Усовершенствованный способ получения 2-[(2e)-2-фтор-2-(3-пиперидинилиден)этил]-1h-изоиндол-1,3(2h)-диона






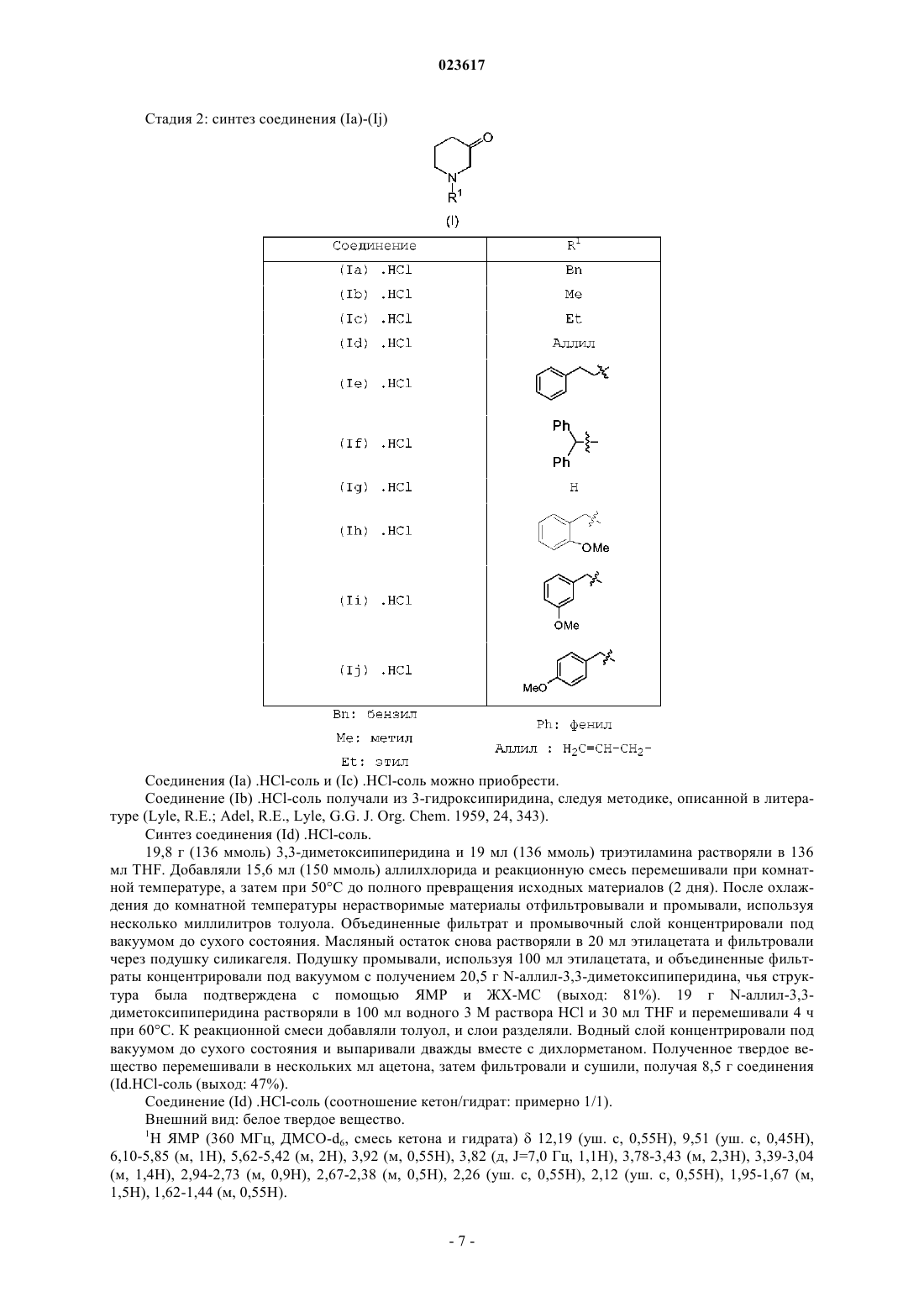
Формула / Реферат
1. Способ получения соединения (1) или его соли присоединения кислоты

который включает следующие стадии:
а) взаимодействие соединения формулы (I) с соединением формулы (II) в инертном растворителе

где R1 представляет собой арилметил или арилэтил;
R2 представляет собой С1-6алкил или фенил;
А представляет собой С1-4алкилоксикарбонил или аминокарбонил;
X представляет собой галоген;
где арил представляет собой фенил,
b) восстановление соединения (III), которое представляет собой смесь (Е)-(III) и (Z)-(III), до соединения (IV), которое представляет собой смесь (E)-(IV) и (Z)-(IV), с последующим выделением соединения (E)-(IV) или его соли в виде осадка

с) взаимодействие соединения формулы (Е)-(IV) с соединением (V) в условиях реакции Мицунобу с получением соединения формулы (VI), которое преобразуют в соединение (1) или его соль присоединения кислоты путем удаления заместителя R1

2. Способ по п.1, где R1 представляет собой арилметил, где арил является фенилом.
3. Способ по п.2, где R2 представляет собой метил, этил, бутил, изобутил или фенил; X представляет собой бром; А представляет собой аминокарбонил или С1-4алкилоксикарбонил, где С1-4алкил является метилом, этилом или трет-бутилом.
4. Способ по любому из пп.1-3, в котором R1 представляет собой арилметил, где арил является фенилом; R2 представляет собой метил; X представляет собой бром; А представляет собой С1-4алкилоксикарбонил, где С1-4алкил является этилом.
5. Способ получения соединения формулы (III), включающий взаимодействие соединения формулы (I) с соединением формулы (II) в инертном растворителе

где R1 представляет собой С1-4алкил, аллил, арилметил или арилэтил;
R2 представляет собой С1-6алкил или фенил;
А представляет собой С1-4алкилоксикарбонил или аминокарбонил;
X представляет собой галоген;
где арил представляет собой фенил или фенил, замещенный одним С1-4алкилокси.
6. Способ по п.5, где R1 представляет собой С1-4алкил, арилметил или аллил, где арил является фенилом.
7. Способ по п.6, где R2 представляет собой метил, этил, бутил, изобутил или фенил; X представляет собой бром; А представляет собой аминокарбонил или С1-4алкилоксикарбонил, где С1-4алкил является метилом, этилом или трет-бутилом.
8. Способ по п.7, в котором R1 представляет собой арилметил, где арил является фенилом; R2 представляет собой метил; X представляет собой бром; А представляет собой С1-4алкилоксикарбонил, где С1-4алкил является этилом.
9. Способ восстановления соединения (III), которое представляет собой смесь (Е)-(III) и (Z)-(III), до соединения (IV), которое представляет собой смесь (Е)-(IV) и (Z)-(IV), с последующим выделением соединения (Е)-(IV) или его соли в виде осадка

где R1 представляет собой арилметил или арилэтил;
А представляет собой С1-4алкилоксикарбонил;
где арил представляет собой фенил.
10. Способ по п.9, где R1 представляет собой арилметил, где арил является фенилом.
11. Способ по п.9, где А представляет собой С1-4алкилоксикарбонил, где С1-4алкил является этилом.
12. Способ по п.10, в котором R1 представляет собой арилметил, где арил является фенилом; А представляет собой С1-4алкилоксикарбонил, где С1-4алкил является этилом.
13. Способ взаимодействия соединения формулы (Е)-(IV) с соединением (V) в условиях реакции Мицунобу с получением соединения формулы (VI), которое может быть преобразовано в соединение (1) или его соль присоединения кислоты, путем удаления заместителя R1

где R1 представляет собой арилметил или арилэтил,
где арил является фенилом.
14. Способ по п.13, в котором R1 представляет собой арилметил, где арил является фенилом.
15. Соединение формулы (Е)-(III)

или его соли присоединения кислоты,
где R1a представляет собой С1-4алкил, аллил, арилметил или арилэтил;
А представляет собой С1-4алкилоксикарбонил или аминокарбонил;
где арил представляет собой фенил или фенил, замещенный одним С1-4алкилокси.
16. Соединение формулы (E)-(III) по п.15, в котором R1a представляет собой арилметил, где арил является фенилом.
17. Соединение формулы (Е)-(III) по п.16, в котором А представляет собой С1-4алкилоксикарбонил.
18. Соединение формулы (E)-(III) по п.17, где А представляет собой С1-4алкилоксикарбонил, где С1-4алкил является этилом.
19. Соединение формулы (Е)-(IV)

или его соль присоединения кислоты,
где R1a представляет собой арилметил или арилэтил;
А представляет собой С1-4алкилоксикарбонил;
где арил представляет собой фенил.
20. Соединение формулы (E)-(IV) по п.19, в котором R1a представляет собой арилметил, где арил является фенилом.
Текст
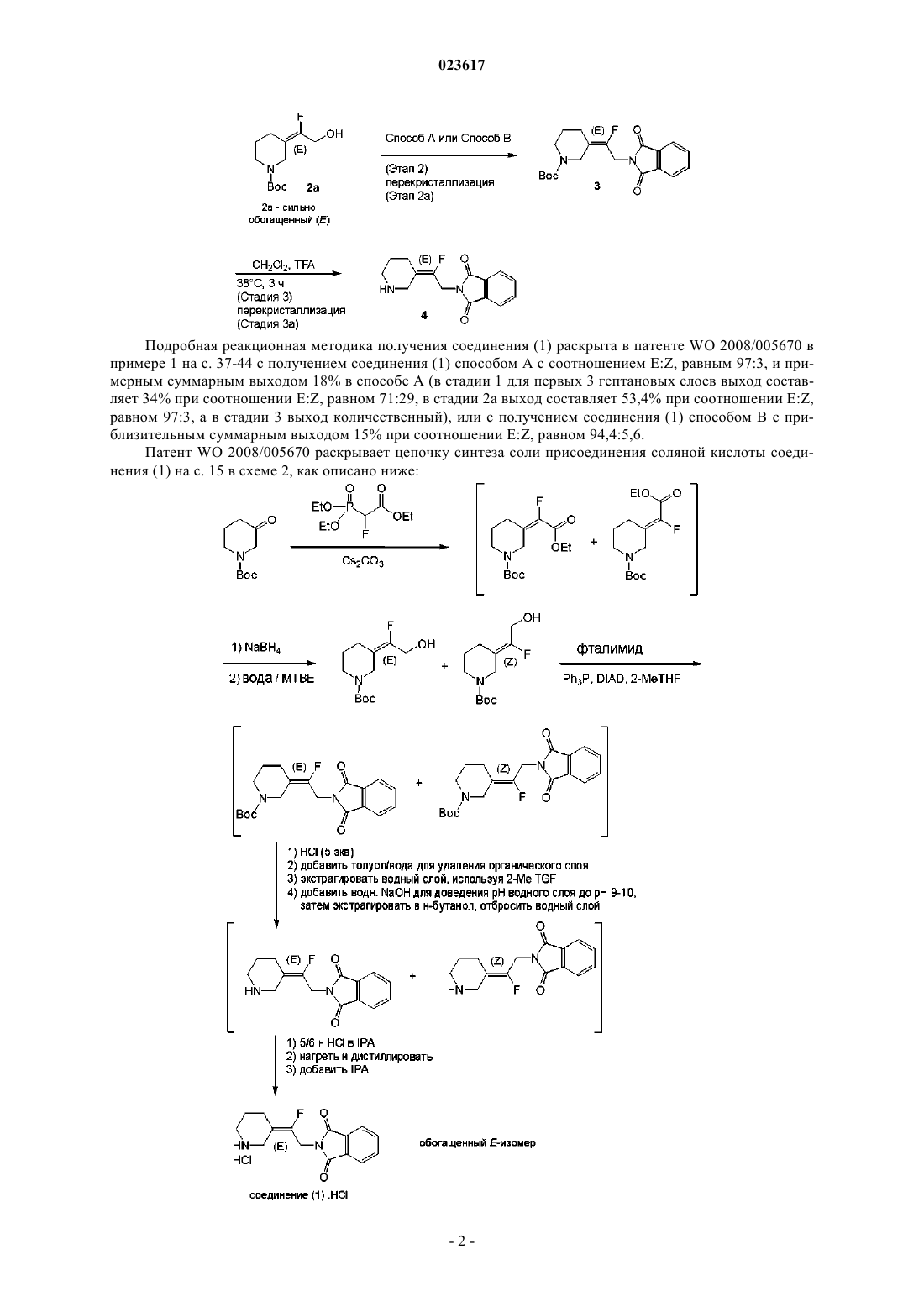
УСОВЕРШЕНСТВОВАННЫЙ СПОСОБ ПОЛУЧЕНИЯ 2-[(2E)-2-ФТОР-2-(3 ПИПЕРИДИНИЛИДЕН)ЭТИЛ]-1H-ИЗОИНДОЛ-1,3(2H)-ДИОНА Изобретение относится к усовершенствованному способу получения 2-[(2E)-2-фтор-2(3-пиперидинилиден)этил]-1H-изоиндол-1,3(2 Н)-диона (соединение (1 или его соли,который является промежуточным соединением в цепочке синтеза антибактериального соединения - 7-[(3 Е)-3-(2-амино-1-фторэтилиден)-1-пиперидинил]-1-хлорпропил-6-фтор-1,4 дигидро-8-метокси-4-оксо-3-хинолинкарбоновой кислоты. Лан Иоланд Лидия, Депре Доминик Поль Мишель (BE) Медведев В.Н. (RU)(71)(73) Заявитель и патентовладелец: ЯНССЕН ФАРМАЦЕВТИКА НВ (BE) Область изобретения Настоящее изобретение относится к усовершенствованному способу получения 2-[(2 Е)-2-фтор-2-(3 пиперидинилиден)этил]-1 Н-изоиндол-1,3(2 Н)-диона или его соли, который является промежуточным соединением в цепочке синтеза антибактериального соединения - 7-[(3 Е)-3-(2-амино-1-фторэтилиден)-1 пиперидинил]-1-хлорпропил-6-фтор-1,4-дигидро-8-метокси-4-оксо-3-хинолинкарбоновой кислоты. Предпосылки изобретенияWO 2006/101603 описывает 7-амино-алкилиденил-гетероциклические хинолины в качестве антимикробных соединений и раскрывает синтез 7-[(3 Е)-3-(2-амино-1-фторэтилиден)-1-пиперидинил]-1 хлорпропил-6-фтор-1,4-дигидро-8-метокси-4-оксо-3-хинолинкарбоновой кислоты, обозначенной как соединение (303) в табл. 1 на с. 20. Здесь и далее для удобства на это соединение ссылаются как на соединение "А".WO 2008/005670 раскрывает однореакторные способы получения замещенных аллильных спиртов,а также экстракционные способы разделения определенных продуктов изомерных спиртов, которые полезны для получения хинолинов, таких как антимикробного соединения 7-[(3 Е)-3-(2-амино-1 фторэтилиден)-1-пиперидинил]-1-хлорпропил-6-фтор-1,4-дигидро-8-метокси-4-оксо-3 хинолинкарбоновой кислоты (т.е. соединения "А"). Важным промежуточным соединением в общей цепочке синтеза указанного антимикробного соединения "А" является 2-[(2 Е)-2-фтор-2-(3-пиперидинилиден)этил]-1 Н-изоиндол-1,3(2 Н)-дион и его соль соляной кислоты: Соединение (1) привносит желаемую Е-стереохимию в общую цепочку синтеза антимикробного соединения "А". Патент WO 2008/005670 раскрывает цепочку синтеза соединения (1) на с. 38, как описано ниже: Подробная реакционная методика получения соединения (1) раскрыта в патенте WO 2008/005670 в примере 1 на с. 37-44 с получением соединения (1) способом А с соотношением E:Z, равным 97:3, и примерным суммарным выходом 18% в способе А (в стадии 1 для первых 3 гептановых слоев выход составляет 34% при соотношении E:Z, равном 71:29, в стадии 2 а выход составляет 53,4% при соотношении E:Z,равном 97:3, а в стадии 3 выход количественный), или с получением соединения (1) способом В с приблизительным суммарным выходом 15% при соотношении E:Z, равном 94,4:5,6. Патент WO 2008/005670 раскрывает цепочку синтеза соли присоединения соляной кислоты соединения (1) на с. 15 в схеме 2, как описано ниже: Подробная реакционная методика получения HCl-соли соединения (1) раскрыта в патенте WO 2008/005670 в примере 4 на с. 49-52 с получением 95% целевого Е-изомера с суммарным выходом 1822%, начиная с N-boc-3-пиперидона. Описанные в WO 2008/005670 реакционные методики получения соединения (1) или его HCl-соли характеризуются недостаточной селективностью реакции Хорнера-Уодсворта-Эммонса, в которой в больших количествах образуется нежелательный Z-изомер. Наличие нежелательного Z-изомера требует дополнительных длительных стадий разделения. Поэтому существует потребность в более эффективном способе получения соединения (1) или егоHCl-соли с меньшим образованием отходов. Патент WO 2010/056633 раскрывает на с. 87 схему синтеза XIV для получения трет-бутил-4-(2 этокси-2-оксоэтилиден)пиперидинил-1-карбоксилата, а на с. 111 схему синтеза XXVI для получения этилового эфира (1-бензил-пиперидин-4-илиден)бромуксусной кислоты. В первом варианте осуществления настоящее изобретение относится к усовершенствованному способу получения соединений формулы (III), имеющего улучшенное соотношение целевого (Е)-изомера и нежелательного (Z)-изомера. В дополнительном осуществлении изобретения соединение (Е)-(III) затем превращают в соединение (1) или его соль присоединения соляной кислоты. Описание изобретения В одном аспекте настоящее изобретение относится к способу получения соединения формулы (III),который характеризуется стадиями взаимодействия соединения формулы (I) с соединением формулы (II) в инертном растворителеX представляет собой галоген; где арил представляет собой фенил или фенил, замещенный одним или двумя С 1-4 алкилокси, галогенами, С 1-4 алкилами, нитрогруппами или ди(С 1-4 алкил)аминогруппами. Инертным растворителем может быть любой растворитель, такой как дихлорметан, ацетонитрил,этилацетат, гептаны, тетрагидрофуран (THF), циклопентил метиловый эфир (СРМЕ), ди-н-бутиловый эфир (DBE), метилциклогексан (МеСу), хлорбензол, фторбензол, N,N-диметилацетамид, N,Nдиметилформамид, толуол или анизол или любая их смесь. На практике инертным растворителем обычно является толуол. Во втором аспекте настоящее изобретение относится к способу восстановления соединения (III),которое представляет собой смесь (Е)-(III) и (Z)-(III), в соединение (IV), которое представляет собой смесь (E)-(IV) и (Z)-(IV), с последующим выделением соединения (E)-(IV) или его соли в виде осадка. Восстановление может быть выполнено с помощью любого известного в данном уровне техники восстановителя, такого как LiBH4, LiAlH4, натрия бис-(2-метоксиэтокси)алюминийгидрид (Red-Al), и силановых восстановителей, таких как триалкилсиланы (например, триметилсилан, триэтилсилан,трис(триметилсилил)силан), трифенилсилан, триалкоксисиланы (например, триэтоксисилан) и полимерные силоксаны, как поли(метилгидросилоксан), в присутствии катализатора. Реакция восстановления может быть проведена в любом подходящем инертном растворителе, таком как толуол. Осаждение соединений (E)-(IV) производится смешиванием соединений (IV) с подходящим растворителем, таким как, например, диизопропиловый эфир или ди-н-бутиловый эфир (DBE), с последующим выделением осажденных соединений (E)-(IV). Растворитель необязательно нагревают после введения соединений (IV), и при охлаждении соединения (E)-(IV) выпадают в осадок. В третьем аспекте настоящее изобретение относится к способу взаимодействия соединения формулы (E)-(IV) с соединением (V) в условиях реакции Мицунобу, таким образом получая соединение формулы (VI), которое может быть преобразовано в соединение (1), необязательно в виде соли присоединения, путем удаления заместителя R1, используя известные в уровне техники способы снятия защиты, такие как, например, гидрогенизация или обработка хлорформиатом. В четвертом аспекте первичную гидроксильную группу в соединении (E)-(IV) сначала преобразуют в уходящую группу Y, такую как галоген или сульфонилоксигруппу, например, метансульфонилокси,бензолсульфонилокси, трифторметансульфонилокси, пара-толуолсульфонилокси, перед проведением реакции с соединением (V) или его солью (например, калиевой солью) и удалением заместителя R1 по описанным выше методикам. В пятом аспекте настоящее изобретение также относится к новым соединениям формулы (Е)-(III) или (E)-(IV) или их солям присоединения кислоты,где R1a представляет собой С 1-4 алкил, арил, арилметил, арилэтил, дифенилметил, аллил или 3 фенилаллил (циннамил); А представляет собой С 1-4 алкилоксикарбонил, гидроксикарбонил, аминокарбонил или цианогруппу; где арил представляет собой фенил или фенил, замещенный одним или двумя С 1-4 алкилокси, гало-4 023617 генами, С 1-4 алкилами, нитрогруппами или ди(С 1-4 алкил)аминогруппами. Как использовано в предшествующих определениях: термин "галоген" является общим для атомов фтора, хлора, брома и йода; С 1-4 алкил обозначает углеводородные радикалы с прямой и разветвленной цепью, содержащие от 1 до 4 атомов углерода, такие как, например, метил, этил, пропил, бутил, 1-метилэтил, 2-метилпропил и т.п.; С 1-6 алкил включает С 1-4 алкил и его более высокие гомологи, содержащие 5 или 6 атомов углерода,такие как, например, 2-метилбутил, пентил, гексил и т.п.; Соли присоединения кислот, упоминаемые здесь и ранее, включают формы солей присоединения кислот, которые могут быть образованы соединением формулы (I). Такие соли присоединения кислот можно легко получить путем обработки основной формы такой соответствующей кислотой. Соответствующие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты,например соляная или бромоводородная кислота, серная, азотная, фосфорная и подобные кислоты; или органические кислоты, такие как, например, уксусная, пропионовая, гликолевая, молочная, пировиноградная, щавелевая (т.е. этандиовая), малоновая, янтарная (т.е. бутандиовая), малеиновая, фумаровая,яблочная, винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, птолуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и подобные кислоты. И наоборот, указанные солевые формы можно превратить путем обработки соответствующим основанием в свободную основную форму. Интересными способами настоящего изобретения являются те, к которым применимы одно или несколько из следующих ограничений:b) R1 представляет собой метил, этил, аллил, фенилэтил, дифенилметил или арилметил, где арил представляет собой фенил или метоксизамещенный фенил; в частности, R1 представляет собой арилметил, где арил является фенилом; илиe) А представляет собой гидроксикарбонил,аминокарбонил,цианогруппу или С 1-4 алкилоксикарбонил, где С 1-4 алкил представляет собой метил, этил или трет-бутил; в частности, А представляет собой С 1-4 алкилоксикарбонил, где С 1-4 алкил является этилом. Химические определения. Термин "изомер" означает соединения, которые имеют одинаковый состав и молекулярную массу,но отличаются физическими и/или химическими свойствами. Такие вещества имеют одинаковое число и тип атомов, но отличаются структурой. Структурное отличие может быть в построении (геометрические изомеры) или может происходить в результате различных пространственных расположений групп в молекуле (стереоизомеры). Термин "стереоизомер" означает изомеры одинакового построения, которые отличаются расположением своих атомов в пространстве. Энантиомеры и диастереомеры являются стереоизомерами, где асимметрично замещенный атом углерода выступает в роли хирального центра. Термин "хиральный" относится к молекуле, которая при наложении на свое зеркальное отображение не совпадает с ним, что говорит об отсутствии оси и плоскости симметрии или центра симметрии. Изомер обозначают как находящийся в конфигурации "Z" (zusammen = "вместе"), если группы с наивысшим приоритетом лежат по одну сторону от базовой плоскости, проходящей через двойную связь,и перпендикулярной плоскости, содержащей связи, соединяющие группы с атомами, соединенными двойной связью; другой изомер обозначают как "Е" (entgegen = "напротив"). Термин "приоритет", используемый для определения Е и Z изомеров в данном документе, относится к правилам, установленным с целью ясного обозначения изомеров, описанных у R.S. Cahn, C.K. Ingold и V. Prelog. Экспериментальная часть. Следующие аббревиатуры будут использованы в последующей части: Пример 1: получение соединений формулы (I). Соединения формулы (I) при их выделении в виде соли присоединения соляной кислоты могут существовать либо в форме кетона (I), либо как форма геминального диола (VII) ("гидрат") или в виде смеси их обоих в различном соотношении. Однако во время нейтрализации соли (образования амина) геминальный диол спонтанно теряет молекулу воды, образуя кетон. В следующем ниже тексте термин "HCl-соль соединения (I)" всегда относится либо непосредственно к "HCl-соли соединения (I)", "HCl-соли соединения (VII)", либо к их смеси. Стадия 1: синтез 3,3-диметоксипиперидина. 25 г (125,5 ммоль) N-boc-3-пиперидона растворяли в 125 мл метанола. Добавляли 9,1 мл (138 ммоль) метансульфокислоты и реакционную смесь перемешивали в течение нескольких часов при комнатной температуре, а затем при 50 С до завершения реакции. Реакционную смесь нейтрализовали карбонатом натрия 15,96 г (150,6 ммоль), разбавляли, используя 125 мл толуола, и метанол удаляли под вакуумом. Полученную суспензию далее разбавляли толуолом (125 мл). Твердые вещества отфильтровывали и промывали толуолом (63 мл), а объединенные фильтраты концентрировали под вакуумом. Остаток перегоняли при пониженном давлении (100-105 С, 25 мбар) с получением 7,61 г 3,3 диметоксипиперидина в виде бесцветной жидкости (выход: 42%) (т. кип.: 100-105 С, 25 мбар). 1 Соединения (Ia) .HCl-соль и (Ic) .HCl-соль можно приобрести. Соединение (Ib) .HCl-соль получали из 3-гидроксипиридина, следуя методике, описанной в литературе (Lyle, R.Е.; Adel, R.Е., Lyle, G.G. J. Org. Chem. 1959, 24, 343). Синтез соединения (Id) .HCl-соль. 19,8 г (136 ммоль) 3,3-диметоксипиперидина и 19 мл (136 ммоль) триэтиламина растворяли в 136 мл THF. Добавляли 15,6 мл (150 ммоль) аллилхлорида и реакционную смесь перемешивали при комнатной температуре, а затем при 50 С до полного превращения исходных материалов (2 дня). После охлаждения до комнатной температуры нерастворимые материалы отфильтровывали и промывали, используя несколько миллилитров толуола. Объединенные фильтрат и промывочный слой концентрировали под вакуумом до сухого состояния. Масляный остаток снова растворяли в 20 мл этилацетата и фильтровали через подушку силикагеля. Подушку промывали, используя 100 мл этилацетата, и объединенные фильтраты концентрировали под вакуумом с получением 20,5 г N-аллил-3,3-диметоксипиперидина, чья структура была подтверждена с помощью ЯМР и ЖХ-МС (выход: 81%). 19 г N-аллил-3,3 диметоксипиперидина растворяли в 100 мл водного 3 М раствора HCl и 30 мл THF и перемешивали 4 ч при 60 С. К реакционной смеси добавляли толуол, и слои разделяли. Водный слой концентрировали под вакуумом до сухого состояния и выпаривали дважды вместе с дихлорметаном. Полученное твердое вещество перемешивали в нескольких мл ацетона, затем фильтровали и сушили, получая 8,5 г соединения С ЯМР (91 МГц, ДМСО-d6, сигнал от формы кетона)м.д. 200,80, 126,93, 125,20, 58,49, 57,35,48,69, 36,89, 19,17. 13 С ЯМР (91 МГц, ДМСО-d6, сигнал от формы гидрата)м.д. 127,69, 124,78, 90,20, 58,41, 58,02,51,35, 34,25, 19,72 масс-спектрометрия высокого разрешения: расчетное для C8H14NO (форма кетона,М+Н): 140,1070, найденное: 140,1064, расчетное для C8H16NO2 (форма гидрата, М+Н): 158,1176, найденное: 158,1190. Синтез соединения (Ie) .HCl-соль. 8,0 г (55,1 ммоль) 3,3-диметоксипиперидина и 7,1 мл (60, 6 ммоль) фенилацетальдегида растворяют в 83 мл MeTHF и раствор помещают в инертную атмосферу. Добавляют 2,37 г влажного 5%-ного Pd/C и реакционную смесь перемешивают 5 ч при комнатной температуре при давлении водорода 6 бар. По завершении реакции давление сбрасывают, катализатор отфильтровывают и фильтрат концентрируют под вакуумом до сухого состояния с получением 13,7 г неочищенного 3,3-диметокси-N-фенетилпиперидина(количественный выход сырого продукта). Неочищенный ацеталь растворяют в 66 мл водной 1 М HCl. Водный раствор промывают, используя 55 мл изопропилацетата, затем осторожно концентрируют под вакуумом (температура: 60 С) до сухого состояния. Добавляли 110 мл MIBK и полученную смесь медленно выпаривали под вакуумом. Снова добавляли 110 мл MIBK и смесь кипятили с обратным холодильником в течение 2 ч перед тем, как ее перемешивали в течение ночи при комнатной температуре. Суспензию концентрировали до сухого состояния и остаток перекристаллизовывали из 30 мл ацетонитрила с получением 5,51 г соединения (Ie) .HCl-соль (выход: 42%) в виде светло-желтого твердого вещества. Соединение (1 е): 3,3-диметокси-N-фенилэтилпиперидин: 1H ЯМР (360 МГц, хлороформ-d)7,30-7,23 (м, 2 Н), 7,22-7,14 (м, 3 Н), 3,22 (с, 6 Н), 2,88-2,78 (м, 2 Н),2,67-2,56 (м, 2 Н), 2,49 (д, J=15,4 Гц, 4 Н), 1,67 (уш. с, 4 Н). 13 С ЯМР (91 МГц, хлороформ-d)м.д. 140,2, 128,5, 128,2, 125,8, 98,0, 60,5, 57,4, 53,7, 47,6, 33,2,31,1, 22,2. ЖХ-МС: 21,38 (М-ОМе), 250,34 (М+Н). Соединение (Ie) .HCl-соль (смесь кетона и гидрата): 1 Н ЯМР (360 МГц, ДМСО-d6, сигнал для формы кетона)м.д. 12,14 (уш. с, 1 Н), 7,33-7,39 (м, 2 Н),7,24-7,33 (м, 3 Н), 3,99 (дд, J=15,4, 8,4 Гц, 1 Н), 3,87 (д, J=15,4 Гц, 1 Н), 3,73 (д, J=12,1 Гц, 1 Н), 3,27-3,48 (м,3 Н), 3,07-3,18 (м, 2 Н), 2,44-2,67 (м, 2 Н), 2,24-2,42 (м, 1 Н), 2,07-2,20 (м, 1 Н). 13 С ЯМР (151 МГц, ДМСО-d6, сигнал для формы кетона)м.д. 200,90, 137,10, 128,76, 128,67,126,84, 58,90, 56,66, 51,80, 34,42, 29,43, 19,86. 13 С ЯМР (151 МГц, ДМСО-d6, сигнал для формы гидрата)м.д. 136,98, 128,73, 128,67, 126,84,90,21, 59,14, 56,39, 49,50, 36,88, 29,19, 19,27. Масс-спектрометрия высокого разрешения: расчетное C13H18NO (форма кетона, М+Н): 204,1388,найденное: 204,1395, расчетное для C13H20NO2 (форма гидрата, М+Н): 222,1489, найденное: 222,1551. Соединение (If) .HCl-соль. 5,00 г (34 ммоль) 3,3-диметоксипиперидина растворяли в 69 мл MeTHF. Добавляли 5,71 г (41 ммоль) карбоната калия и 8,51 г (34 ммоль) бромдифенилметана и реакционную смесь кипятили с обратным холодильником в течение ночи. После охлаждения до комнатной температуры неорганические материалы промывали, используя 34 мл воды, к органическому слою добавляли 39 мл водного 1 М раствора HCl и полученную двухфазную смесь кипятили с обратным холодильником, затем концентрировали под вакуумом до сухого состояния. Полутвердый остаток кипятили с обратным холодильником в 39 мл MIBK. После охлаждения до комнатной температуры твердое вещество фильтровали, промывали несколькимиMIBK и сушили, получая 7,48 г соединения (If) .HCl-соль в виде окрашенного твердого вещества (выход 72%). 1H ЯМР (360 МГц, ДМСО-d6)м.д. 13,12 (уш. с, 1 Н), 7,78-8,16 (м, 4 Н), 7,20-7,56 (м, 6 Н), 5,81 (д,J=7,7 Гц, 1 Н), 3,85 (дд, J=13,2, 7,0 Гц, 1 Н), 3,22-3,54 (м, 3 Н), 2,55 (уш. с, 2 Н), 2,50-2,53 (м, 1 Н), 2,12 (уш. с, 1 Н). 13 С ЯМР (101 МГц, ДМСО-d6)м.д. 199,93, 135,81, 134,96, 129,35, 129,22, 128,86, 128,56, 127,94,73,96, 58,62, 49,56, 36,86, 18,54. Масс-спектрометрия высокого разрешения: расчетное C18H20NO (форма кетона, М+Н): 266,1545,найденное: 266,1549, расчетное для C18H22NO2 (форма гидрата, М+Н): 284,1651, найденное: 284,1654. Соединение (Ig) .HCl-соль: 37,6 мл (37,6 ммоль) водного 1 М раствора HCl добавляли к 5 г (25,1 ммоль) N-boc-3-пиперидона и реакционную смесь перемешивали при комнатной температуре несколько часов, прежде чем ее концентрировали под вакуумом. Остаток перекристаллизовывали из изопропанола, получая 3,40 г соединения(Ig) .HCl-соль в виде твердого вещества (выход: 63%). 1 Соединение (Ih) .HCl-соль. Смесь 800 мг (5,51 ммоль) 3,3-диметоксипиперидина и 750 мг (5,51 ммоль) 2-метоксибензальдегида в 8,3 мл метанола перемешивали при давлении водорода 1 бар в присутствии 59 мг (0,055 ммоль) 10%ного (вес./вес.) Pd/C. После завершения реакции (несколько часов) реакционную смесь продували азотом, фильтровали и к фильтрату добавляли 6,6 мл водной 1 М HCl. Смесь концентрировали под вакуумом до сухого состояния. Остаток повторно растворяли в 4,4 мл горячего ацетона. К горячему раствору добавляли 4,4 мл MIBK, смесь кипятили с обратным холодильником несколько часов, затем охлаждали до комнатной температуры. Суспензию фильтровали, и собранное твердое вещество промывали несколькими миллилитрами MIBK, затем сушили под вакуумом с получением 950 мг соединения (In). HCl в виде грязно-белого твердого вещества (выход: 68%). Отношение кетон/гидрат составляло примерно 95/5. 1 Н ЯМР (360 МГц, ДМСО-d6, сигналы, относящиеся к кетону)м.д. 11,88 (уш. с, 1 Н), 7,63 (дд,J=7,5, 1,6 Гц, 1 Н), 7,35-7,55 (м, 1 Н), 7,14 (д, J=7,7 Гц, 1 Н), 7,03 (тд, J=7,3, 0,7 Гц, 1 Н), 4,35 (уш. с, 2 Н),3,90 (дд, J=15,5, 9,5 Гц, 1 Н), 3,86 (с, 3 Н), 3,59 (д, J=15,0 Гц, 1 Н), 3,50 (д, J=11,7 Гц, 1 Н), 3,26 (кв., J=9,9 Гц,1 Н), 2,25-2,62 (м, 3 Н), 2,00-2,16 (м, 1 Н). 13 С ЯМР (91 МГц, ДМСО-d6, сигналы, относящиеся к форме кетона)м.д. 200,64, 148,10, 133,37,131,56, 120,47, 116,91, 111,47, 58,91, 55,69, 53,05, 48,90, 36,71, 19,25. 13 С ЯМР (91 МГц, ДМСО-d6, сигналы, относящиеся к форме гидрата)м.д. 148,10, 133,37, 131,56,120,47, 116,91, 111,47, 90,18, 58,91, 55,69, 51,28, 41,06, 37,48, 19,93. Масс-спектрометрия высокого разрешения: расчетное C13H18NO2 (форма кетона, М+Н): 220,1332,найденное: 220,1326, расчетное для C13H20NO3 (форма гидрата, М+Н): 238,1438, найденное: 238,1463. Соединение (Ii) .HCl-соль. Смесь 800 мг (5,51 ммоль) 3,3-диметоксипиперидина и 750 мг (5,51 ммоль) 3-метоксибензальдегида в 8,3 мл метанола перемешивали при давлении водорода 1 бар в присутствии 59 мг (0,055 ммоль) 10%ного (вес./вес.) Pd/C. После завершения реакции (несколько часов) реакционную смесь продували азотом, фильтровали и к фильтрату добавляли 6,6 мл водной 1 М HCl. Смесь концентрировали под вакуумом до сухого состояния. Остаток суспендировали в 4,4 мл горячего ацетона. Добавляли 4,4 мл MIBK и ацетон отгоняли. Суспензию затем охлаждали до комнатной температуры и фильтровали. Собранное твердое вещество промывали несколькими миллилитрами MIBK, затем сушили под вакуумом с получением 1,09 г соединения (Ih) .HCl в виде грязно-белого твердого вещества (выход: 77%). Масс-спектрометрия высокого разрешения: расчетное C13H18NO2 (форма кетона, М+Н): 220,1332,найденное: 220,1312, расчетное для C13H20NO3 (форма гидрата, М+Н): 238,1438, найденное: 238,1444. Отношение кетон/гидрат: примерно 95/5. 1 Н ЯМР (360 МГц, ДМСО-d6, сигналы, относящиеся к кетону)м.д. 12,20 (уш. с, 1 Н), 7,30-7,42 (м,2 Н), 7,17 (д, J=7,7 Гц, 1 Н), 7,02 (дд, J=8,2, 2,4 Гц, 1 Н), 4,38 (уш. с, 2 Н), 3,86 (дд, J=15,0, 8,8 Гц, 1 Н), 3,79 Соединение (IIa). 74 г (400 ммоль) этилбромфторацетата медленно добавляли к 400 мл (400 ммоль) 1 М раствору триметилфосфина в толуоле и реакционную смесь перемешивали в течение ночи при комнатной температуре. Соединение (IIa) отфильтровывали, промывали 80 мл толуола и сушили под вакуумом при 40 С. Выход: 90,97 г (87% выход). Внешний вид: белое твердое вещество. 1F ЯМР (376 МГц, хлороформ-d)м.д. -212,30 (дд, J=65,2, 43,9 Гц, 1F). Соединение (IIb). 74 г (400 ммоль) этилбромфторацетата медленно добавляли к 58,8 мл (400 ммоль) триэтилфосфина,растворенного в 400 мл толуола. Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем несколько часов при 0 С, прежде чем отфильтровывали соединение (IIb), промывали 80 мл толуола и сушили. Выход: 102 г (84%). Внешний вид: белое твердое вещество (т. пл.: 135-210 С). 1F-HMP (377 МГц, CDCl3)-209,53 (д, 2JP-F=55, 6 Гц). 31 Р-ЯМР (162 МГц, CDCl3)35,05 (д, 2JP-F=64,6 Гц). Соединение (IIc) (соединение описано в литературе: Thenappan, A.; Burton, D.J. J. Org. Chem. 1990,55, 2311-2317). 3,71 г (20 ммоль) этилбромфторацетата добавляли к раствору 5 мл (20 ммоль) трибутилфосфина в 50 мл этилацетата. Раствор перемешивали в течение ночи при комнатной температуре, прежде чем его концентрировали под вакуумом с получением неочищенного соединения (IIc) в виде полутвердого соединения (выход неочищенного продукта: количественный). 1F ЯМР (376 МГц, хлороформ-d)м.д. -208,67 (дд, J=56,1, 43,4 Гц, 1F). Соединение (IId). 4,57 г (24,7 ммоль) этилбромфторацетата добавляли к раствору 5 г (24,7 ммоль) трибутилфосфина в 25 мл THF. Раствор перемешивали 4 дня при комнатной температуре, прежде чем его концентрировали под вакуумом. Остаток сушили под высоким вакуумом с образованием твердого вещества. Твердое вещество повторно суспендировали в нескольких миллилитрах толуола, фильтровали, промывали и сушили под вакуумом с получением 1,07 г соединения (IId) (выход: 11%). Внешний вид: белое твердое вещество (т. пл.: 101 С). 1F-ЯМР (377 МГц, CDCl3)-123,75 (д, 2JP-F=56,4 Гц). 31 Р-ЯМР (162 МГц, CDCl3)37,82 (д, 2JP-F=56,7 Гц). Масс-спектрометрия высокого разрешения (катион фосфония): расчетное для C16H33FO2P (катион фосфония): 307,2202, найденное: 307,2225. Соединение (IIe) (соединение описано в литературе: Thenappan, A.; Burton, D.J. J. Org. Chem. 1990,55, 2311-2317) 3,52 г (19 ммоль) этилбромфторацетата добавляли к раствору 5 г (19 ммоль) трифенилфосфина в 15 мл дихлорметана. Реакционную смесь перемешивали при комнатной температуре, прежде чем его концентрировали под вакуумом. Остаток повторно суспендировали в горячем этилацетате/изопропаноле (80/20), затем фильтровали после охлаждения до комнатной температуры и сушили под вакуумом с получением 4,54 г соединения (IIe) (выход: 54%). Внешний вид: белое твердое вещество. 1H ЯМР (360 МГц, хлороформ-d)м.д. 9,71 (дд, J=41,7, 5,9 Гц, 1 Н), 8,01 (дд, J=13,2, 8,1 Гц, 6 Н),7,79-7,89 (м, 3 Н), 7,71 (тд, J=7,9, 3,7 Гц, 6 Н), 4,11 (кв., J=7,2 Гц, 2 Н), 1, 00 (т, J=7,1 Гц, 3 Н). 13 С ЯМР (91 МГц, хлороформ-d)м.д. 163,34 (дд, J=21,5, 2,8 Гц, 1 С), 135,70 (д, J=3,5 Гц, 3 С),134,75 (д, J=10,4 Гц, 6 С), 130,34 (д, J=13,1 Гц, 6 С), 114,53-115,85 (м, 3 С), 83,61-86,96 (м, 1 С), 63,60 (с,1 С), 13,56 (с, 1 С). Соединение (IIf). 4,27 г (25 ммоль) метилбромфторацетата добавляли к 25 мл (25 ммоль) 1 М раствора триметилфосфина в толуоле. Реакционную смесь перемешивали в течение ночи при комнатной температуре, фильтровали и собранное твердое вещество промывали несколькими миллилитрами толуола, затем сушили под вакуумом с получением 5,88 г соединения (IIf) в виде белого твердого вещества (выход: 95%). Внешний вид: белое твердое вещество (т. пл.: 110 С). 1F ЯМР (376 МГц, ДМСО-d6)-212,44 (дд, J=64,9, 43,6 Гц, 1F). 31 Р ЯМР (162 МГц, ДМСО-d6)35,75 (д, J=65,2 Гц, 1 Р). Масс-спектрометрия высокого разрешения (катион фосфония): расчетное для C6H13FO2P (катион фосфония): 167,0637, найденное: 167,0645. Соединение (IIg). 10,65 г т-бутил бромфторацетата добавляли к 50 мл (50 ммоль) 1 М раствора триметилфосфина в толуоле и реакционную смесь перемешивали в течение ночи при комнатной температуре, затем в течение нескольких часов при 0 С. Образовавшееся твердое вещество промывали несколькими миллилитрами толуола и сушили под вакуумом. Получали 10,55 г соединения (IIg) (выход: 73%). Внешний вид: белое твердое вещество (т. пл.: 97,8 С). 1H ЯМР (400 МГц, ДМСО-d6)6,69 (дд, J=7,3, 43,6 Гц, 1 Н), 2,14 (д, J=15,4 Гц, 9 Н), 1,51 (с, 9 Н). 13 С ЯМР (101 МГц, ДМСО-d6)161,9 (д, J=21,3 Гц), 86,4, 83,4 (дд, J=200,3, 58,7 Гц), 27,5, 5,7 (д,J=51,4 Гц). 19F ЯМР (376 МГц, ДМСО-d6)-209,98 (дд, J=65, 6, 43,7 Гц, 1F). 31 Р ЯМР (162 МГц, ДМСО-d6)35,17 (д, J=65,2 Гц, 1 Р). Масс-спектрометрия высокого разрешения (катион фосфония): расчетное для C9H19FO2P (катион фосфония): 209,1107, найденное: 209,1111. Соединение (IIh). 100 мл (100 ммоль) 1 М раствора триметилфосфина в толуоле добавляли к раствору 15,60 г (100 ммоль) бромфторацетамида в 150 мл MeTHF. Реакционную смесь перемешивали в течение ночи при комнатной температуре, затем соединение (IIh) отфильтровывали, промывали несколькими миллилитрами толуола и сушили под вакуумом. Выход: 7,60 г (33%). Внешний вид: белое твердое вещество (т. пл.: 166,5 С). 1 Результаты получения соединения (III) приведены в таблице ниже: Соединение (IIIa). 30 г (124 ммоль) 94%-ного (вес./вес.) соединения (Ia) .HCl-соль суспендируют в 124 мл толуола. Добавляют 124 мл воды и 13,14 г карбоната натрия и полученную смесь перемешивают несколько минут при комнатной температуре перед декантированием. Два слоя разделяют и органический слой сушат над сульфатом натрия, затем фильтруют с получением 142 г раствора 16,6% (вес./вес.) соединения (Ia) в толуоле. Полученный таким образом раствор соединения (Ia) добавляют к суспензии 38,8 г (148 ммоль) соединения (IIa) при 0 С. Добавляют 22,5 г (148 ммоль) DBU и реакционную смесь перемешивают в течение ночи при 0 С, прежде чем прекращают реакцию с помощью 62 мл воды. Два слоя разделяют и органический слой промывают, используя 62 мл воды, сушат над сульфатом натрия, затем фильтруют с получением 251,7 г раствора 12,9% (вес./вес.) соединения (IIIa) в толуоле, которые затем используют в следующей стадии (выход: 96%, отношение E/Z: 80/20). Такую же методику использовали в других растворителях и при других температурах. Раствор 50,5 ммоль соединения (IIIa) в толуоле концентрируют под вакуумом и остаток очищают с помощью хроматографии с получением 11,36 г соединения (IIIa) (смесь (Е)- и (Z)-изомера) в виде нестабильного масла. Выход: 81%. 5,56 г моногидрата п-толуолсульфокислоты добавляют к раствору 8,12 г соединения (IIIa) в MIBK и реакционную смесь нагревают для завершения растворения, затем охлаждают до комнатной температуры. Получают 12 г тозилатной соли соединения (IIIa) в виде стабильного твердого вещества после фильтрации, промывания и сушки под вакуумом (отношение E/Z: 85/15). 12 г соединения (IIIa) очищают с помощью препаративной ВЭЖХ на колонке ChiralPak AD с получением 4,16 г соединения (Е)-(IIIa) и 820 мг соединения (Z)-(IIIa). 2,85 г (15,0 ммоль) моногидрата п-толуолсульфокислоты добавляют к 4,16 г (15 ммоль) соединения(Е)-(IIIa), растворенного в 34 мл MIBK. Смесь нагревают, пока не растворится соль, фильтруют горячей и дают охладиться до комнатной температуры. Полученное таким образом твердое соединение отфильтровывают, промывают несколькими миллилитрами MIBK и сушат. Получают 5,23 г соединения (Е)(IIIa).TsOH-соль в виде белого твердого вещества (выход: 78%). 0,57 г (3 ммоль) моногидрата п-толуолсульфокислоты добавляют к 820 мг (2,96 ммоль) соединения(Z)-(IIIa), растворенного в 10 мл MIBK и 3 мл этанола. Смесь нагревают, пока не растворится соль, фильтруют горячей и дают охладиться до комнатной температуры. Полученное таким образом твердое соединение отфильтровывают, промывают несколькими миллилитрами MIBK и сушат. Получают 790 мг соединения (Z)-(IIIa). TsOH-соль в виде белого твердого вещества (выход: 59%). Соединение (Е)-(IIIa). Внешний вид: бледно-желтая жидкость, быстро темнеет. 1F ЯМР (377 МГц, хлороформ-d) 5=-128,28 (с, 1F). Соединение (E)-(IIIa) .TsOH-соль. Внешний вид: белое твердое вещество (т. пл.: 103,5 С). 1F ЯМР (377 МГц, хлороформ-d)-128,47 (с, 1F). Соединение (Z)-(IIIa) .TsOH. Внешний вид: белое твердое вещество (т. пл.: 167,1 С). 1 Н ЯМР (400 МГц, хлороформ-d)м.д. 10,63 (уш. с, 1 Н), 7,74 (д, J=8,3 Гц, 2 Н), 7,52 (д, J=6,8 Гц,2 Н), 7,34-7,42 (м, 1 Н), 7,26-7,34 (м, 2 Н), 7,15 (д, J=7,8 Гц, 2 Н), 4,33 (д, J=5,3 Гц, 2 Н), 4,20-4,28 (м, 3 Н),4,17 (д, J=13,6 Гц, 1 Н), 3,70 (ддд, J=13,6, 7,7, 2,6 Гц, 1 Н), 3,39-3,53 (м, 1 Н), 3,08-3,30 (м, 2 Н), 2,43-2,59 (м,1 Н), 2,35 (с, 3 Н), 1,94-2,09 (м, 1 Н), 1,87 (с, 1 Н), 1,30 (т, J=7,2 Гц, 3 Н).(60,91%), Н (6,34%), N (3,15%). Соединение (IIIb). 1,0 г (6,68 ммоль) соединения (Ib) .HCl-соль, 2,09 г (8,02 ммоль) соединения (IIa) и 924 мг (6,68 ммоль) карбоната калия суспендировали в 13 мл толуола. После охлаждения до 0 С добавляли 1,21 мл(8,02 ммоль) DBU и реакционную смесь перемешивали в течение ночи при 0 С. К реакционной смеси добавляли 13 мл воды и два слоя разделяли после нескольких минут перемешивания. Водный слой экстрагировали, используя 13 мл толуола, и объединенные органические слои сушили над сульфатом натрия и фильтровали с получением 50,6 г 0,94%-ного раствора (вес./вес.) соединения (IIIb) в толуоле (выход insitu: 35%). Раствор концентрировали под вакуумом и остаток очищали путем фильтрации через подушку силикагеля (элюент: ацетон) с получением 404 мг очищенного продукта (выход: 30%). Отношение E/Z: 96/4. Внешний вид: бледно-оранжевое масло. Соединение (Е)-(IIIb): 1F ЯМР (376 МГц, хлороформ-d) 5 м.д. -129,12 (уш. с, 1F). Соединение (IIIc). Соединение (IIIc) получали из соединений (Ic) .HCl-соль и (IIa), используя такую же методику, как и для соединения (IIIa). Выход: 65%, отношение E/Z: 85/15. Внешний вид: бледно-желтая жидкость, быстро темнеет. Соединение (Е)-(IIIc): 1 Н ЯМР (360 МГц, хлороформ-d)м.д. 1,13 (т, J=7,32 Гц, 3 Н), 1,34 (т, J=7,14 Гц, 3 Н), 1,77 (дд,J=6,59, 5,12 Гц, 2 Н), 2,40 (тд, J=6,40, 2,56 Гц, 2 Н), 2,48-2,56 (м, 2 Н), 2,60 (д, J=5,49 Гц, 2 Н), 3,62 (д, J=1,46 Гц, 2 Н), 4,28 (кв., J=7, 32 Гц, 3 Н). Соединение (Z)-(IIIc): 1 Н ЯМР (360 МГц, хлороформ-d)м.д. 1,13 (т, J=7,32 Гц, 3 Н), 1,34 (т, J=7,14 Гц, 3 Н), 1,77 (дд,J=6,59, 5,12 Гц, 2 Н), 2,52 (кв., J=7,32 Гц, 2 Н), 2,56-2,62 (м, 2 Н), 2,73-2,81 (м, 2 Н), 3,21 (д, J=2,93 Гц, 2 Н),4,28 (кв., J=7,32 Гц, 2 Н). Соединение (IIId). Соединение (IIId) получали из соединения (Id) .HCl-соль и (IIa), используя такую же методику, как и для соединения (IIIa). Выход: 40%, отношение E/Z: 90/10. Внешний вид: бледно-желтая жидкость, быстро темнеет. Соединение (E)-(IIId): 1 Н ЯМР (400 МГц, хлороформ-d)м.д. 1,33 (т, J=7,05 Гц, 3 Н), 1,68-1,81 (м, 2 Н), 2,33-2,46 (м, 2 Н),2,50-2,61 (м, 2 Н), 3,02-3,14 (м, 2 Н), 3,57 (д, J=2,01 Гц, 2 Н), 4,27 (д, J=7,05 Гц, 2 Н), 5,12-5,29 (м, 2 Н), 5,775,99 (м, 1 Н). 13 С ЯМР (91 МГц, хлороформ-d)м.д. 14,42 (с, 1 С), 20,91 (с, 1 С), 23,50 (д, J=7,61 Гц, 1 С), 49,69 (д,J=4,84 Гц, 1 С), 50,99 (с, 1 С), 58,28 (с, 1 С), 62,67 (с, 1 С), 125,91 (с, 1 С), 127,17 (с, 1 С), 145,65 (д, J=260,90 Гц, 1 С), 160,47 (д, J=31,80 Гц, 1 С). 19F ЯМР (377 МГц, хлороформ-d)-128,85 (с, 1F). Соединение (IIIe). Соединение (IIIe) получали из соединения (Ie) .HCl-соль и (IIa), используя такую же методику, как и для соединения (IIIa). Выход: 86%, отношение E/Z: 93/7. Внешний вид: бледно-желтая жидкость, быстро темнеет.F ЯМР (377 МГц, хлороформ-d, сигналы, относящиеся к Z-изомеру)-128,85 (с, 1F). Соединение (IIIf). Соединение (IIIf) получали из соединения (If) .HCl-соль и (IIa), используя такую же методику, как и методика для соединения (IIIa). Внешний вид: бледно-желтая жидкость, быстро темнеет. Соединение (IIIg). Соединение (IIIg) получали из соединения (Ig) .HCl-соль и (IIa), используя такую же методику, как и методика для соединения (IIIa). Внешний вид: бледно-желтая жидкость, быстро темнеет. Соединение (IIIh). Соединение (IIIh) получали из соединения (In) .HCl-соль и (IIa), используя такую же методику, как и методика для соединения (IIIa). Внешний вид: бледно-желтая жидкость, быстро темнеет. Соединение (E)-(IIIh): 1(тд, J=7,5, 1,1 Гц, 2 Н), 6,86 (д, J=8,4 Гц, 2 Н), 4,20 (кв., J=7,3 Гц, 2 Н), 3,81 (с, 3 Н), 3,62-3,68 (м, 4 Н), 2,542,63 (м, 2 Н), 2,37 (тд, J=6,5, 2,7 Гц, 2 Н), 1,68-1,79 (м, 2 Н), 1,24 (т, J=7,1 Гц, 3 Н). 13 С ЯМР (91 МГц, хлороформ-d)м.д. 160,97 (д, J=36,7 Гц, 1 С), 157,73 (с, 1 С), 142,06 (д, J=247,7 Гц, 1 С), 130,62 (с, 1 С), 128,07 (с, 1 С), 125,67 (с, 1 С), 120,16 (с, 1 С), 110,29 (с, 1 С), 61,04 (с, 1 С), 55,51 (с,1 С), 55,28 (с, 1 С), 52,76 (с, 1 С), 52,47 (д, J=4,8 Гц, 1 С), 25,00 (д, J=8,3 Гц, 1 С), 24,59 (д, J=2,1 Гц, 1 С),13,95 (с, 1 С). Соединение (IIIk). Соединение (IIIk) получали из соединения (Ia) .HCl-соль и (IIf), используя такую же методику, как и для соединения (IIIa). Внешний вид: бледно-желтая жидкость, быстро темнеет. Соединение (E)-(IIIk): 1 Соединение (E)-(IVa). 437 ммоль соединения (IIIa) в растворе в толуоле охлаждают до 0 С. Добавляют 212 г (682 ммоль) 65%-ного (вес./вес.) раствора натрия бис-(2-метоксиэтокси)алюминийгидрида (Red-Al) в толуоле и реакционную смесь перемешивают 1 ч при 0 С, избыток Red-Al разлагают, используя 77 мл ацетона, и реакционной смеси дают нагреться до комнатной температуры. Добавляют 568 мл воды и 235 мл 50%-ного(вес./вес.) водного раствора гидроксида натрия и полученную смесь нагревают до 50 С перед декантированием. Два слоя разделяют и органический слой промывают водой (437 мл), сушат над сульфатом натрия, фильтруют и концентрируют под вакуумом. К маслянистому остатку добавляют 1170 мл гептанов и полученное твердое вещество отфильтровывают и перекристаллизовывают из 188 мл диизопропилового эфира с получением 53,5 г соединения (E)-(IVa). Выход: 52%, отношение E/Z99/1. Внешний вид: твердое вещество (т. пл.: 92,0 С). 1(Red-Al) в толуоле добавляли к 38 г (10,2 ммоль) 7,8%-ного (вес./вес.) раствора соединения (IIIe) в толуоле, выдержанного при 0 С. Через 1 ч при 0 С избыток Red-Al разлагали, используя 1,2 мл (16,4 ммоль) ацетона и реакционную смесь перемешивали с течение ночи при комнатной температуре. Реак- 16023617 ционную смесь нагревали до 50 С и добавляли 14 мл воды и 5,5 мл (105 ммоль) 50%-ного (вес./вес.) раствора гидроксида натрия. Слои разделяли и органический слой промывали водой (14 мл), сушили над сульфатом натрия, фильтровали и концентрировали под вакуумом. Фильтрат выпаривали под вакуумом до сухого состояния с получением 2,40 г неочищенного соединения (IVe) (ЖХ анализ: 94,1% (вес./вес),отношение E/Z: 92/8, выход: 89%). Неочищенный продукт перекристаллизовывали из 6 мл DBE с получением 1,87 г соединения (E)-(IVe) в виде твердого продукта (выход: 73%, отношение E/Z: 98/2). ЖХ анализ маточного раствора показал, что отношение E/Z составляет 40/60, что является доказательством селективного осаждения соединения (E)-(IVe). Внешний вид: окрашенное твердое вещество (т. пл.: 110,5 С). 1 Соединение (VIa) через мезилат соединения (IIIa). 28,2 г (120 ммоль) соединения (E)-(IVa) и 18,4 мл (132 ммоль) триэтиламина растворяют в 180 мл толуола при 0 С. Медленно добавляют 14,4 г (125,8 ммоль) метансульфонилхлорида и реакционную смесь перемешивают 1 ч при 0 С. Образовавшийся триэтиламин гидрохлорид промывают 120 мл холодной воды, и органический слой сушат над сульфатом натрия, фильтруют и добавляют к 22,2 г (120 ммоль) фталимида калия (соединение (V), M=K) и 2,43 г (6,6 ммоль) TBAI в 60 мл толуола. Реакционную смесь перемешивают в течение ночи при комнатной температуре, затем промывают водой (120 мл),фильтруют и концентрируют под вакуумом. Остаток перекристаллизовывают из 30 мл изопропанола с получением 33 г белого твердого вещества. Выход: 76%. Внешний вид: белое твердое вещество (т. пл.: 125,1 С).F ЯМР (377 МГц, хлороформ-d)м.д. -118,91 (с, 1F). Элементный анализ: расчетный: С (72,51%), Н (5,81%), F (5,21%), N (7,69%), О (8,78%); найденный: С (72,68%), Н (5,84%), N (7, 66%). Соединение (VIa) путем реакции Мицунобу. 8,43 мл (42,5 ммоль) DIAD по каплям добавляли к холодному (-10 С) раствору 10 г (42,5 ммоль) соединения (E)-(IVa), 6,25 г (42,5 ммоль) фталимида и 11,15 г (42,5 ммоль) трифенилфосфина в 85 мл толуола. Через 24 ч при -10 С добавляли воду, нерастворимые вещества отфильтровывали и фильтрат декантировали. Два слоя разделяли и органический слой сушили над сульфатом натрия, фильтровали и концентрировали под вакуумом. Остаток перекристаллизовывали из изопропанола, получая 10,2 г соединения (VIa) (выход: 66%). Спектр ЯМР и масс-спектр идентичны спектрам соединения (VIa), полученного через мезилат соединения (IIIa). Соединение (VIe) через мезилат соединения (IIIe). 0,49 мл (6,32 ммоль) метансульфонилхлорида добавляют за 10 мин к раствору 1,50 г (6,02 ммоль) соединения (IVe) и 0,92 мл (6,62 ммоль) триэтиламина в 9 мл толуола, выдержанного при 0 С. Через 1 ч при 0 С добавляют 6 мл воды. Слои разделяли, органический слой сушили над сульфатом натрия и фильтровали. Полученный таким образом раствор мезилата добавляли за 5 минут к суспензии 1,17 г(6,32 ммоль) фталимида калия (соединение (V), М=K) и 133 мг (0,36 ммоль) TBAI в 3 мл толуола. Реакционную смесь перемешивали при 10 С в течение 2 ч, затем в течение ночи при комнатной температуре. Добавляли 6 мл воды, нерастворимые вещества отфильтровывали, фильтрат декантировали, водный слой отбросили, а органический слой сушили над сульфатом натрия и фильтровали с получением 25,12 г 4,6%-ного (вес./вес.) раствора соединения (VIe) в толуоле (выход in situ: 51%). Раствор концентрировали под вакуумом и остаток перекристаллизовывали из н-бутанола с получением 1,02 г соединения (VIe). Маточный раствор концентрировали под вакуумом и фильтровали через подушку силикагеля (элюент: гептаны-этилацетат 1/1-1/3) с получением еще 0,11 г соединения (VIe). Суммарный выход: 47%. Внешний вид: белое твердое вещество (т. пл.: 97,0 С). 1 Соединение (1) .HCl-соль из соединения (VIa). 9,8 мл (90,6 ммоль) 1-хлорэтил хлорформиата медленно добавляют к раствору 30 г (82,3 ммоль) соединения (Е)-(Va) в 165 мл толуола, выдержанного при 0 С. Реакционную смесь перемешивают в течение 1 ч при комнатной температуре, затем 1 ч при 80 С и фильтруют. 24 мл этанола и 15,35 мл (90,6 ммоль) 6 М раствора HCl в изопропаноле добавляют к фильтрату и полученную смесь кипятят с обратным холодильником 4 ч, затем охлаждают до 0 С. Осадок отфильтровывают, промывают 16 мл ацетона и 16 мл толуола и сушат под вакуумом с получением 21,94 г соединения (1) .HCl-соль: Выход: 86%. ЯМР и МС данные идентичны опубликованным в литературе данным. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения (1) или его соли присоединения кислоты который включает следующие стадии: а) взаимодействие соединения формулы (I) с соединением формулы (II) в инертном растворителе где R1 представляет собой арилметил или арилэтил;X представляет собой галоген; где арил представляет собой фенил,b) восстановление соединения (III), которое представляет собой смесь (Е)-(III) и (Z)-(III), до соединения (IV), которое представляет собой смесь (E)-(IV) и (Z)-(IV), с последующим выделением соединения (E)-(IV) или его соли в виде осадка с) взаимодействие соединения формулы (Е)-(IV) с соединением (V) в условиях реакции Мицунобу с получением соединения формулы (VI), которое преобразуют в соединение (1) или его соль присоединения кислоты путем удаления заместителя R1 2. Способ по п.1, где R1 представляет собой арилметил, где арил является фенилом. 3. Способ по п.2, где R2 представляет собой метил, этил, бутил, изобутил или фенил; X представляет собой бром; А представляет собой аминокарбонил или С 1-4 алкилоксикарбонил, где С 1-4 алкил является метилом, этилом или трет-бутилом. 4. Способ по любому из пп.1-3, в котором R1 представляет собой арилметил, где арил является фенилом; R2 представляет собой метил; X представляет собой бром; А представляет собой С 14 алкилоксикарбонил, где С 1-4 алкил является этилом. 5. Способ получения соединения формулы (III), включающий взаимодействие соединения формулыX представляет собой галоген; где арил представляет собой фенил или фенил, замещенный одним С 1-4 алкилокси. 6. Способ по п.5, где R1 представляет собой С 1-4 алкил, арилметил или аллил, где арил является фенилом. 7. Способ по п.6, где R2 представляет собой метил, этил, бутил, изобутил или фенил; X представляет собой бром; А представляет собой аминокарбонил или С 1-4 алкилоксикарбонил, где С 1-4 алкил является метилом, этилом или трет-бутилом. 8. Способ по п.7, в котором R1 представляет собой арилметил, где арил является фенилом; R2 представляет собой метил; X представляет собой бром; А представляет собой С 1-4 алкилоксикарбонил, где С 14 алкил является этилом. 9. Способ восстановления соединения (III), которое представляет собой смесь (Е)-(III) и (Z)-(III), до соединения (IV), которое представляет собой смесь (Е)-(IV) и (Z)-(IV), с последующим выделением соединения (Е)-(IV) или его соли в виде осадка где R1 представляет собой арилметил или арилэтил; А представляет собой С 1-4 алкилоксикарбонил; где арил представляет собой фенил. 10. Способ по п.9, где R1 представляет собой арилметил, где арил является фенилом. 11. Способ по п.9, где А представляет собой С 1-4 алкилоксикарбонил, где С 1-4 алкил является этилом. 12. Способ по п.10, в котором R1 представляет собой арилметил, где арил является фенилом; А представляет собой С 1-4 алкилоксикарбонил, где С 1-4 алкил является этилом. 13. Способ взаимодействия соединения формулы (Е)-(IV) с соединением (V) в условиях реакции Мицунобу с получением соединения формулы (VI), которое может быть преобразовано в соединение (1) или его соль присоединения кислоты, путем удаления заместителя R1 где R1 представляет собой арилметил или арилэтил,где арил является фенилом. 14. Способ по п.13, в котором R1 представляет собой арилметил, где арил является фенилом. 15. Соединение формулы (Е)-(III) или его соли присоединения кислоты,где R1a представляет собой С 1-4 алкил, аллил, арилметил или арилэтил; А представляет собой С 1-4 алкилоксикарбонил или аминокарбонил; где арил представляет собой фенил или фенил, замещенный одним С 1-4 алкилокси. 16. Соединение формулы (E)-(III) по п.15, в котором R1a представляет собой арилметил, где арил является фенилом. 17. Соединение формулы (Е)-(III) по п.16, в котором А представляет собой С 1-4 алкилоксикарбонил. 18. Соединение формулы (E)-(III) по п.17, где А представляет собой С 1-4 алкилоксикарбонил, где С 14 алкил является этилом. 19. Соединение формулы (Е)-(IV) или его соль присоединения кислоты,где R1a представляет собой арилметил или арилэтил; А представляет собой С 1-4 алкилоксикарбонил; где арил представляет собой фенил. 20. Соединение формулы (E)-(IV) по п.19, в котором R1a представляет собой арилметил, где арил является фенилом.
МПК / Метки
МПК: C07F 9/24, C07D 401/04, C07D 211/70
Метки: усовершенствованный, 2-[(2e)-2-фтор-2-(3-пиперидинилиден)этил]-1h-изоиндол-1,3(2h)-диона, способ, получения
Код ссылки
<a href="https://eas.patents.su/22-23617-usovershenstvovannyjj-sposob-polucheniya-2-2e-2-ftor-2-3-piperidinilidenetil-1h-izoindol-132h-diona.html" rel="bookmark" title="База патентов Евразийского Союза">Усовершенствованный способ получения 2-[(2e)-2-фтор-2-(3-пиперидинилиден)этил]-1h-изоиндол-1,3(2h)-диона</a>
Полиморфная форма iii n-[2-(диэтиламино)этил]-5-[(5-фтор-1,2-дигидро-2-оксо-3н-индол-3-илиден)метил]-2,4-диметил-1н-пиррол-3-карбоксамида и способ ее получения

Номер патента: 20067
Опубликовано: 29.08.2014
Авторы: Штригель Ханс-Гюнтер, Бёзе Роланд, Латц Рюдигер, Мохамад Несрин
МПК: C07D 403/06
Метки: n-[2-(диэтиламино)этил]-5-[(5-фтор-1,2-дигидро-2-оксо-3н-индол-3-илиден)метил]-2,4-диметил-1н-пиррол-3-карбоксамида, форма, полиморфная, способ, получения
Формула / Реферат:
1. Полиморфная форма III Сунитиниба, характеризующаяся пиками на XRPD диаграмме при 6,3±0,2; 22,2±0,2 и 26,4±0,2 градусах 2-тета.2. Полиморфная форма III Сунитиниба по п.1, характеризующаяся следующими характеристиками:кристаллографическая система: моноклинная;пространственная группа: Р 21/n;параметры элементарной ячейки:а = 4,97560(10) Å;b = 28,1365(6) Å;с= 14,5880(3) Å;β = 93,5130(10)°;объем ячейки V: 2038,42(7)...
Способ получения (-)-(s)-3-[1-(диметиламино)этил]фенил-n-этил-n-метилкарбамата

Номер патента: 6967
Опубликовано: 30.06.2006
Авторы: Симек Станислав, Гайичек Йосеф, Степанкова Гана
МПК: C07C 271/44, C07C 215/50, C07C 269/00...
Метки: способ, получения, s)-3-[1-(диметиламино)этил]фенил-n-этил-n-метилкарбамата
Формула / Реферат:
1. Способ получения (-)-(S)-3-[1-(диметиламино)этил]фенил-N-этил-N-метилкарбамата, то есть ривастигмина формулы II или его гидротартрата формулы I отличающийся тем, что метоксиацетофенон формулы VI подвергают восстановительному аминированию с получением соединения формулы V которое затем О-дезалкилируют с получением рацемического амина формулы IV который далее разделяют путем взаимодействия с оптически активной кислотой, после чего...
Усовершенствованный способ получения производных норбензоморфана с ценными фармацевтическими свойствами

Номер патента: 3059
Опубликовано: 26.12.2002
Авторы: Грауэрт Маттиас, Шнаубельт Йюрген, Бальтес Ханфрид
МПК: C07D 221/26
Метки: ценными, усовершенствованный, способ, получения, свойствами, фармацевтическими, норбензоморфана, производных
Формула / Реферат:
1. Способ получения R-, соответственно S-норбензоморфанов общей формулы 1 в которой R1 может обозначать Н, С1-С8алкил, С1-С8алкокси-, гидроксигруппу или галоген, отличающийся тем, что производное 4-метиленпиперидина общей формулы 2 переводят с помощью кислоты в соответствующую кислотно-аддитивную соль и эту соль подвергают в реакционной среде взаимодействию с галогенидом алюминия (III), предпочтительно с трибромидом алюминия или трихлоридом...
Усовершенствованный способ получения карбапенема

Номер патента: 6098
Опубликовано: 25.08.2005
Авторы: Уилльямс Джон М., Скерлдж Ренато
МПК: C07D 477/20
Метки: карбапенема, получения, способ, усовершенствованный
Формула / Реферат:
1. Способ получения соединения, представленного формулой I или его фармацевтически приемлемой соли, где R1 и R2 независимо означают H, C1-10алкил, C6-10арил или C5-10гетероарил, причем указанные алкил, арил или гетероарил могут быть незамещенными или замещенными 1-3 группами, выбранными из группы, состоящей из галогена, гидроксигруппы, цианогруппы, ацила, ациламиногруппы, аралкоксигруппы, алкилсульфонила, арилсульфонила,...
Усовершенствованный способ получения невирапина

Номер патента: 7497
Опубликовано: 27.10.2006
Авторы: Ло Юн С., Гаптон Бернард Франклин, Босуэлл Роберт Ф.Джр.
МПК: C07D 213/78, A61K 31/551, C07D 213/80...
Метки: способ, усовершенствованный, невирапина, получения
Формула / Реферат:
1. Способ получения невирапина, заключающийся в том, что (а) подвергают взаимодействию 2-гало-3-пиридинкарбонитрил формулы где X обозначает атом фтора, хлора, брома или йода, предпочтительно хлора или брома, с циклопропиламином с получением 2-(циклопропиламино)-3-пиридинкарбонитрила; (б) гидролизуют 2-(циклопропиламино)-3-пиридинкарбонитрил с получением 2-(циклопропиламино)-3-пиридинкарбоновой кислоты; (в) выделяют...
Предыдущий патент: Антагонисты trpv4
Следующий патент: Производные (гет)арил-п-хинона для лечения митохондриальных болезней
Случайный патент: Держатель интраокулярной линзы