Новые промежуточные продукты и их использование для получения n, n’-мостиковых бисиндолилмалеимидов
Номер патента: 639
Опубликовано: 29.12.1999
Авторы: Крумрич Кристин А., Фол Маргарет М., Виннероски Леонард Л.







Формула / Реферат
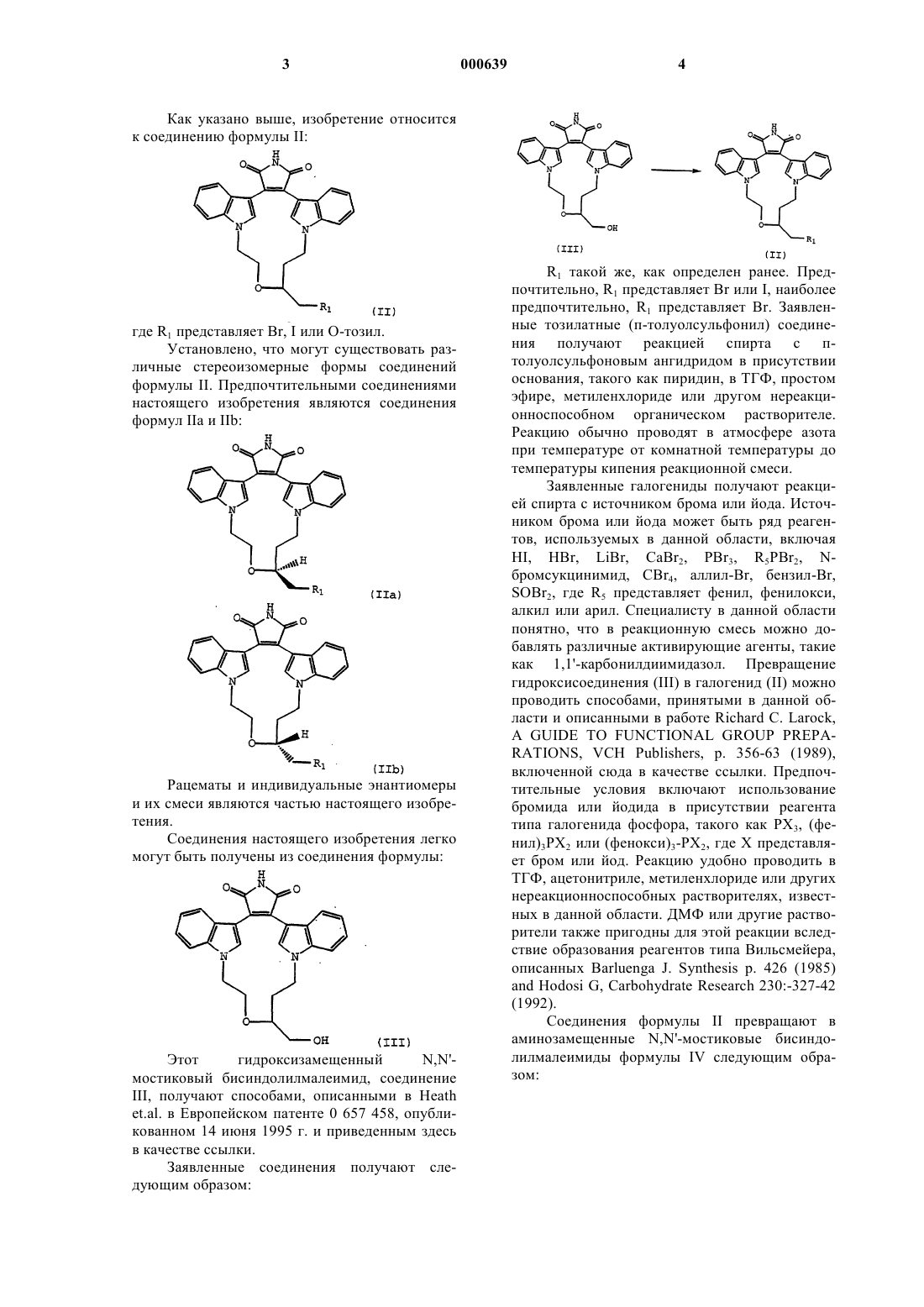
1. Соединение формулы

где R1 представляет Вr, I или O-тозил.
2. Соединение по п.1 формулы

3. Соединение по п.1 или 2, где R1 представляет Вr или I.
4. Фармацевтическая композиция, содержащая соединение по любому из пп.1-3 и один или несколько фармацевтически приемлемых носителей, разбавителей или наполнителей.
5. Способ получения аминозамещенного N,N'-мостикового бисиндолилмалеимида формулы:

где R2 представляет -N(СF3)(СН3), -NH(CF3) или -NR3R4, где R3 и R4, независимо, представляют водород, C1-C4 алкил, фенил, бензил или вместе с азотом, к которому они присоединены, образуют насыщенное или ненасыщенное 5- или 6-членное кольцо, который включает реакцию соединения по любому из пп.1-3 с амином в нереакционноспособном полярном растворителе.
6. Способ по п.5, где соединение представляет собой

и R1 представляет Вr или I.
7. Способ по п.6, где амин представляет НN(СН3)2 или Н2N(СН3).
8. Способ получения N,N'-мостикового бисиндолилмалеимида формулы (V)

который включает реакцию соединения формулы

где R1 представляет Вr или I;
с НN(СН3)2 в нереакционноспособном полярном растворителе.
9. Способ получения соединения по любому из пп.1-3, который включает превращение соединения формулы

в соединение по любому из пп.1-3.
10. Способ по п.9, где способ осуществляют в присутствии РХ3 (фенил)3РХ2 или (фенокси)3РХ2, где Х представляет бром или йод.
11. Способ по п.10, где способ осуществляют в присутствии (фенокси)3РВr2.
12. Способ получения соединения формулы (V)

который включает
а) реакцию соединения формулы (IIIa)

с РХ3 (фенил)3РХ2 или (фенокси)3РХ2, где Х представляет бром или йод, с образованием соединения формулы (IIа)

где R1 представляет Вr или I; и
b) реакцию соединения формулы (IIа) с НN(СН3)2 в нереакционноспособном полярном растворителе.
13. Способ по п.12, где R1 представляет Вr.
14. Способ по п.13, который дополнительно включает реакцию соединения формулы (V) с СН3SО3Н.
15. Соединение формулы

где R1 представляет Br, I или O-тозил, полученное способом по любому из пп.9-11.
Текст
Повсеместная природа изоферментов протеинкиназы С и их важная роль в физиологии являются стимулом для получения высоко селективных ингибиторов ПКС. Вследствие очевидности связи определенных изоферментов с болезненными состояниями, разумно предположить, что ингибирующие соединения, которые селективны к одному или двум изоферментам протеинкиназы С относительно других изоферментов ПКС и других протеинкиназ, будут превосходными терапевтическими средствами. Такие соединения демонстрируют более высокую эффективность и низкую токсичность благодаря их специфичности действия. Класс N,N'-мостиковых бисиндолилмалеимидов, селективных для изофермента ПКС,был описан Heath et.al. (Европейский патент 0 657 458, опубликованный 14 июня 1995 г.). Предпочтительным соединением в этом N,N'мостиковом ряду является соединение формулы чения аминозамещенных N,N'-мостиковых бисиндолилмалеимидов при более низких температурах и за более короткое время реакции, что приводит к более высокому выходу с меньшим содержанием побочных продуктов. Таким образом, этот промежуточный продукт пригоден для получения N,N'-мостиковых бисиндолилмалеимидов с высоким выходом без нежелательных токсичных примесей. Кроме того, заявленные соединения полезны в качестве ингибиторов ПКС, селективных для изоферментов. Как таковые эти соединения полезны для лечения состояний, связанных с сахарным диабетом и его осложнениями, ишемии, воспаления, нарушения деятельности центральной нервной системы,сердечнососудистого заболевания, дерматологической болезни и рака. Настоящее изобретение относится к соединениям формулы II: где R представляет амино, алкиламино или диалкиламино. Неаth et.al. приводят в качестве примера ряд этих аминозамещенных N,N'мостиковых бисиндолилмалеимидов, полученных следующим образом: где R1 представляет Br, I или O-тозил. Настоящее изобретение далее относится к фармацевтической композиции, содержащей соединение формулы II и один или несколько фармацевтически приемлемых носителей, разбавителей или наполнителей. Следующим аспектом изобретения является способ использования соединения формулыII для получения аминозамещенных N,N'мостиковых бисиндолилмалеимидов формулы I,который включает реакцию соединения формулы II с амином в нереакционноспособном, полярном растворителе. В настоящем изобретении, как описано и заявлено здесь, нижеследующие термины и аббревиатуры определяются следующим образом. Термин "C1-C4 алкил" представляет алкильную группу с циклической, неразветвленной или разветвленной цепью, содержащую от одного до четырех атомов углерода, такую как метил, этил, н-пропил, изопропил, циклопропил,н-бутил, изобутил, втор-бутил, трет-бутил и тому подобное. Термин "арил" представляет замещенный или незамещенный бензил, фенил или нафтил. Термин "амин", используемый здесь, относится к -N(СF3)(СН 3), -NН(СF3) или -NR3R4, гдеR3 и R4, независимо, представляют водород, C1C4 алкил, фенил, бензил или в комбинации с азотом, к которому они присоединены, образуют насыщенное или ненасыщенное 5- или 6 членное кольцо. К сожалению, было обнаружено, что Омезилатная функциональная группа, используемая для получения аминозамещенных N,N'мостиковых бисиндолилмалеимидов, токсична и является нежелательной примесью при получении аминозамещенных N,N'-мостиковых бисиндолилмалеимидов. Для гарантии того, что Oмезилатный промежуточный продукт удален из конечного продукта, нужно применять дорогие методики очистки. Настоящее изобретение предлагает ключевой промежуточный продукт для синтеза аминозамещенных N,N'-мостиковых бисиндолилмалеимидов. Этот новый промежуточный продукт легко превращается в аминозамещенныеN,N'-мостиковые бисиндолилмалеимиды без прохождения через стадию образования Омезилатного промежуточного продукта. Этот промежуточный продукт также значительно более реакционноспособен. Промежуточный продукт предпочтительно используют для полу 3 где R1 представляет Br, I или O-тозил. Установлено, что могут существовать различные стереоизомерные формы соединений формулы II. Предпочтительными соединениями настоящего изобретения являются соединения формул IIа и IIb: Рацематы и индивидуальные энантиомеры и их смеси являются частью настоящего изобретения. Соединения настоящего изобретения легко могут быть получены из соединения формулы:et.al. в Европейском патенте 0 657 458, опубликованном 14 июня 1995 г. и приведенным здесь в качестве ссылки. Заявленные соединения получают следующим образом:R1 такой же, как определен ранее. Предпочтительно, R1 представляет Вr или I, наиболее предпочтительно, R1 представляет Br. Заявленные тозилатные (п-толуолсульфонил) соединения получают реакцией спирта с птолуолсульфоновым ангидридом в присутствии основания, такого как пиридин, в ТГФ, простом эфире, метиленхлориде или другом нереакционноспособном органическом растворителе. Реакцию обычно проводят в атмосфере азота при температуре от комнатной температуры до температуры кипения реакционной смеси. Заявленные галогениды получают реакцией спирта с источником брома или йода. Источником брома или йода может быть ряд реагентов, используемых в данной области, включаяHI, HBr, LiBr, СаВr2, РВr3, R5PBr2, Nбромсукцинимид, СВr4, аллил-Вr, бензил-Вr,SOBr2, где R5 представляет фенил, фенилокси,алкил или арил. Специалисту в данной области понятно, что в реакционную смесь можно добавлять различные активирующие агенты, такие как 1,1'-карбонилдиимидазол. Превращение гидроксисоединения (III) в галогенид (II) можно проводить способами, принятыми в данной области и описанными в работе Richard С. Larock,A GUIDE TO FUNCTIONAL GROUP PREPARATIONS, VCH Publishers, p. 356-63 (1989),включенной сюда в качестве ссылки. Предпочтительные условия включают использование бромида или йодида в присутствии реагента типа галогенида фосфора, такого как РХ 3, (фенил)3 РХ 2 или (фенокси)3-РХ 2, где Х представляет бром или йод. Реакцию удобно проводить в ТГФ, ацетонитриле, метиленхлориде или других нереакционноспособных растворителях, известных в данной области. ДМФ или другие растворители также пригодны для этой реакции вследствие образования реагентов типа Вильсмейера,описанных Barluenga J. Synthesis p. 426 (1985) где R1 представляет Вr, I или O-тозил и R2 представляет -N(СF3)(СН 3), -NH(CF3) или -NR3R4,где R3 и R4, независимо, представляют водород,C1-C4 алкил, фенил, бензил или вместе с азотом,к которому они присоединены, образуют насыщенное или ненасыщенное 5- или 6-членное кольцо. R2, предпочтительно, представляетN(СН 3). Способ использования соединения II для образования соединения IV включает реакцию соединения II с амином формулы HN(CF3)(СН 3),HNH(CF3) или НNR3R4, где R3 и R4, независимо,представляют водород, C1-C4 алкил, фенил, бензил или вместе с азотом, к которому они присоединены, образуют насыщенное или ненасыщенное 5- или 6-членное кольцо, в нереакционноспособном, полярном апротонном растворителе. Реакцию предпочтительно проводят в растворе ДМФ, смеси ТГФ:вода или диметилацетамида при температурах в диапазоне от 0 С до температуры кипения реакционной смеси. Реакцию обычно завершают за время приблизительно от 1 до 20 ч. Предпочтительно, реакцию проводят при температуре от комнатной температуры до 50 С. Соединение IV можно очистить от реакционной смеси с использованием стандартных методов, но, предпочтительно, его кристаллизуют непосредственно из реакционной смеси. Очень неожиданно, что использование заявленного промежуточного продукта для получения аминозамещенных N,N'-мостиковых бисиндолилмалеимидов приводит к более высокому выходу и позволяет избежать токсичных примесей. Заявленный промежуточный продукт неожиданно оказался более реакционноспособным, чем известный мезилатный промежуточный продукт. Реакционная способность различных уходящих групп по отношению к НN(СН 3)2 представлена в таблице. Относительная реакционная способность, предсказанная в настоящей области, описана CAREY AND SUNDBERG,Part A, 3rd Edition, page 291 (1990). Таблица Скорости реакции с НN(СН 3)2 ГруппаKrel (Скорость реакции уходящих групп с НN(СН 3)2) п-толуолсульфонат 2,2 х 10-2 МsО 5,5 х 10-3 Сl 9,8 х 10-4 6 Данные таблицы показывают, что тозил,бромид и йодид неожиданно реакционноспособны, особенно бромид и йодид, которые от 8 до 36 раз более реакционноспособны, чем известный мезилатный промежуточный продукт. Эта повышенная реакционная способность относительно МsО наблюдалась также с H2NCH3,Н 2 Н(бензил). Повышение реакционной способности приводит к возможности использовать более низкую температуру реакции, которая завершается за более короткий период времени. Использование заявленного промежуточного продукта приводит также к уменьшению примесей в продукте. При использовании известногоO-мезилпромежуточного продукта в реакции для получения аминозамещенныхN,N'мостиковых бисиндолилмалеимидов приводит к получению примесей в количестве от 15 до 30% вследствие получения побочных продуктов реакцией по карбонильной группе малеимида. При использовании заявленного промежуточного продукта количество примеси составляет менее 5%, что представляет собой значительное улучшение. Как указано ранее, было обнаружено, чтоO-мезилатная функциональная группа, используемая для получения аминозамещенных N,N'мостиковых бисиндолилмалеимидов, токсична и является нежелательной примесью при получении аминозамещенных N,N'-мостиковых бисиндолилмалеимидов. Для гарантии того, что Oмезилатный промежуточный продукт удален из конечного продукта, нужно применять дорогие методики очистки. Следовательно, дополнительным преимуществом настоящих промежуточных продуктов и способа получения аминозамещенных N,N'-мостиковых бисиндолилмалеимидов с использованием заявленных промежуточных продуктов является возможность избежать трудных стадий очистки для удаления токсичных примесей. Предпочтительными соединениями, полученными с использованием заявленных промежуточных продуктов, являются следующие соединения: (S)-13-[(диметиламино)метил]-10,11,14,15-тетрагидро-4,9:16,21-диметено-1 Н,13 Ндибензо[е,k]пирроло[3,4-h][1,4,13]оксадиазациклогексадецин-1,3(2 Н)-дион, особенно в виде мезилатной соли;[1,4,13]оксадиазациклогексадецин-1,3(2 Н)-дион; моногидрохлорид (S)-13-[(пирролидино)метил]10,11,14,15-тетрагидро-4,9:16,-21-диметено 1 Н,13 Н-дибензо[е,k]пирроло[3,4-h][1,4,13]оксадиазациклогексадецин-1,3(2 Н)-диона и (S)-13[бензиламинометил]-10,-11,14,15-тетрагидро 4,9:16,21-диметено-1 Н,13 Н-дибензо[е,k]пирроло[3,4-h][1,4,13]оксадиазациклогексадецин 1,3(2 Н)-дион. Предпочтительные монозамещенные амины формулы IV можно получить непосредст 7 венно из заявленных соединений. Прямой способ получения этих соединений с высоким выходом невозможен при использовании мезилатного промежуточного продукта. Соединения формулы IV получают в виде свободного основания и, предпочтительно, превращают в фармацевтически приемлемую соль способами, принятыми в данной области. Предпочтительные соли включают гидрохлоридную и мезилатную соль. Следующие примеры и получения представлены только для дальнейшей иллюстрации изобретения. Объем данного изобретения не истолковывается как состоящий только из следующих примеров. В следующих примерах и примерах получения точка плавления, спектр ядерного магнитного резонанса, масс-спектр,жидкостная хроматография высокого давления на силикагеле, N,N'-диметилформамид, тетрагидрофуран и этилацетат обозначены следующими аббревиатурами: т. пл., ЯМР, МС, ЖХВД,ДМФ, ТГФ, и EtOAc, соответственно. Термины"ЯМР" и "МС" указывают, что спектр согласуется с желаемой структурой. Пример получения 1. Метансульфонат 3-(2-[(метилсульфонил)окси]этокси]-4-трифенилметокси)-1 бутанола. Тритилхлорид (175,2 г, 0,616 моль) растворяли в 500 мл CH2Cl2 в атмосфере N2. Добавляли триэтиламин (71,9 г, 100 мл, 0,710 моль) и затем добавляли R,S-глицидный спирт (50,0 г,0,648 моль) и реакционный раствор нагревали для мягкого кипячения с обратным холодильником (42 С) в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры и экстрагировали два раза 250 мл водного насыщенного раствора хлорида аммония и затем 250 мл солевого раствора. Водные слои обратно экстрагировали 100 мл СН 2 Сl2 и органический слой сушили (МgSO4) и выпаривали в вакууме, получая тритилглицидный спирт в виде масла, которое кристаллизовали из этанола, получая 104,4 г(54%) тритилглицидного спирта в виде твердого вещества. 1 М раствор бромида винилмагния (50 мл,50 ммоль, 2,0 экв.) в ТГФ охлаждали до 20 С в атмосфере N2 и добавляли каталитическое количество йодида меди (0,24 г, 1,26 ммоль, 0,05 экв.). Полученную смесь перемешивали при 20 С в течение 5 мин и затем по каплям в течение 15 мин при 20 С добавляли раствор тритилглицидного спирта (7,91 г, 25,0 ммоль) в 40 мл сухого ТГФ. Реакционную смесь перемешивали в течение 3 ч при 20 С и затем давали ей нагреться до комнатной температуры и перемешивали в течение 15 мин. Реакцию останавливали охлаждением реакционной смеси до 30 С и медленно добавляли 125 мл водного насыщенного раствора хлорида аммония. Полученную смесь экстрагировали 200 мл этилацетата. 8 Органический слой затем экстрагировали водным раствором 0,93 г (2,50 ммоль, 0,1 экв.) дигидрата двунатриевой соли этилендиаминтетрауксусной кислоты (ЭДТК) в 125 мл деионизированной воды для удаления любых металлов. Водные слои обратно экстрагировали 50 мл этилацетата и объединенные органические слои промывали 100 мл солевого раствора, сушили(MgS04) и выпаривали в вакууме, получая масло, которое фильтровали через диоксид кремния(76 г), используя 1,2 л смеси гексан/этилацетат,3/1. Фильтрат выпаривали в вакууме, получая 9,07 г 1-(трифенилметокси)-4-пентен-2-ола в виде легкого масла желтого цвета (100%). 60% суспензию гидрида натрия в минеральном масле (6,13 г, 0,153 моль, 1,5 экв.) суспендировали в 175 мл сухого ТГФ и при комнатной температуре в суспензию добавляли 1(трифенилметокси)-4-пентен-2-ол. Полученную смесь перемешивали при 45 С в течение 1,5 ч и затем через шприц добавляли 17,7 мл (0,204 ммоль, 2,0 экв.) свежеперегнанного аллилбромида. Реакционную смесь нагревали до 45 С в течение 1 ч. Реакцию можно контролировать ТСХ или ЖХВД. Реакционную смесь охлаждали до 0 С и медленно добавляли 400 мл водного насыщенного раствора хлорида аммония для гашения избытка основания. Полученную смесь экстрагировали 800 мл этилацетата и органический слой промывают 500 мл воды. Водные слои обратно экстрагировали 100 мл этилацетата и объединенные органические слои промывали 200 мл солевого раствора, сушили (MgSO4) и выпаривали в вакууме, получая 41,5 г (100%) 1,1',1"-2-(2-пропенилокси)-4-пентенил]окси]метилидин]трис[бензола] в виде желтого масла. 1,1',1"-2-(2-Пропенилокси)-4-пентенил]окси]метилидин]-трис[бензол] (39,3 г, 0,102 моль) растворяли в растворе 390 мл безводного метилового спирта и 60 мл CH2Cl2 и охлаждали до температуры от 50 до 40 С при барботировании N2 через вязкий реакционный раствор. Затем через реакционную смесь при температуре от 50 до 40C в течение 80 мин барботировали озон до тех пор, пока реакционная смесь не стала бледно-голубой. Полученной реакционной смеси давали нагреться до 0 С в атмосфере N2 и затем для остановки реакции медленно добавляли раствор борогидрида натрия (23,15 г, 0,612 моль, 6 экв.) в смеси 85 мл этанола/85 мл воды при поддержании температуры реакции ниже 10 С. Реакционную смесь перемешивали при охлаждении на ледяной бане в течение 30 мин и затем давали нагреваться до комнатной температуры и перемешивали в течение ночи. Поднимали температуру до 31 С нагреванием. Реакционную смесь разбавляли 400 мл водного насыщенного раствора хлорида аммония и экстрагировали 800 мл этилацетата. Органический слой промывали 400 мл воды и водные слои 9 обратно экстрагировали 150 мл этилацетата. Объединенный органический слой промывали 200 мл солевого раствора, сушили (MgSO4) и выпаривали в вакууме, получая мутное масло. Это масло кристаллизовали в виде 3 порций из смеси гексан/этилацетат, 2/1, получая 28,9 г 3(2-гидроксиэтокси)-4-(трифенилметокси)-1 бутанола (72%). 3-(2-Гидроксиэтокси)-4-(трифенилметокси)-1-бутанол (14,0 г, 35,7 ммоль) растворяли в 140 мл СН 2 Сl2, охлаждали до 0 С в атмосфереN2 и добавляли триэтиламин (10,8 г, 14,9 мл,0,107 моль, 3,0 экв.). Затем по каплям при 5 С добавляли метансульфонил-хлорид (11,0 г, 7,46 мл, 96,4 ммоль, 2,7 экв.). Полученную реакционную смесь разбавляли дополнительным количеством CH2Cl2 (300 мл) и промывали 200 мл воды и 200 мл водного насыщенного раствора хлорида аммония. Водные слои обратно экстрагировали 50 мл CH2Cl2 и объединенный органический слой промывали 100 мл солевого раствора, сушили (MgSO4) и выпаривали в вакууме,получая 18,4 г (94%) метансульфоната 3-(2[(метилсульфонил)окси]этокси]-4-трифенилметокси)-1-бутанола в виде белого твердого вещества. Пример получения 2.(S)-Тритилглицидный спирт. Тритилхлорид (2866 г, 10,3 моль) растворяли в 7 л CH2Cl2 в атмосфере азота. Добавляли триэтиламин (1189 г, 1638 мл, 11,8 моль) и затем добавляли (R)-(+)-глицидный спирт (795,0 г,10,6 моль) с использованием 1 л CH2Cl2 в качестве жидкости для промывки. Реакционный раствор нагревали для мягкого кипячения с обратным холодильником (42 С) в течение 3-4 ч. Реакционную смесь охлаждали до комнатной температуры и затем добавляли 3 л солевого раствора. Органический слой сушили (600 гNa2SO4) и выпаривали в вакууме, получая указанное в заголовке соединение в виде масла,которое кристаллизовали из этанола, получая 2354 г (70%) указанного в заголовке соединения в виде твердого вещества. Пример получения 3. Метансульфонат (S)-3-[2-[(метилсульфонил)окси]этокси]-4-(трифенилметокси)-1-бутанола. 1 М раствор бромида винилмагния (5,76 л,5,76 моль, 1,96 экв.) в ТГФ охлаждали до 20 С в атмосфере N2 и добавляли каталитическое количество йодида меди (28,2 г, 0,148 моль, 0,05 экв.). Полученную смесь перемешивали при 20 С в течение 5 мин и затем при 20 С в течение 1,5 ч по каплям добавляли раствор (S)тритилглицидного спирта (929,0 г, 2,94 моль) в 3,2 л сухого ТГФ. Реакционную смесь перемешивали в течение 1 ч при 20 С. Реакцию останавливали охлаждением реакционной смеси до 30 С и медленно добавляли 5 л водного насыщенного раствора хлорида аммония. Органиче 000639 10 ский слой затем экстрагировали два раза 1 л 10% (мас./объмы) раствора дигидрата двунатриевой соли этилендиаминтетрауксусной кислоты (ЭДТК) для удаления любых металлов. Органический слой промывали 2 л солевого раствора, сушили (MgS04) и выпаривали в вакууме,получая 1061 г (96%) (S)-1-О-трифенилметил-4 гидроксипентанола в виде масла. 60% суспензию гидрида натрия в минеральном масле (268,9 г, 6,72 моль, 1,5 экв.) суспендировали в 2,8 л сухого ТГФ в атмосфере N2 и при комнатной температуре добавляли раствор (S)-1-О-трифенилметил-4-гидроксипентанола (1543 г, 4,48 моль) в 5,6 л сухого ТГФ. Полученную смесь перемешивали при комнатной температуре в течение 1,5 ч и затем в течение 20 мин добавляли 770 мл (8,89 моль, 2,0 экв.) свежеперегнанного аллилбромида. Реакционную смесь нагревали до 45 С в течение 1-2 ч. Реакционную смесь охлаждали до 15-20 С и медленно добавляли 2 л водного насыщенного раствора хлорида аммония для гашения избытка основания. Получаемую смесь разбавляли 1 л этилацетата и 1 л воды и органический слой отделяли. Водный слой обратно экстрагировали 500 мл этилацетата и объединенные органические слои сушили (MgSO4) и выпаривали в вакууме, получая 1867 г (98%) (S)-1,1',1- 2-(2 пропенилокси)-4-пентенил]окси]метилидин]трис[бензола] в виде желтого масла.(S)-1,1',1-2-(Пропенилокси)-4 пентенил]окси]метилидин]трис[бензол] (1281 г,3,33 моль) растворяли в растворе 4 л безводного метилового спирта и 3,6 л CH2Cl2 и охлаждали до температуры от 50 до 40 С при барботировании N2 через вязкий реакционный раствор N2. К реакционной смеси добавляли индикатор судан III и через реакционную смесь при температуре от 50 до 35 С в течение 13 ч барботировали озон до тех пор, пока цвет реакционной смеси не изменялся от персикового до бледнозелено-желтого цвета. Получаемой реакционной смеси давали нагреться до 0 С в атмосфере N2 и затем медленно, в течение 40 мин, добавляли к раствору борогидрид натрия (754 г, 19,9 моль, 6 экв.) в смеси 2,5 л этанола/2,5 л вода при поддержании температуры реакции ниже 30 С. Реакционную смесь затем оставляли для перемешивания при комнатной температуре в течение ночи. Реакцию можно контролировать ЖХВД. Реакционную смесь охлаждали до 10-15C и медленно добавляли к 4 л водного насыщенного раствора хлорида аммония при 20 С. Гашеную реакционную смесь затем фильтровали и твердую часть промывали 3 л CH2Cl2. Органический слой отделяли и промывали 3 л водного насыщенного раствора хлорида аммония и водные слои обратно экстрагировали 1 л CH2Cl2. Объединенный органический слой сушили (МgSO4) и выпаривали в вакууме, получая 1361 г (100%)(S)-3-(2-Гидроксиэтокси)-4-(трифенилметокси)-1-бутанол (500 г, 1,27 моль) растворяли в 4,8 г CH2Cl2, охлаждали до 0 С в атмосфере N2 и добавляли триэтиламин (386,4 г, 532 мл, 3,81 моль, 3,0 экв.). Затем по каплям в течение 30 мин при 5 С добавляли метансульфонилхлорид (396,3 г, 286 моль, 3,46 моль, 2,7 экв.). Полученную реакционную смесь перемешивали при температуре от 0 до 5 С в течение 1-2 ч и контролировали ЖХВД. Реакционную смесь разбавляли дополнительным CH2Cl2 и промывали два раза 2 л воды и 2 л водного насыщенного раствора хлорида аммония. Водные слои обратно экстрагировали 1 л CH2Cl2 и объединенный органический слой сушили (MgSO4) и выпаривали в вакууме, получая сырой твердый продукт, который перекристаллизовали из смеси гептан/этилацетат (1/1), получая 615 г (88%) метансульфоната (S)-3-[2[(метилсульфонил)окси]этокси]-4-(трифенилметокси)-1-бутанола в виде трех порций в форме твердого вещества. ЯМР. МС. Пример получения 4.(S)-10,11,14,15-Тетрагидро-13-(гидроксиметил)-4,9:16,21-диметено-1H,13 Н-дибензо[е,k]пирроло[3,4-h][1,4,13]оксадиазациклогексадецин-1,3(2 Н)-дион. 2,3-Бис-(1H-индол-3-ил)-N-метилмалеимид (114,7 г, 0,336 моль) и метансульфонат (S)3-[2[(метилсульфонил)окси]этокси]-4-(трифенилметокси)-1-бутанола (220,0 г, 0,401 моль, 1,2 экв.) растворяли в 4,3 л ДМФ. Этот раствор реагентов затем медленно, в течение 70 ч (приблизительно 1 мл/мин), добавляли при 50 С к суспензии карбоната цезия (437,8 г, 1,34 моль, 4,0 экв.) в 7 л ДМФ. Через 70-72 ч реакционную смесь охлаждали и фильтровали и ДМФ удаляли в вакууме, получая остаток, который растворяли в 4,6 л CH2Cl2. Органический слой экстрагировали 1,15 л водного 1 н НСl и затем 4,6 л солевого раствора. Объединенные водные слои обратно экстрагировали 1,1 л CH2Cl2. Объединенный органический слой сушили (Na2SO4) и фильтровали. Большую часть растворителя удаляли в вакууме, и полученный раствор фильтровали через 2 кг силикагеля, используя 15,14018,925 л дополнительного СН 2 Сl2 для исходного материала. Растворитель удаляли в вакууме и полученное твердое вещество, окрашенное в пурпурный цвет, растирали в 7 объемах ацетонитрила (на основе массы сырого (S)10,11,14,15-тетрагидро-2-метил-13-[(трифенилметокси)метил]-4,9:16,21-диметено-1H,13 Ндибензо[е,k]пирроло[3,4-h][1,4,13]оксадиазациклогексадецин-1,3(2 Н)-диона), получая 150,2 г 12 сушки (87% чистота по данным ЖХВД по сравнению со стандартом).(S)-10,11,14,15-Тетрагидро-2-метил-13[(трифенилметокси)-метил]-4,9:16,21-диметено 1 Н,13 Н-дибензо[е,k]пирроло[3,4-h]-[1,4,13]оксадиазациклогексадецин-1,3(2 Н)-дион (32,7 г, 46,9 ммоль) суспендировали в 1,6 л этанола и 1,6 л водного 10 н КОН. Полученную смесь нагревали для мягкого кипячения с обратным холодильником (78 С) в течение 19 ч. Большая часть твердого соединения растворялась при достижении температуры кипения. Реакционный раствор охлаждали до температуры от 10 до 15 С и при 15 С медленно добавляли водную 10 н НСl (1,2 л) для установления кислотности рН=1. При подкислении происходило образование красной суспензии. Реакционную смесь разбавляли 500 мл CH2Cl2 и перемешивали в течение 20 мин и фильтровали для удаления большей части солей. Соли промывали дополнительнымCH2Cl2 (l,5 л) и фильтрат экстрагировали два раза 1 л воды. Объединенные водные слои обратно экстрагировали 1 л CH2Cl2 и органический слой сушили (МgSO4). Растворитель удаляли в вакууме, получая 36,0 г (100%) (S)10,11,14,15-тетрагидро-13-[(трифенилметокси)метил]-4,9:16,21-диметено-13 Н-дибензо[е,k]фуро[3,4-h][1,4,13]оксадиазациклогексадецин-1,3-диона в виде твердого вещества пурпурного цвета (80% чистота по данным ЖХВД по сравнению со стандартом).(S)-10,11,14,15-Тетрагидро-13-[(трифенилметокси)метил]-4,9:16,21-диметено-13 Н-дибензо[е,k]фуро[3,4-h][1,4,13]оксадиазациклогексадецин-1,3-дион (36,0 г, предположительно 46,9 ммоль) растворяли в 320 мл сухого ДМФ в атмосфере N2 и обрабатывали предварительно смешанным раствором 1,1,1,3,3,3-гексаметилдисилазана (99 мл, 75,7 г, 0,469 моль, 10 экв.) и метанола (9,5 мл, 7,51 г, 0,235 моль, 5 экв.). Полученный раствор нагревали при 45 С в течение 7 ч. Реакцию контролируют ЖХВД. Большую часть ДМФ удаляли в вакууме и полученный остаток экстрагировали в 200 мл этилацетата и промывали 200 мл воды и два раза 100 мл водного 5% раствора LiCl. Водные слои обратно экстрагировали 100 мл этилацетата. Объединенный органический слой промывали 200 мл насыщенного водного раствора хлорида аммония. Объединенный органический слой сушили (МgSO4) и выпаривали в вакууме,получая 35,9 г (100%) сырого (S)-10,11,14,15 тетрагидро-13-[(трифенилметокси)метил]4,9:16,21-диметено-1 Н,13 Н-дибензо[е,k]пирроло[3,4-h][1,4,13]оксадиазациклогексадецин 1,3(2 Н)-диона в виде твердого вещества пурпурного цвета. 13 и охлаждали до 25 С в атмосфере азота. В реакционный раствор в течение приблизительно 12 мин при 0 С барботировали безводный газообразный НСl. Полученной суспензии давали нагреться до комнатной температуры и перемешивали в течение 1 ч. Реакцию контролируют ЖХВД. Суспензию фильтровали и твердую часть промывали 200 мл CH2Cl2. Твердую часть сушили в вакуумном сушильном шкафу при 50 С, получая 18,6 г (90%) (S)-10,11,-14,15 тетрагидро-13-(гидроксиметил)-4,9:16,21-диметено-1H,13 Н-дибензо[е,k]пирроло[3,4h][1,4,13]оксадиазациклогексадецин-1,3-(2 Н)диона в виде пурпурного твердого вещества(93% чистота, определенная по площади ЖХВД). Пример 1.(S)-10,11,14,15-Тетрагидро-13[бром(метил)]-4,9:16,21-диметено-1 Н,13 Ндибензо[е,k]пирроло[3,4-h][1,4,13]оксадиазациклогексадецин-1,3(2 Н)-дион. Бром (2,0 экв.) и пиридин (0,1 экв.) помещали в метиленхлорид (10 объемов) и раствор охлаждали до 5 С. Бром титровали трифенилфосфитом (2,0 экв.). Раствор превращался из желтого в прозрачный, когда был израсходован весь бром. Во второй реактор загружали (S)10,11,14,15-тетрагидро-13-[гидрокси(метил)]4,9:16,21-диметено-1 Н,13 Н-дибензо[е,k]пирроло[3,4-h][1,4,13]оксадиазациклогексадецин 1,3(2 Н)-дион (1,0 экв.) в метиленхлориде (10 объемов). Суспензию охлаждали до 5 С. Затем к суспензии пирролодиона добавляли раствор дибромида трифенилфосфита и реакционной смеси давали нагреться до комнатной температуры и перемешивали в течение 12-16 ч до завершения реакции (0,4% соединения III по данным ЖХВД). Суспензию концентрировали в вакууме при комнатной температуре в течение 2 ч, затем реакцию останавливали 1 объемом деионизированной воды и перемешивали в течение 15 мин. В реакционную суспензию для осаждения продукта добавляли толуол (40 объемов). После перемешивания при 10 С в течение 1 ч продукт выделяли фильтрованием и промывали дважды толуолом (5 объемов), деионизированной водой (5 объемов) и в конце промывали 5 объемами толуола. Целевой бромид сушили в вакуумной сушилке при 50 С. Выход 85-90%(примеси 1-2%). Для дальнейшего снижения количества примесей продукт повторно суспендируют в системе растворителей, такой как ацетон:вода,метанол:вода, изопропанол:вода, или этилацетате. Предпочтительно продукт повторно суспендируют в смеси ТГФ:вода с отношением от 1:1 до 5:1 (ТГФ:вода). Пример 2. 14 зо[е,k]пирроло[3,4-h][1,4,13]оксадиазациклогексадецин-1,3(2 Н)-дион. К раствору (S)-10,11,14,15-тетрагидро-13[бром(метил)]-4,9:-16,21-диметено-1 Н,13 Ндибензо[е,k]пирроло[3,4-h][1,4,13]оксадиазациклогексадецин-1,3(2 Н)-диона (1,0 экв.) в N,Nдиметилформамиде (17 объемов) добавляли диметиламин (10,73 кг, 12 экв.). Реакционный сосуд герметизировали и нагревали при 45 С в течение 9 ч. Реакционную смесь затем охлаждали до комнатной температуры и перемешивали в течение 12-16 ч. Для образования свободного основания к реакционной смеси добавлялиN,N-диметилформамида в вакууме до получения 5-7 объемов к реакционной смеси при 60 С в течение 1 ч добавляли МеОН (30 объемов) также при 60 С. Реакционную смесь затем охлаждали до комнатной температуры и перемешивали в течение ночи, затем далее охлаждали до 0-10 С. Продукт выделяли фильтрованием и промывали МеОН (3 объема). Материал сушили в вакуумной сушилке при 50 С до постоянной массы. Выход 85-92%. Другими растворителями, которые использовали в этой реакции, являются ТГФ/вода и диметилацетамид, вследствие низкой растворимости исходного материала и продуктов реакцию следует проводить в полярном апротонном растворителе. Исследовали другие основания (см. ниже). Для превращения соли НВr в свободное основание in situ исследовали другие основания (см. ниже), но наиболее эффективными основаниями были 6 н NaOH, 12 н NaOH и К 2 СО 3. Пример 3.N,Nдиметилформамида (20 объемов). К раствору добавляли йодид натрия (3,0 г, 19,4 ммоль, 10 экв.) и реакционную смесь перемешивали и нагревали при 50 С в течение 36 ч. После охлаждения реакционной смеси до комнатной температуры продукт выделяли добавлением воды (50 мл, 50 объемов). Продукт осаждался в виде пурпурного твердого вещества, который перекристаллизовывали из смеси ТГФ:Н 2O, 5:1, получая 0,87 г (81%) указанного в заголовке соединения. Пример 4.(S)-10,11,14,15-Тетрагидро-13-[гидрокси(метил]-4,9:16,21-диметено-1H,13 Н-дибен 15 зо[е,k]пирроло[3,4-h][1,4,13]оксадиазациклогексадецин-1,3(2 Н)-дион (1,0 г, 2,27 ммоль) растворяли в 20 мл дихлорметана (20 объемов). К раствору добавляли толуолсульфоновый ангидрид (2,22 г, 6,80 ммоль, 3,0 экв,) и пиридин (0,72 г, 9,08 ммоль, 4,0 экв.) и реакционную смесь перемешивали и нагревали с обратным холодильником при 42 С в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры и разбавляли 40 мл дихлорметана. Органическую фазу промывали 50 мл 1 н соляной кислоты и 50 мл солевого раствора. Водные слои обратно экстрагировали 30 мл дихлорметана и метиленхлорид в объединенных органических слоях заменяли на этанол. Продукт осаждали в виде пурпурного твердого вещества и фильтровали, получая 1,25 г (выход 93%) указанного в заголовке соединения. Пример 5.(S)-13-[(Монометиламино)метил]10,11,14,15-тетрагидро-4,9:16,-21-диметено 1 Н,13 Н-дибензо[е,k]пирроло[3,4-h][1,4,13]оксадиазациклогексадецин-1,3(2 Н)-дион. Метиламин (37,1 г, 1,19 моль, 20 экв.) растворяли в 600 мл N,N-диметилацетамида, поддерживая температуру ниже 23 С. К раствору добавляли соединение примера 1 (30 г, 0,595 моль). Реакционную смесь перемешивали при комнатной температуре в течение 24 ч в герметичном сосуде. Для акцептирования НВr, образованного в реакции, добавляли триэтиламин(8,3 мл, 0,0595 моль, 1 экв.) и реакционную смесь перемешивали дополнительно 30 мин и затем охлаждали до 4 С и медленно добавляли воду (450 мл), поддерживая температуру реакции ниже 25 С. При добавлении воды образовалась суспензия, которую перемешивали 1 ч и фильтровали, дополнительно используя 200 мл воды для промывания отфильтрованного твердого продукта. Твердый продукт сушили в вакуумном сушильном шкафу при 50 С, получая 25,03 г (93%) указанного в заголовке соединения. Пример 6. Моногидрохлорид(S)-13-[(пирролидино)метил]-10,11,14,15-тетрагидро-4,9:16,21 диметено-1 Н,13 Н-дибензо[е,k]пирроло[3,4h][I,4-13]оксадиазациклогексадецин-1,3(2 Н)диона. Соединение примера 1 (1,0 г, 1,0 экв.) растворяли в 5 мл N,N-диметилацетамида и добавляли пирролидин (1,6 мл, 10 экв.). Реакционную смесь нагревали при 45 С в течение 9 ч, затем охлаждали до комнатной температуры. К красной суспензии добавляли 12 н NaOH (0,17 мл,1,0 экв.) и смесь перемешивали при комнатной температуре в течение 2 ч, получая красный раствор. Растворитель удаляли в вакууме, и масло разбавляли метиленхлоридом (100 мл) и промывали насыщенным хлоридом аммония 16 из которого указанное в заголовке соединение осаждали в виде красного твердого вещества при добавлении метил-трет-бутилового эфира. Твердое вещество отделяли фильтрованием и сушили в вакуумном сушильном шкафу при 50 С в течение ночи, получая 0,8 г (81%) продукта. Пример 7.(S)-13-[Бензиламинометил]-10,11,14,15 тетрагидро-4,9:16,21-диметено-1H,13 Н-дибензо[е,k]пирроло[3,4-h][1,4,13]оксадиазациклогексадецин-1,3 (2 Н)-дион. Соединение примера 1 растворяли в 20 объемах N,N-диметилацетамида и в виде одной порции добавляли бензиламин (6,0 экв.). Реакционную смесь перемешивали и нагревали при 80 С в течение 24 ч в герметизированном сосуде. Реакционную смесь охлаждали до комнатной температуры и для акцептирования НВr добавляли триэтиламин (1 экв.) и реакционную смесь перемешивали в течение дополнительных 30 мин. Добавляли этилацетат и органический слой промывали насыщенным раствором хлорида натрия. Этилацетат раствора заменяли на этанол, получая красную суспензию, которую фильтровали, получая указанное в заголовке соединение в виде красного твердого вещества с выходом 79%. Пример 8. Кинетическое исследование.R = NHBn (Соединение Н) Для использования в кинетических исследованиях готовили 2 молярный раствор диметиламина в N,N-диметилацетамиде (ДМА). Каждое из соединений А (0,25 г, 0,45 ммоль), В(0,236 г, 0,45 ммоль) и Е (0,271 г, 0,45 ммоль) растворяли в ДМА (20 мл/г, 4-6 мл) и в каждую реакционную смесь добавляли 2 молярный раствор диметиламина в ДМА (4,5 мл, 9,0 ммоль,20 экв.). Реакционные растворы закупоривали,перемешивали при 23 С и отбирали образцы для анализа ЖХВД на протяжении времени исследования. Использовали колонку Zorbax SBCN 4,6 мм х 25 см с подвижной фазой: изократная смесь 50:50 ТГФ/вода, эабуференная 0,1% трифторуксусной кислотой, при скорости потока 1 мл/мин и УФ-детектировании при 233 нм 17 9,3 мин, Rt соединения С = 9,0 мин, Rt соединения D = 6,2 мин, Rt соединения Е = 10,6 мин). Концентрации в реакционных смесях определяли, исходя из показателя ответа, который получали для каждого соединения из линейного уравнения калибровочной кривой, составленной по трем точкам (концентрации 0,1 мг/мл, 0,05 мг/мл и 0,025 мг/мл соответствующая площадь ответов). Образцы реакционных смесей (0,1 мл) разводили до 25 мл в мерной колбе перед проведением анализа ЖХВД и концентрации(мг/мл) пересчитывали в молярные концентрации (ммоль/мл). Строили график зависимости натурального log концентрации диметиламина,деленной на концентрацию соединения А, В, С,D или Е от времени. Наклон линии, полученный на каждом графике, делили на разность между исходной концентрацией амина и исходными концентрациями соединений для получения констант скорости реакции второго порядка(единицы L М-1 ч-1). Результаты приведены в таблице. Пример 9. Сравнительное изучение скоростей реакции мезилата и бромида с диметиламином, метиламином и бензиламином. Для использования в кинетических исследованиях готовили 2 молярный раствор метиламина и диметиламина в N,N-диметилацетамиде(1,00 г, 1,98 ммоль) объединяли в N,Nдиметилацетамиде (20 мл/г для реакций с метиламином и диметиламином и 36 мл/г для реакции с бензиламином) и добавляли 2 молярный раствор метиламина в ДМА (19,8 мл, 39,7 ммоль, 20 экв.) или 2 молярный раствор диметиламина в ДМА (19,8 мл, 39,7 ммоль, 20 экв.) или бензиламина (4,25 г, 39,7 ммоль, 20 экв.). Реакционные растворы закупоривали, перемешивали при 23 С и для анализа ЖХВД отбирали образцы на протяжении времени исследования. Использовали колонку Zorbax SB-CN 4,6 мм х 25 см с подвижной фазой: изократная смесь 50:50 ТГФ/вода, забуференная 0,1% трифторуксусной кислотой, при скорости потока 1 мл/мин и УФ-детектировании при 233 нм (Rt соединения D = 11,4 мин, Rt соединения В = 19,9 мин). Концентрации в реакционных смесях определяли, исходя из показателя ответа, который получали для соединений В и D из линейного уравнения калибровочной кривой, составленной по трем точкам (концентрации 0,1 мг/мл, 0,05 мг/мл и 0,025 мг/мл соответствующая площадь ответов). Образцы реакционных смесей (0,1 мл) разводили до 25 мл в мерной колбе перед проведением анализа ЖХВД и концентрации(мг/мл) пересчитывали в молярные концентрации (ммоль/мл). Строили график зависимости натуральный log концентраций аминов, деленной на концентрации соединений, от времени. Наклон линии, полученный на каждом графике,делили на разность между исходной концентра 000639 18 цией амина и исходными концентрациями соединений для получения констант скорости реакции второго порядка (единицы L М-1 ч-1). Метиламино- и бензиламинопроизводные получали непосредственно из заявленного бромидного промежуточного продукта с высоким выходом. Мезилатный промежуточный продукт не давал непосредственно метиламино- и бензиламинопроизводное с высоким выходом. Как указано выше, соединения по настоящему изобретению дополнительно активны в качестве ингибиторов, селективных для протеинкиназы С. Активность соединений была определена с помощью анализа кальцийкальмодулинзависимой протеинкиназы, анализа казеин-протеинкиназы II, анализа каталитической субъединицы сАМР-зависимой протеинкиназы и анализа протеин-тирозин-киназы, описанными Heath et.al. в ЕП 0 657 458, опубликованном 14 июня 1995 г., введен сюда в качестве ссылки. Заявленные соединения активны и селективны для изофермента в этих анализах,имея величину IC50 менее 10 мкМ. Соединения с такой фармакологической активностью полезны для лечения состояний, в патологии которых играет роль протеинкиназа С. Такие состояния, известные в данной области, включают: сахарный диабет и его осложнения (включая ретинопатию, невропатию и нефропатию), ишемию, воспаление, нарушения деятельности центральной нервной системы, сердечно-сосудистую болезнь, болезнь Альцгеймера, дерматологическую болезнь и рак. Соединения формулы II перед введением,предпочтительно, изготовляют в виде готовой препаративной формы. Следовательно, еще одним объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение формулы II и один или несколько фармацевтически приемлемых носителей, разбавителей или наполнителей. Настоящие фармацевтические композиции получают известными методами, используя хорошо известные и легко доступные ингредиенты. При приготовлении композиций настоящего изобретения активный ингредиент обычно смешивают с носителем или разбавляют носителем или заключают в носитель, который может быть в форме капсулы, саше, бумаги или другого контейнера. Когда носитель служит в качестве разбавителя, он может быть твердым, полутвердым или жидким материалом, который действует в качестве наполнителя или среды для активного ингредиента. Таким образом, композиции могут быть в форме таблеток, пилюль, лепешек,саше, облаток, эликсиров, суспензий, эмульсий,растворов, сиропов, аэрозоля (как твердого или в жидкой среде), мягких или твердых желатиновых капсул, суппозиториев, стерильных инъецируемых растворов и стерильных упакованных порошков. 19 Некоторые примеры подходящих носителей, наполнителей и разбавителей включают лактозу, декстрозу, сахарозу, сорбит, маннит,крахмалы, аравийскую камедь, фосфат кальция,альгинаты, трагакант, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, водный сироп,метилцеллюлозу, метил- и пропилгидроксибензоаты, тальк, стеарат магния и минеральное масло. Готовые препаративные формы могут дополнительно включать смазывающие агенты,смачивающие агенты, эмульгирующие и суспендирующие агенты, консервирующие агенты,подслащивающие вещества и ароматизирующие агенты. Композиции изобретения можно изготовить таким образом, чтобы обеспечить быстрое, длительное или замедленное высвобождение активного ингредиента после введения пациенту. Фармацевтически эффективное количество соединения представляет количество, способное ингибировать активность ПКС у млекопитающих. Типичная суточная доза будет составлять от около 0,01 мг/кг до около 20 мг/кг активного соединения. Предпочтительные дозы составляют от около 0,01 до около 10 мг/кг. Композиции предпочтительно изготовляют в единичной дозированной форме, причем каждая доза содержит от около 1 до около 500 мг, обычно от около 5 до около 300 мг, активного ингредиента. Однако должно быть понятно, что вводимая терапевтическая доза будет определяться врачом в зависимости от конкретных условий,включающих состояние, которое лечат, выбор вводимого соединения и выбор способа введения, и, следовательно, указанный выше диапазон доз никоим образом не предназначен для ограничения объема данного изобретения. Термин "единичная дозированная форма" относится к физически дискретным единицам, пригодным в качестве единичных доз для людей и других млекопитающих, причем каждая единица содержит заданное количество активного материала, вычисленное для обеспечения желаемого терапевтического эффекта, в комбинации с подходящим фармацевтическим носителем. Следующие примеры готовых препаративных форм являются только иллюстративными и никоим образом не предназначены для ограничения объема изобретения. Готовая препаративная форма 1. Твердые желатиновые капсулы получают с использованием следующих ингредиентов: Активный ингредиент Крахмал, высушенный Стеарат магния Всего Указанные выше ингредиенты смешивают и наполняют в твердые желатиновые капсулы в количестве 200 мг. 20 Готовая препаративная форма 2. Таблетку получают с использованием указанных ниже ингредиентов: Активный ингредиент Целлюлоза, микрокристаллическая Диоксид кремния, пылеобразный Стеариновая кислота Всего Компоненты смешивают и прессуют для образования таблеток, каждая по 100 мг. Готовая препаративная форма 3. Таблетки, каждая из которых содержит 10 мг активного ингредиента, изготовляют следующим образом: Активный ингредиент Крахмал Микрокристаллическая целлюлоза Поливинилпирролидон(в виде 10% раствора в воде) Натрийкарбоксиметилкрахмал Стеарат магния Тальк Всего Активный ингредиент, крахмал и целлюлозу пропускают через сито меш 45 по шкале США и тщательно смешивают. Раствор поливинилпирролидона смешивают с получаемым порошком и затем пропускают через сито меш 14 по шкале США. Полученные таким образом гранулы сушат при 50 С и пропускают через сито меш 18 по шкале США. Натрийкарбоксиметилкрахмал, стеарат магния и тальк, предварительно пропущенные через сито меш 60 США, затем добавляют к гранулам, которые после смешивания прессуют на аппарате для получения таблеток, получая таблетки, каждая по 100 мг. Готовая препаративная форма 4. Капсулы, каждая из которых содержит 40 мг лекарственного средства, приготовляют следующим образом: Активный ингредиент Крахмал Микрокристаллическая целлюлоза Стеарат магния Всего Активный ингредиент, целлюлозу, крахмал и стеарат магния смешивают, пропускают через сито меш 45 по шкале США и наполняют в твердые желатиновые капсулы на 200 мг. В вышеприведенном описании были описаны принципы, предпочтительные осуществления изобретения и способы использования на практике настоящего изобретения. Однако изобретение, которое должно быть здесь защищено, не должно быть истолковано как ограниченное конкретными описанными формами, поскольку они должны рассматриваться как иллю 21 стративные, а не как ограничительные. Специалисты в данной области могут осуществить варианты и изменения без отклонения от сущности изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы и R1 представляет Вr или I. 7. Способ по п.6, где амин представляет НN(СН 3)2 или Н 2N(СН 3). 8. Способ получения N,N'-мостикового бисиндолилмалеимида формулы (V) где R1 представляет Вr, I или O-тозил. 2. Соединение по п.1 формулы который включает реакцию соединения формулы: 3. Соединение по п.1 или 2, где R1 представляет Вr или I. 4. Фармацевтическая композиция, содержащая соединение по любому из пп.1-3 и один или несколько фармацевтически приемлемых носителей, разбавителей или наполнителей. 5. Способ получения аминозамещенного-NR3R4, где R3 и R4, независимо, представляют водород, C1-C4 алкил, фенил, бензил или вместе с азотом, к которому они присоединены, образуют насыщенное или ненасыщенное 5- или 6 членное кольцо, который включает реакцию соединения по любому из пп.1-3 с амином в нереакционноспособном полярном растворителе. 6. Способ по п.5, где соединение представляет собой где R1 представляет Вr или I; с НN(СН 3)2 в нереакционноспособном полярном растворителе. 9. Способ получения соединения по любому из пп.1-3, который включает превращение соединения формулы в соединение по любому из пп.1-3. 10. Способ по п.9, где способ осуществляют в присутствии РХ 3 (фенил)3 РХ 2 или (фенокси)3 РХ 2, где Х представляет бром или йод. 11. Способ по п.10, где способ осуществляют в присутствии (фенокси)3 РВr2. 12. Способ получения соединения формулы (V) который включает: а) реакцию соединения формулы (IIIa)b) реакцию соединения формулы (IIа) с НN(СН 3)2 в нереакционноспособном полярном растворителе. 13. Способ по п.12, где R1 представляет Вr. 14. Способ по п.13, который дополнительно включает реакцию соединения формулы (V) с СН 3SО 3 Н. 15. Соединение формулы с РХ 3 (фенил)3 РХ 2 или (фенокси)3 РХ 2, где Х представляет бром или йод, с образованием соединения формулы (IIа): где R1 представляет Br, I или O-тозил, полученное способом по любому из пп.9-11. где R1 представляет Вr или I; и
МПК / Метки
МПК: C07D 487/08
Метки: продукты, бисиндолилмалеимидов, получения, новые, промежуточные, использование, n'-мостиковых
Код ссылки
<a href="https://eas.patents.su/13-639-novye-promezhutochnye-produkty-i-ih-ispolzovanie-dlya-polucheniya-n-n-mostikovyh-bisindolilmaleimidov.html" rel="bookmark" title="База патентов Евразийского Союза">Новые промежуточные продукты и их использование для получения n, n’-мостиковых бисиндолилмалеимидов</a>
Промежуточные продукты для ассиметричного синтеза (-)6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2н-3,1-бензоксазин-2-она и способ их получения

Номер патента: 107
Опубликовано: 27.08.1998
Авторы: Томпсон Эндрю С., Ясуда Нобуёси, Грабовский Эдвард Дж.Дж., Корли Эдвард Г.
МПК: C07C 213/00, C07D 265/18
Метки: ассиметричного, 6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2н-3,1-бензоксазин-2-она, получения, продукты, синтеза, способ, промежуточные
Формула / Реферат:
1. 6-Хлор-2-[(R)-циклопропилэтинилгидрокси-трифторметил] амин общей формулы в которой Р представляет собой группу, защищающую аминогруппу. 2. Соединение по п.1, представляющее собой N-(4-метоксибензил)-6-хлор-2-[(R)-циклопропилэтинилгидрокси-трифторметил]-метиланилин формулы 3. Способ получения соединения по п.1, заключающийся в том, что осуществляют стадии: а) получение смеси избытка (1R, 2S)-N-замещенного норэфедрина формулы в...
Конъюгаты соединения, содержащего сульфгидрильную группу, и производного жирной кислоты, способ получения конюгатов, промежуточные соединения для их получения, способы повышения абсорбции и пролонгированного сохранения в крови и тканях млекопитающего соединения, содержащего сульфгидрильную группу

Номер патента: 584
Опубликовано: 29.12.1999
Авторы: Шен Вей Чанг, Икрами Хуссейн М.
МПК: A61K 31/44, C07H 19/048, C07D 213/70...
Метки: сульфгидрильную, сохранения, повышения, млекопитающего, производного, способы, промежуточные, пролонгированного, конюгатов, получения, способ, абсорбции, группу, конъюгаты, крови, содержащего, тканях, кислоты, жирной, соединения
Формула / Реферат:
1. Соединение общей формулы VI где Р является фрагментом соединения, содержащего сульфгидрильную группу, выбранного из группы, включающей пептиды, белки или олигонуклеотиды; R1 представляет собой водород, низший алкил или арил; R2 представляет собой фрагмент, содержащий липидную группу; а R3 представляет собой гидроксил, фрагмент, содержащий липидную группу или аминокислотную последовательность, включающую 1 или 2 аминокислоты и...
Новые противосудорожные i-ar(алк)ил-имидазолин-2-оны, содержащие в 4-положении двузамещенный остаток амина, и способ их получения

Номер патента: 535
Опубликовано: 28.10.1999
Авторы: Ланкау Ханс-Йоахим, Шефер Харри, Менцер Манфред, Гевальд Карл, Унферферт Клаус
МПК: C07D 233/88, A61K 31/415
Метки: содержащие, двузамещенный, новые, противосудорожные, амина, способ, получения, 4-положении, i-ar(алк)ил-имидазолин-2-оны, остаток
Формула / Реферат:
1. Новые соединения общей формулы 1 где Х = водород, С1-С4-алкил, С1-С4-алкокси, трифторметил, галоген, R1 или R2 = С1-С4-алкил, циклоалкил, гетероалкил или R1 и R2 вместе обозначают группу алкилена с 2-6 атомами углерода, в которой группа -СН2- может быть замещена кислородом, азотом или серой. 2. Соединения по п.1, представляющие собой: 1-фенил-4-морфолино-имидазолин-2-он, 1-(4-метокси)-4-пиперидино-имидазолин-2-он, ...
Новые производные стафилокиназы

Номер патента: 121
Опубликовано: 27.08.1998
Автор: Дезире Жозе Коллен
МПК: C07K 14/31, C12N 15/31, A61K 38/16...
Метки: производные, новые, стафилокиназы
Формула / Реферат:
1. Производные стафилокиназы, имеющие аминокислотную последовательность, представленную на фиг.1, в которой одна или более аминокислот в одном или более подчеркнутых кластеров заменены другой аминокислотой. 2. Производные стафилокиназы по п.1, имеющие аминокислотную последовательность, представленную на фиг.1, в которой одна или более аминокислот заменены аланином со снижением таким образом реактивности этих производных с панелью моноклональных...
Cпособ получения замещенных фенолов и способ получения витамина е с их использованием

Номер патента: 28
Опубликовано: 26.02.1998
Авторы: Ансель Жан-Эрик, Бьенейм Юг, Мейллян Пьер
МПК: B01J 31/24, C07C 39/19, A61K 31/355...
Метки: cпособ, получения, использованием, витамина, способ, замещенных, фенолов
Формула / Реферат:
1. Способ получения замещенных фенолов путем конденсации в однофазной среде фенола общей формулы где R обозначает один или несколько одинаковых или различных радикалов, выбранных из группы, включающей водород, гидроксильную группу и C1-C6 алкил, с производным бутадиена в присутствии катализатора на основе Rd+1, фосфинового соединения и основания, отличающийся тем, что в качестве производного бутадиена используют соединение, содержащее по...
Предыдущий патент: Синергетическая фунгицидная композиция, содержащая соединение-аналог стробилурина
Следующий патент: Панелевидный микрофон
Случайный патент: Несущий каркас для информационной и/или рекламной поверхности