Новые соединения пирролидина и тиазолидина, способ их получения и фармацевтические композиции, которые их содержат
Номер патента: 9292
Опубликовано: 28.12.2007
Авторы: Арле Элизабет, Комбетт Мюриелль, Де Нантей Гийом, Бенуа Ален







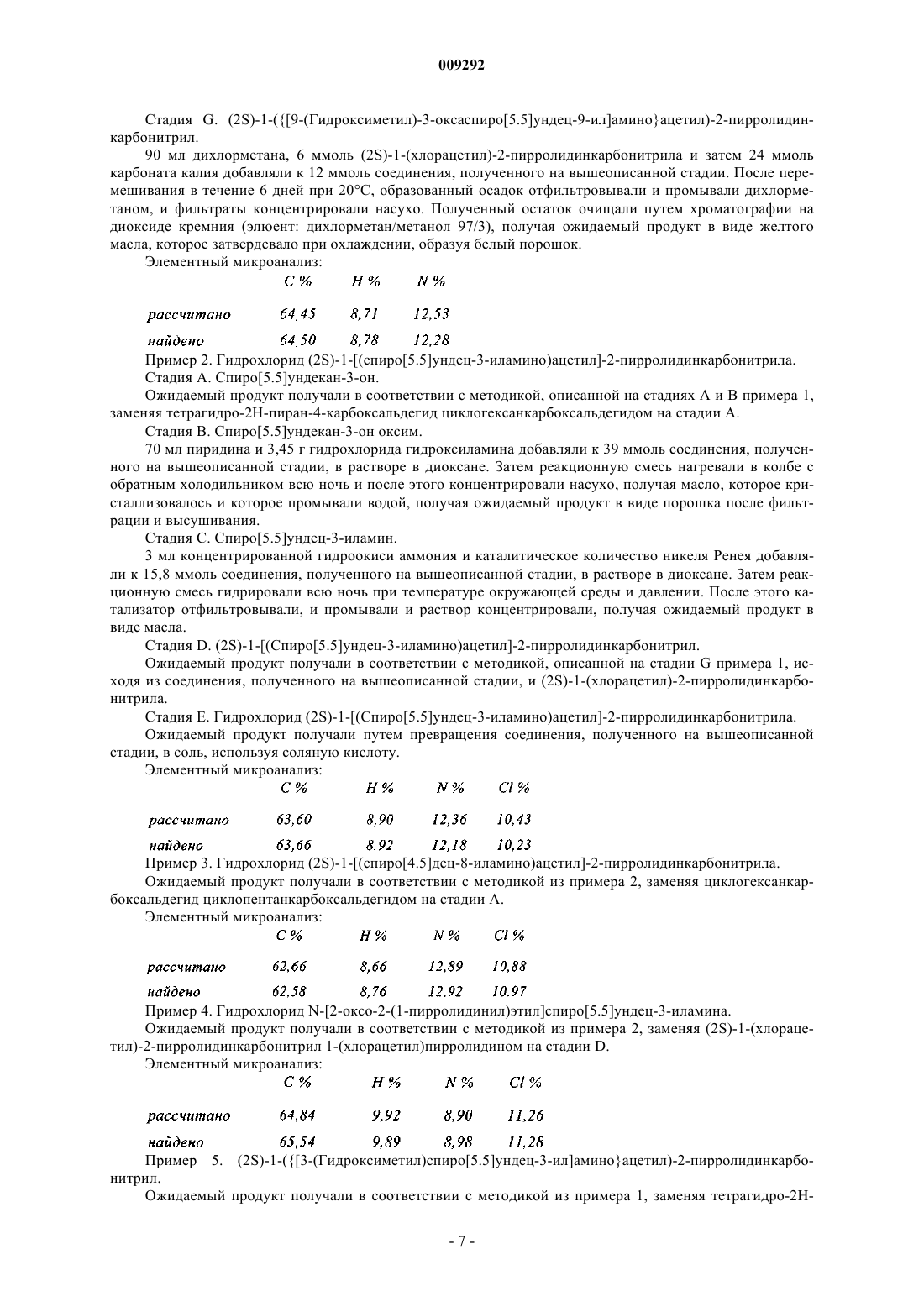
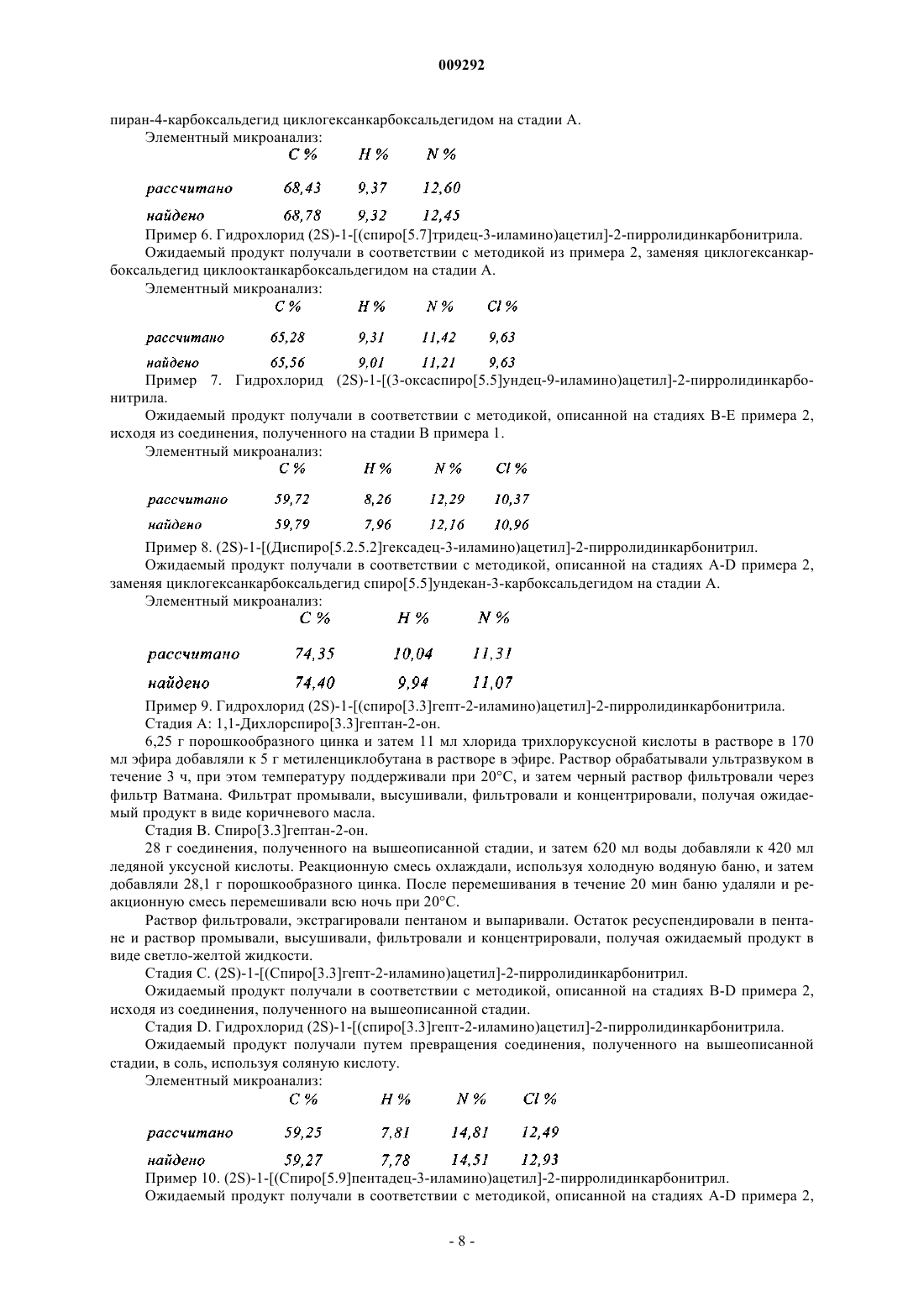
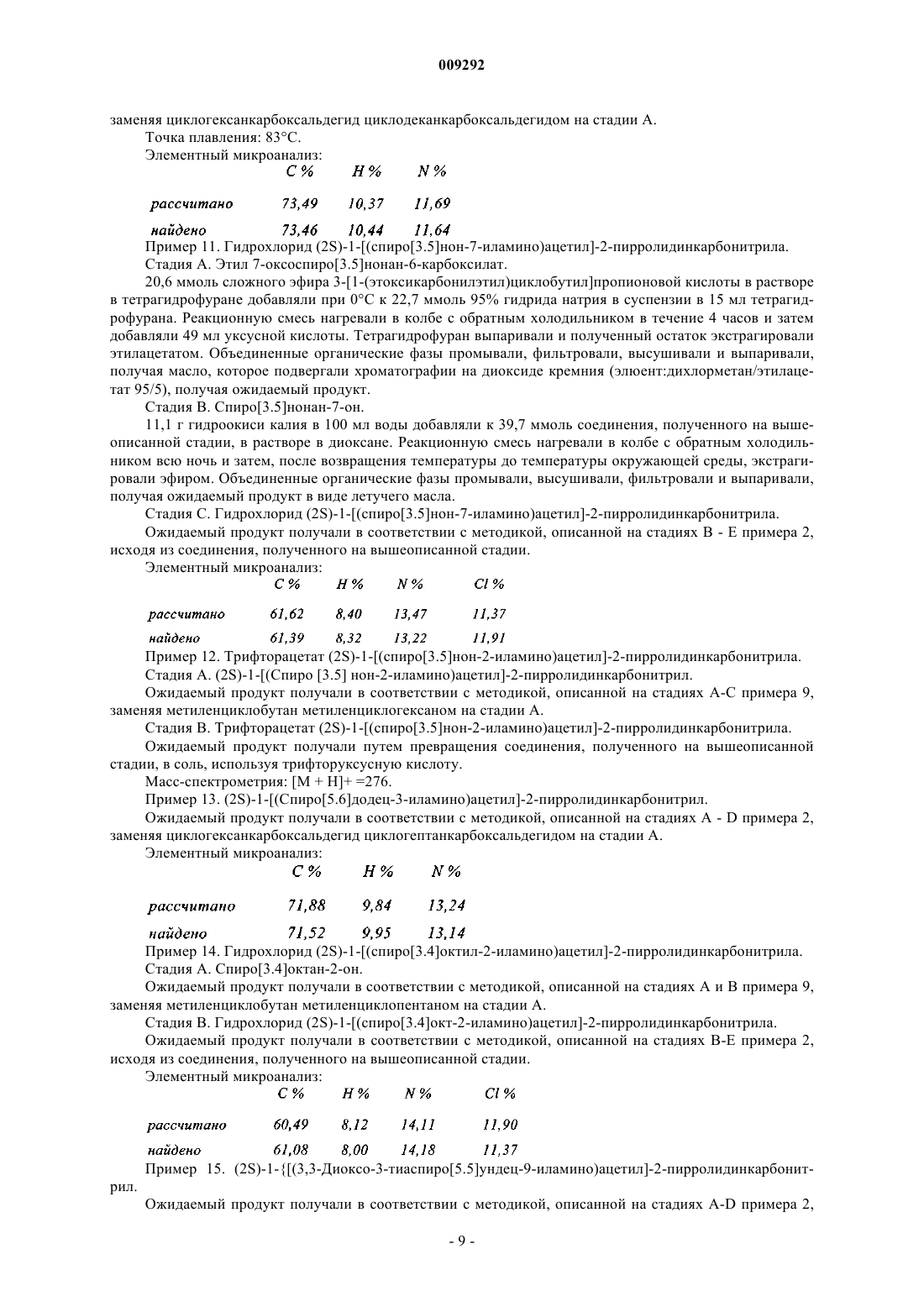
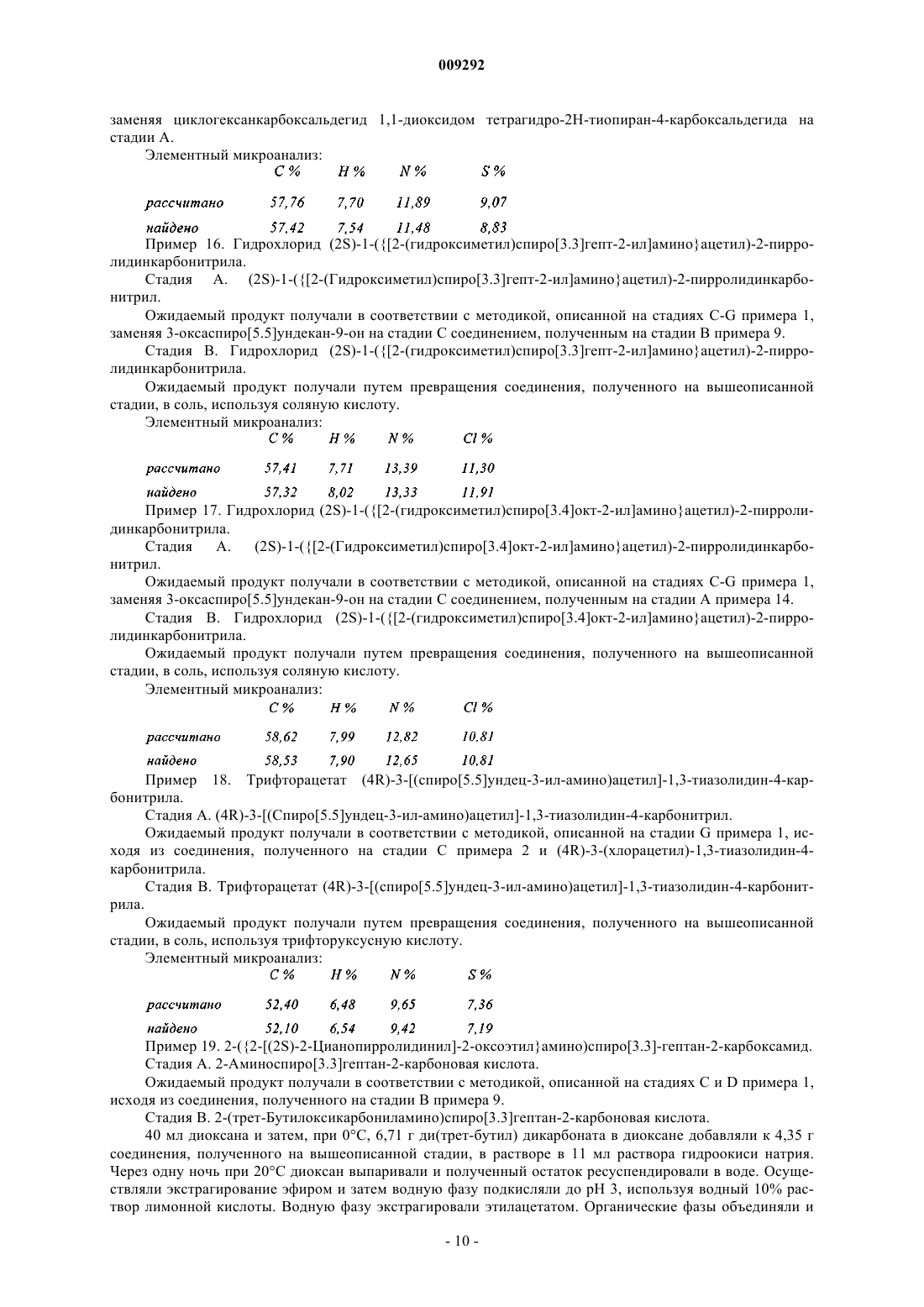







Формула / Реферат
1. Соединение пирролидина или тиазолидина формулы (I)

в которой
X1 представляет собой атом или группу, выбранную из CR4aR4b, О, S(O)q1 и NR5,
где R4a и R4b, которые могут быть одинаковыми или разными, каждый представляет собой атом водорода или С1-С6-алкильную группу,
или R4a и R4b вместе с атомом углерода, к которому они присоединены, образуют С3-С7-циклоалкильную группу,
q1представляет собой 0 или 2,
и R5 представляет собой атом водорода или C1-С6-алкильную группу, замещенную гидроксигруппой,
m1 представляет собой 0 или целое число от 1 до 4 включительно,
m2 представляет собой целое число от 1 до 4 включительно,
n1 и n2, которые могут быть одинаковыми или разными, каждый представляет собой целое число от 1 до 3 включительно,
R1 представляет собой атом водорода, карбамоил или С1-С6-алкил, замещенный гидроксигруппой,
R2 представляет собой атом водорода,
Ak представляет собой С1-С4-алкиленовую цепь,
р представляет собой 1,
R3 представляет собой атом водорода или цианогруппу,
Х2 и Х3, которые могут быть одинаковыми или разными, каждый представляет собой или S(O)q2 группу, в которой q2 представляет собой О, или CR6aR6b группу, в которой R6a и R6b, которые могут быть одинаковыми или разными, каждый представляет собой атом водорода или атом галогена, или R6a представляет собой атом водорода, а R6b представляет собой гидроксигруппу,
его изомеры, если они существуют, и его соли присоединения с фармацевтически приемлемой кислотой.
2. Соединение формулы (I) по п.1, в которой X1 представляет собой атом кислорода или -СН2- группу.
3. Соединение формулы (I) по п.1 и 2, в которой m1 и m2 каждый представляет собой 1 или 2.
4. Соединение формулы (I) по любому из пп.1-3, в которой n1 и n2 каждый представляет собой 1 или 2 и являются одинаковыми.
5. Соединение формулы (I) по п.1, в которой

представляет собой группу, выбранную из


6. Соединение формулы (I) по любому из пп.1-5, в которой R2 представляет собой атом водорода.
7. Соединение формулы (I) по любому из пп.1-6, в которой Ak представляет собой -СН2- группу.
8. Соединение формулы (I) по любому из пп.1-7, в которой р представляет собой 1.
9. Соединение формулы (I) по любому из пп.1-8, в которой R3 представляет собой цианогруппу.
10. Соединение формулы (I) по любому из пп.1-9, в которой Х2 представляет собой CR6aR6b группу.
11. Соединение формулы (I) по любому из пп.1-10, в которой Х3 представляет собой CR6aR6b группу.
12. Соединение формулы (I) по любому из пп.9, 10 или 11, где конфигурация углерода, несущего группу R3, представляет собой (S)-конфигурацию.
13. Соединение формулы (I) по п.9, в которой Х2 или Х3 представляет собой группу S(O)q2 и конфигурация углерода, несущего группу R3, представляет собой (R)-конфигурацию.
14. Соединение формулы (I) по п.1, выбранное из следующих соединений
(2S)-1-({[9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]амино}ацетил)-2-пирролидинкарбонитрила, и его солей присоединения с фармацевтически приемлемой кислотой;
(2S)-1-({[3-(гидроксиметил)спиро[5.5]ундец-3-ил]амино}ацетил)-2-пирролидинкарбонитрила, и его солей присоединения с фармацевтически приемлемой кислотой;
(2S)-1-({[2-(гидроксиметил)спиро[3.4]окт-2-ил]амино}ацетил)-2-пирролидинкарбонитрила, и его солей присоединения с фармацевтически приемлемой кислотой;
(4R)-3-[(спиро[5.5]ундец-3-иламино)ацетил]-1,3-тиазолидин-4-карбонитрила, и его солей присоединения с фармацевтически приемлемой кислотой;
2-({2-[(2S)-2-цианопирролидинил]-2-оксоэтил}амино)спиро[3.3]гептан-2-карбоксамида, и его солей присоединения с фармацевтически приемлемой кислотой;
(2S)-1-({[(2-(гидроксиметил)-7-оксаспиро[3.5]нон-2-ил]амино}ацетил)-2-пирролидинкарбонитрила, и его солей присоединения с фармацевтически приемлемой кислотой;
(2S,4S)-4-фтор-1-({[9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]амино}ацетил)-2-пирролидинкарбонитрила, его (2S,4R) изомера, его (2R,4R) изомера и солей присоединения с фармацевтически приемлемой кислотой;
(2S)-4,4-дифтор-1-({[9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]амино}ацетил)-2-пирролидинкарбонитрила, и его солей присоединения с фармацевтически приемлемой кислотой;
и (4R)-3-({[9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]амино}ацетил)-1,3-тиазолидин-4-карбонитрила, и его солей присоединения с фармацевтически приемлемой кислотой.
15. Способ синтеза соединений пирролидина или тиазолидина формулы (I) по п.1, исходя из соединения формулы (III)

в которой X1, m1, m2, n1, n2 и R1 имеют значения, указанные в п.1, которое подвергают реакции с соединением формулы (IV)

в которой Ak, р, R3, Х2 и Х3 имеют значения, указанные в п.1, и Y1 представляет собой уходящую группу, с получением соединения формулы (I)

в которой X1, m1, m2, n1, n2, R1, Ak, p, R3, Х2 и Х3 имеют значения, указанные в п.1, которое очищают в соответствии с обычным способом очистки, необязательно разделяют на оптические изомеры в соответствии с обычным способом разделения и превращают, если это является желательным, в соли присоединения с фармацевтически приемлемой кислотой.
16. Фармацевтическая композиция, обладающая активностью ингибитора дипептидил-пептидазы IV, содержащая в качестве активного компонента соединение по любому из пп.1-14 в сочетании с одним или несколькими фармацевтически приемлемыми, инертными, нетоксичными носителями.
17. Применение соединения по любому из пп.1-14 при приготовлении лекарственных средств для использования в качестве ингибиторов дипептидил-пептидазы IV.
18. Применение соединения по любому из пп.1-14 при приготовлении лекарственных средств для использования при лечении непереносимости глюкозы и нарушений, связанных с гипергликемией.
19. Применение соединения по любому из пп.1-14 при приготовлении лекарственных средств для использования при лечении диабета II типа.
Текст
009292 Настоящее изобретение относится к новым соединениям пирролидина и тиазолидина, к способу их получения, фармацевтическим композициям, которые их содержат, и к их применению в качестве ингибиторов дипептидил-пептидазы IV (DPP IV). Дипептидил-пептидаза IV представляет собой мембранную сериновую протеазу, которая находится в различных тканях человека и вовлечена в различные патологии. В частности, было показано, что DPP IV отвечает за инактивацию GLP-1 (глюкагон-подобный пептид-1). GLP-1 является важным стимулятором секреции инсулина в поджелудочной железе и поэтому оказывает прямое положительное влияние на уровень глюкозы в крови. Следовательно, ингибирование DPP IV является чрезвычайно перспективным подходом при лечении непереносимости глюкозы и нарушений, связанных с гипергликемией, таких, например, как инсулин-независимый диабет (диабет II типа) или ожирение. В литературе уже были описаны ингибиторы DPP IV, в частности амидные соединения в патентной заявке ЕР 0490379 и в журнале Adv. Exp. Med. Biol. 1997, 421, 157-160, карбаматные соединения в патентной заявке DE 19826972, -аминосоединения в патентной заявке ЕР 1258476 и сульфоновые соединения в патентной заявке ЕР 1 245 568. Соединения по изобретению обладают ингибирующими свойствами по отношению к дипептидилпептидазе IV, что делает их особенно пригодными для лечения непереносимости глюкозы и нарушений,связанных с гипергликемией. В частности, настоящее изобретение относится к соединениям формулы (I) в которой X1 представляет собой атом или группу, выбранную из CR4aR4b,O, S(O)q1 и NR5,в которой R4a и R4b, которые могут быть одинаковыми или разными, каждый представляет собой атом водорода или линейную или разветвленную C1-С 6- алкильную группу,или R4a и R4b вместе с атомом углерода, к которому они присоединены, образуют С 3-С 7 циклоалкильную группу,q1 представляет собой 0, 1 или 2, иR5 представляет собой атом водорода или линейную или разветвленную С 1-С 6-алкильную группу,необязательно замещенную гидроксигруппой,m1 представляет собой 0 или целое число от 1 до 4 включительно,m2 представляет собой целое число от 1 до 4 включительно,n1 и n2, которые могут быть одинаковыми или разными, каждый представляет собой целое число от 1 до 3 включительно,R1 представляет собой атом водорода или группу, выбранную из карбокси, линейного или разветвленного C1-С 6-алкоксикарбонила, карбамоила, необязательно замещенного 1 или 2 линейными или разветвленными C1-C6-алкильными группами, и линейного или разветвленного C1-С 6-алкила, необязательно замещенного гидрокси группой или аминогруппой, необязательно замещенной 1 или 2 линейными или разветвленными С 1-С 6-алкильными группами,R2 представляет собой атом водорода или линейную или разветвленную C1-С 6-алкильную группу,Ak представляет собой линейную или разветвленную С 1-С 4-алкиленовую цепь, необязательно замещенную одним или несколькими атомами галогена, предпочтительно атомами фтора,р представляет собой 0, 1 или 2,R3 представляет собой атом водорода или цианогруппу,Х 2 и Х 3, которые могут быть одинаковыми или разными, каждый представляет собой или S(O)q2 группу, в которой q2 представляет собой 0, 1 или 2, илиCR6aR6b группу, в которой R6a и R6b, которые могут быть одинаковыми или разными, каждый представляет собой атом водорода или атом галогена, предпочтительно атом фтора, или R6a представляет собой атом водорода и R6b представляет собой гидрокси группу,к их оптическим изомерам, если они существуют, и к солям присоединения с фармацевтически приемлемой кислотой. Среди фармацевтически приемлемых кислот можно отметить, но не ограничиваясь только ими, соляную кислоту, бромисто-водородную кислоту, серную кислоту, фосфорную кислоту, уксусную кислоту,трифторуксусную кислоту, молочную кислоту, пировиноградную кислоту, малоновую кислоту, янтарную кислоту, глутаровую кислоту, фумаровую кислоту, винную кислоту, малеиновую кислоту, лимонную кислоту, аскорбиновую кислоту, метансульфоновую кислоту, камфорную кислоту, щавелевую кислоту.X1 предпочтительно представляет собой атом кислорода или -СН 2- группу;m1 и m2 предпочтительно каждый представляет собой 1 или 2;n1 и n2 предпочтительно каждый представляет собой 1 или 2, и предпочтительно являются одинаковыми. предпочтительно представляет собой группу, выбранную изR2 предпочтительно представляет собой атом водорода;Ak предпочтительно представляет собой -СН 2- группу; р предпочтительно представляет собой 1;R3 предпочтительно представляет собой цианогруппу. В этом случае, конфигурация углерода, который с ней связан, предпочтительно представляет собой (S)-конфигурацию, если Х 2 и Х 3 каждый представляет собой CR6aR6b группу, и (R)-конфигурацию, если Х 2 или Х 3 представляет собой S(O)q2 группу. Х 2 и Х 3 предпочтительно каждый представляет собой CR6aR6b группу. Предпочтительными соединениями формулы (I) являются(2S)-1-([9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]аминоацетил)-2-пирролидинкарбонитрил,и его соли присоединения с фармацевтически приемлемой кислотой;(2S)-1-([3-(гидроксиметил)спиро[5.5]ундец-3-ил]аминоацетил)-2-пирролидинкарбонитрил, и его соли присоединения с фармацевтически приемлемой кислотой;(2S)-1-([2-(гидроксиметил)спиро[3.4]окт-2-ил]аминоацетил)-2-пирролидинкарбонитрил, и его соли присоединения с фармацевтически приемлемой кислотой;(4R)-3-[(спиро[5.5]ундец-3-иламино)ацетил]-1,3-тиазолидин-4-карбонитрил, и его соли присоединения с фармацевтически приемлемой кислотой; 2-2-[(2S)-2-цианопирролидинил]-2-оксоэтиламино)спиро[3.3]гептан-2-карбоксамид, и его соли присоединения с фармацевтически приемлемой кислотой;(2S)-1-([(2-(гидроксиметил)-7-оксаспиро[3.5]нон-2-ил]аминоацетил)-2-пирролидинкарбонитрил,и его соли присоединения с фармацевтически приемлемой кислотой;(2S)-4,4-дифтор-1-([9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]аминоацетил)-2-пирролидинкарбонитрил, и его соли присоединения с фармацевтически приемлемой кислотой; и (4R)-3-([9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]аминоацетил)-1,3-тиазолидин-4-карбонитрил, и его соли присоединения с фармацевтически приемлемой кислотой. Изобретение также относится к способу получения соединений формулы (I) исходя из соединения формулы (II) в которой Х 1, m1, m2, n1 и n2 имеют значения, указанные для формулы (I), которое превращают в соединение формулы (III) в которой Х 1, m1, m2, n1, n2 и R1 имеют значения, указанные для формулы (I), которое подвергают реакции с соединением формулы (IV) в которой Ak, р, R3, Х 2 и Х 3 имеют значения, указанные для формулы (I), и Y1 представляет собой уходящую группу, получая соединения формулы (Ia), частный случай соединений формулы (I), в которой R2 представляет собой атом водорода в которой X1, m1,m2, n1, n1, R1, Ak, p, R3, Х 2 и Х 3 имеют значения, указанные для формулы (I), которое, если является желательным получить соединения формулы (I), в которой R2 не представляет собой атом водорода, подвергают реакции с соединением формулы (V) в которой Y2 представляет собой уходящую группу и R'2 представляет собой линейную или разветвленную C1-С 6-алкильную группу, получая соединения формулы (Ib), частный случай соединений формулы (I), в которой R2 представляет собой линейную или разветвленную C1-С 6-алкильную группу в которой Х 1, m1, m2, n1, n2, R1, Ak, p, R3, Х 2 и Х 3 имеют значения, указанные для формулы (I), и R'2 имеет значение, указанное выше, где соединения формулы (I/a) и (I/b), которые представляют собой совокупность соединений формулы (I), очищают в соответствии с обычным способом очистки, необязательно разделяют на их оптические изомеры в соответствии с обычным способом разделения, и превращают, если это является желательным, в соли присоединения с фармацевтически приемлемой кислотой. Соединения формулы (IV) могут быть получены в соответствии со способом, описанным в J. Med.Chem. 2002, том. 45(12), 2362-2365. Если является желательным получить соединения формулы (I), в которой R1 представляет собой атом водорода, то соединение формулы (II) подвергают реакции с гидроксиламином, получая соединение формулы (VI) в которой Х 1, m1, m2, n1 и n2 имеют значения, указанные для формулы (I), которое затем гидрируют до соединения формулы (IIIa), частного случая соединений формулы (III), в которой R1 представляет собой атом водорода в которой Х 1, m1, m2, n1 и n2 имеют значения, указанные для формулы (I). Если является желательным получить соединения формулы (I), в которой R1 представляет собой гидроксиметильную, карбокси или алкоксикарбонильную группу, то соединение формулы (II) подвергают реакции с карбонатом аммония и цианидом калия, получая соединение формулы (VII): в которой Х 1, m1, m2, n1 и n2 имеют значения, указанные для формулы (I), которое нагревают с сульфатом бария, получая соединение формулы (IIIb), частный случай соединений формулы (III), в которой в которой X1, m1, m2, n1 и n2 имеют значения, указанные для формулы (I), которое эстерифицируют до соединения формулы (IIIc), частного случая соединений формулы (I), в которой R1 представляет собой алкилксикарбонильную группу в которой X1, m1, m2, n1 и n2 имеют значения, указанные для формулы (I), и R7 представляет собой линейную или разветвленную C1-С 6-алкильную группу, при восстановлении которого получают соединение формулы (IIId), частный случай соединений формулы (I), в которой R1 представляет собой гидроксиметильную группу в которой X1, m1, m2, n1 и n2 имеют значения, указанные для формулы (I). Если является желательным получить соединения формулы (I), в которой R1 представляет собой 1 гидрокси-1-метилэтильную группу, то соединение формулы (IIIс) после защиты функциональной аминогруппы подвергают реакции с йодидом метилмагния, получая, после снятия защиты с функциональной аминогруппы, соединение формулы (IIIe), частный случай соединений формулы (III), в которой R1 представляет собой 1-гидрокси-1-метилэтильную группу в которой X1, m1, m2, n1 и n2 имеют значения, указанные для формулы (I). Если является желательным получить соединения формулы (I), в которой R1 представляет собой карбамоильную группу, то соединение формулы (IIIb) после защиты функциональной аминогруппы подвергают реакции с аммиаком в присутствии связывающего средства, получая, после снятия защиты с функциональной аминогруппы, соединение формулы (IIIf), частный случай соединений формулы (III), в которой R1 представляет собой карбамоильную группу в которой X1, m1, m2, n1 и n2 имеют значения, указанные для формулы (I). Соединения формулы (II) могут быть получены исходя из соединения формулы (VIII) в которой X1, m1, m2 и n2 имеют значения, указанные выше, и n'1 представляет собой 0, 1 или 2, которое помещают в присутствие основания, такого как гидрид натрия, получая соединение формулы (IX) в которой X1, m1, m2, n'1 и n2 имеют значения, указанные выше,при взаимодействии которого с гидроокисью калия получают соединение формулы (II). Соединения формулы (IIа), частный случай соединений формулы (II), в которой n1 и n2 являются одинаковыми и каждый представляет собой 1, также могут быть получены, исходя из соединения формулы (X) в которой X1, m1 и m2 имеют значения, указанные для формулы (I),которое подвергают реакции с хлоридом трихлоруксусной кислоты и цинком, получая соединение формулы (XI) в которой X1, m1 и m2 имеют значения, указанные для формулы (I), при восстановлении которого получают соединение формулы (IIa) в которой Х 1, m1 и m2 имеют значения, указанные для формулы (I). Соединения формулы (IIb), частный случай соединений формулы (II), в которой n1 и n2 являются одинаковыми и каждый представляет собой 2, также могут быть получены, исходя из соединения формулы (XII) : в которой Х 1, m1 и m2 имеют значения, указанные для формулы (I), которое подвергают реакции с метилвинилкетоном, получая соединение формулы (XIII) в которой X1, m1 и m2 имеют значения, указанные для формулы (I), при каталитической гидрогенизации которого получают соединение формулы (IIb)-5 009292 в которой Х 1, m1 и m2 имеют значения, указанные для формулы (I). Соединения согласно настоящему изобретению являются новыми, а также обладают ценными фармакологическими свойствами. Они обладают ингибирующими свойствами по отношению к дипептидилпептидазе IV, что делает их пригодными для лечения непереносимости глюкозы и нарушений, связанных с гипергликемией, таких как диабет II типа или ожирение. Настоящее изобретение также относится к фармацевтическим композициям, которые содержат в качестве активного компонента по крайней мере одно соединение формулы (I) с одним или несколькими подходящими, инертными, нетоксичными наполнителями. Среди фармацевтических композиций согласно изобретению особенно можно отметить те, которые являются пригодными для перорального, парентерального (внутривенного, внутримышечного или подкожного) или назального введения, таблетки или драже, подъязычные таблетки, желатиновые капсулы, лепешки, суппозитории, составы для инъекций,суспензии для питья. Подходящая дозировка адаптируется в соответствии с природой и тяжестью расстройства, путем введения, а также возрастом и весом пациента и любыми сопутствующими видами лечения. Дозировка находится в диапазоне от 0,5 мг до 2 г в сутки на одно или несколько введения. Последующие примеры иллюстрируют изобретение Используемые исходные материалы являются известными продуктами или продуктами, которые получают в соответствии с известными способами. Структуры соединений, описанных в примерах, были определены согласно обычным спектрофотометрическим способам (инфракрасная спектрометрия, ядерный магнитный резонанс, масс-спектрометрия). Под соединением, которое имеет (2RS)-конфигурацию, подразумевают рацемическую смесь соединений с конфигурациями (2R) и (2S). Пример 1. (2S)-1-([9-(Гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]аминоацетил)-2-пирролидинкарбонитрил. Стадия А. 3-Оксаспиро[5.5]ундец-7-ен-9-он. 0,5 мл серной кислоты добавляли к 317 ммоль тетрагидро-2 Н-пиран-4-карбоксальдегида и 317 ммоль метилвинилкетона в 500 мл бензола. Затем реакционную смесь нагревали в колбе с обратным холодильником в течение 3 ч, после этого воду удаляли, используя аппарат Дина-Старка, и затем добавляли дополнительно 317 ммоль метилвинилкетона и продолжали нагревать в течение 3 ч. Затем смесь промывали, высушивали и после этого концентрировали, и полученный остаток перегоняли, получая ожидаемый продукт. Стадия В. 3-Оксаспиро[5.5]ундекан-9-он. 147,4 ммоль соединения, полученного на вышеописанной стадии, гидрировали в течение 15 ч, при температуре окружающей среды и давлении 4,6 бар, в 100 мл этилацетата в присутствии каталитического количества 10% Pd/C. Затем катализатор отфильтровывали и промывали этилацетатом, и концентрировали, получая ожидаемый продукт. Стадия С. 11-Окса-1,3-диазадиспиро[4.2.5.2]пентадекан-2,4-дион. 165 мл водного 60% этанола и 480 ммоль карбоната аммония добавляли к 300 ммоль соединения,полученного на вышеописанной стадии. Затем реакционную смесь нагревали до 55 С, после этого добавляли 5,3 г цианида калия в 40 мл воды в течение 5 мин и смесь перемешивали в течение 2 ч при 55 С. После этого этанол выпаривали и затем смесь фильтровали; осадок промывали водой и ацетоном, и затем высушивали, получая ожидаемый продукт в виде хлопьевидного белого твердого вещества. Стадия D. Гидрохлорид 9-амино-3-оксаспиро[5.5]ундекан-9-карбоновой кислоты. В автоклав объемом 1 л добавляли 177,8 ммоль сульфата бария к 88,9 ммоль соединения, полученного на вышеописанной стадии, в 335 мл воды. Затем смесь нагревали всю ночь при 160 С и после этого охлаждали на ледяной бане. Образованный карбонат бария отфильтровывали и промывали водой, и в водную фазу барботировали углекислый газ. Затем водную фазу снова фильтровали, и после этого фильтрат концентрировали насухо, получая ожидаемый продукт в виде порошка. Стадия Е. Гидрохлорид метил 9-амино-3-оксаспиро[5.5]ундекан-9-карбоксилата. 100 мл метанола добавляли к 61,3 ммоль соединения, полученного на вышеописанной стадии. Полученную суспензию охлаждали до 5 С и затем по каплям добавляли 184 ммоль тионилхлорида. Затем реакционную смесь перемешивали в течение 1 ч при 20 С и после этого в течение 2 ч в колбе с обратным холодильником, и потом выпаривали насухо, получая ожидаемый продукт в виде порошка. Стадия F. (9-Амино-3-оксаспиро[5.5]ундец-9-ил)метанол. 6,8 г литийалюмминийгидрида и затем 59,7 ммоль соединения, полученного на вышеописанной стадии, в виде основания в растворе в 75 мл тетрагидрофурана, добавляли при 0 С к 50 мл тетрагидрофурана. Затем реакционную смесь перемешивали в течение 30 мин при 0 С и потом всю ночь при 20 С. После этого смесь охлаждали до 5 С и затем добавляли 6,8 мл воды, 6,8 мл 15% раствора гидроокиси натрия и 3 х 6,8 мл воды. Смесь интенсивно перемешивали в течение 20 мин и потом фильтровали через целит. Осадок промывали эфиром и фильтрат высушивали, и затем выпаривали, получая ожидаемый продукт в виде твердого вещества.-6 009292 Стадия G. (2S)-1-([9-(Гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]аминоацетил)-2-пирролидинкарбонитрил. 90 мл дихлорметана, 6 ммоль (2S)-1-(хлорацетил)-2-пирролидинкарбонитрила и затем 24 ммоль карбоната калия добавляли к 12 ммоль соединения, полученного на вышеописанной стадии. После перемешивания в течение 6 дней при 20 С, образованный осадок отфильтровывали и промывали дихлорметаном, и фильтраты концентрировали насухо. Полученный остаток очищали путем хроматографии на диоксиде кремния (элюент: дихлорметан/метанол 97/3), получая ожидаемый продукт в виде желтого масла, которое затвердевало при охлаждении, образуя белый порошок. Элементный микроанализ: Пример 2. Гидрохлорид (2S)-1-[(спиро[5.5]ундец-3-иламино)ацетил]-2-пирролидинкарбонитрила. Стадия А. Спиро[5.5]ундекан-3-он. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А и В примера 1,заменяя тетрагидро-2 Н-пиран-4-карбоксальдегид циклогексанкарбоксальдегидом на стадии А. Стадия В. Спиро[5.5]ундекан-3-он оксим. 70 мл пиридина и 3,45 г гидрохлорида гидроксиламина добавляли к 39 ммоль соединения, полученного на вышеописанной стадии, в растворе в диоксане. Затем реакционную смесь нагревали в колбе с обратным холодильником всю ночь и после этого концентрировали насухо, получая масло, которое кристаллизовалось и которое промывали водой, получая ожидаемый продукт в виде порошка после фильтрации и высушивания. Стадия С. Спиро[5.5]ундец-3-иламин. 3 мл концентрированной гидроокиси аммония и каталитическое количество никеля Ренея добавляли к 15,8 ммоль соединения, полученного на вышеописанной стадии, в растворе в диоксане. Затем реакционную смесь гидрировали всю ночь при температуре окружающей среды и давлении. После этого катализатор отфильтровывали, и промывали и раствор концентрировали, получая ожидаемый продукт в виде масла. Стадия D. (2S)-1-[(Спиро[5.5]ундец-3-иламино)ацетил]-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадии G примера 1, исходя из соединения, полученного на вышеописанной стадии, и (2S)-1-(хлорацетил)-2-пирролидинкарбонитрила. Стадия Е. Гидрохлорид (2S)-1-[(Спиро[5.5]ундец-3-иламино)ацетил]-2-пирролидинкарбонитрила. Ожидаемый продукт получали путем превращения соединения, полученного на вышеописанной стадии, в соль, используя соляную кислоту. Элементный микроанализ: Пример 3. Гидрохлорид (2S)-1-[(спиро[4.5]дец-8-иламино)ацетил]-2-пирролидинкарбонитрила. Ожидаемый продукт получали в соответствии с методикой из примера 2, заменяя циклогексанкарбоксальдегид циклопентанкарбоксальдегидом на стадии А. Элементный микроанализ: Пример 4. Гидрохлорид N-[2-оксо-2-(1-пирролидинил)этил]спиро[5.5]ундец-3-иламина. Ожидаемый продукт получали в соответствии с методикой из примера 2, заменяя (2S)-1-(хлорацетил)-2-пирролидинкарбонитрил 1-(хлорацетил)пирролидином на стадии D. Элементный микроанализ: Пример 5. (2S)-1-([3-(Гидроксиметил)спиро[5.5]ундец-3-ил]аминоацетил)-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой из примера 1, заменяя тетрагидро-2 Н-7 009292 пиран-4-карбоксальдегид циклогексанкарбоксальдегидом на стадии А. Элементный микроанализ: Пример 6. Гидрохлорид (2S)-1-[(спиро[5.7]тридец-3-иламино)ацетил]-2-пирролидинкарбонитрила. Ожидаемый продукт получали в соответствии с методикой из примера 2, заменяя циклогексанкарбоксальдегид циклооктанкарбоксальдегидом на стадии А. Элементный микроанализ: Пример 7. Гидрохлорид (2S)-1-[(3-оксаспиро[5.5]ундец-9-иламино)ацетил]-2-пирролидинкарбонитрила. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях В-Е примера 2,исходя из соединения, полученного на стадии В примера 1. Элементный микроанализ: Пример 8. (2S)-1-[(Диспиро[5.2.5.2]гексадец-3-иламино)ацетил]-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А-D примера 2,заменяя циклогексанкарбоксальдегид спиро[5.5]ундекан-3-карбоксальдегидом на стадии А. Элементный микроанализ: Пример 9. Гидрохлорид (2S)-1-[(спиро[3.3]гепт-2-иламино)ацетил]-2-пирролидинкарбонитрила. Стадия А: 1,1-Дихлорспиро[3.3]гептан-2-он. 6,25 г порошкообразного цинка и затем 11 мл хлорида трихлоруксусной кислоты в растворе в 170 мл эфира добавляли к 5 г метиленциклобутана в растворе в эфире. Раствор обрабатывали ультразвуком в течение 3 ч, при этом температуру поддерживали при 20 С, и затем черный раствор фильтровали через фильтр Ватмана. Фильтрат промывали, высушивали, фильтровали и концентрировали, получая ожидаемый продукт в виде коричневого масла. Стадия В. Спиро[3.3]гептан-2-он. 28 г соединения, полученного на вышеописанной стадии, и затем 620 мл воды добавляли к 420 мл ледяной уксусной кислоты. Реакционную смесь охлаждали, используя холодную водяную баню, и затем добавляли 28,1 г порошкообразного цинка. После перемешивания в течение 20 мин баню удаляли и реакционную смесь перемешивали всю ночь при 20 С. Раствор фильтровали, экстрагировали пентаном и выпаривали. Остаток ресуспендировали в пентане и раствор промывали, высушивали, фильтровали и концентрировали, получая ожидаемый продукт в виде светло-желтой жидкости. Стадия С. (2S)-1-[(Спиро[3.3]гепт-2-иламино)ацетил]-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях В-D примера 2,исходя из соединения, полученного на вышеописанной стадии. Стадия D. Гидрохлорид (2S)-1-[(спиро[3.3]гепт-2-иламино)ацетил]-2-пирролидинкарбонитрила. Ожидаемый продукт получали путем превращения соединения, полученного на вышеописанной стадии, в соль, используя соляную кислоту. Элементный микроанализ: Пример 10. (2S)-1-[(Спиро[5.9]пентадец-3-иламино)ацетил]-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А-D примера 2,-8 009292 заменяя циклогексанкарбоксальдегид циклодеканкарбоксальдегидом на стадии А. Точка плавления: 83 С. Элементный микроанализ: Пример 11. Гидрохлорид (2S)-1-[(спиро[3.5]нон-7-иламино)ацетил]-2-пирролидинкарбонитрила. Стадия А. Этил 7-оксоспиро[3.5]нонан-6-карбоксилат. 20,6 ммоль сложного эфира 3-[1-(этоксикарбонилэтил)циклобутил]пропионовой кислоты в растворе в тетрагидрофуране добавляли при 0 С к 22,7 ммоль 95% гидрида натрия в суспензии в 15 мл тетрагидрофурана. Реакционную смесь нагревали в колбе с обратным холодильником в течение 4 часов и затем добавляли 49 мл уксусной кислоты. Тетрагидрофуран выпаривали и полученный остаток экстрагировали этилацетатом. Объединенные органические фазы промывали, фильтровали, высушивали и выпаривали,получая масло, которое подвергали хроматографии на диоксиде кремния (элюент:дихлорметан/этилацетат 95/5), получая ожидаемый продукт. Стадия В. Спиро[3.5]нонан-7-он. 11,1 г гидроокиси калия в 100 мл воды добавляли к 39,7 ммоль соединения, полученного на вышеописанной стадии, в растворе в диоксане. Реакционную смесь нагревали в колбе с обратным холодильником всю ночь и затем, после возвращения температуры до температуры окружающей среды, экстрагировали эфиром. Объединенные органические фазы промывали, высушивали, фильтровали и выпаривали,получая ожидаемый продукт в виде летучего масла. Стадия С. Гидрохлорид (2S)-1-[(спиро[3.5]нон-7-иламино)ацетил]-2-пирролидинкарбонитрила. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях В - Е примера 2,исходя из соединения, полученного на вышеописанной стадии. Элементный микроанализ: Пример 12. Трифторацетат (2S)-1-[(спиро[3.5]нон-2-иламино)ацетил]-2-пирролидинкарбонитрила. Стадия А. (2S)-1-[(Спиро [3.5] нон-2-иламино)ацетил]-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А-С примера 9,заменяя метиленциклобутан метиленциклогексаном на стадии А. Стадия В. Трифторацетат (2S)-1-[(спиро[3.5]нон-2-иламино)ацетил]-2-пирролидинкарбонитрила. Ожидаемый продукт получали путем превращения соединения, полученного на вышеописанной стадии, в соль, используя трифторуксусную кислоту. Масс-спектрометрия: [M + H]+ =276. Пример 13. (2S)-1-[(Спиро[5.6]додец-3-иламино)ацетил]-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А - D примера 2,заменяя циклогексанкарбоксальдегид циклогептанкарбоксальдегидом на стадии А. Элементный микроанализ: Пример 14. Гидрохлорид (2S)-1-[(спиро[3.4]октил-2-иламино)ацетил]-2-пирролидинкарбонитрила. Стадия А. Спиро[3.4]октан-2-он. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А и В примера 9,заменяя метиленциклобутан метиленциклопентаном на стадии А. Стадия В. Гидрохлорид (2S)-1-[(спиро[3.4]окт-2-иламино)ацетил]-2-пирролидинкарбонитрила. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях В-Е примера 2,исходя из соединения, полученного на вышеописанной стадии. Элементный микроанализ: Пример 15. (2S)-1-[(3,3-Диоксо-3-тиаспиро[5.5]ундец-9-иламино)ацетил]-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А-D примера 2,-9 009292 заменяя циклогексанкарбоксальдегид 1,1-диоксидом тетрагидро-2H-тиопиран-4-карбоксальдегида на стадии А. Элементный микроанализ: Пример 16. Гидрохлорид (2S)-1-([2-(гидроксиметил)спиро[3.3]гепт-2-ил]аминоацетил)-2-пирролидинкарбонитрила. Стадия А. (2S)-1-([2-(Гидроксиметил)спиро[3.3]гепт-2-ил]аминоацетил)-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях С-G примера 1,заменяя 3-оксаспиро[5.5]ундекан-9-он на стадии С соединением, полученным на стадии В примера 9. Стадия В. Гидрохлорид (2S)-1-([2-(гидроксиметил)спиро[3.3]гепт-2-ил]аминоацетил)-2-пирролидинкарбонитрила. Ожидаемый продукт получали путем превращения соединения, полученного на вышеописанной стадии, в соль, используя соляную кислоту. Элементный микроанализ:(2S)-1-([2-(Гидроксиметил)спиро[3.4]окт-2-ил]аминоацетил)-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях С-G примера 1,заменяя 3-оксаспиро[5.5]ундекан-9-он на стадии С соединением, полученным на стадии А примера 14. Стадия В. Гидрохлорид (2S)-1-([2-(гидроксиметил)спиро[3.4]окт-2-ил]аминоацетил)-2-пирролидинкарбонитрила. Ожидаемый продукт получали путем превращения соединения, полученного на вышеописанной стадии, в соль, используя соляную кислоту. Элементный микроанализ: Пример 18. Трифторацетат (4R)-3-[(спиро[5.5]ундец-3-ил-амино)ацетил]-1,3-тиазолидин-4-карбонитрила. Стадия А. (4R)-3-[(Спиро[5.5]ундец-3-ил-амино)ацетил]-1,3-тиазолидин-4-карбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадии G примера 1, исходя из соединения, полученного на стадии С примера 2 и (4R)-3-(хлорацетил)-1,3-тиазолидин-4 карбонитрила. Стадия В. Трифторацетат (4R)-3-[(спиро[5.5]ундец-3-ил-амино)ацетил]-1,3-тиазолидин-4-карбонитрила. Ожидаемый продукт получали путем превращения соединения, полученного на вышеописанной стадии, в соль, используя трифторуксусную кислоту. Элементный микроанализ: Пример 19. 2-(2-[(2S)-2-Цианопирролидинил]-2-оксоэтиламино)спиро[3.3]-гептан-2-карбоксамид. Стадия А. 2-Аминоспиро[3.3]гептан-2-карбоновая кислота. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях С и D примера 1,исходя из соединения, полученного на стадии В примера 9. Стадия В. 2-(трет-Бутилоксикарбониламино)спиро[3.3]гептан-2-карбоновая кислота. 40 мл диоксана и затем, при 0 С, 6,71 г ди(трет-бутил) дикарбоната в диоксане добавляли к 4,35 г соединения, полученного на вышеописанной стадии, в растворе в 11 мл раствора гидроокиси натрия. Через одну ночь при 20 С диоксан выпаривали и полученный остаток ресуспендировали в воде. Осуществляли экстрагирование эфиром и затем водную фазу подкисляли до рН 3, используя водный 10% раствор лимонной кислоты. Водную фазу экстрагировали этилацетатом. Органические фазы объединяли и- 10009292 промывали соляным раствором, высушивали и концентрировали насухо, получая ожидаемый продукт в виде белой пасты. Стадия С. 2-(трет-Бутилоксикарбониламино)спиро[3.3]гептан-2-карбоксамид. 811 мг N-гидроксисукцинимида и 1,45 г дициклогексилкарбодиимида добавляли к 1,8 г соединения,полученного на вышеописанной стадии, в растворе в тетрагидрофуране. Через одну ночь при 20 С реакционную смесь фильтровали, и в фильтрат барботировали газообразный аммиак. Через одну ночь при 20 С реакционную смесь снова фильтровали и концентрировали насухо, получая ожидаемый продукт в виде белого порошка. Стадия D. Гидрохлорид 2-аминоспиро[3.3]гептан-2-карбоксамида. 1,84 г соединения, полученного на вышеописанной стадии, растворяли в этилацетате и затем барботировали газообразной HCl при 0 С в течение 10 минут. Появлялся осадок. Через 30 мин при 0 С осадок отфильтровали и промывали этилацетатом, и затем высушивали, получая ожидаемый продукт в виде белого порошка. Стадия Е. 2-(2-[(2S)-2-Цианопирролидинил]-2-оксоэтиламино)спиро[3.3]гептан-2-карбоксамид. Ожидаемый продукт получали в соответствии с методикой, описанной на стадии G примера 1, исходя из соединения, полученного на вышеописанной стадии, и (2S)-1-(хлорацетил)-2-пирролидинкарбонитрила. Элементный микроанализ: Пример 20. Бис(трифторацетат) (2S)-1-[(3-азаспиро[5.5]ундец-9-иламино)ацетил]-2-пирролидинкарбонитрила. Стадия А. 3-Азаспиро[5.5]ундекан-9-он. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А и В примера 1,заменяя тетрагидро-2 Н-пиран-4-карбоксальдегид бензил 4-формил-1-пиперидинкарбоксилатом на стадии А. Стадия В. трет-Бутил 9-оксо-3-азаспиро[5.5]ундекан-3-карбоксилат. Ожидаемый продукт получали путем взаимодействия соединения, полученного на вышеописанной стадии, с ди-трет-бутил дикарбонатом в соответствии с методикой, описанной на стадии в примера 19. Стадия С. трет-Бутил 9-(2-[(2S)-2-цианопирролидинил]-2-оксоэтиламино)-3-азаспиро-[5.5]ундекан-3-карбоксилат. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях В-D примера 2,исходя из соединения, полученного на вышеописанной стадии. Стадия D. Бис(трифторацетат) (2S)-1-[(3-азаспиро[5.5]ундец-9-иламино)ацетил]-2-пирролидинкарбонитрила. Ожидаемый продукт получали путем взаимодействия соединения, полученного на вышеописанной стадии, с 30 эквивалентами трифторуксусной кислоты в дихлорметане при 0-5 С в течение 1 ч. Смесь концентрировали насухо. Соединение очищали путем препаративной ВЭЖХ. Масс-спектрометрия: [М + H]+ = 305 Пример 21. (2S)-1-([3-(Гидроксиметил)-9,9-диметилспиро[5.5]ундец-3-ил]аминоацетил)-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой из примера 1, заменяя тетрагидро-2 Нпиран-4-карбоксальдегид 4,4-диметилциклогексанкарбоксальдегидом на стадии А. Элементный микроанализ: Пример 22. Гидрохлорид (2S)-1-([2-(1-гидрокси-1-метилэтил)спиро[3.3]гепт-2-ил]аминоацетил)2-пирролидинкарбонитрила. Стадия А. Метил 2-аминоспиро[3.3]гептан-2-карбоксилат. Ожидаемый продукт получали в соответствии с методикой, описанной на стадии Е примера 1, исходя из соединения, полученного на стадии А примера 19. Стадия В. Метил 2-(трет-бутилоксикарбониламино)спиро[3.3]гептан-2-карбоксилат. 5,8 мл триэтиламина и затем, при 0 С, 9,02 г ди(трет-бутил) дикарбоната в дихлорметане, добавляли к 8,5 г соединения, полученного на вышеописанной стадии, в растворе в дихлорметане. После перемешивания в течение ночи при 20 С, реакционную смесь промывали водным 10% раствором лимонной кислоты и затем водой. Органическую фазу высушивали, фильтровали и концентрировали, получая ожидаемый продукт в виде оранжевого масла.- 11009292 Стадия С. 2-(2-трет-Бутилоксикарбониламиноспиро[3.3]гепт-2-ил)-2-пропанол. Добавляли к 50 мл эфира 42 мл 3 М раствора йодида метилмагния в эфире и затем по каплям при 0 С 5 г соединения, полученного на вышеописанной стадии, в растворе в эфире. После перемешивания в течение 1 ч при 0 С добавляли нашатырь и затем реакционную смесь перемешивали в течение 1 ч при 20 С. Водную фазу экстрагировали этилацетатом и затем объединенные органические фазы промывали,высушивали и концентрировали насухо, получая ожидаемый продукт в виде масла, которое кристаллизовалось. Стадия D. Трифторацетат 2-(2-аминоспиро[3.3]гепт-2-ил)-2-пропанола. 40 мл трифторуксусной кислоты добавляли при 0 С к 4,8 г соединения, полученного на вышеописанной стадии, в растворе в дихлорметане. После перемешивания в течение 30 мин при 0 С реакционную смесь выпаривали насухо и остаток несколько раз ресуспендировали толуолом, затем толуол выпаривали, получая ожидаемый продукт в виде бесцветного масла. Стадия Е. (2S)-1-([2-(1-Гидрокси-1-метилэтил)спиро[3.3]гепт-2-ил]аминоацетил)-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадии G примера 1, исходя из соединения, полученного на вышеописанной стадии, и (2S)-1-(хлорацетил)-2-пирролидинкарбонитрила. Стадия F. Гидрохлорид (2S)-1-([2-(1-гидрокси-1-метилэтил)спиро[3.3]гепт-2-ил]аминоацетил)-2 пирролидинкарбонитрила. Ожидаемый продукт получали путем превращения соединения, полученного на вышеописанной стадии, в соль, используя соляную кислоту. Элементный микроанализ:(2S)-1-[(3-тиаспиро[5.5]ундец-9-иламино)ацетил]-2 пирролидинкарбонитрила. Стадия А. 3-Тиаспиро[5.5] ундекан-9-он оксим 3,3-диоксид. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А и В примера 2,заменяя циклогексанкарбоксальдегид 1,1-диоксидом тетрагидро-2 Н-тиопиран-4-карбоксальдегида на стадии А. Стадия В. 3-Тиаспиро[5.5]ундекан-9-амин. 4 г соединения, полученного на вышеописанной стадии, порциями добавляли к 7,9 г литийалюминийгидрида в суспензии в 200 мл тетрагидрофурана. Затем реакционную смесь нагревали в колбе с обратным холодильником в течение 12 ч и после этого гидролизовали путем добавления 8 мл воды, 8 мл 15% раствора гидроокиси натрия и 16 мл воды. Затем полученные соли отфильтровывали и после этого фильтрат концентрировали насухо, получая указанный в заглавии продукт. Стадия С. (2S)-1-[(3-Тиаспиро[5.5]ундец-9-иламино)ацетил]-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадии G примера 1, исходя из соединения, полученного на вышеописанной стадии, и (2S)-1-(хлорацетил)-2-пирролидинкарбонитрила. Стадия D. Гидрохлорид (2S)-1-[(3-тиаспиро[5.5]ундец-9-иламино)ацетил]-2-пирролидинкарбонитрила. Ожидаемый продукт получали путем превращения соединения, полученного на вышеописанной стадии, в соль, используя соляную кислоту. Элементный микроанализ: Пример 24. Бис(трифторацетат) (2S)-1-([3-(2-гидроксиэтил-3-азаспиро[5.5]ундец-9-ил]аминоацетил)-2-пирролидинкарбонитрила. Стадия А. 3-Азаспиро[5.5]ундекан-9-он. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А и В примера 1,заменяя тетрагидро-2 Н-пиран-4-карбоксальдегид бензил 4-формил-1-пиперидинкарбоксилатом на стадии А. Стадия В. 3-(2-Гидроксиэтил)-3-азаспиро[5.5]ундекан-9-он. Ожидаемый продукт получали путем алкилирования соединения, полученного на вышеописанной стадии, 2-бромэтанолом в присутствии карбоната калия.- 12009292 Стадия С. (2S)-1-([3-(2-Гидроксиэтил)-3-азаспиро[5.5]ундец-9-ил]аминоацетил)-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях В-D примера 2,исходя из соединения, полученного на вышеописанной стадии. Стадия D. Бис(трифторацетат) (2S)-1-([3-(2-гидроксиэтил)-3-азаспиро[5.5]ундец-9-ил]аминоацетил)-2-пирролидинкарбонитрила. Ожидаемый продукт получали путем превращения соединения, полученного на вышеописанной стадии, в соль, используя трифторуксусную кислоту. Элементный микроанализ:(2S)-1-([2-(Гидроксиметил)-7-оксаспиро[3.5]нон-2-ил]аминоацетил)-2-пирролидинкарбонитрил. Стадия А. 7-Оксаспиро[3.5]нонан-2-он. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А и В примера 9,заменяя метиленциклобутан 4-метилентетрагидро-2 Н-пираном на стадии А. Стадия В. (2S)-1-([2-(Гидроксиметил)-7-оксаспиро[3.5]нон-2-ил]аминоацетил)-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях С-G примера 1,исходя из соединения, полученного на вышеописанной стадии. Точка плавления: 103 С. Элементный микроанализ: Пример 26. (2S)-1-([2-(Гидроксиметил)спиро[3.5]нон-2-ил]аминоацетил)-2-пирролидинкарбонитрил. Стадия А. Спиро[3.5]нонан-2-он. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А и В примера 9,заменяя метиленциклобутан метиленциклогексаном на стадии А. Стадия В. (2S)-1-([2-(Гидроксиметил)спиро[3.5]нон-2-ил]аминоацетил)-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях С-G примера 1,исходя из соединения, полученного на вышеописанной стадии. Элементный микроанализ: Пример 27. Трифторацетат (2S)-1-[(7-оксаспиро[3.5]нон-2-ил)амино)ацетил]-2-пирролидинкарбонитрила. Стадия А. (2S)-1-[(7-Оксаспиро[3.5]нон-2-иламино)ацетил]-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А-С примера 9,заменяя метиленциклобутан 4-метилентетрагидро-2 Н-пираном на стадии А. Стадия В. Трифторацетат (2S)-1-[(7-оксаспиро[3.5]нон-2-иламино)ацетил]-2-пирролидинкарбонитрила. Ожидаемый продукт получали путем превращения соединения, полученного на вышеописанной стадии, в соль, используя трифторуксусную кислоту. Элементный микроанализ: Пример 28. (2S)-1-[(Спиро[2.5]окт-6-иламино)ацетил]-2-пирролидинкарбонитрил. Стадия А. Спиро[2.5] октан-6-он. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А и В примера 11, заменяя 3-[1-(этоксикарбонилэтил)циклобутил]пропионовую кислоту 3-[1-(этоксикарбонилэтил) циклопропил]пропионовой кислотой на стадии А. Стадия В. (2S)-1-[(Спиро[2.5]окт-6-иламино)ацетил]-2-пирролидинкарбонитрил.- 13009292 Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А-D примера 2,исходя из соединения, полученного на вышеописанной стадии. Элементный микроанализ: Пример 29. (2S,4S)-4-Фтор-1-([9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]аминоацетил)-2 пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной в примере 1, заменяя (2S)-1(хлорацетил)-2-пирролидинкарбонитрил (2S,4S)-1-(хлорацетил)-4-фтор-2-пирролидинкарбонитрилом на стадии G. Элементный микроанализ: Пример 30. (2S)-4,4-Дифтор-1-([9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]аминоацетил)-2 пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной в примере 1, заменяя (2S)-1(хлорацетил)-2-пирролидинкарбонитрил (2S)-1-(хлорацетил)-4,4-дифтор-2-пирролидинкарбонитрилом на стадии G. Элементный микроанализ: Пример 31. (9-[2-Оксо-2-(1-пирролидинил)этил]амино-3-оксаспиро[5.5]ундец-9-ил)метанол. Ожидаемый продукт получали в соответствии с методикой, описанной в примере 1, заменяя (2S)1(хлорацетил)-2-пирролидинкарбонитрил 1-(хлорацетил)пирролидином на стадии G. Элементный микроанализ: Пример 32. (4R)-3-([9-(Гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]аминоацетил)-1,3-тиазолидин-4-карбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной в примере 1, заменяя (2S)-1(хлорацетил)-2-пирролидинкарбонитрил (4R)-3-(хлорацетил)-1,3-тиазолидин-4-карбонитрилом на стадии Пример 33. (2S,4S)-4-Гидрокси-1-([9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]аминоацетил)2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной в примере 1, заменяя (2S)-1(хлорацетил)-2-пирролидинкарбонитрил (2S,4S)-1-(хлорацетил)-4-гидрокси-2-пирролидинкарбонитрилом на стадии G. Пример 34. Трифторацетат (2S,4R)-4-фтор-1-([9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил] аминоацетил)-2-пирролидинкарбонитрила. Ожидаемый продукт получали в соответствии с методикой, описанной в примере 1, заменяя (2S)-1(хлорацетил)-2-пирролидинкарбонитрил (2S,4R)-1-(хлорацетил)-4-фтор-2-пирролидинкарбонитрилом на стадии G. Элементный микроанализ:- 14009292 Ожидаемый продукт получали в соответствии с методикой, описанной в примере 1, заменяя (2S)-1(хлорацетил)-2-пирролидинкарбонитрил (3S)-1-(хлорацетил)-3-фторпирролидином на стадии G. Пример 36. (9-[2-Оксо-2-(3,3,4,4-тетрафтор-1-пирролидинил)этил]амино-3-оксаспиро[5.5]ундец 9-ил)метанол. Ожидаемый продукт получали в соответствии с методикой, описанной в примере 1, заменяя (2S)-1(хлорацетил)-2-пирролидинкарбонитрил 1-(хлорацетил)-3,3,4,4-тетрафторпирролидином на стадии G. Элементный микроанализ:(2S)-1-([7-(Гидроксиметил)спиро[3.5]нон-7-ил]аминоацетил)-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной в примере 1, заменяя 3 оксаспиро[5.5]ундекан-9-он на стадии С соединением, полученным на стадии В примера 11. Пример 38.(2S)-1-([8-(Гидроксиметил)спиро[4.5]дец-8-ил]аминоацетил)-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой из примера 1, заменяя тетрагидро-2 Нпиран-4-карбоксальдегид циклопентанкарбоксальдегидом на стадии А. Пример 39.(2S)-1-[(2,2-Диоксо-2-тиаспиро[3.5]нон-7-ил)амино]ацетил-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадиях А-D примера 2,заменяя циклогексанкарбоксальдегид 1,1-диоксидом 3-тиетан-карбоксальдегида на стадии А. Пример 40. Гидрохлорид (2S)-1-[3-(спиро[5.5]ундец-3-иламино)пропаноил]-2-пирролидинкарбонитрила. Стадия А. (2S)-1-[3-(Спиро[5.5]ундец-3-иламино)пропаноил]-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой, описанной на стадии G примера 1, исходя из соединения, полученного на стадии С примера 2 и (2S)-1-(3-хлорпропионил)-2 пирролидинкарбонитрила. Стадия В. Гидрохлорид (2S)-1-[3-(спиро[5.5]ундец-3-иламино)пропаноил]-2-пирролидинкарбонитрила. Ожидаемый продукт получали путем превращения соединения, полученного на вышеописанной стадии, в соль, используя соляную кислоту. Масс-спектрометрия: [М + Н]+ = 318. Пример 41. Трифторацетат (2S)-1-2RS)-2-[9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]амино пропаноил)-2-пирролидинкарбонитрила. Ожидаемый продукт получали в соответствии с методикой из примера 1, заменяя (2S)-1(хлорацетил)-2-пирролидинкарбонитрил (2S)-1-[(2RS)-2-бромпропаноил]-2-пирролидинкарбонитрилом на стадии G. Масс-спектрометрия: [М + H]+ = 350. Пример 42. (2R)-1-([9-(Гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]аминоацетил)-2-пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой из примера 1, заменяя (2S)-1(хлорацетил)-2-пирролидинкарбонитрил (2R)-1-(хлорацетил)-2-пирролидинкарбонитрилом на стадии G. Элементный микроанализ: Пример 43. (2RS)-4,4-Дифтор-1-([9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]аминоацетил)-2 пирролидинкарбонитрил. Ожидаемый продукт получали в соответствии с методикой из примера 1, заменяя (2S)-1(хлорацетил)-2-пирролидинкарбонитрил (2RS)-1-(хлорацетил)-4,4-дифтор-2-пирролидинкарбонитрилом на стадии G. Элементный микроанализ: Пример 44. (2R)-4,4-Дифтор-1-([9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]аминоацетил)-2 пирролидинкарбонитрил. Ожидаемый продукт получали путем разделения при помощи препаративной хиральной ВЭЖХ ра- 15009292 цемического соединения из примера 43. Точка плавления: 128 С. Фармакологическое исследование соединений по изобретению Пример 45. Ингибирование дипептидил-пептидазы IV в почке поросенка. Получение мембранной фракции почки поросенка. Ткань почки поросенка (2-3-х месячный большой белый поросенок) гомогенизировали (5 г ткани в 15 мл трис-буфера (114 мМ, рН 7,8-8 и затем центрифугировали при 150000 х g в течение 30 минут при 4 С. Осадок от центрифугирования ресуспенировали в 15 мл буфера и повторного центрифугировали при 150000 х g в течение 30 мин при 4 С. После перемешивания полученный осадок от центрифугирования растворяли в 15 мл буфера с 60 мМ н-октил-р-глюкопиранозидом в течение 30 мин при температуре окружающей среды. После центрифугирования при 150000 х g в течение 30 мин при 4 С супернатант диализировали (MWCO 12-14000) против 114 мМ трис рН 7,8-8 и затем разделяли на аликвоты при 80 С. Измерение активности дипептидил пептидазы IV (DPP IV). Активность фермента измеряли путем расщепления хромогенного субстрата, глицил-пролил-nпитроанилида (Gly-Pro-пHA), получая Gly-Pro и пНА (n-нитроанилин); последний определяется путем измерения поглощения при 405 нм. Активность измеряли при отсутствии (контроль) или в присутствии 10 мкл ингибиторов, используя 10 мкл препарата из почки поросенка (0,81 милиединиц, 1 единица = 1 мкмоль пНА продукта/минуту при 37 С). Инкубирование начинали путем добавления субстрата (250 мкл), растворенного в трис-буфере (114 мМ) рН 7,8-8, в течение 60 мин при 37 С. Для продуктов, растворенных в диметилсульфоксиде, конечная концентрация последнего не превышала 0,37%. Результаты выражали в процентах по сравнению с контрольными значениями и определяли значения IC50 (эффективная доза, которая ингибирует активность на 50% по сравнению с контролем), используя нелинейный анализ между 0 и 100% (GraphPad Prism версия 4.01 для Windows). Каждое измерение осуществляли в четырех повторах и определение каждого значения IC50 осуществляли от 1 до 5 раз. Полученные результаты (среднее геометрическое IC50) для соединений по изобретению приведено в следующей таблице: Вышеприведенные результаты свидетельствуют о том, что соединения по изобретению являются эффективными ингибиторами DPP IV. Пример 46. Фармацевтическая композиция. Состав для приготовления 1000 таблеток, каждая содержит дозу 10 мг ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение пирролидина или тиазолидина формулы (I)X1 представляет собой атом или группу, выбранную из CR4aR4b, О, S(O)q1 и NR5,где R4a и R4b, которые могут быть одинаковыми или разными, каждый представляет собой атом водорода или С 1-С 6-алкильную группу,или R4a и R4b вместе с атомом углерода, к которому они присоединены, образуют С 3-С 7 циклоалкильную группу,q1 представляет собой 0 или 2,и R5 представляет собой атом водорода или C1-С 6-алкильную группу, замещенную гидроксигруппой,m1 представляет собой 0 или целое число от 1 до 4 включительно,m2 представляет собой целое число от 1 до 4 включительно,n1 и n2, которые могут быть одинаковыми или разными, каждый представляет собой целое число от 1 до 3 включительно,R1 представляет собой атом водорода, карбамоил или С 1-С 6-алкил, замещенный гидроксигруппой,R2 представляет собой атом водорода,Ak представляет собой С 1-С 4-алкиленовую цепь,р представляет собой 1,R3 представляет собой атом водорода или цианогруппу,Х 2 и Х 3, которые могут быть одинаковыми или разными, каждый представляет собой или S(O)q2 группу, в которой q2 представляет собой О, или CR6aR6b группу, в которой R6a и R6b, которые могут быть одинаковыми или разными, каждый представляет собой атом водорода или атом галогена, или R6a представляет собой атом водорода, а R6b представляет собой гидроксигруппу,его изомеры, если они существуют, и его соли присоединения с фармацевтически приемлемой кислотой. 2. Соединение формулы (I) по п.1, в которой X1 представляет собой атом кислорода или -СН 2 группу. 3. Соединение формулы (I) по п.1 и 2, в которой m1 и m2 каждый представляет собой 1 или 2. 4. Соединение формулы (I) по любому из пп.1-3, в которой n1 и n2 каждый представляет собой 1 или 2 и являются одинаковыми. 5. Соединение формулы (I) по п.1, в которой 6. Соединение формулы (I) по любому из пп.1-5, в которой R2 представляет собой атом водорода. 7. Соединение формулы (I) по любому из пп.1-6, в которой Ak представляет собой -СН 2- группу. 8. Соединение формулы (I) по любому из пп.1-7, в которой р представляет собой 1. 9. Соединение формулы (I) по любому из пп.1-8, в которой R3 представляет собой цианогруппу. 10. Соединение формулы (I) по любому из пп.1-9, в которой Х 2 представляет собой CR6aR6b группу. 11. Соединение формулы (I) по любому из пп.1-10, в которой Х 3 представляет собой CR6aR6b груп 12. Соединение формулы (I) по любому из пп.9, 10 или 11, где конфигурация углерода, несущего группу R3, представляет собой (S)-конфигурацию. 13. Соединение формулы (I) по п.9, в которой Х 2 или Х 3 представляет собой группу S(O)q2 и конфигурация углерода, несущего группу R3, представляет собой (R)-конфигурацию. 14. Соединение формулы (I) по п.1, выбранное из следующих соединений(2S)-1-([9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]аминоацетил)-2-пирролидинкарбонитрила, и его солей присоединения с фармацевтически приемлемой кислотой;(2S)-1-([3-(гидроксиметил)спиро[5.5]ундец-3-ил]аминоацетил)-2-пирролидинкарбонитрила, и его солей присоединения с фармацевтически приемлемой кислотой;(2S)-1-([2-(гидроксиметил)спиро[3.4]окт-2-ил]аминоацетил)-2-пирролидинкарбонитрила, и его солей присоединения с фармацевтически приемлемой кислотой;(4R)-3-[(спиро[5.5]ундец-3-иламино)ацетил]-1,3-тиазолидин-4-карбонитрила, и его солей присоединения с фармацевтически приемлемой кислотой; 2-(2-[(2S)-2-цианопирролидинил]-2-оксоэтиламино)спиро[3.3]гептан-2-карбоксамида, и его солей присоединения с фармацевтически приемлемой кислотой;(2S)-1-([(2-(гидроксиметил)-7-оксаспиро[3.5]нон-2-ил]аминоацетил)-2-пирролидинкарбонитрила,и его солей присоединения с фармацевтически приемлемой кислотой;(2S)-4,4-дифтор-1-([9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]аминоацетил)-2-пирролидинкарбонитрила, и его солей присоединения с фармацевтически приемлемой кислотой; и (4R)-3-([9-(гидроксиметил)-3-оксаспиро[5.5]ундец-9-ил]аминоацетил)-1,3-тиазолидин-4-карбонитрила, и его солей присоединения с фармацевтически приемлемой кислотой. 15. Способ синтеза соединений пирролидина или тиазолидина формулы (I) по п.1, исходя из соединения формулы (III) в которой X1, m1, m2, n1, n2 и R1 имеют значения, указанные в п.1, которое подвергают реакции с соединением формулы (IV) в которой Ak, р, R3, Х 2 и Х 3 имеют значения, указанные в п.1, и Y1 представляет собой уходящую группу, с получением соединения формулы (I) в которой X1, m1, m2, n1, n2, R1, Ak, p, R3, Х 2 и Х 3 имеют значения, указанные в п.1, которое очищают в соответствии с обычным способом очистки, необязательно разделяют на оптические изомеры в соответствии с обычным способом разделения и превращают, если это является желательным, в соли присоединения с фармацевтически приемлемой кислотой. 16. Фармацевтическая композиция, обладающая активностью ингибитора дипептидил-пептидазыIV, содержащая в качестве активного компонента соединение по любому из пп.1-14 в сочетании с одним или несколькими фармацевтически приемлемыми, инертными, нетоксичными носителями. 17. Применение соединения по любому из пп.1-14 при приготовлении лекарственных средств для использования в качестве ингибиторов дипептидил-пептидазы IV. 18. Применение соединения по любому из пп.1-14 при приготовлении лекарственных средств для использования при лечении непереносимости глюкозы и нарушений, связанных с гипергликемией. 19. Применение соединения по любому из пп.1-14 при приготовлении лекарственных средств для использования при лечении диабета II типа.
МПК / Метки
МПК: C07D 409/12, A61K 31/40, C07D 405/12, A61K 31/4025, A61K 31/427, A61K 31/426, C07D 207/04, C07D 277/06, A61P 3/10
Метки: которые, пирролидина, соединения, содержат, фармацевтические, композиции, новые, получения, способ, тиазолидина
Код ссылки
<a href="https://eas.patents.su/19-9292-novye-soedineniya-pirrolidina-i-tiazolidina-sposob-ih-polucheniya-i-farmacevticheskie-kompozicii-kotorye-ih-soderzhat.html" rel="bookmark" title="База патентов Евразийского Союза">Новые соединения пирролидина и тиазолидина, способ их получения и фармацевтические композиции, которые их содержат</a>
Новые соединения 4-оксо-4, 6, 7, 8-тетрагидропирроло[1, 2-а]пиразин-6-карбоксамида, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 8688
Опубликовано: 29.06.2007
Авторы: Бенуа Ален, Валле Мари-Одиль, Рюпэн Ален, Парментье Жан-Жилль, Вербёрен Тони, Глоанек Филипп, Де Нантей Гийом
МПК: A61K 31/4985, A61P 7/02, C07D 487/04...
Метки: соединения, которые, фармацевтические, способ, содержат, 2-а]пиразин-6-карбоксамида, 8-тетрагидропирроло[1, новые, получения, композиции, 4-оксо-4
Формула / Реферат:
1. Соединения 4-оксо-4,6,7,8-тетрагидропирроло[1,2-а]пиразин-6-карбоксамида формулы (I) в которой представляет собой 1-оксидопиридильную группу, замещенную остатком молекулы в любом из положений 2, 3 и 4, m и n каждый равен 1, R1 представляет собой атом водорода, R2 и R3, которые могут быть одинаковыми или разными, каждый представляет собой атом, выбранный из водорода и линейного или разветвленного (С1-С6)алкила, R4 и R5 каждый представляет...
Новые соединения тиадиазина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 9240
Опубликовано: 28.12.2007
Авторы: Лестаж Пьер, Пиротт Бернар, Бовери Стефан, Грэндорж Эмманюэль, Ренар Пьер, Де Тюллио Паскаль, Франкотт Пьер, Данобер Лоранс, Кенар Даньель-Анри
МПК: A61K 31/549, C07D 513/04
Метки: которые, фармацевтические, соединения, новые, способ, тиадиазина, композиции, содержат, получения
Формула / Реферат:
1. Соединения тиадиазина формулы в которой A вместе с двумя атомами углерода, к которым он присоединен, образует тиенильную группу, выбранную из групп A1, A2, A3 где X представляет собой атом серы, Ra и Rb, которые могут быть одинаковыми или разными, каждый независимо друг от друга, представляет собой атом водорода, линейную или разветвленную C1-C6алкильную группу или атом галогена, представляет собой простую или двойную связь, R1...
Новые соединения пиридопиримидинона, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 9043
Опубликовано: 26.10.2007
Авторы: Пфайффер Брюно, Лансело Жан-Шарль, Роль Сильвен, Ренар Пьер, Кэньяр Даньель-Анри, Копп Марина
МПК: A61K 31/505, C07D 471/04
Метки: соединения, получения, новые, содержат, пиридопиримидинона, композиции, которые, способ, фармацевтические
Формула / Реферат:
1. Соединения формулы (I) в которой R1 и R2, которые являются одинаковыми или разными, представляют собой атом водорода или алкильную группу или вместе с атомом азота, к которому они присоединены, образуют гетероцикл, R3 представляет собой атом галогена, алкоксигруппу, необязательно замещенную арильную группу или группу NR'1R'2, в которой R'1 и R'2, которые являются одинаковыми или разными, представляют собой атом водорода или алкильную группу...
Новые соединения фенилнафталина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 8250
Опубликовано: 27.04.2007
Авторы: Делагранж Филипп, Пуассоннье-Дюрьё Софи, Беннежан Каролин, Ренар Пьер, Йоус Саид, Лезьё Даньель
МПК: A61K 31/16, A61P 25/00, A61K 31/40...
Метки: новые, содержат, получения, которые, композиции, фенилнафталина, соединения, фармацевтические, способ
Формула / Реферат:
1. Соединения формулы (I) в которой А представляет собой группировку в которой R1 представляет собой линейную или разветвленную C1-С6-алкильную группу или С3-С8-циклоалкильную группу и R2 представляет собой атом водорода, и дополнительно возможно, что R1 и R2 вместе образуют линейную или разветвленную алкиленовую цепь, содержащую от 3 до 6 атомов углерода, R3 представляет собой линейную или разветвленную C1-С6-алкоксигруппу, R4 представляет...
Новые соединения бензотиазина и бензотиадиазина, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 8797
Опубликовано: 31.08.2007
Авторы: Корди Алекс, Лестаж Пьер, Десо Патрик
МПК: A61K 31/549, C07D 513/04, A61P 25/28...
Метки: фармацевтические, новые, получения, способ, бензотиадиазина, композиции, бензотиазина, которые, содержат, соединения
Формула / Реферат:
1. Соединения формулы (I) где R1 представляет собой атом водорода, атом галогена или линейную или разветвленную C1-С6-алкильную группу, R1a представляет собой атом водорода или линейную или разветвленную C1-С6-алкильную группу, R2 представляет собой атом водорода, атом галогена или гидроксильную группу, А представляет собой CR4R5-группу или NR4-группу, R3 представляет собой атом водорода, линейную или разветвленную C1-С6-алкильную группу или...
Предыдущий патент: Применение эффекторов глутаминил- и глутаматциклаз
Следующий патент: Пестициды на основе сульфониламинопиразолов
Случайный патент: Система для подъёма и опускания рубашки резервуара