Производные индола, обладающие противоопухолевой активностью
Номер патента: 13542
Опубликовано: 30.06.2010
Авторы: Дзунино Франко, Баттистуцци Джанфранко, Ди Марцо Мария, Пизано Клаудио, Джаннини Джузеппе, Весчи Лоредана, Марци Мауро
















Формула / Реферат
1. Соединение формулы (I)

где А представляет собой

A1 соответствует А, где R, R' и R1 являются одинаковыми для обоих колец индола;
X представляет собой насыщенный или ненасыщенный (алкенилен или алкинилен), неразветвленный или разветвленный (С3-С10)алкилен, необязательно замещенный ОН;
Q представляет собой SH, COCONR2R3, COCF3 или
Q представляет собой CONHOG

где G представляет собой либо Н, либо гликозил;
R2, R3 являются одинаковыми или разными и представляют собой Н или (С1-С4)алкил;
R1 выбран из группы, состоящей из Н, (С1-С4)алкила, (C6-С12)арила, (С6-С12)арил-(C1-С4)алкилена, (C1-С4)алканоила и (C1-С4)алкил-(С6-С10)арилена;
R и R', одинаковые или разные, выбраны из группы, состоящей из -Н;
насыщенного или ненасыщенного, неразветвленного или разветвленного (С1-С10)алкила, необязательно замещенного (С3-С10)гетероарилом или (С3-С10)гетероциклил-(С1-С4)алкиленом, где гетероцикл содержит по меньшей мере один гетероатом, выбранный из N, О или S, или группой -NR5R6, где R5, R6 являются одинаковыми или разными и представляют собой Н, неразветвленный или разветвленный (C1-С4)алкил, (С1-С4)алканоил;
OR4, где R4 представляет собой Н, (C1-С4)алкил, мезил, тозил, (С1-С4)алканоил, гликозил;
галогена, азида, нитро, нитрила и NR5R6.
2. Соединение по п.1, где X представляет собой насыщенный или ненасыщенный неразветвленный С2-С10алкилен.
3. Соединение по п.1, где Q представляет собой CONHOG.
4. Соединение по п.1 или 3, где G, R1, R2 и R3 являются одинаковыми и представляют собой Н.
5. Соединение по п.1, где R и R' являются одинаковыми и выбраны из группы, состоящей из Н, (C1-С3)алкиламина и (C1-С3)алкилморфолина.
6. Соединение формулы (I) по п.1, которое выбрано из группы, состоящей из
гидроксиамида 5,5-бис-(1Н-индол-3-ил)пентановой кислоты,
гидроксиамида 6,6-бис-(1Н-индол-3-ил)гексановой кислоты,
гидроксиамида 7,7-бис-(1Н-индол-3-ил)гептановой кислоты,
гидроксиамида 7,7-бис-(4-фтор-1Н-индол-3-ил)гептановой кислоты,
гидроксиамида 7,7-бис-(5-метил-1Н-индол-3-ил)гептановой кислоты,
гидроксиамида 7,7-бис-(7-этил-1Н-индол-3-ил)гептановой кислоты,
гидроксиамида 7,7-бис-(7-метокси-1Н-индол-3-ил)гептановой кислоты,
гидроксиамида 7,7-бис-(5-морфолин-4-илметил-3Н-индол-3-ил)гептановой кислоты,
гидроксиамида 7-(1Н-индол-3-ил)-7-(1Н-индол-2-ил)гептановой кислоты,
7,7-бис-(1H-индол-3-ил)гептановой кислоты,
гидроксиамида 8,8-бис-(1Н-индол-3-ил)октановой кислоты.
7. Способ получения соединений по любому из предыдущих пунктов, у которых А представляет собой A1, включающий димеризацию исходного производного индола в присутствии альдегида или ацеталя.
8. Применение соединения по любому из пп.1-6 в качестве лекарственного средства.
9. Применение соединения по любому из пп.1-6 для изготовления лекарственного средства с противоопухолевой активностью.
10. Применение соединения по любому из пп.1-6 для изготовления лекарственного средства для лечения опухолевой патологии, в которой опухоль обнаруживала резистентность к противоопухолевым лекарственным средствам, ранее применяемым для ее лечения, в котором указанное соединение формулы (I) оказывает химиосенсибилизирующее действие на опухоль, резистентную к указанным лекарственным средствам.
11. Применение по п.9 или 10, где опухоль выбрана из группы, состоящей из саркомы, карциномы, карциноида, опухоли кости, нейроэндокринной опухоли, лимфоидного лейкоза, острого промиелоцитарного лейкоза, миелоидного лейкоза, моноцитарного лейкоза, мегакариобластного лейкоза и болезни Ходжкина.
12. Применение по п.9 или 10, в котором соединение формулы (I) комбинируют с одним или несколькими известными противоопухолевыми агентами.
13. Применение по п.12, в котором известным противоопухолевым агентом является полностью транс-изомер ретиноевой кислоты.
14. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы (I) по любому из пп.1-6 и по меньшей мере один фармацевтически приемлемый эксципиент и/или разбавитель.
15. Способ получения композиции по п.13, включающий смешивание соединения(й) по любому из пп.1-6 с подходящим эксципиентом(ами) и/или разбавителем(ями).
16. Способ лечения млекопитающего, страдающего заболеванием по любому из пп.9-11, включающий введение терапевтически эффективного количества соединения по любому из пп.1-6.
Текст
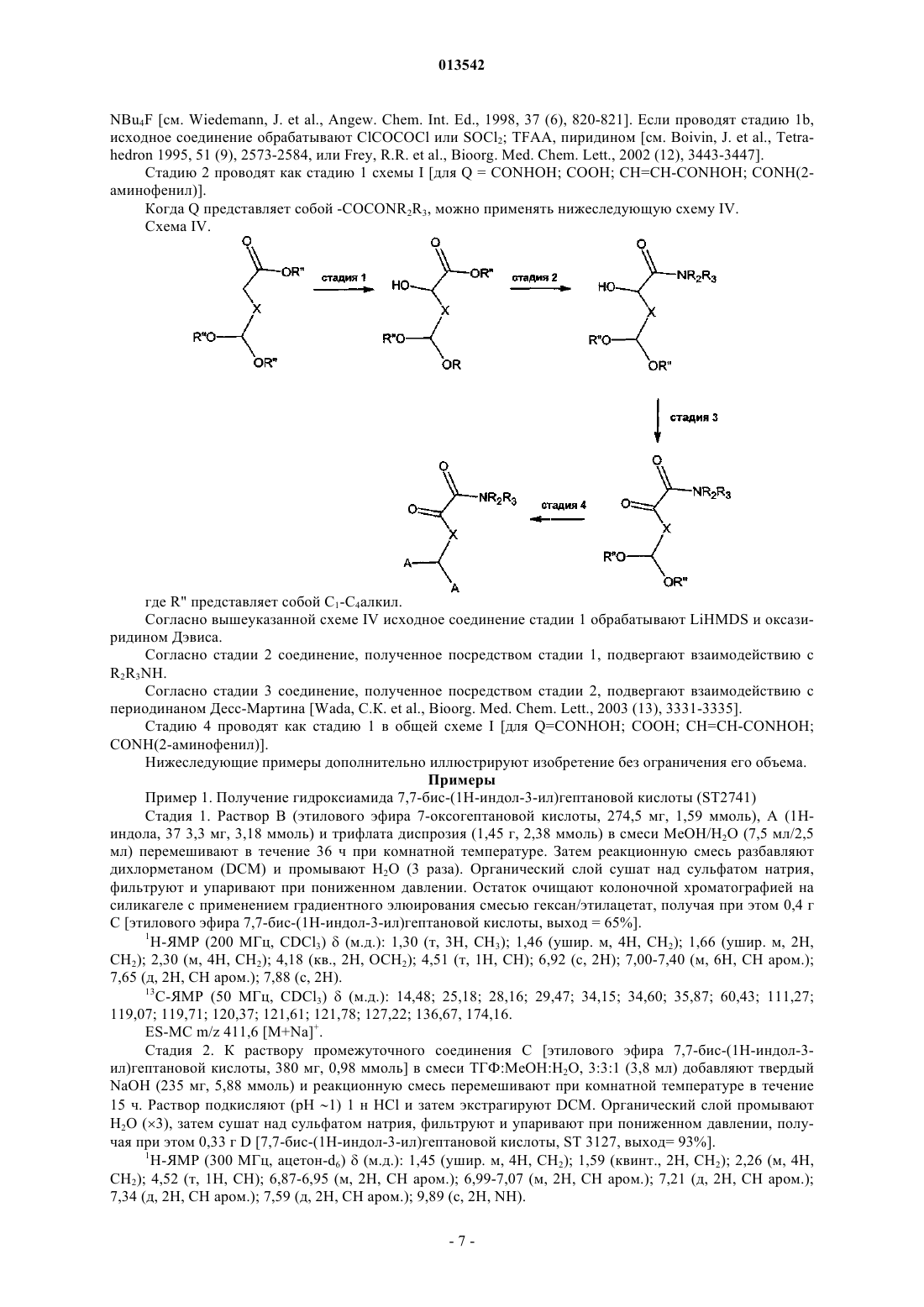
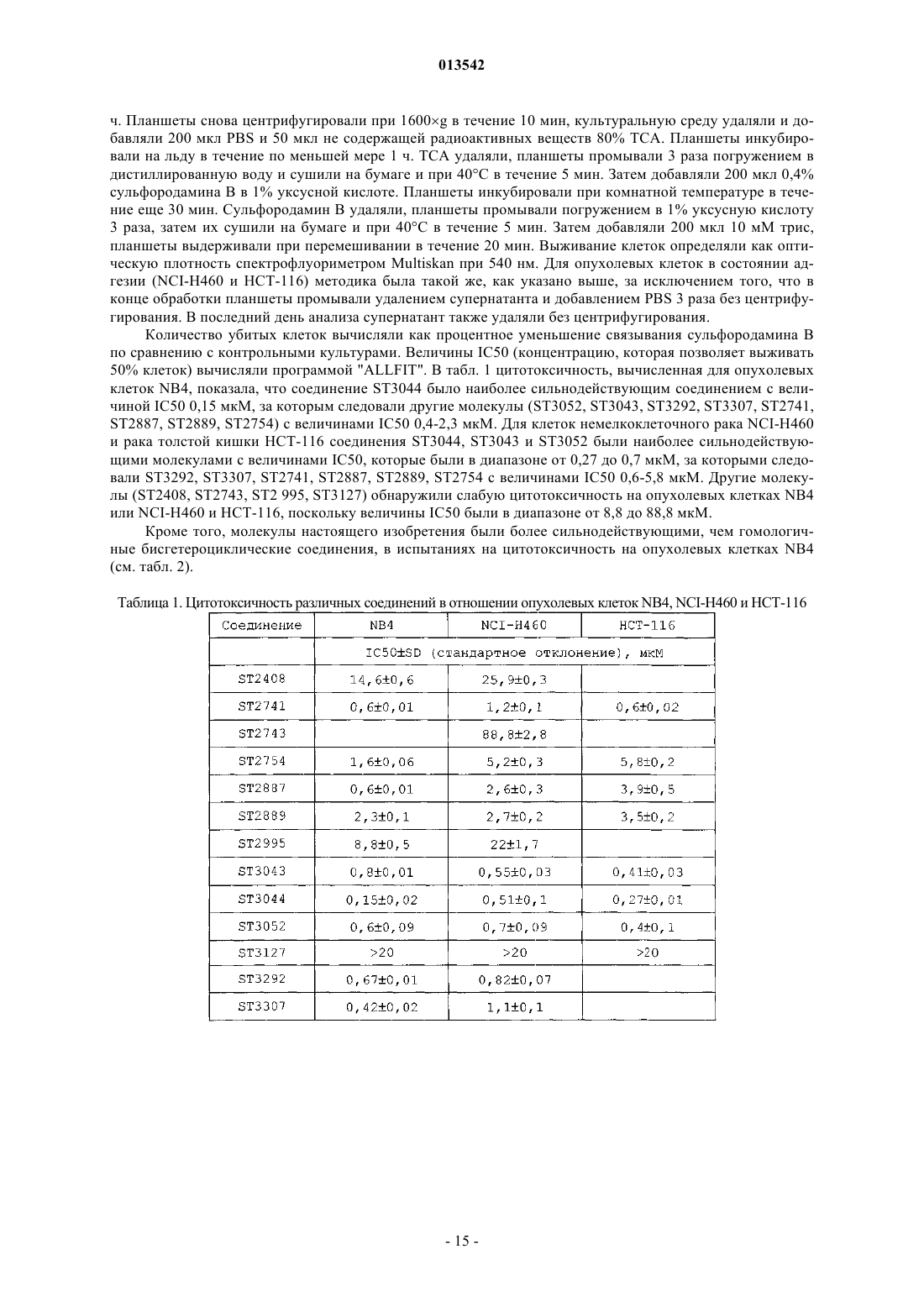
013542 Настоящее изобретение относится к производным индола, обладающим противоопухолевой активностью, а также фармацевтическим композициям, содержащим вышеуказанные соединения, для лечения опухолей. Терапию опухолей в настоящее время проводят хирургическим вмешательством, лучевой терапией и химиотерапией. Недостатки последней терапии в основном обусловлены токсичностью цитотоксических лекарственных средств, действие которых обычно не ограничивается раковыми клетками, и приобретенной резистентностью раковых клеток к некоторым из наиболее широко применяемых лекарственных средств, которая снижает эффективность продолжительной терапии. Удаление первичной опухоли хирургией не всегда возможно и в любом случае не предотвращает образование большинства метастазирующих опухолей, таких как, например, рак молочной железы или меланома, распространяющихся на другие органы-мишени. Очевидной становилась маловероятность того, что терапия метастазирующих опухолей приведет к полному излечению пациента; поэтому лечение цитотоксическими лекарственными средствами теперь рассматривают в качестве паллиативы и метода пролонгирования жизни, а не в качестве целебного метода. Постоянное лечение лекарственным средством, обладающим низкой токсичностью, может быть предпочтительным при направленной доставке для подавления развития заболевания. В течение последних лет разработка лекарственных средств для лечения рака двигалась от общепринятых цитотоксических химиотерапий к более основанному на механизме направленной доставки подходу для достижения общей цели остановки роста опухоли. Быстрый прогресс в исследовании хроматина и понимании эпигенетического регулирования обеспечил плетору потенциальных мишеней для вмешательства в раковое заболевание. Гистондеацетилазы (HDAC) принимают широкое участие в регуляции роста и транскрипции клеток, и ингибирование активности HDAC с применением небольших молекул вызывает апоптоз в опухолевых клетках. Теперь известно, что ингибиторы гистондеацетилазы являются сильнодействующими индукторами остановки роста, дифференциации или апоптотической гибели клеток в различных трансформированных клетках в культуре и у имеющих опухоли животных (Marks, P.A., Current Opinions inOncology, 2001, Nov. 13(6): 477-83; Marks, P., Nat. Rev. Cancer 2001 Dec. 1 (3): 194-202). С другой стороны, как и предполагалось ранее, другим очень важным и воспринимаемым с большим интересом аспектом онкологической терапии является начало резистентности к применяемому лекарственному средству обработанных опухолевых клеток. Клетки, которые развивают резистентность к лекарственному средству, часто способны проявлять резистентность к действиям многих других противоопухолевых лекарственных средств, даже если они не являются химически родственными или имеют другой механизм действия (Annu. Rev. Med. 1991, 42: 277-286; Drugs of Future 1997, 22: 653-660). Ряд опухолей, таких как, например, опухоли коркового вещества надпочечника, толстой кишки, почек и тощей кишки и карцинома печени, проявляют резистентность к лекарственному средству сразу, с самого начала лечения противоопухолевыми лекарственными средствами (Barrows, L. R. Antineoplasticand immunoactive Drugs, 1995; 75; 1236-1262). В других случаях опухолевые клетки приобретают резистентность, до некоторой степени аналогичную резистентности бактерий к антибиотикам. Этот тип резистентности имеет генетические или эпигенетические изменения; эти изменения позволяют дочерним клеткам пролиферировать в окружающей среде, в которой присутствует противоопухолевый агент. Независимо от причины резистентности, она приводит к неэффективности при длительном противоопухолевом лечении. В заявке на патент WO 99/00381 описаны производные бисиндола с антиметастатической активностью. В заявке на патент WO 02/36561, зарегистрированной на имя заявителя, описаны бисгетероциклические соединения, полезные в качестве противоопухолевых агентов. В заключение, в области онкологии существует значительная потребность в новых соединениях,обладающих противоопухолевой и/или химиосенсибилизирующей активностью, т.е. соединениях, которые являются активными против резистентных к лекарственным средствам опухолей и/или способны делать известные противоопухолевые лекарственные средства активными против опухолей, против которых они были неэффективными вследствие начала вышеуказанных условий резистентности к лекарственным средствам. Описание изобретения Авторами изобретения обнаружено, что класс производных индола обладает всем необходимым,что является существенным для такой противоопухолевой, антиметастатической и химиосенсибилизирующей активности. Обнаружено, что соединения настоящего изобретения являются более сильнодействующими, чем гомологичные соединения, описанные в заявке на патент WO 02/36561, при ингибировании пролиферации клеток промиелоцитарного лейкоза NB4 (см. результаты, представленные в табл. 1 и 2 в разделе примеров). Эти неожиданные антипролиферативные результаты, обнаруженные для этого нового класса соединений, обуславливают противораковую потенциальную возможность применения их для лечения-1 013542 раковых пациентов, резистентных к применяемым в настоящее время терапиям. Поэтому основной задачей настоящего изобретения являются производные индола приведенной ниже формулы (I), которые являются полезными агентами в качестве противоопухолевых, антиметастатических и химиосенсибилизирующих средств.A1 представляет собой либо Н, либо А, где R, R' и R1 являются одинаковыми для обоих колец индоX представляет собой насыщенный или ненасыщенный (алкенилен или алкинилен), неразветвленный или разветвленный (С 2-С 10)алкилен, необязательно замещенный ОН, или (C6-C12)арил, (С 3 С 10)гетероарил, содержащий по меньшей мере один гетероатом, выбранный из N, О или S, где в циклических группах по меньшей мере один -СН- необязательно заменен С-галогеном или С-(C1-С 3)алкилом;Q представляет собой SH, COCONR2R3, COCF3 или Q представляет собой CONHOG, СООН, где G представляет собой либо Н, либо гликозил;R2, R3 являются одинаковыми или разными и представляют собой Н или (C1-С 4)алкил;R и R', одинаковые или разные, выбраны из группы, состоящей из -Н; насыщенного или ненасыщенного, неразветвленного или разветвленного (С 1-С 10)алкила, необязательно замещенного (С 3-С 10)гетероарилом или (С 3-С 10)гетероциклил-(C1-С 4)алкиленом, где гетероцикл содержит по меньшей мере один гетероатом, выбранный из N, О или S, или группой -NR5R6A где R5, R6 являются одинаковыми или разными и представляют собой Н, неразветвленный или разветвленный (C1 С 4)алкил, (C1-С 4)алканоил;OR4, где R4 представляет собой Н, (C1-С 4)алкил, мезил, тозил, (C1-С 4)алканоил, гликозил; галогена, азида, нитро, нитрила и NR5R6. Настоящее изобретение включает в себя также таутомеры, геометрические изомеры, оптически активные формы, как энантиомеры, диастереомеры, и рацематные формы, а также фармацевтически приемлемые соли соединений формулы (I). Предпочтительными фармацевтически приемлемыми солями формулы (I) являются кислотноаддитивные соли, образованные с фармацевтически приемлемыми кислотами, подобные гидрохлоридным, гидробромидным, сульфатным или бисульфатным, фосфатным или гидрофосфатным, ацетатным,бензоатным, сукцинатным, фумаратным, малеатным, лактатным, цитратным, тартратным, глюконатным,метансульфонатным, бензолсульфонатным и пара-толуолсульфонатным солям. Понятно, что в объеме настоящего изобретения примеры неразветвленной или разветвленной (C1 С 4)алкильной группы включают в себя метил, этил, пропил и бутил и их возможные изомеры, такие как,например, изопропил, изобутил и трет-бутил. Термин алкилен относится к двухвалентному углеводородному радикалу с неразветвленной или разветвленной цепью. Используемые в описании примеры алкилена включают в себя, но не ограничиваются перечисленным, метилен, этилен, пропан-1,3-диил, пропан-1,2-диил и тому подобное. Примерами (С 6-С 12)арильной или (С 6-С 12)арил-(С 1-С 4)алкильной группы являются фенил, 1-или 2 нафтил, антраценил, бензил, 2-фенилэтил, 1-фенилэтил, 3-фенилпропил, 2-антраценилпропил, 1-2 013542 антраценилпропил, нафтилметил, 2-нафтилэтил, 1-нафтилэтил, 3-нафтилпропил, 2-нафтилпропил, 1 нафтилпропил. Используемый в описании термин (С 3-С 6)гетероцикло или термин(С 3-С 6)гетероциклил относится к одновалентному радикалу от трех- до шестичленного неароматического кольца, содержащего один или несколько гетероатомных замен, независимо выбранных из S, О или N, и имеющего нуль-пять ненасыщенных связей. Примеры гетероциклила, используемого в описании, включают в себя, но не ограничиваются перечисленным, тетрагидрофурил, пиранил, 1,4-диоксанил, 1,3-диоксанил, пиперидинил, пирролидинил, тетрагидротиопиранил, тетрагидротиофенил и тому подобное. Используемый в описании термин (С 3-С 6)гетероциклилен относится к двухвалентному радикалу трех-шестичленного неароматического гетероциклического кольца, содержащего один или несколько гетероатомов, независимо выбранных из S, О или N, и имеющего нуль-пять ненасыщенных связей. Примеры гетероциклилена, используемого в описании, включают в себя, но не ограничиваются перечисленным, тетрагидрофуран-2,5-диил, пиран-2,4-диил, 1,4-диоксан-2,3-диил, 1,3-диоксан-2,4-диил, пиперидин-2,4-диил, пиперидин-1,4-диил, пирролидин-1,3-диил и тому подобное. Термин галоген означает фтор, хлор, бром и йод. Примерами гликозильного остатка являются 6-D-галактозил и 6-D-глюкозил. Согласно независимо предпочтительным вариантам осуществления изобретения X представляет собой насыщенный или ненасыщенный (алкенилен или алкинилен), неразветвленный (С 2-С 10)алкилен;G, R1, R2 и R3 представляют собой водород; и R и R' являются одинаковыми или разными и представляют собой Н, неразветвленный (С 1 С 10)алкил, необязательно замещенный (С 3-С 12)гетероарилом, содержащим по меньшей мере один гетероатом, выбранный из N, О или S, или группой -NR5R6 или OR4, где R4 представляет собой (C1-С 4)алкил. Некоторыми из наиболее предпочтительных соединений согласно изобретению являются следующие соединения: 3-[4-бис-(1 Н-индол-3-ил)метил]фенил-N-гидроксиакриламид (ST2887),гидроксиамид 5,5-бис-(1 Н-индол-3-ил)пентановой кислоты (ST2743),гидроксиамид 6,6-бис-(1 Н-индол-3-ил)гексановой кислоты (ST2754),гидроксиамид 7,7-бис-(1 Н-индол-3-ил)гептановой кислоты (ST2741),гидроксиамид 7,7-бис-(4-фтор-1 Н-индол-3-ил)гептановой кислоты (ST3052),гидроксиамид 7,7-бис-(5-метил-1 Н-индол-3-ил)гептановой кислоты (ST3044),гидроксиамид 7,7-бис-(7-этил-1 Н-индол-3-ил)гептановой кислоты (ST3126),гидроксиамид 7,7-бис-(7-метокси-1 Н-индол-3-ил)гептановой кислоты (ST3043),гидроксиамид 8,8-бис-(1 Н-индол-3-ил)октановой кислоты (ST2889),N-гидрокси-4,4-бис-(lH-индол-3-ил)бутирамид (ST2408),N-гидрокси-6-(1 Н-индол-3-ил)гексанамид (ST2995),гидроксиамид 7,7-бис-(5-морфолин-4-илметил-3 Н-индол-3-ил)гептановой кислоты (ST3307),гидроксиамид 7-(1 Н-индол-3-ил)-7-(1 Н-индол-2-ил)гептановой кислоты (ST3292) и 7,7-бис-(1 Н-индол-3-ил)гептановая кислота (ST3127). Полученные экспериментальные результаты (указываются в разделе, озаглавленном Примеры) показывают, что соединения формулы (I), как по отдельности, так и в комбинации с другими известными противоопухолевыми лекарственными средствами, являются полезными агентами для лечения опухолей. Следующим объектом настоящего изобретения являются соединения общей формулы (I) и их применение в области медицины. Следующим объектом настоящего изобретения является фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы (I) и по меньшей мере один фармацевтически приемлемый эксципиент и/или разбавитель. Следующим объектом настоящего изобретения являются соединения общей формулы (I) и способ их получения. Следующим объектом настоящего изобретения является фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы (I), применяемая для лечения опухолевой патологии, в которой опухоль выбрана из группы, состоящей из саркомы, карциномы, карциноида, опухоли костей, нейроэндокринной опухоли, лимфоидного лейкоза, острого промиелоцитарного лейкоза,миелоидного лейкоза, моноцитарного лейкоза, мегакариобластного лейкоза и болезни Ходжкина. Следующим объектом настоящего изобретения является фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы (I), применяемая для лечения опухолевой-3 013542 патологии, в которой опухоль обнаруживает резистентность к антибиотическим лекарственным средствам, ранее применяемым для ее лечения, в которой указанное соединение формулы (I) оказывает химиосенсибилизирующее действие на опухоль, резистентную к указанным лекарственным средствам. Следующим объектом настоящего изобретения является фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы (I) в комбинации с одним или несколькими известными противоопухолевыми агентами, в которой противоопухолевое соединение выбрано из группы, состоящей из алкилирующих агентов, ингибиторов топоизомеразы, антитубулиновых агентов, интеркалирующих соединений, антиметаболитов, природных продуктов, таких как винкаалкалоиды, эпиподофиллотоксинов, антибиотиков, ферментов, таксанов и цитодифференцирующих соединений. Среди цитодифференцирующих противоопухолевых агентов предпочтительным является полностью трансизомер ретиноевой кислоты. Следующим объектом настоящего изобретения является способ получения фармацевтических композиций, описываемых ранее, включающий в себя смешивание соединения(й) формулы (I) с подходящим эксципиентом(ами) и/или разбавителем(ями). Следующим объектом настоящего изобретения является применение соединения формулы (I) для получения лекарственного средства для лечения опухолевой патологии. Следующим объектом настоящего изобретения является применение соединения формулы (I) для получения лекарственного средства для лечения опухолевой патологии, в которой опухоль обнаруживает лекарственную резистентность к противоопухолевым лекарственным средствам, ранее применяемым для ее лечения, в которой указанное соединение формулы (I) оказывает химиосенсибилизирующее действие на резистентность опухоли к указанным лекарственным средствам. Следующим объектом настоящего изобретения является применение соединения формулы (I) в комбинации с одним или несколькими известными противоопухолевыми агентами для получения лекарственного средства для лечения опухолевых патологий. Следующим объектом настоящего изобретения является применение соединения формулы (I) в комбинации с полностью транс-изомером ретиноевой кислоты для получения лекарственного средства для лечения острого промиелоцитарного лейкоза. Следующим объектом настоящего изобретения является способ лечения млекопитающего, страдающего опухолевой патологией, включающий в себя введение терапевтически эффективного количества соединения(й) формулы (I). Терапевтически эффективным количеством является количество, эффективное для достижения желательного результата лечения у подвергаемого лечению субъекта. Фармацевтические композиции могут содержать подходящие фармацевтически приемлемые носители, биологически совместимые наполнители, подходящие для введения животному (например, физиологический солевой раствор), и в конечном счете включающие в себя вспомогательные средства (подобные эксципиентам, стабилизаторам или разбавителям), которые облегчают изготовление из активных соединений препаратов, которые можно применять в фармации. Фармацевтические композиции можно изготовлять любым приемлемым способом, который удовлетворяет требованиям способа введения. В литературе описано применение биоматериалов и других полимеров для доставки лекарственного средства, а также различных методик и моделей для обоснованности определенного способа введения. Могут быть также полезными модификации соединений изобретения для улучшения проникновения их через гематоэнцефалический барьер. Можно применять любой приемлемый способ введения, его определяет специалист в данной области. Например, введение может быть проведено различными парентеральными путями, такими как подкожный, внутривенный, внутрикожный, внутримышечный, внутрибрюшинный, интраназальный, чрескожный, пероральный или трансбуккальный путь. Парентеральным введением может быть инъекция болюса или постепенная перфузия на протяжении определенного времени. Препараты для парентерального введения включают в себя стерильные водные или неводные растворы, суспензии и эмульсии, которые могут содержать вспомогательные агенты или эксципиенты, известные в данной области, их можно получить согласно обычным методам. Кроме того, можно вводить суспензию активных соединений, при необходимости инъекцией суспензий в масле. Подходящие липофильные растворители или наполнители включают в себя жирные масла, например кунжутное масло, или синтетические эфиры жирных кислот, например кунжутное масло, или синтетические эфиры жирных кислот, например этилолеат или триглицериды. Водные суспензии для инъекции, которые могут содержать вещества, повышающие вязкость суспензии, включают в себя, например, натриевую соль карбоксиметилцеллюлозы, сорбит и/или декстран. Суспензия может необязательно содержать стабилизаторы. Фармацевтические композиции включают в себя подходящие растворы для введения инъекцией и содержат от приблизительно 0,01 до 99%, предпочтительно от приблизительно 20 до 75% активного соединения, вместе с эксципиентом. Композиции, которые можно вводить ректально, включают в себя суппозитории.-4 013542 Понятно, что вводимая доза будет зависеть от возраста, пола, здоровья и массы реципиента, типа совместного лечения, если его применяют, частоты лечения и природы требуемого действия. Доза будет разработана для отдельного субъекта на основе знания и определения специалиста в данной области. Общую дозу, требуемую для каждого лечения, можно вводить в виде нескольких доз или в виде одной дозы. Фармацевтическую композицию настоящего изобретения можно вводить как таковую или в сочетании с другими терапиями, направленными для лечения данного состояния, или направленными для лечения других симптомов состояния. Суточная доза активного ингредиента обычно находится между 0,01 и 100 мг на килограмм массы тела. Соединения настоящего изобретения можно вводить пациенту внутривенно в фармацевтически приемлемом носителе, таком как физиологический солевой раствор. Можно применять стандартные методы внутриклеточной доставки пептидов, например доставку посредством липосом. Такие методы являются хорошо известными специалистам в данной области. Препараты данного изобретения являются применимыми для парентерального введения, такого как внутривенное, подкожное, внутримышечное и внутрибрюшинное введение. Как хорошо известно в области терапии, дозы для любого пациента зависят от многих факторов,включающих в себя размер пациента, площадь поверхности тела, возраст, конкретное вводимое соединение, пол, время и путь введения, общее здоровье и другие, одновременно вводимые лекарственные средства. Все ссылки, цитированные в описании, включены в качестве ссылки во всей их полноте, в том числе все данные, таблицы, фигуры и текст, представленный в цитированных ссылках. Кроме того, полные содержания ссылок, цитированных в ссылках, цитированных в описании, также включены в качестве ссылки во всей их полноте. Ссылка на стадии известных способов, стадии общепринятых способов, известные способы или общепринятые способы не является во всяком случае допущением, что любой аспект, описание или вариант осуществления настоящего изобретения описан, указан или предложен в рассматриваемой области. После ознакомления с признаками способов и продуктов, описанных в настоящей заявке, можно легко обосновать необходимость и тип дополнительных стадий просмотром известного уровня техники,а также неограничивающих нижеследующих фигур и примеров, описывающих основные подробности и некоторые применения изобретения. Соединения формулы (I) можно получить из легко доступных исходных веществ с применением нижеследующих общих способов и методик. Будет понятно, что, когда приводятся типичные или предпочтительные экспериментальные условия (т.е. температуры, время реакции, моли реагентов, растворители и т.д.), можно применять также другие экспериментальные условия, если не оговорено особо. Оптимальные условия реакции могут варьировать в зависимости от конкретных применяемых реагентов или растворителей, но такие условия может определить специалист в данной области стандартными методиками оптимизации. Способ получения соединений формулы (I), в которой А представляет собой А 1, включает в себя димеризацию исходного производного индола в присутствии альдегида или ацеталя. Соединения настоящего изобретения можно получить, например, согласно нижеследующей общей схеме. Общая схема I. [для Q = CONHOH; COOH; CH=CH-CONHOH; CONH(2-аминофенил)]R" представляет собой С 1-С 4 алкил. Общая методика для стадии 1. Стадию 1 проводят в органических, полярных или неполярных, протонных или апротонных растворителях или их смесях, т.е. дихлорметане, дихлорэтане, ацетонитриле, метаноле, этаноле, диметилфор-5 013542 мамиде, воде, уксусной кислоте. Реагент В подвергают взаимодействию в виде свободного альдегида или ацеталя и реакцию катализируют органическими, неорганическими кислотами или кислотами Льюиса,т.е. HCl (Herbert R. et al., J. Chem. Soc, 1969, 1505), трифторуксусной кислотой [Tominaga Y. et al., Heterocycles, 2001, 55 (8), 1447-1450], уксусной кислотой (Wang Q.M. et al., Synlett, 1995, 12, 1267-1268), трифлатом диспрозия [Chen D. et al., Tetrahedron Lett, 1996, 37 (26), 4467-4470], перхлоратом лития [Yadav J.S.et al, Synthesis, 2001, (5), 783-787]. Применяют температуры от -10 до 150 С. Соединения С можно очищать флэш-хроматографией на силикагеле. Общая методика для стадии 2. Стадию 2 обычно проводят в смеси воды и органических растворителей, таких как метанол, этанол,с применением органического основания, такого как NaOH, LiOH. После перемешивания при комнатной температуре реакционную смесь подкисляют 1 н HCl и соединения D очищают экстракцией органическими растворителями, т.е. дихлорметаном, этилацетатом. Общая методика для стадии 3. Стадию 3 проводят в органических, полярных или неполярных, апротонных растворителях или их смесях, т.е. дихлорметане, дихлорэтане, диметилформамиде, тетрагидрофуране, диоксане. Можно применять различные агенты конденсации и/или активации, такие как DCC, EDC, HOBt, HATU, PyBOP[Lloyd-Williams P., Albericio F. and Giralt E., Chemical Approaches to the synthesis of peptides and proteins,(1997) и ссылки], и реакцию катализируют применением органического основания, такого как Nметилморфолин, N,N-диизопропилэтиламин, триэтиламин, DBU [Sandanayaka V.P. et al., Tetrahedron Lett,2001, 42 (28), 4605-4607. Bailen M.A. et al., Tetrahedron Lett, 2001, 42 (30), 5013-5016]. Применяют температуры от -10 до 100 С. Соединения F очищают хроматографией на силикагеле или высокоэффективной хроматографией на обращенной фазе (ОФ-ВЭЖХ). Общая методика для стадии 4. Если Z промежуточного соединения F представляет собой O-бензильную группу, взаимодействие проводят в органических растворителях, т.е. метаноле, с применением давлений водорода от 15 до 60 фунт/кв. дюйм и различных катализаторов, таких как 10% Pd/C, 5 или 10% Pd/BaSO4 [Nikam S.S. et al.,Tetrahedron Lett, 1995, 36 (2), 197-200]. Конечные продукты G очищают выпариванием растворителя и в конце колоночной хроматографией на силикагеле или высокоэффективной хроматографией с обращенной фазой. Когда Q представляет собой -SH, можно применять нижеследующую схему II. Схема II. [для Q = -SH] Согласно вышеуказанной схеме II исходное соединение стадии 1 (которое соответствует соединению С схемы I) обрабатывают NaBH4. Согласно стадии 2 а соединение, полученное посредством стадии 1, подвергают взаимодействию с реагентом Лавессона [см. Rajagopalan, S. et al., Synth. Comm. 1997, 27(1), 187-194]. Согласно стадии 2b соединение, полученное посредством стадии 1, подвергают взаимодействию с CH3COSH, DEAD, TPP [см. Wisniewski, К. et al., Bioorg. Med. Chem. Lett., 1999 (9), 2801-2804]. В стадии 3 соединения, полученные посредством стадии 2 или стадии 2b, обрабатывают NaOH, получая при этом соответствующие соединения формулы I. Когда Q представляет собой -COCF3, можно применять нижеследующую схему III. Схема III. где W представляет собой А или Y и R" представляет собой C1-С 4 алкил. Согласно вышеуказанной схеме III исходное соединение стадии 1 а обрабатывают (CH3)3SiCF3 иNBu4F [см. Wiedemann, J. et al., Angew. Chem. Int. Ed., 1998, 37 (6), 820-821]. Если проводят стадию 1b,исходное соединение обрабатывают ClCOCOCl или SOCl2; TFAA, пиридином [см. Boivin, J. et al., Tetrahedron 1995, 51 (9), 2573-2584, или Frey, R.R. et al., Bioorg. Med. Chem. Lett., 2002 (12), 3443-3447]. Стадию 2 проводят как стадию 1 схемы I [для Q = CONHOH; СООН; CH=CH-CONHOH; CONH(2 аминофенил)]. Когда Q представляет собой -COCONR2R3, можно применять нижеследующую схему IV. Схема IV. где R" представляет собой С 1-С 4 алкил. Согласно вышеуказанной схеме IV исходное соединение стадии 1 обрабатывают LiHMDS и оксазиридином Дэвиса. Согласно стадии 2 соединение, полученное посредством стадии 1, подвергают взаимодействию сR2R3NH. Согласно стадии 3 соединение, полученное посредством стадии 2, подвергают взаимодействию с периодинаном Десс-Мартина [Wada, С.К. et al., Bioorg. Med. Chem. Lett., 2003 (13), 3331-3335]. Стадию 4 проводят как стадию 1 в общей схеме I [для Q=CONHOH; СООН; CH=CH-CONHOH;CONH(2-аминофенил)]. Нижеследующие примеры дополнительно иллюстрируют изобретение без ограничения его объема. Примеры Пример 1. Получение гидроксиамида 7,7-бис-(1 Н-индол-3-ил)гептановой кислоты (ST2741) Стадия 1. Раствор В (этилового эфира 7-оксогептановой кислоты, 274,5 мг, 1,59 ммоль), А (1 Ниндола, 37 3,3 мг, 3,18 ммоль) и трифлата диспрозия (1,45 г, 2,38 ммоль) в смеси МеОН/Н 2 О (7,5 мл/2,5 мл) перемешивают в течение 36 ч при комнатной температуре. Затем реакционную смесь разбавляют дихлорметаном (DCM) и промывают H2O (3 раза). Органический слой сушат над сульфатом натрия,фильтруют и упаривают при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле с применением градиентного элюирования смесью гексан/этилацетат, получая при этом 0,4 г С [этилового эфира 7,7-бис-(1H-индол-3-ил)гептановой кислоты, выход = 65%]. 1 Н-ЯМР (200 МГц, CDCl3)(м.д.): 1,30 (т, 3 Н, СН 3); 1,46 (ушир. м, 4 Н, СН 2); 1,66 (ушир. м, 2 Н,СН 2); 2,30 (м, 4 Н, СН 2); 4,18 (кв., 2 Н, ОСН 2); 4,51 (т, 1 Н, СН); 6,92 (с, 2 Н); 7,00-7,40 (м, 6 Н, СН аром.); 7,65 (д, 2 Н, СН аром.); 7,88 (с, 2 Н). 13 С-ЯМР (50 МГц, CDCl3)(м.д.): 14,48; 25,18; 28,16; 29,47; 34,15; 34,60; 35,87; 60,43; 111,27; 119,07; 119,71; 120,37; 121,61; 121,78; 127,22; 136,67, 174,16.ES-MC m/z 411,6 [M+Na]+. Стадия 2. К раствору промежуточного соединения С [этилового эфира 7,7-бис-(1 Н-индол-3 ил)гептановой кислоты, 380 мг, 0,98 ммоль] в смеси ТГФ:МеОН:Н 2 О, 3:3:1 (3,8 мл) добавляют твердыйNaOH (235 мг, 5,88 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 15 ч. Раствор подкисляют (рН 1) 1 н HCl и затем экстрагируют DCM. Органический слой промывают Н 2 О (3), затем сушат над сульфатом натрия, фильтруют и упаривают при пониженном давлении, получая при этом 0,33 г D [7,7-бис-(1 Н-индол-3-ил)гептановой кислоты, ST 3127, выход= 93%]. 1ES-MC m/z: 359,2 [М-Н]- и 383,2 [M+Na]+. Стадия 3. К раствору промежуточного соединения D [7,7-бис-(1 Н-индол-3-ил)гептановой кислоты,140 мг, 0,388 ммоль] и РуВОР (259,5 мг, 0,465 ммоль) в DCM (1 мл) добавляют NMM (0,213 мл, 1,94 ммоль) и Е (гидрохлорид О-бензилгидроксиламина, 74,3 мг, 0,465 ммоль). Раствор перемешивают при комнатной температуре в течение 2,5 ч, затем разбавляют этилацетатом и промывают Н 2 О (3). Органический слой сушат над сульфатом натрия, фильтруют и упаривают при пониженном давлении. Остаток очищают препаративной ТСХ на силикагеле с применением смеси гексан/этилацетат, 7:3, в качестве системы элюентов, получая при этом 0,12 г F (бензилоксиамида 7,7-бис-(1 Н-индол-3-ил)гептановой кислоты, выход =70%]. 1ES-MC m/z: 464,4 [М-Н]- и 488,5 [M+Na]+. Стадия 4. Палладий на активированном угле (10%, кат.) добавляют к раствору промежуточного соединения F [бензилоксиамида 7,7-бис-(1 Н-индол-3-ил)гептановой кислоты, 79 мг, 0,170 ммоль] в МеОН(4 мл). После 6 ч перемешивания в атмосфере водорода (15 фунт/кв. дюйм) реакционную смесь фильтруют и фильтрат выпаривают при пониженном давлении. Остаток очищают препаративной хроматографией с обращенной фазой (колонка Lichrosorb RP-18, 7 мкм, элюенты: CH3CN/H2O, 1:1, поток=10 мл/мин), получая при этом 50,0 мг G [гидроксиамида 7,7-бис-(lH-индол-3-ил)гептановой кислоты, ST 2741, выход= 78%]. 1 Н-ЯМР (300 МГц, ацетон-d6)(м.д.): 1,42 (ушир. 4 Н, СН 2); 1,57 (квинт., 2 Н, СН 2); 2,06 (ушир. 2 Н,СН 2); 4,48 (т, 1 Н, СН); 6,89 (м, 2 Н, СН аром.); 6,99 (м, 2 Н, СН аром.); 7,20 (с, 2 Н, СН аром.); 7,33 (д, 2 Н,СН аром.); 7,57 (д, 2 Н, СН аром.); 9,92 (ушир. 2 Н, NH). 13 С-ЯМР (75 МГц, ацетон-d6)(м.д.): 24,66; 31,65; 33,22; 34,84 (некоторые сигналы под сигналом растворителя); 110,37; 117,32; 118,45; 118,89; 120,09; 120,80; 120,94; 126,50; 136,28; 169,81.ES-MC m/z: 374,4 [М-Н]-. Пример 2. Получение гидроксиамида 5,5-бис-(1 Н-индол-3-ил)пентановой кислоты (ST2743) Стадия 1. Раствор А (1 Н-индола, 1,12 г, 9,75 ммоль), В [метилового эфира 5,5-диметоксипентановой кислоты; 521,3 мг, 2,96 ммоль] и трифлата диспрозия (2,64 г, 4,33 ммоль) в смеси МеОН/Н 2 О (6 мл/4 мл) подвергают взаимодействию, как в примере 1 (при 60 С), получая при этом 0,74 г С [метилового эфира 5,5-бис-(1 Н-индол-3-ил)пентановой кислоты, выход= 72%]. 1ES-MC m/z: 369,5 [M+Na]+. Стадия 2. Промежуточное соединение С [метиловый эфир 5,5-бис-(1 Н-индол-3-ил)пентановой кислоты, 495 мг, 1,43 ммоль] и твердый NaOH (4 57 мг, 11,4 ммоль) в смеси MeOH/DCM (3 мл/1 мл) подвергают взаимодействию, как в примере 1, получая при этом 0,45 г D [5,5-бис-(1 Н-индол-3 ил)пентановой кислоты, выход=95%]. 1 Н-ЯМР (200 МГц, CDCl3)(м.д.): 1,78 (м, 2 Н, СН 2); 2,28 (кв., 2 Н, СН 2); 2,41 (т, 2 Н, СН 2); 4,53 (т,1 Н, СН); 6,95 (с, 2 Н); 7,00-7,36 (м, 6 Н, СН аром.); 7,63 (д, 2 Н, СН аром.); 7,89 (с, 2 Н, NH). 13 С-ЯМР (50 МГц, CDCl3)(м.д.): 23,58; 33,96; 34,18; 35,17; 111,29; 119,14; 119,61; 119,76; 121,71; 121,85; 122,04; 127,07; 136,64; 179,85.ES-MC m/z: 331,4 [М-Н]-. Стадия 3. Раствор промежуточного соединения D [5,5-бис-(lH-индол-3-ил)пентановой кислоты, 202 мг, 0,608 ммоль], РуВОР (406,2 мг, 0,73 ммоль), NMM (0,33 мл, 3,04 ммоль) и Е (гидрохлорида Обензилгидроксиламина, 116,5 мг, 0,73 ммоль) в DCM (4 мл) подвергают взаимодействию в таких же условиях, как в примере 1, получая при этом 0,16 г F [бензилоксиамида 5,5-бис-(1 Н-индол-3-ил)пентановой кислоты, выход = 60%]. 1(т, 1 Н, СН); 4,86 (с, 2 Н, СН 2 О); 6,97-7,22 (м, 6 Н, СН аром.); 7,27-7,43 (ушир. 2 Н, СН аром.); 7,56 (д, 2 Н,СН аром.); 8,08 (с, 2 Н, NH). ES-MC m/z: 436,4 [М-Н]- и 460,3 [M+Na]+. Стадия 4. Взаимодействие промежуточного соединения F [бензилоксиамида 5,5-бис-(1 Н-индол-3 ил)аентановой кислоты, 132 мг, 0,30 ммоль] проводят, как в примере 1, получая при этом 0,10 г G [гид-8 013542 роксиамида 5,5-бис-(1 Н-индол-3-ил)пентановой кислоты, ST 274 3, выход = 97%]. 1 Н-ЯМР (300 МГц, CD3OH)(м.д.): 1,69 (ушир. квинт., 2 Н, СН 2); 2,22 (ушир. м, 4 Н, СН 2); 4,44 (т,1 Н, СН); 6,86 (м, 2 Н, СН аром.); 7,00 (т, 2 Н, СН аром.); 7,20 (м, 4 Н, СН аром.); 7,27 (д, 2 Н, СН аром.); 7,47 (д, 2 Н, СН аром.). 13 С-ЯМР (75 МГц, CD3OH)(м.д.): 25,80; 35,22; 36,45; 36,62; 112,11; 119,18; 120,24; 120,42; 122,08; 122,84; 128,41; 138,40; 179,45.ST 2754 синтезируют, как описано в примере 1. Стадия 1. Раствор А (1 Н-индола, 740 мг, 6,32 ммоль), В [этилового эфира 6-оксогексановой кислоты, 500 мг, 3,16 ммоль) и трифлата диспрозия (2,89 г, 4,74 ммоль) в смеси МеОН/Н 2 О (13 мл/8 мл) подвергают взаимодействию, как в примере 1 (при 65 С), получая при этом 0,60 г С [этилового эфира 6,6 бис-(1 Н-индол-3-ил)гексановой кислоты, выход = 50%]. 1ES-MC m/z: 375,6 [М+Н]+. Стадия 2. Промежуточное соединение С [этиловый эфир 6,6-бис-(lH-индол-3-ил)гексановой кислоты, 300 мг, 0,80 ммоль] и твердый NaOH (400 мг, 9,6 ммоль) в смеси MeOH/DCM (4 мл/0,5 мл) подвергают взаимодействию, как в примере 1, получая при этом 0,27 г D [6,6-бис-(1 Н-индол-3-ил)гексановой кислоты, выход=97%]. 1 Н-ЯМР (200 МГц, CDCl3)(м.д.): 1,48 (м, 2 Н, СН 2); 1,71 (м, 2 Н, СН 2); 2,29 (м, 4 Н, СН 2); 4,50 (т,1 Н, СН); 6,90-7,40 (м, 8 Н, СН аром.); 7,63 (д, 2 Н, СН аром.); 7,90 (с, 2 Н, NH). 13 С-ЯМР (50 МГц, CDCl3)(м.д.): 15,35; 24,91; 27,82; 34,01; 35,44; 111,20; 119,15; 119,70; 120,24; 121,54; 121,87; 127,17; 136,69; 179,48.ES-MC m/z: 345,5 [М-Н]-. Стадия 3. Раствор промежуточного соединения D [6,6-бис-(1 Н-индол-3-ил)гексановой кислоты, 232 мг, 0,69 ммоль], РуВОР (422,2 мг, 0,76 ммоль), NMM (0,38 мл, 3,45 ммоль) и Е (гидрохлорида Обензилгидроксиламина, 121,1 мг, 0,76 ммоль) в DCM (3 мл) подвергают взаимодействию в таких же условиях, как в примере 1, получая при этом 0,19 г F [бензилоксиамида 6,6-бис-(lH-индол-3-ил)гексановой кислоты, выход= 62%]. 1ES-MC m/z: 452,7 [М+Н]+. Стадия 4. Взаимодействие промежуточного соединения F [бензилоксиамида 6,6-бис-(1 Н-индол-3 ил)гексановой кислоты, 162,7 мг, 0,36 ммоль] проводят, как в примере 1, получая при этом 67 мг G [гидроксиамида 6,6-бис-(1 Н-индол-3-ил)гексановой кислоты, ST 2754, выход= 54%]. 1 Н-ЯМР (200 МГц, CD3OH)(м.д.): 1,44 (м, 2 Н, СН 2); 1,69 (м, 2 Н, СН 2); 2,05 (т, 2 Н, СН); 2,25 (кв.,2 Н, СН 2); 4,44 (т, 1 Н, СН); 6,89 (т, 2 Н, СН аром.); 7,05 (м, 4 Н, СН аром.); 7,31 (д, 2 Н, СН аром.); 7,50 (д,2 Н, СН аром.). 13 С-ЯМР (50 МГц, CD3OH)(м.д.): 25,03; 27,28; 32,05; 33,35; 34,75; 110,05; 117,65; 118,25; 119,77; 120,08; 120,69; 126,39; 136,20; 179,45.ES-MC m/z: 360,3 [М-Н]- и 384,3 [M+Na]+. Пример 4. Получение N-гидрокси-4,4-бис- (1 Н-индол-3-ил) бутирамида (ST2408) Стадия 1. Раствор А (1 Н-индола, 3,1 г, 26,5 ммоль), В [4-оксомасляной кислоты; 4,8 мл, 7,69 ммоль] и трифлата диспрозия (1,55 г, 2,5 ммоль) в смеси 95% EtOH/H2O (32 мл/16 мл) подвергают взаимодействию, как в примере 1, получая при этом 0,36 г С [метилового эфира 5,5-бис-(1 Н-индол-3-ил)пентановой кислоты, выход= 15%]. 1 Н-ЯМР (200 МГц, ДМСО-d6)(м.д.): 2,28 (м, 2 Н, СН 2); 2,48 (кв., 2 Н, СН 2); 4,48 (т, 1 Н, СН); 6,807,16 (м, 6 Н, СН аром.); 7,36 (д, 2 Н, СН аром.); 7,56 (д, 2 Н, СН аром.); 9,20 (с, 2 Н). ES-MC m/z: 317,4 [МН]-. Стадия 3. Раствор промежуточного соединения D [4,4-бис-(lH-индол-3-ил)масляной кислоты, ST 1961, 220 мг, 0,69 ммоль], РуВОР (392 мг, 0,70 ммоль), TEA (0,40 мл, 2,77 ммоль) и Е (гидрохлорида Обензилгидроксиламина, 113 мг, 0,70 ммоль) в DCM (40 мл) подвергают взаимодействию в таких же условиях, как в примере 1, получая при этом 0,20 г F [N-бензилокси-4,4-бис-(1 Н-индол-3-ил)бутирамида, выход= 68%].ES-MC m/z: 422,3 [М-Н]- и 424,5 [М+Н]+. Стадия 4. Взаимодействие промежуточного соединения F, [N-бензилокси-4,4-бис-(1H-индол-3 ил)бутирамида, 200 мг, 0,47 ммоль] проводят, как в примере 1, получая при этом 0,11 г G [N-гидрокси 4,4-бис-(1 Н-индол-3-ил)бутирамида, ST 2408, выход=71%]. 1ES-MC m/z: 332,4 [М-Н]-. Пример 5. Получение гидроксиамида 8,8-бис-(1 Н-индол-3-ил)октановой кислоты (ST2889) Стадия 1. Раствор В (этилового эфира 8-оксооктановой кислоты, 920,0 мг, 4,94 ммоль), А (1 Ниндола, 1,16 мг, 9,88 ммоль) и трифлата диспрозия (4,5 г, 7,41 ммоль) в смеси МеОН/Н 2 О (21 мл/7 мл) подвергают взаимодействию в таких же условиях, как в примере 1 (16 ч при 50 С), получая при этом 0,4 г С [этилового эфира 8,8-бис-(1 Н-индол-3-ил)октановой кислоты, выход = 23%]. 1 Н-ЯМР (200 МГц, CDCl3)(м.д.): 1,31 (т, 3 Н, СН 3); 1,42 (ушир. 4 Н, СН 2); 1,64 (м, 2 Н, СН 2); 2,30(м, 4 Н, СН 2); 4,18 (кв., 2 Н, ОСН 2); 4,51 (т, 1 Н, СН); 6,95 (с, 2 Н); 7,10 (т, 2 Н, СН аром.); 7,21 (т, 2 Н, СН аром.); 7,33 (д, 2 Н, СН аром.); 7,66 (д, 2 Н, СН аром.); 7,90 (с, 2 Н, NH). 13 С-ЯМР (50 МГц, CDCl3)(м.д.): 14,39; 25,13; 28,26; 29,24; 29,53; 34,12; 34,52; 35,94; 60,35; 111,25; 119,08; 119,76; 120,51; 121,60; 121,80; 127,29; 136,73; 174,18. ES-MC m/z: 401,3 [М-Н]- и 425,5 [M+Na]+. Стадия 2. Взаимодействие промежуточного соединения С [этилового эфира 8,8-бис-(1 Н-индол-3 ил)октановой кислоты, 340 мг, 0,846 ммоль] и твердого LiOH (177,5 мг, 4,23 ммоль) в смеси МеОН/Н 2 О(2,9 мл/0,3 мл) проводят, как в примере 1, получая при этом 0,3 г D [8,8-бис-(1 Н-индол-3-ил)октановой кислоты, выход=95%]. 1ES-MC m/z: 373,0 [М-Н]- и 397,4 [M+Na]+. Стадия 3. В колбе реагент Е (гидрохлорид гидроксиламина, 56,0 мг, 0,80 ммоль) и DBU (122,6 мг,0,80 ммоль) растворяют в ДМФА (0,8 мл) и образовавшийся раствор добавляют к раствору промежуточного соединения D [8,8-бис-(1 Н-индол-3-ил)октановой кислоты, 200 мг, 0,537 ммоль], HATU (224,6 мг,0,591 ммоль) и DIEA (187,1 мкл, 1,07 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 30 мин и затем разбавляют этилацетатом. Органический слой промывают Н 2 О (3), сушат над сульфатом натрия, фильтруют и упаривают при пониженном давлении. Остаток очищают колоночной хроматографией на силикагеле с применением градиентного элюирования смесью DCM/MeOH,получая при этом 0,12 г F (гидроксиамида 8,8-бис-(1 Н-индол-3-ил)октановой кислоты, ST 2889, выход = 57%). 1 Н-ЯМР (300 МГц, ДМСО-d6)(м.д.): 1,14-1,28 (ушир. м, 6 Н, СН 2); 1,41 (квинт., 2 Н, СН 2); 1,87 (т,2 Н, СН 2); 2,13 (ушир. 2 Н, СН 2); 4,32 (т, 1 Н, СН); 6,83 (т, 2 Н, СН аром.); 6,96 (т, 2 Н, СН аром.); 7,18 (с,2 Н, СН аром.); 7,26 (д, 2 Н, СН аром.); 7,46 (д, 2 Н, СН аром.); 8,59 (с, 1 Н); 10,26 (с, 1 Н); 10,68 (с, 2 Н, NH). 13 С-ЯМР (50 МГц, ДМСО-d6)(м.д.): 25,83; 28,48; 29,35; 29,48; 32,93; 34,09; 35,73; 110,95; 118,51; 119,57; 119,65; 121,21; 122,52; 127,37; 137,14; 169,80.ES-MC m/z: 388,2 [М-Н]- и 412,7 [M+Na]+. Пример 6. Получение гидроксиамида 7,7-бис-(7-метокси-1 Н-индол-3-ил)гептановой кислоты (ST3043) Стадия 1. Раствор В (этилового эфира 7-оксогептановой кислоты, 58 мг, 0,34 ммоль), А (7-метокси 1 Н-индола, 100 мг, 0,68 ммоль) и трифторуксусной кислоты (0,03 ммоль) в CH3CN (1 мл) перемешивают в течение 72 ч при комнатной температуре, затем растворитель выпаривают при пониженном давлении. Соединение С [этиловый эфир 7,7-бис-(7-метокси-1 Н-индол-3-ил)гептановой кислоты] не выделяют и характеризуют ES-MC (m/z: 449, 6 [М+Н]+). Стадия 2. Сырое промежуточное соединение С [этиловый эфир 7,7-бис-(7-метокси-1 Н-индол-3 ил)гептановой кислоты], полученное на стадии 1, подвергают взаимодействию с твердым LiOH (43 мг,1,02 ммоль) в смеси МеОН/ТГФ/Н 2 О, 3:3:1 (2 мл), как описано в примере 1, получая при этом продукт D[7,7-бис-(7-метокси-1 Н-индол-3-ил)гептановую кислоту], который не выделяют и характеризуют ES-MC(m/z: 419,4 [М-Н]-). Стадия 3. Сырое промежуточное соединение D [7,7-бис-(7-метокси-1 Н-индол-3-ил)гептановую ки- 10013542 слоту] стадии 2 подвергают взаимодействию с РуВОР (265,2 мг, 0,51 ммоль), DIEA (0,30 мл, 1,7 ммоль) и Е (гидрохлоридом О-бензилгидроксиламина, 271,3 мг, 1,7 ммоль) в DCM (2 мл), как в примере 1, получая при этом F [бензилоксиамид 7,7-бис-(7-метокси-1 Н-индол-3-ил)гептановой кислоты]; ES-MC (m/z: 526,8 [М+Н]+). Стадия 4. Промежуточное соединение F [бензилоксиамид 7,7-бис-(7-метокси-1 Н-индол-3 ил)гептановой кислоты], полученное на стадии 3, растворяют в МеОН и добавляют 10% Pd/BaSO4 (кат.). Раствор механически перемешивают в атмосфере водорода (55 фунт/кв. дюйм) в течение ночи. Реакционную смесь фильтруют и фильтрат выпаривают при пониженном давлении. Твердый остаток очищают препаративной ОФ-ВЭЖХ (колонка Lichrosorb RP-18, 7 мкм, элюенты: CH3CN/H2O + 0,1% TFA, 60:40),получая при этом 60,0 мг G [гидроксиамида 7,7-бис-(7-метокси-1 Н-индол-3-ил)гептановой кислоты, ST 3043, общий выход = 40%]. 1ST 3044 синтезируют, как в примере 6. Стадия 1. Реагент А (5-метил-1 Н-индол, 100 мг, 0,762 ммоль) и В [этиловый эфир 7-оксогептановой кислоты, 65,6 мг, 0,381 ммоль] подвергают взаимодействию, получая при этом С [этиловый эфир 7,7 бис-(5-метил-1 Н-индол-3-ил) гептановой кислоты], который не выделяют и характеризуют ES-MC (m/z: 417,6 [М+Н]+). Стадия 2. Промежуточное соединение D [7,7-бис-(5-метил-1 Н-индол-3-ил)гептановую кислоту], невыделенное, характеризуют ES-MC (m/z: 387,4 [М-Н]-). Стадия 3. Промежуточное соединение F [бензилоксиамид 7,7-бис-(5-метил-1 Н-индол-3 ил)гептановой кислоты] очищают флэш-хроматографией на силикагеле с градиентным элюированием смесью гексан/этилацетат и характеризуют ES-MC (m/z: 494,7 [М+Н]+). Стадия 4. Продукт G [гидроксиамид 7,7-бис-(5-метил-1 Н-индол-3-ил)гептановой кислоты, ST3044] очищают фильтрованием реакционной смеси и выпариванием растворителя при пониженном давленииST 3052 синтезируют, как в примере 6. Стадия 1. Реагент А (4-фтор-1 Н-индол, 100 мг, 0,74 ммоль) и В [этиловый эфир 7-оксогептановой кислоты, 63,7 мг, 0,37 ммоль] подвергают взаимодействию, получая при этом С [этиловый эфир 7,7-бис(4-фтор-1 Н-индол-3-ил)гептановой кислоты], который не выделяют и характеризуют ES-MC (m/z: 425,6[М+Н]+). Стадия 2. Промежуточное соединение D [7,7-бис-(4-фтор-1 Н-индол-3-ил)гептановую кислоту] не выделяют и характеризуют ES-МС (m/z: 395,4 [М-Н]-). Стадия 3. Промежуточное соединение F [бензилоксиамид 7,7-бис-(4-фтор-1 Н-индол-3 ил)гептановой кислоты] грубо очищают флэш-хроматографией на силикагеле с градиентным элюированием смесью гексан/этилацетат и характеризуют ES-MC (m/z: 502,7 [М+Н]+). Стадия 4. Продукт G [гидроксиамид 7,7-бис-(4-фтор-1 Н-индол-3-ил)гептановой кислоты, ST 3052] очищают фильтрованием реакционной смеси, выпариванием растворителя и препаративной ОФ-ВЭЖХ- 11013542 Пример 9. Получение 3-4-[бис-(1 Н-индол-3-ил) метил]фенил-N-гидроксиакриламида (ST2887) Стадия 1. Взаимодействие А (1 Н-индола, 234 мг, 2 ммоль) и В [3-(4-формилфенил) акриловой кислоты, 176 мг, 1 ммоль] проводят, как в примере 1, получая при этом кислоту С [3-4-[бис-(1 Н-индол-3 ил)метил]фенилакриловую кислоту], применяемую в неочищенном виде для стадии 2. 1ES-MC m/z: 405,71 [М-Н]-. Пример 10. Получение (2-аминофенил)амида 7,7-бис- (1 Н-индол-3-ил)гептановой кислоты (ST3071) Стадия 3. Промежуточное соединение D [7,7-бис-(1 Н-индол-3-ил)гептановую кислоту, 60 мг, 0,167 ммоль], полученное на стадии 2 в примере 1, растворяют в DCM (1 мл), затем добавляют РуВОР (95,3 мг,0,184 ммоль) и DIEA (87 мкл, 0,50 ммоль). После перемешивания 5 мин образовавшийся раствор добавляют к раствору Е (2-аминофениламина; 86 мг, 0,335 ммоль) в DCM (1 мл) и реакционную смесь перемешивают при комнатной температуре в течение 2 ч. Затем растворитель удаляют при пониженном давлении и остаток растворяют в этилацетате. Раствор промывают 0,5 н HCl, насыщенным растворомNaHCO3 и водой (2). Органический слой сушат над сульфатом натрия, фильтруют и упаривают при пониженном давлении. Остаток грубо очищают флэш-хроматографией на силикагеле с применением смеси гексан/этилацетат в качестве системы элюентов, получая при этом F [(2-аминофенил)амид 7,7-бис-(1 Ниндол-3-ил)гептановой кислоты, ST 3071, 50 мг, выход= 67%]. 1ES-MC m/z: 451,2 [М+Н]+ и 449,4 [М-Н]-. Пример 11. Получение N-гидрокси-3-(1 Н-индол-3-ил)акриламида (ST2913) Реагент Е (гидрохлорид гидроксиламина, 112,0 мг, 1,61 ммоль) и DBU (254,0 мг, 1,61 ммоль) растворяют в ДМФА (0,5 мл) и образовавшийся раствор добавляют к суспензии D [3-(lH-индол-3 ил)акриловой кислоты, 151 мг, 0,806 ммоль], HATU (337,1 мг, 0,887 ммоль) и DIEA (281,0 мкл, 1,61 ммоль) в смеси ДМФА/DCM (1,5 мл/2 мл). Реакционную смесь обрабатывают, как описано в стадии 3 примера 3, и конечной очисткой препаративной ОФ-ВЭЖХ (колонка Lichrosorb RP-18, 7 мкм; элюенты:ES-MC m/z: 201,1 [М-Н]- и 224,86 [M+Na]+. Пример 12. Получение N-гидрокси-6-(1 Н-индол-3-ил)гексанамида (ST2995) Стадия 3. В колбе реагент Е (гидрохлорид гидроксиламина, 120,8 мг, 1,74 ммоль) и DIEA (302,6 мкл, 1,74 ммоль) растворяют в ДМФА (0,5 мл) и образовавшийся раствор добавляют к раствору D [6-(lHиндол-3-ил)гексановой кислоты, 200 мг, 0,869 ммоль, HATU (363 мг, 0,956 ммоль) и DIEA (302,6 мкл,1,74 ммоль) в ДМФА (2 мл). Реакционную смесь перемешивают при комнатной температуре в течение 2 ч и затем разбавляют этилацетатом. Органический слой промывают 1 н HCl, 3% NaHCO3, насыщенным раствором соли (2), сушат над сульфатом натрия, фильтруют и упаривают при пониженном давлении. Осаждение из DCM, фильтрование и конечные промывания осадка DCM дают 0,17 г F [гидроксиамида 8,8-бис-(1 Н-индол-3-ил)октановой кислоты, ST 2995, выход 80%). 1ES-MC m/z: 245,2 [М-Н]- и 247,3 [М+Н]+ и 269,2 [M+Na]+. Пример 13. Получение гидроксиамида 7,7-бис-(7-этил-1 Н-индол-3-ил)гептановой кислоты (ST3126) Стадия 1. Раствор В (этилового эфира 7-оксогептановой кислоты, 90 мг, 0,516 ммоль), А (7-этил 1 Н-индола, 100,2 мг, 0,688 ммоль) и трифлата диспрозия (420 мг, 0,688 ммоль) в AcCN (1,2 мл) подвергают взаимодействию, как в примере 1 (при комнатной температуре в течение 2 дней), получая при этом С [этиловый эфир 7,7-бис-(7-этил-1H-индол-3-ил)гептановой кислоты], который не выделяют и характеризуют ES-MC (m/z: 445,8 [М+Н]+). Стадия 2. Сырое промежуточное соединение С [этиловый эфир 7,7-бис-(7-этил-1 Н-индол-3 ил)гептановой кислоты], полученное на стадии 1, подвергают взаимодействию, как описано в примере 6,получая при этом продукт D [7,7-бис-(7-этил-1 Н-индол-3-ил)гептановую кислоту], который не выделяют и характеризуют ES-МС (m/z: 415,4 [М-Н]-). Стадия 3. Неочищенное промежуточное соединение D [7,7-бис-(7-этил-1 Н-индол-3-ил)гептановую кислоту], полученное на стадии 2, подвергают взаимодействию, как описано в примере 6, получая при этом F [бензилоксиамид 7,7-бис-(7-этил-1 Н-индол-3-ил)гептановой кислоты], который не выделяют и характеризуют ES-МС (m/z: 522,8 [М+Н]+). Стадия 4. Неочищенное промежуточное соединение F [бензилоксиамид 7,7-бис- (7-этил-1 Н-индол 3-ил)гептановой кислоты], полученное на стадии 3, подвергают взаимодействию, как описано в примере 6. Очистка препаративной ОФ-ВЭЖХ (колонка Lichrosorb RP-18, 7 мкм; элюенты: CH3CN/H2O, 60:40) дает 100,0 мг G [гидроксиамида 7,7-бис-(7-этил-1 Н-индол-3-ил)гептановой кислоты, ST3126, общий выход= 67%]. 1ES-MC m/z: 430,4 [М-Н]- и 454,0 [M+Na]+. Пример 14. Получение гидроксиамида 7,7-бис-(5-морфолин-4-илметил-3 Н-индол-3-ил)гептановой кислоты (ST3307) Стадия 1. Раствор В (этилового эфира 7-оксогептановой кислоты, 2,7 г, 15,68 ммоль) и А (5 морфолин-4-илметил-3 Н-индола, 2,8 г, 12,95 ммоль) в ледяной СН 3 СООН (20 мл) подвергают взаимодействию при кипячении с обратным холодильником в течение ночи, получая при этом этиловый эфир 7,7-бис-(5-морфолин-4-илметил-3 Н-индол-3-ил)гептановой кислоты, который не выделяют и характеризуют ES-MC (m/z: 587,4 [М+Н]+ и 609,4 [М+23]+. Стадия 2. Неочищенное промежуточное соединение С, этиловый эфир 7,7-бис-(5-морфолин-4 илметил-3 Н-индол-3-ил)гептановой кислоты, полученное на стадии 1, подвергают взаимодействию, как описано в примере 6, получая при этом продукт D, 7,7-бис-(5-морфолин-4-илметил-3 Н-индол-3 ил)гептановая кислота, который не выделяют и характеризуют ES-MC (m/z: 557,6 [М-Н]-). Стадия 3. Неочищенное промежуточное соединение D, 7,7-бис-(5-морфолин-4-илметил-3 Н-индол 3-ил)гептановая кислота, полученное на стадии 2, подвергают взаимодействию, как описано в примере 6,получая при этом F, бензилоксиамид 7,7-бис-(5-морфолин-4-илметил-3 Н-индол-3-ил)гептановой кислоты, который не выделяют и характеризуют ES-MC (m/z: 664,9 [М+Н]+). Стадия 4. Неочищенное промежуточное соединение F, бензилоксиамид 7,7-бис-(5-морфолин-4 илметил-3 Н-индол-3-ил)гептановой кислоты, полученное на стадии 3, подвергают взаимодействию, как описано в примере 6. Очистка препаративной ОФ-ВЭЖХ (колонка Lichrosorb RP-18, 7 мкм, элюенты: СН 3 ОН + 0,1% TFA/H2O + 0,1% TFA=45:55) дает 100,0 мг G [гидроксиамида 7,7-бис-(5-морфолин-4 илметил-3 Н-индол-3-ил)гептановой кислоты, ST3307, общий выход = 40%]. 1(ST3292) Во время синтеза ST2741, пример 1, гидроксиамид 7-(1 Н-индол-3-ил)-7-(1 Н-индол-2-ил)гептановой кислоты получают в качестве примеси. Продукт очищают и характеризуют. 1 Н-ЯМР (300 МГц, ДМСО-d6)(м.д.): 1,28 (ушир. м, 4 Н, 2 СН 2); 1,44 (ушир. м, 2 Н, СН 2); 1,43 Биологические результаты Изучения цитотоксичности Для испытания действий соединений на рост клеток применяли клетки промиелоцитарного лейкозаNB4 человека, клетки немелкоклеточной карциномы NCI-H460 и клетки карциномы толстой кишки НСТ-116 человека. Клетки опухолей NB4 и NCI-H460 выращивали в среде RPMI 1640, содержащей 10% фетальную бычью сыворотку (GIBCO), тогда как клетки опухоли НСТ-116 выращивали в среде McCoy 5A, содержащей 10% фетальную бычью сыворотку (GIBCO). Опухолевые клетки засевали в 96-луночные планшеты для культур тканей (Corning) приблизительно при 10% уровне конфлюэнтности и давали им возможность прикрепляться и восстанавливаться в течение по меньшей мере 24 ч. К каждой лунке затем добавляли варьируемые концентрации лекарственных средств для вычисления их величин IC50 (концентрация, которая позволяет выживать 50% клеток). Планшеты инкубировали в течение 24 ч при 37 С. В конце обработки для опухолевых клеток NB4 в суспензии применяли следующую методику: культуральную среду отделяли центрифугированием планшетов при 1600g в течение 10 мин и супернатант удаляли. Добавляли 250 мкл PBS, затем планшеты центрифугировали при 1600g в течение 10 мин, супернатант удаляли. Добавляли 200 мкл/лунку культуральной среды RPMI 1640, содержащей 10% FCS, и планшеты инкубировали при 37 С в течение еще 48- 14013542 ч. Планшеты снова центрифугировали при 1600g в течение 10 мин, культуральную среду удаляли и добавляли 200 мкл PBS и 50 мкл не содержащей радиоактивных веществ 80% ТСА. Планшеты инкубировали на льду в течение по меньшей мере 1 ч. ТСА удаляли, планшеты промывали 3 раза погружением в дистиллированную воду и сушили на бумаге и при 40 С в течение 5 мин. Затем добавляли 200 мкл 0,4% сульфородамина В в 1% уксусной кислоте. Планшеты инкубировали при комнатной температуре в течение еще 30 мин. Сульфородамин В удаляли, планшеты промывали погружением в 1% уксусную кислоту 3 раза, затем их сушили на бумаге и при 40 С в течение 5 мин. Затем добавляли 200 мкл 10 мМ трис,планшеты выдерживали при перемешивании в течение 20 мин. Выживание клеток определяли как оптическую плотность спектрофлуориметром Multiskan при 540 нм. Для опухолевых клеток в состоянии адгезии (NCI-H460 и НСТ-116) методика была такой же, как указано выше, за исключением того, что в конце обработки планшеты промывали удалением супернатанта и добавлением PBS 3 раза без центрифугирования. В последний день анализа супернатант также удаляли без центрифугирования. Количество убитых клеток вычисляли как процентное уменьшение связывания сульфородамина В по сравнению с контрольными культурами. Величины IC50 (концентрацию, которая позволяет выживать 50% клеток) вычисляли программой "ALLFIT". В табл. 1 цитотоксичность, вычисленная для опухолевых клеток NB4, показала, что соединение ST3044 было наиболее сильнодействующим соединением с величиной IC50 0,15 мкМ, за которым следовали другие молекулы (ST3052, ST3043, ST3292, ST3307, ST2741,ST2887, ST2889, ST2754) с величинами IC50 0,4-2,3 мкМ. Для клеток немелкоклеточного рака NCI-H460 и рака толстой кишки НСТ-116 соединения ST3044, ST3043 и ST3052 были наиболее сильнодействующими молекулами с величинами IC50, которые были в диапазоне от 0,27 до 0,7 мкМ, за которыми следовали ST3292, ST3307, ST2741, ST2887, ST2889, ST2754 с величинами IC50 0,6-5,8 мкМ. Другие молекулы (ST2408, ST2743, ST2 995, ST3127) обнаружили слабую цитотоксичность на опухолевых клетках NB4 или NCI-H460 и НСТ-116, поскольку величины IC50 были в диапазоне от 8,8 до 88,8 мкМ. Кроме того, молекулы настоящего изобретения были более сильнодействующими, чем гомологичные бисгетероциклические соединения, в испытаниях на цитотоксичность на опухолевых клетках NB4(см. табл. 2). Таблица 1. Цитотоксичность различных соединений в отношении опухолевых клеток NB4, NCI-H460 и НСТ-116- 15013542 Таблица 2. Цитотоксичность гомологичных бисгетероциклических соединений в отношении опухолевых клеток NB4A1 соответствует А, где R, R' и R1 являются одинаковыми для обоих колец индола;X представляет собой насыщенный или ненасыщенный (алкенилен или алкинилен), неразветвленный или разветвленный (С 3-С 10)алкилен, необязательно замещенный ОН;G представляет собой либо Н, либо гликозил;R2, R3 являются одинаковыми или разными и представляют собой Н или (С 1-С 4)алкил;R и R', одинаковые или разные, выбраны из группы, состоящей из -Н; насыщенного или ненасыщенного, неразветвленного или разветвленного (С 1-С 10)алкила, необязательно замещенного (С 3-С 10)гетероарилом или (С 3-С 10)гетероциклил-(С 1-С 4)алкиленом, где гетероцикл содержит по меньшей мере один гетероатом, выбранный из N, О или S, или группой -NR5R6, где R5, R6 являются одинаковыми или разными и представляют собой Н, неразветвленный или разветвленный (C1 С 4)алкил, (С 1-С 4)алканоил;OR4, где R4 представляет собой Н, (C1-С 4)алкил, мезил, тозил, (С 1-С 4)алканоил, гликозил; галогена, азида, нитро, нитрила и NR5R6. 2. Соединение по п.1, где X представляет собой насыщенный или ненасыщенный неразветвленный С 2-С 10 алкилен. 3. Соединение по п.1, где Q представляет собой CONHOG. 4. Соединение по п.1 или 3, где G, R1, R2 и R3 являются одинаковыми и представляют собой Н. 5. Соединение по п.1, где R и R' являются одинаковыми и выбраны из группы, состоящей из Н, (C1- 16013542 С 3)алкиламина и (C1-С 3)алкилморфолина. 6. Соединение формулы (I) по п.1, которое выбрано из группы, состоящей из гидроксиамида 5,5-бис-(1 Н-индол-3-ил)пентановой кислоты,гидроксиамида 6,6-бис-(1 Н-индол-3-ил)гексановой кислоты,гидроксиамида 7,7-бис-(1 Н-индол-3-ил)гептановой кислоты,гидроксиамида 7,7-бис-(4-фтор-1 Н-индол-3-ил)гептановой кислоты,гидроксиамида 7,7-бис-(5-метил-1 Н-индол-3-ил)гептановой кислоты,гидроксиамида 7,7-бис-(7-этил-1 Н-индол-3-ил)гептановой кислоты,гидроксиамида 7,7-бис-(7-метокси-1 Н-индол-3-ил)гептановой кислоты,гидроксиамида 7,7-бис-(5-морфолин-4-илметил-3 Н-индол-3-ил)гептановой кислоты,гидроксиамида 7-(1 Н-индол-3-ил)-7-(1 Н-индол-2-ил)гептановой кислоты,7,7-бис-(1H-индол-3-ил)гептановой кислоты,гидроксиамида 8,8-бис-(1 Н-индол-3-ил)октановой кислоты. 7. Способ получения соединений по любому из предыдущих пунктов, у которых А представляет собой A1, включающий димеризацию исходного производного индола в присутствии альдегида или ацеталя. 8. Применение соединения по любому из пп.1-6 в качестве лекарственного средства. 9. Применение соединения по любому из пп.1-6 для изготовления лекарственного средства с противоопухолевой активностью. 10. Применение соединения по любому из пп.1-6 для изготовления лекарственного средства для лечения опухолевой патологии, в которой опухоль обнаруживала резистентность к противоопухолевым лекарственным средствам, ранее применяемым для ее лечения, в котором указанное соединение формулы (I) оказывает химиосенсибилизирующее действие на опухоль, резистентную к указанным лекарственным средствам. 11. Применение по п.9 или 10, где опухоль выбрана из группы, состоящей из саркомы, карциномы,карциноида, опухоли кости, нейроэндокринной опухоли, лимфоидного лейкоза, острого промиелоцитарного лейкоза, миелоидного лейкоза, моноцитарного лейкоза, мегакариобластного лейкоза и болезни Ходжкина. 12. Применение по п.9 или 10, в котором соединение формулы (I) комбинируют с одним или несколькими известными противоопухолевыми агентами. 13. Применение по п.12, в котором известным противоопухолевым агентом является полностью транс-изомер ретиноевой кислоты. 14. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы (I) по любому из пп.1-6 и по меньшей мере один фармацевтически приемлемый эксципиент и/или разбавитель. 15. Способ получения композиции по п.13, включающий смешивание соединения(й) по любому из пп.1-6 с подходящим эксципиентом(ами) и/или разбавителем(ями). 16. Способ лечения млекопитающего, страдающего заболеванием по любому из пп.9-11, включающий введение терапевтически эффективного количества соединения по любому из пп.1-6.
МПК / Метки
МПК: A61K 31/405, A61P 35/00, C07D 209/18
Метки: производные, активностью, обладающие, индола, противоопухолевой
Код ссылки
<a href="https://eas.patents.su/18-13542-proizvodnye-indola-obladayushhie-protivoopuholevojj-aktivnostyu.html" rel="bookmark" title="База патентов Евразийского Союза">Производные индола, обладающие противоопухолевой активностью</a>
Производные камптотецина, обладающие противоопухолевой активностью

Номер патента: 3605
Опубликовано: 26.06.2003
Авторы: Пенко Серджо, Карминати Паоло, Мерлини Лючио, Цунино Франко
МПК: A61K 31/435, C07D 491/22
Метки: противоопухолевой, камптотецина, активностью, производные, обладающие
Формула / Реферат:
1. Соединения формулы I в которой R1 обозначает группу -C(R5)=N-O(n)R4, в которой R4 обозначает нормальный или разветвленный C1-C8алкил, нормальную или разветвленную C1-C8алкенильную группу, C3-C10циклоалкил, (C3-C10)циклоалкилнормальный или разветвленный (C1-C8)алкил, (C6-C14)арил, (C6-C14)арилнормальный или разветвленный (C1-C6)алкил, гетероциклическую группу или гетероциклонормальный или разветвленный-(C1-C8)алкил, где гетероциклическая...
2-(1h-индол-3-ил)-2-оксоацетамиды, обладающие противоопухолевой активностью

Номер патента: 4925
Опубликовано: 28.10.2004
Авторы: Мента Эрнесто, Пескалли Николетта
МПК: C07D 405/12, A61P 35/00, A61K 31/404...
Метки: 2-(1h-индол-3-ил)-2-оксоацетамиды, противоопухолевой, обладающие, активностью
Формула / Реферат:
1. Соединения формулы I где R1, R2 и R5 независимо представляют собой водород или C1-C6алкильную группу; R3 представляет собой водород, C1-C4алкил, аралкил, возможно замещенный фенил; R4 представляет собой водород, прямой или разветвленный C1-C8алкил, C5-C6циклоалкил; аралкил; гетероаралкил; X представляет собой одну или более чем одну группу, максимально четыре, независимо выбранную из водорода; C1-C6алкила; C1-C6галогеналкила;...
Производные хиноксалин-2,3-диона и их фармацевтически приемлемые соли, фармацевтические композиции, обладающие антагонистической активностью в отношении рецептора глютамата и противосудорожной активностью, способы лечения пациентов, страдающих от удара или заболеваний, при помощи этих соединений.

Номер патента: 762
Опубликовано: 24.04.2000
Авторы: Никам Шам, Корнберг Брайэн Эдвард, Рафферти Майкл Фрэнсис
МПК: C07D 241/44
Метки: противосудорожной, удара, способы, антагонистической, отношении, соединений, соли, композиции, рецептора, глютамата, фармацевтические, страдающих, активностью, пациентов, заболеваний, фармацевтически, лечения, хиноксалин-2,3-диона, этих, помощи, производные, приемлемые, обладающие
Формула / Реферат:
1. Производные хиноксалин-2,3-диона общей формулы I где R - группа - NR4R5, где R4 и R5 независимо друг от друга означают водород, низший алкил, незамещенный или замещенный низшим алкилом, низший циклоалкил, который может содержать 1 или 2 атома кислорода в качестве гетероатома, R1 и R2 независимо друг от друга означают водород или нитрогруппу, R3 - низший алкил, при этом радикалы R3 и R-CH2- могут каждый находиться в положениях 5 или...
Производные азобициклооктанов и нонанов обладающие активностью в ингибировании dpp – iv

Номер патента: 8166
Опубликовано: 27.04.2007
Авторы: Араньи Петер, Сабо Тибор, Батори Шандор, Т.Надь Лайош, Урбан-Сабо Каталин, Капуи Зольтан, Балаж Ласло, Боронкаи Эва, Варга Мартон, Шушан Эдит, Бата Имре
МПК: A61K 31/46, C07D 207/06, A61P 3/10...
Метки: ингибировании, обладающие, азобициклооктанов, производные, нонанов, активностью
Формула / Реферат:
1. Соединения общей формулы (I) в которой R означает азотсодержащий ароматический фрагмент с одним или двумя циклами, состоящий из одного или двух ароматических циклов, предпочтительно пиридила, пиридазинила, пиримидинила, пиразинила, имидазолила, пиразолила, тиазолила, изотиазолила, оксазолила, изоксазолила, оксадиазолила, хинолинила, изохинолинила, циннолинила, фталазинила, хиназолинила, хиноксалинила, бензимидазолила, индазолила,...
Производные триазола, обладающие противогрибковой активностью, предназначенные для человека и животных

Номер патента: 3189
Опубликовано: 27.02.2003
Авторы: Альбини Энрико, Наполетано Маоро, Скиоппакасси Джованна, Фраире Кристина
МПК: C07D 249/08, A61P 31/10, A61K 31/4196...
Метки: обладающие, активностью, животных, противогрибковой, человека, предназначенные, производные, триазола
Формула / Реферат:
1. Соединение, выбранное из группы, содержащей (2R,3S) 1-(1H-1,2,4-триазолил)-2-(2,4-дифторфенил)-3-этил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол, (2R,3S) 1-(1H-1,2,4-триазолил)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол, (2R,3R) 1-(1H-1,2,4-триазолил)-2-(2,4-дихлорфенил)-3-метил-4-(1,1,2,2-тетрафторэтилтио)-2-бутанол, (2R,3S) 1-(1H-1,2,4-триазолил)-2-(2,4-дифторфенил)-3-метил-4-(1,1,2,2-тетрафторэтокси)-2-бутанол, (2R)...
Предыдущий патент: Способ повышения плодовитости здоровых самок позвоночных животных и кормовая композиция
Следующий патент: Способ получения полиморфной формы i гидросульфата (s)-(+)-метил-a-(2-хлорфенил)-6,7-дигидротиено[3,2-c] пиридин-5(4н)-ацетата
Случайный патент: Способ очистки ствола скважины и разжижающий флюид