Циклопентадиенильно-флуоренильные лиганды с углеродными мостиками








Формула / Реферат
1. Способ получения каталитического компонента общей формулы
Ra2C(3,6-Rb2-Flu)(2-Rc-4-Rd-C5H2)MQ2,
где Ra2C является мостиком с одним углеродным атомом и либо оба Ra представляют собой водород, либо один Ra является водородом, а другой Ra является незамещенным или замещенным фенилом, либо оба Ra являются замещенными фенильными группами;
Rb, Rc и Rd независимо выбирают из Н или алкила, имеющего от 1 до 12 атомов углерода, или арильных групп, замещенных или незамещенных, при ограничении, что они не все одновременно являются водородом;
М является металлом 4 группы Периодической системы и
Q является галогеном или алкилом, имеющим от 1 до 12 атомов углеродов, и, при ограничении, что
когда Rc является алкильной группой и один Ra является незамещенной фенильной группой, другой Ra является водородом;
когда Rc является алкильной группой и один Ra является замещенной фенильной группой, другой Ra может быть водородом либо такой же или другой замещенной фенильной группой, и заместители представляют собой электроноакцепторные группы;
когда Rc является водородом, каждый Ra независимо выбирают из Н или незамещенной или замещенной фенильной группы;
при этом указанный способ включает:
а) реакцию нуклеофильного присоединения в растворителе группы (Ra2C-2-Rc-4-Rd-фульвен) с группой [3,6- Rb2-Flu]-[M']+, где М' является Li;
б) гидролиз и отделение полученного лиганда;
в) депротонирование лиганда стадии б) с помощью R'M" для получения дианионного лиганда, где R' является алкилом, имеющем от 1 до 6 атомов углерода, и М"является Li, Na или K;
г) реакцию солевого обмена в растворителе дианионного лиганда стадии в) с MQ4;
д) выделение каталитического компонента.
2. Способ по п.1, в котором заместители на фенильных группах, если присутствуют, находятся в положении 4 для одного заместителя или в положениях 3 и 5 для двух заместителей.
3. Способ по п.1 или 2, в котором оба Rb являются одинаковыми и представляют собой трет-бутил.
4. Способ по любому из предшествующих пунктов, в котором Rc является метилом в положении 2 и Rd является трет-бутилом в положении 4.
5. Способ по любому из предшествующих пунктов, в котором растворителем стадии а) является Et2O.
6. Способ по п.5, в котором растворителем стадии г) также является Et2O.
7. Способ по любому из предшествующих пунктов, в котором М" является Li.
8. Металлоценовый каталитический компонент, полученный способом по любому из пп.1-7.
9. Применение металлоценового каталитического компонента, полученного способом по любому из пп.1-7, в сочетании с подходящим активирующим агентом для получения этилен-пропиленового каучука с содержанием этилена от 8 до 15 мас.% и среднемассовой молекулярной массой по меньшей мере 500 кДа.
10. Применение металлоценового каталитического компонента, полученного способом по любому из пп.1-7, в сочетании с подходящим активирующим агентом для получения изотактического полипропилена, имеющего среднемассовую молекулярную массу более 500 кДа, температуру плавления более 150°С и mmmm более 95.
Текст
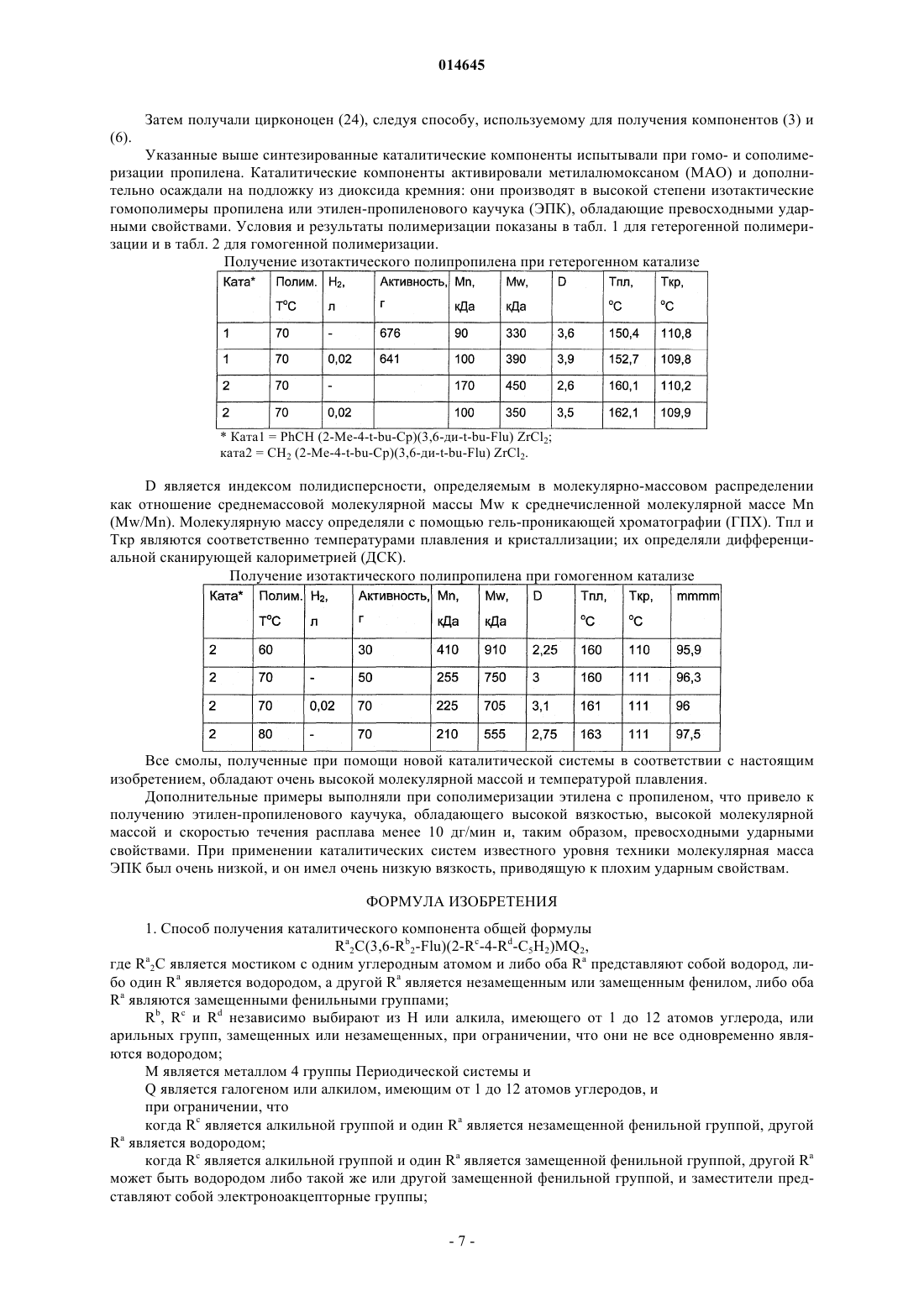
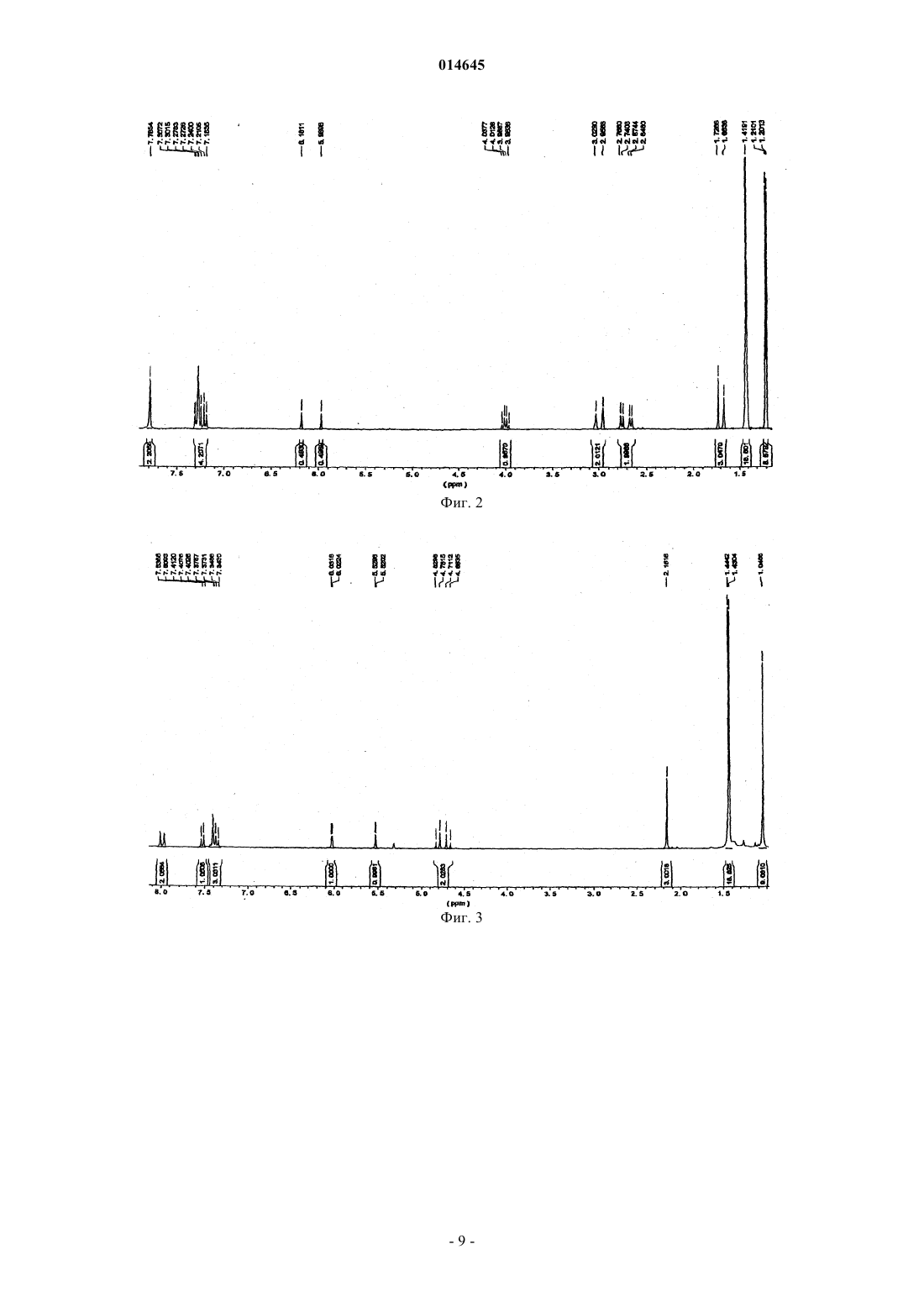
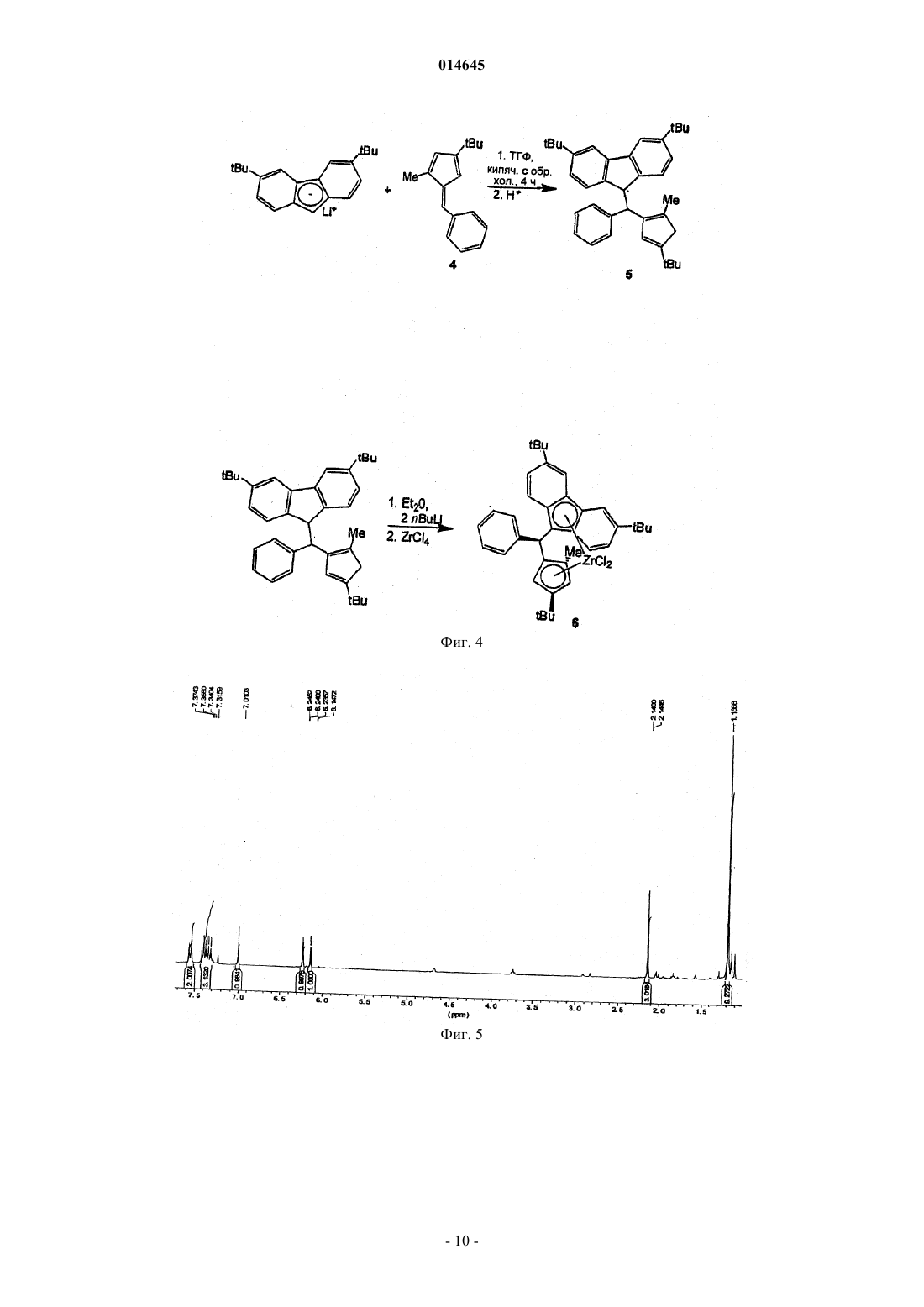
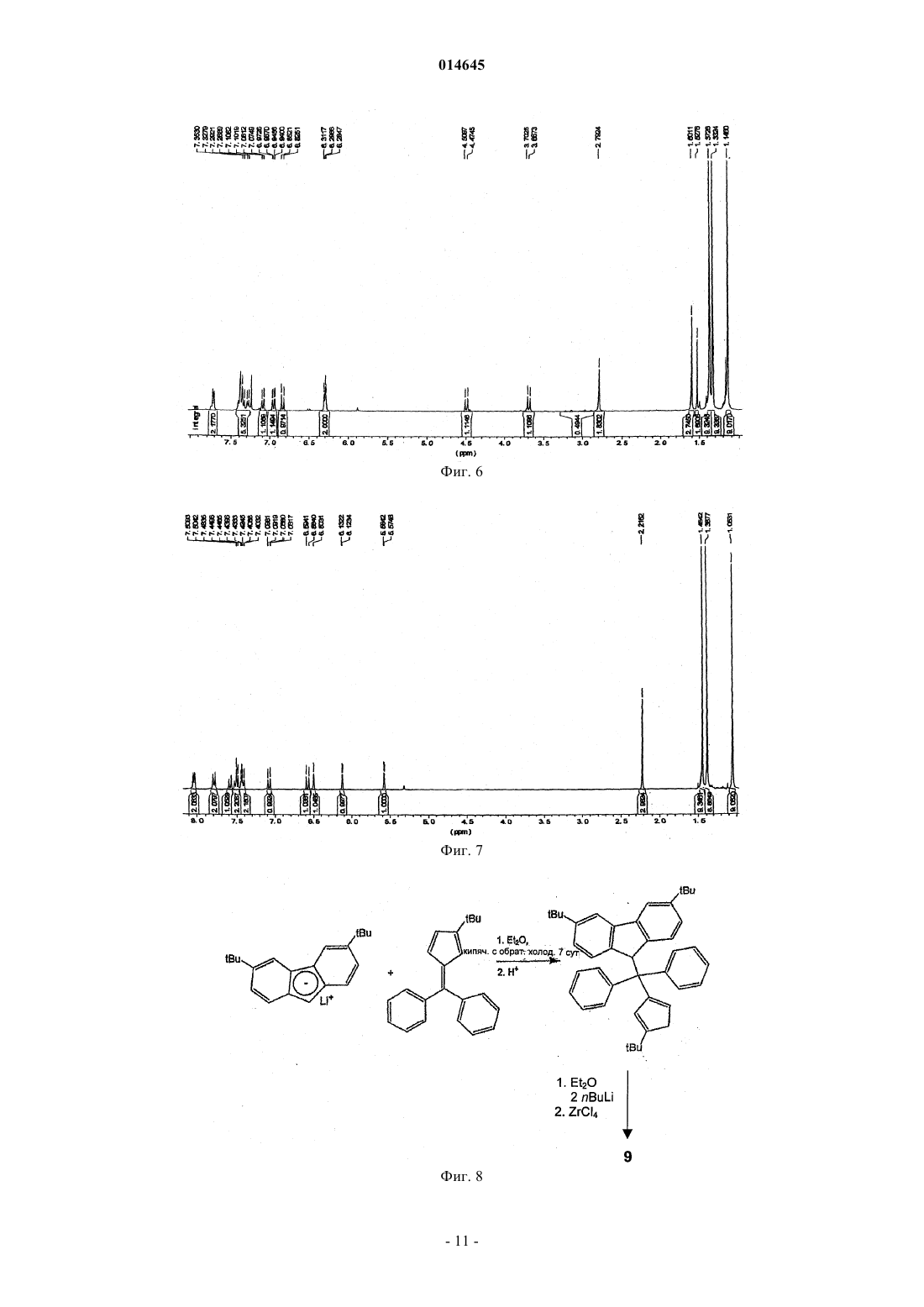
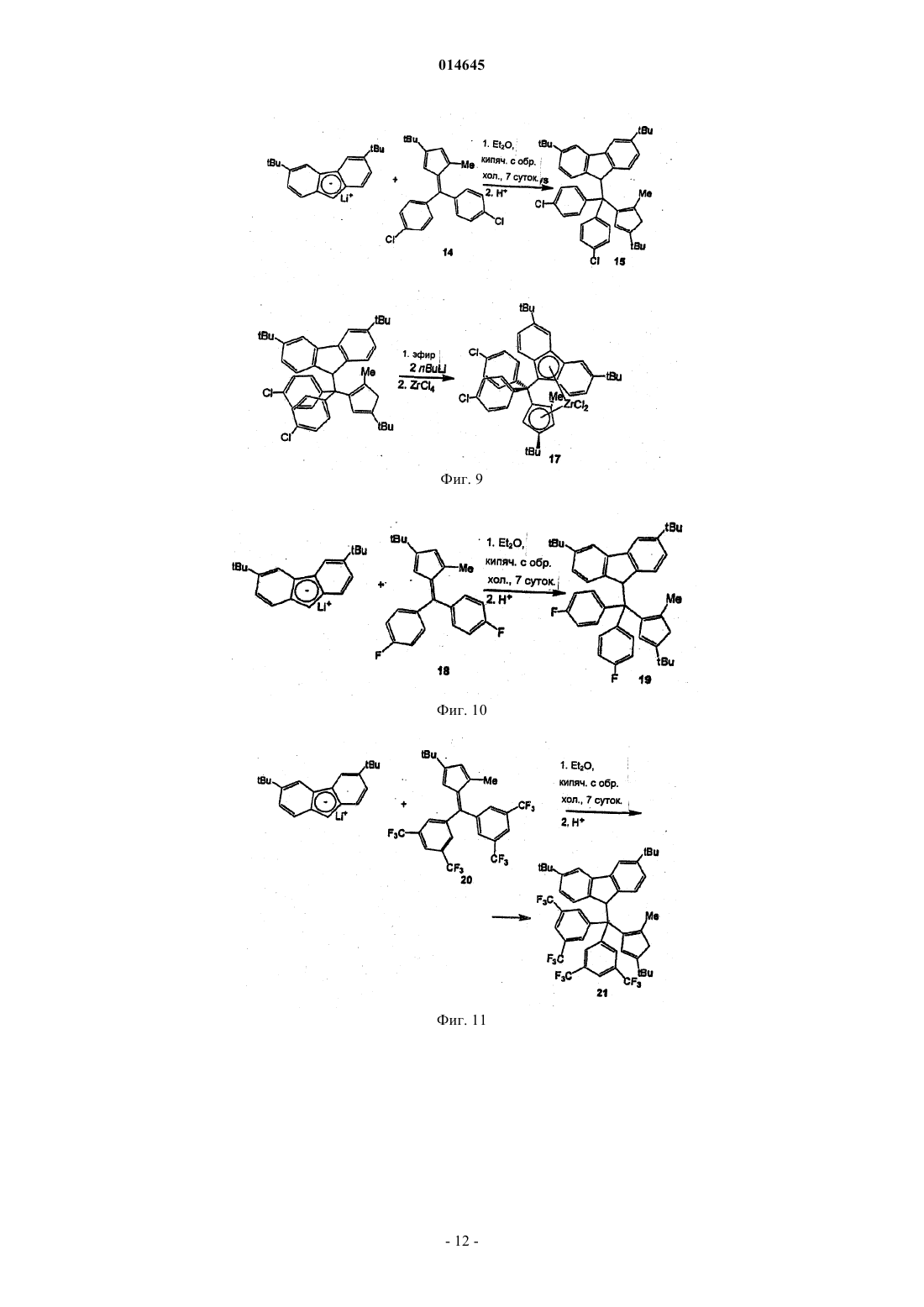
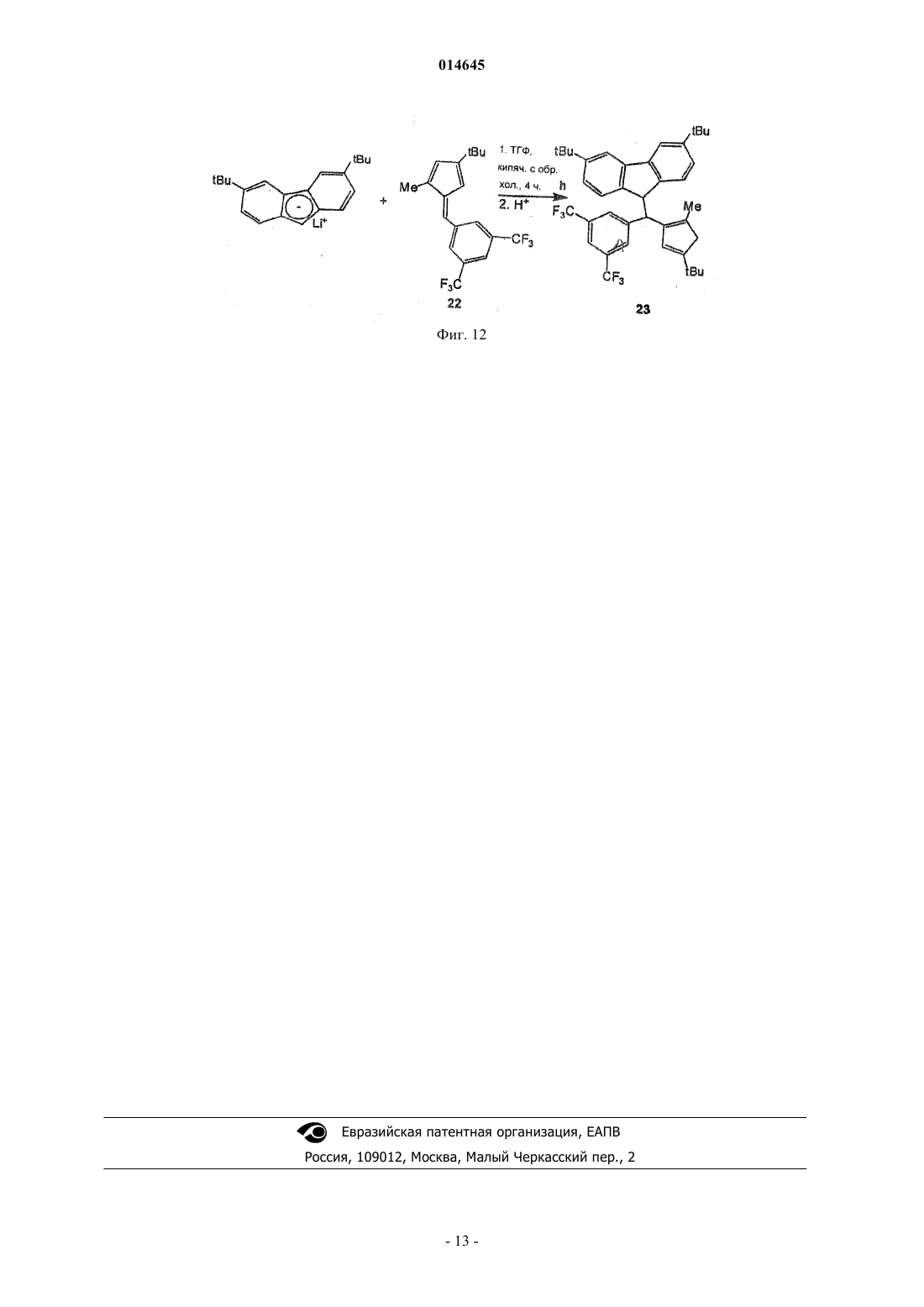
В настоящем изобретении раскрывают эффективные способы получения замещенных циклопентадиенильно-флуоренильных каталитических компонентов, содержащих мостики с одним атомом углерода.(71)(73) Заявитель и патентовладелец: ТОТАЛ ПЕТРОКЕМИКАЛС РИСЕРЧ ФЕЛЮЙ (BE) 014645 Настоящее изобретение относится к новым способам получения металлоценовых каталитических компонентов на основе циклопентадиенильно-флуоренильных лигандов с углеродными мостиками. Представляется возможным разработать каталитические системы для получения различных типов полимеров, таких как изотактические, синдиотактические или атактические. Однако желательно, чтобы выбранный катализатор обеспечивал получение преимущественно изотактического или синдиотактического полимера с очень небольшим количеством атактического полимера. Известны металлоценовые катализаторы с симметрией С 2 или С 1 для синтеза изотактических полиолефинов. Например, с помощью типичных цирконоценов бис-инденильного типа с симметрией С 2 можно синтезировать изотактический полипропилен с высокой молекулярной массой и высокой температурой плавления. Однако получение такого металлоценового катализатора является дорогостоящим и трудоемким. Более того, конечный катализатор состоит из смеси рацемических и мезоизомеров, часто в неблагоприятном соотношении. Мезостереоизомеры необходимо отделять, чтобы избежать образования атактического полипропилена в ходе реакции полимеризации. ЕР-А-0426644 относится к синдиотактическим сополимерам олефинов, таким как пропилен, получаемый с использованием в качестве каталитического компонента изопропил(флуоренил)(циклопентадиенил)цирконий дихлорида. Синдиотактичность, как показали измерения количества синдиотактических пентад, rrrr составляла 73-80%. ЕР 747406 относится к полимеризации олефинового мономера для получения синдиотактического/изотактического блок-сополимера полиолефина, в частности блок-сополимера полипропилена. Компонентом катализатора полимеризовации являлся 3-триметилсилилциклопентадиенил-9-флуоренил цирконий или гафний дихлорид, содержащий изопропилиденовый или дифенилметилиденовый мостик. В ЕР-А-577581 раскрывают синтез синдиотактических полипропиленов с помощью металлоценовых катализаторов, содержащих флуоренильные группы, замещенные в положениях 2 и 7, и незамещенное циклопентадиениловое кольцо. В ЕР-А-0419677 описан синтез синдиотактического пропилена с целью получения композиций смол, имеющих высокую жесткость при формовании. Металлоценовые катализаторы, такие как изопропил(циклопентадиенил-1-флуоренил цирконий дихлорид, использовали при синтезе полипропилена. Однако молекулярная масса, температура плавления и синдиотактичность таких продуктов были сравнительно низкими. Существует потребность в разработке новых каталитических систем, способных обеспечить полимеры с улучшенными свойствами, и эффективных способов их получения. Целью настоящего изобретения является обеспечение каталитических систем для получения полимеров, имеющих высокую молекулярную массу. Также целью настоящего изобретения является получение полимеров, имеющих высокую температуру плавления. Другой целью настоящего изобретения является получение ударопрочных полимеров, имеющих улучшенные ударные свойства. Еще одной целью настоящего изобретения является получение каталитических систем, способных обеспечить такие улучшенные полимеры. Таким образом, в настоящем изобретении обеспечивают способ получения каталитического компонента в соответствии с основной формулойRa2C(3,6-Rb2-Flu)(2-Rc-4-Rd-C5H2)MQ2,a где R 2C является мостиком с одним атомом углерода и каждый Ra независимо выбирают из водорода или незамещенной или замещенной ароматической группы, предпочтительно фенильной группы;Rb, Rc и Rd независимо выбирают из водорода, или алкила, имеющего от 1 до 12 атомов углерода,или арильных групп, замещенных или незамещенных, при ограничении, что все они одновременно не являются водородом; М является металлом 4 группы Периодической системы иQ является галогеном или алкилом, имеющим от 1 до 12 атомов углеродов,и при ограничении, что,если Rc является алкильной группой и один Ra является незамещенной ароматической группой, другой Ra является водородом,если Rc является алкильной группой и один Ra является замещенной ароматической группой, другой Ra может быть водородом, или той же самой, или другой замещенной ароматической группой, и заместители представляют собой электроноакцепторные группы,если Rc является водородом, каждый Ra независимо выбирают из водорода или незамещенной или замещенной ароматической группы,указанный способ включает: а) реакцию нуклеофильного присоединения в растворителе группы (Ra2C-2-Rc-4-Rd-фульвен) с группой [3,6- Rb2-Flu]-[M']+; б) гидролиз и отделение полученного лиганда; в) депротонирование лиганда стадии (б) с помощью R'M" для получения дианион лиганда, где R'-1 014645 является алкилом, имеющим от 1 до 6 атомов углерода, и М" является Li, Na или K; г) реакцию солевого обмена в растворителе дианион лиганда стадии (в) с MQ4; д) выделение каталитического компонента. В предпочтительном воплощении в соответствии с настоящим изобретением в мостике Ra2C одинR является незамещенным фенилом, а другой Ra является водородом. В другом предпочтительном воплощении оба Ra в мостике являются замещенными фенильными группами. Заместители на фенильных группах предпочтительно являются электроноакцепторными группами, которые можно выбирать из галогена, более предпочтительно F или Cl, или из СХ 3, в котором X является галогеном, более предпочтительно F, предпочтительно F, или из NO2. Заместители на фенильных группах могут быть расположены в положении 4 в случае одного заместителя или в положениях 3 и 5 для двух заместителей. Предпочтительно оба фенила имеют одинаковую схему замещения Предпочтительно оба Rb являются одинаковыми и являются алкилом, имеющим от 1 до 6 атомов углерода, более предпочтительно они оба являются трет-бутилом. Предпочтительно Rc является Н или метилом. Предпочтительно Rd является алкилом, имеющим от 1 до 6 атомов углерода, более предпочтительd но R является трет-бутилом. Предпочтительно М является Zr, Hf или Ti, более предпочтительно М является Zr. Предпочтительно Q является галогеном или метилом, более предпочтительно Q является хлором. Предпочтительно М" является Li. Растворители стадий (а) и (г) могут быть одинаковыми или различными и представляют собой углеводороды, предпочтительно выбираемые из пентана, толуола, тетрагидрофурана (ТГФ) или диэтилового эфира (Et2O). Предпочтительно они являются одинаковыми и представляют собой Et2O. Не углубляясь в теорию, полагают, что Et2O стабилизирует переходное состояние нуклеофильной реакции присоединения, включая зафиксированные в объеме реагенты. Реакцию стадии (а) проводят при температуре от 0 до 60 С, предпочтительно при комнатной температуре в течение периода времени от 6 до 24 ч, предпочтительно приблизительно в течение 12 ч. Любой известный в технике активирующий агент, имеющий ионизирующее действие, может быть использован для активации металлоценового компонента. Например, его можно выбрать из алюмосодержащих или борсодержащих соединений. Алюмосодержащие соединения включают алюмоксан,алкилалюминий и/или кислоты Льюиса. Алюмоксаны наиболее предпочтительны и могут содержать олигомерные линейные и/или циклические алкилалюмоксаны, представленные формулой для олигомерных, циклических алюмоксанов,где n является целым числом от 1 до 40, предпочтительно от 10 до 20;m является целым числом от 3 до 40, предпочтительно от 3 до 20 иR является C1-C8-алкильной группой и предпочтительно метилом. Подходящие борсодержащие активирующие агенты, которые можно использовать, включают трифенилкарбений боронат, такой как тетракис-пентафторфенилборатотрифенилкарбений, описанный в ЕР-А-0427696, или активирующие агенты, основная формула которых [L'-H]+[BAr1Ar2X3X4], описанные в ЕР-А-0277004 (с. 6, строка 30 - с. 7, строка 7).-2 014645 Каталитическая система также может быть нанесенной. Носитель, если присутствует, может быть пористым минеральным оксидом. Преимущественно его выбирают из диоксида кремния, оксида алюминия и их смесей. Предпочтительно, чтобы это был диоксид кремния. В качестве альтернативы используют активирующий носитель. Каталитическую систему по настоящему изобретению можно использовать при полимеризации этилена и альфа-олефинов. Предпочтительно ее используют для получения в высокой степени изотактических гомополимеров пропилена, имеющих высокую среднемассовую молекулярную массу, по меньшей мере 500 кДа, предпочтительно по меньшей мере 700 кДа, высокую температуру плавления более 150 С, предпочтительно более 160 С. Каталитическую систему также можно использовать для получения этиленпропиленового каучука(ЭПК), имеющего содержание этилена от 8 до 15 мас.%, высокую среднемассовую молекулярную массу по меньшей мере 500 кДа, предпочтительно по меньшей мере 700 кДа, индекс текучести расплава (ИТР) от 2 до 10 дг/мин. Индекс текучести расплава измеряют, следуя стандартной методике испытаний Американского общества по испытанию материалов (ASTM) D1238, при нагрузке 2,16 кг и температуре 230 С. ЭПК, полученный в настоящем изобретении, характеризуется превосходными ударными свойствами. Его используют во всех областях, где требуются эластомеры с превосходными термопластичными свойствами. Краткий перечень чертежей На фиг. 1 представлена схема реакции получения комплекса Н 2 С(3,6-tBu2Flu)(3-tBu-5-Me-Cp)ZrCl2 (3). На фиг. 2 представлен 1 Н ЯМР-спектр лиганда 3,6-ди-трет-бутил-9-[(3-трет-бутил-5 метилциклопента-1,4-диен-1-ил)метил]-9 Н-флуорен (2). На фиг. 3 представлен 1 Н ЯМР-спектр комплекса H2C(3,6-tBu2Flu)(3-tBu-5-Me-Cp)ZrCl2 (3). На фиг. 4 представлена схема реакции получения комплекса PhHC(3,6-tBu2Flu)(3-tBu-5-Me-Cp)ZrCl2 (6). На фиг. 5 представлен 1 Н ЯМР-спектр 6-фенил-2-метил-4-трет-бутилфульвена (4). На фиг. 6 представлен 1 Н ЯМР-спектр лиганда 3,6-ди-трет-бутил-9-[(4-трет-бутил-2 метилциклопента-1,4-диенил)фенилэтил]-9 Н-флуорена (5). На фиг. 7 представлен 1 Н ЯМР-спектр комплекса PhHC(3,6-tBu2Flu)(3-tBu-5-Me-Cp)ZrCl2 (6). На фиг. 8 представлена схема реакции получения комплекса Ph2C(3,6-tBu2-Flu)(3-tBu-Cp)ZrCl2 (9). На фиг. 9 представлена схема реакции получения комплекса (p-Cl-Ph)2C(3,6-tBu2Flu)(3-tBu-5-MeCp)ZrCl2 (17). На фиг. 10 представлена схема реакции получения лиганда 3,6-ди-трет-бутил-9-(4-трет-бутил-2 метилциклопента-1,4-диен-1-ил)[бис-(4-флуоренил)]метил-9 Н-флуорена (19). На фиг. 11 представлена схема реакции получения лиганда 9-[бис-(3,5-бис(трифторметил)фенил](4-трет-бутил-2-метилциклопента-1,4-диен-1-ил)метил]-3,6-ди-трет-бутил-9 Нфлуорена (21). На фиг. 12 представлена схема реакции получения лиганда 9-3,5-бис(трифторметил)фенил](4 трет-бутил-2-метилциклопента-1,4-диен-1-ил) метил]-3,6-ди-трет-бутил-9 Н-флуорена (23). Примеры Все эксперименты выполняли в атмосфере очищенного аргона, используя стандартные методики Шленка или в защитной камере с перчатками. Растворители перегоняли над Na/бензофеноном (тетрагидрофуран (ТГФ), диэтиловый эфир (Et2O и над сплавом Na/K (толуол, пентан в атмосфере азота, их полностью дегазировали и хранили в атмосфере азота. Дейтерированные растворители (бензол-d6, толуол-d6, ТГФ-d8, 99,5% D, Deutero GmbH) транспортировали в вакууме от сплава Na/K в накопительные трубки. Перед использованием хлороформ-d1 и CD2Cl2 выдерживали над гидридом кальция и транспортировали в вакууме перед использованием. Предшественники 3,6,6'-триметилфульвен, 2-метил-4-третбутилциклопентадиенил (смесь изомеров) и 1-метил-3-трет-бутилциклопентадиенил литий получали по известным методикам и охарактеризовывали с помощью 1 Н ЯМР-спектроскопии. 1-третБутилциклопентадиенил (смесь изомеров) получали по методике, описанной в работе Мура и Джина Кинга (Moore W.R. and Jean King В., J. Org. Chem., 36, 1882, 1971). Синтез H2C(3,6-tBu2Flu)(2-Me-4-tBu-Cp)ZrCl2 (3). Схема представлена на фиг. 1. а) Синтез 3,6-ди-трет-бутил-9-[(2-метил-4-трет-бутилциклопента-1,4-диен-1-ил)метил 1-9 Нфлуорена (2). К раствору 3,2 г (16,73 ммоль) 6-диметиламино-2-метил-4-трет-бутилфульвена в 50 мл ТГФ добавляли при комнатной температуре, 50 мл раствора 3,6-ди-трет-бутил-флуоренил-лития, полученного из 4,65 г (16,70 ммоль) 3,6-дитрет-бутилфлуорена и 6,70 мл (16,70 ммоль) 2,5 М раствора н-бутиллития. Реакционную смесь перемешивали в течение 12 ч при комнатной температуре. Затем добавляли 1,17 г(30,79 ммоль) LiAlH4 и полученную смесь кипятили с обратным холодильником в течение последующих 12 ч, затем с осторожностью быстро охлаждали 50 мл насыщенного раствора NH4Cl, разбавленного в 100 мл диэтилового эфира. Органический слой отделяли, промывали дважды 200 мл воды и осушали надCaCl2. Все летучие компоненты удаляли в вакууме. Неочищенный продукт желтого цвета очищали хроматографией на колонке с силикагелем, чтобы получить 4,27 г (10,02 ммоль) конечного продукта 2 с вы-3 014645 ходом 60%. Данный продукт являлся смесью 1:1 изомеров по положению двойной связи в циклопентадиенильном кольце. 1 Н ЯМР (CDCl3, 300 МГц, 25 С) спектр представлен на фиг. 2 и характеризуется следующим образом:7,78 (s, 4H, Flu); 7,40-7,10 (m, 8H, Flu); 7,08 (dd, 1 Н, Flu); 6,18 (s, 1H, Cp); 5,97(d, 1H, Cp); 4,00 (q,2H, 3J=14,7 Гц, 9-Flu); 3,00 (m, 4H, CH2, Cp); 2,70 (m, 4H, CH2); 1,73; 1,66 (s, 3H, CH3); 1,42 (s, 36H,CCH3-Flu); 1,21 (s, 18H, ССН 3-Flu). Анализ. Рассчитано для С 32 Н 42: С, 90,08; Н, 9,92. Найдено: С, 91,01; Н, 9,99. б) Синтез комплекса H2C(3,6-tBu2Flu)(2-Me-4-tBu-Cp)ZrCl2 (3). К раствору 1,67 г (3,91 ммоль) 3,6-ди-трет-бутил-9-[(2-метил-4-трет-бутил.-циклопента-1,4-диен-1 ил)метил]-9 Н-флуорена (2) в 40 мл Et2O добавляли 3,10 мл (7,82 ммоль) 2,5 М раствора бутиллития в гексане при температуре 0 С. Реакционную смесь перемешивали в течение 4 ч и растворитель выпаривали при пониженном давлении. Затем в защитной камере с перчатками добавляли 0,91 г (3,90 ммоль) безводного ZrCl4 с последующим добавлением 50 мл пентана. Полученную реакционную смесь розового цвета перемешивали при комнатной температуре в течение ночи. Реакционную смесь отфильтровывали и фильтрат выпаривали в вакууме. Добавляли порцию гексана, приблизительно 30 мл, и полученный прозрачный раствор выдерживали при температуре -30 С в течение ночи с получением осадка - розового микрокристаллического порошка комплекса 3. Вторую партию продукта 3 массой 1,48 г (2,52 ммоль) получали из маточного раствора при охлаждении с выходом 64%. Кристаллы, пригодные для рентгеноструктурного анализа, получали путем медленного концентрирования из смеси 3:7 CH2Cl2/гексан. 1 Н ЯМР (CD2Cl2, 300 МГц, 25 С) спектр представлен на фиг. 3 и характеризуется следующим образом:8,01 (s, 1H, Flu); 7,97 (s, 1H, Flu); 7,52 (s, 1 Н, Flu); 7,40 (m, 2H, Flu); 7,37 (m, 1H, Flu); 6,02 (d, 1H,4C ЯМР (CD2Cl2, 75 МГц, 25 С):150,1; 150,0; 146,4; 127,9; 127,6; 124,8; 124,1; 124,0; 123,3; 121,8; 120,7; 120,6; 120,2; 119,7; 118,2; 102,4; 97,0; 70,3; 35,6; 33,3; 32,2; 32,1; 32,0; 30,0; 22,5(CH2); 16,1. Анализ. Рассчитано для C32H40Cl2Zr: С, 65,50; H, 6,87; Cl 12,08 (молекулярная масса Mr=586,79 кг/моль). Найдено: С, 66,12; Н, 6,99. Синтез комплекса Ph(H)C(3,6-tBu2Flu)(2-Me-4-tBu-Cp)ZrCl2 (6). Схема синтеза представлена на фиг. 4. а) Синтез 6-фенил-2-метил-4-трет-бутилфульвена (4). К раствору 1,94 г (14,24 ммоль) 1-метил-3-трет-бутилциклопентадиена (смесь изомеров) в 50 мл диэтилового эфира добавляли 5,7 мл (14,24 ммоль) 2,5 М раствора бутиллития в гексане при температуре 0 С. Реакционную смесь перемешивали в течение 2 ч и по каплям добавляли раствор 1,44 мл(14,24 ммоль) бензальдегида в 30 мл простого эфира. Реакционная смесь становилась оранжевой. После 2 ч медленно добавляли 50 мл концентрированного раствора NH4Cl. Указанную смесь перемешивали в течение ночи. Отделяли органический слой, осушали над MgSO4 и удаляли все летучие в вакууме. Оранжевый остаток перекристаллизовывали из метанола при температуре 30 С с получением 1,0 г(4,46 ммоль) соединения (4) с выходом 31%. 1 Н ЯМР (CDCl3, 300 МГц, 25 С) спектр представлен на фиг. 5 и характеризуется следующим образом:7,57 (m, 2H, Ph); 7,40 (m, 3 Н, Ph); 7,01 (s, 1H, =CHPh); 6,24 (t, 1H, СН); 6,14 (d, 1H, СН); 2,15 (s, 3 Н,СН 3); 1,19 (s, 9H, ССН 3). 13 С ЯМР (CD2Cl2, 75 МГц, 25 С):160,4; 145,9; 137,4; 135,5; 131,9; 129,5; 128,4; 128,2; 128,0; 127,2; 125,5; 110,5; 32,1; 29,5; 12,6. б) Синтез 3,6-ди-трет-бутил-9-[(4-трет-бутил-2-метилциклопента-1,4-диенил)фенилэтил]-9 Нфлуорена (5). К раствору 1,75 г (7,80 ммоль) соединения (4) в 20 мл ТГФ добавляли при комнатной температуре 30 мл раствора 3,6-ди-трет-бутилфлуорениллития, полученного реакцией 1,95 г (7,00 ммоль) 3,6-ди-третбутилфлуорена с 2,80 мл (7,00 ммоль) 2,5 М раствора н-бутиллития. Реакционную смесь перемешивали в течение 4 ч при температуре окружающей среды (приблизительно 25 С) и затем быстро охлаждали в 50 мл насыщенного раствора NH4Cl и разбавляли 50 мл диэтилового эфира. Органический слой отделяли, дважды промывали 200 мл воды и осушали над CaCl2. Все летучие удаляли в вакууме и остаток растворяли в горячем MeOH. Раствор охлаждали до -30 С и формировался белый осадок. Последний фильтровали и промывали холодным метанолом (-30 С) и сушили в вакууме в течение ночи с получением 2,30 г (4,57 ммоль) конечного продукта (5) с выходом 65%. Продукт содержит 20% изомера 3,6-ди-третбутил-9-[(4-трет-бутил-2-метилциклопента-1,3-диен-1-ил)(фенил)метил]-9 Н-флуорена. 1 Н ЯМР (CDCl3, 300 МГц, 25 С) спектр представлен на фиг. 6 и характеризуется следующим образом:7,70 (dd, 2H, Flu); 7,35 (m, 5H, Ph); 7,08 (dd, 1 Н, Flu); 6,95 (dd, 1H, Flu); 6,83 (d, 1H, Flu); 6,29 (t, 2H,Flu); 4,48 (d, 1H, 3J=10,6 Гц, CHPh); 3,68 (d, 1H, 3J=10,6 Гц, 9-Flu); 2,79 (s, 2H, CH2, Cp); 1,60 (s, 3H, CH3);C ЯМР (CD2Cl2, 75 МГц, 25 С):155,6; 150,0; 149,9; 144,2; 143,9; 143,6; 141,4; 141,3; 139,9; 139,8; 134,6; 128,9; 128,7; 128,6; 126,4; 125,6; 125,3; 124,7; 123,5; 123,4; 115,9; 115,8; 51,0; 50,9; 49,4; 44,0; 34,9; 33,2; 31,8; 31,1; 13,4. с) Синтез комплекса Ph(H)C(3,6-tBu2Flu)(2-Me-4-tBu-Cp)ZrCl2 (6). К раствору 1,025 г (2,04 ммоль) соединения 5 в 40 мл Et2O добавляли 1,67 мл (4,08 ммоль) 2,5 М раствора бутиллития в гексане при температуре 0 С. Реакционную смесь перемешивали в течение 4 ч и затем в защитной камере с перчатками добавляли 0,475 г (2,04 ммоль) безводного ZrCl4. Полученную реакционную смесь розового цвета перемешивали при комнатной температуре в течение ночи. Затем растворитель выпаривали в вакууме и 40 мл гексана конденсировали при пониженном давлении. Полученную смесь отфильтровывали и фильтрат выпаривали в вакууме с получением 1,18 г (1,78 ммоль) порошка розового цвета неочищенного комплекса (6) с выходом 88%. К остатку розового цвета добавляли новое количество гексана объемом 20 мл. По истечении периода времени от 1 до 2 ч формировался розовый осадок. Его отделяли декантацией, промывали дважды 10 мл холодного гексана и сушили в вакууме с получением 0,53 г (8,80 ммоль) дихлорцирконоцена (6) с выходом 40%. Кристаллы, пригодные для рентгеноструктурного анализа, получали путем медленного концентрирования из смеси 1:9 CH2Cl2/гексан. 1 Н ЯМР (CD2Cl2, 300 МГц, 25 С) спектр представлен на фиг. 7 и характеризуется следующим образом:8,03 (dd, 2H, Flu); 7,78 (d, 2H); 7,58 (d, 1 Н); 7,48 (m, 2H); 7,43 (m, 2H); 7,08 (dd, 1H, Flu); 6,57 (d, 1H,Flu); 6,50 (s, 1H, Cp); 6,12 (d, 1H, 4J=2,6 Гц, Cp); 5,57 (d, 1H, 4J=2,6 Гц, CHPh); 2,21 (s, 3H, CH3); 1,45 (s,9H, ССН 3-Flu); 1,38 (s, 9H, CCH3-Flu); 1,05 (s, 9H, CCH3-Flu). 13C ЯМР (CD2Cl2, 75 МГц, 25 С):150,2; 150,0; 147,1; 140,1; 129,2; 128,9; 128,6; 128,4; 128,2; 127,6; 126,7; 125,9; 125,3; 124,8; 122,8; 122,5; 121,5; 120,0; 119,5; 119,3; 116,9; 103,2; 100,9; 74,6; 40,2; 35,5; 35,4; 33,2; 32,0; 29,7; 15,8. Анализ. Рассчитано для C38H44Cl2Zr: С, 68,85; H, 6,69; Cl 10,70 (Mr=662,885 кг/моль). Найдено: С, 69,01; Н, 7,37. Синтез комплекса Ph2C(3,6-tBu2-Flu)(4-tBu-C5H3)ZrCl2 (9). Схема представлена на фиг. 8. а) Синтез комплекса Ph2C(3,6-tBu2FluH)(4-tBu-C5H4) (8). Известно, что реакция стерически затрудненных стабилизированных 6,6-дифенилфульвенов с флуорениланионом протекает медленно и требует продолжительного и существенного нагрева. Реакция между 6,6'-дифенил-4-трет-бутил-фульвеном (7) и флуорениланионом проявляла зависимость от природы растворителя. Наилучшие результаты показал диэтиловый эфир: реакция протекала в течение 5-7 дней при температуре от 60 до 70 С с получением требуемого продукта (8) с выходом 21%. б) Синтез комплекса Ph2C(3,6-tBu7-Flu)(4-tBu-C5H3)ZrCl2 (9). Проводили реакцию солевого обмена между полученным in situ лигандом (8) - дианионом и ZrCl4. Реакция протекала при комнатной температуре с сопутствующим осаждением LiCl. После обычной отработки реакционную смесь в виде раствора в гексане выдерживали в течение одного месяца при комнатной температуре с получением красных микрокристаллов комплекса (9) с выходом 46%. 1 Н ЯМР спектроскопия комплекса (9) показала асимметричную структуру в растворе, аналогичную одному из описанных комплексов (3) и (6). Синтез (p-Cl-Ph)2C(3,6-ди-трет-бутил-9-флуоренил)(2-Ме-4-трет-бутилциклопентадиенил)цирконий дихлорида (17). Схема представлена на фиг. 9. а) Синтез комплекса 6,6'-бис-(4-хлор-фенил)-4-трет-бутил-2-метилфульвена (14). К раствору 2,27 г (16,66 ммоль) смеси изомеров 1-метил-3-трет-бутилциклопентадиена в 150 мл тетрагидрофурана добавляли 6,67 мл (16,66 ммоль) 2,5 М раствора бутиллития в гексане при температуре 0 С. Реакционную смесь перемешивали в течение 2 ч и добавляли по капле раствор 4,4'-дихлорбензофенона массой 4,18 г (16,66 ммоль) в 50 мл ТГФ. Реакционная смесь изменила цвет на оранжевый. Спустя 4 ч медленно добавляли 50 мл концентрированного раствора NH4Cl. Смесь перемешивали в течение ночи. Удаляли органический слой, осушали над MgSO4 и все летучие удаляли в вакууме. Органический остаток перекристаллизовывали из горячего метанола при температуре 25 С, чтобы получить 3,7 г (10,02 ммоль) 6,6'-бис(4-хлор-фенил)-2-метил-4-трет-бутилфульвена, с выходом 60%. 1 Н ЯМР (CDCl3, 400 МГц, 25 С) характеризуется следующим образом:7,32 (m, 4 Н, Ph); 7,18 (m,4 Н, Ph); 6,37 (s, 1H, Ср); 5,66 (s, 1H, Ср); 1,53 (s, 3 Н, СН 3); 1,17(s, 9H, ССН 3). Анализ. Рассчитано для C23H22Cl2: С, 74,80; Н, 7,00. Найдено: С, 74,85; Н, 7,10.Et2O добавляли при комнатной температуре 30 мл раствора 3,6-ди-трет-бутил-флуоренил-лития в Et2O,полученного из 1,0 г (3,59 ммоль) 3,6-ди-трет-бутилфлуорена и 1,44 мл (3,59 ммоль) 2,5 М раствора н-бутиллития в гексане. Реакционную смесь перемешивали в течение 5 дней при кипячении с обратным холодильником и затем быстро охлаждали в 50 мл насыщенного раствора NH4Cl, разбавленного 50 мл диэтилового эфира. Отделяли органический слой, промывали дважды 200 мл воды и осушали над CaCl2. Все летучие удаляли в вакууме. Остаток промывали в МеОН, затем в холодном пентане при температуреC ЯМР (ТГФ-d8, 75 МГц, 90 С)151,0; 150,1; 149,8; 144,7; 144,3; 143,5; 143,3; 143,2; 141,8; 135,0; 133,7; 133,1; 131,9; 131,5; 130,1; 129,6; 128,9; 127,1; 126,5; 125,3; 123,9; 116,5; 116,3; 116,2; 57,1; 55,4; 41,3; 35,2; 32,5; 31,9; 31,8; 28,9; 28,7; 28,3. Анализ. Рассчитано для C44H48Cl2: С, 81,58; Н, 7,47. Найдено: С, 82,04; Н, 8,55. Затем получали цирконоцен (17) посредством реакции с безводным ZrCl4, следуя той же схеме, что показана для получения комплекса (3) или комплекса (6). В качестве альтернативы каждая фенильная группа в бифенильном мостике может быть замещена фтором в положении, как показано на фиг. 10, или двумя CF3 соответственно в положениях 3 и 5, как показано на фиг. 11. Синтез (3,5-бис-(трифторметил)фенил)СН(3,6-ди-трет-бутилэфлуоренил)(2-Ме-4-трет-бутилциклопентадиенил)цирконий дихлорида (24). а) Синтез 6-(3,5-бис-(трифторметил)фенил)-3-трет-бутил-5-метилфульвена (22). К раствору 2,81 г (20,63 ммоль) смеси изомеров 1-метил-3-трет-бутилциклопентадиенила в 150 мл диэтилового эфира добавляли 8,20 мл (20,63 ммоль) 2,5 М раствора бутиллития в гексане при температуре 0 С. Реакционную смесь перемешивали в течение 2 ч и добавляли по капле раствор 5,0 г (20,65 ммоль) 3,5-бис-(трифторметил)бензальдегида в 50 мл простого эфира. Реакционная смесь изменила цвет на красный. Спустя 2 ч медленно добавляли 50 мл концентрированного раствора NH4Cl. Смесь перемешивали в течение ночи. Удаляли органический слой, осушали над MgSO4 и все летучие удаляли в вакууме. Органический остаток перекристаллизовывали из метанола при температуре -30 С, чтобы получить 2,60 г (7,22 ммоль) соединения (22), с выходом 35%. 1 Н ЯМР (CDCl3, 300 МГц, 25 С) характеризуется следующим образом:7,92 (s, 2 Н, Ph); 7,79 (s, 1 Н,Ph); 6,95 (s, 1 Н, =CHPh); 6,25 (t, 1 Н, СН); 5,92 (d, 1H, СН); 2,11 (s, 3 Н, СН 3); 1,15 (s, 9 Н, ССН 3). 19F ЯМР (CDCl3, 282 МГц, 25 С):-62,6. Анализ. Рассчитано для C34H32F2: С, 62,98; Н, 5,56. Найдено: С, 63,67; Н, 5,98. б) Синтез 9-3,5-бис-(трифторметил)фенил](4-трет-бутил-2-метилциклопента-1,4-диен-1 ил)метил]-3,6-ди-трет-бутил-9 Н-флуорена (23). Схема получения компонента (23) представлена на фиг. 12. К раствору 2,60 г (7,21 ммоль) соединения 22 в 25 мл ТГФ добавляли при комнатной температуре 30 мл раствора 3,6-ди-трет-бутилфлуорениллития, полученного из 2,00 г (7,20 ммоль) 3,6-ди-третбутилфлуорена и 2,90 мл (7,21 ммоль) 2,5 М раствора н-бутиллития. Реакционную смесь перемешивали в течение 4 ч при комнатной температуре и затем быстро охлаждали в 50 мл насыщенного раствора NH4Cl,разбавленного 50 мл диэтилового эфира. Отделяли органический слой, промывали дважды в 200 мл воды и осушали над CaCl2. Все летучие удаляли в вакууме. Остаток очищали хроматографией на колонке с силикагелем, используя гексан в качестве элюента, чтобы получить 0,2 г (0,31 ммоль) конечного соединения 23, в виде смеси двух изомеров при соотношении величин 2:3, с выходом 4%. 1 Н ЯМР (CDCl3, 300 МГц, 25 С) характеризуется следующим образом:7,70 (m, 3H, Ph); 7,20-6,30-6 014645 Затем получали цирконоцен (24), следуя способу, используемому для получения компонентов (3) и(6). Указанные выше синтезированные каталитические компоненты испытывали при гомо- и сополимеризации пропилена. Каталитические компоненты активировали метилалюмоксаном (МАО) и дополнительно осаждали на подложку из диоксида кремния: они производят в высокой степени изотактические гомополимеры пропилена или этилен-пропиленового каучука (ЭПК), обладающие превосходными ударными свойствами. Условия и результаты полимеризации показаны в табл. 1 для гетерогенной полимеризации и в табл. 2 для гомогенной полимеризации. Получение изотактического полипропилена при гетерогенном катализеD является индексом полидисперсности, определяемым в молекулярно-массовом распределении как отношение среднемассовой молекулярной массы Mw к среднечисленной молекулярной массе Mn(Mw/Mn). Молекулярную массу определяли с помощью гель-проникающей хроматографии (ГПХ). Тпл и Ткр являются соответственно температурами плавления и кристаллизации; их определяли дифференциальной сканирующей калориметрией (ДСК). Получение изотактического полипропилена при гомогенном катализе Все смолы, полученные при помощи новой каталитической системы в соответствии с настоящим изобретением, обладают очень высокой молекулярной массой и температурой плавления. Дополнительные примеры выполняли при сополимеризации этилена с пропиленом, что привело к получению этилен-пропиленового каучука, обладающего высокой вязкостью, высокой молекулярной массой и скоростью течения расплава менее 10 дг/мин и, таким образом, превосходными ударными свойствами. При применении каталитических систем известного уровня техники молекулярная масса ЭПК был очень низкой, и он имел очень низкую вязкость, приводящую к плохим ударным свойствам. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения каталитического компонента общей формулыRa2C(3,6-Rb2-Flu)(2-Rc-4-Rd-C5H2)MQ2,a где R 2C является мостиком с одним углеродным атомом и либо оба Ra представляют собой водород, либо один Ra является водородом, а другой Ra является незамещенным или замещенным фенилом, либо обаRa являются замещенными фенильными группами;Rb, Rc и Rd независимо выбирают из Н или алкила, имеющего от 1 до 12 атомов углерода, или арильных групп, замещенных или незамещенных, при ограничении, что они не все одновременно являются водородом; М является металлом 4 группы Периодической системы иQ является галогеном или алкилом, имеющим от 1 до 12 атомов углеродов, и при ограничении, что когда Rc является алкильной группой и один Ra является незамещенной фенильной группой, другойR является водородом; когда Rc является алкильной группой и один Ra является замещенной фенильной группой, другой Ra может быть водородом либо такой же или другой замещенной фенильной группой, и заместители представляют собой электроноакцепторные группы;-7 014645 когда Rc является водородом, каждый Ra независимо выбирают из Н или незамещенной или замещенной фенильной группы; при этом указанный способ включает: а) реакцию нуклеофильного присоединения в растворителе группы (Ra2C-2-Rc-4-Rd-фульвен) с группой [3,6- Rb2-Flu]-[M']+, где М' является Li; б) гидролиз и отделение полученного лиганда; в) депротонирование лиганда стадии б) с помощью R'M" для получения дианионного лиганда, гдеR' является алкилом, имеющим от 1 до 6 атомов углерода, и М"является Li, Na или K; г) реакцию солевого обмена в растворителе дианионного лиганда стадии в) с MQ4; д) выделение каталитического компонента. 2. Способ по п.1, в котором заместители на фенильных группах, если присутствуют, находятся в положении 4 для одного заместителя или в положениях 3 и 5 для двух заместителей. 3. Способ по п.1 или 2, в котором оба Rb являются одинаковыми и представляют собой трет-бутил. 4. Способ по любому из предшествующих пунктов, в котором Rc является метилом в положении 2 иR является трет-бутилом в положении 4. 5. Способ по любому из предшествующих пунктов, в котором растворителем стадии а) являетсяEt2O. 6. Способ по п.5, в котором растворителем стадии г) также является Et2O. 7. Способ по любому из предшествующих пунктов, в котором М" является Li. 8. Металлоценовый каталитический компонент, полученный способом по любому из пп.1-7. 9. Применение металлоценового каталитического компонента, полученного способом по любому из пп.1-7, в сочетании с подходящим активирующим агентом для получения этилен-пропиленового каучука с содержанием этилена от 8 до 15 мас.% и среднемассовой молекулярной массой по меньшей мере 500 кДа. 10. Применение металлоценового каталитического компонента, полученного способом по любому из пп.1-7, в сочетании с подходящим активирующим агентом для получения изотактического полипропилена, имеющего среднемассовую молекулярную массу более 500 кДа, температуру плавления более 150 С и mmmm более 95.
МПК / Метки
МПК: C07F 17/00, C08F 4/64, C08F 10/00
Метки: углеродными, лиганды, мостиками, циклопентадиенильно-флуоренильные
Код ссылки
<a href="https://eas.patents.su/14-14645-ciklopentadienilno-fluorenilnye-ligandy-s-uglerodnymi-mostikami.html" rel="bookmark" title="База патентов Евразийского Союза">Циклопентадиенильно-флуоренильные лиганды с углеродными мостиками</a>
Имплантаты с функционализированными углеродными поверхностями

Номер патента: 9836
Опубликовано: 28.04.2008
Авторы: Бэн Андреас, Майер Бернхард, Асгари Соэйл, Ратенов Йорг, Кунстманн Йюрген
МПК: A61F 2/02, A61L 27/30, A61L 27/50...
Метки: углеродными, поверхностями, функционализированными, имплантаты
Формула / Реферат:
1. Способ изготовления медицинских имплантатов, включающий следующие стадии: а) изготовление медицинского имплантата по крайней мере с одним углеродсодержащим слоем по крайней мере на части поверхности имплантата; б) создание пористости углеродсодержащего слоя; в) введение в углеродсодержащий слой веществ, выбранных из фармакологически активных веществ, линкеров, микроорганизмов, растительных или животных клеток, включая человеческие клетки, или...
Лиганды ванилоидных рецепторов

Номер патента: 10380
Опубликовано: 29.08.2008
Авторы: Чен Нинг, Норман Марк Х., Ванг Хаи-Линг, Петтус Липинг Х., Жу Джиаванг, Никси Томас, Кэйтон Джоди, Домингэз Селиа, Рзаса Роберт Майкл, Огнянов Вассил И., Дохерти Элизабет М., Чакрабарти Партха П., Фэлси Джеймс Ричард, Халм Кристофер, Фотш Кристофер Х., Стек Маркиан
МПК: C07D 239/38, C07D 401/12, C07D 239/34...
Метки: лиганды, ванилоидных, рецепторов
Формула / Реферат:
1. Соединение общей формулы где J обозначает О или S; Rd обозначает Н; R1 обозначает: (i) фенил, необязательно замещенный одним или более заместителями, выбранными из группы, включающей -С(СН3)3, СН3, CF3, F, Cl, Br, CN, ОН, -OCH3, -C(O)H, -CH2OH, -OCF3, NO2, NH2, -О(СН2)2морфолин, -ОСН2ОСН3, пиперидинил, -OBz, -SMe, пирролидинил, пиридинил, -OSO2CF3, -NHC(O)CH3, фенил, фторфенил, -СН=СН2, -NHC(O)OC(CH3)3, -C(O)OMe, -NHSO2Me, -NHSO2Ph, ...
Азабициклические лиганды рецепторов 5нт1

Номер патента: 3400
Опубликовано: 24.04.2003
Автор: Брайт Джин Майкл
МПК: A61P 25/00, A61K 31/495, C07D 471/04...
Метки: рецепторов, 5нт1, азабициклические, лиганды
Формула / Реферат:
1. Соединение формулы где R3, R4 и Z независимо выбраны из водорода, галогена (например хлора, фтора, брома или иода), (C1-C4)алкила, возможно замещенного атомами фтора в количестве от одного до трех, (C1-C4)алкокси, возможно замещенного атомами фтора в количестве от одного до трех, и (C1-C4)алкокси(C1-C4)алкила, где каждая алкильная группировка может быть замещена атомами фтора в количестве от одного до трех; W представляет собой...
Лиганды рецепторов 5-нт и их применение

Номер патента: 7183
Опубликовано: 25.08.2006
Авторы: Уэлч Уиллард Маккауэн, Чьянг Юан-Чинг Фиби, Новомайсл Уильям Альберт
МПК: A61P 25/28, A61K 31/506, C07D 239/46...
Метки: рецепторов, лиганды, применение, 5-нт
Формула / Реферат:
1. Соединение формулы (IA) где X и Y представляют собой CR, a Z представляет собой N или X представляет собой N, a Y и Z представляют собой CR, где R для каждого случая представляет собой водород; W представляет собой окси; по меньшей мере один из R1a, R1b, R1d и R1e независимо выбран из группы, состоящей из галогена, нитро, амино, циано, -C(O)NH2 и (C1-C4)алкила, либо R1a и R1b, взятые вместе, образуют пяти- или шестичленное ароматическое или...
Аминоарильные сульфонамидные производные как функциональные 5-нт6 лиганды

Номер патента: 13875
Опубликовано: 30.08.2010
Авторы: Камбхампати Рама Састри, Кандикере Нагарадж Вишвоттам, Рамакришна Венката Сатья Нироги, Кота Сринивасулу, Джасти Венкатесварлу, Вишвакарма Сантош, Ширсатх Викас Шрикришна
МПК: A61K 31/4523, A61K 31/4468, C07D 401/12...
Метки: функциональные, аминоарильные, производные, 5-нт6, лиганды, сульфонамидные
Формула / Реферат:
1. Соединение, имеющее формулу (I)или его фармацевтически приемлемая соль,где Ar представляет собой любую одну группу, выбранную из фенила, нафтила, моноциклического или бициклического гетероарила, каждый из которых может быть дополнительно замещен одним или более независимыми заместителями, и такие заместители определены как R1R представляет собой атом водорода, (C1-С3)алкильную или гало(C1-С3)алкильную группу;R1 и R4независимо представляют...
Предыдущий патент: Способы защиты от апоптоза с применением липопептидов
Следующий патент: Макроциклические ингибиторы вируса гепатита с
Случайный патент: Установка для грохочения материалов