Способ и промежуточные соединения для получения ингибиторов интегразы







Формула / Реферат
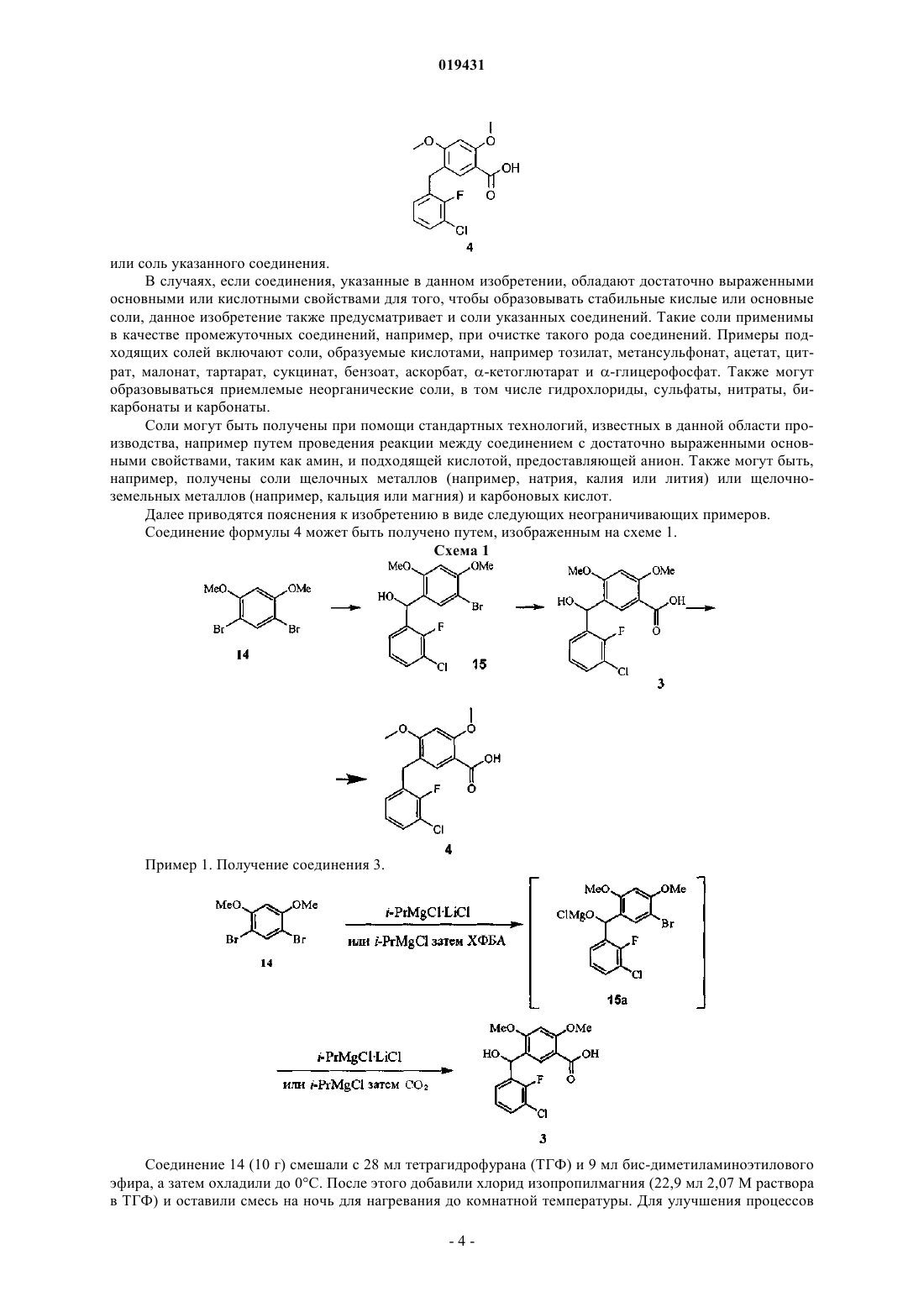
1. Способ получения соединения формулы 4
или соли указанного соединения из соединения формулы 15
или соли указанного соединения путем проведения стадий замещения атома брома карбоксильной группой и замещения гидроксильной группы атомом водорода.
2. Способ по п.1, отличающийся тем, что соединение формулы 4 или его соль получают путем металлирования соединения формулы 15 или его соли и обработки диоксидом углерода с получением соединения формулы 3
или соли указанного соединения, после чего соединение 3 преобразуют путем замены гидроксильной группы на атом водорода с получением соединения формулы 4.
3. Способ по п.2, отличающийся тем, что соединение формулы 15 или его соль представляет собой соль формулы 15а
4. Способ по п.1, отличающийся тем, что соединение формулы 15 преобразуют в соединение формулы 16
путем замены гидроксильной группы на атом водорода с получением соединения формулы 16, которое затем металлируют и обрабатывают диоксидом углерода с получением соединения формулы 4.
5. Соединение формулы 15
или соль указанного соединения.
6. Соединение по п.5, которое представляет собой соль формулы 15а
7. Способ получения соединения формулы 15
или соли указанного соединения, включающий преобразование соответствующего соединения формулы 14
в соединение формулы 15 или соль указанного соединения, причем соединение формулы 14 преобразуют в соединение формулы 15 или соль указанного соединения путем металлирования соединения формулы 14 и обработки его 3-хлор-2-фторбензальдегидом с получением соединения формулы 15 или соли указанного соединения.
8. Способ получения соединения формулы 3
или соли указанного соединения, согласно которому соединение формулы 15 преобразуют в соединение формулы 3 путем металлирования соединения формулы 15 и обработки его диоксидом углерода с получением соединения формулы 3.
9. Соединение формулы 16
Текст
СПОСОБ И ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ ДЛЯ ПОЛУЧЕНИЯ ИНГИБИТОРОВ ИНТЕГРАЗЫ Согласно изобретению предложены способы синтеза и синтезированные промежуточные соединения, которые можно использовать для получения соединений 4-оксихинолона, обладающих полезными свойствами в отношении ингибирования интегразы. Приоритет изобретения Заявка на данный патент испрашивает приоритет согласно 35 Кодексу Законов США 119 (е) на основании предварительной заявки на патент США 60/971395, поданной 11 сентября 2007 г., содержание которой полностью включено в настоящее изобретение. Уровень техники В публикации международной заявки на патент WO 2004/046115 представлены некоторые соединения 4-оксохинолона, применяемые в качестве ингибиторов интегразы ВИЧ. Указано, что данные соединения могут быть использованы в качестве агентов против ВИЧ. В публикации международной заявки на патент WO 2005/113508 представлены некоторые специфические кристаллические формы одного из таких соединений 4-оксохинолона, 6-(3-хлор-2 фторбензил)-1-[(S)-1-гидроксиметил-2-метилпропил]-7-метокси-4-оксо-1,4-дигидрохинолон-3 карбоновой кислоты. Указано, что данные специфические кристаллические формы обладают повышенной физической и химической устойчивостью по сравнению с другими физическими формами указанного соединения. В настоящее время существует потребность в усовершенствовании способов получения соединений 4-оксихинолона, представленных в публикациях международной заявки на патент WO 2004/046115 и международной заявки на патент WO 2005/113508. В частности, существует необходимость в создании новых способов синтеза, более простых и менее дорогостоящих в реализации, которые обеспечивают больший выход или которые исключают применение токсичных или дорогостоящих реагентов. Краткое описание изобретения В настоящем изобретении предложены новые способы синтеза и синтезированные промежуточные соединения, пригодные для получения соединений 4-оксихинолона, представленных в публикациях международной заявки на патент WO 2004/046115 и международной заявки на патент WO 2005/113508. Так, согласно одному из вариантов реализации настоящего изобретения предложен способ получения соединения формулы 4 или соли указанного соединения из соединения формулы 15 или соли указанного соединения путем проведения стадии замещения атома брома карбоксильной группой и стадии замещения гидроксильной группы атомом водорода. В другом варианте реализации данного изобретения предложено соединение формулы 15 или соль указанного соединения. Согласно другому варианту реализации данного изобретения предложен способ получения соединения формулы 15 или соли указанного соединения, включающий преобразование соответствующего соединения формулы 14 в соединение формулы 15 или соль указанного соединения. Согласно другому варианту реализации данного изобретения предложено соединение формулы 15 а которое может быть использовано в качестве промежуточного соединения для получения соединений 4 оксихинолона. Согласно другому варианту реализации данного изобретения предложено соединение формулы 16 которое может быть использовано в качестве промежуточного соединения для получения соединений 4 оксихинолона. Изобретение также охватывает другие способы синтеза и синтезированные промежуточные соединения, описанные в данном изобретении, которые могут быть использованы для получения соединений 4-оксихинолона. Подробное описание изобретения В настоящем описании используются следующие определения (если не указано иное): галоген обозначает фтор, хлор, бром или йод; алкил обозначает как линейные группы, так и группы с разветвленной цепью, но ссылка на индивидуальный радикал типа пропила относится только к радикалу с линейным строением, в то время как изомер с разветвленной цепью, такой как изопропил, упоминается отдельно. Специалистам очевидно, что соединение, имеющее хиральный центр, может существовать и может быть выделено в оптически активной и рацемической формах. Некоторые соединения могут проявлять полиморфизм. Следует понимать, что настоящее изобретение включает в себя способы получения любых рацемических, оптически активных, полиморфных, таутомерных или стереоизомерных форм или смеси указанных форм соединения, описанного в настоящем изобретении, так как в данной области техники хорошо известны способы получения оптически активных форм (например, путем разделения рацемической формы с использованием методик рекристаллизации, путем синтеза из оптически активных исходных веществ, путем хирального синтеза или путем хроматографического разделения с использованием хиральной неподвижной фазы). Перечисленные ниже конкретные и предпочтительные значения для радикалов, заместителей и диапазоны приводятся только для пояснений и не исключают других определенных значений или других значений в пределах указанных диапазонов для радикалов и заместителей. В частности, C1-C6-алкил может представлять собой метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, пентил, 3-пентил или гексил. Конкретным значением для Ra является метил. Конкретным значением для Rb является метил. Конкретным значением для Rc является 1-имидазолил. Конкретным значением для R является этил. Согласно одному из вариантов реализации соединение формулы 4 или соль указанного соединения получают путем металлирования соединения формулы 15 или его соли и обработки его диоксидом углерода с получением соединения формулы 3 или соли указанного соединения, после чего соединение формулы 3 преобразуют в соединение формулы 4. Соединение формулы 15 или соль указанного соединения могут представлять собой, например,соль формулы 15 а Согласно другому варианту реализации данного изобретения соединение формулы 15 преобразуют в соединение формулы 16 которое впоследствии подвергается металлированию и обработке диоксидом углерода с получением соединения формулы 4. Следует понимать, что стадия замещения атома брома карбоксильной группой представляет собой реакцию карбоксилирования. Данную стадию можно осуществить подходящим способом при помощи металлирования, например путем обработки хлоридом изопропилмагния или комплексом из хлорида изопропилмагния и хлорида лития, с последующей обработкой диоксидом углерода. Также следует понимать, что стадия замещения гидроксильной группы атомом водорода представляет собой реакцию дегидроксилирования. Данную стадию можно осуществить путем обработки триалкилсиланом, таким как триэтилсилан, особенно легко в присутствии трифторуксусной кислоты. В другом варианте реализации данного изобретения соединение формулы 15 или соль указанного соединения преобразуют в соединение формулы 3 или соль указанного соединения. Например, соединение формулы 15 или его соль могут быть преобразованы в соединение формулы 3 или его соль путем металлирования соединения формулы 15 или его соли (например, путем обработки хлоридом изопропилмагния) и обработки диоксидом углерода, получая соединение формулы 3 или его соль. В другом варианте реализации данного изобретения соединение формулы 3 или его соль преобразуют в соединение формулы 4 или соль указанного соединения. В случаях, если соединения, указанные в данном изобретении, обладают достаточно выраженными основными или кислотными свойствами для того, чтобы образовывать стабильные кислые или основные соли, данное изобретение также предусматривает и соли указанных соединений. Такие соли применимы в качестве промежуточных соединений, например, при очистке такого рода соединений. Примеры подходящих солей включают соли, образуемые кислотами, например тозилат, метансульфонат, ацетат, цитрат, малонат, тартарат, сукцинат, бензоат, аскорбат, -кетоглютарат и -глицерофосфат. Также могут образовываться приемлемые неорганические соли, в том числе гидрохлориды, сульфаты, нитраты, бикарбонаты и карбонаты. Соли могут быть получены при помощи стандартных технологий, известных в данной области производства, например путем проведения реакции между соединением с достаточно выраженными основными свойствами, таким как амин, и подходящей кислотой, предоставляющей анион. Также могут быть,например, получены соли щелочных металлов (например, натрия, калия или лития) или щелочноземельных металлов (например, кальция или магния) и карбоновых кислот. Далее приводятся пояснения к изобретению в виде следующих неограничивающих примеров. Соединение формулы 4 может быть получено путем, изображенным на схеме 1. Схема 1 Соединение 14 (10 г) смешали с 28 мл тетрагидрофурана (ТГФ) и 9 мл бис-диметиламиноэтилового эфира, а затем охладили до 0C. После этого добавили хлорид изопропилмагния (22,9 мл 2,07 М раствора в ТГФ) и оставили смесь на ночь для нагревания до комнатной температуры. Для улучшения процессов превращения добавляли дополнительный хлорид изопропилмагния, после чего добавляли 3-хлор-2 фторбензальдегид (ХФБА) (4,4 мл). После помешивания при комнатной температуре в течение 2 ч к смеси добавляли 38,6 г 14%-ного (мас.) раствора комплекса хлорид изопропилмагния с хлоридом лития в ТГФ. После перемешивания прикомнатной температуре в течение ночи в реакционную смесь добавлялиCO2 в виде пузырьков газа. Когда превращение было завершено, pH реакции изменили в кислую сторону(3) при помощи 2 М соляной кислоты. Фазы разделяли и экстрагировали органическую фазу при помощи этилацетата. Смешанные органические слои промывали насыщенным водным раствором хлорида натрия. Затем органическую фазу концентрировали, а продукт осаждали путем добавления МТБЭ (метил-трет-бутиловый эфир). Смесь фильтровали, конечный продукт высушивали на воздухе для получения соединения 3. 1 Н ЯМР (ДМСО-d6, 400 МГц)12,15 (br s, 1H), 7,81 (s, 1H), 7,42 (t, J=7,2 Гц, 1H), 7,26 (t, J=6,8 Гц,1H), 7,15 (t, J=7,8 Гц, 1H), 6,77 (s, 1H), 6,09 (d, J=4,7 Гц, 1H), 5,90 (d, J=4,9 Гц, 1H), 3,84 (s, 3H), 3,80 (s,3H). Пример 2. Получение соединения 4. К трифторуксусной кислоте (33,13 г), заранее охлажденной в ледяной бане, добавили триэтилсилан(6,83 г). К смеси добавили соединение 3 (10 г) при поддержании температуры ниже 15C. После перемешивания при комнатной температуре в течение 2 ч добавляли МТБЭ для осаждения продукта. Смесь фильтровали, а продукт промывали в дополнительном количестве МТБЭ. После высушивания выделили 9,12 г соединения 4. 1 Н ЯМР (ДМСО-d6, 400 МГц)12,11 (br s, 1H), 7,47 (s, 1H), 7,42-7,38 (m, 1H), 7,14-7,08 (m, 2H), 6,67(s, 1H), 3,87-3,84 (m, 8 Н). Существует и другой вариант получения соединения 4, приведенный ниже. К трифторуксусной кислоте (49,02 г), заранее охлажденной в ледяной бане, добавляли триэтилсилан (7,50 г). К смеси добавляли соединение 3 (14,65 г) при поддержании температуры ниже 15C. После перемешивания в течение 1 ч к смеси добавили раствор 17,63 г ацетата натрия в 147 мл метанола. Смесь нагревали в сосуде с обратным холодильником в течение 3 ч, а затем охладили до 0C. Раствор отфильтровали, продукт дополнительно отмыли метанолом. После высыхания выделили 12,3 г соединения 4 бис-Диметиламиноэтиловый эфир (2,84 г) растворяли в 42 мл ТГФ и охлаждали на ледяной бане. К смеси постепенно добавляли хлорид изопропилмагния (8,9 мл 2 М раствора в ТГФ), а затем медленно постепенно добавляли соединение 14 (5 г, раствор в 5 мл ТГФ). Смеси давали нагреться до комнатной температуры, а затем оставляли для перемешивания на ночь. Затем добавили 2,1 мл 3-хлор-2 фторбензальдегида. После перемешивания в течение примерно 1 ч смесь довели 2 н. HCl до pH7. Продукт экстрагировали этилацетатом и высушивали органическую фазу над сульфатом натрия. Растворитель заменяли на гептан для выпадения продукта в осадок, а затем внесли смесь гептана и метил-третбутилового эфира (4:1) для формирования суспензии. После фильтрации твердую фазу суспензировали в толуоле, отфильтровали и высушили под действием вакуума для получения соединения 15. 1 Н ЯМР (CD3CN, 400 МГц)7,47 (s, 1H), 7,41-7,35 (m, 2H), 7,15 (t, J=7,4 Гц, 1H), 6,66 (s, 1H), 6,21 Соединение 14 (5 г), хлорид изопропилмагния (8,9 мл 2 М раствора в ТГФ) и ТГФ (56 мл) смешали при комнатной температуре, а затем нагревали до 50C в течение 5 ч. После охлаждения до комнатной температуры и перемешивания в течение ночи добавляли по каплям 2,1 мл 3-хлор-2-фторбензальдегида для образования суспензии. После перемешивания в течение ночи из смеси путем фильтрования выделяли твердую фазу, которую отмывали в метил-трет-бутиловом эфире для получения соединения 15 а. Пример 5. Получение соединения 16. Триэтилсилан (1,2 мл) смешивали с трифторуксусной кислотой (2,3 мл), предварительно охлажденной на ледяной бане. К смеси добавляли соединение 15 (1,466 г), поддерживая при этом температуру ниже 5C. После перемешивания в течение примерно 2 ч к смеси добавляли лед для быстрого охлаждения. Продукт выделяли при помощи дихлорметана, органическую фазу отмывали водным растворомNaHCO3. Органическую фазу высушивали над Na2SO4 и концентрировали до сухого состояния. Продукт очищали при помощи колоночной хроматографии с силикагелем. Получили 1,341 г соединения 16. 1 Н ЯМР (CDCl3, 400 МГц)7,20 (t, J=7,0 Гц, 1H), 6,99-6,91 (m, 3H), 6,46 (s, 1H), 3,91 (s, 3H), 3,81 (s,5H). Соединение формулы 16 может быть карбоксилировано для получения соединения формулы 4 способом, аналогичным описанному в примере 1. Пример 6. Альтернативный вариант получения соединения формулы 3. Соединение 14 смешивали с безводной смесью тетрагидрофуран:диоксан (5:0,9), после чего перемешивали в атмосфере с высоким содержанием азота до получения однородного раствора. Раствор охлаждали до -3C и добавляли к нему 1,3 экв. i-PrMgClLiCl в ТГФ. Реакционную смесь перемешивали при 0C до образования моносоединения Гриньяра (определяется по анализу ВЭЖХ). После этого добавляли раствор 1,1 экв. 3-хлор-2-фторбензилальдегида в ТГФ. Смесь оставляли перемешиваться при 0C до образования соединения 15, что определяется посредством ВЭЖХ. Затем добавляли дополнительную порцию раствора i-PrMgClLiCl в ТГФ (2,5 экв.) и нагревали смесь до примерно 20C. После завершения процесса перехода ко второму промежуточному соединению Гриньяра реакционную смесь охлаждали до 3C. Затем при температуре примерно 5C в реакционную смесь вводили безводный CO2 (г). Реакционную смесь нагревали примерно до 20C. После завершения реакции карбоксилирования (по данным ВЭЖХ) реакционную смесь охлаждали приблизительно до 10C, а затем добавляли воду для быстрого охлаждения, после чего вносили концентрированную соляную кислоту для доведения pH до значения не больше 3. Затем реакционную смесь нагревали приблизительно до 20C. Фазы разделяли. Для органической фазы меняют растворитель на смесь изопропилового спирта и воды и полученную суспензию охлаждают примерно до 0C. Продукт отделяют фильтрованием, отмывают смесью изопропилового спирта и воды и высушивают при температуре около 40C для получения соединения 3. Пример 7. Альтернативный вариант получения соединения формулы 4. В реактор вносили трифторуксусную кислоту (10 экв.) и охлаждали ее до 0C. Поддерживая температуру на уровне менее 15C, добавляли триэтилсилан (15 экв.) и смесь тщательно перемешивали. К смеси по порциям добавляли соединение 3, при этом продолжали поддерживать температуру на уровне менее 15C. Когда, по данным ВЭЖХ, реакция находилась на завершающем этапе, соединение 4 осаждали за счет добавления раствора 5 экв. ацетата натрия в метаноле (13 объемов), при этом температура поддерживалась на уровне не более 45C. Суспензию нагревали в сосуде с обратным холодильником, затем перемешивали в течение 2-3 ч. После этого суспензию охлаждали приблизительно до 0C и перемешивали при этой температуре в течение 2-3 ч. Продукт отделяли фильтрованием, промывали метанолом и высушивали при температуре около 40C для получения соединения 4. Все публикации, патенты и патентные документы включены в настоящий патент посредством ссылки, как если бы каждый из указанных источников индивидуально был бы включен посредством ссылки. Изобретение было описано со ссылкой на различные конкретные и предпочтительные варианты реализации и методики. Тем не менее, следует понимать, что возможно внесение различных вариаций и модификаций при сохранении сущности и объема изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Способ получения соединения формулы 4 или соли указанного соединения из соединения формулы 15 или соли указанного соединения путем проведения стадий замещения атома брома карбоксильной группой и замещения гидроксильной группы атомом водорода. 2. Способ по п.1, отличающийся тем, что соединение формулы 4 или его соль получают путем металлирования соединения формулы 15 или его соли и обработки диоксидом углерода с получением соединения формулы 3 или соли указанного соединения, после чего соединение 3 преобразуют путем замены гидроксильной группы на атом водорода с получением соединения формулы 4. 3. Способ по п.2, отличающийся тем, что соединение формулы 15 или его соль представляет собой соль формулы 15 а 4. Способ по п.1, отличающийся тем, что соединение формулы 15 преобразуют в соединение формулы 16 путем замены гидроксильной группы на атом водорода с получением соединения формулы 16, которое затем металлируют и обрабатывают диоксидом углерода с получением соединения формулы 4. 5. Соединение формулы 15 или соль указанного соединения. 6. Соединение по п.5, которое представляет собой соль формулы 15 а 7. Способ получения соединения формулы 15 или соли указанного соединения, включающий преобразование соответствующего соединения формулы 14 в соединение формулы 15 или соль указанного соединения, причем соединение формулы 14 преобразуют в соединение формулы 15 или соль указанного соединения путем металлирования соединения формулы 14 и обработки его 3-хлор-2-фторбензальдегидом с получением соединения формулы 15 или соли указанного соединения. 8. Способ получения соединения формулы 3 или соли указанного соединения, согласно которому соединение формулы 15 преобразуют в соединение формулы 3 путем металлирования соединения формулы 15 и обработки его диоксидом углерода с получением соединения формулы 3. 9. Соединение формулы 16
МПК / Метки
МПК: C07C 43/23, C07D 215/56, C07C 43/225, C07C 41/30
Метки: получения, соединения, способ, промежуточные, интегразы, ингибиторов
Код ссылки
<a href="https://eas.patents.su/9-19431-sposob-i-promezhutochnye-soedineniya-dlya-polucheniya-ingibitorov-integrazy.html" rel="bookmark" title="База патентов Евразийского Союза">Способ и промежуточные соединения для получения ингибиторов интегразы</a>
Способ и промежуточные соединения для получения ненуклеозидных ингибиторов ревертазы вируса вич-1

Номер патента: 8149
Опубликовано: 27.04.2007
Авторы: Ким Дзиюн, Бусакка Карл А., Эрикссон Магнус С.
МПК: C07D 221/00, C07D 243/00, C07D 471/14...
Метки: соединения, ингибиторов, получения, ревертазы, вич-1, способ, вируса, промежуточные, ненуклеозидных
Формула / Реферат:
1. Способ получения соединения формулы I в которой R2 выбран из группы, включающей Н, F, Cl, C1-C4алкил, C3-C4диклоалкил и CF3, R4 обозначает Н или Me, R5 обозначает Н, Me или Et, при условии, что оба R4 и R5 одновременно не обозначают Me и, если R4 обозначает Me, то R5 не может обозначать Et, R11 обозначает Me, Et, циклопропил, пропил, изопропил или циклобутил и Q выбран из группы, включающей заключающийся в том, что (а) исходное соединение...
Трициклические соединения – ингибиторы интегразы вич, способ их получения (варианты), фармацевтическая композиция на их основе, способ ее получения и способы их использования в лечении болезней

Номер патента: 11399
Опубликовано: 27.02.2009
Авторы: Шахерер Лаура Н., Ким Чанг Ю., Фардис Мерайя, Джин Хаолун, Чен Сяову, Чен Джеймс М.
МПК: C07D 498/20, C07D 471/04, C07D 487/04...
Метки: фармацевтическая, ингибиторы, интегразы, вич, способы, соединения, композиция, способ, основе, лечении, трициклические, использования, варианты, получения, болезней
Формула / Реферат:
1. Соединение формулы где А1 независимо выбирают из ряда: C(R2)2, CR2OR, CR2OC(=O)R, C(=O), C(=S), CR2SR, C(=NR); А2 независимо выбирают из ряда: О, S, NR, C(R2)2-C(R3)2, C(R2)=C(R3), C(=O)C(R3)2, NR-C(R3)2, N=C(R3), N=N или C(R2)=N; Q означает N или CR4; L выбирают из ряда: связь, О, S, S-S, S(=O), S(=O)2, S(=O)2NR, NR, N-OR, C1-С12алкилен, замещенный C1-C12алкилен, С2-С12алкенилен, замещенный С2-С12алкенилен, С2-С12алкинилен, замещенный...
Нафтильные соединения, промежуточные соединения для их получения, применение нафтильных соединений, способ снижения холестерина

Номер патента: 1600
Опубликовано: 25.06.2001
Авторы: Палковиц Алан Д., Кроуелл Томас А., Брайант Генри У., Джонс Чарльз Д.
МПК: A61P 19/10, A61K 31/33, C07C 47/546...
Метки: соединения, промежуточные, нафтильных, нафтильные, соединений, получения, холестерина, снижения, применение, способ
Формула / Реферат:
1. Соединение формулы I где R1 является -Н, -ОН, -O(С1-С4-алкилом), -ОСОАr, где Аr является фенилом или замещенным фенилом, -O(СО)ОАr; где Аr является фенилом или замещенным фенилом, OCO(С1-С6-алкилом), -O(СО)O(С1-С6-алкилом) или -ОSО2(С4-С6-алкилом); R2 является -Н, -F, -Cl, -ОН, -О(С1-С4-алкилом), -ОСОАr, где Аr является фенилом или замещенным фенилом, -O(СО)ОАr, где Аr является фенилом или замещенным фенилом, -OCO(С1-С6-алкилом),...
Фармацевтические промежуточные соединения в синтезе ингибиторов ацетилхолинэстеразы (асе) и их применение

Номер патента: 15450
Опубликовано: 31.08.2011
Авторы: Верешшне Пандур Ангела, Мезеи Тибор, Шимиг Дьюла, Порч-Маккаи Марта, Луцакш Дьюла
МПК: C07C 269/04, C07D 209/42, C07C 271/22...
Метки: фармацевтические, применение, синтезе, ингибиторов, асе, ацетилхолинэстеразы, соединения, промежуточные
Формула / Реферат:
1. Соединения общей формулы (I)где R1представляет собой ароматическую углеводородную группу, содержащую одно или более ароматическое кольцо, которое может быть замещено одним или более одинаковыми или различными заместителями, выбранными из галогена, C1-C6-алкила, гидрокси, амино, или R1представляет собой прямую или разветвленную, насыщенную, алифатическую углеводородную группу, содержащую от 1 до 6 атомов углерода;R2 представляет собой прямую...
Конъюгаты соединения, содержащего сульфгидрильную группу, и производного жирной кислоты, способ получения конюгатов, промежуточные соединения для их получения, способы повышения абсорбции и пролонгированного сохранения в крови и тканях млекопитающего соединения, содержащего сульфгидрильную группу

Номер патента: 584
Опубликовано: 29.12.1999
Авторы: Икрами Хуссейн М., Шен Вей Чанг
МПК: C07H 19/048, C07D 213/70, A61K 31/44...
Метки: получения, пролонгированного, жирной, сохранения, повышения, соединения, содержащего, группу, абсорбции, способ, тканях, млекопитающего, сульфгидрильную, промежуточные, конъюгаты, конюгатов, производного, кислоты, способы, крови
Формула / Реферат:
1. Соединение общей формулы VI где Р является фрагментом соединения, содержащего сульфгидрильную группу, выбранного из группы, включающей пептиды, белки или олигонуклеотиды; R1 представляет собой водород, низший алкил или арил; R2 представляет собой фрагмент, содержащий липидную группу; а R3 представляет собой гидроксил, фрагмент, содержащий липидную группу или аминокислотную последовательность, включающую 1 или 2 аминокислоты и...
Предыдущий патент: Устройство для хранения и транспортировки конусовидных преформ пластмассовых контейнеров
Следующий патент: Жидкие композиции lh
Случайный патент: Устройство для закрепления пакера