Эффективный синтез 1,4-дигидро 2н-3,1-бензоксазин-2-она
Номер патента: 1784
Опубликовано: 27.08.2001
Авторы: Тилльер Ричард Д., Грабовски Эдвард Дж.Дж., Фрей Лайза Ф.







Формула / Реферат
1. Способ получения 1,4-дигидро-2Н-3,1-бензоксазин-2-она формулы

включающий следующие стадии:
1) добавление арилхлорформиата к перемешиваемой смеси аминоспирта формулы

в органическом растворителе с основанием при температуре примерно от 0 до 25шС, в атмосфере инертного газа для получения промежуточного карбамата формулы

где R представляет собой арильную боковую цепь хлорформиата, которую определяют как фенил или нафтил, необязательно замещенный одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена (F, Cl, Вr, I), СF3, CO2C1-C6-алкила и NO2;
2) перемешивание реакционной смеси при температуре примерно от 20 до 25шС в течение примерно от 1 до 6 ч для завершения образования промежуточного карбамата;
3) гашение реакции водой или водным основанием для получения двухфазного раствора, содержащего 1,4-дигидро-2Н-3,1-бензоксазин-2-он в фазе органического растворителя;
4) перемешивание двухфазной смеси при температуре примерно от 20 до 50шС в течение примерно от 1 до 6 ч для завершения циклизации до 1,4-дигидро-2Н-3,1-бензоксазин-2-она; и
5) выделение 1,4-дигидро-2Н-3,1-бензоксазин-2-она из органической фазы.
2. Способ по п.1, где основание определяют как твердое вещество или раствор KОН, NaOH, LiOH, K2СО3, Na2CO3, Li2СО3, KНСО3, NaHCO3, LiНСО3 или комбинацию указанных оснований.
3. Способ по п.2, где органический растворитель выбирают из группы, состоящей из метил-t-бутилового эфира, толуола, тетрагидрофурана, ацетонитрила, диметилацетамида, N-метилпирролидинона или комбинации указанных растворителей.
4. Способ по п.3, где арильную группу арилхлорформиата определяют как фенилхлорформиат, в котором фенил необязательно замещен одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена (F, Cl, Вr, I), СF3 и NO2.
5. Способ по п.4, где основание определяют как твердое вещество или раствор KОН, NaOH, K2СО3, Nа2СО3, KНСО3, NаНСО3 или комбинацию указанных оснований.
6. Способ по п.5, где арильную группу арилхлорформиата определяют как 4-нитрофенилхлорформиат.
7. Способ получения 1,4-дигидро-2Н-3,1-бензоксазин-2-она формулы

включающий следующие стадии:
1) добавление 4-нитрофенилхлорформиата порциями к перемешиваемой смеси аминоспирта формулы

в метилтретбутиловом эфире с водным раствором KНСО3 при температуре около 25шС в атмосфере азота при поддержании рН между 8,5 и 4 для получения промежуточного карбамата формулы

2) перемешивание реакционной смеси при температуре примерно от 20 до 25шС около 2 ч для завершения образования промежуточного карбамата;
3) гашение реакции водным KОН до рН около 11 и добавление воды для получения двухфазной смеси, содержащей 1,4-дигидро-2Н-3,1-бензоксазин-2-он в фазе органического растворителя;
4) выделение 1,4-дигидро-2Н-3,1-бензоксазин-2-она из органической фазы.
8. Способ получения 1,4-дигидро-2Н-3,1-бензоксазин-2-она формулы

включающий следующие стадии:
1) добавление арилхлорформиата к перемешиваемой смеси аминоспирта формулы

в органическом растворителе при температуре примерно от 0 до 25шС в атмосфере инертного газа для получения промежуточного карбамата формулы

где R представляет собой арильную боковую цепь хлорформиата, которую определяют как фенил или нафтил, необязательно замещенный одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена (F, Cl, Вr, I), СF3, СO2C1-C6-алкила и NO2;
2) перемешивание реакционной смеси при температуре примерно от 20 до 25шС примерно от 1 до 6 ч для завершения образования промежуточного карбамата;
3) гашение реакции водным основанием для получения двухфазного раствора, содержащего 1,4-дигидро-2Н-3,1-бензоксазин-2-он в фазе органического растворителя;
4) перемешивание двухфазной смеси при температуре примерно от 20 до 50шС в течение примерно от 1 ч до 6 ч для завершения циклизации до 1,4-дигидро-2Н-3,1-бензоксазин-2-она; и
5) выделение 1,4-дигидро-2Н-3,1-бензоксазин-2-она из органической фазы.
9. Способ по п.8, где основание определяют как твердое вещество или раствор KОН, NaOH, LiOH, K2СО3, Nа2СО3, Li2СО3, KНСО3, NаНСО3, LiНСО3 или комбинацию указанных оснований.
10. Способ по п.9, где органический растворитель выбирают из группы, состоящей из метил-t-бутилового эфира, толуола или комбинации указанных растворителей.
11. Способ по п.10, где арильную группу арилхлорформиата определяют как фенилхлорформиат, в котором фенил необязательно замещен одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена (F, Cl, Вr, I), СF3 и NO2.
12. Способ по п.11, где основание определяют как твердое вещество или раствор KОН, NaOH, K2СО3, Nа2СО3, KНСО3, NаНСО3 или комбинацию указанных оснований.
13. Способ по п.12, где арильную группу арилхлорформиата определяют как 4-нитрофенилхлорформиат.
14. Способ получения 1,4-дигидро-2Н-3,1-бензоксазин-2-она формулы

включающий следующие стадии:
1) добавление алкилхлорформиата к перемешиваемой смеси аминоспирта формулы

в первом органическом растворителе с первым основанием при температуре примерно от 0 до 25шС в атмосфере инертного газа для получения промежуточного карбамата формулы

где R представляет собой алкильную боковую цепь хлорформиата, который определяют как C1-C10-алкил, необязательно замещенный одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена (F, Cl, Вr, I), СF3, C3-C7циклоалкила, СО2C1-C6-алкила и NO2;
2) перемешивание реакционной смеси при температуре примерно от 20 до 25шС в течение примерно от 1 до 30 ч для завершения образования промежуточного карбамата;
3) выделение органической фазы, содержащей алкилкарбамат;
4) отгонка в вакууме примерно от 90 до 95% первого органического растворителя и добавление противорастворителя для выделения твердого алкилкарбамата;
5) добавление второго органического растворителя к твердому алкилкарбамату для получения раствора алкилкарбамата;
6) взаимодействие раствора алкилкарбамата со вторым основанием в интервале температур примерно от 20 до 25шС в течение примерно от 2 до 30 ч для получения 1,4-дигидро-2Н-3,1-бензоксазин-2-она;
7) гашение реакционной смеси кислотой для получения двухфазного раствора, содержащего 1,4-дигидро-2Н-3,1-бензоксазин-2-он в фазе органического растворителя;
8) выделение 1,4-дигидро-2Н-3,1-бензоксазин-2-она из органической фазы.
15. Способ по п.14, где первое основание определяют как тверфюх вещество или раствор KОН, NaOH, LiOH, K2СО3, Na2CO3, Li2CO3, KНСО3, NаНСО3, LiНСО3 или комбинацию указанных оснований.
16. Способ по п.15, где первый органический растворитель выбирают из группы, состоящей из метил-t-бутилового эфира, толуола, тетрагидрофурана, ацетонитрила или комбинации указанных растворителей.
17. Способ по п.16, где противорастворитель представляет собой гептан.
18. Способ по п.17, где второй органический растворитель выбирают из группы, состоящей из метил-t-бутилового эфира, толуола, тетрагидрофурана, C1-C6-алканола или комбинации указанных растворителей.
19. Способ по п.18, где второе основание определяют как твердое вещество или раствор KOC1-C6-алкила, NaOC1-C6-алкила, LiOC1-C6-алкила, KC1-C6-алкила, NaC1-C6-алкила, LiC1-C6-алкила, KHMDS, NaHMDS, LiHMDS, LDA или комбинацию указанных оснований.
20. Способ по п.19, где кислоту выбирают из группы состоящей из НСl, НNО3, Н2SO4 и СН3СО2Н.
21. Способ по п.20, включающий дополнительно стадию кристаллизации алкилкарбамата, полученного на стадии 4, из смесей толуола-гептана или метил-t-бутилового эфира-гептана с образованием желаемого кристаллического алкилкарбамата.
22. Способ по п.21, где алкилхлорформиат определяют как метилхлорформиат или этилхлорформиат.
23. Способ по п.22, где органический растворитель (или смесь растворителей) выбирают из группы, состоящей из метил-t-бутилового эфира; метил-t-бутилового эфира и тетрагидрофурана; и метил-t-бутилового эфира и этанола.
24. Способ по п.23, где основание определяют как твердое вещество или раствор KНСО3 и KОН, KНСО3 и K2СО3 или KНСО3 и LiOtBu.
25. Способ по п.14, где R, представляющий собой алкильную боковую цепь хлорформиата, является незамещенным C1-C10-алкилом.
Текст
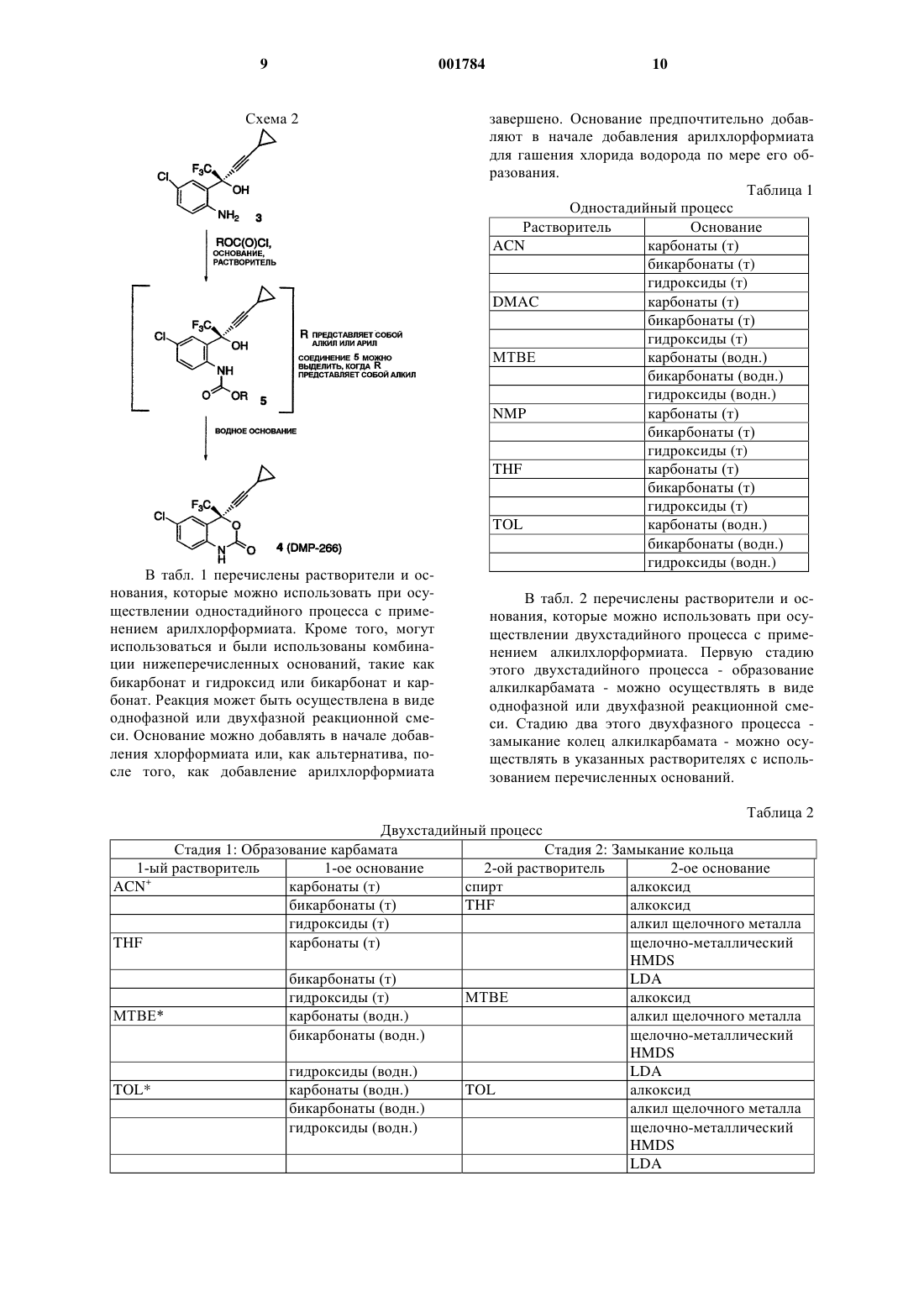
1 Ключевой стадией в синтезе ингибитора обратной транскриптазы (ревертазы), (-)-6-хлор 4-циклопропилэтинил-4-трифторметил-1,4 дигидро-2 Н-3,1-бензоксазин-2-она, известного также под названием DMP-266, является циклизация аминоспирта с помощью фосгена. Синтез DMP-266 и структурно подобных ингибиторов обратной транскриптазы описан в патенте США 5519021 и в соответствующей заявке РСТ WO 95/20389, опубликованной 3 августа 1995 г. Кроме того, асимметричный синтез энантиомерного бензоксазинона путем добавления высокоэнантиоизбирательного ацетилинида и последовательность циклизации описаны Томпсоном и др. (Thompson et al.) вTetrahedron Letters 1995, 36, 8937-8940, а также в международной публикации WO 96/37457 от 28 ноября 1996 г. Настоящее изобретение описывает эффективный способ циклизации аминоспирта формулы В этом способе циклизации применяется арил- или алкилхлорформиат и не используется фосген, который представляет собой токсичный и очень опасный газ, требующий особого обращения. Настоящее изобретение касается эффективного способа циклизации аминоспирта формулы 2 Настоящее изобретение описывает эффективный способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она формулы включающий в себя следующие стадии: 1) добавление арилхлорформиата к перемешиваемой смеси аминоспирта формулы в органическом растворителе с основанием при температуре примерно от 0 до 25 С в атмосфере инертного газа для получения промежуточного карбамата формулы в которой R представляет собой арильную боковую цепь хлорформиата; 2) перемешивание реакционной смеси при температуре примерно от 20 до 25 С в течение примерно от 1 до 6 ч для завершения образования промежуточного карбамата; 3) гашение реакции водой или водным основанием для получения двухфазного раствора,содержащего 1,4-дигидро-2 Н-3,1-бензоксазин-2 он в фазе органического растворителя; 4) перемешивание двухфазной смеси при температуре примерно от 20 до 50 С в течение примерно от 1 до 6 ч для завершения циклизации до 1,4-дигидро-2 Н-3,1-бензоксазин-2-она, и 5) выделение 1,4-дигидро-2 Н-3,1-бензоксазин-2-она из органической фазы. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она, стадии которого перечислены выше, в котором арильная группа арилхлорформиата определяется как фенил или нафтил,который необязательно замещен одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена (F, Cl, Вr, I), СF3,СО 2 С 1-С 6-алкила и NO2. Пример, включающий арилхлорформиаты, применяющиеся в данном способе, определяется как фенилхлорформиат, в котором фенил необязательно замещен одним,двумя или тремя заместителями, выбранными из группы, состоящей из галогена (F, Cl, Вr, I), СF3 и NO2. Предпочтительный арилхлорформиат,применяющийся в данном способе, представляет собой 4-нитрофенилхлорформиат. 3 Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она, стадии которого перечислены выше, в котором основание определяется как твердое вещество или раствор КОН, NaOH,LiOH, К 2 СО 3, Na2CO3, Li2 СО 3, КНСО 3, NаНСО 3,LiНСО 3 или комбинация указанных оснований. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она, стадии которого перечислены выше, в котором предпочтительное основание определяется как твердое вещество или раствор КОН, NaOH, К 2 СО 3, Nа 2 СО 3, КНСО 3,NаНСО 3 или комбинация указанных оснований. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она, стадии которого перечислены выше, в котором органический растворитель выбран из группы, состоящей из метил-tбутилового эфира, толуола, тетрагидрофурана,ацетонитрилдиметилацетамида, N-метилпирролидинона или комбинации этих растворителей. Предпочтительным вариантом осуществления настоящего изобретения является способ получения 1,4-дигидро-2 Н-3,1-бензоксазин-2 она формулы включающий в себя следующие стадии: 1) добавление 4-нитрофенилхлорформиата порциями к перемешиваемой смеси аминоспирта формулы в метилтретбутиловом эфире с водным раствором КНСО 3 при температуре около 25 С в атмосфере азота, при поддерживании значения рН между 8,5 и 4, чтобы получить промежуточный карбамат формулы 2) перемешивание реакционной смеси при температуре примерно от 20 до 25 С в течение примерно 2 ч для завершения образования промежуточного карбамата; 3) гашение реакции водным КОН до рН около 11 и добавление воды для получения двухфазной смеси, содержащей 1,4-дигидро-2 Н 001784 включающий в себя следующие стадии: 1) добавление арилхлорформиата к перемешиваемой смеси аминоспирта формулы в органическом растворителе при температуре примерно от 0 до 25 С в атмосфере инертного газа для получения промежуточного карбамата формулы в которой R представляет собой арильную боковую цепь хлорформиата; 2) перемешивание реакционной смеси при температуре примерно от 20 до 25 С в течение примерно от 1 до 6 ч для завершения образования промежуточного карбамата; 3) гашение реакции водным основанием для получения двухфазного раствора, содержащего 1,4-дигидро-2 Н-3,1-бензоксазин-2-она, в фазе органического растворителя; 4) перемешивание двухфазной смеси при температуре примерно от 20 до 50 С в течение примерно от 1 до 6 ч для завершения циклизации до 1,4-дигидро-2 Н-3,1-бензоксазин-2-она; и 5) выделение 1,4-дигидро-2 Н-3,1-бензоксазин-2-она из органической фазы. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она, стадии которого перечислены выше, в котором арильная группа арилхлорформиата определяется как фенил или нафтил,который необязательно замещен одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена (F, Cl, Вr, I), СF3,СО 2 С 1-С 6-алкила и NO2. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она, стадии которого перечислены выше, в котором основание определяется как твердое вещество или раствор КОН, NaOH, 5LiOH, К 2 СО 3, Nа 2 СО 3, Li2 СО 3, КНСО 3, NaHCO3,LiНСО 3 или комбинация указанных оснований. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она, стадии которого перечислены выше, в котором органический растворитель выбран из группы, состоящей из метил-tбутилового эфира, толуола или комбинации указанных растворителей. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазина-2-она, стадии которого перечислены выше, в котором арильная группа арилхлорформиата определяется как фенилхлорформиат, в котором необязательно замещен одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена (F, Cl,Вr, I), СF3 и NO2. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она, стадии которого перечислены выше, в котором основание определяется как твердое вещество или раствор КОН, NaOH,К 2 СО 3, Nа 2 СО 3, КНСО 3, NaHCO3 или комбинация указанных оснований. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она, стадии которого перечислены выше, в котором арильная группа арилхлорформиата определяется как 4-нитрофенилхлорформиат. Другой предмет настоящего изобретения представляет собой способ получения 1,4 дигидро-2 Н-3,1-бензоксазин-2-она формулы включающий в себя следующие стадии: 1) добавление алкилхлорформиата к перемешиваемой смеси аминоспирта формулы в первом органическом растворителе с первым основанием при температуре примерно от 0 до 25 С в атмосфере инертного газа для получения промежуточного карбамата формулы в которой R представляет собой алкильную боковую цепь хлорформиата; 6 2) перемешивание реакционной смеси при температуре примерно от 20 до 25 С в течение примерно от 1 до 30 ч для завершения образования промежуточного карбамата; 3) выделение органической фазы, содержащей алкилкарбамат; 4) дистилляция примерно от 90 до 95% первого органического растворителя в вакууме и добавление противорастворителя для выделения твердого алкилкарбамата; 5) добавление второго органического растворителя к твердому алкилкарбамату для получения раствора алкилкарбамата; 6) осуществление реакции раствора алкилкарбамата со вторым основанием в интервале температур примерно от 20 до 25 С в течение примерно от 2 до 30 ч для получения 1,4 дигидро-2 Н-3,1-бензоксазин-2-она; 7) гашение реакционной смеси кислотой для получения двухфазного раствора, содержащего 1,4-дигидро-2 Н-3,1-бензоксазин-2-он в фазе органического растворителя; 8) выделение 1,4-дигидро-2 Н-3,1-бензоксазин-2-она из органической фазы. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она, стадии которого перечислены выше, в котором алкильная группа алкилхлорформиата определяется как C1-С 10-алкил,который необязательно замещен одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена (F, Cl, Вr, I), СF3, С 3 С 7-циклоалкила, СO2 С 1-С 6-алкила и NO2. Примерами алкилхлорформиатов, используемых в способе получения 1,4-дигидро-2 Н-3,1-бензоксазин-2-она, стадии которого перечислены выше, являются метил- или этилхлорформиат. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она, стадии которого перечислены выше, в котором первое основание определяется как твердое вещество или раствор КОН,NaOH, LiOH, К 2 СО 3, Na2CO3, Li2 СО 3, КНСО 3,NаНСО 3, LiНСО 3 или комбинация указанных оснований. Способ получения 1,4-дигидро-2 Н 3,1-бензоксазин-2-она, стадии которого перечислены выше, в котором второе основание определяется как твердое вещество или растворKOC1-С 6-алкила, NaOC1-C6-алкила, LiOC1-C6 алкила, KC1-C6-алкила, NaC1-C6-алкила, LiC1C6-алкила, KHMDS, NaHMDS, LiHMDS, LDA или комбинация указанных оснований. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она, стадии которого перечислены выше, в котором первый органический растворитель выбран из группы, состоящей из метил-t-бутилового эфира, толуола, тетрагидрофурана, ацетонитрила или комбинации вышеуказанных растворителей. Способ получения 1,4 дигидро-2 Н-3,1-бензоксазин-2-она, стадии которого перечислены выше, в котором противорастворитель представляет собой гептан. Способ получения 1,4-дигидро-2 Н-3,1-бензоксазин 2-она, стадии которого перечислены выше, в 7 котором второй органический растворитель выбран из группы, состоящей из метил-tбутилового эфира, толуола, тетрагидрофурана,C1-C6-алканола или комбинации вышеуказанных растворителей. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она, стадии которого перечислены выше, в котором кислота выбрана из группы,состоящей из НСl, НNО 3, H2SO4 и СН 3 СO2 Н. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она, стадии которого перечислены выше, включающий в себя следующую дополнительную стадию: кристаллизация алкилкарбамата, полученного на стадии 4, из толуолагептана или метил-t-бутилового эфира-гептана,для получения кристаллического алкилкарбамата. Еще одним предметом настоящего изобретения является алкилкарбамат формулы в которой R представляет собой C1-C10-алкил,который необязательно замещен одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена (F, Cl, Вr, I), СF3, С 3 С 7-циклоалкила, СО 2C1-C6-алкила и NO2. В данном описании термин алкил включает в себя алкильные группы с указанным количеством атомов углерода, имеющие либо прямую, либо разветвленную, либо циклическую конфигурацию, которые необязательно замещены заместителем, выбранным из группы,состоящей из галогена (F, Cl, Вr, I), СF3, СО 2C1C6-алкила, С 3-С 7-циклоалкила и NO2. Примеры алкила включают в себя метил, этил, пропил,изопропил, бутил, втор- и трет-бутил, пентил,гексил, гептил, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, норборнил и им подобные. Термин арил определяется как фенильное или нафтильное кольцо, которое необязательно замещено одним, двумя или тремя заместителями, с любым имеющимся количеством атомов углерода, выбранными из группы, состоящей из галогена (F, Cl, Вr, I), СF3,СО 2C1-C6-алкила и NO2. Под термином атмосфера инертного газа понимают атмосферу аргона или азота, предпочтительно азота. На схеме 1 показаны ключевые стадии синтеза (-)-6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2 Н-3,1-бензоксазин-2 она (DMP-266). Стадия хирального добавления позволяет осуществлять энантиоизбирательное добавление циклопропилацетилинида через трифторметилкетон соединения 1. Получают РМВ-защищенный аминоспирт 2, а затем эту 8 защиту снимают и получают аминоспирт 3. Затем этот аминоспирт подвергают циклизации с помощью хлорформиата и основания, в результате чего получают DMP-266. Схема 1 Циклизация аминоспирта 3 для получения 1,4-дигидро-2 Н-3,1-бензоксазин-2-она 4 представлена ниже на схеме 2. Эта реакция может осуществляться в виде одностадийного процесса или, в качестве альтернативы, в виде двухстадийного процесса с возможным выделением промежуточного карбамата 5, в зависимости от используемого хлорформиата. Показано, что арилхлорформиаты образуют менее устойчивые карбаматы, которые при обработке их водным основанием циклизируются до желаемого продукта в течение одностадийного процесса. В противоположность этому, алкилхлорформиат обеспечивает получение алкилкарбамата, ключевого промежуточного продукта, который можно выделить и очистить перед осуществлением стадии циклизации. Основываясь на стабильности алкилкарбаматов, разработали практичный двухстадийный способ получения DMP266, который включает в себя образование промежуточного алкилкарбамата 5, вслед за чем осуществляют циклизацию этого карбамата, в результате чего получают желаемый продукт 4. В табл. 1 перечислены растворители и основания, которые можно использовать при осуществлении одностадийного процесса с применением арилхлорформиата. Кроме того, могут использоваться и были использованы комбинации нижеперечисленных оснований, такие как бикарбонат и гидроксид или бикарбонат и карбонат. Реакция может быть осуществлена в виде однофазной или двухфазной реакционной смеси. Основание можно добавлять в начале добавления хлорформиата или, как альтернатива, после того, как добавление арилхлорформиата 10 завершено. Основание предпочтительно добавляют в начале добавления арилхлорформиата для гашения хлорида водорода по мере его образования. Таблица 1 Одностадийный процесс Растворитель ОснованиеTOL карбонаты (водн.) бикарбонаты (водн.) гидроксиды (водн.) В табл. 2 перечислены растворители и основания, которые можно использовать при осуществлении двухстадийного процесса с применением алкилхлорформиата. Первую стадию этого двухстадийного процесса - образование алкилкарбамата - можно осуществлять в виде однофазной или двухфазной реакционной смеси. Стадию два этого двухфазного процесса замыкание колец алкилкарбамата - можно осуществлять в указанных растворителях с использованием перечисленных оснований. Таблица 2 Двухстадийный процесс Стадия 1: Образование карбамата Стадия 2: Замыкание кольца 1-ый растворитель 1-ое основание 2-ой растворитель 2-ое основаниеACN+ карбонаты (т) спирт алкоксид бикарбонаты (т)LDA гидроксиды (т) МТВЕ алкоксид МТВЕ карбонаты (водн.) алкил щелочного металла бикарбонаты (водн.) щелочно-металлическийTOL алкоксид бикарбонаты (водн.) алкил щелочного металла гидроксиды (водн.) щелочно-металлический+ использование ацетонитрила в качестве первого основания может потребовать альтернативной процедуры выделения, такой как дистилляция ацетонитрила и охлаждение, чтобы вызвать кристаллизацию карбамата в противоположность дистилляции большинства органических первых растворителей, а затем добавление противорастворителя для того, чтобы кристаллизовать карбамат.двухфазные реакционные смеси. т означает твердый, когда используют твердое вещество в безводной реакционной смеси, а по окончании реакции ее гасят водой. водн. означает водный раствор основания.(спирт); диметилацетамид (DMAC); метил-tбутиловый эфир (МТВЕ); N-метилпирролидинон (NMP); тетрагидрофуран (THF) и толуол (TOL). Кроме того, для оптимизации реакции можно использовать смеси перечисленных растворителей. Сокращенные наименования оснований: гидроксиды лития, натрия и калия; карбонаты лития, натрия и калия; бикарбонаты лития, натрия и калия; алкил лития, натрия и калия (например, n-бутил лития); гексаметилдисилазид лития, натрия и калия (например, LiHMDS); и диизопропиламид лития (LDA). Следующие ниже примеры служат для иллюстрации настоящего изобретения и ни коим образом не ограничивают его объем. Пример 1. 4 (49%), нитрофенилкарбоната (7%) и нескольких видов примесей с низкой концентрацией(0,3%). Взятие проб из порций и разбавление их, как указано выше, важно для получения воспроизводимых данных. Если пробу разбавили ACN и проанализировали методом ВЭЖХ, то основной пик обусловлен нитрофенилкарбаматом 5. После добавления водного основания он преобразуется в соединение 4. Реакцию погасили путем добавления водного К 2 СО 3 (10 г в 150 мл H2O). Полученную двухфазную смесь энергично перемешивали при 25 С в течение двух часов. Затем добавили МТВЕ (100 мл), разделили слои и органический слой промыли 5%-ным водным К 2 СО 3 (250 мл) и водой (250 мл). В качестве растворителя поочередно использовали этанол (EtOH) или изопропанол (IPA), и продукт кристаллизовался из EtOH или IPA и воды. См. примеры 10 и 11. Пример 2. В трехгорлую круглодонную колбу, оснащенную механической мешалкой, линией азота и термопарой, поместили твердый аминоспирт 3, THF (100 мл) и твердый КНСО 3. Полученную смесь охладили до +5 С, а затем добавили твердый 4-нитрофенилхлорформиат, одной порцией. Эта реакция является экзотермической. Во время растворения/реакции нитрофенилхлорформиата наблюдался подъем температуры на 4 градуса (окончательная температура составила 9 С). Смесь перемешивали при температуре 2025 С в течение двух часов. Из порции взяли пробу и разбавили ее ACN и 5%-ным NаНСО 3, в результате получили желтый однородный раствор. Анализ методом ВЭЖХ при 220 нм показал присутствие нитрофенола (43%), соединения В трехгорлую круглодонную колбу, оснащенную механической мешалкой, линией N2 и термопарой, поместили твердый аминоспирт 12,МТВЕ (100 мл) и водный К 2 СО 3. (10 г в 120 мл Н 2 О, 3 эквивалента). Добавили твердый 4 нитрофенилхлорформиат, одной порцией, при 25 С. Смесь перемешивали при температуре 2025 С в течение двух часов. В этот момент двух 13 фазную смесь нагревали до 50 С более 3 ч, чтобы осуществить превращение нитрофенилкарбоната в нитрофенол. После охлаждения до 25 С разделили слои, и слой МВТЕ экстрагировали с помощью водного К 2 СО 3 (10 г в 150 мл Н 2 О, двумя порциями по 75 мл), а затем водой(250 мл). В этот момент смесь поочередно растворяли в EtOH/IPA и осуществили кристаллизацию, как описано в примерах 10 и 11. Пример 3. В трехгорлую круглодонную колбу, оснащенную механической мешалкой, линией азота и термопарой, поместили твердый аминоспирт 3, МТВЕ (500 мл) и водный КНСО 3 (45 г в 654 мл Н 2 О). Добавили твердый 4-нитрофенилхлорформиат, четырьмя порциями, при 25 С. Во время добавления отслеживали рН. Показатель рН поддерживали во время реакции между 8,5 и 4, а закончили реакцию при рН 8,0. Смесь перемешивали при температуре 20-25 С в течение двух часов. Водный КОН (2N) добавляли в течение 20 мин до тех пор, пока рН водного слоя не достиг 11,0. Разделили слои и к слою МВТЕ добавили рассол (500 мл). Добавляли 0,1N уксусную кислоту до тех пор, пока показатель рН не достиг 6-7. Разделили слои и органическую фазу промыли рассолом (500 мл). В этот момент смесь поочередно растворяли в EtOH/IPA и осуществили кристаллизацию, как описано в примерах 10 и 11. Пример 5. В трехгорлую круглодонную колбу, оснащенную механической мешалкой, линией азота и термопарой, поместили твердый аминоспирт 3, МТВЕ (500 мл) и водный КНСО 3 (45 г в 654 мл H2O). Добавили твердый 4-нитрофенилхлорформиат, четырьмя порциями, при 25 С. Во время добавления следили за показателем рН. Показатель рН поддерживали во время реакции между 8,5 и 4, а закончили реакцию при рН 8,0. Смесь перемешивали при температуре 20-25 С в течение двух часов. В течение 20 мин добавляли водный КОН (2N) до тех пор, пока рН водного слоя не достиг 11,0. Слои отделили друг от друга, и слой МВТЕ промыли буфером с рН 7 (500 мл) и рассолом (500 мл). В этот момент смесь поочередно растворяли в EtOH/IPA и осуществили кристаллизацию, как описано в примерах 10 и 11. Пример 4.ClCO2Me МТВЕ Вода Рассол Гептан Молеку лярная масса 289,7 100,12 94,50 Количе- ЭквиваПлотммоль ство лентов ность Трехгорлую круглодонную колбу на 500 мл оснастили установленной через верх мешалкой, датчиком температуры и линией азота. В колбу поместили аминоспирт 3 (35,38 ммоль,10,25 г) и МТВЕ (100 мл), в результате чего получилась легкая суспензия. Добавили воду (100 мл), а потом бикарбонат калия (2 эквивалента,7,08 г). С помощью шприца ввели метилхлорформиат (2 эквивалента, 5,46 мл) и полученную двухфазную смесь энергично перемешивали при 20-25 С. Отбирали пробы и анализировали их с помощью метода ВЭЖХ до тех пор, пока осталось 0,5% аминоспирта 3 (примерно 8,5 ч). Разделили слои и органический слой промыли рассолом (100 мл) и высушили над сульфатом магния. После фильтрации осуществили поочередную обработку растворителями (при температуре 50-60 С в условиях вакуума) смеси МТВЕ и гептана (содержащей примерно 5% МТВЕ по объему, что измерялось с помощью 1H ЯМР) от общего объема в 102 мл (10 мл/г исходного материала). Во время поочередной обработки растворителями успешно осуществ 15 лялась кристаллизация карбамата 5. После выдержки суспензии при температуре 20-25 С в течение около 30 мин материал профильтровали. Твердое вещество промыли маточными растворами, а затем гептаном, в объеме одного фильтровального осадка. Выделили сухой метилкарбамат 5, выход которого составил 92% ПлотКоличе- Эквиваммоль ность ство лентов 32,55 32,55 Трехгорлую круглодонную колбу на 500 мл оснастили установленной через верх мешалкой, датчиком температуры и линией азота. Метилкарбамат 5 (32,55 ммоль, 11,32 г) растворили в МТВЕ (170 мл). Добавили LiOtBu (1 эквивалент, 32,6 мл), и реакционная смесь сразу же превратилась в суспензию. Эта суспензия со временем становилась все более жидкой и через 30 мин превратилась в прозрачный желтый раствор. Реакционную смесь выдержали при 2025 С. Реакцию сопровождали отбором проб и анализом их с помощью метода ВЭЖХ. Через 8 ч осталось 1% метилкарбамата 5. Примерно через 16 ч осталось 0,3% метилкарбамата 5. Реакцию погасили с помощью 0,5NНСl (150 мл). Разделили слои и органический слой промыли рассолом (150 мл), высушили над сульфатом магния и профильтровали. Анализ методом ВЭЖХ показал, что выход соединения 4 составил 96,0%. Материал подвергли поочередной обработке растворителями в препарате Стадия А: получение карбамата 5 Процедура повторяет ту, что описана в примере 5, стадия А, за исключением того, что в качестве растворителя использовали толуол, а карбамат выделяли из смеси толуола и гептана. Стадия В: получение соединения с замкнутым кольцом 4 Процедура повторяет ту, что описана в примере 5, стадия В, за исключением того, что в качестве растворителя использовали толуол, а продукт 4 выделяли путем кристаллизации из смеси толуола и гептана.ClCO2Et МТВЕ Вода Рассол Гептан Трехгорлую круглодонную колбу на 500 мл оснастили установленной через верх мешалкой, датчиком температуры и линией азота. В колбу поместили аминоспирт 3 (35,38 ммоль,10,25 г) и МТВЕ (100 мл), в результате получилась легкая суспензия. Добавили воду (100 мл),а потом бикарбонат калия (2 эквивалента, 7,08 г). С помощью шприца ввели этилхлорформиат(2 эквивалента, 6,76 мл) и полученную двухфазную смесь энергично перемешивали при 2025 С. Отбирали пробы и анализировали их с помощью метода ВЭЖХ до тех пор, пока осталось 2% аминоспирта 3 (примерно 30 ч). Разделили слои, органический слой промыли рассолом (100 мл) и высушили над сульфатом магния. После фильтрации осуществили поочередную обработку растворителями (при температуре 50-60 С в условиях вакуума) в смеси МТВЕ и гептана (содержащей примерно 5% МТВЕ, что измерялось с помощью 1H ЯМР) от общего объема в 102 мл (10 мл/г исходного материала). Во время поочередной обработки растворителями легко кристаллизовался этилкарбамат 5. После выдержки суспензии при температуре 20-25 С в течение около 30 мин материал профильтровали. Твердое вещество промыли маточными растворами, а затем гептаном в объеме одного фильтровального осадка. Сухой этилкарбамат 5 выделили с выходом 92% (11,77 г). 1-2% было потеряно с маточными растворами. Стадия В: получение соединения с замкнутым кольцом 4 ПлотКоличе- Эквиваммоль ность ство лентов 32,55 32,55 Трехгорлую круглодонную колбу на 500 мл оснастили установленной через верх мешалкой, датчиком температуры и линией азота. Этилкарбамат 5 (32,55 ммоль, 11,32 г) растворили в МТВЕ (170 мл). Добавили LiOtBu (1 эквивалент, 32,6 мл), и реакционная смесь сразу же превратилась в суспензию. Эта суспензия со временем становилась все более жидкой и через 30 мин превратилась в прозрачный желтый раствор. Реакционную смесь выдержали при 2025 С. Реакцию сопровождали отбором проб и анализом их с помощью метода ВЭЖХ. Через 26 ч осталось 1% этилкарбамата 5. Реакцию погасили с помощью 0,5N НСl (150 мл). Разделили слои и органический слой промыли рассолом (150 мл), высушили над сульфатом магния и профильтровали. Анализ методом ВЭЖХ показал, что выход соединения 4 составил 96,0%. Материал подвергли поочередной обработке растворителями в препарате EtOH для осуществления EtOH-водной кристаллизации. См. примеры 10 и 11. Прмер 8. Стадия А: получение карбамата 5 Процедура повторяет ту, что описана в примере 7, стадия А, за исключением того, что в качестве растворителя использовали толуол, а карбамат выделяли из смеси толуола и гептана. Стадия В: получение соединения с замкнутым кольцом 4 Процедура повторяет ту, что описана в примере 7, стадия В, за исключением того, что в качестве растворителя использовали толуол, а продукт 4 выделяли путем кристаллизации из смеси толуола и гептана. Пример 9. 15 2,5 МnBuLi МТВЕ Буферный раствор с рН 7 Рассол МолекуЭквиПлотКоличеммоль лярная валенность ство масса тов 361,75 32,55 11,77 г 32,55 13,0 мл 1 170 мл 150 мл 150 мл Трехгорлую круглодонную колбу на 500 мл оснастили установленной через верх мешалкой, датчиком температуры и линией азота. Этилкарбамат 5 (32,55 ммоль, 11,32 г) растворили в МТВЕ (170 мл). Добавили nBuLi (1 эквивалент, 13,0 мл) и реакционную смесь выдержали при 20-25 С. Реакцию сопровождали отбо 18 ром проб и анализом их с помощью метода ВЭЖХ. Через 30 ч осталось 1% этилкарбамата 5. Реакцию погасили с помощью буферного раствора с рН 7 (150 мл). Разделили слои и органический слой промыли рассолом (150 мл), высушили над сульфатом магния и профильтровали. Анализ методом ВЭЖХ показал, что выход соединения 4 составил 96,0%. Материал подвергли поочередной обработке растворителями в препарате EtOH для осуществления EtOHводной кристаллизации. См. примеры 10 и 11. Пример 10. Процесс кристаллизации, контролируемый путем добавления противорастворителя. 400 г исходного материала DMP-266 растворяют в 2,400 л этанола. Раствор фильтруют,чтобы удалить посторонние вещества. К раствору добавляют 2,088 л деионизированной (ДИ) воды в течение 30-60 мин. Затем к раствору добавляют 20 г затравочного DMP-266. Затравочный слой выдерживают в течение 1 ч. Для размешивания суспензий предпочтительно использовать мешалки модели Интермиг. Если требуется (что определяется наличием чрезмерно длинных кристаллов или слишком густой суспензии), суспензию подвергают мокрому размолу в течение 15-60 с. К суспензии добавляют 1,512 л ДИ воды, в течение 4-6 ч. Если требуется (что определяется наличием чрезмерно длинных кристаллов или слишком густой суспензии), суспензию подвергают мокрому размолу в течение 15-60 с во время добавления воды. Суспензию выдерживают в течение от 1 до 3 ч, а затем охлаждают до 10 С в течение 3 ч. Суспензию выдерживают в течение от 2 до 16 ч до тех пор, пока концентрация продукта в надосадочном слое станет постоянной. Суспензию фильтруют, чтобы выделить влажный кристаллический фильтровальный осадок. Этот влажный фильтровальный осадок промывают 40% раствором этанола в воде в количестве, равном 1-2 объемам слоя, а затем дважды промывают ДИ водой, по 2 л в каждой порции. Промытый влажный фильтровальный осадок высушивают в условиях вакуума при 50 С. Пример 11. Полупериодический процесс донной кристаллизации. 400 г исходного материала DMP-266 растворяют в 2,400 л этанола. Путем размешивания 20 г DMP-266 в 0,3 л 40%-ого (по объему) раствора этанола в воде получают придонную суспензию. Растворенную порцию и 3,6 л ДИ воды одновременно добавляют к придонной суспензии с постоянной скоростью в течение 6 ч, чтобы поддерживать постоянный состав растворителя в кристаллизаторе. Для размешивания во время кристаллизации предпочтительно использовать мешалки модели Интермиг. Во время этого добавления суспензию подвергают мокрому размолу, когда длина кристаллов становится чрезмерно большой или когда суспензия становится слишком густой. Суспензию охлаж 19 дают до около 10 С в течение 3 ч. Суспензию выдерживают в течение от 2 до 16 ч до тех пор,когда концентрация продукта в надосадочном слое станет постоянной. Суспензию фильтруют,чтобы выделить влажный кристаллический фильтровальный осадок. Этот влажный фильтровальный осадок промывают 40% раствором этанола в воде в количестве, равном 1-2 объемам слоя, а затем дважды промывают ДИ водой,по 2 л в каждой порции. Промытый влажный фильтровальный осадок высушивают в условиях вакуума при 50 С. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она формулы включающий следующие стадии: 1) добавление арилхлорформиата к перемешиваемой смеси аминоспирта формулы в органическом растворителе с основанием при температуре примерно от 0 до 25 С, в атмосфере инертного газа для получения промежуточного карбамата формулы где R представляет собой арильную боковую цепь хлорформиата, которую определяют как фенил или нафтил, необязательно замещенный одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена (F,Cl, Вr, I), СF3, CO2C1-C6-алкила и NO2; 2) перемешивание реакционной смеси при температуре примерно от 20 до 25 С в течение примерно от 1 до 6 ч для завершения образования промежуточного карбамата; 3) гашение реакции водой или водным основанием для получения двухфазного раствора,содержащего 1,4-дигидро-2 Н-3,1-бензоксазин-2 он в фазе органического растворителя; 4) перемешивание двухфазной смеси при температуре примерно от 20 до 50 С в течение примерно от 1 до 6 ч для завершения циклизации до 1,4-дигидро-2 Н-3,1-бензоксазин-2-она; и 5) выделение 1,4-дигидро-2 Н-3,1-бензоксазин-2-она из органической фазы. 20 2. Способ по п.1, где основание определяют как твердое вещество или раствор KОН,NaOH, LiOH, K2 СО 3, Na2CO3, Li2 СО 3, KНСО 3,NaHCO3, LiНСО 3 или комбинацию указанных оснований. 3. Способ по п.2, где органический растворитель выбирают из группы, состоящей из метил-t-бутилового эфира, толуола, тетрагидрофурана, ацетонитрила, диметилацетамида, Nметилпирролидинона или комбинации указанных растворителей. 4. Способ по п.3, где арильную группу арилхлорформиата определяют как фенилхлорформиат, в котором фенил необязательно замещен одним, двумя или тремя заместителями,выбранными из группы, состоящей из галогена(F, Cl, Вr, I), СF3 и NO2. 5. Способ по п.4, где основание определяют как твердое вещество или раствор KОН,NaOH, K2 СО 3, Nа 2 СО 3, KНСО 3, NаНСО 3 или комбинацию указанных оснований. 6. Способ по п.5, где арильную группу арилхлорформиата определяют как 4 нитрофенилхлорформиат. 7. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она формулы включающий следующие стадии: 1) добавление 4-нитрофенилхлорформиата порциями к перемешиваемой смеси аминоспирта формулы в метилтретбутиловом эфире с водным раствором KНСО 3 при температуре около 25 С в атмосфере азота при поддержании рН между 8,5 и 4 для получения промежуточного карбамата формулы 2) перемешивание реакционной смеси при температуре примерно от 20 до 25 С около 2 ч для завершения образования промежуточного карбамата; 3) гашение реакции водным KОН до рН около 11 и добавление воды для получения двухфазной смеси, содержащей 1,4-дигидро-2 Н 21 3,1-бензоксазин-2-он в фазе органического растворителя; 4) выделение 1,4-дигидро-2 Н-3,1 бензоксазин-2-она из органической фазы. 8. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она формулы включающий следующие стадии: 1) добавление арилхлорформиата к перемешиваемой смеси аминоспирта формулы в органическом растворителе при температуре примерно от 0 до 25 С в атмосфере инертного газа для получения промежуточного карбамата формулы где R представляет собой арильную боковую цепь хлорформиата, которую определяют как фенил или нафтил, необязательно замещенный одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена (F,Cl, Вr, I), СF3, СO2C1-C6-алкила и NO2; 2) перемешивание реакционной смеси при температуре примерно от 20 до 25 С примерно от 1 до 6 ч для завершения образования промежуточного карбамата; 3) гашение реакции водным основанием для получения двухфазного раствора, содержащего 1,4-дигидро-2 Н-3,1-бензоксазин-2-он в фазе органического растворителя; 4) перемешивание двухфазной смеси при температуре примерно от 20 до 50 С в течение примерно от 1 до 6 ч для завершения циклизации до 1,4-дигидро-2 Н-3,1-бензоксазин-2-она; и 5) выделение 1,4-дигидро-2 Н-3,1-бензоксазин-2-она из органической фазы. 9. Способ по п.8, где основание определяют как твердое вещество или раствор KОН,NaOH, LiOH, K2 СО 3, Nа 2 СО 3, Li2 СО 3, KНСО 3,NаНСО 3, LiНСО 3 или комбинацию указанных оснований. 10. Способ по п.9, где органический растворитель выбирают из группы, состоящей из метил-t-бутилового эфира, толуола или комбинации указанных растворителей. 22 11. Способ по п.10, где арильную группу арилхлорформиата определяют как фенилхлорформиат, в котором фенил необязательно замещен одним, двумя или тремя заместителями,выбранными из группы, состоящей из галогена(F, Cl, Вr, I), СF3 и NO2. 12. Способ по п.11, где основание определяют как твердое вещество или раствор KОН,NaOH, K2 СО 3, Nа 2 СО 3, KНСО 3, NаНСО 3 или комбинацию указанных оснований. 13. Способ по п.12, где арильную группу арилхлорформиата определяют как 4 нитрофенилхлорформиат. 14. Способ получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она формулы включающий следующие стадии: 1) добавление алкилхлорформиата к перемешиваемой смеси аминоспирта формулы в первом органическом растворителе с первым основанием при температуре примерно от 0 до 25 С в атмосфере инертного газа для получения промежуточного карбамата формулы где R представляет собой алкильную боковую цепь хлорформиата, который определяют какC1-C10-алкил, необязательно замещенный одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена (F, Cl,Вr, I), СF3, C3-C7 циклоалкила, СО 2C1-C6-алкила и NO2; 2) перемешивание реакционной смеси при температуре примерно от 20 до 25 С в течение примерно от 1 до 30 ч для завершения образования промежуточного карбамата; 3) выделение органической фазы, содержащей алкилкарбамат; 4) отгонка в вакууме примерно от 90 до 95% первого органического растворителя и добавление противорастворителя для выделения твердого алкилкарбамата; 5) добавление второго органического растворителя к твердому алкилкарбамату для получения раствора алкилкарбамата; 6) взаимодействие раствора алкилкарбамата со вторым основанием в интервале температур примерно от 20 до 25 С в течение примерно от 2 до 30 ч для получения 1,4-дигидро-2 Н-3,1 бензоксазин-2-она; 7) гашение реакционной смеси кислотой для получения двухфазного раствора, содержащего 1,4-дигидро-2 Н-3,1-бензоксазин-2-он в фазе органического растворителя; 8) выделение 1,4-дигидро-2 Н-3,1-бензоксазин-2-она из органической фазы. 15. Способ по п.14, где первое основание определяют как твердое вещество или растворKОН, NaOH, LiOH, K2 СО 3, Na2CO3, Li2CO3,KНСО 3, NаНСО 3, LiНСО 3 или комбинацию указанных оснований. 16. Способ по п.15, где первый органический растворитель выбирают из группы, состоящей из метил-t-бутилового эфира, толуола,тетрагидрофурана, ацетонитрила или комбинации указанных растворителей. 17. Способ по п.16, где противорастворитель представляет собой гептан. 18. Способ по п.17, где второй органический растворитель выбирают из группы, состоящей из метил-t-бутилового эфира, толуола,тетрагидрофурана, C1-C6-алканола или комбинации указанных растворителей. 19. Способ по п.18, где второе основание определяют как твердое вещество или растворKOC1-C6-алкила, NaOC1-C6-алкила, LiOC1-C6 алкила, KC1-C6-алкила, NaC1-C6-алкила, LiC1C6-алкила, KHMDS, NaHMDS, LiHMDS, LDA или комбинацию указанных оснований. 20. Способ по п.19, где кислоту выбирают из группы состоящей из НСl, НNО 3, Н 2SO4 и СН 3 СО 2 Н. 21. Способ по п.20, включающий дополнительно стадию кристаллизации алкилкарбамата,полученного на стадии 4, из смесей толуолагептана или метил-t-бутилового эфира-гептана с образованием желаемого кристаллического алкилкарбамата. 22. Способ по п.21, где алкилхлорформиат определяют как метилхлорформиат или этилхлорформиат. 23. Способ по п.22, где органический растворитель (или смесь растворителей) выбирают из группы, состоящей из метил-t-бутилового эфира; метил-t-бутилового эфира и тетрагидрофурана; и метил-t-бутилового эфира и этанола. 24. Способ по п.23, где основание определяют как твердое вещество или раствор KНСО 3 и KОН, KНСО 3 и K2 СО 3 или KНСО 3 и LiOtBu. 25. Способ по п.14, где R, представляющий собой алкильную боковую цепь хлорформиата,является незамещенным C1-C10-алкилом.
МПК / Метки
МПК: C07D 265/18, C07C 271/28
Метки: 2н-3,1-бензоксазин-2-она, эффективный, синтез, 1,4-дигидро
Код ссылки
<a href="https://eas.patents.su/13-1784-effektivnyjj-sintez-14-digidro-2n-31-benzoksazin-2-ona.html" rel="bookmark" title="База патентов Евразийского Союза">Эффективный синтез 1,4-дигидро 2н-3,1-бензоксазин-2-она</a>
Промежуточные продукты для ассиметричного синтеза (-)6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2н-3,1-бензоксазин-2-она и способ их получения

Номер патента: 107
Опубликовано: 27.08.1998
Авторы: Грабовский Эдвард Дж.Дж., Корли Эдвард Г., Томпсон Эндрю С., Ясуда Нобуёси
МПК: C07C 213/00, C07D 265/18
Метки: получения, синтеза, 6-хлор-4-циклопропилэтинил-4-трифторметил-1,4-дигидро-2н-3,1-бензоксазин-2-она, продукты, промежуточные, способ, ассиметричного
Формула / Реферат:
1. 6-Хлор-2-[(R)-циклопропилэтинилгидрокси-трифторметил] амин общей формулы в которой Р представляет собой группу, защищающую аминогруппу. 2. Соединение по п.1, представляющее собой N-(4-метоксибензил)-6-хлор-2-[(R)-циклопропилэтинилгидрокси-трифторметил]-метиланилин формулы 3. Способ получения соединения по п.1, заключающийся в том, что осуществляют стадии: а) получение смеси избытка (1R, 2S)-N-замещенного норэфедрина формулы в...
Синтез гидроксисульфона и родственных соединений.

Номер патента: 1246
Опубликовано: 25.12.2000
Авторы: Матр Дэвид Дж., Муди Дэвид, Сохар Пол, Блэкер Эндрю Дж.
МПК: C07D 495/04
Метки: соединений, родственных, гидроксисульфона, синтез
Формула / Реферат:
1. Способ получения соединения формулы IV, имеющего структурную формулу где R представляет водород, С1-4 алкил или С1-4 алкокси-С1-4 алкил, включающий добавление ангидрида к первому раствору, содержащему растворитель и соединение формулы I где R определен выше, при поддержании температуры от примерно -5 до примерно 50шС, с получением второго раствора, содержащего соединение структурной формулы II где R определен выше, добавление ко...
Синтез бисиндолилмалеимидов

Номер патента: 1392
Опубликовано: 26.02.2001
Авторы: Фол Маргарет Мэри, Виннероски Леонард Л.
МПК: C07D 207/50, A61P 9/00
Метки: бисиндолилмалеимидов, синтез
Формула / Реферат:
1. Способ получения соединения формулы где: R1 и R2 независимо являются необязательно замещенным 3-индолилом, включающий взаимодействие необязательно замещенного индол-3-ацетамида формулы с необязательно замещенным индолил-3-глиоксильным реагентом формулы где R3 является I, Сl, или Вr, или OR4; R4 является С1-С4-алкилом; и R11 является СН3 или Н; в присутствии основания, достаточно сильного для депротонирования амида и метилена в...
Стереоспецифический синтез хиральных 1-арил- и 1-гетероарил-2-замещенных этил-2-аминов

Номер патента: 1364
Опубликовано: 26.02.2001
Авторы: Грондар Люк, Казимир Жан-Поль, О'брайен Майкл К, Пауэрс Мэттью Р., Леон Патрик, Робин Даниэль
МПК: C07C 303/38, C07D 203/24, A61P 9/12...
Метки: этил-2-аминов, стереоспецифический, хиральных, 1-арил, синтез, 1-гетероарил-2-замещенных
Формула / Реферат:
1. Способ стереоспецифического синтеза [(1-необязательно замещенный арил)- или (1-необязательно замещенный гетероарил)]-2-замещенного этил-2-амина, имеющего хиральный атом в положении 2, включающий взаимодействие 2-амино-2-замещенного этилового спирта, имеющего хиральный атом в положении 2, с [(необязательно замещенный арил)- или (тригалогенметил)сульфонил]-галогенидом или ангидридом в присутствии основания с образованием [(N-арилсульфонил)- или...
Тригидрат мезилата 5-(2-(4-(1,2-бензизотиазол-3-ил)пиперазин-1-ил)этил)-6-хлор-1,3-дигидро-2н-индол-2-она, фармацевтическая композиция и способ лечения психического расстройства

Номер патента: 1180
Опубликовано: 30.10.2000
Авторы: Роуз Кэрол Э., Буш Фрэнк Р.
МПК: A61K 31/497, C07D 417/12, A61P 25/06...
Метки: 5-(2-(4-(1,2-бензизотиазол-3-ил)пиперазин-1-ил)этил)-6-хлор-1,3-дигидро-2н-индол-2-она, мезилата, расстройства, композиция, фармацевтическая, способ, психического, лечения, тригидрат
Формула / Реферат:
1. Тригидрат мезилата 5-(2-(4-(1,2-бензизотиазол-3-ил)пиперазин-1-ил)этил)-6-хлор-1,3-дигидро-2Н-индол-2-она. 2. Фармацевтическая композиция для лечения психического расстройства, содержащая такое количество соединения по п.1, которое эффективно при лечении указанного психического расстройства, и фармацевтически приемлемый носитель. 3. Способ лечения психического расстройства у млекопитающего, при котором указанному млекопитающему вводят такое...
Предыдущий патент: Соединения и способ получения замещенных 4-фенил-4-цианоциклогексановых кислот
Следующий патент: Антивирусный препарат для инъекций
Случайный патент: Флотационная машина