Способ получения пиразоло[4,3-d]пиримидин-7-он-3- пиридилсульфонильных соединений и их промежуточных соединений
Номер патента: 3145
Опубликовано: 27.02.2003
Авторы: Леветт Филип Чарльз, Девриз Кейт Майкл, Негри Джоанна Тереза, Вуд Альберт Шо




















Формула / Реферат
1. Способ получения соединения формулы (I)

где R представляет собой C1-C6алкил, необязательно замещенный одним или двумя заместителями, выбранными из C3-C5циклоалкила, OH, C1-C4алкокси, бензилокси, NR5R6, фенила, фуранила и пиридинила; C3-C6циклоалкил; 1-(C1-C4алкил)пиперидинил; тетрагидрофуранил или тетрагидропиранил, и где указанные C1-C6алкильные или C1-C4алкоксильные группы необязательно замещены галогеналкилом;
R1 (который может быть связан с любым азотом пиразольного кольца) представляет собой C1-C3алкил, необязательно замещенный фенилом, Het или N-связанной гетероциклической группой, выбранной из пиперидинила и морфолинила, и где указанная фенильная группа необязательно замещена C1-C4алкилом, который необязательно замещен галогеналкилом или галогеналкокси; или C1-C4алкокси; или галогеном, или CN;
R2 представляет собой C1-C6алкил;
и Het представляет собой С-связанную 6-членную гетероциклическую группу, содержащую один или два атома азота, необязательно в виде его моно-N-оксида, или C-связанную 5-членную гетероциклическую группу, содержащую два или три атома азота, где любая из указанных гетероциклических групп является необязательно замещенной C1-C4алкилом или C1-C4алкокси или NHR7, где R7 представляет собой H, C1-C4алкил или C1-C4алканоил;
R3 и R4 вместе с атомом азота, к которому они присоединены, образуют 4-R8-пиперазинильную группу, необязательно замещенную одной или двумя C1-C4алкильными группами, и необязательно в виде ее 4-N-оксида;
R5 и R6, каждый независимо, выбраны из H и C1-C4алкила, необязательно замещенного C3-C5циклоалкилом или C1-C4алкокси, или вместе с атомом азота, к которому они присоединены, образуют азетидинильную, пирролидинильную, пиперидинильную или морфолинильную группу;
R8 представляет собой H; C1-C4алкил, необязательно замещенный одним или двумя заместителями, выбранными из OH, NR5R6, CONR5R6, фенила, необязательно замещенного C1-C4алкокси, бензодиоксолила и бензодиоксанила; C3-C6алкенил;
пиридинил или пиримидинил;
причем указанный способ включает взаимодействие соединения формулы (II), (III) или (IV) в присутствии -OR и агента, улавливающего гидроксид, или, в случае соединений формулы (IV), взаимодействие в присутствии вспомогательного основания и агента, улавливающего гидроксид (т.е. -OR заменяют на вспомогательное основание), где X представляет собой уходящую группу и R1-R4 такие, как определены выше.



2. Способ по п.1 получения соединения формул (IA) и (IB)


включающий взаимодействие соединения формулы (IIA), (IIIA) или (IVA), и (IIB), (IIIB) или (IVB) соответственно






в присутствии -OR и агента, улавливающего гидроксид, или, альтернативно, в случае соединений формул (IVA) и (IVB) взаимодействие в присутствии -OR и вспомогательного основания, где OR в случае образования соединения (IA) представляет собой CH3O(CH2)2O-, и OR в случае образования соединения (IB) представляет собой (R)-CH3OCH2CH(CH3)O-, и где X в формулах (IIA)-(IIIA) и в формулах (IIB)-(IIIB) представляет собой уходящую группу.
3. Способ по п.1 или 2, где агентом, улавливающим гидроксид, является сложный эфир.
4. Способ по любому из пп.1-3, где агентом, улавливающим гидроксид, является сложный эфир формулы
TOC(O)W,
где OT представляет собой OR, или остаток объемного спирта, или ненуклеофильного спирта, или TOH представляет собой спирт, который может быть азеотропно удален в процессе реакции;
и C(O)W представляет собой остаток карбоновой кислоты.
5. Способ по любому из пп.1-4, где X выбран из группы, состоящей из арилсульфонилокси, C1-C4алкилсульфонилокси, нитро- или галогензамещенного бензолсульфонилокси, C1-C4перфторалкилсульфонилокси, необязательно замещенного ароилокси, C1-C4перфторалканоилокси, C1-C4алканоилокси, галогена; диазония; C1-C4первичного и вторичного алкокси, оксония, перхлорилокси, C1-C4алкилсульфонилокси с остатком четвертичного аммония, галогенсульфонилокси, галония и диарилсульфониламино.
6. Способ по п.5, где X представляет собой C1-C6алкокси.
7. Способ по любому из пп.1-6, где -OR присутствует вместе со вспомогательным основанием.
8. Способ по п.7, где вспомогательное основание выбрано из группы, состоящей из стерически затрудненного основания, гидрида металла, оксида металла, карбоната металла и бикарбоната металла.
9. Способ по п.8, где стерически затрудненным основанием является металлическая соль стерически затрудненного спирта или амина.
10. Способ по п.9, где реакцию проводят в инертном растворителе или в ROH или в смеси обоих.
11. Способ по п.10, где растворитель является выбранным из группы, состоящей из ROH, вторичного или третичного C4-C12алканола, C3-C12циклоалканола, третичного C4-C12циклоалканола, вторичного или третичного (C3-C7 циклоалкил)C2-C6алканола, C3-C9алканона, 1,2-диметоксиэтана, 1,2-диэтоксиэтана, диглима, тетрагидрофурана, 1,4-диоксана, толуола, ксилола, хлорбензола, 1,2-дихлорбензола, ацетонитрила, диметилсульфоксида, сульфолана, диметилформамида, N-метилпирролидин-2-она, пиридина, и их смеси.
12. Способ по п.11, где растворитель представляет собой ROH.
13. Способ по любому из пп.2-12, где соединение (IA) образуется в результате взаимодействия соединений (IIA) или (IIIA)
a) с 2-метоксиэтанолом и вспомогательным основанием необязательно в инертном растворителе и в присутствии указанного улавливающего агента; или
b) с ZO(CH2)2OCH3 и вспомогательным основанием в 2-метоксиэтаноле или инертном растворителе, или в обоих в присутствии указанного улавливающего агента; или
c) с ZO(CH2)2OCH3 и 2-метоксиэтанолом, или инертным растворителем или обоими в присутствии указанного улавливающего агента, и где Z является металлом.
14. Способ по любому из пп.2-12, где соединение (IB) образуется в результате взаимодействия соединения (IIB) или (IIIB)
a) с (R)-CH3OCH2CH(CH3)OH и вспомогательным основанием, необязательно в инертном растворителе и в присутствии указанного улавливающего агента; или
b) ё (R)-CH3OCH2CH(CH3)OZ и вспомогательным основанием в (R)-CH3OCH2CH(CH3)OH или инертном растворителе, или в обоих в присутствии указанного улавливающего агента; или
c) с (R)-CH3OCH2CH(CH3)OZ и (R)-CH3OCH2CH(CH3)OH, или инертным растворителем, или с обоими в присутствии указанного улавливающего агента, и где Z является металлом.
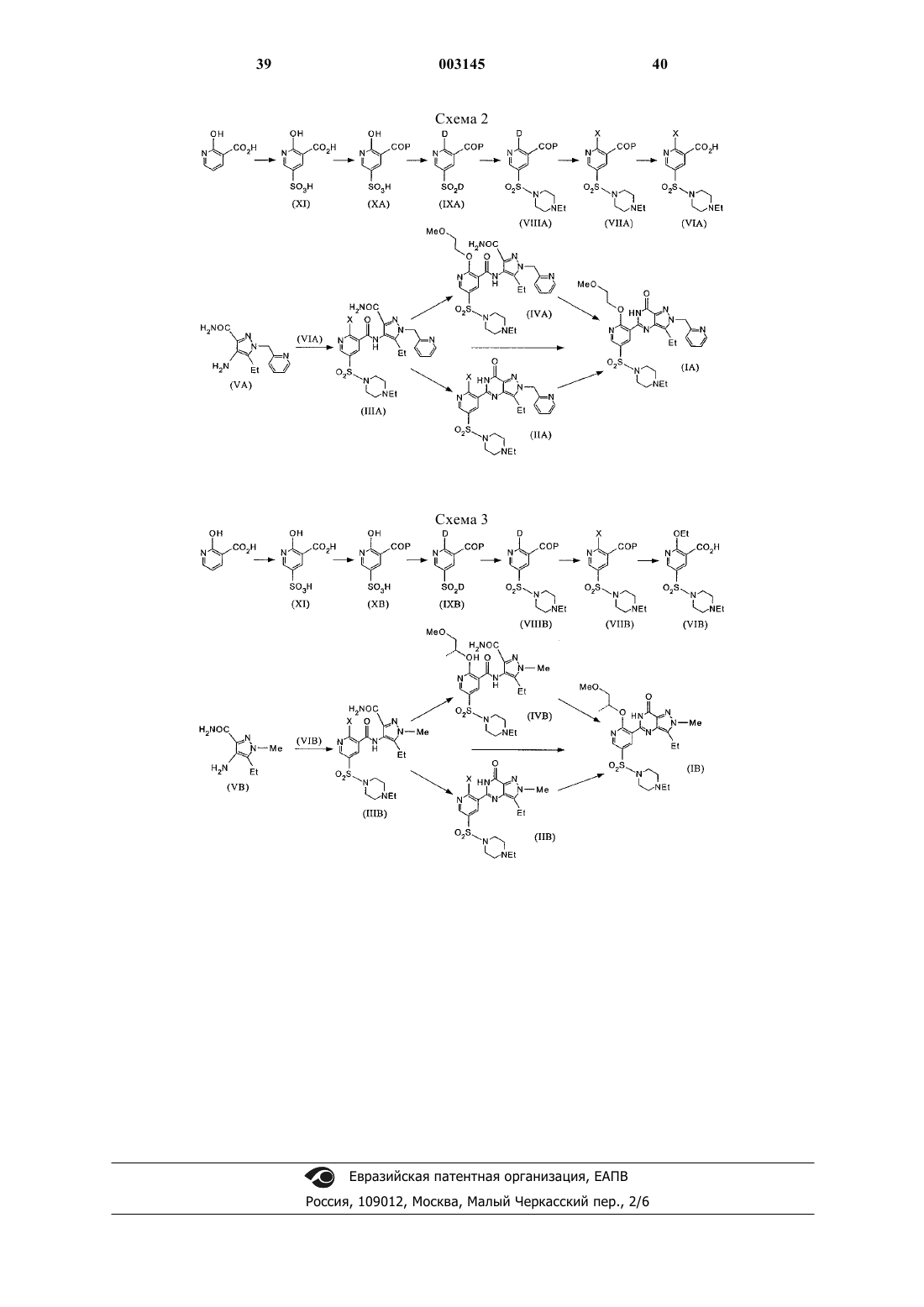
15. Способ по любому из пп.1-14, где соединение формулы (III) образуется в результате сочетания соединения формулы (V) с соединением формулы (VI) или его солью.
16. Способ по п.15, где соединение формулы (VI) или его соль получают из соединения формулы (VII)


(a) который в случае соединения формулы (VI), где X представляет собой арилсульфонилокси, C1-C4алкилсульфонилокси, C1-C4перфторалкилсульфонилокси, ароилокси, C1-C4перфторалканоилокси, C1-C4алканоилокси, C1-C4алкилсульфонилокси с остатком четвертичного аммония или галогенсульфонилокси, включает взаимодействие соединения формулы (VII), где Y и V представляют собой OH, в присутствии соответствующего сульфонирующего, арилирующего или ацилирующего агента X;
(b) который в случае соединения формулы (VI), где X представляет собой Cl, включает взаимодействие соединения формулы (VII), где Y представляет собой Cl, и V представляет собой P, и P является защитной группой, с агентом, удаляющим защитные группы;
(c) который в случае соединения формулы (VI), где X представляет собой диазоний, включает взаимодействие соединения формулы (VII), где Y представляет собой NH2, V представляет собой OH, с азотистой кислотой;
(d) который в случае соединения формулы (VI), где X представляет собой (диарилсульфонил)амино, включает взаимодействие соединения формулы (VII), где Y=NH2 и V=OH, в присутствии соответствующего сульфонирующего агента;
(e) который в случае соединения формулы (VI), где X представляет собой OR, который является C1-6алкокси, включает взаимодействие соединения формулы (VII), где V представляет собой P, где P представляет собой защитную группу и Y представляет собой C1-6первичный или вторичный алкокси, с агентом, удаляющим защитные группы.
17. Способ по п.16, где соединение формулы (VII) получают из соединения формулы (VIII)

где D представляет собой Cl или Br и P представляет собой защитную группу,
(a) который в случае соединений формулы (VII), где Y представляет собой OH и V представляет собой OH, включает взаимодействие соединения формулы (VIII) с гидролизующим агентом и необязательно дополнительным удаляющим защитную группу агентом, если P не удаляется под действием гидролизующего агента;
(b) который в случае соединений формулы (VII), где Y представляет собой NH2 и V представляет собой OH, включает взаимодействие соединения формулы (VIII) с аммонирующим агентом с образованием промежуточного соединения формулы (VII), где Y представляет собой NH2 и V представляет собой P (защитную группу), и взаимодействие указанного промежуточного соединения (VII) с агентом, удаляющим защитные группы; и
(c) который в случае соединений формулы (VII), где Y представляет собой OR, который представляет собой C1-6алкокси, и V представляет собой P, включает взаимодействие соединения формулы (VIII) в присутствии -OR;
18. Способ по п.17, где соединение формулы (VIII) получают из соединения формулы (IX) в присутствии амина NH(R3)(R4), где R3 и R4 такие, как определены в п.1

где D и P такие, как определены в п.17.
19. Способ по п.18, где соединение формулы (IX) получают взаимодействием соединения формулы (X) с хлорирующим или бромирующим агентом

где P такой, как определен в п.17.
20. Способ по п.19, где соединение формулы (X) получают из соединения формулы (XI) в присутствии агента, образующего защитную группу (P) на карбоновой кислоте

21. Способ по п.20, где соединение формулы (XI) получают из 2-гидроксиникотиновой кислоты или ее соли в присутствии SO3 в растворителе.
22. Способ по п.21, где SO3 находится в апротонном растворителе, минеральной кислоте или жидкой карбоновой кислоте.
23. Способ по любому из пп.16-22, где соединения формулы (VI), (VII) и (VIII) представляют собой (VIA), (VIIA) и (VIIIA) соответственно



где X представляет собой C1-6алкокси, D и P такие, как определены в пп.16 и 17, и соединения (VIA) и (VIIA) образуются в соответствии с пп.16(e) и 17(c) соответственно, и соединение (VIIIA) образуется в соответствии с п.18 в результате взаимодействия соединения (IX) с N-этилпиперазином или его солью.
24. Способ по п.23, где X представляет собой OEt, и соединение (VIA) образуется в результате взаимодействия соединения (VIIA) с агентом, удаляющим защитные группы, и соединение (VIIA) образуется в результате взаимодействия соединения (VIIIA) в присутствии OEt.
25. Способ по п.23 или 24, где соединение (X) образуется в результате взаимодействия соединения (XI) или его соли с этанолом, с образованием защитной группы OEt.
26. Способ по любому из пп.23-25, где две или более последовательных стадий образования (VIA), (VIIA) или (VIIIA) проводятся в толуоле и объединяются вместе в одну объединенную стадию без выделения промежуточного соединения.
27. Способ по любому из пп.1-26 получения соединения формулы (I), (IA) или (IB), включающий в качестве исходного соединения коммерчески доступную 2-гидроксиникотиновую кислоту или ее соль, и взаимодействие ее в присутствии SO3 в растворителе, с образованием соединения формулы (XI).
28. Соединение (IVA)

где R имеет значения, указанные в п.1.
29. Соединение (IVB)

где R имеет значения, указанные в п.1.
Текст
1 Данное изобретение относится к ряду пиразоло[4,3-d]пиримидин-7-он-3-пиридилсульфонильных соединений формулы I (как определено ниже) и их промежуточным соединениям. Причем большое значение имеет то, что большинство рассматриваемых соединений являются ингибиторами циклический гуанозин-3',5'монофосфат фосфодиэстеразы типа 5 (cGMPPDE5) и имеют применение в ряде терапевтических областей (таких как эректильная дисфункция у самцов). Отдельными соединениями,представляющими интерес, являются 1-этил-45-[3-этил-6,7-дигидро-7-оксо-2-(2-пиридилметил)-2 Н-пиразоло[4,3-d]пиримидин-5-ил]-6-(2 метоксиэтокси)-3-пиридилсульфонилпиперазин (далее в данном документе соединение формулы IA) и (R)-1-этил-4-[5-(3-этил-6,7 дигидро-2-метил-7-оксо-2 Н-пиразоло[4,3-d]пиримидин-5-ил)-6-(2-метокси-1-метилэтокси)-5 пиридилсульфонил]пиперазин (далее в данном документе соединение формулы IB). Альтернативным названием (IB) является (+)-3-этил-5-[5(4-этилпиперазин-1-илсульфонил)-2-(2-метокси 1(R)-метилэтокси)пиридин-3-ил]-2-метил-2,6 дигидро-7 Н-пиразоло[4,3-d]пиримидин-7-он. Способы получения многих соединений формулы I раскрыты в WO 98/49166 и PCT/IB 99/00519 (опубликован как WO 99/54333). В частности, примеры 4 и 118 PCT/IB 99/00519 раскрывают способ получения соединений IA иIB. В соответствии с первым аспектом данного изобретения предоставляется способ получения соединения формулы (I) или его соли где R представляет собой C1-C6 алкил, необязательно замещенный одним или двумя заместителями, выбранными из С 3-С 5 циклоалкила, ОН,C1-C4 алкокси, бензилокси, NR5R6, фенила, фуранила и пиридинила; С 3-С 6 циклоалкил; 1-(C1C4 алкил)пиперидинил; тетрагидрофуранил или тетрагидропиранил, и где указанные C1-C6 алкильные или C1-C4 алкоксильные группы необязательно замещены галогеналкилом;R1 (который может быть связан с любым азотом пиразольного кольца) представляет собой C1-С 3 алкил, необязательно замещенный фенилом, Het или N-связанной гетероциклической группой, выбранной из пиперидинила и морфолинила, и где указанная фенильная группа необязательно замещена: C1-C4 алкилом, который необязательно замещен галогеналкилом или галогеналкокси; или C1-C4 алкокси; или галогеном, или CN; 2 и Het представляет собой С-связанную 6 членную гетероциклическую группу, содержащую один или два атома азота, необязательно в виде его моно-N-оксида, или С-связанную 5 членную гетероциклическую группу, содержащую два или три атома азота, где любая из указанных гетероциклических групп является необязательно замещенной C1-C4 алкилом или C1C4 алкокси или NHR7, где R7 представляет собой Н, C1-C4 алкил или C1-C4 алканоил.R3 и R4 вместе с атомом азота, к которому они присоединены, образуют 4-R8-пипepaзинильнyю группу, необязательно замещенную одной или двумя C1-C4 алкильными группами, и необязательно в виде ее 4-N-оксида;R8 представляет собой Н; C1-C4 алкил, необязательно замещенный одним или двумя заместителями, выбранными из ОН, NR5R6,CONR5R6, фенила, необязательно замещенногоC1-C4 алкокси, бензодиоксолила и бензодиоксанила; C3-C6 алкенил; пиридинил или пиримидинил; причем указанный способ включает взаимодействие соединения формулы (II), (III) или(IV) в присутствии -OR и агента, улавливающего гидроксид, или, в случае соединений формулы (IV), взаимодействие в присутствии вспомогательного основания и агента, улавливающего гидроксид (т.е. -OR заменяют на вспомогательное основание), где Х представляет собой уходящую группу и R1-R4 такие, как определены выше. В приведенном выше определении, если не указано иначе, алкильные, алкоксильные и алкенильные группы, имеющие три или более атомов углерода, и алканоильные группы,имеющие четыре или более атомов углерода,могут быть линейными или разветвленными. Термин атом галогена включает Cl, Br, F и I. Галогеналкил и галогеналкокси включают СF3 и ОСF3 соответственно. Соединения формулы (I) могут содержать один или несколько хиральных центров и, следовательно, могут существовать в виде стереоизомеров, т.е. в виде энантиомеров или диастереоизомеров, а также в виде их смесей. Данное изобретение относится к образованию как инди 3 видуальных стереоизомеров соединений формулы (I), так и любой их смеси. В первом и втором предпочтительных воплощениях данного изобретения получают соединения формул (IA) и (IB) Соответственно в предпочтительном аспекте данного изобретения предоставляется способ получения соединения формулы (IA) или включающий взаимодействие соединения формулы (IIA), (IIIA) или (IVA), и (IIB), (IIIB) или в присутствии -OR и агента, улавливающего гидроксид, или, альтернативно, в случае соединений формул (IVA) и (IVB), взаимодействие в присутствии агента, улавливающего гидроксид,и вспомогательного основания, где OR в случае образования соединения (IA) представляет собой СН 3 О(СН 2)2 О-, и OR в случае образования соединения (IB) представляет собой(R)-СН 3 ОСН 2 СН(СН 3)O-,и где Х в формулах от (IIА) до (IIIA) и в формулах от (IIB) до (IIIB) представляет собой уходящую группу. 4 Промежуточные соединения общей формулы (IV) и, более конкретно, (IVA) и (IVB), в том случае, когда являются новыми, образуют дальнейшие аспекты данного изобретения. Как результат применения агента, улавливающего гидроксид, особым преимуществом настоящего способа по сравнению с предыдущим уровнем техники (PCT/IB 99/00519) является то, что может быть получен более высокий выход конечного продукта (соединений формулы (I, IA, IB) и промежуточных соединений (VI,VIA, VIB). В предпочтительном воплощении соединение формулы (I) получают с хорошим выходом без выделения промежуточного соединения. Наиболее предпочтительным является образование соединений формулы (I) из промежуточных соединений формулы (III), так как стадия циклизации (от III до II) и нуклеофильное замещение Х на -OR (от II до I) могут быть проведены в одном сосуде. Кроме того, этот способ может проводиться при атмосферном давлении,тогда как стадия циклизации 2-х стадийного способа требует повышенного давления, где ХН является низшим алканолом, например метанолом, этанолом, 1-пропанолом и 2-пропанолом. Например, образование соединения (IIА) из(IIIA) в примере 1B PCT/IB 99/00519 протекает при высоком давлении, тогда как теперь мы можем получать (IA) из (IIIA) при атмосферном давлении. В следующем аспекте данного изобретения предоставляется способ получения соединений формулы (II) (более конкретно IIА и IIB), включающий циклизацию соединений формулы (III)(более конкретно IIIA и IIIB) в присутствии указанного агента, улавливающего гидроксид. Опять же, преимущество этой стадии состоит в повышенном выходе и качестве, которые обеспечиваются применением агента, улавливающего гидроксид. Конечно, технология улавливающего агента может использоваться для получения соединений формулы (IV) (более конкретно IVA иIVB) из соединения формулы (III) (более конкретно IIIA и IIIB, соответственно) в присутствии -OR, преимущественно приблизительно до 1 молярного эквивалента -OR (по отношению к соединению (III. Если используется существенно больше 1 эквивалента -OR, то реакция будет протекать до соединения (I) (более конкретно IA или IB). Предпочтительно агентом, улавливающим гидроксид, является сложный эфир. Более предпочтительно указанным агентом, улавливающим гидроксид, является сложный эфир формулыTOC(O)W,где ОТ представляет собой OR, или остаток объемного спирта, или ненуклеофильного спирта, или ТОН представляет собой спирт, который 5 может быть азеотропно удален в процессе реакции; и C(O)W представляет собой остаток карбоновой кислоты. Для дальнейшего улучшения выходов конечного продукта и уменьшения примесей,предпочтительно, чтобы C(O)W являлось остатком стерически затрудненной карбоновой кислоты и/или карбоновой кислоты, которая не содержит енолизуемый протон (например, пивалевая кислота). Например, если Х представляет собой OEt в соединении (IIА) и (IIIA), сложноэфирным улавливающим агентом может быть этилацетат(т.е. ОТ=Х и C(O)W представляет собой остаток уксусной кислоты), более предпочтительно,этилпивалат (ОТ=Х и C(O)W представляет собой остаток пивалевой кислоты - т.е. карбоновой кислоты, не содержащей енолизуемый протон), 2-метоксиэтилацетат (OT=OR и C(O)W представляет собой остаток уксусной кислоты),или наиболее предпочтительно 2-метоксиэтилпивалат (OT=OR и C(O)W представляет собой остаток карбоновой кислоты, не содержащей енолизуемый протон). Предпочтительно Х выбран из группы, состоящей из необязательно замещенного арилсульфонилокси, предпочтительно фенилсульфонилокси, более предпочтительно, паразамещенного арила(фенила), замещенного группой, такой как C1-C4 алкил, например птолуолсульфонилокси; C1-C4 алкилсульфонилокси; например метансульфонилокси; нитро- или галогензамещенного бензолсульфонилокси, предпочтительно паразамещенного, например п-бромбензолсульфонилокси или п-нитробензолсульфонилокси;C1-C4 алкилсульфонилокси с остатком четвертичного аммония; галогенсульфонилокси,например фторсульфонилокси, и других фторированных уходящих групп; и диарилсульфониламино, например дитозила (NTs2). Наиболее предпочтительно для образования соединений формулы (I), более конкретно(IA) и (IB), если Х представляет собой C1-C6 алкокси (преимущественно этокси), так как это способствует более простому и дешевому образованию соединений, например см. схемы 1, 2 и 3 далее в данном документе. Однако преимуществом использования лабильных уходящих групп, таких как хлор или фтор, может быть то, что инертный растворитель в этом случае может использоваться пред 003145 6 почтительнее, чем ROH (которые часто являются более дорогими). Таким образом, будет требоваться только количество -OR (такой как из(вытесняя уходящую группу посредством нуклеофильного замещения), и как основание (вызывая циклизацию).OR может образовываться в растворе, например, из соли ZOR (где Z является катионом),такой как соль металла. Более конкретно, соль щелочного (такого как натрий или калий) или щелочно-земельного металла -OR в подходящем растворителе является источником -OR в растворе. Например, 2-метоксиэтоксид натрия в подходящем растворителе с промежуточным соединением (IIА) или (IIIA) образует соединение (IA). В другом воплощении -OR образуется(т.е. основания, отличающегося от -OR). Однако в другой системе ZOR может быть использован в реакционной системе со вспомогательным основанием. Полагают, что растворителем, в котором протекает данная реакция, может быть ROH или инертный растворитель (или смесь обоих). Под инертным растворителем мы подразумеваем растворитель, который не образует нуклеофила в условиях реакции, или, если нуклеофил образуется, то он существует в достаточно затрудненной или нереакционноспособной форме, так что он, по существу, не является конкурентом в реакции замещения. Если ROH используется в качестве источника -OR, то другой растворитель не является существенно необходимым, но в качестве сорастворителя может использоваться(вспомогательный) инертный растворитель (т.е. растворитель, отличающийся от ROH). Подходящими растворителями являются следующие:ROH, вторичный или третичный C4-C12 алканол, C3-C12 циклоалканол, третичный C4-C12 циклоалканол, вторичный или третичный (C3-C7 циклоалкил)C2-C6 алканол, C3-C9 алканон, 1,2-диметоксиэтан, 1,2-диэтоксиэтан, диглим, тетрагидрофуран, 1,4-диоксан, толуол, ксилол, хлорбензол, 1,2-дихлорбензол, ацетонитрил, диметилсульфоксид, сульфолан, диметилформамид,N-метилпирролидин-2-он, пиридин и их смеси. Более предпочтительно растворитель представляет собой ROH, третичный C4-C12 алканол, третичный C4-C12 циклоалканол, третичный (C3-C7 циклоалкил)C2-C6 алканол, C3-C9 алканон, 1,2-диметоксиэтан, 1,2-диэтоксиэтан,диглим, тетрагидрофуран, 1,4-диоксан, толуол,ксилол, хлорбензол, 1,2-дихлорбензол, ацетонитрил, диметилсульфоксид, сульфолан, диметилформамид, N-метилпирролидин-2-он, пиридин и их смеси. Наиболее предпочтительно растворитель представляет собой ROH, это означает, что -OR образуется in situ, как в присутствии вспомога 7 тельного основания. Для соединений (IA) и (IB) растворитель представляет собой предпочтительно СН 3 О(СН 2)2OН и (R)-СН 3 ОСН 2 СН(СН 3) ОН соответственно. В способе данного изобретения может использоваться широкий ряд вспомогательных оснований. Обычно основания не конкурируют в существенной степени с -OR в нуклеофильном замещении Х (т.е. они не проявляют нуклеофильного характера) поскольку они являются подходящим образом стерически затрудненными. Предпочтительно вспомогательное основание выбирают из группы, состоящей из стерически затрудненного основания, гидрида металла, оксида металла, карбоната металла и бикарбоната металла. Стерически затрудненное основание преимущественно представляет собой соль металла и стерически затрудненного спирта или амина. Более предпочтительно вспомогательные основания в соответствии с данным изобретением выбирают из группы, состоящей из солей металлов стерически затрудненного спирта или амина, такого как вторичный или третичный C4C12 алканол, C3-C12 циклоалканол и вторичный или третичный (C3-C8 циклоалкил)C1-C6 алканол,N-(вторичный или третичный C3-C6 алкил)-N(первичный, вторичный или третичный C3-C6 алкил)амин,N-(C3-C8 циклоалкил)-N-(первичный, вторичный или третичный C3-C6 алкил)амин, ди(C3-C8 циклоалкил)амин или гексаметилдисилазан; 1,5-диазабицикло[4,3,0]нон-5-ена и 1,8 диазабицикло-[5,4,0]ундец-7-ена; гидрида, оксида, карбоната и бикарбоната металлов. Еще более предпочтительно, если вспомогательные основания выбраны из группы, состоящей из солей металлов стерически затрудненного спирта или амина, такого как третичный C4-C12 алканол, C3-C12 циклоалканол и третичный (C3-C8 циклоалкил) C1-C6 алканол, N(вторичный или третичный C3-C6 алкил)-N(первичный, вторичный или третичный C3C6 алкил)амин, N-(C3-C8 циклоалкил)-N-(первичный, вторичный или третичный C3-C6 алкил)амин, ди(C3-C8 циклоалкил)амин или гексаметилдисилазан; 1,5-диазабицикло[4,3,0]нон-5-ена и 1,8 диазабицикло-[5,4,0]ундец-7-ена. Еще более предпочтительно, если вспомогательное основание выбрано из стерически затрудненных оснований предыдущего параграфа (т.е. все они, за исключением гидрида,оксида, карбоната и бикарбоната металлов). Наиболее предпочтительно вспомогательным основанием являются соли металлов третичного C4-C6 спирта, такие как соли щелочных или щелочно-земельных металлов (например,Na/K) т-бутанола или т-амилового спирта, или 8 основанием является гексаметилдисилазон калия (KHMDS). Наиболее предпочтительно, вспомогательным основанием является соль щелочного металла т-бутанола (например, т-бутоксид калия). Предпочтительно металлы соли ZOR и вспомогательного основания независимо выбраны из щелочных металлов (литий, натрий,калий, рубидий, цезий) или щелочно-земельных металлов (бериллий, магний, кальций, стронций, барий). Более предпочтительно этим металлом является натрий или калий. Для получения максимальных выходов далее предпочтительно использовать, по меньшей мере, около 1 молярного эквивалента вспомогательного основания и -OR в соответствии с данным изобретением. Если -OR также функционирует в качестве основания (т.е. вспомогательное основание отсутствует), то предпочтительно,чтобы присутствовало, по меньшей мере, около 2 эквивалентов -OR. Подходящим является присутствие, по меньшей мере, приблизительно 1 эквивалента улавливающего агента (предпочтительно, по меньшей мере, приблизительно 2 эквивалента). В особенно предпочтительном воплощении данного изобретения соединения формулы(I) могут быть предоставлены с качеством, подходящим для клинических испытаний, что позволяет избежать необходимости в дальнейших стадиях очистки. Температура реакции соединений (III) и(IV) до (I) (как соответственное образование соединений IA и IB) предпочтительно составляет, по меньшей мере, около 80 С, более предпочтительно приблизительно от 80 до 130 С,еще более предпочтительно приблизительно от 100 до 130 С и наиболее предпочтительно приблизительно от 115 до 125 С. Эти температуры также являются применимыми для превращения соединений (II) в (I), хотя температура в этом случае вероятно может также быть ниже (например, около 60 С), так как не происходит циклизации. Температура реакции, необходимая для достижения эффекта превращения соединений формул (II) и (III) в соединения формулы (I) зависит от растворителя, природы -OR и X. Если Х представляет собой алкокси и ROH является растворителем, предпочтительно ХН (такой какC1-C6 алкокси) удаляют азеотропно (конечно,конфигурация реакционного сосуда должна подходить для перегонки азеотропной смеси) сROH путем проведения реакции при азеотропной температуре ХН и ROH. Таким образом могут быть дополнительно улучшены выход и качество конечного продукта. Например, (если Х является алкокси) превращение соединения(IIА), (IIIA) или (IVA) в (IA) предпочтительно проводят при азеотропной температуре спирта 9 Предпочтительными воплощениями данного изобретения являются 1. синтез соединения (IA) путем взаимодействия соединения (IIА) или (IIIA):a) с 2-метоксиэтанолом и вспомогательным основанием, необязательно в инертном растворителе и в присутствии указанного улавливающего агента; илиb) с ZО(СН 2)2OСН 3 и вспомогательным основанием в 2-метоксиэтаноле или инертном растворителе или в обоих, в присутствии указанного улавливающего агента; илиc) с ZO(СН 2)2 ОСН 3 и 2-метоксиэтанолом или инертным растворителем или обоими, в присутствии указанного улавливающего агента. Предпочтительно улавливающий агент представляет собой CH3O(CH2)2OC(O)W илиCH3OC(O)W, где C(O)W является остатком карбоновой кислоты (предпочтительно стерически затрудненным). 2. синтез соединения (IB) путем взаимодействия соединения (IIB) или (IIIB):a) с (R)-СН 3 ОСН 2 СН(СН 3)ОН и вспомогательным основанием, необязательно в инертном растворителе и в присутствии указанного улавливающего агента; илиb) с (R)-СН 3 ОСН 2 СН(СН 3)ОZ и вспомогательным основанием в (R)-СН 3 ОСН 2 СН(СН 3) ОН или инертном растворителе или в смеси обоих, в присутствии указанного улавливающего агента; или с) с (R)-СН 3 ОСН 2 СН(СН 3)ОZ и (R)СН 3 ОСН 2 СН(СН 3)ОН или инертным растворителем или с обоими, в присутствии указанного улавливающего агента. Предпочтительно улавливающий агент представляет собой(R)-СН 3 ОСН 2 СН(СН 3) ОС(O)W или CH3OC(O)W, где C(O)W является остатком карбоновой кислоты (предпочтительно стерически затрудненным). Для получения максимальных выходов в соответствии с данным изобретением далее предпочтительно использовать, по меньшей мере, примерно 1 молярный эквивалент вспомогательного основания и -OR. Если -OR также функционирует в качестве основания (т.е. вспомогательное основание отсутствует), то предпочтительно, чтобы присутствовало, по меньшей мере, приблизительно 2 эквивалента -OR. Таким образом для получения максимальных выходов соединений (IA) и (IB), подходящим является, если присутствует, по меньшей мере,1 эквивалент улавливающего агента (предпочтительно, по меньшей мере, приблизительно 2 эквивалента). Относительно пункта 1 а), приведенного выше, предпочтительно, чтобы присутствовало, по меньшей мере, приблизительно 2 молярных эквивалента основания и, по меньшей мере, приблизительно 1 молярный эквивалент улавливающего агента по отношению к субстрату (более предпочтительно, по меньшей мере, приблизительно 2,2 и 2,5 соответственно). 10 Для пункта 1b), приведенного выше, предпочтительно, чтобы присутствовало, по меньшей мере, приблизительно 1 молярный эквивалент вспомогательного основания, улавливающего агента и ZО(СН 2)2OСН 3 по отношению к субстрату (более предпочтительно, по меньшей мере, приблизительно 1,2 эквивалента вспомогательного основания и, по меньшей мере, приблизительно 2,5 эквивалента улавливающего агента). Для пункта 1 с), приведенного выше,предпочтительно, чтобы присутствовало, по меньшей мере, приблизительно 2 молярных эквивалента ZО(СН 2)2OСН 3 и, по меньшей мере,приблизительно 1 эквивалент улавливающего агента по отношению к субстрату (более предпочтительно, по меньшей мере, приблизительно 2 и 2,5 эквивалента соответственно). Соединения общей формулы (III), (IIIA) и(IIIB) могут быть получены из легко доступных исходных веществ, например, с помощью способов, изображенных на следующих схемах реакций. Схема реакции 1 приведена для соединений общей формулы (I), схема 2 приведена для соединения (IA) и схема 3 приведена для соединения (IB). В соответствии со схемой 1, промежуточное соединение формулы (VI) или его соль образуется из соединения формулы (VIII), причем уточненный способ зависит от уходящей группы X. Для соединений формулы (VI) или их солей, где Х=арилсульфонилокси, C1-C4 алкилсульфонилокси, C1-C4 перфторалкилсульфонилокси, арилокси, C1-C4 перфторалканоилокси, C1C4 алканоилокси, C1-C4 алкилсульфонилокси или галогенсульфонилокси с остатком четвертичного аммония, соединение (VI) может быть получено из соединения (VII) (где Y=OH и V=OH) и соответствующего дериватизирующего агента,более конкретно, соответствующего сульфонирующего агента, такого как арилсульфонилгалогенид, C1-C4 алкилсульфонилгалогенид, C1-C4 перфторалкилсульфонилгалогенид, C1-C4 алкилсульфонилгалогенид с остатком четвертичного аммония, галогенсульфонилгалогенид, или соответствующего арилирующего агента, такого как арилгалогенид, или соответствующего ацилирующего агента, такого как C1-C4 перфторалканоилгалогенид, или C1-C4 алканоилгалогенид), например, в соответствующем растворителе. Предпочтительно вышеупомянутый галогенидный заместитель представляет собой хлорид, и предпочтительно также, чтобы реакция проходила в присутствии основания. Соединения формулы (VII) (где Y=OH и V=OH) могут быть образованы из соединений (VIII) и гидролизующего агента, предпочтительно гидроксидного основания (идеально 2 молярных эквивалента), более предпочтительно гидроксида металла, такого как гидроксид натрия, в соответствующем растворителе, таком как вода. Металл гидроксидного основания может быть та 11 ким, как определено ранее для Z (в ZOR). Это также применимо для других реакций схем 1, 2 и 3, приведенных ниже, где используют гидроксидное основание/гидролизующий агент. Хотя гидролизующий агент обычно действует, вводя гидроксид в D и в Р, в пределах сферы данного изобретения могут иметь место некоторые защитные группы, которые могут не гидролизоваться, в этом случае могут использоваться разные агенты для удаления защитных групп. Соединения формулы (VI), где Х=хлор, могут быть образованы из (VII), где Y=Сl и V=P (такой какOEt) (т.е. формула VIII) и агента, удаляющего защитные группы. Предпочтительно агент, удаляющий защитные группы, использующийся здесь в соответствии с данным изобретением, представляет собой гидролизующий агент, более предпочтительно гидроксидный нуклеофил, преимущественно гидроксидное основание (идеально 1 молярный эквивалент), такое как гидроксид натрия, предпочтительно, в соответствующем растворителе, таком как вода. Соединения формулы (VI), где Х=диазоний, могут быть образованы из (VII) (гдеY=NH2, V=OH) и азотистой кислоты. Соединения формулы (VII) (где Y=NH2, V=OH) могут быть образованы из соединений формулы (VII)(где Y=NH2, V=P, например OEt) и агента, удаляющего защитные группы. Промежуточное соединение (VII) (Y=NH2, V=P, например OEt) образуется из (VIII) и аммонирующего агента,такого как аммиак, в соответствующем растворителе, таком как вода. Соединения формулы (VI), где Х=(диарилсульфонил)амино, могут быть образованы из(VII) (где Y=NH2, V=OH) и соответствующего дериватизирующего агента, предпочтительно соответствующего сульфонирующего агента,такого как арилсульфонилгалогенид, предпочтительно арилсульфонилхлорид (идеально, по меньшей мере, 2 молярных эквивалента), предпочтительно в присутствии основания (идеально 2 его молярных эквивалента), такого как триэтиламин, в соответствующем растворителе. Соединения формулы (VI), где Х являетсяOR, который представляет собой C1-6 (предпочтительно C1-C4) предпочтительно первичную или вторичную алкоксигруппу, могут быть образованы из (VII) (где Y=C1-6 (предпочтительноC1-C4) первичная или вторичная алкоксигруппа и V=P (такой как OEt), и агента, удаляющего защитные группы. Соединения формулы (VII)(где Y=C1-6 (предпочтительно C1-C4 первичная или вторичная алкоксигруппа, V=P (например,OEt) могут быть образованы путем взаимодействия соединения формулы (VIII) в присутствииOR, которое представляет собой соответствующий алкоксид (С 1-6, более предпочтительноC1-C4) более предпочтительно первичный или вторичный алкоксид), такой как этоксид натрия,предпочтительно в соответствующем раствори 003145 12 теле, таком как толуол. Наиболее предпочтительно Р=Х (где Х представляет собой алкокси),так как это позволяет избежать процессов переэтерификации. Соединения формулы (VIII) могут быть образованы из соединений формулы (IX) путем взаимодействия с амином NH(R3)(R4) необязательно в присутствии дополнительного основания (т.е. отличающегося от NH(R3)(R4, предпочтительно, в соответствующем растворителе,таком как толуол. "D" в соединениях (VIII) и(IX) представляет собой Сl или Вr. Если используется дополнительное основание, то оно или не взаимодействует с сульфонилхлоридным фрагментом (такое как оксид, карбонат или бикарбонат металла), или оно взаимодействует с сульфонилхлоридным фрагментом таким образом, чтобы активировать его для нуклеофильной атаки (например, третичный амин, такой как триэтиламин). Амин NH(R3)(R4) может также действовать как основание, в этом случае предпочтительно, чтобы присутствовало более одного эквивалента, более предпочтительно приблизительно 2 эквивалента (или более). Соединения формулы (IX) могут быть образованы из соединений формулы (X) в присутствии хлорирующего или бромирующего агента, такого как тионилхлорид или тионилбромид,более предпочтительно в присутствии галогенирующего катализатора, еще более предпочтительно тионилхлорид или тионилбромид в присутствии диметилформамида. Тионилхлорид/ бромид может также действовать как растворитель, но более предпочтительно реакцию проводят в соответствующем другом растворителе,таком как толуол. В таком случае требуются только стехиометрические количества тионилхлорида/бромида, предпочтительно как, по меньшей мере, 2 молярных эквивалента, более предпочтительно, по меньшей мере, 5 молярных эквивалента. Является возможным проводить четырехстадийное превращение (X) в (VI) (более конкретно (ХА) в (VIA) и (ХВ) в (IVB в одну объединенную стадию, без выделения промежуточного соединения, используя везде такой же растворитель (далее в данном документе "растворитель объединенной стадии"). Таким образом,если Х представляет собой алкокси, стадии (X)(VI) могут быть объединены вместе с применением одного растворителя, такого как не смешиваемый с водой инертный органический растворитель. Более предпочтительно, углеводородный растворитель (такой как толуол, ксилолы, анизол, хлорбензол, гексан, гептан, октан,нонан, декан, циклогексан, метилциклогексан),или простые эфиры (такие как дибутиловый эфир, дифениловый эфир), или кетоны (такие как метилизобутилкетон, метилэтилкетон) или сложные эфиры (такие как этилацетат, бутилацетат) или диметилформамид. Еще более предпочтительно, углеводородный растворитель(такой как толуол, ксилолы, анизол, хлорбензол,октан, нонан, декан, метилциклогексан), или простые эфиры (такие как дибутиловый эфир,дифениловый эфир), или сложные эфиры (такие как этилацетат, бутилацетат). Еще более предпочтительно, когда растворителем объединенной стадии является толуол. Промежуточное соединение формулы (X) образуется из соединения формулы (XI) или его соли в присутствии защитного формирующего агента, который образует защитную группу (Р) для карбоновой кислоты (т.е. с образованием группы -СОР). Предпочтительно, упомянутый защитный формирующий агент является этерифицирующим агентом, образующим сложный эфир карбоновой кислоты (где, например, Р является алкокси и защитный формирующий агент является спиртом), такой как эфир C1-6 карбоновой кислоты, который существует на протяжении всей схемы реакции и гидролизуется в основных условиях до функциональной группы карбоновой кислоты соединения (VI). Наиболее предпочтительно, если этерифицирующим агентом является этанол. Может быть уместным дополнительный растворитель, такой как толуол. Промежуточное соединение формулы (XI) образуется из 2-гидроксиникотиновой кислоты или ее соли в присутствии SО 3 (идеально, по меньшей мере, 1 молярного эквивалента SО 3),например, при использовании SО 3 в апротонном растворителе (например, в нитробензоле, нитрометане, 1,4-диоксане, дихлорметане), или в минеральной кислоте в качестве растворителя(например, в серной кислоте), или в жидкой карбоновой кислоте в качестве растворителя(например, в уксусной кислоте), или в THF, или в гептане. Еще более предпочтительно, если сульфонирующим агентом является олеум (SО 3 в серной кислоте), такой как приблизительно 20-30% олеум.(III) образуется в результате взаимодействия промежуточного соединения (VI) и соединения (V) в присутствии конденсирующего агента, такого как N,N'-карбонилдиимидазол, и подходящего растворителя, такого как этилацетат. Соединения общей формулы (V) получают в соответствии со способом, показанным в примере 1a-1f далее в данном документе (т.е. для образования соединения VA). Схема 2 иллюстрирует предпочтительное воплощение для образования соединения (VIA),где Х представляет собой алкокси (и Y в соединении VIIA представляет X), более предпочтительно, C1-6 первичную или вторичную алкоксигруппу, такую как этокси. Однако для других уходящих групп применим способ схемы 1. В соответствии со схемой 2, промежуточное соединение формулы (VIA) образуется из соединения формулы (VIIA) путем удаления защитной группы Р с помощью агента, удаляю 003145 14 щего защитные группы, преимущественно путем омыления в присутствии гидролизующего агента (как определено выше для схемы 1), такого как гидроксид натрия, предпочтительно в соответствующем растворителе, таком как вода и толуол. Промежуточное соединение формулы(VIIA) образуется из соединения формулы(VIIIA) в присутствии соответствующего C1-6 алкоксида (такого как первичный или вторичный алкоксид), предпочтительно алкоксида металла,где металл (Z) является таким, как определено ранее для ZOR, такого как этоксид натрия,предпочтительно в соответствующем растворителе, таком как толуол или ХН, где Х является таким, как определено ранее в данном документе. "D" в соединениях (VIIIA) и (IXA) представляет собой Сl или Вr. Промежуточное соединение формулы(VIIIA) образуется из соединения формулы(IXA) образуется из соединения формулы (ХА) в присутствии хлорирующего или бромирующего агента, как определено для такой же стадии в схеме 1, наиболее предпочтительно тионилхлорида или бромида/диметилформамида. Промежуточное соединение формулы(ХА) образуется из соединения формулы (XI) в присутствии агента, образующего защитную группу (Р) для карбоновой кислоты (т.е. образующего группу -СОР), как определено ранее в данном документе. Наиболее предпочтительно,если этерифицирующим агентом является этанол. Может быть уместным дополнительный растворитель, такой как толуол. Промежуточное соединение формулы (XI) образуется из 2-гидроксиникотиновой кислоты с использованием сульфонирующего агента (как определено ранее в данном документе), такой как 20-30% олеум. Опять же, четырехстадийное превращение(ХА) в (VIA) может быть объединено, (как установлено ранее в данном документе), без выделения промежуточного соединения, с использованием такого же растворителя. Здесь можно непосредственно применять список растворителей, приведенный для схемы 1. Наиболее предпочтительно, если растворителем является толуол. Например, после образования соединения(IXA), избыток хлорирующего/бромирующего агента может быть удален с помощью азеотропной перегонки при азетропной температуре указанного агента и растворителя объединенной стадии. После образования соединения (VIIIA),образовавшиеся HBr/HCl (т.е. HD) соли могут быть отмыты (в водной среде) или отфильтрованы из реакционного сосуда и остаток водного 15 растворителя удаляют с помощью азеотропной перегонки с некоторым количеством растворителя объединенной стадии. При образовании соединения (VIIA), если алкоксид, используемый для введения X, растворен в растворителе(таком как этанол), то этот растворитель может быть опять удален с помощью азеотропной перегонки с некоторым количеством растворителя объединенной стадии перед образованием соединения (VIA) для облегчения выделения. Если используется твердый алкоксид, то последняя стадия азеотропной отгонки не требуется. Наиболее предпочтительно, если растворителем для любой объединенной стадии схемы 1, и особенно схем 2 и 3, является толуол.(IIIA) образуется в результате взаимодействия промежуточного соединения (VIA) и 4 амино-5-этил-1-(2-пиридилметил)-1 Н-пиразол 3-карбоксамида (соединение VA) в присутствии конденсирующего агента, такого как гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида и желательно также в присутствии основания и/или катализатора. В одном примере конденсирующей системы, вначале активируется функциональная группа карбоновой кислоты(VIA) при использовании молярного эквивалента такого реагента, как N,N'-карбонилдиимидазол (в качестве конденсирующего агента) в подходящем растворителе, например этилацетате, при температуре, варьирующей приблизительно от комнатной до 80 С, с последующим взаимодействием промежуточного соединения имидазолида с (IXA) при приблизительно от 35 до 80 С. В другом примере, промежуточное соединение (VIA) может быть конденсировано с пиразолом (VA) в присутствии 1-гидроксибензотриазола, триэтиламина и гидрохлорида 1(3-диметиламинопропил)-3-этилкарбодиимида. Соединение (IB) (схема 3), где Х также представляет собой C1-6 алкокси (предпочтительно первичную или вторичную алкокси группу, такую как этокси), может быть образовано аналогично соединению (IA). Реагенты и т.д. для схемы 2 могут быть также непосредственно применены для схемы 3. Понятно, что соли соединений схем 1-3 могут быть образованы в соответствии с данным изобретением путем превращения соответствующего соединения в его соль (либо in situ,либо в отдельной стадии). Например, могут быть образованы основно-аддитивные соли соединений формул (VI) и (XI), и они являются соответствующими данному изобретению. Также в соответствии с данным изобретением могут быть образованы кислотно-аддитивные соли соединения формулы (I). В качестве иллюстрации, кислотноаддитивные соли соединения формулы (I) (более конкретно IA и IB) могут быть образованы в результате взаимодействия соединения формулы (I) с эквимолярным или избыточным количеством кислоты. Затем эта соль может быть оса 003145 16 ждена из раствора и выделена путем фильтрации, или растворитель может быть удален с помощью обычных средств. Типичные соли, которые могут применяться в схемах 1-3, приведены в PCT/IB 99/00519. Примером солей соединенийIA и IB являются тозилат и безилат соответственно. Подходящие защитные группы для использования в соответствии с данным изобретением могут быть обнаружены в "ProtectingGroups" под редакцией P.J. Kocienski, Thieme,New York, 1994 -см. особенно главу 4, стр. 118154 для защиты карбоксильной группы; и "Protective Groups in Organic Synthesis" 2-ое издание,T.W. GreeneP.G.M. Wutz, Wiley-Interscience(1991) - см. особенно главу 5 для защиты карбоксильной группы. Данное изобретение далее будет иллюстративно описано со ссылкой на следующие примеры. Пример 1.(1a) Этил 3-этил-1 Н-пиразол-5-карбоксилат. Этанольный раствор этоксида натрия (21% мас./мас.; 143 мл, 0,39 моль) добавляют по каплям при перемешивании, к охлаждаемому во льду раствору диэтилоксалата (59,8 мл, 0,44 моль) в абсолютном этаноле (200 мл) в атмосфере азота, и полученный раствор перемешивают в течение 15 мин. Затем добавляют по каплям бутан-2-он (39 мл, 0,44 моль), охлаждающую баню удаляют, реакционную смесь перемешивают в течение 18 ч при комнатной температуре и затем в течение 6 ч при 40 С, затем вновь вводят охлаждающую баню. Затем добавляют по каплям ледяную уксусную кислоту (25 мл, 0,44 моль), полученный раствор перемешивают в течение 30 мин при 0 С, добавляют по каплям гидразингидрат (20 мл, 0,44 моль), затем реакционную смесь оставляют нагреваться до комнатной температуры и поддерживают так в течение 18 ч, перед упариванием при пониженном давлении. Остаток распределяют между дихлорметаном (300 мл) и водой (100 мл), затем органическую фазу отделяют, промывают водой(2 х 100 мл), сушат (Nа 2SO4) и концентрируют при пониженном давлении, получая указанное в заголовке соединение (66,0 г).(CDCl3): 1,04 (3 Н, т), 1,16 (3 Н, т), 2,70(1b) 3-Этил-1 Н-пиразол-5-карбоновая кислота. Водный раствор гидроксида натрия (10 М; 100 мл, 1,0 моль) добавляют по каплям к перемешиваемой суспензии соединения, указанного в заголовке примера (4 а) (66,0 г, 0,39 моль), в метаноле, и полученный раствор нагревают при температуре кипения с обратным холодильником в течение 4 ч. Охлажденную реакционную смесь концентрируют при пониженном давлении до са. 200 мл, разбавляют водой (200 мл) и 17 эту смесь промывают толуолом (3 х 100 мл). Полученную водную фазу подкисляют концентрированной соляной кислотой до рН 4 и белый осадок собирают и сушат отсасыванием, получая указанное в заголовке соединение (34,1 г).(DMSOd6): 1,13 (3 Н, т), 2,56 (2 Н, кв),6,42(1 Н, с).(1 с) 3-Этил-4-нитро-1 Н-пиразол-5-карбоновая кислота. Дымящуюся серную кислоту (17,8 мл) добавляют по каплям при перемешивании к охлаждаемой во льду дымящейся азотной кислоте(16,0 мл), полученный раствор нагревают до 50 С, порциями добавляют 3-этил-1 Н-пиразол 5-карбоновую кислоту в течение 30 мин, при этом поддерживая температуру реакции ниже 60 С. Полученный раствор нагревают в течение 18 ч при 60 С, оставляют охлаждаться, затем выливают на лед. Указанное в заголовке соединение получают в виде коричневого твердого вещества (64%).(DMSOd6): 1,18 (3 Н, т), 2,84 (2 Н, м), 13,72(1d) 3-Этил-4-нитро-1 Н-пиразол-5-карбоксамид. Раствор соединения, указанного в заголовке примера (1 с) (15,4 г, 0,077 моль), в тионилхлориде (75 мл) нагревают при температуре кипения с обратным холодильником в течение 3 ч и затем охлажденную реакционную смесь упаривают при пониженном давлении. Остаток подвергают азеотропной перегонке с тетрагидрофураном (2 х 50 мл) и затем суспендируют в тетрагидрофуране (50 мл), затем при перемешивании суспензию охлаждают во льду и обрабатывают газообразным аммиаком в течение 1 ч. Добавляют воду (50 мл) и полученную смесь упаривают при пониженном давлении, получая твердое вещество, которое, после перетирания с водой и высушиванием отсасыванием, предоставляется как указанное в заголовке соединение в виде белого твердого вещества (90%).(DMSOd6): 1,17 (3 Н, т), 2,87 (2 Н, м), 7,40(1 е) 5-Этил-4-нитро-1-(2-пиридилметил)1H-пиразол-3-карбоксамид. Карбонат цезия (1,414 кг, 4,34 моль) добавляют к суспензии соединения, указанного в заголовке примера (1d) (800 г, 4,34 моль), в ацетонитриле (51) и смесь нагревают до 60 С. Добавляют 2-хлорметилпиридин (664,7 г, 5,23 моль) и реакционную смесь нагревают при 70 С в течение 7 ч, затем добавляют воду (9,5 л) и реакционную смесь охлаждают до 10 С. Гранулирование этой смеси дает осадок, который фильтруют и сушат, получая 3-этил-4-нитро-1(пиридин-2-ил)метилпиразол-5-карбоксамид(367 г). К фильтрату добавляют хлорид натрия(4 х 1,75 л). Объединенные органические экс 003145 18 тракты упаривают для удаления приблизительно 10 л растворителя, к горячему раствору (6976 С) добавляют в течение 35 мин толуол (5,6 л) и смесь оставляют охлаждаться. Полученную суспензию гранулируют при 10 С в течение 30 мин, фильтруют, твердое вещество промывают смесью этилацетат:толуол (50:50) (600 мл) и сушат (60 С), получая указанное в заголовке соединение (624 г, 52%) в виде светлокоричневого твердого вещества.(DMSOd6): 1,08 (3 Н, т), 3,02 (2 Н, кв), 5,53(1f) 4-Амино-5-этил-1-(2-пиридилметил)1H-пиразол-3-карбоксамид (соединение VA). Смесь катализатора Линдлара (2 г) и соединения, указанного в заголовке примера (1 е)(20 г, 72,7 ммоль), в этаноле (160 мл) гидрируют в течение 48 ч при 345 кПа (50 psi (фунтов/кв. дюйм и 50 С, затем охлаждают и фильтруют. Фильтрат объединяют с IMS, промывают (50 мл) фильтровальную прокладку и концентрируют при пониженном давлении до объема 100 мл. Оставшийся этанол удаляют путем упаривания, и заменяют на этилацетат до тех пор, пока не будет достигнута температура головной фракции, равная 77 С. Охлажденную смесь гранулируют при 4 С, фильтруют и сушат, получая указанное в заголовке соединение (13,17 г, 73%) в виде светло-коричневого твердого вещества,(58,1 кг) при 50 С в течение 1 ч. Это вызывает повышение температуры до 82 С. Реакционную смесь нагревают дополнительно до 140 С. После поддерживания этой температуры в течение 12 ч содержимое реактора охлаждают до 15 С и фильтруют. Отфильтрованную массу затем повторно суспендируют в acetone (33 кг) при комнатной температуре, фильтруют и сушат, получая указанное в заголовке соединение (35,3 кг,83%) в виде белого твердого вещества. Температура разложения 273 С.(DMSOd6): 7,93 (1H, д), 8,42 (1 Н, д). m/z(2,5 л) при перемешивании и нагревают до 80 С. Через 30 мин 0,5 л растворителя отгоняют, затем заменяют свежим этанолом (0,5 л) и снова нагревают до 80 С. После дополнительных 60 мин 1,0 л растворителя отгоняют, затем заме 19 няют свежим этанолом (1,0 л) и снова нагревают до 80 С. После дополнительных 60 мин отгоняют 1,0 л растворителя, реакционную смесь охлаждают до 22 С и перемешивают в течение 16 ч. Осажденный продукт фильтруют, промывают этанолом (0,5 л) и сушат при 50 С под вакуумом, получая указанное в заголовке соединение (416 г, 74%) в виде белого твердого вещества. Температура разложения 237 С.(DMSOd6): 1,25 (3 Н, т), 4,19 (2 Н, кв), 7,66(1,0 мл) при пeремешивании. Реакционную смесь затем нагревают при температуре кипения с обратным холодильником в течение 2,5 ч. Основную массу тионилхлорида удаляют под вакуумом, остальной тионилхлорид удаляют путем азеотропной перегонки с толуолом, получая неочищенное указанное в заголовке соединение(1 Н, д), 9,09 (1 Н, д). Оно непосредственно принимает участие в следующей стадии.(1j) Этил 2-xлор-5-(4-этил-1-пиперазинилсульфонил)никотиноат (cоединение VIIIA). Неочищенный этил 2-хлор-5-хлорсульфоникотиноат (IXA) (30,7 г, 0,1 моль предположительно) растворяют в этилацетате (150 мл) при премешивании, затем охлаждают во льду. К этому раствору осторожно, в течение 30 мин,поддерживая внутреннюю температуру ниже 10 С, добавляют раствор N-этилпиперазина(11,4 г, 0,1 моль) и триэтиламина (22,5 г, 0,22 моль) в этилацетате (50 мл). После завершения добавления реакционную смесь оставляют нагреваться до 22 С и перемешиваться в течение 1 ч. Твердое вещество отфильтровывают и оставшийся фильтрат концентрируют под вакуумом,получая неочищенное указанное в заголовке соединение (37,1 г, 103%) в виде неочищенной желтой смолы.(СDСl3): 1,10 (3 Н, т), 1,42 (3 Н, м), 2,50(10,2 г, 0,15 моль) добавляют порциями, поддерживая температуру ниже 20 С. Реакционную смесь затем перемешивают при температуре(180 мл). Фильтрат затем нагревают до 40 С в течение 1 ч. Этанол (180 мл) затем отгоняют при атмосферном давлении и оставшийся водный раствор оставляют охлаждаться до температуры окружающей среды. Осажденный продукт затем отфильтровывают, промывают водой и сушат под вакуумом при 50 С, получая указанное в заголовке соединение (12,6 г, 34%) в виде светло-коричневого твердого вещества. Т.пл. 66-68 С.(CDCl3): 1,04 (3 Н, т), 1,39 (3 Н, т), 1,45VIA: X=OEt). Этил 2-этокси-5-(4-этил-1-пиперазинилсульфонил)никотиноат (VIIA) (10,2 г, 0,0275 моль) растворяют в толуоле (50 мл) и к нему добавляют раствор гидроксида натрия (1,1 г,0,0275 моль) в воде (20 мл). Эту двухфазную смесь затем энергично перемешивают при температуре окружающей среды в течение ночи. Водную фазу отделяют и доводят до рН=5,6 путем добавления концентрированной соляной кислоты. Осажденный продукт суспендируют при охлаждении льдом в течение 15 мин,фильтруют, промывают водой и сушат под вакуумом при 50 С, получая указанное в заголовке соединение в виде не совсем белого твердого вещества. Т.пл. 206-207 С.(CDCl3): 1,25 (3 Н, т), 1,39 (3 Н, т), 2,82[М+Н]+, 100%. Рассчитано для C14H22N3 О 5S: 344). Эта стадия (1l) уже раскрыта в способе получения 23 PCT/IB 99/00519 (в данном документе включена в качестве ссылки) и полученный выход составляет 88%.(1m) N-[3-карбамоил-5-этил-1-(2-пиридилметил)-1H-пиразол-4-ил]-2-этокси-5-(4-этил-1 пиперазинилсульфонил)никотинамид (cоединение IIIA: X=OEt). В реакционный сосуд загружают 2-этокси 5-(4-этил-1-пиперазинилсульфонил)никотиновую кислоту (VIA) (0,875 кг, 2,55 моль), затем этилацетат (7 л, 8 мл/г) и 2 мл/г отгоняют при атмосферном давлении для обеспечения сухой реакционной системы. Суспензию охлаждают до комнатной температуры в атмосфере азота и одной порцией добавляют карбонилдиимидазол(0,43 кг, 2,65 моль). Суспензию нагревают до 35 С и выдерживают в течение получаса. Реакционную смесь далее нагревают до 45-50 С и выдерживают в течение еще получаса. Реакционную смесь затем нагревают при температуре 21 кипения с обратным холодильником, перемешивают при температуре кипения с обратным холодильником в течение одного часа. После подтверждения завершения образования имидазола реакционную смесь охлаждают до 45-50 С в атмосфере азота и одной порцией загружают 4-амино-5-этил-1-(2-пиридилметил)-1 Нпиразол-3-карбоксамид (VA) (0,59 кг, 2,42 моль), затем повторно нагревают с обратным холодильником и дополнительно 1 мл/г отгоняют при атмосферном давлении. Реакционную смесь перемешивают при температуре кипения с обратным холодильником в течение 16 ч. Реакционную смесь охлаждают до 10-15 С и гранулируют в течение 1 ч. Реакционную суспензию фильтруют и промывают (этилацетатом),сушат под вакуумом при 50 С, получая указанное в заголовке соединение (1,252 кг, 90,7%) в виде белого твердого вещества. Т.пл. 178-179 С.(СDСl3): 1,04 (3 Н, т), 1,06 (3 Н, т), 1,59(1n) 4-6-Этокси-5-[3-этил-6,7-дигидро-7 оксо-2-(2-пиридилметил)-2 Н-пиразоло[4,3-d] пиримидин-5-ил]-3-пиридилсульфонил-1 этилпиперазин (cоединение IIА: X=OEt). В сосуд загружают раствор этоксида калия(86 г, 0,25 моль, 24% мас./мас. в этаноле) и добавляют этанол (235 мл). К реакционной смеси добавляют этилацетат (10,8 г) при условиях окружающей среды. К смеси растворителей добавляют одной порцией N-[3-карбамоил-5-этил 1-(2-пиридилметил)-1 Н-пиразол-4-ил]-2-этокси 5-(4-этил-1-пиперазинилсульфонил)никотинамид (IIIA) (70 г, 0,122 моль) и реакционную смесь перемешивают при условиях окружающей среды. Реакционную смесь нагревают в герметично закрытом сосуде до температуры 120 С, получая внутреннее давление, составляющее приблизительно 50-60 p.s.i, затем давление увеличивают до 80 p.s.i путем применения давления азота, и реакционную смесь перемешивают в течение 8 ч. Через 8 ч реакционную смесь охлаждают и затем отгоняют этанол при атмосферном давлении до объема, составляющего приблизительно 720 мл (3 мл/г). К этанольному раствору добавляют этилацетат (840 мл) и затем отгоняют этанол при атмосферном давлении до объема, составляющего 1920 мл (8 мл/г). Добавляют разбавленный водный раствор соляной кислоты для доведения рН реакционной смеси приблизительно от рН=13 до рН=8. Его добавляют в течение 30 мин. Эту смесь перемешивают в течение 5 мин, нагревают до 50 С и фазы разделяют. К этилацетатному слою добавляют воду (140 мл), перемешивают, нагревают до 50 С и фазы разделяют. Этилацетатную фазу охлаждают от 50 до 0-5 С в течение 2 ч и 22 перемешивают в течение еще 1 ч, твердое вещество отфильтровывают, промывают этилацетатом (3) 0-5 С и сушат под вакуумом при 60 С,получая указанное в заголовке соединение(83%) в виде белого твердого вещества. Т.пл. 178-180 С.(CDCl3): 1,02 (3 Н, т), 1,30 (3 Н, т), 1,58(1 о) 1-Этил-4-5-[3-этил-6,7-дигидро-7 оксо-2-(2-пиридилметил)-2 Н-пиразоло[4,3-d]пиримидин-5-ил]-6-(2-метоксиэтокси)-3-пиридилсульфонилпиперазин (соединение IA из соединения IIА: X=OEt). 4-6-Этокси-5-[3-этил-6,7-дигидро-7-оксо 2-(2-пиридилметил)-2 Н-пиразоло[4,3-d]пиримидин-5-ил]-3-пиридилсульфонил-1-этилпиперазин (IIА) (100 г, 0,18 моль) добавляют одной порцией к 2-метоксиэтанолу (600 мл) и реакционную смесь перемешивают в условиях окружающей среды в течение 30 мин, получая гетерогенную смесь. К этой смеси добавляют 2 метоксиэтилацетат (42,8 г, 0,36 моль). К реакционной смеси добавляют одной порцией при перемешивании т-бутоксид калия (30,52 г, 0,27 моль), было обнаружено, что это вызывало увеличение температуры на 20-30 С. Реакционную смесь перемешивают до тех пор, пока температура не перестанет меняться. Реакционную смесь нагревают до температуры кипения с обратным холодильником (115-125)С и поддерживают при температуре кипения с обратным холодильником в течение 15 мин. Температура кипения с обратным холодильником должна составлять 123-125 С, если она не равна этой температуре, растворитель отгоняют до тех пор,пока температура не установится в нужном интервале и поддерживают эту температуру кипения с обратным холодильником в течение 4 ч. Реакционную смесь затем охлаждают до 60 С и оставшийся растворитель отгоняют под вакуумом до объема 3 мл/г. Этот вязкий раствор перемешивают и охлаждают до температуры окружающей среды. К вязкому раствору добавляют воду (533 мл) и перемешивают, получая гомогенный раствор. Воду (266 мл) и концентрированную соляную кислоту (25,82 г) предварительно смешивают и добавляют в течение 1 ч для доведения рН реакционной смеси приблизительно от рН=13 до рН=8. Смесь охлаждают от температуры окружающей среды до 0-5 С и перемешивают в течение дополнительного часа,отфильтровывают твердое вещество, промывают водой (200 мл) и сушат под вакуумом при 60 С, получая указанное в заголовке соединение(98,12 г, 92,7%) в виде белого твердого вещества. Т.пл. 157-158 С.(11,41 г, 0,02 моль) добавляют в атмосфере азота одной порцией к 2-метоксиэтанолу (45 мл) и реакционную смесь перемешивают при температуре окружающей среды в течение 10 мин. Этилпивалат (6,5 г, 0,05 моль) затем вводят в 2 метоксиэтанол (5 мл). К реакционной смеси при перемешивании добавляют порциями в течение 10 мин трет-бутоксид калия (5,4 г, 0,048 моль),как обнаружено, это происходит в экзотермическом режиме (20-50 С). Реакционную смесь перемешивают до тех пор, пока температура не перестанет изменяться. Реакционная смесь представляет собой прозрачный бледно-желтый раствор. Реакционную смесь нагревают при температуре кипения с обратным холодильником (115-125 С) и поддерживают при температуре кипения с обратным холодильником в течение 5 ч. Оставшийся растворитель затем отгоняют под вакуумом до объема 2 мл/г. Вязкий раствор перемешивают и охлаждают до температуры окружающей среды. К вязкому раствору добавляют воду (70 мл, 6 мл/г) и перемешивают,получая гомогенный раствор. Добавляют разбавленный водный раствор соляной кислоты для доведения рН реакционной смеси приблизительно от рН=13 до рН=8. Это добавление производят медленно, в течение одного часа. Осажденную полученную суспензию охлаждают во льду и гранулируют в течение 1 ч, фильтруют,промывают водой и сушат под вакуумом при 50 С, получая указанное в заголовке соединение(9,7 г, 83,3%) в виде белого твердого вещества. Т.пл. 157-158 С.(CDCl3): 1,02 (3 Н, т), 1,30 (3 Н, т), 2,40(1 Н, д), 8,62 (1 Н, д), 8,97 (1 Н, д). m/z (Обнаружено: 583 [М+Н]+, 100%. Рассчитано для С 27 Н 35N8O5S: 583). Вышеописанные реакции могут быть также повторены с различными агентами, улавливающими гидроксиды. Если этилпивалат (2,5 молярных эквивалента) используют для 2 003145 24 метоксиэтилацетата (2,5 молярных эквивалента) или 2-метоксинивалата (2,5 молярных эквивалента), полученный выход составляет 76,3% и 84,8% соответственно. Пример 3. 1-Этил-4-5-[3-этил-6,7-дигидро-7-оксо-2-(2-пиридилметил)-2 Н-пиразоло[4,3d]пиримидин-5-ил]-6-(2-метоксиэтокси)-3 пиридилсульфонилпиперазин (cоединение IА) из N-[3-карбамоил-5-этил-1-(2-пиридилметил)1 Н-пиразол-4-ил]-2-этокси-5-(4-этил-1-пиперазинилсульфонил)никотинамид (соединение IIIA:N-[3-карбамоил-5-этил-1-(2-пиридилметил)-1 Н-пиразол-4-ил]-2-этокси-5-(4-этил-1 пиперазинилсульфонил)никотинамид (IIIA) (500 г, 0,876 моль) добавляют в атмосфере азота одной порцией к 2-метоксиэтанолу (2,5 л, 5 мл/г) и реакционную смесь перемешивают при температуре окружающей среды в течение 10 мин. К реакционной смеси при перемешивании порциями добавляют трет-бутоксид калия (236 г,2,103 моль) в течение 10 мин, как обнаружено,это происходит в экзотермическом режиме (2050 С). Реакционную смесь перемешивают до тех пор, пока температура не перестанет изменяться. Реакционная смесь представляет собой прозрачный бледно-желтый раствор. Этилпивалат (285 г, 2,190 моль) затем вводят в 2 метоксиэтанол (0,3 л). Реакционную смесь нагревают при температуре кипения с обратным холодильником (115-125 С) и поддерживают при температуре кипения с обратным холодильником в течение 6 ч. Оставшийся растворитель затем отгоняют под вакуумом до объема 2 мл/г. Вязкий раствор перемешивают и охлаждают до температуры окружающей среды. К вязкому раствору добавляют воду (3 л, 6 мл/г) и перемешивают, получая гомогенный раствор. Добавляют разбавленный водный раствор соляной кислоты для доведения рН реакционной смеси приблизительно от рН=13 до рН=8. Это добавление производят медленно, в течение 1 ч. Затем добавляют метилизобутилкетон (3 л) и смесь нагревают до 55 С. Нижний водный слой отделяют, в то время как оставшийся органический слой промывают теплой водой (500 мл). Берут органический слой и отгоняют 1 л под вакуумом. Оставшийся раствор охлаждают до 50 С, полученный осадок гранулируют в течение 1 ч, фильтруют, промывают метилизобутилкетоном (1 л) и сушат под вакуумом при 50 С, получая указанное в заголовке соединение(400,3 г, 78,4%) в виде белого твердого вещества. Т.пл. 157-158 С.(CDCl3): 1,02 (3 Н, т), 1,30 (3 Н, т), 2,40 25 Пример 4. 2-Этокси-5-(4-этил-1-пиперазинилсульфонил)никотиновая кислота (соединение VIA) - объединенный способ в толуоле из этил 2-гидрокси-5-сульфоникотиноата (соединение ХА, Х=ОЕt). В особенно предпочтительном воплощении данного изобретения соединение ХА образуется с помощью объединенного способа, посредством этого дополнительно уменьшается общее число стадий, с образованием соединенияIA из коммерчески доступных исходных веществ. Этил 2-гидрокси-5-сульфоникотиноат(1,77 л) и затем добавляют тионилхлорид (1,06 кг, 8,93 моль) и диметилформамид (71,3 мл). Затем суспензию при перемешивании нагревают при температуре кипения с обратным холодильником в течение 3 ч, получая желтый раствор. Тионилхлорид (2,87 л) затем отгоняют при непрерывном замещении толуолом (2,15 л). Бледно-желтый раствор затем охлаждают до 10 С и добавляют при перемешивании по каплям раствор N-этилпиперазина (198,9 г, 1,66 моль) и триэтиламина (392,2 г, 3,88 моль) в толуоле (700 мл) в течение 90 мин, поддерживая температуру реакционной смеси ниже 10 С. Реакционную смесь перемешивают при температуре окружающей среды в течение 18 ч, затем промывают водой (2 х 700 мл) и насыщенным солевым раствором (2 х 350 мл). Толуольную фазу азеотропно сушат путем отгонки 1750 мл, при непрерывной замене на сухой толуол (1750 мл). Оставшийся коричневый раствор охлаждают до 10 С и порциями добавляют этоксид натрия(178,0 г), поддерживая температуру ниже 10 С. Реакционную смесь затем перемешивают при 10 С в течение 1 ч, затем оставляют нагреваться до температуры окружающей среды и перемешивают в течение 18 ч. Гидроксид натрия (34,9 г) растворяют в воде (1,5 л), затем добавляют к толуольной смеси и 2-фазовую смесь энергично перемешивают в течение 18 ч при 40 С. После охлаждения до температуры окружающей среды водную фазу отделяют. К ней добавляют концентрированную соляную кислоту до получения рН=3, что вызывает осаждение светлокоричневого твердого вещества, которое гранулируют в течение 2 ч при охлаждении во льду. Осадок фильтруют, промывают водой (300 мл) и сушат под вакуумом при 50 С, получая указанное в заголовке соединение (338,4 г, 57,4%) в виде не совсем белого твердого вещества. Т.пл. 206-207 С.(CDCl3): 1,25 (3 Н, т), 1,39 (3 Н, т), 2,82 26 1-пиперазинилсульфонил)-2-(2-метоксиэтокси) никотинамид (соединение IVА). 5-(4-Этил-1-пиперазинилсульфонил)-2-(2 метокси)никотиновую кислоту (37,3 г, 0,1 моль) получают из свободного основания способа получения 29 PCT/IB/00519 и загружают, с последующим загружением этилацетата (7 л, 8 мл/г), в реакционный сосуд и 2 мл/г этилацетата отгоняют при атмосферном давлении, для гарантированного получения сухой реакционной системы. Взвесь охлаждают до комнатной температуры в атмосфере азота и одной порцией добавляют карбонилдиимидазол (16,87 г, 0,104 моль). Взвесь нагревают до 35 С и выдерживают в течение 30 мин. Далее реакционную смесь нагревают до 45-50 С и выдерживают в течение еще 30 мин. Реакционную смесь затем нагревают при температуре кипения с обратным холодильником, перемешивают при нагревании при температуре кипения с обратным холодильником в течение 1 ч. После подтверждения завершения образования имидазола реакционную смесь охлаждают до 40-43 С в атмосфере азота и затем одной порцией загружают 4-амино-5-этил-1-(2 пиридилметил)-1 Н-пиразол-3-карбоксамид(способ получения 40 PCT/IB 99/00519; 0,59 кг,2,42 моль), перед повторным нагреванием при температуре кипения с обратным холодильником, и дополнительно отгоняют 1 мл/г при атмосферном давлении. Реакционную смесь перемешивают при нагревании при температуре кипения с обратным холодильником в течение 20 ч. Реакционную смесь упаривают, получая масло, которое кристаллизуется при стоянии в течение ночи. Полученное твердое вещество растворяют в 5 мл/г дихлорметана и промывают 5 мл/г воды. Органический слой упаривают, получая указанное в заголовке соединение (42 г,70% выход), в виде белого кристаллического твердого вещества. Т.пл. 145-148 С.(CDCl3): 1,02 (3 Н, т), 1,07 (3 Н, т), 1,64N-[3-карбамоил-5-этил-1-(2 пиридилметил)-1 Н-пиразол-4-ил]-5-(4-этил-1 пиперазинилсульфонил)-2-(2-метоксиэтокси) никотинамид (4,1 г, 6,8 ммоль), затем 3-метил-3 пентанол (21 мл, 5 мл/г), т-бутоксид калия (1,5 г, 13,6 ммоль) и метоксиэтилпивалат (2,18 г,13,6 моль) для того, чтобы образовать раствор, и затем нагревают при температуре кипения с обратным холодильником. После 20 ч нагрева 27 ния при температуре кипения с обратным холодильником жидкостная хроматография образца показала, что образовано 70% указанного в заголовке соединения (в соответствии со ссылкой на стандарт указанного в заголовке соединения,структура которого подтверждена с помощью(6 а) 5-Этил-1-метил-4-нитро-1 Н-пиразол 3-карбоксамид. Взвесь 3-этил-4-нитро-1 Н-пиразол-5-карбоксамида (100 г, 0,54 моль) в ацетоне (1 л) при комнатной температуре обрабатывают карбонатом калия (150 г, 1,08 моль) в воде (0,7 л), получая желтый раствор. Этот раствор затем обрабатывают метилйодидом (37 мл, 0,58 моль) и реакционную смесь оставляют перемешиваться в течение приблизительно 48 ч. Реакционную смесь фильтруют и жидкости разделяют, водный слой отбрасывают и органическую фазу концентрируют до осаждения твердого вещества. Взвесь охлаждают, гранулируют, фильтруют и промывают ацетоном (100 мл). Полученное твердое вещество сушат под вакуумом, получая белое твердое вещество, 47,5 г, 39%. Т.пл.=188 С.(DMSOd6): 1,19 (3 Н, т), 2,95 (2 Н, кв), 3,85(6b) 4-Aмино-5-этил-1-метил-1 Н-пиразол 3-карбоксамид (соединение VB). 5-Этил-1-метил-4-нитро-1 Н-пиразол-3 карбоксамид (1,0 кг, 5,05 моль) добавляют к (15 л) IMS и смесь гидрируют при комнатной температуре, 55 p.s.i. в течение ночи над 5% Pd/C(15% мас./мас.). Катализатор отфильтровывают и фильтрат упаривают, получая твердое вещество, которое суспендируют в изопропиловом спирте (750 мл), нагревают при температуре кипения с обратным холодильником, охлаждают до температуры окружающей среды, фильтруют и промывают изопропиловым спиртом (1 л). Твердое вещество сушат в вакууме в течение ночи, получая 529 г, 84,5% от не совсем белого до розового твердого вещества, Т.пл.=155 С.(CDCl3): 1,19 (3 Н, т), 2,60 (2 Н, кв), 3,76(6 с) N-(3-Карбамоил-5-этил-1-метил-1 Нпиразол-4-ил)-2-этокси-5-(4-этил-1-пиперазинилсульфонил)никотинамид (соединение IIIB). В реакционный сосуд загружают 2-этокси 5-(4-этил-1-пиперазинилсульфонил)никотиновую кислоту (17,3 г, 0,05 моль), затем этилацетат (137 мл) и 2 мл/г отгоняют при атмосферном давлении для получения сухой системы. Взвесь охлаждают до комнатной температуры в атмо 003145 28 сфере азота и одной порцией добавляют 1,1 карбонилдиимидазол (8,51 г, 52,0 ммоль). Взвесь нагревают до 35 С и выдерживают в течение получаса. Реакционную смесь нагревают до 45-50 С и выдерживают в течение дополнительных 30 мин. Реакционную смесь затем нагревают при температуре кипения с обратным холодильником, перемешивают при нагревании при температуре кипения с обратным холодильником в течение 0,5 ч. После подтверждения завершения образования имидазола реакционную смесь охлаждают до 40-43 С в атмосфере азота и одной порцией загружают 4-амино-5 этил-1-метил-1 Н-пиразол-3-карбоксамид (7,98 г,47,5 ммоль), перед повторным нагреванием при температуре кипения с обратным холодильником. Отгоняют еще 1 мл/г при атмосферном давлении. Реакционную смесь перемешивают при температуре кипения с обратным холодильником в течение 6 ч. Реакционную взвесь охлаждают и гранулируют при 10-15 С в течение одного часа, фильтруют и промывают 1,5 мл/г(2% воды в этилацетате). Твердое вещество сушат в вакууме в течение ночи при 50 С, получая 21,2 г, 90,5%, белого кристаллического твердого вещества. Т.пл.=180 С.(CDCl3): 1,04 (3 Н, т), 1,24 (3 Н, т), 1,59(R)-2-гидрокси-3-метоксипропан (200 мл) перемешивают в реакционном сосуде в атмосфере азота при температуре окружающей среды. Трет-бутоксид калия (10,9 г, 97,2 ммоль) добавляют порциями в течение 2 мин. Добавляют этилпивалат (15,4 мл, 0,1 моль) и реакционную смесь нагревают до 120 С. Растворитель непрерывно отгоняют, добавляя при этом дополнительное количество этилпивалата (0,3 моль) и(R)-2-гидрокси-3-метоксипропана для того, чтобы поддержать объем растворителя равным 200 мл. Приблизительно через девять часов реакци 29 онную смесь охлаждают до температуры окружающей среды и разбавляют CH2Cl2 (200 мл). рН доводят до 8 путем медленного добавления 6 М НСl (водн.). К полученной суспензии добавляют воду (200 мл) и органическую фазу отделяют. Водную фазу экстрагируют СН 2 Сl2 (2 х 150 мл) и объединенные органические фазы переносят в отдельный сосуд. Летучий органический растворитель удаляют и заменяют этилметилкетоном (200 мл) и затем поддерживают при 50 С в течение 30 мин. К этому раствору добавляют по каплям бензолсульфоновую кислоту (7,7 г,48,6 ммоль) в виде раствора в этилметилкетоне(80 мл). Реакционную смесь перемешивают при 80 С в течение 90 мин, после чего этилметилкетон (200 мл) отгоняют. Полученную взвесь охлаждают до температуры окружающей среды и гранулируют в течение ночи. Фильтрация, промывание охлажденным во льду этилметилкетоном (50 мл) и высушивание под вакуумом дают безилатную соль (R)-1-этил-4-[3-(3-этил-6,7 дигидро-2-метил-7-оксо-2 Н-пиразоло-[4,3-d] пиримидин-5-ил)-2-(2-метокси-1-метилэтокси)5-пиридилсульфонил]пиперазина, 24,0 г, 79% в виде бледно-желтого твердого вещества,Т.пл.=212-214 С.(СDСl3): 1,16 (3 Н, т), 1,25 (3 Н, т), 1,31(1 Н, с, шир.). m/z Обнаружено: 517,91 [М-Н]+,(рассчитано для C23H33N7O5S: 519,63, соль фрагментируется до свободного основания в МС). Таким образом, выход конечного продукта(IA) при использовании агента, улавливающего гидроксид, является очень хорошим. Кроме того, в соответствии с предпочтительным воплощением данного изобретения может быть непосредственно предоставлено вещество, подходящее для клинического применения. Кроме того, в соответствии с данным изобретением, промежуточные соединения VII и VI(более конкретно VIIA, VIA и VIIB, VIB) могут быть получены из коммерчески доступных исходных веществ (2-гидроксиникотиновой кислоты) с лучшим выходом, чем в соответствующей последовательности реакций PCT/IB 99/00519. Например, соединение VII (где Р и Х представляют собой OEt) образуется с выходом 14,5% в способе получения 18 PCT/IB 99/00519(т.е. в результате последовательности реакций способов получений 1, 3, 5, 7 и 18), тогда как такое же соединение в соответствии с настоящим изобретением получают с выходом 23%(см. примеры от 1g до 1k). Более предпочтительно, если все реакции, участвующие в получении соединений VII и VI, или их часть, могут быть объединены вместе в соответствии с данным изобретением, с получением еще большего выхода. Таким образом, соединение VI (где Х 30 представляет собой OEt) получают с выходом 35% (см. пример 4 данного документа). Кроме того, схема реакции настоящего изобретения является более безопасной и более дешевой в исполнении, и в случае объединенного способа также включает меньше стадий (и требует меньше времени). В предпочтительном аспекте соединения формулы (I), (IА) и (IB) получают из никотиновой кислоты в соответствии со схемами 1-3. Таким образом, в предпочтительном аспекте данного изобретения предоставляется способ получения соединения формулы (I), (IA) и (IB), этот способ начинается со взаимодействия 2-гидроксиникотиновой кислоты или ее соли в присутствии SО 3 в растворителе с образованием соединения формулы (XI). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы где R представляет собой C1-C6 алкил, необязательно замещенный одним или двумя заместителями, выбранными из С 3-C5 циклоалкила, ОН,C1-C4 алкокси, бензилокси, NR5R6, фенила, фуранила и пиридинила; С 3-С 6 циклоалкил; 1-(C1C4 алкил)пиперидинил; тетрагидрофуранил или тетрагидропиранил, и где указанные C1-С 6 алкильные или C1-C4 алкоксильные группы необязательно замещены галогеналкилом;R1 (который может быть связан с любым азотом пиразольного кольца) представляет собой C1-C3 алкил, необязательно замещенный фенилом, Het или N-связанной гетероциклической группой, выбранной из пиперидинила и морфолинила, и где указанная фенильная группа необязательно замещена C1-C4 алкилом, который необязательно замещен галогеналкилом или галогеналкокси; или C1-C4 алкокси; или галогеном, или CN;R2 представляет собой C1-C6 алкил; и Het представляет собой С-связанную 6 членную гетероциклическую группу, содержащую один или два атома азота, необязательно в виде его моно-N-оксида, или С-связанную 5 членную гетероциклическую группу, содержащую два или три атома азота, где любая из указанных гетероциклических групп является необязательно замещенной C1-C4 алкилом или C1C4 алкокси или NHR7, где R7 представляет собой Н, C1-C4 алкил или C1-C4 алканоил;R3 и R4 вместе с атомом азота, к которому они присоединены, образуют 4-R8-пипepaзи 31 нильнyю группу, необязательно замещенную одной или двумя C1-C4 алкильными группами, и необязательно в виде ее 4-N-оксида;R8 представляет собой Н; C1-C4 алкил, необязательно замещенный одним или двумя заместителями, выбранными из ОН, NR5R6,CONR5R6, фенила, необязательно замещенногоC1-C4 алкокси, бензодиоксолила и бензодиоксанила; C3-C6 алкенил; пиридинил или пиримидинил; причем указанный способ включает взаимодействие соединения формулы (II), (III) или(IV) в присутствии -OR и агента, улавливающего гидроксид, или, в случае соединений формулы (IV), взаимодействие в присутствии вспомогательного основания и агента, улавливающего гидроксид (т.е. -OR заменяют на вспомогательное основание), где Х представляет собой уходящую группу и R1-R4 такие, как определены выше. 2. Способ по п.1 получения соединения формул (IA) и (IB) включающий взаимодействие соединения формулы (IIА), (IIIA) или (IVA), и (IIB), (IIIB) или в присутствии -OR и агента, улавливающего гидроксид, или, альтернативно, в случае соединений формул (IVA) и (IVB) взаимодействие в присутствии -OR и вспомогательного основания, где OR в случае образования соединения(IA) представляет собой СH3 О(СН 2)2O-, и OR в случае образования соединения (IB) представляет собой (R)-СН 3 ОСН 2 СН(СН 3)O-, и где Х в формулах (IIA)-(IIIA) и в формулах (IIB)-(IIIB) представляет собой уходящую группу. 3. Способ по п.1 или 2, где агентом, улавливающим гидроксид, является сложный эфир. 4. Способ по любому из пп.1-3, где агентом, улавливающим гидроксид, является сложный эфир формулыTOC(O)W где ОТ представляет собой OR, или остаток объемного спирта, или ненуклеофильного спирта, или ТОН представляет собой спирт, который может быть азеотропно удален в процессе реакции; и C(O)W представляет собой остаток карбоновой кислоты. 5. Способ по любому из пп.1-4, где Х выбран из группы, состоящей из арилсульфонилокси, C1-C4 алкилсульфонилокси, нитро- или галогензамещенного бензолсульфонилокси, C1C4 перфторалкилсульфонилокси, необязательно замещенного ароилокси, C1-C4 перфторалканоилокси, C1-C4 алканоилокси, галогена; диазония;C1-C6 алкокси. 7. Способ по любому из пп.1-6, где -OR присутствует вместе со вспомогательным основанием. 8. Способ по п.7, где вспомогательное основание выбрано из группы, состоящей из стерически затрудненного основания, гидрида металла, оксида металла, карбоната металла и бикарбоната металла. 9. Способ по п.8, где стерически затрудненным основанием является металлическая соль стерически затрудненного спирта или амина. 10. Способ по п.9, где реакцию проводят в инертном растворителе или в ROH или в смеси обоих. 11. Способ по п.10, где растворитель является выбранным из группы, состоящей из ROH, 003145 34 вторичного или третичного C4-C12 алканола, С 3C12 циклоалканола, третичного C4-C12 циклоалканола, вторичного или третичного (C3-C7 циклоалкил)С 2-С 6 алканола, C3-C9 алканона, 1,2-диметоксиэтана, 1,2-диэтоксиэтана, диглима, тетрагидрофурана, 1,4-диоксана, толуола, ксилола,хлорбензола, 1,2-дихлорбензола, ацетонитрила,диметилсульфоксида, сульфолана, диметилформамида, N-метилпирролидин-2-она, пиридина, и их смеси. 12. Способ по п.11, где растворитель представляет собой ROH. 13. Способ по любому из пп.2-12, где соединение (IA) образуется в результате взаимодействия соединений (IIА) или (IIIA)a) с 2-метоксиэтанолом и вспомогательным основанием необязательно в инертном растворителе и в присутствии указанного улавливающего агента; илиb) с ZО(СН 2)2OСН 3 и вспомогательным основанием в 2-метоксиэтаноле или инертном растворителе, или в обоих в присутствии указанного улавливающего агента; илиc) с ZО(СН 2)2OСН 3 и 2-метоксиэтанолом,или инертным растворителем, или обоими в присутствии указанного улавливающего агента,и где Z является металлом. 14. Способ по любому из пп.2-12, где соединение (IB) образуется в результате взаимодействия соединения (IIB) или (IIIB)a) с (R)-СН 3 ОСН 2 СН(СН 3)ОН и вспомогательным основанием, необязательно в инертном растворителе и в присутствии указанного улавливающего агента; илиb) с (R)-СН 3 ОСН 2 СН(СН 3)OZ и вспомогательным основанием в (R)-СН 3 ОСН 2 СН(СН 3) ОН или инертном растворителе, или в обоих в присутствии указанного улавливающего агента; илиc) с (R)-СН 3 ОСН 2 СН(СН 3)OZ и (R)СН 3 ОСН 2 СН(СН 3)ОН, или инертным растворителем, или с обоими в присутствии указанного улавливающего агента, и где Z является металлом. 15. Способ по любому из пп.1-14, где соединение формулы (III) образуется в результате сочетания соединения формулы (V) с соединением формулы (VI) или его солью. 16. Способ по п.15, где соединение формулы (VI) или его соль получают из соединения формулы (VII)(VI), где Х представляет собой арилсульфонилокси, C1-C4 алкилсульфонилокси, C1-C4 перфторалкилсульфонилокси, ароилокси, C1-C4 перфторалканоилокси, C1-C4 алканоилокси, C1C4 алкилсульфонилокси с остатком четвертичного аммония или галогенсульфонилокси, включает взаимодействие соединения формулы (VII),где Y и V представляют собой ОН, в присутствии соответствующего сульфонирующего, арилирующего или ацилирующего агента X;(VI), где Х представляет собой Сl, включает взаимодействие соединения формулы (VII), где(VI), где Х представляет собой диазоний, включает взаимодействие соединения формулы (VII),где Y представляет собой NH2, V представляет собой ОН, с азотистой кислотой;(VI), где Х представляет собой (диарилсульфонил)амино, включает взаимодействие соединения формулы (VII), где Y=NH2 и V=OH, в присутствии соответствующего сульфонирующего агента;(VI), где Х представляет собой OR, который является C1-6 алкокси, включает взаимодействие соединения формулы (VII), где V представляет собой Р, где Р представляет собой защитную группу и Y представляет собой C1-6 первичный или вторичный алкокси, с агентом, удаляющим защитные группы. 17. Способ по п.16, где соединение формулы (VII) получают из соединения формулы где D представляет собой Сl или Вr и Р представляет собой защитную группу,(a) который в случае соединений формулы(VII), где Y представляет собой ОН и V представляет собой ОН, включает взаимодействие соединения формулы (VIII) с гидролизующим агентом и необязательно дополнительным удаляющим защитную группу агентом, если Р не 36 удаляется под действием гидролизующего агента;(VII), где Y представляет собой NH2 и V представляет собой ОН, включает взаимодействие соединения формулы (VIII) с аммонирующим агентом с образованием промежуточного соединения формулы (VII), где Y представляет собой NH2 и V представляет собой Р (защитную группу), и взаимодействие указанного промежуточного соединения (VII) с агентом, удаляющим защитные группы; и(VII), где Y представляет собой OR, который представляет собой C1-6 алкокси, и V представляет собой Р, включает взаимодействие соединения формулы (VIII) в присутствии -OR; 18. Способ по п.17, где соединение формулы (VIII) получают из соединения формулы (IX) в присутствии амина NH(R3)(R4), где R3 и R4 такие, как определены в п.1 где D и Р такие, как определены в п.17. 19. Способ по п.18, где соединение формулы (IX) получают взаимодействием соединения формулы (X) с хлорирующим или бромирующим агентом где Р такой, как определен в п.17. 20. Способ по п.19, где соединение формулы (X) получают из соединения формулы (XI) в присутствии агента, образующего защитную группу (Р) на карбоновой кислоте 21. Способ по п.20, где соединение формулы (XI) получают из 2-гидроксиникотиновой кислоты или ее соли в присутствии SО 3 в растворителе. 22. Способ по п.21, где SО 3 находится в апротонном растворителе, минеральной кислоте или жидкой карбоновой кислоте. 23. Способ по любому из пп.16-22, где соединения формулы (VI), (VII) и (VIII) представляют собой (VIA), (VIIA) и (VIIIА) соответственно 38 единения (XI) или его соли с этанолом, с образованием защитной группы OEt. 26. Способ по любому из пп.23-25, где две или более последовательных стадий образования (VIA), (VIIA) или (VIIIA) проводятся в толуоле и объединяются вместе в одну объединенную стадию без выделения промежуточного соединения. 27. Способ по любому из пп.1-26 получения соединения формулы (I), (IA) или (IВ),включающий в качестве исходного соединения коммерчески доступную 2-гидроксиникотиновую кислоту или ее соль, и взаимодействие ее в присутствии SO3 в растворителе, с образованием соединения формулы (XI). 28. Соединение (IVA)(VIIIA) образуется в соответствии с п.18 в результате взаимодействия соединения (IX) с Nэтилпиперазином или его солью. 24. Способ по п.23, где Х представляет собой OEt, и соединение (VIA) образуется в результате взаимодействия соединения (VIIA) с агентом, удаляющим защитные группы, и соединение (VIIA) образуется в результате взаимодействия соединения (VIIIA) в присутствии(X) образуется в результате взаимодействия со где R имеет значения, указанные в п.1. 29. Соединение (IVB)
МПК / Метки
МПК: C07D 401/12, C07D 401/14, C07D 487/04
Метки: пиридилсульфонильных, пиразоло[4,3-d]пиримидин-7-он-3, соединений, промежуточных, способ, получения
Код ссылки
<a href="https://eas.patents.su/21-3145-sposob-polucheniya-pirazolo43-dpirimidin-7-on-3-piridilsulfonilnyh-soedinenijj-i-ih-promezhutochnyh-soedinenijj.html" rel="bookmark" title="База патентов Евразийского Союза">Способ получения пиразоло[4,3-d]пиримидин-7-он-3- пиридилсульфонильных соединений и их промежуточных соединений</a>
Способ получения циклопропанкарбоновых кислот и их промежуточных соединений

Номер патента: 683
Опубликовано: 28.02.2000
Авторы: Колинн-Андерсен Ханс, Винкельманн Иб, Клемменсен Пер Дауселль
МПК: C07D 307/93, C07C 61/35
Метки: промежуточных, получения, соединений, кислот, циклопропанкарбоновых, способ
Формула / Реферат:
1. Способ получения соединений общей формулы I где R' представляет Н, а два атома водорода циклопропанового кольца находятся в цис-положении по отношению друг к другу, включающий взаимодействие между соединением общей формулы II и соединением СF3-CClХ2, где Х представляет атом галогена, в частности атом хлора или брома, в инертной среде в присутствии Zn и при подходящей температуре от 0 до 150шС, предпочтительно от 20 до 100шС, в...
Способ получения 5-[2-этокси-5-(4-метилпиперазин-1-илсульфонил)-фенил]-1-метил-3-н-пропил,- 1,6-дигидро-7н-пиразоло[4,3-d]пиримидин-7-она

Номер патента: 102
Опубликовано: 27.08.1998
Авторы: Вуд Элберт Шо, Данн Питер Джеймз
МПК: C07D 487/04
Метки: 1,6-дигидро-7н-пиразоло[4,3-d]пиримидин-7-она, способ, получения, 5-[2-этокси-5-(4-метилпиперазин-1-илсульфонил)-фенил]-1-метил-3-н-пропил
Формула / Реферат:
1. Способ получения 5-[2-этокси-5-(4-метилпиперазин-1-ил-сульфонил)-фенил]-1-метил-3-н-пропил-1,6-дигидро-7Н-пиразоло-[4,3-d] пиримидин-7-она формулы (I) отличающийся тем, что соединение формулы (II) подвергают реакции циклизации в щелочной, нейтральной или кислой среде. 2. Способ по п.1, отличающийся тем, что циклизацию осуществляют в присутствии основания, предпочтительно в растворителе, возможно в присутствии перекиси водорода или...
Способы получения промежуточных соединений для пестицидов.

Номер патента: 955
Опубликовано: 28.08.2000
Авторы: Клавель Жан-Луи, Вилкинсон Джон Херри, Робертс Девид Ален, Хоукинс Девид Вилльям
МПК: C07D 231/38, C07C 253/00
Метки: пестицидов, способы, соединений, получения, промежуточных
Формула / Реферат:
1. Способ получения соединения формулы (I) где R представляет собой алкил с нормальной или разветвленной цепью, содержащий от 1 до 18 атомов углерода, или его соли, который включает реакцию цианоацетата формулы (II) RO2C-CH2CN (II) где R определен выше, с солью цианисто-водородной кислоты и формальдегидом или источником последнего. 2. Способ по п.1, отличающийся тем, что соль цианисто-водородной кислоты представляет собой соль...
Получение промежуточных соединений эндотелина реакцией ассиметрического сопряженного присоединения с использованием хиральной добавки.

Номер патента: 2056
Опубликовано: 24.12.2001
Авторы: Ксу Фенг, Щаен Дэвид М., Тилльер Ричард Д.
МПК: C07D 213/55, A61K 31/44, C07B 37/02...
Метки: соединений, реакцией, присоединения, эндотелина, использованием, сопряженного, хиральной, получение, ассиметрического, добавки, промежуточных
Формула / Реферат:
1. Способ получения соединения формулы I где представляет а) 5- или 6-членный гетероциклил, содержащий одну, две или три двойные связи, по крайней мере одну двойную связь, и 1, 2 или 3 гетероатома, выбранных из О, N и S, незамещенный или замещенный одним, двумя или тремя заместителями, выбранными из группы, состоящей из ОН, CO2R4, Br, Cl, F, I, СF3, N(R5)2, C1-C8 алкокси, C1-C8 алкила, C2-C8 алкенила, C2-C8 алкинила или C3-C8 циклоалкила,...
(1r, 2s, 4r)-(-)-2-[n,n-(диметиламиноэтокси)]-2-фенил-1,7,7-триметилбицикло [2,2,1]гептан высокой степени чистоты, его фармацевтически приемлемые кислотные аддитивные соли, способ получения этих соединений и их применение, а также лекарственные средства, содержащие одно или более из этих соединений

Номер патента: 2164
Опубликовано: 24.12.2001
Авторы: Шимиг Дьюла, Мезеи Тибор, Немет Норберт, Надь Калман, Суладьи Янош, Будаи Золтан, Сабо Тибор, Порч-Маккаи Марта, Лукач Дьюла, Краснаи Дьёрдь
МПК: C07C 217/12, A61K 31/13, A61P 25/04...
Метки: получения, 4r)-(-)-2-[n,n-(диметиламиноэтокси)]-2-фенил-1,7,7-триметилбицикло, соединений, степени, одно, этих, лекарственные, приемлемые, аддитивные, средства, соли, применение, высокой, фармацевтически, более, способ, кислотные, 2,2,1]гептан, содержащие, чистоты, также
Формула / Реферат:
1. (1R,2S,4R)-(-)-2-[N,N-(диметиламиноэтокси)]-2-фенил-1,7,7-триметилбицикло-[2,2,1]гептан по формуле и его фармацевтически приемлемые кислотные аддитивные соли, отличающиеся тем, что они содержат не более 0,2% (1R,3S,4R)-3-[2-N,N-(диметиламиноэтил)]-1,7,7-триметилбицикло[2,2,1]гептан-2-она по формуле или его фармацевтически приемлемой кислотной аддитивной соли. 2....
Предыдущий патент: Способ лечения диабета тиазолидиндионом и метформином
Следующий патент: Новые цианоиндольные соединения, являющиеся ингибиторами повторного захвата серотонина, способ их получения и фармацевтические композиции, содержащие их
Случайный патент: Способ транспортировки природного газа по магистральному трубопроводу