Производные 4-(2-фенилоксифенил)пиперидина или -1,2,3,6- тетрагидропиридина в качестве ингибиторов обратного захвата серотонина














Формула / Реферат
1. Производное 4-(2-фенилоксифенил)пиперидина или 1,2,3,6-тетрагидропиридина общей формулы I

где пунктирная линия ---- означает простую связь или двойную связь;
R1, R2, R3, R4, R5 независимо выбраны из водорода, галогена, C1-6-алкила, C1-6-алкилокси, галоген-С1-6-алкила; или R2 и R3 вместе образуют гетероцикл, конденсированный с фенильным кольцом, выбранный из  a
a
R1, R4, R5 имеют определенные выше значения;
R6, R7, R8, R9 независимо представляют собой водород;
или его соль.
2. Соединение по п.1, где R1 выбран из водорода, галогена, C1-6-алкила, C1-6-алкилокси, галоген-С1-6-алкила.
3. Соединение по любому из пп.1 и 2, где R2 выбран из водорода, галогена, C1-6-алкила, C1-6-алкилокси, галоген-С1-6-алкила.
4. Соединение по любому из пп.1-3, где R3 выбран из водорода, галогена, C1-6-алкила, C1-6-алкилокси, галоген-С1-6-алкила.
5. Соединение по любому из пп.1 и 2, где R2 и R3 вместе образуют гетероцикл, конденсированный с фенильным кольцом, выбранный из 
6. Соединение по любому из пп.1-5, где R4 выбран из водорода, галогена, C1-6-алкила, C1-6-алкилокси, галоген-С1-6-алкила.
7. Соединение по любому из пп.1-6, где R5 выбран из водорода, галогена, C1-6-алкила, C1-6-алкилокси, галоген-С1-6-алкила.
8. Соединение по любому из пп.1-7, где R6 является водородом.
9. Соединение по любому из пп.1-8, где R7 является водородом.
10. Соединение по любому из пп.1-9, где R8 является водородом.
11. Соединение по любому из пп.1-10, где R9 является водородом.
12. Соединение по любому из пп.1-11, где пунктирная линия означает простую связь.
13. Соединение по любому из пп.1-11, где пунктирная линия означает двойную связь.
14. Соединение по п.1, причем указанное соединение представляет собой
4-[2-(2,4-диметилфенокси)фенил]-1,2,3,6-тетрагидропиридин,
4-[2-(4-хлорфенокси)фенил]-1,2,3,6-тетрагидропиридин,
4-[2-(4-фтор-2-метилфенокси)фенил]-1,2,3,6-тетрагидропиридин,
4-[2-(4-фторфенокси)фенил]-1,2,3,6-тетрагидропиридин,
4-[2-(4-метилфенокси)фенил]-1,2,3,6-тетрагидропиридин,
4-[2-(4-метоксифенокси)фенил]-1,2,3,6-тетрагидропиридин,
4-[2-(2,4-диметилфенокси)фенил]пиперидин,
4-[2-(4-хлорфенокси)фенил]пиперидин,
4-[2-(4-фтор-2-метилфенокси)фенил]пиперидин,
4-[2-(4-фторфенокси)фенил]пиперидин,
4-[2-(4-метилфенокси)фенил]пиперидин,
4-[2-(4-хлор-2-метилфенокси)фенил]пиперидин,
4-[2-(3-хлор-2-метилфенокси)фенил]пиперидин,
4-[2-(2-хлор-4-метилфенокси)фенил]пиперидин,
4-[2-(2,4-дихлорфенокси)фенил]пиперидин,
4-[2-(бензо[1,3]диоксол-5-илокси)фенил]пиперидин,
4-[2-(4-метокси-2-метилфенокси)фенил]пиперидин,
4-[2-(3,4-дихлорфенокси)фенил]пиперидин,
4-[2-(3,4-диметилфенокси)фенил]пиперидин,
4-[2-(2,3,4,5-тетраметилфенокси)фенил]пиперидин,
4-[2-(4-трифторметилфенокси)фенил]пиперидин,
4-[2-(4-метоксифенокси)фенил]пиперидин,
4-[2-(2-хлор-4-метоксифенокси)фенил]пиперидин,
4-[2-(3,4-диметоксифенокси)фенил]пиперидин,
4-[2-(4-хлор-3-трифторметилфенокси)фенил]пиперидин,
или его фармацевтически приемлемую соль.
15. Фармацевтическая композиция, содержащая соединение по любому из пп.1-14 или его фармацевтически приемлемую кислотно-аддитивную соль и по меньшей мере один фармацевтически приемлемый носитель или разбавитель.
16. Применение соединения по любому из пп.1-14 или его фармацевтически приемлемой кислотно-аддитивной соли для получения лекарственного средства для лечения аффективных расстройств, таких как депрессия, состояние тревожности, в том числе общее состояние тревожности, состояние социальной тревожности, посттравматический шок, навязчивое компульсивное расстройство, паническое расстройство, приступы паники, специфические фобии, социальная фобия и агорафобия.
17. Способ лечения аффективных расстройств, таких как депрессия, состояние тревожности, в том числе общее состояние тревожности, состояние социальной тревожности, посттравматический шок, навязчивое компульсивное расстройство, паническое расстройство, приступы паники, специфические фобии, социальная фобия и агорафобия, у животных, включая человека, который включает введение терапевтически эффективного количества соединения по любому из пп.1-14 или его фармацевтически приемлемой кислотно-аддитивной соли.
Текст
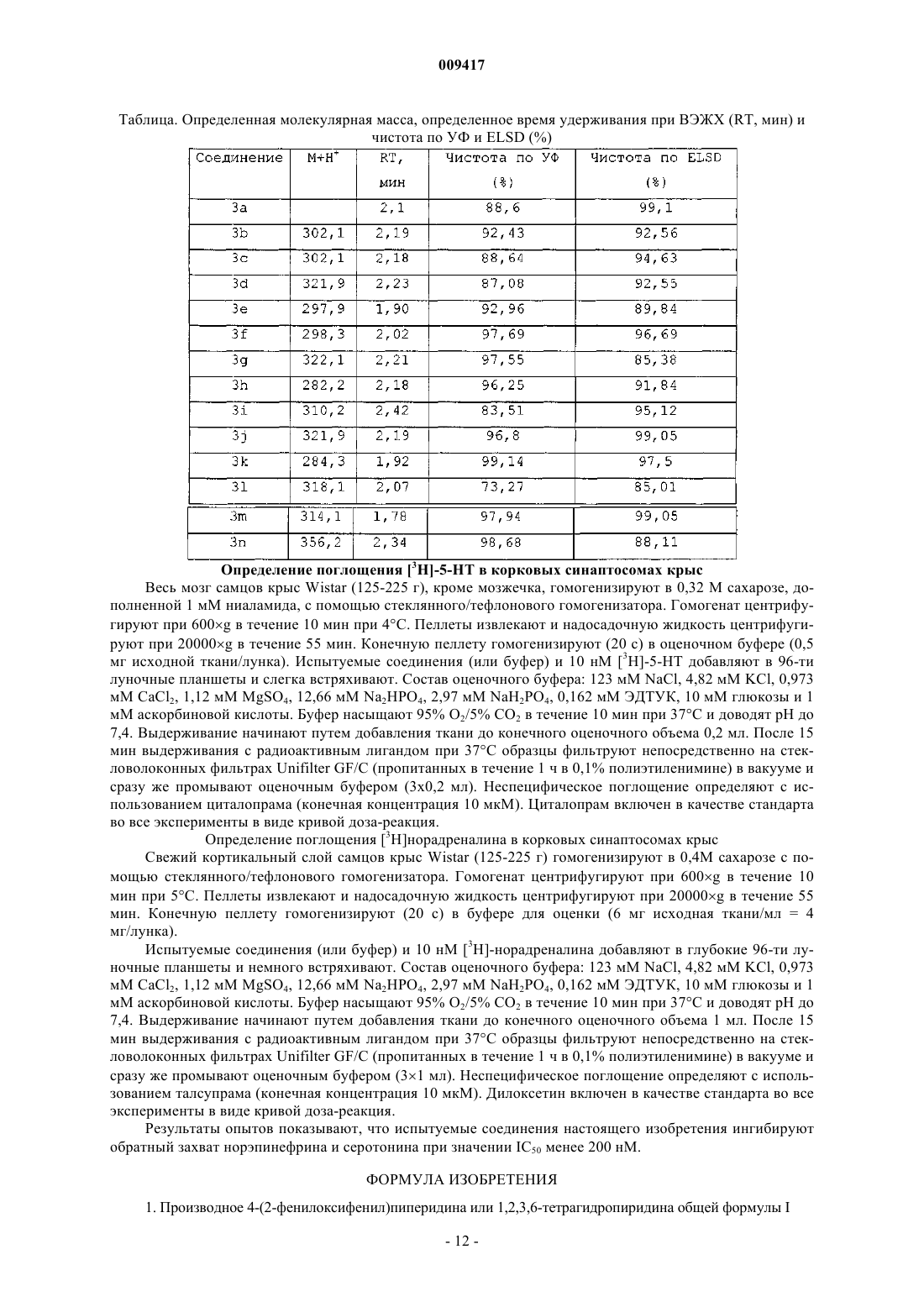
009417 Настоящее изобретение относится к новым соединениям, которые являются ингибиторами обратного захвата серотонина и фактически эффективны при лечении депрессии и состояния тревожности. Область техники Селективные ингибиторы обратного захвата серотонина (далее называемые SSRI) стали первым объектом выбора терапевтических средств при лечении депрессии, некоторых форм состояний тревожности и социальных фобий, так как они эффективны, хорошо переносимы и имеют благоприятный профиль безопасности по сравнению с классическими трициклическими антидепрессантами. Однако клинические исследования депрессии показывают, что имеет место значительное отсутствие реакции на SSRI до 30%. Другим, часто не принимаемым во внимание фактором при лечении антидепрессантами является соблюдение больным режима приема лекарственных средств, которое имеет сильное глубокое влияние на мотивацию пациента продолжать медикаментозное лечение. Во-первых, существует задержка в терапевтическом действии SSRI. Иногда симптомы даже ухудшаются в течение первых недель лечения. Во-вторых, обычным для SSRI побочным эффектом является нарушение сексуальной функции. Без рассмотрения этих проблем реальный прогресс в медикаментозном лечении депрессии и заболеваний, связанных с состоянием тревожности, по всей видимости, маловероятен. В клинических исследованиях оценено комбинированное влияние ингибирования обратного захвата серотонина и ингибирования поглощения норэпинефрина на депрессию таких соединений, как дулоксетин (Wong, Duloxetine (LY-248686): an inhibitor of serotonin and noradrenaline uptake and an antidepressant drug candidate. - Expert Opinion on Investigational Drugs, 1998, 7, 10, 1691-1699) и венлафаксин (KlanA et al., Venlafaxine in depressed outpatients. Psychopharmacology Bulletin, 1991, 27, 141-144). Настоящее изобретение предлагает новые соединения, которые обладают комбинированным эффектом ингибирования обратного захвата серотонина и ингибирования поглощения норэпинефрина при лечении аффективных заболеваний, таких как депрессия, состояния тревожности, в том числе общего состояния тревожности, состояние социальной тревожности, посттравматический шок, навязчивое компульсивное расстройство, паническое расстройство, приступы паники, специфические фобии, социальная фобия и аторафобия. Сущность изобретения Настоящее изобретение предлагает соединения общей формулы I где пунктирная линия, R1, R2, R3, R4, R5, R6, R7, R8 и R9 имеют значения, определенные ниже. Изобретение предлагает фармацевтическую композицию, содержащую соединение, описанное выше, или его фармацевтически приемлемую кислотно-аддитивную соль и по меньшей мере один фармацевтически приемлемый носитель или разбавитель. Изобретение предлагает применение соединения, описанного выше, или его фармацевтически приемлемой кислотно-аддитивной соли для получения лекарственного средства для лечения аффективных заболеваний, таких как депрессия, состояния тревожности, в том числе общего состояния тревожности,состояние социальной тревожности, посттравматический шок, навязчивое компульсивное расстройство,паническое расстройство, приступы паники, специфические фобии, социальная фобия и агорафобия. Изобретение также предлагает способ лечения аффективных заболеваний, таких как депрессия, состояния лекарственного средства, в том числе общего состояния тревожности, состояние социальной тревожности, посттравматический шок, навязчивое компульсивное расстройство, паническое расстройство, приступы паники, специфические фобии, социальная фобия и агорафобия у животных, в том числе у человека, который включает введение терапевтически эффективного количества соединения, описанного выше, или его фармацевтически приемлемой кислотно-аддитивной соли. Определение заместителей Галоген означает фтор, хлор, бром или йод. Определение C1-6-алкил относится к разветвленным или неразветвленным алкильным группам, содержащим от одного до шести атомов углерода, включая, но, не ограничиваясь только ими, метил, этил,1-пропил, 2-пропил, 1-бутил, 2-бутил, 2-метил-2-пропил и 2-метил-1-пропил. Определения C1-6-алкилокси- и галоген-С 1-6-алкил означают такие группы, в которых C1-6-алкил имеет определенные выше значения. Галоген означает галоген.-1 009417 Описание изобретения Настоящее изобретение относится к производным 4-(2-фенилоксифенил)пиперидина или -1,2,3,6 тетрагидропиридина, которые являются ингибиторами обратного захвата серотонина и, по существу,эффективны при лечении, например, депрессии и состояний тревожности. В частности, пиперидины также являются хорошими ингибиторами поглощения норэпинефрина. Таким образом, настоящее изобретение относится к производному 4-(2-фенилоксифенил)пиперидина или -1,2,3,6-тетрагидропиридина общей формулы I где пунктирная линияозначает простую связь или двойную связь;R1, R2, R3, R4, R5 независимо выбраны из водорода, галогена, C1-6-алкила, C1-6-алкилокси, галогенС 1-6-алкила; илиa R1, R4, R5 имеют определенные выше значения; или к его соли. В одном из вариантов соединения формулы I заместитель R1 выбран из водорода, галогена, C1-6 алкила, C1-6-алкилокси, галоген-С 1-6-алкила. Дополнительной иллюстрацией без ограничения изобретения является вариант, где заместитель R1 представляет собой водород; в другом варианте R1 представляет собой C1-6-алкил, такой как метил; в еще одном варианте R1 представляет собой галоген, такой как фтор или хлор. В еще одном варианте соединения формулы I заместитель R2 выбран из водорода, галогена, C1-6 алкила, C1-6-алкилокси, галоген-С 1-6-алкила. Дополнительной иллюстрацией без ограничения изобретения является вариант, где заместитель R2 представляет собой водород; в другом варианте R2 представляет собой C1-6-алкокси, такую как метокси; в еще одном варианте R2 представляет собой галоген-С 1-6 алкил, такой как трифторметил; в еще одном варианте заместитель R2 представляет собой C1-6-алкил,такой как метил; в еще одном варианте R2 представляет собой галоген, такой как хлор. В еще одном варианте соединения формулы I заместитель R3 выбран из водорода, галогена, C1-6 алкила, C1-6-алкилокси, галоген-С 1-6-алкила. Дополнительной иллюстрацией без ограничения изобретения является вариант, где заместитель R3 представляет собой водород; в другом варианте заместитель R3 представляет собой C1-6-алкил, такой как метил; в еще одном варианте заместитель R3 представляет собой C1-6-алкокси, такую как метокси; в еще одном варианте R3 представляет собой галоген, такой как хлор или фтор; в еще одном варианте R3 представляет собой галоген-С 1-6-алкил, такой как трифторметил. В еще одном варианте осуществления соединения формулы I заместители R2 и R3 вместе образуют гетероцикл, конденсированный с фенильным кольцом, выбранный из В еще одном варианте соединения формулы I заместитель R4 выбран из водорода, галогена, C1-6 алкила, C1-6-алкилокси, галоген-С 1-6-алкила. Дополнительной иллюстрацией без ограничения изобретения является вариант, где заместитель R4 представляет собой водород; в другом варианте R4 представляет собой C1-6-алкокси, такую как метокси; в еще одном варианте R4 представляет собой галоген-С 1-6 алкил, такой как трифторметил; в еще одном варианте R4 представляет собой C1-6-алкил, такой как метил; в еще одном варианте R4 представляет собой галоген, такой как хлор. В еще одном варианте соединения формулы I заместитель R5 выбран из водорода, галогена, C1-6 алкила, C1-6-алкилокси, галоген-С 1-6-алкила. Дополнительной иллюстрацией без ограничения изобретения является вариант, где заместитель R5 представляет собой водород; в другом варианте заместитель R5 представляет собой C1-6-алкил, такой как метил; в еще одном варианте заместитель R5 представляет собой галоген, такой как хлор или фтор. В еще одном варианте соединения формулы I пунктирная линияуказывает на простую связь. В еще одном варианте соединения формулы I пунктирная линияуказывает на двойную связь. Обычно соединение формулы I имеет по меньшей мере один заместитель в фенильном кольце, выбранный из любого из заместителей R1-R5, который отличается от водорода, например, 1, 2, 3 или 4 заместителя в фенильном кольце, выбранные из любого из заместителей R1-R5, который(ые) отличае(ю)тся от водорода, а остальные заместители представляют собой водород. Для иллюстрации этих вариантов без ограничения изобретения далее приведены некоторые типичные варианты соединений. Таким образом, в еще одном варианте соединения формулы I присутствует один заместитель, который представляет собой R2, определенный выше, за исключением водорода. В еще одном варианте со-2 009417 единения формулы I присутствует один заместитель, который представляет собой R3, определенный выше, за исключением водорода. В еще одном варианте соединения формулы I присутствуют два заместителя, представляющие собой R1 и R2, где R1 и R3 определены выше, за исключением водорода. В еще одном варианте соединения формулы I присутствуют два заместителя, представляющие собой R2 и R3, гдеR2 и R3 определены выше, за исключением водорода, в этой связи заместители R2 и R3 могут вместе образовывать гетероцикл, определенный выше. В каждом из упомянутых выше вариантов остальные заместители представляют собой водород. В еще одном варианте соединения формулы I указанное соединение выбирают из следующих соединений: 4-[2-(2,4-диметилфенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(4-хлорфенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(4-фтор-2-метилфенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(4-фторфенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(4-метилфенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(4-метоксифенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(2,4-диметилфенокси)фенил]пиперидин,4-[2-(4-хлорфенокси)фенил]пиперидин,4-[2-(4-фтор-2-метилфенокси)фенил]пиперидин,4-[2-(4-фторфенокси)фенил]пиперидин,4-[2-(4-метилфенокси)фенил]пиперидин,4-[2-(4-хлор-2-метилфенокси)фенил]пиперидин,4-[2-(2-хлор-4-метилфенокси)фенил]пиперидин,4-[2-(2,4-дихлорфенокси)фенил]пиперидин,4-[2-(бензо[b]тиофен-5-илокси)фенил]пиперидин,4-[2-(бензо[1,3]диоксол-5-илокси)фенил]пиперидин,4-[2-(4-метокси-2-метилфенокси)фенил]пиперидин,4-[2-(3,4-дихлорфенокси)фенил]пиперидин,4-[2-(3,4-диметилфенокси)фенил]пиперидин,4-[2-(2,3,4,5-тетраметилфенокси)фенил]пиперидин,4-[2-(4-трифторметилфенокси)фенил]пиперидин,4-[2-(4-метоксифенокси)фенил]пиперидин,4-[2-(2-хлор-4-метоксифенокси)фенил]пиперидин,4-[2-(3,4-диметоксифенокси)фенил]пиперидин,4-[2-(4-хлор-3-трифторметилфенокси)фенил]пиперидин или их фармацевтически приемлемой соли. Каждое из этих соединений считается конкретным вариантом осуществления изобретения и может быть объектом отдельных пунктов. Как упоминалось выше, большинство из испытанных соединений обладает комбинированным эффектом ингибирования обратного захвата серотонина и ингибирования поглощения норэпинефрина, однако несколько соединений, выбранных из числа следующих: 4-[2-(2,4-диметилфенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(4-хлорфенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(4-фтор-4-метилфенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(4-фторфенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(4-метилфенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(4-метоксифенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(3,4-диметилфенокси)фенил]пиперидин,4-[2-(2,3,4,5-тетраметилфенокси)фенил]пиперидин,4-[2-(3,4-диметоксифенокси)фенил]пиперидин в проведенных в данной работе испытаниях действительно проявляют ингибирование обратного захвата серотонина, но не показывают ингибирования поглощения норэпинефрина. Настоящее изобретение также включает соли соединений настоящего изобретения, обычно фармацевтически приемлемые соли. Такие соли включают фармацевтически приемлемые кислотноаддитивные соли, фармацевтически приемлемые соли металлов, аммонийные соли, алкилированные аммонийные соли. Кислотно-аддитивные соли включают соли неорганических кислот, а также органических кислот. Типичными примерами подходящих неорганических кислот являются хлористо-водородная, бромисто-водородная, йодисто-водородная, фосфорная, серная, сульфаминовая, азотная кислоты и т.д. Типичными примерами подходящих органических кислот являются муравьиная, уксусная, трихлоруксусная, трифторуксусная, пропионовая, бензойная, коричная, лимонная, фумаровая, гликолевая, итаконовая,молочная, метансульфоновая, малеиновая, яблочная, малоновая, миндальная, щавелевая, пикриновая,пировиноградная, салициловая, янтарная, метансульфоновая, этансульфоновая, винная, аскорбиновая,-3 009417 памовая, бисметиленсалициловая, этандисульфоновая, глюконовая, цитраконовая, аспарагиновая, стеариновая, пальмитиновая, этилендиаминтетрауксусная, гликолевая, п-аминобензойная, глютаминовая,бензолсульфоновая, п-толуолсульфоновые кислоты, теофиллинуксусные кислоты, а также 8 галогентеофиллины, например 8-бромтеофиллин и др. Примерами солей с металлами являются соли лития, натрия, калия, магния и т.д. Примерами аммонийных и алкилированных аммонийных солей являются аммониевая, метил-, диметил-, триметил-, этил-, гидроксиэтил-, диэтил-, н-бутил-, втор-бутил-, трет.-бутил-, тетраметиламмониевые соли и т.д. Кроме того, соединения настоящего изобретения могут существовать в несольватированной, а также в сольватированной формам с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.д. В общем случае для целей настоящего изобретения сольватированные формы считаются эквивалентными несольватированным формам. Соединения настоящего изобретения могут содержать один или несколько асимметричных центров,и подразумевается, что любой из оптических изомеров (то есть, энантиомеров или диастереомеров), которые выделены, чистые или частично очищенные оптические изомеры и их любые смеси, в том числе рацемические смеси, входят в объем настоящего изобретения. Рацемические формы могут быть разделены на их оптические антиподы известными способами,например, путем разделения диастереомерных солей с оптически активной кислотой и высвобождения оптически активного аминного соединения обработкой основанием. Другие способы разделения рацематов на их оптические антиподы основаны на хроматографировании на оптически активной матрице. Рацемические соединения настоящего изобретения могут быть разделены на их оптические антиподы, например, с помощью фракционной кристаллизации d- или l-солей (тартратов, манделатов или камфорсульфонатов). Соединения настоящего изобретения также могут быть разделены за счет образования диастереомерных производных. Могут быть использованы другие способы разделения оптических изомеров, известные специалистам в данной области. Такие способы осуждаются в публикации J. Jagues, A. Collet, S. Wilen "Enantiomers, Racemates and Resolutions", Jonh Wiley and Sons, New York (1981). Оптически активные соединения также могут быть получены из оптически активных исходных материалов или с помощью стереоселективного синтеза. Кроме того, когда в молекуле присутствует двойная связь или полностью или частично ненасыщенная циклическая система, могут существовать геометрические изомеры. Подразумевается, что любой из геометрических изомеров, который выделен, чистые или частично очищенные геометрические изомеры или их смеси входят в объем настоящего изобретения. Аналогично, молекулы, имеющие связь с ограниченным вращением, могут образовывать геометрические изомеры. Они также входят в объем настоящего изобретения. Кроме того, некоторые соединения настоящего изобретения могут существовать в различных таутомерных формах, и подразумевается, что все таутомерные формы, которые соединения способны образовывать, входят в объем настоящего изобретения. Изобретение также включает пролекарства соединений настоящего изобретения, которые при введении подвергаются химическому превращению посредством метаболических процессов до превращения в фармакологически активные вещества. В общем случае такие пролекарства будут представлять собой функциональные производные соединений общей формулы (I), которые легко превращаются invivo в требуемые соединения формулы (I). Обычные методики выбора и способы получения подходящих пролекарственных форм описаны, например, в публикации "Design of Prodrugs", ed. H. Bundgaard, Elsevier, 1985. Изобретение также включает активные метаболиты соединений настоящего изобретения. Как упоминалось выше, соединения формулы (I) являются ингибиторами обратного захвата серотонина и, следовательно, могут быть использованы для лечения, в том числе для профилактики, аффективных расстройств, таких как депрессия, состояния тревожности, включая общее состояние тревожности, паническое расстройство и навязчивое компульсивное расстройство. Таким образом, в первом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы I и фармацевтически приемлемый носитель или разбавитель. Композиция может включать один или несколько вариантов соединения формулы I, которые описаны выше. В варианте осуществления фармацевтической композиции соединение формулы I присутствует в количестве приблизительно от 0,001 до 100 мг/кг массы тела в день. Настоящее изобретение также относится к применению соединения формулы I для получения лекарственного средства для лечения заболевания или расстройства, где положительный эффект оказывает ингибитор обратного захвата серотонина. Лекарственное средство может включать один или несколько вариантов соединения формулы I, которые описаны выше. В частности, настоящее изобретение также относится к применению соединения формулы I для получения лекарственного средства для лечения аффективных расстройств.-4 009417 В еще одном варианте осуществления настоящее изобретение также относится к применению соединения формулы I для получения лекарственного средства для лечения депрессии. В еще одном варианте осуществления настоящее изобретение также относится к применению соединения формулы I для получения лекарственного средства для лечения состояний тревожности. В еще одном варианте осуществления настоящее изобретение также относится к применению соединения формулы I для получения лекарственного средства для лечения общего состояния тревожности. В еще одном варианте осуществления настоящее изобретение также относится к применению соединения формулы I для получения лекарственного средства для лечения социальной тревожности. В еще одном варианте осуществления настоящее изобретение также относится к применению соединения формулы I для получения лекарственного средства для лечения посттравматического шока. В еще одном варианте осуществления настоящее изобретение также относится к применению соединения формулы I для получения лекарственного средства для лечения навязчивого компульсивного расстройства. В еще одном варианте осуществления настоящее изобретение также относится к применению соединения формулы I для получения лекарственного средства для лечения панического расстройства. В еще одном варианте осуществления настоящее изобретение также относится к применению соединения формулы I для получения лекарственного средства для лечения приступов паники. В еще одном варианте осуществления настоящее изобретение также относится к применению соединения формулы I для получения лекарственного средства для лечения специфических фобий. В еще одном варианте осуществления настоящее изобретение также относится к применению соединения формулы I для получения лекарственного средства для лечения социальной фобии. В еще одном варианте осуществления настоящее изобретение также относится к применению соединения формулы I для получения лекарственного средства для лечения агорафобии. В еще одном аспекте настоящее изобретение относится к способу лечения заболевания или расстройства, выбранного из группы, включающей аффективное расстройство, такое как депрессия, состояние тревожности, в том числе общее состояние тревожности, состояние социальной тревожности, посттравматический шок, навязчивое компульсивное расстройство, паническое расстройство, приступы паники, специфические фобии, социальная фобия и агорафобия, у животных, включая человека, который включает введение пациенту, нуждающемуся в лечении, терапевтически эффективного количества соединения формулы I. В еще одном аспекте настоящее изобретение относится к способу получения соединения формулыI, который включает: а) снятие защитной группы или отщепление от полимерной подложки соединения формулы II где пунктирная линия, заместители R1-R9 имеют ранее определенные значения, и R' представляет собой трет-бутильную, метильную, этильную, аллильную или бензильную группу, или R'OCO является нанесенной на твердую подложку карбаматной группой; илиb) дегидратацию и необязательно одновременное снятие защитной группы соединения формулы III где заместители R1-R9 имеют ранее определенные значения, и R" представляет собой или водород, или-5 009417 карбаматную группу R'OCO, где R' представляет собой трет-бутильную, метильную, этильную, аллильную или бензильную группу, или R'OCO является нанесенной на твердую подложку карбаматной группой; или с) восстановление двойной связи в соединении формулы IV где заместители R1-R9 имеют ранее определенные значения. Фармацевтические композиции Соединения настоящего изобретения могут быть введены отдельно или в комбинации с фармацевтически приемлемыми носителями или наполнителями, или в разовых или многократных дозах. Фармацевтические композиции настоящего изобретения могут быть получены с фармацевтически приемлемыми носителями или разбавителями, а также с любыми другими известными адъювантами и наполнителями в соответствии с обычными методиками, такими как описанные в публикации "Remington: The Science and Practice of Pharmacy", 19 Edition, Gennaro, Ed., Mack Publishing Co., Easton, PA, 1995. Фармацевтические композиции могут быть специально получены для введения любым подходящим способом, таким как пероральный, ректальный, назальный, легочный, местный (в том числе, буккальный и подъязычный), трансдермальный, интрацистернальный, внутрибрюшинный, вагинальный и парентеральный (в том числе подкожный, внутримышечный, интратекальный, внутривенный и интрадермальный) способ, причем пероральный способ является предпочтительным. Следует отметить, что предпочтительный способ введения будет зависеть от общего состояния и возраста пациента, который нуждается в лечении, природы состояния, подвергающегося лечению, и выбранного активного ингредиента. Фармацевтические композиции для перорального введения включают твердые дозированные лекарственные формы, такие как капсулы, таблетки, драже, лепешки, порошки и гранулы. Когда это допустимо, твердые лекарственные формы могут быть получены с покрытиями, такими как энтеросолюбильные покрытия, или они могут быть получены так, чтобы обеспечить контролируемое высвобождение активного ингредиента, такое как постепенное или пролонгированное высвобождение, в соответствии со способами, известными в данной области. Жидкие дозированные лекарственные формы для перорального введения включают растворы,эмульсии, суспензии, сиропы и эликсиры. Фармацевтические композиции для парентерального введения включают стерильные водные и неводные инъецируемые растворы, дисперсии, суспензии или эмульсии, а также стерильные порошки, восстанавливаемые в стерильные инъецируемые растворы и дисперсии перед применением. Инъецируемые препараты-депо, как подразумевается, также входят в объем настоящего изобретения. Другими подходящими формами введения являются свечи, спреи, мази, кремы, гели, средства для ингаляции, дермальные пластыри, имплантаты и др. Типичная пероральная доза находится в интервале приблизительно от 0,001 до 100 мг/кг массы тела в день, предпочтительно приблизительно от 0,01 до 50 мг/кг массы тела в день, и наиболее предпочтительно приблизительно от 0,05 до 10 мг/кг массы тела в день, которую вводят в одной или в нескольких дозировках, например от 1 до 3 дозировок. Точная доза будет зависеть от частоты и способа введения,пола, возраста, массы и общего состояния пациента, нуждающегося в лечении, природы и сложности состояния, подвергающегося лечению, и любых сопутствующих заболеваний, которые требуют лечения,а также от других факторов, очевидных для специалиста в данной области. Препараты обычно могут быть получены в стандартных дозированных лекарственных формах способами, которые известны специалистам в данной области. Типичная стандартная дозированная лекарственная форма для перорального введения один или несколько раз в день, например, от 1 до 3 раз в день,может содержать от 0,01 до приблизительно 1000 мг, предпочтительно приблизительно от 0,05 до 500 мг,и более предпочтительно приблизительно от 0,5 до 200 мг. В случае парентеральных способов, таких как внутривенный, интратекальный, внутримышечный и подобные способы введения, дозы обычно составляют приблизительно половину дозы, используемой при пероральном введении. Соединения настоящего изобретения обычно используют в виде свободного вещества или в виде-6 009417 его фармацевтически приемлемой соли. Одним из примеров является кислотно-аддитивная соль соединения, находящего применение в виде свободного основания. Когда соединение формулы (I) содержит свободное основание, такую соль получают обычным способом реакцией раствора или суспензии свободного основания формулы (I) с химическим эквивалентом фармацевтически приемлемой кислоты. Типичные примеры обсуждались выше. Для парентерального введения могут быть использованы растворы новых соединений формулы (I) в стерильном водном растворе, водном пропиленгликоле, водном витамине Е или кунжутном или арахисовом масле. Такие водные растворы должны быть соответствующим образом забуферены, если это необходимо, а жидкий разбавитель вначале должен быть сделан изотоничным с помощью достаточного количества рассола или глюкозы. Водные растворы особенно подходят для внутривенного, внутримышечного, подкожного и внутрибрюшинного введения. Все используемые стерильные водные среды легко доступны по стандартным методикам, известным специалистам в данной области. Подходящими фармацевтическими носителями являются инертные твердые разбавители или наполнители, стерильный водный раствор и различные органические растворители. Примерами твердых носителей являются лактоза, магнезия, сахароза, циклодекстрин, тальк, желатин, агар-агар, пектин, аравийская камедь, стеарат магния, стеариновая кислота и низшие алкиловые простые эфиры целлюлозы. Примерами жидких носителей являются очищенная патока, арахисовое масло, оливковое масло, фосфолипиды, жирные кислоты, амины жирных кислот, полиоксиэтилен и вода. Аналогично носитель или разбавитель могут включать любой постепенно высвобождаемый материал, известный в данной области,такой как моностеарат глицерина или дистеарат глицерина, отдельно или смешанный с воском. Фармацевтические композиции, полученные смешением новых соединений формулы (I) и фармацевтически приемлемых носителей, затем легко вводятся в различные дозированные лекарственные формы, подходящие для описанных способов введения. Обычно препараты могут быть получены в стандартной дозированной лекарственной форме с помощью способов, известных в области фармакологии. Препараты настоящего изобретения, подходящие для перорального введения, могут быть представлены в виде отдельных единиц, таких как капсулы или таблетки, каждая из которых содержит заранее определенное количество активного ингредиента, и которые могут содержать подходящий наполнитель. Кроме того, перорально активные препараты могут иметь форму порошка или гранул, раствора или суспензии в водной или неводной жидкости, или эмульсии масло-в-воде или вода-в-масле. Если в случае перорального введения используется твердый носитель, препарат может представлять собой таблетку, помещенную в твердую желатиновую капсулу в порошкообразной или пеллетированной форме, или он может находиться в форме пастилки или лепешки. Количество твердого носителя будет меняться в широких пределах, но обычно составляет приблизительно от 25 мг до 1 г. Если используется жидкий носитель, препарат может иметь форму сиропа, эмульсии, мягкой желатиновой капсулы или стерильной инъецируемой жидкости, такой как суспензия или раствор в водной или неводной жидкости. Соединения настоящего изобретения получают следующими общими способами или как описано в экспериментальной части данного патента: а) снятием защитной группы или отщеплением от полимерной подложки соединения формулы II: где заместители R1-R9 имеют ранее определенные значения, и R' представляет собой трет.-бутильную,метильную, этильную, аллильную или бензильную группу, или R'OCO является нанесенной на твердую подложку карбаматной группой, такой как связанный со смолой Ванга (Wang resin) карбаматный линкер; илиb) дегидратацией и необязательно одновременным снятием защитной группы соединения формулы III где заместители R1-R9 имеют ранее определенные значения, и R" представляет собой или водород или карбаматную группу R'OCO, где R' представляет собой трет.-бутильную, метильную, этильную, аллильную или бензильную группу, или R'OCO является нанесенной на твердую подложку карбаматной группой, такой как связанный со смолой Ванга карбаматный линкер; или с) восстановлением двойной связи в соединении формулы IV где заместители R1-R9 имеют ранее определенные значения. Снятие защитной группы в соответствии со способом а) проводят по стандартным методикам, известным специалистам в данной области и подробно описанным в учебнике Protective Groups in OrganicSynthesis, T.W. Greene, P.G.M. Wuts, Wiley Interscience (1991), ISNB 0471623016. Отщепление от полимерной подложки, например, от связанного со смолой Ванга карбаматного линкера, в соответствии со способом а) может быть проведено по описанным в литературе методикам (Zaragoza Tetrahedron Lett.,1995, 36, 8677-8678, и Conti et al., Tetrahedron Lett., 1997, 38, 2915-2918). Исходные материалы формулы II могут быть получены путем удаления гидроксильной группы соединений формулы III рядом способов, известных специалистам в данной области, например, путем использования триэтилсилана в трифторуксусной кислоте и диэтилэфирата трифторида бора (см. Encyclopaedia of Reagents for Organic Synthesis, vol. 7, Paquette, ed., John Wiley and Sons, Chichester, 1995, 51225123). Исходные материалы формулы II, которые представляют собой пиперидины, могут быть получены путем восстановления двойной связи соответствующих тетрагидропиридинов стандартными методиками гидрирования, такими как, например, каталитическое гидрирование при низком давлении ( 3 атм) в аппарате Парра. Реакцию дегидратации и необязательного одновременного снятия защитной группы соединения формулы III в соответствии со способом b) проводят аналогично способу, который описан в публикацииPalmer et al., J.Med. Chem., 1997, 40, 1982-1989. Исходные материалы формулы III получают из соответствующих должным образом замещенных 1 бром-2-феноксибензолов формулы VI (где заместители R1-R9 имеют значения, определенные ранее, a G представляет собой атом брома или йода) путем обмена металл-водород с последующим добавлением подходящего электрофила формулы V (где R' имеет ранее определенные значения) способом, аналогичным описанному в публикации Palmer et al., J.Med. Chem., 1997, 40, 1982-1989. Должным образом замещенные 1-бром-2-феноксибензолы получают реакцией должным образом замещенных фенолов (натриевую соль фенолов получают in situ путем использования гидрида натрия) с должным образом замещенными 1-бром-2-фторбензолами в диметилфорамиде (ДМФА) при повышенной температуре. Диариловые эфиры также могут быть получены с помощью различных модификаций этого способа (см., например, Schimittlinger et al., J. Org. Chem., 1993, 58, 3229-3230; Beugelmans et al.,Tetrahedron Lett., 1994, 35, 5649-5652; Sawyer et al., J. Org. Chem., 1998, 63, 6338-6343) в условиях Ульмана или через арилирование фенолов арилбороновыми кислотами (Evans et al., Tetrahedron Lett., 1998, 39,2937-2940). Фенолы и 1-бром-2-фторбензолы являются коммерчески доступными. Восстановление двойной связи в соответствии со способом с) обычно проводят путем каталитического гидрирования при низком давлении ( 3 атм) в аппарате Парра. Исходные материалы формулы IV могут быть получены из соединений формулы II путем снятия защитной группы, как описано выше. Примеры Аналитические данные ЖХ-МС получены на приборе РЕ Sciex API 150EX, оборудованном источником ионного впрыска, и системы Shimadzu LC-8A/SLC-10A LC. Колонка: колонка 30x4,6 мм Waters(100:0,05) и В = вода/ацетонитрил/трифторуксусная кислота (5:95:0,03). Способ: элюирование с линейным градиентом от 90% А до 100% В за 4 мин при скорости потока 2 мл/мин. Чистоту определяют путем интегрирования следов УФ (254 нм) и ELSD. Время удерживания (RT) выражено в минутах. Препаративную очистку с помощью ЖХ-МС проводят на том же приборе. Колонка: 50x20 мм YMCODS-A с размером частиц 5 мкм. Способ: элюирование с линейным градиентом от 80% А до 100% В за 7 минут и при скорости потока 22,7 мл/мин. Сбор фракций проводят с помощью МС-детектирования с разделением потока. Реакции, проводимые в микроволновой печи, проводят в SmithSynthesizer фирмы PersonalChemistry, работающий при 2450 МГц. Получение промежуточных соединений Получение замещенных 2-бром-(замещенная фенокси)бензолов 1-Бром-2-(2,4-диметилфенокси)бензол Раствор 2,4-диметилфенола (2,4 г) в сухом диметилформамиде (ДМФА) (10 мл) добавляют по каплям к смеси гидрида натрия (1,0 г, 60% в минеральном масле) и сухого ДМФА (25 мл), и полученную смесь перемешивают при 100 С в течение 30 мин. К полученной смеси добавляют 1-бром-2-фторбензол(3,5 г) в сухом ДМФА (5 мл) и полученную смесь перемешивают при 150 С в течение 6 ч. Смесь затем выливают на смесь лед/вода и водную фазу экстрагируют диэтиловым эфиром. Объединенную органическую фазу промывают 2 н. гидроксидом натрия, сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток очищают быстрой хроматографией на силикагеле (элюент: гептан), получают сырой продукт(1,2 г, чистота 70%). Сырой продукт используют на следующей стадии без дополнительной очистки. Следующие соединения получены аналогичным способом: 1-бром-2-(4-хлорфенокси)бензол 1-бром-2-(4-фтор-2-метилфенокси)бензол 1-бром-2-(4-фторфенокси)бензол 1-бром-2-(4-метилфенокси)бензол 1-бром-2-(4-метоксифенокси)бензол Получение алкил-4-[2-(замещенная фенокси)замещенный фенил]-4-гидроксипиперидин-1-карбоксилатов трет.-Бутил-4-[2-(2,4-диметилфенокси)фенил]-4-гидроксипиперидин-1-карбоксилат Раствор 1-бром-2-(2,4-диметилфенокси)бензола (0,7 г) в сухом тетрагидрофуране (ТГФ) (3 мл) добавляют к раствору н-BuLi (1,6 М в гексане, 2 мл) в сухом ТГФ (15 мл) при -78 С. Полученную смесь перемешивают при -78 С в течение 1 ч и затем добавляют раствор трет.-бутил-4-оксопиперидин-1 карбоксилата (1,0 г) в сухом ТГФ (3 мл). Смесь перемешивают 16 ч при комнатной температуре и затем выливают в насыщенный раствор хлорида аммония. Водную фазу экстрагируют диэтиловым эфиром и-9 009417 объединенную органическую фазу промывают водой и рассолом, затем сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток очищают быстрой хроматографией на силикагеле (элюент: гептан/этилацетат, 4:1), получают сырой продукт (0,55 г). Сырой продукт используют на следующей стадии без дополнительной очистки. Следующие соединения получены аналогичным способом: этил-4-[2-(2,4-диметилфенокси)фенил]-4-гидроксипиперидин-1-карбоксилат этил-4-[2-(4-хлорфенокси)фенил]-4-гидроксипиперидин-1-карбоксилат трет.-бутил-4-[2-(4-фтор-2-метилфенокси)фенил]-4-гидроксипиперидин-1-карбоксилат этил-4-[2-(4-фтор-2-метилфенокси)фенил]-4-гидроксипиперидин-1-карбоксилат этил-4-[2-(4-фторфенокси)фенил]-4-гидроксипиперидин-1-карбоксилат трет.-бутил-4-[2-(4-метилфенокси)фенил]-4-гидроксипиперидин-1-карбоксилат этил-4-[2-(4-метилфенокси)фенил]-4-гидроксипиперидин-1-карбоксилат трет.-бутил-4-[2-(4-метоксифенокси)фенил]-4-гидроксипиперидин-1-карбоксилат. Получение этил-4-(2-метоксифенил)пиперидин-1-карбоксилата К 26 ммолям 4-(2-метоксифенил)пиперидина (Maybridge) в 100 мл сухого дихлорметана добавляют 28,6 ммолей триэтиламина и 78 ммолей этилхлорформиата при 0 С. Раствор перемешивают при комнатной температуре в течение ночи, промывают дважды 0,5 М HCl (125 мл), затем сушат над MgSO4 и упаривают. Продукт достаточно чистый для использования на следующей стадии. Получение этил-4-(2-гидроксифенил)пиперидин-1-карбоксилата К 24 ммолям этил-4-(2-метоксифенил)пиперидин-1-карбоксилата в 150 мл сухого дихлорметана добавляют 48 ммолей BBr3 при 0 С. Раствор перемешивают при комнатной температуре в течение ночи,промывают дважды 0,5 М HCl (125 мл), затем сушат над MgSO4 и упаривают. Продукт достаточно чистый для использования на следующей стадии. Соединения настоящего изобретения Получение 4-[2-(замещенная фенокси)замещенный фенил]-1,2,3,6-тетрагидропиридинов 1 а. 4-[2-(2,4-Диметилфенокси)фенил]-1,2,3,6-тетрагидропиридин Смесь трет.-бутил-4-[2-(2,4-диметилфенокси)фенил]-4-гидроксипиперидин-1-карбоксилата (0,5 г) и смеси уксусной кислоты и конц. соляной кислоты (3:1) кипятят с обратным холодильником 16 ч. Смесь охлаждают, выливают в щелочную воду и экстрагируют этилацетатом. Объединенную органическую фазу сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток очищают быстрой хроматографией на силикагеле (элюент: этилацетат/метанол/триэтиламин, 8:2:1), получают целевое соединение (11 мг,выход 3%). ЖХ/МС (m/z) 280 (МН+); RT=2,16; чистота (УФ, ELSD): 85, 97%. Следующие соединения получены аналогичным образом: 1b. 4-[2-(4-Хлорфенокси)фенил]-1,2,3,6-тетрагидропиридин Из этил-4-[2-(4-хлорфенокси)фенил]-4-гидроксипиперидин-1-карбоксилата. ЖХ/МС (m/z) 286 (МН+); RT=2,10; чистота (УФ, ELSD): 85, 95%; выход: 33 мг (6%). 1 с. 4-[2-(4-Фтор-2-метилфенокси)фенил]-1,2,3,6-тетрагидропиридин Из трет.-бутил-4-[2-(4-фтор-2-метилфенокси)фенил]-4-гидроксипиперидин-1-карбоксилата. ЖХ/МС (m/z) 284 (МН+); RT=2,08; чистота (УФ, ELSD): 97, 99%; выход: 100 мг (21%). 1d. 4-[2-(4-Фторфенокси)фенил]-1,2,3,6-тетрагидропиридин Из этил-4-[2-(4-фторфенокси)фенил]-4-гидроксипиперидин-1-карбоксилата. ЖХ/МС (m/z) 270 (МН+); RT=1,93; чистота (УФ, ELSD): 87, 97%; выход: 45 мг (11%). 1 е. 4-[2-(4-Метилфенокси)фенил]-1,2,3,6-тетрагидропиридин Из трет.-бутил-4-[2-(4-метилфенокси)фенил]-4-гидроксипиперидин-1-карбоксилата. ЖХ/МС (m/z) 266 (МН+); RT=2,04; чистота (УФ, ELSD): 98, 99%; выход: 250 мг (24%). 1f. 4-[2-(4-Метоксифенокси)фенил]-1,2,3,6-тетрагидропиридин Из трет.-бутил-4-[2-(4-метоксифенокси)фенил]-4-гидроксипиперидин-1-карбоксилата. ЖХ/МС (m/z) 282 (МН+); RT=1,95; чистота (УФ, ELSD): 79, 99%; выход: 14,7 мг (19%). Получение 4-[2-(замещенная фенокси)замещенный фенил]пиперидинов Способ А 2 а. 4-[2-(2,4-Диметилфенокси)фенил]пиперидин Смесь этил-4-[2-(2,4-диметилфенокси)фенил]-4-гидроксипиперидин-1-карбоксилата (0,6 г), дихлорметана (25 мл), триэтилсилана (1 мл), трифторуксусной кислоты (0,1 мл) и диэтилэфирата трифторида бора (0,2 мл) перемешивают при комнатной температуре 16 ч. Полученную смесь выливают в щелочную воду и затем экстрагируют этилацетатом. Объединенную органическую фазу сушат (MgSO4),фильтруют и концентрируют в вакууме (0,4 г). Остаток растворяют в смеси конц. соляной кислоты и уксусной кислоты (1:3) (25 мл) и кипятят с обратным холодильником 16 ч. Смесь выливают в щелочную воду и затем экстрагируют этилацетатом. Объединенную органическую фазу сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток очищают быстрой хроматографией на силикагеле (элюент: этилацетат/метанол/триэтиламин, 8:2:2), получают целевое соединение (10,6 мг, выход 3%). ЖХ/МС (m/z) 282 (МН+); RT=2,22; чистота (УФ, ELSD): 67, 83%. Следующие соединения получены аналогичным образом:(0,1 ммоль) смешивают в 0,5 мл 1-метилпирролидин-2-она с 0,12 ммолями соответствующего арилбромида или йодида. Добавляют в качестве катализатора CuI (0,037 ммоль) и сосуд запаивают перед тем,как нагревать его 1 ч в микроволновой печи при 220 С. Из образца удаляют растворитель и добавляют раствор КОН в воде (3,7 ммоль), диоксан и этанол (99,9%), смесь нагревают при 130 С в течение 1 ч в микроволновой печи. Затем к образцу добавляют воду и твердый NaCl, экстрагируют этилацетатом. Органическую фазу упаривают и сырой продукт очищают препаративной ЖХ-МС. Выделенные продукты наносят на колонки SCX и свободные амины передают на испытание в виде растворов в ДМСО. Описанным способом получают следующие соединения, для которых определенная молекулярная масса, определенное время удерживания при ВЭЖХ (RT, мин) и чистота по УФ и ELSD (%) представлены в таблице. 3 а. 4-[2-(4-хлор-2-метилфенокси)фенил]пиперидин 3b. 4-[2-(3-хлор-2-метилфенокси)фенил]пиперидин 3 с. 4-[2-(2-хлор-4-метилфенокси)фенил]пиперидин 3d. 4-[2-(2,4-дихлорфенокси)фенил]пиперидин 3 е. 4-[2-(бензо[1,3]диоксол-5-илокси)фенил]пиперидин 3f. 4-[2-(4-метокси-2-метилфенокси)фенил]пиперидин 3g. 4-[2-(3,4-дихлорфенокси)фенил]пиперидин 3h. 4-[2-(3,4-диметилфенокси)фенил]пиперидин 3i. 4-[2-(2,3,4,5-тетраметилфенокси)фенил]пиперидин 3j. 4-[2-(4-триметилфенокси)фенил]пиперидин 3k. 4-[2-(4-метоксифенокси)фенил]пиперидин 3l. 4-[2-(2-хлор-4-метоксифенокси)фенил]пиперидин 3m. 4-[2-(3,4-диметоксифенокси)фенил]пиперидин 3n. 4-[2-(4-хлор-3-трифторметилфенокси)фенил]пиперидин- 11009417 Таблица. Определенная молекулярная масса, определенное время удерживания при ВЭЖХ (RT, мин) и чистота по УФ и ELSD (%) Определение поглощения [3 Н]-5-НТ в корковых синаптосомах крыс Весь мозг самцов крыс Wistar (125-225 г), кроме мозжечка, гомогенизируют в 0,32 М сахарозе, дополненной 1 мМ ниаламида, с помощью стеклянного/тефлонового гомогенизатора. Гомогенат центрифугируют при 600g в течение 10 мин при 4 С. Пеллеты извлекают и надосадочную жидкость центрифугируют при 20000g в течение 55 мин. Конечную пеллету гомогенизируют (20 с) в оценочном буфере (0,5 мг исходной ткани/лунка). Испытуемые соединения (или буфер) и 10 нМ [3 Н]-5-НТ добавляют в 96-ти луночные планшеты и слегка встряхивают. Состав оценочного буфера: 123 мМ NaCl, 4,82 мМ KCl, 0,973 мМ CaCl2, 1,12 мМ MgSO4, 12,66 мМ Na2HPO4, 2,97 мМ NaH2PO4, 0,162 мМ ЭДТУК, 10 мМ глюкозы и 1 мМ аскорбиновой кислоты. Буфер насыщают 95% О 2/5% CO2 в течение 10 мин при 37 С и доводят рН до 7,4. Выдерживание начинают путем добавления ткани до конечного оценочного объема 0,2 мл. После 15 мин выдерживания с радиоактивным лигандом при 37 С образцы фильтруют непосредственно на стекловолоконных фильтрах Unifilter GF/C (пропитанных в течение 1 ч в 0,1% полиэтиленимине) в вакууме и сразу же промывают оценочным буфером (3x0,2 мл). Неспецифическое поглощение определяют с использованием циталопрама (конечная концентрация 10 мкМ). Циталопрам включен в качестве стандарта во все эксперименты в виде кривой доза-реакция. Определение поглощения [3 Н]норадреналина в корковых синаптосомах крыс Свежий кортикальный слой самцов крыс Wistar (125-225 г) гомогенизируют в 0,4 М сахарозе с помощью стеклянного/тефлонового гомогенизатора. Гомогенат центрифугируют при 600g в течение 10 мин при 5 С. Пеллеты извлекают и надосадочную жидкость центрифугируют при 20000g в течение 55 мин. Конечную пеллету гомогенизируют (20 с) в буфере для оценки (6 мг исходная ткани/мл = 4 мг/лунка). Испытуемые соединения (или буфер) и 10 нМ [3 Н]-норадреналина добавляют в глубокие 96-ти луночные планшеты и немного встряхивают. Состав оценочного буфера: 123 мМ NaCl, 4,82 мМ KCl, 0,973 мМ CaCl2, 1,12 мМ MgSO4, 12,66 мМ Na2HPO4, 2,97 мМ NaH2PO4, 0,162 мМ ЭДТУК, 10 мМ глюкозы и 1 мМ аскорбиновой кислоты. Буфер насыщают 95% О 2/5% СО 2 в течение 10 мин при 37 С и доводят рН до 7,4. Выдерживание начинают путем добавления ткани до конечного оценочного объема 1 мл. После 15 мин выдерживания с радиоактивным лигандом при 37 С образцы фильтруют непосредственно на стекловолоконных фильтрах Unifilter GF/C (пропитанных в течение 1 ч в 0,1% полиэтиленимине) в вакууме и сразу же промывают оценочным буфером (31 мл). Неспецифическое поглощение определяют с использованием талсупрама (конечная концентрация 10 мкМ). Дилоксетин включен в качестве стандарта во все эксперименты в виде кривой доза-реакция. Результаты опытов показывают, что испытуемые соединения настоящего изобретения ингибируют обратный захват норэпинефрина и серотонина при значении IC50 менее 200 нМ. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Производное 4-(2-фенилоксифенил)пиперидина или 1,2,3,6-тетрагидропиридина общей формулы I где пунктирная линияозначает простую связь или двойную связь;R1, R2, R3, R4, R5 независимо выбраны из водорода, галогена, C1-6-алкила, C1-6-алкилокси, галогенС 1-6-алкила; или R2 и R3 вместе образуют гетероцикл, конденсированный с фенильным кольцом, выбранный изR1, R4, R5 имеют определенные выше значения;R6, R7, R8, R9 независимо представляют собой водород; или его соль. 2. Соединение по п.1, где R1 выбран из водорода, галогена, C1-6-алкила, C1-6-алкилокси, галоген-С 1-6 алкила. 3. Соединение по любому из пп.1 и 2, где R2 выбран из водорода, галогена, C1-6-алкила, C1-6 алкилокси, галоген-С 1-6-алкила. 4. Соединение по любому из пп.1-3, где R3 выбран из водорода, галогена, C1-6-алкила, C1-6 алкилокси, галоген-С 1-6-алкила. 5. Соединение по любому из пп.1 и 2, где R2 и R3 вместе образуют гетероцикл, конденсированный с фенильным кольцом, выбранный из 6. Соединение по любому из пп.1-5, где R4 выбран из водорода, галогена, C1-6-алкила, C1-6 алкилокси, галоген-С 1-6-алкила. 7. Соединение по любому из пп.1-6, где R5 выбран из водорода, галогена, C1-6-алкила, C1-6 алкилокси, галоген-С 1-6-алкила. 8. Соединение по любому из пп.1-7, где R6 является водородом. 9. Соединение по любому из пп.1-8, где R7 является водородом. 10. Соединение по любому из пп.1-9, где R8 является водородом. 11. Соединение по любому из пп.1-10, где R9 является водородом. 12. Соединение по любому из пп.1-11, где пунктирная линия означает простую связь. 13. Соединение по любому из пп.1-11, где пунктирная линия означает двойную связь. 14. Соединение по п.1, причем указанное соединение представляет собой 4-[2-(2,4-диметилфенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(4-хлорфенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(4-фтор-2-метилфенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(4-фторфенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(4-метилфенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(4-метоксифенокси)фенил]-1,2,3,6-тетрагидропиридин,4-[2-(2,4-диметилфенокси)фенил]пиперидин,4-[2-(4-хлорфенокси)фенил]пиперидин,4-[2-(4-фтор-2-метилфенокси)фенил]пиперидин,4-[2-(4-фторфенокси)фенил]пиперидин,4-[2-(4-метилфенокси)фенил]пиперидин,4-[2-(4-хлор-2-метилфенокси)фенил]пиперидин,4-[2-(3-хлор-2-метилфенокси)фенил]пиперидин,4-[2-(2-хлор-4-метилфенокси)фенил]пиперидин,4-[2-(2,4-дихлорфенокси)фенил]пиперидин,4-[2-(бензо[1,3]диоксол-5-илокси)фенил]пиперидин,4-[2-(4-метокси-2-метилфенокси)фенил]пиперидин,4-[2-(3,4-дихлорфенокси)фенил]пиперидин,4-[2-(3,4-диметилфенокси)фенил]пиперидин,4-[2-(2,3,4,5-тетраметилфенокси)фенил]пиперидин,4-[2-(4-трифторметилфенокси)фенил]пиперидин,4-[2-(4-метоксифенокси)фенил]пиперидин,4-[2-(2-хлор-4-метоксифенокси)фенил]пиперидин,4-[2-(3,4-диметоксифенокси)фенил]пиперидин,4-[2-(4-хлор-3-трифторметилфенокси)фенил]пиперидин,- 13009417 или его фармацевтически приемлемую соль. 15. Фармацевтическая композиция, содержащая соединение по любому из пп.1-14 или его фармацевтически приемлемую кислотно-аддитивную соль и по меньшей мере один фармацевтически приемлемый носитель или разбавитель. 16. Применение соединения по любому из пп.1-14 или его фармацевтически приемлемой кислотноаддитивной соли для получения лекарственного средства для лечения аффективных расстройств, таких как депрессия, состояние тревожности, в том числе общее состояние тревожности, состояние социальной тревожности, посттравматический шок, навязчивое компульсивное расстройство, паническое расстройство, приступы паники, специфические фобии, социальная фобия и агорафобия. 17. Способ лечения аффективных расстройств, таких как депрессия, состояние тревожности, в том числе общее состояние тревожности, состояние социальной тревожности, посттравматический шок, навязчивое компульсивное расстройство, паническое расстройство, приступы паники, специфические фобии, социальная фобия и агорафобия, у животных, включая человека, который включает введение терапевтически эффективного количества соединения по любому из пп.1-14 или его фармацевтически приемлемой кислотно-аддитивной соли.
МПК / Метки
МПК: C07D 211/22, A61K 31/4418, A61K 31/451, C07D 407/12, A61P 25/00, C07D 211/70
Метки: обратного, тетрагидропиридина, качестве, серотонина, захвата, 1,2,3,6, 4-(2-фенилоксифенил)пиперидина, ингибиторов, производные
Код ссылки
<a href="https://eas.patents.su/15-9417-proizvodnye-4-2-feniloksifenilpiperidina-ili-1236-tetragidropiridina-v-kachestve-ingibitorov-obratnogo-zahvata-serotonina.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 4-(2-фенилоксифенил)пиперидина или -1,2,3,6- тетрагидропиридина в качестве ингибиторов обратного захвата серотонина</a>
Производные аминоиндана в качестве ингибиторов обратного захвата серотонина и захвата норэпинефрина

Номер патента: 7655
Опубликовано: 29.12.2006
Авторы: Брегнедаль Петер, Пюшль Аск, Келер Ян, Бегесе Клаус Петер
МПК: A61K 31/135, A61P 25/22, A61K 31/357...
Метки: серотонина, обратного, аминоиндана, захвата, ингибиторов, норэпинефрина, производные, качестве
Формула / Реферат:
1. Соединение аминоиндана, имеющее формулу I где X означает -O-, -S- или -CR4R5-; Y означает -CR6R7-, -CR6R7-CR8R9- или -CR6=CR7-; или X и Y вместе образуют группу -CR4=CR5- или -CR4=CR5-CR6R7-; и U означает -O-, -S- или -CR10R11 или X означает -O-, -S- или -CR4R5-; и Y и U вместе образуют группу -CR6=CR7-, -CR6=CR7-CR10R11- или -CR6R7-CR10=CR11-; или X, и Y, и U вместе образуют -CR4=CR5-CR6=CR7-; R1 и R2 независимо выбраны из водорода,...
Производные бензо[3,4] циклобута [1,2-c] пиррола и их применение в качестве ингибиторов обратного захвата серотонина

Номер патента: 3302
Опубликовано: 24.04.2003
Авторы: Риве Жан-Мишель, Гумен Бертран, Миллан Марк, Дессанж Эме, Декейн Анн, Пеглион Жан-Луи
МПК: A61K 31/403, A61P 25/24, C07D 209/58...
Метки: применение, производные, ингибиторов, качестве, бензо[3,4, 1,2-c, серотонина, циклобута, пиррола, захвата, обратного
Формула / Реферат:
1. Соединение формулы (I), имеющее цис-соединение колец в которой R1, R2 и R3, которые могут быть одинаковыми или различными, каждый независимо от других, представляет группу, выбранную из атома водорода, атома галогена, линейной или разветвленной (C1-C6)алкоксигруппы, линейной или разветвленной (C1-C6)тригалогеналкильной группы, или два из R1, R2 и R3 в смежных положениях представляют (C1-C2)алкилендиоксигруппу; R4 представляет группу,...
Производные фенил-гетероциклильных эфиров в качестве ингибиторов обратного захвата серотонина

Номер патента: 8936
Опубликовано: 26.10.2007
Авторы: Миддлтон Доналд Стюарт, Хепворт Дейвид, Адам Мейвис Дайан, Хауард Гарри Ральф Мл., Джаймер Джеффри Эдвард, Эндрюс Марк Дейвид, Стоуби Алан
МПК: C07D 213/65, A61K 31/44, A61P 25/24...
Метки: эфиров, ингибиторов, качестве, серотонина, фенил-гетероциклильных, захвата, производные, обратного
Формула / Реферат:
1. Соединение общей формулы (I), его фармацевтически приемлемые соли, сольваты или полиморфы где L и U, которые могут быть одинаковыми или разными, представляют собой -N- или -С(Н)-; M представляет собой -N-, -N+(-O-)- или -C(R4)-; Q представляет собой -N- или -C(R4)-; кольцо А содержит 1 или 2 атома азота, в том случае, когда M представляет собой -N+(O-)-, тогда кольцо А не содержит других атомов азота; R1 и R2, которые могут быть одинаковыми...
Производные 4-(2-фенилсульфанилфенил)-1,2,3,6-тетрагидропиридина в качестве ингибиторов повторного поглощения серотонина

Номер патента: 9282
Опубликовано: 28.12.2007
Авторы: Руланд Томас, Банг-Андерсен Бенни, Юль Карстен, Келер Ян, Йергенсен Мортен, Пюшль Аск, Андерсен Ким
МПК: A61K 31/4409, A61P 25/00, C07D 211/70...
Метки: 4-(2-фенилсульфанилфенил)-1,2,3,6-тетрагидропиридина, повторного, производные, серотонина, поглощения, качестве, ингибиторов
Формула / Реферат:
1. Соединение, выбранное из группы, состоящей из 4-[2-(4-фторфенилсульфанил)фенил]-1,2,3,6-тетрагидропиридина, 4-[2-(4-хлорфенилсульфанил)фенил]-1,2,3,6-тетрагидропиридина, 4-[2-(4-метоксифенилсульфанил)-5-метилфенил]-1,2,3,6-тетрагидропиридина, 4-(5-метил-2-п-толилсульфанилфенил)-1,2,3,6-тетрагидропиридина, 4-[2-(4-фторфенилсульфанил)-5-метилфенил]-1,2,3,6-тетрагидропиридина, 4-[2-(4-хлорфенилсульфанил)-5-метилфенил]-1,2,3,6-тетрагидропиридина,...
Производные биариловых эфиров, полезные в качестве ингибиторов обратного захвата моноамина

Номер патента: 5671
Опубликовано: 28.04.2005
Авторы: Хауард Гарри Ралф Младший, Адам Мейвис Дайан
МПК: A61K 31/13, C07C 217/58, A61P 25/24...
Метки: эфиров, захвата, производные, полезные, качестве, биариловых, ингибиторов, моноамина, обратного
Формула / Реферат:
1. Соединение формулы где n и m независимо выбраны из единицы и двух; R1 и R2 независимо выбраны из водорода и (C1-C4)алкила; R3 и R4 независимо выбраны из водорода и (C1-C4)алкила; каждый X независимо выбран из фенила и 5- или 6-членного гетероцикла, содержащего от 1 до 3 гетероатомов, выбранных из N, O и S, где каждый X может быть дополнительно замещен водородом, галогено, (C1-C4)алкилом, оксо, амино и группировкой NR5(C=O)(C1-C4)алкил, где...