Способ синтеза ивабрадина и его аддитивных солей с фармацевтически приемлемой кислотой







Формула / Реферат
1. Способ синтеза ивабрадина формулы (I)

который отличается тем, что соединение формулы (VI)

подвергают реакции лактамизации в присутствии связующего компонента, выбранного из оксалилхлорида, тионилхлорида, N,N-дициклогексилкарбодиимида (DCC), 1-этил-3-(3-диметиламинопропил)карбодиимида (EDCI), N,N-карбонилдиимидазола (CDI), циклического ангидрида 1-пропанфосфоновой кислоты (Т3Р) и 1-(метилсульфонил)-1Н-бензотриазола, и основания, в органическом растворителе, с получением ивабрадина формулы (I), который может быть превращен в его аддитивные соли с фармацевтически приемлемой кислотой, выбранной из соляной, бромисто-водородной, серной, фосфорной, уксусной, трифторуксусной, молочной, пировиноградной, малоновой, янтарной, глутаровой, фумаровой, винной, малеиновой, лимонной, аскорбиновой, щавелевой, метансульфоновой, бензолсульфоновой и камфорной кислот, и в его гидраты.
2. Способ синтеза по п.1, который отличается тем, что связующий компонент, который используют для осуществления реакции лактамизации соединения формулы (VI), представляет собой тионилхлорид.
3. Способ синтеза по п.2, который отличается тем, что количество тионилхлорида, которое используют для осуществления реакции лактамизации соединения формулы (VI), находится в интервале 1-5 экв. включительно.
4. Способ синтеза по любому из пп.1-3, который отличается тем, что основание, которое используют для осуществления реакции лактамизации соединения формулы (VI), выбирают из триэтиламина, диизопропилэтиламина и пиридина.
5. Способ синтеза по п.4, который отличается тем, что основание, которое используют для осуществления реакции лактамизации соединения формулы (VI), представляет собой триэтиламин.
6. Способ синтеза по любому из пп.1-5, который отличается тем, что органический растворитель, который используют для осуществления реакции лактамизации соединения формулы (VI), выбирают из дихлорметана, тетрагидрофурана, ацетонитрила, ацетона и толуола.
7. Способ синтеза по п.6, который отличается тем, что органический растворитель, который используют для осуществления реакции лактамизации соединения формулы (VI), представляет собой дихлорметан.
8. Способ синтеза по любому из пп.1-7, который отличается тем, что реакцию лактамизации соединения формулы (VI) осуществляют при температуре между 0 и 40°C включительно.
9. Способ синтеза по п.1, который отличается тем, что соединение формулы (VI) получают исходя из соединения формулы (VII)

которое используют для осуществления реакции с соединением формулы (VIII)

в которой X представляет собой атом галогена, мезилатную группу или тозилатную группу,
в присутствии основания, в органическом растворителе, с получением соединения формулы (IX)

которое гидролизируют посредством действия основания в смеси органического растворителя и воды, с образованием соединения формулы (VI)

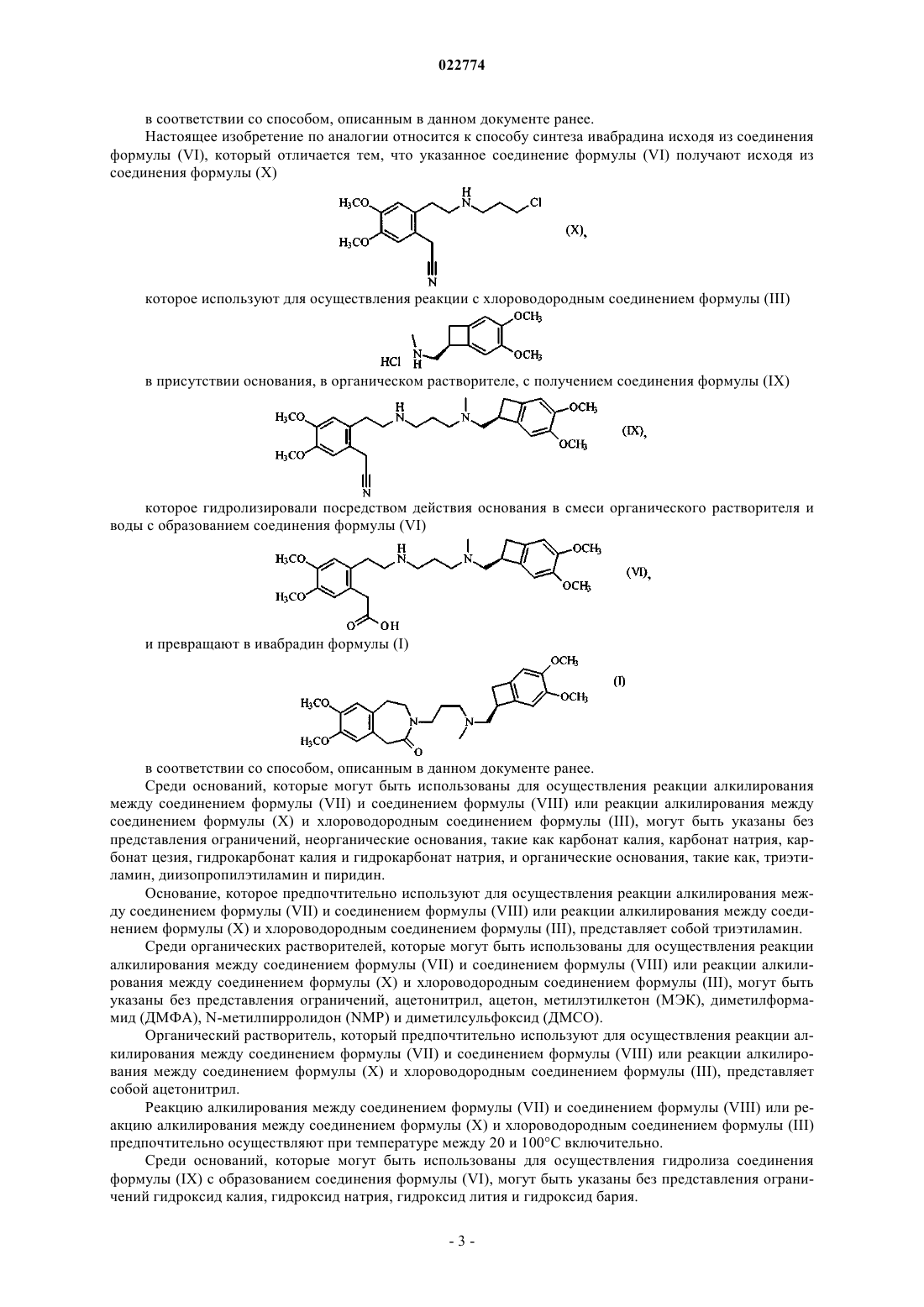
10. Способ синтеза по п.1, который отличается тем, что соединение формулы (VI) получают исходя из соединения формулы (X)

которое используют для осуществления реакции с хлороводородным соединением формулы (III)

в присутствии основания, в органическом растворителе, с получением соединения формулы (IX)

которое гидролизируют посредством действия основания в смеси органического растворителя и воды, с образованием соединения формулы (VI)

11. Способ синтеза по любому п.9 или 10, который отличается тем, что основание, которое используют для осуществления реакции алкилирования между соединением формулы (VII) и соединением формулы (VIII) или реакции алкилирования между соединением формулы (X) и хлороводородным соединением формулы (III), выбирают из карбоната калия, карбоната натрия, карбоната цезия, гидрокарбоната калия, гидрокарбоната натрия, триэтиламина, диизопропилэтиламина и пиридина.
12. Способ синтеза по п.11, который отличается тем, что основание, которое используют для осуществления реакции алкилирования между соединением формулы (VII) и соединением формулы (VIII) или реакции алкилирования между соединением формулы (X) и хлороводородным соединением формулы (III), представляет собой триэтиламин.
13. Способ синтеза по любому из пп.9-12, который отличается тем, что органический растворитель, который используют для осуществления реакции алкилирования между соединением формулы (VII) и соединением формулы (VIII) или реакции алкилирования между соединением формулы (X) и хлороводородным соединением формулы (III), выбирают из ацетонитрила, ацетона, метилэтилкетона (МЭК), диметилформамида (ДМФА), N-метилпирролидона (NMP) и диметилсульфоксида (ДМСО).
14. Способ синтеза по п.13, который отличается тем, что органический растворитель, который используют для осуществления реакции алкилирования между соединением формулы (VII) и соединением формулы (VIII) или реакции алкилирования между соединением формулы (X) и хлороводородным соединением формулы (III), представляет собой ацетонитрил.
15. Способ синтеза по любому из пп.9-14, который отличается тем, что реакцию алкилирования между соединением формулы (VII) и соединением формулы (VIII) или реакцию алкилирования между соединением формулы (X) и хлороводородным соединением формулы (III) осуществляют при температуре между 20 и 100°C включительно.
16. Способ синтеза по любому из пп.9-15, который отличается тем, что основание, которое используют для осуществления гидролиза соединения формулы (IX) с образованием соединения формулы (VI), выбирают из гидроксида калия, гидроксида натрия, гидроксида лития и гидроксида бария.
17. Способ синтеза по п.16, который отличается тем, что основание, которое используют для осуществления гидролиза соединения формулы (IX) с образованием соединения формулы (VI), представляет собой гидроксид натрия.
18. Способ синтеза по любому из пп.9-17, который отличается тем, что органический растворитель, который используют для осуществления гидролиза соединения формулы (IX) с образованием соединения формулы (VI), представляет собой спиртовой растворитель.
19. Способ синтеза по п.18, который отличается тем, что спиртовой растворитель, который используют для осуществления гидролиза соединения формулы (IX) с образованием соединение формулы (VI), выбирают из метанола, этанола, изопропанола и бутанола.
20. Способ синтеза по п.19, который отличается тем, что спиртовой растворитель, который используют для осуществления гидролиза соединения формулы (IX) с образованием соединения формулы (VI), представляет собой этанол.
21. Способ синтеза по любому из пп.9-20, который отличается тем, что гидролиз соединения формулы (IX) с образованием соединения формулы (VI) осуществляют при температуре между 0 и 110°C включительно.
22. Соединение формулы (VI)

23. Соединение формулы (IX)

24. Соединение формулы (X)

Текст
СПОСОБ СИНТЕЗА ИВАБРАДИНА И ЕГО АДДИТИВНЫХ СОЛЕЙ С ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМОЙ КИСЛОТОЙ Способ синтеза ивабрадина формулы (I) и его аддитивных солей с фармацевтически приемлемой кислотой.(71)(73) Заявитель и патентовладелец: ЛЕ ЛАБОРАТУАР СЕРВЬЕ (FR) Настоящее изобретение относится к способу синтеза ивабрадина формулы (I) или 3-3-(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил 7,8-диметокси-1,3,4,5-тетрагидро-2H-3-бензазепин-2-она, его аддитивной соли с фармацевтически приемлемой кислотой, и его гидратов. Ивабрадин и его аддитивные соли с фармацевтически приемлемой кислотой, и в особенности его гидрохлорид, обладают очень ценными фармакологическими и терапевтическими свойствами, особенно свойствами, которые регулируют брадикардию, что делает эти соединения полезными при лечении или профилактике различных клинических ситуаций ишемии миокарда, таких как стенокардия, инфаркт миокарда, и связанных с ними нарушений ритма, а также при различных патологиях, связанных с нарушением ритма, в особенности, суправентрикулярных нарушениях ритма, и при сердечной недостаточности. Получение и терапевтическое применение ивабрадина и его аддитивных солей с фармацевтически приемлемой кислотой, и, в особенности, его гидрохлорида, были описаны в европейском патенте EP 0534859. В патенте описан синтез гидрохлорида ивабрадина исходя из соединения формулы (II) которое разделяют с получением соединения формулы (III) которое используют для осуществления реакции с соединением формулы (IV) с получением соединения формулы (V) каталитическое гидрирование, которого обеспечивает ивабрадин, который затем превращают в его гидрохлорид. Недостаток такого способа синтеза в том, что это обеспечивает ивабрадин с выходом только 1%. В связи с фармацевтической ценностью данного соединения является важным иметь возможность получать его посредством эффективного метода синтеза с обеспечением ивабрадина с хорошим выходом. Настоящее изобретение относится к способу синтеза ивабрадина формулы (I) который отличается тем, что соединение формулы (VI) подвергают реакции лактамизации в присутствии связующего компонента и основания в органиче-1 022774 ском растворителе с получением ивабрадина формулы (I), который может быть превращен в его аддитивные соли с фармацевтически приемлемой кислотой, выбранной из соляной, бромисто-водородной,серной, фосфорной, уксусной, трифторуксусной, молочной, провиноградной, малоновой, янтарной, глутаровой, фумаровой, винной, лимонной, аскорбиновой, щавелевой, метансульфоновой, бензолсульфоновой и камфорной кислот, и в его гидраты. Среди связующих компонентов, которые могут быть использованы для реакции для лактамизации соединения формулы (VI), могут быть указаны без представления ограничения следующие реагенты: оксалилхлорид,тионилхлорид,N,N-дициклогексилкарбодиимид(DCC),1-этил-3-(3 диметиламинопропил)карбодиимид (EDCI), N,N-карбонилдиимидазол (CDI), циклический ангидрид 1 пропанфосфоновой кислоты (Т 3 Р) и 1-(метилсульфонил)-1 Н-бензотриазол. Связующий компонент, который предпочтительно используют для реакции для лактамизации соединения формулы (VI), представляет собой тионилхлорид. Количество тионилхлорида, которое предпочтительно используют для осуществления реакции для лактамизации соединения формулы (VI), находится в интервале между 1 и 5 экв. включительно. Среди оснований, которые могут быть использованы для реакции для лактамизации соединения формулы (VI), могут быть указаны без представления ограничения триэтиламин, диизопропилэтиламин и пиридин. Основание, которое предпочтительно используют для осуществления реакции для лактамизации соединения формулы (VI), представляет собой триэтиламин. Среди органических растворителей, которые могут быть использованы для реакции для лактамизации соединения формулы (VI), могут быть указаны без представления ограничения дихлорметан, тетрагидрофуран, ацетонитрил, ацетон и толуол. Органический растворитель, который предпочтительно используют для осуществления реакции для лактамизации соединения формулы (VI), представляет собой дихлорметан. Реакцию для лактамизации соединения формулы (VI) предпочтительно осуществляют при температуре между 0 и 40C включительно. Настоящее изобретение по аналогии относится к способу синтеза ивабрадина исходя из соединения формулы (VI), который отличается тем, что указанное соединение формулы (VI) получают исходя из соединения формулы (VII) которое используют для осуществления реакции с соединением формулы (VIII) где X представляет собой атом галогена, мезилатную группу или тозилатную группу, в присутствии основания, в органическом растворителе, с получением соединения формулы (IX) которое гидролизировали посредством действия основания в смеси органического растворителя и воды с образованием соединения формулы (VI) в соответствии со способом, описанным в данном документе ранее. Настоящее изобретение по аналогии относится к способу синтеза ивабрадина исходя из соединения формулы (VI), который отличается тем, что указанное соединение формулы (VI) получают исходя из соединения формулы (X) которое используют для осуществления реакции с хлороводородным соединением формулы (III) в присутствии основания, в органическом растворителе, с получением соединения формулы (IX) которое гидролизировали посредством действия основания в смеси органического растворителя и воды с образованием соединения формулы (VI) в соответствии со способом, описанным в данном документе ранее. Среди оснований, которые могут быть использованы для осуществления реакции алкилирования между соединением формулы (VII) и соединением формулы (VIII) или реакции алкилирования между соединением формулы (X) и хлороводородным соединением формулы (III), могут быть указаны без представления ограничений, неорганические основания, такие как карбонат калия, карбонат натрия, карбонат цезия, гидрокарбонат калия и гидрокарбонат натрия, и органические основания, такие как, триэтиламин, диизопропилэтиламин и пиридин. Основание, которое предпочтительно используют для осуществления реакции алкилирования между соединением формулы (VII) и соединением формулы (VIII) или реакции алкилирования между соединением формулы (X) и хлороводородным соединением формулы (III), представляет собой триэтиламин. Среди органических растворителей, которые могут быть использованы для осуществления реакции алкилирования между соединением формулы (VII) и соединением формулы (VIII) или реакции алкилирования между соединением формулы (X) и хлороводородным соединением формулы (III), могут быть указаны без представления ограничений, ацетонитрил, ацетон, метилэтилкетон (МЭК), диметилформамид (ДМФА), N-метилпирролидон (NMP) и диметилсульфоксид (ДМСО). Органический растворитель, который предпочтительно используют для осуществления реакции алкилирования между соединением формулы (VII) и соединением формулы (VIII) или реакции алкилирования между соединением формулы (X) и хлороводородным соединением формулы (III), представляет собой ацетонитрил. Реакцию алкилирования между соединением формулы (VII) и соединением формулы (VIII) или реакцию алкилирования между соединением формулы (X) и хлороводородным соединением формулы (III) предпочтительно осуществляют при температуре между 20 и 100C включительно. Среди оснований, которые могут быть использованы для осуществления гидролиза соединения формулы (IX) с образованием соединения формулы (VI), могут быть указаны без представления ограничений гидроксид калия, гидроксид натрия, гидроксид лития и гидроксид бария. Основание, которое предпочтительно используют для осуществления гидролиза соединения формулы (IX) с образованием соединения формулы (VI), представляет собой гидроксид натрия. Органический растворитель, который предпочтительно используют для осуществления гидролиза соединения формулы (IX) с образованием соединения формулы (VI), представляет собой спиртовой растворитель. Среди спиртовых растворителей, которые могут быть использованы для осуществления гидролиза соединения формулы (IX) с образованием соединения формулы (VI), могут быть указаны без представления ограничений, метанол, этанол, изопропанол и бутанол. Спиртовой растворитель, который предпочтительно используют для осуществления гидролиза соединения формулы (IX) с образованием соединения формулы (VI), представляет собой этанол. Гидролиз соединения формулы (IX) с образованием соединения формулы (VI) предпочтительно осуществляют при температуре между 0 и 110C включительно. Соединения формул (VI), (IX) и (X), а также 3-(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен 7-ил]метил(метил)амино]-1-пропанол,этилN-2-[2-(цианометил)-4,5-диметоксифенил]этил-аланинат оксалат и (2-2-[(3-гидроксипропил)амино]этил-4,5-диметоксифенил)ацетонитрил, представляют собой новые соединения для применения в качестве промежуточных соединений синтеза в химической или фармацевтической промышленности, в особенности, в синтезе ивабрадина, его аддитивных солей с фармацевтически приемлемой кислотой и его гидратов, и, таким образом, являются неотъемлемой частью настоящего изобретения. Список используемых сокращений: ДМФА: диметилформамид; ДМСО: диметилсульфоксид; ЯМР: ядерный магнитный резонанс; т.п.: температура плавления; ТГФ: тетрагидрофуран. ЯМР спектры были записаны на прибор Bruker при 400 МГц для протонный спектров и при 100 МГц для углеродных спектров. Химические сдвиги выражаются в м.д. (внутренний стандарт: ТМС). Следующие сокращения используются для описания пиков: синглет (s), дублет (d), дублет дублетов(dd), триплет (t), квадруплет (q), мультиплет (m). Примеры, поданные ниже, иллюстрируют изобретение. Способ получения А: N-[2-(3,4-диметоксифенил)этил]-2,2,2-трифторацетамид. К раствору 2-(3,4-диметоксифенил)этанамина (50 г, 276 ммоль) в 350 мл этилацетата перемешивали при комнатной температуре добавляли, по каплях, раствор трифторуксусного ангидрида (46.1 мл, 330 ммоль) в 40 мл этилацетата. После взаимодействия соединений в течение 1 ч при комнатной температуре, смесь гидролизировали с помощью 100 мл воды. Органическую фазу промывали с помощью смеси вода/триэтиламин (100 мл/38.5 мл) и 100 мл насыщенного водного раствора NaCl и затем сушили надMgSO4 и подвергали сушке с получением 65.8 г твердого вещества бежевого цвета, которое соответствует соединению. Выход: 86%; т.п.: 93C. 1Cq) - 157.5 м.д. (С=О, 2J(19F-13C)=37 Hz). Способ получения В: N-2-[2-(хлорметил)-4,5-диметоксифенил]этил-2,2,2-трифторацетамид. В трехгорлой колбе смешивали N-[2-(3,4-диметоксифенил)этил]-2,2,2-трифторацетамид (35 г, 126 ммоль) и 37%-ный водный формальдегид (776 мл, 1.014 моль) в 120 мл дихлорметана при 0C. К полученной двухфазной смеси медленно добавляли при 0C 345 мл 37%-ного водного раствора соляной кислоты и нагревали при 40C. После взаимодействия соединений в течение 3 ч смесь гидролизировали с помощью 250 мл воды и промывали водную фаза посредством дихлорметана (2100 мл). Органические фазы объединяли, сушили над MgSO4 и осуществляли сушку in vacuo с получением меренги бежевого цвета (38.2 г). Полученное вещество рекристаллизировали из толуола с получением 31.5 г порошка белого цвета, который соответствует целевому соединению. Выход: 77%; т.п.: 140C. 1 Способ получения С: N-2-[2-(цианометил)-4,5-диметоксифенил]этил-2,2,2-трифторацетамид. В трехгорлой колбе перемешивали при комнатной температуре суспензию цианида натрия (9.8 г,200 ммоль) в 160 мл ДМСО. К суспензии добавляли по каплях раствор N-2-[2-(хлорметил)-4,5 диметоксифенил]этил-2,2,2-трифторацетамида (26 г, 798 ммоль) в 80 мл ДМСО. После взаимодействия соединений в течение 1 ч 30 мин при комнатной температуре смесь гидролизировали с помощью 300 мл воды и смесь выделяли посредством дихлорметана (3150 мл). Органические фазы объединяли и промывали с помощью 10%-ного водного раствора NaOAc (150 мл) и насыщенного водного раствора NaCl(4150 мл), затем сушили их над MgSO4 и осуществляли сушку in vacuo. Полученное вещество рекристаллизировали из толуола (66 мл) с получением 12.8 г порошка белого цвета, который соответствует целевому соединению. Выход: 51%; т.п.: 131C. 1N-2-[2-(цианометил)-4,5-диметоксифенил]этил-2,2,2 трифторацетамида (20.5 г, 64.8 ммоль), этанола (160 мл), карбоната калия (13.2 г, 97.2 ммоль) и воды (40 мл). После взаимодействия соединений в течение 1 ч 30 мин смесь выделяли посредством добавления 200 мл дихлорметана и 100 мл насыщенного водного раствора NaCl. Водную фазу выделяли с помощью 100 мл дихлорметана. Органические фазы объединяли, промывали их с помощью насыщенного водного раствора NaCl (100 мл), сушили их над MgSO4 и осуществляли сушку in vacuo с получением 10 г масла желтого цвета, которое соответствует целевому соединению. Выход: 70%; т.п.:78C. 1[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]-Nметилметанамин гидрохлорида (20 г, 82 ммоль), триэтиламина (34.2 мл, 246 ммоль) и 3-бром-1 пропанола (14.8 г, 107 ммоль) в 100 мл ТГФ с 5%-ным ДМФА. После взаимодействия соединений в течение 24 ч смесь гидролизировали с помощью 80 мл воды и выделяли с помощью 80 мл дихлорметана. Органическую фазу промывали с помощью насыщенного водного раствора NaCl (560 мл), сушили надMgSO4 и осуществляли сушку с получением 23.7 г масла желтого цвета, которое соответствует целевому соединению. Выход: 96%. 1 Н ЯМР (CDCl3, 400 МГц): 1.66 м.д. (2 Н, m) - 2.31 м.д. (3H, s) - 2.50 до 2.70 м.д. (5 Н, m) - 3.21 м.д.(CH) - 134.8 м.д., 138.5 м.д., 140.4 м.д. и 149.9 м.д. (4 Cq). Способ получения F: 3-хлор-N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-Nметил-1-пропанамин. К смеси 3-(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)-амино]-1 пропанола (20.8 г, 78.4 ммоль) и триэтиламина (11 мл, 78.9 ммоль) в 200 мл дихлорметан добавляли при комнатной температуре 8.65 мл тионилхлорида (157 ммоль). После взаимодействия соединений в течение 3 ч при 40C смесь гидролизировали с помощью 150 мл воды и 30 мл водного раствора 1 н. NaOH. Значение рН водной фазы устанавливали на 10 с помощью водного раствора 10 н. NaOH и выделяли с помощью 50 мл дихлорметана. Органические фазы объединяли и промывали их с помощью насыщенного водного раствора Na2CO3 (100 мл), сушили их над MgSO4 и осуществляли сушку in vacuo с получением 19.8 г масла коричневого цвета, которое соответствует целевому соединению. Выход: 89%. 1 Н ЯМР (CDCl3, 400 МГц): 1.90 м.д. (2 Н, m) - 2.28 м.д. (3H, s) - 2.65 м.д. (3H, m) -2.68 м.д. (2 Н, m) 3.19 м.д. (1H, dd) - 3.54 м.д. (1H, m) - 3.56 м.д. (2 Н, t) - 3.78 м.д. (6 Н, s) - 6.62 м.д. (1H, s) - 6.66 м.д. (1H, s). 13 С ЯМР (CDCl3, 100 МГц): 30.2 м.д. (CH2) - 35.0 м.д. (CH2) - 40.6 м.д. (CH) - 42.6 м.д. (CH3) - 43.1 м.д. (CH2) - 54.8 м.д. (CH2) - 56.2 м.д. (CH3) - 56.3 (CH3) - 62.0 м.д. (CH2) -106.8 м.д. (CH) - 107.4 м.д. Способ получения G: 3-хлор-N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-Nметил-1-пропанамин оксалат. К раствору 3-хлор-N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-N-метил-1 пропанамина (19.8 г, 69.8 ммоль) в 60 мл этилацетата нагревали при температуре, которая устанавливается при кипячении с обратным холодильником, и добавляли раствор щавелевой кислоты (6.91 г, 76.7 ммоль) в 60 мл этанола. Осадок от смесь появлялся во время взаимодействия при нагревании с обратным холодильником. После охлаждения до комнатной температуры смесь фильтровали и промывали с помощью 20 мл этанола с получением 17.35 г порошка коричневого цвета, который соответствует целевому соединению. Выход: 67%; т.п.:154C. 1 Н ЯМР (ДМСО. 400 МГц): 2.14 м.д. (2 Н, m) - 2.75 м.д. (3H, s) - 2.88 м.д. (1 Н, dd) - 3.12 м.д. (2 Н,m) - 3.16 м.д. (1H, dd) - 3.28 м.д. (1 Н, dd) - 3.41 м.д. (1H, dd) - 3.69 до 3.75 м.д. (9 Н, m) - 6.79 м.д. (1H, s) 6.86 м.д. (1 Н, s) - 9.21 м.д. (2OH). 13 С ЯМР (ДМСО, 100 МГц): 26.8 м.д. (CH2) - 35.0 м.д. (CH2) - 37.5 м.д. (CH) - 40.1 м.д. (CH2) - 42.6 м.д. (CH2) - 53.1 м.д. (CH2) - 55.8 м.д. (CH3) - 55.9 м.д. (CH3) - 58.6 м.д. (CH2) - 107.6 м.д. (CH) - 108.0 м.д.(CH) - 134.2 м.д., 135.7 м.д., 149.3 м.д. и 150.2 м.д. (4 Cq)-164.4 м.д. (С=О). Способ получения Н: этил N-2-[2-(цианометил)-4,5-диметоксифенил]этилаланинат оксалат. Смесь [2-(2-аминоэтил)-4,5-диметоксифенил]ацетонитрила (6.5 г, 29.5 ммоль) и этилакрилата (3.9 мл, 36 ммоль, 1.2 экв.) в 120 мл этанола перемешивали в течение 20 ч при комнатной температуре. Реакционную смесь подвергали сушке in vacuo; затем неочищенную реакционную смесь растворяли в смеси этилацетата (133 мл) и этанола (13 мл) и нагревали с обратным холодильником в присутствии щавелевой кислоты (2.52 г, 28 ммоль, 0.95 экв.). Осадок от смеси появлялся во время взаимодействия при температуре, которая устанавливается при кипячении с обратным холодильником. После охлаждения до комнатной температуры смесь фильтровали и промывали с помощью 19 мл этилацетата с получением порошка белого цвета (9 г), который соответствует целевому соединению. Выход: 74%; т.п.: 218C. 1H ЯМР (CDCl3, 400 МГц): 1.19 м.д. (3H, t) - 2.74 м.д. (2 Н, t) - 2.88 м.д. (2 Н, m) -3.06 м.д. (2 Н, m) 3.17 м.д. (2 Н, t) - 3.74 м.д. (3H, s) - 3.75 м.д. (3H, s) - 3.93 м.д. (2 Н, s) - 4.09 м.д. (2 Н, квадруплет) - 6.88 м.д. (1H, s) - 6.97 м.д. (1 Н, s). 13 С ЯМР (CDCl3, 100 МГц): 14.00 м.д. (CH3) - 19.86 м.д. (CH2) - 28.28 м.д. (CH2) - 30.39 м.д. (CH2) 42.25 м.д. (CH2) - 47.19 м.д. (CH2) - 55.60 м.д. (CH3) - 55.62 м.д. (CH3) -60.53 м.д. (CH2) - 113.13 м.д. (CH) 113.74 м.д. (CH) - 119.36 м.д. (Cq) - 121.40 м.д. (Cq) - 127.70 м.д. (Cq) - 147.73 м.д. (Cq) - 148.42 м.д. (Cq) 164.65 м.д. (Cq) - 170.29 м.д. (Cq). Способ получения I: (2-2-[(3-гидроксипропил)амино]этил-4,5-диметоксифенил)ацетонитрил. К суспензии 10.8 г NaBH4 (284 ммоль, 11 экв.) в 110 мл ТГФ добавляли, в несколько разных моментов на протяжении периода времени, этил N-2-[2-(цианометил)-4,5-диметоксифенил]этилаланинат оксалат (10.6 г, 25.9 ммоль). Перемешивание длилось 30 мин при комнатной температуре, затем 23.1 мл метанола (570 ммоль, 22 экв.) добавляли по каплях. Смесь нагревали в течение 16 ч при 60C и затем гидролизировали с помощью 100 мл 5 н. соляной кислоты. Затем добавляли 100 мл дихлорметана и 200 мл деминерализованной воды. После разделения фаз добавляли 50 мл раствора 10 н. гидроксида натрия(pH10) к водной фазе, и выделение осуществляли с помощью 370 мл дихлорметана. Органические фазы объединяли и промывали с помощью 275 мл насыщенного водного раствора NaCl, затем сушили над MgSO4. После осуществления сушки in vacuo получали 6.15 г бесцветного масла, которое соответствует целевому соединению. Выход: 85%. 1(CH) - 119.54 м.д. (Cq) - 120.92 м.д. (Cq) - 131.27 м.д. (Cq) - 147.03 м.д. (Cq) - 148.23 м.д. (Cq). Способ получения J: (2-2-[(3-хлорпропил)амино]этил-4,5-диметоксифенил)ацетонитрил. К смеси 2-(2-[(3-гидроксипропил)амино]этил-4,5-диметоксифенил)ацетонитрила (1.7 г, 6.1 ммоль) и триэтиламина (2.5 мл, 18.3 ммоль, 3 экв.) в 16 мл дихлорметана посредством вливания по каплях добавляли раствор, который состоит из 885 мкл тионилхлорида (12.2 ммоль, 2 экв.) и 1 мл дихлорметана. Смесь нагревали в течение 2 ч при 40C и затем, как только смесь охлаждалась до комнатной температуры, гидролизировали с помощью 15 мл деминерализованной воды. После перемешивания в течение ночи при комнатной температуре добавляли 3 мл водного раствора 10 н. гидроксида натрия (pH10). После разделения фаз органическую фазу удаляли и выдерживали. Водную фазу выделяли помощью 20 мл дихлорметана. Органические фазы объединяли и промывали с помощью 25 мл насыщенного водного раствора NaCl и затем сушили над MgSO4. После осуществления сушки in vacuo получали 1.5 г масла красного цвета, которое соответствует целевому соединению.H ЯМР (CDCl3, 400 МГц): 1.91 м.д. (2 Н, квинтуплет) - 2.75 м.д. (2 Н, m) - 2.77 м.д. (2 Н, m) - 2.83 м.д. (2 Н, m) - 3.59 м.д. (2 Н, t) - 3.71 м.д. (2 Н, s) - 3.86 м.д. (3H,s) - 3.87 м.д. (3H, s) - 6.71 м.д. (1H, s) - 6.84 м.д. (1 Н, s). 13 С ЯМР (CDCl3, 100 МГц): 21.11 м.д. (CH2) - 32.73 м.д. (CH2) - 33.16 м.д. (CH2) - 43.04 м.д. (CH2) 46.88 м.д. (CH2) - 50.45 м.д. (CH2) - 56.03 м.д. (CH3) - 56.08 м.д. (CH3) -112.17 м.д. (CH) -113.08 м.д. (CH) 118.20 м.д. (CH) - 120.02 м.д. (Cq) - 130.19 м.д. (Cq) -147.72 м.д. (Cq) - 148.74 м.д. (Cq). Пример 1: 2-[2-(3-(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропиламино)этил]-4,5-диметоксифенилацетонитрил. Первый вариант. Смесь 3-хлор-N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-N-метил-1 пропанамин оксалата (15 г, 40.1 ммоль) и 85 мл водного раствора 1 н. NaOH в 150 мл дихлорметана перемешивали в течение 1 ч при комнатной температуре. Смесь разделяли и органическую фазу сушили надMgSO4 и подвергали сушке in vacuo с получением 11.3 г масла оранжевого цвета, которое соответствует 3-хлор-N-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил-N-метил-1-пропанамину. Полученное выше вещество перемешивали при комнатной температуре в присутствии йодида калия (1.46 г,8.78 ммоль) в 200 мл ацетонитрила. К полученной смеси последовательно добавляли триэтиламин (5.6 мл, 40.2 ммоль) и затем [2-(2-аминоэтил)-4,5-диметоксифенил]ацетонитрил (8.82 г, 40.1 ммоль) растворяли в 50 мл ацетонитрила. После взаимодействия соединений в течение 24 ч при 60C добавляли 150 мл воды и смесь выделяли посредством добавления дихлорметана (150 мл). Органическую фазу промывали с помощью 185 мл воды и 15 мл 37%-ного водного раствора соляной кислоты. Водную фазу собирали,добавляли 185 мл насыщенного водного раствора NaHCO3 и полученную фазу выделяли с помощью 150 мл дихлорметана. Органическую фазу сушили над MgSO4 и подвергали сушке с получением 14.1 г масла оранжевого цвета, которое соответствует целевому соединению. Выход: 75%. Второй вариант. Смесь (2-2-[(3-хлорпропил)амино]этил-4,5-диметоксифенил)ацетонитрила (870 мг, 2.93 ммоль),1-[(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]-N-метилметанамин гидрохлорида (714 мг,2.93 ммоль) и триэтиламина (1.25 мл, 8.97 ммоль, 3 экв.) в 10 мл ацетонитрила нагревали при температуре, которая устанавливается при кипячении с обратным холодильником, в течение 6 ч. После охлаждения до комнатной температуры смесь фильтровали in vacuo. Фильтрат сушили над MgSO4 и подвергали сушке in vacuo с получением меренги коричневого цвета (0.9 г, 66%), которая соответствует целевому соединению. Выход: 66%. 1(CH) - 113.6 м.д. (CH) - 119.0 м.д., 120.8 м.д., 126.7 м.д., 134.1 м.д., 134.2 м.д., 148.5 м.д., 149.2 м.д., 149.9 м.д. и 150.9 м.д. (9 Cq). Пример 2: 2-[2-(3-(7S)-3,4-диметоксибицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропиламино)этил]-4,5-диметоксифенилуксусная кислота. 1.3 г полученного вещества из примера 1 (2.77 ммоль) и 1.11 г пеллет NaOH (27.7 ммоль) нагревали при температуре, которая устанавливается при кипячении с обратным холодильником, в 6.2 мл этанола и 14.6 мл воды. После нагревания с обратным холодильником в течение 6 ч реакционную смесь гидролизировали с помощью 10 мл воды и добавляли 20 мл дихлорметана. Органическую фазу выделяли с помощью 20 мл воды. Водные фазы объединяли и промывали с помощью 15 мл дихлорметана. ЗначениеpH промытой водной фазы устанавливали на 7 с помощью 37%-ного водного раствора соляной кислоты и затем осуществляли сушку in vacuo. Полученное твердое вещество желтого цвета разбавляли в 40 мл ацетона при комнатной температуре. Полученную суспензию фильтровали in vacuo. Фильтрационные жидкости подвергали сушке in vacuo с получением 0.7 г меренги желтого цвета, которая соответствует целевому соединению. Выход: 52%. 1 м.д. (8 С q) - 176.9 м.д. (С=О). Пример 3: 3-3-(7S)-3,4-диметоксицикло[4.2.0]окта-1,3,5-триен-7-ил]метил(метил)амино]пропил-7,8-диметокси-1,3,4,5-тетрагидро-2H-3-бензазепин-2-он. В трехгорлой колбе смешивали 0.7 г полученного вещества из примера 2 (1.44 ммоль) и 0.4 мл триэтиламина (2.88 ммоль) в 14 мл дихлорметана. Смесь охлаждали при 5C и добавляли по каплях 0.16 мл тионилхлорида (2.16 ммоль). Перемешивали в течение 1 ч при 30C и затем смесь гидролизировали с помощью 12 мл водного раствора 1 н. NaOH. Органическую фазу последовательно промывали с помощью 10 мл воды и затем 10 мл насыщенного водного раствора NaCl. Органическую фазу сушили надMgSO4 и осуществляли сушку in vacuo с получением 0.5 г масла оранжевого цвета, которое соответствует целевому соединению. Выход: 74%. 1(CH) - 112.9 м.д. (CH) - 122.4 м.д., 126.4 м.д., 126.4 м.д., 133.8 м.д., 146.1 м.д., 146.8 м.д., 148.4 м.д. и 148.9 м.д. (8 С q) - 171.2 м.д. (С=О). ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ синтеза ивабрадина формулы (I) который отличается тем, что соединение формулы (VI) подвергают реакции лактамизации в присутствии связующего компонента, выбранного из оксалилхлорида,тионилхлорида,N,N-дициклогексилкарбодиимида(DCC),1-этил-3-(3 диметиламинопропил)карбодиимида (EDCI), N,N-карбонилдиимидазола (CDI), циклического ангидрида 1-пропанфосфоновой кислоты (Т 3 Р) и 1-(метилсульфонил)-1 Н-бензотриазола, и основания, в органическом растворителе, с получением ивабрадина формулы (I), который может быть превращен в его аддитивные соли с фармацевтически приемлемой кислотой, выбранной из соляной, бромисто-водородной,серной, фосфорной, уксусной, трифторуксусной, молочной, пировиноградной, малоновой, янтарной,глутаровой, фумаровой, винной, малеиновой, лимонной, аскорбиновой, щавелевой, метансульфоновой,бензолсульфоновой и камфорной кислот, и в его гидраты. 2. Способ синтеза по п.1, который отличается тем, что связующий компонент, который используют для осуществления реакции лактамизации соединения формулы (VI), представляет собой тионилхлорид. 3. Способ синтеза по п.2, который отличается тем, что количество тионилхлорида, которое используют для осуществления реакции лактамизации соединения формулы (VI), находится в интервале 1-5 экв. включительно. 4. Способ синтеза по любому из пп.1-3, который отличается тем, что основание, которое используют для осуществления реакции лактамизации соединения формулы (VI), выбирают из триэтиламина,диизопропилэтиламина и пиридина. 5. Способ синтеза по п.4, который отличается тем, что основание, которое используют для осуществления реакции лактамизации соединения формулы (VI), представляет собой триэтиламин. 6. Способ синтеза по любому из пп.1-5, который отличается тем, что органический растворитель,который используют для осуществления реакции лактамизации соединения формулы (VI), выбирают из дихлорметана, тетрагидрофурана, ацетонитрила, ацетона и толуола. 7. Способ синтеза по п.6, который отличается тем, что органический растворитель, который используют для осуществления реакции лактамизации соединения формулы (VI), представляет собой дихлорметан. 8. Способ синтеза по любому из пп.1-7, который отличается тем, что реакцию лактамизации соединения формулы (VI) осуществляют при температуре между 0 и 40C включительно. 9. Способ синтеза по п.1, который отличается тем, что соединение формулы (VI) получают исходя из соединения формулы (VII) которое используют для осуществления реакции с соединением формулы (VIII) в которой X представляет собой атом галогена, мезилатную группу или тозилатную группу,в присутствии основания, в органическом растворителе, с получением соединения формулы (IX) которое гидролизируют посредством действия основания в смеси органического растворителя и воды, с образованием соединения формулы (VI) 10. Способ синтеза по п.1, который отличается тем, что соединение формулы (VI) получают исходя из соединения формулы (X) которое используют для осуществления реакции с хлороводородным соединением формулы (III) в присутствии основания, в органическом растворителе, с получением соединения формулы (IX) которое гидролизируют посредством действия основания в смеси органического растворителя и воды, с образованием соединения формулы (VI) 11. Способ синтеза по любому п.9 или 10, который отличается тем, что основание, которое используют для осуществления реакции алкилирования между соединением формулы (VII) и соединением формулы (VIII) или реакции алкилирования между соединением формулы (X) и хлороводородным соединением формулы (III), выбирают из карбоната калия, карбоната натрия, карбоната цезия, гидрокарбоната калия, гидрокарбоната натрия, триэтиламина, диизопропилэтиламина и пиридина. 12. Способ синтеза по п.11, который отличается тем, что основание, которое используют для осуществления реакции алкилирования между соединением формулы (VII) и соединением формулы (VIII) или реакции алкилирования между соединением формулы (X) и хлороводородным соединением формулы (III), представляет собой триэтиламин. 13. Способ синтеза по любому из пп.9-12, который отличается тем, что органический растворитель,который используют для осуществления реакции алкилирования между соединением формулы (VII) и соединением формулы (VIII) или реакции алкилирования между соединением формулы (X) и хлороводородным соединением формулы (III), выбирают из ацетонитрила, ацетона, метилэтилкетона (МЭК), диметилформамида (ДМФА), N-метилпирролидона (NMP) и диметилсульфоксида (ДМСО). 14. Способ синтеза по п.13, который отличается тем, что органический растворитель, который используют для осуществления реакции алкилирования между соединением формулы (VII) и соединением формулы (VIII) или реакции алкилирования между соединением формулы (X) и хлороводородным соединением формулы (III), представляет собой ацетонитрил. 15. Способ синтеза по любому из пп.9-14, который отличается тем, что реакцию алкилирования между соединением формулы (VII) и соединением формулы (VIII) или реакцию алкилирования между соединением формулы (X) и хлороводородным соединением формулы (III) осуществляют при температуре между 20 и 100C включительно. 16. Способ синтеза по любому из пп.9-15, который отличается тем, что основание, которое используют для осуществления гидролиза соединения формулы (IX) с образованием соединения формулы (VI),выбирают из гидроксида калия, гидроксида натрия, гидроксида лития и гидроксида бария. 17. Способ синтеза по п.16, который отличается тем, что основание, которое используют для осуществления гидролиза соединения формулы (IX) с образованием соединения формулы (VI), представляет собой гидроксид натрия. 18. Способ синтеза по любому из пп.9-17, который отличается тем, что органический растворитель,который используют для осуществления гидролиза соединения формулы (IX) с образованием соединения формулы (VI), представляет собой спиртовой растворитель. 19. Способ синтеза по п.18, который отличается тем, что спиртовой растворитель, который используют для осуществления гидролиза соединения формулы (IX) с образованием соединение формулы (VI),выбирают из метанола, этанола, изопропанола и бутанола. 20. Способ синтеза по п.19, который отличается тем, что спиртовой растворитель, который используют для осуществления гидролиза соединения формулы (IX) с образованием соединения формулы (VI),представляет собой этанол. 21. Способ синтеза по любому из пп.9-20, который отличается тем, что гидролиз соединения формулы (IX) с образованием соединения формулы (VI) осуществляют при температуре между 0 и 110C включительно. 22. Соединение формулы (VI)
МПК / Метки
МПК: C07D 223/16, C07C 255/33, C07C 211/09
Метки: фармацевтически, аддитивных, ивабрадина, способ, кислотой, синтеза, приемлемой, солей
Код ссылки
<a href="https://eas.patents.su/11-22774-sposob-sinteza-ivabradina-i-ego-additivnyh-solejj-s-farmacevticheski-priemlemojj-kislotojj.html" rel="bookmark" title="База патентов Евразийского Союза">Способ синтеза ивабрадина и его аддитивных солей с фармацевтически приемлемой кислотой</a>
Новый способ синтеза ивабрадина и его аддитивных солей с фармацевтически приемлемой кислотой

Номер патента: 19373
Опубликовано: 31.03.2014
Авторы: Пегльон Жан-Луи, Кеньяр Паскаль
МПК: C07D 223/16, A61K 31/55, A61P 9/00...
Метки: кислотой, ивабрадина, приемлемой, солей, аддитивных, новый, фармацевтически, синтеза, способ
Формула / Реферат:
1. Способ синтеза ивабрадина формулы (I)отличающийся тем, что соединение формулы (VI)взаимодействует с тиолом в органическом растворителе с получением гемитиоацеталя формулы (VII)где R представляет собой замещенную или незамещенную, линейную или разветвленную алкильную группу, замещенную или незамещенную арильную группу, замещенную или незамещенную бензильную группу или группу CH2CO2Et,который подвергают реакции циклизации для получения...
Способ синтеза ивабрадина и его солей присоединения с фармацевтически приемлемой кислотой

Номер патента: 19465
Опубликовано: 31.03.2014
Авторы: Серкиз Бернар, Лерестиф Жан-Мишель, Пеглион Жан-Луи, Дессинже Эмея, Лекув Жан-Пьер
МПК: A61P 9/06, A61K 31/55, A61P 9/10...
Метки: кислотой, фармацевтически, синтеза, присоединения, ивабрадина, приемлемой, способ, солей
Формула / Реферат:
1. Способ синтеза соединения формулы (VI) в его рацемической или оптически активной формегде А представляет собой Н2С-СН2 или НС=СН, который отличается тем, что соединение формулы (VII) в рацемической или оптически активной формегде X представляет собой атом галогена, мезилатную группу или тозилатную группу, подвергают реакции алкилирования с соединением формулы (VIII)где А имеет значения, указанные выше, в присутствии основания в органическом...
Новый способ получения функционализированных бензоциклобутенов и их применение в синтезе ивабрадина и его аддитивных солей с фармацевтически приемлемой кислотой

Номер патента: 17503
Опубликовано: 30.01.2013
Авторы: Пегльон Жан-Луи, Шомонте Манон, Пиккарди Рикардо, Бодуан Оливье, Одик Никола
МПК: C07C 213/02, C07C 213/08, C07C 213/10...
Метки: ивабрадина, получения, солей, новый, аддитивных, применение, приемлемой, синтезе, функционализированных, способ, фармацевтически, кислотой, бензоциклобутенов
Формула / Реферат:
1. Способ получения соединений формулы (IV)где R1, R2, R3 и R4, которые могут быть идентичными или различными, каждый, представляют собой атом водорода, линейную или разветвленную (C1-C6)алкильную группу, линейную или разветвленную (C1-C6)алкоксигруппу, атом фтора, атом хлора, защищенную аминогруппу, защищенную гидроксильную группу, алкоксикарбонильную группу, в которой алкоксильная группа является линейной или разветвленной (C1-C6), или...
Способ синтеза ивабрадина и его солей присоединения с фармацевтически приемлемой кислотой

Номер патента: 16405
Опубликовано: 30.04.2012
Авторы: Серки Бернар, Пеглион Жан-Луис, Лекув Жан-Пьер, Дессинье Эмея, Лерестиф Жан-Мишель
МПК: C07D 223/16, C07C 47/47
Метки: солей, способ, синтеза, ивабрадина, фармацевтически, приемлемой, кислотой, присоединения
Формула / Реферат:
1. Способ синтеза соединения формулы (VI) в рацемической или оптически активной формегде А представляет собой Н2С-СН2 или НС=СН,который отличается тем, что соединение формулы (VII) в рацемической или оптически активной формеподвергают реакции восстановительного аминирования с соединением формулы (VIII)где А имеет значение, указанное выше, в присутствии восстановителя,в органическом растворителе или смеси органических растворителей.2. Способ...
Новый способ синтеза ивабрадина и его солей присоединения с фармацевтически приемлемой кислотой

Номер патента: 16353
Опубликовано: 30.04.2012
Авторы: Серки Бернар, Дессинье Эмея, Пеглион Жан-Луи
МПК: C07D 223/16
Метки: присоединения, синтеза, фармацевтически, солей, кислотой, новый, приемлемой, способ, ивабрадина
Формула / Реферат:
1. Способ синтеза соединения формулы (VIII) в рацемической или оптически активной формеотличающийся тем, что соединение формулы (II) в рацемической или оптически активной формереагирует с соединением формулы (IX)в присутствии соли переходного металла или лантаноида, в растворителе, для получения соединения формулы (X) в рацемической или оптически активной формекоторое преобразуют в соединение формулы (VIII) при действии донора водорода.2. Способ...
Предыдущий патент: Дезинфицирующий состав
Следующий патент: Устойчивый к смещению микроэлектрод, пучок микроэлектродов и массив микроэлектродов для имплантации в мягкую ткань человека или животного
Случайный патент: Шина с коронной зоной, содержащей слой резиновой смеси с очень высоким модулем упругости