Комбинированная терапия раковых заболеваний с помощью соединений – ингибиторов hsp90
Номер патента: 22119
Опубликовано: 30.11.2015
Авторы: Фоли Кевин Пол, Блэкман Рональд К., Пройа Дэвид



















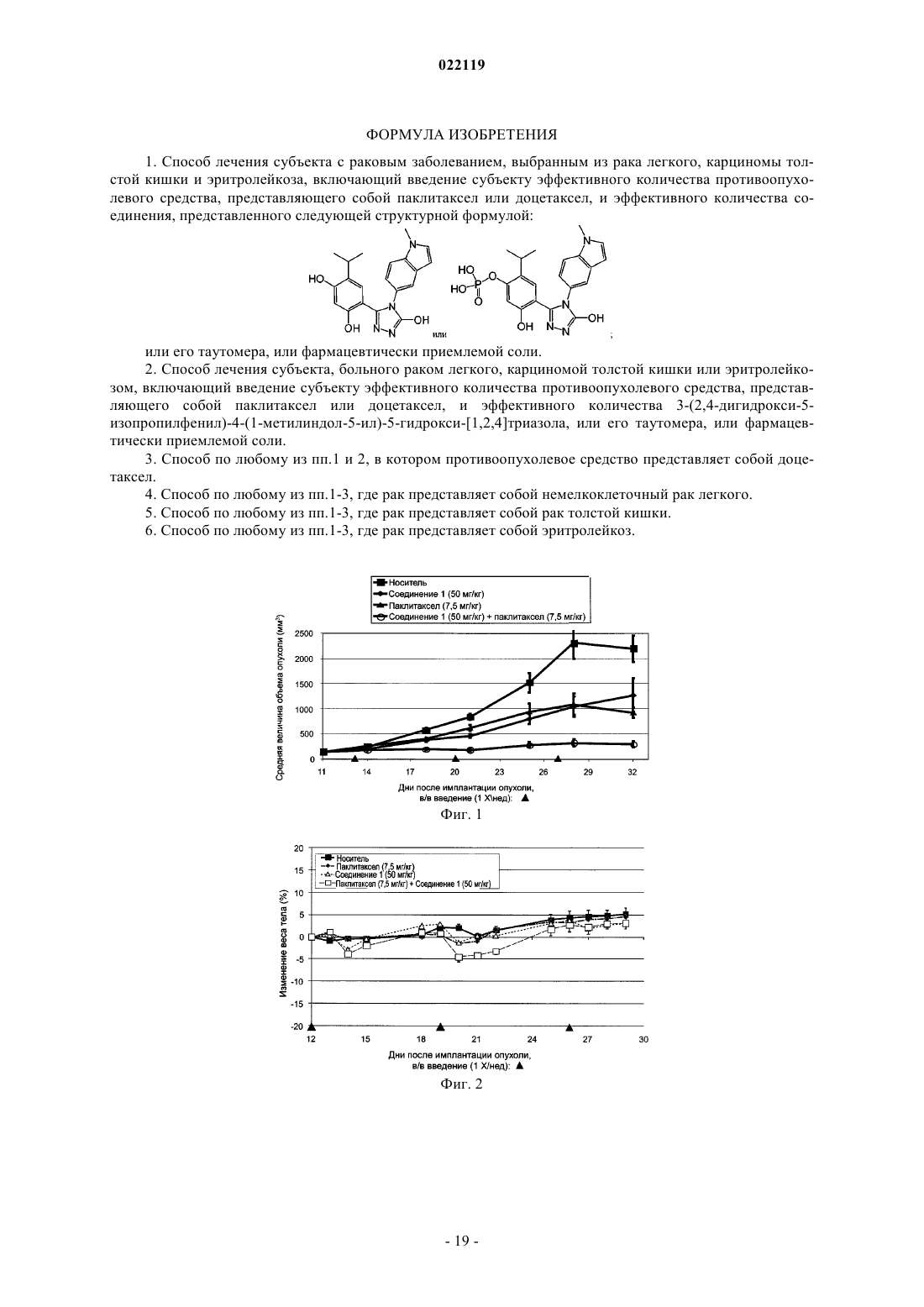
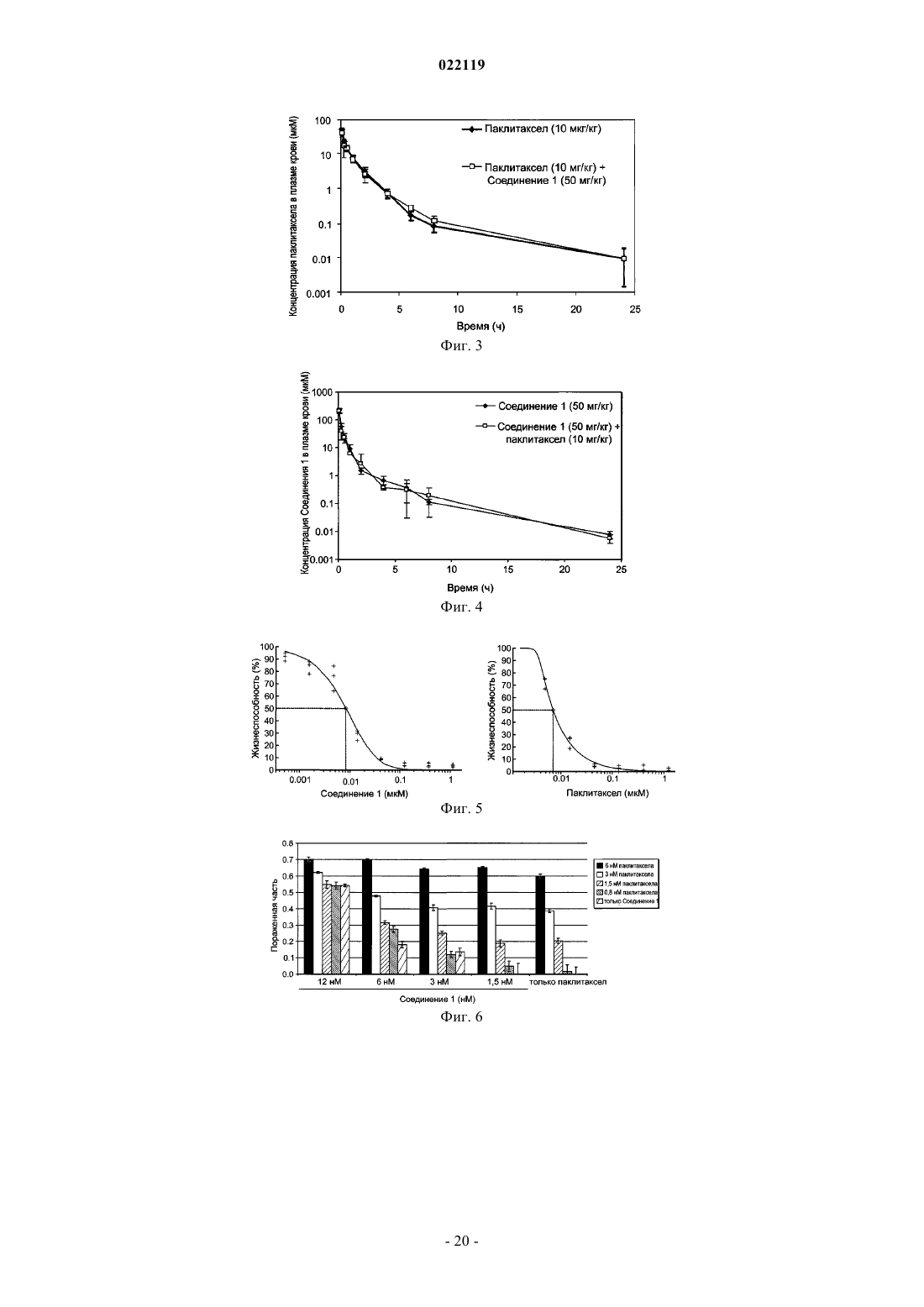
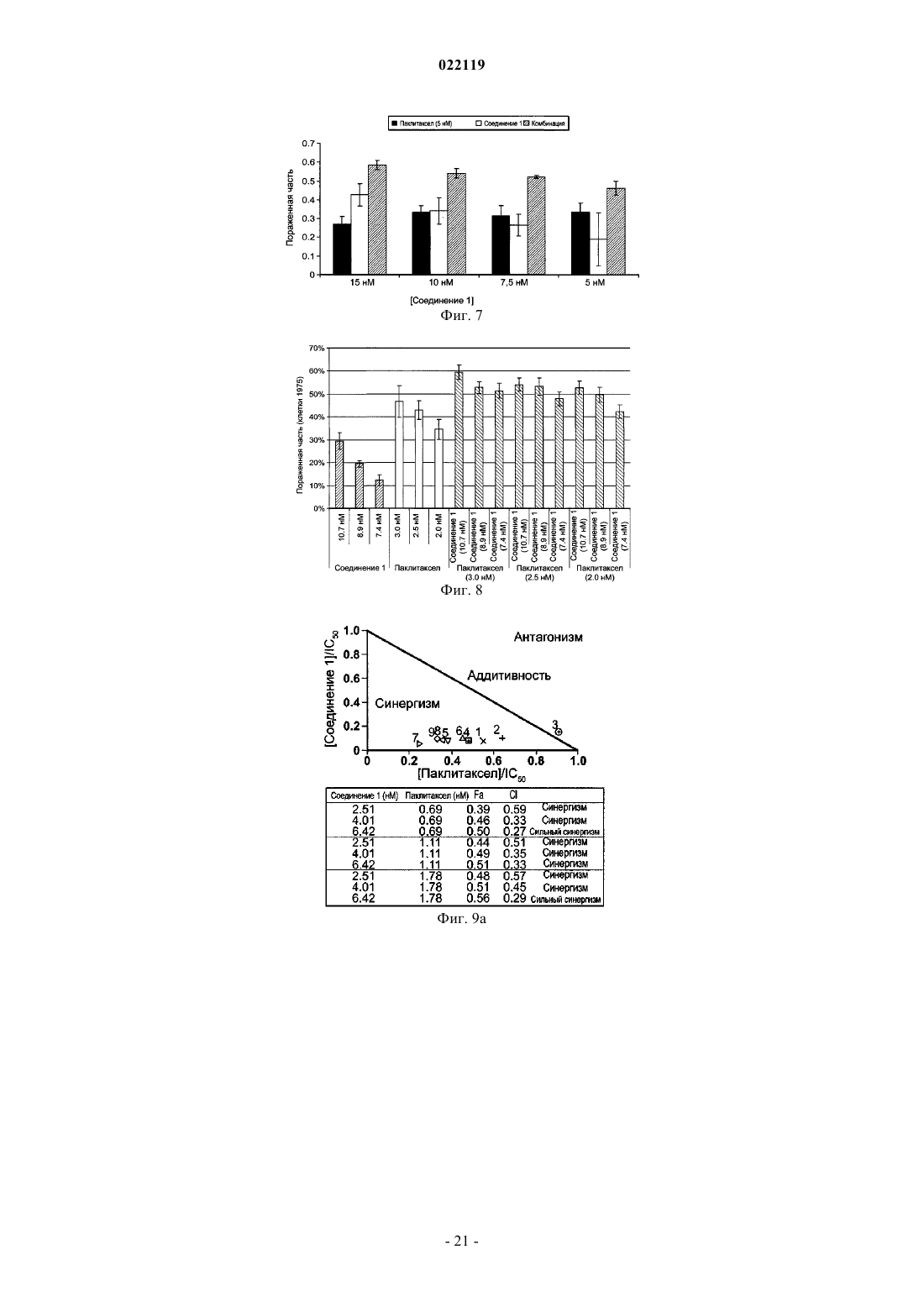
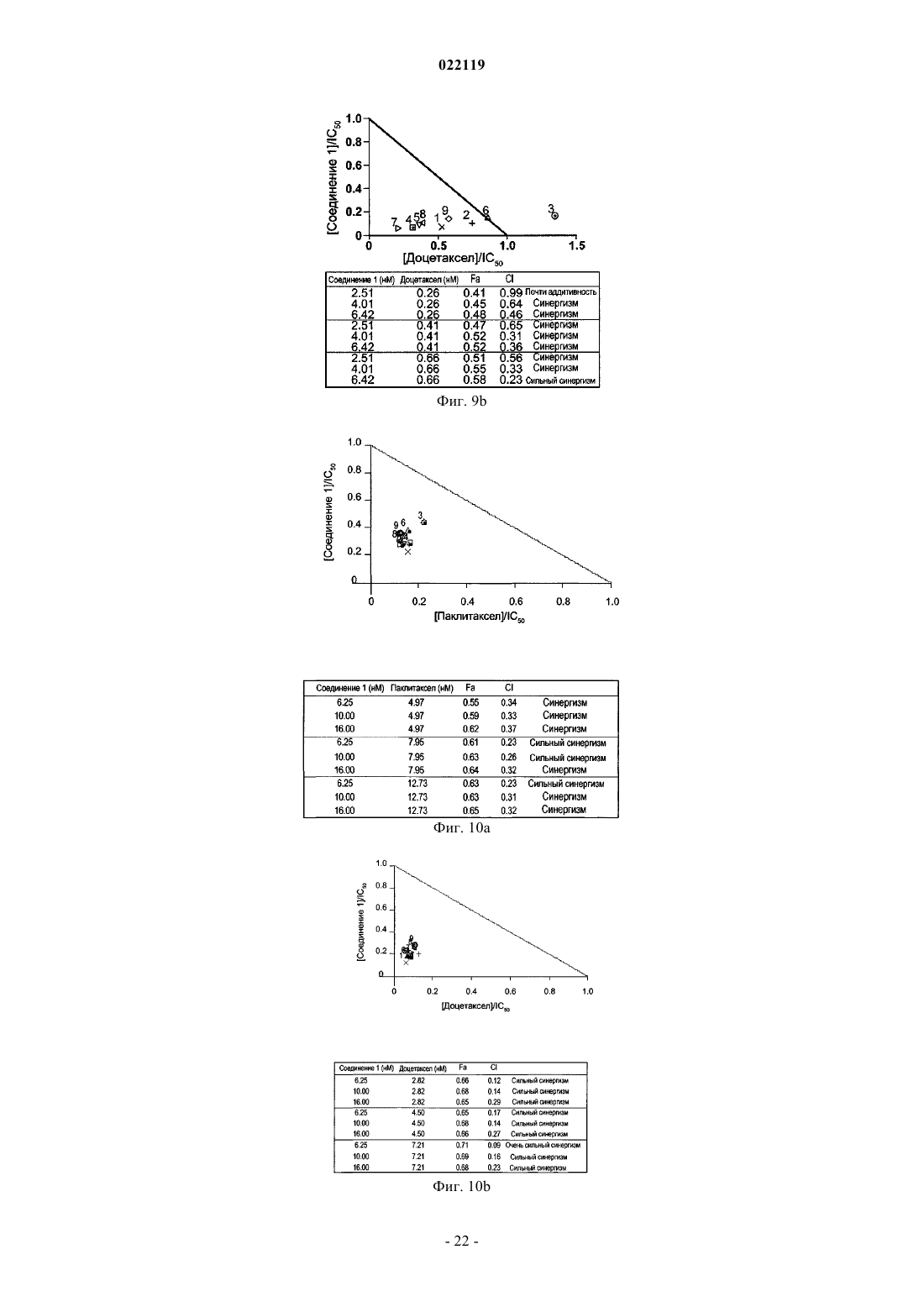
Формула / Реферат
1. Способ лечения субъекта с раковым заболеванием, выбранным из рака легкого, карциномы толстой кишки и эритролейкоза, включающий введение субъекту эффективного количества противоопухолевого средства, представляющего собой паклитаксел или доцетаксел, и эффективного количества соединения, представленного следующей структурной формулой:

или его таутомера, или фармацевтически приемлемой соли.
2. Способ лечения субъекта, больного раком легкого, карциномой толстой кишки или эритролейкозом, включающий введение субъекту эффективного количества противоопухолевого средства, представляющего собой паклитаксел или доцетаксел, и эффективного количества 3-(2,4-дигидрокси-5-изопропилфенил)-4-(1-метилиндол-5-ил)-5-гидрокси-[1,2,4]триазола, или его таутомера, или фармацевтически приемлемой соли.
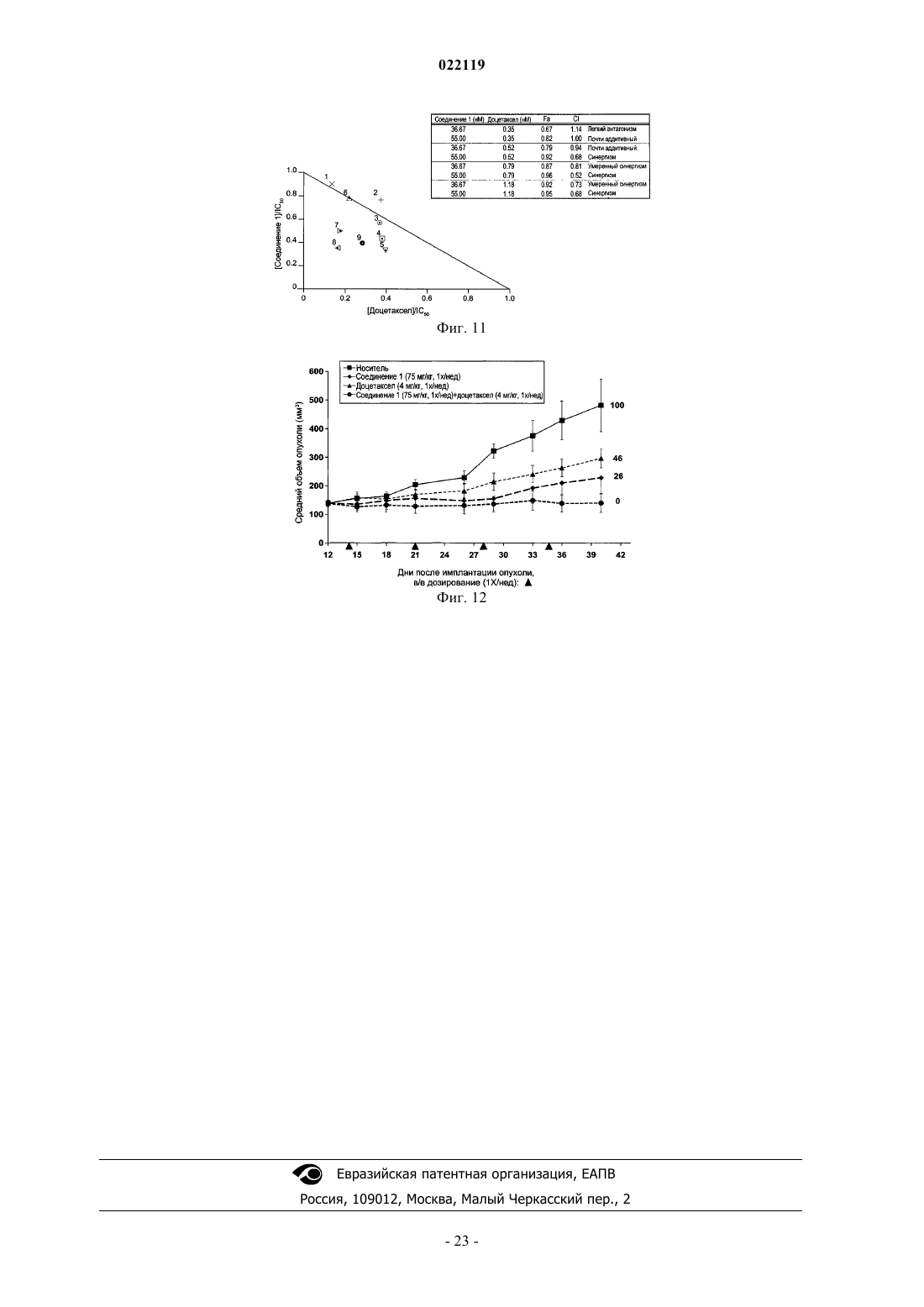
3. Способ по любому из пп.1 и 2, в котором противоопухолевое средство представляет собой доцетаксел.
4. Способ по любому из пп.1-3, где рак представляет собой немелкоклеточный рак легкого.
5. Способ по любому из пп.1-3, где рак представляет собой рак толстой кишки.
6. Способ по любому из пп.1-3, где рак представляет собой эритролейкоз.
Текст
КОМБИНИРОВАННАЯ ТЕРАПИЯ РАКОВЫХ ЗАБОЛЕВАНИЙ С ПОМОЩЬЮ СОЕДИНЕНИЙ - ИНГИБИТОРОВ HSP90 Изобретение относится к способу лечения субъекта, больного раком, выбранным из рака легкого,карциномы толстой кишки и эритролейкоза, включающий введение субъекту эффективного количества противоопухолевого средства, представляющего собой паклитаксел или доцетаксел, и эффективного количества соединения, представленного следующей структурной формулой: или его таутомера, или фармацевтически приемлемой соли. Данное изобретение претендует на приоритет предварительной заявки США на патент 61/279 330, поданной 10 сентября 2009 г., описание которой полностью включено в данное изобретение посредством отсылки. Данное изобретение претендует на приоритет предварительной заявки США на патент 61/335778, поданной 01 ноября 2010 г., описание которой полностью включено в данное изобретение посредством отсылки. Сведения о предшествующем уровне техники Рак продолжает оставаться основной причиной смерти людей в Соединенных Штатах Америки и во всем мире. Следовательно, существует постоянная необходимость в разработке методов лечения раковых заболеваний. Сущность изобретения Теперь было установлено, что комбинации некоторых триазолоновых соединений, являющихся ингибиторами HSP90 (белка теплового шока) и таксанов, являются неожиданно эффективными при лечении субъектов с немелкоклеточным раком легкого, раком толстой кишки и эритролейкозом без увеличения риска появления побочных эффектов. Способ комбинированной терапии, описанный в данном изобретении, демонстрирует неожиданную биологическую активность при лечении рака при минимальном появлении побочных эффектов. Данное изобретение использует триазолоновые соединения, которые ингибируют активностьHSP90 и пригодны для лечения рака легкого (например, немелкоклеточного рака легкого), рака толстой кишки и эритролейкоза в комбинации с таксаном. Способ лечения субъекта с немелкоклеточным раком легкого (например, немелкоклеточным раком легкого), раком толстой кишки и эритролейкозом включает стадию введения субъекту ингибитора HSP90, описанного в данном изобретении, и таксана. Согласно одному варианту введение ингибитора HSP90 и таксана осуществляется одновременно. Согласно другому варианту введение ингибитора HSP90 и таксана осуществляется последовательно. Согласно любому из этих вариантов таксан представляет собой доцетаксел, паклитаксел или Abraxane. Согласно любому из этих вариантов ингибитор HSP90 представляет собой соединение, представленное в табл. 1 (соединение 1) или 2 (соединение 1 а). Согласно одному варианту данного изобретения оно включает применение ингибитора HSP90, описанного в данном изобретении, для изготовления лекарственного средства для лечения рака легкого (например, немелкоклеточного рака легкого), рака толстой кишки и эритролейкоза в комбинации с таксаном. Согласно некоторым вариантам комбинированная терапия, использующая ингибитор HSP90, описанный в данном изобретении, вместе с другими химиотерапевтическими агентами, доцетакселом или паклитакселом, может помочь предотвратить или ослабить развитие рака легкого, резистентного ко многим препаратам (например, немелкоклеточного рака легкого), рака толстой кишки и эритролейкоза у млекопитающего. Согласно этому варианту соединения по изобретению могут обеспечить уменьшение эффективного количества второго химиотерапевтического агента таксана, а именно доцетаксела или паклитаксела, вводимого млекопитающему, так как ингибитор HSP90 будет ингибировать развитие рака легкого, резистентного ко многим препаратам (например, немелкоклеточного рака легкого), рака толстой кишки или эритролейкоза. Согласно другому варианту таксан представляет собой доцетаксел, паклитаксел или Abraxane. Таксаны, паклитаксел и доцетаксел широко применяются при лечении немелкоклеточного рака легкого на развитой стадии (NSCLC). Для изучения комбинации ингибиторов HSP90 по изобретению, таких как соединение 1, используемых в сочетании с таксанами, были проведены in vitro исследования на клетках NCI-H1975. Использовался метод Chau и Talalay, было установлено, что соединение 1 в комбинации или с паклитакселом или с доцетакселом приводило к получению комбинационных индексов в синергическом интервале (0,23-0,65 CI) (см. пример 2). In vivo в модели ксенографта NCI-H1975, комбинация соединения 1 и паклитаксела обеспечила большую эффективность, чем любой из этих агентов в отдельности (% величин Т/С составил 7, 38 и 55 для комбинации, паклитаксела и соединения 1 соответственно)(см. пример 1). Синергизм между соединением 1 и паклитакселом не был обусловлен изменениями в фармакокинетике каждого агента, и комбинационная терапия не привела к дополнительной токсичности. Синергизм был также обнаружен в случае карциномы клеток НТ 29 и эритролейкоза клеток HEL92.1.7(пример 3). Эти результаты показывают, что соединение 1 является высокоактивным ингибитором Hsp90, который проявляет широкий спектр противораковой активности in vitro и in vivo на доклинических моделяхNSCLC, рака толстой кишки и эритролейкоза. Ингибиторы HSP90 по изобретению, такие как соединение 1, также проявляют синергизм с паклитакселом и доцетакселом. Краткое описание чертежей На фиг. 1 показаны результаты исследования на ксенографте SCID у мыши, которое проводилось для определения действия комбинации соединения 1 и паклитаксела на скорость in vivo роста человеческих клеток NSCLC для тестирования клеточной линии NCI-H1975. Животным с опухолями (8 мышей в группе) внутривенно вводили один раз в неделю всего 3 дозы (острия стрелок) одного носителя, одного соединения 1, одного паклитаксела или комбинации соединения 1 и паклитаксела, вводимых совместно. Каждые 3-4 дня измеряли средние величины объема опухолей в каждой группе. Введение дозы соединения 1, равной 50 мг /кг, умеренно ингибировало рост опухолей, на день 32 величина % Т/С составляла 55. Введение паклитаксела с дозой, равной 7,5 мг/кг веса, умеренно ингибировало рост опухолей, на день 32 величина % Т/С составила 38. Совместное введение 50 мг/кг соединения 1 и 7,5 мг/кг паклитаксела значительно замедляло рост опухолей, на день 32 величина % Т/С составляла 7. Эффективность, наблюдаемая в группе при комбинационном лечении, была значительно выше, чем при лечении каждым из агентов в отдельности (Р 0,05; однопроходный метод ANOVA). Чрезмерной токсичности не наблюдалось в группе, получавшей комбинацию соединения 1 и паклитаксела, средняя величина изменения веса животных на день 29 (последний день измерений) по сравнению с началом исследования составила+3,1% (+/-1,2 SEM), а в группе, которой вводили носитель, эта величина составила +5,1% (+/-1,4 SEM). Фиг. 2 показывает, что комбинированное лечение, результаты которого показаны на фиг. 1, не приводит к дополнительной токсичности по сравнению с применением агентов в отдельности, наблюдалось только минимальное действие на кумулятивное изменение средней величины веса животных в течение исследования. На фиг. 3 показано, что соединение 1 не влияет на концентрацию паклитаксела в плазме крови у голых мышей линии CD-I. Планки погрешностей опыта показаны +/-. На фиг. 4 показано, что паклитаксел не влияет на концентрацию соединения 1 в плазме крови у голых мышей линии CD-I. На фиг. 5 показаны величины половинной максимальной ингибирующей концентрации (IC50) для паклитаксела и соединения 1, определенные при трехкратном серийном разведении, начиная с исходной концентрации, равной 1 мкМ. Величина IC50 для соединения 1 в клетках H1975 была рассчитана при 15 нМ; для паклитаксела величина IC50 составила 7 нМ. На фиг. 6 показаны результаты совместного лечения комбинацией паклитаксела и соединения 1 в клетках H1975. На фиг. 7 показан результат применения схемы совместного лечения: вначале вводят паклитаксел и через 24 ч в клетки H1975 вводят соединение 1. На фиг. 8 показано количество клеток H1975 в, погибших под действием соединения 1 (соединение 1), паклитаксела или комбинации двух лекарств при указанных концентрациях. На фиг. 9 а, b показаны результаты проведения метода Chau и Talalay (4) со средним значением выборки и применения компьютерной программы CalcuSyn 2.0 (Biosoft, Cambridge, UK) для изучения взаимодействия между соединением 1 и паклитакселом или доцетакселом. Каждое лекарство в различных концентрациях добавлялось в клетки NCI-H1975 на 72 ч и затем измерялась жизнеспособность при помощи метода с применением alamarBlue(Invitrogen) (A). На фиг. 9 а показана изоболограмма и приведены комбинационные индексы для соединения 1 в комбинации с паклитакселом с применением клеток NCI-H1975. Данные, приведенные ниже красной линии в изоболограмме, показывают наличие синергизма, в то время как данные, приведенные выше красной линии, показывают антагонизм между двумя лекарствами. Величины комбинационных индексов(CI)1 показывают наличие синергизма, в то время как CI1 показывает наличие антагонизма между двумя лекарствами. Похожие результаты наблюдались и в случае клеток НСС 827 (данные не показаны). Было установлено, что соединение 1 является сильным синергистом для паклитаксела (В). На фиг. 9b показаны построенная изоболограмма и величины комбинационных индексов для соединения 1 в комбинации с доцетакселом с применением клеток NCI-H1975, как и выше. Было установлено, что соединение 1 является сильным синергистом для доцетаксела. На фиг. 10 показаны построенные нормализованные изоболограммы (наверху) и величины CI (внизу) при совместном лечении паклитакселом (А) или доцетакселом (В) вместе с соединением 1 в случае клеток HEL92.1.7. На фиг. 11 показаны построенные нормализованные изоболограммы (слева) и величины CI (справа) при совместном лечении доцетакселом вместе с соединением 1 в случае клеток НТ 29. На фиг. 12 приведен график, показывающий влияние комбинации 75 мг/кг соединения 1 и 4 мг/кг доцетаксела, вводимой 1 Х/нед в модель NCI-HCC827 NSCLC. Величины % Т/С на день 40 показаны справа. Погрешности опыта представляют + или -0,5 SEM. Сведения, подтверждающие возможность осуществления изобретения Если иное не оговорено, термины, которые применяются ниже, означают следующее. Применяемые в данном изобретении термины "субъект", "пациент" и "млекопитающее" являются взаимозаменяемыми. Термины "субъект" и "пациент" относятся к животному (например, к птице, такой как цыпленок, куропатка или индейка, или к млекопитающему), предпочтительно к млекопитающему,включающему не-приматов (например, корову, свинью, лошадь, овцу, кролика, морскую свинку, крысу,кошку, собаку и мышь) и приматов (например, мартышку, шимпанзе и человека), и более предпочтительно к человеку. Согласно одному из вариантов субъект представляет собой животное, не относящееся к человеческому роду, такое как сельскохозяйственное животное (например, лошадь, корову, свинью или овцу) или домашнее животное (например, собаку, кошку, морскую свинку или кролика). Согласно предпочтительному варианту субъект является человеком. Применяемый в данном изобретении термин "соединение (соединения) по изобретению" и похожие термины относятся к соединению из табл. 1 или 2 или к их фармацевтически приемлемой соли. Некоторые из описанных здесь способов могут быть особенно эффективными при лечении субъектов, у которых раковое заболевание стало "резистентным к лекарствам" или "резистентным ко многим лекарствам". Раковое заболевание, которое первоначально реагировало на введение противоракового лекарства, становится устойчивым к действию противоракового лекарства, когда противораковое лекарство больше не является эффективным при лечении субъекта, который болен раком. Например, многие опухоли вначале будут реагировать на лечение с помощью противоракового лекарства путем уменьшения размера или даже наступления ремиссии, а затем начинают сопротивляться действию лекарства. "Резистентные к лекарствам" опухоли характеризуются возобновлением роста и/или вновь появляются после кажущейся ремиссии, несмотря на введение увеличенных доз противоракового лекарства. Раковые заболевания, которые стали резистентными к двум или более противораковым лекарствам, называются"резистентными ко многим лекарствам". Например, для ракового заболевания является типичным становиться резистентным к трем или более противораковым лекарствам, чаще к пяти или более противораковым лекарствам и иногда к десяти или более противораковым лекарствам. Применяемый в данном изобретении термин "фармацевтически приемлемая соль" относится к соли,полученной из соединения, имеющего кислую функциональную группу, такую как карбоксильная группа, и фармацевтически приемлемого неорганического или органического основания. Подходящие основания включают, но без ограничения, гидроокиси щелочных металлов, такие как гидроокиси натрия, калия и лития; гидроокиси щелочно-земельных металлов, такие как гидроокиси кальция и магния; гидроокиси других металлов, такие как гидроокиси алюминия и цинка; аммиак и органические амины, такие как незамещенные и гидроксизамещенные моно-, ди- и тризамещенные амины; дициклогексиламин; трибутиламин; пиридин; N-метил-, N-этиламин; диэтиламин; триэтиламин; моно-, ди- или три(низший алкиламины), содержащие 2-гидроксильные группы, такие как моно-, ди- или три(2-гидроксиэтил)амин, 2 гидрокси-трет-бутиламин или три(гидроксиметил)метиламин, N,N-ди-низший алкил-N-(гидроксинизший алкил)амин, такой как N,N-диметил-N-(2-гидроксиэтил)амин или три-(2-гидроксиэтил)амин; Nметил-D-глюкамин; и аминокислоты, такие как аргинин, лизин и т.п. Термин "фармацевтически приемлемая соль" относится также к соли, полученной из соединения, имеющего основную функциональную группу, такую как функциональная аминогруппа, и фармацевтически приемлемой неорганической или органической кислоты. Подходящие кислоты включают, но без ограничения, гидросерную кислоту, лимонную кислоту, уксусную кислоту, щавелевую кислоту, соляную кислоту (HCl), бромисто-водородную кислоту (HBr), йодисто-водородную кислоту (HI), азотную кислоту, двусернистую кислоту, фосфорную кислоту, изоникотиновую кислоту, олеиновую кислоту, дигалловую кислоту, пантотеновую кислоту,сахарную кислоту, молочную кислоту, салициловую кислоту, винную кислоту, дивинную кислоту, аскорбиновую кислоту, янтарную кислоту, малеиновую кислоту, бензосульфоновую кислоту, фумаровую кислоту, глюконовую кислоту, глюкароновую кислоту, муравьиновую кислоту, бензойную кислоту, глутаминовую кислоту, метансульфокислоту, этансульфокислоту, бензосульфокислоту, памовую кислоту и п-толуолсульфокислоту. Фармацевтически приемлемый носитель может содержать инертные ингредиенты, которые не ингибируют ненужным образом биологическую активность соединения(-ий). Фармацевтически приемлемые носители должны быть совместимыми, то есть нетоксичными, невоспалительными, неиммуногенными, и не должны вступать в другие нежелательные реакции после введения субъекту. Можно использовать стандартные методы получения фармацевтических составов, такие как описанные в Remington, J.P., Remington's Pharmaceutical Sciences (Mack Pub. Co., 17th ed., 1985). Подходящие фармацевтические носители для парентерального введения включают, например, стерильную воду, физиологический раствор, раствор бактериостатический (раствор, содержащий около 0,9% мг/мл бензилового спирта), физиологический раствор с фосфатным буфером, раствор Хэнка, раствор Рингера с лактатом и т.п. Способы инкапсулирования композиций, такие как нанесение покрытия на основе твердого желатина или циклодекстрана, известны из уровня техники; см. BAKER, ET AL., Controlled Release of Biological ActiveAgents, (John Wiley and Sons, 1986). Применяемый в данном изобретении термин "эффективное количество" относится к количеству со-3 022119 единения по изобретению, которое является достаточным для снижения или ослабления серьезности,продолжительности, развития или начала появления болезни или нарушения, задержки начала появления болезни или нарушения, замедления или остановки прогрессирования болезни или нарушения, развития болезни или нарушения, для появления регрессии болезни или нарушения, для предотвращения или задержки рецидива, развития, начала появления или развития симптома, связанного с болезнью или нарушением или для усиления или улучшения терапевтического эффекта (эффектов) от применения другой терапии. Точное количество соединения, вводимого субъекту, будет зависеть от способа введения, типа и степени серьезности болезни или состояния и от характеристик субъекта, таких как общее состояние здоровья, возраст, пол, вес и переносимость лекарств. Например, в случае пролиферативного заболевания или нарушения определение эффективного количества будет также зависеть от степени, серьезности и типа клеточной пролиферации. Специалист в данной области сможет определить соответствующие дозы в зависимости от этих и других факторов. В случае совместного введения с другими терапевтическими агентами, например совместного введения с противораковым агентом, "эффективное количество" любого дополнительного терапевтического агента (агентов) будет зависеть от типа применяемого лекарства. Для одобренных терапевтических агентов известны подходящие дозы и они могут быть адаптированы специалистом в данной области в соответствии с состоянием субъекта, типа состояния субъекта,типа состояния (состояний), подвергающихся лечению, и количества используемого соединения по изобретению. В тех случаях, когда количество четко не указано, можно предположить, какое должно быть эффективное количество. Неограничивающие примеры эффективного количества соединения по изобретению приведены ниже. Согласно конкретному варианту изобретение предусматривает способ лечения,управления или улучшения состояния при NSCLC, раке толстой кишки или эритролейкозе или ослабления одного или более симптомов этих болезней, при этом указанный способ включает введение субъекту, нуждающемуся в этом, дозы, равной по меньшей мере 150 мкг/кг, по меньшей мере 250 мкг/кг, по меньшей мере 500 мкг/кг, по меньшей мере 1 мкг/кг, по меньшей мере 5 мкг/кг, по меньшей мере 10 мкг/кг, по меньшей мере 25 мкг/кг, по меньшей мере 50 мкг/кг, по меньшей мере 75 мкг/кг, по меньшей мере 100 мкг/кг, по меньшей мере 125 мкг/кг, по меньшей мере 150 мкг/кг или по меньшей мере 200 мкг/кг или более, одного или более соединений по изобретению один раз каждый день, один раз каждые два дня, один раз каждые 3 дня, один раз каждые 4 дня, один раз каждые 5 дней, один раз каждые 6 дней,один раз каждые 7 дней, один раз каждые 8 дней, один раз каждые 10 дней, один раз каждые две недели,один раз каждые три недели или один раз в месяц. Дневная доза может быть введена в виде одной дозы. Альтернативно, дневная доза может быть разделена на части (обычно равные части), которые вводят два раза, три раза, четыре раза или более в день. Дозирование терапевтического агента, отличающегося от соединения по изобретению, который вводили или вводят для лечения, управления или улучшения состояния при раке легкого (например,NSCLC), раке толстой кишки или эритролейкозе или для ослабления одного или более симптомов этих болезней, может применяться при комбинированной терапии согласно данному изобретению. Предпочтительно, чтобы доза каждого отдельного терапевтического агента, используемого в такой комбинированной терапии, была меньше, чем доза отдельного терапевтического агента, применяемого в отдельности для лечения, управления или улучшения состояния при наличии болезни или состояния или для ослабления одного или более симптомов этих болезней. Рекомендуемые дозы терапевтических агентов, применяемых в настоящее время для лечения,управления или улучшения состояния при наличии болезни или состояния или для ослабления одного или более симптомов этих болезней, можно найти в любом из источников в уровне техники; см., например, GOODMANGILMAN'S The Pharmacological Basis of Basis of Therapeutics 9th ed (Hardman, et al,Eds., NY:Mc-Graw-Hill (1996; Physician's Desk Reference 57th ed. (Medical Economics Co., Inc., Montvale,NJ (2003. Применяемые в данном изобретении термины "лечить", "лечение" и "применяемый для лечения" относятся к уменьшению или ослаблению прогрессирования, степени тяжести и/или продолжительности болезни или расстройства, задержке начала болезни или расстройства или к ослаблению одного или более симптомов (предпочтительно одного или более явных симптомов) болезни или расстройства, возникающим при введении одного или более терапевтических агентов (например, одного или более терапевтических агентов, таких как соединение по изобретению). Термины "лечить", "лечение" и "применяемый для лечения" охватывают также уменьшение риска развития болезни или состояния и задержку или ингибирование рецидива болезни или состояния. Согласно конкретным вариантам термины "лечить", "лечение" и "применяемый для лечения" относятся к ослаблению по меньшей мере одного определяемого признака болезни или состояния, такому как рост опухоли, необязательно видимого для пациента. Согласно другим вариантам термины "лечить", "лечение" и "применяемый для лечения" относятся к ингибированию прогрессирования болезни или состояния, например рака легкого (например, NSCLC), рака толстой кишки или эритролейкоза, или физическому путем стабилизации явного признака, или к физиологическому путем стабилизации физического параметра, или к тому и другому. Согласно другим вариантам термины "лечить", "лечение" и "применяемый для лечения" пролиферативных болезни или состояния относятся к уменьшению или стабилизации размера опухоли или количества раковых клеток и/или задержке образования опухолей. Применяемые в данном изобретении термины "терапевтический агент" и "терапевтические агенты" относятся к любому агенту (агентам), которые могут быть использованы при лечении болезни или состояния, например рака легкого (например, NSCLC), рака толстой кишки или эритролейкоза, или одного или более их симптомов. Согласно некоторым вариантам термин "терапевтический агент" относится к соединению по изобретению. Согласно некоторым другим вариантам термин "терапевтический агент" не относится к соединению по изобретению. Предпочтительно, если терапевтический агент представляет собой агент, который известен как пригодный для лечении болезни или состояния, например рака легкого (в том числе, NSCLC), рака толстой кишки или эритролейкоза или одного или более их симптомов. Применяемый в данном изобретении термин "синергическая" относится к комбинации соединения по изобретению и другого терапевтического агента, которые, взятые вместе, являются более эффективными, чем аддитивное их действие при индивидуальном их применении. Синергическое действие комбинации терапий (например, комбинации терапевтических агентов) позволяет применять более низкие дозы одного или более терапевтических агентов и/или реже вводить указанный агент (агенты) субъекту,страдающему от болезни или состояния, например рака легкого (в том числе NSCLC), рака толстой кишки или эритролейкоза. Возможность применять более низкие дозы одного или более терапевтических агентов и/или реже вводить указанный агент (агенты) снижает токсичность, связанную с введением указанного агента субъекту без снижения эффективности указанной терапии при лечении болезни или состояния. Кроме того, синергическое действие может привести к повышению эффективности агентов при предотвращении, управлении или лечении болезни или состояния, например рака легкого (в том числеNSCLC), рака толстой кишки или эритролейкоза. Наконец, синергическое действие комбинационной терапии может помочь избежать возникновения нежелательных побочных эффектов, связанных с применением терапевтических агентов в отдельности или уменьшить их проявление. Применяемый в данном изобретении термин "в комбинации" относится к применению более чем одного терапевтического агента. Термин "в комбинации" не ограничивает порядок, в котором указанные терапевтические агенты вводятся субъекту, больному раком легкого (например, NSCLC), раком толстой кишки или эритролейкозом. Первый терапевтический агент, такой как соединение по изобретению, может вводиться субъекту, больному раком легкого (например, NSCLC), раком толстой кишки или эритролейкозом, до (например, за 5,15, 30, 45 мин, 1, 2, 4, 6, 12, 24, 48, 72, 96 ч, 1, 2, 3, 4, 5, 6, 8 или 12 нед), одновременно с или после введения второго терапевтического агента (например, через 5, 15, 30, 45 мин, 1, 2, 4, 6, 12, 24, 48, 72, 96 ч, 1, 2,3, 4, 5, 6, 8 или 12 нед), такого как противораковый агент. Применяемые в данном изобретении термины "терапии" и "терапия" могут относиться к любому протоколу (протоколам), способу (способам) и/или агенту (агентам), которые могут быть введены для предотвращения, лечения, управления или уменьшения интенсивности рака легкого (например, NSCLC),рака толстой кишки или эритролейкоза. Применяемый в данном изобретении термин "протокол" включает схемы дозирования и порядок дозирования. Протоколы охватывают методы применения и включают терапевтические протоколы. Применяемый в данном изобретении термин "по существу", относящийся к композиции, включающей соединение, означает, что композиция содержит более примерно 80 вес.%, более предпочтительно более примерно 90 вес.%, даже более предпочтительно более примерно 95 вес.% и наиболее предпочтительно более примерно 97 вес.% этого соединения. Данное изобретение станет более понятным из следующего ниже подробного его описания и иллюстративных примеров, которые не ограничивают данное изобретение. Таксан может представлять собой паклитаксел, который вводят внутривенно с недельной дозой, составляющей примерно 94 мкмол/м 2 (80 мг/м 2). Таксаны, применяемые согласно данному изобретению, включают паклитаксел (например, Taxol) и аналог паклитаксела доцетаксел. Паклитаксел представляет собой хорошо известное противораковое лекарство, которое может действовать путем ускорения и стабилизации образования микротрубочек. Многие аналоги паклитаксела известны, включая доцетаксел, называемый также "Taxotere". Паклитаксел и доцетаксел имеют следующие структурные формулы: Согласно другому варианту соединение представляет собой 3-(2,4-дигидрокси-5-изопропилфенил)4-(1-метилиндол-5-ил)-5-гидрокси [1,2,4]триазол или его фармацевтически приемлемую соль.i) Примеры соединений по изобретению. Примеры соединений по изобретению, включая их таутомеры и фармацевтически приемлемые соли, приведены ниже в табл. 1. Таблица 1 Соединения, применяемые при осуществлении способов, описанных в данном изобретении, могут быть получены в соответствии со способами, описанными в заявке США на патент 2006-0167070 и в заявке WO 2009/ 023211, описание которых включено полностью в данное изобретение посредством отсылок. Данное изобретение предусматривает способы лечения, управления или уменьшения интенсивности рака легкого (например, NSCLC), рака толстой кишки или эритролейкоза или одного или более симптомов этих болезней, при этом такие способы включают введение субъекту, который нуждается в этом,одного или более соединений по изобретению и одного или более других терапевтических агентов (например, одного или более терапевтических агентов, которые применяются в настоящее время, уже применялись раньше или о которых известно, что они подходят для этой цели или которые разрабатываются для применения при лечении или для уменьшения интенсивности рака легкого (например, NSCLC), рака толстой кишки или эритролейкоза или одного или более симптомов этих болезней, связанных с раком легкого (например, NSCLC), раком толстой кишки или эритролейкозом. Терапевтические агенты для комбинированной терапии по изобретению могут вводиться последовательно или совместно. Согласно конкретному варианту комбинированная терапия по изобретению включает применение одного или более соединений по изобретению и по меньшей мере один другой вид терапии, которая имеет тот же самый механизм действия, что и указанные соединения. Согласно другому варианту комбинированная терапия по изобретению включает применение одного или более соединений по изобретению и по меньшей мере один другой вид терапии, которая имеет другой механизм действия, чем указанные соединения. Согласно некоторым вариантам комбинированная терапия по изобретению усиливает терапевтическое действие одного или более соединений по изобретению, действуя вместе с другими соединениями, приводя к появлению аддитивного или синергического эффекта Согласно некоторым вариантам комбинированная терапия по изобретению приводит к уменьшению проявления побочных эффектов, связанных с методами лечения. Согласно некоторым вариантам комбинированная терапия по изобретению позволяет снизить эффективную дозу одного или более терапевтических агентов. Терапевтические агенты для комбинированной терапии могут вводиться субъекту, предпочтительно человеку, в составе одной и той же фармацевтической композиции. По альтернативным вариантам терапевтические агенты для комбинированной терапии могут вводиться субъекту совместно в составе разных фармацевтических композиций. Терапевтические агенты могут вводиться субъекту одним и тем же путем или разными путями. Согласно конкретному варианту фармацевтическая композиция по изобретению, содержащая одно или более соединений по изобретению, вводится субъекту, предпочтительно человеку, для предотвращения, лечения, управления или уменьшения интенсивности пролиферативного нарушения, такого как раковая болезнь, или одного или более симптомов этого нарушения. Согласно данному изобретению фармацевтические композиции по изобретению могут также включать один или более других агентов, применяющихся, применявшихся ранее или, как известно, являющихся подходящими для лечения или уменьшения интенсивности рака легкого (например, NSCLC), рака толстой кишки или эритролейкоза или одного или более симптомов этих болезней. Настоящее изобретение предусматривает способы управления, лечения или уменьшения интенсивности рака легкого (например, NSCLC), рака толстой кишки или эритролейкоза или одного или более симптомов этих болезней у субъекта, не поддающегося (или полностью, или частично) лечению рака легкого (например, NSCLC), рака толстой кишки или эритролейкоза существующими агентами, при этом указанные способы включают введение указанному субъекту эффективной дозы одного или более терапевтических агентов. Данное изобретение предусматривает также способы управления, лечения или уменьшения интенсивности рака легкого (например, NSCLC), рака толстой кишки или эритролейкоза или одного или более симптомов этих болезней путем введения одного или более соединений по изобретению в комбинации с любым другим (другими) агентом (агентами) пациентам, которые не поддаются лечению другими терапевтическими агентами, но которые больше не принимают больше эти другие агенты. Соединения по изобретению и/или другие терапевтические агенты могут вводиться субъекту любым путем, известным специалисту в данной области. Примеры способов введения включают, но без ограничения, парентеральный, например внутривенный, интрадермальный, подкожный, оральный (например, ингаляцию), интраназальный, трансдермальный (топический), чресслизистый и ректальный методы введения. Данное изобретение предусматривает композиции для лечения и уменьшения интенсивности рака легкого (например, NSCLC), рака толстой кишки или эритролейкоза. Согласно конкретному варианту композиция включает одно или более соединений по изобретению или фармацевтически приемлемую соль такого соединения. Согласно другому варианту композиция включает один или более терапевтических агентов, отличающихся от соединения по изобретению, или их фармацевтические соли. Согласно еще одному варианту данного изобретения композиция по изобретению включает одно или более соединений по изобретению или его фармацевтически приемлемую соль (соли) и один или более других терапевтических агентов. Согласно другому варианту композиция включает соединение по изобретению или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или эксципиент. Согласно предпочтительному варианту композиция по изобретению представляет собой фармацевтическую композицию в виде единичных доз. Фармацевтические композиции и лекарственные формы по изобретению включают один или более активных ингредиентов в соответствующих количествах и получены таким образом, что могут применяться для лечения рака легкого (например, NSCLC), рака толстой кишки или эритролейкоза. Предпочтительные фармацевтические композиции и лекарственные формы содержат соединение формулы (I) или (Ia), его фармацевтически приемлемую соль, возможно, в сочетании с одним или более дополнительными активными агентами. Фармацевтическая композиция по изобретению получается в таком виде, который совместим с намеченным путем введения. Примеры способов введения включают, но без ограничения, парентеральный,например внутривенный, интрадермальный, подкожный, оральный (например, ингаляцию), интраназальный, трансдермальный (топический), чресслизистый и ректальный методы введения. Согласно конкретному варианту композицию получают согласно принятым методам в виде фармацевтической композиции, адаптированной для внутривенного, подкожного, внутримышечного, орального, интраназального или топического введения людям. Согласно предпочтительному варианту фармацевтическая композиция получается общепринятым способом для подкожного введения людям. Единичные стандартные лекарственные формы по изобретению пригодны для орального, трансмукозального (например, назального, подъязычного, вагинального, трансбуккального или ректального),парентерального (например, подкожного, внутривенного введения, инъекции в виде болюса, внутримышечного или интраартериального) или трансдермального введения пациенту. Примеры лекарственных форм включают, но без ограничения, таблетки, каплеты, капсулы, такие как мягкие эластичные желатиновые капсулы, крахмальные капсулы, пастилки, леденцы, дисперсии, суппозитории, мази, горячие компрессы (примочки), пасты, порошки, обертки, кремы, пластыри, растворы, накладки, аэрозоли (например, назальные спреи или ингаляционные составы), гели, жидкие лекарственные формы, пригодные для орального или трансмукозального введения пациенту, включая суспензии (например, водные или неводные жидкие суспензии, эмульсии масло-в-воде или жидкие эмульсии вода-в-масле), растворы и эликсиры, жидкие лекарственные формы, пригодные для парентерального введения пациенту; и стерильные твердые вещества (например, кристаллические или аморфные твердые формы), которые могут быть воссозданы с получением жидких лекарственных форм, пригодных для парентерального введения пациенту. Состав, форма и вид лекарственных форм по изобретению обычно будет зависеть от их применения. Например, лекарственная форма, подходящая для введения через слизистую оболочку, может содержать меньшее количество активного (активных) ингредиента(-ов), чем оральная форма, применяемая для лечения по тому же назначению. Этот аспект изобретения хорошо понятен специалисту в данной области; см., например, Remington's Pharmaceutical Sciences (1990) 18th ed, Mack Publishing, Easton PA. Типичные фармацевтические композиции и лекарственные формы содержат один или более эксципиентов. Подходящие эксципиенты хорошо известны специалистам в области фармации, а неограничивающие примеры подходящих эксципиентов приведены в данном изобретении. Пригодность или непригодность эксципиента для введения в фармацевтическую композицию или лекарственную форму будет зависеть от различных факторов, хорошо известных из уровня техники, включая, но без ограничения,метод, которым лекарственная форма будет введена пациенту. Например, оральные лекарственные формы, такие как таблетки, могут содержать эксципиенты, которые не пригодны для применения в парентеральных лекарственных формах. Пригодность конкретного эксципиента может также зависеть от вида конкретных активных ингредиентов в лекарственной форме. Например, разложение некоторых активных ингредиентов может быть ускорено некоторыми эксципиентами, такими как лактоза, или под действием воды. Активные ингредиенты, которые содержат первичные или вторичные аминогруппы (например, N-десметилвенлафаксин иN,N-дидесметилвенлафаксин) особенно чувствительны к такому ускоренному разложению. Соответственно данное изобретение охватывает фармацевтические композиции и лекарственные формы, которые содержат небольшое количество лактозы, если она вообще содержится. Применяемый в данном изобретении термин "не содержащая лактозы" означает, что количество содержащейся лактозы, если она есть вообще, является недостаточным для существенного увеличения скорости разложения активного ингредиента. Композиции, не содержащие лактозы, могут включать эксципиенты, которые хорошо известны из уровня техники и перечислены, например, в U.S. Pharmocopoeia (USP) SP (XXI)/NF (XVI). В общем,композиции, не содержащие лактозы, содержат активные ингредиенты, связующее/наполнитель и смазывающее вещество в фармацевтически совместимых и фармацевтически приемлемых количествах. Предпочтительные лекарственные формы, не содержащие лактозы, включают активные ингредиенты,микрокристаллическую целлюлозу, предварительно желатинизированный крахмал и стеарат магния. Данное изобретение охватывает также безводные фармацевтические композиции и лекарственные формы, содержащие активные ингредиенты, так как вода может способствовать разложению некоторых соединений. Например, добавление воды (например, 5% воды) широко применяется в фармацевтике как средство имитирования условий длительного хранения для того, чтобы определить характеристики составов, такие как длительность хранения или стабильность составов со временем; см., например, Jens Т.Carstensen (1995) Drug Stability: PrinciplesPractice, 2d. Ed., Marcel Dekker, NY, NY, 379-80. Действительно, вода и нагревание ускоряют разложение некоторых соединений. Таким образом, действие воды на состав может иметь большое значение, так как влага и/или влажность обычно имеют место во время изготовления, обработки, упаковки, хранения, перевозки и применения составов. Безводные фармацевтические композиции и лекарственные формы по изобретению могут быть получены с использованием безводных ингредиентов или ингредиентов с низким содержанием влаги и в условиях с низким содержанием влаги или с низкой влажностью. Фармацевтические композиции и лекарственные формы по изобретению, которые включают лактозу и по меньшей мере один активный ингредиент, содержащий первичные и вторичные аминогруппы, являются предпочтительно безводными,если ожидается значительный контакт с влагой и/или влажностью во время приготовления, упаковки и/или хранения. Безводная фармацевтическая композиция по изобретению должна быть получена и должна храниться таким образом, чтобы сохранялась ее безводная природа. Соответственно безводные композиции предпочтительно упаковывать с применением материалов, которые, как известно, защищают от действия воды, чтобы их можно было включить в подходящие наборы. Примеры таких упаковок включают, но без ограничения, герметично запаянную фольгу, пластики, контейнеры для единичных доз (например, ампулы), блистеры и контурные упаковки ленточного типа для штучных изделий. Далее, данное изобретение охватывает фармацевтические композиции и лекарственные формы, которые содержат одно или более соединений, которые снижают скорость, с которой активный ингредиент будет разлагаться. Такие соединения, которые в данном изобретении называются "стабилизаторами",включают, без ограничения, антиоксиданты, такие как аскорбиновая кислота, буферы для регулирования рН или буферные соли. Фармацевтические композиции по изобретению, которые пригодны для орального введения, могут быть в виде дискретных лекарственных форм, таких как, но без ограничения, таблетки (например, жевательные таблетки), каплеты, капсулы и жидкости (например, ароматизированные сиропы). Такие лекарственные формы содержат заданные количества активных ингредиентов и могут быть получены фармацевтическими способами, хорошо известными специалистам; см., в общем, Remington's PharmaceuticalSciences (1990) 18th ed., Mack Publishing, Easton PA. Типичные оральные лекарственные формы по изобретению получают путем соединения активного ингредиента(-ов) в смеси по меньшей мере с одним эксципиентом согласно общеизвестным методам смешения, принятым в фармации. Эксципиенты могут быть самыми разными в зависимости от формы препарата, который желательно вводить. Например, эксципиенты, пригодные для применения в оральных жидкостях или в аэрозоле, включают, но без ограничения, воду, гликоли, масла, спирты, ароматизирующие агенты, консерванты и красители. Примеры эксципиентов, подходящих для применения в твердых оральных лекарственных формах (например, в порошках, таблетках, капсулах и каплетах), включают, но без ограничения, крахмалы, сахара, микрокристаллическую целлюлозу, разбавители, гранулирующие агенты, смазывающие агенты, связующие и дезинтегрирующие агенты. Из-за легкости их применения таблетки и капсулы представляют собой наиболее удобную оральную лекарственную форму, в этом случае используют твердые эксципиенты. Если желательно, на таблетки может быть нанесено покрытие с помощью стандартных методов с применением или без применения воды. Такие лекарственные формы могут быть приготовлены любым из фармацевтических методов. В общем, фармацевтические композиции и лекарственные формы получают путем равномерного и тщательного смешения активных ингредиентов с жидкими носителями, мелко измельченными твердыми носителями или и с теми, и с другими и последующего формования продукта с получением нужных форм, если это необходимо. Например, согласно данному изобретению таблетку можно получить прессованием или формованием. Прессованные таблетки могут быть изготовлены путем прессования в подходящем устройстве активных ингредиентов в свободно текучей форме, такой как порошок или гранулы, возможно вместе с эксципиентом. Формованные таблетки можно изготовить путем формования в подходящем устройстве смеси порошкообразного соединения, увлажненного инертным жидким разбавителем. Примеры эксципиентов, которые могут быть использованы в оральных лекарственных формах,включают, но без ограничения, связующие, наполнители, дезинтегранты и смазывающие вещества. Связующие, которые пригодны для использования в фармацевтических композициях и лекарственных формах, включают, но без ограничения, кукурузный крахмал, картофельный крахмал или другие крахмалы,желатин, природные и синтетические камеди, такие как смола акации, альгинат натрия, альгиновую кислоту, другие альгинаты, порошкообразный трагакант, гуаровую смолу, целлюлозу и ее производные(например, этилцеллюлозу, ацетат целлюлозы, кальциевую соль карбоксиметилцеллюлозы, натриевую соль карбоксиметилцеллюлозы), поливинилпирролидон, метилцеллюлозу, предварительно желатинизированный крахмал, гидроксипропилметилцеллюлозу (например,2208, 2906, 2910), микрокристаллическую целлюлозу и смеси указанных ингредиентов. Подходящие виды микрокристаллической целлюлозы включают, но без ограничения, вещества,продаваемые под названиями AVICEL-PH-101, AVICEL-PH-103, AVICEL RC-581, AVICEL-PH-105 (доступные в FMC Corporation, American Viscose Division, Avicel Sales, Marcus Hook, PA) и их смеси. Один конкретный вид связующего представляет собой смесь микрокристаллической целлюлозы и натриевой соли карбоксиметилцеллюлозы, продаваемую под маркой AVICEL RC-581. Подходящие безводные эксципиенты или добавки или эксципиенты и добавки с низким содержанием влаги включают AVICEL-PH103J и Starch 1500 LM. Примеры наполнителей, которые пригодны для использования в фармацевтических композициях и лекарственных формах, описанных в данном изобретении, включают, но без ограничения, тальк, карбонат кальция (например, гранулы или порошок), целлюлозу микрокристаллическую, порошкообразную целлюлозу, декстраты, каолин, маннит, кремниевую кислоту, сорбит, крахмал, предварительно желатинизированный крахмал и их смеси. Связующее или наполнитель в фармацевтических композициях по изобретению обычно содержатся в количестве от примерно 50 до примерно 99 вес.% в расчете на фармацевтическую композицию или лекарственную форму. Дезинтегранты применяются в композициях по изобретению для получения таблеток, которые распадаются под действием окружающей среды, содержащей воду. Таблетки, которые содержат слишком много дезингегранта, могут распадаться при хранении, в то время таблетки, которые содержат слишком мало дезингегранта, могут не распадаться с достаточной скоростью или не распадаться в желательных условиях. Таким образом, для получения твердой оральной лекарственной формы по изобретению нужно использовать достаточное количество дезинтегранта, которое не является ни слишком большим, ни слишком маленьким, чтобы отрицательно влиять на высвобождение активных ингредиентов. Количество применяемого дезинтегранта меняется в зависимости от вида состава и может быть легко определено обычным специалистом в данной области. Типичные фармацевтические композиции по изобретению содержат от примерно 0,5 до примерно 15 вес.% дезинтегранта, предпочтительно от примерно 1 до примерно 5 вес.% дезинтегранта. Дезинтегранты, которые пригодны для использования в фармацевтических композициях и лекарственных формах, описанных в данном изобретении, включают, но без ограничения, агар-агар, альгиновую кислоту, карбонат кальция, микрокристаллическую целлюлозу, натриевую соль кросс-кармелозы, кросповидон, калия полиакрилин (полиакрилат калия), натриевую соль гликолята крахмала, картофельный крахмал или тапиоковый крахмал, другие крахмалы, предварительно желатинизированный крахмал, глины, другие альгины, другие виды целлюлозы, камеди и их смеси. Смазывающие вещества, которые пригодны для использования в фармацевтических композициях и лекарственных формах, описанных в данном изобретении, включают, но без ограничения, стеарат кальция, стеарат магния, минеральное масло, легкое минеральное масло, глицерин, сорбит, маннит, полиэтиленгликоль, другие гликоли, стеариновую кислоту, лаурилсульфат натрия, тальк, гидрированное растительное масло (например, арахисовое масло, масло семян хлорка, подсолнечное масло, кунжутное масло,оливковое масло, кукурузное масло и соевое масло), стеарат цинка, этилолеат, этиллаурат, агар и их смеси. Дополнительные примеры смазывающих веществ включают, например, силоидный силикагель(AEROSIL 200, производимый в W.R. Grace Co. of Baltimore, MD), коагулированный аэрозоль синтетической двуокиси кремния (продаваемый Degussa Co. of Plano, TX), CAB-O-SIL (пирогенная двуокись кремния, продаваемая Cabot Co. of Boston, MA) и их смеси. Если смазывающие вещества применяются вообще, их количество обычно составляет менее 1 вес.% в расчете на фармацевтическую композицию или лекарственную форму, в которые они вводятся. Активные ингредиенты по изобретению могут вводиться в средствах с контролируемым высвобождением или в системах доставки, которые хорошо известны специалисту в данной области. Примеры включают, но без ограничения, средства, описанные в патентах США 3845770, 3916899, 3536809,3598123, 4008719, 5674533, 5059595, 5591767, 5120548, 5073543, 5639476, 5354556 и 5733566, описания которых полностью включены в данное изобретение посредством отсылок. Такие лекарственные формы могут быть использованы для обеспечения медленного или контролируемого высвобождения одного или более активных ингредиентов, при этом применяют, например, гидроксипропилметилцеллюлозу, другие полимерные матрицы, гели, проницаемые мембраны, осмотические системы, многослойные покрытия,микрочастицы, липосомы, микросферы или их комбинации для обеспечения желаемого профиля высвобождения в различном виде. Подходящие составы с контролируемым высвобождением, известные специалистам в данной области, включая составы, описанные в данном изобретении, могут быть легко выбраны для использования с активными ингредиентами по изобретению. Таким образом, данное изобретение охватывает единичные стандартные лекарственные формы, подходящие для орального введения,такие как таблетки, капсулы, гелькапы и каплеты, которые адаптированы для обеспечения контролируемого высвобождения. Все фармацевтические продукты с контролируемым высвобождением имеют общую цель улучшения лекарственной терапии по сравнению с результатами, достигаемыми при применении составов с неконтролируемым высвобождением. В идеале при применении препарата с контролируемым высвобождением медицинское лечение характеризуется минимальным количеством лекарственного вещества, используемого для лечения или контроля состояния за минимальное время. Преимущества составов с контролируемым высвобождением включают продолжительную активность лекарства, уменьшенную частоту дозирования и повышенную комфортность для пациента. Большинство составов с контролируемым высвобождением предназначены для первоначального высвобождения количества лекарства (активного ингредиента), которое быстро обеспечивает желаемый терапевтический эффект, и постепенного и непрерывного высвобождения других количеств лекарства для поддержания этого уровня терапевтического или профилактического эффекта в течение продолжительного времени. Для того чтобы поддерживать этот постоянный уровень лекарства в организме, лекарство должно высвобождаться из лекарственной формы со скоростью, которая обеспечивает замену количества лекарства, подвергшегося метаболизму и выделившегося из организма. Контролируемое высвобождение активного ингредиента может быть имитировано созданием различных условий, включая, но без ограничения, величину рН, температуру, ферменты, воду или другие физиологические условия или соединения. Конкретный состав с пролонгированным высвобождением согласно изобретению содержит терапевтическое или профилактическое количество соединения любой из формул (I) или (Ia) или соединения, или его фармацевтически приемлемую соль в виде сфероидов,которые включают также микрокристаллическую целлюлозу и, возможно, гидроксипропилметилцеллюлозу, с покрытием на основе смеси этилцеллюлозы и гидроксипропилметилцеллюлозы. Такие составы с пролонгированным высвобождением могут быть получены в соответствии с патентом США 6274171,описание которого полностью включено в данное изобретение посредством отсылки. Конкретный состав с контролируемым высвобождением по изобретению содержит от примерно 6 до примерно 40 вес.% соединения любой из формул (I) или (Ia) или соединения, приведенного в табл. 1 или 2, или его фармацевтически приемлемую соль, сольват, гидрат, клатрат или пролекарство, от примерно 50 до примерно 94 вес.% микрокристаллической целлюлозы, NF, и, возможно, от примерно 0,25 до примерно 1 вес.% гидроксипропилметилцеллюлозы, USP, причем сфероиды, из которых состоит состав, покрыты пленкой, состоящей из этилцеллюлозы и гидроксипропилметилцеллюлозы. Парентеральные лекарственные формы, описанные в данном изобретении, могут вводиться пациенту различными путями, включая, но без ограничения, подкожное, внутривенное (в том числе инъекцию болюса), внутримышечное и интраартериальное введение. Чтобы их введение позволяло обычно обойти природные защитные реакции пациентов на загрязняющие вещества, парентеральные лекарственные формы предпочтительно являются стерильными или способны к стерилизации перед введением пациенту. Примеры парентеральных лекарственных форм включают, но без ограничения, растворы, готовые для инъекции, сухие продукты, готовые для растворения или суспендирования в фармацевтичеки приемлемом носителе для инъекции, суспензии, готовые для инъекции, и эмульсии. Подходящие носители, которые могут быть использованы для приготовления парентеральных лекарственных форм по изобретению, хорошо известны специалистам в данной области. Примеры включают, но без ограничения, воду для инъекции, USP; водные носители, включая, но без ограничения, хлористый натрий для инъекции, раствор Рингера для инъекции, декстрозу для инъекции, декстрозу и хлорид натрия для инъекции и раствор Рингера, содержащий лактат; смешивающиеся с водой носители, такие как, но без ограничения, этиловый спирт, полиэтиленгликоль и пропиленгликоль; и неводные носители, такие как, но без ограничения, кукурузное масло, хлопковое масло, арахисивое масло, кунжутное масло, этилолеат, изопропилмиристат и бензилбензоат. В парентеральные лекарственные формы по изобретению могут быть также введены соединения,которые повышают растворимость одного или более активных ингредиентов, описанных в данном изобретении. Трансдермальные, топические и трансмукозальные лекарственные формы по изобретению включают, но без ограничения, офтальмологические растворы, спреи, аэрозоли, кремы, лосьоны, мази, гели,растворы, эмульсии, суспензии или другие формы, которые известны специалисту в данной области; см.,например, Remington's Pharmaceutical Sciences (19801990) 16th and 18th eds., Mack Publishing, EastonPA and Introduction to Pharmaceutical Dosage Forms (1985) 4th ed., LeaFebiger, Philadelphia. Лекарственные формы, пригодные для лечения слизистых тканей в ротовой полости, могут быть приготовлены таким образом, чтобы они были в виде полоскания для ротовой полости или оральных гелей. Кроме того,трансдермальные лекарственные формы включают, но без ограничения, пластыри типа "пластырьрезервуар" или "пластырь матричного типа", которые можно накладывать на кожу и носить в течение определенного времени, нужного для проникновения желательного количества активных ингредиентов. Подходящие эксципиенты (например, носители и разбавители) и другие вещества, которые могут быть использованы для получения трансдермальных, топических и трансмукозальных лекарственных форм,охваченных данным изобретением, хорошо известны специалистам в данной области, и их выбор зависит от конкретной ткани, на которую будет наноситься данная фармацевтическая композиция. С учетом этого в качестве типичных эксципиентов можно назвать, но без ограничения, воду, ацетон, этанол, этиленгликоль, пропиленгликоль, бутан-1,3-диол, изопропилмиристат, изопропилпальмитат, минеральное масло и их смеси с получением лосьонов, настоек, кремов, эмульсий, гелей или мазей, которые являются нетоксичными и фармацевтически приемлемыми. Увлажнители и смачивающие средства также могут быть добавлены в фармацевтические композиции и лекарственные формы, если это желательно. Примеры таких дополнительных ингредиентов хорошо известны из уровня техники; см., например, Remington'sPharmaceutical Sciences (19801990) 16th and 18th eds., Mack Publishing, Easton PA. В зависимости от конкретной ткани, которая должна подвергаться лечению, дополнительные ингредиенты могут быть использованы в сочетании с лечением активными агентами по изобретению или до проведения этого лечения или после этого лечения. Например, ускорители проницаемости можно применять для способствования доставке активных ингредиентов в ткань. Подходящие ускорители проницаемости включают, но без ограничения, ацетон; различные спирты, такие как этанол, олеиловый спирт и тетрагидрофуриловый спирт; алкилсульфоксиды, такие как диметилсульфоксид; диметилацетамид; диметилформамид; полиэтиленгликоль; пирролидоны, такие как поливинилпирролидон; различные виды коллидонов (повидон, поливидон); мочевину и различные растворимые в воде или не растворимые в воде эфиры сахаров, такие как Tween 80 (полисорбат 80) и Span 60 (моностеарат сорбитана). Величина рН фармацевтической композиции, или лекарственной формы, или ткани, на которую наносится фармацевтическая композиция или лекарственная форма, может также регулироваться для улучшения доставки одного или более активных ингредиентов. Точно так же для улучшения доставки можно регулировать полярность носителя-растворителя, его ионную силу или тоничность. Соединения,такие как стеараты, также могут быть добавлены к фармацевтическим композициям или лекарственным формам для благоприятного изменения гидрофильности или липофильности одного или более активных ингредиентов, чтобы улучшить доставку. В этом отношении стеараты могут служить в качестве липидного носителя для состава, в качестве эмульгирующего агента или поверхностно-активного вещества и в качестве агента, улучшающего доставку или проницаемость. Для регулирования свойств полученной композиции могут быть также использованы различные соли, гидраты или сольваты активных ингредиентов. Количество соединения или композиции по изобретению, которое будет эффективным для предотвращения лечения, управления или уменьшения интенсивности пролиферативных расстройств, таких как рак, или одного или более симптомов этих расстройств, будет меняться в зависимости от природы, степени серьезности болезни или состояния и пути, которым вводится активный ингредиент. Частота введения и дозировки также будут меняться в зависимости от факторов, специфичных для каждого пациента,от конкретного вида терапии (например, применения терапевтических или профилактических агентов),- 11022119 степени серьезности нарушения, болезни или состояния, пути введения, а также возраста, веса тела, реакции пациента и предыдущей медицинской истории пациента. Эффективные дозы могут быть экстраполированы из кривых зависимости ответной реакции от дозы, построенных для испытуемых in vitro модели или животной модели. Подходящие схемы введения лекарств могут быть выбраны специалистом в данной области при рассмотрении таких факторов и в соответствии, например, со схемами лечения,описанными в литературе и рекомендованными в Physician's Desk Reference (57th ed., 2003). Примерные дозы для малых молекул выражаются в миллиграммах или микрограммах малой молекулы на килограмм веса субъекта или образца (например, от примерно 1 мкг до примерно 500 мг на 1 кг,от примерно 100 мкг до примерно 5 мг на 1 кг или от примерно 1 мкг до примерно 50 мкг на 1 кг). В общем, величины рекомендуемых доз соединения по изобретению при условиях, описанных в данном изобретении, находятся в интервале от примерно 0,01 до примерно 1000 мг в день, причем доза может быть однократной один раз в день и предпочтительно она вводится в виде разделенных доз в течение дня. Согласно одному из вариантов дневную дозу вводят два раза в день в виде равных частей. Конкретно, дневная доза может быть равной от примерно 5 до примерно 500 мг в день, более конкретно,от примерно 10 до примерно 200 мг в день. Лечение следует начинать с более низкой дозы, возможно,равной от примерно 1 до примерно 25 мг, и затем дозу увеличивают, если это необходимо, до величины от примерно 200 до примерно 1000 мг в день, вводя ее в виде однократной дозы в день или в виде разделенных доз в зависимости от общей ответной реакции пациента. В некоторых случаях может быть необходимо применять величины доз активного ингредиента, которые находятся за пределами указанного выше интервала, что является очевидным для специалиста в данной области. Кроме того, следует отметить, что клиницист или лечащий врач будут знать, как и когда следует прервать, отрегулировать схему лечения или прекратить лечение в соответствии с ответной реакцией отдельного пациента. Для различных пролиферативных нарушений можно применять различные терапевтически эффективные количества, что хорошо известно специалисту в данной области. Точно так же описанные выше интервалы доз и схемы лечения охватывают количества, достаточные для профилактики, управления,лечения или уменьшения интенсивности таких пролиферативных нарушений, но недостаточные для того, чтобы вызвать, или достаточные для того, чтобы уменьшить неблагоприятное действие соединений по изобретению. Кроме того, когда пациенту вводят многократные дозы соединения по изобретению,при этом не все дозы должны быть одинаковыми. Например, доза, вводимая пациенту, может быть увеличена для усиления профилактического или терапевтического эффекта, обеспечиваемого соединением,или может быть уменьшена для снижения проявления одного или более побочных эффектов, которые появляются у конкретного пациента. Согласно конкретному варианту доза композиции по изобретению или соединения по изобретению,вводимая пациенту для профилактики, управления, лечения или уменьшения интенсивности пролиферативных нарушений, таких как раковое заболевание, или одного или более симптомов таких нарушений,составляет 150, предпочтительно 250, 500 мкг/кг, 1, 5, 10, 25, 50, 75, 100, 125, 150 или 200 мг/кг веса пациента или более. Согласно другому варианту доза композиции по изобретению или соединения по изобретению, вводимая пациенту для профилактики, управления, лечения или уменьшения интенсивности пролиферативных нарушений, таких как раковое заболевание, или одного или более симптомов таких нарушений, является стандартной дозой, составляющей от 0,1 до 20 мг, от 0,1 до 15 мг, от 0,1 до 12 мг, от 0,1 до 10 мг, от 0,1 до 8 мг, от 0,1 до 7 мг, от 0,1 до 5 мг, от 0,1 до 2,5 мг, от 0,25 до 20 мг, от 0,25 до 15 мг, от 0,25 до 12 мг, от 0,25 до 10 мг, от 0,25 до 8 мг, от 0,25 до 7 мг, от 0,25 до 5 мг, от 0,5 до 2,5 мг, от 1 до 20 мг, от 1 до 15 мг, от 1 до 12 мг, от 1 до 10 мг, от 1 до 8 мг, от 1 до 7 мг, от 1 до 5 мг или от 1 до 2,5 мг. Единичная доза может вводиться 1-4 или 1 раз в день или один раз каждые 2-6 или 7 дней или один раз в неделю, один раз каждые две недели, один раз каждые три недели или один раз в месяц. Согласно одному из вариантов таксан (например, паклитаксел или доцетаксел) вводится по схеме один раз каждые 3 недели и ингибитор HSP (например, соединение 1) вводится в первую неделю и во вторую неделю (после чего дают пациенту неделю отдыха) перед началом нового цикла. Альтернативно,вводят лекарство один раз каждые три недели, начиная с дозы, равной 60 мг/м 2, затем ее увеличивают до 75 мг/м 2 для паклитаксела или доцетаксела. Согласно другому альтернативному варианту применяют схему: 3/1 нед отдыха, начиная с дозы, равной 30 мг/м 2, и увеличивая ее до 35 мг/м 2 для паклитаксела или доцетаксела. Количество ингибитора HSP 90 регулируется в соответствии с переносимостью пациентом и эффективностью лечения, как описано выше. Согласно другому варианту паклитаксел вводят или один раз в неделю (типичная доза равна 90 мг/м 2, интервал доз составляет 70-100 мг/м 2). Или же его вводят один раз каждые три недели. При введении паклитаксела один раз каждые три недели интервал доз составляет от 175 до 225 мг/м 2. Доза ингибитора HSP 90 обычно является однократной (например, 200 мг/м 2) или менее в зависимости от переносимости пациентом, как описано выше. Согласно еще одному варианту доцетаксел вводят один раз каждые три недели (величина дозы равна 75 мг/м 2, интервал доз составляет 60-100 мг/м 2). Его можно также вводить еженедельно, интервал доз составляет 30-40 мг/м 2. Доза ингибитора HSP 90 обычно является однократной (например, 200 мг/м 2) или менее в зависимости от переносимости пациентом, как описано выше. Или же цикл лечения включает еженедельное лечение в течение 2 нед с последующим отдыхом в течение одной недели. Этот цикл повторяют каждые 3 нед. Ингибитор HSP90 вводится (с дозой 150 или 200 мг/м 2) в дни 1 и 8 каждого цикла, а доцетаксел вводится (с дозой, равной 60 или 75 мг/м 2) в день 1 каждого цикла. Цикл лечения повторяется каждые три недели. Согласно еще одной альтернативе субъектам вводят 200 мг/м 2 ингибитора HSP90 с последующим введением доцетаксела с дозой 25, 30 или 35 мг/м 2 в течение трех последовательных недель и затем дают больному неделю перерыва. Затем цикл лечения повторяют. Профилактические или терапевтические агенты, отличающиеся от соединений по изобретению, которые применялись или применяются для профилактики, управления, лечения или уменьшения интенсивности пролиферативных нарушений, таких как раковое заболевание, или одного или более симптомов таких нарушений, могут быть использованы в комбинированной терапии согласно данному изобретению. Предпочтительно, если в комбинированной терапии согласно данному изобретению используются дозы меньшие, чем те, которые применялись или применяются для профилактики, управления, лечения или уменьшения интенсивности пролиферативных нарушений, таких как раковое заболевание, или одного или более симптомов таких нарушений. Рекомендуемые дозы агентов, которые применяются в настоящее время для профилактики, управления, лечения или уменьшения интенсивности пролиферативных нарушений, таких как раковое заболевание, или одного или более симптомов таких нарушений,можно найти в любом источнике в уровне техники, включая, но без ограничения, справочники Hardmanet al., eds., 1996, GoodmanGilman's The Pharmacological Basis Of Basis Of Therapeutics 9th Ed, Mc-GrawHill, New York; Physician's Desk Reference (PDR), 57th Ed., 2003, Medical Economics Co., Inc., Montvale, NJ,содержание которых полностью включено в данное изобретение посредством отсылок. Согласно некоторым вариантам, когда соединения по изобретению вводятся в комбинации с другими терапевтическими агентами, эти агенты (например, профилактические или терапевтические агенты) вводятся с интервалом менее 5, менее 30 мин, с интервалом в 1 ч, интервалом около 1 ч, с интервалом от примерно 1 до примерно 2 ч, от примерно 2 до примерно 3 ч, от примерно 3 до примерно 4 ч, с интервалом от примерно 4 до примерно 5 ч, с интервалом от примерно 5 до примерно 6 ч, с интервалом от примерно 6 до примерно 7 ч, с интервалом от примерно 7 до примерно 8 ч, с интервалом от примерно 8 до примерно 9 ч, с интервалом от примерно 9 до примерно 10 ч, с интервалом от примерно 10 до примерно 11 ч, с интервалом от примерно 11 до примерно 12 ч, с интервалом от примерно 12 до примерно 18 ч, с интервалом от примерно 18 до примерно 24 ч, с интервалом от примерно 24 до примерно 36 ч, с интервалом от примерно 36 до 48 ч, с интервалом от примерно 48 до 52 ч, с интервалом от примерно 52 до 60 ч, с интервалом от примерно 60 до примерно 72 ч, с интервалом от примерно 72 до 84 ч, с интервалом от примерно 84 до 96 ч или с интервалом от примерно 96 до примерно 120 ч. Согласно одному из вариантов два или более агентов (например, профилактические или терапевтические агенты) вводятся во время одного и того же визита пациента. Согласно некоторым вариантам одно или более соединений по изобретению и один или более других агентов (например, терапевтические агенты) вводятся циклами. Циклическая терапия включает введение первого лекарства (например, первого профилактического или терапевтического агента) в течение определенного периода времени с последующим введением второго лекарства (например, второго профилактического или терапевтического агента) в течение определенного периода времени с последующим введением третьего лекарства (например, третьего профилактического или терапевтического агента) в течение определенного периода времени и так далее, и повторение этого последовательного введения, то есть цикла, для того, чтобы снизить развитие резистентности к одному из агентов, избежать или уменьшить появление побочных эффектов от применения одного из агентов и/или повысить эффективность лечения. Согласно некоторым вариантам введение одного и того же соединения по изобретению можно повторить и эти моменты введения могут быть разделены по меньшей мере 1, 2, 3, 5, 10, 15, 30, 45 днями, 2 мес, 75 днями, 3 или 6 мес. Согласно другим вариантам введение одного и того же профилактического или терапевтического агента можно повторять и эти моменты введения могут быть разделены по меньшей мере 1, 2, 3, 5, 10, 15, 30, 45 днями, 2 мес, 75 днями, 3 или 6 мес. Согласно конкретному варианту данное изобретение предусматривает способ профилактики, лечения, управления или уменьшения интенсивности пролиферативных нарушений, таких как раковое заболевание, или одного или более симптомов таких нарушений, причем этот способ включает введение субъекту, нуждающемуся в этом, дозы, равной по меньшей мере 150 мкг/кг, предпочтительно по меньшей мере 250 мкг/кг, по меньшей мере 500 мкг/кг, по меньшей мере 1 мг/кг, по меньшей мере 5 мг/кг, по меньшей мере 10 мг/кг, по меньшей мере 25 мг/кг, по меньшей мере 50 мг/кг, по меньшей мере 75 мг/кг,по меньшей мере 100 мг/кг, по меньшей мере 125 мг/кг, по меньшей мере 150 мг/кг или по меньшей мере 200 мг/кг или более одного или более соединений по изобретению один раз каждый день, предпочтительно один раз каждые 2 дня, один раз каждые 3 дня, один раз каждые 4 дня, один раз каждые 5 дней,один раз каждые 6 дней, один раз каждые 7 дней, один раз каждые 8 дней, один раз каждые 10 дней, один раз каждые две недели, один раз каждые три недели или один раз в месяц. Или же доза может быть разделена на части (обычно равные части), которые вводят два, три, четыре или более раз в день. Данное изобретение далее иллюстрируется примерами, которые никоим образом не ограничивают это изобретение. Пример 1. Комбинация соединения 1 и паклитаксела обладает повышенной противоопухолевой активностью в отношении человеческих клеток опухоли в модели SCID мышиного ксенографта с клеткамиNCI-H1975 NSCLC. Линия человеческих клеток немелкоклеточного рака легкого (NSCLC), NCI-H1975 (АТСС CRL5908) была получена из American Type Culture Collection (ATCC; Manassas, Virginia, USA). Клеточную линию культивировали в среде для роста, приготовленной из 50% модифицированной по методу Дульбекко среды Игла (с большим содержанием глюкозы), 50% среды RPMI Media 1640 (4,5 г/л глюкозы),10% фетальной телячьей сыворотки (FBS), 10 мМ HEPES, 1% 100 Х пенициллина-стрептомицина, 1% 100 Х пирувата натрия и 1% 100 Х среды MEM, содержащей несущественные аминокислоты. FBS была получена из АТСС, а все другие реагенты получали из Invitrogen Corporation (Carlsbad, California, USA). Клетки, которые были криосохранены в жидком азоте, быстро размораживали при температуре 37 С и переносили в колбу с тканевой культурой, содержащей среду для роста, и затем выдерживали при температуре 37 С в инкубаторе в присутствии 5% СО 2. Для увеличения клеточной массы линии NCI-H1975 культуры разводили 1:5 каждые 3 дня, когда клетки в колбе с площадью культивирования, равной 175 см 2, становились конфлюэнтными на 85%. Культуры пересевали путем промывки 10 мл физиологического раствора с комнатной температурой, буферированного фосфатным буфером (PBS), и затем диссоциации клеток путем добавления 5 мл IX трипсина-ЭДТА и инкубирования при температуре 37 С до тех пор, пока клетки не начали отделяться от поверхности колбы. Для инактивации трипсина добавляли 5 мл среды для роста и затем содержимое колбы подвергали центрифугированию для гранулирования клеток. Надосадочную жидкость отсасывали и гранулы клеток вновь суспендировали в 10 мл среды для роста,после чего определяли количество клеток, используя гемоцитометр. Клетки высевали в колбы с площадью культивирования 175 см 2, содержащие 50 мл среды для роста, и инкубировали при температуре 37 С в инкубаторе в присутствии 5% СО 2. Когда в колбах достигалась 85% конфлюэнтность клеток, описанный выше процесс пересева повторяли до получения достаточного количества клеток для имплантации мышам. Самок мышей CB17/Icr-Prkdcscid/Crl (линия SCID) в возрасте от шести до семи недель получали изCharles River Laboratories (Wilmington, Massachusetts, USA). Животных помещали по 4-5 штук в клетку в микроизоляторы с циклом 12 ч/12 ч свет/темнота, животные проходили акклиматизацию в течение по меньшей мере 1 нед до использования, их кормили нормальным лабораторным кормом ad libitum. При имплантации возраст животных составлял от семи до восьми недель. Для имплантации опухолевых клеток NCI-H1975 мышам линии SCID клетки собирали, как описано выше, промывали их в PBS и снова суспендировали при концентрации 510(7) кл/мл в 50% не дополненной среды и 50% Matrigel BasementMembrane Matrix (354234; BD Biosciences; Bedford, Massachusetts, USA). С помощью иглы калибра 27 и шприца объемом 1 см 3 инъекцией подкожно в подвздошную область мышам линии SCID вводили 510(6) клеток NCI-H1975 в 0,1 мл клеточной суспензии. Затем опухолям давали развиться in vivo до тех пор, пока большинство из них не достигло объема,равного 95-195 мм 3, на что потребовалось 1 1/2 недель после имплантации для модели NCI-H1975. Для исследования отбирали животных с продолговатыми, очень небольшими или большими опухолями, которые характеризовались устойчивой скоростью роста. Объем опухоли (V) определяли с помощью штангенциркуля, измеряя ширину (W), длину (L) и толщину (Т) опухоли, используя следующую формулу:V=0.5236(LWТ). Животных рандомизировали в группы для лечения таким образом, чтобы средняя величина объема опухолей в каждой группе была в начале дозирования похожей. Величины % Т/С, как меру эффективности, определяли следующим образом:(iii) T=изменение средней величины объема опухоли между началом дозирования и концом исследования;(iv) C=изменение средней величины объема опухоли между началом дозирования и концом исследования;(v) T0=средний объем опухоли в начале дозирования. Для получения состава соединения 1, паклитаксела или их комбинации в DRD готовили исходные растворы испытуемого вещества путем растворения соответствующего количества соединения в диметилсульфоксиде (DMSO) под действием ультразвука в ультразвуковой водяной бане. Исходные растворы получали еженедельно, хранили при температуре -20 С и для дозирования разбавляли каждый день. Готовили также раствор 20% Cremophore RH40 (полиоксиэтилированное (40) гидрогенированное касторовое масло; BASF Corp., Aktiengesellschaft, Ludwigshafen, Germany) в 5% декстрозы в воде (Abbott Laboratories, North Chicago, Illinois, USA) путем нагревания вначале 100% Cremophore RH40 при температуре 50-60 С до получения жидкого и прозрачного состава, разбавления 1:5 при помощи 100% D5W, повторного нагревания снова до получения прозрачного раствора и затем тщательного перемешивания. Этот раствор можно хранить при комнатной температуре до 3 мес перед применением. Для приготовления составов DRD для ежедневного дозирования исходные растворы в DMSO разбавляли 1:10 20% Cremophore RH40. Конечный состав DRD для дозирования содержал 10% DMSO, 18% Cremophore RH40, 3,6% декстрозы, 68,4% воды и соответствующее количество испытуемого соединения. Животным вводили этот состав внутривенно (i.v.) в количестве 10 мл на 1 кг веса животного 1 день в каждую неделю. Лечение соединением 1 с дозой 50 мг/кг веса умеренно ингибировало рост опухолевых клеток NCIH1975 у мышей линии SCID с величиной % Т/С, равной 55. Подобно этому лечение паклитакселом с дозой, равной 7,5 мг/кг веса, умеренно ингибировало рост опухолевых клеток NCI-H1975 у мышей линииSCID с величиной % Т/С, равной 38. В противоположность этому совместное введение комбинации 50 мг/кг веса соединения 1 и 7.5 мг/кг паклитаксела приводило к резкому ингибированию роста опухолевых клеток NCI-H1975 у мышей линии SCID с величиной % Т/С, равной 7. Эффективность действия этой комбинации была значительно выше, чем эффективность, наблюдаемая или в группе животных, которым вводили один агент (Р 0.05; однопроходный метод ANOVA). Результаты показаны на фиг. 1. Этот эффект не был связан с чрезмерной токсичностью, так как среднее изменение веса у животных в этой группе, получивших комбинацию соединения 1 и паклитаксела на день 29 (последний день измерений) относительно начала исследования, составило +3,1% (+/-1.2 SEM), по сравнению с величиной, равной +5.1%(+/-1,4 SEM) в группе, получившей носитель. Результаты показаны на фиг. 2. Для изучения возможных взаимодействий между лекарствами соединением 1 и паклитакселом проводили in vivo фармакокинетическое исследование. Самки мышей Crl:CD-1-nuBR (голых) в возрасте от семи до восьми недель были получены из Charles River Laboratories (Wilmington, Massachusetts, USA). Животных помещали по 4-5 штук в клетку в микроизоляторы с циклом 12 ч/12 ч свет/темнота, животные проходили акклиматизацию в течение по меньшей мере 1 нед до использования, их кормили нормальным лабораторным кормом ad libitum. Животным (3/в момент времени) вводили i.v. один раз 50 мг/кг веса одного соединения 1, 10 мг/кг веса одного паклитаксела или комбинацию 50 мг/кг веса соединения 1 и 10 мг/кг веса паклитаксела. Отбирали образцы крови в разные моменты времени (0,083, 0,25, 0,5, 1, 2,4, 6, 8, 24 ч), получали плазму крови и методом HPLC определяли концентрации соединения 1 и паклитаксела. Как видно из табл. 3, соединение 1 не оказывает значительного влияния на период полужизни в плазме (t1/2), величину пика концентрации в плазме (Cmax) и общее пребывание в плазме (AUCinf) паклитаксела. Результаты показаны на фиг. 3. Точно так же паклитаксел не оказывает значительного действия на период полужизни в плазме (t1/2), величину пика концентрации в плазме (Cmax) и общее пребывание в плазме (AUCinf) соединения 1. Результаты показаны на фиг. 4. Таблица 3 Пример 2. In vitro комбинированный анализ соединения 1 и паклитаксела. А. Клетки и клеточная культура. Человеческие клетки карциномы немелкоклеточного рака легкого (H1975) из коллекции AmericanType Culture Collection выращивали в модифицированной по методу Дульбекко среде Игла, содержащей 4 мМ L-глутамина, антибиотики (100 м.е./ мл пенициллина и 100 мкг/мл стрептомицина) и 10% фетальной телячьей сыворотки фирмы Sigma Aldrich. Клетки субкультивировали при отношении от 1:3 до 1:6 от двух до трех раз в неделю. Кривые роста клеток получали при использовании 96-луночных планшетов со стенками черного цвета и с прозрачным дном для обеспечения фазы логарифмического роста во время четырехдневного анализа, описанного ниже. Для этого клетки высевали с несколькими различными величинами плотности на день 0 и определяли величину общего чистого прироста на день 4 в сравнении с днем 0 с помощью витального красителя alamarBlue. Было определено, что оптимальное количество в этом 4-дневном исследовании составляет 2000 кл/лунку. В. Изучение комбинации паклитаксела и соединения 1. Величина половинной максимальной ингибирующей концентрации (IC50) для паклитаксела и соединения 1 была определена с помощью трехкратных серийных разведений соединения, начиная с наибольшей концентрации, равной 1 мкМ. После действия любого лекарства в течение 72 ч определяли жизнеспособность клеток с помощью витального красителя alamarBlue, эти данные использовали для определения величин IC50 при помощи компьютерной программы XLFit software (ID Business Solutions). Величина IC50 для соединения 1 в клетках H1975 рассчитывалась при длине волны 15 нМ; для паклитак- 15022119 села величину IC50 определяли при длине волны, равной 7 нМ (фиг. 5). Затем использовали комбинацию паклитаксела и соединения и анализировали ее действие методом медианного теста получаемого эффекта. Лекарства соединяли или при их эквипотентном отношении,основываясь на мольных концентрациях IC50 для каждого агента, или в неравных отношениях. Клетки инкубировали в течение 3 дней в присутствии комбинации лекарств. Определяли фракцию выживших клеток в сравнении с контрольным образцом, используя метод анализа выживаемости с применением витального красителя alamarBlue. Комбинационный индекс (CI) был определен с помощью медианного теста с использованием компьютерной программы CalcuSyn 2.0 (CalcuSyn, Inc.) для концентраций комбинации, 20-70% клеток были поражены (а именно убиты). Величины комбинационного индекса более 1,равные 1 или менее 1, показывают наличие антагонизма, аддитивности и синергизма соответственно. Как показано на фиг. 6 и в табл. 4, комбинация соединения 1 и паклитаксела оказалась синергичной, когда концентрации обоих соединений были менее чем IC50, но более чем IC20. Таблица 4 Величины комбинационного индекса при совместном лечении паклитакселом и соединением 1 Химиотерапевтические агенты, такие как паклитаксел и другие таксаны, проявляют цитотоксическое действие во время митоза и поэтому необходим рост клеток во время этой фазы клеточного цикла,чтобы этот эффект проявлялся. Из еще неопубликованной работы об изучении свойств соединения 1, а также из ранее опубликованных работ по изучению других ингибиторов Hsp90 (Hexner, E.О. et al., Blood 111 (12), 5663 (2008, видно, что эти данные позволяют предположить, что ингибирование функцииHsp90 приводит к остановке клеточного цикла. Соответственно был проведен опыт для изучения влияния последовательной обработки клеток соединениями (вначале паклитакселом и затем соединением 1) на эффективность комбинации по сравнению с совместным их введением. Клетки H1975 были обработаны 5 нМ паклитаксела в течение 24 ч при 37 С, промыты для удаления лекарства и затем обработаны различными количествами соединения 1 в течение 24 ч. Клетки снова промывали средой, инкубировали еще в течение 24 ч и затем проводили определение жизнеспособности при помощи витального красителя alamarBlue. На фиг. 7 и в табл. 5 показано, что обработка паклитакселом перед введением соединения 1 опять привела к появлению синергического эффекта Таблица 5 Величины комбинационных индексов при последовательном введении паклитаксела и соединения 1 Пример 3. In vitro анализ комбинации соединения 1 и таксанов. А. Материалы и методы. Клеточные линии. Человеческие клетки карциномы немелкоклеточного рака NCI-H1975 из коллекции American TypeCulture Collection выращивали в модифицированной по методу Дульбекко среде Игла, содержащей 4 мМL-глутамина, антибиотики (100 м.е./ мл пенициллина и 100 мкг/мл стрептомицина) и 10% фетальной телячьей сыворотки фирмы Sigma Aldrich. Человеческие клетки HEL92.1.7 эритролейкоза (из коллекции АТСС) выращивали в среде RPMI, содержащей 2 мМ L-глутамина, антибиотики (100 м. ед./мл пенициллина и 100 мкг/мл стрептомицина) и 10% фетальной телячьей сыворотки. Человеческие клетки рака толстой кишки НТ 29 (АТСС) были выращены в модифицированной среде McCoy's, содержащей 10% фетальной телячьей сыворотки и антибиотики (100 м.е./мл пенициллина и 100 мкг/мл стрептомицина). Все клетки выдерживали при температуре 37 С, в атмосфере с 5% СО 2, и пассажи проводили при отношении от 1:3 до 1:6 от двух до трех раз в неделю. Определение жизнеспособности клеток. Жизнеспособность клеток определяли при помощи витального красителя alamarBlue. Вкратце, это метод осуществляли следующим образом. Клетки помещали в 96-луночные планшеты в трех повторах в количестве 2000 кл/лунку (H1975) или 5000 кл/лунку (HEL92.1.7) и инкубировали при температуре 37 С в присутствии 5% СО 2 в течение 24 ч до добавления лекарства или носителя (0,3% DMSO) к культуральной среде. Через 72 ч в лунки добавляли 10 мкл/лунку alamarBlue и проводили инкубацию еще в течение 3 ч при температуре 37 С в присутствии 5% СО 2. Измеряли интенсивность флуоресценции (560EX/590EM нМ) при помощи считывающего устройства для планшетов SpectraMax microplate reader (Molecular Devices),полученные данные использовали для определения жизнеспособности клеток, нормализовав их по контрольному образцу. В. Изучение действия комбинации паклитаксела и соединения 1. Величина половинной максимальной ингибирующей концентрации (IC50) для таксанов (доцетаксела или паклитаксела) или соединения 1 была определена с помощью трехкратных серийных разведений соединения, начиная с наибольшей концентрации, равной 1 мкМ. После действия любого лекарства в течение 72 ч определяли жизнеспособность клеток с помощью витального красителя alamarBlue, эти данные использовали для определения величин IC50 при помощи компьютерной программы XLFit software (ID Business Solutions), они приведены в табл. 6. Таблица 6 Величины IC50 (нМ) для соединения 1 таксанов (н/о=не определяли) Изучение действия комбинации таксанов и соединения 1 проводили на основе IC50 для каждого агента. Конкретно, серийные разведения в отношении 1:1,2, начиная с IC50 паклитаксела или доцетаксела, смешивали с подобными разведениями на основе IC50 соединения 1. Комбинации лекарств, а также каждое отдельное лекарство инкубировали с клетками в течение 3 дней и определяли фракцию выживших клеток по сравнению с контрольным образцом с помощью витального красителя alamarBlue. Комбинационные индексы (CI) определяли, применяя метод медианного анализа с помощью компьютерной программы CalcuSyn 2.0 (CalcuSyn, Inc.). Величины комбинационного индекса более 1, равные 1 или менее 1 показывают наличие антагонизма, аддитивности и синергизма соответственно. Таблица 7 Описание величин комбинационных индексов На фиг. 8 показан типичный набор данных, полученных при проведении опыта с комбинацией лекарств. В этом случае результаты показывают количество клеток, пораженных действием соединения 1,паклитаксела или комбинации этих двух лекарств на примере клеток H1975. Эти данные затем применяли для построения линейной кривой с помощью компьютерной программы CalcuSyn и получения значений комбинационных индексов. На фиг. 9 а и 9b показано, что комбинация соединения 1 с таксаном (или с доцетакселом, или с паклитакселом) приводила к большей гибели клеток Н 1975 NSCLC, чем это ожидалось по их аддитивному действию при применении соответствующих доз (величины комбинационных индексов 1). Следовательно, эти комбинации являются синергическими. Когда в случае клеток эритролейкоза HEL92.1.7 применяли комбинацию любого из таксанов с соединением 1, наблюдался синергизм от сильного до очень сильного (см. фиг. 10). Доцетаксел приводил к появлению синергизма при введении вместе с соединением 1 в клетки НТ 29 карциномы толстой кишки (фиг. 11). Взятые вместе, эти данные показывают, что соединение 1 и таксаны действуют синергически при гибели раковых клеток. Пример 4. Комбинация соединения 1 и доцетаксела проявляет повышенную противоопухолевую активность в отношении человеческих опухолевых клеток в мышиной модели ксенографта SCID Mouse XenograftModel с клетками НСС 827 NSCLC. Мышам линии SCID имплантировали человеческие клетки НСС 827(EGFRDelE726-A750) немелкоклеточного рака легкого (NSCLC) точно так же, как описано в примере 1. Опухолям давали развиваться in vivo до тех пор, пока большинство из них не достигло объема, равного 95195 мм, и затем один раз в неделю вводили один носитель, 10 мл с концентрацией 75 мг/кг веса животного соединения 1, 4 мг/кг доцетаксела (приготовленного так же, как паклитаксел) или комбинацию этих двух агентов. Как видно на фиг. 12, комбинация 75 мг/кг соединения 1 плюс 4 мг/кг доцетаксела проявляла повышенную эффективность в сравнении с действием каждого агента в отдельности, с величинами% Т/С, равными 0, в отличие от величин, равных 46 и 26 для одного доцетаксела и одного соединения 1 соответственно. Этот эффект не связан с чрезмерной токсичностью и взаимодействие между лекарствами, соединением 1 и доцетакселом не наблюдалось (данные не приведены). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ лечения субъекта с раковым заболеванием, выбранным из рака легкого, карциномы толстой кишки и эритролейкоза, включающий введение субъекту эффективного количества противоопухолевого средства, представляющего собой паклитаксел или доцетаксел, и эффективного количества соединения, представленного следующей структурной формулой: или его таутомера, или фармацевтически приемлемой соли. 2. Способ лечения субъекта, больного раком легкого, карциномой толстой кишки или эритролейкозом, включающий введение субъекту эффективного количества противоопухолевого средства, представляющего собой паклитаксел или доцетаксел, и эффективного количества 3-(2,4-дигидрокси-5 изопропилфенил)-4-(1-метилиндол-5-ил)-5-гидрокси-[1,2,4]триазола, или его таутомера, или фармацевтически приемлемой соли. 3. Способ по любому из пп.1 и 2, в котором противоопухолевое средство представляет собой доцетаксел. 4. Способ по любому из пп.1-3, где рак представляет собой немелкоклеточный рак легкого. 5. Способ по любому из пп.1-3, где рак представляет собой рак толстой кишки. 6. Способ по любому из пп.1-3, где рак представляет собой эритролейкоз.
МПК / Метки
МПК: A61K 45/06, A61P 35/00, A61K 31/4196, A61K 31/337
Метки: раковых, соединений, ингибиторов, помощью, комбинированная, заболеваний, терапия, hsp90
Код ссылки
<a href="https://eas.patents.su/24-22119-kombinirovannaya-terapiya-rakovyh-zabolevanijj-s-pomoshhyu-soedinenijj-ingibitorov-hsp90.html" rel="bookmark" title="База патентов Евразийского Союза">Комбинированная терапия раковых заболеваний с помощью соединений – ингибиторов hsp90</a>
Комбинированная терапия с использованием производных 1- аминоциклогексана и ингибиторов ацетилхолинэстеразы

Номер патента: 8863
Опубликовано: 31.08.2007
Автор: Мебиус Ханс-Йорг
МПК: A61K 31/13, A61K 31/16, A61K 31/435...
Метки: использованием, ингибиторов, терапия, ацетилхолинэстеразы, аминоциклогексана, производных, комбинированная
Формула / Реферат:
1. Фармацевтический продукт для лечения деменции, связанной с нарушением центральной нервной системы, где лечение индуцирует улучшение когнитивной функции, содержащий терапевтически эффективные дозы производного 1-аминоциклогексана, выбранного из мемантина, нерамексана и их пролекарств, солей, изомеров, аналогов и производных, и ингибитора ацетилхолинэстеразы, выбранного из донепезила, ривастигмина, такрина, галантамина и физостигмина. 2....
Комбинированная противоопухолевая терапия, включающая применение производных замещенного акрилоилдистамицина и ингибиторов топоизомераз i и ii

Номер патента: 6684
Опубликовано: 24.02.2006
Авторы: Берия Итало, Джерони Мария Кристина Роза, Коцци Паоло
МПК: A61K 31/4025, A61K 45/00, A61P 35/00...
Метки: производных, замещенного, топоизомераз, ингибиторов, акрилоилдистамицина, включающая, применение, комбинированная, противоопухолевая, терапия
Формула / Реферат:
1. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель или наполнитель и в качестве активного ингредиента производное акрилоилдистамицина формулы (I) где R1 означает атом брома или хлора; R2 означает группу формулы (II) где m означает 0; n означает 4; r означает 0; X и Y, оба, означают CH-группу; B является группой где R5, R6 и R7, одинаковые или разные, означают водород или C1-C4алкил; или его фармацевтически...
Комбинированная терапия заболеваний нижних мочевыводящих путей с использованием лигандов α2δ и нестероидных противовоспалительных препаратов (нпвп)

Номер патента: 17171
Опубликовано: 30.10.2012
Авторы: Гварнери Лучано, Леонарди Амедео, Ангелико Патриция
МПК: A61P 13/10, A61K 45/06
Метки: использованием, мочевыводящих, лигандов, нпвп, противовоспалительных, заболеваний, нестероидных, нижних, препаратов, терапия, alpha;2&delta, путей, комбинированная
Формула / Реферат:
1. Применение лиганда α2δ-субъединицы (A2d) кальциевых каналов, выбранного из габапентина, прегабалина, (1R,5R,6S)-6-аминометил-6-карбоксиметилбицикло[3.2.0]гептана, 3-(1-аминометилциклогексилметил)-4Н-1,2,4-оксадиазол-5-она, 5-(1-аминометилциклогексилметил)-1Н-тетразола, (3S,4S)-1-аминометил-1-карбоксиметил-3,4-диметилциклопентана, (1α,3α,5α)-3-аминометил-3-карбоксиметилбицикло[3.2.0]гептана,...
Комбинированная терапия астмы и copd

Номер патента: 21917
Опубликовано: 30.09.2015
Авторы: Усберти Франческа, Цамбелли Энрико, Бонелли Сауро
МПК: A61K 45/06, A61K 31/167, A61K 31/40...
Метки: астмы, терапия, комбинированная
Формула / Реферат:
1. Фармацевтическая композиция, содержащая:(а) гликопиррония бромид в дозировке в диапазоне 0,5-100 мкг на активацию и(б) формотерол или его соль в дозировке в диапазоне 1-25 мкг на активацию;растворенные в HFA (гидрофторалкановом) пропелленте и сорастворителе, отличающаяся тем, что указанная композиция содержит 1 М HCl в количестве в диапазоне 0,1-0,3 мкг/мкл.2. Композиция по п.1, где диапазон 1 М HCl составляет 0,15-0,28 мкг/мкл.3....
Комбинированная терапия для лечения психозов

Номер патента: 1317
Опубликовано: 26.02.2001
Авторы: Толлефсон Гари Д., Перри Кеннет В., Баймастер Фрэнклин П.
МПК: A61K 31/551
Метки: лечения, психозов, терапия, комбинированная
Формула / Реферат:
1. Способ лечения пациента, страдающего от психоза или подверженного психозу, острому маниакальному синдрому, состояниям легкой тревожности и страха или депрессии в сочетании с эпизодами психотического характера, включающий введение указанному пациенту эффективного количества первого компонента, который представляет собой атипическое нейролептическое средство, в сочетании с эффективным количеством второго компонента, который представляет собой...
Предыдущий патент: Ингибиторы протеазы hcv
Следующий патент: Аналоги гетероциклил пиразолопиримидина в качестве ингибиторов jak
Случайный патент: Пестицидные тиазолилоксизамещенные фениламидиновые производные