Лекарственное средство на основе оксикодона














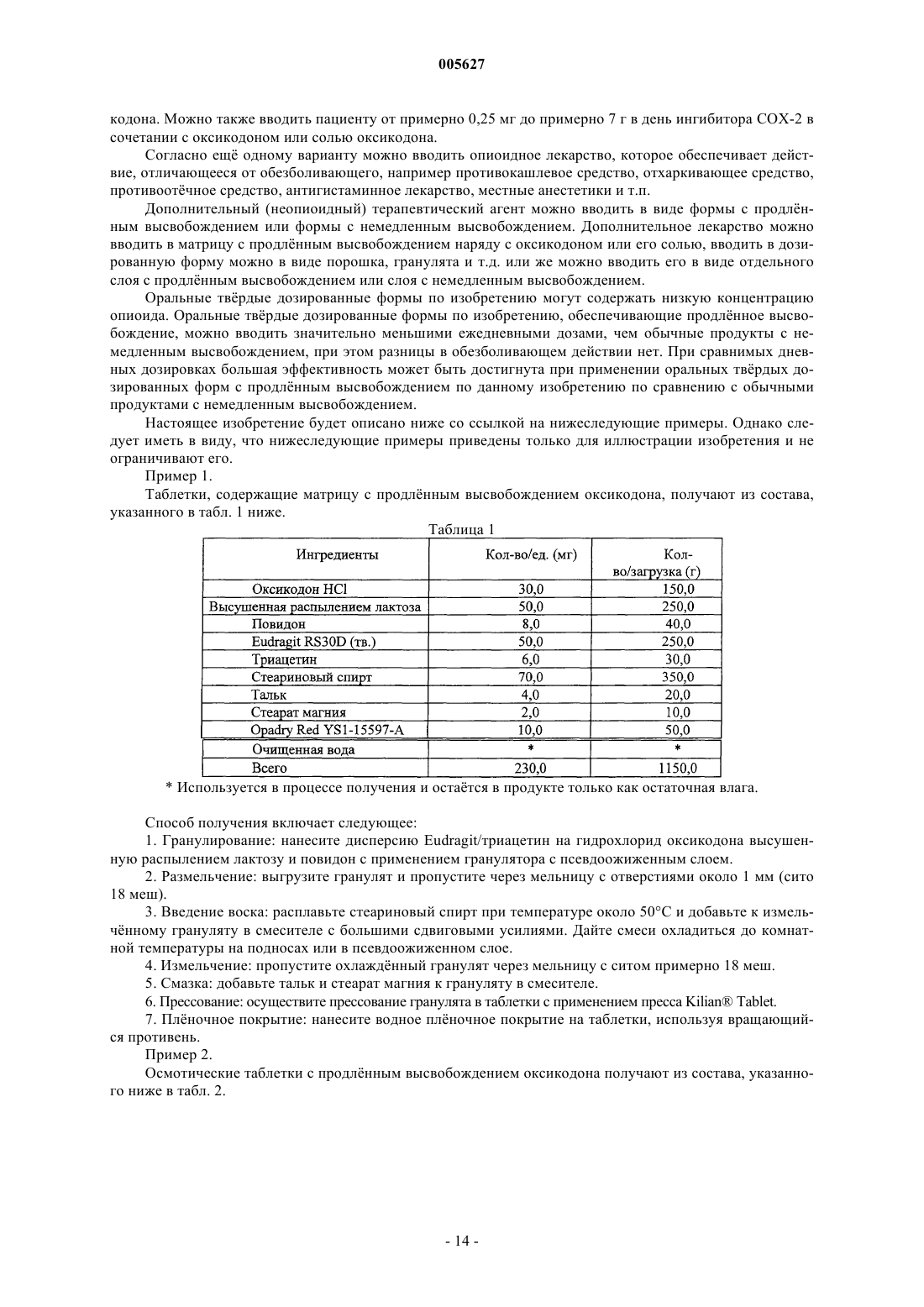
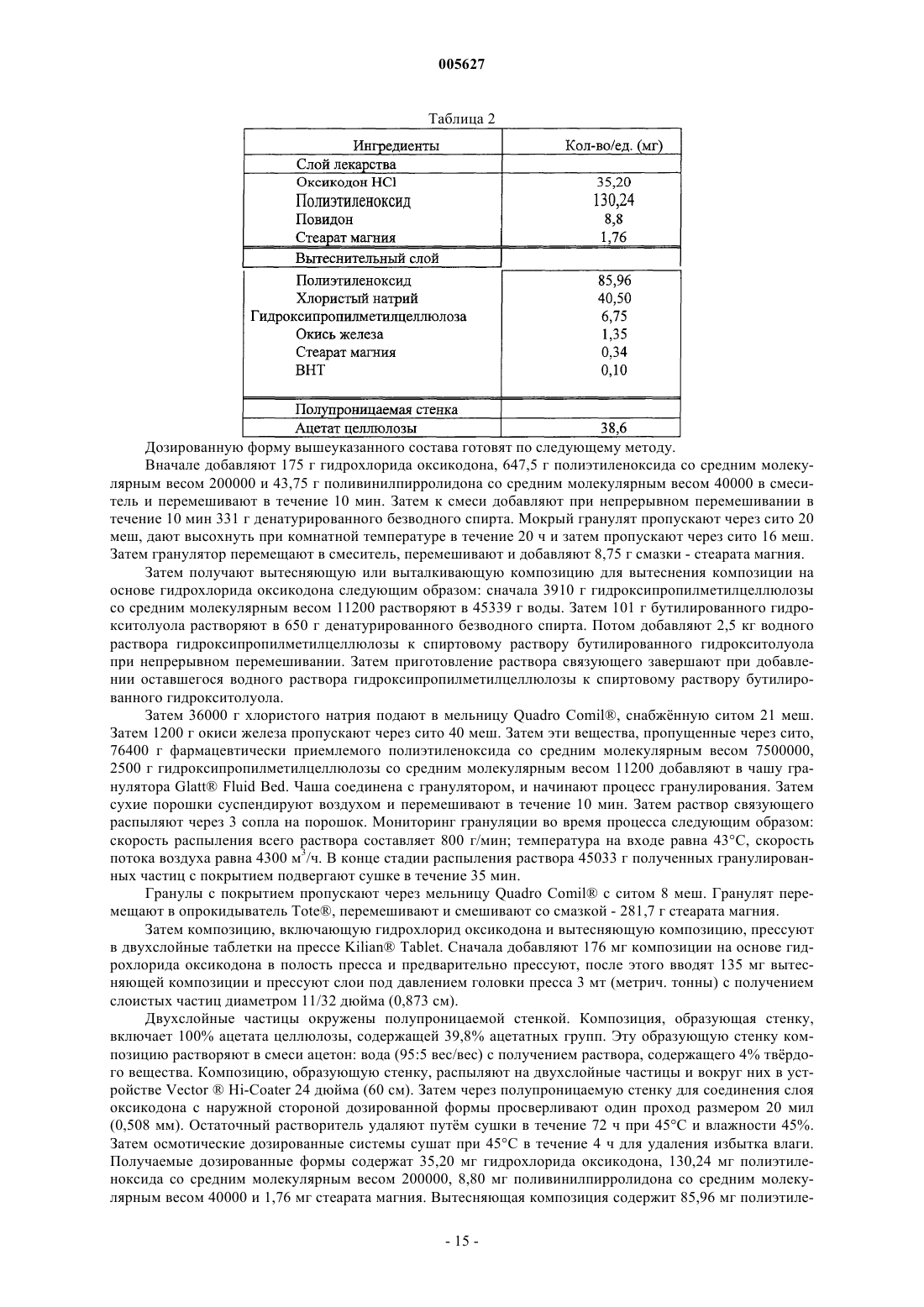


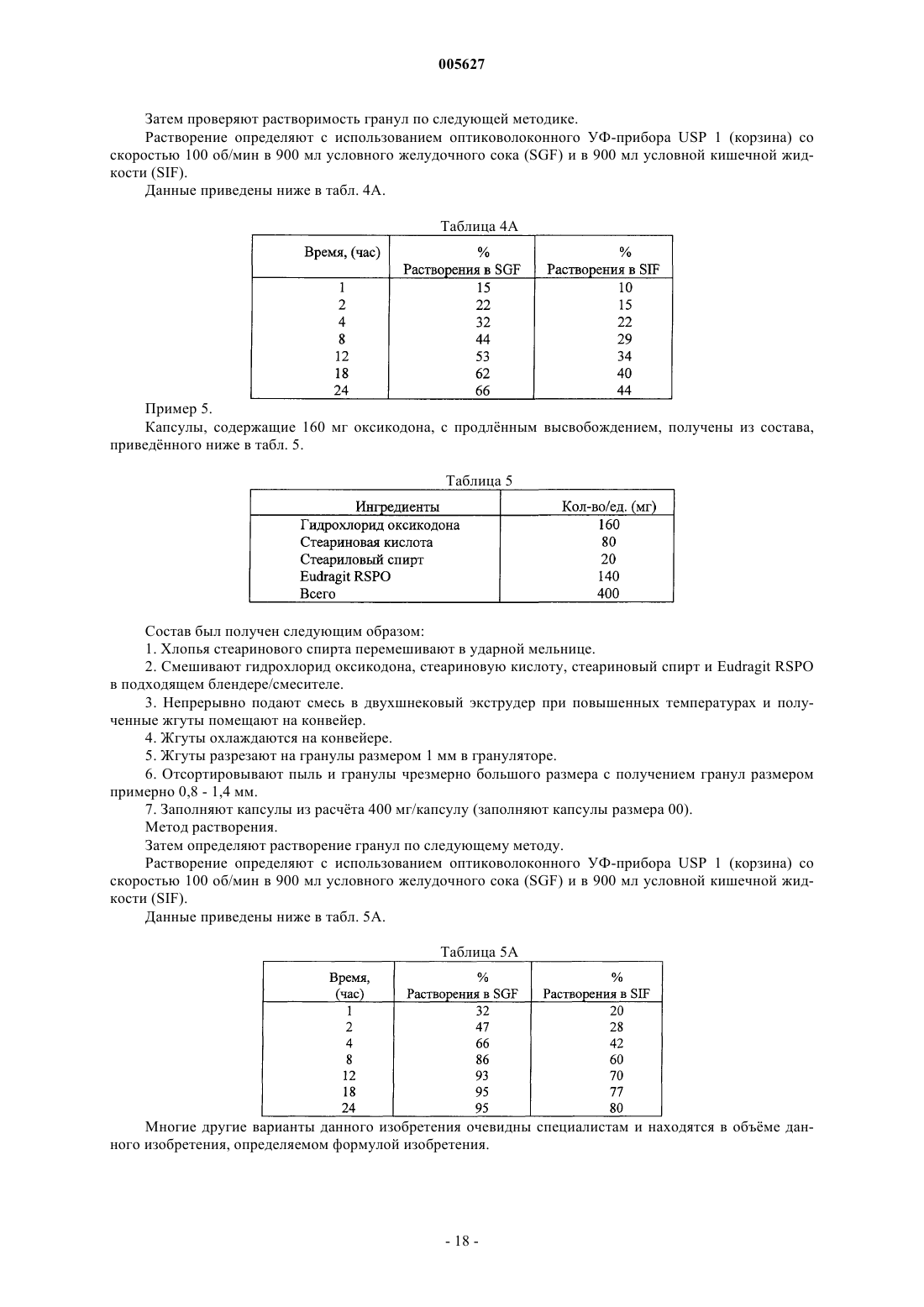


Формула / Реферат
1. Оральная дозированная форма с продлённым высвобождением для введения по схеме один раз в день, включающая фармацевтически приемлемую матрицу, содержащую анальгетически эффективное количество оксикодона или его фармацевтически приемлемой соли и вещество, способствующее продлённому высвобождению, причём указанная дозированная форма обеспечивает обезболивающий эффект в течение по меньшей мере 24 ч после перорального введения в стационарном состоянии людям и указанная дозированная форма обеспечивает среднюю величину отношения C24/Cmax оксикодона, равную 0,6-1,0 после перорального введения в стационарном состоянии указанным пациентам.
2. Оральная дозированная форма с продлённым высвобождением для введения по схеме один раз в день, включающая множество фармацевтически приемлемых матриц, содержащих анальгетически эффективное количество оксикодона или его соли и вещество, способствующее продлённому высвобождению, причём указанная дозированная форма обеспечивает обезболивающий эффект в течение по меньшей мере 24 ч после перорального введения в стационарном состоянии людям и среднюю величину отношения C24/Cmax оксикодона, равную 0,6 - 1,0 после перорального введения в стационарном состоянии указанным пациентам.
3. Дозированная форма по п.1 или 2, отличающаяся тем, что она обеспечивает среднюю величину Tmax оксикодона, равную от 2 до 17 ч, от 8 до 16 ч, от 12 до 16 ч и от 6,5 до 17 ч после введения в стационарном состоянии людям.
4. Дозированная форма по п.1 или 3, отличающаяся тем, что матрица является практически гомогенной.
5. Дозированная форма по п.4, отличающаяся тем, что она содержится в желатиновой капсуле или сформована в виде таблетки.
6. Дозированная форма по п.2 или 3, отличающаяся тем, что указанное множество матриц содержится в желатиновой капсуле и/или указанное множество матриц содержится в виде таблетки.
7. Дозированная форма по любому из пп.1-6, отличающаяся тем, что указанный оксикодон или его фармацевтически приемлемая соль содержится в количестве от 5 до 640 мг.
8. Дозированная форма по п.7, отличающаяся тем, что указанная фармацевтически приемлемая соль оксикодона является гидрохлоридом оксикодона.
9. Дозированная форма по любому из пп.1-7, отличающаяся тем, что она обеспечивает среднюю величину отношения C24/Cmax, равную 0,7-1,0, 0,7-0,99 или 0,8-0,95 после введения в стационарном состоянии указанным пациентам.
10. Дозированная форма по любому из пп.1-9, отличающаяся тем, что она обеспечивает in vitro скорость высвобождения оксикодона или его фармацевтически приемлемой соли, измеренную по методу USP Basket при скорости 100 об./мин в 900 мл водного буфера при pH от 1,6 до 7,2 при 37шC, равную от 0 до 40% через 1 ч, от 8 до 70% через 4 ч, от 20 до 80% через 8 ч, от 30 до 95% через 12 ч, от 35 до 95% через 18 ч и более 50% через 24 ч.
11. Оральная дозированная форма с продлённым высвобождением, включающая
(а) двухслойное ядро, содержащее
(i) слой лекарства, включающий анальгетически эффективное количество оксикодона или его фармацевтически приемлемой соли и
(ii) вытеснительный слой, включающий осмополимер, и
(б) полупроницаемую стенку, окружающую двухслойное ядро, содержащую проход, расположенный в указанной стенке для высвобождения оксикодона или его фармацевтически приемлемой соли; причём указанная дозированная форма обеспечивает обезболивающий эффект в течение по меньшей мере 24 ч после перорального введения в стационарном состоянии людям и среднюю величину отношения C24/Cmax оксикодона, равную 0,6-1,0, после перорального введения в стационарном состоянии указанным пациентам.
12. Дозированная форма по п.11, отличающаяся тем, что она обеспечивает среднюю величину Tmax оксикодона, равную от 2 до 17 ч или от 8 до 16 ч, от 12 до 16 ч после перорального введения в стационарном состоянии.
13. Дозированная форма по п.11 или 12, отличающаяся тем, что указанный вытеснительный слой дополнительно содержит осмагент, предпочтительно выбранный из группы, состоящей из осмотических солей и осмотических карбогидратов.
14. Дозированная форма по любому из пп.11-13, отличающаяся тем, что оксикодон или его фармацевтически приемлемая соль содержится в количестве от 5 до 640 мг.
15. Дозированная форма по п.14, отличающаяся тем, что указанная фармацевтически приемлемая соль оксикодона является гидрохлоридом оксикодона.
16. Дозированная форма по любому из пп.11-14, отличающаяся тем, что она обеспечивает среднюю величину отношения C24/Cmax, равную 0,7-0,99 или 0,8-0,95, после введения в стационарном состоянии.
17. Дозированная форма по любому из пп.11-16, отличающаяся тем, что она обеспечивает in vitro скорость высвобождения оксикодона или его фармацевтически приемлемой соли, измеренную по методу USP Basket при скорости 100 об./мин в 900 мл водного буфера при pH от 1,6 до 7,2 при 37шC, равную от 0 до 40% через 1 ч, от 8 до 70% через 4 ч, от 20 до 80% через 8 ч, от 30 до 95% через 12 ч, от 35 до 95% через 18 ч и более 50% через 24 ч.
18. Применение любой из дозированных форм по пп.1-17 для изготовления фармацевтического препарата для лечения боли для обеспечения обезболивающего эффекта в течение по меньшей мере 24 ч и средней величины отношения C24/Cmax оксикодона, равной 0,6-1,0 у пациентов после введения в стационарном состоянии указанным пациентам.
Текст
Область, к которой относится изобретение Настоящее изобретение относится к лекарственным средствам с продлнным высвобождением, содержащим оксикодон или его фармацевтически приемлемую соль, которая пригодна для введения пациенту. Уровень техники Опиоидные лекарственные средства с продлнным высвобождением для введения один раз в день описаны в патентах США 5 478 577, 5 672 360, 5 958 459, 6 103 261, 6 143 332, 5 965 161, 5 958 452 и 5 968 551. Сущность и цели изобретения Цель данного изобретения заключается в создании лекарственного средства на основе оксикодона,пригодного для введения один раз в день для эффективного снятия боли. Целью предпочтительных вариантов данного изобретения является получение фармацевтически приемлемой дозированной формы для перорального введения оксикодона с целью анальгезирующей терапии, действующей после его сравнительно короткого полупериода существования в организме в течение продолжительного времени и имеющей продолжительность облегчения боли, равную по меньшей мере 24 ч. Вышеуказанные и другие цели достигаются при осуществлении данного изобретения, которое касается дозированной формы, включающей анальгетически эффективное количество оксикодона или его фармацевтически приемлемой соли и вещество, поддерживающее высвобождение лекарства, причм дозированная форма обеспечивает обезболивающий эффект в течение по меньшей мере примерно 24 ч после орального введения в стационарном состоянии людям, и дозированная форма обеспечивает среднее отношение оксикодона С 24/Сmax, равное 0,6-1,0, после перорального введения в стационарном состоянии пациентам. Согласно некоторым вариантам изобретения дозированная форма после введения пациентам обеспечивает среднюю величину Тmах оксикодона in vivo, которое возникает через примерно 2-17 ч (например, примерно 2-8 ч) после введения в стационарном состоянии дозированной формы. Согласно некоторым вариантам изобретения среднее Тmах оксикодона in vivo возникает через примерно 6,5-17 ч, через примерно 8-16 ч, через примерно 10-16 ч или через примерно 12-16 ч после введения в стационарном состоянии дозированной формы. Согласно некоторым вариантам изобретения дозированная форма обеспечивает обезболивающий эффект в течение примерно 24 ч после введения дозированной формы людям в стационарном состоянии и обеспечивает среднее отношение С 24/Сmах для оксикодона, равное 0,6-1,0 после введения пациентам в стационарном состоянии. Согласно некоторым вариантам данного изобретения дозированная форма обеспечивает обезболивающее действие в течение по меньшей мере примерно 24 ч после введения человеку в стационарном состоянии и среднюю величину отношения С 24/Сmах для оксикодона, равную 0,6-1,0 или 0,7-1,0 после введения пациентам в стационарном состоянии. Согласно некоторым вариантам данного изобретения дозированная форма обеспечивает in vitro скорость высвобождения оксикодона или его фармацевтически приемлемой соли (при измерении методом USP Basket при скорости 100 об/мин в 900 мл водного буфера при рН в интервале 1,6-7,2 при 37 С),равную 0-40% за 1 ч, от примерно 8 до примерно 70% за 4 ч, от примерно 20 до примерно 80% через 8 ч,от примерно 30 до примерно 95% через 12 ч, от примерно 35 до примерно 95% через 18 ч и более 50% через 24 ч. Согласно некоторым предпочтительным вариантам оральная дозированная форма с продлнным высвобождением по данному изобретению обеспечивает содержание оксикодона, которое эффективно в течение 24 ч, характеризующееся величиной W50 для оксикодона, равной 4-24 ч после введения в стационарном состоянии. Согласно некоторым вариантам величина W50 равна по меньшей мере 4 ч, предпочтительно по меньшей мере 12 ч и более предпочтительно по меньшей мере 18 ч после введения в стационарном состоянии. Согласно некоторым вариантам оральная дозированная форма с продлнным высвобождением по данному изобретению содержит матрицу, которая включает вещество, обеспечивающее продлнное высвобождение, и оксикодон или его фармацевтически приемлемую соль. Согласно некоторым вариантам матрица прессуется в таблетку и может иметь покрытие, которое в добавление к веществу в матрице, обеспечивающему продлнное высвобождение, может регулировать высвобождение оксикодона или его фармацевтически приемлемой соли из препарата для того, чтобы содержание активного ингредиента в крови поддерживалось в терапевтически эффективном интервале в течение продолжительного периода времени. Согласно некоторым альтернативным вариантам матрица является инкапсулированной. Согласно некоторым вариантам оральная дозированная формас продлнным высвобождением по данному изобретению содержит несколько фармацевтически приемлемых матриц, поддерживающих высвобождение лекарства, содержащих оксикодон или его фармацевтически приемлемую соль, при этом-1 005627 дозированная форма поддерживает содержание оксикодона в плазме крови в терапевтически эффективном материале в течение продолжительного периода времени при введении пациентам. Предпочтительно, чтобы составы, приготовленные согласно данному изобретению, были в виде таблеток, капсул или других подходящих единичных дозированных форм. Согласно некоторым вариантам оральная дозированная форма с продлнным высвобождением по данному изобретению является осмотической дозированной формой, которая содержит ядро из однослойного или двухслойного материала, включающего оксикодон или его фармацевтически приемлемую соль; вспенивающийся полимер; полупроницаемую мембрану, окружающую ядро; и проход, расположенный в полупроницаемой мембране, для продлнного высвобождения оксикодона или его фармацевтически приемлемой соли, для того, чтобы содержание активного ингредиента в крови поддерживалось в терапевтически эффективном интервале в течение продолжительного периода времени после введения пациентам. Согласно некоторым вариантам оральная дозированная форма с продлнным высвобождением по данному изобретению содержит практически гомогенное ядро, включающее оксикодон или его фармацевтически приемлемую соль и вспенивающийся полимер; полупроницаемую мембрану, окружающую ядро; проход, расположенный в полупроницаемой мембране для продлнного высвобождения оксикодона или его фармацевтически приемлемой соли, для того, чтобы содержание активного ингредиента в крови поддерживалось в терапевтически эффективном интервале в течение продолжительного периода времени при введении пациентам. Согласно некоторым вариантам данного изобретения предусмотрен способ лечения состояний, связанных с ощущением боли, у пациентов, нуждающихся в таком лечении, причм этот способ включает введение пациенту эффективного количества оксикодона или его фармацевтически приемлемой соли в виде дозированной формы с продлнным высвобождением, описанной выше. Согласно некоторым вариантам данное изобретение относится к применению дозированной формы с продлнным высвобождением, содержащей фармацевтически приемлемую матрицу, включающую оксикодон или его фармацевтически приемлемую соль и вещество, способствующее продлнному выделению, для изготовления обезболивающего препарата для орального введения человеку по схеме один раз в день, чтобы обеспечить обезболивающий эффект в течение по меньшей мере 24 ч, и среднее отношение С 24/Сmах для оксикодона, равное 0,6-1,0 после введения в стационарном состоянии указанному пациенту. Согласно некоторым вариантам изобретение направлено на применение оральной дозированной формы с продлнным высвобождением, содержащей двухслойное ядро, включающее слой лекарства,представляющего собой анальгетически эффективное количество оксикодона или его фармацевтически приемлемой соли; и вытесняющий слой, содержащий осмотический полимер; и полупроницаемую стенку, окружающую двухслойное ядро, содержащую расположенный в ней проход для высвобождения указанного оксикодона или его фармацевтически приемлемой соли; для изготовления обезболивающего препарата для перорального введения человеку для обеспечения обезболивающего действия в течение по меньшей мере 24 ч после перорального введения в стационарном состоянии человеку и среднего отношения С 24/Сmах для оксикодона, равного 0,6-1,0 после введения в стационарном состоянии пациенту. Согласно некоторым вариантам изобретение относится к применению дозированной формы с продлнным высвобождением, содержащей несколько матриц с продлнным высвобождением, включающих оксикодон или его фармацевтически приемлемую соль и вещество, обеспечивающее продлнное высвобождение, для изготовления обезболивающего препарата для перорального введения человеку для обеспечения обезболивающего действия в течение по меньшей мере 24 ч после перорального введения в стационарном состоянии человеку и среднего отношения С 24/Сmах для оксикодона, равного 0,6-1,0 после введения в стационарном состоянии пациенту. Обозначение Сmах, используемое в данной заявке, означает максимальную концентрацию лекарства в плазме, достигаемую в интервале вводимых доз. Обозначение С 24, используемое в данной заявке, означает концентрацию лекарства в плазме через 24 ч после введения. Обозначение Тmах, используемое в данной заявке, означает период времени после введения дозированной формы до достижения максимальной концентрации лекарства в плазме в интервале вводимых доз. Обозначение W50 для целей данного изобретения означает период времени, в течение которого концентрация в плазме равна или превышает величину, составляющую 50% от пиковой концентрации. Этот параметр определяется путм линейной интерполяции наблюдаемых данных и представляет собой разницу во времени между достижением первого (или единственного) подъма кривой и последнего (или единственного) наклона профиля кривой в плазме. Термин отношение С 24/Сmах используется для целей данного изобретения для обозначения отношения концентрации лекарства в плазме через 24 ч после введения к максимальной концентрации лекарства в плазме, достигаемой в используемом интервале доз. Выражение способ USP Basket означает метод Basket'a, описанный в US Pharmacopoeia XXII-2 005627 Термин стационарное состояние означает, что количество лекарства, достигающего системы,приблизительно равно количеству лекарства, выходящего из системы. Таким образом, в стационарном состоянии из тела пациента лекарство выделяется примерно с той же скоростью, с какой лекарство попадает в систему пациента путм абсорбции потоком крови. Термин полупроницаемая стенка для целей данного изобретения означает, что стенка является проницаемой для жидкости, поступающей извне, например, водной или биологической жидкости в окружающей среде, включая желудочно-кишечный тракт, но не проницаема для лекарства. Термин вспенивающийся полимер для целей данного изобретения означает полимер, который под действием водной или биологической жидкости абсорбирует жидкость, что приводит к увеличению его массы. Термин средний для целей данного изобретения означает фармакокинетический параметр (например, Тmах), представляющий собой арифметическую среднюю величину, измеренную у определнного количества пациентов. Выражение фармацевтически приемлемая соль включает, но не ограничивается этим, соли металлов, такие как натриевая соль, калиевая соль, цезиевая соль и т.п.; соли щелочно-земельных металлов,такие как соль кальция, соль магния и т.п.; соли органических аминов, такие как соль триэтиламина, соль пиридина, соль пиколина, соль этаноламина, соль триэтаноламина, соль дициклогексиламина, соль N,N'дибензилэтилендиамина и т.п.; соли неорганических кислот, такие как гидрохлорид, гидробромид, сульфат, фосфат и т.п.; соли органических кислот, такие как формат, ацетат, трифторацетат, малеат, фумарат,тартрат и т.п.; сульфонаты, такие как метансульфонат, бензолсульфонат, п-толуолсульфонат и т.п.; соли аминокислот, такие как аргинат, аспарагинат, глутамат и т.п. Описание изобретения Согласно некоторым вариантам изобретения дозированная форма с продлнным высвобождением обеспечивает in vitro скорость высвобождения оксикодона или его фармацевтически приемлемой соли,измеренную способом USP Basket'а при скорости 100 об/мин в 900 мл водного буфера при рН в интервале 1,6-7,2 при 37 С, равную от 0 до примерно 40% через 1 ч, от примерно 8 до примерно 70% через 4 ч,от примерно 20 до примерно 80% через 8 ч, от примерно 30 до примерно 95% через 12 ч, от примерно 35 до примерно 95% через 18 ч и более, чем примерно 50% через 24 ч. Согласно некоторым вариантам данного изобретения промежуток времени, в течение которого количество оксикодона в крови (после введения в стационарном состоянии) больше или превышает 75% от величины максимальной концентрации в крови (Т 0,75 Сmах), может составлять 4 ч или более, предпочтительно 6 ч или более. Согласно некоторым вариантам промежуток времени, в течение которого содержание оксикодона в крови достигает максимального значения (Тmax), составляет примерно от 2 до 17 ч,предпочтительно от примерно 6,5 до примерно 17 ч, более предпочтительно от примерно 8 до примерно 16 ч и даже, более предпочтительно, от примерно 10 до примерно 16 ч или от примерно 12 до примерно 16 ч после введения в стационарном состоянии дозированной формы. Согласно некоторым вариантам дозированная форма обеспечивает отношение С 24/Сmax после введения в стационарном состоянии, равное 0,6-1,0, 0,7-0,99 или 0,8-0,95. Согласно другим вариантам изобретения дозированная форма обеспечивает отношение С 24/Сmax после введения в стационарном состоянии,равное 0,7-1,0, 0,72-0,99 и 0,74-0,95. Согласно некоторым вариантам данного изобретения дозированная форма обеспечивает отношение С 24/Сmax после введения в стационарном состоянии, равное 0,6-1,0, 0,7-0,99 и 0,8-0,95 и (Тmах), равное от примерно 6,5 до примерно 17 ч, от примерно 8 до примерно 16 ч, от примерно 10 до примерно 16 ч или от примерно 12 до примерно 16 ч. В других вариантах данного изобретения после введения в стационарном состоянии дозированная форма обеспечивает отношение С 24/Сmax, равное 0,7-1,0, 0,72-0,99 и 0,740,95 и (Тmах), равное от примерно 2 до примерно 17 ч. Согласно некоторым вариантам изобретения одновременный прим пищи увеличивает или уменьшает степень абсорбции оксикодона незначительно. Оральная дозированная форма с поддерживаемым высвобождением по изобретению включает от примерно 1 до примерно 640 мг оксикодона или его фармацевтически приемлемой соли (например, гидрохлорида оксикодона). Предпочтительно, чтобы дозированная форма с поддерживаемым высвобождением по изобретению включала от примерно 5 до примерно 500 мг оксикодона или его фармацевтически приемлемой соли, более предпочтительно, от примерно 10 до примерно 320 мг оксикодона или его фармацевтически приемлемой соли и, ещ более предпочтительно, от примерно 10 до примерно 160 мг оксикодона или его фармацевтически приемлемой соли. Согласно другим вариантам дозированная форма с поддерживаемым высвобождением по изобретению содержит от примерно 10 до примерно 160 мг гидрохлорида оксикодона или эквивалентное количество оксикодона или другой фармацевтически приемлемой его соли, отличающейся от гидрохлорида осикодона. Данное изобретение предусматривает также способ введения от примерно 1 до примерно 640 мг оксикодона или его фармацевтически приемлемой соли по схеме один раз в день пациенту, нуждающемуся в ослаблении боли, в соответствии с фармакокинетическими параметрами, описанными в данной заявке.-3 005627 Предпочтительно этот способ включает введение от примерно 5 до примерно 500 мг оксикодона или его фармацевтически приемлемой соли. Способ введения согласно данному изобретению особенно пригоден для лечения острой и хронической боли, особенно боли, связанной с терминальным заболеванием, таким как рак, хронической боли в спине и послеоперационных болей. Дозированная форма Согласно некоторым вариантам изобретения оральная дозированная форма включает обеспечивающее продлнное высвобождение лекарства вещество, которое введено в матрицу наряду с оксикодоном или его фармацевтически приемлемой солью, для обеспечения продлнного высвобождения оксикодона. Вещество, обеспечивающее продлнное высвобождение, может быть гидрофобным или гидрофильным, как это желательно. Оральная дозированная форма согласно данному изобретению может быть получена в виде гранул, сферических частиц, матриксных мультичастиц и т.д., которые содержат оксикодон или его фармацевтически приемлемую соль в матрице с продлнным высвобождением, которая может быть в виде спрессованной таблетки или инкапсулирована. Оральная дозированная форма по изобретению может также включать другие фармацевтически приемлемые ингредиенты (например, разбавители, связующие, красители, смазки и т.д.). Согласно некоторым вариантам оральная дозированная форма по данному изобретению может быть осмотической дозированной формой, включающей выталкивающую или вытесняющую композицию в виде одного из слоев двухслойного ядра для вытеснения оксикодона или его фармацевтически приемлемой соли из дозированной формы, и полупроницаемую стенку, окружающую ядро, причм эта стенка имеет по меньшей мере один выход или проход для доставки оксикодона из дозированной формы. Иначе, ядро осмотической дозированной формы может представлять собой однослойное ядро, содержащее полимер, способствующий высвобождению, и оксикодон или его фармацевтически приемлемую соль. Предпочтительно, чтобы дозированные формы согласно данному изобретению обеспечивали обезболивающий эффект в течение по меньшей мере 24 ч после введения. Получение матриц с продлнным высвобождением Согласно одному предпочтительному варианту данного изобретения носитель, обеспечивающий продлнное высвобождение, может быть включн в матрицу наряду с оксикодоном или его фармацевтически приемлемой солью, такая матрица обеспечивает продлнное высвобождение оксикодона. Не ограничивающий перечень веществ, обеспечивающих продлнное высвобождение, которые могут быть введены в матрицу, обеспечивающую продлнное высвобождение, включают гидрофильные и/или гидрофобные вещества, такие как смолы, эфиры, целлюлозы, акриловые смолы, вещества белкового происхождения, воска, шеллак и масла, такие как гидрированное касторовое масло и гидрированное растительное масло. Однако, в соответствии с данным изобретением может быть использовано любое фармацевтически приемлемое гидрофобное или гидрофильное вещество, обеспечивающее продлнное высвобождение, которое способно обеспечить продлнное высвобождение оксикодона или его фармацевтически приемлемой соли. Предпочтительными полимерами, обеспечивающими продлнное высвобождение, являются алкилцеллюлозы, такие как этилцеллюлоза, полимеры и сополимеры акриловой и метакриловой кислот, и простые эфиры целлюлозы, особенно гидроксиалкилцеллюлозы (особенно гидроксипропилцеллюлоза) и карбоксиалкилцеллюлозы. Предпочтительные полимеры и сополимеры акриловой и метакриловой кислот включают полимер метилметакрилата, сополимеры метилметакрилата, полиэтоксиэтилметакрилаты, полиэтилакрилат, полимер триметиламмоинийэтилметакрилат, полицианоэтилметакрилат, сополимер аминоалкилметакрилата, полиакриловую кислоту, полиметакриловую кислоту, сополимер алкиламинометакриловой кислоты, полиметилметакрилат, полиангидрид метакриловой кислоты, полиметакрилат, полиакриламид, ангидрид полиметакриловой кислоты и сополимеры глицидилметакрилата. В некоторых предпочтительных случаях в матрице используют смеси любых вышеуказанных веществ, обеспечивающих продлнное высвобождение. Матрица может также содержать связующее. В этих случаях предпочтительно, чтобы связующее способствовало продлнному высвобождению оксикодона или его фармацевтически приемлемой соли из матрицы. Если в состав включено дополнительное гидрофобное связующее, оно предпочтительно выбирается из природных и синтетических восков, жирных кислот, алифатических спиртов и их смесей. Примерами являютсяпчелиный воск, карнаубский воск, стеариновая кислота и стеариновый спирт. Этот перечень не является исчерпывающим. Согласно некоторым предпочтительным вариантам в состав матрицы включается комбинация двух или более гидрофобных связующих. Предпочтительными гидрофобными связующими, которые могут быть применены согласно данному изобретению, являются усваиваемые длинноцепные (C8-C50, особенно C12-C40) замещнные или незамещнные углеводороды, такие как жирные кислоты, алифатические спирты, глицериновые эфиры жирных кислот, минеральные и растительные масла,природные и синтетические воска и полиалкиленгликоли. Предпочтительными являются углеводороды,-4 005627 имеющие температуру плавления между 25 и 90 С. В некоторых вариантах изобретения из длинноцепочечных углеводородных связующих веществ предпочтительными являются жирные (алифатические) спирты. Оральная дозированная форма может содержать до 80% (по весу) по меньшей мере одного перевариваемого длинноцепочечного углеводорода. В некоторых случаях гидрофобное связующее может представлять собой природные или синтетические воска, жирные спирты (такие как лауриловый, миристиловый, стеариловый, цетиловый или,предпочтительно, цетостеариловый), жирные кислоты, включая, но не ограничиваясь этим, эфиры жирных кислот, глицериды жирных кислот (моно-, ди- и триглицериды), гидрированные жиры, углеводороды, обычные воска, стеариновую кислоту, стеариновый спирт и гидрофобные и гидрофильные вещества,содержащие углеводородные цепи. Подходящие воска включают, например, пчелиный воск, гликовоск,касторовый воск и карнаубский воск. Для целей данного изобретения воскоподобным веществом считается любое вещество, которое является тврдым при комнатной температуре и имеет точку плавления от примерно 30 С до примерно 100 С. В некоторых предпочтительных случаях дозированная форма включает матрицу с продлнным высвобождением, содержащую оксикодон или его фармацевтически приемлемую соль и по меньшей мере одну растворимую в воде гидроксиалкилцеллюлозу, по меньшей мере один С 12-С 36-, предпочтительно, С 14-С 22-алифатический спирт и, возможно, по меньшей мере один полиалкиленгликоль. Гидроксиалкилцеллюлоза, предпочтительно, представляет собой гидрокси(С 1 С 6)алкилцеллюлозу, например, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу и, особенно, гидроксиэтилцеллюлозу. Количество по меньшей мере одной гидроксиалкилцеллюлозы в данной оральной дозированной форме может быть определено, наряду с другими факторами, исходя из требуемой точной скорости высвобождения оксикодона или соли оксикодона. Алифатический спирт может быть, например, лауриловым спиртом, миристиловым спиртом или стеариловым спиртом. В особенно предпочтительных вариантах данной оральной дозированной формы,по меньшей мере один, алифатический спирт представляет собой цетиловый спирт или цетостеариловый спирт. Количество алифатического спирта в данной оральной дозированной форме может быть определено, как указано выше, исходя из требуемой точной скорости высвобождения оксикодона или соли оксикодона. Оно может также зависеть от того, содержится или отсутствует в оральной дозированной форме по меньшей мере один полиалкиленгликоль. В отсутствие по меньшей мере одного полиалкиленгликоля оральная дозированная форма предпочтительно содержит от примерно 20 до примерно 50% (по весу) алифатического спирта. Когда в оральной дозированной форме содержится полиалкиленгликоль, общее количество алифатического спирта и полиалкиленгликоля составляет от примерно 20 до примерно 50% (по весу) в расчте на вес дозированной формы. Согласно одному предпочтительному варианту, отношение количества, например по меньшей мере одной гидроксиалкилцеллюлозы или акриловой смолы к количеству по меньшей мере одного алифатического спирта/полиалкиленгликоля определяет в значительной степени скорость высвобождения оксикодона или соли оксикодона из препарата. Согласно другим вариантам отношение количества гидроксиалкилцеллюлозы к количеству алифатического спирта/полиалкиленгликоля составляет, предпочтительно, от 1:1 до 1:4, особенно предпочтительным является отношение от 1:2 до 1:3. Согласно некоторым вариантам полиалкиленгликоль может быть, например, полипропиленгликолем или полиэтиленгликолем, который является предпочтительным. Средний молекулярный вес по меньшей мере одного полиалкиленгликоля равен предпочтительно от 1000 до 15000, предпочтительно 1500-12 000. Другая подходящая матрица с продлнным высвобождением включает алкилцеллюлозу (особенно этилцеллюлозу), С 12-С 36-алифатический спирт и, возможно, полиалкиленгликоль. Кроме того, матрица с продлнным высвобождением может также содержать подходящие количества других материалов, например, разбавителей, смазывающих агентов, связующих, добавок, способствующих гранулированию, красящих веществ, ароматизаторов и агентов, облегчающих скольжение, которые являются известными в фармацевтике. Для того, чтобы облегчить приготовление тврдой оральной дозированной формы с продлнным высвобождением согласно данному изобретению, предусмотрен также способ получения тврдой оральной дозированной формы с продлнным высвобождением, включающий оксикодон или его соль в матрице с продлнным высвобождением. Введение в матрицу можно осуществить, например, путм:(a) образования гранул, содержащих по меньшей мере одно гидрофобное и/или гидрофильное вещество, описанное выше (например, водорастворимую гидроксиалкилцеллюлозу), вместе с оксикодоном или его фармацевтически приемлемой солью;(b) смешения гранул по меньшей мере с одним гидрофобным и/или гидрофильным веществом по меньшей мере с одним С 12-С 36-алифатическим спиртом и(c) возможно, прессования и формования гранул. Гранулы могут быть получены любым методом, известным специалистам в области получения фармпрепаратов. Например, по одному предпочтительному способу гранулы можно получить мокрым гранулированием смеси гидроксиалкилцеллюлоза/оксикодон или его соль с водой. Согласно наиболее-5 005627 предпочтительному варианту количество воды, добавленной на стадии мокрого гранулирования, предпочтительно в 1,5-5 раз, особенно в 1,75-3,5 раза превышает сухой вес оксикодона или его соли. Матрица с продлнным высвобождением может быть также получена, например, гранулированием из расплава или экструзией из расплава. Обычно метод гранулирования из расплава включает плавление обычно тврдого гидрофобного связующего, например воска, и введение в расплав порошка лекарства. Для получения дозированной формы с продлнным высвобождением может быть необходимым введение гидрофобного вещества, обеспечивающего продлнное высвобождение, например, этилцеллюлозы или не растворимого в воде акрилового полимера в расплавленный воск, являющийся гидрофобным связующим. Примеры препаратов с продлнным высвобождением, полученных методом гранулирования из расплава, описаны, например, в патенте США 4861598. Дополнительное гидрофобное связующее может содержать одно или более не растворимых в воде воскоподобных термопластичных веществ, возможно смешанных с одним или несколькими воскоподобными веществами, являющимися менее гидрофобными, чем вышеуказанные водонерастворимые воскоподобные вещества. Для того, чтобы достигнуть продлнного высвобождения, индивидуальные воскоподобные вещества в составе должны быть практически не расщепляемыми и не растворимыми в желудочно-кишечных жидкостях в течение начальных фаз высвобождения. Подходящими не растворимыми в воде воскоподобными связующими могут быть такие вещества, растворимость которых в воде меньше 1:5 000 (вес/вес). Получение подходящей матрицы, экструдированной из расплава, может, например, включать стадии смешения оксикодона или его фармацевтически приемлемой соли с веществом, обеспечивающим продлнное высвобождение, и предпочтительно со связующим для получения гомогенной смеси. Затем гомогенную смесь нагревают до температуры, достаточной, по меньшей мере, для размягчения смеси,чтобы е можно было экструдировать. Полученную гомогенную смесь затем экструдируют, например, с применением двухшнекового экструдера для получения жгутов. Экструдат, предпочтительно, охлаждают и режут на мультичастицы любым приспособлением, известным из уровня техники. Мультичастицы матрицы затем делят на единичные дозы. Экструдат, предпочтительно, имеет диаметр от примерно 0,1 до примерно 5 мм и обеспечивает продлнное высвобождение оксикодона или его фармацевтически приемлемой соли в течение по меньшей мере примерно 24 ч. Можно получать экструдированные из расплава составы по данному изобретению путм непосредственной подачи в экструдер гидрофобного, обеспечивающего продлнное высвобождение вещества,оксикодона или его соли и, необязательно, связующего; нагревания гомогенной смеси; экструдирования гомогенной смеси с получением жгутов; охлаждения жгутов, выполненных из гомогенной смеси; резки жгутов на мультичастицы матрицы, имеющие размер от примерно 0,1 до примерно 12 мм; и распределения указанных частиц на единичные дозы. Согласно этому аспекту изобретения реализуется непрерывный процесс. В полученные экструзией из расплава матрицы можно вводить пластификаторы, такие как описаны выше. Пластификатор предпочтительно вводить в количестве от примерно 0,1 до примерно 30% от веса матрицы. Если желательно, в матрицы с продлнным высвобождением согласно данному изобретению могут добавляться другие фармацевтические эксципиенты, например тальк, моно- или полисахариды,красящие вещества, ароматизаторы, смазочные агенты и т.п. Вводимые количества зависят от желаемых свойств. Диаметр отверстия или выходного канала может регулироваться для изменения толщины экструдированных жгутов. Кроме того, выпускное отверстие экструдера может не быть круглым; оно может быть продолговатым, прямоугольным и т.д. Выходящие жгуты могут быть переработаны в частицы с применением горячего проволочного устройства, ножа и т.д. Расплав матрицы, являющейся системой, состоящей из множества частиц, после экструзии может быть, например, в виде гранул, сферических частиц или шариков в зависимости от формы выпускного отверстия в экструдере. Для целей данного изобретения термины экструдированные из расплава многочисленные частицы матрицы, экструдированная из расплава система частиц матрицы или экструдированные из расплава мультичастицы матрицы будут относиться к множеству частиц, предпочтительно,примерно одного размера и/или формы, содержащих один или более активных агентов и один или более эксципиентов, предпочтительно включающих гидрофобный материал, обеспечивающий продлнное высвобождение. Предпочтительно, чтобы экструдированные из расплава частицы матрицы имели длину от примерно 0,1 до примерно 12 мм и имели диаметр, равный от примерно 0,1 до примерно 5 мм. Кроме того, следует иметь в виду, что экструдированные из расплава мультичастицы с указанными размерами могут иметь любую геометрическую форму. В некоторых случаях экструдат может быть просто разрезан на частицы нужной длины и распределн в единичные дозы терапевтически активного агента без осуществления стадии сферонизации. Согласно одному предпочтительному варианту получают оральные дозированные формы, которые содержат эффективное количество экструдированных из расплава мультичастиц матрицы внутри капсулы. Например, множество экструдированных из расплава мультичастиц матрицы может быть помещено-6 005627 в желатиновую капсулу в количестве, достаточном для обеспечения эффективной дозы с продлнным высвобождением после заглатывания и контакта с желудочно-кишечной жидкостью. Согласно другому варианту подходящее количество экструдата прессуется в таблетку для перорального введения с применением обычного оборудования стандартным методом. Методы получения и составы для изготовления таблеток (полученных прессованием и формованием), капсул (тврдых и мягких желатиновых) и пилюль описаны также в Remington's Pharmaceutical Sciences (Arthur Osol, editor),1553-1593 (1980). Согласно ещ одному предпочтительному варианту экструдат может быть сформован в таблетки,как описано в патенте США 4 957 681 (Klimesch et al.). Мультичастицы матрицы с продлнным высвобождением, таблетки или капсулы могут быть также снабжены покрытием с продлнным высвобождением, например покрытием, описанным далее. Такие покрытия предпочтительно включают достаточное количество гидрофобного и/или гидрофильного вещества, способствующего продлнному высвобождению лекарства, с получением увеличения веса от примерно 2 до примерно 25%, хотя покрытие может быть и толще, например, в зависимости от желаемой скорости высвобождения. Дозированные формы согласно данному изобретению могут дополнительно включать комбинации мультичастиц матрицы, полученных экструзией из расплава и содержащих оксикодон или его фармацевтически приемлемую соль. Кроме того, дозированные формы могут также содержать некоторое количество немедленно высвобождающихся терапевтически активных оксикодона или его фармацевтически приемлемой соли для достижения быстрого терапевтического эффекта. Немедленно высвобождающиеся оксикодон или его фармацевтически приемлемая соль могут вводиться, например, в виде отдельных мультичастиц в желатиновой капсуле или же могут содержать на своей поверхности покрытие на основе, например, экструдированных из расплава мультичастиц матрицы. Профиль продлнного высвобождения экструдированных из расплава составов по изобретению может быть изменн, например, путм изменения количества вещества, обеспечивающего продлнное высвобождение, путм изменения количества пластификатора по отношению к другим компонентам матрицы, путм изменения количества гидрофобного вещества, путм включения дополнительных ингредиентов или эксципиентов, путм изменения способа получения формы и т.д. Согласно другим вариантам изобретения экструдированные из расплава препараты готовят без включения оксикодона или его фармацевтически приемлемой соли, которые добавляются к экструдату впоследствии. Такие препараты обычно будут содержать оксикодон или его фармацевтически приемлемую соль, смешанные с экструдированным веществом матрицы, смесь затем таблетируют и получают состав с замедленным высвобождением. Такие препараты имеют преимущество, например, когда терапевтически активный агент, включнный в состав, является чувствительным к нагреванию, необходимому для размягчения гидрофобного материала и/или замедлителя. Типичные системы для изготовления экструдатов из расплава, подходящие для применения согласно данному изобретению, включают подходящий приводной двигатель экструдера, обеспечивающий различную скорость и постоянное моментно-силовое управление, органы управления пуском и остановом и омметр. Кроме того, эта система включает пульт контроля температуры, который содержит датчики температуры, охлаждающие средства и индикаторы температуры по всей длине экструдера. Кроме того, указанная система включает экструдер, например, двухшнековый экструдер, который состоит из двух вращающихся в противоположных направлениях перемешивающихся шнеков, заключнных в цилиндр или бочку, содержащие отверстие или головку на выходе. Исходные материалы подаются через бункерный питатель и передвигаются вдоль бочки при помощи шнеков и подаются через головку с получением жгутов, которые затем попадают на конвейер, такой как непрерывная движущаяся лента, для охлаждения и затем направляются в гранулятор или другое подходящее устройство с целью превращения экструдированных жгутов в систему мультичастиц матрицы. Гранулятор может состоять из валиков,фиксированного ножа, вращающегося режущего устройства и т.п. Подходящие устройства и системы доступны у таких дистрибьюторов, как C.W. Brabender Instruments Inc., South Hackensack, New Jersey. Специалистам ясно, какие другие подходящие устройства могут быть применены. Дальнейший аспект данного изобретения относится к получению экструдированных из расплава мультичастиц матрицы, описанных выше, способом, который предусматривает регулирование количества воздуха, включнного в экструдированный продукт. Регулирование количества воздуха, включнного в экструдат, может изменять скорость высвобождения оксикодона или его фармацевтически приемлемой соли. Далее, согласно ещ одному аспекту изобретения экструдированный из расплава продукт получают способом, который приводит к практическому исключению воздуха во время стадии экструзии. Это можно осуществить, например, применяя экструдер Лейстрица, имеющий вакуумное приспособление. Экструдированные мультичастицы матрицы, полученные по изобретению с применением экструдера Лейстрица под вакуумом, обеспечивают экструдированный из расплава продукт, имеющий другие физи-7 005627 ческие свойства. В частности, экструдат является практически не содержащим пор, что видно при увеличении, например, при применении сканирующего электронного микроскопа, позволяющего получитьSEM (сканирующую электронную микрофотографию). Такие практически непористые составы могут обеспечить более быстрое высвобождение терапевтически активного агента по сравнению с таким же продуктом, полученным без применения вакуума. SEMs мультичастиц матрицы, полученных в экструдере под вакуумом, показывают очень ровную поверхность, и мультичастицы являются более крупными,чем мультичастицы, полученные без применения вакуума. Было установлено, что в, по меньшей мере,некоторых продуктах применение экструзии под вакуумом обеспечивает получение экструдированных мультичастиц матрицы, которые в большей степени зависят от величины рН, чем продукт, полученный без вакуума. Экструдированный из расплава продукт может быть также получен с применением двухшнекового экструдера Вернера-Пфляйдерера. Согласно некоторым вариантам к грануляту или мультичастицам матрицы добавляют сферодиизирующий агент и затем осуществляют сферодиизацию для получения сферических частиц с продлнным высвобождением лекарства. Сферические частицы затем можно снабжать покрытием с продлнным высвобождением такими способами, как описанные выше. Сферодиизирующие агенты, которые можно использовать для получения мультичастиц матрицы по изобретению, включают любые сферодиизирующие агенты. Предпочтительными являются производные целлюлозы, особенно предпочтительна микрокристаллическая целлюлоза. Подходящей микрокристаллической целлюлозой является, например, продукт, продаваемый как Avicel РН 101 (Trademark, FMCCorporation). Сферодиизирующий агент предпочтительно включать в количестве от примерно 1 до примерно 99% от веса мультичастиц матрицы. Согласно некоторым вариантам кроме активного ингредиента и сферодиизирующего агента сферические частицы могут также содержать связующее. Подходящие связующие, такие как низковязкие водорастворимые полимеры, хорошо известны специалистам в области фармацевтики. Однако, предпочтительными являются водорастворимые гидрокси-н.алкилцеллюлозы, такие как гидроксипропилцеллюлоза. Сферические частицы могут содержать дополнительно (или альтернативно) водонерастворимый полимер, особенно акриловый полимер, акриловый сополимер, такой как сополимер метакриловой кислоты с этилакрилатом, или этилцеллюлозу. Согласно некоторым вариантам покрытие с продлнным высвобождением наносится на сфероиды, гранулы или мультичастицы матрицы с продлнным высвобождением. В таких случаях покрытие с продлнным высвобождением может включать нерастворимое в воде вещество, такое как (а) воск или его смесь с алифатическим спиртом или (б) шеллак или зеин. Покрытие предпочтительно наносить из водной дисперсии гидрофобного вещества, обеспечивающего продлнное высвобождение. В некоторых случаях на сфероиды, гранулы или мультичастицы матрицы с продлнным высвобождением, содержащие оксикодон или его фармацевтически приемлемую соль и носитель, необходимо наносить достаточное количество покрытия из водной дисперсии, например, алкилцеллюлозы или акрилового полимера для получения увеличения веса от примерно 2 до примерно 50%, например, от примерно 2 до примерно 25%, для того чтобы получить препарат с продлнным высвобождением. Это покрытие может быть в меньшей или большей степени зависимым, например, от желаемой скорости высвобождения,наличия пластификатора в водной дисперсии или метода его введения. Для покрытия сфероидов, гранул или мультичастиц матрицы хорошо подходят целлюлозные материалы и полимеры, включая алкилцеллюлозы, способствующие продлнному высвобождению. Например, таким примером, предпочтительно, является в качестве алкилцеллюлозы этилцеллюлоза,хотя специалисту ясно, что могут быть использованы другие целлюлозные и/или алкилцеллюлозные полимеры, в отдельности или в любой комбинации, для получения всего или части гидрофобного покрытия по изобретению. Одной из коммерчески доступных дисперсий является водная дисперсия этилцеллюлозы Aquacoat(FMC Corp., Philadelphia Pennsylvania, USA). Aquacoat получается путм растворения этилцеллюлозы в не смешивающемся с водой органическом растворителе и последующего эмульгирования в воде в присутствии поверхностно-активного вещества и стабилизатора. После гомогенизации с получением субмикронных капель органический растворитель выпаривают под вакуумом, получая псевдолатекс. Во время стадии получения пластификатор в псевдолатекс не вводится. Поэтому перед использованием дисперсии для нанесения покрытия необходимо тщательно смешать Aquacoat с подходящим пластификатором. Другой водной дисперсией этилцеллюлозы является коммерчески доступная Surelease (ColorconInc., West Point, Pennsylvania, USA). Этот продукт получают путм введения пластификатора в дисперсию на стадии е получения. Горячий расплав полимера, пластификатора (дибутилсебацината) и стабилизатора (олеиновая кислота) получают в виде гомогенной смеси, которую затем разбавляют щелочным раствором, чтобы получить водную дисперсию, которую можно сразу наносить на сфероиды, гранулы и мультичастицы матрицы с продлнным высвобождением.-8 005627 Согласно другим предпочтительным вариантам данного изобретения материал с продлнным высвобождением представляет собой фармацевтически приемлемый акриловый полимер, включая, но не ограничиваясь этим, сополимеры акриловой и метакриловой кислоты, сополимеры метилметакрилата,этоксиэтилметакрилатов, цианоэтилметакрилата, полиакриловую кислоту, полиметакриловую кислоту,сополимер алкиламида метакриловой кислоты, полиметилметакрилат, полиметакрилат, сополимер полиметилметакрилата, полиакриламид, сополимер аминоалкилметакрилаты поли(ангидрид метакриловой кислоты) и сополимеры глицидилметакрилата. Согласно некоторым предпочтительным вариантам акриловый полимер состоит из одного или более сополимеров аммонийметакрилата. Сополимеры аммонийметакрилата хорошо известны и описаны вNational Formulary (NF) XVII как полностью заполимеризованные сополимеры эфиров акриловой и метакриловой кислот с низким содержанием групп четвертичного аммония. Для того, чтобы получить желательный профиль растворения, может быть необходимым применять два или более сополимеров аммонийметакрилата, имеющих разные физические свойства, такие как разные молярные отношения групп четвертичного аммония к нейтральным эфирным группам метакрилатов. Согласно данному изобретению для получения покрытий, зависимых от рН, могут быть использованы некоторые полимеры типа эфиров полиметакриловой кислоты. Например, существует семейство сополимеров, полученных из диэтиламиноэтилметакрилата и других нейтральных метакриловых эфиров,также известные как сополимеры метакриловой кислоты или полимеры метакрилатов, коммерчески доступные под маркой Eudragit, Rohm GMBH and Co. Kg Darmstadt, Germany. Имеется несколько различных видов Eudragit. Например, Eudragit E представляет собой пример сополимера метакриловой кислоты, который набухает и растворяется в кислой среде. Eudragit L является сополимером метакриловой кислоты, который не набухает при рН 5,7 и растворяется при рН 6. Eudragit S не набухает при рН 6,5 и растворяется при рН 7. Eudragit RL и Eudragit RS набухают в воде, количество воды, абсорбированное этими полимерами, зависит от рН, однако дозированные формы с покрытием на основе EudragitRL и RS не являются зависимыми от рН. Согласно некоторым предпочтительным вариантам акриловое покрытие получено на основе смеси двух акриловых лаков, коммерчески доступных в фирме Rohm под маркой Eudragit RL30D и EudragitRS30D, соответственно. Eudragit RL30D и Eudragit RS30D являются сополимерами эфиров акриловой и метакриловой кислот с низким содержанием групп четвертичного аммония, мольное отношение аммониевых групп к оставшимся нейтральным эфирным группам метакрилатов равно 1:20 в Eudragit RL30D и 1:40 в Eudragit RS30D. Средний молекулярный вес составляет около 150 000. Обозначение RL (высокая проницаемость) и RS (низкая проницаемость) относятся к характеристикам проницаемости этих агентов. Смеси Eudragit RL/RS не растворяются в воде в пищеварительных жидкостях. Однако покрытия на их основе набухают в водных растворах и пищеварительных жидкостях и проницаемы для них. Дисперсии Eudragit RL/RS по данному изобретению могут быть смешаны в любом желательном соотношении для получения препарата с продлнным высвобождением, характеризующегося желательным профилем растворения. Желательные препараты с продлнным высвобождением могут быть получены, например с покрытием, содержащим ингибитор (замедлитель), полученным на основе 100% Eudragit RL, 50% Eudragit RL и 50% Eudragit RS и 10% Eudragit RL : 90% Eudragit RS. Конечно,специалисту очевидно, что можно также использовать другие акриловые полимеры, такие как, например,Eudragit L. В тех случаях, когда покрытие получено из водной дисперсии гидрофобного вещества, способствующего продлнному высвобождению, физические свойства покрытия с продлнным высвобождением можно улучшить за счт включения эффективного количества пластификатора в состав водной дисперсии гидрофобного вещества. Например, вследствие того, что этилцеллюлоза имеет довольно высокую температуру стеклования и не образует гибкие плнки в обычных условиях, предпочтительно перед использованием этилцеллюлозного состава с продлнным высвобождением для получения покрытия вводить в этилцелллюлозу пластификатор. Обычно количество пластификатора, вводимого в раствор для нанесения покрытия, рассчитывается в зависимости от концентрации плнкообразователя, например, чаще всего это количество равно от примерно 1 до примерно 50% от веса плнкообразователя. Однако концентрация пластификатора может быть определена только после тщательного изучения поведения конкретного раствора для покрытия и метода его нанесения. Примерами подходящих пластификаторов для этилцеллюлозы являются водонерастворимые пластификаторы, такие как дибутилсебацинат, диэтилфталат, триэтилцитрат, трибутилцитрат и триацетин, хотя могут быть использованы и другие водонерастворимые пластификаторы (такие как ацетилированные моноглицериды, фталаты, касторовое масло и прочее). Для водных дисперсий этилцеллюлозы по изобретению особенно предпочтительным пластификатором является триэтилцитрат. Примеры подходящих пластификаторов для акриловых полимеров согласно изобретению включают, но не ограничиваются этим, эфиры лимонной кислоты, такие как триэтилцитрат NF XVI, трибутилцитрат, дибутилфталат и, возможно, 1,2-пропиленгликоль. Оказались подходящими для увеличения эластичности плнок на основе акриловых полимеров, например, растворов Eudragit RL/RS, и другие пла-9 005627 стификаторы, такие как полиэтиленгликоли, пропиленгликоль, диэтилфталат, касторовое масло и триацетин. Особенно предпочтительным пластификатором для водных дисперсий этилцеллюлозы по изобретению является триэтилцитрат. Согласно некоторым вариантам сфероиды, гранулы или мультичастицы матрицы с продлнным высвобождением, содержащие покрытие или не содержащие его, включающие оксикодон или соль оксикодона, отверждаются до тех пор, пока не достигается точка, при которой сфероиды, гранулы или мультичастицы матрицы с продлнным высвобождением обеспечивают стабильное растворение. Эта конечная точка может быть определена при сравнении профиля (кривой) растворения дозированной формы сразу же после отверждения с профилем (кривой) растворения дозированной формы после выдержки в ускоренных условиях хранения, например, по меньшей мере, в течение одного месяца при температуре, равной 40 С, и относительной влажности, равной 75%. Отвержднные составы подробно описаны в патентах США 5 273 760, 5 286 493, 5 500 227, 5 580 578, 5 639 476, 5 681 585 и 6 024 982. Другие примеры составов и покрытий с продлнным высвобождением, которые могут быть использованы согласно данному изобретению, описаны в патентах США 5 324 351, 5 356 467 и 5 472 712. Помимо вышеуказанных ингредиентов сфероиды, гранулы или мультичастицы матрицы могут также содержать подходящие количества других компонентов, например, разбавителей, смазок, связующих,агентов, способствующих гранулированию, красителей, ароматизаторов и добавок, улучшающих скольжение, которые известны в фармацевтике, в количествах до примерно 50% от веса состава, если это желательно. Количества этих добавок в составе должны быть достаточными для обеспечения желательного эффекта. Конкретные примеры фармацевтически приемлемых носителей и эксципиентов, которые можно использовать для изготовления оральных дозированных форм, можно найти в Handbook of PharmaceuticalExcipients, American Pharmaceutical Association (1986), включнной в данное описание в качестве ссылки. Далее, было установлено, что добавление небольших количеств талька в состав покрытия с продлнным высвобождением уменьшает тенденцию водной дисперсии к слипанию во время получения дозированной формы, тальк действует как полирующий агент. Осмотические дозированные формы с продлнным высвобождением Дозированные формы с продлнным высвобождением согласно настоящему изобретению могут быть также получены в виде осмотических дозированных препаратов. Осмотические дозированные формы предпочтительно включают двухслойное ядро, включающее слой лекарства и слой для доставки или выталкивающий слой, при этом двухслойное ядро окружено полупроницаемой стенкой и может иметь по меньшей мере один проход, расположенный в этой стенке. Термин проход, используемый для целей данного изобретения, включает апертуру, отверстие,канал, пору, пористый элемент, через которые оксикодон или соль оксикодона могут прокачиваться,диффундировать или мигрировать через волокно, капилляр, пористый слой, пористую вставку, микропористый элемент, пористую композицию. Этот проход может также содержать соединение, которое выходит или вымывается из стенки в жидкой среде с получением, по меньшей мере, одного прохода. Примеры соединений, образующих проход, включают вымываемые полигликолевую кислоту, полимолочную кислоту, содержащиеся в стенке, желатиноподобное волокно, удаляемый водой поливиниловый спирт, выщелачиваемые соединения, такие как удаляемые жидкостью порообразующие полисахариды, кислоты, соли или окиси. Проход может образоваться при вымывании соединения из стенки, такого как сорбит, сахароза, лактоза, мальтоза или фруктоза, с образованием поры-прохода, обеспечивающего продлнное высвобождение. Проход может иметь любую форму, например круглую, треугольную,квадратную и эллиптическую, способствуя продлнному высвобождению оксикодона или его соли из дозированной формы. Дозированные формы могут быть изготовлены с одним или более проходами, расположенными на расстоянии друг от друга на одной или более сторонах дозированной формы. Получение проходов и оборудование для такого получения описаны в патентах США 3 845 770, 3 916 899,4 063 064 и 4 088 864. Проходы с размерами и формами, обеспечивающими продлнное высвобождение, адаптированные как пористый материал для высвобождения, образованный вымыванием водой, описаны в патентах США 4 200 098 и 4 285 987. Согласно некоторым вариантам двухслойное ядро содержит слой с оксикодоном или его солью и вытесняющий или выталкивающий слой. Иногда слой лекарства может содержать также по меньшей мере один полимерный гидрогель. Полимерный гидрогель может иметь средний молекулярный вес от примерно 500 до примерно 6 000 000. Примеры полимерных гидрогелей включают, но не ограничиваются этим, полимер мальтодекстрина формулы (С 6 Н 12O5)nН 2 О, где n равно 3-7500, и полимер мальтодекстрина со среднечисленным молекулярным весом 500-1250000; полиалкиленоксид, например полиэтиленоксид и полипропиленоксид со средневесовым молекулярным весом 50000-750000, более предпочтительны полиэтиленоксиды со средневесовыми молекулярными весами 100000, 200000, 300000 или 400000; щелочную карбоксиалкилцеллюлозу, где щелочной металл представляет собой натрий или калий, алкил является метилом, этилом,- 10005627 пропилом или бутилом, со средневесовым молекулярным весом 10000-175000; и сополимер этилена с акриловыми кислотами, включая метакриловую и акриловую кислоту, со среднечисленным молекулярным весом 10000-500000. В некоторых случаях слой для доставки лекарства или выталкивающий слой состоит из осмотического полимера. Примеры осмотических полимеров включают, но не ограничиваются этим, полимер,выбранный из группы, состоящей из полиалкиленоксида и карбоксиалкилцеллюлозы. Полиалкиленоксид имеет средневесовой молекулярный вес, равный 1 000 000-10 000 000. Полиалкиленоксид может быть выбран из группы, состоящей из полиметиленоксида, полиэтиленоксида, полипропиленоксида, полиэтиленоксида, имеющего средний молекулярный вес, равный 1000000, полиэтиленоксида со средним молекулярным весом 5000000, полиэтиленоксида со средним молекулярным весом 7000000, сшитого полиметиленоксида со средним молекулярным весом 1000000 и полипропиленоксида со средним молекулярным весом 1200000. Типичная осмотическая карбоксиалкилцеллюлоза включает натриевую соль карбоксиметилцеллюлозы, калиевую соль карбоксиметилцеллюлозы, натриевую соль карбоксиэтилцеллюлозы, литиевую соль карбоксиметилцеллюлозы, натриевую соль карбоксиэтилцеллюлозы, карбоксиалкилгидроксиалкилцеллюлозу, карбоксиметилгидроксиэтилцеллюлозу, карбоксиэтилгидроксиэтилцеллюлозу и карбоксиметилгидроксипропилцеллюлозу. Осмотические полимеры, используемые для изготовления вытеснительного слоя, проявляют градиент осмотического давления при прохождении через полупроницаемую стенку. Осмотические полимеры всасывают жидкость в дозированную форму, набухая и расширяясь при этом с образованием осмотического гидрогеля (известного также как осмогель), при этом они вытесняют оксикодон или его фармацевтически приемлемую соль из осмотической дозированной формы. Вытесняющий слой может также включать одно или несколько осмотически эффективных соединений, известных также как осмагенты и как осмотические эффективные растворнные вещества. Они впитывают окружающую жидкость, например, из желудочно-кишечного тракта, попадающую в дозированную форму, и вносят вклад в кинетику доставки лекарства вытеснительным слоем. Примеры осмотически активных соединений включают соединение, выбранное из группы, состоящей из осмотических солей и осмотических карбогидратов. Примеры конкретных осмагентов включают, но не ограничиваются этим, хлористый натрий, хлористый калий, сульфат магния, фосфат лития, хлорид лития, фосфат натрия, сульфат калия, сульфат натрия, фосфат калия, глюкозу, фруктозу и мальтозу. Вытеснительный слой может включать гидроксипропилалкилцеллюлозу, имеющую среднечисленный молекулярный вес, равный 9000-450000. Гидроксипропилалкилцеллюлозу выбирают из группы, состоящей из гидроксипропилметилцеллюлозы, гидроксипропилэтилцеллюлозы, гидроксипропилизопропилцеллюлозы, гидроксипропилбутилцеллюлозы и гидроксипропилпентилцеллюлозы. Вытеснительный слой может содержать нетоксичный пигмент или краситель. Примеры пигментов или красителей включают, но не ограничиваются этим, красящие вещества, указанные в Food and DragAdministration Colorant (FDC), такими как FDC 1 голубой, FDC 4 красный, красная окись железа, жлтая окись железа, двуокись титана, углеродная сажа и индиго. Вытеснительный слой может также содержать для предотвращения окисления ингредиентов антиоксидант. Некоторые примеры антиоксидантов включают, но не ограничиваются этим, соединение, выбранное из группы, состоящей из аскорбиновой кислоты, аскорбилпальмитата, бутилированного гидроксианизола, смеси 2- и 3-трет-бутил-4-гидроксианизолов, бутилированного гидрокситолуола, изоаскорбата натрия, дигидрогваретиновой кислоты, сорбитата калия, бисульфата натрия, метабисульфата натрия, собиновой кислоты, аскорбата калия, витамина Е, 4-хлор-2,6-дитрет-бутилфенола, альфатокоферола и пропилгаллата. Согласно другим вариантам дозированная форма включает гомогенное ядро, содержащее оксикодон или его фармацевтически приемлемую соль, фармацевтически приемлемый полимер (например, полиэтиленоксид), может включать дезинтегрант (например, поливинилпирролидон), может содержать ускоритель абсорбции (например, жирную кислоту, поверхностно-активное вещество, хелатирующий агент, соль жлчной кислоты и т.д.). Гомогенное ядро окружено полупроницаемой стенкой, содержащей проход (как описано выше) для высвобождения оксикодона или его фармацевтически приемлемой соли. В некоторых случаях полупроницаемая стенка содержит соединение, выбранное из группы, состоящей из сложного эфира целлюлозы, простого эфира целлюлозы и сложно-простого эфира целлюлозы. Полимеры стенки включают полимер, выбранный из группы, состоящей из ацилата целлюлозы, диацилата целлюлозы, триацилата целлюлозы, ацетата целлюлозы, диацетата целлюлозы, триацетата целлюлозы, ди- и триалкенилатов целлюлозы и моно-, ди- и триалкинилатов целлюлозы. Целлюлозы, использованные согласно данному изобретению, имеют среднечисленный молекулярный вес, равный 20000-7500000. Дополнительные полупроницаемые полимеры по изобретению включают ацетальдегид ацетата диметилцеллюлозы, этилкарбамат ацетата целлюлозы, метилкарбамат ацетата целлюлозы, пропилкарбамат диацетата целлюлозы, диэтиламиноацетат ацетата целлюлозы, полупроницаемый полиамид, полупроницаемый полиуретин, полупроницаемый сульфированный полистирол, полупроницаемый сшитый поли- 11005627 мер, полученный соосаждением полианиона и поликатиона, как описано в патентах США 3173876,3276586, 3541005, 3541006 и 3546876, полупроницаемые полимеры, как описанные Loeb и Sourirajan в патенте 3133132, полупроницаемые сшитые полистиролы, полупроницаемый сшитый поли(стиролсульфонат натрия), полупроницаемый сшитый поли(винилбензилтриметиламмоний хлорид) и полупроницаемые полимеры с проницаемостью по жидкости, равной 2,5x10-8 - 2,5x10-2 (см 2 /ч.атм), выраженной как разница гидростатического или осмотического давления при прохождении через полупроницаемую стенку. Другие полимеры, используемые по данному изобретению, известны из патентов США 3845770, 3916899 и 4160020, из Handbook of Common Polymers, Scott J. R. и W. J. Roff, 1971, CRCPress, Cleveland, Ohio. Согласно некоторым вариантам предпочтительно, чтобы полупроницаемая стенка была нетоксичной,инертной и сохраняла физическую и химическую целостность в течение срока отпуска лекарства. Иногда дозированная форма содержит связующее. Примером связующего является, но не ограничивается этим, терапевтически приемлемый виниловый полимер со средневязкостным молекулярным весом 5000-350000, выбранный из группы, состоящей из поли-н-виниламида, поли-н-винилацетамида, поливинилпирролидона, известного также как поли-н-винилпирролидон, поли-н-винилкапролактона, поли-н-винил-5-метил-2-пирролидона и сополимеров н-винилпирролидона с соединением, выбранным из группы, состоящей из винилацетата, винилового спирта,винилхлорида, винилфторида, винилбутирата, виниллаурата и винилстеарата. Другие связующие включают,например, смолу акации, крахмал, желатин и гидроксипропилалкилцеллюлозу со средним молекулярным весом,равным 9200 - 250000. Согласно некоторым вариантам дозированная форма содержит смазывающий агент, который может быть использован во время изготовления дозированной формы, чтобы предотвратить налипание на стенку головки или поверхность штампа. Примеры смазывающих агентов включают, но не ограничиваются этим, стеарат магния, стеарат натрия, стеариновую кислоту, стеарат кальция, олеат магния, олеиновую кислоту, олеат калия, каприловую кислоту, стеарилфумарат натрия и пальмитат магния. Согласно некоторым предпочтительным вариантам данного изобретения оно относится к терапевтической композиции, содержащей 1-640 мг оксикодона или его фармацевтически приемлемой соли, 25500 мг полиалкиленоксида со средним молекулярным весом 150000 - 500000, 1-50 мг поливинилпирролидона со средним молекулярным весом 40000 и 0-7,5 мг смазывающего агента. Согласно другому аспекту данное изобретение относится также к способу введения 1-640 мг оксикодона или его фармацевтически приемлемой соли путм перорального прима 1-640 мг оксикодона или его фармацевтически приемлемой соли пациентом, причем оксикодон или его соль вводятся из дозированной формы, содержащей полупроницаемую стенку, проницаемую для водно-биологической жидкости и непроницаемой для оксикодона или его фармацевтически приемлемой соли, при этом указанная полупроницаемая стенка окружает внутреннее пространство, включающее композицию на основе оксикодона и вытесняющую композицию, а указанная композиция на основе оксикодона содержит 1-640 мг оксикодона или его фармацевтически приемлемой соли, 25-500 мг полиалкиленоксида со средним молекулярным весом 150000-500000, 1-50 мг поливинилпирролидона со средним молекулярным весом 40000 и 07,5 мг смазывающего агента, а вытесняющая композиция содержит 15-250 мг полиалкиленоксида со средним молекулярным весом 3000000-7500000, 0-7,5 мг осмагента, 1-50 мг гидроксиалкилцеллюлозы, 010 мг окиси железа, 0-10 мг смазывающего агента и 0-10 мг антиоксиданта, в полупроницаемой стенке имеется проход для доставки оксикодона или его фармацевтически приемлемой соли из дозированной формы за счт всасывания жидкости через полупроницаемую стенку в дозированную форму, что вызывает распределение оксикодона или его соли и расширение вытеснительной композиции и вытеснение композиции на основе оксикодона или его соли через проход, при этом за счт действия дозированной формы оксикодон или его соль доставляются в организм с терапевтически эффективной дозой со скоростью, регулируемой в течение протяжнного промежутка времени. Дозированные формы по изобретению могут содержать одно или насколько покрытий, пригодных для регулирования высвобождения или для защиты состава. Согласно одному из вариантов покрытия наносят для того, чтобы получить зависимое от значения рН или не зависимое от значения рН высвобождение, например, при действии желудочно-кишечной (GI) жидкости. Когда желательно получить покрытие, зависимое от значения рН, покрытие получают таким образом, чтобы обеспечить оптимальное высвобождение независимо от изменений рН окружающей жидкости, например в GI-тракте. Другие предпочтительные варианты включают зависимое от значения рН покрытие, которое высвобождает оксикодон или его фармацевтически приемлемую соль в нужных областях GI-тракта, например, в желудке или в тонкой кишке, при этом обеспечивается профиль абсорбции, который способен привести к, по меньшей мере, примерно 12-часовому и, предпочтительно, примерно 24-х часовому или более длительному периоду обезболивающего действия у пациента. Можно также получить композиции, которые высвобождают часть дозы в одной нужной области GI-тракта, например в желудке, и высвобождают остальную часть дозы в другой области GI-тракта, например в тонкой кишке. Составы по изобретению с покрытиями, зависимыми от рН, могут также обладать эффектом повторяющегося действия, когда на незащищнное лекарство наносят энтеропокрытие и лекарство высвобождается в желудке, в то время как оставшаяся часть, будучи защищенной энтеропокрытием, высвобожда- 12005627 ется далее в желудочно-кишечном тракте. Покрытия, зависящие от рН, которые могут быть использованы согласно данному изобретению, включают вещество, обеспечивающее продлнное высвобождение,такое как, например, шеллак, ацетатфталат целлюлозы (САР), поливинилацетатфталат (PVAP), фталат гидроксипропилметилцеллюлозы и сополимеры эфиров метакриловой кислоты, зеин и т.п. В некоторых случаях в составы согласно данному изобретению включают эффективное количество оксикодона или его фармацевтически приемлемой соли в виде формы немедленного высвобождения. Эта форма с немедленным высвобождением оксикодона или его соли включается в количестве, эффективном для уменьшения времени, требующегося для достижения максимальной концентрации оксикодона в крови (например, в плазме), то есть Тmах уменьшается. За счт включения такого эффективного количества немедленно высвобождающегося оксикодона или его соли в состав единичной дозы можно уменьшить проявление довольно сильных болей у пациентов. В таких случаях эффективное количество оксикодона или соли оксикодона в виде формы с немедленным высвобождением может быть нанесено в виде покрытия на таблетки по изобретению. Например, когда длительное высвобождение оксикодона или соли оксикодона из состава обусловлено наличием покрытия с продлнным высвобождением, слой с немедленным высвобождением можно наносить на покрытие с продлнным высвобождением. С другой стороны,слой с немедленным высвобождением может быть нанесен на поверхность таблеток, в то время как оксикодон или соль оксикодона вводится в состав матрицы с продлнным высвобождением. Специалистам очевидно, что существуют и другие альтернативные способы введения немедленно высвобождающегося оксикодон или его соли в состав. Такие альтернативы охватываются нижеследующей формулой изобретения. Согласно ещ одному варианту дозированные формы с продлнным высвобождением по изобретению кроме оксикодона или соли оксикодона могут дополнительно включать неопиоидное лекарство,которое может или не может действовать синергически вместе с оксикодоном или его солью. Такие неопиоидные лекарства, предпочтительно, оказывают дополнительное обезболивающее действие и включают, например, аспирин, ацетаминофен, нестероидные противовоспалительные средства (NSAIDS), например ибупрофен, кетопрофен и т.п.; антагонисты рецептора N-метил-D-аспартат (NMDA), например морфинаны, такие как декстрометорфан, декстрорфан или кетамин; ингибиторы циклооксигеназы-II (ингибиторы СОХ-II) и/или антагонисты рецептора глицина. Согласно некоторым вариантам данное изобретение позволяет применять меньшие дозы оксикодона или соли оксикодона путм включения дополнительного неопиоидного анальгетика, такого как, например, NSAID или СОХ-2. За счт применения меньших количеств одного или обоих лекарств можно уменьшить побочные эффекты, связанные с эффективным лечением боли у людей. Подходящие нестероидные противовоспалительные агенты включают ибупрофен, диклофенак, напроксен, беноксапрофен, флурбипрофен, фенопрофен, флубуфен, кетопрофен, индопрофен, пиропрофен,карпрофен, оксапрозин, прамопрофен, муропрофен, триоксапрофен, супрофен, аминопрофен, тиапрофеновую кислоту, флупрофен, буклоксовую кислоту, индометацин, сулиндак, толметин, зомепирак, тиопинак, зидометацин, ацеметацин, фентиазак, клиданак, окспинак, мефенамовую кислоту, меклофенамовую кислоту, флуфенамовую кислоту, нифлумовую кислоту, толфенамовую кислоту, дифлуризал, флуфенисал, пироксикам, судоксикам, изоксикам, их фармацевтически приемлемые соли, их смеси и т.п. Специалистам известны подходящие дозы этих лекарств. Антагонисты рецептора N-метил-D-аспартата (NMDA) хорошо известны и охватывают, например,морфинаны, такие как декстрометорфан или декстрорфан, кетамин или их фармацевтически приемлемые соли. Для целей данного изобретения термин антагонист NMDA охватывает также лекарства, которые,по меньшей мере частично, ингибируют основную часть внутриклеточной последовательности активации рецептора NMDA, например, ганглиозид, такой как GM1 или GT1b, фенотиазин, такой как трифлуоперазин или нафталин-сульфонамид, такой как N-(6-аминогексил)-5-хлор-1-нафталинсульфонамид. Утверждается, что эти лекарства ингибируют развитие толерантности к и/или зависимости от аддиктивных лекарств, например наркотических анальгетиков, таких как морфин, кодеин и т.д. (патенты США 5 321 012 и 5 556 838) и лечат хроническую боль (патент США 5 502 058). Антагонист NMDA может содержаться один или в сочетании с местным анестезирующим средством, таким как лидокаин, как описано в указанных патентах. Лечение хронической боли с применением антагонистов рецепторов глицина и идентификация таких лекарств описаны в патенте США 5514680. Из уровня техники известны ингибиторы СОХ-2, известно, что многие химические структуры ингибируют циклооксигеназу-2. Ингибиторы СОХ-2 описаны, например, в патентах США 5616601,5604266, 5593994, 5550142, 5536752, 5521213, 5475995, 5639780, 5604253, 5552422, 5510368, 5436265,5409944 и 5130311. Предпочтительные ингибиторы СОХ-2 включают целекоксиб (SC-58635), DUP-697,флосулид (CGP-28238), мельксикам, 6-метокси-2-нафтилуксусную кислоту (6-MNA), МК-966 (известный также как Vioxx), набуметон (пролекарство 6-MNA), нимесулид, NS-398, SC-5766, SC-58215, Т-614 или их комбинации. Дозы ингибитора СОХ-2 порядка от примерно 0,005 мг до примерно 140 мг на кг веса пациента в день являются терапевтически эффективными в комбинации с оксикодоном или солью окси- 13005627 кодона. Можно также вводить пациенту от примерно 0,25 мг до примерно 7 г в день ингибитора СОХ-2 в сочетании с оксикодоном или солью оксикодона. Согласно ещ одному варианту можно вводить опиоидное лекарство, которое обеспечивает действие, отличающееся от обезболивающего, например противокашлевое средство, отхаркивающее средство,противоотчное средство, антигистаминное лекарство, местные анестетики и т.п. Дополнительный (неопиоидный) терапевтический агент можно вводить в виде формы с продлнным высвобождением или формы с немедленным высвобождением. Дополнительное лекарство можно вводить в матрицу с продлнным высвобождением наряду с оксикодоном или его солью, вводить в дозированную форму можно в виде порошка, гранулята и т.д. или же можно вводить его в виде отдельного слоя с продлнным высвобождением или слоя с немедленным высвобождением. Оральные тврдые дозированные формы по изобретению могут содержать низкую концентрацию опиоида. Оральные тврдые дозированные формы по изобретению, обеспечивающие продлнное высвобождение, можно вводить значительно меньшими ежедневными дозами, чем обычные продукты с немедленным высвобождением, при этом разницы в обезболивающем действии нет. При сравнимых дневных дозировках большая эффективность может быть достигнута при применении оральных тврдых дозированных форм с продлнным высвобождением по данному изобретению по сравнению с обычными продуктами с немедленным высвобождением. Настоящее изобретение будет описано ниже со ссылкой на нижеследующие примеры. Однако следует иметь в виду, что нижеследующие примеры приведены только для иллюстрации изобретения и не ограничивают его. Пример 1. Таблетки, содержащие матрицу с продлнным высвобождением оксикодона, получают из состава,указанного в табл. 1 ниже. Таблица 1 Используется в процессе получения и остатся в продукте только как остаточная влага. Способ получения включает следующее: 1. Гранулирование: нанесите дисперсию Eudragit/триацетин на гидрохлорид оксикодона высушенную распылением лактозу и повидон с применением гранулятора с псевдоожиженным слоем. 2. Размельчение: выгрузите гранулят и пропустите через мельницу с отверстиями около 1 мм (сито 18 меш). 3. Введение воска: расплавьте стеариновый спирт при температуре около 50 С и добавьте к измельчнному грануляту в смесителе с большими сдвиговыми усилиями. Дайте смеси охладиться до комнатной температуры на подносах или в псевдоожиженном слое. 4. Измельчение: пропустите охлажднный гранулят через мельницу с ситом примерно 18 меш. 5. Смазка: добавьте тальк и стеарат магния к грануляту в смесителе. 6. Прессование: осуществите прессование гранулята в таблетки с применением пресса Kilian Tablet. 7. Плночное покрытие: нанесите водное плночное покрытие на таблетки, используя вращающийся противень. Пример 2. Осмотические таблетки с продлнным высвобождением оксикодона получают из состава, указанного ниже в табл. 2. Дозированную форму вышеуказанного состава готовят по следующему методу. Вначале добавляют 175 г гидрохлорида оксикодона, 647,5 г полиэтиленоксида со средним молекулярным весом 200000 и 43,75 г поливинилпирролидона со средним молекулярным весом 40000 в смеситель и перемешивают в течение 10 мин. Затем к смеси добавляют при непрерывном перемешивании в течение 10 мин 331 г денатурированного безводного спирта. Мокрый гранулят пропускают через сито 20 меш, дают высохнуть при комнатной температуре в течение 20 ч и затем пропускают через сито 16 меш. Затем гранулятор перемещают в смеситель, перемешивают и добавляют 8,75 г смазки - стеарата магния. Затем получают вытесняющую или выталкивающую композицию для вытеснения композиции на основе гидрохлорида оксикодона следующим образом: сначала 3910 г гидроксипропилметилцеллюлозы со средним молекулярным весом 11200 растворяют в 45339 г воды. Затем 101 г бутилированного гидрокситолуола растворяют в 650 г денатурированного безводного спирта. Потом добавляют 2,5 кг водного раствора гидроксипропилметилцеллюлозы к спиртовому раствору бутилированного гидрокситолуола при непрерывном перемешивании. Затем приготовление раствора связующего завершают при добавлении оставшегося водного раствора гидроксипропилметилцеллюлозы к спиртовому раствору бутилированного гидрокситолуола. Затем 36000 г хлористого натрия подают в мельницу Quadro Comil, снабжнную ситом 21 меш. Затем 1200 г окиси железа пропускают через сито 40 меш. Затем эти вещества, пропущенные через сито,76400 г фармацевтически приемлемого полиэтиленоксида со средним молекулярным весом 7500000,2500 г гидроксипропилметилцеллюлозы со средним молекулярным весом 11200 добавляют в чашу гранулятора Glatt Fluid Bed. Чаша соединена с гранулятором, и начинают процесс гранулирования. Затем сухие порошки суспендируют воздухом и перемешивают в течение 10 мин. Затем раствор связующего распыляют через 3 сопла на порошок. Мониторинг грануляции во время процесса следующим образом: скорость распыления всего раствора составляет 800 г/мин; температура на входе равна 43 С, скорость потока воздуха равна 4300 м 3/ч. В конце стадии распыления раствора 45033 г полученных гранулированных частиц с покрытием подвергают сушке в течение 35 мин. Гранулы с покрытием пропускают через мельницу Quadro Comil с ситом 8 меш. Гранулят перемещают в опрокидыватель Tote, перемешивают и смешивают со смазкой - 281,7 г стеарата магния. Затем композицию, включающую гидрохлорид оксикодона и вытесняющую композицию, прессуют в двухслойные таблетки на прессе Kilian Tablet. Сначала добавляют 176 мг композиции на основе гидрохлорида оксикодона в полость пресса и предварительно прессуют, после этого вводят 135 мг вытесняющей композиции и прессуют слои под давлением головки пресса 3 мт (метрич. тонны) с получением слоистых частиц диаметром 11/32 дюйма (0,873 см). Двухслойные частицы окружены полупроницаемой стенкой. Композиция, образующая стенку,включает 100% ацетата целлюлозы, содержащей 39,8% ацетатных групп. Эту образующую стенку композицию растворяют в смеси ацетон: вода (95:5 вес/вес) с получением раствора, содержащего 4% тврдого вещества. Композицию, образующую стенку, распыляют на двухслойные частицы и вокруг них в устройстве VectorHi-Coater 24 дюйма (60 см). Затем через полупроницаемую стенку для соединения слоя оксикодона с наружной стороной дозированной формы просверливают один проход размером 20 мил(0,508 мм). Остаточный растворитель удаляют путм сушки в течение 72 ч при 45 С и влажности 45%. Затем осмотические дозированные системы сушат при 45 С в течение 4 ч для удаления избытка влаги. Получаемые дозированные формы содержат 35,20 мг гидрохлорида оксикодона, 130,24 мг полиэтиленоксида со средним молекулярным весом 200000, 8,80 мг поливинилпирролидона со средним молекулярным весом 40000 и 1,76 мг стеарата магния. Вытесняющая композиция содержит 85,96 мг полиэтиле- 15005627 ноксида со средним молекулярным весом 7500000, 40,50 мг хлористого натрия, 6,75 гидроксипропилметилцеллюлозы, 1,35 мг красной окиси железа, 0,34 мг стеарата магния, 0,10 мг бутилированного гидрокситолуола. Полупроницаемая стенка содержит 38,6 мг ацетата целлюлозы, содержащего 39,8% ацетатных групп. Дозированная форма содержит один проход размером 20 мил (0,508 мм). Пример 3. Осмотические таблетки с продлнным высвобождением оксикодона получают из состава, приведнного ниже в табл. 3. Таблица 3 Дозированная форма вышеуказанного состава была получена следующим образом. Сначала в смеситель с планетарной мешалкой помещают 1728 г гидрохлорида оксикодона, 3852 г полиэтиленоксида со средним молекулярным весом 200 000 и 360 мг поливинилпирролидона со средним молекулярным весом 40000. Затем сухие вещества перемешивают в течение 10 мин. Затем к смеси медленно добавляют 1616 г денатурированного безводного этилового спирта при непрерывном перемешивании в течение 15 мин. Затем только что полученный мокрый гранулят пропускают через сито 20 меш,дают высохнуть при комнатной температуре в течение 20,5 ч и пропускают через сито 16 меш. Потом гранулят перемещают в смеситель с планетарной мешалкой, перемешивают и добавляют 59,8 г смазывающего агента - стеарата магния. Далее готовят вытесняющую композицию: сначала готовят раствор связующего растворением 3910 г гидроксипропилметилцеллюлозы, имеющей средний молекулярный вес, равный 11200, в 45339 г воды. После этого 101 г бутилированного гидрокситолуола растворяют в 650 г денатурированного безводного спирта. При непрерывном перемешивании к раствору бутилированного гидрокситолуола добавляют примерно 2,5 кг раствора гидроксипропилметилцеллюлозы в воде. Приготовление раствора связующего завершают добавлением оставшегося раствора гидроксипропилметилцеллюлозы в воде опять при непрерывном перемешивании. Затем 36000 г хлористого натрия пропускают через мельницу Quadro Comil, используемую для уменьшения размеров частиц хлористого натрия. Для сортировки размеров частиц их также пропускают через другую мельницу с ситом 21 меш. Затем 1200 г окиси железа пропускают через сито 40 меш. После этого все эти отсортированные вещества, 76400 г фармацевтически приемлемого полиэтиленоксида со средним молекулярным весом 7000000, 2520 г гидроксипропилметилцеллюлозы со средним молекулярным весом 11200 помещают в бункер гранулятора Glatt Fluid Bed. Бункер соединен с гранулятором и начинают гранулирование. Затем сухие порошки суспендируют потоком воздуха и перемешивают в течение 10 мин. Затем раствор связующего распыляют через три сопла на порошок. При распылении раствора связующего в течение 10 с каждые 1,5 мин встряхивают фильтровальные мешки для предотвращения возможного осаждения полимера из-за слипания. После окончания распыления 45033 г полученных гранулированных частиц с покрытием подвергают сушке в течение 35 мин. Затем устройство отключают и гранулы с покрытием удаляют из гранулятора. Гранулы с покрытием отсортировывают в Quadro Comil на сите 8 меш. Гранулят подают в Tote Tumbler, перемешивают, вводят 281,7 г стеарата магния в качестве смазки. Затем композицию на основе гидрохлорида оксикодона и вытесняющую композицию прессуют с получением двухслойных таблеток, применяя пресс Kilian Tablet. Вначале в полость пресса загружают- 16005627 434 мг композиции на основе гидрохлорида оксикодона и предварительно прессуют, затем добавляют вытеснительную композицию и прессуют слои под давлением примерно 3 метр.т с получением овальных слоистых частиц размером 0,700 дюйма (1,78 см) х 0,375 дюйма (0,95 см). Двухслойные частицы заключают в полупроницаемую оболочку. Композиция оболочки содержит 95% целлюлозы, содержащей 39,8% ацетильных групп, и 5% полиэтиленгликоля с молекулярным весом 3350. Эту композицию растворяют в смеси ацетон:вода (95:5 вес/вес) с получением раствора, содержащего 4% тврдых веществ. Композицию оболочки наносят распылением на двухслойные частицы в устройстве 24" Vector Hi Coater. Затем в полупроницаемой стенке для соединения слоя лекарство с внешней стороны дозирующей системы в этой стенке просверливают два канала размером 30 мил (0,762 мм). Остаточный растворитель удаляют путм сушки в течение 4 ч при 50 С и влажности 50%. После этого осмотические дозированные формы сушат в течение 4 ч при 50 С для удаления избытка влаги. Дозированная форма, полученная этим способом, содержит 28,8% гидрохлорида оксикодона, 64,2% полиэтиленоксида со средним молекулярным весом 200000, 6% поливинилпирролидона со средним молекулярным весом 40000 и 1% стеарата магния. Вытесняющая композиция включает 63,675% полиэтиленоксида, имеющего средний молекулярный вес 7000000, 30% хлористого натрия, 5% гидроксипропилметилцеллюлозы со средним молекулярным весом 11200, 1% окиси железа, 0,075% бутилированного гидрокситолуола и 0,25% стеарата магния. Полупроницаемая оболочка содержит 95 вес.% ацетата целлюлозы, содержащего 39,8% ацетильных групп, и 5,0 вес.% полиэтиленгликоля со средним молекулярным весом 3350. Дозированная форма имеет два канала, 30 мил (0,762 мм) и характеризуется средней скоростью высвобождения гидрохлорида оксикодона, равной примерно 5 мг/ч. Другие варианты дозированной формы могут содержать от 65 до 100 вес.% целлюлозного полимера, выбранного из группы, состоящей из сложного эфира целлюлозы, диэфира целлюлозы, триэфира целлюлозы, простого эфира целлюлозы, сложно-простого эфира целлюлозы, ацилата целлюлозы, диацилата целлюлозы, триацетата целлюлозы, ацетатбутирата целлюлозы и т.п. Композиция стенки может также содержать от 0 до 40 вес.% простого эфира целлюлозы, выбранного из группы, состоящей из гидроксипропилцеллюлозы и гидроксипропилметилцеллюлозы и от 0 до 20 вес.% полиэтиленгликоля. Общий вес всех компонентов, образующий стенку, равен 100 вес.%. Полупроницаемые полимеры, используемые для изготовления стенки дозированной формы, описаны в патентах США 3845770, 3916899,4008719, 4036228 и 4111021. В других предпочтительных вариантах стенка содержит селективно проницаемый простой эфир целлюлозы - этилцеллюлозу. Этилцеллюлоза содержит этоксигруппу со степенью замещения от примерно 1,4 до 3,0, что эквивалентно содержанию этоксигрупп, равному 40-50%, и величине вязкости 7-100 сП или больше. Более конкретно, композиция стенки содержит 45-85% этилцеллюлозы, от 5 до 30 вес.% гидроксипропилцеллюлозы и от 5 до 30 вес.% полиэтиленгликоля, общее количество всех компонентов,образующих стенку, составляет 100 вес.%. Согласно ещ одному варианту композиция стенки содержит 45-80% этилцеллюлозы, от 5 до 30 вес.% гидроксипропилцеллюлозы, от 2 до 20 вес.% поливинилпирролидона, общее количество всех компонентов, образующих стенку, равно 100 вес.%. Пример 4. Капсулы, содержащие 10 мг оксикодона, с продлнным высвобождением, получают из состава,приведнного ниже в табл. 4. Таблица 4 Вышеуказанный состав готовят следующим образом: 1. Хлопья стеаринового спирта перемешивают в ударной мельнице. 2. Смешивают гидрохлорид оксикодона, стеариновую кислоту, стеариновый спирт и Eudragit RSPO в подходящем блендере/смесителе. 3. Непрерывно подают смесь в двухшнековый экструдер при повышенных температурах и полученные жгуты помещают на конвейер. 4. Жгуты охлаждаются на конвейере. 5. Жгуты разрезают на гранулы размером 1 мм в грануляторе. 6. Отсортировывают пыль и гранулы чрезмерно большого размера с получением гранул размером примерно 0,8 - 1,4 мм. 7. Заполняют капсулы из расчта 120 мг/капсулу (заполняют капсулы размера 2).- 17005627 Затем проверяют растворимость гранул по следующей методике. Растворение определяют с использованием оптиковолоконного УФ-прибора USP 1 (корзина) со скоростью 100 об/мин в 900 мл условного желудочного сока (SGF) и в 900 мл условной кишечной жидкости (SIF). Данные приведены ниже в табл. 4 А. Таблица 4 А Состав был получен следующим образом: 1. Хлопья стеаринового спирта перемешивают в ударной мельнице. 2. Смешивают гидрохлорид оксикодона, стеариновую кислоту, стеариновый спирт и Eudragit RSPO в подходящем блендере/смесителе. 3. Непрерывно подают смесь в двухшнековый экструдер при повышенных температурах и полученные жгуты помещают на конвейер. 4. Жгуты охлаждаются на конвейере. 5. Жгуты разрезают на гранулы размером 1 мм в грануляторе. 6. Отсортировывают пыль и гранулы чрезмерно большого размера с получением гранул размером примерно 0,8 - 1,4 мм. 7. Заполняют капсулы из расчта 400 мг/капсулу (заполняют капсулы размера 00). Метод растворения. Затем определяют растворение гранул по следующему методу. Растворение определяют с использованием оптиковолоконного УФ-прибора USP 1 (корзина) со скоростью 100 об/мин в 900 мл условного желудочного сока (SGF) и в 900 мл условной кишечной жидкости (SIF). Данные приведены ниже в табл. 5 А. Таблица 5 А Многие другие варианты данного изобретения очевидны специалистам и находятся в объме данного изобретения, определяемом формулой изобретения.- 18005627 ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Оральная дозированная форма с продлнным высвобождением для введения по схеме один раз в день, включающая фармацевтически приемлемую матрицу, содержащую анальгетически эффективное количество оксикодона или его фармацевтически приемлемой соли и вещество, способствующее продлнному высвобождению, причм указанная дозированная форма обеспечивает обезболивающий эффект в течение по меньшей мере 24 ч после перорального введения в стационарном состоянии людям и указанная дозированная форма обеспечивает среднюю величину отношения С 24/Сmах оксикодона, равную 0,6-1,0 после перорального введения в стационарном состоянии указанным пациентам. 2. Оральная дозированная форма с продлнным высвобождением для введения по схеме один раз в день, включающая множество фармацевтически приемлемых матриц, содержащих анальгетически эффективное количество оксикодона или его соли и вещество, способствующее продлнному высвобождению, причм указанная дозированная форма обеспечивает обезболивающий эффект в течение по меньшей мере 24 ч после перорального введения в стационарном состоянии людям и среднюю величину отношения С 24/Сmах оксикодона, равную 0,6 - 1,0 после перорального введения в стационарном состоянии указанным пациентам. 3. Дозированная форма по п.1 или 2, отличающаяся тем, что она обеспечивает среднюю величину Тmах оксикодона, равную от 2 до 17 ч, от 8 до 16 ч, от 12 до 16 ч и от 6,5 до 17 ч после введения в стационарном состоянии людям. 4. Дозированная форма по п.1 или 3, отличающаяся тем, что матрица является практически гомогенной. 5. Дозированная форма по п.4, отличающаяся тем, что она содержится в желатиновой капсуле или сформована в виде таблетки. 6. Дозированная форма по п.2 или 3, отличающаяся тем, что указанное множество матриц содержится в желатиновой капсуле и/или указанное множество матриц содержится в виде таблетки. 7. Дозированная форма по любому из пп.1-6, отличающаяся тем, что указанный оксикодон или его фармацевтически приемлемая соль содержится в количестве от 5 до 640 мг. 8. Дозированная форма по п.7, отличающаяся тем, что указанная фармацевтически приемлемая соль оксикодона является гидрохлоридом оксикодона. 9. Дозированная форма по любому из пп.1-7, отличающаяся тем, что она обеспечивает среднюю величину отношения С 24/Сmах, равную 0,7-1,0, 0,7-0,99 или 0,8-0,95 после введения в стационарном состоянии указанным пациентам. 10. Дозированная форма по любому из пп.1-9, отличающаяся тем, что она обеспечивает in vitro скорость высвобождения оксикодона или его фармацевтически приемлемой соли, измеренную по методуUSP Basket при скорости 100 об./мин в 900 мл водного буфера при рН от 1,6 до 7,2 при 37 С, равную от 0 до 40% через 1 ч, от 8 до 70% через 4 ч, от 20 до 80% через 8 ч, от 30 до 95% через 12 ч, от 35 до 95% через 18 ч и более 50% через 24 ч. 11. Оральная дозированная форма с продлнным высвобождением, включающая(i) слой лекарства, включающий анальгетически эффективное количество оксикодона или его фармацевтически приемлемой соли и(б) полупроницаемую стенку, окружающую двухслойное ядро, содержащую проход, расположенный в указанной стенке для высвобождения оксикодона или его фармацевтически приемлемой соли,причм указанная дозированная форма обеспечивает обезболивающий эффект в течение по меньшей мере 24 ч после перорального введения в стационарном состоянии людям и среднюю величину отношения С 24/Сmах оксикодона, равную 0,6-1,0, после перорального введения в стационарном состоянии указанным пациентам. 12. Дозированная форма по п.11, отличающаяся тем, что она обеспечивает среднюю величину Тmах оксикодона, равную от 2 до 17 ч или от 8 до 16 ч, от 12 до 16 ч после перорального введения в стационарном состоянии. 13. Дозированная форма по п.11 или 12, отличающаяся тем, что указанный вытеснительный слой дополнительно содержит осмагент, предпочтительно выбранный из группы, состоящей из осмотических солей и осмотических карбогидратов. 14. Дозированная форма по любому из пп.11-13, отличающаяся тем, что оксикодон или его фармацевтически приемлемая соль содержится в количестве от 5 до 640 мг. 15. Дозированная форма по п.14, отличающаяся тем, что указанная фармацевтически приемлемая соль оксикодона является гидрохлоридом оксикодона. 16. Дозированная форма по любому из пп.11-14, отличающаяся тем, что она обеспечивает среднюю величину отношения С 24/Сmax, равную 0,7-0,99 или 0,8-0,95, после введения в стационарном состоянии. 17. Дозированная форма по любому из пп.11-16, отличающаяся тем, что она обеспечивает in vitro скорость высвобождения оксикодона или его фармацевтически приемлемой соли, измеренную по методуUSP Basket при скорости 100 об/мин в 900 мл водного буфера при рН от 1,6 до 7,2 при 37 С, равную от 0 до 40% через 1 ч, от 8 до 70% через 4 ч, от 20 до 80% через 8 ч, от 30 до 95% через 12 ч, от 35 до 95% через 18 ч и более 50% через 24 ч. 18. Применение любой из дозированных форм по пп.1-17 для изготовления фармацевтического препарата для лечения боли для обеспечения обезболивающего эффекта в течение по меньшей мере 24 ч и средней величины отношения С 24/Сmах оксикодона, равной 0,6-1,0 у пациентов после введения в стационарном состоянии указанным пациентам.
МПК / Метки
МПК: A61K 9/22
Метки: средство, оксикодона, лекарственное, основе
Код ссылки
<a href="https://eas.patents.su/21-5627-lekarstvennoe-sredstvo-na-osnove-oksikodona.html" rel="bookmark" title="База патентов Евразийского Союза">Лекарственное средство на основе оксикодона</a>
Модифицированные аминокислоты и лекарственное средство на их основе

Номер патента: 4037
Опубликовано: 25.12.2003
Авторы: Рудольф Клаус, Энгель Вольфхард, Энцерот Михаель, Винен Вольфганг, Эберлайн Вольфганг, Пипер Хельмут, Халлермайер Герхард, Доодс Хенри
МПК: C07D 401/12, C07K 5/06, A61P 3/10...
Метки: модифицированные, основе, аминокислоты, средство, лекарственное
Формула / Реферат:
1. Модифицированные аминоксилоты общей формулы (I) в которой R означает неразветвленный алкил с 1-3 атомами углерода, который на конце замещен циклоалкилом с 5-7 атомами углерода, одной или двумя фенильными группами, 1-нафтилом, 2-нафтилом или 4-бифенилилом, причем указанные ароматические остатки дополнительно могут быть замещены атомом фтора, хлора или брома, метилом, метоксигруппой, аминогруппой или ацетиламиногруппой, 2-пирролилом,...
Производные аминоциклоалкилпирролидона и антибактериальное лекарственное средство на их основе

Номер патента: 3321
Опубликовано: 24.04.2003
Авторы: Такахаси Хисаси, Такемура Макото, Мияути Сатору, Кимура Кенити, Кимура Юити, Оки Хитоси
МПК: A61K 31/473, C07D 401/04, A61P 31/04...
Метки: аминоциклоалкилпирролидона, производные, антибактериальное, лекарственное, основе, средство
Формула / Реферат:
1. Соединение, представленное формулой (I) где заместитель R1 представляет собой атом водорода; заместитель R2 представляет собой атом водорода; заместитель R3 представляет собой атом водорода; каждый из заместителей R4 и R5 независимо друг от друга представляет собой атом водорода, атом галогена, алкильную группу, содержащую от 1 до 6 атомов углерода, или алкоксигруппу, содержащую от 1 до 6 атомов углерода, которая может быть замещена...
Антибактериальное средство и антисептик, средство для чистки зубов, косметическое средство, средство для ванн, пищевой продукт или напиток на его основе

Номер патента: 2969
Опубликовано: 26.12.2002
Авторы: Ву Хуа-Канг, Икаи Кацусиге, Еноки Тацудзи, Сагава Хироаки, Кобаяси Ейдзи, Кояма Нобуто, Нисияма Ейдзи, Като Икуносин
МПК: A01N 35/06, A23L 1/30, A61K 7/00...
Метки: пищевой, антисептик, напиток, продукт, чистки, основе, косметическое, зубов, антибактериальное, средство, ванн
Формула / Реферат:
1. Антибактериальное средство, включающее 4,5-дигидрокси-2-циклопентен-1-он, представленный формулой [1] и/или его оптически активное производное. 2. Антисептик, отличающийся тем, что в качестве активного ингредиента(ов) он содержит антибактериальное средство по п.1. 3. Средство для чистки зубов, отличающееся тем, что в качестве активного ингредиента(ов) оно содержит антибактериальное средство по п.1. 4. Косметическое средство, отличающееся...
Лекарственное средство с контролированным высвобождением

Номер патента: 2677
Опубликовано: 29.08.2002
Авторы: Стоянов Симеон Иванов, Павлова Валентина Георгиева, Стефанова Евтимия Иванова, Кафеджийски Стефан Крумов, Цветков Пламен Кубратов
МПК: A61P 9/00, A61K 31/522, A61K 9/22...
Метки: лекарственное, высвобождением, средство, контролированным
Формула / Реферат:
Лекарственное средство с контролированным высвобождением на базе пентоксифилина, характеризующееся тем, что пентоксифилин и вспомогательные вещества находятся в соотношении 2:1 и содержит, мг: пентоксифилин от 100 до 500, гидроксипропилметилцеллюлоза от 20 до 60, лактоза моногидрат от 50 до 100, монокристаллическая целлюлоза от 10 до 55, стеарат магния от 2 до 10 и коллоидный диоксид кремния от 1 до 8.
Тиенотриазолодиазепиновые соединения и лекарственное средство

Номер патента: 1732
Опубликовано: 27.08.2001
Авторы: Суеока Хироюки, Кобаяси Харухито, Комацу Хироцугу, Ехара Сюдзи
МПК: A61K 31/5517, C07D 495/14, A61P 1/04...
Метки: лекарственное, средство, тиенотриазолодиазепиновые, соединения
Формула / Реферат:
1. Тиенотриазолодиазепиновое соединение формулы (1) где Х представляет собой галоген; R1 представляет собой алкил, имеющий 1-4 атома углерода; R2 представляет собой алкил, имеющий 1-4 атома углерода; а равен 1; R3 представляет собой алкил, имеющий 1-4 атома углерода, гидроксиалкил, имеющий 1-4 атома углерода, алкокси, имеющий 1-4 атома углерода, фенил, необязательно имеющий заместитель, или пиридил, необязательно имеющий заместитель, его...
Предыдущий патент: Покрытая оболочкой разлагающаяся жевательная резинка с улучшенным сроком хранения и способ ее получения
Следующий патент: Состав для уменьшения гидравлического сопротивления
Случайный патент: Способ получения водорода и электрической энергии при риформинге биоэтанола с использованием топливных элементов и с нулевым выделением загрязнителей