Кристаллические формы eto2c-ch2-(r)cgl-aze-pab-oh
Номер патента: 3264
Опубликовано: 27.02.2003
Авторы: Лундблад Анита, Хедстрём Лена, Эдвардссон Даниель, Петтерссон Урсула













Формула / Реферат
1. По существу, кристаллическая форма EtO2C-CH2-(R)Cgl-Aze-Pab-OH или его фармацевтически приемлемой соли.
2. Соединение по п.1, которое находится в безводной форме.
3. Соединение по п.2, которое находится не в форме соли.
4. Соединение по любому из пп.1-3, которое содержит не более 2 маc.% воды.
5. Соединение по любому из пп.1-4, характеризуемое кривой дифференциальной сканирующей калориметрии, на которой при скорости нагрева 5шC/мин в закрытой чашке с микроотверстием под током азота проявляется эндотермический эффект с экстраполированной начальной температурой около 150шC, температурой пика около 151шC и соответственным тепловым эффектом около 113 Дж/г, за которым следует экзотермический эффект в интервале 190-280шС, и/или порошковой дифракционной картиной, характеризуемой пиками со значениями d 12,0, 10,0, 8,2, 7,2, 6,0, 5,5, 5,0, 4,92, 4,85, 4,80, 4,42, 4,22, 4,11, 4,06, 3,99, 3,78, 3,72, 3,62, 3,34, 3,11, 3,10 и 3,03 А, и/или как представлено на фиг. 1.
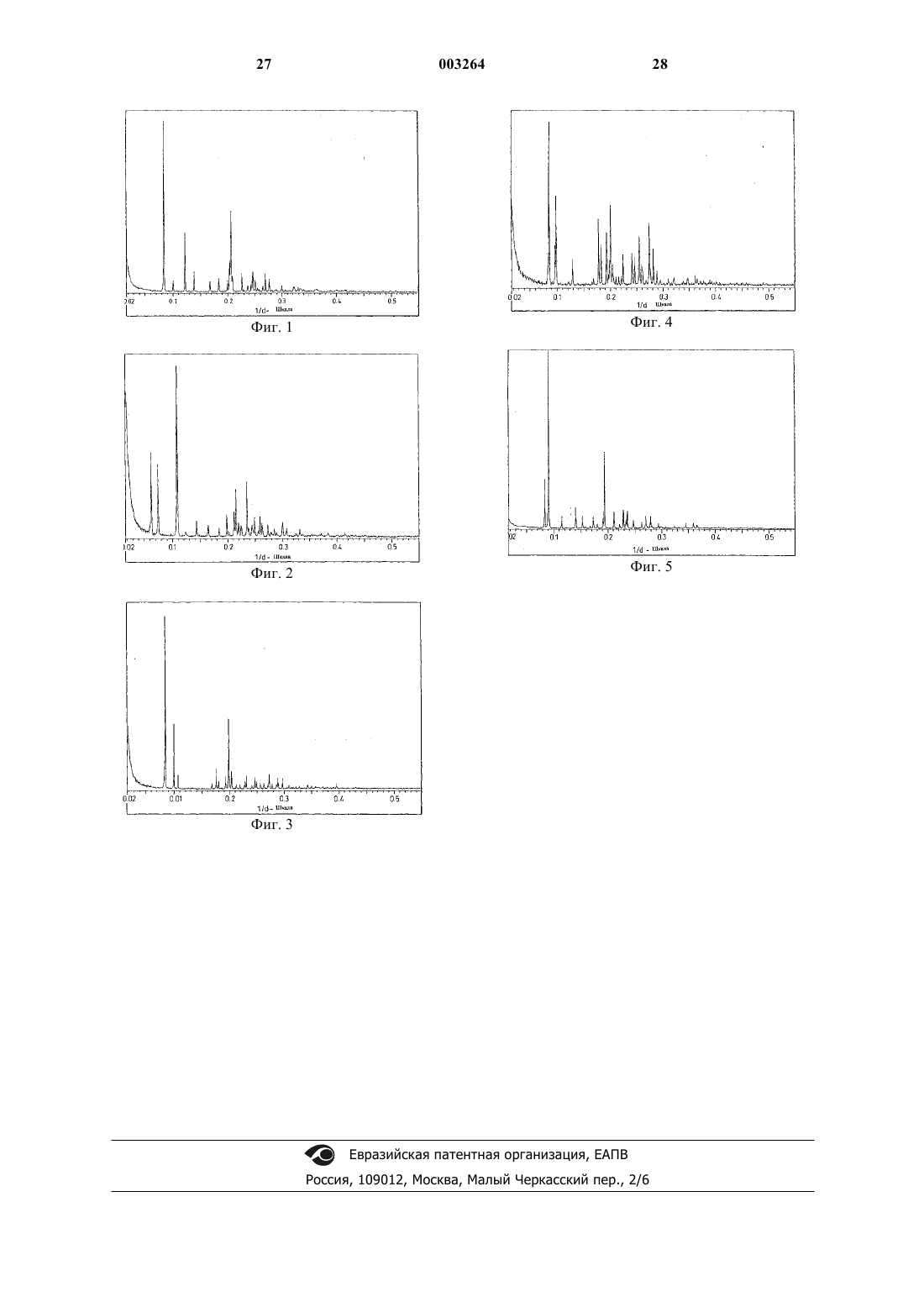
6. Соединение по любому из пп.1-4, характеризуемое кривой дифференциальной сканирующей калориметрии, на которой при скорости нагрева 5шC/мин в закрытой чашке с микроотверстием под током азота проявляется эндотермический эффект с экстраполированной начальной температурой около 169шC, температурой пика около 170шC и соответственным тепловым эффектом около 142 Дж/г, за которым следует экзотермический эффект в интервале 180-280шC, и/или порошковой дифракционной картиной, характеризуемой пиками со значениями d 12,0, 11,1, 8,8, 7,2, 6,8, 6,6, 6,5, 6,4, 6,0, 5,8, 5,6, 5,3, 5,2, 4,75, 4,52, 4,39, 4,31, 4,29, 4,25, 4,06, 4,00, 3,82, 3,73, 3,71, 3,69, 3,66, 3,59, 3,55, 3,41, 3,37, 3,34, 3,29, 3,25, 3,22, 3,10, 3,03, 3,00, 2,91, 2,78, 2,73, 2,62, 2,51, 2,46, 2,40, 2,38, 2,34, 2,29, 2,26 А, и/или как представлено на фиг. 5.
7. Соединение по п.1, которое находится в форме моногидрата.
8. Соединение по п.7, которое находится не в форме соли.
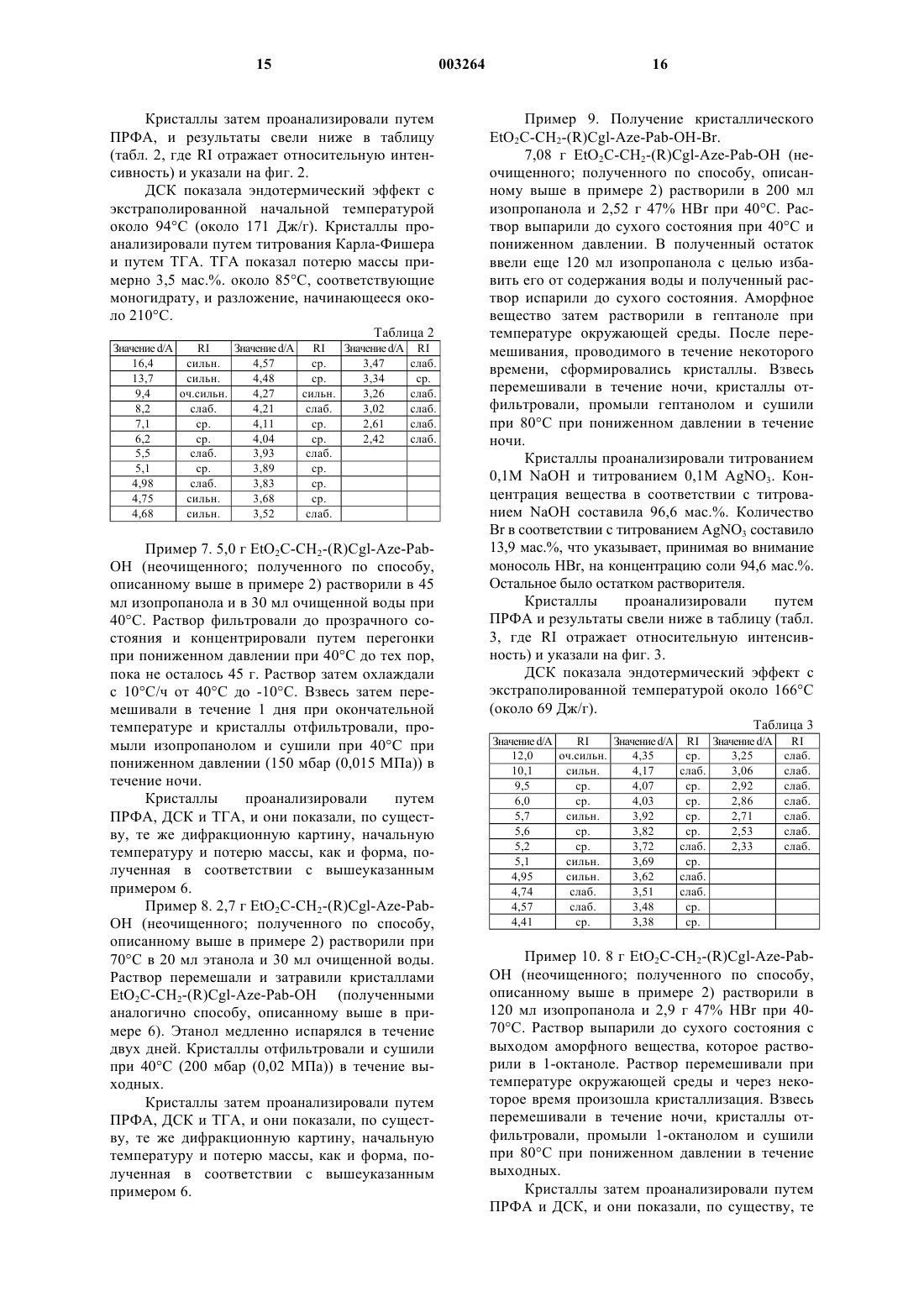
9. Соединение по п.7 или 8, характеризуемое кривой дифференциальной сканирующей калориметрии, на которой при скорости нагрева 5шC/мин в закрытой чашке с микроотверстием под током азота проявляется эндотермический эффект с экстраполированной начальной температурой около 94шC, температурой пика около 109шC и соответственным тепловым эффектом около 171 Дж/г, за которым следует экзотермический эффект в интервале 170-290шC, и/или порошковой дифракционной картиной, характеризуемой пиками со значениями d 16,4, 13,7, 9,4, 8,2, 7,1, 6,2, 5,5, 5,1, 4,98, 4,75, 4,68, 4,57, 4,48, 4,27, 4,21, 4,11, 4,04, 3,93, 3,89, 3,83, 3,68, 3,52, 3,47, 3,34, 3,26, 3,02, 2,61 и 2,42 А, и/или как представлено на фиг. 2.
10. Соединение по любому из пп.1, 2 или 7, которое находится в форме соли гидробромида.
11. Соединение по п.10, характеризуемое кривой дифференциальной сканирующей калориметрии, на которой при скорости нагрева 5шC/мин в закрытой чашке с микроотверстием под током азота проявляется эндотермический эффект с экстраполированной начальной температурой около 166шC, температурой пика около 167шC и соответственным тепловым эффектом около 69 Дж/г, за которым следует экзотермический эффект в интервале 170-220шC, и/или порошковой дифракционной картиной, характеризуемой пиками со значениями d 12,0, 10,1, 9,5, 6,0, 5,7, 5,6, 5,2, 5,1, 4,95, 4,74, 4,57, 4,41, 4,35, 4,17, 4,07, 4,03, 3,92, 3,82, 3,72, 3,69, 3,62, 3,51, 3,48, 3,38, 3,25, 3,06, 2,92, 2,86, 2,71, 2,53 и 2,33 А, и/или как представлено на фиг. 3.
12. Соединение по любому из пп.1, 2 или 7, которое находится в форме соли метансульфоната.
13. Соединение по п.12, характеризуемое кривой дифференциальной сканирующей калориметрии, на которой при скорости нагрева 5шC/мин в закрытой чашке с микроотверстием под током азота проявляется эндотермический эффект с экстраполированной начальной температурой около 134шC, температурой пика около 137шC и соответственным тепловым эффектом около 93 Дж/г, за которым следует экзотермический эффект в интервале 140-220шC, и/или порошковой дифракционной картиной, характеризуемой пиками со значениями d 12,0,11,7,10,4, 10,2, 8,3, 7,8, 6,0, 5,6, 5,5, 5,2, 5,1, 5,0, 4,98, 4,90, 4,75, 4,63, 4,54, 4,46, 4,15, 4,06, 3,92, 3,84, 3,74, 3,65, 3,56, 3,47, 3,39, 3,22, 3,12, 2,95, 2,88, 2,76, 2,74, 2,69, 2,65, 2,54, 2,52, 2,49, 2,27, 2,21, 2,04 и 2,02 А, и/или как проиллюстрировано на фиг. 4.
14. Способ получения соединения по любому из пп.1-13, включающий кристаллизацию EtO2C-CH2-(R)Cgl-Aze-Pab-OH или его фармацевтически приемлемой соли из растворителя, выбранного из группы, включающей ацетаты, низшие алкиловые спирты, алифатические и ароматические углеводороды, простые диалкиловые эфиры, диалкилкетоны, ацетонитрил, водные растворители или их смеси.
15. Способ по п.14, в котором растворитель выбирают из группы, включающей C1-6 алкилацетаты, линейные или разветвленные C1-6 алкиловые спирты, C6-12 алифатические углеводороды, C6-10 ароматические углеводороды, простые ди-C1-6 алкиловые эфиры, ди-C1-6 алкилкетоны, ацетонитрил, воду или их смеси.
16. Способ по п.15, в котором растворитель выбирают из группы, включающей этилацетат, бутилацетат, этанол, изопропанол, изооктан, н-гептан, толуол, простой диизопропиловый эфир, ацетон, метилизобутилкетон, ацетонитрил, воду или их смеси.
17. Способ получения соединения по любому из пп.2-6, включающий способ по любому из пп.14-16, в котором растворитель, по существу, не содержит воды.
18. Способ получения соединения по любому из пп.2-4 или 6, включающий способ по любому из пп.14-16, в котором растворитель содержит воду.
19. Способ получения соединения по любому из пп.7-9, включающий способ по любому из пп.14-16, в котором растворитель содержит воду.
20. Соединение, которое можно получить по способу согласно любому из пп.14-19.
21. Соединение по любому из пп.1-13 или 20 для применения в качестве фармацевтического препарата.
22. Соединение по любому из пп.1-13 или 20 для применения в качестве пролекарства.
23. Фармацевтический препарат, включающий соединение по любому из пп.1-13 или 20, в смеси с фармацевтически приемлемыми вспомогательным средством, разбавителем или носителем.
24. Применение соединения по любому из пп.1-13 или 20 для производства лекарственного средства для лечения состояния, при котором требуется или желательно ингибирование тромбина.
25. Способ лечения состояния, при котором требуется или желательно ингибирование тромбина, при котором пациенту, нуждающемуся в таком лечении, вводят терапевтически эффективное количество соединения по любому из пп.1-13 или 20.
Текст
1 Данное изобретение относится к новым твердым формам лекарства, к содержащим их фармацевтическим композициям и способам их получения. При разработке рецептур лекарственных композиций важно, чтобы лекарственное вещество было в форме, удобной для обращения и обработки. Это важно не только с точки зрения получения коммерчески жизнеспособного способа производства, но также и с точки зрения последующего производства фармацевтических препаратов (например, форм пероральной дозировки, такой как таблетки), включающих активное соединение. Далее при производстве пероральных лекарственных композиций важно обеспечение надежного, воспроизводимого и постоянного профиля концентрации лекарства в плазме после его введения пациенту. Это особенно важно при производстве композиций, включающих агенты противотромботического действия. Химическая стабильность, стабильность твердого состояния и "хранение" активных ингредиентов также являются очень важными факторами. Лекарственное вещество и содержащие его композиции должны быть приспособлены к эффективному хранению в течение значительных периодов времени, не претерпевая ощутимых изменений физико-химических характеристик активной компоненты (например,ее химического состава, плотности, гигроскопичности и растворимости). Кроме того, важно также иметь возможность предоставить лекарство в такой форме,которая настолько химически чиста, насколько возможно. Аморфные материалы могут представлять значительные проблемы в этом отношении. Например, с такими материалами обычно сложно общаться и составлять из них рецептуры, так как они имеют ненадежную растворимость и обычно оказываются нестабильными и химически загрязненными. Специалисту будет понятно, что если лекарство легко получить в стабильной кристаллической форме, то вышеописанные проблемы могут быть решены. Таким образом, при производстве жизнеспособных с коммерческой точки зрения и фармацевтически приемлемых лекарственных композиций важно при любой возможности предоставлять лекарство, по существу, в кристаллической и стабильной форме. Нужно заметить, однако, что эта цель не всегда достижима. Действительно, обычно невозможно предсказать, исходя только из молекулярной структуры, каким будет кристаллизационное поведение соединения либо как такового, либо в форме соли. Это можно определить только эмпирически. В международной заявке на патент WO 97/23499 описан ряд соединений, которые, как 2 было обнаружено, полезны как пролекарства ингибиторы тромбина, где ингибиторы тромбина имеют общую формулуRaO2C-CH2-(R)Cgl-Aze-Pab-H,a где R представляет Н, бензил или C1-6 алкил,Cgl представляет циклогексилглицин,Aze представляет S-азетидин-2-карбоновую кислоту, а Раb-Н представляет 4-аминометиламидинбензол. Сами активные ингибиторы тромбина описаны в более ранней международной заявке на патент WO 94/29336.WO 97/23499 также содержит особое описание соединенияEtO2C-CH2-(R)Cgl-Aze-Pab-OH,где Pab-OH представляет 4-аминометилбензоламидоксим. Способ получения данного соединения описан в примере 17 WO 97/23499, где соединение очищают путем препаративнойRPLC (жидкостной хроматографии с обратимой фазой) и выделяют в аморфной форме. В WO 97/23499 не описано, возможно ли представить EtO2C-CH2-(R)Cgl-Aze-Pab-OH в кристаллической форме. Кроме того, не предоставлено никакой информации насчет того, как можно получить данное соединение в такой форме. С удивлением мы обнаружили, что EtO2CCH2-(R)Cgl-Aze-Pab-OH можно получить в одной или более формах, которые по своей природе являются, по существу, кристаллическими. Таким образом, согласно первому аспекту изобретения, предложен EtO2C-CH2-(R)Cgl-AzePab-OH или его фармацевтически приемлемая соль, по существу, в кристаллической форме(здесь далее называемые "соединения по изобретению"). Несмотря на то, что мы обнаружили, что возможно получить ЕtO2 С-СН 2-(RCgl-Aze-PabOH и его соли в формах, которые более чем на 80% кристаллические, под "по существу, кристаллическими" мы понимаем более 10% (например, более 20%), предпочтительно более 30% и более предпочтительно более 40% кристалличности. Степень кристалличности (%) специалист может определить с использованием порошкового рентгенофазового анализа(ПРФА/XRPD). Можно также использовать другие технологии, такие как ЯMP (NMR) твердых веществ, ИК-спектроскопия с преобразованием Фурье (FT-IR), спектроскопия комбинационного рассеяния, дифференциальная сканирующая калориметрия (ДСК/DSC) и микрокалориметрия. Подходящие, фармацевтически приемлемые соли, которые можно упомянуть, включают неорганические и органические, кислотные и основные соли присоединения, такие как водородные галоидные соли (например, соли НВr),соли карбоновой кислоты, низшие алкансульфонатные соли (например, линейный или раз 3 ветвленный C1-6 алкансульфонат, предпочтительно C1-3 алкансульфонат и особенно этан- и метансульфонатные соли) и соли аммония и амина. Можно также упомянуть толуолсульфонатные соли. Для полного списка солей, которые можно упомянуть, см. Berge и др., J. Pharm.Sci., 66, 1 (1977). Однако мы предпочитаем,чтобы соединения по изобретению не были в форме соли. Соединения по изобретению могут быть в форме сольвата (сюда мы включаем и гидраты) или в другом подобном виде. Мы с удивлением обнаружили, что соединения по изобретению имеют улучшенную стабильность по сравнению с EtO2C-CH2-(R)CglAze-Pab-OH, полученным, как описано в WO 97/23499. Согласно следующему аспекту изобретения, предложена, таким образом, стабильная форма EtO2C-CH2-(R)Cgl-Aze-Pab-OH или его фармацевтически приемлемой соли. Термин "стабильность", как он здесь определен, включает химическую стабильность и стабильность твердого состояния. Под "химической стабильностью" мы понимаем, что соединение или соль можно хранить в изолированной форме или в форме препарата, в котором оно представлено в смеси с фармацевтически приемлемыми носителями,разбавителями или вспомогательными средствами (например, в форме пероральной дозировки, такой как таблетка, капсула и т.д.), при нормальных условиях хранения, с незначительной степенью химической деградации или разложения. Под "стабильностью твердого состояния" мы понимаем, что соединение или соль можно хранить в изолированной твердой форме или в форме твердого препарата, в котором оно представлено в смеси с фармацевтически приемлемыми носителями, разбавителями или вспомогательными средствами (например, в форме пероральной дозировки, такой как таблетка, капсула и т.д.), при нормальных условиях хранения с незначительной степенью изменения твердого состояния (например, кристаллизации, перекристаллизации, фазовых переходов в твердом состоянии, гидратации, дегидратации, сольватации и десольватации). Примеры "нормальных условий хранения" включают температуру между -80 и +50 С(предпочтительно между 0 и 40 С и более предпочтительно комнатную температуру от 15 до 30 С), давление между 0,1 и 2 бар (0,01 и 0,2 МПа) (предпочтительно атмосферное давление),относительную влажность между 5 и 95%(предпочтительно от 10 до 75%) и/или освещение 460 люкс УФ/видимого света в течение длительных периодов времени (т.е. выше или равных шести месяцам). Можно обнаружить, что при таких условиях соединения по изобретению химически деградированы/разложены или 4 трансформированы в твердой фазе менее чем на 15%, более предпочтительно менее чем на 10% и особенно менее чем на 5%, по обстановке. Специалисту будет понятно, что вышеуказанные верхние и нижние границы температуры,давления и относительной влажности представляют собой крайние значения нормальных условий хранения и что определенные комбинации этих крайних значений не будут осуществляться при нормальном хранении (например, температура 50 С и давление 0,1 бар (0,01 МПа. Соединения по изобретению можно с успехом получать путем кристаллизации EtO2CCH2-(R)Cgl-Aze-Pab-OH или соли EtO2C-CH2(R)Cgl-Aze-Pab-OH. В соответствии со следующим аспектом изобретения предложен способ получения соединения по изобретению, включающий кристаллизацию ЕtO2 С-CH2-(R)Cgl-Aze-Pab-OH или его фармацевтически приемлемой соли. Можно кристаллизоватьEtO2C-CH2(R)Cgl-Aze-Pab-OH и его фармацевтически приемлемые соли в присутствии системы растворителя или в ее отсутствие (например, кристаллизация может проходить из расплава, при сверхкритических условиях или может быть достигнута путем сублимации). Однако мы предпочитаем, чтобы кристаллизация проходила из соответствующей системы растворителя. Мы обнаружили, что можно с успехом кристаллизовать EtO2C-CH2-(R)Cgl-Aze-Pab-OH и его фармацевтически приемлемые соли путем кристаллизации, следующей за растворением соединения или рассматриваемой соли, или более предпочтительно из растворов, экстрагированных из реакционных растворов, в которых образовалось рассматриваемое соединение или соль или особо предпочтительно из реакционных растворов, в пределах которых образовались рассматриваемые соединение или соль. Система растворителя может быть гетерогенной или гомогенной и может, таким образом,включать один или более органический растворитель, такой как алкилацетаты (например, линейные или разветвленные C1-6 алкилацетаты,такие как этилацетат, изопропилацетат и бутилацетат), низшие (например, линейные или разветвленные C1-6) алкиловые спирты (например,этанол, изопропанол), алифатичекие (например,C6-12, такие как C7-12, алифатические) углеводороды (например, изооктан и н-гептан) и ароматические углеводороды (например толуол), диалкилкетоны (например, ацетон, метилизобутилкетон), ацетонитрил и простые диалкиловые эфиры (например, простой диизопропиловый эфир) и/или водные растворители, такие как вода. Можно использовать смеси любых вышеуказанных растворителей. Различные кристаллические формы могут иметь различную растворимость в различных органических растворителях при любой данной температуре. В этом отношении вышеуказанные 5 растворители можно использовать как "антирастворители" (т.е. растворитель, в котором соединения по изобретению слабо растворимы) и таким образом можно способствовать процессу кристаллизации. Если кристаллизация происходит из реакционного растворителя, в котором сформировался EtO2C-CH2-(R)Cgl-Aze-Pab-OH или его соль, или из растворителя после экстракции, то подходящие растворители, таким образом,включают алкилацетаты (такие как этилацетат),толуол, метилизобутилкетон, низшие алкиловые спирты (такие как этанол) и так далее. Кристаллизация соединений по изобретению из соответствующей системы растворителя может быть достигнута путем получения перенасыщенности в системе растворителя, включающей EtO2C-CH2-(R)Cgl-Aze-Pab-OH или его соль (например, путем охлаждения, путем испарения растворителя и/или путем добавления антирастворителя (т.е. растворителя, в котором соединения по изобретению слабо растворимы(например, изооктана, н-гептана, диизопропилового простого эфира, толуола, ацетона, или путем уменьшения растворимости вещества путем добавления соли (такой как NaCl или триэтиламин НСl). Температура кристаллизации и время кристаллизации зависят от концентрации соединения в растворе и от используемой системы растворителя. Кристаллизацию также можно инициировать и/или воздействовать на нее путем затравливания или без затравливания кристаллами соответствующего кристаллического соединения по изобретению и/или путем регулирования рН. Соединения по изобретению можно получить в форме сольвата (под этим мы также понимаем форму гидрата, такого как моногидрат) или в какой-либо другой форме (например, в безводной форме). Термин "безводная форма" в данном контексте также включает "несольватированную форму". Чтобы обеспечить получение безводной формы, растворитель, из которого происходит кристаллизация, предпочтительно либо до, либо после процесса кристаллизации, должен быть высушен с целью уменьшения содержания воды до уровня ниже критического, который предпочтительно не превышать во время кристаллизации. Растворитель можно сушить во время процесса кристаллизации, например, путем уменьшения содержания воды в смеси соединения, предназначенного к кристаллизации, и соответствующей системы органический растворитель/водный растворитель (например путем увеличения количества присутствующего органического растворителя и/или удаления воды путем образования азеотропа с последующей перегонкой). Несмотря на это, мы обнаружили,что для обеспечения формирования определен 003264 6 ных соединений по изобретению, находящихся в безводной форме, такая сушка растворителя не является необходимой. Чтобы обеспечить получение моногидрата,в растворителе, из которого происходит кристаллизация, должна присутствовать вода. Содержание воды во время кристаллизации нужно поддерживать предпочтительно выше критического уровня, указанного выше."Критический уровень" воды зависит от таких факторов как температура, концентрация в растворе соединения, предназначенного для кристаллизации, профиля примесей и используемой системы растворителя, но может быть определен не в рамках изобретения. Таким образом, в соответствии со следующим аспектом изобретения, предложено соединение по изобретению в безводной форме и соединение по изобретению в форме моногидрата. Кристаллическую безводную форму можно получить путем кристаллизации EtO2C-CH2(R)Cgl-Aze-Pab-OH из одного или более органических растворителей (таких как этилацетат,бутилацетат, ацетон, этанол, изопропанол, изооктан, простой диизопропиловый эфир), воды или их смесей, которые могут быть уже высушены, и/или их можно сушить во время процесса кристаллизации так, чтобы содержание воды было ниже вышеуказанного критического уровня. Таким образом, безводную форму можно получить путем кристаллизации из системы растворителя, являющейся, по существу, безводной. Под "по существу безводной" мы понимаем, что содержание воды в системе растворителя ниже того, которое бы привело к формированию самое большое 10% моногидрата для любой данной системы растворителя и набора условий кристаллизации. Наоборот, кристаллический моногидрат можно получить путем кристаллизации EtO2CCH2-(R)Cgl-Aze-Pab-OH из системы растворителя, включающей воду или смесь воды с одним или более органическим растворителем, включая органические растворители, способные к растворению воды (например, этилацетат, этанол, изопропанол). Кристаллический моногидрат можно также получить путем растворения вещества в водном растворе (например, воде или в смесях воды со спиртом, таким как этанол или изопропанол) с низким рН с последующим добавлением слабого основания до тех пор, пока рН не превысит(предпочтительно как раз превысит) значение приблизительно от 5 до 6 (при комнатной температуре). Кристаллический моногидрат можно также получить через другие кристаллические формы(такие как безводная форма). Этого достигают,если критическое содержание воды, о котором говорится выше, превышено во время кристал 7 лизации. Подобным образом можно получить кристаллическую безводную форму из кристаллического моногидрата путем понижения содержания воды во время процесса кристаллизации до уровня ниже вышеуказанного критического. Кристаллизуется ли безводная форма или моногидрат, это связано с кинетикой и условиям равновесия соответствующих форм в определенных условиях. Таким образом, как может понять специалист, получаемая кристаллическая форма зависит и от кинетики и от термодинамики способа кристаллизации. При определенных термодинамических условиях (система растворителя, температура, давление и концентрация соединения по изобретению) одна кристаллическая форма может быть более стабильна, чем другая (или в действительности любая другая). Однако кристаллические формы, имеющие относительно низкую термодинамическую стабильность, могут быть благоприятными с точки зрения кинетики. Таким образом, кроме того, и кинетические факторы, такие как время, профиль примесей, перемешивание, наличие или отсутствие затравок и т.д., также могут влиять на то, какие появляются формы. Таким образом,обсуждаемые здесь способы специалист может принять как соответствующие целям получения различных кристаллических форм. Кристаллический моногидрат также можно получать путем вымывания других кристаллических форм (например, безводной формы) в воде или смеси воды с одним или несколькими органическими растворителями (такими как этанол или изопропанол). Полученную взвесь предпочтительно следует затравить кристаллами кристаллического моногидрата с целью обеспечения того, что произойдет соответствующее превращение. Как и ранее, существует критическое содержание воды, зависящее от используемой системы растворителя и температуры. Таким образом, мы обнаружили, что соединения по изобретению одной кристаллической формы могут быть превращены посредством перекристаллизации в другие кристаллические формы. В соответствии со следующим аспектом изобретения, предложен способ превращения одной кристаллической формы соединения по изобретению в другую, включающий перекристаллизацию соединения по изобретению из соответствующей системы растворителя. Соединения по изобретению, находящиеся в безводной форме, содержат не более 3%,предпочтительно 2%, более предпочтительно 1% и более предпочтительно 0,5 маc.% воды,независимо от того, связана эта вода или нет(кристаллизационная вода или любая другая). Гидраты содержат не менее 0,5 моль воды на моль EtO2C-CH2-(R)Cgl-Aze-Pab-OH. 8 Предпочтительными соединениями по изобретению являются те, которые находятся в безводной форме. В соответствии со следующим аспектом изобретения, предложено соединение по изобретению, содержащее не более 3 маc.%, предпочтительно не более 2 маc.% воды,независимо от того, связана эта вода, или нет(кристаллизационная вода или любая другая). Если соединение по изобретению нужно получить в форме кислотной или основной соли присоединения, то к кристаллизационной смеси до начала кристаллизации можно добавить соответствующее количество соответствующей кислоты или основания. Альтернативно, после добавления соли можно выпарить весь растворитель(и) и полученную кристаллическую и/или аморфную форму соли можно повторно растворить в соответствующей системе растворителя,такой как этилформиат или алкиловый спирт,такой как н-гептанол или н-октанол с последующим перемешиванием полученного раствора с целью стимулирования кристаллизации. Предпочтительные соли присоединения включают кислотные соли присоединения, такие как соли гидробромида и метансульфоната. Между прочим, далее описано получение и характеристика безводной и моногидратной форм соединений по изобретению. Различные кристаллические формы соединений по изобретению (например, безводные формы и моногидраты) можно легко охарактеризовать с использованием методов порошкового рентгенофазового анализа (XRPD/ПРФА), например, как описано здесь далее. С целью обеспечения получения кристаллических форм, как здесь описано, без других кристаллических форм, описанных здесь, кристаллизацию предпочтительно проводят путем затравливания ядрами и/или затравочными кристаллами требуемой кристаллической формы при полном отсутствии ядер и/или затравочных кристаллов других кристаллических форм, здесь описанных. Это относится практически к каждой определенной кристаллической форме, получение которой описано здесь далее в примерах. Соединения по изобретению можно выделить, применяя технологии, хорошо известные специалистам, например, декантацией, фильтрацией или центрифугированием. Соединения можно сушить, используя стандартные технологии. Специалисту будет понятно, что температура сушки и время сушки могут повлиять на свойства твердого состояния соединений (или солей), находящихся в форме сольватов, таких как гидраты (например, дегидратацию можно проводить при повышенных температурах и/или пониженном давлении). К примеру, при формировании кристаллического моногидрата существует критическая влажность, ниже которой не следует проводить сушку, так как может быть потеряна кристаллиза 9 ционная вода и может произойти превращение в твердой фазе, т.е. кристаллизационная вода будет потеряна, если кристаллы сушить при высоких температурах или при очень низких давлениях в течение более длительного периода времени. Мы обнаружили, что применяя способ кристаллизации, как здесь описано, можно получать соединения по изобретению с химической чистотой выше, чем у EtO2C-CH2-(R)CglAze-Pab-OH или соли, которая будет выделена сначала. Дальнейшую очистку соединений по изобретению можно проводить с использованием технологий, хорошо известных специалистам в данной области. К примеру, примеси можно удалить путем перекристаллизации из соответствующей системы растворителя (например,этилацетата, изопропанола, изооктана, этанола,воды или смеси этих растворителей). Подходящие температуры и время кристаллизации зависят от концентрации соединения или соли в растворе и от используемой системы растворителя. После кристаллизации или перекристаллизации соединений по изобретению, как указано выше, полученное соединение или соль находится в форме, имеющей указанные здесь ранее повышенные химическую стабильность и стабильность твердого состояния. Фармацевтическое приготовление и медицинское применение В соответствии с изобретением соединения по изобретению можно вводить перорально,внутривенно, подкожно, трансбуккально, ректально, дермально, интраназально, трахеально,бронхиально, любым другим парентеральным путем или путем ингаляции, в форме фармацевтического средства, включающего соединение по изобретению в фармацевтически приемлемой лекарственной форме. Однако мы предпочитаем, чтобы соединение по изобретению было в такой форме, в какой оно подходит для перорального введения. В зависимости от расстройства и пациента,подлежащего лечению, а также от способа введения, соединения можно назначать в различных дозах (смотри ниже). Соединения по изобретению можно дополнительно обрабатывать перед составлением соответствующего фармацевтического препарата. Например, кристаллическую форму можно молоть или измельчать на частицы меньшего размера. В соответствии со следующим аспектом изобретения, предложен фармацевтический препарат, включающий соединение по изобретению в смеси с фармацевтически приемлемым вспомогательным средством, разбавителем или носителем. Количество соединения по изобретению,используемого в таком препарате, будет зависеть от состояния и пациента, подлежащего ле 003264 10 чению, а также от применяемого соединения(ий), но может быть определено не в рамках изобретения. Соединения по изобретению полезны, так как они участвуют в обмене веществ человека после введения с образованием соединений,обладающих фармацевтической активностью. Они, таким образом, указаны как фармацевтические и, в частности, как пролекарства из формакологически-активных соединений. В частности, соединения по изобретению,несмотря на то, что они не воздействуют на тромбин сам по себе, они участвуют в обмене веществ человека с образованием сильнодействующих ингибиторов тромбина, например, как указано в тестах, описанных в WO 97/23499. Предполагается, что соединения по изобретению будут полезны при лечении состояний, при которых необходимо или желательно ингибирование тромбина, включая состояния, описанные в WO 97/23499, на описание которого в данном документе имеется ссылка. Подходящими дозами соединения по изобретению при терапевтическом лечении и/или профилактике млекопитающих, в частности людей, является назначение пациентам в интервале от 2 до 200 мг в день при пероральном введении и от 1 до 100 мг в день при парентеральном введении, и/или от 0,001 до 20 мг/кг, предпочтительно от 0,01 до 5 мг/кг веса тела в день при пероральном введении и от 0,0005 до 10 мг/кг,предпочтительно от 0,005 до 2,5 мг/кг веса тела при парентеральном введении. В соответствии со следующим аспектом изобретения предложен способ лечения состояния, при котором необходимо или желательно ингибирование тромбина, при котором пациенту, нуждающемуся в таком лечении, вводят терапевтически эффективное количество соединения по изобретению. Чтобы избежать сомнений, под "лечением" мы понимаем терапевтическое лечение, а также профилактику состояний. Соединения по изобретению имеют то преимущество, что они находятся в форме, предусматривающей более удобное обращение. Далее, соединения по изобретению имеют то преимущество, что их можно произвести в формах, имеющих повышенную химическую стабильность и стабильность твердого состояния(включая более низкую гигроскопичность). Таким образом, соединения могут быть стабильны при хранении в течение длительных периодов времени. Соединения по изобретению могут также иметь то преимущество, что их можно кристаллизовать с хорошим выходом, более высокой чистотой, меньшим временем, более удобным способом и по более низкой цене, чем формы 11 Изобретение проиллюстрировано, но не ограничено следующими примерами со ссылкой на прилагаемые чертежи, где на фиг. 1 показана порошковая рентгеновская дифрактограмма кристаллической формыEtO2C-CH2-(R)Cgl-Aze-Pab-OH,полученной способом по примеру 1; на фиг. 2 - порошковая рентгеновская дифрактограмма кристаллической формы моногидрата EtO2C-CH2-(R)Cgl-Aze-Pab-OH, полученного способом по примеру 6; на фиг. 3 - порошковая рентгеновская дифрактограмма кристаллической формы гидробромида EtO2C-CH2-(R)Cgl-Aze-Pab-OH, полученного способом по примеру 9; на фиг. 4 - порошковая рентгеновская дифрактограмма кристаллической формы метансульфоната EtO2C-CH2-(R)Cgl-Aze-Pab-OH, полученного способом по примеру 12; на фиг. 5 - порошковая рентгеновская дифрактограмма кристаллической формы EtO2CCH2-(R)Cgl-Aze-Pab-OH, полученной способом по примеру 14. Общие методики Порошковый ренгенофазовый анализ(ПРФА/XRPD) проводили на образцах, полученных по стандартным способам, например описанным в Giacovazzo, С. и др. (1995), Fundamentals of Crystallography, Oxford UniversityYork. Рентгеновский анализ проводили с использованием дифрактометра Siemens D5000 и/или Philips X'pert MPD. Дифференциальную сканирующую калориметрию (ДСК/DSC) проводили с использованием прибора Mettler DSC820 по стандартным способам, например описанным в Нhne,G.W.H. и др. (1996), Differential Scanning Calorimetry, Springer, Berlin. Термогравиметрический анализ (ТГА/Mettler Toledo TGA850. Формы, полученные в соответствии с нижеуказанными примерами показали "по существу" те же дифракционные картины ПРФА и/или ДСК, и/или термограммы ТГА, как и в других примерах, описанных ниже, когда из соответствующих картин/термограмм (с учетом экспериментальной ошибки) ясно, что сформирована та же кристаллическая форма. Таким образом, начальные температуры ДСК могут варьироваться в интервале 5 С (например, 2 С), а значения межплоскостных расстояний по ПРФА могут варьироваться в интервале 2 в последнем десятичном знаке.(аморфного; полученного путем сублимационной сушки продукта, полученного по способу,описанному в примере 17 международной заявки на патент WO 97/23499) растворили в смеси простой диизопропиловый эфир:изопропанол(14 мл; 1:1). Сначала сформировали гомогенный раствор, который затем оставили на 24 ч при комнатной температуре. Фильтрация и сушка(при температуре окружающей среды) дали 160 мг кристаллического продукта. Кристаллы проанализировали путем ПРФА и результаты свели ниже в таблицу (табл. 1, где RI отражает относительную интенсивность) и указали на фиг. 1. Элементарную ячейку определили из данных рентгенографии монокристалла. Она была триклинной, с симметрией Р 1, Z=1 и со следующими параметрами: а=5,149(1) А,b=10,466(1) А, с=12,317(1) А, =80,32(1),=79,78(1), =75,57(1), и V=627,2(2) A3. ДСК показала эндотермический эффект с экстраполированной начальной температурой около 150 С (около 113 Дж/г). ТГА показал уменьшение массы примерно на 0,6 маc.%, около 150 С. Таблица 1 Значение d/AEtO2C-CH2-(R)Cgl-Aze-Pab-OH из экстрагированного раствора. 70 мл этилацетата загрузили в 140,3 г экстрагированного раствора EtO2C-CH2-(R)Сgl-АzеРаb-ОН (полученного путем синтеза, аналогичного способу, описанному в WO 97/23499 с использованием этанола в качестве растворителя,гашением ацетоном, концентрированием полученного раствора и экстрагированием этилацетатом). Раствор, полученный таким образом,включал этилацетат, этанол и воду (8,7 маc.%). Раствор концентрировали и сушили путем последовательной перегонки, проведенной при 250 мбар (0,025 МПа) и температуре бани 60 С. 70 мл этилацета загрузили в первоначальный раствор, который концентрировали до тех пор,пока не осталось 46 г раствора. Затем 103 мл этилацетата загрузили в раствор, который снова концентрировали до тех пор, пока не осталось 70,5 г. Добавили затравки из кристаллов, полученных аналогично способу, описанному в при 13 мере 3, и полученную суспензию перемешивали в течение 3 ч при 40 С, охлаждали до 20 С в течение 5 ч, затем до 5 С в течение 2,5 ч и, наконец, до -5 С в течение 1 ч. Взвесь затем перемешивали при конечной температуре в течение ночи и кристаллы отфильтровали, промыли этилацетатом и сушили при 40 С при пониженном давлении в течение ночи. Кристаллы проанализировали путем ПРФА, ДСК и ТГА, и они показали, по существу, те же дифракционную картину, начальную температуру и потерю массы, как и форма, полученная в соответствии с вышеуказанным примером 1. Пример 3. Перекристаллизация EtO2CCH2-(R)Cgl-Aze-Pab-OH. 3,0 г EtO2C-CH2-(R)Cgl-Aze-Pab-OH (неочищенного; полученного по способу, описанному выше в примере 2) растворили в 49,5 мл изопропанола и 4,5 мл очищенной воды при 60 С. Раствор концентрировали и сушили путем последовательной перегонки, проводимой при 100 мбар (0,01 МПа) и температуре бани 60 С. Раствор сначала концентрировали до тех пор,пока не осталось 17,5 г. Затем в полученный продукт загрузили 30 мл изопропанола и раствор концентрировали до тех пор, пока не осталось 20 г. В раствор загрузили 1,8 г изопропанола и затравочные кристаллы EtO2C-CH2-(R)СglАzе-Pab-OH (полученного по способу, аналогичному способу, описанному ниже в примере 5). Суспензию перемешивали в течение 3 ч при 40 С, а затем охлаждали до 20 С в течение 5 ч,до 5 С в течение 2,5 ч и до -5 С в течение 1 ч. Взвесь затем перемешивали при конечной температуре в течение ночи. Кристаллы отфильтровали, промыли изопропанолом и сушили при пониженном давлении в течение ночи. Кристаллы проанализировали путем ПРФА, ДСК и ТГА, и они показали, по существу, те же дифракционную картину, начальную температуру и потерю массы, как и форма, полученная в соответствии с вышеуказанным примером 1. Пример 4. 4,0 г EtO2C-CH2-(R)Cgl-Aze-PabOH (неочищенного; полученного по способу,описанному выше в примере 2) растворили в 2 мл очищенной воды, 16 мл этанола и 45 мл этилацетата при 55 С. Раствор концентрировали и сушили путем последовательной перегонки,проводимой при 250 мбар (0,025 МПа) и температуре бани 55 С. Раствор сначала концентрировали, до тех пор, пока не осталось 30 г. Затем в полученный продукт загрузили 32 мл этилацетата, и раствор концентрировали до тех пор,пока не осталось 28 г. Добавили дополнительно 16 мл этилацетата и раствор концентрировали до тех пор, пока не осталось 33 г. К раствору добавили затравочные кристаллы ЕtO2 С-CH2(R)Cgl-Aze-Pab-OH (полученного по вышеуказанному примеру 3). Суспензию перемешивали в течение 3 ч при 40 С, а затем охлаждали до-5 С в течение 1 ч. Взвесь затем перемешивали при окончательной температуре в течение ночи. Кристаллы отфильтровали, промыли этилацетатом и сушили при 40 С при пониженном давлении в течение ночи. Кристаллы затем проанализировали путем ПРФА, ДСК и ТГА, и они показали, по существу, те же дифракционную картину, начальную температуру и потерю массы, как и форма, полученная в соответствии с вышеуказанным примером 1. Пример 5. 3,0 г EtO2C-CH2-(R)Cgl-Aze-PabOH (неочищенного; полученного по способу,описанному выше в примере 2) растворили в 4,5 мл очищенной воды и 49,5 мл изопропанола при 55-60 С. Раствор концентрировали и сушили путем последовательной перегонки, проводимой при 100 мбар (0,01 МПа) и температуре бани 60 С. Раствор сначала концентрировали до тех пор, пока не осталось 16 г. Раствор перемешивали при 40 С, затем в полученный продукт ввели 18 мл изооктана, и в раствор добавили затравочные кристаллы EtO2C-CH2-(R)Cgl-AzePab-OH (полученного по вышеуказанному примеру 3). Суспензию перемешивали в течение 3 ч при 40 С, а затем охлаждали до 15 С в течение 6 ч. Взвесь затем перемешивали при окончательной температуре в течение ночи. Кристаллы отфильтровали, промыли смесью изопропанол:изооктан (1:1) и сушили при 40 С при пониженном давлении в течение ночи. Кристаллы затем проанализировали путем ПРФА, ДСК и ТГА, и они показали, по существу, те же дифракционную картину, начальную температуру и потерю массы, как и форма, полученная в соответствии с вышеуказанным примером 1. Кристаллизация моногидрата EtO2C-CH2(R)Cgl-Aze-Pab-OH. Пример 6. 3,0 г EtO2C-CH2-(R)Cgl-Aze-PabOH (неочищенного; полученного по способу,описанному выше в примере 2) растворили в 1,5 мл очищенной воды, 10,5 мл этанола и 36 мл этилацетата при 55 С. Раствор концентрировали путем перегонки при 250 мбар (0,025 МПа) и температуре бани 55 С, до тех пор, пока не осталось 18 г. 30 мл этилацетата затем загрузили в раствор, который затем концентрировали до тех пор, пока не осталось 21 г. К раствору добавили 3,7 г этилацетата, 0,9 г очищенной воды и затравочные кристаллы EtO2C-CH2-(R)Cgl-Aze-PabOH (полученного по вышеуказанному примеру 3). Суспензию перемешивали в течение 3 ч при 40 С, а затем охлаждали до 20 С в течение 5 ч,до 5 С в течение 2,5 ч и до -5 С в течение 1 ч. Взвесь затем перемешивали при окончательной температуре в течение еще одного дня. Кристаллы отфильтровали, промыли этилацетатом и сушили при 40 С при пониженном давлении Кристаллы затем проанализировали путем ПРФА, и результаты свели ниже в таблицу(табл. 2, где RI отражает относительную интенсивность) и указали на фиг. 2. ДСК показала эндотермический эффект с экстраполированной начальной температурой около 94 С (около 171 Дж/г). Кристаллы проанализировали путем титрования Карла-Фишера и путем ТГА. ТГА показал потерю массы примерно 3,5 маc.%. около 85 С, соответствующие моногидрату, и разложение, начинающееся около 210 С. Таблица 2 Значение d/A Пример 7. 5,0 г EtO2C-CH2-(R)Cgl-Aze-PabOH (неочищенного; полученного по способу,описанному выше в примере 2) растворили в 45 мл изопропанола и в 30 мл очищенной воды при 40 С. Раствор фильтровали до прозрачного состояния и концентрировали путем перегонки при пониженном давлении при 40 С до тех пор,пока не осталось 45 г. Раствор затем охлаждали с 10 С/ч от 40 С до -10 С. Взвесь затем перемешивали в течение 1 дня при окончательной температуре и кристаллы отфильтровали, промыли изопропанолом и сушили при 40 С при пониженном давлении (150 мбар (0,015 МПа в течение ночи. Кристаллы проанализировали путем ПРФА, ДСК и ТГА, и они показали, по существу, те же дифракционную картину, начальную температуру и потерю массы, как и форма, полученная в соответствии с вышеуказанным примером 6. Пример 8. 2,7 г EtO2C-CH2-(R)Cgl-Aze-PabOH (неочищенного; полученного по способу,описанному выше в примере 2) растворили при 70 С в 20 мл этанола и 30 мл очищенной воды. Раствор перемешали и затравили кристалламиEtO2C-CH2-(R)Cgl-Aze-Pab-OH (полученными аналогично способу, описанному выше в примере 6). Этанол медленно испарялся в течение двух дней. Кристаллы отфильтровали и сушили при 40 С (200 мбар (0,02 МПа в течение выходных. Кристаллы затем проанализировали путем ПРФА, ДСК и ТГА, и они показали, по существу, те же дифракционную картину, начальную температуру и потерю массы, как и форма, полученная в соответствии с вышеуказанным примером 6.EtO2C-CH2-(R)Cgl-Aze-Pab-OH-Br. 7,08 г EtO2C-CH2-(R)Cgl-Aze-Pab-OH (неочищенного; полученного по способу, описанному выше в примере 2) растворили в 200 мл изопропанола и 2,52 г 47% НВr при 40 С. Раствор выпарили до сухого состояния при 40 С и пониженном давлении. В полученный остаток ввели еще 120 мл изопропанола с целью избавить его от содержания воды и полученный раствор испарили до сухого состояния. Аморфное вещество затем растворили в гептаноле при температуре окружающей среды. После перемешивания, проводимого в течение некоторого времени, сформировались кристаллы. Взвесь перемешивали в течение ночи, кристаллы отфильтровали, промыли гептанолом и сушили при 80 С при пониженном давлении в течение ночи. Кристаллы проанализировали титрованием 0,1 М NaOH и титрованием 0,1 М АgNО 3. Концентрация вещества в соответствии с титрованием NaOH составила 96,6 маc.%. Количество Вr в соответствии с титрованием АgNО 3 составило 13,9 маc.%, что указывает, принимая во внимание моносоль НВr, на концентрацию соли 94,6 маc.%. Остальное было остатком растворителя. Кристаллы проанализировали путем ПРФА и результаты свели ниже в таблицу (табл. 3, где RI отражает относительную интенсивность) и указали на фиг. 3. ДСК показала эндотермический эффект с экстраполированной температурой около 166 С Пример 10. 8 г EtO2C-CH2-(R)Cgl-Aze-PabOH (неочищенного; полученного по способу,описанному выше в примере 2) растворили в 120 мл изопропанола и 2,9 г 47% НВr при 4070 С. Раствор выпарили до сухого состояния с выходом аморфного вещества, которое растворили в 1-октаноле. Раствор перемешивали при температуре окружающей среды и через некоторое время произошла кристаллизация. Взвесь перемешивали в течение ночи, кристаллы отфильтровали, промыли 1-октанолом и сушили при 80 С при пониженном давлении в течение выходных. Кристаллы затем проанализировали путем ПРФА и ДСК, и они показали, по существу, те 17 же дифракционную картину и начальную температуру, как и форма, полученная в соответствии с вышеуказанным примером 9. Пример 11. 1 г EtO2C-CH2-(R)Cgl-Aze-PabOH (неочищенного; полученного по способу,описанному выше в примере 2) растворили в 22 мл изопропанола и 0,36 г 47% НВr при 45 С. Раствор фильтровали до прозрачного состояния и концентрировали путем перегонки при пониженном давлении до тех пор, пока не осталось 3,85 г. Раствор перемешали при 40 С и медленно добавили 6 мл толуола. Раствор затравили кристаллами соли НВг (полученной аналогично способу, описанному выше в примере 9). Когда суспензия стала белой, добавили еще 9 мл толуола. Взвесь перемешивали в течение ночи,кристаллы отфильтровали, промыли толуолом и сушили при 40 С при пониженном давлении. Кристаллы затем проанализировали путем ПРФА и ДСК, и они показали, по существу, те же дифракционную картину и начальную температуру, как и форма, полученная в соответствии с вышеуказанным примером 9. Пример 12. Получение кристаллическогоEtO2C-CH2-(R)Cgl-Aze-Pab-OCH3SO3H. 10 г EtO2C-CH2-(R)Cgl-Aze-Pab-OH (неочищенного; полученного по способу, описанному выше в примере 2) растворили в 500 мл изопропанола и 2,03 г метансульфоновой кислоты (1 экв.) при 50 С. Раствор выпарили до сухого состояния при 50 С при пониженном давлении с образованием аморфного твердого материала. 1,5 г этого вещества растворили в 15 мл этилформиата, и раствор перемешивали при температуре окружающей среды в течение ночи. Полученные кристаллы отфильтровали, промыли этилформиатом и сушили при 40 С при пониженном давлении в течение ночи. Кристаллы проанализировали путем титрования 0,1 МNaOH, путем двухфазного титрования. На основе предположения о формировании мономезилата была рассчитана концентрация (99,9%). Кристаллы проанализировали путем ПРФА, и результаты сведены ниже в таблицу(табл. 4, где RI отражает относительную интенсивность) и указаны на фиг. 4. ДСК показала эндотермический эффект с экстраполированной начальной температурой около 134 С (около 93 Дж/г). Пример 13. 3,0 г аморфного вещества из первой части вышеуказанного примера 12 растворили в 15 мл октанола. Раствор перемешивали при температуре окружающей среды в течение ночи. Полученные кристаллы отфильтровали, промыли октанолом и сушили при 40 С при пониженном давлении в течение ночи. Кристаллы проанализировали путем ПРФА и ДСК, и они показали, по существу, те же дифракционную картину и начальную температуру, как и форма, полученная в соответствии с вышеуказанным примером 12. Пример 14. Получение дополнительной кристаллической формы EtO2C-CH2-(R)Cgl-AzePab-OH из EtO2C-CH2-(R)Cgl-Aze-NН-СH2 С 6 Н 4-СN. 0,0029 г ЭДТК/EDTA растворили в 16,4 г водного гидроксиламина (50 маc.%) и 110 мл этанола, а затем загрузили в стеклянный реактор с рубашкой, который поддерживали при температуре 32 С. 118,5 г раствора глицина, N-[1 циклогексил-2-4-[циано]фенил]метил]амино] карбонил]-1-азетидинил]-2-оксоэтил-, сложного этилового эфира, (S-(R,S (EtO2C-CH2(R)Cgl-Aze-NH-СН 2-С 6 Н 4-СN) в бутилацетате(приблизительная концентрация 36 маc.%) загрузили в реактор. После 18-часового перемешивания раствор/суспензия стал прозрачным. Была взята проба для проверки превращения с использованием жидкостной хроматографии высокого разрешения (ЖХВР/HPLC). Реакцию погасили путем добавления 14,4 мл ацетона с последующим добавлением 307 мл бутилацетата. Температуру рубашки установили на 70 С и суспензию перемешивали в течение 1 ч при температуре 70 С для растворения осадка. Температуру термостатической бани затем установили на 40 С и раствор перемешивали дополнительно 78 ч. Частицы все еще присутствовали. Осадок отфильтровали. Фильтрат загрузили в стеклянный реактор с рубашкой и перемешивали при 40 С. Начали программу экстракции. Все экстракции проводили при 40 С. 110 млNА 2 СО 3/NаСl (10 маc.%/10 маc.%) загрузили в реактор, раствор перемешивали в течение 10-20 мин. Перемешивание затем прекратили, дав возможность фазам разделиться. Водную фазу удалили. Добавили 12,5 г этанола и дополнительные 110 мл водного раствора NА 2 СО 3/NаСl(10 маc.%/10 маc.%); раствор перемешивали в течение нескольких минут и затем оставили фазы разделяться. Водную фазу удалили и добавили 12,5 г этанола и 110 мл водного раствораNА 2 СО 3/NаСl (10 маc.%/10 маc.%). Полученный раствор перемешивали в течение нескольких минут и оставили две фазы разделяться. Водную фазу удалили. 4 г этанола и 31 мл воды добавили к органической фазе, которую перемешива 19 ли. РН привели к 7 путем добавления HCl (8 маc.%) Раствор перемешали, оставили две фазы разделяться и удалили водную фазу. 52,9 г экстракционного раствора, включающего около 4 гEtO2C-CH2-(R)Cgl-Aze-Pab-OH, загрузили в круглодонную колбу. 4 мл смеси 35:65 (по объему) бутилацетата и этанола загрузили в ту же колбу. Растворитель испаряли с использованием испарителя барабанного типа при давлении 100200 мбар (0,01-0,02 МПа) и температуре бани 73 С до тех пор, пока не осталось 53,1 г раствора. Затем добавили еще 4 мл вышеуказанной смеси растворителя и испаряли еще растворитель до тех пор, пока не осталось 54,74 г раствора. Эту процедуру повторили несколько раз: добавляли 4 мл, раствор концентрировали путем испарения до тех пор, пока не оставалось 56,2 г,добавляли 4 мл, раствор снова концентрировали до тех пор, пока не оставалось 56,04 г, загружали дополнительно 4 мл, снова концентрировали раствор, до тех пор, пока не оставалось 33,43 г. Взяли 2,2 г раствора для анализа состава раствора путем ГХ/GC и определения содержания воды путем титрования Карла-Фишера. Содержание воды составило 1,0 маc.%. 0,34 мл воды загрузили в колбу, которую поместили в баню термостата, поддерживаемую при 40 С. Раствор перемешивали с использованием тефлоновой мешалки в течение 3 ч и затем охлаждали до 20 С в течение 5 ч, до 5 С в течение 2,5 ч и до-5 С в течение 1 ч. Раствор затем перемешивали в течение 2 ч при -5 С. Раствор был все еще практически прозрачным. Раствор затем охлаждали до -20 С в течение 1,5 ч и перемешивали при той же температуре в течение еще 20 ч. Сформировалась очень вязкая белая взвесь. Кристаллы отфильтровали путем вакуумной фильтрации и затем сушили в течение ночи при 150 мбар ( 0,015 МПа) и 40 С. Кристаллы затем проанализировали путем ГХ, ПРФА, ТГА, ДСК и титрования КарлаФишера. Результаты ПРФА свели ниже в таблицу (табл. 5, где RI отражает относительную интенсивность) и указали на фиг. 5. Таблица 5 Значение d/A 12,0 11,1 8,8 7,2 6,8 6,6 6,5 6,4 6,0 5,8 5,6 5,3 5,2 4,75 4,52 4,39 20 ДСК показала эндотермический эффект с экстраполированной начальной температурой около 169 С (около 142 Дж/г). ТГА показал потерю массы около 0,7 маc.% около 170 С. Элементарную ячейку определили из данных по рентгенографии монокристалла. Она была ромбической, с симметрией P212121, Z=4, и со следующими параметрами: а=7,753(1) А,b=14,331(1) А, с=22,276(1) А, ===90, иEtO2C-CH2-(R)Cgl-Aze-NH-CH2-C6H4-CN в смеси этилацетат/очищенная вода (концентрация растворенного вещества около 23% мас./общ.об., концентрация воды около 10 мас.% от общей массы), загрузили 50 мл этанола. Температуру раствора привели к 39 С и загрузили в реактор смесь 0,61 мг ЭДТК/EDTA и 3,62 г гидроксиламина (водного; 50 маc.%). Через 1 ч 50 мин добавили затравочные кристаллы безводной формы EtO2C-CH2-(R)Cgl-Aze-PabOH (полученного аналогично способу, описанному выше в примере 14). Суспензию перемешивали в течение 21 ч. Затем в реактор загрузили 3,18 г ацетона и перемешивали суспензию в течение дополнительных 30 мин. Затем суспензию нагревали до 60 С в течение 6 ч. Температуру поддерживали при 60 С в течение 6 ч и затем охладили путем снижения температуры рубашки со скоростью 10 С/ч до тех пор, пока температура не стала близка к -10 С. Затем кристаллы подвергали старению в течение дополнительных 2 ч. Кристаллы отделили от маточного раствора путем фильтрации, промыли 20 мл этанола, затем следующими 10 мл этанола, а затем сушили при 40 С и при пониженном давлении. Кристаллы затем проанализировали путем ПРФА, ДСК и ТГА. Анализы показали, по существу, ту же картину ПРФА, термограммы ДСК и потери массы, как и форма, полученная в соответствии с вышеуказанным примером 14. Пример 16. К раствору примерно 4,5 гEtO2C-CH2-(R)Cgl-Aze-NH-CH2-C6H4-CN в смеси этилацетат/очищенная вода (концентрация растворенного вещества около 24 мас.%/ общ.об., концентрация воды около 10 мас.% на общую массу), загрузили 9 мл этилацетата и 11,2 мл этанола. Температуру раствора привели к 41 С и загрузили в круглодонную колбу смесь 0,3 мг ЭДТК/EDTA и 1,78 г гидроксиламина(водного; 50 маc.%). Через 1 ч 40 мин добавили затравочные кристаллы безводной формы(полученного аналогично способу, описанному выше в примере 14). Суспензию перемешивали в течение 22 ч. Затем в круглодонную колбу загрузили 2,79 г ацетона и перемешивали суспензию дополнительно в течение 19 ч. Кристаллы отделили от маточного раствора путем фильтрации,промыли 210 мл этанола, а затем сушили при 40 С и при пониженном давлении. 21 Кристаллы проанализировали путем ПРФА, ДСК и ТГА. Анализы показали, по существу, ту же картину ПРФА, термограммы ДСК и потери массы, как и форма, полученная в соответствии с вышеуказанным примером 14. Пример 17. К раствору примерно 30 кгEtO2C-CH2-(R)Cgl-Aze-NH-CH2-C6H4-CN в 130 л смеси этилацетат/вода загрузили 151 кг этанола. Температуру раствора привели к 38 С и загрузили в реактор смесь 2,1 г ЭДТК/EDTA и 12,0 кг гидроксиламина (водного; 51 маc.%). Через приблизительно 1,5-2 ч добавили 171 г затравочных кристаллов безводной формы EtO2CCH2-(R)Cgl-Aze-Pab-OH (полученного аналогично способу, описанному выше в примере 14). Суспензию перемешивали в течение примерно 23 ч. Затем в реактор загрузили 19 кг ацетона и перемешивали суспензию в течение еще 30 мин. Затем суспензию нагрели до 73 С до растворения всех кристаллов. Раствор охладили путем уменьшения температуры рубашки при 10 С/ч. Когда температура была около 61 С добавили дополнительно 172 г тех же затравочных кристаллов и продолжили охлаждение до температуры, близкой к -10 С. Кристаллы подвергали старению в течение еще 11 ч. Кристаллы отделили от маточного раствора путем центрифугирования, промыли 90 кг изопропанола, а затем сушили при 40 С при пониженном давлении. Кристаллы проанализировали путем ПРФА, ДСК и ТГА. Анализы показали, по существу, ту же картину ПРФА, термограммы ДСК и потери массы, как и форма, полученная в соответствии с вышеуказанным примером 14. Пример 18. К раствору приблизительно 5 гEtO2C-CH2-(R)Cgl-Aze-NH-CH2-C6H4-CN в смеси этилацетат/вода (концентрация растворенного вещества около 26 мас.%/общ.об., концентрация воды около 10 мас.% на общую массу) загрузили 12,5 мл этанола. Температуру раствора привели к 41 С и в круглодонную колбу загрузили смесь 0,3 мг ЭДТК/EDTA и 7,49 г гидроксиламина (водного; 50 маc.%). Через 1 ч 40 мин добавили затравочные кристаллы безводной формы EtO2C-CH2-(R)Cgl-Aze-Pab-OH (полученного аналогично способу, описанному выше в примере 14). Суспензию перемешивали в течение 22 ч. Затем в круглодонную колбу загрузили 2,79 г ацетона и перемешивали суспензию в течение еще 19 ч. Кристаллы отделили от маточного раствора путем фильтрации, промыли 210 мл этанола, а затем сушили при 40 С при пониженном давлении. Кристаллы проанализировали путем ПРФА, ДСК и ТГА. Анализы показали, по существу, ту же картину ПРФА, термограммы ДСК и потери массы, как и форма, полученная в соответствии с вышеуказанным примером 14. Пример 19. К раствору приблизительно 9 гEtO2C-CH2-(R)Cgl-Aze-NH-CH2-C6H4-CN в смеси этилацетат/очищенная вода (концентрация растворенного вещества около 23 мас.%/ 22 общ.об., концентрация воды около 10 мас.% на общую массу), загрузили 56 мл этанола. Температуру раствора привели к 50 С и в круглодонную колбу загрузили смесь 0,6 мг ЭДТК/EDTA и 3,49 г гидроксиламина (водного; 50 маc.%). Через 1 ч 40 мин добавили 50 мг затравочных кристаллов безводной формы EtO2C-CH2(R)Cgl-Aze-Pab-OH (полученного аналогично способу, описанному выше в примере 14). Суспензию перемешивали в течение 18 ч. Затем в круглодонную колбу загрузили 3,1 г ацетона и перемешивали суспензию в течение еще 30 мин. Суспензию затем нагревали до 75 С в течение 0,5 ч. Через 40 мин температура бани снизилась до 25 С (то есть суспензию кристаллов охладили путем естественного охлаждения). Через 21 ч кристаллы отделили от маточного раствора путем фильтрации, а затем сушили при 40 С при пониженном давлении. Кристаллы проанализировали путем ПРФА, ДСК и ТГА. Анализы показали, по существу, ту же картину ПРФА, термограммы ДСК и потери массы, как и форма, полученная в соответствии с вышеуказанным примером 14. Пример 20. Перекристаллизация EtO2CCH2-(R)Cgl-Aze-Pab-OH, полученного аналогично способам примеров с 14 по 19. 2,0 г неочищенного EtO2C-CH2-(R)CglAze-Pab-OH (полученного аналогично способам, описанным выше в примерах с 14 по 19) растворили в смеси 13 мл изопропанола, 5 мл очищенной воды и 0,65 г этанола при 70 С. Раствор перемешали при 40 С и охлаждали от 70 до 55 С в течение 1,5 ч. Затем добавили 2 мл ацетона и продолжили охлаждение от 55 до 30 С в течение 5 ч и от 30 до 0 С в течение 3 ч. Суспензию затем перемешивали в течение 340 мин при окончательной температуре. Кристаллы отфильтровали и промыли 4 мл изопропанола и после этого сушили при 40 С при пониженном давлении. Кристаллы проанализировали путем ПРФА и ДСК и ТГА. Анализы показали, по существу, ту же картину ПРФА , ДСК термограммы и потери массы, как и форма, полученная в соответствии с вышеуказанным примером 14. Пример 21. 2,0 г неочищенного EtO2CCH2-(R)Cgl-Aze-Pab-OH (полученного аналогично способам, описанным выше в примерах с 14 по 19) растворили в смеси 1,5 мл изопропанола, 4,8 мл очищенной воды и 0,001 г гидроксида натрия, растворенного в 0,2 мл очищенной воды при 70 С. Раствор перемешивали в течение 1,5 ч при 70 С и после этого охлаждали до 0 С в течение 7 ч. Суспензию перемешивали в течение 55 мин при окончательной температуре. Кристаллы отфильтровали и промыли 5 мл изопропанола и затем сушили при 40 С при пониженном давлении. Кристаллы проанализировали путем ПРФА, ДСК и ТГА. Анализы показали, по существу, ту же картину ПРФА , ДСК термограм 23 мы и потери массы, как и форма, полученная в соответствии с вышеуказанным примером 14. Пример 22. 1,5 г неочищенного EtO2CCH2-(R)Cgl-Aze-Pab-OH (полученного аналогично способам, описанным выше в примерах с 14 по 19) растворили в смеси 10,13 мл изопропанола, 3,75 мл очищенной воды и 1,13 мл этилацетата при 70 С. Раствор затем охлаждали до 55 С и после этого до 0 С в течение 5,5 ч. Суспензию перемешивали в течение 695 мин при окончательной температуре. Кристаллы отфильтровали и промыли 3 мл изопропанола и затем сушили при 40 С при пониженном давлении. Кристаллы проанализировали путем ПРФА, ДСК и ТГА. Анализы показали, по существу, ту же картину ПРФА , ДСК термограммы и потери массы, как и форма, полученная в соответствии с вышеуказанным примером 14. Пример 23. 31,0 кг неочищенного EtO2CCH2-(R)Cgl-Aze-Pab-OH (полученного аналогично способам, описанным выше в примерах с 14 по 19) растворили в смеси 158 кг изопропанола и 78 кг очищенной воды при 71 С. Дополнительно 24 кг изопропанола добавили после фильтрации до осветления. Раствор затем охлаждали до 57 С и начали кристаллизацию путем добавления 78 г затравочных кристаллов (полученных аналогично способу, описанному выше в примере 14). Суспензию перемешивали во время ее охлаждения при 10 С/ч до 0 С. Суспензию затем перемешивали в течение 11 ч при окончательной температуре. После центрифугирования взвеси кристаллы промыли 84 кг изопропанола и после этого сушили при 40 С при пониженном давлении. Кристаллы проанализировали путем ПРФА, ДСК и ТГА. Анализы показали, по существу, ту же картину ПРФА , ДСК термограммы и потери массы, как и форма, полученная в соответствии с вышеуказанным примером 14. Пример 24. 3,0 г неочищенного EtO2CCH2-(R)Cgl-Aze-Pab-OH (полученного аналогично способам, описанным выше в примерах с 14 по 19) растворили в смеси 22,5 мл изопропанола, 7,5 мл очищенной воды и 37,1 мг соляной(32%) при 70 С. Раствор перемешивали в течение 3 ч при 70 С и затем охлаждали до 55 С в течение 0,5 ч, после чего кристаллизация началась без затравливания. Суспензию затем охлаждали до 0 С в течение 5,5 ч. Суспензию перемешивали в течение 740 мин при окончательной температуре. Кристаллы отфильтровали и промыли 7,5 мл изопропанола и затем сушили при 40 С при пониженном давлении. Кристаллы проанализировали путем ПРФА, ДСК и ТГА. Анализы показали, по существу, ту же картину ПРФА, ДСК термограммы и потери массы, как и форма, полученная в соответствии с вышеуказанным примером 14. Пример 25. 3,0 г EtO2C-CH2-(R)Cgl-AzePab-OH (полученного аналогично способам, 003264 24 описанным выше в примерах с 14 по 19) растворили в смеси 22,5 мл изопропанола и 7,5 мл очищенной воды при 70 С. Раствор перемешивали в течение 3 ч при 70 С и затем охлаждали до 55 С в течение 0,5 ч, после чего кристаллизация началась без затравливания. Суспензию затем охлаждали до 0 С в течение 5,5 ч. Суспензию перемешивали в течение 760 мин при окончательной температуре. Кристаллы отфильтровали и промыли 7,5 мл изопропанола и затем сушили при 40 С при пониженном давлении. Кристаллы проанализировали путем ПРФА, ДСК и ТГА. Анализы показали, по существу, ту же картину ПРФА , ДСК термограммы и потери массы, как и форма, полученная в соответствии с вышеуказанным примером 14. Сокращения оч.сильн.=очень сильный сильн.=сильный ср.=средний слаб.=слабый оч.слаб.=очень слабый ФОРМУЛА ИЗОБРЕТЕНИЯ 1. По существу, кристаллическая формаEtO2C-CH2-(R)Cgl-Aze-Pab-OH или его фармацевтически приемлемой соли. 2. Соединение по п.1, которое находится в безводной форме. 3. Соединение по п.2, которое находится не в форме соли. 4. Соединение по любому из пп.1-3, которое содержит не более 2 маc.% воды. 5. Соединение по любому из пп.1-4, характеризуемое кривой дифференциальной сканирующей калориметрии, на которой при скорости нагрева 5 С/мин в закрытой чашке с микроотверстием под током азота проявляется эндотермический эффект с экстраполированной начальной температурой около 150 С, температурой пика около 151 С и соответственным тепловым эффектом около 113 Дж/г, за которым следует экзотермический эффект в интервале 190-280 С,и/или порошковой дифракционной картиной,характеризуемой пиками со значениями d 12,0,10,0, 8,2, 7,2, 6,0, 5,5, 5,0, 4,92, 4,85, 4,80, 4,42,4,22, 4,11, 4,06, 3,99, 3,78, 3,72, 3,62, 3,34, 3,11,3,10 и 3,03 , и/или как представлено на фиг. 1. 6. Соединение по любому из пп.1-4, характеризуемое кривой дифференциальной сканирующей калориметрии, на которой при скорости нагрева 5 С/мин в закрытой чашке с микроотверстием под током азота проявляется эндотермический эффект с экстраполированной начальной температурой около 169 С, температурой пика около 170 С и соответственным тепловым эффектом около 142 Дж/г, за которым следует экзотермический эффект в интервале 180-280 С,и/или порошковой дифракционной картиной,характеризуемой пиками со значениями d 12,0,11,1, 8,8, 7,2, 6,8, 6,6, 6,5, 6,4, 6,0, 5,8, 5,6, 5,3, 25 5,2, 4,75, 4,52, 4,39, 4,31, 4,29, 4,25, 4,06, 4,00,3,82, 3,73, 3,71, 3,69, 3,66, 3,59, 3,55, 3,41, 3,37,3,34, 3,29, 3,25, 3,22, 3,10, 3,03, 3,00, 2,91, 2,78,2,73, 2,62, 2,51, 2,46, 2,40, 2,38, 2,34, 2,29, 2,26, и/или как представлено на фиг. 5. 7. Соединение по п.1, которое находится в форме моногидрата. 8. Соединение по п.7, которое находится не в форме соли. 9. Соединение по п.7 или 8, характеризуемое кривой дифференциальной сканирующей калориметрии, на которой при скорости нагрева 5 С/мин в закрытой чашке с микроотверстием под током азота проявляется эндотермический эффект с экстраполированной начальной температурой около 94 С, температурой пика около 109 С и соответственным тепловым эффектом около 171 Дж/г, за которым следует экзотермический эффект в интервале 170-290 С, и/или порошковой дифракционной картиной, характеризуемой пиками со значениями d 16,4, 13,7,9,4, 8,2, 7,1, 6,2, 5,5, 5,1, 4,98, 4,75, 4,68, 4,57,4,48, 4,27, 4,21, 4,11, 4,04, 3,93, 3,89, 3,83, 3,68,3,52, 3,47, 3,34, 3,26, 3,02, 2,61 и 2,42 , и/или как представлено на фиг. 2. 10. Соединение по любому из пп.1, 2 или 7, которое находится в форме соли гидробромида. 11. Соединение по п.10, характеризуемое кривой дифференциальной сканирующей калориметрии, на которой при скорости нагрева 5 С/мин в закрытой чашке с микроотверстием под током азота проявляется эндотермический эффект с экстраполированной начальной температурой около 166 С, температурой пика около 167 С и соответственным тепловым эффектом около 69 Дж/г, за которым следует экзотермический эффект в интервале 170-220 С, и/или порошковой дифракционной картиной, характеризуемой пиками со значениями d 12,0, 10,1, 9,5,6,0, 5,7, 5,6, 5,2, 5,1, 4,95, 4,74, 4,57, 4,41, 4,35,4,17, 4,07, 4,03, 3,92, 3,82, 3,72, 3,69, 3,62, 3,51,3,48, 3,38, 3,25, 3,06, 2,92, 2,86, 2,71, 2,53 и 2,33, и/или как представлено на фиг. 3. 12. Соединение по любому из пп.1, 2 или 7, которое находится в форме соли метансульфоната. 13. Соединение по п.12, характеризуемое кривой дифференциальной сканирующей калориметрии, на которой при скорости нагрева 5 С/мин в закрытой чашке с микроотверстием под током азота проявляется эндотермический эффект с экстраполированной начальной температурой около 134 С, температурой пика около 137 С и соответственным тепловым эффектом около 93 Дж/г, за которым следует экзотермический эффект в интервале 140-220 С, и/или порошковой дифракционной картиной, характеризуемой пиками со значениями d 12,0,11,7,10,4,10,2, 8,3, 7,8, 6,0, 5,6, 5,5, 5,2, 5,1, 5,0, 4,98, 4,90,4,75, 4,63, 4,54, 4,46, 4,15, 4,06, 3,92, 3,84, 3,74, 003264 26 3,65, 3,56, 3,47, 3,39, 3,22, 3,12, 2,95, 2,88, 2,76,2,74, 2,69, 2,65, 2,54, 2,52, 2,49, 2,27, 2,21, 2,04 и 2,02 , и/или как проиллюстрировано на фиг. 4. 14. Способ получения соединения по любому из пп.1-13, включающий кристаллизациюEtO2C-CH2-(R)Cgl-Aze-Pab-OH или его фармацевтически приемлемой соли из растворителя,выбранного из группы, включающей ацетаты,низшие алкиловые спирты, алифатические и ароматические углеводороды, простые диалкиловые эфиры, диалкилкетоны, ацетонитрил,водные растворители или их смеси. 15. Способ по п.14, в котором растворитель выбирают из группы, включающей С 1-6 алкилацетаты, линейные или разветвленные C1-6 алкиловые спирты, С 6-12 алифатические углеводороды, С 6-10 ароматические углеводороды, простые ди-С 1-6 алкиловые эфиры, ди-С 1-6 алкилкетоны, ацетонитрил, воду или их смеси. 16. Способ по п.15, в котором растворитель выбирают из группы, включающей этилацетат, бутилацетат, этанол, изопропанол, изооктан, н-гептан, толуол, простой диизопропиловый эфир, ацетон, метилизобутилкетон, ацетонитрил, воду или их смеси. 17. Способ получения соединения по любому из пп.2-6, включающий способ по любому из пп.14-16, в котором растворитель, по существу, не содержит воды. 18. Способ получения соединения по любому из пп.2-4 или 6, включающий способ по любому из пп.14-16, в котором растворитель содержит воду. 19. Способ получения соединения по любому из пп.7-9, включающий способ по любому из пп.14-16, в котором растворитель содержит воду. 20. Соединение, которое можно получить по способу согласно любому из пп.14-19. 21. Соединение по любому из пп.1-13 или 20 для применения в качестве фармацевтического препарата. 22. Соединение по любому из пп.1-13 или 20 для применения в качестве пролекарства. 23. Фармацевтический препарат, включающий соединение по любому из пп.1-13 или 20, в смеси с фармацевтически приемлемыми вспомогательным средством, разбавителем или носителем. 24. Применение соединения по любому из пп.1-13 или 20 для производства лекарственного средства для лечения состояния, при котором требуется или желательно ингибирование тромбина. 25. Способ лечения состояния, при котором требуется или желательно ингибирование тромбина, при котором пациенту, нуждающемуся в таком лечении, вводят терапевтически эффективное количество соединения по любому из пп.1-13 или 20.
МПК / Метки
МПК: A61K 38/55, C07K 5/062, A61P 7/02
Метки: кристаллические, формы, eto2c-ch2-(r)cgl-aze-pab-oh
Код ссылки
<a href="https://eas.patents.su/15-3264-kristallicheskie-formy-eto2c-ch2-rcgl-aze-pab-oh.html" rel="bookmark" title="База патентов Евразийского Союза">Кристаллические формы eto2c-ch2-(r)cgl-aze-pab-oh</a>
Способ получения 2-метил-тиенобензодиазепина и его кристаллические формы

Номер патента: 149
Опубликовано: 29.10.1998
Авторы: Хендриксен Барри А., Баннелл Чарлз А., Ларсен Самуел Д.
МПК: C07D 495/04, A61K 31/55
Метки: способ, получения, формы, кристаллические, 2-метил-тиенобензодиазепина
Формула / Реферат:
1. Полиморфная форма II оланзапина, имеющая порошковую рентгенограмму, представленную следующими межплоскостными расстояниями: d (A) 10,2689 8,577 7,4721 7,125 6,1459 6,071 5,4849 5,2181 5,1251 4,9874 4,7665 4,7158 4,4787 4,3307 4,2294 4,141 3,9873 3,7206 3,5645 3,5366 3,3828 3,2516 3,134 3,0848 3,0638 3,0111 2,8739 2,8102 2,7217 2,6432 2,6007 2. Форма II по п.1, которая является по существу чистой. 3. Форма II по п.2, которая содержит...
Кристаллические формы (r)-(+)-(n)[[3-[1-бензоил-3-(3,4-дихлорфенил) пиперидин-3-ил]проп-1-ил]-4-фенилпиперидин-4-ил]-n-метилацетамида (осанетана) и способ их получения

Номер патента: 3205
Опубликовано: 27.02.2003
Авторы: Моннье Оливье, Гросклод Патрик, Алькад Ален, Анн-Аршар Гий, Рош Жером
МПК: C07D 211/58, A61K 31/445
Метки: кристаллические, способ, получения, формы, осанетана, r)-(+)-(n)[[3-[1-бензоил-3-(3,4-дихлорфенил, пиперидин-3-ил]проп-1-ил]-4-фенилпиперидин-4-ил]-n-метилацетамида
Формула / Реферат:
1. Способ получения кристаллического (R)-(+)-(N)-[[3-[1-бензоил-3-(3,4-дихлорфенил)пиперидин-3-ил]проп-1-ил]-4-фенилпиперидин-4-ил]-N-метилацетамида (осанетана) путем кристаллизации аморфного осанетана, содержащего менее 20% примесей, из смеси этанол/вода или из изопропилового спирта с получением кристаллической формы 1 или из смеси этанол/простой изопропиловый эфир/вода с получением кристаллической формы 2. 2. Способ по п.1, отличающийся тем,...
Новые кристаллические формы противовирусного соединения бензимидазола, способ их получения,их применение, фармацевтическая композиция и способ лечения

Номер патента: 2970
Опубликовано: 26.12.2002
Авторы: Картер Барри Хауард, Сиклз Барри Риддл, Хьюанг Льян-Фенг, Риззолио Мишел Кэтрин, Шмитт Эрик Аллен, Ланкастер Роберт Уильям, Лонг Стейси Тодд, Гловер Бобби Нил
МПК: C07H 19/052, A61P 31/12, A61K 31/7056...
Метки: новые, композиция, применение, кристаллические, противовирусного, способ, получения,их, бензимидазола, лечения, формы, соединения, фармацевтическая
Формула / Реферат:
1. Кристаллическая форма II 5,6-дихлор-2-(изопропиламино)-1-b-L-рибофуранозил-1Н-бензимидазола, характеризующаяся картиной дифракции рентгеновских лучей на порошке, выраженной в терминах углов 2 тета и полученной при помощи дифрактометра с графитовым монохроматором с использованием рентгеновского СuKa-излучения, где указанная картина дифракции рентгеновских лучей на порошке включает в себя углы 2 тета в пяти или более чем пяти позициях,...
Кристаллические формы (1s)-[1-альфа-(2s*, 3r*)-9-альфа]-6, 10-диоксо-n-(2-этокси-5-оксотетрагидро-3-фуранил)-9-[[(1-изохинолил) карбонил] амино]-октагидро-6h-пиридазино[1,2-а][1,2] диазепин-1-карбоксамида

Номер патента: 2761
Опубликовано: 29.08.2002
Авторы: Роньон Валери, Годар Жан-Ив
МПК: C07D 487/04, A61K 31/5025
Метки: 3r*)-9-альфа]-6, 1s)-[1-альфа-(2s, формы, карбонил, 10-диоксо-n-(2-этокси-5-оксотетрагидро-3-фуранил)-9-[[(1-изохинолил, диазепин-1-карбоксамида, кристаллические, амино]-октагидро-6h-пиридазино[1,2-а][1,2
Формула / Реферат:
1. Кристаллическая форма безводного (1S)-[1-альфа-(2S*,3R*)-9-альфа]-6,10-диоксо-N-(2-этокси-5-оксотетрагидро-3-фуранил)-9-[[(1-изохинолил)карбонил]амино]октагидро-6Н-пиридазино[1,2-а][1,2]диазепин-1-карбоксамида (форма А), представляющая собой триклинную кристаллическую систему и имеющая степень кристалличности альфа 91,38; бета 93,62; гамма 90,43 и пространственную группу симметрии P1; Z2. 2. Кристаллическая форма безводного...
Стабильные готовые формы инсулина

Номер патента: 2880
Опубликовано: 31.10.2002
Авторы: Фрэнк Брюс Хилл, Доббинс Майкл Аллен, Ли Шун, Ребхан Дон Мари, Дефелиппис Майкл Розарио
МПК: A61K 38/28, A61P 3/10
Метки: стабильные, формы, инсулина, готовые
Формула / Реферат:
1. Готовая форма в виде раствора, включающая a) физиологически толерантный буфер, выбранный из группы, состоящей из Трис и аргинина; b) мономерный аналог человеческого инсулина, выбранный из группы, состоящей из человеческого инсулина, в котором Pro в положении В28 замещен Asp, Lys, Leu, Val или Ala и в котором Lys в положении В29 представляет собой Lys или замещен Pro; AlaB26-человеческий инсулин, дез-(В28-В30)человеческий инсулин и...
Предыдущий патент: Способ изготовления многослойной печатной платы и предназначенная для этого композиционная фольга
Следующий патент: Вращающийся стабилизатор энергетической системы
Случайный патент: Способ обнаружения и определения типа папилломавируса человека с использованием амплификации-гибридизации