Производные 2h-1-бензопирана и содержащая их фармацевтическая композиция
Номер патента: 6158
Опубликовано: 27.10.2005
Авторы: Амари Габриеле, Дельканале Маурицио, Чивелли Маурицио, Армани Элизабетта, Гальбиати Элизабетта







Формула / Реферат
1. Производные 2H-1-бензопирана общей формулы

где X представляет собой H, галогено или O-R3, где
R3 представляет собой H, C1-4алкил или C1-5алканоил,
Y представляет собой H, C1-4алкил или C1-5алканоил,
Z представляет собой H или O-R4, где R4 представляет собой H, C1-4алкил, или C1-5алканоил,
m равно 1 или 2,
n равно 0 или 1,
-----представляет собой одинарную или двойную связь между атомами в положениях 3-4 или 4-4a, когда n равно 1, двойная связь может быть альтернативно экзоциклической или эндоциклической, образуя соответственно 4-бензилиденхроман при m=1 или 4-бензил-хром-3-ен при m=2, когда n равно 0,
-----представляет собой одинарную или двойную связь в 3-4, и Z не является H,
и их фармацевтически приемлемые соли.
2. Производные 2H-1-бензопирана по п.1,
где X представляет собой H или C1-C4алкокси,
Y представляет собой H или C1-C5алканоил,
Z представляет собой H,
m равно 2,
n равно 1,
связь между атомами в положении 3-4 представляет собой двойную связь, а связь между атомами в положении 4-4a представляет собой одинарную связь.
3. Соединение по пп.1 и 2, которое представляет собой 3-фенил-4-(4-(2-(N-пиперидинил)этокси)бензил)-7-гидроксихром-3-ен, его сложные эфиры и соли.
4. Соединение по пп.1 и 2, которое представляет собой 3-(4-метокси)фенил-4-[[4-[2-(1-пиперидинил)этокси]фенил]метил]-2H-1-бензопиран-7-ол, его сложные эфиры и соли.
5. Фармацевтические композиции, содержащие в качестве активного ингредиента по меньшей мере одно соединение по пп.1-4 вместе с фармацевтически приемлемыми эксципиентами.
6. Применение соединений по пп.1-4 для приготовления лекарства, полезного при предупреждении и лечении ряда постменопаузальных патологий, в частности остеопороза, коронарной болезни сердца и эстроген-зависимого рака человека.
Текст
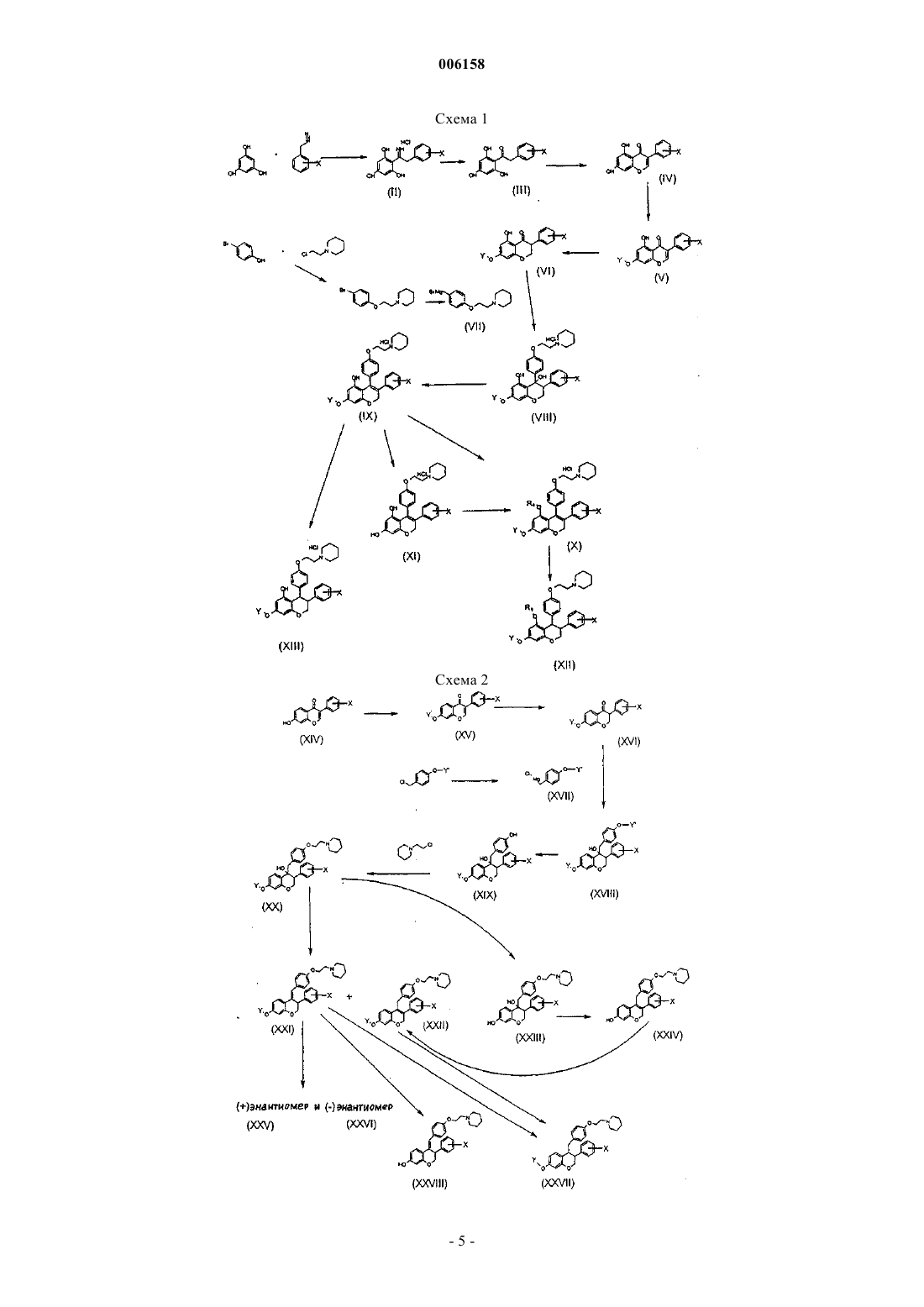
006158 Настоящее изобретение относится к производным 2 Н-1-бензопирана, способам их получения и их фармацевтическим композициям. Точнее, изобретение относится к производным 2 Н-1-бензопирана общей формулы где R1 и R2 независимо представляют собой Н, алкил, галогеноалкил, алкенил или галогеноалкенил, или вместе с атомом азота, с которым они связаны, могут образовывать 4-8-членное гетероциклическое кольцо,X представляет собой Н, алкил, арил, нитро, галогено, O-R3, где R3 представляет собой Н, алкил,арил, алканоил, арилоил,X1 представляет собой Н, алкил или алкокси, и, когда X и X1 представляют собой алкил, они могут образовывать вместе с атомом углерода, с которым они связаны, конденсированное ароматическое кольцо с образованием -нафталенила,Y представляет собой Н, алкил, алканоил, арилоил, алкиламинокарбонил, алкилоксикарбонил,Z представляет собой Н, O-R4, где R4 представляет собой Н, алкил, алканоил, арилоил,m равно 1, 2,n равно 0, 1,р равно 2-6,представляет собой одинарную или двойную связь между атомами в положениях 3-4 или 4-4 а: когда n равно 1, двойная связь может быть альтернативно экзоциклической или эндоциклической, образуя соответственно 4-бензилиденхроман при m=1 или 4-бензил-хром-3 ен при m=2; когда n равно 0, эндоциклическая связь 3-4 может быть одинарной или двойной связью, и,когда n равно 0, а эндоциклическая связь 3-4 представляет собой одинарную связь, то Z не является Н. Соединения по изобретению могут существовать в форме солей. Соединения, несущие основные группы, могут существовать в форме солей присоединения органических или неорганических кислот. Соединения, несущие кислотные группы, могут существовать в форме солей присоединения щелочного или щелочно-земельного металла или органических аминов. Когда в соединениях формулы (I) связь между 3 и 4 положениями бензопиранового кольца представляет собой одинарную связь, два атома углерода в 3 и 4 положениях кольца могут быть асимметрическими, и соединения могут существовать в виде чистого энантиомера, смеси диастереоизомеров или рацематов. Алкил предпочтительно представляет собой С 1-С 4 алкил, например, метил, этил, н-пропил, изопропил, трет-бутил. Галогеноалкил предпочтительно представляет собой С 1-С 4 алкил, замещенный одним - пятью атомами фтора или хлора, например, трифторметил. Алкенил предпочтительно представляет собой С 2-С 4 алкенил, например, этенил или аллил. Галогеноалкенил предпочтительно представляет собой С 2-С 4 алкенил, замещенный одним - тремя атомами галогена. Арил предпочтительно представляет собой фенил или нафтил, возможно замещенный одним - тремя заместителями, которые являются одинаковыми или различными, выбранными из атомов галогена,алкильных, алкокси-, нитро-, циано-, трифторметильных, амино-, гидроксигрупп. Алканоил предпочтительно представляет собой С 1-С 4 алканоильную группу, например группу формил, ацетил или пропионил. Арилоил предпочтительно представляет собой бензоил, возможно замещенный одним - тремя заместителями, которые являются одинаковыми или различными, выбранными из атомов галогена, групп алкил, алкокси, нитро, циано, трифторметил, амино, гидрокси. Предпочтительные соединения формулы (I) представляют собой соединения, где R1 и R2, взятые вместе с атомом азота, образуют пиперидиновое или пирролидиновое кольцо, Y представляет собой водород или группу алканоил,Z представляет собой водород, гидрокси или алкокси, n равно 0 или 1, р равно 2, X и X1 представляют собой водород, или один представляет собой водород, а другой представляет собой галоген или группу OR3, где R3 является таким, как указано выше, предпочтительно в пара-положении.-1 006158 В предпочтительном воплощении изобретение относится к производным 2 Н-1-бензопирана формулы (IA) где X представляет собой Н, С 1-С 4 алкокси, и Y представляет собой Н, С 1-С 4 алкил, С 1-С 5 алканоил,арилоил. Арилоил предпочтительно представляет собой бензоил, возможно замещенный одним - тремя заместителями, которые являются одинаковыми или различными, выбранными из атомов галогена, групп алкил, алкокси, нитро, циано, трифторметил, амино, гидрокси. Соединения могут существовать в форме солей присоединения органических или неорганических кислот или солей присоединения щелочного или щелочно-земельного металла или органических аминов. Соединения формулы (I) принадлежат к классу селективных модуляторов рецепторов эстрогена(SERM), которые связываются и взаимодействуют с рецептором эстрогена, но которые действуют в определенных тканях, таких как кость, в качестве агонистов эстрогена, а в других тканях, и в частности в груди и матке, в качестве антагонистов эстрогена.SERM сохраняют благоприятные воздействия эстрогена без некоторых его побочных действий. Уровень техники В WO 94/20098 (Zymogenetics) описаны 3,4-диарил-2 Н-1-бензопираны для применения при снижении костной потери и, в частности, костной потери, связанной с остеопорозом. Особенно предпочтительным соединением для применения в рамках изобретения Zymogenetics является 1-энантиомер центхромана, впоследствии разработанный фирмой Novo Nordisk в виде левормелоксифена (levormeloxifene) для лечения остеопороза. В дальнейшем левормелоксифен используют в качестве эталонного соединения. Производные замещенного бензопирана и тиобензопирана, обладающие потенциальной антиэстрогенной активностью, были описаны позднее в WO 98/25916 и WO 99/65893 (СС Research Laboratories). Соединения по изобретению отличаются по своей химической структуре от бензопиранов, раскрытых в предшествующем уровне техники- наличием двойной связи между атомами в положении 3-4 или 4-4 а, альтернативно эндо- или экзоциклической;- замещением атома углерода в 4 положении бензопиранового кольца третаминоалкилоксибензильным или бензилиденильным радикалом;- наличием гидроксигруппы в 5 положении, когда заместитель на атоме углерода в 4 положении представляет собой трет-аминоалкилоксифенильный радикал. Эти различия в структуре бензопирана и, в частности, наличие метиленового или метилиденового мостика между атомом углерода в положении 4 бензопиранового кольца и атома углерода фенильной группы представляют основное значение для фармакологической активности соединений по изобретению. Ралоксифен, нестероидное производное бензотиофена, который описан в Jones C.D. et al. J. Med.Chem. 27, 1057-1059, 1984, оказывающий тканеспецифичное эстроген-агонистическое и антагонистическое действие, разрешен для клинического применения в США и некоторых европейских странах. Соединения по изобретению также выигрывают при сравнении с ралоксифеном. Другие синтетические соединения с данным возможным спектром активностей, включая трифенилэтилен и дигидронафталин, были описаны как SERM. Однако было показано, что разработка многих из этих соединений в качестве лекарственных средств является проблематичной вследствие чрезмерного раздражения ими ткани матки. Новые SERM по изобретению оказывают значительные благоприятные воздействия на костную ткань и уровни липидов в сыворотке, и антагонистические воздействия, связанные с низкой степенью внутренней эстрогенной активности на репродуктивную ткань. Вследствие своих тканеспецифичных эстроген-агонистических и антагонистических свойств соединения по изобретению могут быть использованы в терапии, в частности, для предупреждения и лечения ряда постменопаузальных патологий, в частности остеопороза, коронарной болезни сердца и эстроген-зависимого рака человека. Согласно настоящему изобретению также предложен способ получения соединения формулы (I)R1 и R2 независимо представляют собой Н, алкил, галогеноалкил, алкенил или галогеноалкенил, или вместе с атомом азота, с которым они связаны, могут образовывать 4-8-членное гетероциклическое кольцо,X представляет собой Н, алкил, арил, нитро, галогено, O-R3, где R3 представляет собой Н, алкил,арил, алканоил, арилоил,X1 представляет собой Н, алкил или алкокси, и, когда X и X1 представляют собой алкил, они могут образовывать вместе с атомом углерода, с которым они связаны, конденсированное ароматическое кольцо с образованием -нафталенила,Y представляет собой Н, алкил, алканоил, арилоил,Z представляет собой Н, O-R4, где R4 представляет собой Н, алкил, алканоил, арилоил,m равно 1,2,n равно 0, 1,р равно 2-6,представляет собой одинарную или двойную связь между атомами в положениях 3-4 или 4-4 а: когда n равно 1, двойная связь может быть альтернативно экзоциклической или эндоциклической, образуя соответственно 4-бензилиденхроман при m=1 или 4-бензил-хром-3-ен при m=2; когда n равно 0, эндоциклическая связь 3-4 может быть одинарной или двойной связью, и, когда n равно 0, а эндоциклическая связь 3-4 представляет собой одинарную связь, то Z не является Н. На схемах (1) и (2) представлены способы получения 2-(N-пиперидинил)этоксисоединений формулы (I). Эти схемы реакций могут также быть использованы для других трет-аминоалкилоксианалогов формулы (I). Соединения формулы (I), где Z=OR4, могут быть получены согласно схеме (I) посредством взаимодействия 1,3,5-тригидроксибензола с замещенным фенилацетонитрилом в апротонном растворителе, насыщенном газообразным HCl, с получением кетиминов формулы (II), которые могут быть гидролизованы в кипящей воде с получением соответствующих кетонов формулы (III), которые могут быть подвергнуты циклизации с использованием диметилацеталя N,N-диметилформамида и диэтилэфирата трифторида бора в диметилформамиде (ДМФ) с получением соединений формулы (IV). Соединения формулы (IV) могут быть защищены в положении 7 с помощью, например, соответствующего ангидрида с растворителем или без него при температуре от 0 С до точки кипения растворителя или чистого ангидрида, или с помощью соответствующего галогенида в апротонном растворителе и в присутствии основания, с получением эфира формулы (V), который должен быть стабильным, когда его подвергают реакции Гриньяра. Полученные соединения формулы (V) можно гидрировать в присутствии подходящего катализатора с получением соединений формулы (VI). Добавление реактивов Гриньяра формулы (VII), полученных из 4-(трет-аминоалкилокси)-1 бромбензола и Мg в апротонном растворителе, к кетонам формулы (VI) при температуре от -70 С до точки кипения растворителя приводит к получению спирта (VIII), который может быть дегидратирован,например, в этанольном растворе концентрированной HCl с получением соединений формулы (IX). Соединения формулы (IX) могут быть алкилированы или ацилированы с помощью алкил- или ацилгалогенида в соответствующем растворителе и в присутствии основания или могут быть ацилированы с помощью ацилангидрида при высокой температуре и в отсутствие растворителя с получением соединений формулы (X), где Y (отличный от R4) представляет собой защитную группу, выбранную из групп, которые устойчивы к реакции Гриньяра. С соединений формулы (IX) защита может быть удалена,например, при помощи концентрированной HCl или концентрированной НВr при высокой температуре или, когда Y=ацил, при помощи K2 СО 3 в водном метаноле при комнатной температуре с получением соответствующих свободных фенолов формулы (XI), которые могут быть диалкилированы, например, с помощью алкилгалогенида в соответствующем растворителе и в присутствии основания или могут быть диацилированы, например, с помощью ацилгалогенида в соответствующем растворителе и в присутствии основания или с помощью ацилангидрида с растворителем или без него с получением соединений-3 006158 формулы (X), где Y представляет собой R4, но является отличным от Н; соединения формулы (X) могут быть гидрированы в подходящем растворителе и в присутствии катализатора с получением соответствующих соединений формулы (XII), где Y представляет собой R4 или Y отличен от R4, и где Y может быть отличен от групп, выбранных в качестве защитной группы в реакции Гриньяра. Соединения формулы (IX) могут быть также гидрированы с получением соединений формулы(XIII). Соединения формулы (I), где Z представляет собой Н, можно получить, следуя схеме (2). Соединение (XIV) защищали по фенольной гидроксигруппе при помощи соответствующей защитной группировки (Y), выбранной из тех, которые описаны в литературе для защиты фенолов (смотри,например, T.W. Greene, P.G. Wuts "Protective groups in organic synthesis", 3rd Ed, John WileySons, Inc.,1999, pages 246-292) с получением соединения (XV). Защитная группа должна быть выбрана из групп,которые стабильны в условиях последующих стадий. Соединение (XV) затем избирательно гидрировали по алкеновой двойной связи, в некотором смысле чтобы избежать восстановления кетона до спиртов,посредством каталитического гидрирования с использованием соответствующего катализатора (содержащего Pd, Pt, Rh, Ru или другие переходные металлы) в соответствующем растворителе (спирты, тетрагидрофуран (ТГФ), ацетон, диоксан, этилацетат, смеси спирты-вода и так далее) или посредством химического восстановления гидридом в соответствующем апротонном растворителе с получением соединения (XVI). Соединение (XVI) подвергали взаимодействию с соединением (XVII) (полученным посредством взаимодействия защищенного 4-гидроксибензилхлорида с Мg в подходящем растворителе, таком как диэтиловый эфир, ТГФ или другие эфирные растворители при температуре между комнатной и температурой кипения растворителя) с получением соединения формулы (XVIII). Защитная группа (Y') для 4-гидроксибензилхлорида должна быть выбрана из тех же групп, которые описаны выше, но, как правило, отличных от Y, выбранных для соединения (XV). Избирательное удаление этой второй защитной группы (Y') привело к получению соединения (XIX). Соединения формулы (XIX) могут быть алкилированы галогеноалкиламином в подходящем растворителе и в присутствии основания с получением соединений формулы (XX). Соединения формулы (XX) могут быть дегидратированы (например, водной HCl в ацетонитриле при комнатной температуре) с получением смеси соединений формул (XXI) и (XXII), или с соединений формулы (XX) может быть удалена защита (когда Y=ацил) в мягких условиях, например, при помощиK2 СО 3 в водном метаноле при комнатной температуре с получением соединений формулы (XXIII). (XXI) могут быть выделены из смеси посредством кристаллизации, тогда как (XXII) могут быть очищены посредством хроматографических методов. Чистые энантиомеры соединений формулы (XXI) могут быть получены посредством хиральной хроматографии с получением соединений формулы (XXV) и (XXVI). Соединения формулы (XXI) и (XXII) могут быть гидрированы с получением соединений формулы(XXVII). Соединения формулы (XXIII) могут быть дегидратированы, например, водной HCl в ацетонитриле с получением соединений формулы (XXIV), которые могут быть ацилированы или алкилированы, например, ацил- или алкилхлоридом в подходящем растворителе и в присутствии основания с получением соединений формулы (XXII), в которых Y может отличаться от группы, выбранной в качестве защитной группы в реакции Гриньяра. При соответствующем выборе Y возможно получить соединение (XXIV) непосредственно из соединения (XX) путем кислотного удаления защитной группы Y и одновременно гидролиза третичного спирта. Соединения формулы (XXI), где Y=ацил, могут быть гидролизованы, например, при помощи K2 СО 3 в водном метаноле с получением соответствующих фенолов формулы (XXVIII), и они могут быть алкилированы или ацилированы с помощью алкилирующего или ацилирующего агента с получением соединений формулы (XXI), в которых Y может отличаться от группы, выбранной в качестве защитной группы в реакции Гриньяра. Предпочтительная защитная группа представляет собой пивалоил-(2,2-диметилпропаноил).-5 006158 Следующие примеры дополнительно иллюстрируют изобретение. Пример 1. Получение гидрохлорида 2,4,6-тригидроксифенилбензилкетимина Раствор флороглюцина (60 г) в диэтиловом эфире (400 мл), охлажденный при 5 С, добавляли к раствору фенилацетонитрила (50,1 г) и диэтилэфирата трифторида бора (4,7 мл) в HCl/ETOAc5 М (800 мл), полученном посредством барботирования газообразного HCl в охлажденном этилацетате. Раствор хранили при 5 С в течение 3 дней. Кристаллический желтый осадок отфильтровывали,промывали диэтиловым эфиром (50 мл) и использовали без дополнительной очистки. Получали 95 г продукта (выход = 80%). Тонкослойная хроматография (ТСХ): хлороформ/метанол = 90/10; Rf = 0,7. Пример 2. Получение 2,4,6-тригидроксифенилбензилкетона Гидрохлорид 2,4,6-тригидроксифенилбензилкетимина (95 г) суспендировали в воде ( 2000 мл) и нагревали при 90 С при перемешивании в течение 1 ч. После охлаждения получали кристаллический продукт. Твердое вещество отфильтровывали, промывали водой (200 мл) и сушили под вакуумом при 60 С. Получали 70 г продукта (выход = 85%). ТСХ: хлороформ/метанол = 90/10; Rf = 0,8. Пример 3. Получение 5,7-дигидроксиизофлавона Раствор диэтилэфирата трифторида бора (228 мл) и диметилацеталя N,N-диметилформамида (84 г) в N,N-диметилформамиде (715 мл) добавляли к раствору соединения из примера 2 (55 г) в N,Nдиметилформамиде (1400 мл), нагретому при 50 С. Смесь нагревали при 95 С в течение 60 мин. Темно-оранжевый раствор охлаждали при комнатной температуре, выливали в холодную воду (10 л) и оставляли без перемешивания в течение ночи. Твердое розовое вещество отфильтровывали, промывали водой (1 л) и сушили под вакуумом при 70 С. Получали 40,3 г продукта (выход = 70,4%). ТСХ: гексан/этилацетат = 70/30; Rf = 0,55. Пример 4. Получение 7-пивалоилокси-5-гидроксиизофлавона Соединение из примера 3 (10 г) и пивалиновый ангидрид (50 мл) нагревали при 120 С при перемешивании магнитной мешалкой в закрытой колбе в течение 4 ч. Густой красный раствор хранили при комнатной температуре без перемешивания в течение ночи. Осадок отфильтровывали, промывали петролейным эфиром и сушили под вакуумом при 45 С. Получали 8,9 г твердого розового вещества (выход = 65%). ТСХ: метиленхлорид/метанол = 95/5; Rf = 0,8. Пример 5. Получение 3-фенил-5-гидрокси-7-пивалоилоксихроман-4-она К раствору соединения из примера 4 (8,7 г) в ацетоне (300 мл) добавляли 5% Pd/C (9 г) (содержание воды = 50%) и гидрировали при 40 ф/кв. дюйм (275,8 кПа) в аппарате Парра в течение 2,5 ч. Катализатор отфильтровывали на подушке из целита и фильтрат упаривали досуха. Маслянистый остаток растворяли в диэтиловом эфире, сушили (Nа 2SO4) и снова упаривали. Получали 8 г оранжевого масла (выход = 90%). Его использовали без дополнительной очистки. К раствору бромфенола (10 г) в диметилформамиде (160 мл) добавляли K2 СО 3 (20,7 г) и нагревали при 100 С при перемешивании в течение 10 мин. Гидрохлорид N-(2-хлорэтил)пиперидина (9,6 г) добавляли в течение 10 мин. Смесь нагревали и перемешивали в течение 2 ч, затем охлаждали при комнатной температуре, выливали в воду (300 мл) и экстрагировали этилацетатом (300 мл). Органическую фазу экстрагировали 3% HCl (2x200 мл). После алкалинизации водную фазу экстрагировали этилацетатом (300 мл); выпаривание растворителя привело к получению 13 г продукта в виде оранжевого масла. ТСХ: метиленхлорид/метанол/уксусная кислота = 70/20/10; Rf = 0,7. Пример 7. Получение 4-(2-(N-пиперидинил)этокси)фенилмагнийбромида Магний (2,1 г) загружали в четырехгорлую колбу на 500 мл, снабженную конденсатором, капельной воронкой и магнитной мешалкой, в атмосфере N2. Добавляли 5 мл раствора соединения из примера 6 (11,8 г) в свежедистиллированном тетрагидрофуране (ТГФ) (50 мл), затем смесь нагревали до температуры дефлегмации. Оставшийся раствор по каплям добавляли в кипящую реакционную смесь в течение 40 мин. Смеси давали достичь комнатной температуры, затем мутный серый раствор использовали на следующей стадии. Пример 8. Получение гидрохлорида 3-фенил-4-(4-(2-(N-пиперидинил)этокси)фенил)-4,5 дигидрокси-7-пивалоилоксихромана Раствор Гриньяра, полученный на предыдущей стадии, быстро добавляли к раствору соединения из примера 5 (2,6 г) в безводном ТГФ (20 мл) при перемешивании. Температуру повышали до 40 С. После перемешивания в течение 1,5 ч добавляли воду (10 мл) и смесь упаривали досуха под вакуумом. Неочищенный продукт растворяли в этилацетате (300 мл) и нерастворимое вещество отфильтровывали. Органический раствор промывали 0,25 н. HCl (200 мл) (растворимость соли гидрохлорида продукта выше в этилацетате, чем в воде) и концентрировали до небольшого объема, пока не появлялся осадок. После обработки ультразвуком твердое вещество отфильтровывали и сушили на воздухе. Получали 1,6 г продукта в виде белого порошка (выход 36%). ТСХ: метиленхлорид/метанол = 90/10; Rf = 0,3. Точка плавления: 220-221 С. ЯМР: соответствует. Пример 9. Получение гидрохлорида 3-фенил-4-(4-(2-(N-пиперидинил)этокси)фенил)-5-гидрокси-7 пивалоилоксихром-3-ена Соединение из примера 8 (0,71 г) растворяли в кипящем этиловом спирте (40 мл). Раствор охлаждали при комнатной температуре и добавляли 37% HCl (0,2 мл). После выдерживания при комнатной температуре в течение 2 ч раствор упаривали досуха, и твердое пенистое вещество кристаллизовалось из ацетона. Полученное твердое белое вещество отфильтровывали, промывали ацетоном и сушили под вакуумом при 35 С. Получали 550 мг (выход 80%). Точка плавления: 229-230 С. 470 мг соединения из примера 9 суспендировали в ацетоне (50 мл), добавляли Н 2 О (2 мл) и прозрачный раствор гидрировали в аппарате Парра при 40 ф/кв. дюйм (275,8 кПа) с использованием 5% Pd/C(2 г; 50% Н 2 О) в течение 3 ч. После отфильтровывания катализатора раствор упаривали и полученное белое твердое пенистое вещество растворяли в небольшом количестве МеОН и осаждали посредством добавления Et2O. Получали 230 мг соединения. ТСХ: метиленхлорид/метанол/триэтиламин = 90/10/0,5 Точка плавления: 260-270 С (с разложением). ЯМР: соответствует. Пример 11. Получение гидрохлорида 3-фенил-4-(4-(2-(N-пиперидинил)этокси)фенил)-5,7 дигидроксихром-3-ена 370 мг соединения из примера 9 растворяли в горячем ТГФ (20 мл), затем добавляли 130 мг LiAlH4 и смесь перемешивали в течение 30 мин. Осторожно добавляли воду (170 мг), серое твердое вещество отфильтровывали и раствор подкисляли HCl/EtOAc. Смесь упаривали, остаток растворяли в ЕtOН (2 мл) и обрабатывали ультразвуком. В осадок выпадало белое твердое вещество. Его отфильтровывали, промывали Et2O и сушили при 50 С под вакуумом. Получали 210 мг соединения. Точка плавления: 248-251 С (с разложением). ТСХ: метиленхлорид/метанол/триэтиламин = 95/5/1; Rf = 0,25. ЯМР: соответствует. Пример 12. Получение 4',7-дипивалоилокси-5-гидроксиизофлавона Генистеин (10 г) и пивалиновый ангидрид (75 мл), имеющиеся в продаже или полученные известными способами, нагревали при 110 С в течение 22 ч, затем коричневый раствор охлаждали и оставляли при комнатной температуре в течение ночи. Образовывалось небольшое количество твердого вещества; петролейный эфир (150 мл) добавляли к смеси, затем полученное в большом количестве твердое вещество отфильтровывали, промывали петролейным эфиром и сушили под вакуумом при 45 С. Получали 11,6 г соединения, указанного в заголовке. ТСХ: петролейный эфир/ЕtOАс = 85/15; Rf = 0,5. Соединение из примера 12 (10 г) растворяли в ацетоне (750 мл) и гидрировали при 40 ф/кв. дюйм(275,8 кПа), используя 5% Pd/C (10 г; 50% содержания воды) в качестве катализатора. Через 3 ч катализатор отфильтровывали и раствор упаривали до получения масла, которое кристаллизовалось из петролейного эфира (150 мл). Получали 8,7 г соединения, указанного в заголовке. ТСХ: петролейный эфир/ЕtOАс = 85/15; Rf = 0,5. Пример 14. Получение гидрохлорида 3-(4-пивалоилоксифенил)-4-(4-(2-(N-пиперидинил)этокси)фенил)-4,5-дигидрокси-7-пивалоилоксихромана Соединение, указанное в заголовке, получали при помощи того же метода, что описан в примере 8,при котором соединение из примера 13 используют вместо соединения из примера 5. ТСХ: хлороформ/метанол/30% NH4OH = 95/5/0,1; Rf = 0,35. Пример 15. Получение гидрохлорида 3-(4-пивалоилоксифенил)-4-(4-(2-(N-пиперидинил)этокси)фенил)-5-гидрокси-7-пивалоилоксихром-3-ена Соединение, указанное в заголовке, получали при помощи того же метода, что описан в примере 9,при котором гидрохлорид 3-(4-пивалоилоксифенил)-4-(4-(2-(N-пиперидинил)этокси)фенил)-4,5 дигидрокси-7-пивалоилоксихромана используют вместо соединения из примера 8. ТСХ: хлороформ/метанол/30% NH4OH = 95/5/0,1; Rf = 0,4. Точка плавления: 243-245 С (с разложением). Пример 16. Получение гидрохлорида 3-(4-пивалоилоксифенил)-4-(4-(2-(N-пиперидинил)этокси)фенил)-5-гидрокси-7-пивалоилоксихромана Соединение, указанное в заголовке, получали при помощи того же метода, что описан в примере 10,при котором соединение из примера 15 используют вместо соединения из примера 9. ТСХ: хлороформ/метанол/30% NH4OH = 95/5/0,1; Rf = 0,45. Точка плавления: 205-207 С (с разложением). Соединение из примера 15 (210 мг) растворяли в ДМФ (4 мл), затем добавляли K2 СО 3 (300 мг) и 2 иодпропан (200 мг) и смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли Н 2 О (5 мл), выпавший в осадок продукт отфильтровывали, промывали Н 2 О и очищали посредством флэш-хроматографии на силикагеле (элюент: метиленхлорид/метанол = 95/5; Rf = 0,7). Полученный продукт растворяли в ацетоне (2 мл) и добавляли к нему небольшой избыток HCl вEtOAc для получения кристаллической соли гидрохлорида. Получали 7 мг соединения, указанного в заголовке. Точка плавления: 237-239 С. ЯМР: соответствует. Пример 18. Получение 4'-метокси-7-пивалоилокси-5-гидроксиизофлавона Биоханин А (20 г), имеющийся в продаже или полученный известными способами, суспендировали в пивалиновом ангидриде (100 мл) и смесь нагревали при 120 С при перемешивании в течение 4 ч. Коричневый раствор оставляли при комнатной температуре в течение ночи, выпавший в осадок продукт отфильтровывали, промывали петролейным эфиром и сушили при 70 С под вакуумом. Получали 19,6 г соединения, указанного в заголовке. ТСХ: метиленхлорид/метанол = 95/5; Rf = 0,8. Пример 19. Получение 3-(4-метоксифенил)-5-гидрокси-7-пивалоилоксихроман-4-она Соединение, указанное в заголовке, получали при помощи того же метода, что описан в примере 5,при котором 4'-метокси-7-пивалоилокси-5-гидроксиизофлавон используют вместо соединения из примера 4. ТСХ: петролейный эфир/ЕtOАс = 95/5; Rf = 0,5. Пример 20. Получение гидрохлорида 3-(4-метоксифенил)-4-(4-(2-(N-пиперидинил)этокси)фенил)4,5-дигидрокси-7-пивалоилоксихромана Соединение, указанное в заголовке, получали при помощи того же метода, что описан в примере 8,при котором 3-(4-метоксифенил)-5-гидрокси-7-пивалоилоксихроман-4-он используют вместо соединения из примера 5. ТСХ: хлороформ/метанол/30% NH4OH = 95/5/0,1; Rf = 0,3. Соединение, указанное в заголовке, получали при помощи того же метода, что описан в примере 9,при котором гидрохлорид 3-(4-метоксифенил)-4-(4-(2-(N-пиперидинил)этокси)фенил)-4,5-дигидрокси-7 пивалоилоксихромана используют вместо соединения из примера 8. ТСХ: хлороформ/метанол/30% NH4OH = 95/5/0,1; Rf = 0,35. Точка плавления: 214-216 С (с разложением). ЯМР: соответствует. Пример 22. Получение гидрохлорида 3-(4-метоксифенил)-4-(4-(2-(N-пиперидинил)этокси)фенил)5,7-дигидроксихром-3-ена Соединение, указанное в заголовке, получали при помощи того же метода, что описан в примере 11,при котором соединение из примера 21 используют вместо соединения из примера 9. ТСХ: хлороформ/метанол/30% NH4OH = 95/5/1; Rf = 0,3. Точка плавления: 198-200 С (с разложением). Пример 23. Получение 7-пивалоилоксиизофлавона К раствору 7 г 7-гидроксиизофлавона в 190 мл N,N-диметилформамида добавляли 2 г гидрида натрия (80% в парафиновом масле). Смесь перемешивали в течение 10 мин, затем по каплям добавляли 4,2 г пивалоилхлорида в течение 1 мин при сильном перемешивании. После выдерживания при комнатной температуре в течение 20 мин смесь выливали в воду (400 мл), осадок отфильтровывали, промывали водой (1000 мл) и растворяли в хлороформе (800 мл). Органический раствор сушили над сульфатом натрия и упаривали досуха. Получали 7,6 г твердого белого вещества. ТСХ элюент: петролейный эфир/этилацетат = 85/15; Rf = 0,6. Пример 24. Получение 3-фенил-7-пивалоилоксихроман-4-она К раствору 15,7 г соединения из примера 23 в 770 мл 1,4-диоксана добавляли 8 г влажного 5% палладия на углероде и гидрировали в аппарате Парра при 40 ф/кв. дюйм (275,8 кПа). Реакцию контролировали посредством ТСХ для избежания дальнейшего восстановления до 4-гидроксипроизводных. Через 2 ч катализатор отфильтровывали на подушке из целита и промывали 1,4-диоксаном (200 мл). Раствор упаривали досуха и остаточное масло (21,35 г) кристаллизовали посредством его растворения в 115 мл этилацетата, добавления 635 мл петролейного эфира и хранения при -20 С в течение ночи. Получали 8,53 г продукта. Кристаллизация маточного раствора в этиловом спирте давала вторую порцию 1,3 г чистого продукта. ТСХ: петролейный эфир/этилацетат = 80/20; Rf = 0,85. Соединение, указанное в заголовке, получали при помощи того же метода, что описан в примере 8,при котором соединение из примера 24 используют вместо соединения из примера 5. ТСХ: метиленхлорид/метанол = 90/10; Rf = 0,4. Пример 26. Получение гидрохлорида 3-фенил-4-(4-(2-(N-пиперидинил)этокси)фенил)-4-гидрокси-7 пивалоилоксихром-3-ена Соединение, указанное в заголовке, получали при помощи того же метода, что описан в примере 9,при котором соединение из примера 25 используют вместо соединения из примера 8. ТСХ: метиленхлорид/метанол/30% NH4OH = 95/5/1; Rf = 0,4. Точка плавления: 233-235 С (с разложением). Пример 27. Получение 3-фенил-5-гидрокси-7-метоксихроман-4-она Соединение, указанное в заголовке, получали при помощи того же метода, что описан в примере 5,при котором 5-гидрокси-7-метоксиизофлавон используют вместо соединения из примера 4. Пример 28. Получение гидрохлорида 3-фенил-4-(4-(2-(N-пиперидинил)этокси)фенил)-4,5 дигидрокси-7-метоксихромана Соединение, указанное в заголовке, получали при помощи того же метода, что описан в примере 8,при котором 3-фенил-5-гидрокси-7-метоксихроман-4-он используют вместо соединения из примера 5. ТСХ: метиленхлорид/метанол = 90/10; Rf = 0,25. Пример 29. Получение гидрохлорида 3-фенил-4-(4-(2-(N-пиперидинил)этокси)фенил)-5-гидрокси-7 метоксихром-3-ена Соединение, указанное в заголовке, получали при помощи того же метода, что описан в примере 9,при котором соединение из примера 28 используют вместо соединения из примера 8. Соединение из примера 9 (550 мг) растворяли в ДМФ (30 мл), затем добавляли раствор K2 СО 3 (400 мг) в Н 2 О (3,5 мл) и раствор СН 3I (0,9 мл) в ДМФ (9,5 мл) и смесь перемешивали в течение 5 ч, гасили водой (200 мл) и экстрагировали ЕtOАс (200 мл). Органическую фазу промывали рассолом (2x200 мл),сушили над сульфатом натрия и упаривали под вакуумом. Желтое масло очищали посредством флэшхроматографии на силикагеле (элюент: CH2Cl2/MeOH/NEt3 = 95/5/0,1; Rf = 0,4). Полученный продукт преобразовывали в соль гидрохлорид посредством добавления HCl/EtOAc к раствору свободного основания в ЕtOАс. Получали 190 мг белого аморфного твердого вещества. ТСХ: метиленхлорид/метанол/NEt3 = 95/5/0,1; Rf = 0,4. ЯМР: соответствует. Пример 31. Получение гидрохлорида 3-(4-фторфенил-4-(4-(2-(N-пиперидинил)этокси)фенил)-5 гидрокси-7-пивалоилоксихром-3-ена Соединение, указанное в заголовке, получали следуя методам, описанным в примерах 1-9, при которых исходное вещество представляло собой 4-фторфенилацетонитрил вместо фенилацетонитрила по примеру 1. ТСХ: метиленхлорид/метанол/30% NH4OH = 95/5/1; Rf = 0,3. Точка плавления: 198-200 С (с разложением). ЯМР: соответствует. Пример 32. Получение цис-3-фенил-4-(4-бензилоксибензил)-4-гидрокси-7-пивалоилоксихромана Магний (4,9 г) загружали в атмосфере N2 в четырехгорлую колбу на 500 мл, снабженную конденсатором, капельной воронкой и магнитной мешалкой. 4-Бензилоксибензилхлорид (14,7 г) растворяли в 190 мл свежедистиллированного ТГФ и загружали в капельную воронку. 10 мл этого раствора по каплям добавляли на магний при комнатной температуре и смесь сильно нагревали до тех пор, пока сильное вспенивание не станет видно на поверхности магния. Колбу немедленно помещали в масляную баню при 75 С и добавляли оставшийся раствор в течение 40 мин. Смеси давали достичь комнатной температуры, затем мутный серый раствор переносили в атмосфере N2 в колбу на 500 мл, снабженную термометром, хлоркальциевой трубкой, магнитной мешалкой и капельной воронкой. Раствор Гриньяра немедленно использовали на следующей стадии или хранили в морозильной камере (Выход 100%, подсчитанный взвешиванием непрореагировавшего магния). Раствор соединения из примера 24 (10 г) в свежедистиллированном ТГФ (100 мл) по каплям добавляли в вышеуказанный раствор, охлажденный при-20 С в течение 30 мин. Желто-оранжевой смеси давали достичь комнатной температуры, перемешивали в течение 2 ч, охлаждали при 0 С и добавляли воду (8 мл) при перемешивании. Образовавшееся твердое вещество удаляли, раствор упаривали досуха и растирали с ЕtOАс/петролейным эфиром = 15/85 (2x300 мл). После фильтрации твердого вещества (соли Мg и другие побочные продукты) раствор упаривали и- 13006158 маслянистый остаток кристаллизовали из ЕtOН (200 мл). Получали 7,0 г твердого белого вещества; вторую порцию (1,4 г) чистого цис-3-фенил-4-(4 бензилоксибензил)-4-гидрокси-7-пивалоилоксихромана получали посредством флэш-хроматографии Соединение из примера 32 (6,9 г) растворяли в МеОН (800 мл) и гидрировали в аппарате Парра с использованием 14 г влажного 5% Pd/C при 40 ф/кв. дюйм (275,8 кПа) в течение 45 мин. После отфильтровывания катализатора на подушке из целита раствор упаривали при пониженном давлении и маслянистый остаток использовали без дополнительной очистки на следующей стадии. ТСХ: ЕtOАс/петролейный эфир = 30/70; Rf = 0,5. Пример 34. Получение цис-3-фенил-4-(4-(2-(N-пиперидинил)этокси)бензил)-4-гидрокси-7 пивалоилоксихромана К раствору соединения из примера 33 (5,3 г) в ацетоне (170 мл) добавляли гидрохлорид N-(2 хлорэтил)пиперидина (2,48 г) и K2 СО 3 (3,38 г), затем смесь нагревали с обратным холодильником при перемешивании в течение 16 ч. Твердое вещество отфильтровывали, раствор упаривали и маслянистый остаток растворяли в кипящем гексане (500 мл). После отфильтровывания нерастворимого вещества горячий раствор охлаждали при комнатной температуре и обрабатывали ультразвуком (или сильно перемешивали) до наступления кристаллизации. После хранения при -20 С твердое вещество отфильтровывали и сушили под вакуумом при 50 С. Получали 3,2 г твердого белого вещества. Маточные растворы могут быть подвергнуты флэш-хроматографии (CH2Cl2/MeOH/NEt3 = 95/5/0,1) с получением дополнительных 1,2 г продукта. ТСХ: метиленхлорид/метанол/NEt3 = 95/5/0,1; Rf = 0,4. Пример 35. Получение гидрохлорида рац-3-фенил-4-Е-(4-(2-(N-пиперидинил)этокси)бензилиден-7 пивалоилоксихромана Раствор соединения из примера 34 (4,2 г) в CH3CN (300 мл), охлажденный при -20 С, добавляли к холодному (-20 С) раствору 37% HCl (3 мл) в CH3CN (300 мл) и оставляли при -20 С в течение ночи. Раствору давали достичь комнатной температуры и контролировали посредством высокоэффективной жидкостной хроматографии (ВЭЖХ). Когда соединение из примера 34 было полностью израсходовано, растворитель выпаривали под вакуумом, остаток растворяли в ацетоне и упаривали снова для получения твердого белого пенистого вещества, которое растворяли в ЕtOАс и кристаллизовали посредством обработки ультразвуком или посредством перемешивания. ТСХ: ЕtOАс/н-ВutOН/Н 2 О/СН 3 СООН = 50/10/10/10; Rf = 0,3. Точка плавления: 214-216 С. ЯМР: соответствует. Пример 36. Разделение энантиомеров соединения из примера 35 Соединение из примера 35 подвергали разделению посредством препаративной ВЭЖХ. Условия препаративного разделения перечислены ниже:: 254 нм Загрузка: 25 мг, растворенные в 0,5 мл подвижной фазы Энантиомерную чистоту двух энантиомеров определяли посредством аналитической хиральной хроматографии; условия перечислены ниже: Колонка: Chiralcel OJ-R, 5 мкм, 4,6x150 мм Элюент: Ацетонитрил/0,01 М NaH2PO4 + 1 мл HClO4 (рН=5,6) = 65/35 Скорость потока: 0,5 мл/мин: 254 нм Время удерживания (+) изомера: 5,6 мин Время удерживания (-) изомера: 6,2 мин Два изомера собирали в виде свободного основания: Их превращали в соль гидрохлорид посредством барботирования газообразного HCl в эфирном растворе свободных оснований. Раствор K2 СО 3 (440 мг) в Н 2 О (2,5 мл) добавляли к раствору соединения из примера 35 (440 мг) в МеОН (50 мл) и перемешивали в течение ночи. Добавляли 1 н. HCl (6,3 мл), затем раствор частично упаривали до образования белой эмульсии. После охлаждения до 0 С твердое вещество отделяли, фильтровали, промывали водой и сушили на воздухе. Получали 320 мг белого аморфного твердого вещества. ТСХ: ЕtOАс/н-ВutOН/Н 2 О/СН 3 СООН = 50/10/10/10; Rf = 0,2. ЯМР: соответствует. Пример 38. Получение цис-3-фенил-4-(4-(2-(N-пиперидинил)этокси)бензил)-4,7-дигидроксихромана Смесь соединения из примера 34 (12,5 г), МеОН (800 мл), K2 СО 3 (12,5 г) и Н 2 О (40 мл) перемешивали в течение 4 ч, растворитель упаривали до 50 мл и добавляли Н 2 О (500 мл) при перемешивании магнитной мешалкой. Твердое белое вещество отделяли и собирали посредством фильтрации, промывали водой и сушили при 45 С. Получали 9,2 г соединения, указанного в заголовке. ТСХ: метиленхлорид/метанол/30% NH4OH = 93/7/0,7; Rf = 0,35. Точка плавления: 95-103 С. ЯМР: соответствует. Пример 39. Получение 3-фенил-4-(4-(2-(N-пиперидинил)этокси)бензил)-7-гидроксихром-3-ена Раствор 37% HCl (3 мл) в ацетонитриле (40 мл) добавляли к кипящему раствору соединения из примера 38 (9 г) в ацетонитриле (300 мл). Через 20 мин раствор быстро охлаждали до 0 С, добавляли К 2 СО 3(9,0 г), перемешивали в течение 3 ч и выпавшее в осадок свободное основание вместе с K2 СО 3 отфильтровывали, промывали водой и сушили под вакуумом при 40 С. Пивалоилхлорид (0,5 г) добавляли к смеси соединения из примера 39 (0,5 г) и K2 СО 3 (0,5 г) в ацетонитриле (40 мл); смесь перемешивали в течение 5 ч, соли отфильтровывали, затем добавляли HCl в ЕtOАс (небольшой избыток) и раствор упаривали досуха с получением твердого пенистого вещества,которое растирали с этиловым эфиром, отфильтровывали и сушили при комнатной температуре. Получали 400 мг соединения, указанного в заголовке в виде белого твердого аморфного вещества. ТСХ: ЕtOАс/н-ВutOН/Н 2 О/СН 3 СООН = 50/10/10/10; Rf = 0,4. ЯМР: соответствует. Пример 41. Получение гидрохлорида 3-фенил-4-(4-(2-(N-пиперидинил)этокси)фенил)-5,7 диацетилоксихром-3-ена Соединение из примера 11 (210 мг, полученное как в примере 11) растворяли в уксусном ангидриде(2 г) при 130 С и перемешивали в течение 30 мин. Раствор охлаждали до комнатной температуры, добавляли лед (2 г), разбавляли водой (50 мл), обрабатывали водным K2 СО 3 и экстрагировали ЕtOАс (50 мл). Органический слой сушили над сульфатом натрия, фильтровали, подкисляли небольшим избыткомHCl/EtOAc и упаривали досуха. Образовавшееся желтое масло кристаллизовалось из ацетона/Еt2 О с получением 100 мг соединения, указанного в заголовке, в виде светло-желтого порошка. ТСХ: метиленхлорид/метанол/NEt3 = 95/5/0,1; Rf = 0,55. Точка плавления: 154-156 С (с разложением). ЯМР: соответствует. Пример 42. Получение гидрохлорида 3-фенил-4-(4-(2-(N-пиперидинил)этокси)бензил)-7 изобутироилоксихром-3-ена Соединение, указанное в заголовке, получали, следуя методам, описанным в примере 40, при которых изобутироилхлорид используют вместо пивалоилхлорида. ТСХ: ЕtOАс/н-ВutOН/Н 2 О/СН 3 СООН = 50/10/10/10; Rf = 0,4. Точка плавления: 152-155 С. ЯМР: соответствует. Пример 43. Получение метансульфоната 3-фенил-4-(4-(2-(N-пиперидинил)этокси)бензил)-7 гидроксихром-3-ена (1:1) (растворимой соли соединения из примера 39) 2,7 г соединения из примера 39 растворяли в 120 мл горячего ацетона, затем добавляли раствор 512 мг метансульфоновой кислоты в 30 мл ацетона и раствор перемешивали магнитной мешалкой до наступления кристаллизации. После выдерживания при -20 С в течение 2 ч твердое вещество отфильтровывали, промывали Et2O и сушили под вакуумом при 40 С. Получали 2,9 г твердого белого вещества. Точка плавления: 170-175 С (с разложением).- 16006158 Растворимость этой соли в воде при комнатной температуре составляла более 1 мг/мл. Пример 44. Получение 3-фенил-4-(4-(2-(N-пиперидинил)этокси)бензил)-7-гидроксихром-3-ена посредством изомеризации соединения из примера 37 25 мл 1%-ного раствора концентрированной HCl в CH3CN добавляли к суспензии соединения из примера 37 (660 мг) в CH3CN/H2O (150/5 мл), нагретой при 80 С. Реакцию контролировали посредством ВЭЖХ (колонка: Select В RP C18, элюент: CH3CN-MeOH = 80-20 (70%), буфер (30%), скорость потока 0,8 мл/мин,= 254 нм). После 15 минутного нагревания при 80 С реакция заканчивалась: образовывалось 85% продукта (время удерживания = 4,8 мин), и 11% реагента оставалось непрореагировавшим. Реакция дальше не протекала ни при дальнейшем подведении тепла, ни при добавлении большего количества кислоты. Реакционную смесь подготавливали и продукт выделяли, как описано в примере 39. Получали 500 мг продукта (выход 75%). Пример 45. Получение 3-(4-метоксифенил)-4-оксо-4 Н-1-бензопиран-7-илового эфира 2,2 диметилпропионовой кислоты Формононетин (90 г; 335 ммоль) суспендировали в ацетонитриле, добавляли K2 СО 3 и смесь перемешивали в течение приблизительно 20 мин при комнатной температуре. Пивалоилхлорид (59,85 г; 496 ммоль) по каплям добавляли в смесь, затем реакционную смесь перемешивали еще в течение 15 мин и гасили водой. Твердое вещество отфильтровывали, промывали водой, растворяли в СНСl3, раствор сушили и упаривали досуха с получением соединения, указанного в заголовке, которое можно кристаллизовать из горячего толуола. Получали 102 г белого кристаллического порошка. Точка плавления: 159-161 С. ЯМР, МС (масс-спектроскопия) и ИК (инфракрасная спектроскопия) соответствуют структуре. Пример 46. Получение 3,4-дигидро-3-(4-метоксифенил)-4-оксо-2 Н-1-бензопиран-7-илового эфира 2,2-диметилпропионовой кислоты Соединение из примера 45 растворяли в диоксане и гидрировали в аппарате Парра при 35 ф/кв. дюйм (241,325 кПа) с использованием 5% Pd/BaSO4 в качестве катализатора. Через 3 ч катализатор отфильтровывали, раствор упаривали до небольшого объема и гасили водой с получением осадка, который отфильтровывали и растворяли в горячем 95% этаноле. После выдерживания в течение ночи в холодильнике белый продукт отфильтровывали и промывали петролейным эфиром с получением 66 г соединения, указанного в заголовке. Точка плавления: 119-121 С. ЯМР, МС и ИК соответствуют структуре. Пример 47. Получение 3,4-дигидро-4-гидрокси-3-(4-метоксифенил)-4-4-(фенилметокси)фенил]метил]-2 Н-1-бензопиран-7-илового эфира 2,2-диметилпропионовой кислоты Магниевую стружку (15,4 г; 635 ммоль) загружали в колбу на 2 л; 4-бензилоксибензилхлорид (49,6 г; 211,6 ммоль) растворяли в безводном ТГФ и этот раствор по каплям добавляли в течение 1 ч на магниевую стружку в атмосфере инертного газа, поддерживая умеренную дефлегмацию. Смесь охлаждали до -20 С, затем по каплям добавляли раствор соединения из примера 46 (60 г; 169,3 ммоль) в безводном ТГФ (300 мл) в течение 15 мин. После того, как смесь оставляли для достижения комнатной температуры, добавляли воду (50 мл), твердое вещество отфильтровывали и раствор упаривали досуха. Этилацетат (200 мл) добавляли к остаточному маслу и смеси давали отстояться при -20 С в течение 1 ч. Твердое вещество отфильтровывали и раствор упаривали досуха. Остаточное масло кристаллизовалось из смеси 80/20 95% этанола и петролейного эфира с получением 60,8 г (110 ммоль) соединения, указанного в заголовке. Точка плавления: 104-111 С. ЯМР, МС и ИК соответствуют структуре. Пример 48. Получение 3,4-дигидро-4-гидрокси-4-[(4-гидроксифенил)метил]-3-(4-метоксифенил)- 17006158 2 Н-1-бензопиран-7-илового эфира 2,2-диметилпропионовой кислоты Соединение из примера 47 (60,8 г; 110 ммоль) растворяли в этилацетате, добавляли 5% Pd/C и смесь гидрировали. Катализатор отфильтровывали и раствор упаривали досуха, получая твердое пенистое вещество(50,88 г; 110 ммоль), которое использовали без какой-либо дополнительной очистки. ЯМР, МС и ИК соответствуют структуре. Пример 49. Получение 3,4-дигидро-4-гидрокси-3-(4-метоксифенил)-4-4-[2-(1-пиперидинил)этокси]фенил]метил]-2 Н-1-бензопиран-7-илового эфира 2,2-диметилпропионовой кислоты Смеси соединения из примера 48 (50 г; 108,1 ммоль), гидрохлорида N-(2-хлорэтил)пиперидина (20 г; 108,7 ммоль) и K2 СО 3 в кипящем ацетоне (860 мл) давали прореагировать. Твердое вещество отфильтровывали и раствор упаривали досуха, получая масло (62 г; 108,1 ммоль),которое использовали без какой-либо дополнительной очистки. ЯМР, МС и ИК соответствуют структуре. Пример 50. Получение 3,4-дигидро-4-гидрокси-3-(4-метоксифенил)-4-4-[2-(1 пиперидинил)этокси]фенил]метил]-2 Н-1-бензопиран-4,7-диола К раствору соединения из примера 49 (60 г; 104,6 ммоль) в метаноле добавляли K2 СО 3, растворенный в воде, и давали прореагировать при комнатной температуре в течение 3 ч. Растворитель упаривали до 300 мл, и смесь гасили водой, и перемешивали в течение ночи. Твердое вещество отфильтровывали,промывали водой и сушили под вакуумом при комнатной температуре, получая 36 г неочищенного продукта, который кристаллизовался из кипящего ацетонитрила с получением 28 г соединения. Точка плавления: 107,5-108,5 С. ЯМР, МС и ИК соответствуют структуре. Пример 51. Получение гидрохлорида 3-(4-метокси)фенил-4-4-[2-(1-пиперидинил)этокси]фенил]метил]-2 Н-1-бензопиран-7-ола Соединение из примера 50 (24 г; 49 ммоль) растворяли в горячем ацетонитриле (550 мл), добавляли 37% HCl (4,9 мл) и раствор перемешивали при температуре дефлегмации в течение 45 мин. Смеси давали достичь комнатной температуры и выкристаллизовавшееся твердое вещество отфильтровывали и промывали диэтиловым эфиром. Получали 17,3 г (34 ммоль) соединения, указанного в заголовке. Точка плавления: 199-205 С (с разложением). ЯМР, МС и ИК соответствуют структуре. Проверяли фармакологическую активность соединений по изобретению. В следующих примерах представлены результаты, полученные при испытании соединений 3-фенил-4(4-(2-(N-пиперидинил)этокси)бензил)-7-гидроксихром-3-ена (соединения из примера 39, которое используют в виде свободного основания или соли метансульфоната) и гидрохлорида 3-(4-метокси)фенил-4-4-[2-(1 пиперидинил)этокси]фенил]-метил]-2 Н-1-бензопиран-7-ола (соединения из примера 51), на которые ссылаются в дальнейшем как на соединение I (пример 39) и соединение II (пример 51).- 18006158 Исследование in vitro - - и -рецепторы эстрогена человека (ER- и ER-) Эксперимент проводили, как описано в Obourn JD et al, Biochemistry 32, 6229, 1993. Соединение I по изобретению связывается с ER- и ER- человека, демонстрируя высокое сродство. Кi связывания составляла 0,0410,011 и 0,1570,028 нМ, соответственно. По сравнению с хорошо известным SERM ралоксифеном, его сродство к ER- было сходным (Ki = 0,071 0,008 нМ), тогда как сродство к ER- было в 10 раз выше (Ki = 1,620,348 нМ). Соединение II по изобретению связывается с еще более высоким сродством с рецепторами эстрогена человека ER- и ER-. Кi связывания составляла 0,0170,002 и 0,0990,005 нМ, соответственно. По сравнению с хорошо известным SERM ралоксифеном (Ki ER- = 1,620,348, ER- = 0,0710,008 нМ), сродство к ER- было в 16 раз выше, а сродство ER- было сходным. Исследования in vivo - анализ неполовозрелых самок крыс Антиутеротрофическая активность Эксперименты проводили, как описано в Eppenberger U. et. al., Am. J. Clin. Oncol. 14 (suppl. 2), S5S14, 1991. У неполовозрелых самок крыс, которых обрабатывали 17-этинилэстрадиолом (Е 2) в количестве 0,05 мг/кг перорально в течение 3 дней, значительно увеличилась сырая масса матки (170-220%) по сравнению с контролями, которых обрабатывали только носителем. Эта концентрация Е 2 была наименьшей оказывающей действие, близкое к максимальному, и была выбрана, исходя из предварительных экспериментов доза-ответ. Соединения I и II, вводимые перорально, ингибировали утеротрофическое действие Е 2 дозозависимым образом. Полный антагонизм наблюдали у соединения I при 1-10 мг/кг/день (ЕС 50 (концентрация соединения, при которой наблюдается эффект, равный 50% от максимального) = 0,33 мг/кгдень),тогда как эталонное соединение левормелоксифен оказывало только частичное антагонизирующее действие на стимуляцию матки с помощью Е 2 (40-45% ингибирование при 1 мг/кг/день). Полный антагонизм наблюдали у соединения II при 0,1-1 мг/кгдень. Сравнение антиэстрогенного действия (ED50 = доза, требуемая для создания 50% снижения роста, стимулированного эстрадиолом) показало, что соединение II по изобретению было примерно в 25 раз более действенным, чем ралоксифен(ED50 = 0,016 и 0,39 мг/кгдень, соответственно). Утеротрофическая активность Соединение I и ралоксифен при 0,1 мг/кг/день слегка увеличивали массу матки по сравнению с контрольными крысами, которых обрабатывали носителем (17 и 32%, соответственно, значительно меньше,чем 0,1 мг/кг/день Е 2); однако масса матки при 0,01, 1 и 10 мг/кг/день соединения I и ралоксифена незначительно отличалась от контрольных крыс. Масса матки при 0,01, 0,1, 1 и 10 мг/кгдень соединения II и ралоксифена незначительно отличалась от контрольных крыс. В соответствии с ранее опубликованными данными (М. Salman et al., J. Med. Chem. 1986, 29, 18011803), левормелоксифен при тех же уровнях доз значительно увеличивал сырую массу матки дозозависимым образом, и максимальная агонистическая активность, 60-65% от активности Е 2, проявлялась при 1 мг/кг. Авторы изобретения могут сделать вывод, что при анализе неполовозрелых самок крыс предпочтительные соединения по изобретению оказались сильнодействующими и эффективными в антагонизации эстрогенной стимуляции матки вплоть до уровня контрольных крыс, которых обрабатывали носителем,без значительной эстрогенной активности. Соединение II оказалось более действенным, чем ралоксифен, в антагонизации эстрогенной стимуляции матки. По сравнению с этим, левормелоксифен был частичным агонистом, проявляя при 1 мг/кгдень максимальную агонистическую активность, 60-65% от активности Е 2. Фармакологический профиль заявленных соединений у неполовозрелых крыс оказывается совершенно отличным по сравнению со структурно родственным соединением левормелоксифеном, показывая, что структурные различия между соединениями по изобретению и соединениями, раскрытыми в предшествующем уровне техники, являются существенными при снижении воздействий, стимулирующих матку. Действие на модели крыс, подвергнутых овариэктомии (OVX) Эксперименты проводили в соответствии с Kaln D.N., Bone Mineral 15, 175-192, 1991 и Grese ТА et.al. J. Med. Chem. 40, 146-167, 1997. Четырехнедельный анализ OVX крыс Действие тестируемых соединений оценивали также на 9-11-месячных OVX крысах, которым давали лекарство в течение 4 недель после операции, и сравнивали с OVX и имитационным контролями. Тканеспецифичное эстроген-агонистическое действие исследовали, используя массу матки, гистологию матки, активность пероксидазы эозинофилов матки (ЕРО), минеральную плотность кости (BMD) и уровни холестерина в сыворотке в качестве конечных точек.- 19006158 Введение в течение 4 недель не сопровождалось никакими явными признаками токсичности. Действие на минеральную плотность кости (BMP) (поясничный отдел позвоночника L1-4) Минеральную плотность кости (BMD) измеряли посредством DEXA (двухэнергетической рентгеновской абсорбциометрии), используя прибор Hologic QDR-1000 plus, оборудованный программой, специально предназначенной для измерений мелких животных. Режим сверхвысокого разрешения (межстрочный интервал 0,0254 см и разрешение 0,0127 см) использовали с коллиматором диаметром 0,63 мм. Это оборудование обеспечивает интегрированное измерение как трубчатой, так и губчатой кости.In vivo DEXA измерения проводили непосредственно перед операцией (исходное сканирование) и через 4 недели после операции. Исследуемая анатомическая область представляла собой поясничный отдел позвоночника L1-4. Всем животным перед сканированием давали наркоз смесью кетамина и п-промазина. Для каждого сканирования крысу укладывали на спину, причем положение позвоночника было параллельно горизонтальной оси столика денситометра. Поясничный отдел позвоночника сканировали, используя тазовые кости в качестве ориентира; анализ этого участка выполняли посредством разделения позвоночного и межпозвоночного пространств при помощи программы с высоким разрешением подобласти и включения только целевого позвонка в глобальную область интереса. Процент защиты подсчитывали, используя следующую формулу:%защиты = [(% изменения BMDтестируемого соединения -% изменения BMDOVX контроля)/(% изменения BMD имитационного контроля- % изменения BMDOVX контроля)]100. В первом эксперименте определяли сберегающие действия соединения I в отношении кости. Через 4 недели после операции наблюдали значительно более низкий процент изменений BMD от исходного значения у OVX крыс по сравнению с крысами имитационного контроля (-9,390,60 и -0,110,75%, соответственно, р 0,01). Как показано в литературе, 0,1 мг/кг 17-этинилэстрадиола (Е 2) частично предупреждали костную потерю (50% защиты). Обработка 0,1-, 1 и 10 мг/кг/день соединения I снижала костную потерю, так как их % измененияBMD от исходного значения был значительно выше по сравнению с OVX крысами (40,46 и 47% защиты,соответственно, р 0,01). Во втором эксперименте определяли сберегающие действия соединения II в отношении кости. Через 4 недели после операции наблюдали значительно более высокую костную потерю от исходного значения у OVX крыс по сравнению с крысами имитационного контроля (% изменения BMD -5,16% и +1,28%, соответственно, р 0,01). Как показано в литературе, 0,1 мг/кг 17-этинилэстрадиола (Е 2) предупреждал костную потерю(95% защиты). Обработка 0,1 и 1 мг/кгдень тестируемого соединения снижала костную потерю дозо-зависимым образом, так как их % изменения BMD от исходного значения был значительно выше по сравнению сOVX крысами (60 и 119% защиты, соответственно, р 0,01). Сберегающие действия тестируемых соединений в отношении кости были сравнимы с действиями,достигнутыми при использовании Е 2 0,1 мг/кгдень, показывая, что они являются полными агонистами эстрогена в отношении кости. Ралоксифен и левормелоксифен сравнивали с соединением II: их защитное действие было значительным (50-60% защиты при 1 и 10 мг/кгдень), но значительно более низким по сравнению с соединением по изобретению. Действие на уровни холестерина в сыворотке 0,1-10 мг/кг/день соединения I дозозависимым образом уменьшал общий холестерин в сыворотке уOVX крыс с половиной максимальной эффективности ED50 0,12 мг/кг и максимальным снижением холестерина, наблюдаемым при 10 мг/кг (67% ингибирование относительно OVX контроля). 0,1 и 1 мг/кгдень соединения II дозозависимым образом уменьшал общий холестерин в сыворотке у OVX крыс (75 и 81% ингибирование, соответственно). Соединение II было таким же эффективным, как и Е 2 при снижении уровней холестерина в сыворотке по отношению к контролям, тогда как ралоксифен и левормелоксифен проявили себя значительно менее действенными. Действие на матку У OVX крыс, которых обрабатывали соединениями по изобретению в дозах 0,1, 1 и 10 мг/кгдень,масса матки была значительно ниже, чем у обоих имитационных контролей и OVX крыс, которых обрабатывали Е 2. Степень индукции пероксидазы эозинофилов матки может быть полезным показателем для ответных реакций роста под действием эстрогена (Lyttle C.R. and DeSombre E.R. Proc. Natl. Acad. Sci USA 74,3162-3166, 1977). Тестируемые соединения не оказывали значительного действия на активность пероксидазы эозинофилов по сравнению с OVX матками, тогда как левормелоксифен значительно увеличивал этот параметр. Более того, введение тестируемых соединений не было связано со значительной стимуляцией эпи- 20006158 телия матки. Вместо этого эстроген и левормелоксифен, которые вызывали полное исчезновение атрофии эпителия, поддерживали гистологию эндометрия на уровнях имитационных контролей. Соединения I и II по изобретению, вводимые в дозах 0,1, 1 и 10 мг/кг/день и 0,1 и 1 мг/кг/день, соответственно, в течение 4 недель пероральным способом, значительно снижали костную потерю, уменьшая уровни холестерина в сыворотке. Этих защитных действий достигали при дозе с минимальной стимуляцией матки. Взятые вместе, представленные результаты показывают, что производные бензопирана по изобретению обладают интересной SERM (селективная модуляция рецепторов эстрогена) активностью, выигрывающей по тканевой селективности при сравнении со структурно родственными эталонными соединениями, раскрытыми в предшествующем уровне техники. Исследование in vitro - пролиферация клеток MCF-7 В этом тесте соединение I сравнивали с двумя продуктами, имеющимися в продаже, ралоксифеном и тамоксифеном. Клетки опухоли получали из Американской Коллекции Типовых Культур (АТСС НТВ-22). Используемые культуральные среды представляли собой минимальную поддерживающую среду с 10% фетальной бычьей сыворотки, и в среды добавляли 1% антибиотика, представляющего собой противогрибковое средство. Аликвоты 100 мкл суспензии клеток (5 х 103/лунка) помещали в 96-луночные микротитрационные планшеты в атмосфере 5% СО 2 при 37 С. Через 24 ч 100 мкл среды для выращивания 2 мкл тестируемого раствора, митомицин или носитель (ДМСО (диметилсульфоксид), конечная концентрация 0,4%) добавляли соответственно на лунку в двух повторностях для дополнительного 72-часового инкубирования. В конце инкубирования 20 мкл 75% реагента alamarBlue добавляли в каждую лунку для другого 6 часового инкубирования перед определением жизнеспособности клеток при помощи интенсивности флуоресценции (возбуждение при 530 нм и эмиссия при 590 нм). Соединение I и два соединения сравнения, ралоксифен и тамоксифен, проявили значительное ингибирование роста относительно контрольной группы, которую обрабатывали носителем, в концентрациях между 1 и 100 мкМ. Кроме того, в условиях этого эксперимента тамоксифен и соединение I были более действенными (ингибирующая концентрация IC50 = 6,0 и 6,5 мкМ, соответственно), чем ралоксифен (IC50 = 11 мкМ). Общее ингибирование роста наблюдали при 11 мкМ для тамоксифена и соединения I или 18 мкМ для ралоксифена, тогда как 50% летальные концентрации (LC50) составляли 22 (тамоксифен), 20 (соединение I) и 29 (ралоксифен) мкМ. Представленные результаты подтверждают, что соединения по изобретению действуют как сильные и селективные тканеспецифичные агонисты и антагонисты эстрогена. Тестируемые соединения обладают лучшим фармакологическим профилем, чем структурно родственное эталонное соединение левормелоксифен, производное бензопирана, и выигрывают при сравнении с ралоксифеном, одним из первых SERM, разработанных для клинического применения. С учетом их терапевтического применения соединения по изобретению можно подходящим образом комбинировать с обычными фармацевтически приемлемыми эксципиентами для приготовления фармацевтических композиций для парентерального, перорального, назального, ректального, подкожного или чрескожного введения в соответствии с общепринятыми способами. Специалист в данной области техники может приготовить указанные соединения в виде препаратов соответствующим образом и в соответствии с принятыми технологиями, такими как описанные в где X представляет собой Н, галогено или O-R3, где R3 представляет собой Н, С 1-4 алкил или С 1-5 алканоил,Y представляет собой Н, С 1-4 алкил или С 1-5 алканоил,Z представляет собой Н или O-R4, где R4 представляет собой Н, С 1-4 алкил, или С 1-5aлканоил,m равно 1 или 2,n равно 0 или 1,представляет собой одинарную или двойную связь между атомами в положениях 3-4 или 4-4 а,- 21006158 когда n равно 1, двойная связь может быть альтернативно экзоциклической или эндоциклической, образуя соответственно 4-бензилиденхроман при m=1 или 4-бензил-хром-3-ен при m=2, когда n равно 0,представляет собой одинарную или двойную связь в 3-4 и Z не является Н,и их фармацевтически приемлемые соли. 2. Производные 2 Н-1-бензопирана по п.1, гдеX представляет собой Н или С 1-С 4 алкокси,Y представляет собой Н или С 1-С 5 алканоил,Z представляет собой Н,m равно 2,n равно 1,связь между атомами в положении 3-4 представляет собой двойную связь, а связь между атомами в положении 4-4 а представляет собой одинарную связь. 3. Соединение по пп.1 и 2, которое представляет собой 3-фенил-4-(4-(2-(N-пиперидинил)этокси)бензил)-7-гидроксихром-3-ен, его сложные эфиры и соли. 4. Соединение по пп.1 и 2, которое представляет собой 3-(4-метокси)фенил-4-4-[2-(1 пиперидинил)этокси]фенил]метил]-2 Н-1-бензопиран-7-ол, его сложные эфиры и соли. 5. Фармацевтические композиции, содержащие в качестве активного ингредиента по меньшей мере одно соединение по пп.1-4 вместе с фармацевтически приемлемыми эксципиентами. 6. Применение соединений по пп.1-4 для приготовления лекарства, полезного при предупреждении и лечении ряда постменопаузальных патологий, в частности остеопороза, коронарной болезни сердца и эстроген-зависимого рака человека.
МПК / Метки
МПК: C07D 405/12, A61K 31/453, A61P 5/32
Метки: 2h-1-бензопирана, содержащая, производные, композиция, фармацевтическая
Код ссылки
<a href="https://eas.patents.su/23-6158-proizvodnye-2h-1-benzopirana-i-soderzhashhaya-ih-farmacevticheskaya-kompoziciya.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 2h-1-бензопирана и содержащая их фармацевтическая композиция</a>
Новые производные арилглицинамида, способ их получения и фармацевтическая композиция , содержащая эти соединения

Номер патента: 2201
Опубликовано: 28.02.2002
Авторы: Юнг Биргит, Шнорренберг Герд, Шромм Курт, Доллингер Хорст, Шпек Георг, Эссер Франц
МПК: A61K 31/395, C07D 295/14, A61P 9/00...
Метки: соединения, способ, арилглицинамида, эти, получения, содержащая, производные, композиция, фармацевтическая, новые
Формула / Реферат:
1. Производные арилглицинамида общей формулы (I) или их фармацевтически приемлемые соли, где Ar означает незамещенный или 1-5-кратнозамещенный фенил или незамещенный или 1-2-кратнозамещенный нафтил, причем заместители фенила и нафтила независимо друг от друга являются галогеном (фтором, хлором, бромом, йодом), алкилом с 1-4 атомами углерода, O-(С1-С4)алкилом, трифторметилом, трифторметокси или NR12R13, где R12 и R13 независимо друг от друга...
Гибридный полипептид &beta 1 &alpha-интерферона человека, его мутантные формы и производные и содержащая их фармацевтическая композиция

Номер патента: 5005
Опубликовано: 28.10.2004
Авторы: Ранкель Лаура, Витти Эдриан, Хочман Паула, Брикельмэйер Маргот
МПК: C07K 14/565, A61P 31/12, C12N 15/62...
Метки: содержащая, полипептид, композиция, beta, человека, формы, мутантные, фармацевтическая, производные, alpha-интерферона, гибридный
Формула / Реферат:
1. Полипептид, содержащий аминокислотную последовательность, выбранную из группы, состоящей из SEQ ID NO: 45-60, причем каждый полипептид также содержит шарнирный, CH2- и CH3-домены иммуноглобулина. 2. Полипептид по п.1, в котором иммуноглобулин представляет собой иммуноглобулин человека. 3. Полипептид по п.1, в котором иммуноглобулин представляет собой IgG. 4. Полипептид по п.3, в котором IgG представляет собой IgG1. 5. Полипептид по п.4,...
Пирролидинил-, пиперидинил- или гомопиперидинилзамещенные производные, способ их получения, их применение в медицине, содержащая их фармацевтическая композиция и способ ее получения

Номер патента: 4648
Опубликовано: 24.06.2004
Авторы: Вирсхюэрен Вим Гастон, Вигеринк Пит Том Берт Поль, Ван Эмелен Кристоф, Де Брюин Марсель Франс Леопольд
МПК: A61K 31/505, C07D 405/14, A61P 9/00...
Метки: способ, медицине, композиция, пиперидинил, содержащая, производные, получения, применение, гомопиперидинилзамещенные, пирролидинил, фармацевтическая
Формула / Реферат:
1. Соединение формулы (I) его стереохимически изомерная форма или его фармацевтически приемлемая кислотно-аддитивная соль, в которых R1, R2 и R3 каждый представляет водород; -Z1-Z2- представляет двухвалентный радикал формулы -O-CH(R4)-CR2-CH2- (a-4), R4 представляет водород; Alk представляет C1-6алкандиил, необязательно замещенный гидроксигруппой; представляет двухвалентный радикал формулы в которой m равно 0 или 1; R6...
Фармацевтическая композиция, содержащая выбранные гидраты карбоната лантана

Номер патента: 270
Опубликовано: 25.02.1999
Авторы: Пауэлл Найджел Энтони, Меррер Барри Энтони
МПК: A61K 33/24
Метки: гидраты, выбранные, фармацевтическая, карбоната, композиция, лантана, содержащая
Формула / Реферат:
1. Фармацевтическая композиция для лечения гиперфосфатемии, включающая карбонат лантана формулыLa2(C03)3 • хН20, где х имеет значение от 3 до 6, в смеси или в ассоциации с фармацевтически приемлемым разбавителем или носителем.2. Композиция по п. 1, в которой значение х в карбонате лантана составляет от 3,5 до 5.3. Композиция по п. 2, в которой значение х в карбонате лантана составляет от 3,8 до 4,5.4. Композиция по любому из пп. 1-3,...
Бета- карболиновые соединения, способ их получения и фармацевтическая композиция, их содержащая

Номер патента: 4140
Опубликовано: 26.02.2004
Авторы: Декейн Анн, Миллан Марк, Бутэн Жан, Брион Жан-Даниель, Пуассонне Гийом, Парментье Жан-Жилль, Гольдстейн Соло
МПК: A61P 25/00, A61K 31/437, C07D 471/04...
Метки: карболиновые, получения, фармацевтическая, содержащая, соединения, способ, beta, композиция
Формула / Реферат:
1. Соединения формулы (I) в которой --- представляет простую или двойную связь, способную необязательно сообщать ароматический характер кольцу, несущему ее, R1 представляет группу, выбранную из водорода, прямого или разветвленного (C1-C6)алкила, -R6-арила, -R6-циклоалкила, -R6-гетероцикла, где группы R6 представляют прямую или разветвленную (C1-C6)алкиленовую группу, -CO2R7, где R7 представляет прямую или разветвленную (C1-C6)алкильную группу,...
Предыдущий патент: Лекарственная форма силимарина и способ ее получения
Следующий патент: Катализатор для очистки выхлопного газа дизельного двигателя
Случайный патент: Система для тушения огня