Фениламино-изоникотинамидные соединения
Номер патента: 18539
Опубликовано: 30.08.2013
Авторы: Гоутопоулос Андреас, Ю Хенри, Аскью Бенни С., Лю-Буджалски Лесли




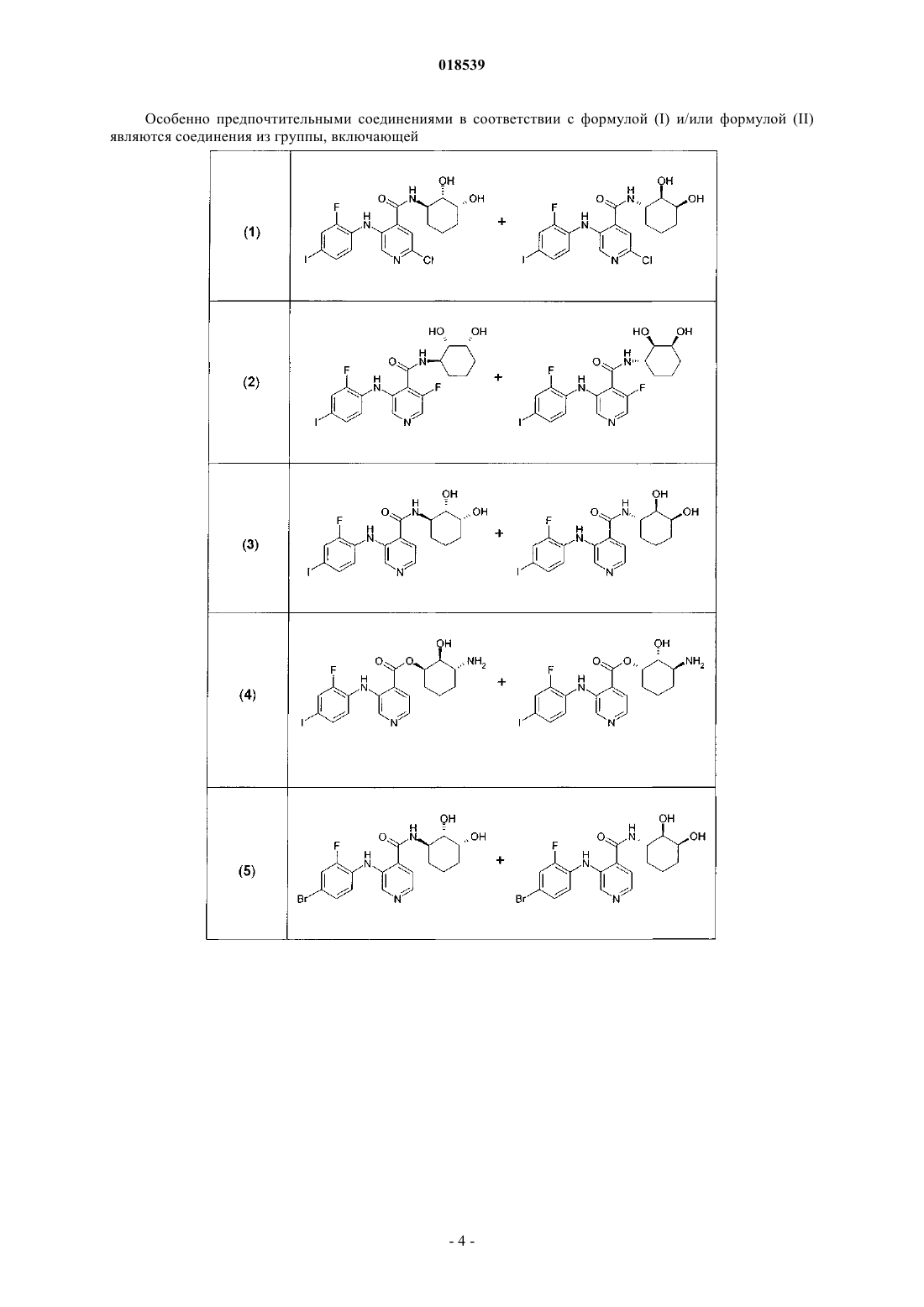

















Формула / Реферат
1. Соединение формулы (I)

и его фармацевтически приемлемые соли,
где X представляет собой NH или О;
R1 представляет собой водород, метил, этил, н-пропил, изопропил, SH или Hal;
R2 представляет собой водород, метокси, этокси, ацетилен, циано, SH или Hal;
R3, R4 независимо выбирают из водорода, SH или Hal;
R5, R6 независимо выбирают из ОН, SH или NH2;
Hal представляет собой F, Cl, Br или I.
2. Соединение в соответствии с п.1, где радикалы, не указанные подробно, имеют значения, указанные для формулы (I) в соответствии с п.1, но где
в подформуле (IA)
X представляет собой NH;
R1 представляет собой Hal, метил или этил;
R2 представляет собой водород, Hal, метокси или ацетилен;
R3 представляет собой водород или Hal;
R4 представляет собой водород или Hal;
R5, R6 представляют собой ОН;
Hal представляет собой F, Cl, Br или I;
в подформуле (IB)
X представляет собой NH;
R1 представляет собой Hal;
R2 представляет собой водород или Hal;
R3 представляет собой водород или Hal;
R4 представляет собой водород или Hal;
R5, R6 представляют собой ОН;
Hal представляет собой F, Cl, Br или I;
в подформуле (IC)
X представляет собой NH;
R1 представляет собой F, Cl, метил или этил;
R2 представляет собой водород, I, Br, метокси или ацетилен;
R3 представляет собой водород или Hal;
R4 представляет собой водород или Hal;
R5, R6 представляют собой ОН;
Hal представляет собой F, Cl, Br или I;
в подформуле (ID)
X представляет собой NH;
R1 представляет собой F, Cl, метил или этил;
R2 представляет собой водород, I, Br, метокси или ацетилен;
R3 представляет собой водород или F;
R4 представляет собой водород или Cl;
R5, R6 представляют собой ОН;
в подформуле (IE)
X представляет собой NH;
R1 представляет собой F или Cl;
R2 представляет собой I или Br;
R3 представляет собой водород или F;
R4 представляет собой водород или Cl;
R5, R6 представляют собой ОН;
в подформуле (IF)
X представляет собой NH;
R1 представляет собой F или Cl;
R2 представляет собой I или Br;
R3 представляет собой водород или F;
R4 представляет собой водород или Cl;
R5, R6 представляют собой ОН;
в подформуле (IG)
X представляет собой NH;
R1 представляет собой F или Cl;
R2 представляет собой I или Br;
R3 представляет собой водород;
R4 представляет собой водород;
R5, R6 представляют собой ОН; и
в подформуле (IH)
X представляет собой NH;
R1 представляет собой F;
R2 представляет собой I;
R3 представляет собой водород или F;
R4 представляет собой водород или Cl;
R5, R6 представляют собой ОН,
и его фармацевтически приемлемые соли.
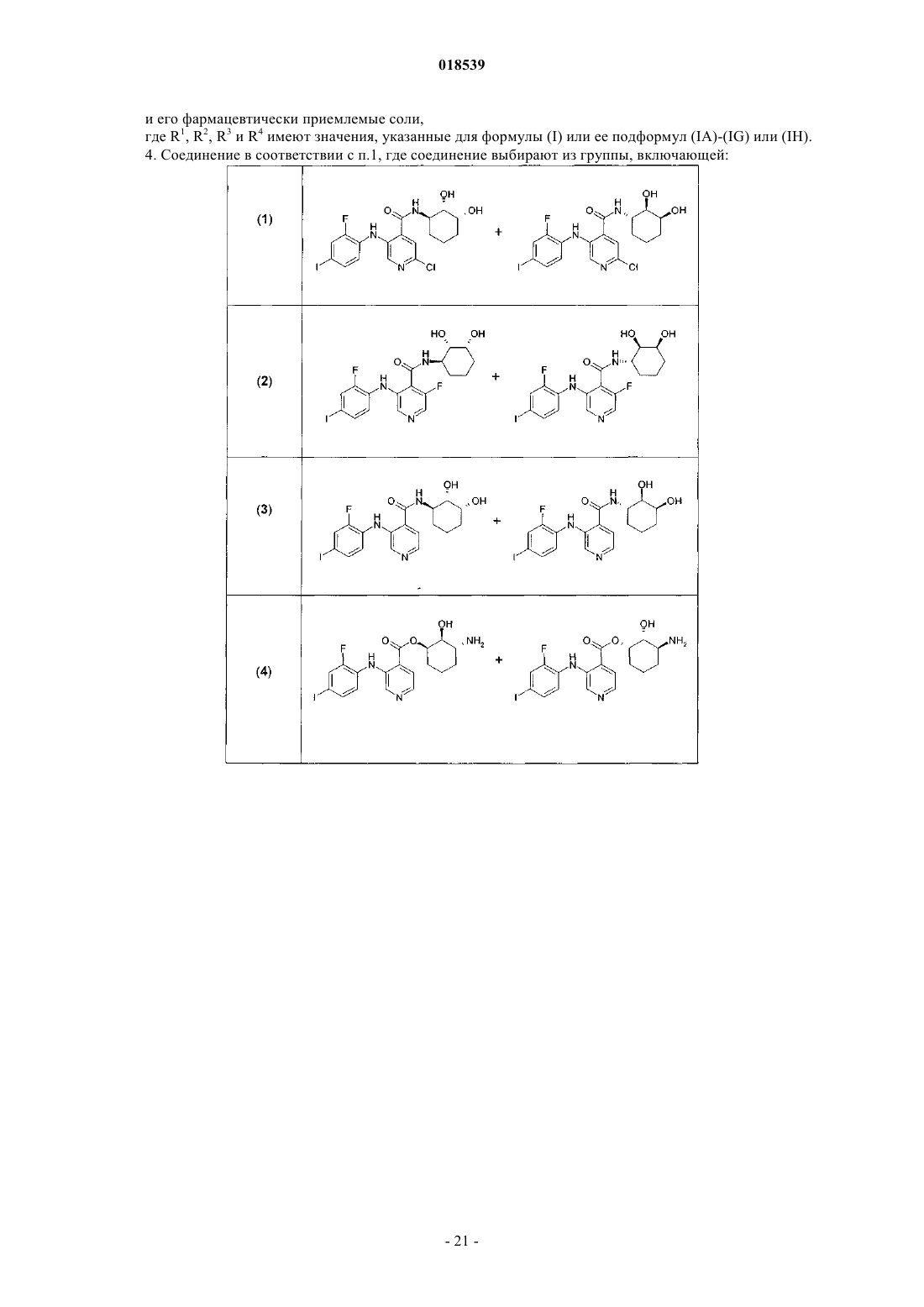
3. Соединение в соответствии с п.1 или 2, которое соответствует формуле (II)

и его фармацевтически приемлемые соли,
где R1, R2, R3 и R4 имеют значения, указанные для формулы (I) или ее подформул (IA)-(IG) или (IH).
4. Соединение в соответствии с п.1, где соединение выбирают из группы, включающей:


и их фармацевтически приемлемые соли.
5. Фармацевтическая композиция, которая содержит соединение в соответствии с любым из пп.1-4, или его фармацевтически приемлемую соль, в качестве активного компонента совместно с фармацевтически приемлемым носителем.
6. Применение соединения в соответствии с любым из пп.1-4 или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения гиперпролиферативных заболеваний, связанных с гиперактивностью MEK, а также заболеваний, модулируемых MEK каскадом у млекопитающих.
7. Применение в соответствии с п.6, где заболевание выбирают из группы, включающей злокачественное новообразование, воспаление, панкреатит или заболевание почек, боль, доброкачественную гиперплазию кожи, рестеноз, предстательной железы, заболевания, связанные с васкулогенезом или ангиогенезом, опухолевым ангиогенезом, заболевания кожи, выбранные из псориаза, экземы и склеродермы, диабет, диабетическую ретинопатию, ретинопатию недоношенных, возрастную дегенерацию желтого пятна, гемангиому, глиому, меланому и саркому Капоши.
8. Комплект (набор), состоящий из отдельных пакетов эффективного количества соединения в соответствии с одним или несколькими пп.1-4 или его фармацевтически приемлемой соли и эффективного количества другого противоракового терапевтического средства, выбранного из группы, включающей ингибиторы митоза, алкилирующие средства, антиметаболиты, интеркалирующие антибиотики, ингибиторы факторов роста, ингибиторы клеточного цикла, фермента, ингибиторы типоизомеразы, модификаторы биологического отклика, антигормональные средства, ингибиторы ангиогенеза, антиандрогены, бевацизумаб, CD40-специфические антитела, chTNT-1/B, денозумаб, занолимумаб, IGFIR-специфические антитела, линтузумаб, эдреколомаб, WX G250, ритуксимаб, тицилимумаб, трастузумаб и цетуксимаб.
Текст
(71)(73) Заявитель и патентовладелец: МЕРК ПАТЕНТ ГМБХ (DE) Изобретение обеспечивает новые соединения в соответствии с формулой (I) их приготовление и применение для лечения гиперпролиферативных заболеваний, таких как злокачественное новообразование, рестеноз и воспаление. Область техники, к которой относится изобретение Изобретение относится к группе замещенных фениламино-изоникотинамидных соединений, пригодных для лечения гиперпролиферативных заболеваний, таких как злокачественное новообразование и воспалительные нарушения, у млекопитающих. Также описано применение таких соединений для лечения гиперпролиферативных заболеваний у млекопитающих, в особенности людей, и фармацевтические композиции, содержащие такие соединения. Краткое изложение релевантного уровня техникиRas/Raf/MEK/ERK метаболический путь является центральным путем передачи сигналов, который передает сигналы от различных рецепторов клеточной поверхности к транскрипционным факторам в ядре, которые регулируют экспрессию генов. Этот путь часто обозначается как путь MAP киназы, гдеMAPK обозначает митоген-активированную протеинкиназу, указывая на то, что этот путь может стимулироваться митогенами, цитокинами и факторами роста (Steelman и др., Leukemia 2004, 18, 189-218). В зависимости от стимула и типа клетки, этот путь может передавать сигналы, которые приводят к предотвращению или индукции апоптоза или осуществлению клеточного цикла. Показано, чтоRas/Raf/MEK/ERK путь является чрезвычайно важным для пролиферации клеток и предотвращения апоптоза. Аберрантная активация этого пути обычно наблюдается в злокачественно трансформированных клетках. Амплификация ras прото-онкогенов и активирующие мутации, которые приводят к экспрессии конститутивно активных Ras белков, наблюдается приблизительно в 30% всех злокачественных новообразований человека (Stirewalt и др., Blood 2001, 97, 3589-95). Мутированные, онкогенные формыRas были обнаружены в 50% рака ободочной кишки и 90% рака поджелудочной железы, а также при многих других типах злокачественных новообразований (Kohl и др., Science 1993, 260, 1834-1837). Влияния Ras на пролиферацию и опухолегенез был описан на иммортализованных клеточных линиях (McCubrey и др., Int J. Oncol 1995, 7, 295-310). bRaf мутации были идентифицированы более чем в 60% злокачественной меланомы (Davies, H и др., Nature 2002, 417, 949-954). Принимая во внимание высокий уровень мутаций, который обнаружен в Ras, этот путь всегда рассматривался как основная мишень для терапевтического вмешательства (Chang и др., Leukemia 2003, 17, 1263-93).Ras/Raf/MEK/ERK путь передачи сигналов может регулировать пролиферацию путем нижерасположенных мишеней - транскрипционных факторов, включая NF-kKB, CREB, Ets-1, АР-1 и с-Мус. ERK может непосредственно фосфорилировать Ets-1, АР-1 и с-Мус, что приводит к их активации. Альтернативно, ERK может фосфорилировать и активировать нижерасположенную целевую киназу RSK, которая затем фосфорилирует и активирует транскрипционные факторы, такие как CREB. Эти транскрипционные факторы индуцируют экспрессию генов, которые важны для осуществления клеточного цикла, например Cdk, циклины, факторы роста, и для предотвращения апоптоза, например антиапоптотическийBcl-2 и цитокины. В целом, обработка клеток факторами роста приводит к активации ERK, что вызывает пролиферацию и, в некоторых случаях, дифференциацию (Lewis и др., Adv. Cancer Res, 1998, 74, 49-139). Семейство MEK генов состоит из пяти генов: MEK1, MEK2, MEK3, MEK4 и MEK5. Это семейство киназ с двойной специфичностью обладает как серин/треонин, так и тирозинкиназной активностью. Структура MEK включает амино-концевой отрицательный регуляторный домен и карбокси-концевой домен связывания MAP киназы, который необходим для связывания и активации ERK. Делеция регуляторного MEK1 домена приводит к конститутивной MEK1 и ERK активации (Steelman и др., Leukemia 2004, 18, 189-218).MEK1 представляет собой белок из 393 аминокислот с молекулярной массой 44 кДа (Crews и др.,Science 1992, 258, 478-80). MEK1 умеренно экспрессируется при эмбриональном развитии и повышена в тканях взрослых с наивысшими уровнями, определяемыми в ткани головного мозга. Для активацииMEK1 необходимо фосфорилирование S218 и S222 и замещение этих остатков либо аспарагиновой кислотой (D) или глутаминовой кислотой (Е) приводит к повышению активности и образованию очагов вNIH3T3 клетках (Huang и др., Mol Biol Cell, 1995, 6, 237-45). Конститутивная активность MEK1 в первичной клеточной культуре способствует старению и индуцирует р 53 и p16INK4a, и противоположное наблюдается в иммортализированных клетках или клетках, в которых отсутствует либо р 53 или p16INK4a(Lin и др., Genes Dev, 1998, 12, 3008-3019). Конститутивная активность MEK1 ингибирует NF-kKB транскрипцию путем отрицательной регуляции р 38 MAPK активностями (Carter и др., J. Biol Chem 2000, 275,27858-64). Основными физиологическими субстратами MEK являются представители ERK (киназа, регулируемая внеклеточными сигналами) или MAPK (митоген-активируемая протеинкиназа) семейства белков. Аберрантная экспрессия MEK1 была обнаружена при многих различных типах злокачественных новообразований, и мутированные формы MEK1 будут трансформировать фибробласты, гемопоэтические клетки и клетки других типов. Конститутивная активация MEK1 приводит к трансформации клеток. Следовательно, MEK1 представляет собой перспективную мишень для фармакологического вмешательства при пролиферативных и воспалительных заболеваниях (Lee и др., Nature 1994, 372, 739-746; Dudley и др., Proc. Natl. Acad. Sci.USA 1995, 92, 7686-7689). Были разработаны пригодные ингибиторы MEK, которые проявляли потенциальные терапевтические преимущества в некоторых исследованиях. Например, было показано, что низкомолекулярные ин-1 018539 гибиторы MEK ингибируют рост опухоли на ксенотрансплантатах у "голых" мышей (Yeh, Т. и др., Proceedings of the American Association of Cancer Research 2004, 45, реф. 3889 и Lee, P. и др., Proceedings ofthe American Association of Cancer Research 2004, 45, реф. 3890). MEK ингибиторы проходят клинические испытания, т.е. ARRY142886 (Wallace, E. и др., Proceedings of the American Association of Cancer Research 2004, 45, Abs 3891; Adjei, А.А. и др., Journal of Clinical Oncology 2008, 26, 2139-2146; Shannon, A.M. и др.,Molecular Cancer Therapeutics 2007, 6, 3414S-3415S Part 2), PD-0325901 (Swanton C., Johnston S. IDDBWO 03/077855; WO 03/077914; WO 2004/005284; WO 2004/056789, WO 2006/045514, WO 2008/076415,WO 2007/121269 и WO 2007/121481. Описание изобретения Объектом настоящего изобретения является обеспечение новых MEK ингибиторов, пригодных для лечения гиперпролиферативных заболеваний, связанных с гиперактивностью MEK, а также заболеваний,модулируемых MEK каскадом, таких как злокачественное новообразование и воспаление у млекопитающих с улучшенными фармакологическими свойствами как относительно их активностей, так и относительно их растворимости, метаболического клиренса и характеристик биодоступности. Таким образом, настоящее изобретение обеспечивает новые, замещенные фениламиноизоникотинамидные соединения и их фармацевтически приемлемые соли, которые являются ингибиторами MEK и пригодны для лечения вышеуказанных заболеваний. Соединения определяются формулой (I) и их фармацевтически приемлемые соли,где X представляет собой NH или О;R3, R4 независимо выбирают из водорода, SH или Hal;R5, R6 независимо выбирают из ОН, SH или NH2;Hal представляет собой F, Cl, Br или I. Предпочтительными являются соединения подформул (IA), (IB), (IC), (ID), (IE), (IF), (IG) и (IH) формулы (I) и их фармацевтически приемлемые соли, где в подформуле (IA)R3 представляет собой водород или Hal;R4 представляет собой водород или Hal;R2 представляет собой водород или Hal;R3 представляет собой водород или Hal;R4 представляет собой водород или Hal;R3 представляет собой водород или Hal;R4 представляет собой водород или Hal;R3 представляет собой водород или F;R4 представляет собой водород или Cl;R3 представляет собой водород или F;R4 представляет собой водород или Cl;R3 представляет собой водород или F;R4 представляет собой водород или Cl;R3 представляет собой водород или F;R4 представляет собой водород или Cl;R5, R6 представляют собой ОН. Более предпочтительная группа соединений формулы (I) и ее подформул (IA)-(IG) и (IH) соответствует формуле (II) в которой R1, R2, R3 и R4 имеют значения, указанные для формулы (I) или ее предпочтительных подформул (IA)-(IG) или (IH). Особенно предпочтительными соединениями в соответствии с формулой (I) и/или формулой (II) являются соединения из группы, включающей или их фармацевтически приемлемые соли. Если две структуры представлены со знаком "+" между ними, то это указывает на 1:1 рацемическую смесь двух энантиомеров. Соединения в соответствии с настоящим изобретением могут находиться в форме пролекарственного соединения. "Пролекарственное соединение" обозначает производное, которое превращается в биологически активное соединение в соответствии с настоящим изобретением в физиологических условиях в живом организме, например, путем окисления, восстановления, гидролиза или других, каждое из которых осуществляется ферментативно, или без задействования ферментативной среды. Примерами пролекарств являются соединения, где аминогруппа в соединении согласно настоящему изобретению ацилирована, алкилирована или фосфорилирована, например ейкозаноиламино, аланиламино, пивалоилоксиметиламино, или где гидроксильная группа ацилирована, алкилирована, фосфорилирована или превращена в борат, например ацетилокси, пальмитоилокси, пивалоилокси, сукцинилокси, фумарилокси, аланилокси, или где карбоксильная группа эстерифицирована или амидирована, или где сульфгидрильная группа образует дисульфидный мостик с молекулой носителя, например пептидом, которая доставляет лекарственное средство к мишени и/или в цитозоль клетки. Эти соединения могут быть получены из соединений согласно настоящему изобретению в соответствии с хорошо известными методами. Другими примерами пролекарств являются соединения, где карбоксилат в соединении согласно настоящему изобретению, например, превращен в алкил-, арил-, холин-, амино, ацилоксиметиловый сложный эфир, линоленоил-сложный эфир. Метаболиты соединений согласно настоящему изобретению также охватываются объемом настоящего изобретения. Если встречается таутомерия, например кето-енольная таутомерия, соединений согласно настоящему изобретению или их пролекарств, то индивидуальные формы, например кето- или енольная форма,заявляются отдельно и совместно в виде смесей в любом соотношении. Аналогичное применяется для стереоизомеров, например энантиомеров, цис-/транс-изомеров, конформеров и т.д. Если это является желательным, то изомеры могут быть разделены с помощью методов, хорошо известных в данной области техники, например путем жидкостной хроматографии. Аналогичное применяется для энантиомеров, например, путем применения хиральных неподвижных фаз. Дополнительно,энантиомеры могут быть выделены путем их превращения в диастереомеры, т.е. сочетания в энантио-5 018539 мерно чистым вспомогательным соединением, последующего разделения полученных диастереомеров и отщепления вспомогательного остатка. Альтернативно, любой энантиомер соединения согласно настоящему изобретению может быть получен при стереоселективном синтезе, используя оптически чистые исходные вещества. Соединения в соответствии с настоящим изобретением могут находиться в форме фармацевтически приемлемой соли или сольвата. Термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические основания или кислоты и органические основания или кислоты. В тех случаях, когда соединения в соответствии с настоящим изобретением содержат одну или несколько кислотных или основных групп, то изобретение также охватывает их соответствующие фармацевтически или токсикологически приемлемые соли, в частности их фармацевтически используемые соли. Таким образом, соединения в соответствии с настоящим изобретением, которые содержат кислотные группы, могут быть представлены в солевой форме, и могут использоваться в соответствии с изобретением, например в виде солей щелочных металлов, солей щелочно-земельных металлов или в виде солей аммония. Более предпочтительные примеры таких солей включают соли натрия, соли калия, соли кальция, соли магния или соли с аммиаком или органическими аминами, такими как, например, этиламин, этаноламин, триэтаноламин или аминокислотами. Соединения согласно настоящему изобретению содержат одну или несколько основных групп, т.е. групп, которые могут быть протежированы, могут быть представлены в солевой форме и могут использоваться в соответствии с изобретением в форме их солей присоединения с неорганическими или органическими кислотами. Примерами подходящих кислот являются соляная, бромисто-водородная,фосфорная, серная, азотная, метансульфоновая, п-толуолсульфоновая, нафталиндисульфоновые кислоты,щавелевая, уксусная, винная, молочная, салициловая, бензойная, муравьиная, пропионовая, пивалиновая,диэтилуксусная, малоновая, янтарная, пимелиновая, фумаровая, малеиновая, яблочная, сульфаминовая,фенилпропионовая, глюконовая, аскорбиновая, изоникотиновая, лимонная, адипиновая и другие кислоты, известные специалисту в данной области техники. Если соединения в соответствии с настоящим изобретением одновременно содержат кислотные и основные группы в молекуле, то изобретение также включает дополнительно к указанным солевым формам внутренние соли или бетаины (цвиттер-ионы). Соответствующие соли могут быть получены с помощью обычных методов, которые известны специалисту в данной области техники, например, путем их контактирования с органической или неорганической кислотой или основанием в растворителе или диспергаторе или путем обмена анионами или обмена катионами с другими солями. Настоящее изобретение также включает все соли соединений в соответствии с настоящим изобретением, которые, вследствие их низкой физиологической совместимости, не могут быть непосредственно пригодны для применения в лекарственных средствах, но которые могут применяться, например, в качестве промежуточных продуктов для химических реакций или для приготовления фармацевтически приемлемых солей. Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим соединение согласно настоящему изобретению, или его фармацевтически приемлемую соль в качестве активного компонента совместно с фармацевтически приемлемым носителем."Фармацевтическая композиция" обозначает один или несколько активных компонентов и один или несколько инертных компонентов, которые составляют носитель, а также любой другой продукт, которая образуется, непосредственно или опосредованно, при комбинации, комплексообразовании или агрегации любых двух или более компонентов, или при диссоциации одного или более компонентов, или при других типах реакций или взаимодействий одного или более компонентов. Следовательно, фармацевтические композиции согласно настоящему изобретению охватывают любую композицию, полученную при смешивании соединения согласно настоящему изобретению и фармацевтически приемлемого носителя. Фармацевтическая композиция согласно настоящему изобретению дополнительно может содержать одно или несколько других соединений в качестве активных компонентов, такие как одно или несколько дополнительных соединений согласно настоящему изобретению, или пролекарственное соединение или другие MEK ингибиторы. Фармацевтические композиции включают композиции, пригодные для перорального, ректального,местного, парентерального (включая подкожное, внутримышечное и внутривенное), глазного (офтальмического), легочного (назальной или буккальной ингаляции) или назального введения, хотя наиболее подходящий путь для любого данного случая будет зависеть от природы и тяжести состояний, подвергаемых лечению, и природы активного компонента. Они подходяще могут быть представлены в единичной дозированной форме и могут быть приготовлены с помощью любого из способов, хорошо известных в области фармацевтики. В одном варианте осуществления указанные соединения и фармацевтическая композиция предназначены для лечения злокачественного новообразования, такого как рак головного мозга, легкого, плоских клеток, мочевого пузыря, желудка, поджелудочной железы, молочной железы, головы, шеи, почки,почки, яичников, предстательной железы, ободочной и прямой кишки, пищевода, яичек, женских половых органов, щитовидной железы, меланомы, гемобластозов, таких как острый миелолейкоз, множественная миелома, хронический миелолейкоз, лейкоз миелоидных клеток, или любого другого типа солид-6 018539 ных или жидких опухолей. В другом варианте осуществления указанная фармацевтическая композиция предназначена для лечения незлокачественного гиперпролиферативного нарушения, такого как доброкачественная гиперплазия кожи (например, псориаз), рестеноз, поликистозное заболевание почек или предстательной железы (например, доброкачественная гипертрофия предстательной железы (ВРН, и также для лечения воспалительных и аутоиммунных заболеваний, таких как ревматоидный артрит, болезнь Крона, астма, неспецифический язвенный колит, синдром раздраженного кишечника, рассеянный склероз, красная волчанка и другие. В другом варианте осуществления указанная фармацевтическая композиция предназначена для лечения генетических состояний, которые характеризуются повышенной регуляцией MEK/ERK пути, такие как синдром Costello, синдром Noonan, кардиофациокожный синдром и другие. Изобретение также относится к применению соединений в соответствии с изобретением для приготовления лекарственного средства для лечения гиперпролиферативных заболеваний, связанных с гиперактивностью MEK, а также заболеваний, модулируемых MEK каскадом у млекопитающих, или нарушений, опосредуемых аберрантной пролиферацией, таких как злокачественное новообразование и воспаление. Изобретение также относится к соединению или фармацевтической композиции для лечения панкреатита или заболевания почек (включая пролиферативный гломерулонефрит и заболевание почек, индуцированное диабетом) или боли у млекопитающего, которая включает терапевтически эффективное количество соединения согласно настоящему изобретению или его фармацевтически приемлемой соли,пролекарства или гидрата и фармацевтически приемлемый носитель. Изобретение также относится к соединению или фармацевтической композиции для предотвращения имплантации бластоцитов у млекопитающего, которая включает терапевтически эффективное количество соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата и фармацевтически приемлемый носитель. Изобретение также относится к соединению или фармацевтической композиции для лечения заболевания, связанного с васкулогенезом или ангиогенезом у млекопитающего, которая включает терапевтически эффективное количество соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата и фармацевтически приемлемый носитель. В одном варианте осуществления указанное соединение или фармацевтическая композиция предназначены для лечения заболевания, выбранного из группы, включающей опухолевый ангиогенез, хроническое воспалительное заболевание, такое как ревматоидный артрит, воспалительное заболевание кишечника, атеросклероз, заболевания кожи, такие как псориаз, экзема и склеродерма, диабет, диабетическую ретинопатию, ретинопатию недоношенных, возрастную дегенерацию желтого пятна, гемангиому,глиому, меланому, саркому Капоши и рак яичников, молочной железы, легкого, поджелудочной железы,предстательной железы, ободочной кишки и эпидермиоидный рак. Изобретение также относится к применению для лечения гиперпролиферативного нарушения у млекопитающего, которое включает введение указанному млекопитающему терапевтически эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата. В одном варианте осуществления указанное применение относится к лечению злокачественного новообразования, такого как рак головного мозга, легкого, плоских клеток, мочевого пузыря, желудка,поджелудочной железы, молочной железы, головы, шеи, почки, почки, яичников, предстательной железы, ободочной и прямой кишки, пищевода, яичек, женских половых органов или щитовидной железы. В другом варианте осуществления указанное применение относится к лечению незлокачественных гиперпролиферативных нарушений, таких как доброкачественная гиперплазия кожи (например, псориаз), рестеноз или предстательной железы (например, ВРН). Изобретение также относится к применению для лечения гиперпролиферативного нарушения у млекопитающего, которое включает введение указанному млекопитающему терапевтически эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата, в комбинации с противоопухолевым средством, выбранным из группы,включающей ингибиторы митоза, алкилирующие средства, антиметаболиты, интеркалирующие антибиотики, ингибиторы факторов роста, антиангиогенные средства, ингибиторы клеточного цикла, ингибиторы ферментов, ингибиторы типоизомеразы, модификаторы биологического отклика, антигормональные средства, ингибиторы ангиогенеза и антиандрогены или иммуномодуляторы. Изобретение также относится к применению для лечения панкреатита или заболевания почек либо боли у млекопитающего, который включает введение указанному млекопитающему терапевтически эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата. Изобретение также относится к применению для предотвращения имплантации бластоцитов у млекопитающего, которое включает введение указанному млекопитающему терапевтически эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата. Изобретение также относится к применению для лечения заболеваний, связанных с васкулогенезом или ангиогенезом, у млекопитающего, которое включает введение указанному млекопитающему терапевтически эффективного количества соединения согласно настоящему изобретению или его фармацевтически приемлемой соли, пролекарства или гидрата. В одном варианте осуществления указанный способ предназначен для лечения заболевания, выбранного из группы, включающей опухолевый ангиогенез,хроническое воспалительное заболевание, такое как ревматоидный артрит, атеросклероз, воспалительное заболевание кишечника, заболевания кожи, такие как псориаз, экзема и склеродерма, диабет, диабетическую ретинопатию, ретинопатию недоношенных, возрастную дегенерацию желтого пятна, гемангиому,глиому, меланому, саркому Капоши и рак яичников, молочной железы, легкого, поджелудочной железы,предстательной железы, ободочной кишки и эпидермиоидный рак. Пациенты, которых можно подвергать лечению с применением соединений согласно настоящему изобретению или фармацевтически приемлемых солей, пролекарств и гидратов указанных соединений, в соответствии со способами согласно настоящему изобретению включают, например, пациентов, у которых был поставлен диагноз псориаз,рестеноз, атеросклероз, ВРН, рак легкого, рак костей, хронические миеломоноцитарные лейкозы, рак поджелудочной железы, рак кожи, рак головы и шеи, кожная или внутриглазная меланома, рак матки,рак яичников, рак прямой кишки, рак анальной области, рак желудка, рак ободочной кишки, рак молочной железы, опухоли яичек, женских половых органов (например, саркомы матки, карциномы фаллопиевых труб, карцинома эндометрия, карцинома шейки матки, карцинома влагалища или карцинома наружных женских половых органов), болезнь Ходжкина, рак пищевода, рак тонкого кишечника, рак эндокринной системы (например, рак щитовидной железы, паращитовидной железы или надпочечников), саркомы мягких тканей, рак уретры, рак пениса, рак предстательной железы, хронический или острый лейкоз, солидные опухоли у детей, лимфоцитарные лимфомы, рак мочевого пузыря, рак почки или уретры(например, почечно-клеточный рак, карцинома почечной лоханки), или новообразования центральной нервной системы (например, первичная лимфома ЦНС, опухоли оси позвоночника, глиомы ствола мозга или аденомы гипофиза). Настоящее изобретение также относится к соединению или фармацевтической композиции для ингибирования атипичного клеточного роста у млекопитающего, которая включает количество соединения согласно настоящему изобретению, или его фармацевтически приемлемой соли, или сольвата, или пролекарства, в комбинации с количеством другого противоракового терапевтического средства, где количества соединения, соли, сольвата или пролекарства, и химиотерапевтического средства совместно эффективны для ингибирования атипичного клеточного роста. В настоящее время в данной области известны многие противораковые терапевтические средства. В одном варианте осуществления противораковое терапевтическое средство представляет собой химиотерапевтическое средство, выбранное из группы,включающей ингибиторы митоза, алкилирующие средства, антиметаболиты, интеркалирующие антибиотики, ингибиторы факторов роста, ингибиторы клеточного цикла, фермента, ингибиторы типоизомеразы, модификаторы биологического отклика, антигормональные средства, ингибиторы ангиогенеза и антиандрогены. В другом варианте осуществления противораковое терапевтическое средство представляет собой антитело, выбранное из группы, включающей бевацизумаб, CD40-специфические антитела,chTNT-1/B, денозумаб, занолимумаб, IGF1R-специфические антитела, линтузумаб, эдреколомаб,WX G250, ритуксимаб, тицилимумаб, трастузумаб и цетуксимаб. Соединения по изобретению могут использоваться в способе ингибирования атипичного клеточного роста у млекопитающего или лечения гиперпролиферативного нарушения, который включает введение млекопитающему количества соединения согласно настоящему изобретению, или его фармацевтически приемлемой соли, или сольвата, или пролекарства, в комбинации с лучевой терапией, где количества соединения, соли, сольвата или пролекарства представлены в комбинации с лучевой терапией, эффективны для ингибирования атипичного клеточного роста или лечения гиперпролиферативного нарушения у млекопитающего. Техники введения лучевой терапии известны из уровня техники и эти техники можно использовать в комбинированной терапии, раскрытой в настоящем изобретении. Введение соединения согласно изобретению в этой комбинированной терапии можно определить, как описано в настоящем изобретении. Полагают, что соединения в соответствии с настоящим изобретением могут придавать атипичным клеткам большую чувствительность к лечению с применением лучевой терапии для уничтожения и/или ингибирования роста таких клеток. Также соединения по изобретению могут использоваться в способе сенсибилизации атипичных клеток у млекопитающего к лечению с применением лучевой терапии, который включает введение млекопитающему количества соединения согласно настоящему изобретению, или его фармацевтически приемлемой соли, или сольвата, или пролекарства, где количество является эффективным для сенсибилизации атипичных клеток к лечению с помощью лучевой терапии. Количество соединения, соли или сольвата в этом методе можно определить в соответствии с методами для установления эффективных количеств таких соединений, описанных в настоящем изобретении. Соединения по изобретению могут использоваться в способе ингибирования атипичного клеточного роста у млекопитающего, который включает количество соединения согласно настоящему изобретению, или его фармацевтически приемлемой соли, или сольвата, его пролекарства, или его производного, меченного радиоактивным изотопом, и количество одного или нескольких веществ, выбранных из антиангиогенных средств, ингибиторов переда-8 018539 чи сигналов и антипролиферативных средств. При практическом использовании соединения в соответствии с настоящим изобретением можно комбинировать в качестве активного компонента в тщательной смеси с фармацевтическим носителем в соответствии с общепринятыми техниками приготовления лекарственных средств. Носитель может иметь различные формы в зависимости от формы препарата, желательного для введения, например перорального или парентерального (включая внутривенное). Для приготовления композиций для пероральных дозированных форм можно использовать любые обычные фармацевтические среды, такие как, например, вода, гликоли, масла, спирты, ароматизаторы, консерванты, красители и другие. В случае пероральных жидких препаратов можно использовать любые обычные фармацевтические среды, такие как,например, суспензии, эликсиры и растворы; или носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие средства, замасливатели, связующие, дезинтеграторы и другие. В случае твердых пероральных препаратов композиция может находиться в таких формах, как,например, порошки, твердые и мягкие капсулы и таблетки, при этом твердые пероральные препараты являются преимущественными относительно жидких препаратов. В связи с простотой их введения таблетки и капсулы являются наиболее благоприятными пероральными дозируемыми единичными формами, в этих случаях обязательно используются твердые фармацевтические носители. Если это является желательным, то таблетки могут быть покрыты оболочкой с помощью стандартных водных или неводных техник. Такие композиции и препараты должны содержать по меньшей мере 0,1% активного соединения. Процент активного соединения в этих композициях, очевидно, может изменяться и подходяще может составлять от приблизительно 2 до приблизительно 60% от веса единицы. Количество активного соединения в таких терапевтически пригодных композициях будет таким, чтобы получить эффективную дозу. Активные соединения также можно вводить интраназально,например в виде жидких капель или спрея. Таблетки, пилюли, капсулы и другие формы также могут содержать связующее, такое как трагакантовую камедь, гуммиарабик, кукурузный крахмал или желатин; наполнители, такие как дикальций фосфат; дезинтегрирующее средство, такое как кукурузный крахмал, картофельный крахмал, альгиновую кислоту; замасливатель, такой как стеарат магния; и подсластитель, такой как сахароза, лактоза или сахарин. Если единичная дозированная форма представляет собой капсулу, то она может содержать дополнительно к материалам вышеописанного типа жидкий носитель, такой как жирное масло. Различные другие материалы могут быть представлены в виде покрытий или для модификации физической формы дозированной единицы. Например, таблетки могут быть покрыты с помощью шеллака,сахара или обоих веществ. Сироп или эликсир может содержать дополнительно к активному компоненту сахарозу в качестве подсластителя, метил и пропилпарабены в качестве консервантов, краситель и ароматизатор, такой как вишневый или апельсиновый ароматизатор. Соединения согласно настоящему изобретению также могут вводиться парентерально. Растворы или суспензии этих активных соединений могут быть приготовлены в подходящей воде, смешаны с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии также могут быть приготовлены в глицерине, жидких полиэтиленгликолях и их смесях в маслах. При обычных условиях хранения и применения эти препараты содержат консервант для предотвращения роста микроорганизмов. Фармацевтические формы, подходящие для инъекций, включают стерильные водные растворы или дисперсии и стерильные порошки для экстемпоральных препаратов стерильных инъекционных растворов или дисперсий. Во всех случаях, форма должна быть стерильной и должна быть жидкой до такой степени, чтобы ее легко можно было вводить с помощью шприца. Она должна быть стабильной в условиях изготовления и хранения и должна быть защищена от загрязняющего действия микроорганизмов,таких как бактерии и грибы. Носитель может переставлять собой растворитель или дисперсионную среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), их подходящие смеси и растительные масла. Любой подходящий путь введения может применяться для обеспечения млекопитающего, в особенности человека, эффективной дозой соединения согласно настоящему изобретению. Например, можно применять пероральный, ректальный, местный, парентеральный, глазной, легочный, назальный и другие пути. Дозированные формы включают таблетки, лепешки, дисперсии, суспензии, растворы, капсулы,кремы, мази, аэрозоли и другие. Предпочтительно соединения согласно настоящему изобретению вводят перорально. Эффективная дозировка применяемого активного компонента может изменяться в зависимости от конкретного применяемого соединения, способа введения, состояния, подвергаемого лечению, и тяжести состояния, подвергаемого лечению. Такие дозировки легко могут быть установлены специалистом в данной области техники. При лечении или предотвращении злокачественного новообразования, воспаления или других пролиферативных заболеваний, для которых показаны соединения согласно настоящему изобретению,обычно удовлетворительные результаты получают, если соединения в соответствии с настоящим изобретением вводят в суточной дозе от приблизительно 0,01 до приблизительно 100 мг на 1 кг веса тела жи-9 018539 вотного, предпочтительно представлено в виде единичной суточной дозы. Для наиболее крупных млекопитающих общая суточная доза составляет от приблизительно 0,1 до приблизительно 1000 мг, предпочтительно от приблизительно 0,2 до приблизительно 50 мг. Для взрослого человека весом 70 кг общая суточная доза обычно будет составлять от приблизительно 0,2 до приблизительно 200 мг. Эта схема дозирования может регулироваться для обеспечения оптимального терапевтического ответа. Изобретение также относится к комплекту (набору), состоящему из отдельных пакетов эффективного количества соединения в соответствии изобретением или его фармацевтически приемлемой соли и эффективного количества другого противоракового терапевтического средства, выбранного из группы,включающей ингибиторы митоза, алкилирующие средства, антиметаболиты, интеркалирующие антибиотики, ингибиторы факторов роста, ингибиторы клеточного цикла, ферменты, ингибиторы типоизомеразы, модификаторы биологического отклика, антигормональные средства, ингибиторы ангиогенеза,антиандрогены, бевацизумаб, CD40-специфические антитела, chTNT-1/В, денозумаб, занолимумаб,IGF1R-специфические антитела, линтузумаб, эдреколомаб, WX G250, ритуксимаб, тицилимумаб, трастузумаб и цетуксимаб. Комплект включает подходящие контейнеры, такие как коробки, отдельные бутылки, пакеты или ампулы. Комплект может содержать, например, отдельные ампулы, каждая из которых содержит эффективное количество соединения в соответствии с изобретением и/или его фармацевтически пригодных производных, сольватов и стереоизомеров, включая их смеси во всех соотношениях, и эффективного количества дополнительного активного компонента лекарственного средства в растворенной или лиофилизированной форме. Некоторые сокращения, которые могут использоваться в настоящем патенте, имеют следующие значения. Сокращения. Соединения в соответствии с настоящим изобретением могут быть приготовлены в соответствии с процедурами из следующих схем и примеров, используя подходящие материалы, и дополнительно иллюстрируются следующими специфическими примерами. Кроме того, при использовании процедур, описанных в настоящем патенте, в сочетании с типичными знаниями для квалифицированного специалиста в данной области техники, легко могут быть приготовлены дополнительные соединения согласно настоящему изобретению, заявленные в настоящем изобретении. Тем не менее, соединения, иллюстрируемые в примерах, не должны рассматриваться как составляющие только класс, охватываемый изобретением. Примеры дополнительно иллюстрируют детали приготовления соединений согласно настоящему изобретению. Для квалифицированных специалистов в данной области техники будет очевидным, что для приготовления этих соединений можно использовать известные модификации условий и процессов следующих препаративных процедур. Соединения, описанные в настоящем патенте, обычно выделяют в виде их фармацевтически прием- 11018539 лемых солей, таких как соли, описанные выше. Основания со свободным амином, соответствующие выделенным солям, могут быть получены путем нейтрализации с подходящим основанием, таким как водный гидрокарбонат натрия, карбонат натрия, гидроксид натрия и гидроксид калия, и экстрагирования выделенного в свободном состоянии основания со свободным амином в органическом растворителе, с последующим упариванием. Основание со свободным амином, выделенное таким образом, в дальнейшем может быть превращено в другую фармацевтически приемлемую соль путем растворения в органическом растворителе, с последующим добавлением подходящей кислоты и последующего упаривания,осаждения или кристаллизации. Иллюстрации приготовления соединений согласно настоящему изобретению представлены на следующих схемах. Если на схемах специально не указано иначе, то переменные имеют значения, аналогичные указанным выше. Настоящее изобретение также относится к способам приготовлений соединения формул (I), (II),подформула (IA)-(IH), а также соединений, представленных в табл. 1, в соответствии с нижеописанными схемами и примерами обработки. Схема 1 иллюстрирует общий путь синтеза, используемый для синтеза всех примеров 2.1-2.20 согласно формуле (I). Первым этапом является реакция конденсации между замещенным анилином (11) и 3-фтор-изоникотиновой кислотой, или ее производным (12), получая соответствующие промежуточные кислоты (13). Промежуточные кислоты, в свою очередь, подвергают реакции с подходящими аминами или спиртами, получая амиды или сложные эфиры (14) соответственно. В тех случаях, когда присутствует защитная группа (например, ацетонидная группа, или другая), включают подходящую стадию снятия защиты на завершающей стадии, получая соединения (15). Схема 1 Схема 2 иллюстрирует синтез предпочтительного аминдиольного промежуточного соединения, или его диол-защищенного аналога. Синтез начинают, исходя из рацемического циклогекс-2-енола (16), сначала с ацетилированием. Затем ацетильную группу (17) замещают стереоспецифическим образом в катализируемой палладием реакции с фталимидом калия в присутствии лиганда Trost (как описано Trost и др. в J. Am. Chem. Soc. 1994, 116, 4089-4090), получая соединение (18). После этого двойную связь синдигидроксилировали путем обработки с тетроксидом осмия, получая диол (19), затем либо путем непосредственного удаления фталимидо группы (получая амин 27), или сначала путем защиты диола в виде ацетонида (20) и затем удаления фталимидо группы, получая амин (21). Схема 2 На схеме 3 иллюстрируется синтез рацемических аминодиолов. Как описано в (1) Nishikawa, N.;Waring, M.J.; Winter, J.J.G.; Helliwell, M.; Newcombe, N.J.; Stemp, G. J. Org. Chem. 2002, 7946-7956, синтез начинали с перегруппировки Overman рацемического циклогекс-2-енола (16) (посредством трихлорацетимидатного промежуточного продукта), получая трихлорацетамид (22), который затем синдигидроксилировали путем обработки с тетроксидом осмия, получая диолы 23-26. Полученную диастереомерную смесь четырех аминодиолов (23-26) разделяют посредством ахиральной хроматографии на две рацемические пары (23/24 и 25/26), с которых затем можно снять защиту, получая соответствующие рацемические аминодиолы (27/28 и 29/30). Схема 3 Примеры Примеры, представленные ниже, предназначены для иллюстрации предпочтительных вариантов осуществления изобретения и никоим образом не предназначены для ограничения объема описания или формулы изобретения. 1. Анализы. Аналитическую ЖХ/МС осуществляли, используя два следующих метода. Метод A: Discovery С 18, 5 мкм, 330 мм колонку использовали при скорости потока 400 мкл/мин,пробоотборная петля 5 мкл, подвижная фаза: (А) вода с 0,1% муравьиной кислотой, подвижная фаза,(В) метанол с 0,1% муравьиной кислотой; время удерживания представлено в минутах. Подробности метода: (I) прогоняли на Quaternary Pump G1311 А (Agilent) с УФ/Вид. детектором на диодной матрицеG1315B (Agilent) и Finnigan LCQ Duo MS детектором в ESI+ режиме с УФ-определением при 254 и 280 нм с градиентом 15-95% (В) в 3,2-минутном линейном градиенте (II), выдерживая в течение 1,4 мин при 95% (В) (III), снижая от 95-15% (В) в 0,1-минутном линейном градиенте (IV), выдерживая в течение 2,3 мин при 15% (В). Метод В: Waters Symmetry С 18, 3,5 мкм, 4,675 мм колонка при скорости потока 1 мл/мин, пробоотборная петля 10 мкл, подвижная фаза (А) представляет собой воду с 0,05% ТФУ, подвижная фаза (В) представляет собой ACN с 0,05% ТФУ; время удерживания представлены в минутах. Подробности методов: (I) прогоняли на Binary Pump G1312A (Agilent) с УФ/Вид. детектором на диодной матрице G1315B(III), снижая от 20-85% (В) в 0,2-минутном линейном градиенте (IV), выдерживая в течение 3,8 мин при 20% (В). Аналитическая хиральная ВЭЖХ. Аналитическую хиральную ВЭЖХ осуществляли, используя колонку ChiralPak AD-H (2504,6 мм) от Daicel Chemical Industries, Ltd. на системе Agilent 1100 Series. В методе использовали вводимый объем 5,0 мкл, со скоростью потока 1 мл/мин 100% метанола в течение 15 мин при 25 С и УФ-обнаружении при 254 и 280 нм. Препаративная ВЭЖХ. Препаративную ВЭЖХ осуществляли, используя либо колонку Waters Atlantis dC18 OBD 10 мкМ(30250 мм) или колонку Waters Sunfire Prep C18 OBD 10 мкМ (30250 мм). Колонки использовали при скорости потока 60 мл/мин на Waters Prep LC 4000 System, оборудованной пробоотборной петлей (10 мл) и ISCO UA-6 УФ/Вид. детектором. Подвижная фаза поступала из двух резервуаров растворителей, содержащих (А) воду и (В) ацетонитрил ВЭЖХ-степени. При обычном препаративном прогоне использовали линейный градиент (например, 0-60% растворителя В в течение 60 мин). 2. Химический синтез. 2.1. Циклогекс-2-енил ацетат (17) К раствору 2-циклогексен-1-ола (16) (45,5 ммоль, 5,02 мл) в пиридине (0,27 моль, 22 мл) при 0 С добавляли по каплям уксусный ангидрид и реакционную смесь перемешивали при 0 С в течение получаса. Ледяную баню удаляли и смесь перемешивали при комнатной температуре в течение ночи. Смесь разводили с помощью EtOAc, промывали HCl (1 н.), насыщенным NaHCO3, солевым раствором и концентрировали в вакууме, получая 6 г (94%) неочищенного продукта 17, который использовали на следующей стадии без дополнительной очистки. ТСХ с 20% EtOAc/гексан, образование пятен с 5% H2SO4/EtOH,Rf для (16): 0,4, Rf для (17): 0,8. 2.2. 2-[(1R)-Циклогекс-2-ен-1-ил]-1 Н-изоиндол-1,3(2 Н)-дион (18) К смеси фталимида калия (160 ммоль, 29,6 г), N,N-(1S,2S)циклогексан-1,2-диил-бис-[2(дифенилфосфино)бензамида] (лиганд Trost) (6 ммоль, 4,14 г), [Pd(п-аллил)Cl]2 (2 ммоль, 0,73 г) и бромида тетрагексиламмония (216 ммоль, 94 г) в безводном ДХМ (40 мл) в атмосфере N2, добавляли 2 циклогексен-1-ил ацетат (17) (13 г неочищенного) в 40 мл безводного ДХМ. Смесь перемешивали при температуре окружающей среды в течение ночи. После этого реакцию закаливали водой (250 мл) и экстрагировали с помощью ДХМ (3250 мл). Органические компоненты объединяли и промывали солевым раствором (400 мл), высушивали над Na2SO4 и концентрировали, получая неочищенный продукт в виде желтого масла. При перекристаллизации из МеОН получали 9,024 г продукта (чистота: 100% с помощью ВЭЖХ, выход 50%). Маточную жидкость очищали путем флэш-хроматографии (10% EtOAc/гексаны),получая 8,8 г (18) (чистота 86% с помощью ВЭЖХ, выход 48%). ЯМР и МС данные соответствуют публикации: Trost, В.М.; Bunt, R.С. J. Am. Chem. Soc. 1994, 4089-4090. 2.3. 2-[(1R,2S,3R)-2,3-дигидроксициклогексил]-1 Н-изоиндол-1,3(2 Н)-дион (19)(0,2 ммоль, 50 мг). Реакцию перемешивали в течение 4 ч. Смесь концентрировали и добавляли насыщенный сульфит натрия (40 мл). Затем его сразу экстрагировали с помощью EtOAc три раза. Объединенные органические слои промывали водой и солевым раствором, высушивали над MgSO4 и концентрировали,получая неочищенный продукт в виде белого твердого вещества (4,94 г, выход 95%). Хиральная ВЭЖХ[8,53 мин]. Аналитические данные соответствуют опубликованным данным (Donohoe, J.Т.; и др., J. Org. К раствору 2-[(1R,2S,3R)-2,3-дигидроксициклогексил]-1 Н-изоиндол-1,3(2 Н)-диона (19) (50,0 г,191,4 ммоль) и 2,2-метоксипропана (500 мл, 4,07 моль) в ацетоне (500 мл) добавляли каталитическое количество п-толуолсульфоновой кислоты и полученную реакционную смесь перемешивали при комнат- 14018539 ной температуре в течение 3 ч. Реакцию закаливали с помощью насыщенного раствора Na2CO3 для доведения рН до 10 и растворитель удаляли. К остатку добавляли воду (750 мл) и экстрагировали с помощью этилацетата (2750, 500 мл). Органические компоненты объединяли, высушивали над Na2SO4 и концентрировали, получая желательный продукт в виде белого твердого вещества (55,8 г, 96,8% выход). ЖХ/МС [метод В: rt 6,16 мин; m/z: 302 (М+1)]. 2.5. (3aS,4R,7aR)-2,2-диметилгексагидро-1,3-бензодиоксол-4-амин (21)(20) (191,4 ммоль, 50,00 г), гидразина (308,4 ммоль, 15,0 мл) в этаноле (500 мл) нагревали в колбе с обратным холодильником в течение 4 ч. За осуществлением реакции наблюдали с помощью ВЭЖХ. ВЭЖХ анализ указывал на то, что реакция не завершилась (86% превращение). К реакционной смеси дополнительно добавляли 3 мл гидразина и нагревали в колбе с обратным холодильником в течение 2 ч. Реакцию охлаждали до 0 С и фильтровали. Осадок на фильтре промывали IPA и высушивали, получая желательный продукт 21 (9,52 г, выход 21%, весовая чистота: 84% на основании ЯМР). Фильтрат концентрировали и остаток ресуспендировали в IPA (около 200 мл). Побочный продукт кристаллизовали и отфильтровывали, из фильтрата удаляли летучие компоненты и высушивали, получая вторую партию желательного продукта 21 (28,46 г, 64% выход). ТСХ (80% МеОН/EtOAc с 1 каплей уксусной кислоты, пятна образовались путем погружения в нингидрин). 2.6. 3-[(2-Фтор-4-йодфенил)амино]изоникотиновая кислота Смесь 2-фтор-4-йод-фениламина (20,0 г, 84,38 ммоль) в безводном тетрагидрофуране (80 мл) охлаждали до -65 С в инертной атмосфере, затем медленно добавляли 1,0 М бис-(триметилсилил)амид лития(255 мл, 255 ммоль) с такой скоростью, чтобы внутреннюю температуру поддерживать ниже -55 С. После завершения добавления густую взвесь перемешивали в течение 30 мин и затем обрабатывали с помощью 3-фтор-изоникотиновой кислоты (8,0 г, 56,69 ммоль). Смесь перемешивали при комнатной температуре в течение 4 дней и после этого вливали в водный 2,0 н. гидроксид натрия (1000 мл) и этилацетата (250 мл). Слои разделяли и органические компоненты снова экстрагировали с помощью водного гидроксида натрия (21000 мл). рН объединенных водных фракций доводили до 2 с помощью концентрированной соляной кислоты, что вызывало осаждение твердого вещества. Материал фильтровали, промывали водой (300 мл) и высушивали в высоком вакууме при 40 С в течение 18 ч, получая продукт (19,05 г,53,19 ммоль, 94%) в виде желтого твердого вещества. 2.7. N-[(3aS,4R,7aR)-2,2-диметилгексагидро-1,3-бензодиоксол-4-ил]-3-[(2-фтор-4-йодфенил)амино]изоикотинамид (31) Суспензию 3-[(2-фтор-4-йодфенил)амино]изоникотиновой кислоты (10,46 г, 29,2 ммоль),(3aS,4R,7aR)-2,2-диметилгексагидро-1,3-бензодиоксол-4-амина (21) (5,00 г, 29,2 ммоль), 1 гидроксибензотриазола (4,41 г, 29,2 ммоль) и EDCl (5,6 г, 29,2 ммоль) в ДМФА (100 мл) перемешивали при комнатной температуре в течение ночи. Реакцию закаливали с помощью воды (150 мл) и экстрагировали с помощью этилацетата (150 мл). Образовалась эмульсия и ее собирали, фильтровали и фильтрат объединяли с органическим слоем. Органический слой промывали насыщенным раствором NaHCO3(150 мл) и водой (2150 мл), солевым раствором и высушивали над Na2SO4 и концентрировали, получая коричневую пену. Неочищенное вещество очищали путем кристаллизации из IPA. Маточную жидкость концентрировали и очищали путем флэш-хроматографии, получая желательный продукт (12 г, выход 80%). ЖХ/МС [метод A: rt: 7,35 мин; m/z: 512 (М+1)]. 2.8. (N-[(1R,2S,3R)-2,3-дигидроксициклогексил]-3-[(2-фтор-4-йодфенил)амино]изоникотинамид) (8) 17,62 мл 2 М HCl в простом диэтиловом эфире добавляли к коричневому раствору N-[(3aR,4S,7aS)2,2-диметилгексагидро-1,3-бензодиоксол-4-ил]-3-[(2-фтор-4-йодфенил)амино]изоникотинамида(6,65 ммоль, 3,15 г) в МеОН (64 мл) и полученную реакционную смесь перемешивали при комнатной температуре в течение 5 ч. Реакционную смесь концентрировали до приблизительно 30 мл и образовывалось желтое твердое вещество, фильтровали, получая продукт в виде гидрохлоридной соли. К соли добавляли 10 мл МеОН и добавляли гидроксид аммония до получения рН 10. Образовывалось белое твердое вещество и твердое вещество (1,83 г, выход 58%) собирали путем фильтрации, промывали водой. Фильтрат концентрировали и получали вторую партию в виде белого твердого вещества (0,56 г, выход 18%). ЖХ/МС [метод В: rt: 5,83 мин; m/z: 472 (М+1)]. Хиральная ВЭЖХ [7,06 мин]. 2.9. 2,2,2-Трихлор-N-циклогекс-2-ен-1-илацетамид (22)DBU (41,88 г, 0,275 моль) и смесь охлаждали до -20 С. К этому раствору добавляли по каплям трихлорацетонитрил (47,66 г, 0,330 моль). Реакцию перемешивали в течение 3 ч при -20 С и закаливали с помощью водного раствора хлорида аммония. Органическую фазу отделяли и высушивали над карбонатом калия. Растворитель удаляли в вакууме, получая промежуточный циклогекс-2-ен-1-ил 2,2,2 трихлорэтанимидоат. Его растворяли в толуоле (100 мл) и обрабатывали карбонатом калия (30 г). Смесь нагревали в колбе с обратным холодильником в течение 12 ч. После охлаждения смесь фильтровали через целит и фильтрат упаривали, получая 9 г (20%) (22) в виде твердого вещества. ЖХ/МС: Обнаруженная масса (m/z, МС, 242,9). Метод: А - 0,1% НСООН, В - ACN (70%), поток - 0,8 мл/мин. Колонка: К раствору 2,2,2-трихлор-N-циклогекс-2-ен-1-илацетамида (22) (4,5 г, 0,0182 моль) и N-метил морфолин-N-оксида (7,5 г, 0,055 моль) в ацетоне (100 мл) добавляли воду (25 мл), затем добавляли каталитическое количество тетроксида осмия (0,1 г, твердое вещество). Реакционную смесь перемешивали при комнатной температуре в течение 14 ч и закаливали с помощью насыщенного раствора сульфита натрия(20 мл). Смесь дополнительно перемешивали в течение 20 мин и затем растворитель удаляли в вакууме и остаток очищали путем хроматографии, используя петролейный эфир/этилацетат (3/7) в качестве элюента, получая 3,3 г (60%) рацемического диола в виде твердого вещества. ЖХ/МС: Обнаруженная масса Смесь 2,2,2-трихлор-N-[(1RS,2SR,3RS)-2,3-дигидроксициклогексил]ацетамида (23 и 24) (2,3 г) и 5 М водной HCl (30 мл) нагревали в колбе с обратным холодильником в течение 10 ч и упаривали при пониженном давлении, получая 1,2 г (86%) указанного в заглавии соединения в виде вязкой жидкости. МС: масса (m/z, МС, 131,9), ВЭЖХ 98%. Метод: А - Вода, В - CAN, поток - 0,8 мл/мин. Колонка: С 18 XDB,2504,6 мм, SC/276. Rt (мин): 2,715. 1 Н ЯМР (CD3OD, 400 МГц)1,43-1,91 (6 Н, m), 2,98-3,02 (1 Н, m), 3,31-3,42 (1 Н, m), 3,84 (1 Н, m). 3-Фтор-5-(2-фтор-4-йод-фениламино)изоникотиновую кислоту (119 мг, 0,32 ммоль) и 1,1'карбонил-бис-(1 Н-имидазол) (67 мг, 0,41 ммоль) суспендировали в ДМСО (2 мл). Смеси позволяли перемешиваться в течение ночи (12 ч) при комнатной температуре. Затем добавляли 3-аминоциклогексан 1,2-диол (рацемический 27/28) (40 мг, 0,31 ммоль) и триэтиламин (0,07 мл, 0,49 ммоль). Смесь перемешивали дополнительно в течение 6 ч при комнатной температуре. После завершения реакции реакционную смесь обрабатывали водным гидроксидом натрия (1 мл, 1,0 М) и перемешивали при комнатной температуре в течение 4 ч. Раствор нейтрализовали до рН 7 с помощью конц. HCl. После этого смесь упаривали для удаления большего количества воды. Полученную смесь очищали путем препаративной ВЭЖХ,получая продукт (2). ЖХ/МС [метод A: rt: 4,89 мин; m/z: 490 (М+1)]. 2.13. 3-(2-Фтор-4-йод-фениламино)изоникотиновую кислоту (300 мг, 0,84 ммоль) и 1,1'-карбонил-бис(1 Н-имидазол) (177 мг, 1,1 ммоль) суспендировали в ДМСО (4 мл). Смеси позволяли перемешиваться в течение ночи (12 ч) при комнатной температуре. После этого добавляли 3-аминоциклогексан-1,2-диол(рацемический 27/28) (110 мг, 0,84 ммоль). Смесь перемешивали дополнительно в течение 12 ч при комнатной температуре. Смесь разделяли путем препаративной ВЭЖХ, получая два продукта, рацемический амид (3) и рацемические сложные эфиры (4). ЖХ/МС [метод A: rt: 6,01 мин; m/z: 472 (М+1)]. Хиральная ВЭЖХ [7,10 мин, 13,12 мин]. 2.14. 2-Хлор-5-(2-фтор-4-йод-фениламино)изоникотиновую кислоту (75 мг, 0,19 ммоль) и 1,1'-карбонилбис-(1 Н-имидазол) (40 мг, 0,25 ммоль) суспендировали в ДМСО (2,5 мл). Смеси позволяли перемешиваться в течение ночи (12 ч) при комнатной температуре. После этого добавляли 3-аминоциклогексан 1,2-диол (рацемический 27/28) (32 мг, 0,19 ммоль) и триэтиламин (0,05 мл, 0,38 ммоль). Смесь перемешивали в течение ночи при комнатной температуре. Смесь очищали путем препаративной ВЭЖХ, получая продукт ЖХ/МС [метод В: rt: 5,359 мин; m/z: 506 (М+1)]. 2.16. 3-(4-Бром-2-фторфениламино)-N-1RS,2SR,3RS)-2,3-дигидроксициклогексил)изоникотинамид (5) Получен по общей методике для (1), ЖХ/МС [метод В: rt: 5,602 мин; m/z: 425 (М+1)]. Получен по общей методике для (1), ЖХ/МС [метод В: rt: 6,163 мин; m/z: 441 (M+1)]. 2.18. N-1R,2S,3R)-2,3-Дигидроксициклогексил)-3-(2-фторфениламино)изоникотинамид (10) В запечатанную трубку загружали (N-[(1R,2S,3R)-2,3-дигидроксициклогексил]-3-[(2-фтор-4 йодфенил)амино]изоникотинамид) (8) (42 мг, 0,09 ммоль), борогидрид натрия (34 мг, 0,89 ммоль), дихлорид палладия (7,9 мг, 0,04 ммоль), гидроксид натрия (17,8 мг, 0,45 ммоль), ТГФ-воду (1:1, 3 мл). Смесь перемешивали при комнатной температуре в течение 2 дней. Смесь фильтровали и фильтрат концентрировали. Полученный остаток подвергали флэш-хроматографии, получая продукт. ЖХ/МС [метод Получен по общей методике для (1) за исключением того, что вместо рацемической смеси диолов использовали хирально чистый 3-аминоциклогексан-1,2-диол (27). ЖХ/МС [метод В: rt: 5,19 мин; m/z: 426,1 (М+1)]. 2.20. N-1S,2R,3S)-2,3-дигидроксициклогексил)-3-(2-фтор-4-йодфениламино)изоникотинамид (7) Получен по общей методике для (1) за исключением того, что вместо рацемической смеси диолов использовали хирально чистый 3-аминоциклогексан-1,2-диол (28). ЖХ/МС [метод A: rt: 4,54 мин; m/z: 472,3 (М+1)]. 3. Биологическая активность. 3.1. Исследование фермента MEK-1 (LANCE-HTRF). Активность соединений в соответствии с настоящим изобретением можно определить с помощью следующей процедуры: за ингибированием активности MEK1 киназы человека наблюдали с помощью гомогенного анализа на основе флуоресценции. В анализе использовали резонансный перенос энергии флуоресценции с разрешением во времени к пробе для фосфорилирования ERK1 с помощью MEK1. Анализ осуществляли в низкообъемных микротитровальных планшетах на 96 лунок. В общем объеме 15 мкл соединения инкубировали с 100 нМ MEK1, 15 мкМ АТР, 300 нМ ERK2, применяя буфер, содержащий 20 мМ TRIS/HCl, 10 мМ MgCl2, 100 мкМ NaVO4, 1 мМ DTT и 0,005% Твин 20 (рН 7,4). Через два часа, добавляли 5 нМ европий-анти-PY20 (Perkin Elmer) и 50 нМ анти-GST-аллофикоцианина (CisBio) в буфере, содержащем 50 мМ EDTA и 0,05% BSA и реакцию инкубировали в течение 1 ч в темноте. Флуоресценцию с временным разрешением измеряли с помощью LJL-Analyst (Molecular Devices) при длине волны возбуждения 340 нм и длине волны эмиссии 665 нм. Конечная концентрация ДМСО составляла 2%. Для оценки ингибирующего потенциала соединений определяли значения IC50, как показано в таблице далее. 3.2. Исследование пролиферации опухолевых клеток (ATP Lite). Клетки С 26 ободочной кишки мышей, А 375 меланомы человека и MiaPaCa-2 поджелудочной железы человека помещали в белые планшеты Corning на 96 лунок (1500 клеток/лунку для С 26, и 2000 клеток/лунку для А 375, и MiaPaCa-2) и культивировали в течение ночи при 37 С в 5% CO2. Ингибиторы серийно разводили в 100% ДМСО и после этого добавляли к клеткам для достижения конечной концентрации 0,25% ДМСО. Клетки инкубировали в течение 4 дней в присутствии тестируемых соединений в среде для роста клеток (DMEM с 10% фетальной бычьей сывороткой, 2 мМ глутамином для С 26, и MiaPaCa-2, и RPMI с 10% фетальной бычьей сывороткой, 2 мМ глутамином для А 375). Количественно определяли пролиферацию клеток с помощью набора для определения пролиферации клеток ATP lite (Packard). Ингибирование пролиферации клеток представлено в таблице далее. В столбцах 2-5 представлена концентрация соединений, необходимая для индуцирования гибели 50% клеток (IC50 в мкМ) эндометрия человека. 3.3. Исследования эффективности in vivo (модели ксенотрансплантатов у мышей). Самцам "голых" (nu/nu) мышей подкожно инъецировали выше правой передней лапы определенное количество клеток опухолевых клеточных линий человека, таких как Colo-205, A375 или MiaPaCa2. Опухоли измеряли с помощью штангенциркуля через одну неделю после имплантации клеток. Измеряли длину опухоли (д) и ширину (ш) и объем опухоли рассчитывали с помощью уравнения дш 2/2. Животных разделяли на группы таким образом, чтобы в каждой группе средний объем опухоли составлял 150200 мм 3, и начинали обработки с применением соединений (обозначали как День 0). Для каждого животного измеряли объем опухоли и вес тела в Дни 0, 4, 6, 8, 10, 12 и 14. Объем опухоли и процент веса тела анализировали с помощью двойного дисперсионного анализа повторных измерений (Two-Way RepeatedMeasures Analysis of Variance (RM-ANOVA с последующим многократным попарным сравнением полученных результатов согласно Fisher для средних значений леченных групп. Соединения в этом варианте осуществления изобретения являются эффективными на этих моделях опухолей и приводят к дозозависимому ингибированию роста опухоли, включая регресс опухоли или опухоль. Например, соединение(9) вызывает ингибирование роста опухоли на 98,5% на модели ксенотрансплантата Colo-205 при ежедневном введении в дозе 1,5 мг/кг при пероральном пути введения. Соединение (8) на аналогичной модели вызывает ингибирование роста опухоли на 100,69% (TGI) при введении в дозе 50 мкг/кг/сутки при пероральном пути введения и ингибирование роста опухоли на 115,33% при введении в дозе 150 мкг/кг/сутки. На модели MiaPaCa2 соединение (8) обеспечивает 100,97% TGI при введении в дозе 33 мкг/кг/сутки и 110,74% TGI при введении в дозе 50 мкг/кг/сутки. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) и его фармацевтически приемлемые соли,где X представляет собой NH или О;R3, R4 независимо выбирают из водорода, SH или Hal;R5, R6 независимо выбирают из ОН, SH или NH2;Hal представляет собой F, Cl, Br или I. 2. Соединение в соответствии с п.1, где радикалы, не указанные подробно, имеют значения, указанные для формулы (I) в соответствии с п.1, но где в подформуле (IA)R3 представляет собой водород или Hal;R4 представляет собой водород или Hal;R2 представляет собой водород или Hal;R3 представляет собой водород или Hal;R4 представляет собой водород или Hal;R3 представляет собой водород или Hal;R4 представляет собой водород или Hal;R3 представляет собой водород или F;R4 представляет собой водород или Cl;R3 представляет собой водород или F;R4 представляет собой водород или Cl;R3 представляет собой водород или F;R4 представляет собой водород или Cl;R3 представляет собой водород или F;R4 представляет собой водород или Cl;R5, R6 представляют собой ОН,и его фармацевтически приемлемые соли. 3. Соединение в соответствии с п.1 или 2, которое соответствует формуле (II) и его фармацевтически приемлемые соли,где R1, R2, R3 и R4 имеют значения, указанные для формулы (I) или ее подформул (IA)-(IG) или (IH). 4. Соединение в соответствии с п.1, где соединение выбирают из группы, включающей: и их фармацевтически приемлемые соли. 5. Фармацевтическая композиция, которая содержит соединение в соответствии с любым из пп.1-4,или его фармацевтически приемлемую соль, в качестве активного компонента совместно с фармацевтически приемлемым носителем. 6. Применение соединения в соответствии с любым из пп.1-4 или его фармацевтически приемлемой соли для приготовления лекарственного средства для лечения гиперпролиферативных заболеваний, связанных с гиперактивностью MEK, а также заболеваний, модулируемых MEK каскадом у млекопитающих. 7. Применение в соответствии с п.6, где заболевание выбирают из группы, включающей злокачественное новообразование, воспаление, панкреатит или заболевание почек, боль, доброкачественную гиперплазию кожи, рестеноз, предстательной железы, заболевания, связанные с васкулогенезом или ангиогенезом, опухолевым ангиогенезом, заболевания кожи, выбранные из псориаза, экземы и склеродермы,диабет, диабетическую ретинопатию, ретинопатию недоношенных, возрастную дегенерацию желтого пятна, гемангиому, глиому, меланому и саркому Капоши. 8. Комплект (набор), состоящий из отдельных пакетов эффективного количества соединения в соответствии с одним или несколькими пп.1-4 или его фармацевтически приемлемой соли и эффективного количества другого противоракового терапевтического средства, выбранного из группы, включающей ингибиторы митоза, алкилирующие средства, антиметаболиты, интеркалирующие антибиотики, ингибиторы факторов роста, ингибиторы клеточного цикла, фермента, ингибиторы типоизомеразы, модификаторы биологического отклика, антигормональные средства, ингибиторы ангиогенеза, антиандрогены,бевацизумаб, CD40-специфические антитела, chTNT-1/B, денозумаб, занолимумаб, IGFIRспецифические антитела, линтузумаб, эдреколомаб, WX G250, ритуксимаб, тицилимумаб, трастузумаб и цетуксимаб. Евразийская патентная организация, ЕАПВ Россия, 109012, Москва, Малый Черкасский пер., 2
МПК / Метки
МПК: C07D 213/81, C07D 213/79, A61K 31/44, A61P 35/00, A61P 29/00
Метки: соединения, фениламино-изоникотинамидные
Код ссылки
<a href="https://eas.patents.su/23-18539-fenilamino-izonikotinamidnye-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Фениламино-изоникотинамидные соединения</a>
Способ получения 2-(n-фениламино) бензойных кислот

Номер патента: 4209
Опубликовано: 26.02.2004
Авторы: Мэйгэно Джавьер, Чен Майкл Хьюэй Гу
МПК: C07C 227/08
Метки: кислот, способ, бензойных, получения, 2-(n-фениламино
Формула / Реферат:
1. Способ получения 2-(N-фениламино)бензойных кислот, включающий в себя стадию взаимодействия бензойной кислоты, имеющей формулу I и анилина, имеющего формулу II с гексаметилдисилазидом щелочного металла в количестве 3 эквивалента или более по отношению к бензойной кислоте, с образованием 2-(N-фениламино)бензойной кислоты, имеющей формулу III где каждый R независимо представляет собой водород, галоген, C1-C6алкил, -OC1-C6алкил, CN или NO2,...
4-(n-фениламино)хиназолины/-хинолины в качестве ингибиторов тирозинкиназы

Номер патента: 9300
Опубликовано: 28.12.2007
Авторы: Солка Флавио, Химмельсбах Франк, Юнг Биргит
МПК: C07D 239/94, A61P 35/00, A61K 31/517...
Метки: ингибиторов, тирозинкиназы, качестве
Формула / Реферат:
1. Бициклические гетероциклы общей формулы в которой Ra обозначает атом водорода, Rb обозначает фенил, в котором фенильное ядро замещено остатками R1-R3, где R1 и R2 могут иметь идентичные или различные значения и обозначают атом водорода, фтора, хлора, брома или иода, C1-4алкильную группу, C1-4алкоксигруппу, С2-3алкинильную группу, арилметоксигруппу, гетероарилоксигруппу, гетероарилметоксигруппу и R3 обозначает атом водорода, Rc обозначает...
Соли е-2-метокси-n-(3- (4- (3- метилпиридин -3- илокси) фениламино) хиназолин -6- ил-аллил)ацетамида, их получение и их применение против рака

Номер патента: 7412
Опубликовано: 27.10.2006
Авторы: Ричтер Дэниел Тайлер, Кэт Джон Чарлз
МПК: A61K 31/505, C07D 401/12
Метки: хиназолин, метилпиридин, рака, получение, ил-аллил)ацетамида, фениламино, против, илокси, соли, применение, е-2-метокси-n-(3
Формула / Реферат:
1. Сукцинат Е-2-метокси-N-(3-{4-[3-метил-4-(6-метилпиридин-3-илокси)фениламино]хиназолин-6-ил}аллил)ацетамида. 2. Соединение по п.1, в котором сукцинатом является сесквисукцинат Е-2-метокси-N-(3-{4-[3-метил-4-(6-метилпиридин-3-илокси)фениламино]хиназолин-6-ил}аллил)ацетамида. 3. Способ лечения рака у млекопитающих, который включает введение указанному млекопитающему такого количества сесквисукцината...
Кристаллическая кальцевая соль [r-(r*,r*)]-2-(4-фторфенил)-β,δ- дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1н-пирролгептановой кислоты

Номер патента: 9795
Опубликовано: 28.04.2008
Авторы: Ергов Александр, Фаустманн Иржи
МПК: C07D 207/327
Метки: соль, кристаллическая, кислоты, дигидрокси-5-(1-метилэтил)-3-фенил-4-[(фениламино)карбонил]-1н-пирролгептановой, кальцевая, r-(r*,r*)]-2-(4-фторфенил)-β,&delta
Формула / Реферат:
1. Соединение, представляющее кристаллическую форму Fa кальций аторвастатина, имеющее картину дифракции рентгеновских лучей, включающую следующие значения 2q, измеренные с использованием излучения CuK-a: примерно 10,2, примерно 11,3 или примерно 18,9. 2. Соединение по п.1, имеющее спектр 13C ЯМР в твердом состоянии вещества, включающий по меньшей мере один химический сдвиг при примерно 20,7, примерно 23,3, примерно 24,5 или примерно 26,1 м.д. 3....
Соединения-ингибиторы дипептидилпептидазы-iv, способы их получения, а также фармацевтические композиции, содержащие указанные соединения в качестве активного ингредиента

Номер патента: 12591
Опубликовано: 30.10.2009
Авторы: Ким Геун Тае, Хан Хее Оон, Лим Донгчул, Ли Чанг-Сеок, Йеом Зи-Хо, Бу Сеонг Чеол, Кох Дзонг Сунг, Ким Киоунг-Хее, Хур Гвонг-Чеунг, Ким Сунгсуб, Ким Сунг Хо, Хонг Санг Йонг, Ким Дзи Янг, Квон Ох Хван, Ким Мин-Дзунг, Йим Хиеон Дзоо, Йео Донг-Дзун, Ким Хие Дзин, Коо Ки Донг
МПК: A61K 31/452, A61P 3/10, A61K 31/444...
Метки: фармацевтические, указанные, получения, качестве, также, ингредиента, дипептидилпептидазы-iv, соединения, соединения-ингибиторы, содержащие, композиции, способы, активного
Формула / Реферат:
1. Соединение формулы (1) или его фармацевтически приемлемая соль где (А) А выбран из группы, состоящей из заместителей следующих формул со (2) по (7): где R1 представляет собой водород или С1-С4алкил, необязательно замещенный галогеном или гидроксилом; и X представляет собой углерод или азот; где R2 представляет собой водород или С1-С4алкил, необязательно замещенный галогеном или гидроксилом; где R3 представляет собой водород или...
Предыдущий патент: Солнцезащитное остекление, обладающее повышенным коэффициентом светопропускания
Следующий патент: Способ проходки шахтного ствола в рыхлых грунтах, залегающих с поверхности земли
Случайный патент: Способ и устройство для получения металлического порошка