Промежуточные продукты для ассиметричного синтеза (-)6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2н-3,1-бензоксазин-2-она и способ их получения
Номер патента: 107
Опубликовано: 27.08.1998
Авторы: Грабовский Эдвард Дж.Дж., Корли Эдвард Г., Ясуда Нобуёси, Томпсон Эндрю С.





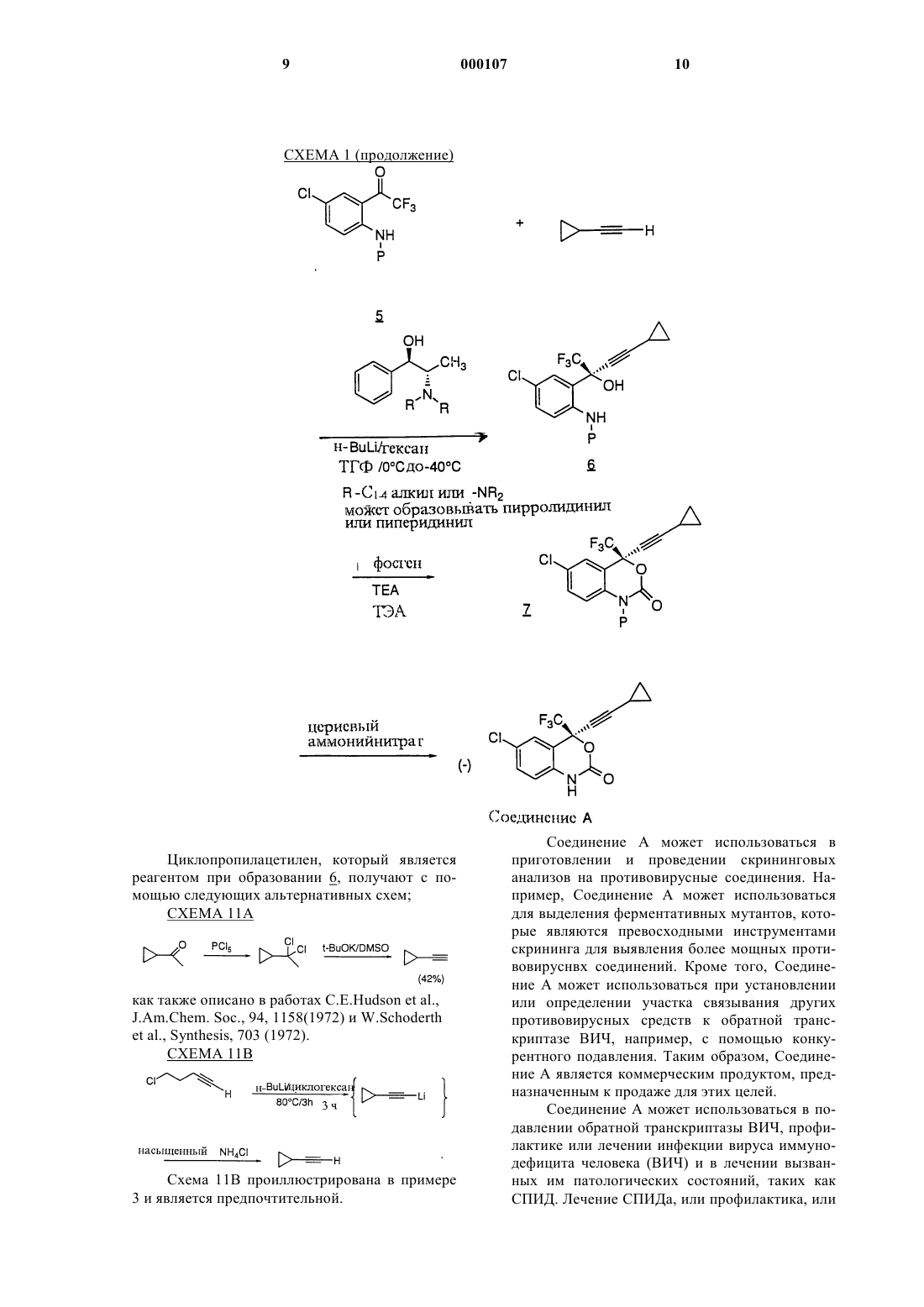

Формула / Реферат
1. 6-Хлор-2-[(R)-циклопропилэтинилгидрокси-трифторметил] амин общей формулы

в которой Р представляет собой группу, защищающую аминогруппу.
2. Соединение по п.1, представляющее собой N-(4-метоксибензил)-6-хлор-2-[(R)-циклопропилэтинилгидрокси-трифторметил]-метиланилин формулы

3. Способ получения соединения по п.1, заключающийся в том, что осуществляют стадии:
а) получение смеси избытка (1R, 2S)-N-замещенного норэфедрина формулы

в которой R представляет собой С1-4 алкил или -NR2 образует пирролидинил или пиперидинил;
с избытком циклопропилацетилена и избытком литирующего вещества, выбранного из н-бутиллития, втор-бутиллития или трет-бутиллития при температуре от приблизительно -78°С до приблизительно 10°С в апротонном растворителе;
(b) смешивание со смесью, полученной на стадии (а), приблизительно одного эквивалента реагента формулы

в которой Р представляет собой защищающую аминогруппу, и выдержку полученной реакционной смеси при температуре от приблизительно -78°С до приблизительно -20°С;
(с) тушение реакции путем добавления источника протонов;
(d) образование желаемого соединения.
4. Способ по п.3, отличающийся тем, что при получении соединения по п.2, осуществляют стадии:
а) получение смеси избытка (1R, 2S)-N-замещенного норэфедрина формулы

в которой R представляет собой С1-4 алкил или -NR2 образует пирролидинил или пиперидинил;
с избытком циклопропилацетилена и избытком литирующего вещества, выбранного из н-бутиллития, втор-бутиллития или трет-бутиллития, при температуре от приблизительно -78°С до приблизительно 10°С в апротонном растворителе;
(b) смешивание со смесью, полученной на стадии (а), приблизительно одного эквивалента N-(4-метоксибензил)-6-хлор-2-(2-трифтор-1-оксоэтил)-анилина формулы

и выдержку полученной реакционной смеси при температуре от приблизительно -78°С до приблизительно -20°С;
(с) тушение реакции путем добавления источника протонов;
(d) образование желаемого соединения.
5. Способ по п.3 получения соединения по п.1, отличающийся тем, что осуществляют стадии:
а) получение смеси избытка (1R, 2S)-N-пирролидинил-норэфедрина формулы

с избытком циклопропилацетилена и избытком литирующего вещества, выбранного из н-бутиллития, втор-бутиллития или трет-бутиллития, при температуре приблизительно -15°С в апротонном растворителе;
(b) смешивание со смесью, полученной на стадии (а), приблизительно одного эквивалента реагента формулы

в которой Р представляет собой защищающую аминогруппу; и выдержку полученной реакционной смеси при температуре приблизительно 40°С;
(с) тушение реакции путем добавления источника протонов;
(d) образование желаемого соединения.
6. Способ по п.5, отличающийся тем, что при получении соединения по п.2 осуществляют стадии:
(а) получение смеси избытка (1R, 2S)-N-пирролидинил норэфедрина формулы

с избытком циклопропилацетилена и избытком н-бутиллития при температуре приблизительно -15°С в апротонном растворителе;
(b) смешивание со смесью, полученной на стадии (а), приблизительно одного эквивалента N-(4-метоксибензил)-6-хлор-2-(2-трифтор-1-оксоэтил)-анилина формулы

и выдержку полученной реакционной смеси при температуре приблизительно -40°C;
(с) тушение реакции путем добавления источника протонов;
(d) образование желаемого соединения.
7. Способ по любому из пп.3-4, отличающийся тем, что проводят дополнительно стадию нагревания между стадией (а) и стадией (b), при этом смесь, полученную на стадии (а), нагревают до температуры от приблизительно -10°С до приблизительно 10°С в течение, по меньшей мере, 5 мин, затем охлаждают до температуры от приблизительно -78°С до приблизительно -20°С перед стадией (b).
8. Способ по любому из пп.5-6, отличающийся тем, что проводят дополнительно стадию нагревания между стадией (а) и стадией (b), при этом смесь, полученную на стадии (а), нагревают до температуры от приблизительно -10°С до приблизительно 0°С в течение от приблизительно 10 мин до приблизительно 60 мин, затем охлаждают до температуры, по меньшей мере, приблизительно -40°С перед стадией (b).
Текст
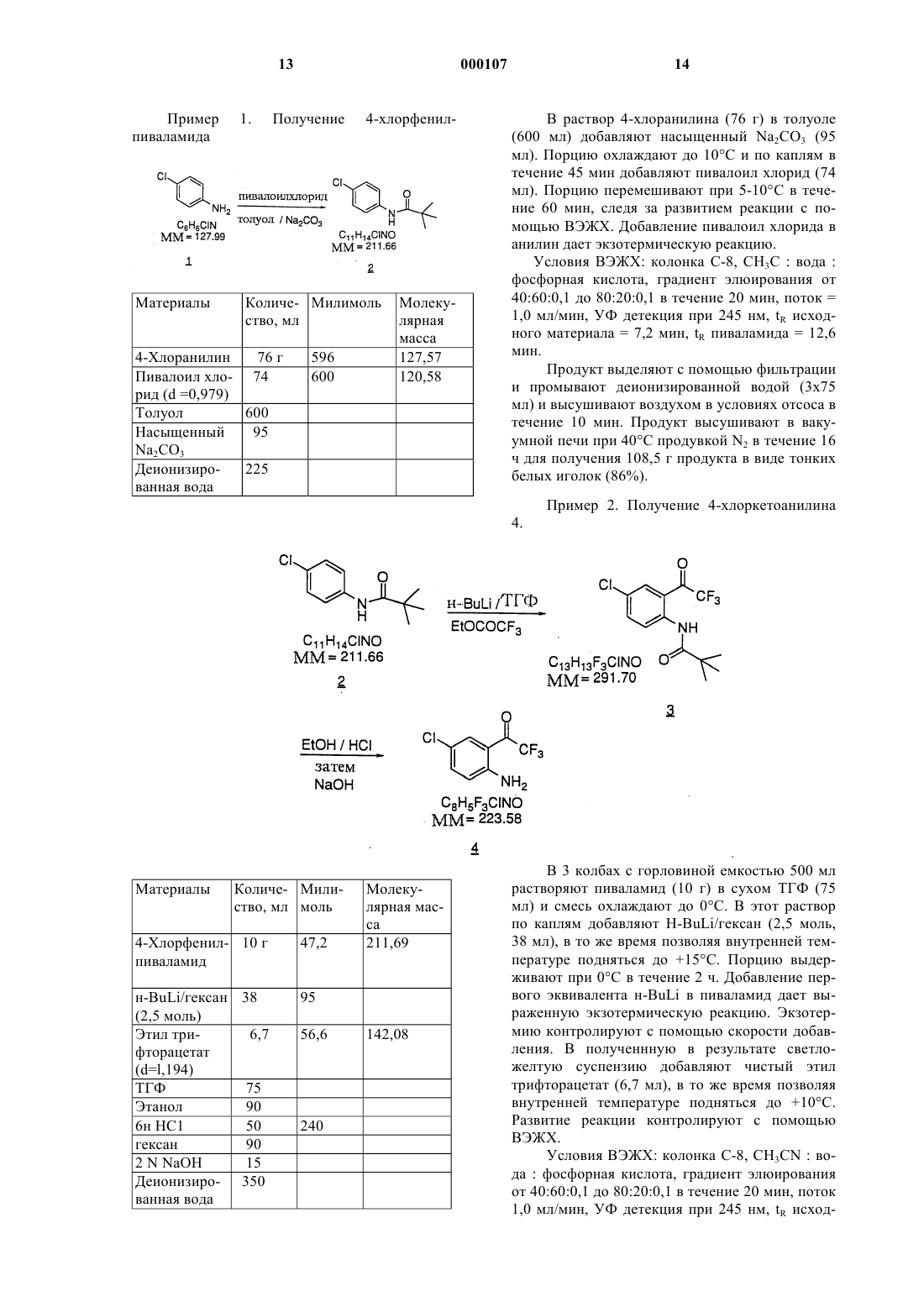
1 Настоящая заявка относится к заявке компании Мерк 187931 В, которая является частичным продолжением заявки 187931 А, в свою очередь являющейся частичным продолжением заявки компании Мерк 18793, поданной 7 августа 1992 г. U.S.S.N. 07/926.607 и 19345. Называемый ретровирусом вирус иммунодефицита человека (ВИЧ) является этиологическим фактором сложного заболевания, которое включает прогрессирующее разрушение иммунной системы (синдром приобретенного иммунодефицита; СПИД) и дегенерацию центральной и периферической нервной системы. Ранее этот вирус был известен под названиямиLAV, HTLV-111 или ARV. Общим признаком репликации ретровируса является обратная транскрипция генома РНК, вирусно закодированной обратной транскриптазой для генерирования копий ДНК последовательностей ВИЧ,этапа, требуемого при репликации вирусов. Известно, что некоторые соединения, например азидотимидин или АЗТ, являются ингибиторами обратной транскриптазы и представляют собой эффективные средства для лечения СПИДа и подобных ему заболеваний. Определение нуклеотидной последовательности ВИЧ показывает наличие гена роl в одной открытой рамке считывания [Ratner, L.etal., Nature, 313,277 (1985)]. Гомология аминокислотной последовательности предоставляет доказательство, что последовательность ро 1 кодирует обратную транскриптазу, эндонуклеазу и протеазу ВИЧ [ Toh, H. et al., EMBO J., 4,1267 (1985); Power, M.D. et al., Science, 231,1567 (1986); Pearl, L.H. et al., Nature, 329.351(1987)]. Заявители демонстрируют в значительной степени усовершенствованный синтез ингибитора обратной транскриптазы ВИЧ структуры называемого (-) 6-хлор-4-циклопропилэтинил-4 трифторметил-1,4-дигидро-2 Н-3,1-бензоксазин 2-оном, далее именуемого "Соединение А". Это соединение обладает высокой активностью даже против обратной транскриптазы ВИЧ, устойчивой к другим противовирусным соединениям СПИДа. Заявители изобрели асимметричный синтез Соединения А. По ранее предложенным способам требовалось получение рацемата в качестве предпоследнего продукта, и они давали более низкий общий выход продукта. Настоящее изобретение относится к прямому синтезу оптически активного Соединения А с помощью хирального присоединения к кетоновому проме 000107 2 жуточному продукту для получения третичного спирта с энантиомерным избытком, превышающим 95%. Кроме того, является неожиданным, что в результате реакции ацетилида с трифторметилкетоном образуется оптически активное химическое соединение. В настоящем изобретении это достигается хиральным аминоспиртом для опосредования реакции присоединения по асимметричному пути. Заявители также открыли, что нагревание (с последующим охлаждением) смеси литиированного аминоспирта и циклопропилацетилена перед добавлением трифторкетона, способствует повышению энантиомерного избытка с приблизительно 85% при типичном проведении реакции приблизительно до более 95%. Необычно высокие уровни оптической активности (95% энантиомерный избыток) делают этот способ выгодным и практичным. Раскрыт усовершенствованный синтез (-) 6-хлор-4-циклопропилэтинил-4-трифторметил 1,4-дигидро-2 Н-3,1-бензоксазин-2-она,включающий хиральное присоединение к кетоновому промежуточному продукту для получения третичного спирта. Соединение может использоваться в подавлении обратной транскриптазы ВИЧ (и его устойчивых разновидностей), профилактике инфекции ВИЧ, лечении инфекции ВИЧ и в лечении СПИДа и/или связанного со СПИДом комплекса, в виде соединений, фармацевтически приемлемых солей (в подходящих случаях), ингредиентов фармацевтической композиции, отдельно или в комбинации с другими противовирусными средствами, противоинфекционными средствами, иммуномодуляторами,антибиотиками или вакцинами. Раскрыты также способы лечения СПИДа, способы профилактики инфекции ВИЧ и способы лечения инфекции ВИЧ. В этом изобретении раскрыт способ асимметричного синтеза хирального соединения структуры:(в которой Р представляет собой группу, защищающую аминогруппу), включающего стадии: а) получение смеси избытка (1R, 2S)-Nзамещенного норэфедрина структуры:NR2 образует пирролидинил или пиперидинил; с избытком циклопропилацетилена и избытком литирующего вещества, выбранного из нбутиллития или втор-бутиллития, или третбутиллития в диапазоне температуры от приблизительно -78 С до приблизительно 10 С в апротонном растворителе;b) смешивание со смесью, полученной на стадии (а), приблизительно одного эквивалента реагента структуры: в которой Р представляет собой группу, защищающую аминогруппу, и выдержку полученной реакционной смеси при температуре от приблизительно -78 С до приблизительно -20 С; с) тушение реакции с помощью добавления источника протонов;d) образование желаемого соединения. Один из вариантов реализации этого изобретения представляет собой способ асимметричного синтеза хирального соединения N-(4 метоксибензил)-6-хлор-2-[(R)-циклопропилэтинилгидрокси-трифторметил]-метиланилина структуры: включающий стадии: а) получение смеси избытка (1R, 2S)-Nзамещенного норэфедрина структуры:NR2 образует пирролидинил или пиперидинил; с избытком циклопропилацетилена и избытком литирующего вещества, выбранного из н-бутиллития или втор-бутиллития, или трет-бутиллития в диапазоне температуры от приблизительно-78 С до приблизительно 10 С в апротонном растворителе,b) смешивание со смесью, полученной на стадии (а), приблизительно одного эквивалента реагента N-(4-метоксибензил)-6-хлор-2-(2-трифтор-1-оксоэтил)-анилина структуры: и сохранение полученной реакционной смеси при температуре от приблизительно -78 С до приблизительно -20 С; с) тушение реакции с помощью добавления источника протонов;d) образование желаемого соединения. Другой вариант реализации того же изобретения представляет собой способ асимметричного синтеза хирального соединения структуры:(в которой Р представляет собой группу, защищающую аминогруппу), включающий стадии: а) получение смеси избытка (1R, 2S)-Nпирролидинил норэфедрина структуры: с избытком циклопропилацетилена и избытком литирующего вещества, выбранного из нбутиллития, или втор-бутиллития, или третбутиллития при температуре приблизительноd) смешивание со смесью стадии (а) приблизительно одного эквивалента реагента структуры: в которой Р представляет собой группу, защищающую аминогруппу, и выдержку полученной реакционной смеси при температуре приблизительно -40 С; с) тушение реакции путем добавления источника протонов;d) получение желаемого соединения. Другой вариант реализации того же изобретения представляет собой способ асиммет 5 ричного синтеза хирального соединения N-(4 метоксибензил)-6-хлор-2-[(R)-циклопропилэтинилгидрокси-трифторметил)-метиланилина] структуры: 6 дующим охлаждением) между стадией (а) и стадией (b), то есть, смесь стадии (а) нагревают до температуры от приблизительно -10 С до приблизительно 0 С в течение от приблизительно 10 мин до приблизительно 60 мин, затем охлаждают до температуры от приблизительно -40 С перед добавлением трифторкетона. Это изобретение также охватывает соединение структуры: включающий стадии: а) получение смеси избытка (1R, 2S)-Nпирролидинил норэфедрина структуры:b) смешивание со смесью стадии (а) приблизительно одного эквивалента реагента N-(4 метоксибензил)-6-хлор-2-(2-трифтор-1-оксоэтил)-анилина структуры: и выдержку полученной реакционной смеси при температуре приблизительно -40 С; с) гашение реакции с помощью добавления источника протонов;d) получение желаемого соединения с выходом 85%, энантиомерный избыток 95%. Любой из указанных выше способов может быть модифицирован для улучшения энантиомерного избытка с помощью дополнительной стадии нагревания (с последующим охлаждением) между стадией (а) и стадией (b), то есть,смесь стадии (а) нагревают до температуры от приблизительно -10 С до приблизительно 10 С,по меньшей мере, в течение 5 мин, затем охлаждают до температуры от приблизительно -78 С до -20 С перед добавлением трифторкетона. Если температура смеси стадии (а) уже находится на уровне от приблизительно -10C до приблизительно 10 С, подъем температуры может не улучшить энантиомерный избыток. Одной предпочтительной модификацией для улучшения энантиомерного избытка является дополнительно стадия нагревания (с после в которой Р представляет собой группу,защищающую аминогруппу. Другое соединение, охватываемое этим изобретением, представляет собой N-(4 метоксибензил)-6-хлор-2-[(R)-циклопропилэтинилгидрокси-трифторметил]-метиланилин структуры: Сначала избыток (1R, 2S)-N-замещенного норэфедрина смешивается в апротонном растворителе с избытком циклопропилацетилена, и кислотные протоны удаляются с помощью литирования с избытком н-бутиллития, или вторбутиллития, или трет-бутиллития. Полученная реакционная смесь выдерживается при температуре в диапазоне от приблизительно -78 С до приблизительно 10 С, предпочтительно при приблизительно -15 С. Если полученная смесь нагревается до температуры от приблизительно 10 С до приблизительно 10C, продукт 6 в конечном итоге получается с большим энантиомерным избытком, обычно приблизительно 95% вместо приблизительно 85% или менее. Некоторые аспекты этого изобретения исключают этот этап нагревания, другие включают его. В способах настоящего изобретения Р представляет собой любую подходящую аминозащитную группу и включает, но не ограничивается, бензил, незамещенный или замещенный С 1-4 алкилом; параметоксибензил; паранитробензил; парахлорбензил; 2,4-дихлорбензил; 2,4 диметоксибензил; 4-метилсульфинилбензил; 9 антрилметил; дифенилметил или аминозащитной группой является параметоксибензил. Как только реакция начинается добавлением приблизительно одного эквивалента кетона 5, трифторкетона, полученная реакционная смесь выдерживается при температуре в диапазоне от приблизительно -78 С до приблизительно -20 С, предпочтительно при приблизительно -40 С. Реакция проводится в апротонном растворителе или эфирном растворителе. Примеры апротонных растворителей включают тетрагидрофуран, диоксан, Еt2O, бензол, ДМЭ, фенилметил, н-октан, н-гексан и циклогексан или их смеси. Предпочтительным растворителем является тетрагидрофуран. Время инкубации этой реакции составляет, по меньшей мере, 2-3 мин после добавления кетона. В этот момент реакционная смесь гасится с помощью добавления источника протонов в водной среде обычно слабой кислотой. Подходит любой такой источник протонов, например,предпочтительным источником протонов является 1 М лимонная кислота. Другим является 1 М уксусная кислота. Хиральный продукт 6 очищается с помощью обычных методик. Соединения настоящего изобретения могут иметь асимметричные центры и могут встречаться, кроме случаев, когда это специально отмечено, в виде рацематов, рацемических смеCXЕMA 1 8 сей или в виде отдельных диастереомеров или энантиомеров, причем все изомерные формы включены в настоящее изобретение. Термин(+/-) предназначен охватывать (+) оптические изомеры или (-) оптические изомеры или их смеси. Если любая переменная величина (например R) встречается более одного раза в любом ингредиенте или в формуле 1, его определение в каждом положении независимо от его определения в каждом другом положении. Также комбинацию заместителей и/или переменных величин допустимы, только если такие комбинации приводят к получению устойчивых соединений. За исключением отмеченных случаев используемый здесь термин "алкил" подразумевает включение как разветвленно-, так и прямоцепочечных насыщенных алифатических углеводородных групп, имеющих определенное количество атомов углерода; если количество атомов углерода не определено, термин "алкил" подразумевает включение от 1 до 4 атомов углерода как разветвленно-, так и прямоцепочечных насыщенных алифатических углеводородных групп. Используемые здесь термины "галоген" или "гало" обозначают фтор, хлор, бром и иод. Соединение А может быть синтезировано с помощью следующего способа. Циклопропилацетилен, который является реагентом при образовании 6, получают с помощью следующих альтернативных схем; СХЕМА 11 А как также описано в работах C.E.Hudson et al.,J.Am.Chem. Soc., 94, 1158(1972) и W.Schoderth Соединение А может использоваться в приготовлении и проведении скрининговых анализов на противовирусные соединения. Например, Соединение А может использоваться для выделения ферментативных мутантов, которые являются превосходными инструментами скрининга для выявления более мощных противовируснвх соединений. Кроме того, Соединение А может использоваться при установлении или определении участка связывания других противовирусных средств к обратной транскриптазе ВИЧ, например, с помощью конкурентного подавления. Таким образом, Соединение А является коммерческим продуктом, предназначенным к продаже для этих целей. Соединение А может использоваться в подавлении обратной транскриптазы ВИЧ, профилактике или лечении инфекции вируса иммунодефицита человека (ВИЧ) и в лечении вызванных им патологических состояний, таких как СПИД. Лечение СПИДа, или профилактика, или 11 лечение инфекции ВИЧ определено как включающее, но не ограничивающееся лечением широкого диапазона состояний ВИЧ инфекции: СПИД, ССК (связанный со СПИДом комплекс),как клинически выраженные, так и бессимптомные, и истинная или возможная подверженность воздействию ВИЧ. Например, Соединение А может использоваться в лечении инфекции ВИЧ после подозреваемой в прошлом подверженности воздействию ВИЧ вследствие, например,переливания крови, обменного переливания биологических жидкостей, укусов, случайных уколов иглой или контакт с кровью пациента во время операции. Особым преимуществом Соединения А является его мощное подавляющее действие на обратную транскриптазу ВИЧ, являющуюся устойчивой к другим противовирусным средствам, таким как L-697.661, которое представляет собой 3-([(4,7-дихлор-1,3-бензоксазол-2-ил) метил] амино)-5-этил-6-метилпиридин-2(1 Н)-он,или L-696.229, которое представляет собой 3-[2(1,3-бензоксазол-2-ил)этил]-5-этил-6-метилпиридин-2(1 Н)-он, или АЗТ. Для этих целей Соединение А может вводиться внутрь, парентерально (включая подкожные инъекции, внутривенные, внутримышечные, внутригрудинные инъекции или инфузионные методики), с помощью ингаляционного аэрозоля или ректально в композициях дозированных лекарственных форм, содержащих обычные нетоксичные фармацевтически приемлемые носители, адъюванты и растворители. Таким образом, в соответствии с настоящим изобретением, кроме того, предоставляется способ лечения и фармакологическая композиция для лечения ВИЧ инфекции и СПИДа. Лечение включает введение нуждающемуся в таком лечении пациенту фармацевтической композиции, включающей фармацевтический носитель и терапевтически эффективное количество соединения настоящего изобретения. Эти фармацевтические композиции могут быть в форме перорально вводимых суспензий или таблеток; стерильных инъецируемых препаратов, например в виде стерильных инъецируемых водных или масляных суспензий или свечей. При пероральном введении в виде суспензии эти композиции получаются в соответствии с методиками, хорошо известными в области приготовления фармацевтических композиций,и могут содержать микрокристаллическую целлюлозу для придания объема, альгиновую кислоту или альгинат натрия в качестве суспензирующего вещества, метилцеллюлозу в качестве усилителя вязкости и подслащивающие/ароматизирующие вещества, известные в этой области. В виде таблеток немедленного высвобождения эти композиции могут содержать микрокристаллическую целлюлозу, дикальций фосфат, крахмал, магний стеарат и лак 000107 12 тозу и/или другие наполнители, связывающие вещества, разбавители, разрыхлители, растворители и смазывающие вещества, известные в этой области. При введении с помощью интраназального аэрозоля или ингаляции эти композиции получаются в соответствии с методиками, хорошо известными в области приготовления фармацевтических композиций, и могут готовиться в виде солевых растворов с использованием бензилового спирта или других подходящих консервантов, промоторов всасывания для усиления биодоступности, фторуглеродов и/или других солюбилизирующих или диспергирующих веществ, известных в этой области. Композиции растворов или суспензий для инъекций могут быть получены в соответствии с известными способами с использованием нетоксичных, приемлемых для парентерального введения разбавителей или растворителей, таких как маннитол, 1,3-бутандиол, вода, раствор Рингера или изотонический раствор хлористого натрия, или подходящих диспергирующих и суспензирующих веществ, таких как стерильные, низковязкие, нелетучие масла, включая синтетические моно- или диглицериды, и жирные кислоты, включая олеиновую кислоту. При ректальном введении в форме свечей эти композиции могут быть получены с помощью смешивания лекарственного препарата с подходящим не раздражающим наполнителем,таким как масло какао, синтетические глицеридные эфиры или полиэтиленгликоли, которые являются твердыми при обычных температурах,но становятся текучими и/или растворяются в полости прямой кишки для высвобождения лекарственного препарата. Соединение А можно вводить перорально людям в диапазоне доз от 1 до 100 мг/кг массы тела дробными дозами. Одним из предпочтительных диапазонов доз является от 0,1 до 20 мг/кг массы тела дробными дозами. Для комбинированной терапии с нуклеозидными аналогами предпочтительным диапазоном доз является от 0,1 до 20 мг/кг массы тела для соединений этого изобретения, вводимых перорально дробными дозами, и от 50 мг до 5 г/кг массы тела для нуклеозидных аналогов, вводимых перорально дробными дозами. Однако следует понимать, что определенный уровень дозы и частота введения для любого конкретного пациента может варьироваться и будет зависеть от множества факторов, включая активность конкретного применяемого соединения, метаболическую устойчивость и продолжительность действия этого соединения, возраст, массу тела, общее состояние здоровья, пол, пищевой рацион,способ и время введения, скорость выведения,комбинацию лекарственных препаратов, тяжесть состояния в конкретном случае и подвергаемого лечению носителя вируса.(600 мл) добавляют насыщенный Na2CO3 (95 мл). Порцию охлаждают до 10 С и по каплям в течение 45 мин добавляют пивалоил хлорид (74 мл). Порцию перемешивают при 5-10 С в течение 60 мин, следя за развитием реакции с помощью ВЭЖХ. Добавление пивалоил хлорида в анилин дает экзотермическую реакцию. Условия ВЭЖХ: колонка С-8, CH3C : вода : фосфорная кислота, градиент элюирования от 40:60:0,1 до 80:20:0,1 в течение 20 мин, поток = 1,0 мл/мин, УФ детекция при 245 нм, tR исходного материала = 7,2 мин, tR пиваламида = 12,6 мин. Продукт выделяют с помощью фильтрации и промывают деионизированной водой (3 х 75 мл) и высушивают воздухом в условиях отсоса в течение 10 мин. Продукт высушивают в вакуумной печи при 40 С продувкой N2 в течение 16 ч для получения 108,5 г продукта в виде тонких белых иголок (86%). Пример 2. Получение 4-хлоркетоанилина 4. В 3 колбах с горловиной емкостью 500 мл растворяют пиваламид (10 г) в сухом ТГФ (75 мл) и смесь охлаждают до 0 С. В этот раствор по каплям добавляют H-BuLi/гексан (2,5 моль,38 мл), в то же время позволяя внутренней температуре подняться до +15 С. Порцию выдерживают при 0 С в течение 2 ч. Добавление первого эквивалента н-BuLi в пиваламид дает выраженную экзотермическую реакцию. Экзотермию контролируют с помощью скорости добавления. В полученнную в результате светложелтую суспензию добавляют чистый этил трифторацетат (6,7 мл), в то же время позволяя внутренней температуре подняться до +10 С. Развитие реакции контролируют с помощью ВЭЖХ. Условия ВЭЖХ: колонка С-8, CH3CN : вода : фосфорная кислота, градиент элюирования от 40:60:0,1 до 80:20:0,1 в течение 20 мин, поток 1,0 мл/мин, УФ детекция при 245 нм, tR исход 15 ного материала = 12,6 мин, tR кетопиваламида = 11,6 мин. Обычно выход составляет 85 А% продукта и 10-15 А% не вступившего в реакцию пиваламида. Реакцию гасят с помощью добавления 6 N НСl (10 мл) и деионизированной воды (20 мл). ВЭЖХ в этот момент показывает выход 13,1 г (90%) продукта. Раствор концентрируют в вакууме до объема 50 мл и промывают этанолом(50 мл) для удаления гексана и ТГФ. В порцию добавляют 6N НС 1 (40 мл) и смесь нагревают до дефлегмации (80 С) в течение 1 ч. Количественный анализ с помощью ВЭЖХ показывает выход 85-90 А% кетоанилина и 10 А% не вступившего в реакцию пиваламида. Таким образом, ацилированный материал подвергается гидролизу, тогда как не вступивший в реакцию пиваламид остается неизменным. В этот момент выход количественного анализа составил 7,78 г (74 %). Порцию концентрируют в вакууме до объема 50 мл, во время чего образуется осадок (предположительно соль НС 1 продукта). Дистилляцию прекращают и порцию охлаждают до 0 С. После выдержки в течение 1 ч порцию фильтруют и промывают гексаном(3 х 30 мл). Промывания гексаном удаляют не вступивший в реакцию пиваламид из продукта. Твердое вещество контролируется с помощью ВЭЖХ для гарантии его полного удаления в этот момент. Фильтрат и промывочная жидкость обычно содержат 1,2-1,5 г продукта (812%). Большая часть потери продукта происходит в водном фильтрате. Соль высушивают в вакуумной печи при 40 С в течение 16 ч для получения 10,4 г твердого вещества, которое имеет чистоту 71,4% по массе (70% выход). Соль суспензируют в деионизированной воде (260 мл) и нейтрализуют 2NNaOH (15 мл) до рН приблизительно 6-7. Ввиду разложения продукта очень важно не доводить рН до уровня выше 9,0. Полученное в результате светло-желтое твердое вещество выделяют с помощью фильтрации и промывают деионизированной водой (2 х 25 мл). Продукт высушивают в вакуумной печи при 40 С в течение 16 ч для получения 6 г кетоанилина, который имеет чистоту 96,6% по массе (54% выход). Дальнейшую очистку продукта проводят с помощью перекристаллизации из гексана. Пример 3. Получение N-4-метоксибензилкетоанилина 5. Количест- Мили- Молекуво моль лярная масса 15,5 г 69,5 223,58 Р-метоксибен- 10,9 г зилхлорид Молекулярное сито 4 ангстрем Толуол В колбу емкостью 250 мл загружают кетоанилин (15,5 г), активированное молекулярное сито 4 ангстрем (50 г) и толуол (75 мл). Смесь перемешивают при 23 С под N2 в течение 24 ч. Количественный анализ с помощью ВЭЖХ выявляет смесь продукта и исходного материала в соотношении приблизительно 1:1. Условия ВЭЖХ: колонка С-8, CH3CN : вода : фосфорная кислота, изократическое элюирование при 65:35:0,1 в течение 20 мин, поток = 1,0 мл/мин, УФ детекция при 260 нм, tR толуола= 5,7 мин, tR исходного кетоанилина = 6,5 мин,tR продукта = 15,0 мин. Обычно выход составляет 25 А% толуола. В реакционную смесь загружают свежее молекулярное сито (40 г) и перемешивают в течение еще 3 дней при 23 С. Реакцию считают завершенной, когда остается меньше 2 А% исходного материала. Альтернативно вместо сита для удаления НСl из системы может использоваться основная окись алюминия или силикагель. Смесь фильтруют через целит и промывают ацетоном (7 х 75 мл) до тех пор, пока из целита не смоется желтый цвет. Фильтрат концентрируют для получения 27 г желто-оранжевого масла, которое при стоянии затвердевает. Твердое вещество очищают с помощью растворения в горячих гексанах (100 мл). Порцию охлаждают до комнатной температуры, затем до 0 С в водно-ледяной бане. После выдержки в течение 1,5 ч порцию фильтруют и промывают холодными гексанами(2 х 10 мл). Порцию высушивают воздухом с отсосом в течение 10 мин, затем высушивают в вакуумной печи при 40 С в течение 2 ч. Это дает 20,5 г (86%) ярко-желтого порошка. Коли- Мили- Молекулярчество, моль ная масса мл 10 г 98 102,57NH4Cl В раствор 5-хлор-1-пентина в циклогексане (80 мл) при 0 С под N2 добавляют нбутиллитий в циклогексане (2,0 моль, 122 мл). Смесь нагревают до 75 С в течение 5 ч. Добавление н-бутиллития в алкин дает экзотермическую реакцию, температуру поддерживают ниже +5 С с помощью водно-ледяной бани. Развитие этапа циклизации контролируют с помощью ВЭЖХ. Реакцию считают завершенной, когда выход количественного анализа составляет 90%. Условия ВЭЖХ: фенильная колонка,CH3CN : вода : фосфорная кислота, изократическое элюирование при 50:50:0,1 в течение 20 мин, поток = 1,0 мл/мин, УФ детекция при 195 нм, tR исходного материала = 7,5 мин, tR циклопропилацетилена = 6,0 мин. Продукт имеет фактор реакции, который в 20 раз больше, чем исходный материал. Как только заканчивается этап циклизации, реакционную смесь охлаждают до 0 С и гасят насыщенным NH4Cl. Количественный анализ органической фазы с помощью ВЭЖХ показал 5,5 г циклопропилацетилена (выход 85%). Продукт очищают с помощью фракционной дистилляции через колонку 6" х 0,5", упакованную 4 мм стеклянными бусинами. Собирают фракцию с точкой кипения от 45 до 75 С. Это дает 4,2 г (65%) циклопропилацетилена в виде бесцветного масла. Материалы Количество, М. моль Молекумл лярная масса Кетон 10 г 29,1 343 13,1 г 64 205 1R, 2S Nпирролидинилнорэфедрин циклопропилацетилен(KF=20 мкг/мл) 1 М лимон 90 ная кислота Этилацетат 75 Пирролидинилнорэфедрин (13,1 г) растворяют в ТГФ (55 мл) и смесь охлаждают до-15 С. В смесь при -15 С под N2 по каплям добавляют чистый циклопропилацетилен (5,3 мл) и н-бутиллитий (50 мл). Смесь выдерживают при 5 С в течение 30 мин, затем охлаждают до-55 С. Добавление н-бутиллития вызывает экзотермическую реакцию, и температуру поддерживают в диапазоне от -5 до 0 С с помощью скорости добавления. Кетон (10 г) растворяют в ТГФ (25 мл) под N2 и добавляют в анионную смесь в течение 15 мин, позволяя внутренней температуре подняться до -40 С во время добавления. Полученный в результате светлый оранжевый раствор выдерживают при -40 С в течение 60 мин и реакцию гасят с помощью добавления 1 М лимонной кислоты (45 мл) и этилацетата (75 мл). Реакционную смесь нагревают 19 до окружающей температуры и разделяют слои. Органические слои промывают 1 М лимонной кислотой (45 мл). Проводят количественный анализ реакционной смеси с помощью ВЭЖХ на процент превращения и содержания продукта, энантиомерный избыток. Условия ВЭЖХ: колонка С-8, CH3CN : вода : фосфорная кислота; изократическое элюирование 65:35:0,1 в течение 20 мин, поток = 1,0 мл/мин, УФ детекция при 252 нм, tR исходного материала = 12,8 мин, tR продукта = 10,3 мин. Условия хиральной ВЭЖХ: колонка, содержащая в качестве стационарной фазы амилозу,гексан : изопропанол 85:15 изократическое элюирование, поток = 1,0 мл/мин, УФ детекция при 252 нм, tR исходного материала = 4,9 мин, tR главного энантиомера = 5,5 мин, tR второстепенного энантиомера = 25,0 мин. Энантиомерный избыток составляет 96,5% и реакционное превращение составляет 93% (6 А% исходного материала). Выход количественного анализа составляет 85%. Продукт очищают с помощью смешивания в смеси гептан: толуол в соотношении 10:1 (65 мл) при 23 С в течение 18 ч. Продукт выделяют с помощью фильтрации и высушивания в печи при 35 С для получения 9,5 г Количест- Мили- Молекуво, мл моль лярная масса 3,2 г 7,8 409 4,6 8,9EtOAc 45 Гексаны 30 1 М лимонная 40 кислота Насыщенный 25 рассол Аминоспирт растворяют в ТГФ (15 мл) и охлаждают до -10 С под N2. В смесь добавляют триэтиламин (5,4 мл) и фосген в толуоле (4,6 мл). Добавление фосгена вызывает экзотермическую реакцию, и температуру поддерживают ниже 20 С с помощью скорости добавления. 20 Развитие реакции контролируют с помощью ВЭЖХ, и она обычно заканчивается через 15 мин. Условия хиральной ВЭЖХ: колонка С-8,СН 3 СN : вода : фосфорная кислота, градиентное элюирование от 50:50:0,1 до 90:10:0,1 в течение 20 мин, поток = 1,5 мл/мин, УФ детекция при 252 нм, tR исходного материала = 14,6 мин, tR продукта = 16,0 мин. Реакционную смесь охлаждают до 0 С и реакцию гасят ледяной холодной водой (15 мл) и этилацетатом (20 мл). Для разрушения любых эмульсий используют насыщенный рассол. Органический слой удаляют, а водный экстрагируют этилацетатом (15 мл). Комбинированные органические слои промывают 1 М лимонной кислотой (40 мл) и насыщенным рассолом (25 мл). Органический слой высушивают(Na2SO4 ) и концентрируют в вакууме для получения 3,8 г коричневого масла. Продукт кристаллизуют из смеси гексан : этилацетат в соотношении 5:1 (25 мл), охлаждают до 0 С, выдерживают в течение 1 ч и фильтруют. Слежавшийся осадок промывают холодной смесью гексан - этилацетат в соотношении 5:1 (2 х 5 мл). Слежавшийся осадок высушивают воздухом с отсосом для получения 2,9 г (85%) светло-оранжевого твердого вещества. Пример 7. Получение Соединения А.CH3CN 15 Этилацетат 30 Деионизи- 30 рованная вода Насыщен- 10 ный рассол Защищенное п-метоксибензилом Соединение А растворяют в CH3CN (15 мл). В этот раствор добавляют раствор цериевого аммонийнитрата (4,4 г) в воде (5 мл). Обычно реакция заканчивается через 2 ч при 23 С по данным ВЭЖХ. Условия хиральной ВЭЖХ: колонка С 8, CH3CN : вода : фосфорная кислота, градиент 21 ное элюирование от 50:50:0,1 до 90:10:0,1 в течение 20 мин, поток = 1,5 мл/мин, УФ детекция при 252 нм, tR исходного материала = 16,0 мин,tR продукта = 9,0 мин. Реакционную смесь разбавляют деионизированной водой (5 мл) и концентрируют приблизительно до 1/2 объема. Продукт экстрагируют из полученного в результате водного слоя этилацетатом (2 х 15 мл). Комбинированный органический слой промывают деионизированной водой (2 х 10 мл) и рассолом (10 мл). Органический слой концентрируют в вакууме для получения желтой смолы. Продукт выделяют с помощью хроматографии фильтрацией через силикагель. КОЛИЧЕСТВЕННЫЙ АНАЛИЗ ОБРАТНОЙ ТРАНСКРИПТАЗЫ Анализ производит количественное определение включения тритиевого дезоксигуанозин монофосфата рекомбинантной обратной транскриптазой ВИЧ (ОТрВИЧ) (или другой ОТ) в осаждаемую кислотой кДНК при величинах константы Михаэлиса дГТФ(dGТР) поли (С)x олиго d(G)12-18. Ингибиторы настоящего изобретения подавляют это включение. Количественные анализы проводили в растворе, содержащем 55 ммоль Трис (рН 8.2) - 30 ммоль КСl - 30 ммоль MgCl2 - 1 ммоль дитиотретиола - 20 мкг r(C):d(G)12-18(Pharmacia) на мл 8 ммоль [3H](dGTP)( New England Nuclear) 0,01% Тритон X-100-50 ммоль этиленгликольбис(-аминоэтилэфир)-N,N,N',N'-тетрауксусной кислоты (EGTA) - 1 мг бычьего сывороточного альбумина на мл. После 60 мин инкубации при 37 С осаждаемый кислотой материал собирают на стеклянноволоконные фильтры с помощью полуавтоматического сборщика клеток. Экстракты бактериальных клеток, содержащие ОТ,разводят до пределов линейного диапазона количественного определения, и активность определяют в присутствии и отсутствии ингибитора. Очищенный гетеродимер ОТ ВИЧ-1, вырабатываемый в E.coli, также служил в качестве контроля. Результаты определяют как концентрацию ингибитора, которая дает подавление на 50% (ИК 50 массы), в наномолях/литр. Соединение А дало ИК 50 2 нмоля. Для количественного определения двойного мутанта (дм) при анализе применяют ОТ А 17. ОТ А 17 устойчива к различным аминопиридонам, как описано в работе Nunberg , J.H. et al.,J.Virol., 65, 4887 (1991). Результаты измеряются как ИК 50 (IС 50) сухого вещества в наномолях/литр. Соединение А дает ИК 50 сухого вещества 85 наномоль. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ КЛЕТОЧНОГО РАСПРОСТРАНЕНИЯ Подавление распространения ВИЧ в клеточной культуре измеряют в соответствии с методикой Nunberg, J.H. et al., J.Virol., 65, 4887(1991). При этом количественном определении Т-лимфодиные клетки МТ-4 инфицируют ВИЧ 000107 22 1 (дикого типа, если нет других указаний) с помощью использования предварительно определенного инокулята, и культуры инкубируют в течение 24 ч. В это время при непрямой иммунофлюоресценции положительный результат дают 1% клеток. Затем клетки интенсивно промывают и распределяют в 96-ячеечные кульутральные планшеты. Серийные двукратные разведения ингибитора добавляют в ячейки и культивацию продолжают в течение еще 3 дней. Через 4 дня после инфекции 100% в контрольных культурах были инфицированы. Накопление р 24 ВИЧ-1 прямо коррелирует с распространением вируса. Ингибирующую концентрацию клеточной культуры определяют как концентрацию ингибитора в ммолях/литр, которая снижает распространение инфекции, по меньшей мере, на 95% или ИКК 95(CIC95). Хотя приведенное выше описание излагает принципы настоящего изобретения с примерами, приведенными с целью иллюстрации, следует понимать, что практическое использование изобретения охватывает все обычные варианты,приспособления или модификации как находящиеся в пределах диапазона следующей формулы изобретения и ее эквивалентов. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. 6-Хлор-2-[(R)-циклопропилэтинилгидрокси-трифторметил] амин общей формулы 3. Способ получения соединения по п.1,заключающийся в том, что осуществляют стадии: а) получение смеси избытка (1R, 2S)-Nзамещенного норэфедрина формулы-NR2 образует пирролидинил или пиперидинил; с избытком циклопропилацетилена и избытком литирующего вещества, выбранного из н-бутиллития, втор-бутиллития или трет-бутиллития при температуре от приблизительно -78 С до приблизительно 10 С в апротонном растворителе;(b) смешивание со смесью, полученной на стадии (а), приблизительно одного эквивалента реагента формулы в которой Р представляет собой защищающую аминогруппу, и выдержку полученной реакционной смеси при температуре от приблизительно -78 С до приблизительно -20 С;(с) тушение реакции путем добавления источника протонов;(d) образование желаемого соединения. 4. Способ по п.3, отличающийся тем, что при получении соединения по п.2, осуществляют стадии: а) получение смеси избытка (1R, 2S)-Nзамещенного норэфедрина формулы-NR2 образует пирролидинил или пиперидинил; с избытком циклопропилацетилена и избытком литирующего вещества, выбранного из н-бутиллития, втор-бутиллития или трет-бутиллития,при температуре от приблизительно -78 С до приблизительно 10 С в апротонном растворителе;(b) смешивание со смесью, полученной на стадии (а), приблизительно одного эквивалента 24 и выдержку полученной реакционной смеси при температуре от приблизительно -78 С до приблизительно -20 С;(с) тушение реакции путем добавления источника протонов;(d) образование желаемого соединения. 5. Способ по п.3 получения соединения по п.1, отличающийся тем, что осуществляют стадии: а) получение смеси избытка (1R, 2S)-Nпирролидинил-норэфедрина формулы(b) смешивание со смесью, полученной на стадии (а), приблизительно одного эквивалента реагента формулы в которой Р представляет собой защищающую аминогруппу; и выдержку полученной реакционной смеси при температуре приблизительно 40 С;(с) тушение реакции путем добавления источника протонов;(d) образование желаемого соединения. 6. Способ по п.5, отличающийся тем, что при получении соединения по п.2 осуществляют стадии:(а) получение смеси избытка (1R, 2S)-Nпирролидинил норэфедрина формулы(b) смешивание со смесью, полученной на стадии (а), приблизительно одного эквивалента и выдержку полученной реакционной смеси при температуре приблизительно -40C;(с) тушение реакции путем добавления источника протонов;(d) образование желаемого соединения. 7. Способ по любому из пп.3-4, отличающийся тем, что проводят дополнительно стадию нагревания между стадией (а) и стадией (b), при этом смесь, полученную на стадии (а), нагрева 26 ют до температуры от приблизительно -10 С до приблизительно 10 С в течение, по меньшей мере, 5 мин, затем охлаждают до температуры от приблизительно -78 С до приблизительно-20 С перед стадией (b). 8. Способ по любому из пп.5-6, отличающийся тем, что проводят дополнительно стадию нагревания между стадией (а) и стадией (b), при этом смесь, полученную на стадии (а), нагревают до температуры от приблизительно -10 С до приблизительно 0 С в течение от приблизительно 10 мин до приблизительно 60 мин, затем охлаждают до температуры, по меньшей мере,приблизительно -40 С перед стадией (b).
МПК / Метки
МПК: C07C 213/00, C07D 265/18
Метки: 6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2н-3,1-бензоксазин-2-она, получения, синтеза, промежуточные, ассиметричного, продукты, способ
Код ссылки
<a href="https://eas.patents.su/14-107-promezhutochnye-produkty-dlya-assimetrichnogo-sinteza-6-hlor-4-ciklopropiletinil-4-triftormetil-14-digidro-2n-31-benzoksazin-2-ona-i-sposob-ih-polucheniya.html" rel="bookmark" title="База патентов Евразийского Союза">Промежуточные продукты для ассиметричного синтеза (-)6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2н-3,1-бензоксазин-2-она и способ их получения</a>
Трициклические соединения, способ их получения, способы получения оптически активных или рацемических производных колхицина и тиохолкицина с использованием трициклических соединений и промежуточныепродукты синтеза

Номер патента: 93
Опубликовано: 25.06.1998
Авторы: Диолез Кристиан, Брион Франсис, Миддендорп Мишель, Мари Кристиан, Шаппер Бернадетт, Мазюри Алан, Пронин Дидье, Тороманофф Эдмон
МПК: C07C 43/21, C07D 317/44
Метки: тиохолкицина, синтеза, промежуточныепродукты, соединения, рацемических, производных, получения, способ, колхицина, способы, трициклические, активных, трициклических, использованием, оптически, соединений
Формула / Реферат:
1. Трициклические соединения общей формулы I в которой либо а) оба R1 и R2 представляют собой алкильную группу, a R3 представляет собой атом водорода или группу A-SO2-, либо б) оба R2 и R3 представляют собой атом водорода или алкил, a R1 представляет собой группу A-SO2-, либо в) все три: R1, R2 и R3 представляют собой атом водорода или все три представляют собой алкил, либо г) R1 представляет собой группу А-SO2- или атом водорода, a...
Способ получения 5-[2-этокси-5-(4-метилпиперазин-1-илсульфонил)-фенил]-1-метил-3-н-пропил,- 1,6-дигидро-7н-пиразоло[4,3-d]пиримидин-7-она

Номер патента: 102
Опубликовано: 27.08.1998
Авторы: Данн Питер Джеймз, Вуд Элберт Шо
МПК: C07D 487/04
Метки: получения, способ, 5-[2-этокси-5-(4-метилпиперазин-1-илсульфонил)-фенил]-1-метил-3-н-пропил, 1,6-дигидро-7н-пиразоло[4,3-d]пиримидин-7-она
Формула / Реферат:
1. Способ получения 5-[2-этокси-5-(4-метилпиперазин-1-ил-сульфонил)-фенил]-1-метил-3-н-пропил-1,6-дигидро-7Н-пиразоло-[4,3-d] пиримидин-7-она формулы (I) отличающийся тем, что соединение формулы (II) подвергают реакции циклизации в щелочной, нейтральной или кислой среде. 2. Способ по п.1, отличающийся тем, что циклизацию осуществляют в присутствии основания, предпочтительно в растворителе, возможно в присутствии перекиси водорода или...
Cпособ получения замещенных фенолов и способ получения витамина е с их использованием

Номер патента: 28
Опубликовано: 26.02.1998
Авторы: Ансель Жан-Эрик, Бьенейм Юг, Мейллян Пьер
МПК: A61K 31/355, B01J 31/24, C07C 39/19...
Метки: cпособ, замещенных, использованием, получения, фенолов, способ, витамина
Формула / Реферат:
1. Способ получения замещенных фенолов путем конденсации в однофазной среде фенола общей формулы где R обозначает один или несколько одинаковых или различных радикалов, выбранных из группы, включающей водород, гидроксильную группу и C1-C6 алкил, с производным бутадиена в присутствии катализатора на основе Rd+1, фосфинового соединения и основания, отличающийся тем, что в качестве производного бутадиена используют соединение, содержащее по...
Способ получения циклопропилацетилена.

Номер патента: 74
Опубликовано: 25.06.1998
Авторы: Корли Эдвард Г., Томпсон Эндрю С., Хантингтон Марта
МПК: C07C 13/04
Метки: получения, способ, циклопропилацетилена
Формула / Реферат:
1. Способ получения циклопропилацетилена, включающий следующие стадии: (а) смешивание, по крайней мере, около 1,0 эквивалента сильного основания, выбранного из группы, включающей н-бутиллитий, амид натрия, диэтиламид натрия, гидрид натрия, гидрид калия, бис(триметилсилил) амид натрия, бис(триметилсилил)амид калия, диизопропиламид лития, втор-бутиллитий, трет-бутиллитий и тетраметилпиперидид лития, в апротонном растворителе с одним эквивалентом...
Трициклические диазепины в качестве антагонистов вазопрессина, способ их получения и способ лечения с использованием трициклических диазепинов

Номер патента: 56
Опубликовано: 30.04.1998
Авторы: Венкатесан Аранапакан Мудумбай, Гросу Джордж Теодор, Олбрайт Джей Дональд
МПК: A61K 31/55, C07D 487/04
Метки: получения, трициклические, трициклических, диазепинов, лечения, использованием, антагонистов, качестве, диазепины, способ, вазопрессина
Формула / Реферат:
1. Соединение, выбранное среди соединений с общей формулой I: где Y является радикалом, выбранным из -(СН2)n-, где n является целым числом, 0 или 1, и А-В является радикалом, выбранным из где m является целым числом от 1 до 2; и радикал: представляет: (1) фенил или замещенный фенил, необязательно замещенный одним или двумя заместителями, выбранными из (C1-С3) низшего алкила, галогена, амина, (C1-С3) низшего алкокси или (C1-С3)...
Предыдущий патент: Способ каталитического газофазного окисления этилена
Следующий патент: Способ получения окислов алкиленов путем эпоксидирования олефинов ароматическими пероксикарбоновыми кислотами
Случайный патент: Дрожжевой автолизат, способ его приготовления и применение, вкусоароматическая добавка для пищевых продуктов на его основе