Соединения, обладающие антибактериальной активностью в отношении clostridium
Номер патента: 21567
Опубликовано: 30.07.2015
Авторы: Люни Насер, Гийемон Жером Эмиль Жорж, Рабуассон Пьер Жан-Мари Бернар
















Формула / Реферат
1. Соединение формулы (I)

включая любые его стереохимические изомерные формы и таутомеры, где
R1 и R2, каждый независимо, выбраны из водорода, галогена, гидрокси, C1-6алкила, полигалоген-C1-6алкила, C1-6алкилокси или полигалоген-C1-6алкилокси;
R3 представляет собой гидрокси, амино, моно- или ди(С1-4алкил)амино;
R4 представляет собой водород или C1-4алкил;
X представляет собой азот или CR5, где R5 представляет собой водород, галоген или C1-4алкил;
при условии, что, когда R3 представляет собой гидрокси, X представляет собой СН и R4 представляет собой водород;
или его фармацевтически приемлемая соль присоединения кислоты.
2. Соединение по п.1 или его фармацевтически приемлемая соль присоединения кислоты, имеющие (R)-конфигурацию в положении 4 хроманильного фрагмента.
3. Соединение по п.1 или 2 или его фармацевтически приемлемая соль присоединения кислоты, отличающиеся тем, что R1 и R2, каждый, представляют собой галоген.
4. Соединение по п.1 или его фармацевтически приемлемая соль присоединения кислоты, отличающиеся тем, что R1 и R2, каждый, представляют собой бром и находятся в положении 6 и 8 хроманильного фрагмента.
5. Соединение по п.1 или 3 или его фармацевтически приемлемая соль присоединения кислоты, отличающиеся тем, что R1 и R2, каждый, представляют собой бром и находятся в положении 6 и 8 хроманильного фрагмента, a R3 представляет собой гидрокси.
6. Соединение по п.1, отличающееся тем, что указанное соединение представляет собой

или

7. Фармацевтическая композиция, имеющая антибактериальную активность, содержащая фармацевтически приемлемый носитель и терапевтически активное количество соединения по любому из пп.1-6 или его фармацевтически приемлемой соли присоединения кислоты.
8. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли присоединения кислоты в качестве лекарственного средства.
9. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли присоединения кислоты для лечения бактериальных инфекций.
10. Применение по п.9, отличающееся тем, что бактериальная инфекция представляет собой инфекцию, вызываемую бактериями Clostridium.
11. Способ лечения бактериальной инфекции у теплокровных субъектов, включающий введение теплокровному субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по любому из пп.1-6 или его фармацевтически приемлемой соли присоединения кислоты.
12. Способ по п.11, отличающийся тем, что бактериальная инфекция представляет собой инфекцию, вызываемую бактериями Clostridium.
13. Способ получения соединения формулы (I), согласно которому:
а) промежуточное соединение формулы (II) N-алкилируют промежуточным соединением формулы (III), где заместители R1, R2, R3, R4 и X такие, как определено в п.1

или b) соединения формулы (I) преобразовывают друг в друга согласно известным реакциям преобразования; или, при необходимости, соединение формулы (I) преобразовывают в фармацевтически приемлемую соль присоединения кислоты или, наоборот, соль присоединения кислоты соединения формулы (I) переводят в форму свободного основания при помощи щелочи; и, при необходимости, получают их стереохимически изомерные формы.
Текст
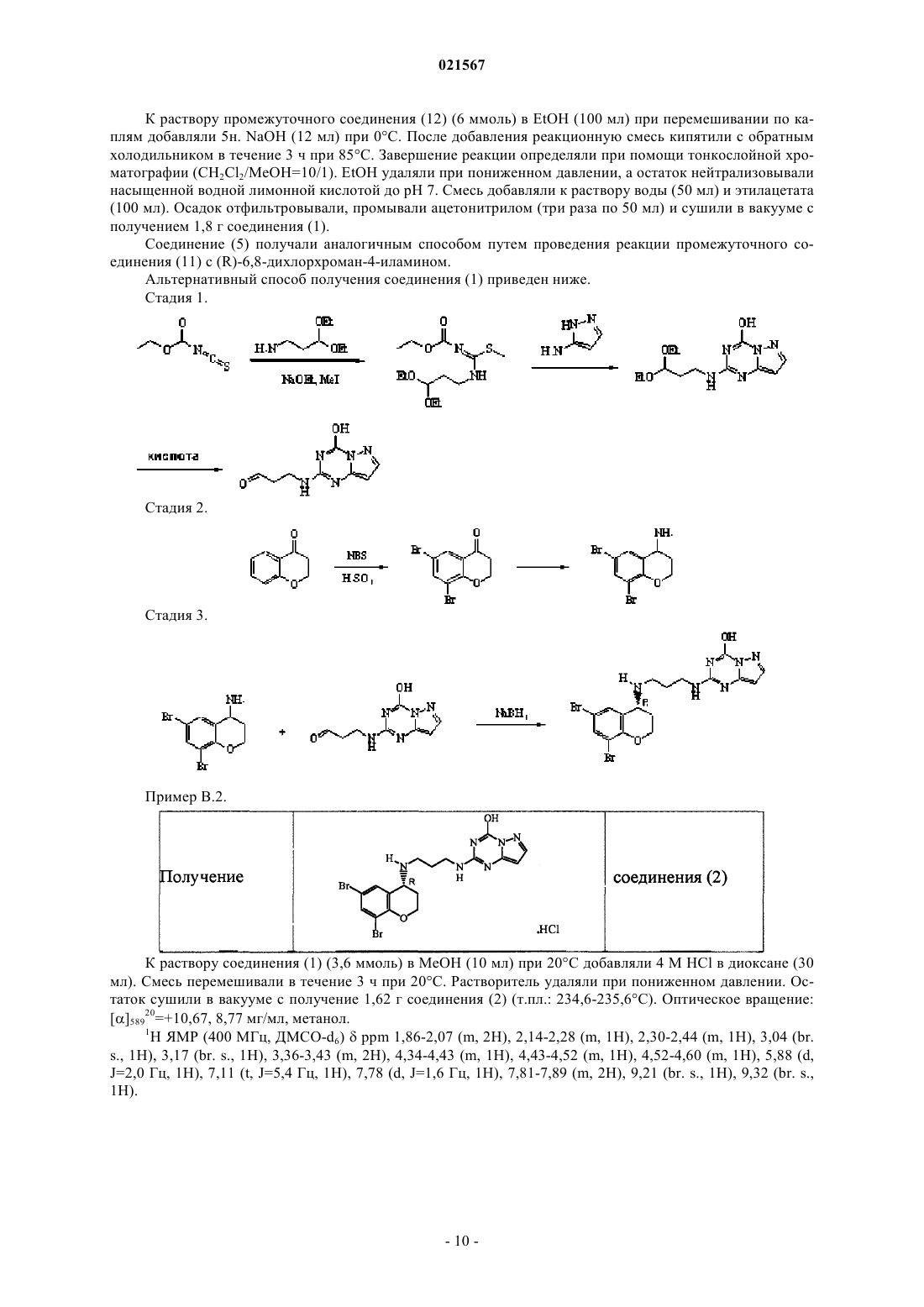
СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АНТИБАКТЕРИАЛЬНОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ CLOSTRIDIUM Гийемон Жером Эмиль Жорж (FR),Рабуассон Пьер Жан-Мари Бернар,Люни Насер (BE) Лыу Т.Н. (RU) Настоящее изобретение относится к новым соединениям формулы (I), обладающим антибактериальной активностью в отношении бактерий Clostridium, в частности Clostridiumperfringens, фармацевтическим композициям, содержащим указанные соединения, и химическим способам получения указанных соединений(71)(73) Заявитель и патентовладелец: ЭЛАНКО ЭНИМАЛ ХЕЛС АЙРЛЭНД ЛИМИТЕД (IE) Настоящее изобретение относится к новым соединениям формулы (I), обладающим антибактериальной активностью в отношении бактерий Clostridium, в частности Clostridium perfringens, фармацевтическим композициям, содержащим указанные соединения, и химическим способам получения указанных соединений.Clostridium представляет собой род спорообразующих грамположительных бактерий, которые растут в анаэробных условиях, включающий более 100 видов. Существует четыре основных вида, вызывающих заболевания у людей и других теплокровных животных: С. botulinum - организм, продуцирующий токсины в пищевых продуктах или ранах, вызывающий ботулизм; С. difficile, который может вызывать псевдомембранный колит, токсический мегаколон и диареи, связанные с приемом антибиотиков; С.tetani, который является возбудителем столбняка; и С. perfringens. С. perfringens распространен повсеместно в окружающей среде и обнаружен в почве, пыли, сырьевых ингредиентах, таких как используемые в пищевой промышленности специи, и в кишечнике человека и животных. Данная бактерия вырабатывает более 15 различных токсинов, а инфекции, вызываемые С.Perfringens, могут приводить к пищевому отравлению, обусловленному С. perfringens типа А, энтеротоксемии, некротическому энтериту и газовой гангрене. В птицеводстве инфекции С. perfringens могут вызывать проблемы с кишечником у бройлеров со значительными негативными экономическими последствиями. Поскольку применение антибиотиков в пищевой промышленности жестко регламентируется, существует потребность в альтернативных антибактериальных соединениях. ВWO 2008/039640 предложено соединение 5-[3-R)(+)-6,8-дибром-хроман-4 иламино)пропиламино]-4 Н-тиено[3,2-b]пиридин-7-он, также известное как REP3123, и раскрыта его антибактериальная активность в отношении Clostridium difficile. Исследования антибактериальной активности соединения REP3123 in vitro показывают, что указанное соединение обладает активностью в отношении бактерий рода Clostridium, однако REP3123 также обладает антибактериальной активностью в отношении широкого спектра бактерий, которые присутствуют в кишечнике. Такая антибактериальная активность широкого спектра в отношении грамположительных бактерий оказывает негативное влияние на кишечную флору. Следовательно, существует потребность в антибактериальных соединениях, обладающих активностью в отношении бактерий родаClostridium, но имеющих узкий спектр активности в отношении грамположительных бактерий и не оказывающих негативного влияния на кишечную флору. Настоящее изобретение относится к соединению формулы (I) включая любые его стереохимические изомерные формы и таутомеры, гдеR4 представляет собой водород или C1-4 алкил;X представляет собой азот или CR5, где R5 представляет собой водород, галоген или C1-4 алкил; при условии, что, когда R3 представляет собой гидрокси, X представляет собой СН и R4 представляет собой водород; или его фармацевтически приемлемой соли присоединения кислоты, или сольвату. Указанное условие направлено на исключение соединений, обладающих малой или не обладающих антибактериальной активностью в отношений бактерий рода Clostridium. В вышеприведенных определениях: галоген представляет собой фтор, хлор, бром и йод;C1-4 алкил представляет собой линейные или разветвленные углеводородные радикалы, содержащие от 1 до 4 атомов углерода, такие как, например, метил, этил, пропил, бутил, 1-метилэтил, 2-метилпропил и т.п.; подразумевается, что C1-6 алкил включает C1-4 алкил и его высшие гомологи, содержащие 5 или 6 атомов углерода, такие как, например, 2-метилбутил, пентил, гексил и т.п.; полигалоген-C1-6 алкил представляет собой полигалогензамещенный C1-6 алкил, в частности возможные изомерные формы, которые способны образовывать соединения формулы (I). Если не оговорено иное, химическое обозначение соединений подразумевает смесь всех возможных стереохимически изомерных форм, причем указанные смеси включают все диастереомеры и энантиомеры основной молекулярной структуры. Более конкретно, стереогенные центры могут иметь R- или S-конфигурацию; заместители в двухвалентных циклических (частично) насыщенных радикалах могут иметь цис- или трансконфигурацию. Стереохимически изомерные формы соединений формулы (I), несомненно, находятся в рамках настоящего изобретения. Абсолютные стереохимические конфигурации соединений формулы (I) и промежуточных соединений, используемых при их получении, могут быть легко определены специалистом в данной области техники при помощи известных методов, таких как, например, рентгеновская дифракция. Некоторые из соединений формулы (I) могут также существовать в таутомерной форме. Предполагается, что такие формы, хотя прямо и не указанные в приведенной выше формуле, также находятся в рамках настоящего изобретения. Например, для соединения формулы (I), где R3 представляет собой гидрокси, соответствующая кето-форма может представлять собой наиболее распространенный таутомер Кроме того, некоторые соединения формулы (I) и некоторые промежуточные соединения, используемые при их получении, могут проявлять полиморфизм. Следует понимать, что настоящее изобретение охватывает любые полиморфные формы, обладающие свойствами, подходящими для лечения состояний,указанных выше. Фармацевтически приемлемые соли присоединения кислоты, как указано выше, включают формы терапевтически активных нетоксичных солей присоединения кислоты, которые способны образовывать соединения формулы (I). Эти фармацевтически приемлемые соли присоединения кислоты можно легко получать путем обработки основной формы подходящей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например соляная или бромисто-водородная кислота, серная, азотная, фосфорная и подобные кислоты; или органические кислоты,такие как, например, уксусная, пропионовая, гликолевая, молочная, пировиноградная, щавелевая (т.е. этандиовая), малоновая, янтарная (т.е. бутандиовая кислота), малеиновая, фумаровая, яблочная, винная,лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая,салициловая, п-аминосалициловая, памовая и подобные кислоты. И наоборот, указанные формы солей можно преобразовать в щелочную форму путем обработки соответствующим основанием. Соединения формулы (I) могут существовать в сольватированной и несольватированной формах. Термин "сольват" в настоящем описании применяется для описания ассоциаций молекул, включающих соединение настоящего изобретения и одну или более молекул фармацевтически приемлемых растворителей, например, воды или этанола. Термин "гидрат" используется в том случае, если указанный растворитель представляет собой воду. Соединения формулы (I) содержат по меньшей мере один асимметричный атом углерода, как показано ниже, где асимметричный атом углерода обозначен Один вариант реализации настоящего изобретения относится к соединениям формулы (R)-(I), которые определены как соединения формулы (I), имеющие (R)-конфигурацию в положении 4 хроманильного фрагмента Представляющими интерес соединениями формулы (I) являются такие соединения формулы (I), для которых применимы следующие ограничения:b) R1 и R2, каждый, представляют собой бром и находятся в 6- и 8-положениях хроманильного фрагмента; илиj) X представляет собой CR5, где R5 представляет собой галоген, в частности хлор. Первая группа соединений представляет собой такие соединения формулы (R)-(I), в которых R1 и 2R , каждый, представляют собой бром и находятся в 6- и 8-положениях хроманильного фрагмента, a R3 представляет собой гидрокси. Вторая группа соединений представляет собой такие соединения формулы (R)-(I), в которых X представляет собой азот, R1 и R2, каждый, представляют собой бром и находятся в 6- и 8-положениях хроманильного фрагмента, a R3 представляет собой гидрокси. Третья группа соединений представляет собой такие соединения формулы (R)-(I), в которых R1 и 2R , каждый, представляют собой бром и находятся в 6- и 8-положениях хроманильного фрагмента и в которых R3 представляет собой амино. Соединения формулы (I) можно получать путем восстановительного N-алкилирования промежуточного соединения формулы (II) промежуточным соединением формулы (III) в соответствии с известными способами восстановительного N-алкилирования. Указанное восстановительное N-алкилирование можно осуществлять в инертном растворителе, таком как, например, дихлорметан, тетрагидрофуран (ТГФ), толуол или их смесь, и в присутствии восстанавливающего агента, такого как, например, боргидрид, например боргидрид натрия, цианоборгидрид натрия или триацетоксиборгидрид. Также может быть удобно использовать водород в качестве восстанавливающего агента в комбинации с подходящим катализатором, таким как, например, палладий-наугле или платина-на-угле. В случае использования водорода в качестве восстанавливающего агента, может быть предпочтительно добавлять к реакционной смеси осушитель, такой как, например, третбутоксид алюминия. В целях предотвращения дальнейшего нежелательного гидрирования определенных функциональных групп в реагентах и продуктах реакции может быть эффективно добавлять в реакционную смесь соответствующий каталитический яд, например, тиофен или хинолин-серу. Для повышения скорости реакции температуру можно увеличивать в диапазоне между комнатной температурой и температурой кипения реакционной смеси и, необязательно, можно увеличивать давление водорода. При получении соединений формулы (I), в которых R3 представляет собой амино или моно(С 1-4 алкил)амино, с использованием описанного выше способа N-алкилирования может быть целесообразно защищать указанную функциональную аминогруппу. Защитные группы для функциональных аминогрупп известны в данной области и удаляются после проведения N-алкилирования. Также соединения формулы (I), в которых R3 представляет собой гидрокси, можно получать при использовании описанного выше способа N-алкилирования, в результате чего функциональные гидроксигруппы защищаются известными защитными группами. Соединения формулы (I-а), определяемые как соединения формулы (I), в которых R3 представляет собой гидрокси, можно получать путем гидролиза промежуточных соединений формулы (IV) в щелочных условиях. Промежуточные соединения формулы (IV) можно получать в соответствии с описанным выше способом N-алкилирования Исходные материалы и некоторые промежуточные соединения представляют собой известные соединения и являются коммерчески доступными или могут быть получены в соответствии с обычными способами, известными в данной области техники. Соединения формулы (I), полученные вышеописанными способами, можно синтезировать в виде рацемических смесей энантиомеров, каждый из которых можно отделить от другого посредством известных процедур разделения. Те соединения формулы (I), которые получают в виде рацематов, можно перевести в соответствующие формы диастереомерных солей путем реакции с подходящей хиральной кислотой. Далее указанные формы диастереомерных солей разделяют, например, путем селективной или фракционной кристаллизации, и энантиомеры высвобождают при помощи щелочи. Альтернативный способ разделения энантиомерных форм соединений формулы (I) включает жидкостную хроматографию с использованием хиральной неподвижной фазы. Указанные чистые стереохимически изомерные формы также можно получать из соответствующих чистых стереохимически изомерных форм соответствующих исходных материалов, при условии, что реакция протекает стереоспецифически. Если необходим конкретный стереоизомер, предпочтительно синтезировать указанное соединение путем стереоспецифических способов получения. В этих способах преимущественно используют энантиомерно чистые исходные материалы. Соединения формулы (II), включающие любые стереохимически изомерные формы и их таутомеры, а также их фармацевтически приемлемые соли обладают антибактериальной активностью, в частности, в отношении бактерий рода Clostridium, в частности Clostridium perfringens, продемонстрированную в фармакологических примерах. Соответственно, настоящее изобретение также относится к соединениям формулы (I) для применения в качестве лекарственных средств, особенно для применения для лечения бактериальных инфекций,в частности инфекций, вызванных бактериями рода Clostridium, более конкретно, инфекций, вызванныхClostridium perfringens. Кроме того, соединения согласно настоящему изобретению можно применять для получения лекарственных средств для лечения бактериальных инфекций, в частности, вызванных бактериями рода Clostridium, более конкретно, инфекций, вызываемых Clostridium perfringens. Кроме того, настоящее изобретение относится к способу лечения бактериальных инфекций, в частности, вызываемых бактериями рода Clostridium, более конкретно Clostridium perfringens, у теплокровного субъекта, включающему введение теплокровному субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. Инфекции, вызванные бактериями Clostridium perfringens, представляют собой, например, пищевое отравление, обусловленное С. perfringens типа А, энтеротоксемию, некротический энтерит и газовую гангрену. Термин "лечение", используемый в настоящем изобретении, относится к лечебному, паллиативному и профилактическому лечению, включая обращение вспять, облегчение, подавление прогрессирования или предотвращение заболевания, нарушения или состояния, к которым применяются эти термины, или одного или более симптомов такого заболевания, нарушения или состояния. Теплокровные животные в данном изобретении включают как людей, так и животных, таких как сельскохозяйственные животные (например, овца, крупный рогатый скот, свиньи, козы или лошади),домашние животные (например, собаки, кошки или морские свинки), а также дикие животные, содержащиеся в неволе, и птицы (например, домашние птицы). Кроме того, настоящее изобретение относится к фармацевтически приемлемым композициям,включающим по меньшей мере один фармацевтически приемлемый носитель и терапевтически эффективное количество соединения формулы (I). Термин "терапевтически эффективное количество соединения формулы (I)", используемый в настоящем изобретении, означает количество соединения формулы (I), которое вызывает биологическую или медицинскую ответные реакции у теплокровных животных, которых добивается врач или ветеринар и которые включают облегчение симптомов состояния, подвергающегося лечению. Терапевтически эффективное количество можно определять при помощи обычных способов оптимизации, и оно зависит от конкретного состояния, подвергающегося лечению, состояния теплокровного животного, способа введения, состава, от назначений лечащего врача, а также от прочих факторов, очевидных для специалистов в данной области техники. Терапевтически эффективное количество может быть достигнуто путем многократного дозирования. Кроме того, согласно настоящему изобретению предложены фармацевтические композиции, содержащие по меньшей мере один фармацевтически приемлемый носитель и терапевтически эффективное количество соединения формулы (I). При применении у теплокровных животных, включая людей, соединения формулы (I) можно вводить отдельно, но, как правило, их вводят в смеси с фармацевтически или ветеринарно приемлемым разбавителем или носителем, выбранным с учетом способа введения и стандартной фармацевтической практики. Например, их можно вводить перорально, в том числе сублингвально, в виде таблеток, содержащих такие вспомогательные вещества, как крахмал или лактоза, или в виде капсул или суппозиториев,как отдельно, так и в смеси со вспомогательными веществами, или в форме эликсиров, растворов или суспензий, содержащих ароматизаторы или красители. Соединения формулы (I) можно включать в капсулы, таблетки или пилюли для доставки в толстую или двенадцатиперстную кишку посредством отсроченного растворения указанных капсул, таблеток или пилюль в течение определенного времени после перорального введения. Соединения формулы (I) можно вводить парентерально, например внутривенно,внутримышечно или подкожно. Для парентерального введения соединения формулы (I) лучше всего использовать в форме стерильного водного раствора или суспензии, которые могут содержать другие вещества, например, достаточное для приготовления изотонического с кровью раствора количества соли или глюкозы. Соединения формулы (I) можно вводить местно в виде стерильных кремов, гелей, составов для наливания или точечного нанесения, суспензий, лосьонов, мазей, присыпок, спреев, повязок или кожных пластырей, содержащих лекарственное средство. Например, соединения формулы (I) можно включать в крем, состоящий из водной или масляной эмульсии полиэтиленгликолей или жидкого парафина; или их также можно включать в мазь, состоящую из мягкой парафиновой основы белого воска; также они могут представлять собой гидрогель с целлюлозой, или производными полиакрилата, или другими модификаторами вязкости; также они могут представлять собой сухой порошок, жидкий спрей или аэрозоль с бутан/пропановой смесью, гидрофторалкановыми (ГФА) или хлорфторуглеродными (ХФУ) пропеллентами; также они могут представлять собой повязки, содержащие лекарственное средство, например, в виде сетчатых перевязок с белым мягким парафином, марлевых повязок, пропитанных полиэтиленгликолями, повязок с гидрогелем, гидроколлоидом, альгинатом или пленочных повязок. Соединения формулы (I) также можно вводить внутриглазным способом в виде глазных капель с соответствующими буферами, модификаторами вязкости (например, производными целлюлозы), консервантами (например, бензалкония хлорид (БАХ и агентами для регулировки предела прочности (например, хлорид натрия). Такие составы хорошо известны в данной области. Все такие составы также могут содержать соответствующие стабилизаторы и консерванты. Для ветеринарного применения соединения можно вводить в виде подходящего приемлемого состава в соответствии с нормальной ветеринарной практикой, а ветеринар будет определять режим дозирования и способ введения, которые будут наиболее подходящими для конкретного животного. Для местного применения можно применять дезинфицирующий раствор, спрей, порошок, присыпку, составы для наливания, составы для точечного нанесения, эмульгируемый концентрат, инъекционную жидкость, шампуни, ошейники, метки или жгуты. Такие составы готовят обычным способом в соответствии со стандартной ветеринарной и фармацевтической практикой. Таким образом, капсулы, пилюли или таблетки можно получать путем смешивания активного ингредиента с подходящим тонко измель-5 021567 ченным разбавителем или носителем, дополнительно содержащим разрыхлитель и/или связывающее вещество, такое как крахмал, лактоза, тальк или стеарат магния. Пропитывающий состав можно получать путем диспергирования активных ингредиентов в водном растворе вместе с диспергирующими или увлажняющими агентами, а инъекционные составы можно получать в виде стерильного раствора или эмульсии. Составы для наливания или точечного нанесения можно получать путем растворения активных ингредиентов в приемлемом жидком носителе, таком как бутилдиголь, жидкий парафин или нелетучий эфир с добавлением или без добавления летучего компонента, такого как изопропанол. Альтернативно, составы для наливания, составы для точечного нанесения или составы в форме спреев можно получать путем инкапсулирования, с тем чтобы обеспечить присутствие остаточного количества активного агента на поверхности животного. Эти составы будут меняться по отношению к весу активного соединения в зависимости от вида животного, подвергающегося лечению, тяжести и типа инфекции и типа и массы тела животного. Составы, включающие соединение формулы (I), можно вводить непрерывно, в частности, для профилактики, известными способами. В качестве альтернативы, комбинации можно вводить с кормом для животных, и с этой целью можно готовить концентрированные кормовые добавки или премиксы для последующего смешивания с обычным кормом для животных. При применении у людей соединения формулы (I) вводят в виде фармацевтически приемлемого состава в соответствии с обычной медицинской практикой. Специалисты в области лечения бактериальных инфекций, в частности инфекций, вызываемыхClostridium, без труда определят терапевтически эффективное количество соединения формулы (I), исходя из результатов исследований, представленных ниже. В целом предполагается, что терапевтически эффективная доза будет составлять от примерно 0,1 до примерно 20 мг/кг массы тела, более предпочтительно от примерно 1 до примерно 10 мг/кг массы тела теплокровного животного, подвергающегося лечению. Может быть целесообразным вводить терапевтически эффективную дозу в форме двух или более частичных доз через определенные интервалы в течение дня. Точная дозировка и частота введения зависят от конкретного используемого соединения формулы(I), конкретного состояния, подвергающегося лечению, тяжести состояния, подвергающегося лечению,возраста, веса и общего физического состояния конкретного теплокровного животного, а также можно брать другие медикаменты и других теплокровных животных, как хорошо известно специалистам в данной области техники. Кроме того, указанное эффективное суточное количество можно снижать или повышать в зависимости от ответа животного на лечение и/или в зависимости от заключения врача или ветеринара, назначающего соединения согласно настоящему изобретению. Диапазоны эффективных суточных количеств, таким образом, являются только рекомендациями. Экспериментальная часть."ДМФ" означает N,N-диметилформамид, "CH2Cl2" означает дихлорметан, "МеОН" означает метанол, "EtOH" означает этанол, "ТЭА" означает триэтиламин, "DPPA" означает дифениловый эфир фосфорилазидной кислоты, "DBU" означает 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-а]азепин, "NaBH(OAc)3" означает триацетоксиборгидрид натрия, "MgSO4" означает сульфат магния, "POCl3" означает фосфорилхлорид, "Na2SO3" означает сульфит натрия, "CH3NH2" означает метанамин, "NaHCO3" означает бикарбонат натрия, "CHCl3" означает трихлорметан, "Na2SO4" означает сульфат натрия, "NH4OH" означает гидроксид аммония, "H2SO4" означает серную кислоту, "NCS" означает 1-хлор-2,5-пирролидиндион,"NaOH" означает гидроксид натрия и "ТГФ" означает тетрагидрофуран. Для ряда соединений температуры плавления (т.пл.) определяли при помощи аппарата для определения температуры плавления WRS-2A, который был приобретен у Shanghai Precision and Scientific Instrument Co. Ltd. Температуры плавления измеряли с линейным нагревом со скоростью 0,2-5,0 С/мин. Значения представлены в виде диапазонов температур плавления. Максимальная температура составляла 300 С. 1H ЯМР. Для ряда соединений снимали 1 Н ЯМР спектры при помощи спектрометров Bruker DPX-300 илиBruker DPX-400 с последовательностями стандартного импульса, работающими на частоте 300 и 400 МГц соответственно, с использованием CHLOROFORM-d (дейтерированный хлороформ, CDCl3) или ДМСО-d6 (дейтерированный ДМСО, диметил-d6 сульфоксид) в качестве растворителей. Химические сдвигипредставляли в частях на миллион (ppm) относительно тетраметилсилана (ТМС), который использовали в качестве внутреннего стандарта. А. Получение промежуточных соединений. Пример А.1. Муравьиную кислоту (81 г) по каплям добавляли при 0 С к ТЭА (1040 ммоль). После перемешива-6 021567 ния в течение 10 мин к реакционной смеси добавляли 6,8-дибром-2,3-дигидро-4 Н-1-бензопиран-4-он(261 ммоль), а также промежуточное соединение (14) (0,5 ммоль) и ДМФ (300 мл) при 25 С. После добавления реакционную смесь перемешивали при 40 С в течение 24 ч. Завершение реакции определяли при помощи тонкослойной хроматографии (петролейный эфир/этилацетат=5/1). Реакционную смесь гасили путем добавления воды (1000 мл) при 0 С. Полученную реакционную смесь экстрагировали этилацетатом (три раза по 1000 мл). Органическую фазу промывали солевым раствором (500 мл), сушили надNa2SO4 и концентрировали с получением неочищенного продукта. Неочищенный продукт промывали трет-бутилметиловым эфиром с получением 78 г промежуточного соединения (1). К раствору промежуточного соединения (1) (253 ммоль) в ТГФ (2000 мл) при перемешивании добавляли DPPA (334 ммоль) при 25 С. После перемешивания в течение 15 мин при 25 С добавляли DBU при 0 С. После добавления реакционную смесь перемешивали при комнатной температуре в течение 12 ч. Полное расходование исходного вещества определяли при помощи тонкослойной хроматографии(петролейный эфир/этилацетат=10:1). Реакционную смесь обрабатывали водой (1000 мл) и экстрагировали с этилацетатом (три раза по 1000 мл). Органическую фазу промывали солевым раствором (1000 мл),сушили над Na2SO4 и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали при помощи хроматографии на силикагеле (петролейный эфир/этилацетат=50/1) с получением 60,3 г промежуточного соединения (2). К смеси промежуточного соединения (2) (181 ммоль) в Н 2 О (80 мл) и ТГФ (800 мл) при 25 С добавляли трифенилфосфин (362 ммоль). После добавления реакционную смесь перемешивали при 25 С в течение 1 ч. Завершение реакции определяли при помощи тонкослойной хроматографии (петролейный эфир/этилацетат=5/1). Реакционную смесь концентрировали и остаток распределяли между этилацетатом(1000 мл) и Н 2 О (1000 мл). После разделения водную фазу экстрагировали этилацетатом (500 мл). Органическую фазу промывали солевым раствором (1000 мл), сушили над Na2SO4 и концентрировали с получением 150 г промежуточного соединения (3). К раствору промежуточного соединения (3) (181 ммоль) в EtOH (1500 мл) при 0 С добавлялиNH4OH (180 мл). Полученную реакционную смесь кипятили с обратным холодильником в течение 3 ч. Завершение реакции определяли при помощи тонкослойной хроматографии (CH2Cl2/MeOH=10/1). Реакционную смесь выпаривали для удаления EtOH и остаток подкисляли добавлением 6 н. HCl до рН 2. Полученную смесь отфильтровывали и полученное твердое вещество промывали этилацетатом (500 мл) с получением 40 г промежуточного соединения (4). Пример А.2. Смесь этилового эфира N-[(1 Н-пиразол-3-иламино)тиокарбонил]карбаминовой кислоты (514 ммоль) в 2 н. водном растворе NaOH (565 ммоль) перемешивали при 15 С в течение 3 ч. Затем смесь подкисляли 2 н. H2SO4. Осадок отфильтровывали, промывали водой (1000 мл) и трет-бутилметиловым эфиром (500 мл). Полученное твердое вещество сушили в вакууме с получением 78 г промежуточного соединения (5) в виде белого твердого вещества. К суспензии промежуточного соединения (5) (464 ммоль, 1 экв.) в EtOH (1600 мл) при перемешивании по каплям добавляли водный 2 н. раствор NaOH (480 мл, 2 экв.), а затем йодметан (511 ммоль, 1,1 экв.) при 0 С. После добавления смесь перемешивали при 15 С в течение 2 ч. Завершение реакции определяли при помощи тонкослойной хроматографии (CH2Cl2/MeOH=10/1). Осадок отфильтровывали, а затем суспендировали в воде (800 мл) и подкисляли 2 н. H2SO4. Суспензию перемешивали при 0 С в течение 5 мин. Осадок отфильтровывали и промывали холодной водой (900 мл). Полученное твердое вещество сушили в вакууме с получением 75 г промежуточного соединения (6) в виде белого твердого вещества. Смесь промежуточного соединения (6) (374 ммоль) и N,N-диметил-4-пиридинамина (1,31 ммоль) вPOCl3 (1500 мл) кипятили с обратным холодильником в течение 2 ч при 100 С. После охлаждения до комнатной температуры избыток POCl3 удаляли под вакуумом, а полученный остаток сушили в течение 2 ч. Неочищенный продукт использовали непосредственно на следующей стадии без дальнейшей очистки с получением 240 г промежуточного соединения (7). Промежуточное соединение (7) растворяли в безводном CH2Cl2 (2000 мл), а затем при 0 С по каплям добавляли N-метиланилин (285 мл) и ТЭА (355 мл). После перемешивания в течение 10 мин смесь нагревали до комнатной температуры и перемешивали в течение ночи. Завершение реакции определяли при помощи тонкослойной хроматографии (CH2Cl2/МеОН=10/1). Реакционную смесь промывали водой(600 мл) и солевым раствором (300 мл). Органический слой сушили над MgSO4, отфильтровывали и концентрировали с получением неочищенного продукта, который в дальнейшем промывали EtOH с получением 82 г промежуточного соединения (8). К раствору промежуточного соединения (8) (303 ммоль) в CH2Cl2 (2000 мл) при перемешивании порциями добавляли 3-хлорпероксибензойную кислоту (1059 ммоль) при 0 С. После добавления реакционную смесь перемешивали при 0 С в течение 1 ч, а затем при 15 С в течение 2 ч. Завершение реакции определяли при помощи тонкослойной хроматографии (петролейный эфир/этилацетат=5/1). Реакционную смесь промывали насыщенным водным Na2SO3 (четыре раза по 600 мл), а затем добавляли насыщенный водный NaHCO3 до рН 7. Водный слой экстрагировали с CH2Cl2 (500 мл). Объединенные органические фазы промывали солевым раствором (800 мл), сушили над Na2SO4 и концентрировали с получением неочищенного продукта. Неочищенный продукт промывали трет-бутилметиловым эфиром (три раза по 500 мл) с получением 87 г промежуточного соединения (9). К раствору промежуточного соединения (9) (287 ммоль) в CHCl3 (2000 мл) при перемешивании порциями добавляли 3,3-диэтокси-1-пропанамин (474 ммоль) при 0 С. После добавления реакционную смесь перемешивали при 0 С в течение 1 ч, а затем при 15 С в течение 2 ч. Завершение реакции опреде-8 021567 ляли при помощи тонкослойной хроматографии (петролейный эфир/этилацетат=5/1). Реакционную смесь промывали насыщенным водным Na2SO3 (четыре раза по 600 мл), а затем добавляли насыщенный водный NaHCO3 до рН 7. Водный слой экстрагировали с CH2Cl2 (500 мл). Объединенные органические фазы промывали солевым раствором (800 мл), сушили над Na2SO4 и концентрировали с получением неочищенного продукта. Неочищенный продукт промывали трет-бутилметиловым эфиром (три раза по 500 мл) с получением 87 г промежуточного соединения (10) (т.пл.: 100,8-103,8 С). К раствору промежуточного соединения (10) (81 ммоль) в ТГФ (450 мл) при температуре ниже 20 С добавляли 12 н. HCl (7,5 мл). После перемешивания в течение 5 мин реакционную смесь перемешивали при 20 С в течение 1 ч. Завершение реакции определяли при помощи тонкослойной хроматографии(CH2Cl2/МеОН=20/1). Добавляли этилацетат (500 мл). Реакционную смесь перемешивали в течение 30 мин. Осадок отфильтровывали, промывали этилацетатом и сушили в вакууме с получением 32 г промежуточного соединения (11). Пример А.3. Смесь N-[(1S,2S)-2-амино-1,2-дифенилэтил]-4-метилбензолсульфонамида (2,13 ммоль), димера дихлор(п-цимол)рутения(II) (2,13 ммоль) и ТЭА (0,6 мл) в 2-пропаноле (21 мл) перемешивали при 80 С в течение 1 ч. После охлаждения до 20 С органический раствор концентрировали под вакуумом. Полученное твердое вещество промывали водой (10 мл) и сушили при пониженном давлении с получением неочищенного продукта, который в дальнейшем перекристаллизовывали из метанола с получением 0,37 г промежуточного соединения (14) в виде ярко-оранжевого твердого вещества. В. Получение продуктов реакции. Пример В.1. К раствору промежуточного соединения (11) (81 ммоль) и промежуточного соединения (4) (81 ммоль) добавляли ТЭА (210,6 ммоль). После перемешивания в течение 1 ч добавляли NaBH(OAc)3 (113 ммоль). Реакционную смесь перемешивали при 20 С в течение 2 ч. Завершение реакции определяли при помощи тонкослойной хроматографии (CH2Cl2/МеОН=10/1). Смесь разбавляли этилацетатом (дважды по 500 мл). Органический слой сушили над Na2SO4, отфильтровывали и концентрировали с получением 55 г неочищенного продукта промежуточного соединения (12) в виде бесцветной маслянистой жидкости, которое использовали на следующей стадии без дальнейшей очистки. К раствору промежуточного соединения (12) (6 ммоль) в EtOH (100 мл) при перемешивании по каплям добавляли 5 н. NaOH (12 мл) при 0 С. После добавления реакционную смесь кипятили с обратным холодильником в течение 3 ч при 85 С. Завершение реакции определяли при помощи тонкослойной хроматографии (CH2Cl2/МеОН=10/1). EtOH удаляли при пониженном давлении, а остаток нейтрализовывали насыщенной водной лимонной кислотой до рН 7. Смесь добавляли к раствору воды (50 мл) и этилацетата(100 мл). Осадок отфильтровывали, промывали ацетонитрилом (три раза по 50 мл) и сушили в вакууме с получением 1,8 г соединения (1). Соединение (5) получали аналогичным способом путем проведения реакции промежуточного соединения (11) с (R)-6,8-дихлорхроман-4-иламином. Альтернативный способ получения соединения (1) приведен ниже. Стадия 1. К раствору соединения (1) (3,6 ммоль) в МеОН (10 мл) при 20 С добавляли 4 М HCl в диоксане (30 мл). Смесь перемешивали в течение 3 ч при 20 С. Растворитель удаляли при пониженном давлении. Остаток сушили в вакууме с получение 1,62 г соединения (2) (т.пл.: 234,6-235,6 С). Оптическое вращение: Раствор промежуточного соединения (12) (10,2 ммоль) в диметилформамиде (300 мл) перемешивали при 80 С в течение 5 мин. NCS (20,4 ммоль) добавляли к смеси в атмосфере азота и реакционную смесь перемешивали в течение 3 ч. Затем ДМФ удаляли при пониженном давлении. Остаток промывали трет-бутилметиловым эфиром (три раза по 50 мл) и отфильтровывали. Твердое вещество сушили при пониженном давлении с получением 2,8 г промежуточного соединения (13).EtOH (5 мл) при 20 С. Смесь перемешивали при 150 С при микроволновом облучении в течение 4 ч.EtOH удаляли при пониженном давлении. Остаток очищали путем препаративной высокоэффективной жидкостной хроматографии (колонка: YMC, 25020 мм, подвижная фаза: 20-50% CH3CN (0,075% об./об.CF3COOH), скорость потока: 25 мл/мин, время завершения: 15 мин). Желаемую фракцию собирали и выпаривали. Водный раствор нейтрализовывали до рН 7 и концентрировали. Твердое вещество отфильтровывали и промывали водой (три раза по 30 мл) с получением 0,065 г соединения (3) (т.пл.: 108,8118,6 С). Пример В.4. Смесь промежуточного соединения (13) (0,97 ммоль) и 2 М NH3 в МеОН (10 мл) перемешивали при 125 С при микроволновом облучении в течение 3 ч. Смесь концентрировали при пониженном давлении. Остаток очищали путем препаративной высокоэффективной жидкостной хроматографии (колонка: YMC,25020 мм, подвижная фаза: 30-60% CH3CN (0,075% об./об. CF3COOH), скорость потока: 25 мл/мин,время завершения: 15 мин). Желаемую фракцию собирали и выпаривали. Водный раствор нейтрализовывали до рН 7 и концентрировали. Твердое вещество отфильтровывали. А затем промывали водой (три раза по 30 мл). Продукт сушили под высоким вакуумом с получением 0,09 г соединения (4) (т.пл.: 78,794,1 С). В табл. 1 приведены соединения, которые получали в соответствии с одним из приведенных выше примеров. С. Аналитическая часть. С.1. ЖХ-МС общая методика А. Общая методика А. ВЭЖХ осуществляли с использованием модуля Agilent 1100, содержащего насос, диодноматричный детектор (ДМД) (использовали длину волны 220 нм), нагреватель колонки и колонку, указанные ниже в соответствующих способах. Поток из колонки делили на Agilent MSD серий G1946C иG1956A. МС детектор настраивали с API ES (электроспрей-ионизация при атмосферном давлении). Масс-спектры получали путем сканирования от 100 до 1000. Напряжение капилляра составляло 2500 В для режима положительной ионизации и 3000 В для режима отрицательной ионизации. Напряжение фрагментации составляло 50 В. Температуру сушильного газа поддерживали на уровне 350 С при потоке 10 л/мин. Способ 1. В дополнение к общему способу А: обращенно-фазовую ВЭЖХ проводили на YMC-Pack ODS-AQ,502,0 мм 5-мкм колонке со скоростью потока 0,8 мл/мин. Использовали две подвижные фазы (подвижная фаза А: вода с 0,1% ТФУ; мобильная фаза Б: ацетонитрил с 0,05% ТФУ). Сначала элюировали 100% А в течение 1 мин. Затем применяли градиент до 40% А и 60% Б в течение 4 мин и элюировали в течение 2,5 мин. Использовали обычные объемы инъекций, равные 2 мкл. Температура печи составляла 50 С(МС полярность: положительная). Способ 2. В дополнение к общей методике А: обращенно-фазовую ВЭЖХ проводили на YMC-Pack ODS-AQ,502,0 мм 5-мкм колонке со скоростью потока 0,8 мл/мин. Использовали две подвижные фазы (подвижная фаза А: вода с 0,1% ТФУ; мобильная фаза Б: ацетонитрил с 0,05% ТФУ). Сначала элюировали 90% А и 10% Б в течение 0,8 мин. Затем применяли градиент до 20% А и 80% Б в течение 3,7 мин и элюировали в течение 3 мин. Использовали обычные объемы инъекций, равные 2 мкл. Температура печи составляла 50 С (МС полярность: положительная). Таблица 2 Аналитические данные - время удерживания (Rt в минутах), (МН)+ пик (свободного основания),ЖХ-МС способ и температура плавления (т.пл. определяется как температура плавления)D.1.1. In vitro способ исследования соединений против С. Perfringens. Плоскодонные стерильные 96-луночные пластиковые титрационные микропланшеты заполняли 100 мкл бульонной среды с сердечно-мозговым экстрактом. Затем добавляли базовый раствор (100 конечной концентрации исследования) соединений объемами 2 мкл в ряд лунок для оценки их влияния на рост бактерий. Необработанные контрольные образцы с инокулятом или без него включали в каждый титрационный микропланшет. В лунки добавляли приблизительно 50000 КОЕ на лунку Clostridium perfringens(штамм 56) в объеме 100 мкл бульонной среды с сердечно-мозговым экстрактом. Культуры инкубировали при 37 С в течение 24 ч в анаэростате. Оптическую плотность (ОП) считывали при помощи управляемого компьютером спектрофотометра (Envision) при длине волны 540 нм. Процент ингибирования роста, достигнутый при помощи соединений, рассчитывали в соответствии со стандартными способами и выражали в виде IC90 (мкг/мл), что соответствует концентрации, ингибирующей 90% роста бактерий.D.1.2. In vitro способ исследования соединений, обладающих антибактериальной активностью в отношении штамма С. difficile. Плоскодонные стерильные 96-луночные пластиковые титрационные микропланшеты заполняли 100 мкл бульонной среды Reinforced Clostridial. Затем добавляли базовый раствор (100 конечной концентрации исследования) соединений объемами 2 мкл в ряд лунок для оценки их влияния на рост бактерий. Необработанные контрольные образцы с инокулятом или без него включали в каждый титрационный микропланшет. В лунки добавляли приблизительно 50000 КОЕ на лунку Clostridium difficile (ATCC9689) в объеме 100 мкл бульонной среды Reinforced Clostridial. Культуры инкубировали при 37 С в течение 24 ч в анаэростате. ОП считывали при помощи управляемого компьютером спектрофотометра (Thermomax) при длине волны 570 нм. Процент ингибирования роста, достигнутый при помощи соединений, рассчитывали в соответствии со стандартными способами и выражали в виде IC90 (мкг/мл), что соответствует концентрации, ингибирующей 90% роста бактерий. Результаты для D.1.1 и D.1.2:REP3123 демонстрировал хорошую активность в отношении как С. Perfringens, так и С. difficile приIC90 0,25 и 0,5-1 мкг/мл соответственно. Соединение (2) согласно настоящему изобретению демонстрировало хорошую активность в отношении как С. Perfringens, так и С. difficile при IC90 0,5 и 2 мкг/мл соответственно.D.1.3. In vitro способ исследования соединений против ряда бактерий. Плоскодонные стерильные 96-луночные пластиковые титрационные микропланшеты заполняли 100 мкл бульонной среды. Затем добавляли базовый раствор (100 конечной концентрации исследования) соединений объемами 2 мкл в ряд лунок для оценки их влияния на рост бактерий. Необработанные контрольные образцы с инокулятом или без него включали в каждый титрационный микропланшет. В лунки добавляли приблизительно 50000 КОЕ на лунку бактерий в объеме 100 мкл бульонной среды. Культуры инкубировали при 37 С. Минимальную концентрацию ингибирования определяли, как наибольшую концентрацию без видимого роста, и выражали в виде IC90 (мкг/мл), что соответствует концентрации,ингибирующей 90% роста бактерий. Среда и условия инкубации. Результаты для D.1.3. Как REP3123, так и соединение (2) согласно настоящему изобретению не продемонстрировали какой-либо активности в отношении грамотрицательных бактерий. IC90 для REP3123 составляла 32, 64,16, 78 и 64 мкг/мл в отношении Е.coli, K. pneumoniae, M. cattarhalis, P. aeruginosa and A. baumanii соответственно. IC90 для соединения (2) настоящего изобретения составляла 31, 64, 16, 75 и 64 мкг/мл в отношении Е.coli, K. pneumoniae, M. cattarhalis, P. aeruginosa и A. baumanii соответственно.REP3123 демонстрировал хорошую активность в отношении всех тестируемых грамположительных бактерий. IC90 для REP3123 составляла 0,25, 0,25, 0,25, 0,25, 1, 0,12 и 0,12 мкг/мл в отношении S.monocytogenes соответственно. Соединение (2) настоящего изобретения не продемонстрировало какой-либо активности в отношении всех исследуемых грамположительных бактерий, за исключением Е. faecalis и L. monocytogenes. Однако активность соединения (2) согласно настоящему изобретению в отношении этих 2 микроорганизмов оставалась намного ниже, чем активность REP3123. IC90 для соединения (2) согласно настоящему изобретению составляла 8-15, 7-13, 15, 1,5, 31, 4 и 2 мкг/мл в отношении S. aureus, резистентного к метициллину S. aureus, S. epidermidis, E. faecalis, S. pneumonia, B. subtilis и L. monocytogenes соответственно. Как REP3123, так и соединение (2) согласно настоящему изобретению не продемонстрировали какой-либо активности в отношении Mycobacterium smegmatis. IC90 REP3123 и соединения (2) настоящего изобретения в отношении М. smegmatis составляла 6-8 и 31 мкг/мл соответственно. Таблица 3 Антибактериальная активность REP3123 и соединения (2) в отношении грамположительных бактерий Сравнение между REP3123 и соединением (2) согласно настоящему изобретению наглядно демонстрирует широкий спектр активности REP3123 в отношении грамположительных бактерий и узкий спектр активности соединения (2). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) включая любые его стереохимические изомерные формы и таутомеры, гдеR4 представляет собой водород или C1-4 алкил;X представляет собой азот или CR5, где R5 представляет собой водород, галоген или C1-4 алкил; при условии, что, когда R3 представляет собой гидрокси, X представляет собой СН и R4 представляет собой водород; или его фармацевтически приемлемая соль присоединения кислоты. 2. Соединение по п.1 или его фармацевтически приемлемая соль присоединения кислоты, имеющие(R)-конфигурацию в положении 4 хроманильного фрагмента. 3. Соединение по п.1 или 2 или его фармацевтически приемлемая соль присоединения кислоты, отличающиеся тем, что R1 и R2, каждый, представляют собой галоген. 4. Соединение по п.1 или его фармацевтически приемлемая соль присоединения кислоты, отли- 14021567 чающиеся тем, что R1 и R2, каждый, представляют собой бром и находятся в положении 6 и 8 хроманильного фрагмента. 5. Соединение по п.1 или 3 или его фармацевтически приемлемая соль присоединения кислоты, отличающиеся тем, что R1 и R2, каждый, представляют собой бром и находятся в положении 6 и 8 хроманильного фрагмента, a R3 представляет собой гидрокси. 6. Соединение по п.1, отличающееся тем, что указанное соединение представляет собой 7. Фармацевтическая композиция, имеющая антибактериальную активность, содержащая фармацевтически приемлемый носитель и терапевтически активное количество соединения по любому из пп.16 или его фармацевтически приемлемой соли присоединения кислоты. 8. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли присоединения кислоты в качестве лекарственного средства. 9. Применение соединения по любому из пп.1-6 или его фармацевтически приемлемой соли присоединения кислоты для лечения бактериальных инфекций. 10. Применение по п.9, отличающееся тем, что бактериальная инфекция представляет собой инфекцию, вызываемую бактериями Clostridium. 11. Способ лечения бактериальной инфекции у теплокровных субъектов, включающий введение теплокровному субъекту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по любому из пп.1-6 или его фармацевтически приемлемой соли присоединения кислоты. 12. Способ по п.11, отличающийся тем, что бактериальная инфекция представляет собой инфекцию,вызываемую бактериями Clostridium. 13. Способ получения соединения формулы (I), согласно которому: а) промежуточное соединение формулы (II) N-алкилируют промежуточным соединением формулы или b) соединения формулы (I) преобразовывают друг в друга согласно известным реакциям преобразования; или, при необходимости, соединение формулы (I) преобразовывают в фармацевтически приемлемую соль присоединения кислоты или, наоборот, соль присоединения кислоты соединения формулы(I) переводят в форму свободного основания при помощи щелочи; и, при необходимости, получают их стереохимически изомерные формы.
МПК / Метки
МПК: A61K 31/53, A61P 31/04, C07D 487/04
Метки: отношении, соединения, clostridium, активностью, обладающие, антибактериальной
Код ссылки
<a href="https://eas.patents.su/17-21567-soedineniya-obladayushhie-antibakterialnojj-aktivnostyu-v-otnoshenii-clostridium.html" rel="bookmark" title="База патентов Евразийского Союза">Соединения, обладающие антибактериальной активностью в отношении clostridium</a>
Трициклические соединения, обладающие активностью в отношении интегринов, в частности, в отношении интегрина альфаvбета 3, способ их получения и промежуточные соединения, используемые в этом способе,их применение в качестве медикаментов и содержащие их фармацевтические композиции.

Номер патента: 2271
Опубликовано: 28.02.2002
Авторы: Карниато Дени, Штильц Ханс-Ульрих, Гурвест Жан-Франсуа, Бодари Сара С., Тетш Жан-Жорж, Венер Фолькмар, Бернар Серж, Кнолле Йохен, Гадек Томас Р., Макдауэлл Роберт С., Питти Роберт М.
МПК: A61P 9/10, A61K 31/19, C07C 281/12...
Метки: применение, композиции, этом, используемые, фармацевтические, активностью, качестве, способ, интегрина, частности, интегринов, отношении, получения, соединения, промежуточные, способе,их, содержащие, обладающие, альфаvбета, медикаментов, трициклические
Формула / Реферат:
1. Соединения общей формулы (I) в которой R1 обозначает группу -О-[А]-[В]-COR6, в которой R6 обозначает -ОН, C1-С6алкокси, -О-СН2-СН(ОН)-СН2OН, [A] обозначает группу C1-С6алкилен, возможно замещенный оксогруппой, [B] обозначает радикал -CH(Z)- или простую связь, Z обозначает группу -NHCO2Rc, или -NHSO2Rc, где Rc обозначает радикал фенил(C1-С4)алкил-, хинолинил или пиридинилимидазолил(C1-С4)алкил-, R2 и R3, одинаковые или разные, обозначают атом...
Бензимидазолоновые соединения, обладающие агонистической активностью в отношении 5-нт4 рецепторов

Номер патента: 9457
Опубликовано: 28.12.2007
Авторы: Коджима Такаси, Игучи Сатору, Соне Хироки, Учида Чикара, Катсу Ясухиро
МПК: A61P 1/04, C07D 401/12, A61K 31/4184...
Метки: обладающие, агонистической, отношении, 5-нт4, активностью, соединения, бензимидазолоновые, рецепторов
Формула / Реферат:
1. Соединение формулы (I) где Het представляет собой собой группу формулы которая является незамещенной или замещена 1-4 заместителями, независимо выбранными из группы, состоящей из заместителей a1; А представляет собой метиленовую группу; В представляет собой ковалентную связь или алкиленовую группу, имеющую от 1 до 5 атомов углерода, и указанная алкиленовая группа является незамещенной или замещена оксогруппой, когда R3 представляет собой...
Новые соединения, обладающие ингибирующей активностью в отношении натрийзависимого транспортёра

Номер патента: 10299
Опубликовано: 29.08.2008
Авторы: Номура Сумихиро, Каваниси Ийдзи, Уита Киитиро
МПК: C07H 19/06
Метки: обладающие, отношении, натрийзависимого, новые, транспортёра, ингибирующей, активностью, соединения
Формула / Реферат:
1. Соединение формулы где кольцо А представляет собой где R1a, R2a, R3a, R1b, R2b и R3b, каждый независимо, представляет собой атом водорода, атом галогена, гидроксигруппу, алкоксигруппу, алкильную группу, галогеналкильную группу, галогеналкоксигруппу, гидроксиалкильную группу, алкоксиалкильную группу, алкоксиалкоксигруппу, алкенильную группу, алкинильную группу, циклоалкильную группу, циклоалкилиденметильную группу, циклоалкенильную группу,...
Новые соединения, обладающие ингибирующей активностью в отношении натрийзависимого транспортера

Номер патента: 15104
Опубликовано: 30.06.2011
Авторы: Номура Сумихиро, Уита Киитиро, Каваниси Ийдзи
МПК: C07H 19/06
Метки: новые, активностью, обладающие, соединения, отношении, натрийзависимого, транспортера, ингибирующей
Формула / Реферат:
1. Соединение формулыгде кольцо А представляет собой ненасыщенное моноциклическое гетероциклическое кольцо или ненасыщенное конденсированное гетеробициклическое кольцо, где группа сахара Х-(сахар) и группа -Y-(кольцо В), обе, присутствуют в одном и том же гетероциклическом кольце указанного конденсированного гетеробициклического кольца и где каждое ненасыщенное моноциклическое гетероциклическое кольцо и ненасыщенное конденсированное...
Производные хиноксалин-2,3-диона и их фармацевтически приемлемые соли, фармацевтические композиции, обладающие антагонистической активностью в отношении рецептора глютамата и противосудорожной активностью, способы лечения пациентов, страдающих от удара или заболеваний, при помощи этих соединений.

Номер патента: 762
Опубликовано: 24.04.2000
Авторы: Рафферти Майкл Фрэнсис, Никам Шам, Корнберг Брайэн Эдвард
МПК: C07D 241/44
Метки: производные, фармацевтически, рецептора, антагонистической, заболеваний, соединений, этих, удара, композиции, фармацевтические, помощи, обладающие, отношении, глютамата, приемлемые, соли, пациентов, способы, лечения, противосудорожной, хиноксалин-2,3-диона, активностью, страдающих
Формула / Реферат:
1. Производные хиноксалин-2,3-диона общей формулы I где R - группа - NR4R5, где R4 и R5 независимо друг от друга означают водород, низший алкил, незамещенный или замещенный низшим алкилом, низший циклоалкил, который может содержать 1 или 2 атома кислорода в качестве гетероатома, R1 и R2 независимо друг от друга означают водород или нитрогруппу, R3 - низший алкил, при этом радикалы R3 и R-CH2- могут каждый находиться в положениях 5 или...
Предыдущий патент: Стабилизированные пищевые продукты с покрытием, приготавливаемые с помощью микроволнового излучения
Следующий патент: Конденсированные гетероароматические пирролидиноны как ингибиторы syk
Случайный патент: Способ и устройство для отделения катодных пластин