, 2 Н); 4,50 (с, 3H); 7,25-7,47 (м, 4 Н). 8g) Изохроман-1-он. К раствору изохромана (5,0 г; 37,3 ммоль) в дихлорметане (465 мл) при перемешивании и при комнатной температуре в течение 15 мин добавляли гомогенную смесь перманганата калия и диоксида марганца (74,60 г) в соотношении 1:3. Полученную суспензию оставляли на 18 ч при комнатной температуре. Суспензию фильтровали через фильтр Бюхнера. Растворитель выпаривали при пониженном давлении, получая 4,42 г неочищенного продукта (80%), который использовали на следующей стадии без дополнительной очистки. 8h) Метил 2-(2-хлорэтил)бензолкарбоксилат. Смесь изохроман-1-она, полученного в примере 8g (3,0 г; 20,2 ммоль), и PCl5 (20,4 ммоль) выдерживали при постоянной температуре 150C в течение 30 мин. Образующийся при этом POCl3 выпаривали и затем добавляли метанол (15 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Растворитель выпаривали, получая неочищенный продукт, который очищали хроматографией на колонке с оксидом кремния, элюируя смесью 1/3 этилацетат/н-гексан. Таким способом получали 2,16 г (54%) чистого продукта в виде масла. 1H ЯМР (CDCl3,м.д.): 3,23 (т, 2 Н); 3,71 (т, 2 Н); 3,95 (с, 3H); 7,32-7,35 (м, 2 Н); 8,04-8,06 (м, 2 Н). 8i) 4-(2-Бромэтил)бензолсульфонилхлорид. К раствору 2-бромэтилбензола (10,0 г; 54 ммоль) в 16 мл хлороформа при температуре 0C по каплям добавляли раствор хлорсульфоновой кислоты (162 ммоль, 18,87 г; 10,82 мл) в 11 мл хлороформа. Реакционную смесь выдерживали 15 мин при 0C и затем 3 ч при комнатной температуре. После этого реакционную смесь выливали на лед и водную фазу экстрагировали хлороформом. Объединенные органические фазы промывали насыщенным раствором хлорида натрия и сушили над безводным сульфатом натрия. Растворитель выпаривали, получая 12,34 г неочищенного продукта, который очищали хроматографией на колонке с оксидом кремния, элюируя хлороформом (выход 76%). Полученный продукт использовали в следующей стадии без дальнейшей очистки. 8j) 4-(2-Бромэтил)бензолсульфонамид. К раствору 4-хлорсульфонил-1-(2-бромэтил)бензола, полученного в примере 8i (4,0 г; 15,2 ммоль), в ТГФ (1,76 мл; 15,2 ммоль), охлажденному до 0C, добавляли раствор гидроксида аммония (1,51 г; 30,4 ммоль, 1,71 мл). Взаимодействие происходило немедленно. Реакционную смесь разбавляли водой и фильтровали через фильтр Gooch, получая 7,0 г неочищенного продукта, который очищали хроматографией на колонке с оксидом кремния, используя хлороформ в качестве элюента. Описанным способом было получено 570 мг (выход 18%) продукта со следующими характеристиками: твердое вещество белого цвета, т.пл. 115-116C (этанол),1 Н ЯМР (CDCl3,м.д.): 3,24 (т, 2 Н); 3,58 (т, 2 Н); 7,37 (д, 2 Н); 7,91 (д, 2 Н). 9) 4-[1-(2-(4-Морфолинил)этил)пиперид-4-ил]метилокси-2-метил-2H-пирроло[3,4-с]хинолин (соединение формулы (I): R1=CH3; R2=H; R3=СН 2-N-морфолин). К раствору продукта, полученного в примере 8 (1,3 ммоль; 380 мг), в абсолютном этаноле (12 мл) добавляли гидрохлорид 4-(2-хлорэтил)морфолина (1,3 ммоль; 240 мг) и бикарбонат натрия (3,64 ммоль; 310 мг). Смесь перемешивали при кипячении с обратным холодильником в течение 3 ч 15 мин. После охлаждения растворитель удаляли при пониженном давлении. Остаток смешивали с водой (50 мл) и экстрагировали этилацетатом (350 мл). Объединенные органические фазы промывали насыщенным раствором NaCl, сушили над сульфатом натрия и упаривали при пониженном давлении, получая 480 мг(м, 2 Н); 3,78 (м, 4 Н); 4,03 (с, 3H); 4,49 (д, 2 Н); 7,32-7,46 (м, 4 Н); 7,78 (м, 1 Н); 7,93 (м, 1 Н). 9 а) 4-[1-(2-(4-Морфолинил)этил)пиперид-4-ил]метилокси-2-метил-2H-пирроло[3,4-с]хинолина гидрохлорид. Раствор хлористого водорода в метаноле получали, добавляя по каплям ацетилхлорид (1,68 ммоль; 120 мг) к 3,2 мл метанола, охлажденного на ледяной бане. Смесь несильно перемешивали в течение нескольких минут, после чего по каплям добавляли раствор амина, полученного в примере 9 (0,76 ммоль; 310 мг), в метаноле (4,2 мл). По окончании добавления смесь перемешивали при 0C в течение 45 мин и затем добавляли безводный этиловый эфир (примерно 65 мл) до осаждения смеси продукта с растворителем. После растирания в течение 2 дней полученную соль отделяли фильтрованием, промывали петролейным эфиром (32 мл) и сушили в вакууме при 45C в течение 1 дня. Описанным способом получали 150 мг (41%) чистого продукта со следующими характеристиками: твердое вещество желтого цвета, т.пл. 190-192C (изопропиловый эфир/изопропанол),1 Н ЯМР (ДМСО-d6,м.д.): 1,71-2,01 (м, 2 Н); 2,16-2,48 (м, 3H); 2,94-3,14 (м, 4 Н); 3,19-3,39 (м, 4 Н); 3,57 (т, J=7,31 Гц, 2 Н); 3,81 (д, J=12,57 Гц, 2 Н); 3,97 (т, J=4,38 Гц, 3H); 4,01 (с, 3H); 4,42 (д, J=5,70 Гц,- 16019300 2 Н); 7,42 (д, J=1,75 Гц, 1 Н); 7,45-7,58 (м, 4 Н); 7,61 (с, 1 Н); 7,78 (д, J=7,75 Гц; 1 Н). 10) 4-[(1-(4-Нитрофенилэтил)пиперид-4-ил)метилокси]-2-метил-2 Н-пирроло[3,4-с]хинолин (соединение формулы (I): R1=CH3; R2=H; R3=CH2-C6H4-(4-NO2. К раствору продукта, полученного в примере 8 (2,2 ммоль; 0,65 г), в ДМФА (5 мл) добавляли 4-нитрофенилэтилбромид (2,6 ммоль; 0,61 г) и K2CO3 (6,6 ммоль; 0,91 г). Смесь перемешивали при 70C в течение 2 ч 45 мин. После охлаждения смесь разбавляли водой (20 мл) и экстрагировали этилацетатом(320 мл). Объединенные органические фазы промывали насыщенным раствором NaCl (350 мл), сушили над сульфатом натрия и упаривали при пониженном давлении, получая неочищенный продукт, который очищали хроматографией на колонке с оксидом кремния (элюируя смесью 10/1 хлороформ/метанол), получая 0,62 г (62%) чистого продукта со следующими характеристиками: твердое вещество желтого цвета, т.пл. 157-159C (толуол/циклогексан),1 Н ЯМР (CDCl3,м.д.): 1,65 (м, 2 Н); 2,00 (м, 3H); 2,19 (м, 2 Н); 2,72 (т, J=8,2 Гц, 2 Н); 3,02 (т,J=8,2 Гц, 2 Н); 3,12 (м, 2 Н); 4,03 (с, 3H); 4,53 (д, 2 Н); 7,32-7,47 (м, 6 Н); 7,78 (м, 1 Н); 7,93 (2 дд, J=8,1 Гц,J=1,1 Гц, 2 Н); 8,20 (д, J=8,7 Гц, 2 Н). 11) 4-[(1-(4-Аминофенилэтил)пиперид-4-ил)метилокси]-2-метил-2H-пирроло[3,4-с]хинолин (соединение формулы (I): R1=CH3; R2=H; R3=CH2-C6H4-(4-NH2. К раствору продукта, полученного в примере 10 (1,4 ммоль; 0,61 г), в этилацетате (100 мл) добавляли 10% палладий на угле (200 мг). Смесь перемешивали в атмосфере Н 2 при комнатной температуре и атмосферном давлении в течение 4 ч. Затем добавляли дополнительную порцию палладия на угле(100 мг) и смесь оставляли в атмосфере Н 2 при комнатной температуре и атмосферном давлении в течение 19 ч, пропуская через нее поток Н 2 каждые 3 ч. Затем смесь фильтровали в вакууме с помощью картриджа Merck RP18 для удаления палладия и удаляли растворитель при пониженном давлении, получая 0,57 г (99%) чистого продукта со следующими характеристиками: твердое вещество желтого цвета, т.пл. 150-152C (толуол/циклогексан),1 Н ЯМР (CDCL3,м.д.): 1,69 (м, 2 Н); 1,98 (м, 3H); 2,21 (м, 2 Н); 2,69 (м, 2 Н); 2,87 (м, 2 Н); 3,20 (м,2 Н); 3,64 (шир.с, 2 Н); 4,04 (с, 3H); 4,53 (д, 2 Н); 6,69 (д, J=8,4 Гц, 2 Н); 7,07 (д, J=8,4 Гц, 2 Н); 7,32-7,47 (м,4 Н); 7,79 (дд, J=7,7 Гц, J=1,2 Гц, 2 Н); 7,93 (2 дд, J=7,7 Гц, J=1,2 Гц,2 Н). 11 а) 4-[(1-(4-Аминофенилэтил)пиперид-4-ил)метилокси]-2-метил-2H-пирроло[3,4-с]хинолина гидрохлорид. Раствор хлористого водорода в метаноле получали, добавляя по каплям ацетилхлорид (1,86 ммоль; 140 мг) к 7,0 мл метанола, охлажденного на ледяной бане. Смесь несильно перемешивали в течение нескольких минут, после чего по каплям добавляли раствор амина, полученного в примере 11 (0,84 ммоль; 350 мг) в метаноле (5,6 мл). По окончании добавления смесь перемешивали при 0C в течение 45 мин и затем добавляли безводный этиловый эфир (примерно 70 мл) до осаждения соли. После растирания в течение 3 ч полученную соль отделяли фильтрованием, промывали этиловым эфиром (32 мл) и сушили в вакууме при 45C в течение 2 дней. Описанным способом получали 390 мг (95%) чистого продукта со следующими характеристиками: твердое вещество желтого цвета, т.пл. 165-167C (изопропиловый эфир/изопропанол),1 Н ЯМР (ДМСО-d6,м.д.): 1,66-1,94 (м, 2 Н); 1,96-2,29 (м, 3H); 2,86-3,89 (м, 8 Н); 4,00 (с, 3H); 4,49(1,79 ммоль; 0,53 г),этил 4-(2-хлорэтил)бензолкарбоксилата, полученного в примере 8 а (8,97 ммоль; 1,91 г), Nal (8,97 ммоль; 1,34 г) и триэтиламина (8,97 ммоль; 0,91 г; 125 мл) в 2-бутаноне (22 мл) кипятили с обратным холодильником при перемешивании в течение 12 ч. После охлаждения смесь выливали в воду (200 мл) и экстрагировали этилацетатом (250 мл). Объединенные органические фазы экстрагировали 1 н. HCl (350 мл),выделяя целевой амин в форме твердого гидрохлорида. Полученное таким образом твердое вещество и кислые фазы объединяли, обрабатывали карбонатом натрия до достижения щелочной реакции и вновь экстрагировали этилацетатом (350 мл). Органическую фазу сушили над безводным сульфатом натрия и удаляли растворитель при пониженном давлении. Полученный неочищенный продукт очищали хроматографией на колонке с оксидом алюминия (используя хлороформ в качестве элюента), получая 430 мг(51%) чистого продукта со следующими характеристиками: коричневое масло,1 Н ЯМР (CDCl3,м.д.): 1,44 (т, 3H); 1,65 (м, 2 Н); 1,98 (м, 3H); 2,18 (м, 2 Н); 2,70 (т, 2 Н); 2,97 (т, 2 Н); 3,14 (м, 2 Н); 4,01 (с, 3H); 4,42 (кв, 2 Н); 4,52 (д, 2 Н); 7,32-7,46 (м, 6 Н); 7,78 (дд, J=8,1 Гц, J=1,1 Гц, 2 Н); 7,92 (дд, J=8,1 Гц, J=1,1 Гц, 2 Н); 8,02 (д, J=8,4 Гц, 2 Н). 12 а) 4-[2-[4-[O-2-Метил-2 Н-пирроло[3,4-с]хинолин-4-ил]метоксипиперид-1-ил]этил]бензойная кислота (соединение формулы (I): R1=CH3; R2=H; R3=CH2-C6H4-(4-CO2H. К раствору продукта, полученного в примере 12 (0,42 ммоль; 0,2 г), в 2,4 мл смеси 1/1 ТГФ/EtOH добавляли 1 н. NaOH (1,02 ммоль, 1,0 мл). Полученную смесь перемешивали при комнатной температуре в течение 17 ч. Затем добавляли 1 н. HCl до достижения рН 7 и отделяли фильтрованием образовавшийся при этом осадок. Описанным способом получали 50 мг (27%) чистого конечного продукта: твердое вещество, т.пл. 155-166C (этанол),1(соединение формулы (I): R1=CH3; R2=H; R3=CH2-C6H4-(4-NHCOCH3. К раствору продукта, полученного в примере 8 (1,5 ммоль; 440 мг), в 2-бутаноне (20 мл) добавляли 4'-ацетамидо-2-бромэтилбензол, полученный в примере 8 с (380 мг; 1,6 ммоль). Смесь кипятили с обратным холодильником в течение 1,5 ч. Затем добавляли триэтиламин (161 мг; 1,6 ммоль; 0,2 мл) и смесь перемешивали в течение 1,5 ч, после чего охлаждали до комнатной температуры, разбавляли водой и экстрагировали 2-бутаноном. Органическую фазу сушили над безводным Na2SO4. В заключение удаляли растворитель при пониженном давлении, получая неочищенный продукт, который очищали хроматографией на колонке с Al2O3, элюируя этилацетатом, и получали 250 мг (40%) продукта со следующими характеристиками: твердое вещество, т.пл. 145-146C (этанол/гексан),1 Н ЯМР (CDCl3,м.д.): 1,64-1,76 (м, 4 Н); 1,99-2,02 (м, 2 Н); 2,23-2,24 (с, 3H); 2,93-2,95 (м, 2 Н); 2,97(м, 2 Н); 3,26 (м, 2 Н); 4,04 (с, 3H); 4,53-4,54 (д, 2 Н); 7,18 (м, 1 Н); 7,22-7,24 (м, 2 Н); 7,35 (м, 1 Н); 7,39 (м,3H); 7,77-7,79 (м, 1 Н); 7,93 (дд, 1 Н). 14) 4-[(1-(4-Метансульфониламидофенилэтил)пиперид-4-ил)метилокси]-2-метил-2 Н-пирроло[3,4 с]хинолин (соединение формулы (I): R1=CH3; R2=H; R3=CH2-C6H4-(4-NHSO2CH3. К раствору продукта, полученного в примере 8 (1,17 ммоль; 345 мг), в 2-бутаноне (14 мл) добавляли 4-(метансульфониламидо) фенилэтилбромид, полученный в примере 8d (360 мг, 1,29 ммоль). Эту смесь кипятили в течение 30 мин, затем добавляли триэтиламин (130 мг; 1,29 ммоль; 0,2 мл) и перемешивали смесь в течение 3 ч. Температуру смеси доводили до комнатной, разбавляли водой и экстрагировали 2-бутаноном. Органическую фазу сушили над безводным Na2SO4. Растворитель выпаривали при пониженном давлении, получая неочищенный продукт, который очищали хроматографией на колонке сAl2O3, элюируя этилацетатом. Описанным способом получали 330 мг продукта (38%) со следующими характеристиками: твердое вещество, т.пл. 110-111C (толуол),1 Н ЯМР (CDCl3,м.д.): 1,31 (с, 1 Н); 1,70 (м, 2 Н); 1,86 (м, 3H); 2,23-2,24 (м, 4 Н); 2,67-2,69 (м, 2 Н); 2,87-2,89 (м, 2 Н); 3,04 (с, 3H); 3,13-3,16 (д, 2 Н); 4,04 (с, 3H); 4,52-4,53 (д, 2 Н); 7,34-7,35 (м, 1 Н); 7,39 (м,1 Н); 7,44 (м, 1 Н); 7,77-7,80 (дд, 1 Н); 7,93 (дд, 1 Н). 15) 4-[(1-(4-Гидроксиметилфенилэтил)пиперид-4-ил)метилокси]-2-метил-2 Н-пирроло[3,4 с]хинолин (соединение формулы (I): R1=CH3; R2=H; R3=CH2-C6H4-(4-CH2OH. К раствору продукта, полученного в примере 8 (3,3 ммоль; 990 мг) в 2-бутаноне (33,5 мл) добавляли 4'-гидроксиметил-2-бромэтилбензол, полученный в примере 8 е (800 мг, 3,7 ммоль). Раствор кипятили в течение 30 мин, затем добавляли триэтиламин (130 мг; 1,29 ммоль; 0,2 мл) и перемешивали смесь в течение 2 ч. Затем добавляли еще одну порцию этиламина (3,3 ммоль; 0,33 г) и еще через 2 ч температуру реакционной смеси доводили до комнатной, разбавляли водой и экстрагировали 2-бутаноном. Органическую фазу сушили над безводным Na2SO4. Растворитель выпаривали при пониженном давлении, получая неочищенный продукт, который очищали хроматографией на колонке с Al2O3, элюируя этилацетатом. Описанным способом получали 0,67 г продукта (48,5%) со следующими характеристиками: твердое вещество, т.пл. 145-146C (бензол/циклогексан),1 Н ЯМР (CDCl3,м.д.): 1,31 (с, 1 Н); 1,64 (м, 2 Н); 1,84 (м, 3H); 1,99 (м, 3H); 2,10-2,15 (м, 3H); 2,642,68 (м, 2 Н); 2,87-2,92 (м, 2 Н); 3,14 (д, 2 Н); 4,03 (с, 3H); 4,53 (д, 2 Н); 4,72 (д, 2 Н); 7,26-7,28 (м, 2 Н); 7,327,38 (м, 5 Н); 7,44 (м, 1 Н); 7,78-7,80 (дд, 1 Н); 7,93 (дд, 1 Н). 16) 4-[(1-(4-Метоксиметилфенилэтил)пиперид-4-ил)метилокси]-2-метил-2 Н-пирроло[3,4-с]хинолин(соединение формулы (I): R1=CH3; R2=H; R3=CH2-C6H4-(4-СН 2 ОСН 3. К раствору продукта, полученного в примере 8 (3,3 ммоль; 990 мг), в 2-бутаноне (33,5 мл) добавляли 4'-метоксиметил-2-бромэтилбензол, полученный в примере 8f (800 мг, 3,7 ммоль). Раствор кипятили с обратным холодильником в течение 30 мин, затем добавляли триэтиламин (130 мг; 1,29 ммоль; 0,2 мл) и перемешивали смесь в течение 1,5 ч. Затем добавляли еще одну порцию этиламина (3,3 ммоль; 0,33 г) и еще через 2 ч температуру реакционной смеси доводили до комнатной, разбавляли водой и экстрагировали 2-бутаноном. Органическую фазу сушили над безводным Na2SO4. Растворитель выпаривали при пониженном давлении, получая неочищенный продукт, который очищали хроматографией на колонке сAl2O3, элюируя этилацетатом. Описанным способом получали 0,67 г продукта (48,5%) со следующими характеристиками:(соединение формулы (I): R1=CH3; R2=H; R3=CH2-C6H4-(2-COOCH3. Смесь продукта, полученного в примере 8 (3,31 ммоль; 1,0 г), и метил 2-(2-бромэтил)бензолкарбоксилата, полученного в примере 8h (3,64 ммоль; 0,74 г), в 2-бутаноне (35 мл) кипятили с обратным холодильником в течение 30 мин. После добавления триэтиламина (4,0 ммоль; 0,4 г; 0,56 мл) реакционную смесь кипятили с обратным холодильником при перемешивании в течение 72 ч. После охлаждения смесь разбавляли водой (50 мл) и затем экстрагировали 2-бутаноном (250 мл). Объединенные органические фазы сушили над безводным сульфатом натрия и удаляли растворитель при пониженном давлении. Полученный таким образом неочищенный продукт очищали хроматографией на колонке с оксидом кремния, элюируя этилацетатом, и получали 450 мг (30%) чистого продукта со следующими характеристиками: желтое масло,1 Н ЯМР (CDCl3,м.д.): 1,28 (м, 1 Н); 1,65-1,68 (м, 2 Н); 1,96-1,98 (м, 3H); 2,26 (м, 2 Н); 2,70-2,74 (м,2 Н); 3,17-3,29 (м, 4 Н); 3,92 (с, 3H); 3,99 (с, 3H); 4,50 (дд, 2 Н); 7,27-7,36 (м, 5 Н); 7,39-7,48 (м, 2 Н); 7,75 (дд,1 Н); 7,88-7,92 (м, 2 Н). 17 а) 2-[2-[4-[ O-2-Метил-2H-пирроло[3,4-с]хинолин-4-ил]метоксипиперид-1-ил]этил]бензойная кислота (соединение формулы (I): R1=CH3; R2=H; R3=CH2-C6H4-(2-COOH. К раствору продукта, полученного в примере 17 (0,74 ммоль; 0,34 г), в 3,4 мл смеси 1/1 ТГФ/EtOH добавляли 1 н. NaOH (1,4 мл). Раствор перемешивали при комнатной температуре в течение 12 ч и затем добавляли ледяную уксусную кислоту до рН 6. Образовавшееся твердое вещество отделяли фильтрованием, получая продукт (с выходом 50%) со следующими характеристиками: твердое вещество, т.пл. 165C (этилацетат),1 Н ЯМР (DMF-d7,м.д.): 1,30 (м, 2 Н); 1,51 (м, 2 Н); 1,79-1,82 (м, 1 Н); 2,51 (м, 5 Н); 2,87-2,92 (м, 2 Н); 3,07-3,10 (м, 2 Н); 3,98 (с, 3H); 4,35 (д, 2 Н); 7,23-7,37 (м, 5 Н); 7,57-7,62 (м, 3H); 7,72 (д, 1 Н); 7,96 (дд, 1 Н). 18) 4-[(1-(4-Сульфонамидофенилэтил)пиперид-4-ил)метилокси]-2-метил-2H-пирроло[3,4-с]хинолин(соединение формулы (I): R1=CH3; R2=H; R3=CH2-C6H4- (4-SO2NH2. Смесь продукта, полученного в примере 8 (3,37 ммоль; 0,99 г), и 4-сульфонамидо-1-(2 бромэтил)бензола, полученного в примере 8j (3,78 ммоль, 1,00 г), в 2-бутаноне (34 мл) кипятили с обратным холодильником в течение 10 мин. После добавления триэтиламина (3,64 ммоль; 0,188 г; 0,52 мл) полученную реакционную смесь кипятили с обратным холодильником при перемешивании в течение 35 мин. После охлаждения смесь разбавляли водой (50 мл) и экстрагировали 2-бутаноном (250 мл). Объединенные органические фазы сушили над безводным сульфатом натрия и удаляли растворитель при пониженном давлении. Получали неочищенный продукт, который очищали хроматографией на колонке с оксидом алюминия, элюируя этилацетатом, и затем на колонке с оксидом кремния, элюируя смесью 10/1 этилацетат/метанол. Описанным способом получали 140 мг (11%) продукта со следующими характеристиками: твердое вещество оранжевого цвета, т.пл. 169C с разложением (толуол),1 Н ЯМР (ДМСО-d6,м.д.): 1,18-1,36 (м, 2 Н); 1,85 (м, 3H); 2,01-2,26 (м, 2 Н); 3,07 (м, 2 Н); 3,71-3,92(м, 2 Н); 3,98 (с, 3H); 4,43 (м, 2 Н); 7,27-7,36 (м, 2 Н); 7,55-7,60 (м, 3H); 7,73 (м, 2 Н); 7,97 (м, 2 Н). Примеры композиций. Пример 1. Таблетки, содержащие в качестве действующего начала соединение 12 а по настоящему изобретению, имели следующий состав, мг: 1 Пример 2. Ампулы, включающие в качестве действующего начала соединение 12 а по настоящему изобретению, заполняли следующим составом: Пример 3. Фармацевтическая композиция в форме гранул, содержащих в качестве действующего начала соединение 12 а по настоящему изобретению, имела следующий состав, мг: где R1 означает атом водорода, линейную или разветвленную алкильную группу, содержащую 1-6 атомов углерода, или алкоксиалкилгруппу, содержащую 1-6 атомов углерода;R2 означает атом водорода, атом галогена или линейную или разветвленную алкильную группу, содержащую 1-3 атома углерода;R3 представляет собой (i) атом водорода; (ii) линейную или разветвленную алкильную группу, содержащую 1-6 атомов углерода; (iii) алкоксиалкильную группу, содержащую 1-6 атомов углерода;(iv) арилалкильную группу, в которой алкильная группа содержит 1-3 атома углерода, а арильная группа представляет собой остаток бензола и может быть замещена одним или двумя заместителями, которые могут быть одинаковыми или различными, выбранными из атома галогена, алкильной группы, содержащей 1-3 атома углерода, алкоксигруппы, содержащей 1-3 атома углерода, гидроксиалкильной группы,содержащей 1-3 атома углерода, -ОН, -NR'R", -NO2, -CF3, -CO2R', R'CON(R")-, R'SO2N(R")- и R'R"NSO2-,где R' и R", которые могут быть одинаковыми или различными, представляют собой атом водорода или алкильную группу, содержащую 1-3 атома углерода; (v) RivRvNCO(CH2)n-, где n означает целое число от 0 до 2 и Riv и Rv, которые могут быть одинаковыми или различными, являются атомами водорода, алкильными группами, содержащими 1-3 атома углерода, арильными группами, выбранными из остатков бензола, необязательно замещенными одним или двумя заместителями, которые могут быть одинаковыми или различными, выбранными из атома галогена, алкильной группы, содержащей 1 атома углерода,-ОН, -NH2; и (vi) C-(CH2)m-, где m означает целое число от 0 до 2 и C является алициклической группой,содержащей от 3 до 7 атомов углерода, или насыщенной 5- или 6-членной гетероциклической группой,содержащей по меньшей мере один гетероатом, выбранный из О и N, необязательно замещенный по атому N алкильной группой, содержащей 1 атом углерода,их кислотно-аддитивные соли с фармацевтически приемлемыми органическими или неорганическими кислотами, а также их основно-аддитивные соли с фармацевтически приемлемыми органическими или неорганическими основаниями. 2. Соединение по п.1, в котором R1 представляет собой атом водорода или линейную или разветвленную алкильную группу, содержащую 1-6 атомов углерода; R2 означает атом водорода или атом галогена и R3 представляет собой (i) атом водорода; (ii) линейную или разветвленную алкильную группу,содержащую 1-6 атомов углерода; (iii) арилалкильную группу, в которой алкильная группа содержит 1-3 атома углерода, а арильная группа представляет собой остаток бензола и может быть замещена одним или двумя заместителями, которые могут быть одинаковыми или различными, выбранными из атома галогена, алкильной группы, содержащей 1-3 атома углерода, алкоксигруппы, содержащей 1-3 атома углерода, гидроксиалкильной группы, содержащей 1-3 атома углерода, -ОН, -NR'R", -NO2, -CO2R',R'CON(R")-, R'SO2N(R")- и R'R"NSO2-, где R' и R", которые могут быть одинаковыми или различными,представляют собой атом водорода или алкильную группу, содержащую 1-3 атома углерода;(iv) RivRvNCO(CH2)n-, где n означает целое число от 0 до 2 и Riv и Rv, которые могут быть одинаковыми или различными, являются атомами водорода, арильными группами, выбранными из остатков бензола; и(v) C-(CH2)m-, где m означает целое число от 0 до 2 и С является насыщенным гетероциклическим фрагментом, выбранным из группы, включающей морфолин, пиперидин, N-метилпиперазин и пирролидин. 3. Соединение по п.1, в котором R1 является линейной или разветвленной алкильной группой, содержащей 1-6 атомов углерода; R2 означает атом водорода и R3 представляет собой (i) линейную или разветвленную алкильную группу, содержащую 1-6 атомов углерода; (ii) арилалкильную группу, в которой алкильная группа содержит 1-3 атома углерода, а арильная группа выбрана из остатков бензола и может быть замещена заместителем, выбранным из алкоксигруппы, содержащей 1-3 атома углерода,гидроксиалкильной группы, содержащей 1-3 атома углерода, -NR'R", -CO2R', R'CON(R")-, R'SO2N(R")- иR'R"NSO2-, где R' и R", которые могут быть одинаковыми или различными, представляют собой атомы водорода или алкильные группы, содержащие 1-3 атома углерода; (iii) RivRvNCO(CH2)n-, где n означает целое число от 0 до 2 и Riv и Rv, которые могут быть одинаковыми или различными, являются атомами водорода или арильными группами, выбранными из остатков бензола; и (iv) C-(CH2)m-, где m означает целое число от 0 до 2 и C представляет собой остаток морфолина или пиперидина. 4. Соединения по п.1, в котором R1, R2 и R3 представлены в следующей таблице: 5. Фармацевтическая композиция, включающая эффективное количество соединения формулы (I) или его кислотно-аддитивной или основно-аддитивной соли по любому из предшествующих пунктов, а также по меньшей мере один фармацевтически приемлемый наполнитель. 6. Фармацевтическая композиция по п.5, которая включает такое количество соединения формулы(I) или его кислотно-аддитивной или основно-аддитивной соли, чтобы обеспечить введение от 0,001 до 100 мг/кг/день соединения формулы (I) в пересчете на соединение в свободной форме. 7. Способ получения соединения формулы (I) или его фармацевтически приемлемой кислотно-аддитивной или основно-аддитивной соли,где R1, R2 и R3 соответствуют определениям, данным в пп.1-4,- 21019300 отличающийся тем, что он включает взаимодействие между соединением формулы (II) где R1, R2 и R3 имеют указанные выше значения и X означает атом галогена. 8. Способ получения по п.7, в котором указанное взаимодействие проводят в органическом растворителе в присутствии органического или неорганического основания. 9. Способ получения по любому из пп.7 и 8, где указанную реакцию проводят в присутствии активирующего агента, выбранного из группы, включающей йодид калия, йодид натрия, йодид цезия, йодид тетрабутиламмония и йодид триметилфениламмония. 10. Способ получения соединения формулы (I) или его фармацевтически приемлемой кислотно-аддитивной или основно-аддитивной соли,где R1, R2 и R3 соответствуют определениям, данным в любом из пп.1-4,отличающийся тем, что он включает взаимодействие между соединением формулы (IV) где R1, R2 и R3 имеют указанные выше значения. 11. Способ получения по п.10, где указанную реакцию проводят в полярном апротонном органическом растворителе в присутствии основания. 12. Способ получения соединения формулы (II)(1) реакцию между соединением формулы (IV) где R1 и R2 имеют указанные выше значения,с получением соединения формулы (VII)(2) реакцию дебензилирования пиперидинового атома азота соединения формулы (VII) с получением соединения формулы (II), где указанную реакцию осуществляют путем гидрирования. 13. Способ получения по п.12, где указанную реакцию (1) проводят в полярном апротонном органическом растворителе, выбранном из группы, включающей кетоны, тетрагидрофуран, диметилформамид, диметилсульфоксид, диоксан и ацетонитрил в присутствии основания, выбранного из группы,включающей гидроксид натрия и гидрид натрия, и указанную реакцию (2) проводят в спиртовом растворителе в атмосфере водорода в присутствии палладия на угле в качестве катализатора. 14. Соединение формулы (II) где R1 является атомом водорода, линейной или разветвленной алкильной группой, содержащей 1-6 атомов углерода, или алкоксиалкильной группой, содержащей 1-6 атомов углерода; иR2 является атомом водорода, атомом галогена или линейной или разветвленной алкильной группой, содержащей 1-3 атома углерода. 15. Соединение формулы (I) или его фармацевтически приемлемая кислотно-аддитивная или основно-аддитивная соль,где R1, R2 и R3 соответствуют определениям, данным в любом из пп.1-4,для фармацевтического применения. 16. Применение соединения формулы (I) или его фармацевтически приемлемой кислотно-аддитивной или основно-аддитивной соли,где R1, R2 и R3 соответствуют определениям, данным в любом из пп.1-4,для получения фармацевтической композиции, предназначенной для лечения расстройств перистальтики кишечника, расстройств центральной нервной системы, недержания мочи и сердечной аритмии. 17. Применение соединения формулы (I) или его фармацевтически приемлемой кислотно-аддитивной или основно-аддитивной соли,где R1, R2 и R3 соответствуют определениям, данным в любом из пп.1-4,для получения фармацевтической композиции, предназначенной для лечения хронической боли. 18. Применение соединения по п.17 для получения фармацевтической композиции для лечения невропатической боли, вызванной диабетом, раком, иммунодефицитом, травмами, ишемией, рассеянным склерозом, ишиалгией, тригеминальной невралгией и постгерпетическим синдромом.
МПК / Метки
МПК: A61P 35/00, A61K 31/4745, A61P 9/00, A61P 1/00, C07D 471/04, A61P 13/02, A61P 25/00
Метки: активностью, серотонинергической, соединения, получения, фармацевтические, содержащие, композиции, способ
Код ссылки
<a href="https://eas.patents.su/25-19300-soedineniya-s-serotoninergicheskojj-aktivnostyu-sposob-ih-polucheniya-i-soderzhashhie-ih-farmacevticheskie-kompozicii.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения с серотонинергической активностью, способ их получения и содержащие их фармацевтические композиции</a>
Трициклические соединения, обладающие активностью в отношении интегринов, в частности, в отношении интегрина альфаvбета 3, способ их получения и промежуточные соединения, используемые в этом способе,их применение в качестве медикаментов и содержащие их фармацевтические композиции.

Номер патента: 2271
Опубликовано: 28.02.2002
Авторы: Питти Роберт М., Тетш Жан-Жорж, Кнолле Йохен, Бернар Серж, Бодари Сара С., Штильц Ханс-Ульрих, Карниато Дени, Венер Фолькмар, Гурвест Жан-Франсуа, Гадек Томас Р., Макдауэлл Роберт С.
МПК: C07C 281/12, A61K 31/19, A61P 9/10...
Метки: частности, композиции, трициклические, используемые, альфаvбета, соединения, этом, фармацевтические, качестве, отношении, применение, активностью, способ, получения, обладающие, интегринов, промежуточные, содержащие, интегрина, медикаментов, способе,их
Формула / Реферат:
1. Соединения общей формулы (I) в которой R1 обозначает группу -О-[А]-[В]-COR6, в которой R6 обозначает -ОН, C1-С6алкокси, -О-СН2-СН(ОН)-СН2OН, [A] обозначает группу C1-С6алкилен, возможно замещенный оксогруппой, [B] обозначает радикал -CH(Z)- или простую связь, Z обозначает группу -NHCO2Rc, или -NHSO2Rc, где Rc обозначает радикал фенил(C1-С4)алкил-, хинолинил или пиридинилимидазолил(C1-С4)алкил-, R2 и R3, одинаковые или разные, обозначают атом...
Соединения 1,2,4,5-тетрагидро-3н-бензазепина, способ их получения и фармацевтические композиции, содержащие эти соединения

Номер патента: 14756
Опубликовано: 28.02.2011
Авторы: Вийенов Николь, Шименти Стефано, Вилен Жан-Поль, Гумен Бертран, Толлон Катрин, Дессинже Эмея, Кеньяр Паскаль, Пеглион Жан-Луи
МПК: A61K 31/55, C07D 223/16, A61P 9/00...
Метки: соединения, получения, эти, фармацевтические, способ, композиции, содержащие, 1,2,4,5-тетрагидро-3н-бензазепина
Формула / Реферат:
1. Соединение формулы (I)где R1представляет собой атом водорода или группу, выбранную из С3-С7 циклоалкила, бензила и линейного или разветвленного C1-C6алкила, алкильной группы, которая может быть насыщенной или ненасыщенной и может быть необязательно замещенной гидроксигруппой или С3-С7 циклоалкильной группой или одним или несколькими атомами галогена,R2, R3, R4 и R5могут быть одинаковыми или различаться, каждый выбирается из атома водорода или...
Новые пирролы, обладающие гиполипидемической и гипохолестеринемической активностью, способ их получения и содержащие их фармацевтические композиции и их применение в медицине

Номер патента: 6858
Опубликовано: 28.04.2006
Авторы: Лохрай Видья Бхушан, Барот Виджай Кумар, Басу Суджай, Равал Преети Саурин, Лохрай Брадж Бхушан, Равал Саурин Химшанкар
МПК: A61K 31/40, A61K 31/4025, A61P 3/06...
Метки: гипохолестеринемической, активностью, новые, медицине, обладающие, пирролы, получения, способ, применение, гиполипидемической, содержащие, композиции, фармацевтические
Формула / Реферат:
1. Соединениz формулы (I) их таутомерные формы, их стереоизомеры, их полиморфы, их фармацевтически приемлемые соли, их фармацевтически приемлемые сольваты, где R2 и R3 представляют собой водород и R1, R4 могут быть одинаковыми или отличными друг от друга и представляют собой водород, галогеналкил, где алкильная группа, как определено ниже, замещена галогеном, выбранным из F, Cl, Вr и иода; пергалогеналкил, нитро, циано, формил или замещенные...
Соединения фенилпиридилпиперазина, способ их получения и фармацевтические композиции, содержащие их

Номер патента: 11165
Опубликовано: 27.02.2009
Авторы: Десо Патрис, Корди Алексис, Лестаж Пьер
МПК: A61P 25/00, A61K 31/496, C07D 213/74...
Метки: соединения, получения, фармацевтические, композиции, содержащие, способ, фенилпиридилпиперазина
Формула / Реферат:
1. Соединения формулы (I) в которой R1 представляет собой NR3SO2R4 группу, где R3 представляет собой атом водорода, R4 представляет собой линейную или разветвлённую (С1-С6)алкильную группу, фенил, необязательно замещенный галогеном, или NR5R6 группу, где R5 и R6 вместе с несущим их атомом азота образуют 6-членное кольцо, в котором один из атомов углерода заменён на атом кислорода, R2 представляет собой линейную или разветвлённую...
Соединения пиримидин-4-она способ их получения и содержащие их фармацевтические композиции

Номер патента: 5402
Опубликовано: 24.02.2005
Авторы: Мюлле Оивье, Лавие Жильбер, Дюбюффе Тьерри, Миллан Марк, Брокко Морисетт, Декен Анн
МПК: A61K 31/519, A61P 25/00, C07D 471/00...
Метки: пиримидин-4-она, соединения, композиции, фармацевтические, получения, способ, содержащие
Формула / Реферат:
1. Соединения формулы (I) в которой R1, R2, R3 и R4, которые могут быть одинаковыми или различными, каждый представляет собой атом водорода, атом галогена или группу, выбираемую из линейного или разветвленного (C1-C6)алкила, линейного или разветвленного (C1-C6)алкокси, линейного или разветвленного (C1-C6)полигалоалкил, гидрокси, циано, нитро и амино, или R1 и R2, R2 и R3 или R3 и R4 совместно с атомами углерода, их несущими, образуют...
Предыдущий патент: Полихромные вещества и их применение
Следующий патент: Фунгицидная композиция для протравливания семян
Случайный патент: 2-,3-,4- или 5-замещенные n-(1- (арил-, гетероарил-или арилалкил)сульфонил) индолы и их применение в терапии