Циклобутановые и метилциклобутановые производные, композиции на их основе и способы их применения
















Формула / Реферат
1. Соединение, которое представляет собой 3-циклобутил-3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил]пропаннитрил или его фармацевтически приемлемую соль.
2. Соединение по п.1, которое представляет собой (R)-3-циклобутил-3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил]пропаннитрил или его фармацевтически приемлемую соль.
3. Соединение по п.1, которое представляет собой (S)-3-циклобутил-3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил]пропаннитрил или его фармацевтически приемлемую соль.
4. Соединение, которое представляет собой 3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-3-(3-метилциклобутил)пропаннитрил или его фармацевтически приемлемую соль.
5. Соединение по п.4, которое представляет собой 3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-3-((транс)-3-метилциклобутил)пропаннитрил или его фармацевтически приемлемую соль.
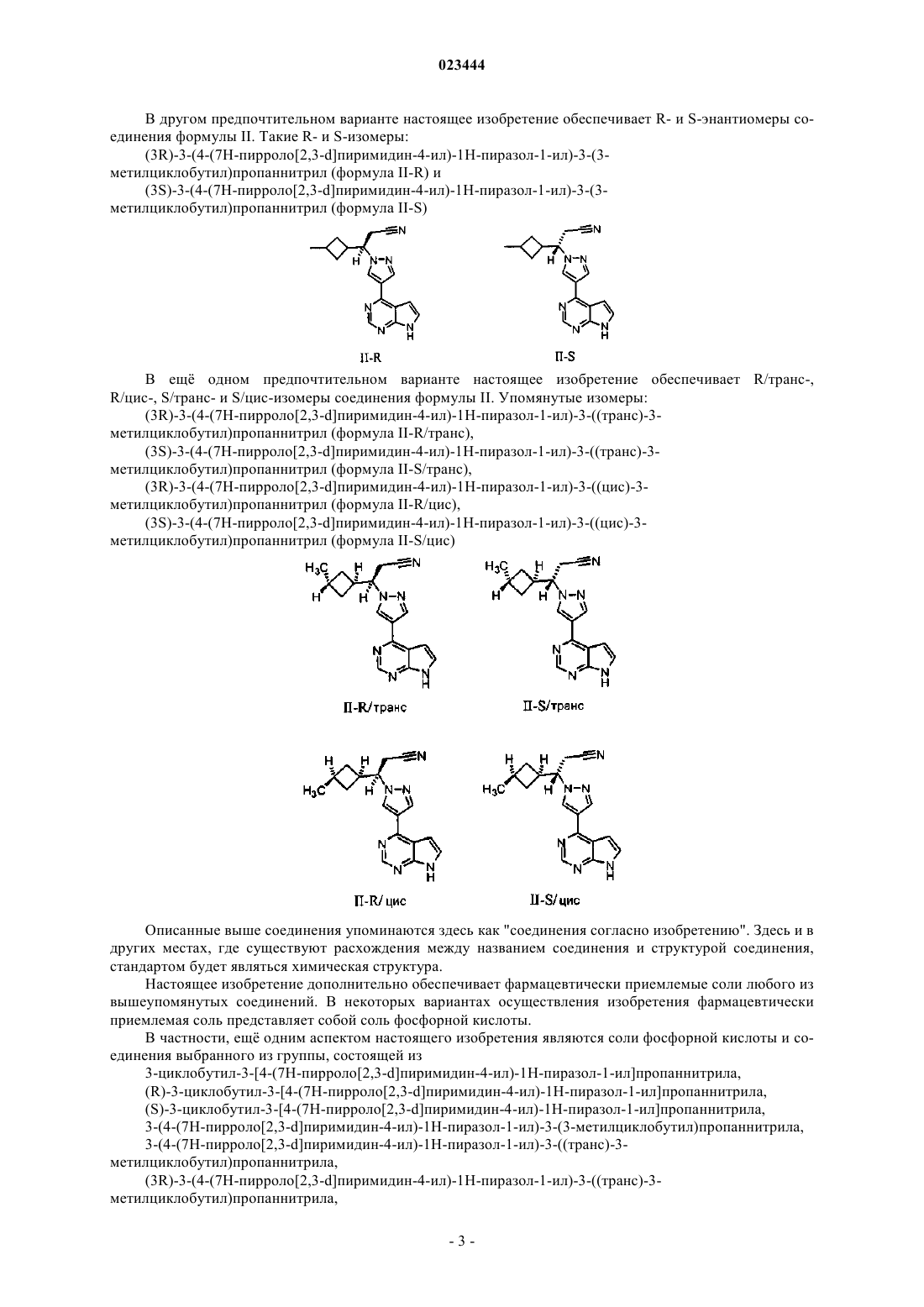
6. Соединение по п.5, которое представляет собой (3R)-3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-3-((транс)-3-метилциклобутил)пропаннитрил или его фармацевтически приемлемую соль.
7. Соединение по п.5, которое представляет собой (3S)-3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-3-((транс)-3-метилциклобутил)пропаннитрил или его фармацевтически приемлемую соль.
8. Соединение по п.4, которое представляет собой 3-(4-(7Н-пирроло[2, 3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-3-((цис)-3-метилциклобутил)пропаннитрил или его фармацевтически приемлемую соль .
9. Соединение по п.8, которое представляет собой (3R)-3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-3-((цис)-3-метилциклобутил)пропаннитрил или его фармацевтически приемлемую соль.
10. Соединение по п.8 которое представляет собой (3S)-3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-3-((цис)-3-метилциклобутил)пропаннитрил или его фармацевтически приемлемую соль.
11. Соль фосфорной кислоты и соединения, выбранного из группы, состоящей из
3-циклобутил-3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил]пропаннитрила,
(R)-3-циклобутил-3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил]пропаннитрила,
(S)-3-циклобутил-3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил]пропаннитрила,
3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-3-(3-метилциклобутил)пропаннитрила,
3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-3-((транс)-3-метилциклобутил)пропаннитрила,
(3R)-3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-3-((транс)-3-метилциклобутил)пропаннитрила,
(3S)-3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-3-((транс)-3-метилциклобутил)пропаннитрила,
3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-3-((цис)-3-метилциклобутил)пропаннитрила,
(3R)-3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-3-((цис)-3-метилциклобутил)пропаннитрила,
(3S)-3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-3-((цис)-3-метилциклобутил)пропаннитрила.
12. Композиция для лечения миелопролиферативного расстройства, содержащая соединение по любому из пп.1-10 или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый носитель.
13. Способ лечения миелопролиферативного расстройства у пациента, включающий в себя введение упомянутому пациенту терапевтически эффективного количества соединения по любому из пп.1-10 или его фармацевтически приемлемой соли.
14. Способ по п.13, в котором упомянутое миелопролиферативное расстройство (MPD) представляет собой истинную полицитемию (PV), эссенциальную тромбоцитемию (ЕТ), первичный миелофиброз (PMF), миелофиброз с миелоидной метаплазией (МММ), хронический миелогенный лейкоз (CML), хронический миеломоноцитарный лейкоз (CMML), гиперэозинофильный синдром (HES), идиопатический миелофиброз (IMF), системный мастоцитоз (SMCD) или миелофиброз, развившийся на фоне истинной полицитемии или эссенциальной тромбоцитемии (Post-PV/ET MF).
Текст
ЦИКЛОБУТАНОВЫЕ И МЕТИЛЦИКЛОБУТАНОВЫЕ ПРОИЗВОДНЫЕ,КОМПОЗИЦИИ НА ИХ ОСНОВЕ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ Настоящее изобретение относится к циклобутановым и метилциклобутановым производным,а также их солям, которые представляют собой ингибиторы Янус-киназ (JAK), применимые при лечении JAK-ассоциированных заболеваний, включая, например, миелопролиферативные расстройства. Кроме того, настоящее изобретение относится к фармацевтической композиции,содержащей циклобутановые или метилциклобутановые производные, которые представляют собой ингибиторы JAK, и к способу лечения миелопролиферативного расстройства у пациента,включающему введение упомянутому пациенту терапевтически эффективного количества циклобутанового или метилциклобутанового производного, представляющего собой ингибиторы(71)(73) Заявитель и патентовладелец: ИНСАЙТ ХОЛДИНГС КОРПОРЕЙШН (US) Область техники, к которой относится изобретение Настоящее изобретение относится к циклобутановым и метилциклобутановым производным, а также их солям, композициям и способам применения. Упомянутые соединения являются ингибиторами Янус-киназы (JAK), применимыми для лечения JAK-ассоциированных заболеваний, включая, например,воспалительные и аутоиммунные расстройства, а также злокачественные опухоли и миелопролиферативные нарушения. Уровень техники Протеинкиназы (РК) представляют собой группу ферментов, которые регулируют различные важные биологические процессы, включая, среди прочего, рост, жизнеобеспечение и дифференциацию клеток, формирование и морфогенез органов, неоваскуляризацию, заживление и регенерацию тканей. Протеинкиназы выполняют свои физиологические функции, катализируя фосфорилирование белков (или субстратов) и модулируя тем самым клеточные активности субстратов в различных биологических обстоятельствах. В дополнение к функциям, выполняемым в нормальных тканях/органах, многие протеинкиназы также играют более специализированные роли при множестве заболеваний человека, включая рак. Подкласс протеинкиназ (также упоминаемых как онкогенные протеинкиназы) в случае разрегулированности может вызывать образование и рост опухоли и вносить дополнительный вклад в сохранение и развитие опухоли. На сегодняшний день онкогенные протеинкиназы представляют собой одну из самых больших и наиболее привлекательных групп белков-мишеней для воздействия на злокачественные опухоли и для разработки лекарственных средств. Семейство Янус-киназ (JAK) играет роль в цитокин-зависимой регуляции пролиферации и функции клеток, вовлеченных в иммунный ответ. В настоящее время в клетках млекопитающих известно четыре члена JAK-семейства: JAK1 (также известная как Янус-киназа-1), JAK2 (также известная как Янускиназа-2), JAK3 (также известная как Янус-киназа лейкоцита; JAKL; L-JAK и Янус-киназа-3) и TYK2(также известная как протеинтирозинкиназа 2). JAK-белки имеют молекулярную массу в диапазоне от 120 до 140 кДа и содержат семь консервативных гомологичных (JH) JAK-доменов; один из которых представляет собой функциональный каталитический киназный домен, а другой представляет собой псевдокиназный домен, потенциально выполняющий регуляторную функцию и/или служащий в качестве сайта связывания со STAT-белками. Блокирование сигнальной трансдукции на уровне JAK-киназ является многообещающим для развития способов лечения воспалительных заболеваний, аутоиммунных заболеваний, миелопролиферативных заболеваний и рака человека, не говоря уже о других заболевания. Также предполагается, что ингибирование JAK-киназ будет давать терапевтические преимущества для пациентов, страдающих от кожных иммунных нарушений, таких как псориаз и сенсибилизация кожи. Таким образом, постоянно требуются новые или усовершенствованные средства, которые ингибируют киназы, такие как Янус-киназы, для разработки новых и более эффективных фармацевтических препаратов для лечения рака и других заболеваний. Описанные здесь соединения, соли и композиции направлены на удовлетворение таких потребностей и других задач. Сущность изобретения Настоящее изобретение относится к соединению,которое представляет собой 3-циклобутил-3-[4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрил или его фармацевтически приемлемую соль. В другом аспекте настоящее изобретение относится к соединению, которое представляет собой 3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-(3-метилциклобутил)пропаннитрил или его фармацевтически приемлемую соль. В ещ одном аспекте настоящее изобретение относится к соли фосфорной кислоты и соединения,выбранного из группы, состоящей из 3-циклобутил-3-[4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрила,(R)-3-циклобутил-3-[4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрила,(S)-3-циклобутил-3-[4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрила,3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-(3-метилциклобутил)пропаннитрила,3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-транс)-3-метилциклобутил)пропаннитрила,(3R)-3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-транс)-3 метилциклобутил)пропаннитрила,(3S)-3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-транс)-3 метилциклобутил)пропаннитрила,3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-цис)-3 метилциклобутил)пропаннитрила,(3R)-3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-цис)-3 метилциклобутил)пропаннитрила,(3S)-3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-цис)-3 метилциклобутил)пропаннитрила. В ещ одном другом аспекте настоящее изобретение относится к композиции для лечения миелопролиферативного расстройства, содержащей любое из указанных выше соединений или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый носитель. Настоящее изобретение также относится к способу лечения миелопролиферативного расстройства у пациента, включающему введение упомянутому пациенту терапевтически эффективного количества любого из указанных выше соединений или его фармацевтически приемлемой соли. Подробное описание изобретения Настоящее изобретение обеспечивает, в частности, JAK-ингибирующее соединение 3-циклобутил 3-[4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрил (формула I) и его фармацевтически приемлемые соли В предпочтительном варианте настоящее изобретение дополнительно обеспечивает соединения:I-R) и (S)-3-циклобутил-3-[4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрил (формула I-S) и их фармацевтически приемлемые соли В другом аспекте настоящее изобретение обеспечивает JAK-ингибирующее соединение 3-(4-(7 Нпирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-(3-метилциклобутил)пропаннитрил (формула II) и его фармацевтически приемлемые соли В предпочтительном варианте настоящее изобретение обеспечивает цис- и транс-изомеры соединения формулы II. Упомянутые цис- и транс-изомеры: 3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-транс)-3 метилциклобутил)пропаннитрил (формула II - транс) и 3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-цис)-3 метилциклобутил)пропаннитрил (формула II - цис) В другом предпочтительном варианте настоящее изобретение обеспечивает R- и S-энантиомеры соединения формулы II. Такие R- и S-изомеры: В ещ одном предпочтительном варианте настоящее изобретение обеспечивает R/транс-,R/цис-, S/транс- и S/цис-изомеры соединения формулы II. Упомянутые изомеры: Описанные выше соединения упоминаются здесь как "соединения согласно изобретению". Здесь и в других местах, где существуют расхождения между названием соединения и структурой соединения,стандартом будет являться химическая структура. Настоящее изобретение дополнительно обеспечивает фармацевтически приемлемые соли любого из вышеупомянутых соединений. В некоторых вариантах осуществления изобретения фармацевтически приемлемая соль представляет собой соль фосфорной кислоты. В частности, ещ одним аспектом настоящего изобретения являются соли фосфорной кислоты и соединения выбранного из группы, состоящей из 3-циклобутил-3-[4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрила,(R)-3-циклобутил-3-[4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрила,(S)-3-циклобутил-3-[4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрила,3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-(3-метилциклобутил)пропаннитрила,3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-транс)-3 метилциклобутил)пропаннитрила,(3R)-3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-транс)-3 метилциклобутил)пропаннитрила,-3 023444(3S)-3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-транс)-3 метилциклобутил)пропаннитрила,3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-цис)-3 метилциклобутил)пропаннитрила,(3R)-3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-цис)-3 метилциклобутил)пропаннитрила,(3S)-3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-цис)-3 метилциклобутил)пропаннитрила. Описанные здесь соединения являются асимметричными (например, содержат один или несколько стереоцентров). Если не указано иначе, в изобретении подразумеваются все стереоизомеры, такие как энантиомеры и диастереомеры. Соединения согласно настоящему изобретению, которые содержат асимметрично замещенные атомы углерода, можно выделять в виде оптически активных или рацемических форм. Способы получения оптически активных форм известны в данной области, такие как разделение рацемических смесей или стереоселективный синтез. Для описанных здесь соединений также могут существовать геометрические изомеры, и все такие стабильные изомеры предусмотрены в настоящем изобретении. Геометрические цис- и транс-изомеры соединений согласно настоящему изобретению описаны и могут быть выделены в виде смеси изомеров или в виде фактически отдельных изомерных форм. Когда соединение, способное к стереоизомерии (например, оптической и/или геометрической изомерии), обозначается по его структуре или названию без ссылки на конкретные R/S или цис/транс-конфигурации,подразумевается, что все такие изомеры предусмотрены. Например, понятно, что формулы I и II, которые изображены выше, должны включать в себя как R-и S-изомеры, так и цис- и транс-изомеры в тех случаях, когда молекулы позволяют такую изомерию. Разделение рацемических смесей или разделение смеси оптических и/или геометрических изомеров можно осуществлять с помощью любого из многочисленных способов, известных в данной области,включая хроматографические способы (например, хиральную колоночную хроматографию) или фракционную кристаллизацию. Соединения согласно изобретению также могут включать таутомерные формы. Таутомерные формы являются результатом перестановки одинарной связи и соседней двойной связи вместе с сопутствующей миграцией протона. Таутомерные формы включают в себя прототропные таутомеры, которые представляют собой изомерные состояния протонирования, имеющие одинаковую эмпирическую формулу и суммарный заряд. Примеры прототропных таутомеров включают в себя кетон-енольные пары,амид-имидокислотные пары, лактам-лактимные пары, амид-имидокислотные пары, енамин-иминные пары и кольцевые формы, где протон может занимать два или более положений в гетероциклической системе, например, 1 Н- и 3 Н-имидазол, 1 Н-, 2 Н- и 4 Н-1,2,4-триазол, 1 Н- и 2 Н-изодол и 1 Н- и 2 Н-пиразол. Соединения и соли согласно настоящему изобретению могут существовать вместе с другими молекулами, такими как молекулы растворителя и воды, образуя при этом гидраты и сольваты. Соединения и соли согласно настоящему изобретению также могут включать в себя все изотопы присутствующих в них атомов. Изотопы включают в себя такие атомы, которые имеют одинаковый атомный номер, но разные массовые числа. Например, изотопы водорода включают тритий и дейтерий. В некоторых вариантах осуществления изобретения соединения согласно изобретению и их соли фактически являются изолированными. Под выражением "фактически являются изолированными" имеется в виду, что соединение по меньшей мере частично или фактически выделено из окружающей среды,в которой оно было образовано или обнаружено. Частичное выделение может включать в себя, например, композицию, обогащенную соединением или солью согласно изобретению. Фактическое выделение может включать в себя композиции, содержащие по меньшей мере приблизительно 50%, по меньшей мере приблизительно 60%, по меньшей мере приблизительно 70%, по меньшей мере приблизительно 80%, по меньшей мере приблизительно 90%, по меньшей мере приблизительно 95%, по меньшей мере приблизительно 97% или по меньшей мере приблизительно 99% по массе соединения согласно изобретению или его соли. Фраза "фармацевтически приемлемые" используется здесь для ссылки на те соединения, соли, материалы, композиции и/или лекарственные формы, которые по результатам тщательной медицинской(клинической) оценки подходят для контакта с тканями человека и животных без повышенной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соизмеримых с оправданным соотношением эффект/риск. Настоящее изобретение также включает в себя фармацевтически приемлемые соли описанных здесь соединений. Применяемая здесь фраза "фармацевтически приемлемые соли" относится к производным описанных здесь соединений, в которых исходное соединение модифицировано путем превращения присутствующего кислотного или основного фрагмента в его солевую форму. Примеры фармацевтически приемлемых солей включают в себя, но не ограничиваются перечисленным, соли минеральных или органических кислот и основных остатков, таких как амины; щелочные или органические соли кислотных остатков, таких как карбоновые кислоты, и т.п. Фармацевтически приемлемые соли согласно настоящему изобретению включают в себя обычные нетоксичные соли исходного соединения, образованные, например, из нетоксичных неорганических или органических кислот. Фармацевтически приемлемые соли согласно настоящему изобретению можно синтезировать из исходного соединения, которое содержит основной или кислотный фрагмент, обычными химическими способами. Обычно такие соли можно получать путем взаимодействия упомянутых соединений в форме свободной кислоты или основания со стехиометрическим количеством подходящего основания или кислоты в воде или в органическом растворителе или в смеси того и другого. Перечни подходящих солей можно найти в публикациях: Remington'sPharmaceutical Science, 66, 2 (1977), каждая из которых включена в настоящую заявку путем ссылки в своем полном объеме. Способы Соединения и соли согласно изобретению могут ингибировать активность одной или нескольких Янус-киназ (JAK). JAK, с которыми настоящие соединения связываются и/или ингибируют, включают в себя любой член семейства JAK. Настоящие соединения ингибируют активности как JAK1, так и JAK2. Поэтому в еще в одном аспекте настоящее изобретение относится к способу лечения миелопролиферативного расстройства у пациента, включающему введение упомянутому пациенту терапевтически эффективного количества любого из описанных выше соединений или его фармацевтически приемлемой соли. Миелопролиферативные расстройства (MPD) могут включать такие заболеваний как истинная полицитемия (PV), эссенциальная тромбоцитемия (ЕТ), миелофиброз с миелоидной метаплазией (МММ),хронический миелогенный лейкоз (CML), хронический миеломоноцитарный лейкоз (CMML), гиперэозинофильный синдром (HES), системный мастоцитоз (SMCD) и т.п. В некоторых вариантах осуществления изобретения миелопролиферативное расстройство представляет собой первичный миелофиброз (PMF) или миелофиброз, развившийся на фоне истинной полицитемии или эссенциальной тромбоцитемии(Post-PV/ET MF). Применяемый здесь термин "контактирование" относится к объединению указанных веществ в системе in vitro или в системе in vivo. Например, "контактирование" JAK с соединением согласно изобретению включает в себя введение соединения согласно настоящему изобретению индивидууму или пациенту, такому как человек, имеющему JAK, а также, например, введение соединения согласно изобретение в образец, содержащий клеточный препарат или очищенный препарат, содержащий JAK. Применяемый здесь термин "индивидуум" или "пациент", применяемые взаимозаменяемо, относится к любому животному, включая млекопитающих, предпочтительно к мышам, крысам, другим грызунам, кроликам, собакам, кошкам, свиньям, крупному рогатому скоту, овцам, лошадям или приматам, и наиболее предпочтительно к человеку. Применяемая здесь фраза "терапевтически эффективное количество" относится к количеству активного соединения или фармацевтического средства, которое вызывает биологический или лекарственный ответ в ткани, системе, у животного, индивидуума или человека, который желает получить исследователь, ветеринар, лечащий врач или другой практикующий врач. Применяемый здесь термин "лечить" или "лечение" относится к одному или нескольким из следующих пунктов: (1) предотвращение заболевания; например, предотвращение заболевания, состояния или расстройства у индивидуума, который может быть предрасположен к заболеванию, состоянию или расстройству, но патология или симптоматика заболевания еще не ощущаются или не проявляются; (2) замедление заболевания; например, замедление заболевания, состояния или расстройства у индивидуума,у которого ощущаются или проявляются патология или симптоматика заболевания, состояния или расстройства; и (3) облегчение заболевания; например, облегчение заболевания, состояния или расстройства у индивидуума, у которого ощущаются или проявляются патология или симптоматика заболевания, состояния или расстройства (т.е. обратимое развитие патологии и/или симптоматики), такое как уменьшение серьезности заболевания. Термин "применение" включает в себя один или несколько следующих вариантов осуществления изобретения, соответственно: применение при лечении расстройства; применение для производства фармацевтических композиций, применяемых при лечении расстройства, например, применение при производстве лекарственного препарата; способы применения соединений согласно изобретению при лечении упомянутых заболеваний; фармацевтические препараты, содержащие соединения согласно изобретению,для лечения упомянутых заболеваний; и соединения согласно изобретению для применения при лечении упомянутых заболеваний; в зависимости от конкретного случая и практической целесообразности, если не указано иначе. В частности, заболевания, подлежащие лечению, и при этом предпочтительные для применения соединения согласно настоящему изобретению, выбраны из заболеваний, ассоциированных с активностью JAK-киназы. Комбинированные терапии Для лечения JAK-ассоциированных заболеваний, расстройств или состояний в комбинации с соединениями или солями согласно настоящему изобретению можно применять одно или несколько дополнительных фармацевтических средств, например, таких как химиотерапевтические препараты, противовоспалительные средства, стероиды, иммуносупрессанты, а также ингибиторы Bcr-Abl, Flt-3, RAF иFAK-киназ, например, такие как препараты, описанные в публикации WO 2006/056399, или другие терапевтические средства. Одно или несколько дополнительных фармацевтических средств можно вводить пациенту одновременно или последовательно. Примеры химиотерапевтического препарата включают в себя протеосомные ингибиторы (например, бортезомиб), талидомид, ревлимид и ДНК-повреждающие средства, такие как мелфалан, доксорубицин, циклофосфамид, винкристин, этопозид, кармустин и т.п. Примеры стероидов включают в себя кортикостероиды, такие как дексаметазон или преднизон. Примеры ингибиторов Bcr-Abl включают в себя соединения и их фармацевтически приемлемые соли, родов и видов, описанных в патенте США 5521184, публикациях WO 04/005281 и WO 2005/123719. Примеры подходящих ингибиторов Flt-3 включают в себя соединения и их фармацевтически приемлемые соли, которые описаны в публикациях WO 03/037347, WO 03/099771 и WO 04/046120. Примеры подходящих ингибиторов RAF включают в себя соединения и их фармацевтически приемлемые соли, которые описаны в публикациях WO 00/09495 и WO 05/028444. Примеры подходящих ингибиторов FAK включают в себя соединения и их фармацевтически приемлемые соли, которые описаны в публикациях WO 04/080980, WO 04/056786, WO 03/024967,WO 01/064655, WO 00/053595 и WO 01/014402. В некоторых вариантах осуществления изобретения одно или несколько соединений согласно изобретению можно применять в комбинации с одним или несколькими другими ингибиторами киназ,включая иматиниб, в частности для лечения пациентов, не поддающихся лечению иматинибом или другими ингибиторами киназ. Фармацевтические препараты и лекарственные формы В случае использования в качестве фармацевтических препаратов соединения и соли согласно изобретению могут вводиться в форме фармацевтических композиций. Такие композиции можно получать способом, хорошо известным в фармацевтической области, и их можно вводить с помощью различных способов введения в зависимости от того, требуется местное или системное лечение, и в зависимости от области, подлежащей лечению. Введение может быть местным (включая трансдермальное, эпидермальное, офтальмологическое и введение через слизистые оболочки, включая интраназальную, вагинальную и ректальную доставку), пульмональное (например, путем ингаляции или инсуффляции порошков или аэрозолей, включая введение с помощью ингалятора; интратрахеальное или интраназальное), пероральное или парентеральное. Парентеральное введение включает в себя внутривенное, внутриартериальное,подкожное, внутрибрюшное, внутримышечное или инъекцию или инфузию; или внутричерепное введение, например, интратекальное или интравентрикулярное введение. Парентеральное введение может быть в форме одноразовой болюсной дозы или может быть, например, в виде непрерывного нагнетания при перфузии. Фармацевтические композиции и препараты для местного введения могут включать в себя трансдермальные пластыри, мази, лосьоны, кремы, гели, капли, суппозитории, спреи, жидкости и порошки. Может быть необходимым или желательным присутствие традиционных фармацевтических носителей, водных, порошкообразных или масляных основ, загустителей и т.п. Также могут применяться кондомы, перчатки, покрытые слоем лекарственного средства, и т.п. Данное изобретение также включает в себя фармацевтические композиции, которые содержат в качестве активного ингредиента одно или несколько описанных выше соединений согласно изобретению в комбинации с одним или несколькими фармацевтически приемлемыми носителями (инертными наполнителями). При изготовлении композиций согласно изобретению активный ингредиент обычно смешивают с инертным наполнителем, разбавляют инертным наполнителем или заключают внутри такого носителя в форме, например, капсулы, порционного пакетика, бумажной упаковки или другого контейнера. Когда инертный наполнитель служит в качестве разбавителя, он может представлять собой твердый, полужидкий или жидкий материал, который действует как связующее, носитель или среда для активного ингредиента. Таким образом, композиции могут быть в форме таблеток, пилюль, порошков, пастилок,порционных пакетиков, крахмальных капсул, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (в твердом виде или в жидкой среде), мазей, содержащих, например, до 10% по массе активного соединения, мягких и жестких желатиновых капсул, суппозиториев, стерильных растворов для инъекций и стерильно упакованных порошков. При получении препарата активное соединение можно измельчать, чтобы обеспечить подходящий размер частиц перед объединением с другими ингредиентами. Если активное соединение является фактически нерастворимым, его можно измельчать до размера частиц менее 200 меш. Если активное соединение является фактически водорастворимым, размер частиц можно регулировать путем измельчения,чтобы обеспечить фактически равномерное распределение в препарате, например, приблизительно 40 меш. Соединения согласно изобретению можно измельчать с применением известных процедур измельчения, таких как мокрое измельчение, чтобы получать размер частиц, подходящий для образования таблеток и препаратов других типов. Тонкодисперсные (в форме наночастиц) препараты соединений согласно изобретению можно получать с помощью способов, известных в данной области, см., например,международную патентную заявку WO 2002/000196. Некоторые примеры подходящих инертных наполнителей включают в себя лактозу, декстрозу, сахарозу, сорбит, маннит, крахмалы, гуммиарабик, фосфат кальция, альгинаты, трагакант, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, паточный сироп и метилцеллюлозу. Препараты могут дополнительно включать в себя смазывающие средства, такие как тальк, стеарат магния и минеральное масло; смачивающие средства; эмульгирующие и суспендирующие средства; консерванты, такие как метил- и пропилгидроксибензоаты; подсластители и ароматизаторы. Композиции согласно изобретению можно получать таким образом, чтобы обеспечить быстрое, замедленное или отсроченное высвобождение активного ингредиента после введения пациенту с помощью процедур, известных в данной области. Композиции можно получать в стандартной лекарственной форме, где каждая доза содержит от приблизительно 5 до приблизительно 1000 мг (1 г), еще чаще - от приблизительно 100 до приблизительно 500 мг активного ингредиента. Термин "стандартные лекарственные формы" относится к физически дискретным элементам, подходящим в качестве однократных доз для человеческих субъектов и других млекопитающих, при этом каждый элемент содержит заданное количество активных материалов, рассчитанное таким образом, чтобы создавать требуемый терапевтический эффект в сочетании с подходящим фармацевтическим инертным наполнителем. Активное соединение может быть эффективным в широком диапазоне дозировок, и обычно его вводят в фармацевтически эффективном количестве. Однако будет понятно, что количество фактически вводимого соединения обычно определяется врачом согласно соответствующим обстоятельствам, включающим в себя состояние, подвергаемое лечению, выбор способа введения, фактически вводимое соединение, возраст, массу тела и реакцию индивидуального пациента, серьезность симптомов, наблюдаемых у пациента и т.п. Для получения твердых композиций, таких как таблетки, основной активный ингредиент смешивают с фармацевтическим инертным наполнителем для образования твердой, предварительно смешанной композиции, содержащей гомогенную смесь соединения согласно настоящему изобретению. Когда ссылаются на такие предварительно смешанные композиции как гомогенные, это означает, что активный ингредиент обычно распределен равномерно на всем протяжении композиции для того, чтобы можно было легко разделить композицию на одинаково эффективные стандартные лекарственные формы, такие как таблетки, пилюли и капсулы. Такую твердую, предварительно смешанную композицию затем делят на стандартные лекарственные формы описанного выше типа, содержащие, например, от приблизительно 0,1 до приблизительно 1000 мг активного ингредиента согласно настоящему изобретению. На таблетки или пилюли согласно настоящему изобретению можно наносить слой покрытия или иным образом компоновать, чтобы обеспечить лекарственную форму, предусматривающую эффект пролонгированного действия. Например, таблетка или пилюля может содержать дозировку внутреннего компонента и дозировку внешнего компонента, при этом второй компонент находится в форме оболочки сверху первого. Два компонента могут разделяться с помощью кишечнорастворимого слоя, который служит для противодействия разрушению в желудке и позволяет внутреннему компоненту проходить в неизменном виде в двенадцатиперстную кишку или иметь отсрочку в высвобождении. Для таких кишечнорастворимых слоев или покрытий можно применять различные материалы, такие материалы включают в себя ряд полимерных кислот и смеси полимерных кислот с такими материалами, как шеллак, цетиловый спирт и ацетат целлюлозы. Жидкие формы для перорального введения или инъекции, в которые можно включать соединения и композиции согласно настоящему изобретению, включают в себя водные растворы, подходящим образом ароматизированные сиропы, водные или масляные суспензии и ароматизированные эмульсии с пищевыми маслами, такими как хлопковое масло, кунжутное масло, кокосовое масло или арахисовое масло, а также эликсиры и подобные фармацевтические носители. Композиции для ингаляции или инсуффляции включают в себя растворы и суспензии в фармацевтически приемлемых, водных или органических растворителях или их смесях, и порошки. Жидкие или твердые композиции могут содержать подходящие фармацевтически приемлемые инертные наполнители, которые описаны выше. В некоторых вариантах осуществления изобретения композиции вводят с помощью перорального введения или назального респираторного способа введения для местного или системного действия. Композиции можно распылять с помощью инертных газов. Распыляемые растворы можно вдыхать непосредственно из распыляющего устройства, или распыляющее устройство можно присоединять к тампонированным дыхательным маскам или к дыхательному устройству периодического действия с положительным давлением. Раствор, суспензию или порошкообразные композиции можно вводить перорально или назально из устройств, которые доставляют препарат подходящим способом. Количество соединения или композиции, вводимое пациенту, будет меняться в зависимости от того, что вводится, цели введения, такой как профилактика или терапия, состояния пациента, способа введения и т.п. При терапевтических применениях композиции можно вводить пациенту, уже страдающему от заболевания, в количестве, достаточном для лечения или по меньшей мере для частичного купирования симптомов заболевания и связанных с ним осложнений. Эффективные дозы будут зависеть от состояния заболевания, подвергаемого лечению, а также от оценки лечащего врача, зависящей от таких факторов, как серьезность заболевания, возраст, масса, общее состояние пациента и т.п. Композиции, вводимые пациенту, могут быть в форме описанных выше фармацевтических композиций. Упомянутые композиции можно стерилизовать с помощью традиционных способов стерилизации или с помощью стерилизующего фильтрования. Водные растворы можно расфасовывать для применения как есть или подвергать лиофилизации, при этом лиофилизованный препарат перед введением объединяют со стерильным водным носителем. рН препаратов, содержащих соединение, обычно будет составлять от 3 до 11, более предпочтительно от 5 до 9 и наиболее предпочтительно от 7 до 8. Будет понятно,что применение определенных вышеупомянутых инертных наполнителей, носителей или стабилизаторов будет приводить к образованию фармацевтических солей. Терапевтическая дозировка соединений согласно настоящему изобретению может меняться, например, согласно конкретному применению, для которого проводится лечение, способу введения соединения, здоровью и состоянию пациента и назначению, прописанному врачом. Доля или концентрация соединения согласно изобретению в фармацевтической композиции может меняться в зависимости от ряда факторов, включая дозировку, химические характеристики (например, гидрофобность) и способ введения. Например, для парентерального введения соединения согласно изобретению можно получать в водном физиологическом буферном растворе, содержащем от приблизительно 0,1 до приблизительно 10% мас./об. соединения. Некоторые типичные диапазоны доз составляют от приблизительно 1 мкг/кг до приблизительно 1 г/кг массы тела в день. В некоторых вариантах осуществления изобретения диапазон доз составляет от приблизительно 0,01 до приблизительно 100 мг/кг массы тела в день. Вероятно, дозировка должна зависеть от таких переменных факторов, как тип и степень развития заболевания или расстройства, общее состояние здоровья конкретного пациента, относительная биологическая эффективность выбранного соединения, состава инертного наполнителя и способа введения. Эффективные дозы можно экстраполировать из кривых зависимости доза-ответ, полученных для систем испытаний in vitro или испытаний на животных моделях. В некоторых вариантах осуществления изобретения соединение согласно изобретению или его фармацевтически приемлемую соль вводят в виде офтальмологической композиции. Соответственно, в некоторых вариантах осуществления изобретения способы включают введение соединения или его фармацевтически приемлемой соли и офтальмологически приемлемого носителя. В некоторых вариантах осуществления изобретения офтальмологическая композиция представляет собой жидкую композицию,полужидкую композицию, офтальмологическую вкладку, пленку, микрочастицы или наночастицы. Офтальмологические композиции подробно описаны в патентной заявке США сер.12/571834, поданной 1 октября 2009 г., которая включена в настоящую заявку путем ссылки. Кроме того, следует принимать во внимание, что некоторые признаки изобретения, которые для ясности описаны в контексте отдельных вариантов осуществления изобретения, также могут обеспечиваться в комбинации с одним вариантом осуществления изобретения. И наоборот, различные признаки изобретения, которые для краткости описаны в контексте одного варианта осуществления изобретения,также могут обеспечиваться отдельно или в любой подходящей подкомбинации. Различные модификации согласно изобретению в дополнение к описанным здесь вариантам осуществления будут очевидны специалистам в данной области из описания. Каждая из ссылок, цитируемых в настоящей заявке, включена в настоящую заявку путем ссылки в ее полном объеме. Изобретение будет описано более подробно с помощью конкретных примеров. Следующие примеры предлагаются с иллюстративными целями и не предназначены ограничивать изобретение каким-либо способом. Специалисты в данной области легко установят различные некритические параметры, которые можно изменять или модифицировать, чтобы получить фактически те же самые результаты. Стадия 1. Циклобутанкарбоксальдегид. К раствору оксалилхлорида (20,6 мл, 0,244 моль) в метиленхлориде (700 мл, 10 моль) при -78 С добавляли раствор диметилсульфоксида (34,6 мл, 0,488 моль) в метиленхлориде (100 мл). Спустя 10 мин добавляли циклобутилметанол (Aldrich, 17,5 г, 0,203 моль) в метиленхлориде (100 мл) и полученную смесь перемешивали при -78 С в течение 30 мин. Затем добавляли раствор триэтиламина (14 0 мл, 1,0 моль) в метиленхлориде (100 мл) и перемешивали смесь в течение 5 ч, давая смеси возможность постепенно нагреться до комнатной температуры. После гашения реакционной смеси водой смесь разделяли. Органический слой промывали водой (2), насыщенным раствором соли, сушили над сульфатом натрия и фильтровали. Фильтрат перегоняли, собирая фракцию 86-92 С, чтобы получить альдегид (18,6 г,54,4%). Стадия 2. 3-Циклобутилакрилонитрил. К раствору 1,0 0 М трет-бутилата калия в тетрагидрофуране (116 мл, 0,116 моль) при 0 С по каплям добавляли раствор диэтилцианометилфосфоната (Aldrich, 19,7 мл, 0,122 моль) в тетрагидрофуране(200 мл). Реакционную смесь нагревали до комнатной температуры и затем снова охлаждали до 0 С. К реакционной смеси добавляли раствор циклобутанкарбоксальдегида (см. стадию 1, 18,6 г, 0,110 моль) в тетрагидрофуране (100 мл). Реакционной смеси давали нагреться до комнатной температуры и перемешивали при комнатной температуре в течение ночи. После гашения водой смесь экстрагировали простым эфиром. Объединенные органические слои промывали водой, насыщенным раствором соли, сушили и выпаривали досуха. Неочищенную смесь очищали на силикагеле, элюируя раствором 0-40% EtOAc в гексане, чтобы получить требуемый продукт (5,30 г, 44,7%). ЖХ-МС: рассчитано для(S)-3-циклобутил-3-[4-(7-[2(триметилсилил)этокси]метил-7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрил. К раствору 4-(1 Н-пиразол-4-ил)-7-[2-(триметилсилил)этокси]метил-7 Н-пирроло[2,3d]пиримидина (см. публикацию патентной заявки США 2007/0135461, 15,6 г, 0,050 моль) в ацетонитриле(124 мл, 2,37 моль) добавляли 3-циклобутилакрилонитрил (5,30 г, 0,050 моль) с последующим добавлением 1,8-диазабицикло[5.4.0]ундец-7-ена (3,70 мл, 0,025 моль). Полученную смесь перемешивали при комнатной температуре в течение ночи, затем выпаривали досуха. Смесь очищали на силикагеле, элюируя раствором 0-60% EtOAc в гексане, чтобы получить требуемый продукт в виде рацемической смеси(16 г, 76%). ЖХ-МС: рассчитано для C22H31N6OSi (M+H)+: m/z=423,2; найдено: 423,0. Рацемическую смесь разделяли с помощью хиральной ВЭЖХ (колонка: ChiralCel OJ-H, 30250 мм, 5 мкм; подвижная фаза: 30% этанола/70% гексанов; скорость потока: 24 мл/мин), чтобы получить два энантиомера. Для хиральной аналитической ВЭЖХ (колонка: ChiralCel OJ-H, 4,6250 мм, 5 мкм; подвижная фаза: 30% этанола/70% гексанов; скорость потока: 0,8 мл/мин): время удерживания первого пика: 6,6 мин; время удерживания второго пика: 8,1 мин. Стадия 4. (R или S)-3-Циклобутил-3-[4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1 ил]пропаннитрил. В 500-мл круглодонную колбу, оборудованную магнитной мешалкой, холодильником и входным отверстием для азота, загружали ацетонитрил (55 мл), воду (4,8 мл) и [R или S) -3-циклобутил-3-[4-(7[2-(триметилсилил)этокси]метил-7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрил(второй пик при разделении на хиральной колонке на стадии 3, 2,8 г, 6,6 ммоль). Добавляли тетрафторборат лития (7,50 г, 0,078 моль). Полученную смесь нагревали с обратным холодильником в течение ночи, охлаждали до комнатной температуры и порциями загружали 3,00 М раствор гидроксида аммония в воде (9,78 мл), в течение 5 мин доводя рН до 9-10. Спустя 30 мин полученную смесь очищали с помощью ОФ-ВЭЖХ (колонка XBridge C18 30100 мм, объем вводимой пробы 5 мл (50 мг/пробу, элюирование с градиентом ацетонитрил/вода, содержащая 0, 15% NH4 OH, при скорости потока 60 мл/мин), чтобы получить требуемый продукт в виде свободного основания (1,51 г, 77,96%). ЖХ-МС: рассчитано дляC16H17N6 (M+H)+: m/z=293,2; найдено: 293,1. 1H ЯМР (300 МГц CD3OD)8,65 (1 Н, с), 8,59 (1 Н, с),8,34 (1 Н, с), 7,50 (1 Н, д, J=3,6 Гц), 6,94 (1 Н, д, J=3,6 Гц), 4,69 (1 Н, м), 3,07-2,97 (3 Н, м), 2,21 (1 Н, м), 1,971,84 (5 Н, м) ч./млн, ее 98,8%. Другой энантиомер можно получать таким же образом, исходя из соединения, соответствующего первому пику, полученному при разделении на хиральной колонке на стадии 3. Пример 2. Соль Стадия 1. 3-Метиленциклобутанкарбоновая кислота. В круглодонную колбу,оборудованную холодильником,добавляли 3-метиленциклобутанкарбонитрил (BePharma, 10,0 г, 0,107 моль). В колбу добавляли раствор гидроксида калия(24,1 г, 0,365 моль) в этаноле (112 мл) и воду (88 мл) и нагревали смесь при 100 С. Спустя приблизительно 2 ч, когда прекращалось выделение аммиака, растворитель выпаривали досуха при пониженном давлении. Твердые вещества растворяли в воде (75 мл), охлаждали на ледяной бане и подкисляли приблизительно до рН 1 концентрированной хлористоводородной кислотой. Полученный верхний слой дважды экстрагировали дихлорметаном. Органические слои объединяли и сушили над безводным сульфатом магния. При удалении органических растворителей получали требуемый неочищенный продукт(100 мл) добавляли оксалилхлорид (Aldrich, 5,33 мл, 62,9 ммоль) с последующим добавлением каталитического количества диметилформамида (DMF). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, затем выпаривали досуха. Неочищенный хлорангидрид растворяли в метиленхлориде (200 мл). К полученному раствору добавляли гидрохлорид N, О-диметилгидроксиламина Aldrich, 6,14 г, 62,9 ммоль) с последующим добавлением по каплям триэтиламина (TEA) (21,9 мл,0,157 моль) при 0 С. Реакционную смесь перемешивали при комнатной температуре в течение ночи и отфильтровывали соль ТЕА-HCl. Фильтрат промывали 1 н раствором HCl, затем водным раствором бикарбоната натрия, насыщенным раствором соли, сушили над сульфатом магния и выпаривали досуха. Неочищенный амид (7,30 г, 89,7%) применяли непосредственно на следующей стадии. ЖХ-МС: рассчитано для C8H14NO2 (M+H)+: m/z=156,l; найдено: 156,3. Стадия 3. 3-Метиленциклобутанкарбальдегид. К суспензии тетрагидроалюмината лития (2,18 г, 57,5 ммоль) в простом эфире (200 мл) при -15 С добавляли по каплям раствор N-метокси-N-метил-3-метиленциклобутанкарбоксамида (стадия 2, 7,14 г,46,0 ммоль) в тетрагидрофуране (75 мл). Реакционную смесь перемешивали при температуре от 0 до -15 С в течение 30 мин, затем гасили водным раствором гидросульфата калия. Полученную смесь экстрагировали простым эфиром. Объединенные органические слои промывали насыщенным раствором соли, сушили над сульфатом магния и выпаривали. Неочищенный продукт (6,70 г, 151,5%) применяли непосредственно на следующей стадии. Стадия 4. 3-(3-Метиленциклобутил)акрилонитрил. К раствору 1,00 М трет-бутилата калия в тетрагидрофуране (48,3 мл, 48,3 ммоль) при 0 С добавляли по каплям раствор диэтилцианометилфосфоната (Aldrich, 8,19 мл, 50,6 ммоль) в тетрагидрофуране(80 мл). Реакционную смесь нагревали до комнатной температуры и затем охлаждали до 0 С. К реакционной смеси добавляли раствор 3-метиленциклобутанкарбальдегида (стадия 3, 4,42 г, 46,0 ммоль) в тетрагидрофуране (40 мл). Реакционной смеси давали нагреться до комнатной температуры и затем перемешивали при комнатной температуре в течение ночи. После гашения водой смесь экстрагировали простым эфиром. Объединенные органические слои промывали водой, насыщенным раствором соли, сушили и выпаривали досуха. Неочищенную смесь (5,90 г, 107,7%) применяли непосредственно на следующей стадии. Стадия 5. 3-(3-Метиленциклобутил)-3-[4-(7-[2-(триметилсилил)этокси]метил-7 Н-пирроло[2,3d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрил. К раствору 4-(1 Н-пиразол-4-ил)-7-[2-(триметилсилил)этокси]метил-7 Н-пирроло[2,3-d]пиримидина (см. публикацию патентной заявки США 2007/0135461, 7,25 г, 23,0 ммоль) в ацетонитриле(57,4 мл) добавляли неочищенный (сырой) 3-(3-метиленциклобутил)акрилонитрил (стадия 4, 2,74 г,23,0 ммоль) с последующим добавлением 1,8-диазабицикло[5.4.0]ундец-7-ена (3,44 мл, 23,0 ммоль). Полученную смесь перемешивали при комнатной температуре в течение уикенда, затем выпаривали досуха. Остаток очищали на силикагеле, элюируя раствором 0-8 0% EtOAc в гексане, чтобы получить требуемый продукт (6,0 г, 60,1%). ЖХ-МС: рассчитано для C23H31N6OSi (M+H)+: m/z=435,2; найдено: 435,4. Стадия 6. (3R или 3S)-3-транс)-3-Метилциклобутил)-3-(4-(7-( (2-(триметилсилил)этокси)метил)7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)пропаннитрил. Смесь из 3-(3-метиленциклобутил)-3-[4-(7-[2-(триметилсилил)этокси]метил-7 Н-пирроло[2,3d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрила (стадия 5, 4,0 г, 9,2 моль) и 100 мл метанола течение 1 ч подвергали гидрированию в присутствии 0,6 г 10% Pd/C с применением баллона с водородом, находящимся под давлением. После удаления катализатора фильтрованием фильтрат упаривали досуха и очищали на силикагеле, элюируя раствором 0-100% EtOAc в гексане, чтобы получить требуемый продукт в виде смеси транс- и цис-изомеров. ЖХ-МС: рассчитано для C23H33N6OSi (M+H)+: m/z=437,3; найдено: 437,4. Продукт дважды подвергали очистке на колонке для хиральной ВЭЖХ. После первого разделения ВЭЖХ (колонка: ChiralCel OD-H, 30250 мм, 5 мкм; подвижная фаза: 15% этанола/85% гексанов; скорость потока: 28 мл/мин) получали две фракции, А и В. Фракция А представляла собой цис/транс-смесь одного из энантиомеров. Время удерживания: 10,51 мин. Фракция В представляла собой цис/транс-смесь другого энантиомера, которая давала два неразделимых пика, соответствующие временам удерживания 13,05 мин и 13,92 мин. Первую фракцию (А) подвергали дополнительному разделению с помощью хиральной ВЭЖХ (колонка: ChiralPak IA, 20250 мм, 5 мкм; подвижная фаза: 10% этанола/90% гексанов; скорость потока: 15 мл/мин), чтобы получить два пика, А 1 и А 2: один пик, соответствующий цис-изомеру, и другой пик, соответствующий транс-изомеру. Согласно хиральной аналитической ВЭЖХ (колонка: ChiralPak IA, 4,6250 мм, 5 мкм; подвижная фаза: 15% этанола/85% гексанов; скорость потока: 1,0 мл/мин): время удерживания первого пика (А 1): 11,79 мин; время удерживания второго пика (А 2): 12,78 мин. Вторую фракцию (В) подвергали разделению с помощью хиральной ВЭЖХ (колонка: ChiralPak IA, 20250 мм, 5 мкм; подвижная фаза: 15% этанола/85% гексанов; скорость потока: 15 мл/мин), чтобы получить два пика, В 1 и В 2 (для каждого пика 800 мг, 19,9%). Позднее с помощью ЯЭО (nOe) было показано, что В 1 соответствует цис-изомеру, а В 2 соответствует транс-изомеру другого энантиомера. Согласно хиральной аналитической ВЭЖХ (колонка: ChiralPak IA, 4,6250 мм, 5 мкм; подвижная фаза: 15% этанола/85% гексанов; скорость потока: 1,0 мл/мин), время удерживания первого пика(В 1) 12,48 мин, и время удерживания второго пика (В 2) 14,16 мин. Стадия 7. (3R или 3S)-3-(4-(7 Н-Пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-транс)-3 метилциклобутил)пропаннитрил. В 500-мл круглодонную колбу, оборудованную магнитной мешалкой, холодильником и входным отверстием для азота, загружали ацетонитрил (9,69 мл), воду (0,84 мл) и 3-транс)-3-метилциклобутил)3-[4-(7-[2-(триметилсилил)этокси]метил-7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1 ил]пропаннитрил (0,60 г, 1,4 моль) (В 2, полученный при хиральном разделении на предыдущей стадии(т.е. пик 2 для второй фракции. Добавляли тетрафторборат лития (1,31 г, 13,7 ммоль). Смесь нагревали с обратным холодильником в течение ночи, затем порциями загружали 7,2 М раствор гидроксида аммония в воде (0,71 мл, 5,1 ммоль) в течение 5 мин при комнатной температуре, доводя рН до 9-10. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Твердое вещество удаляли фильтрованием и фильтрат очищали с помощью ОФ-ВЭЖХ (колонка XBridge C18 30100 мм, объем вводимой пробы 5 мл (50 мг/пробу), элюирование градиентом ацетонитрил/вода, содержащая 0, 15 NH4OH, при скорости потока 60 мл/мин), чтобы получить требуемый продукт в виде свободного основания. ЖХ-МС: рассчитано для C17H19N6 (M+H)+: m/z=3 07,2; найдено: 3 07,4. 1H ЯМР (500 МГц, ДМСО-d6)12,08 (1 Н, с), 8,78 (1 Н, с), 8,68 (1 Н, с), 8,36 (1 Н, с), 7,59 (1 Н, д, J=3,0 Гц), 6,99 (1 Н, д, J=3,0 Гц), 4,78 (1 Н,м), 3,12 (2 Н, м), 2,88 (1 Н, м), 2,30 (1 Н, м), 2,06 (1 Н, м) 1,88 (1 Н, м), 1,74 (1 Н, м), 1,44 (1 Н, м), 1,08 (3 Н, д,J=7,0 Гц) ч./млн, ее 93,3%. Другой энантиомер можно получать таким же способом, исходя из соединения, соответствующего фракции А со стадии 6. Стадия 8. Соль (3R или 3S)-3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-транс)-3 метилциклобутил)пропаннитрила с фосфорной кислотой. К раствору (3R или 3S)-3-транс)-3-метилциклобутил)-3-[4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)1 Н-пиразол-1-ил]пропаннитрила (стадия 7, 0,275 г, 0,8 98 ммоль) в изопропиловом спирте (5,83 мл) добавляли фосфорную кислоту (96,8 мг, 0,987 ммоль) в 1,0 мл изопропанола при 60 С. После перемешивания в течение 1 ч смеси давали охладиться до комнатной температуры. Осадок отфильтровывали и сушили на воздухе, затем промывали простым этиловым эфиром и дополнительно сушили на воздухе, чтобы получить требуемый фосфатный продукт (330 мг, 90,9%). Стадия 1. (3R или 3S)-3-цис)-3-метилциклобутил)-3-(4-(7-2- (триметилсилил)этокси)метил)-7'Нпирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)пропаннитрил. В 500-мл круглодонную колбу, оборудованную магнитной мешалкой, холодильником и входным отверстием для азота, загружали ацетонитрил (8,1 мл), воду (0,70 мл) и (3R или 3S)-3-цис)-3 метилциклобутил)-3-[4-(7-[2-(триметилсилил)этокси]метил-7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Нпиразол-1-ил]пропаннитрил (0,50 г, 1,1 ммоль) (фракция В 1, полученная при хиральном разделении,описанная в примере 2, стадия 6 (т.е. пик 1 второй фракции. Добавляли тетрафторборат лития (1,10 г,11,4 ммоль). Раствор нагревали с обратным холодильником в течение ночи. Затем в раствор порциями загружали раствор гидроксида аммония в воде (7,2 М, 0,59 мл, 4,3 ммоль) в течение 5 мин при комнатной температуре, доводя рН до 9-10. Реакционную смесь перемешивали в течение 2 ч при комнатной температуре. Твердое вещество удаляли фильтрованием и фильтрат очищали с помощью ОФ-ВЭЖХ (колонкаXBridge C18 30100 мм, объем вводимой пробы 5 мл (50 мг/пробу), элюирование с градиентом ацетонитрил/вода, содержащая 0,15% NH4OH при скорости потока 60 мл/мин), чтобы получить требуемый продукт. ЖХ-МС: рассчитано для C17H19N6 (M+H)+: m/z=307,2; найдено: 307,4. 1 Н ЯМР (500 МГц,ДМСО-d6)12,08 (1 Н, с), 8,75 (1 Н, с), 8,68 (1 Н, с), 8,36 (1 Н, с), 7,59 (1 Н, д, J=3,0 Гц), 6,99 (1 Н, д,J=3,0 Гц), 4,66 (1 Н, м), 3,11 (2 Н, м), 2,66 (1 Н, м), 2,20 (2 Н, м), 1,88 (1 Н, м), 1,42 (2 Н, м), 0,97 (3 Н, д,J=6,0 Гц) ч/млн, ее 99,8%. Другой энантиомер можно получать тем же самым способом, исходя из соединения, соответствующего фракции А по примеру 2, стадия 6. Стадия 2. Соль (3R или 3S)-3-(4-(7 Н-пирроло[2,3-d] пиримидин- 4 -ил)-1 Н-пиразол-1-пл)-3-цис)-3 метилциклобутил)пропаннитрила и фосфорной кислоты. К раствору (3R или 3S)-3-цис)-3-метилциклобутил)-3-[4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Нпиразол-1-ил]пропаннитрила (стадия 1, 0,23 г, 0,751 ммоль) в изопропиловом спирте (4,87 мл) добавляли фосфорную кислоту (80,9 мг, 0,83 ммоль) в 1,0 мл изопропанола при 60 С. Смесь перемешивали в течение 2 ч, затем давали ей охладиться до комнатной температуры. Осадок отфильтровывали и сушили на воздухе, затем промывали простым этиловым эфиром и дополнительно сушили на воздухе, чтобы получить требуемый фосфатный продукт (300 мг, 98,8%). 1 Н ЯМР (400 МГц, ДМСО-d6)12,08 (1 Н, с),8,75 (1 Н, с), 8,65 (1 Н, с), 8,34 (1 Н, с), 7,58 (1 Н, д, J=2,4 Гц), 6,97 (1 Н, д, J=2,4 Гц), 4,63 (1 Н, м),3,09 (2 Н, м), 2,64 (1 Н, м), 2,18 (2 Н, м), 1,86 (1 Н, м), 1,40 (2 Н, м), 0,96 (3 Н, д, J=6,4 Гц) ч./млн. Пример А: JAK-киназный анализ in vitro. Описанные здесь соединения подвергали испытанию на ингибиторную активность в отношенииJAK-мишеней согласно следующему анализу in vitro, описанному в публикации Park и др., AnalyticalBiochemistry 1999, 269, 94-104. Каталитические домены человеческих JAK1 (а.к. 837-1142), JAK2 (а.к. 828-1132) и JAK3 (а.к. 781-1124) с N-терминальным His-tag экспрессировали в клетках насекомого с применением бакуловируса и очищали. Каталитическую активность JAK1, JAK2 или JAK3 анализировали путем измерения фосфорилирования биотинилированного пептида. Фосфорилированный пептид обнаруживали с помощью метода гомогенной флуоресценции с временным разрешением (HTRF). Для каждой киназы измеряли значения IC50 соединений в реакционных смесях, которые содержали фермент,АТФ и 500 нМ пептида в 50 мМ Tris-буфере (рН 7,8) с 100 мМ NaCl, 5 мМ DTT и 0,1 мг/мл (0,01%) BSA. Концентрация АТФ в реакционных смесях составляла 90 мкМ для JAK1, 30 мкМ для JAK2 и 3 мкМ дляJAK3. Реакции проводили при комнатной температуре в течение 1 ч и затем останавливали с помощью 20 мкл 45 мМ ЭДТК, 300 нМ SA-APC, 6 нМ Eu-Ру 20 в буфере для анализа (Perkin Elmer, Boston, MA). Связывание с антителом, меченным европием, происходило в течение 40 мин, и HTRF-сигнал измеряли на спектрофотометре для считывания планшетов Fusion (Perkin Elmer, Boston, MA). Установлено, что соединения по примерам 1, 2 и 3 имеют значения IC50 менее 2 нМ в отношении JAK1 и менее 1 нМ в отношении JAK2. Пример В: Клеточные анализы. Одно или несколько описанных здесь соединений испытывали на ингибиторную активность в отношении JAK-мишени согласно по меньшей мере одному из следующих клеточных анализов. Для выращивания линии клеток рака, зависимой от цитокинов и, следовательно, от JAK/STATсигнальной трансдукции, высевали в количестве 6000 клеток на лунку (формат планшета: 96-луночный) в RPMI 1640, 10% FBS и 1 нг/мл соответствующего цитокина. К клеткам в ДМСО/среда (конечная концентрация 0,2% ДМСО) добавляли соединения и инкубировали в течение 72 ч при 37 С и 5% СО 2. Влияние соединения на жизнеспособность клеток оценивали с использованием люминесцентного анализа жизнеспособности клеток CellTiter-Glo (Promega) с последующим подсчетом на считывающем устройстве TopCount (Perkin Elmer, Boston, MA). Потенциальные нецелевые эффекты соединений измеряли в параллельных пробах с использованием линии клеток, не стимулированных JAK, с тем же самым считыванием данных анализа. Все эксперименты проводили в двух параллельных опытах. Упомянутые выше клеточные линии также можно применять для исследования влияния соединений на фосфорилирование JAK-киназ или возможных последующих субстратов, таких как STAT-белки,Akt, Shp2 или Erk. Упомянутые эксперименты можно проводить после удаления цитокинов в течение ночи с последующей короткой, предварительной инкубацией с соединением (2 ч или менее) и цитокиновой стимуляцией в течение приблизительно 1 ч или менее. Затем белки экстрагируют из клеток и анализируют с помощью методов, хорошо известных специалистам в данной области, включая вестернблоттинг или ELISA с использованием антител, которые могут различать фосфорилированный и общий белок. В таких экспериментах можно использовать нормальные клетки или клетки рака, чтобы исследовать активность соединений в отношении биологических механизмов жизнеобеспечения опухолевых клеток или в отношении медиаторов воспалительного заболевания. Например, что касается последнего,цитокины, такие как IL-6, IL-12, IL-23 или IFN, можно применять для стимулирования активации JAK,приводящей к фосфорилированию STAT-белка (белков) и потенциально к транскрипционным профилям(оценивали с помощью набора или технологии qPCR) или продуцированию и/или секреции белков, таких как IL-17. Способность соединений ингибировать упомянутые цитокин-опосредованные эффекты можно измерять с применением методик, хорошо известных специалистам в данной области. Описанные здесь соединения также можно испытывать в клеточных моделях, разработанных для оценки эффективности и активности соединений в отношении мутантных JAK, например, установлено,что мутация JAK2V617F приводит к пролиферативным миелоидным расстройствам. В таких экспериментах часто используют цитокин-зависимые клетки гематологической клеточной линии (например,BaF/3), в которой JAK-киназы дикого типа или мутантные JAK-киназы экспрессируются эктопически(James С. и др., Nature 434:1144-1148; Staerk J. и др., JBC 280:41893-41899). Конечные критерии оценки включают в себя данные о воздействии соединений на жизнеобеспечение клеток, пролиферацию и фосфорилированные JAK, STAT, Akt или Erk-белки. Некоторые из описанных здесь соединений оценивались или могли оцениваться на предмет их ингибирующей активности в отношении пролиферации Т-клеток. В качестве такого анализа можно рассматривать повторный анализ цитокинстимулированной пролиферации (т.е. JAK), а также упрощенный анализ иммуносупрессии или ингибирования иммунной активации. Далее следует короткий план того,как можно проводить такие эксперименты. Мононуклеарные клетки периферической крови (РВМС) получают из образцов цельной человеческой крови с применением (градиентного) способа разделения Ficoll Hypaque, а Т-клетки (фракция 2000) можно получать из РВМС путем элютриации. Свежевыделенные человеческие Т-клетки можно хранить в культуральной среде (RPMI 1640, дополненная 10% фетальной бычьей сыворотки, 100 Ед./мл пенициллина, 100 мкг/мл стрептомицина) при плотности 2106 клеток/мл при 37 С в течение не более 2 дней. Для анализа IL-2-стимулированной клеточной пролиферации Т-клетки сначала в течение 72 ч обрабатывают фитогемагглютинином (РИА) при конечной концентрации 10 мкг/мл. После однократного промывания PBS 60 00 клеток/лунку высевают в 9 6-луночные планшеты и обрабатывают соединениями, взятыми при различных концентрациях, в культуральной среде в присутствии 100 Ед./мл человеческого IL-2 (ProSpec-Tany TechnoGene; Rehovot, Israel). Планшеты инкубируют при 37 С в течение 72 ч и оценивают индекс пролиферации с применением реагентов для люминесцентного анализа CellTiter-Glo, следуя протоколу, который рекомендует производитель (Promega; Madison,WI). Пример С. Противоопухолевая эффективность in vivo. Описанные здесь соединения можно оценивать на ксенотрансплантате человеческой опухоли у иммунодефектных мышей. Например, для подкожной инокуляции SCID-мышей можно использовать туморогенный вариант INA-6-плазмоцитомной клеточной линии (Burger R. и др., Hematol J. 2:42-53, 2001). Животных-опухоленосителей затем можно рандомизировать на группы, которые лечат лекарственным средством или связующим, и можно вводить различные дозы соединений любым из ряда общепринятых способов введения, включая пероральный способ, интраперитонеальный или непрерывную инфузию с применением имплантируемых помп. Рост опухоли во времени контролируют с помощью штангенциркулей. Кроме того, в любой момент после начала лечения можно брать образцы опухоли для анализа,который описан выше (пример В), чтобы оценить эффекты соединения в отношении активности JAK и последующих путей сигналинга. Кроме того, селективность соединения (соединений) можно оценивать с применением ксенотрансплантата опухоли у мышей, которая стимулируется другими известными кина- 13023444 зами (например, Bcr-Abl), в такой модели, как модель опухоли К 562. Пример D. Испытание на мышах с применением реакции контактной (кожной) гиперчувствительности замедленного типа. Описанные здесь соединения также можно испытывать в отношении их эффективности (для ингибирования JAK-мишеней) на мышиной модели испытания гиперчувствительности замедленного типа,стимулированной Т-клетками. Считается, что мышиная реакция контактной (кожной) гиперчувствительности замедленного типа (DTH) представляет собой валидную модель клинического контактного дерматита и других иммунных кожных расстройств, опосредованных Т-лимфоцитами, таких как псориаз (Immunol Today., 1998 Jan; 19(1):37-44). Мышиная DTH связана с псориазом множеством характеристик,включая иммунный инфильтрат, сопутствующее увеличение воспалительных цитокинов и гиперпролиферацию кератиноцитов. Кроме того, многие классы лекарственных средств, которые с клинической точки зрения эффективны при лечении псориаза, также являются эффективными ингибиторами реакцииDTH у мышей (Agents Actions., 1993 Jan; 38 (1-2):11 б-21). На 0 и 1 день мышей Balb/c сенсибилизировали с помощью местного нанесения на их бритый живот антигена 2,4-динитрофторбензола (ДНФБ). На 5 день измеряли толщину ушных раковин с помощью технического микрометра. Данное измерение регистрировали и использовали в качестве контрольного. Затем на обе ушные раковины животных наносили провокационную пробу путем местного нанесения 20 мкл ДНФБ в целом (10 мкл на внутреннюю поверхность ушной раковины и 10 мкл на наружную поверхность ушной раковины) при концентрации 0,2%. Спустя 24-72 ч после провокации ушные раковины снова измеряли. Лечение тестируемыми соединениями проводили на протяжении сенсибилизации и всех фаз провокации (от 1 дня до 7 дня) или перед началом и на протяжении фазы провокации (обычно после полудня от 4 дня до 7 дня). При лечении тестируемые соединения (в разной концентрации) вводили либо системно, либо местно (местное нанесение на ушные раковины). Эффективности тестируемых соединений отмечали по уменьшению данных ушного аллерготеста по сравнению с ситуацией отсутствия лечения. Соединения, вызывающие уменьшение на 20% или более, считались эффективными. В некоторых экспериментах мышей подвергали провокации, но не сенсибилизировали (отрицательный контроль). Ингибирующий эффект (ингибирование активации JAK-STAT-пути) тестируемых соединений можно подтвердить данными иммуногистохимического анализа. Активация JAK-STAT-пути (путей) приводит к образованию и транслокации функциональных транскрипционных факторов. Кроме того,инфлюкс иммунных клеток и повышенная пролиферация кератиноцитов также должны обеспечивать однозначные изменения профилей экспрессии в ушной раковине, которые можно исследовать и выразить в количественной форме. Фиксированные в формалине и залитые парафином участки ушной раковины(взятые после фазы провокации в модели DTH) подвергали иммуногистохимическому анализу с применением антитела, которое специфически взаимодействует с фосфорилированным STAT3 (клон 58 Е 12,Cell Signaling Technologies). Мышиные ушные раковины обрабатывали тестируемыми соединениями,связующим или дексаметазоном (клинически эффективное лечение псориаза) или оставляли без обработки в модели DTH для сравнений. Тестируемые соединения и дексаметазон могут продуцировать подобные транскрипционные изменения как качественно, так и количественно; и как тестируемые соединения, так и дексаметазон могут уменьшать число проникающих клеток. Как системное, так и местное введение тестируемых соединений может оказывать ингибирующие эффекты, т.е. уменьшать число проникающих клеток и ингибировать транскрипционные изменения. Пример Е: Противовоспалительная активность in vivo. Можно проводить оценку описанных здесь соединений на экспериментальных моделях с участием грызунов или на моделях с участием не грызунов, разработанных для того, чтобы воспроизводить отдельную или комплексную воспалительную реакцию. Например, модели артрита грызунов можно применять для оценки терапевтического потенциала соединений, принимаемых с превентивной или терапевтической целью. Такие модели включают в себя, но не ограничиваются перечисленным, коллагениндуцированный артрит мышей или крыс, адьювантный артрит крыс и артрит, индуцированный антителами к коллагену. Аутоиммунные заболевания, включающие в себя, но не ограничивающиеся перечисленным, множественный склероз, сахарный диабет I-типа, увеоретинит, тиреоидит, тяжелую псевдопаралитическую миастению, иммуноглобулиновые нефропатии, миокардит, сенсибилизацию дыхательных путей (астма), волчанку или колит, также можно применять для оценки терапевтического потенциала описанных здесь соединений. Такие модели хорошо адаптированы в научном сообществе и хорошо знакомы специалистам в данной области Current Protocols in Immunology, Vol 3., Coligan J.E. и др., WileyPress.; Methods in Molecular Biology: Vol. 225, Inflammation Protocols., Winyard, P.G. and Willoughby D.A.,Humana Press, 2003). Пример F: Животные модели для лечения синдрома сухого глаза, увеита и конъюнктивита. Соединения можно оценивать с помощью одной или нескольких доклинических моделей синдрома сухого глаза, известных специалистам в данной области, включая, но, не ограничиваясь перечисленным,кроличью модель слезных желез с применением конканавалина А (ConA), мышиную модель синдрома сухого глаза, индуцированного скополамином (подкожное или трансдермальное введение), мышиную модель слезных желез с применением ботулинического токсина или любую модель из ряда моделей спонтанных аутоиммунных расстройств грызунов, которые приводят к дисфункции глазной железы (например, NOD-SCID, MRL/lpr или NZB/NZW) (публикации Barabino и др., Experimental Eye Research 2004, 79, 613-621, и Schrader и др., Developmental Opthalmology, Karger 2008, 41, 298-312, каждая из которых включена в настоящую заявку путем ссылки в полном объеме). Конечные критерии оценки в таких экспериментальных моделях могут включать в себя данные о гистопатологии глазных желез и глаза (роговицы и т.д.) и, по возможности, данные классического теста Ширмера или его модифицированных версий (Barabino и др.), которые определяют продукцию слезной жидкости. Активность можно оценивать путем введения определенных доз с помощью нескольких способов введения (например, системное или местное введение), которое можно начинать до или после измеряемого проявления существующего заболевания. Соединения можно оценивать с помощью одной или нескольких доклинических моделей увеита,известных специалистам в данной области. Такие модели включают в себя, но не ограничиваются перечисленным, модели экспериментального аутоиммунного увеита (EAU) и эндотоксин-индуцированного увеита (EIU). EAU-эксперименты могут проводиться на кроликах, крысах или мышах и могут включать в себя пассивную или активную иммунизацию. Например, можно применять любой из ряда ретинальных антигенов, чтобы сенсибилизировать животных по отношению к соответствующему иммуногену, после чего животным можно вносить в глаза провокационную пробу с использованием того же самого антигена. EIU-модель является более опасной и включает в себя местное или системное введение липополисахарида в сублетальных дозах. Конечные критерии оценки при использовании как EIU-модели, так и дляEAU-модели могут включать в себя среди прочего данные фундоскопического осмотра и гистопатологии. Упомянутые модели рассмотрены в публикации Smith и др. (Immunology and Cell Biology 1998, 76,497-512, которая включена в настоящую заявку путем ссылки в полном объеме). Активность оценивают путем введения определенных доз с помощью нескольких способов введения (например, системного или местного введения), которое можно начинать до или после измеряемого проявления существующего заболевания. В некоторых из перечисленных выше моделей также можно развивать склерит/эписклерит,хориоидит, циклит или ирит и, следовательно, они применимы при исследовании потенциальной активности соединений для терапевтического лечения упомянутых заболеваний. Соединения также можно оценивать с помощью одной или нескольких доклинических моделей конъюнктивита, известных специалистам в данной области. Такие модели включают в себя, но не ограничиваются перечисленным, модели на грызунах с использованием морской свинки, крысы или мыши. Модели на морских свинках включают в себя модели с использованием протоколов активной или пассивной иммунизации и/или иммунной провокации с помощью антигенов, таких как овальбумин или амброзия (рассмотрены в публикации Groneberg D.A. и др., Allergy 2003, 58, 1101-1113, которая включена в настоящую заявку путем ссылки в полном объеме). Общий замысел в крысиных и мышиных моделях аналогичен моделям на морских свинках (также рассмотрены в публикации Groneberg). Активность можно оценивать путем введения определенных доз с помощью нескольких способов введения (например, системного или местного введения), которое можно начинать до или после измеряемого проявления существующего заболевания. Конечные критерии оценки в таких исследованиях могут включать в себя,например, данные гистологического, иммунологического, биохимического или молекулярного анализа глазных тканей, таких как конъюнктива. Различные модификации согласно изобретению в дополнение к описанным здесь вариантам осуществления изобретения будут очевидны специалистам в данной области из приведенного выше описания. Каждая из ссылок, цитируемых в настоящей заявке, включена в настоящую заявку путем ссылки в ее полном объеме. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, которое представляет собой 3-циклобутил-3-[4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)1 Н-пиразол-1-ил]пропаннитрил или его фармацевтически приемлемую соль. 2. Соединение по п.1, которое представляет собой (R)-3-циклобутил-3-[4-(7 Н-пирроло[2,3d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрил или его фармацевтически приемлемую соль. 3. Соединение по п.1, которое представляет собой (S)-3-циклобутил-3-[4-(7 Н-пирроло[2,3d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрил или его фармацевтически приемлемую соль. 4. Соединение, которое представляет собой 3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1 ил)-3-(3-метилциклобутил)пропаннитрил или его фармацевтически приемлемую соль. 5. Соединение по п.4, которое представляет собой 3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Нпиразол-1-ил)-3-транс)-3-метилциклобутил)пропаннитрил или его фармацевтически приемлемую соль. 6. Соединение по п.5, которое представляет собой (3R)-3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)1 Н-пиразол-1-ил)-3-транс)-3-метилциклобутил)пропаннитрил или его фармацевтически приемлемую соль. 7. Соединение по п.5, которое представляет собой (3S)-3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)1 Н-пиразол-1-ил)-3-транс)-3-метилциклобутил)пропаннитрил или его фармацевтически приемлемую соль. 8. Соединение по п.4, которое представляет собой 3-(4-(7 Н-пирроло[2, 3-d]пиримидин-4-ил)-1 Нпиразол-1-ил)-3-цис)-3-метилциклобутил)пропаннитрил или его фармацевтически приемлемую соль. 9. Соединение по п.8, которое представляет собой (3R)-3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)1 Н-пиразол-1-ил)-3-цис)-3-метилциклобутил)пропаннитрил или его фармацевтически приемлемую соль. 10. Соединение по п.8 которое представляет собой (3S)-3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)1 Н-пиразол-1-ил)-3-цис)-3-метилциклобутил)пропаннитрил или его фармацевтически приемлемую соль. 11. Соль фосфорной кислоты и соединения, выбранного из группы, состоящей из 3-циклобутил-3-[4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил]пропаннитрила;(3S)-3-(4-(7 Н-пирроло[2,3-d]пиримидин-4-ил)-1 Н-пиразол-1-ил)-3-цис)-3 метилциклобутил)пропаннитрила. 12. Композиция для лечения миелопролиферативного расстройства, содержащая соединение по любому из пп.1-10 или его фармацевтически приемлемую соль и по меньшей мере один фармацевтически приемлемый носитель. 13. Способ лечения миелопролиферативного расстройства у пациента, включающий в себя введение упомянутому пациенту терапевтически эффективного количества соединения по любому из пп.1-10 или его фармацевтически приемлемой соли. 14. Способ по п.13, в котором упомянутое миелопролиферативное расстройство (MPD) представляет собой истинную полицитемию (PV), эссенциальную тромбоцитемию (ЕТ), первичный миелофиброз(PMF), миелофиброз с миелоидной метаплазией (МММ), хронический миелогенный лейкоз (CML), хронический миеломоноцитарный лейкоз (CMML), гиперэозинофильный синдром (HES), идиопатический миелофиброз (IMF), системный мастоцитоз (SMCD) или миелофиброз, развившийся на фоне истинной полицитемии или эссенциальной тромбоцитемии (Post-PV/ET MF).
МПК / Метки
МПК: A61K 31/519, A61P 35/00, A61P 29/00, A61P 37/00, A61P 27/00, C07D 487/04, A61P 17/00
Метки: циклобутановые, применения, композиции, основе, метилциклобутановые, производные, способы
Код ссылки
<a href="https://eas.patents.su/17-23444-ciklobutanovye-i-metilciklobutanovye-proizvodnye-kompozicii-na-ih-osnove-i-sposoby-ih-primeneniya.html" rel="bookmark" title="База патентов Евразийского Союза">Циклобутановые и метилциклобутановые производные, композиции на их основе и способы их применения</a>
Производные пиперидина, композиции на их основе, способы их применения

Номер патента: 14686
Опубликовано: 30.12.2010
Авторы: Кассайре Джером, Майенфиш Петер, Корси Камилла, Питтерна Томас, Седербаум Фредрик, Моллейре Луи-Пьер
МПК: A01N 43/40, C07D 211/06, C07D 211/68...
Метки: способы, применения, производные, композиции, пиперидина, основе
Формула / Реферат:
1. Способ борьбы с насекомыми, клещами, нематодами или моллюсками или их уничтожения, который включает нанесение на вредителей, на очаг вредителей или на растение, подверженное нашествию вредителей, инсектицидно, акарицидно, нематоцидно или моллюскоцидно эффективного количества соединения формулы Iв которой Y обозначает ординарную связь, С=O или C=S;кольцопредставляет собой бензол, пиридин, пиримидин, пиразин, пиридазин, триазин, пиррол,...
Тиоацетатные соединения, композиции на их основе и способы их применения

Номер патента: 22933
Опубликовано: 31.03.2016
Авторы: Вернье Жан-Мишель, Гуник Эсмир, Оук Самеди
МПК: A61K 31/4965, A61K 31/444, A61K 31/44...
Метки: тиоацетатные, соединения, применения, основе, композиции, способы
Формула / Реферат:
1. Соединение формулыили его фармацевтически приемлемая соль.2. Способ снижения уровня мочевой кислоты в сыворотке у человека, включающий введение человеку эффективного количества соединения по п.1.3. Способ лечения гиперурикемии у человека, включающий введение человеку эффективного количества соединения по п.1.4. Способ лечения гиперурикемии у человека, болеющего подагрой, включающий введение человеку эффективного количества соединения по...
Композиции на основе слитого белка ов и способы их применения

Номер патента: 4790
Опубликовано: 26.08.2004
Авторы: Хект Рэнди Айра, Мэнн Майкл Бенджамин
МПК: A61K 38/17, A61P 3/00, C07K 14/575...
Метки: композиции, слитого, применения, основе, белка, способы
Формула / Реферат:
1. Слитой белок, не обязательно имеющий метионин на N-конце, содержащий константный домен антитела или его часть, слитую с N-концом человеческого белка лептина, который выбран из группы, включающей (а) аминокислотную последовательность 1-146, приведенную в SEQ.ID. No.6; (б) аминокислотную последовательность 1-146, приведенную в SEQ.ID. No.6, имеющую остаток лизина в положении 35 и остаток изолейцина в положении 74; (в) аминокислотную...
Композиции на основе слитого белка ов и способы их применения

Номер патента: 4791
Опубликовано: 26.08.2004
Авторы: Мэнн Майкл Бенджамин, Хект Рэнди Айра
МПК: A61K 38/17, A61P 3/00, C07K 14/575...
Метки: применения, белка, способы, основе, композиции, слитого
Формула / Реферат:
1. Белок с формулой, выбранной из группы R1-R2 и R1-L-R2, где R1 является белком Fc, его производным или аналогом, R2 является белком лептином, его производным или аналогом, а L представляет собой линкер, причем белок Fc, его производное или аналог выбраны из группы, включающей (а) аминокислотные последовательности Fc, приведенные в SEQ.ID. No.9, 12, 15 и 18; (б) аминокислотные последовательности (а), у которых различные аминокислоты...
Композиции на основе производных липопептидных антибиотиков и способы их применения

Номер патента: 10294
Опубликовано: 29.08.2008
Авторы: Джиа Кьюи, Вакович-Сгарби Ширли А., Чен Юхен, Камерон Дэйл Р., Дугорд Доминик, Сгарби Паоло В.М., Бойд Винсент А., Курран Вильям В., Лиз Ричард А., Нодвелл Мэтью, Бордерс Дональд Б.
МПК: A61K 38/12, A61P 31/04, C07K 7/56...
Метки: антибиотиков, липопептидных, применения, производных, композиции, основе, способы
Формула / Реферат:
1. Противомикробное соединение и его фармацевтически приемлемые соли структурной формулы (II) где R1 представляет собой ОН или NH2; L выбран из по меньшей мере одной аминокислоты, по меньшей мере одной замещенной аминокислоты, -R'C(=O)-, -R'OC(=O)(NR')- и -O-PhC(=O)-; R2 выбран из -C(=O)R5, -C(=O)OR5, -C(=O)NHR4, -C(=O)NR4R4, -C(=S)NHR4, -C(=S)NR4R4, -C(=NR4)NHR4 и -C(=NR4)NR4R4; R3 выбран из группы: -OR5, -SR5, NR5R5, -CN, -NO2, -N3,...
Предыдущий патент: Держатель разгрузочной насадки и циклон
Следующий патент: Способ и устройство для оптимизированной передачи сигналов дистанционного управления, в частности, воротами, дверями и шлагбаумами
Случайный патент: Способ борьбы с сорняками в культурах полезных растений