Применение 5-тиазолилметил[(1s,2r)-3-[[(2-амино-6-бензоксазолил)сульфонил](2-метилпропил)амино]-2-гидрокси-1-(фенилметил)пропил]карбамата или его энантиомера для улучшения фармакокинетики ингибиторов протеазы
Номер патента: 16340
Опубликовано: 30.04.2012
Авторы: Ван`т Клостер Гербен Альберт Элетериус, Де Мейер Сандра, Вигеринк Пит Том Берт Поль, Барт Ливен Элвире Колетт, Де Кок Херман Аугустинус











Формула / Реферат
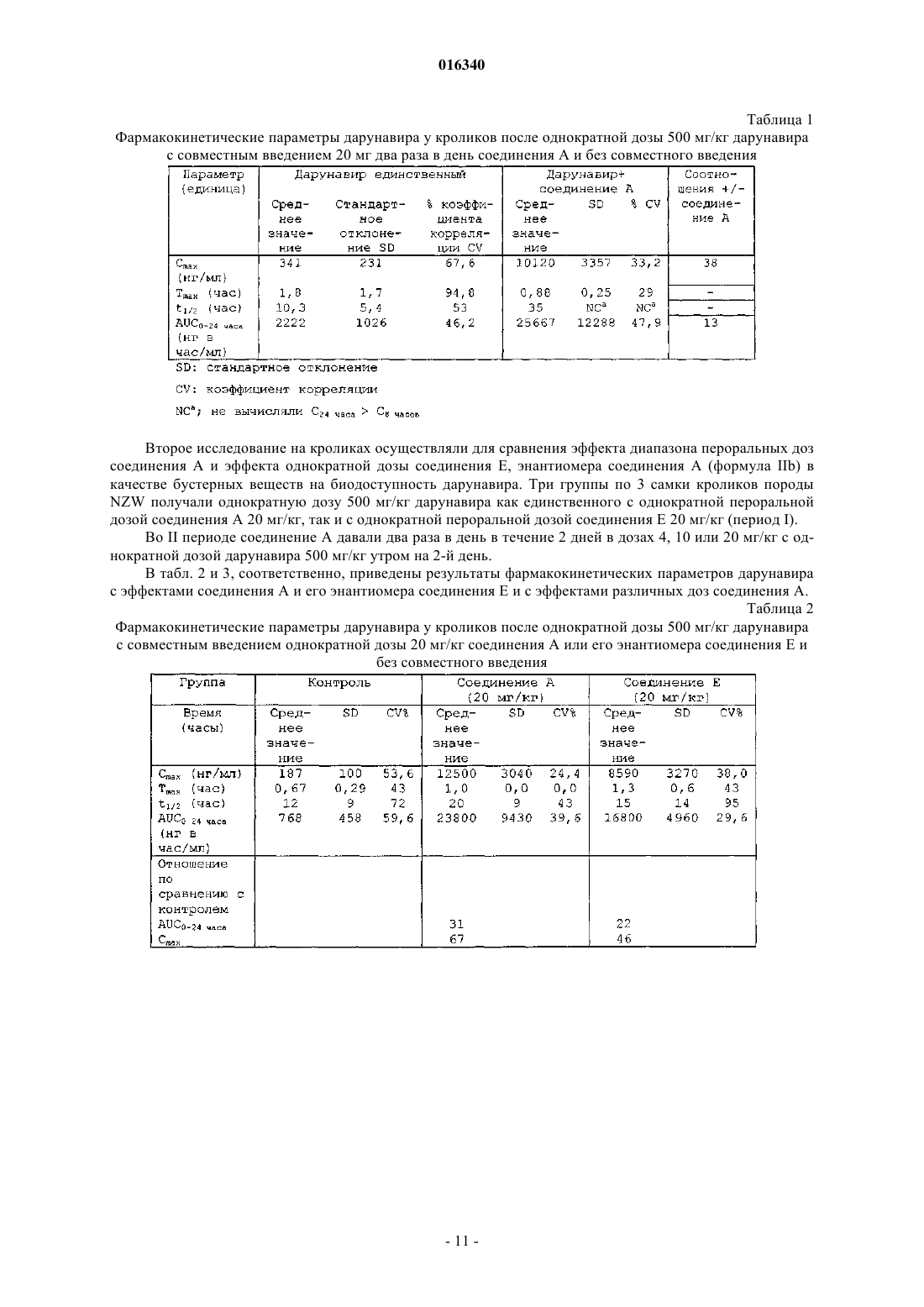
1. Применение соединения, имеющего формулу

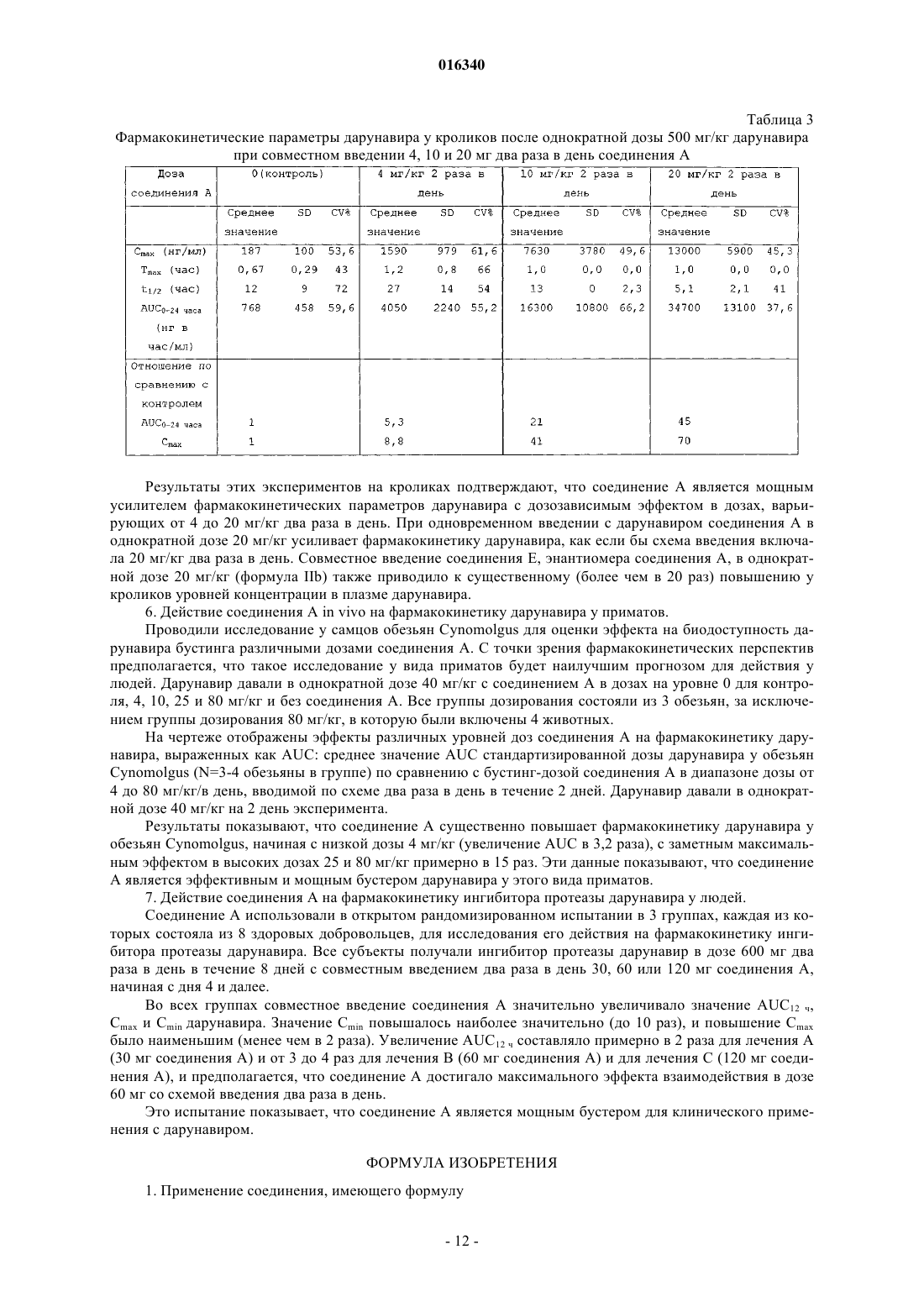
или его энантиомера, имеющего формулу

для получения лекарственного вещества для улучшения фармакокинетики лекарственного средства, где лекарственным средством является ингибитор протеазы.
2. Применение по п.1, в котором ингибитором протеазы является ингибитор ВИЧ-аспарагиновой протеазы.
3. Применение по п.2, в котором ингибитор протеазы является дарунавиром или саквинавиром.
Текст
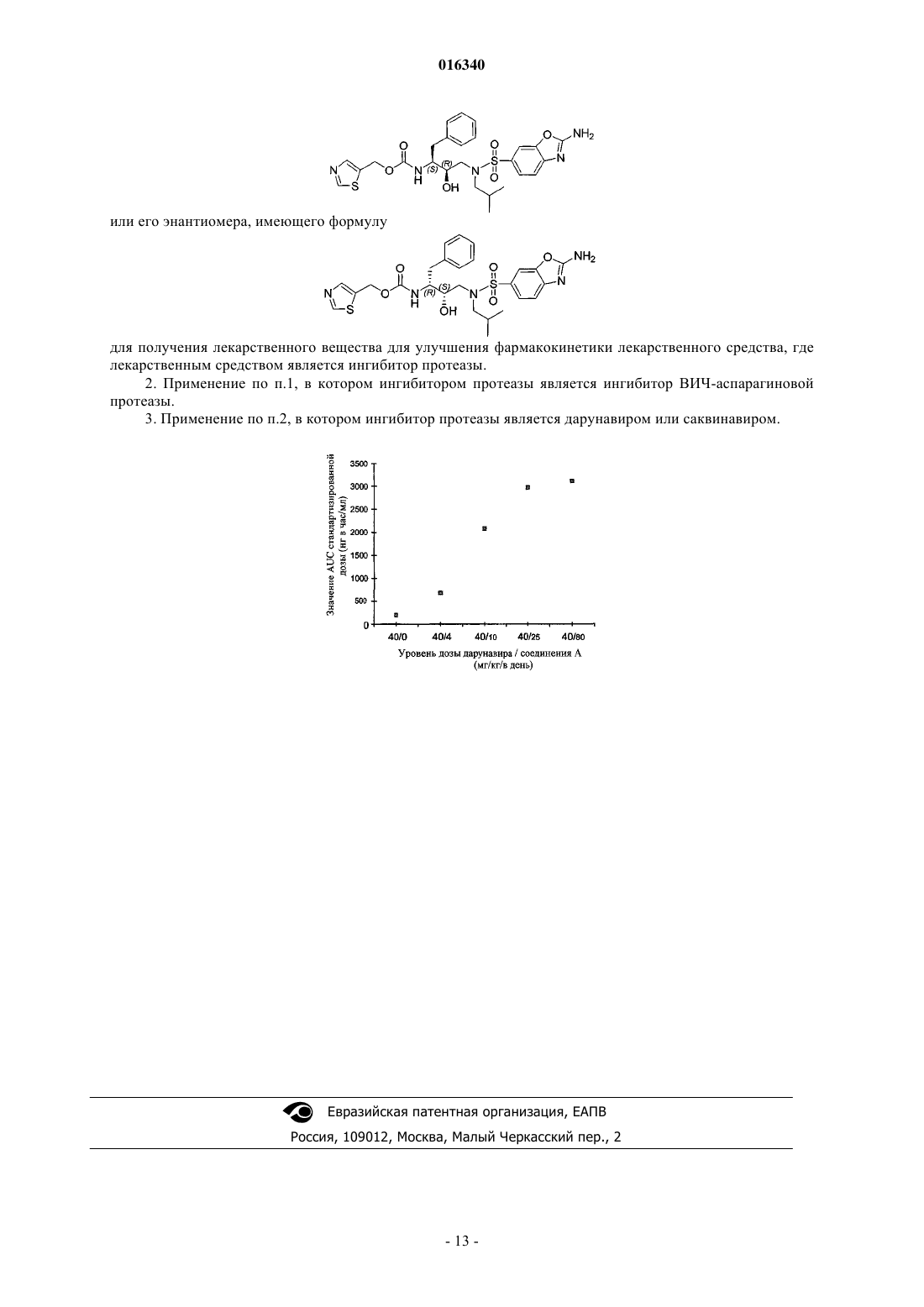
ПРИМЕНЕНИЕ 5-ТИАЗОЛИЛМЕТИЛ[(1S,2R)-3-(2-АМИНО-6 БЕНЗОКСАЗОЛИЛ)СУЛЬФОНИЛ](2-МЕТИЛПРОПИЛ)АМИНО]-2-ГИДРОКСИ-1(ФЕНИЛМЕТИЛ)ПРОПИЛ]КАРБАМАТА ИЛИ ЕГО ЭНАНТИОМЕРА ДЛЯ УЛУЧШЕНИЯ ФАРМАКОКИНЕТИКИ ИНГИБИТОРОВ ПРОТЕАЗЫ Раскрывается применение соединения, имеющего формулу (IIa) или (IIb), определенные ниже, для получения лекарственного вещества для улучшения фармакокинетики лекарственного средства, где лекарственным средством является ингибитор протеазы, предпочтительно в котором ингибитором протеазы является ингибитор ВИЧ-аспарагиновой протеазы и более предпочтительно в котором ингибитор протеазы является дарунавиром или саквинавиром. Ван'Т Клостер Гербен Альберт Элетериус (NL), Вигеринк Пит Том Берт Поль, Де Мейер Сандра, Барт Ливен Элвире Колетт, Де Кок Херман Аугустинус (BE) Медведев В.Н. (RU) 016340 Настоящее изобретение относится к способу улучшения фармакокинетики лекарственных средств,метаболизируемых цитохромом-P450 монооксигеназой. Более конкретно настоящее изобретение относится к способу улучшения фармакокинетики ингибиторов ретровирусной протеазы и, в частности, к улучшению фармакокинетики ингибиторов протеазы вируса иммунодефицита человека (ВИЧ). Вирус, вызывающий синдром приобретенного иммунодефицита (СПИД), известен под различными названиями, включающими в себя T-лимфоцитарный вирус III (HTLV), или лимфаденопатияассоциированный вирус (LAV), или СПИД-ассоциированный вирус (ARV), или вирус иммунодефицита человека (ВИЧ). В настоящее время идентифицированы два различных семейства, то есть ВИЧ-1 и ВИЧ 2. Далее и везде в настоящем документе для общего обозначения этих вирусов будет использован термин ВИЧ. До настоящего времени на рынке существовали различные классы соединений против ВИЧ: нуклеозидные ингибиторы обратной транскриптазы (NRTI), ненуклеозидные ингибиторы обратной транскриптазы (NNRTI), один нуклеотидный ингибитор обратной транскриптазы (NtRTI), один фузионный ингибитор и ингибиторы протеазы (ИП). Стандартом лечения считают тройную схему, и в случае ее эффективности результатом является супрессия вируса до уровня ниже пределов чувствительности современных тестов вирусной нагрузки, что, таким образом, серьезно уменьшает возникновение резистентности и улучшает качество жизни больного. Один из критически важных путей жизненного цикла ретровируса заключается в процессинге полипротеиновых предшественников аспарагиновой протеазой. Например, в ВИЧ белок gag-pol процессирован ВИЧ-протеазой. Для сборки инфекционных вирионов необходим корректный процессинг предшественников полипротеинов аспарагиновой протеазой, что, таким образом, делает аспарагиновую протеазу выгодной целью для противовирусной терапии. В частности, выгодной целью для лечения ВИЧ является ВИЧ-протеаза. Ингибиторы ВИЧ-протеазы стали основой в лечении заболевания ВИЧ, в особенности у больных с продолжительным анамнезом противоретровирусной терапии, и появление ИП (ингибиторов протеазы) привело к большому прорыву в лечении ВИЧ-1 инфекции, существенно снижая заболеваемость и летальность у инфицированных людей. Вместе с тем их продолжительное применение ограничено различными факторами: недостаточное согласие из-за высокой лекарственной нагрузки и ограничений в питании, особенно при схемах введения единственного ИП без совместного введения низких доз ритонавира или при схемах введения двух ИП; побочные эффекты (например, липодистрофия, расстройства метаболизма), серьезно влияющие на качество жизни; и появление ВИЧ-изолятов, которые перестали ингибироваться применяемыми ИП и во многих случаях также являются устойчивыми к другим современным известным ИП в силу высокого уровня перекрестной резистентности в пределах этого класса. Все доступные в настоящее время ингибиторы протеазы (ИП) имеют фармакокинетические профили, ограничивающие их эффективность. Как и многие другие лекарственные средства, ингибиторы протеазы (ИП) и ненуклеозидные ингибиторы обратной транскриптазы (NNRTI) тщательно метаболизируются системой цитохрома P450. Цитохром P450 представляет собой группу находящихся в печени и кишечнике ферментов, имеющих ряд функций в организме человека. Одной из функций является расщепление и выведение лекарств и других химических веществ. Прием двух или более лекарственных средств, метаболизируемых цитохромомP450, может вызвать лекарственное взаимодействие, влияющее на концентрации одного или обоих препаратов, и стать причиной побочных эффектов или нарушения клинической эффективности лекарства(лекарств). Активность цитохрома P450 у разных людей и в разных популяциях является различной. Незначительные генетические изменения могут влиять на количество экспрессируемых специфических ферментов и в этой связи на скорость метаболизма лекарственного средства. Ферменты цитохрома P450,происходящие из определенного гена, называются изоформами. На основе сходства химического состава изоформы разделяют на семейства и субсемейства. Варианты ферментов описывают посредством системы цифр и букв, отражающих их химическую и генетическую структуру. Цитохром P450, субсемейство IIIA (нифедипин-оксидаза), полипептид 4, также называемыйCYP3A4, представляет собой один конкретный метаболический путь, используемый для расщепления и выведения лекарств и других веществ. Множество лекарственных средств, включающих в себя некоторые ингибиторы ВИЧ-протеазы, метаболизируются цитохромом P450-монооксигеназой, что приводит к неблагоприятной фармакокинетике и необходимости более частого введения и превышения наиболее желательных доз. Введение таких лекарственных средств с веществом, ингибирующим метаболизм посредством цитохрома P450 монооксигеназы, будет улучшать фармакокинетику лекарственного средства. В современной клинической практике большинство ингибиторов ВИЧ-протеазы соединяют с ритонавиром для улучшения воздействия и, таким образом, для повышения клинической эффективности. Этот тип применяемого лекарственного взаимодействия называют бустингом. Бустинг также способст-1 016340 вует упрощению схем лечения современными ИП путем снижения лекарственной нагрузки и частоты ежедневного приема. Ритонавир, являющийся сам по себе ИП, в качестве бустера обычно применяется на субтерапевтическом уровне в дозе 100 мг два раза в день. Фармакологическое усиление посредством бустинга ритонавиром опосредуется путем ингибирования цитохрома P450 (CYP) 3A4 и переносчиков лекарственного средства, в частности P-гликопротеина. К сожалению, использование даже низких доз ритонавира для бустинга схем ИП не лишено риска. Токсичность ритонавира, включающая в себя воздействие на желудочно-кишечный тракт, повышает риск гепатотоксичности и обычно вызывает повышение уровня липидов и холестерина в сыворотке.(Sulkowski и др., JAMA, 2000; 283:74-80). Среди этих потенциальных побочных эффектов наибольшее беспокойство вызывает дислипидемия, поскольку она может значительно увеличить риск сердечнососудистых и цереброваскулярных нарушений. Таким образом, в медицине существует большая необходимость в альтернативах ритонавиру в качестве бустера для эффективного и безопасного лечения ВИЧ, при котором альтернативные соединения улучшают фармакокинетический профиль лекарственных средств, метаболизируемых цитохромом P450. Согласно настоящему изобретению было выявлено, что соединения, имеющие формулу улучшают фармакокинетику лекарственного средства, представляющего собой ингибитор протеазы. Химические названия и соответствующие им химические структуры каждого из двух стереоизомеров соединений согласно формуле (II), подходящих для использования в настоящем изобретении, представляют собой 5-тиазолилметил[(1R,2S)-3-(2-амино-6-бензоксазолил)сульфонил](2-метилпропил)амино]-2 гидрокси-1-(фенилметил)пропил]карбамат. В предпочтительном варианте осуществления настоящего изобретения соединения, имеющие формулу (IIa) или (IIb), используют для улучшения фармакокинетики лекарственного средства, при этом указанное лекарственное средство метаболизируется цитохромом P450 или более предпочтительно цитохромом P450-монооксигеназой 3A4. Соединения, имеющие формулу (IIa) или (IIb), также применяются для улучшения фармакокинетики лекарственного средства, при этом указанное лекарственное средство ингибируется действием транспортных белков, таким как действие P-гликопротеина. Соединения, имеющие формулу (IIa) или (IIb),также используют для улучшения фармакокинетики лекарственного средства, при этом указанное лекарственное средство ингибируется действием белков множественной лекарственной устойчивости, связанных с каналом высвобождения, таких как MRP1 или MRP2. Белки множественной лекарственной устойчивости (MRP) составляют транспортное субсемейство АТФ-связывающей кассеты (ABC), как выявлено-2 016340 авторами Borst et al., BBA, 1461, 347-357, 1999). Первым описанным белком был MRP1. Предпочтительные сульфонамидные соединения, используемые в настоящем изобретении, представляют собой соединения, имеющие формулу (IIa) или (IIb), наиболее предпочтительным является соединение, имеющее формулу (IIa), на которое в дальнейшем ссылаются как на соединение A. Соединение A, 5-тиазолилметил[(1S,2R)-3-(2-амино-6-бензоксазолил)сульфонил]-(2-метилпропил) амино]-2-гидрокси-1-(фенилметил)пропил]карбамат, раскрытое в WO 02/092595, обладает активностью против дикого типа ВИЧ-1 in vitro и также обладает активностью против большой группы вирусов, устойчивых к известным в настоящее время ИП. В настоящее время выявлено, что соединения настоящего изобретения, имеющие формулу (IIa) и, в частности, формулу (IIb), более конкретно имеющие формулу (IIa) или (IIb), обладают неожиданными свойствами. Каждое из этих соединений, и в особенности соединение A (формула IIa) и соединение E (формулаIIb), в клинических испытаниях по лечению ВИЧ-инфекций повышает в плазме кроликов уровень дарунавира, нового ингибитора протеазы. Дарунавир, называемый также TMC 114, имеет следующее химическое название: (3R,3aS,6aR)-гексагидрофуро[2,3-b]фуран-3-ил N-[(1S,2R)-1-бензил-2-гидрокси-3-(N1 изобутилсульфаниламидо)пропил]карбамат. Известно, что саквинавир, другой ингибитор протеаз, также является субстратом для метаболизма CYP3A4. Показано, что низкие дозы ритонавира заметно повышают концентрации в плазме саквинавира, что позволяет снизить дозы от 1200 мг три раза в день при его единственном применении, до 1000 мг два раза в день с применением ритонавира в дозе 100 мг два раза в день. С различными уровнями дозы было выявлено, что соединения настоящего изобретения, имеющие формулу (IIa) или (IIb), и наиболее предпочтительно соединение A (IIa), улучшают фармакокинетический профиль саквинавира у здоровых людей-добровольцев. Предпочтительные уровни дозы соединений, имеющих формулу (IIa) или (IIb), варьируют от 10 до 1200 мг/день, предпочтительно от 10 до 800 мг/день (например, 120, 320 или 800 мг/день), более предпочтительно от 20 до 400 мг/день и наиболее предпочтительно от 10 до 150 мг/день. Предпочтительными являются дозы, выбираемые из уровней, состоящих из 40, 60, 80 или 120 мг/день. Везде при использовании термина "улучшение фармакокинетики лекарственного средства" он обозначает (применительно к ситуации, когда лекарственное средство вводят единственным), например,увеличенную биодоступность лекарственного средства, выражаемую в терминах AUC (область под кривой зависимости "концентрации от времени"), повышение уровня в крови вводимого лекарственного средства, более конкретно увеличение падения концентрации (Cmin) или пика (Cmax) в плазме лекарственного средства, или увеличение периода полувыведения рассматриваемого лекарственного средства, где увеличение указанного периода полувыведения составляет, по меньшей мере, однократное превышение периода полувыведения лекарственного средства без бустинга, предпочтительно по меньшей мере в 1,25 раз превышение периода полувыведения лекарственного средства без бустинга, более предпочтительно превышение по меньшей мере в 1,4 раза или в 1,5 раза периода полувыведения лекарственного средства без бустинга и наиболее предпочтительно по меньшей мере в 1,75 раза выше периода полувыведения указанного лекарственного средства без бустинга. Наиболее предпочтительным является увеличение по меньшей мере в 2 раза периода полувыведения лекарственного средства без бустинга. Во избежание сомнений приведенные в настоящей заявке примеры обеспечивают в этом отношении дополнительные указания. Необходимо широко понимать термин "лекарственное средство", включающий в себя, среди прочего, любое соединение, которое метаболизируется цитохромом P450 или ингибируется действием транспортного белка, такого как P-гликопротеин, или ингибируется действием белков множественной лекарственной устойчивости, связанных с каналом высвобождения, таких как MRP1 или MRP2, или является ингибитором протеазы, предпочтительно ингибитором ВИЧ-протеазы. Соединения и препараты, раскрытые в настоящем изобретении, при желании могут находиться в виде так называемого пролекарства. "Пролекарство" означает фармакологически приемлемые производные, такие как сложные эфиры, амиды и фосфаты, таким образом, что получаемый in vivo продукт биотрансформации производного является активным соединением, обозначаемым формулой (IIa) или (IIb),или рассматриваемым лекарственным средством. В целом описание пролекарств авторами Goodman andDrugs", pp 13-15) включено в настоящее изобретение со ссылкой. Пролекарства используемого в настоящем изобретении соединения изготовляют путем модификации функциональных групп, присутствующих в соединении, посредством расщепления модификаций как с помощью стандартных процедур, так иin vivo, до исходного соединения. Пролекарства включают в себя соединения, используемые в настоящем изобретении, в которых гидроксигруппа, например гидроксигруппа на асимметричном атоме углерода, или аминогруппа присоединена к любой группе, которая при введении пролекарства больному отщепляется для образования, соответственно, свободного гидроксила или свободной аминогруппы. Типичные примеры пролекарств описаны, например, в патентах WO 99/33795, WO 99/33815, WO 99/33793, WO 99/33792 и WO 03/090690, включенных в настоящее изобретение в качестве ссылки.-3 016340 По сравнению с исходными соединениями пролекарства отличаются улучшенной растворимостью в воде, увеличенной биодоступностью и легко метаболизируются in vivo в активные ингибиторы. Цель настоящего изобретения состоит в том, что лекарственное средство, подвергнутое бустингу соединениями, имеющими формулу (IIa) или (IIb), является предпочтительно ингибитором протеаз, например ингибитором ВИЧ-протеазы, более конкретно ингибитором аспарагиновой ВИЧ-протеазы. Ингибитор протеазы выбирают из группы, состоящей из дарунавира, ампренавира, фосампренавира, ритонавира, нелфинавира, саквинавира, индинавира, лопинавира, ласинавира, атазанавира, BMS 186316, DPC 681, DPC 684, типранавира, AG 1776, DMP 450, L 756425, PD178390, PNU 140135 или ингибиторов гликозилирования, таких как кастаноспермин, деоксиножиримицин. В частности, ингибитор протеазы выбирают из группы, состоящей из дарунавира, ампренавира, фосампренавира, ритонавира,нелфинавира, саквинавира, индинавира, лопинавира, ласинавира, атазанавира или типранавира. Указанные ингибиторы протеазы хорошо известны специалистам в данной области техники. В качестве примера ласинавир представляет собой амид 5(S)-(трет-бутоксикарбониламино)-4(S)-гидрокси-6 фенил-2(R)-(2,3,4-триметоксифенилметил)гесканоил-N-(2-метоксиэтил)валина. Предпочтительные варианты осуществления для бустинга соединений, имеющих формулу (IIa) или(IIb), являются вариантами, в которых ингибитор протеазы выбирают из группы, состоящей из дарунавира, ампренавира, фосампренавира, ритонавира, нелфинавира, саквинавира, индинавира, лопинавира, ласинавира, атазанавира или типранавира. Наиболее предпочтительные варианты осуществления для бустинга соединений, имеющих формулу (IIa) или (IIb), представляют собой варианты, в которых ингибитор протеазы, соответственно, является дарунавиром или саквинавиром. Наиболее предпочтительным для бустинга соединений, имеющих формулу (IIa), является вариант осуществления, в котором ингибитор протеазы представляет собой дарунавир. Целью настоящего изобретения также является фармацевтическая композиция, содержащая соединение, имеющее формулу (IIa), фармацевтически приемлемый носитель и лекарственное средство, метаболизируемое цитохромом P450. Указанное лекарственное средство в фармацевтической композиции предпочтительно является ингибитором ВИЧ-протеазы, более предпочтительно его выбирают из группы,состоящей из дарунавира, ампренавира, фосампренавира, ритонавира, нелфинавира, саквинавира, индинавира, лопинавира, ласинавира, атазанавира, BMS 186318, DPC 681, DPC 684, типранавира, AG 1776,DMP 450, L 756425, PD 178390, PNU 140135 или ингибиторов гликозилирования, таких как кастаноспермин, деоксиножиримицин. Более предпочтительной является фармацевтическая композиция, в которой указанный ингибитор протеазы представляет собой дарунавир или саквинавир. В наиболее предпочтительной фармацевтической композиции соединения имеют формулу (IIa), и ингибитором протеазы является дарунавир. Соединения, имеющие формулу (IIa) или (IIb) или соответствующие фармацевтические композиции, как указано выше, используют для изготовления лекарственного средства и улучшения фармакокинетики лекарственного средства, предпочтительно для ингибирования активности цитохрома P450 у человека. Целью настоящего изобретения также является использование ингибитора ВИЧ-протеазы, метаболизируемого цитохромом P450, в производстве лекарственного средства для ингибирования активности цитохрома P450 в организме человека, в комбинации с соединениями, содержащими формулу (IIa) или(IIb), является достаточным для улучшения у больного фармакокинетики ингибитора ВИЧ-протеазы по сравнению с фармакокинетикой введения единственного ингибитора ВИЧ-протеазы. Другой целью настоящего изобретения является фармацевтический набор, содержащий фармацевтическую композицию, имеющую соединения формулы (IIa) или (IIb) и наиболее предпочтительно соединения формулы (IIa), фармацевтически приемлемый носитель и лекарственное средство, метаболизируемое цитохромом P450. Лекарственное средство, метаболизируемое цитохромом P450, является ингибитором ВИЧ-протеазы, таким как дарунавир или саквинавир. Также целью настоящего изобретения является способ улучшения фармакокинетики лекарственного средства, которое метаболизируется цитохромом P450, где способ содержит введение в организм человека, нуждающегося в таком лечении, терапевтически эффективного количества комбинации указанного лекарственного средства или его фармацевтически приемлемой соли и соединений, содержащих формулу (IIa) или (IIb) или их фармацевтически приемлемую соль. Другой целью настоящего изобретения является способ ингибирования цитохрома P450, содержащий введение в организм человека, нуждающегося в таком лечении, такого количества соединений, содержащих формулу (IIa) или (IIb) или их фармацевтически приемлемую соль, которое эффективно для ингибирования цитохрома P450. Для терапевтического применения соли соединений формулы (IIa) или (IIb) представляют собой соли, в которых противоион является фармацевтически или физиологически приемлемым. Вместе с тем,соли, имеющие фармацевтически неприемлемый противоион, могут также иметь применение, например,в изготовлении или очистке фармацевтически приемлемого соединения формулы (IIa) или (IIb). Все соли, фармацевтически как приемлемые, так и неприемлемые, включены в объем настоящего изобретения.-4 016340 Фармацевтически приемлемые или физиологически переносимые формы аддитивных солей, которые способны образовывать соединения настоящего изобретения, можно удобно изготовлять с использованием подходящих кислот, таких как, например, неорганические кислоты, такие как галогеноводородные кислоты, например соляная или гидробромистая кислота; серная; геми-серная, азотная; фосфорная и т.п. кислоты; или органические кислоты, такие как, например, уксусная, аспарагиновая кислота, додецилсерная, гептаноевая, гексаноевая, никотиновая, пропаноевая, гидроксиуксусная, молочная, пировиноградная, щавелевая, малоновая, янтарная, малеиновая, фумаровая, яблочная, виннокаменная, лимонная,метансульфоновая, этансульфоновая, бензолсульфоновая, р-толуолсульфоновая, цикламовая, салициловая, р-аминосалициловая, памовая и подобные кислоты. С другой стороны, указанные кислотно-аддитивные формы солей можно превращать обработкой подходящим основанием в форму свободного основания. Соединения формулы (IIa) или (IIb), содержащие кислый протон, также можно превращать в нетоксичную форму их аддитивных солей металлов или аминов путем обработки подходящими органическими и неорганическими основаниями. Подходящие формы основных солей содержат, например, соли аммония, щелочь и соли щелочно-земельных металлов, например лития, натрия, калия, магния, соли кальция и т.п., соли органических оснований, например соли бензатина, N-метила, -D-глюкамина, гидрабамина, и соли аминокислот, такие как, например, аргинин, лизин и т.п. С другой стороны, указанные основные аддитивные формы солей можно превращать обработкой подходящей кислотой в форму свободной кислоты. Термин "соли" также охватывает гидраты и сольвентные аддитивные формы, которые могут образовывать соединения настоящего изобретения. Примерами таких форм являются, например, гидраты,алкоголяты и т.п. Рассматриваемое соединение, используемое в настоящем изобретении, также может существовать в стереохимически изомерической форме, указывая на все возможные соединения, составленные из одних и тех же атомов, связанных той же последовательностью связей, но имеющие различные трехмерные структуры, не являющиеся взаимозаменяемыми. Если не упомянуто или обозначено иначе, химическое обозначение соединений охватывает смесь всех возможных стереохимически изомерических форм, которые могут иметь указанные соединения. Указанная смесь может содержать все диастереоизомеры и/или энантиомеры основной молекулярной структуры указанного соединения. Все стереохимически изомерические формы соединений, используемых в настоящем изобретении, как в чистой форме, так и в смеси друг с другом, входят в объем настоящего изобретения. Чистые стереоизомерические формы соединений и промежуточные продукты, упомянутые в настоящем изобретении, определяют как изомеры, по существу, свободные от других энантиомерических или диастереомерических форм этой же основной молекулярной структуры указанных соединений или промежуточных продуктов. В частности, термин "стереоизомерически чистый" относится к соединениям или к промежуточным продуктам, имеющим избыток стереоизомеров, составляющий по меньшей мере от 90% (т.е. минимум 90% одного изомера и максимум 10% других возможных изомеров) до 100% избытка стереоизомеров (т.е. 100% одного изомера и отсутствие другого), более конкретно к соединениям или промежуточным продуктам, имеющим избыток стереоизомеров от 90 до 100%, более конкретно к имеющим избыток стереоизомеров от 94 до 100% и наиболее конкретно к имеющим избыток стереоизомеров от 97 до 100%. Подобным образом необходимо понимать термины "энантиомерически чистый" и"диастереомерически чистый", но в этом случае они относятся, соответственно, к избытку энантиомеров,избытку диастереомеров рассматриваемой смеси. Применяя известные в данной области техники процедуры, можно получать чистые стереоизомерические формы соединений и промежуточных продуктов, используемых в настоящем изобретении. Например, энантиомеры можно отделять друг от друга селективной кристаллизацией их диастереомерических солей с оптически активными кислотами или основаниями. Их примерами являются виннокаменная кислота, дибензоилвиннокаменная кислота, дитолуоилвиннокаменная кислота и камфосульфоновая кислота. Альтернативно, энантиомеры можно разделять хроматографическими способами, используя хиральные постоянные фазы. Указанные чистые стереохимически изомерические формы также можно получать из соответствующих чистых стереохимически изомерических форм подходящих исходных материалов, при условии, что реакция осуществляется стереоспецифично. Если желательно получение конкретного стереоизомера, предпочтительно синтезировать указанное соединение стереоспецифичными способами изготовления. Предпочтительно использовать для этих способов энантиомерически чистые исходные материалы. Специалисту в данной области техники понятно, что соединения формулы (IIa) или (IIb) содержат два центра асимметрии и, таким образом, могут существовать в виде различных стереоизомерических форм. Абсолютную конфигурацию каждого центра асимметрии, который может присутствовать в соединениях формулы (IIa) или (IIb), можно обозначать стереохимическими дескрипторами R и S, эти примечания R и S соответствуют правилам, описанным в Pure Appl. Chem. 1976, 45, 11-30. То же самое применимо к формуле (II).-5 016340 Предполагается, что настоящее изобретение включает в себя все изотопы атомов, встречающихся в соединениях настоящего изобретения. Изотопы включают в себя атомы, имеющие подобные атомные числа, но разные массовые числа. Согласно общему примеру, изотопы водорода включают в себя без ограничения тритий и дейтерий. Изотопы углерода включают в себя C-13 и C-14. Таким образом, настоящие соединения можно применять у животных, предпочтительно у млекопитающих, и в частности, у людей, в качестве лекарств per se, в смесях друг с другом или в форме фармацевтических препаратов. Настоящее изобретение касается фармацевтических препаратов, которые в дополнение к общепринятым фармацевтически инертным наполнителям и добавкам в качестве активного компонента содержат эффективную дозу соединений формулы (IIa) или (IIb), более предпочтительно (IIa), и лекарственное средство, метаболизируемое цитохромом P450. Фармацевтические препараты можно изготовлять способом, по существу, известным рядовому специалисту в данной области техники. С этой целью соединение формулы (IIa) вместе с одним или более твердыми или жидкими фармацевтическими наполнителями и/или вспомогательными веществами и, если желательно, в комбинации с другими фармацевтическими активными соединениями приводят в подходящую для введения форму или лекарственную форму, которую затем можно использовать в виде фармацевтической продукции в медицине человека или в ветеринарии. Специалист в данной области техники на основе своих специальных знаний знаком с вспомогательными веществами, подходящими для желательной фармацевтической рецептуры. Помимо растворителей также являются полезными гелеобразующие вещества, основы для суппозиториев, вспомогательные вещества для таблетирования и другие активные соединения носителей, антиоксиданты, диспергирующие вещества, эмульгаторы, ароматизирующие корригенты, консерванты, солюбилизаторы, средства для создания эффекта депо, буферные вещества или красители. Фармацевтические препараты, содержащие такие соединения, вводят перорально, парентерально,например внутривенно, ректально, путем ингаляции или местно, и предпочтительный путь введения зависит от отдельного случая, например конкретного течения заболевания, которое необходимо лечить. Предпочтительным является пероральный прием. В лекарственной форме для перорального приема соединения смешивают с подходящими добавками, такими как наполнители, стабилизаторы или инертные разбавители, и посредством общепринятых способов приводят в подходящие для введения формы, такие как таблетки, таблетки, покрытые оболочкой, твердые капсулы, водные, спиртовые или масляные растворы. Примерами подходящих инертных носителей являются гуммиарабик, магнезия, карбонат магния, фосфат калия, лактоза, глюкоза или крахмал, в частности кукурузный крахмал. В этом случае препарат можно изготовлять в виде или сухих, или влажных гранул. Подходящими масляными наполнителями или растворителями являются растительные масла или животные масла, такие как подсолнечное масло или жир печени трески. Подходящими растворителями для водных или спиртовых растворов являются вода, этанол, сахарные растворы или их смеси. В качестве дополнительных вспомогательных веществ для других форм введения также полезными являются полиэтиленгликоли и полипропиленгликоли. Для подкожного или внутривенного введения активные соединения, если желательно, вместе с общепринятыми для этих целей веществами, такими как солюбилизаторы, эмульгаторы или дополнительные вспомогательные вещества, создают в виде раствора, (нано)суспензии или эмульсии. Соединение формулы (IIa) также можно лиофилизировать, и полученные лиофилизаты использовать, например, для производства препаратов для инъекций или инфузий. Подходящими растворителями являются, например, вода, физиологический солевой раствор или спирты, например этанол, пропанол, глицерин, кроме того, также можно упомянуть сахарные растворы, такие как растворы глюкозы или маннитола, или альтернативно, смеси различных растворителей. Подходящие фармацевтические рецептуры для введения в форме аэрозолей или распыляемых растворов представляют собой, например, растворы, суспензии или эмульсии соединения формулы (IIa) или их физиологически приемлемые соли в фармацевтически приемлемом растворителе, такие как этанол,или вода, или смесь таких растворителей. При необходимости рецептура дополнительно может содержать также другие фармацевтические вспомогательные вещества, такие как сурфактанты, эмульгаторы и стабилизаторы, а также пропелленты. Такой препарат обычно содержит активное соединение в концентрации от приблизительно 0,1 до 50%, в частности от приблизительно 0,3 до 3 вес.%. В частности, можно создавать рецептуру соединения настоящего изобретения в фармацевтической композиции, содержащей терапевтически эффективное количество частиц, состоящих из твердой дисперсии, содержащей: (a) соединение формулы (IIa) и (b) один или более фармацевтически приемлемых водорастворимых или водонерастворимых полимеров. Термин "твердая дисперсия" определяет систему в твердом состоянии (в противоположность жидкому или газообразному состоянию), содержащую по меньшей мере два компонента, в которой один компонент более или менее равномерно диспергирован во всем объеме другого компонента или компонентов. Если указанная дисперсия компонентов такова, что вся система химически и физически однородна или гомогенна или, как определяет термодинамика, состоит из одной фазы, такую твердую дис-6 016340 персию называют "твердым раствором". Твердые растворы являются предпочтительными физическими системами, поскольку для организмов, которым их вводят, их компоненты обычно легко биодоступны. Термин "твердая дисперсия" также охватывает дисперсии, менее гомогенные во всем объеме, чем твердые растворы. Такие дисперсии не являются химически и физически однородными во всем объеме или содержат более одной фазы. Водорастворимым полимером в частицах обычно является полимер, имеющий видимую вязкость от 1 до 100 мП при растворении в 2%-м водном растворе при температуре раствора 20C. Предпочтительными водорастворимыми полимерами являются гидроксипропилметилцеллюлоза(ГПМЦ) или гидроксипропилметилцеллюлозы ацетатсукцинат (ГПМЦ-АС). ГПМЦ, имеющая степень замещения метокси- от около 0,8 до около 2,5 и гидроксипропилмолярное замещение от около 0,05 до около 3,0, обычно является водорастворимой. Степень замещения метокси относится к среднему количеству присутствующих метилэфирных групп на ангидроглюкозную единицу молекулы целлюлозы. Молярное замещение гидроксипропила относится к среднему количеству молей пропиленоксида, которые прореагировали с каждой ангидроглюкозной единицей молекулы целлюлозы. Согласно вышеприведенному определению частицы можно изготовлять, начиная с приготовления твердой дисперсии компонентов и затем, необязательно, измельчая или перемалывая эту дисперсию. Для изготовления твердой дисперсии существуют различные способы, включающие в себя вытеснение из расплава, распылительную сушку и выпаривание раствора. Путь введения может зависеть от состояния субъекта, сопутствующего лечения и тому подобного. Вводимая доза соединений настоящего изобретения или их физиологически приемлемой соли (солей) зависит от конкретного случая, и общепринято, что для оптимального эффекта ее необходимо адаптировать к условиям конкретного случая. Таким образом, она, безусловно, зависит от частоты введения и от силы и продолжительности действия соединений, применяемых в каждом случае для лечения или профилактики, а также от характера и тяжести инфекции и симптомов и от пола, возраста, веса, сопутствующего лечения и индивидуальной ответной реакции человека или животного, нуждающегося в лечении, и от того, является ли лечение неотложным или профилактическим. Дозу можно вводить в отдельной лекарственной форме или разделять на несколько, например на две, три или на четыре отдельные дозы. Другой аспект настоящего изобретения касается комплекта или контейнера, содержащего соединение формулы (IIa), необязательно вместе с ингибитором протеазы, таким как саквинавир или дарунавир,в количестве, эффективном для использования в качестве стандарта или реактива в тесте или в исследовании для определения способности потенциального фармацевтического препарата ингибировать ВИЧпротеазу, рост ВИЧ или и то, и другое. Этот аспект может найти использование в программах фармацевтических исследований. Альтернативно, для лечения больных с диагностированным СПИДом/ВИЧинфекцией можно создавать рецептуру соединений, имеющих формулу (IIa), в одной капсуле, таблетке или шприце с ингибитором протеазы, или дарунавиром, или саквинавиром. Примеры 1. Приготовление 5-тиазолилметил-[(1S,2R)-3-(2-амино-6-бензоксазолил)сульфонил](2-метилпропил)амино]-2-гидрокси-1-фенилметил)пропил]карбамата. Обычный способ приготовления 5-тиазолилметил-[(1S,2R)-3-(2-амино-6-бензоксазолил)сульфонил](2-метилпропил)амино]-2-гидрокси-1-фенилметил)пропил]карбамата (соединение 1-4) раскрыт в-7 016340 Смесь 1,15 г тиазол-5-ил-метанола (1-1) и 1,2 г триэтиламина (ТЭА) в 25 мл дихлорметана (ДХМ) перемешивали при комнатной температуре в атмосфере азота. Затем добавляли 2,56 г N,N'дисукцинимидилкарбоната и получаемую смесь перемешивали в течение 10-15 мин. Раствор перемешивали в течение дополнительных 2 ч. Полученный промежуточный продукт (1-2) напрямую использовали в последующей реакции с амином (1-3). Вместо аминов также можно использовать их соли. К 40 мл дихлорметана добавляли 2 г триэтиламина и 5 г амина (1-3), в котором R2 является фенилом; R3 является изобутилом; R4 является водородом и R5 является водородом, и полученную смесь перемешивали при комнатной температуре. После этого по каплям добавляли часть раствора, содержащего 1-2. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь промывали водой и затем высушивали для получения соединения (1-4). Стереоизомеры соединения (1-4) приготовляли аналогичным химическим способом. Проводили анализ соединения (1-4), в котором R2 является фенилом; R3 является изобутилом; R4 является водородом и R5 является водородом, представленного таким образом формулой (IIa). Все реактивы приобретали из коммерческих источников (фирмы Acros, Aldrich или Fluorochem) и использовали как есть. Спектры ЯМР регистрировали на спектрометре Bruker Avance 400 с рабочим режимом 400 МГц для 1H с CDCl3 в качестве растворителя. В каждом случае использовали тетраметилсилан (TMC) как внутренний стандарт. Химические изменения даны в промилле. Множественность обозначена с использованием следующих сокращений: d - двойной, t - тройной, m - множественный и т.д. Масс-спектрометрию с низкой разрешающей способностью (LRMS) выполняли на ионной ловушке(ThermoFinnigan LCQ Deca) или проводили времяпролетную масс-спектрометрию (Waters LCT) с использованием электрораспылительной ионизации (ЭСИ) с режимом определения положительных ионов. Проводили колоночную хроматографию с силикагелем 60, 60-200 мкм (ROCC). Тонкослойную хроматографию с силикагелем 60 F254 проводили на пластине (Merck). Аналитическую высокоэффективную жидкостную хроматографию ВЭЖХ выполняли на системе Waters Alliance 2690 (насос + автосемплер),оборудованной детектором на фотодиодной матрице Waters 996. Для проверки чистоты конечных продуктов использовали следующую хроматографическую систему. Колонка: Waters MS Xterra C18 (3,5 мкм, 4,60 мм 100 мм), мобильная фаза A: 10 мМ CH3COONH4 в H2O, мобильная фаза B: CH3CN. Анализ проводили при 30C с использованием скорости потока 1 мл/мин, применяя следующий градиент: 0 мин: 5% B, 10 мин: 95% B, 12 мин: 95% B. В каждом случае вводили 10 мкл 1 мМ раствора. Время установления равновесия между двумя проходами составляло 3 мин. Пики элюирования выявляли при единственной длине волны (max). Время удерживания обозначено в минутах. Данные для (IIa):-8 016340 Определение чистоты: Rt=7,27 мин, чистота: 96,56%. 2. Действие 5-тиазолилметил-[(1S,2R)-3(2-амино-6-бензоксазолил)сульфонил](2-метилпропил) амино]-2-тидрокси-1-фенилметил)пропил]карбамата на фармакокинетику ингибитора протеазы саквинавира у здоровых добровольцев мужского пола. Раствор 5-тиазолилметил-[(1S,2R)-3-(2-амино-6 бензоксазолил)сульфонил](2-метилпропил)амино]-2-гидрокси-1-фенилметил)пропил]карбамата (в дальнейшем называемый соединением A), представленный химической формулой использовали в фазе I открытого рандомизированного испытания на здоровых субъектах для исследования состояния фармакокинетики соединения A и его действия на фармакокинетику единственной дозы ингибитора протеазы саквинавира. Три группы из 8 здоровых субъектов принимали однократную дозу 1000 мг саквинавира единственного и принимали соединение A. Проводили сравнение фармакокинетики этих двух приемов саквинавира. В 1 день все субъекты во всех группах получили однократную дозу саквинавира 1000 мг. С 4 до 10 дня одна группа здоровых добровольцев (группа 1) получала соединение A в дозе 60 мг 2 раза в день,одна группа получала 160 мг соединения A 2 раза в день (группа 2), и одна группа получала соединениеA в дозе 400 мг 2 раза в день (группа 3). В 9 день все субъекты принимали однократную дозу саквинавира 1000 мг одновременно с соединением A. Полные фармакокинетические профили соединения A определяли на 4 день, на 8 день и на 9 день. Полные фармакокинетические профили саквинавира определяли в 1 день и на 9 день. Результаты исследования приведены в таблице ниже. Фармакокинетические параметры саквинавира (среднее значениеSD (стандартное отклонение при отсутствии (день 1) и в присутствии соединения A (день 9). Эти результаты показывают, что соединение А существенно улучшает фармакокинетику саквинавира, с полным выделением, выраженным в AUC, увеличенным в 20 раз для всех уровней оцениваемых доз соединения A. 3. Ингибирование in vitro цитохрома P450, конкретно CYP3A4. Была изучена константа ингибирования Ki соединения A на опосредованном CYP450 3A4 метаболизме тестостерона в микросомах печени человека. Результаты этого эксперимента показали, что соединение A является мощным ингибитором 6-гидроксилирования тестостерона. Соединение A в экспериментах в качестве ингибитора CYP3A4 - опосредованного метаболизма, проявляло такую же силу действия, как ритонавир, и IC50 составляла от 100 до 25 нм. Режим ингибирования CYP3A4 соединением A можно описать неконкурентной моделью ингибирования с константой ингибирования Ki, составляющей 65 нМ. Было невозможно установить значения Km для метаболизма, используя микросомы человека, крысы и собаки, несмотря на последовательные инкубации при понижающих концентрациях. В самой низкой тестируемой концентрации (50 нм) скорость метаболизма соединения A оставалась сходной со скоростью, наблюдаемой при 10 мкМ. 4. Действие соединения A на транспорт ингибитора протеазы через монослои клеток Caco-2. Транспорт экспериментальных ингибиторов протеазы изучали в выращенных до конфлуэнтности монослоях Caco-2 (Augustijns et al. (1998). Int. J. of Pharm., 166, 45-54). После подтверждения целостности-9 016340 клеточного монослоя экспериментальные ингибиторы ВИЧ-протеазы, как описано в WO 02/083657, соединение B и соединение C (химические структуры приведены ниже), наносили или на апикальную (AP),или на базолатеральную (BL) сторону клеточных монослоев для изучения транспорта в направлении отAP к BL и от BL к AP соответственно. Измеряли эффекты соединения A и ингибитора P-гликопротеина(P-др) верапамила (100 мкМ) на двунаправленный транспорт. Результаты приведены в таблице ниже. Сравнение значений скорости высвобождения (CB) (90 мин) в отсутствие и в присутствии верапамила и соединения A (100 мкМ) для экспериментальных ингибиторов протеазы соединения B и соединения C (30 мкМ) Оба ингибитора протеаз имели очень сопоставимый профиль, поскольку все показали очень высокую направленность переноса при высоком превышении секреторного транспорта над абсорбирующим транспортом при низких концентрациях (3-30 мкМ). Верапамил, который является известным маркерным ингибитором P-др, а также соединение A значительно уменьшают направленность переноса. Верапамил и соединение A с равной мощностью снижают высвобождение, из чего ясно предполагается, что соединение A является ингибитором P-др. 5. Действие in vivo соединения A на фармакокинетику дарунавира у кроликов. Способность соединения A улучшать фармакокинетику нового ИП дарунавира в исследовании для лечения ВИЧ-инфекций оценивали на кроликах. В качестве вида модели были выбраны самки кролика, в силу схожести их профиля метаболизма дарунавира с профилем у людей и в силу того, что они оказались репрезентативной и чувствительной моделью животных для изучения эффекта бустинга на биодоступность дарунавира. Четырем кроликам перорально вводили соединение A в дозе 20 мг/кг в 0 и 6 ч в течение 2 дней подряд. На второй день сразу после приема дозы соединения A в 0 ч перорально давали единственную дозу 500 мг/кг дарунавира. Полученные в результате фармакокинетические параметры дарунавира после перорального введения с соединением A и без него приведены в таблице ниже. Лечение соединением A привело к значительному увеличению фармакокинетических параметров вводимого совместно ингибитора протеазы дарунавира. Наблюдали 38-кратное среднее повышение Cmax дарунавира с соединением A и без него, соответственно со средними значениями 10,1 и 0,34 мкг/мл. Среднее значение AUC0-24 ч дарунавира в присутствии соединения A составляло 25,7 мкг в ч/мл по сравнению со значением введения единственного дарунавира 2,2 мкг в ч/мл. Определение относительной биодоступности дарунавира, вводимого в комбинации с соединениемA, проводили вычислением соотношений значений AUC дарунавира после введения с соединением A и значений AUC после введения единственного дарунавира у тех же животных. Средние фармакокинетические параметры дарунавира, вводимого в комбинации с соединением A,увеличивались в 13 раз.- 10016340 Таблица 1 Фармакокинетические параметры дарунавира у кроликов после однократной дозы 500 мг/кг дарунавира с совместным введением 20 мг два раза в день соединения A и без совместного введения Второе исследование на кроликах осуществляли для сравнения эффекта диапазона пероральных доз соединения A и эффекта однократной дозы соединения E, энантиомера соединения A (формула IIb) в качестве бустерных веществ на биодоступность дарунавира. Три группы по 3 самки кроликов породыNZW получали однократную дозу 500 мг/кг дарунавира как единственного с однократной пероральной дозой соединения A 20 мг/кг, так и с однократной пероральной дозой соединения E 20 мг/кг (период I). Во II периоде соединение A давали два раза в день в течение 2 дней в дозах 4, 10 или 20 мг/кг с однократной дозой дарунавира 500 мг/кг утром на 2-й день. В табл. 2 и 3, соответственно, приведены результаты фармакокинетических параметров дарунавира с эффектами соединения A и его энантиомера соединения E и с эффектами различных доз соединения A. Таблица 2 Фармакокинетические параметры дарунавира у кроликов после однократной дозы 500 мг/кг дарунавира с совместным введением однократной дозы 20 мг/кг соединения A или его энантиомера соединения E и без совместного введения- 11016340 Таблица 3 Фармакокинетические параметры дарунавира у кроликов после однократной дозы 500 мг/кг дарунавира при совместном введении 4, 10 и 20 мг два раза в день соединения A Результаты этих экспериментов на кроликах подтверждают, что соединение A является мощным усилителем фармакокинетических параметров дарунавира с дозозависимым эффектом в дозах, варьирующих от 4 до 20 мг/кг два раза в день. При одновременном введении с дарунавиром соединения A в однократной дозе 20 мг/кг усиливает фармакокинетику дарунавира, как если бы схема введения включала 20 мг/кг два раза в день. Совместное введение соединения E, энантиомера соединения A, в однократной дозе 20 мг/кг (формула IIb) также приводило к существенному (более чем в 20 раз) повышению у кроликов уровней концентрации в плазме дарунавира. 6. Действие соединения A in vivo на фармакокинетику дарунавира у приматов. Проводили исследование у самцов обезьян Cynomolgus для оценки эффекта на биодоступность дарунавира бустинга различными дозами соединения A. С точки зрения фармакокинетических перспектив предполагается, что такое исследование у вида приматов будет наилучшим прогнозом для действия у людей. Дарунавир давали в однократной дозе 40 мг/кг с соединением A в дозах на уровне 0 для контроля, 4, 10, 25 и 80 мг/кг и без соединения A. Все группы дозирования состояли из 3 обезьян, за исключением группы дозирования 80 мг/кг, в которую были включены 4 животных. На чертеже отображены эффекты различных уровней доз соединения A на фармакокинетику дарунавира, выраженных как AUC: среднее значение AUC стандартизированной дозы дарунавира у обезьянCynomolgus (N=3-4 обезьяны в группе) по сравнению с бустинг-дозой соединения A в диапазоне дозы от 4 до 80 мг/кг/в день, вводимой по схеме два раза в день в течение 2 дней. Дарунавир давали в однократной дозе 40 мг/кг на 2 день эксперимента. Результаты показывают, что соединение А существенно повышает фармакокинетику дарунавира у обезьян Cynomolgus, начиная с низкой дозы 4 мг/кг (увеличение AUC в 3,2 раза), с заметным максимальным эффектом в высоких дозах 25 и 80 мг/кг примерно в 15 раз. Эти данные показывают, что соединениеA является эффективным и мощным бустером дарунавира у этого вида приматов. 7. Действие соединения А на фармакокинетику ингибитора протеазы дарунавира у людей. Соединение A использовали в открытом рандомизированном испытании в 3 группах, каждая из которых состояла из 8 здоровых добровольцев, для исследования его действия на фармакокинетику ингибитора протеазы дарунавира. Все субъекты получали ингибитор протеазы дарунавир в дозе 600 мг два раза в день в течение 8 дней с совместным введением два раза в день 30, 60 или 120 мг соединения A,начиная с дня 4 и далее. Во всех группах совместное введение соединения А значительно увеличивало значение AUC12 ч,Cmax и Cmin дарунавира. Значение Cmin повышалось наиболее значительно (до 10 раз), и повышение Cmax было наименьшим (менее чем в 2 раза). Увеличение AUC12 ч составляло примерно в 2 раза для лечения A(30 мг соединения A) и от 3 до 4 раз для лечения B (60 мг соединения A) и для лечения C (120 мг соединения A), и предполагается, что соединение A достигало максимального эффекта взаимодействия в дозе 60 мг со схемой введения два раза в день. Это испытание показывает, что соединение A является мощным бустером для клинического применения с дарунавиром. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Применение соединения, имеющего формулу для получения лекарственного вещества для улучшения фармакокинетики лекарственного средства, где лекарственным средством является ингибитор протеазы. 2. Применение по п.1, в котором ингибитором протеазы является ингибитор ВИЧ-аспарагиновой протеазы. 3. Применение по п.2, в котором ингибитор протеазы является дарунавиром или саквинавиром.
МПК / Метки
МПК: A61K 31/426, A61P 31/18
Метки: улучшения, протеазы, фармакокинетики, энантиомера, 5-тиазолилметил[(1s,2r)-3-[[(2-амино-6-бензоксазолил)сульфонил](2-метилпропил)амино]-2-гидрокси-1-(фенилметил)пропил]карбамата, ингибиторов, применение
Код ссылки
<a href="https://eas.patents.su/14-16340-primenenie-5-tiazolilmetil1s2r-3-2-amino-6-benzoksazolilsulfonil2-metilpropilamino-2-gidroksi-1-fenilmetilpropilkarbamata-ili-ego-enantiomera-dlya-uluchsheniya-farmakokinetiki-ingi.html" rel="bookmark" title="База патентов Евразийского Союза">Применение 5-тиазолилметил[(1s,2r)-3-[[(2-амино-6-бензоксазолил)сульфонил](2-метилпропил)амино]-2-гидрокси-1-(фенилметил)пропил]карбамата или его энантиомера для улучшения фармакокинетики ингибиторов протеазы</a>
(4r,5s,6s,7r)-гексагидро-1-[5-(3-аминоиндазол)метил]-3-бутил-5,6-дигидрокси-4,7-бис[фенилметил]-2н-1,3-диазепин-2-он и его применение в качестве ингибитора вич-протеазы.

Номер патента: 1154
Опубликовано: 30.10.2000
Авторы: Роджерс Джеймс Дэвид, Лэм Патрик Юк-Сун
МПК: A61K 31/5513, C07D 403/06
Метки: ингибитора, вич-протеазы, качестве, применение, 4r,5s,6s,7r)-гексагидро-1-[5-(3-аминоиндазол)метил]-3-бутил-5,6-дигидрокси-4,7-бис[фенилметил]-2н-1,3-диазепин-2-он
Формула / Реферат:
1. Соединение формулы I или его фармацевтически приемлемая соль, или его пролекарственная форма, где пролекарство формулы I является соединением, в котором две гидроксигруппы соединены с образованием эпоксида, -OCH2SCH2O-; -ОС(=O)O-; -OCH2O-; -OC(=S)O-; -ОС(=O)С(=O)О-; -ОС(СН3)2O-; -ОС((СН2)3NН2)(СН3)О-; -ОС(ОСН3) (СН2СН2СН3)О-; или -OS(=O)О- группы. 2. Соединение по п.1, где соединение является соединением формулы I. 3. Фармацевтическая...
N-гидрокси-2-(алкил-, арил- или гетероарилсульфанил, -сульфинил или -сульфонил)-3-замещенные алкилов, арилов или гетероариламидов в качестве ингибиторов матриксных металлопротеиназ

Номер патента: 1742
Опубликовано: 27.08.2001
Авторы: Венкатесан Аранапакам Мудумбай, Гросу Джордж Теодор, Дэвис Жами Мари, Бэйкер Жанни Леа
МПК: A61K 31/164, A61P 5/48, C07D 211/66...
Метки: гетероарилсульфанил, сульфонил)-3-замещенные, металлопротеиназ, арилов, алкилов, матриксных, качестве, n-гидрокси-2-(алкил, сульфинил, арил, гетероариламидов, ингибиторов
Формула / Реферат:
1. Соединения формулы I в которой R1 представляет собой алкил из 1-18 углеродных атомов, необязательно замещенный одной или двумя группами, независимо выбранными из R5; алкенил, содержащий 3-18 углеродных атомов с 1-3 двойными связями, необязательно замещенный одной или двумя группами, независимо выбранными из R5; алкинил, содержащий 3-18 углеродных атомов с 1-3 тройными связями, необязательно замещенный одной или двумя группами,...
Способ восстановительного алкилирования для получения n-(2-(r)-гидрокси-1-(s)-инданил)-2(r)-фенилметил-4(s)-гидрокси-5-(1-(4-(3-пиридилметил)-2-(s)-n’-трет.-бутилкарбоксамидо)-пиперазинил)-пентанамида

Номер патента: 635
Опубликовано: 29.12.1999
Авторы: Аскин Дэвид, Чанчози Стивен Дж., Херрнер Роберт С.
МПК: C07D 401/06
Метки: получения, способ, восстановительного, алкилирования, n-(2-(r)-гидрокси-1-(s)-инданил)-2(r)-фенилметил-4(s)-гидрокси-5-(1-(4-(3-пиридилметил)-2-(s)-n'-трет.-бутилкарбоксамидо)-пиперазинил)-пентанамида
Формула / Реферат:
1. Способ восстановительного алкилирования для получения N-(2-(R)-гидрокси-1-(S)-инданил)-2(R)-фенилметил-4-(S)-гидрокси-5-(1-(4-(3-пиридилметил)-2(S)-N'-(трет.-бутилкарбоксамидо)-пиперазинил))-пентанамида структурной формулы или его гидратов, отличающийся тем, что осуществляют взаимодействие в течение, по крайней мере, 5 мин в подходящем растворителе одного эквивалента соединения структурной формулы с избытком...
Способ получения особо чистого фумарата рац-1-{4-[2-гидрокси-3-(5-хинолилокси)пропил]пиперазин-1-ил}-2,2-дифенилэтан-1-она и особо чистый фумарат рац-1-{4-[2-гидрокси-3-(5-хинолилокси)пропил] пиперазин-1-ил}-2,2-дифенилэтан-1-она

Номер патента: 8687
Опубликовано: 29.06.2007
Авторы: Шнайдер Маттиас, Готтфрид Михаэль, Вайнманн Хильмар
МПК: C07D 215/20
Метки: способ, фумарата, получения, фумарат, особо, рац-1-{4-[2-гидрокси-3-(5-хинолилокси)пропил, пиперазин-1-ил}-2,2-дифенилэтан-1-она, чистый, чистого, рац-1-{4-[2-гидрокси-3-(5-хинолилокси)пропил]пиперазин-1-ил}-2,2-дифенилэтан-1-она
Формула / Реферат:
1. Способ получения особо чистого фумарата рац-1-{4-[2-гидрокси-3-(5-хинолилокси)пропил]пиперазин-1-ил}-2,2-дифенилэтан-1-она, отличающийся тем, что сначала: а) эпокситозилат структурной формулы I подвергают взаимодействию с б) 5-гидроксихинолином формулы II и карбонатом цезия в соответствующем растворителе и при соответствующей температуре с образованием 5-(2,3-эпоксипропокси)хинолина формулы III после чего этот 5-(2,3-эпоксипропокси)хинолин...
N-гидрокси-2- (алкил, арил или гетероарилсульфанил, сульфинил или сульфонил) -3-замещенный алкил, арил или гетероариламиды в качестве ингибиторов матричной металлопротеиназы

Номер патента: 3836
Опубликовано: 30.10.2003
Авторы: Гросу Джордж Теодор, Дэвис Жами Мари, Коул Дерек Сесил, Венкатесан Мудумбай Аранапакам, О'делл Мэтью Джеймс, Ху Байхуа, Бэйкер Жанни Леа, Джекобсон Мэрси Памела
МПК: A61K 31/16, C07D 295/08, C07C 323/60...
Метки: 3-замещенный, ингибиторов, металлопротеиназы, качестве, гетероарилсульфанил, сульфонил, сульфинил, матричной, n-гидрокси-2, арил, гетероариламиды, алкил
Формула / Реферат:
1. Соединение формулы где A представляет собой -S-, -SO- или SO2-; R1 представляет собой нафтил, фенил, гетероциклил, выбранный из группы: тиенил, триазолил, пиридил, пиразинил, имидазолил, необязательно бензоконденсированные и необязательно замещенные C1-C6алкилом, C1-C6алкокси или фенилом, арилокси, бензилокси и гетероарилокси; или R1 представляет собой R11-Y-Ph-Z-, где Y представляет собой O, S, NH или простую связь, Z представляет собой...
Предыдущий патент: Полипропиленовые пленки, полученные раздувом
Следующий патент: Одноцентровые каталитические системы, имеющие скорпионоподобную трехмерную структуру
Случайный патент: Соединения пирроло[2,3-d]пиримидина