Синтез ингибиторов тирозинкиназы брутона












Формула / Реферат
1. Способ получения соединений формул 4, 13 и 14

включающий обработку соединения 3

кислотой, а затем основанием с последующим сочетанием с хлорангидридом.
2. Способ по п.1, где кислота представляет собой соляную кислоту.
3. Способ по п.1, где кислота представляет собой соляную кислоту в диоксане.
4. Способ по п.1, где хлорангидрид представляет собой акрилоилхлорид.
5. Способ по любому из пп.2-4, включающий обработку соединения 3 соляной кислотой в диоксане с последующим концентрированием реакционной смеси досуха и сочетанием с акрилоилхлоридом.
6. Способ по любому из пп.2-5, где соединение 4, 13 или 14 промывают водной лимонной кислотой и солевым раствором.
7. Способ по любому из пп.2-6, где соединение 4, 13 или 14 очищают посредством флэш-хроматографии.
8. Способ по любому из пп.1-7, где соединение 3 существует в R-конфигурации, или S-конфигурации, или в виде их смеси.
9. Способ получения фармацевтически приемлемой соли соединения формулы 4, 13 или 14, включающий получение соединения формулы 4, 13 или 14 способом по любому из пп.2-8 и образование фармацевтически приемлемой соли.
10. Способ по п.9, где образование фармацевтически приемлемой соли включает взаимодействие соединения 4, 13 или 14 с неорганической кислотой.
11. Способ по п.10, где неорганическая кислота представляет собой соляную, бромисто-водородную, серную, азотную, фосфорную или метафосфорную кислоты.
12. Способ по п.9, где образование фармацевтически приемлемой соли включает взаимодействие соединения 4, 13 или 14 с органической кислотой.
13. Способ по п.12, где органическая кислота представляет собой уксусную, пропионовую, капроновую, циклопентанпропионовую, гликолевую, пировиноградную, молочную, малоновую, янтарную, яблочную, малеиновую, фумаровую, трифторуксусную, винную, лимонную, бензойную, 3-(4-гидроксибензоил)бензойную, коричную, миндальную кислоты, метансульфокислоту, этансульфокислоту, 1,2-этандисульфокислоту, 2-гидроксиэтандисульфокислоту, бензолсульфокислоту, толуолсульфокислоту, 2-нафталинсульфокислоту, 4-метилбицикло[2.2.2]окт-2-ен-1-карбоновую, глюкогептоновую кислоты, 4,4'-метилен-бис-(3-гидрокси-2-ен-1-карбоновую кислоту), 3-фенилпропионовую, триметилуксусную, трет-бутилуксусную, лаурилсерную, глюконовую, глутаминовую, гидроксинафтойную, салициловую, стеариновую или муконовую кислоты.
14. Способ по п.1, где соединение 3 получают взаимодействием соединения 2

с трет-бутил-3-гидроксипиперидин-1-карбоксилатом

в условиях реакции Мицунобу.
15. Способ по п.14, где условия реакции Мицунобу включают использование соединенного со смолой трифенилфосфина, диизопропилазодикарбоксилата и тетрагидрофурана.
16. Способ по п.14, где соединение 3 очищают посредством флэш-хроматографии.
Текст
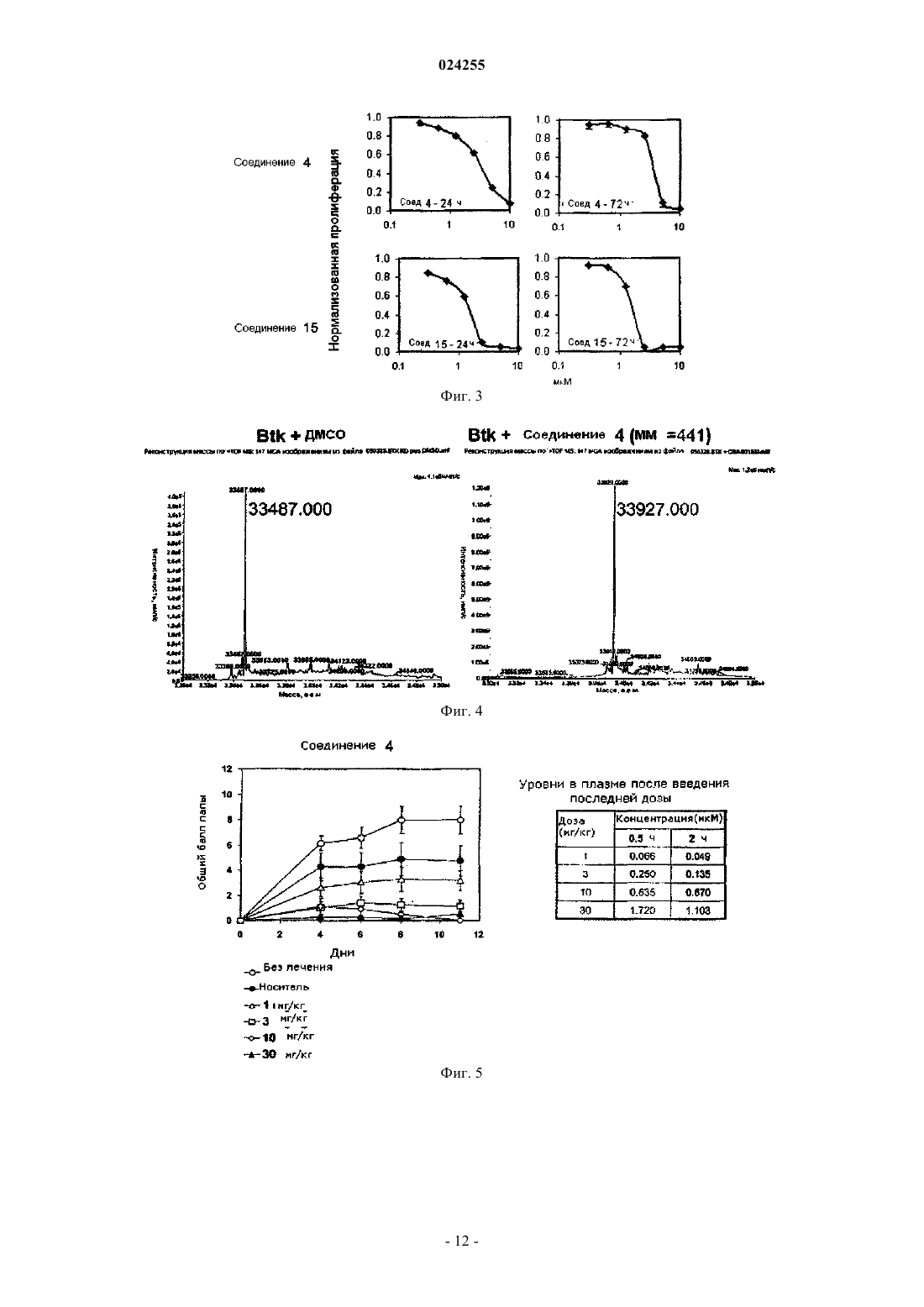
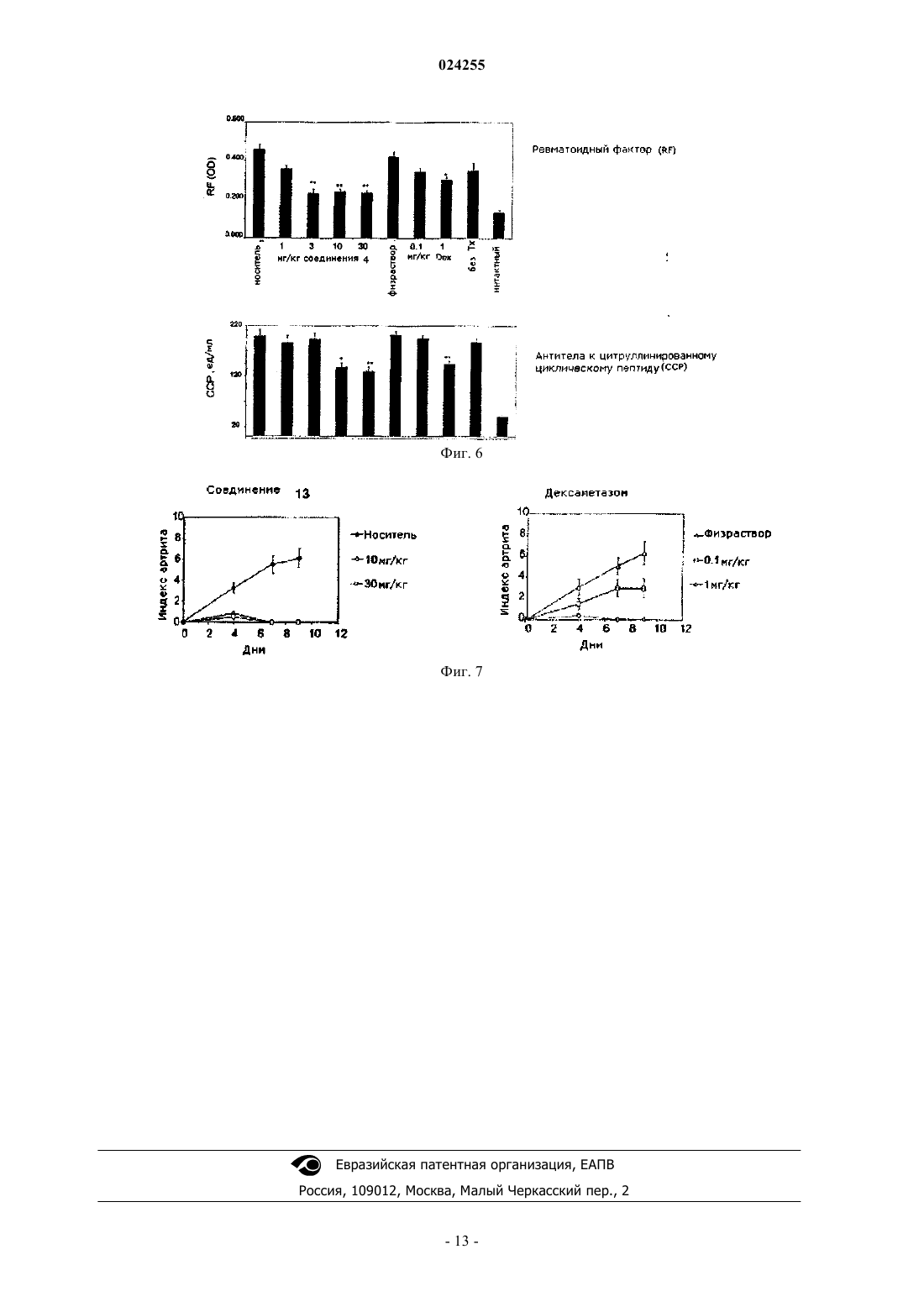
Chemistry, 2001, 66(26), 9056-9062 (реферат) [онлайн] Найдено из БД ACS on STN, CASREACT,СА: 136:151066, схемы 5, 9[он-лайн] Найдено из БД ACS on STN, CASREACT,CA: 143:387217, схема 5 В настоящем изобретении раскрыт способ синтеза ингибиторов тирозинкиназы Брутона. Этот способ включает обработку трет-бутил-3-(4-амино-3-(4-феноксифенил)-1 Н-пиразоло[3,4d]пиримидин-1-ил)пиперидин-1-карбоксилата кислотой, а затем основанием с последующим сочетанием с хлорангидридом. Родственные заявки Заявка на данный патент испрашивает приоритет согласно предварительной заявке на патент США 60/826720, "INHIBITORS OF BRUTON'S TYROSINE KINASE", поданной 22 сентября 2006 г., и предварительной заявке на патент США 60/828590 "INHIBITORS OF BRUTON'S TYROSINE KINASE",поданной 6 октября 2006 г., содержание которых включено в данное изобретение в полном объеме посредством ссылки. Область техники В настоящем изобретении описаны способы получения соединений, представляющих собой ингибиторы тирозинкиназы Брутона. Уровень техники Тирозинкиназа Брутона (Btk), представитель семейства нерецепторных тирозинкиназ Тес, представляет собой ключевой фермент сигнальных каскадов, экспрессирующийся во всех типах гематопоэтических клеток, кроме Т-лимфоцитов и клеток - натуральных киллеров. Btk играет важную роль в сигнальных каскадах В-клеток, связывая стимуляцию рецептора В-клеток (BCR) с последующими внутриклеточными реакциями.Btk является ключевым регулятором развития, активации, передачи сигналов и выживания Вклеток (Kurosaki, Curr Op Imm, 2000, 276-281; Schaeffer и Schwartzberg, Curr Op Imm 2000, 282-288). Кроме того, Btk играет важную роль в ряде других сигнальных каскадах гематопоэтических клеток, например образование ФНО- в макрофагах, опосредуемое Toll-подобными рецепторами (TLR) и рецепторами цитокинов; сигнальный каскад IgE рецептора (Fc-эпсилон-RI) тучных клеток; ингибирование сигнала апоптоза с Fas/APO-1 у лимфоидных В-клеток; и агрегация тромбоцитов, инициируемая коллагеном. См., например, С.A. Jeffries et al. (2003), Journal of Biological Chemistry 278:26258-26264; N.J. Horwood et al. (2003), The Journal of Experimental Medicine 197:1603-1611; Iwaki et al. (2005), Journal of Biological Chemistry 280(48):40261-40270; Vassilev et al. (1999), Journal of Biological Chemistry 274(3):16461656, and Quek et al. (1998), Current Biology 8(20):1137-1140. Краткое изложение сущности изобретения Согласно настоящему изобретению предложен способ получения соединений формул 4, 13 и 14 включающий обработку соединения 3 кислотой, а затем основанием с последующим сочетанием с хлорангидридом. Предпочтительно, когда в указанном выше способе кислота представляет собой соляную кислоту. Также предпочтительно, когда в указанном выше способе кислота представляет собой соляную кислоту в диоксане. Кроме того, предпочтительно, когда в указанном выше способе хлорангидрид представляет собой акрилоилхлорид. Предпочтительно, когда способ по изобретению включает обработку соединения 3 соляной кислотой в диоксане с последующим концентрированием реакционной смеси досуха и сочетанием с акрилоилхлоридом. Также предпочтительно, когда в способе по изобретению соединение 4, 13 или 14 промывают водной лимонной кислотой и солевым раствором. Также предпочтительно, когда в способе по изобретению соединение 4, 13 или 14 очищают посредством флэш-хроматографии. Также согласно настоящему изобретению предложен способ получения фармацевтически приемлемой соли соединения формулы 4, 13 или 14, включающий получение соединения формулы 4, 13 или 14 указанным выше способом по изобретению и образование фармацевтически приемлемой соли. Предпочтительно, когда образование фармацевтически приемлемой соли включает взаимодействие соединения 4, 13 или 14 с неорганической кислотой. Более предпочтительно, когда образование фармацевтически приемлемой соли включает взаимодействие соединения 4, 13 или 14 с неорганической кислотой, представляющей собой соляную кислоту, бромисто-водородную кислоту, серную кислоту, азотную кислоту, фосфорную кислоту или метафосфорную кислоту. Также предпочтительно, когда образование фармацевтически приемлемой соли включает взаимодействие соединения 4, 13 или 14 с органической кислотой. Более предпочтительно, когда образование фармацевтически приемлемой соли включает взаимодействие соединения 4, 13 или 14 с органической кислотой, представляющей собой уксусную, пропионовую, капроновую, циклопентанпропионовую, гликолевую, пировиноградную, молочную, малоновую, янтарную, яблочную, малеиновую, фумаровую, трифторуксусную, винную, лимонную, бензойную,3-(4-гидроксибензоил)бензойную, коричную, миндальную кислоты, метансульфокислоту, этансульфокислоту, 1,2-этандисульфокислоту, 2-гидроксиэтандисульфокислоту, бензолсульфокислоту, толуолсульфокислоту, 2-нафталинсульфокислоту, 4-метилбицикло[2.2.2]окт-2-ен-1-карбоновую кислоту, глюкогептоновую кислоту, 4,4'-метилен-бис-(3-гидрокси-2-ен-1-карбоновую кислоту), 3-фенилпропионовую, триметилуксусную, трет-бутилуксусную, лаурилсерную, глюконовую, глутаминовую, гидроксинафтойную,салициловую, стеариновую или муконовую кислоты. Предпочтительным является способ по изобретению, когда соединение 3 получают взаимодействием соединения 2 в условиях реакции Мицунобу. Предпочтительно, когда условия реакции Мицунобу включают использование соединенного со смолой трифенилфосфина, диизопропилазодикарбоксилата и тетрагидрофурана. Также предпочтительно,когда в указанном выше способе соединение 3 очищают посредством флэш-хроматографии. Терминология Следует заметить, что подробное описание и конкретные примеры, описывающие частные варианты реализации, даны исключительно с целью иллюстрации; на основе данного подробного описания специалисты в данной области смогут определить различные изменения и модификации, соответствующие сущности и объему настоящего описания. Если не определено иначе, все используемые в настоящем изобретении технические и научные термины имеют значения, общепринятые среди специалистов в областях, к которым принадлежит изобретение. В случае, если существует несколько определений, действительным считается описанное в настоящем разделе. Очевидно, что приведенное выше краткое описание и приведенное ниже подробное описание изобретения даны исключительно с иллюстративной целью и ни в коей мере не ограничивают ни один из заявленных объектов. Если не определено иное, использование в настоящем изобретении единственного числа, также может включать множественное число. Если не определено иное, использование в настоящем изобретении союза "или" подразумевает "и/или". Кроме того, использование терминов "к которым относятся", "включая" и их синонимов не является ограничивающим. Используемые в настоящем изобретении названия разделов даны с целью упорядочивания информации, а не с целью ограничения заявленных объектов. Все цитируемые в настоящем изобретении документы или выдержки из документов, включая, но не ограничиваясь ими, патенты, заявки на патенты,статьи, книги, инструкции и научные работы, непосредственно включены в настоящее изобретение посредством ссылок во всей своей полноте для любых целей. Определения стандартных химических терминов можно найти в работах, ссылки на которые приведены в настоящем изобретении, в том числе Carey and Sundberg "ADVANCED ORGANIC CHEMISTRY 4th Ed." Vol. A (2000) и В (2001), Plenum Press, New York. Если отдельно не указано иное, в работе применялись стандартные методы масс-спектрометрии, ЯМР, ВЭЖХ, химии белков, биохимии, технологий рекомбинантных ДНК и фармакологии, в соответствии с известным уровнем техники. Если не даны специальные определения, используемая номенклатура и описанные в настоящем изобретении лабораторные процедуры и методики аналитической химии, химии органического синтеза, медицинской и фармацевтической химии известны из уровня техники. Для химического синтеза, химического анализа, фармацевтического производства, составлений композиций, доставки и лечения пациентов можно применять стандартные методики. К рекомбинантным ДНК, для синтеза олигонуклеотидов, к культуре ткани и для трансформации (например, электропорация, липофекция) можно применять стандартные техники. Реакции и способы очистки можно осуществлять, например, с использованием наборов согласно спецификации производителя или как это принято в соответствующей области или как описано в настоящем изобретении. Изложенные ниже методики и процедуры можно, как правило, осуществлять с использованием распространенных методов, известных из уровня техники, а также описанных в различных источниках, как в общих, так и более частных, на которые даны ссылки, и которые рассмотрены в настоящем описании. Следует заметить, что используемая в настоящем изобретении терминология используется исключительно с целью описания конкретных вариантов реализации и не ограничивает объем описанных способов, который ограничивает только прилагаемая формула изобретения. В данном патенте термин "приемлемый" или "фармацевтически приемлемый" применительно к препарату, композиции или компоненту означает не оказывающий вредного воздействия на общее состояние здоровья субъекта, подвергаемого лечению, или не подавляющий биологическую активность или свойства соединения и относительно нетоксичный. Термин "тирозинкиназа Брутона" данном патенте относится к тирозинкиназе Брутона HomoNo. NP000052). Термины "подавляет/ингибирует", "ингибирующий" или "ингибитор" киназы в данном патенте относится к подавлению/ингибированию ферментативной фосфотрансферазной активности. Краткое описание графических материалов На фиг. 1 представлено сравнение последовательности Btk с другим тирозинкиназами. На фиг. 2 представлены данные, касающиеся ингибирования индуцируемого рецептором В-клеток фосфорилирования фосфолипазы-Су соединением 4 в клетке. В данном примере использовали 2 е 6 клеток Ramos/лунку в бессывороточной среде; клетки предварительно обрабатывали соединением в течение 1,5 ч. Рецептор В-клеток стимулировали антителами к IgM в течение 3 минут; к клеткам немедленно добавляли 10 Х буфер для лизиса, содержащий ДНКазу. Добавляли буфер для образца и сразу же наносили пробы на гель. Образцы исследовали методом Вестерн-блот на содержание фосфорилированной формыBtk и PLC7I, и общей фракции Btk и PLCyl. Блот проявляли с помощью ПЗС ChemiDoc и проводили количественный анализ с помощью ImageQuant. Полосы, соответствующие фосфорилированным формам,нормировали относительно полос, соответствующих общей фракции, и рассчитывали ИК 50. На фиг. 3 представлены данные, демонстрирующие, что соединение 4 и соединение 15 подавляют рост клеток DHL-6. В этом примере использовали 3 Е 4 DHL-6 клетки/лунку в полной среде. Клетки обрабатывали в течение указанного времени соединениемв конечной концентрации 0,1% в ДМСО. Число клеток определяли с помощью системы Alamar Blue согласно стандартному протоколу. На фиг. 4 представлены масс-спектры, показывающие, что соединение 4 ковалентно модифицируетBtk. В данном примере 30 мкМ соединения 4 ингибировали 6-7 мкМ рекомбинантной ВТК (мутант YD, только киназный домен) в течение ночи при комнатной температуре. Комплекс белок-ингибитор обессоливали методом обратно-фазной ВЭЖХ и сразу же исследовали масс-спектрометрии для определения молекулярной массы. 99% рекомбинантного белка Btk было ковалентно модифицировано соединением 4. На фиг. 5 показано подавление развития артрита в модели у мышей соединением 4. На фиг. 6 представлены данные, демонстрирующие, что эффективность соединения 4 связана с уменьшением содержания ревматоидного фактора и антител к цитруллинированному циклическому пептиду в модели CAIA. В данных примерахр 0,01;р 0,001 по сравнению с обработкой наполнителем или солевым раствором. На фиг. 7 представлены данные, касающиеся подавления развития артрита в модели на мышах со-3 024255 единением 13. Данный энантиомер соединения 4 полностью подавлял развитие артрита в модели CAIA при уровнях дозы 10 и 30 мг/кг. Для сравнения представлены данные, касающиеся ингибирования развития артрита, для дексаметазона, в такой же модели на мышах. Упоминание источников Все публикации и заявки на патент, упомянутые в данном описании, включены в данное изобретение посредством ссылки в том же объеме, как если бы конкретно и отдельно было указано, что каждая отдельная публикация или заявка на патент в отдельности включена посредством ссылки. Подробное описание изобретения Соединения. В настоящем изобретении предусмотрено получение соединений, представляющих собой 1-(3-(4-амино-3-(4-феноксифенил)-1 Н-пиразоло[3,4-d]пиримидин-1-ил)пиперидин-1-ил)проп-2-ен 1-он (соединение 4); 1-R)-3-(4-амино-3-(4-феноксифенил)-1 Н-пиразоло[3,4-d]пиримидин-1-ил)пиперидин-1-ил)проп-2 ен-1-он (соединение 13) и 1-S)-3-(4-амино-3-(4-феноксифенил)-1 Н-пиразоло[3,4-d]пиримидин-1-ил)пиперидин-1-ил)проп-2 ен-1-он (соединение 14). Указанные соединения можно применять для лечения пациентов, страдающих зависимыми от тирозинкиназы Брутона или опосредованными тирозинкиназой Брутона патологическими состояниями или заболеваниями, включающими, но не ограниченными перечисленными, рак, аутоиммунные и другие воспалительные заболевания. Получение соединений. Соединение, соответствующее любой из указанных выше формул, можно синтезировать с применением стандартных методов синтеза, известных специалистам в данной области или с применением способов, известных в данной области, в комбинации со способами, описанными в данном изобретении. Добавки, растворители, температурные режимы и другие условия реакции, представленные в настоящем изобретении, могут быть изменены по усмотрению специалиста в данной области. В качестве дополнительного руководства можно также применять следующие способы синтеза. Реакции можно применять в прямой последовательности с получением соединений, описанных в данном изобретении, или их можно применять для синтеза фрагментов, которые впоследствии соединяют способами, описанными в данном изобретении и/или известными в данной области. Синтез соединений. Исходные вещества, применяемые для синтеза соединений, описанных в настоящем изобретении,могут быть синтезированы или могут быть получены из коммерческих источников, таких как, без ограничений, Aldrich Chemical Co. (Milwaukee, Wisconsin), Bachem (Torrance, California) или Sigma ChemicalCo. (St. Louis, Mo). Соединения, описанные в настоящем изобретении, и другие схожие соединения,имеющие другие заместители, могут быть синтезированы с использованием методик и веществ, известных специалистам в данной области, таких как описанные, например, в руководствах March, AdvancedCarbon Compounds, Vol. 1-5 and Supplemental (Elsevier Science Publishers, 1989); Organic Reactions, Vol. 140 (John Wiley and Sons, 1991); Larock's Comprehensive Organic Transformations (VCH Publishers Inc.,1989) (которые включены в настоящее описание посредством ссылки в полном объеме). Другие способы синтеза соединений, описанных в настоящем изобретении, можно найти в международной патентной публикации WO 01/01982901, работах Arnold et al. BioorganicMedicinal Chemistry Letters 10 (2000) 2167-2170; Burchat et al. BioorganicMedicinal Chemistry Letters (2002), 1687-1690. Общие способы получения соединений, таких как описанные в настоящем изобретении, могут быть получены в результате реакций, известных в данной области, и реакции могут быть модифицированы с использованием подходящих реагентов и условий, которые известны специалистам в данной области, для введения различных групп, присутствующих в формулах, представленных в настоящем изобретении. В качестве руководства можно использовать следующие способы синтеза. Продукты реакций могут быть выделены и очищены, при необходимости, с применением стандартных способов, включающих, но не ограниченных перечисленными, фильтрацию, дистилляцию, кристаллизацию, хроматографию и т.п. Такие вещества могут быть охарактеризованы с помощью стандартных способов, позволяющих получить физические константы и спектральные данные и пр. Соединения, описанные в настоящем изобретении, могут быть получены с применением способов синтеза, описанных в настоящем изобретении, в виде одного изомера или смеси изомеров. Неограничивающий пример способа синтеза для получения соединений представлен на схеме 1. В одном воплощении 1 Н-пиразоло[3,4-d]пиримидин-4-амин обрабатывают N-иодсукцинамидом с образованием 3-иод-1 Н-пиразоло[3,4-d]пиримидин-4-амина. Затем проводят катализируемые металлом реакции перекрестного связывания 3-иод-1 Н-пиразоло[3,4-d]пиримидин-4-амина. В одном воплощении в опосредуемой палладием реакции перекрестного связывания соответствующим образом замещенной фенилбороновой кислоты при основных условиях образуется промежуточный продукт 2. Промежуточный продукт 2 сочетают с N-Вос-3-гидроксипиперидином (как неограничивающий пример) посредством реакции Мицунобу с образованием защищенного группой Boc-(трет-бутилоксикарбонил) промежуточного продукта 3. После снятия защитной группы с помощью кислоты, реакции сочетания с, без ограничений, хлорангидридом, таким как, без ограничения, акрилоилхлорид, синтез завершается с образованием соединения 4. С использованием способов синтеза, описанных в настоящем изобретении, а также известных в данной области, ингибиторы тирозинкиназы, такие как описанные в настоящем изобретении, получают с хорошим выходом и чистотой. Соединения, полученные с помощью способов, описанных в настоящем изобретении, очищают стандартными способами, известными в данной области, такими как, например,фильтрация, перекристаллизация, хроматография, дистилляция и их комбинации. Дополнительные формы соединений. Соединения, описанные в настоящем изобретении, могут обладать одним или более стереоцентром,и каждый центр может существовать в конфигурации R или S. Соединения, представленные в настоящем изобретении, включают все диастереомерные, энантиомерные и эпимерные формы, а также соответствующие смеси таких форм. Стереоизомеры могут быть получены, при необходимости, способами, известными в данной области, например, путем разделения стереоизомеров с помощью хиральных хроматографических колонок. Диастереомерные смеси могут быть разделены на отдельные диастереомеры на основании их физико-химических различий известными способами, например, путем хроматографии и/или фракционной кристаллизации. В одном воплощении энантиомеры могут быть разделены с помощью хиральных хроматографических колонок. В других воплощениях энантиомеры могут быть разделены путем преобразования энантиомерной смеси в диастереомерную с помощью реакции с подходящим оптически активным соединением (например, спиртом), разделения диастереомеров и преобразования (например, путем гидролиза) отдельных диастереомеров в соответствующие чистые энантиомеры. Соединения, описанные в настоящем изобретении, включают меченные изотопами соединения, которые идентичны соединениям, указанным в различных формулах и структурах, представленных в настоящем изобретении, за исключением того, что один или более атом заменен атомом, имеющим атомную массу или массовое число, отличное от атомной массы или массового числа, характерного для природной формы соединения. Примеры изотопов, которые могут быть включены в настоящие соединения,включают изотопы водорода, углерода, азота, кислорода, фтора и хлора, такие как, 2 Н, 3 Н, 13 С, 14 С, 15N,18O, 17O, 35S, 18F, 36Cl соответственно. Некоторые меченные изотопами соединения, описанные в настоящем изобретении, например соединения, в которые введены радиоактивные изотопы, такие как 3 Н и 14 С,применимы для анализа тканевой локализации лекарственного средства и/или субстрата. Кроме того,замена на изотопы, такие как дейтерий, т.е. 2 Н, может обеспечивать некоторые терапевтические преимущества, обусловленные большей метаболической стабильностью, например, увеличением периода полураспада in vivo или снижением необходимой дозировки. В дополнительных или дальнейших воплощениях соединения, описанные в настоящем изобретении, метаболизируются при введении в организм, нуждающийся в этом, с образованием метаболита, который затем оказывает желаемый эффект, в том числе желаемый терапевтический эффект. Соединения, описанные в настоящем изобретении, могут быть получены в виде и/или их можно применять в виде фармацевтически приемлемых солей. Типы фармацевтически приемлемых солей,включают следующие, но не ограничены ими: (1) соли присоединения кислот, образованные при взаимодействии соединения в форме свободного основания с фармацевтически приемлемой: неорганической кислотой, такой как соляная, бромисто-водородная, серная, азотная, фосфорная, метафосфорная кислоты и т.п.; или с органической кислотой, такой как уксусная, пропионовая, капроновая, циклопентан пропионовая, гликолевая, пировиноградная, молочная, малоновая, янтарная, яблочная, малеиновая, фумаровая,трифторуксусная, винная, лимонная, бензойная, 3-(4-гидроксибензоил)бензойная, коричная, миндальная кислоты, метансульфокислота, этансульфокислота, 1,2-этандисульфокислота, 2-гидроксиэтандисульфокислота,бензолсульфокислота,толуолсульфокислота,2-нафталинсульфокислота,4 метилбицикло[2.2.2]окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 4,4'-метилен-бис-(3 гидрокси-2-ен-1-карбоновая кислота), 3-фенилпропионовая, триметилуксусная, трет-бутилуксусная, лаурилсерная, глюконовая, глутаминовая, гидроксинафтойная, салициловая, стеариновая, муконовая кислоты и т.п.; (2) соли, образованные либо при замещении кислого протона, присутствующего в родительском соединении, ионом металла, например ионом щелочного металла (например, лития, натрия, калия),иона щелочно-земельного металла (например, магния или кальция), или ионом алюминия; либо при образовании координационной связи с органическим основанием. Приемлемые органические основания включают этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п. Приемлемые неорганические основания включают гидроксид алюминия, гидроксид кальция, гидроксид калия, карбонат натрия, гидроксид натрия и т.п. Соответствующие противоионы фармацевтически приемлемых солей можно исследовать и идентифицировать с использованием различных методов, включая перечисленные, но не ограничиваясь ими: ионнообменную хроматографию, ионную хроматографию, капиллярный электрофорез, индукционное связывание плазмы, атомно-абсорбционную спектроскопию, масс-спектрометрию или любую их комбинацию. Соли извлекают с применением по меньшей мере одной из следующих методик: фильтрация, осаждение с осадителем с последующей фильтрацией, выпариванием растворителя, или, в случае водных растворов, лиофилизацией. Следует понимать, что упоминание соли включает ее аддукты с растворителем или кристаллические формы, в частности, сольваты или полиморфы. Сольваты содержат либо стехиометрическое, либо нестехиометрическое количество растворителя, и часто образуются в ходе процесса кристаллизации с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п. Гидраты образуются в случае, если растворителем является вода, а алкоголяты образуются в случае, когда растворителем является спирт. Полиморфы включают кристаллы с различной структурой одинакового элементного состава соединения. Полиморфы, как правило, имеют различный характер рентгеновской дифракции, различные инфракрасные спектры, температура плавления, различную плотность, твердость, кристаллическую форму, оптические и электрические свойства, стабильность и растворимость. Различные факторы, такие как растворитель для рекристаллизации, степень кристаллизации и температура хранения, могут обуславливать доминирование одной кристаллической формы. Соединения, описанные в настоящем изобретении, могут быть представлены в различных формах,включая перечисленные, но не ограничиваясь ими: бесструктурные формы, молотые формы и наночастицы. Кроме того, описанные в настоящем изобретении соединения включают кристаллические формы,также известные как полиморфы. Полиморфы включают кристаллы с различной структурой и одинаковым элементным составом соединения. Различные факторы, такие как растворитель для рекристаллизации, степень кристаллизации и температура хранения, могут обуславливать доминирование одной из кристаллических форм. Скрининг и определение характеристик фармацевтически приемлемых солей, полиморф и/или сольватов можно осуществлять рядом методов, включая перечисленные, но не ограничиваясь ими: термический анализ, рентгенодифракционный метод, спектроскопию, сорбцию пара и микроскопию. Термические методы анализа направлены на исследование термохимического разложения или термофизических процессов, включая, но не ограничиваясь: полиморфные переходы, и такие методы применяют для анализа связи между полиморфными формами, определения потери в массе, для нахождения температуры стеклования или исследования совместимости с наполнителем. Такие способы включают, без ограничения, дифференциальную сканирующую калориметрию (ДСК), модулирующую дифференциальную сканирующую калориметрию (МДСК), термогравиметрический анализ (ТГА), термогравиметрический и инфракрасный анализ (ТГ/ИК). Кристаллографические методы включают перечисленные, но не ограничиваются ими: монокристаллические и порошковые дифрактометры и синхротронные источники. Различные используемые спектроскопические методы включают перечисленные, но не ограничены ими: определение спектра Рамана (комбинационного рассеяния), FTIR, UVIS и ЯМР (жидкого и твердого состояния). Различные методы микроскопии включают перечисленные, но не ограничены ими: микроскопию в поляризованном свете, сканирующую электронную микроскопию (СЭМ) с рентгеновским анализом методом энергетической дисперсии (EDX), сканирующую электронную микроскопию в режиме естественной среды с EDX(в атмосфере газа или водяного пара), ИК-микроскопию и микроскопию комбинационного рассеяния. Примеры Приведенные ниже частные неограничивающие примеры следует рассматривать только как иллюстрацию, они никоим образом не ограничивают настоящие изобретение. Без более детального пояснения предполагается, что специалист в данной области может осуществить его в полном объеме, основываясь на настоящем описании. Пример 1. Синтез соединений. Получение 4-амино-3-(4-феноксифенил)-1 Н-пиразоло[3,4-d]пиримидина (промежуточное соединение 2). 4-Амино-3-(4-феноксифенил)-1 Н-пиразоло[3,4-d]пиримидин (промежуточный продукт 2) синтезируют, как описано в Международной патентной публикации WO 01/019829. Кратко, 4 феноксибензойную кислоту (48 г) добавляют к тионилхлориду (100 мл) и нагревают в условиях мягкого рефлюкса в течение 1 ч. Тионилхлорид удаляют путем дистилляции, оставшееся масло растворяют в толуоле и летучие вещества удаляют при 80 С/20 мбар. Полученный хлорангидрид растворяют в толуоле(200 мл) и тетрагидрофуране (35 мл). Малононитрил (14,8 г) добавляют, растворяют и перемешивают при -10 С с добавлением диизопропилэтилэтиламина (57,9 г) в толуоле (150 мл), поддерживая температуру ниже 0 С. После 1 ч при 0 С смесь перемешивают при 20 С в течение ночи. Гидрохлорид амина удаляют путем фильтрации и фильтрат высушивают в вакууме. Остаток собирают в этилацетат и отмывают 1,25 М серной кислотой, затем рассолом и высушивают над сульфатом натрия. Испарение растворителей дает полутвердый остаток, который обрабатывают небольшим количеством этилацетата, что дает 4,1 г 1,1-дициано-2-гидрокси-2-(4-феноксифенил)этена в виде белого твердого порошка (т. пл. 160162 С). Фильтрат при выпаривании дает 56,58 (96%) 1,1-дициано-2-гидрокси-2-(4-феноксифенил)этена в виде серо-коричневого твердого вещества, которое является достаточно чистым для дальнейшего применения. 1,1-Дициано-2-гидрокси-2-(4-феноксифенил)этен (56,5 г) в ацетонитриле (780 мл) и метаноле (85 мл) перемешивают под азотом при 0 С с добавлением диизопропилэтиламина (52,5 мл), а затем 2 М триметилсилилдиазометана (150 мл) в тетрагидрофуране (ТГФ). Реакционную смесь перемешивают в течение 2 дней при 20 С и затем добавляют 2 г диоксида кремния (для хроматографии). Коричнево-красный раствор выпаривают в вакууме, остаток растворяют в этилацетате и хорошо промывают водой, а затем солевым раствором, высушивают и выпаривают. Остаток экстрагируют диэтиловым эфиром (3250 мл) и путем декантирования отделяют от нерастворимого масла. Выпаривание эфирных экстрактов дает 22,5 г 1,1-дициано-2-метокси-2-(4-феноксифенил)этена в виде бледно-оранжевого твердого вещества. Нерастворимое масло очищают путем флэш-хроматографии, что дает 15,0 г красно-оранжевого масла. 1,1-Дициано-2-метокси-2-(4-феноксифенил)этен(22,5 г) и 1,1-дициано-2-метокси-2-(4 феноксифенил)этеновое масло (15 г) обрабатывают раствором гидрата гидразина (18 мл) в этаноле (25 мл) и греют на паровой бане в течение 1 ч. Добавляют этанол (15 мл), а затем воду (10 мл). Выпавшее в осадок твердое вещество собирают и промывают смесью этанол:вода (4:1) и затем высушивают на воздухе, что дает 3-амино-4-циано-5-(4-феноксифенил)пиразол в виде бледно-оранжевого твердого вещества. 3-Амино-4-циано-5-(4-феноксифенил)пиразол (29,5 г) суспендируют в формамиде (300 мл) и нагревают под азотом при 180 С в течение 4 ч. Реакционную смесь охлаждают до 30 С и добавляют воду (300 мл). Твердое вещество собирают, хорошо промывают водой, затем метанолом и высушивают на воздухе, что дает 4-амино-3-(4-феноксифенил)-1 Н-пиразоло[3,4-d]пиримидин. Пример 1 а. Синтез 1-(3-(4-амино-3-(4-феноксифенил)-1 Н-пиразоло[3,4-d]пиримидин-1 ил)пиперидин-1-ил)проп-2-ен-1-он (соединение 4) Схема 2 трифенилфосфина (ТРР) (полимерлаб) смешивали с 5 мл тетрагидрофурана (ТГФ). К смеси добавляли трет-бутил-3-гидроксипиперидин-1-карбоксилат (200 мг; 2,0 экв.) с последующим добавлением диизопропилдиазодикарбоксилата (0,099 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи, фильтровали для удаления смолы и реакционную смесь концентрировали и очищали путем флэш-хроматографии (пентанацетат/этил = 1/1), что дает промежуточный продукт 3 (55 мг). Промежуточный продукт 3 (48,3 мг) обрабатывали 1 мл 4 н. HCl в диоксане в течение 1 ч и затем концентрировали досуха. Остаток растворяли в дихлорметане и триэтиламине (0,042 мл) и смешивали с акрилхлоридом (0,010 мл). Реакция была остановлена после 2 ч. Реакционную смесь промывали 5% (по массе) водной лимонной кислотой, а затем солевым раствором. Органическую фазу сушили на MgSO4 и концентрировали. Флэш-хроматография (с CH2Cl2/МеОН = 25/1) давала 22 мг соединения 4 в виде белого твердого вещества. MS (M+1): 441,2. 1 Н-ЯМР (400 МГц): 8.26 (s, 1 Н), 7.65 (m, 2 Н), 7.42 (m, 2 Н), 7.1-7.2 (m, 5 Н), 6.7-6.9 (m, 1 Н), 6.1 (m,1 Н), 5.5-5.7 (m, 1 Н), 4.7 (m, 1 Н), 4.54 (m, 0.5 Н), 4.2 (m, 1 Н), 4.1 (m, 0.5 Н), 3.7 (m, 0.5 Н), 3.2 (m, 1H), 3.0 Синтез соединения 14 осуществляли способом, аналогичным описанному в примере 1 а. ЕМ (рассч.): 440.2; MS (ESI) m/e (М+1 Н)+: 441.5, (М. 1 Н)-: 439.2. Пример 2. Ингибирующая активность в отношении Btk in vitro. Значения Btk IC50 соединений, полученных в настоящем изобретении, определяли как с помощью анализа киназной активности в бесклеточной системе, так и с помощью функционального клеточного анализа BCR-индуцированного потока кальция, как описано ниже. Киназную активность Btk определяли с помощью метода резонансного переноса энергии флуоресценции с временным разрешением (TR-FRET). Измерения проводили в реакционном объеме 50 мкл с использованием 96-луночного аналитического планшета. Фермент киназу, ингибитор, АТФ (для Км киназы) и 1 мкМ пептидного субстрата (Biotin-AVLESEEELYSSARQ-NH2) инкубировали в реакционном буфере, включающем 20 мМ Трис, 50 мМ NaCl, MgCl2 (5-25 мМ в зависимости от киназы), MnCl2(0-10 мМ), 1 мМ DTT, 0,1 мМ ЭДТА, 0,01% бычьего сывороточного альбумина, 0,005% Tween-20 и 10% ДМСО при рН 7,4 в течение 1 ч. Реакцию подавляли добавлением 1,2 экв. ЭДТА (относительно двухвалентного катиона) в 25 мкл 1 буфера Ланса (Perkin-Elmer). Добавляли стрептавидин-АРС (Perkin-Elmer) и меченое европием антитело p-TyrlOO (Perkin-Elmer) в 1 буфере Ланса в объеме 25 мкл, что дало конечные концентрации 100 и 2,5 нМ соответственно и смесь оставили для инкубации в течение 1 ч. Сигнал TR-FRET измеряли на многорежимном спектрофотометре для считывания планшетов с длиной волны возбуждения (Ex) 330 нм и длиной волны детектирования (Em) 615 и 665 нм. Активность определяли как отношение флюоресценции при 665 нм к флюоресценции при 615 нм. Для каждого соединения измеряли ферментативную активность при различных концентрациях соединения. Реакции отрицательного контроля ставили в отсутствие ингибитора шесть раз, также ставили две контрольные реакции без фер-8 024255 мента для определения исходных уровней флюоресценции. Константы ингибирования, Kj (app) определяли с использованием программы BatchKj (Kuzmic et al. (2000), Anal. Biochem. 286:45-50), значения IC50 получали согласно уравнению:II384 флюорометрического пишущего планшетного спектрофотометра (Molecular devices) согласно инструкциям изготовителя. Коротко, активно растущие клетки Рамоса (АТСС) на среде RPM1, с добавлением 10% FBS (Invitrogen) промывали и повторно высевали на питательную среду с низким содержанием сыворотки в количестве приблизительно в 5105 клеток на 100 мкл в 96-луночных планшетах. Исследуемые соединения растворяли в диметилсульфоксиде и разбавляли средой с низким содержанием сыворотки до конечной концентрации в пределах от 0 до 10 мкМ (при коэффициенте разбавления 0,3). Разбавленные соединения затем добавляли в каждую лунку (конечная концентрация диметилсульфоксида составляла 0,01%) и инкубировали при 37 в инкубаторе с атмосферой с 5% CO2 в течение 1 ч. Затем в каждую лунку добавляли 100 мкл чувствительного к кальцию красителя (из набора реактивов Calcium 3,Molecular Devices) и инкубировали в течение еще 1 ч. Обработанные соединением клетки стимулировали козьим антителом к IgM человека (80 мкг/мл;Jackson ImmunoResearch) и считывали сигнал на FlexStation II384 с использованием Ex = 485 нм и Em = 538 нм в течение 200 с. Регистрировали относительные единицы флюоресценции (RFU) и IC50 и анализировали их с использованием встроенной программы SoftMax (Molecular devices). Анализ данных для примеров соединений На необратимое ингибирование Btk этими соединениями указывают две группы свидетельств. Вопервых, после предварительной обработки рекомбинантной Btk соединением ее активность не восстанавливалась после повторной отмывки средой без ингибитора (см., например, J.В. Smaill, et al., J-Med.Chem. 1999, 42, 1803). Во-вторых, наблюдаемый основной пик масс-спектра соответствует молекулярной массе ковалентного комплекса между соединением 4 и Btk в соотношении 1:1 (соединение 4: 440 Да, киназный домен рекомбинантной Btk: 33 487 Да; Комплекс: ожидаемый 33 927 Да, наблюдаемый 33 927 Да). Данные соединения являются очень эффективными ингибиторами активности киназы Btk с IC50 в диапазоне от субнаномолярного до одного разряда в наномолярном диапазоне для in vitro активности киназы. Значения ИК 50 для них, определенные при анализе (клетки Рамоса) потока Са 2+, находятся в диапазоне от 7 до 10 нМ. Наконец, в экспериментах с набором энантиомеров (13 против 14) было установлено, что R-конфигурация немного более является несколько предпочтительной абсолютной стереохимической конфигурацией. Подразумевается, что примеры и варианты реализации описаны в настоящем изобретении только для иллюстративных целей и различные модификации или изменения, которые могут быть предложены специалистами в данной области, находятся в рамках сущности и объема настоящего изобретения и прилагаемой формулы. Все публикации, патенты и патентные заявки, цитируемые в настоящем изобретении,полностью включены в него посредством ссылки для любых целей. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединений формул 4, 13 и 14 включающий обработку соединения 3 кислотой, а затем основанием с последующим сочетанием с хлорангидридом. 2. Способ по п.1, где кислота представляет собой соляную кислоту. 3. Способ по п.1, где кислота представляет собой соляную кислоту в диоксане. 4. Способ по п.1, где хлорангидрид представляет собой акрилоилхлорид. 5. Способ по любому из пп.2-4, включающий обработку соединения 3 соляной кислотой в диоксане с последующим концентрированием реакционной смеси досуха и сочетанием с акрилоилхлоридом. 6. Способ по любому из пп.2-5, где соединение 4, 13 или 14 промывают водной лимонной кислотой и солевым раствором. 7. Способ по любому из пп.2-6, где соединение 4, 13 или 14 очищают посредством флэшхроматографии. 8. Способ по любому из пп.1-7, где соединение 3 существует в R-конфигурации, или Sконфигурации, или в виде их смеси. 9. Способ получения фармацевтически приемлемой соли соединения формулы 4, 13 или 14, включающий получение соединения формулы 4, 13 или 14 способом по любому из пп.2-8 и образование фармацевтически приемлемой соли. 10. Способ по п.9, где образование фармацевтически приемлемой соли включает взаимодействие соединения 4, 13 или 14 с неорганической кислотой. 11. Способ по п.10, где неорганическая кислота представляет собой соляную, бромистоводородную, серную, азотную, фосфорную или метафосфорную кислоты. 12. Способ по п.9, где образование фармацевтически приемлемой соли включает взаимодействие соединения 4, 13 или 14 с органической кислотой. 13. Способ по п.12, где органическая кислота представляет собой уксусную, пропионовую, капроновую, циклопентанпропионовую, гликолевую, пировиноградную, молочную, малоновую, янтарную,яблочную, малеиновую, фумаровую, трифторуксусную, винную, лимонную, бензойную, 3-(4 гидроксибензоил)бензойную, коричную, миндальную кислоты, метансульфокислоту, этансульфокислоту, 1,2-этандисульфокислоту, 2-гидроксиэтандисульфокислоту, бензолсульфокислоту, толуолсульфокислоту, 2-нафталинсульфокислоту, 4-метилбицикло[2.2.2]окт-2-ен-1-карбоновую, глюкогептоновую кислоты, 4,4'-метилен-бис-(3-гидрокси-2-ен-1-карбоновую кислоту), 3-фенилпропионовую, триметилуксусную, трет-бутилуксусную, лаурилсерную, глюконовую, глутаминовую, гидроксинафтойную, салициловую, стеариновую или муконовую кислоты. 14. Способ по п.1, где соединение 3 получают взаимодействием соединения 2 в условиях реакции Мицунобу. 15. Способ по п.14, где условия реакции Мицунобу включают использование соединенного со смолой трифенилфосфина, диизопропилазодикарбоксилата и тетрагидрофурана. 16. Способ по п.14, где соединение 3 очищают посредством флэш-хроматографии.
МПК / Метки
МПК: C07D 487/04
Метки: ингибиторов, тирозинкиназы, синтез, брутона
Код ссылки
<a href="https://eas.patents.su/14-24255-sintez-ingibitorov-tirozinkinazy-brutona.html" rel="bookmark" title="База патентов Евразийского Союза">Синтез ингибиторов тирозинкиназы брутона</a>
Соединения гетероарилпиридона и аза-пиридона в качестве ингибиторов тирозинкиназы брутона

Номер патента: 23263
Опубликовано: 31.05.2016
Авторы: Вей Бинкинг, Янг Венди Б., Ортвайн Даниэль Фред, Кроуфорд Джеймс Джон
МПК: A61K 31/381, A61K 31/4353, A61K 31/4985...
Метки: гетероарилпиридона, аза-пиридона, брутона, качестве, соединения, тирозинкиназы, ингибиторов
Формула / Реферат:
1. Соединение формулы Iили его стереоизомер, таутомер или фармацевтически приемлемая соль,где X1 представляет собой CR1 или N;X2 представляет собой CR2 или N;X3 представляет собой CR3 или N;один или два из X1, X2 и X3 представляют собой N;R1, R2 и R3 независимо выбраны из Н, F, Cl, -NH2, -NHCH3, -N(CH3)2, -ОН, -ОСН3, -ОСН2СН3, -ОСН2СН2ОН и C1-С3алкила;R4 выбран из Н, F, Cl, CN, -СН2ОН, -СН(СН3)ОН, -С(СН3)2ОН, -CH(CF3)OH, -CH2F, -CHF2, -CH2CHF2,...
Ингибиторы тирозинкиназы брутона

Номер патента: 18573
Опубликовано: 30.09.2013
Авторы: Хонигберг Ли, Вернер Эрик, Пан Женгьинг
МПК: A01N 43/90, A61K 31/519
Метки: ингибиторы, тирозинкиназы, брутона
Формула / Реферат:
1. Соединение формулы D, имеющее структуругде La представляет собой O;Ar представляет собой фенил;Y представляет собой незамещенный пиперидинил, незамещенный пирролидинил или незамещенный циклогексил;Z представляет собой C(=O), NHC(=O) или S(=O)2;R7 и R8 представляют собой Н или вместе образуют связь;R6 представляет собой Н,или его фармацевтически приемлемая соль.2. Соединение по п.1, отличающееся тем, что Y представляет собой циклогексил.3....
Ингибиторы тирозинкиназы брутона

Номер патента: 20001
Опубликовано: 30.07.2014
Авторы: Пан Женгьинг, Хонигберг Ли, Вернер Эрик
МПК: A61P 19/02, A61P 29/00, A61K 31/519...
Метки: брутона, тирозинкиназы, ингибиторы
Формула / Реферат:
Данные на сервере публикаций отсутствуют
Ингибиторы тирозинкиназы брутона

Номер патента: 21715
Опубликовано: 31.08.2015
Авторы: Луо Уэнчен, Моди Тарак Д., Вернер Эрик, Чен Вей, Лаури Дэвид Дж., Смит Марк Стивен
МПК: A61P 19/02, A61P 19/10, A61K 31/519...
Метки: брутона, тирозинкиназы, ингибиторы
Формула / Реферат:
1. Соединение, выбранное из:или его фармацевтически приемлемые соль, сольват или таутомерная форма.2. Соединение, имеющее структуру формулы (IV)где Т представляет собой связь;Y и Z, каждый независимо, выбраны из C1-С6-алкила, замещенного по меньшей мере одним R1; илиY и Z вместе с атомом углерода, к которому они присоединены, образуют С2-С10-гетероциклоалкил;когда Y и Z вместе с атомом углерода, к которому они присоединены, образуют...
Хинолиноновые производные в качестве ингибиторов тирозинкиназы

Номер патента: 6711
Опубликовано: 24.02.2006
Авторы: Махаевски Тим, Маккреа Билл, Макбрайд Крис, Пекки Сабина, Джазан Элиза, Ренхауэ Пол, Тэйлор Кларк, Шейфер Синтия
МПК: A61K 31/4704, C07D 405/04, C07D 401/04...
Метки: ингибиторов, хинолиноновые, тирозинкиназы, производные, качестве
Формула / Реферат:
1. Соединение, имеющее структуру I, таутомер соединения, фармацевтически приемлемая соль соединения или фармацевтически приемлемая соль таутомера где Y представляет -NR12R13-группу; Z представляет NR14-группу; R1, R2, R3 и R4 могут быть одинаковыми или различными и независимо выбраны из групп H, Cl, Br, F, I, -CN, -NO2, -OH, -OR15-групп, -NR16R17-групп, замещенных и незамещенных амидинильных групп, замещенных и незамещенных гуанидинильных...
Предыдущий патент: Смесительный барабан для доменной печи
Следующий патент: Способ получения дигидроптеридинонов и их промежуточных продуктов
Случайный патент: Установка риформинга топливного элемента