Адамантилацетамиды как ингибиторы 11-бета гидроксистероиддегидрогеназы
Номер патента: 9710
Опубликовано: 28.02.2008
Авторы: Линдерс Йоаннес Теодорус Мария, Виллемсенс Густаф Хенри Мария, Яроскова Либуше, Гилиссен Роналдус Арнодус Хендрика Йозеф, Ван Дер Векен Луи Йозеф Элизабет, Ванхоф Грета Констанция Петер, Бюйк Кристоф Франсис Роберт Нестор






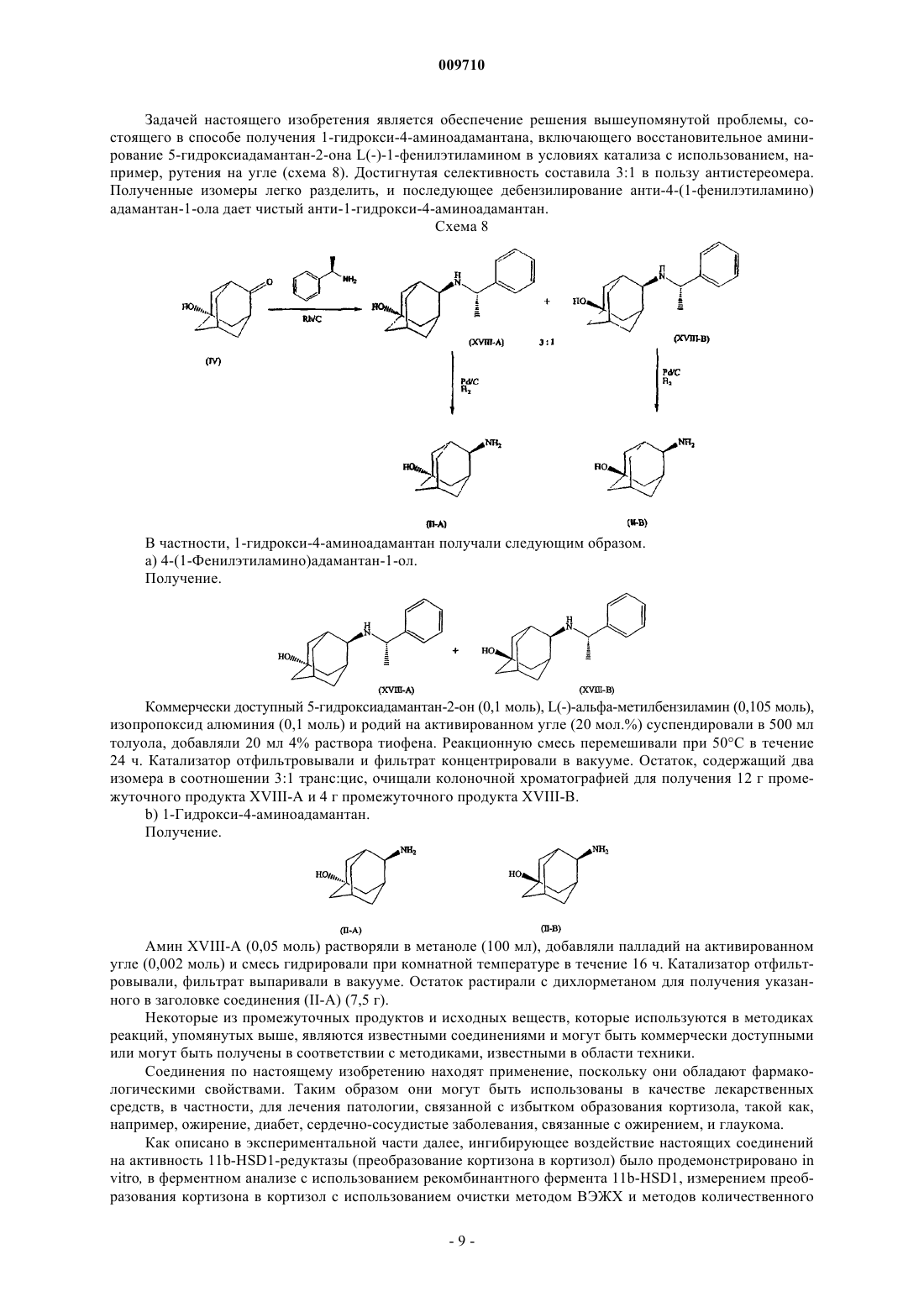
















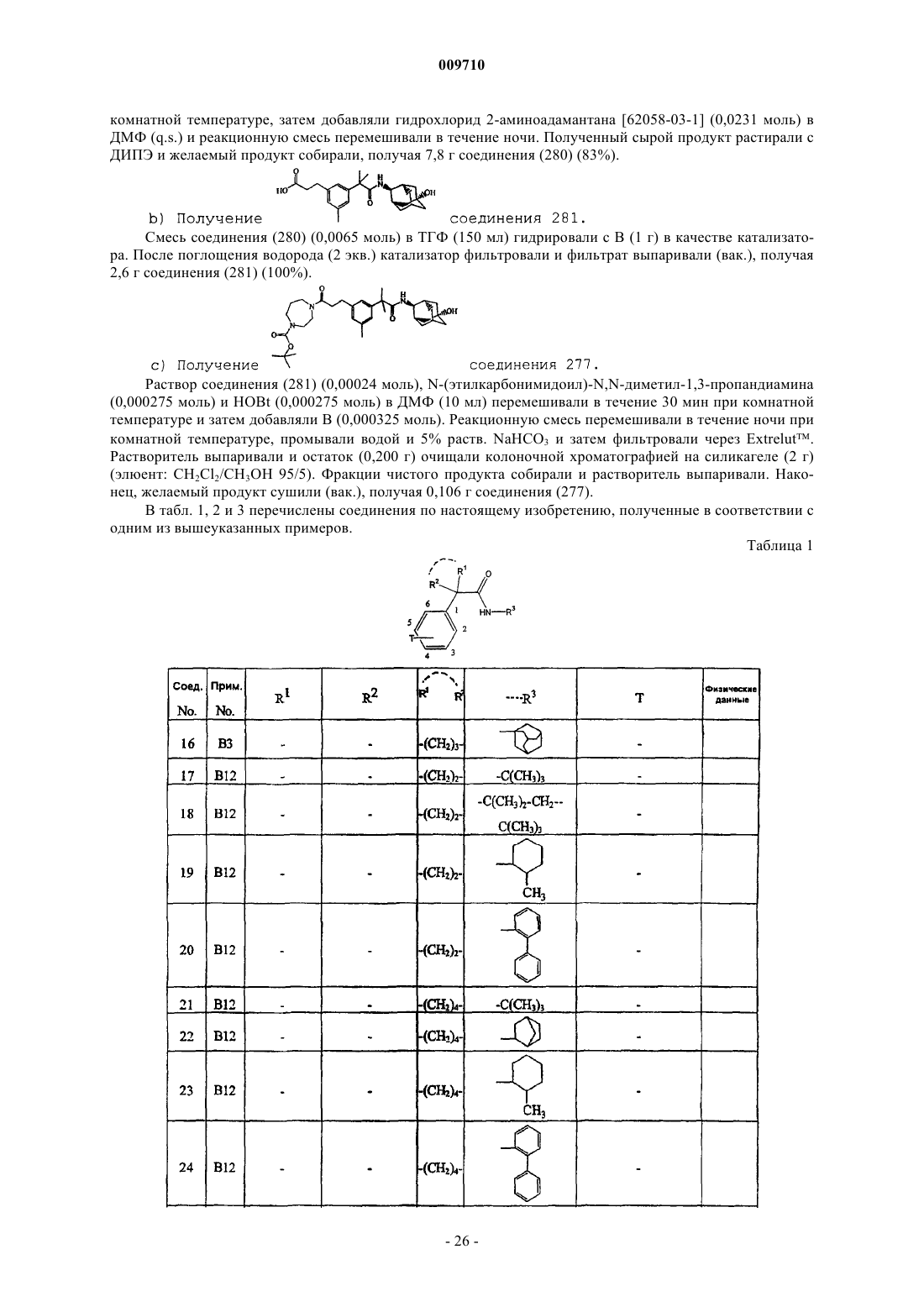
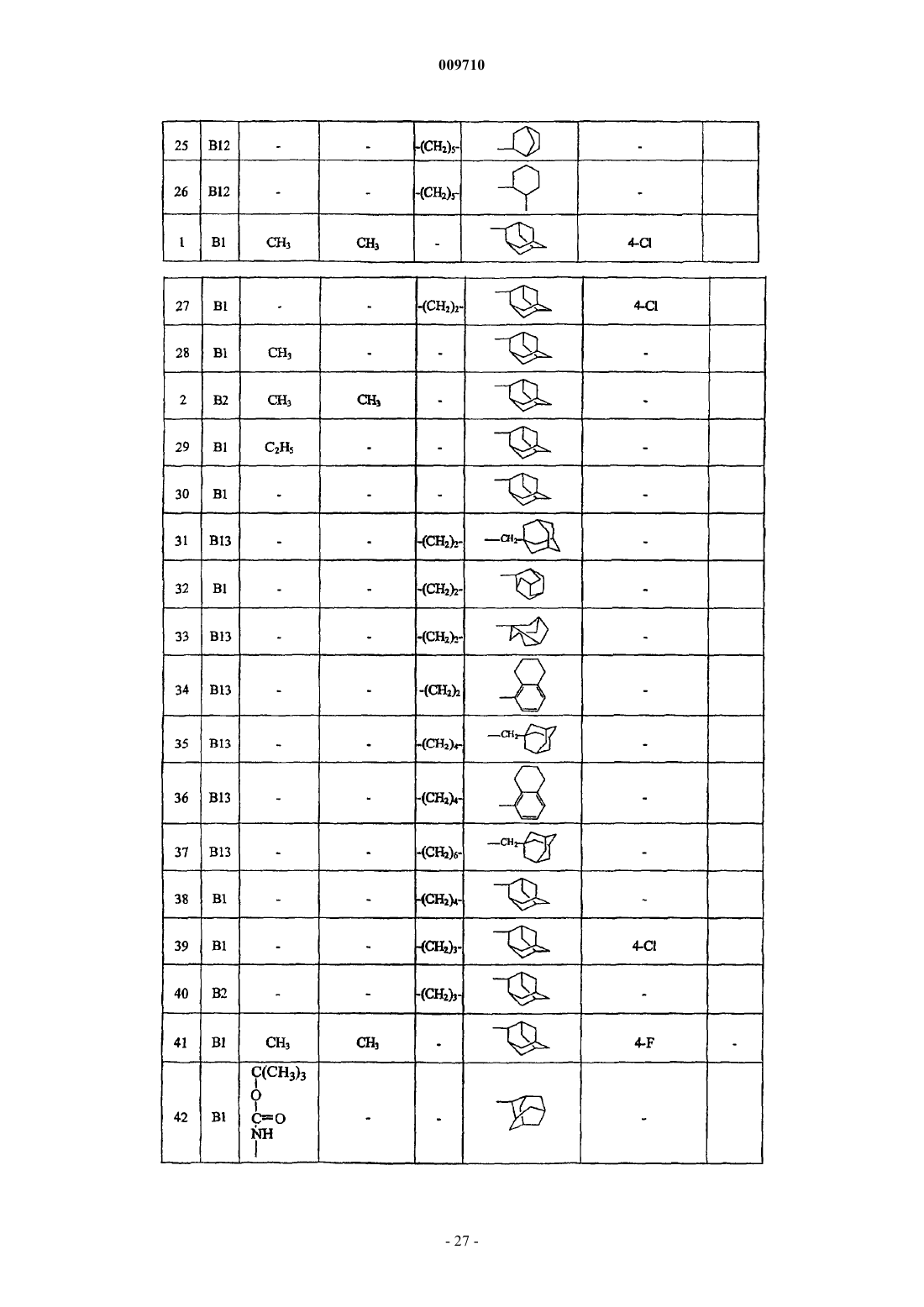
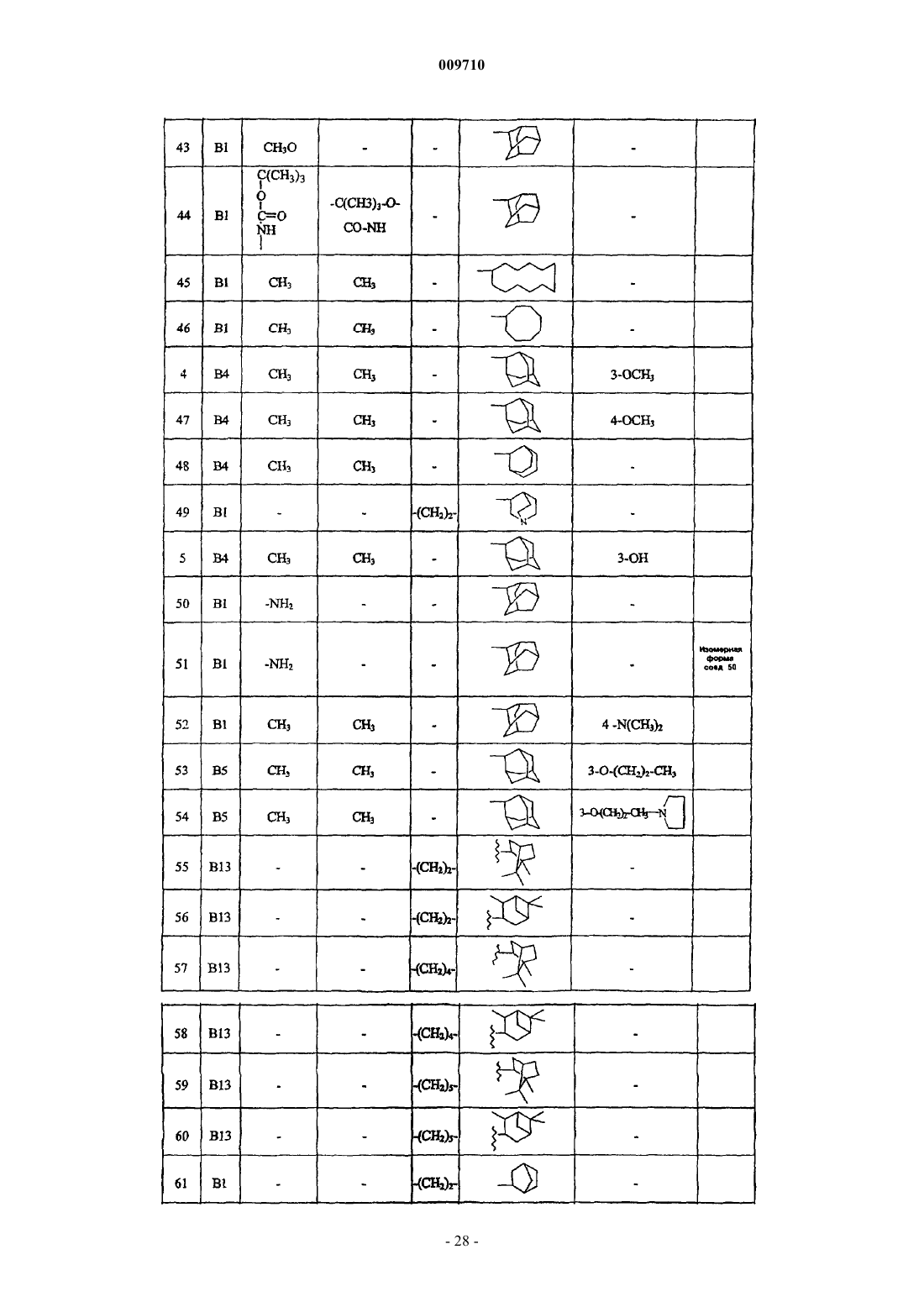
Формула / Реферат
1. Адамантилацетамид, имеющий формулу

его N-оксидные формы, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где
n представляет собой целое число, равное 1;
m представляет собой целое число, равное 0 или 1;
R1 и R2, каждый независимо, представляет собой C1-4алкил; или
R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют С3-6циклоалкил;
R3 представляет собой адамантил, который может необязательно быть замещенным одним или, где возможно, двумя, тремя или более заместителями, выбираемыми из группы, состоящей из галогена, карбонила, гидрокси или 1,3-диоксолила;
R4 представляет собой водород;
Q представляет собой фенил, бензотиофенил или пиридил, где указанный фенил, бензотиофенил или пиридил необязательно и независимо замещены одним или двумя заместителями, выбираемыми из галогена; гидрокси; C1-4алкила; С1-4алкилокси; C1-4алкилокси, замещенного одним или, где возможно, двумя или более заместителями, независимо выбираемыми из гидроксикарбонила, Het2 и NR7R8; С2-4алкенила, замещенного одним заместителем, выбираемым из фенил-C1-4алкилоксикарбонила или Het5-карбонила; и C1-4алкила, замещенного одним или, где возможно, двумя или тремя заместителями, независимо выбираемыми из диметиламина, Het6, Het7-карбонила или гидроксикарбонила;
R7 и R8, каждый независимо, представляет собой C1-4алкил;
L представляет собой C1-4алкил;
Het2 представляет собой моноциклический гетероцикл, выбираемый из пиперазинила или морфолинила, указанный Het2 необязательно замещен одним или, где возможно, двумя или более C1-4алкильными заместителями;
Het5 представляет морфолинил;
Het6 представляет собой моноциклический гетероцикл, выбираемый из пиперазинила или морфолинила;
Het7 представляет собой морфолинил.
2. Соединение по п.1, где
Q представляет собой фенил, указанный фенил необязательно замещен одним или двумя заместителями, выбираемыми из галогена или C1-4алкилокси;
n равно 1;
m равно 0;
R1 и R2 представляют собой C1-4алкил или R1 и R2 вместе с атомом углерода, к которому они присоединены, образуют С3-6циклоалкил;
R4 представляет собой водород;
R3 представляет собой моновалентный радикал, имеющий одну из следующих формул:

необязательно замещенный одним или, где возможно, двумя, тремя или более заместителями, выбираемыми из группы, состоящей из галогена, карбонила или гидрокси.
3. Соединение по любому из пп.1 или 2, где L представляет собой метил.
4. Соединение по п.1, которое представляет собой
(1a,2b,3b,5b,7b)-N-(5-гидрокситрицикло[3,3,1,13,7]дец-2-ил)-a,a-диметилбензолацетамид;
(1a,2b,3b,5b,7b)-N-(5-гидрокситрицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-3-метилбензолацетамид;
(1a,2b,3b,5b,7b)-N-(5-гидрокситрицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-3-метоксибензолацетамид;
(1a,2b,3b,5b,7b)-N-(5-гидрокситрицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-3-гидроксибензолацетамид;
(1a,2b,3b,5b,7b)-N-(5-гидрокситрицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-3,5-диметилбензолацетамид;
(1a,2b,3b,5b,7b)-N-(5-гидрокситрицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-3-(карбоксиметокси)бензолацетамид;
(1a,2b,3b,5b,7b)-N-(5-гидрокситрицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-3-[2-(4-морфолинил)этокси]бензолацетамид;
(1a,2b,3b,5b,7b)-N-(5-фтортрицикло[3,3,1,13,7]дец-2-ил)-a,a-диметилбензолацетамид;
N-(трицикло[3,3,1,13,7]дец-2-ил)-a,a-диметилбензолацетамид;
N-(трицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-3-(карбоксиметокси)бензолацетамид;
N-(трицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-3-[2-(4-морфолинил)этокси]бензолацетамид;
N-(трицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-3,5-диметоксибензолацетамид;
N-(трицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-3-метилбензолацетамид;
N-(трицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-3-метоксибензолацетамид;
N-(трицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-3-гидроксибензолацетамид;
N-(трицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-3,5-диметилбензолацетамид;
N-(трицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-4-фторбензолацетамид;
N-(трицикло[3,3,1,13,7]дец-2-ил)-1-фенилциклопропанкарбоксамид;
N-(трицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-2,6-дифторбензолацетамид;
N-(5-гидрокси-2-адамантил)-2-метил-2-(5-метилпиридин-3-ил)пропанамид;
N-(5-гидрокси-2-адамантил)-2-метил-2-(6-метилпиридин-2-ил)пропанамид;
3-(3-{2-[(5-фтор-2-адамантил)амино]-1,1-диметил-2-оксоэтил}-5-метилфенил)пропионовая кислота;
4-(3-{2-[(5-гидрокси-2-адамантил)амино]-1,1-диметил-2-оксоэтил}-5-метилфенил)бутановая кислота;
или его N-оксид, фармацевтически приемлемая аддитивная соль или стереохимически изомерная форма.
5. Соединение по п.1, которое выбрано из
(1a,2b,3b,5b,7b)-N-(5-гидрокситрицикло[3,3,1,13,7]дец-2-ил)-a,a-диметилбензолацетамида;
(1a,2b,3b,5b,7b)-N-(5-гидрокситрицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-3-метоксибензолацетамида;
(1a,2b,3b,5b,7b)-N-(5-гидрокситрицикло[3,3,1,13,7]дец-2-ил)-a,a-диметил-3-метилбензолацетамида;
или его N-оксида, фармацевтически приемлемой аддитивной соли или стереохимически изомерной формы.
6. Соединение по п.1, которое выбрано из

или его N-оксида, фармацевтически приемлемой аддитивной соли или стереохимически изомерной формы.
7. Фармацевтическая композиция, включающая фармацевтически приемлемый носитель и, в качестве активного ингредиента, эффективное ингибирующее 11b-HSD1 количество соединения по любому из пп.1-6.
8. Способ получения фармацевтической композиции по п.7, отличающийся тем, что фармацевтически приемлемый носитель тщательно смешивают с эффективным ингибирующим 11b-HSD1 количеством соединения по любому из пп.1-6.
9. Применение соединения по любому из пп.1-6 для получения лекарственного средства для лечения висцерального ожирения, чувствительности к инсулину, диабета типа 2, гиперлипидемии и гиперфагии.
Текст
009710 Метаболический синдром представляет собой заболевание, которое распространяется не только в западных странах, но также в Азии и развивающихся странах. Он характеризуется ожирением, в частности центральным или висцеральным ожирением, сахарным диабетом 2 типа, гиперлипидемией, гипертонией, артериосклерозом, ишемической болезнью сердца и, в ряде случаев, хронической почечной недостаточностью (С.Т. Montague et al. (2000), Diabetes, 49, 883-888). Известно, что глюкокортикоиды и 11-HSD1 являются важными факторами в дифференцировке жировых стромальных клеток в зрелые жировые клетки. В висцеральных стромальных клетках пациентов с ожирением уровень мРНК 11-HSD1 увеличен по сравнению с подкожной тканью. Кроме того,повышенная экспрессия в жировой ткани 11-HSD1 у трансгенных мышей связана с повышенным уровнем кортикостерона в жировой ткани, висцеральным ожирением, чувствительностью к инсулину, диабетом 2 типа, гиперлипидемией и гиперфагией (Н. Masuzaki et al (2001), Science, 294, 2166-2170). Таким образом, 11-HSD1 является наиболее вероятно вовлеченной в развитие висцерального ожирения и метаболического синдрома. Ингибирование 11-HSD1 приводит к снижению дифференцировки и повышению пролиферации жировых стромальных клеток. Более того, дефицит глюкокортикоидов (адреналэктомия) усиливает способность инсулина и лептина вызывать потерю аппетита и потерю веса, и такой эффект является обратимым при введении глюкокортикоидов (P.M. Stewart et al. (2002), Trends Endocrin. Metabol., 13, 94-96). Такие данные предполагают, что усиленная реактивация кортизона 11-HSD1 может усиливать ожирение, и ингибирование данного фермента в жировой ткани пациентов, страдающих ожирением, может принести пользу. Ожирение также связано с риском сердечно-сосудистых заболеваний. Существует сильная взаимосвязь между скоростью экскреции кортизола и холестерином ЛПВП у мужчин и женщин с учетом предположения, что глюкокортикоиды регулируют ключевые компоненты возникновения сердечнососудистых заболеваний. Аналогично, ригидность аорты также ассоциирована с висцеральным ожирением у пожилых взрослых пациентов. Глюкокортикоиды и глаукома Глюкокортикоиды увеличивают риск возникновения глаукомы, повышая внутриглазное давление при экзогенном введении и в определенных условиях повышенной продукции, как синдром Кушинга. Индуцированное кортикостероидами повышение внутриглазного давления вызвано повышенным сопротивлением оттоку жидкости из-за индуцированных глюкокортикоидами изменений в трабекулярной сети и ее внутриклеточном матриксе. Zhou et al. (Int. J. Mol. Med. (1998) 1, 339-346) также сообщают, что кортикостероиды увеличивают количество фибронектина, а также коллагена типа I и типа IV в трабекулярной сети культуры органа бычьего переднего сегмента быка. 11-HSD1 экспрессируется в базальных клетках корнеального эпителия и непигментированных эпителиальных клетках. МРНК рецептора глюкокортикоидов определяли только в трабекулярной сети,тогда как в непигментированных эпителиальных клетках присутствовала мРНК рецептора глюкокортикоидов, минералокортикоидов и 11-HSD1. Введение карбеноксолона пациентам приводило к значительному снижению внутриглазного давления (S. Rauz et al. (2001), Invest. Ophtalmol. Vis. Science, 42,2037-2042), предполагая роль ингибиторов HSD1 в лечении глаукомы. Соответственно, основной проблемой настоящего изобретения являлось определение эффективных ингибиторов 11-HSD, с высокой селективностью к 11-HSD1, и их использование для лечения патологий, ассоциированных с избыточным образованием кортизола, таких как ожирение, диабет, сердечнососудистые заболевания, связанные с ожирением, и глаукома. Настоящее изобретение относится к соединениям формулы (I) их N-оксидным формам, фармацевтически приемлемым аддитивным солям и стереохимически изомерным формам, гдеn представляет собой целое число, равное 1;m представляет собой целое число, равное 0 или 1;R3 представляет собой адамантил, который может необязательно быть замещенным одним или, где возможно, двумя, тремя или более заместителями, выбираемыми из группы, состоящей из галогена, карбонила, гидрокси или 1,3-диоксолила;Q представляет собой фенил, бензотиофенил или пиридил, где указанный фенил, бензотиофенил или пиридил необязательно и независимо замещены одним или двумя заместителями, выбираемыми из галогена; гидрокси; С 1-4 алкила; С 1-4 алкилокси; С 1-4 алкилокси, замещенного одним или, где возможно,двумя или более заместителями, независимо выбираемыми из гидроксикарбонила, Het2 и NR7R8; С 2-4 алкенила, замещенного одним заместителем, выбираемым из фенил-С 1-4 алкилоксикарбонила или Het5 карбонила; и С 1-4 алкила, замещенного одним или, где возможно, двумя или тремя заместителями, независимо выбираемыми из диметиламина, Het6, Het7-карбонила или гидроксикарбонила;Het2 представляет собой моноциклический гетероцикл, выбираемый из пиперазинила или морфолинила, указанный Het2 необязательно замещен одним или, где возможно, двумя или более С 1-4 алкильными заместителями;Het6 представляет собой моноциклический гетероцикл, выбираемый из пиперазинила или морфолинила;Het7 представляет собой морфолинил. Как используется в вышеупомянутых определениях и далее по тексту, галоген является общим обозначением для фтора, хлора, брома и йода; C1-4 алкил определяет насыщенные углеводородные радикалы с линейной и разветвленной цепью, имеющие от 1 до 4 атомов углерода, такие как, например, метил,этил, пропил, бутил, 1-метилэтил, 2-метилпропил, 2,2-диметилэтил и подобные; C1-8 алкил определяет насыщенные углеводородные радикалы с линейной и разветвленной цепью, имеющие от 1 до 8 атомов углерода, такие как группы, определяемые для С 1-4 алкила, и пентил, гексил, октил, 2-метилбутил, 2 метилпентил, 2,2-диметилпентил и подобные; С 3-6 циклоалкил является общим обозначением для циклопропила, циклобутила, циклопентила и циклогексила; С 6-12 циклоалкил является общим обозначением для циклогептила и циклооктанила, циклононана, циклодекана, циклоундекана и циклододекана; С 1-4 алкилокси определяет насыщенные углеводородные радикалы с линейной или разветвленной цепью, такие как метокси, этокси, пропилокси, бутилокси, 1-метилэтилокси, 2-метилпропилокси и подобные. Как используется в настоящем описании выше, термины оксо или карбонил относятся к (=O), который образует карбонильный компонент с атомом углерода, к которому он присоединен. Фармацевтически приемлемые аддитивные соли, как упомянуто выше, обозначают терапевтически активные нетоксические формы аддитивных солей с кислотами, которые способны образовывать соединения формулы (I). Последние могут обычно быть получены обработкой щелочной формы соответствующей кислотой. Соответствующие кислоты включают, например, неорганические кислоты, такие как галогеноводородные кислоты, например соляная или бромисто-водородная кислота; серная; азотная; фосфорная и подобные кислоты; или органические кислоты, такие как, например, уксусная, пропионовая, гидроксиуксусная, молочная, пировиноградная, щавелевая, малоновая, янтарная (т.е. бутандиовая кислота), малеиновая, фумаровая, яблочная, винная, лимонная, метансульфоновая, этансульфоновая,бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и подобные кислоты. Фармацевтически приемлемые аддитивные соли, как упомянуто выше, обозначают терапевтически активные нетоксические формы аддитивных солей с основаниями, которые способны образовывать соединения формулы (I). Примерами таких форм аддитивных солей оснований являются, например, соли натрия, калия, кальция, а также соли с фармацевтически приемлемыми аминами, такими как, например,аммиак, алкиламины, бензатин, N-метил-D-глюкамин, гидрабамин, аминокислоты, например аргинин,лизин. Указанные солевые формы могут быть, напротив, преобразованы обработкой соответствующим основанием или кислотой в форму свободной кислоты или основания. Термин аддитивные соли, как используется выше, также включает сольваты, которые способны образовывать соединения формулы (I), а также их соли. Такие сольваты представляют собой, например,гидраты, алкоголяты и подобные. Термин стереохимически изомерные формы, как используется выше, определяет возможные различные изомерные, а также конформационные формы, в которых могут существовать соединения формулы (I). Если не упомянуто или не указано иначе, химическое обозначение соединений обозначает смесь всех возможных стереохимически и конформационно изомерных форм, причем указанные смеси содержат все диастереомеры, энатиомеры и/или конформеры основной молекулярной структуры. Все стереохимически изомерные формы соединений формулы (I) как в чистой форме, так и в смеси друг с другом предназначены для включения в объем настоящего изобретения.N-Оксидные формы соединений формулы (I) обозначают такие соединения формулы (I), где один или несколько атомов азота окислены до так называемого N-оксида. Предпочтительная группа соединений состоит из таких соединений формулы (I), гдеQ представляет собой фенил, указанный фенил необязательно замещен одним или двумя заместителями, выбираемыми из галогена или С 1-4 алкилокси;R3 представляет собой моновалентный радикал, имеющий одну из следующих формул: необязательно замещенный одним или, где возможно, двумя, тремя или более заместителями, выбираемыми из группы, состоящей из галогена, карбонила или гидрокси. Следующими интересными соединениями по изобретению являются такие соединения формулы (I),где L представляет собой метил. Также предпочтительной группой соединений формулы (I) являются следующие соединения:N-(5-гидрокси-2-адамантил)-2-метил-2-(6-метилпиридин-2-ил)пропанамид; 3-(3-2-[(5-фтор-2-адамантил)амино]-1,1-диметил-2-оксоэтил-5-метилфенил)пропионовая кислота; 4-(3-2-[(5-гидрокси-2-адамантил)амино]-1,1-диметил-2-оксоэтил-5-метилфенил)бутановая кислота; или их N-оксиды, фармацевтически приемлемые аддитивные соли или стереохимически изомерные формы. Причем наиболее предпочтительное соединение выбрано из(1,2,3,5,7)-N-(5-гидрокситрицикло[3,3,1,13,7]дец-2-ил)-,-диметил-3-метилбензолацетамида; или их N-оксидов, фармацевтически приемлемых аддитивных солей или стереохимически изомерных форм. Также предпочтительным является соединение, которое выбрано из или его N-оксида, фармацевтически приемлемой аддитивной соли или стереохимически изомерной формы. Амидные соединения по настоящему изобретению могут быть получены любым из нескольких стандартных синтетических способов, обычно используемых специалистом в области органической хи-3 009710 мии и описанных, например, в "Introduction to organic chemistry" Streitweiser and Heathcock - MacmillanPublishing Co., Inc. - second edition - New York - Section 24.7 (part A) p. 753-756. В общем, амиды могут быть получены посредством катализируемого основанием нуклеофильного присоединения соответствующей карбоновой кислоты к соответствующему амину (схема 1) или посредством реакции нуклеофильного замещения, где соответствующий амин реагирует с соответствующим ацилгалогенидом (схема 2), ангидридом или сложным эфиром для получения требуемого амида. При сочетании кислот с аминами используют стандартные конденсирующие реагенты, такие как карбонилдиимидазол (CDI), 1,3-дициклогексилкарбодиимид (DCC) или гидрохлорид 1-этил-3-(3'-диметиламинопропил)карбодиимида (EDCI) в присутствии или в отсутствие гидроксибензотриалзола (HOBt). В целом, присоединение карбоновых кислот формулы (III) к аминам формулы (II) в условиях реакции,катализируемой основанием, приводит к образованию соли амина, которая находится в равновесии с его слабой кислотой и основанием. Для сдвига равновесия в сторону образования амида формулы (I) к реакционной смеси добавляют дегидрогенизирующий агент, такой как карбодиимиды, например DCC и CDI. Схема 1 В альтернативном варианте воплощения карбоновые кислоты преобразуют в соответствующие ацилгалогениды реакцией, например, с тионилхлоридом или оксалилхлоридом. Затем указанный ацилгалогенид (V) присоединяют к амину формулы (II) для получения амида формулы (I) с использованием известных в области техники методик реакции, таких как метод Шоттен-Ваумана. Схема 2 Карбоновые кислоты формулы (III) и амины формулы (II) являются легкодоступными или могут быть получены с использованием способов, которые являются хорошо известными в области техники. Многие соединения являются коммерчески доступными, например, от Aldrich Chemicals, или, когда соединения не являются коммерчески доступными, они могут быть легко получены из доступных предшественников с использованием прямых преобразований, которые хорошо известны в области техники. Например, карбоновые кислоты наиболее часто получают гидролизом нитрилов (схема 3), карбоксилированием металлоорганических соединений или окислением первичных спиртов или альдегидов,см., например, в "Introduction to organic chemistry" Streitweiser и Heathcock - Macmillan Publishing Co., Inc. second edition - New York - Section 19.6, p. 509-511. В частности, карбоновые кислоты формулы (III) получают из соответствующих (гетеро)арилацетонитрилов (VI) преобразованием в диалкильные или спироалкильные производные (VII) с использованием, например, гексаметилдисилазана натрия и метилйодида или дибромбутана (см., например, Trivedi et al., J. Med. Chem. 1993, 36, 3300) с последующим гидролизом в кислых или щелочных условиях до желаемой карбоновой кислоты III. Соответствующими кислотами и-4 009710 основаниями при гидролизе являются, например, H2SO4 и KOH. Реакция гидролиза может быть обычно проведена с использованием микроволнового нагревания. Многие нитрилы формулы (VI) являются коммерчески доступными, или, когда они не являются доступными, они могут быть легко получены из доступных (гетеро)арилметильных производных (X) в известных в области техники условиях, например бромированием с использованием N-бромсукцинамида (NBS) с последующим замещением брома CN с использованием, например, KCN. Схема 3 Во многих случаях карбоновые кислоты, где Q представляет собой бромзамещенный арил (III-A),далее модифицировали в соответствии со схемой реакции 4. На первой стадии бромистый заместитель модифицировали с использованием реакции Хека с акриловыми эфирами, амидами или акрилонитрилом для получения соединений формулы (XII). Восстановление двойной связи и функциональных групп давало замещенные амины формулы (XIV). Схема 4 Для таких соединений формулы (I), где Q представляет собой карбоциклические радикалы, содержащие два цикла, соответствующие бициклические карбоновые кислоты формулы (III-B) синтезировали,например, добавлением триметилсилилцианида к соответствующим кетонам (XV) с последующим кислотным или щелочным гидролизом нитрильных соединений (XVI) с использованием стандартных условий. Кетоны, которые не являются доступными, синтезировали внутримолекулярной циклизацией соответствующих кислот (XVIII) (см. схему 5). Амины формулы (II) обычно получают с использованием известных в области техники методик,см., например, "Introduction to organic chemistry" Streitweiser и Heathcock - Macmillan Publishing Co., Inc. second edition - New York - Section 24.6, p. 742-753, которые включают синтез посредством непрямого алкилирования соответствующих (гетеро)арилгалогенидов, в частности синтезом Габриэля, посредством восстановления соответствующих нитро- или нитрильных соединений, посредством восстановительного аминирования с использованием, например, реакции Eschweiler-Clarke, и в частности посредством восстановления оксимов (IX), которые могут быть получены из альдегидов или кетонов (VIII) реакцией с гидроксиламином (схема 6). В этом последнем случае оксимы восстанавливают алюмогидридом лития или каталитическим гидрогенированием с использованием соответствующего катализатора, такого как никель Ренея, причем указанное восстановление проводят в инертном безводном растворителе, таком как эфир или тетрагидрофуран (ТГФ). Схема 6 Следующие примеры для синтеза соединений формулы (I) с использованием любого из вышеупомянутых синтетических методов представлены в экспериментальной части далее. Когда необходимо или желательно, любая одна или более из следующих дальнейших стадий могут быть проведены в любом порядке:(i) удаление любой оставшейся защитной групп(ы);(ii) преобразование соединения формулы (I) или его защищенной формы в следующее соединение формулы (I) или его защищенную форму;(iii) преобразование соединения формулы (I) или его защищенной формы в N-оксид, соль, четвертичный амин или сольват соединения формулы (I) или его защищенной формы;(iv) преобразование N-оксида, соли, четвертичного амина или сольвата соединения формулы (I) или его защищенной формы в соединение формулы (I) или его защищенную форму;(v) преобразование N-оксида, соли, четвертичного амина или сольвата соединения формулы (I) или его защищенной формы в другой N-оксид, фармацевтически приемлемую аддитивную соль, четвертичный амин или сольват соединения формулы (I) или его защищенной формы;(vi) если соединение формулы (I) получают в виде смеси (R) и (S) энантиомеров, разделение смеси для получения желаемого энантиомера;(vii) если получают соединения формулы (I), где Q состоит из бромзамещенных карбоциклических радикалов, содержащих один или два цикла, возможны различные преобразования, см., например, схему 7, включающую:c) арилирование с использованием условий реакции Хека;d) алкилирование с использованием условий реакции Хека;e) преобразование в нитрил с использованием, например, цианида калия с возможным последующим преобразованием полученного нитрила в амин, который может быть алкилирован или ацилирован в условиях, известных в области техники. Схема 7 Специалист в области техники должен принимать во внимание, что в способах, описанных выше,функциональные группы промежуточных соединений могут нуждаться в блокировании защитными группами. Функциональные группы, которые желательно защитить, включают гидрокси, амино и остаток карбоновой кислоты. Подходящие защитные группы для гидрокси включают триалкилсилиловые группы (например, трет-бутилдиметилсилил, трет-бутилдифенилсилил или триметилсилил), бензил и тетрагидропиранил. Подходящие защитные группы для амино включают трет-бутилоксикарбонил или бензилоксикарбонил. Подходящие защитные группы для карбоновой кислоты включают С(1-6)алкильные или бензиловые эфиры. Защита и деблокирование функциональных групп может иметь место до или после стадии реакции. Применение защитных групп полно описано в "Protective Groups in Organic Chemistry", edited byP.G.M. Wutz, WHey Interscience (1991). Кроме того, N-атомы в соединениях формулы (I) могут быть метилированы способами, известными в области техники, с использованием СН 3-I в подходящем растворителе, таком как, например, 2-пропанон, тетрагидрофуран или диметилформамид. Соединения формулы (I) также могут быть преобразованы друг в друга в соответствии с известными в области техники методиками преобразования функциональных групп, из которых некоторые примеры упомянуты в описании выше. Соединения формулы (I) также могут быть преобразованы в соответствующие N-оксидные формы в соответствии с известными в области техники методиками для преобразования трехвалентного азота в-7 009710 его N-оксид. Указанная реакция N-окисления может обычно проводиться реакцией исходного вещества формулы (I) с 3-фенил-2-(фенилсульфонил)оксазиридином или с соответствующей органической или неорганической перекисью. Соответствующие неорганические перекиси включают, например, перекись водорода, перекиси щелочных металлов или щелочно-земельных металлов, например перекись натрия,перекись калия; соответствующие органические перекиси могут включать пероксикислоты, такие как,например, пероксибензойная кислота или галогензамещенная пероксибензойная кислота, например 3 хлорпероксибензойная кислота, пероксиалкановые кислоты, например пероксиуксусная кислота, алкилгидроперекиси, например т-бутилгидроперекись. Подходящими растворителями являются, например,вода, низшие спирты, например этанол и подобные, углеводороды, например толуол, кетоны, например 2-бутанон, галогенированные углеводороды, например дихлорметан, и смеси таких растворителей. Чистые стереохимически изомерные формы соединений формулы (I) могут быть получены применением методик, известных в области техники. Диастереомеры могут быть разделены физическими методами, такими как селективная кристаллизация, и хроматографическими методами, например противоточным распределением, жидкостной хроматографией и подобными. Некоторые из соединений формулы (I) и некоторые из промежуточных соединений в настоящем изобретении могут содержать асимметричный атом углерода. Чистые стереохимически изомерные формы указанных соединений и указанных промежуточных продуктов могут быть получены применением известных в области техники методик. Например, диастереоизомеры могут быть разделены физическими методами, такими как селективная кристаллизация, или хроматографическими методами, противоточным распределением, жидкостной хроматографией и подобными способами. Энантиомеры могут быть получены из рацемических смесей сначала обработкой указанных рацемических смесей подходящими разделяющим агентами, такими как, например, хиральные кислоты, с получением смесей диастереомерных солей или соединений; затем физическим разделением указанных смесей диастереомерных солей или соединений посредством, например, селективной кристаллизации или хроматографических методов,например жидкостной хроматографии и подобных способов; и, наконец, преобразованием указанных разделенных диастереомерных солей или соединений в соответствующие энантиомеры. Чистые стереохимически изомерные формы могут также быть получены из чистых стереохимически изомерных форм соответствующих промежуточных продуктов и исходных веществ, при условии, что происходящие реакции протекают стереоспецифически. Альтернативный способ разделения энантиомерных форм соединений формулы (I) и промежуточных продуктов включает жидкостную хроматографию, в частности жидкостную хроматографию с использованием хиральной стационарной фазы. Определенная группа энантиомерных промежуточных продуктов для соединений по настоящему изобретению состоит из син- и антиизомера 1-гидрокси-4-аминоадамантана, промежуточного продукта,используемого в синтезе таких соединений формулы (I), где R3 представляет собой необязательно замещенный 2-адамантил. 1-Гидрокси-4-аминоадамантан обычно получают гидроксилированием 2-аминоадамантана, например, с использованием смеси азотной и серной кислоты (Химико-фармацевтический журнал, 1986, 20,810; Журнал органической химии, 1976, 2369). Реакция дает два стереомера 1-гидрокси-4-аминоадамантана в соотношении от 3:1 до 1:1 в пользу синизомера. Так как было обнаружено, что антиизомер имеет улучшенную активность ингибирования HSD1, является желательным иметь способ синтеза, который дает большую селективность в отношении антиизомера. Альтернативно, 1-гидрокси-4-аминоадамантан может быть получен из соответствующего кетона после восстановительного аминирования, т.е. циклический кетон может быть преобразован в амин через образование имина оксима и последующее восстановление двойной связи. Восстановление может быть проведено с использованием алюмогидрида лития, никеля Ренея или благородных металлов, таких как палладий, платина, рутений или родий на угле. Восстановительное аминирование с использованием борогидридов является одностадийной альтернативой (хорошо известный способ, описанный, например, вAdvanced of organic chemistry from March 2003). Селективность восстановления зависит от структуры вещества (кетона) и используемого катализатора. Из-за того, что два изомера 1-гидрокси-4-аминоадамантана, полученные после восстановления оксима или после восстановительного аминирования аммиаком, не определяются ЖХМС или ГХМС, их очень трудно разделить. Реакция сочетания с кислотой формулы (III) дает смесь двух продуктов сочетания формулы (I), которые можно разделить с использованием хроматографии. Однако с целью уменьшения стоимости синтеза и для улучшения выхода антиизомеров может быть желательным отклониться от энантиомерно чистых промежуточных продуктов.-8 009710 Задачей настоящего изобретения является обеспечение решения вышеупомянутой проблемы, состоящего в способе получения 1-гидрокси-4-аминоадамантана, включающего восстановительное аминирование 5-гидроксиадамантан-2-она L(-)-1-фенилэтиламином в условиях катализа с использованием, например, рутения на угле (схема 8). Достигнутая селективность составила 3:1 в пользу антистереомера. Полученные изомеры легко разделить, и последующее дебензилирование анти-4-(1-фенилэтиламино) адамантан-1-ола дает чистый анти-1-гидрокси-4-аминоадамантан. Схема 8 Коммерчески доступный 5-гидроксиадамантан-2-он (0,1 моль), L(-)-альфа-метилбензиламин (0,105 моль),изопропоксид алюминия (0,1 моль) и родий на активированном угле (20 мол.%) суспендировали в 500 мл толуола, добавляли 20 мл 4% раствора тиофена. Реакционную смесь перемешивали при 50 С в течение 24 ч. Катализатор отфильтровывали и фильтрат концентрировали в вакууме. Остаток, содержащий два изомера в соотношении 3:1 транс:цис, очищали колоночной хроматографией для получения 12 г промежуточного продукта XVIII-A и 4 г промежуточного продукта XVIII-B. Амин XVIII-А (0,05 моль) растворяли в метаноле (100 мл), добавляли палладий на активированном угле (0,002 моль) и смесь гидрировали при комнатной температуре в течение 16 ч. Катализатор отфильтровывали, фильтрат выпаривали в вакууме. Остаток растирали с дихлорметаном для получения указанного в заголовке соединения (II-A) (7,5 г). Некоторые из промежуточных продуктов и исходных веществ, которые используются в методиках реакций, упомянутых выше, являются известными соединениями и могут быть коммерчески доступными или могут быть получены в соответствии с методиками, известными в области техники. Соединения по настоящему изобретению находят применение, поскольку они обладают фармакологическими свойствами. Таким образом они могут быть использованы в качестве лекарственных средств, в частности, для лечения патологии, связанной с избытком образования кортизола, такой как,например, ожирение, диабет, сердечно-сосудистые заболевания, связанные с ожирением, и глаукома. Как описано в экспериментальной части далее, ингибирующее воздействие настоящих соединений на активность 11b-HSD1-редуктазы (преобразование кортизона в кортизол) было продемонстрировано invitro, в ферментном анализе с использованием рекомбинантного фермента 11b-HSD1, измерением преобразования кортизона в кортизол с использованием очистки методом ВЭЖХ и методов количественного-9 009710 анализа. Ингибирование 11b-HSD1-редуктазы также было продемонстрировано in vitro, в исследовании клеток, включающем контакт клеток, экспрессирующих 11b-HSD1, с тестируемыми соединениями и оценку воздействия указанных соединений на образование кортизола в клеточной среде таких клеток. Клетки, предпочтительно используемые в анализе по настоящему изобретению, выбирали из группы,состоящей из мышиных фибробластов 3T3-L1, клеток HepG2, клеток почек свиней, в частности клетокLCC-PK1 и гепатоцитов крыс. Соответственно, настоящее изобретение обеспечивает соединения формулы (I) и их фармацевтически приемлемые N-оксиды, аддитивные соли, четвертичные амины и стереохимически изомерные формы для применения в терапии, в частности в лечении или предотвращении заболеваний, опосредованных пролиферацией клеток. Соединения формулы (I) и их фармацевтически приемлемые N-оксиды, аддитивные соли, четвертичные амины и стереохимически изомерные формы могут далее быть обозначены как соединения по изобретению. В свете полезности соединений по изобретению, предлагается способ лечения животного, например млекопитающего, включая человека, страдающего от клеточного пролиферативного расстройства, такого как атеросклероз, рестеноз и рак, который включает введение эффективного количества соединения по настоящему изобретению. Указанный способ включает системное или местное введение эффективного количества соединения по изобретению теплокровным животным, включая людей. Следовательно, задачей настоящего изобретения является обеспечение соединения по настоящему изобретению для применения в качестве лекарственного средства, в частности для применения соединения по настоящему изобретению в получении лекарственного средства для лечения патологии, ассоциированной с избыточным образованием кортизола, такой как, например, ожирение, диабет, сердечнососудистые заболевания, связанные с ожирением, и глаукома. Еще в одном аспекте настоящее изобретение обеспечивает применение соединений по изобретению в получении лекарственного средства для лечения любого из упомянутых выше клеточных пролиферативных расстройств или их признаков. Количество соединения по настоящему изобретению, также обозначаемого в описании как активный ингредиент, которое требуется для достижения терапевтического эффекта, будет, конечно, варьироваться в зависимости от определенного соединения, пути введения, возраста и состояния реципиента и определенного расстройства или заболевания, подлежащего лечению. Подходящая суточная доза составляет от 0,001 до 500 мг/кг массы тела, в частности от 0,005 до 100 мг/кг массы тела. Способ лечения может также включать введение активного ингредиента по схеме от 1 до 4 раз в день. Хотя для активного ингредиента возможно введение отдельно, является предпочтительным представлять его в виде фармацевтической композиции. Соответственно, настоящее изобретение, кроме того, обеспечивает фармацевтическую композицию, включающую соединение по настоящему изобретению, вместе с фармацевтически приемлемым носителем или разбавителем. Носитель или разбавитель должен быть "приемлемым",то есть являться совместимым с другими ингредиентами композиции и не приносить вреда ее потребителям. Фармацевтические композиции по настоящему изобретению могут быть получены любыми способами, хорошо известными в области техники фармации, например с использованием способов, таких как описанные в Gennaro et al. Remington's Pharmaceutical Sciences (18th ed., Mack Publishing Company, 1990,см. особенно часть 8: Pharmaceutical preparations и their Manufacture). Терапевтически эффективное количество определенного соединения в форме основания или аддитивной соли в качестве активного ингредиента тщательно смешивают с фармацевтически приемлемым носителем, который может принимать широкое множество форм, в зависимости от формы препарата, желаемого для введения. Такие фармацевтические композиции находятся желательно в разовых лекарственных формах, подходящих предпочтительно для системного введения, такого как пероральное, подкожное или парентеральное введение; или местного введения, такого как посредством ингаляции, назального спрея, глазных капель или посредством крема, геля, шампуня или подобного. Например, при получении композиций в пероральной лекарственной форме может применяться любая из обычных фармацевтических сред, такая как, например, вода, гликоли, масла, спирты и подобные в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры и растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазывающие вещества, связующие вещества, разрыхляющие вещества и подобные в случае порошков, пилюль, капсул и таблеток. Из-за простоты введения таблетки и капсулы представляют собой наиболее предпочтительные пероральные стандартные лекарственные формы, в таком случае, конечно, применяют твердые фармацевтические носители. Для парентеральных композиций носитель обычно включает стерильную воду, по меньшей мере в большей части, хотя другие ингредиенты, например для улучшения растворимости, также могут быть включены. Например, могут быть получены инъекционные растворы, в которых носитель включает солевой раствор, раствор глюкозы или смесь солевого раствора и раствора глюкозы. Также могут быть получены инъекционные суспензии, в таком случае могут быть использованы соответствующие жидкие носители, суспендирующие агенты и подобные. В композициях, подходящих для подкожного введения, носитель необязательно включает агент, усиливающий проницаемость,и/или подходящий смачиваемый агент, необязательно комбинируемые с подходящими добавками любого вида в минимальных пропорциях, такие добавки не оказывают какого-либо повреждающего воздейст- 10009710 вия на кожу. Указанные добавки могут облегчать введение в кожу и/или могут быть полезными для получения желаемых композиций. Такие композиции могут быть введены различными путями, например в виде чрескожного пластыря,в виде точечной наклейки или в виде мази. Как подходящие композиции для местного нанесения в данном описании могут быть предложены все композиции, обычно применяемые для местного введения лекарственных средств, например кремы, гели, повязки, шампуни, настойки, пасты, мази, бальзамы, порошки и подобные. Нанесение указанных композиций может быть посредством аэрозоля, например, с пропеллентом,таким как азот, диоксид углерода, фреон, или без пропеллента, пульвелизатор, капли, лосьоны или полутвердые вещества, такие как загущенные композиции, которые могут быть нанесены тампоном. В частности, обычно используются полутвердые композиции, такие как бальзамы, кремы, гели, мази и подобные. Является особенно преимущественным создавать упомянутые выше фармацевтические композиции в стандартной лекарственной форме для простоты введения и единообразия дозирования. Стандартная лекарственная форма, как используется в спецификации и формуле изобретения в данном описании, относится к физически отдельным единицам, подходящим в качестве однократных доз, каждая единица содержит заранее определенное количество активного ингредиента, рассчитанное для вызывания желаемого терапевтического эффекта, в связи с требуемым фармацевтическим носителем. Примерами таких стандартных лекарственных форм являются таблетки (включая рифленые или покрытые оболочкой таблетки), капсулы, пилюли, упаковки порошков, пластины, инъекционные растворы или суспензии, чайные ложки, столовые ложки и подобные и их отдельные единицы. С целью усиления растворимости и/или стабильности соединений формулы (I) в фармацевтических композициях может быть полезным использовать -, - или -циклодекстрины или их производные. Также сорастворители, такие как спирты, могут улучшать растворимость и/или стабильность соединений формулы (I) в фармацевтических композициях. При получении водных композиций аддитивные соли соединения по изобретению являются, конечно, более подходящими из-за их повышенной растворимости в воде. Экспериментальная часть В дальнейшем термин "КТ" обозначает комнатную температуру, "ТГФ" - тетрагидрофуран, "АсОН" уксусную кислоту, "EtOH" - этанол, "ДМЭ" - диметиловый эфир, "ДИПЭ" - диизопропиловый эфир, "ТФУ" трифторуксусную кислоту, "EtOAc" - этилацетат, "иPrOH" - изопропанол, "HOBt" - 1-гидрокси-1 Н-бензотриазол, "ДМА" - N,N-диметилацетамид, "ДМФ" - N,N-диметилформамид, "NaГМДС" - N-гексаметилдисилазан натрия, "DPPP" - 1,3-пропандиил-бис-[дифенилфосфин], "EDCI" - моногидрохлорид N'-(этилкарбонимидоил)-N,N-диметил-1,3-пропандиамина, "DAST" - трифторид (диэтиламино)серы и Extrelut является продуктом Merck KgaA (Darmstadt, Germany) и является короткой колонкой, содержащей диатомовую землю. А. Получение промежуточных продуктов. Пример А 1. Оксим бицикло[3,3,1]нонан-2-она [16473-10-2] (1,4 г) растворяли в безводном ТГФ (30 мл) и добавляли раствор алюмогидрида лития (15 мл, 1 М в диэтиловом эфире). Раствор кипятили в колбе с обратным холодильником в течение 16 ч. Добавление воды (0,6 мл), 15% NaOH (0,6 мл) и воды (1,8 мл) с последующей фильтрацией, сушкой фильтрата (MgSO4) и выпариванием давало сырые амины. Остаток растворяли в дихлорметане и экстрагировали 15% лимонной кислотой. Водный слой подщелачивали 1 М KOH и экстрагировали дихлорметаном. Органический слой промывали солевым раствором, сушили и выпаривали для получения смеси аминов 1:1 (0,5 г) промежуточного продукта (1) к промежуточному продукту (2). ЯМР (CDCl3)1,2-2,1 (м, СН), 2,45 (т, 1 Н), 2,9 (м, 1 Н). Пример А 2.(2,3 г, 0,012 моль) (содержащий около 30% дикеталя) растворяли в этаноле и добавляли раствор гидрохлорида гидроксиламина (1,7 г, 0,025 моль) и NaOH (1,0 г) в воде (30 мл). Смесь перемешивали в течение ночи. Летучие фракции выпаривали в вакууме и остаток экстрагировали дихлорметаном. Органический слой промывали солевым раствором, сушили и выпаривали для получения промежуточного продукта оксима (3) (2,4 г). ЯМР (ДМСО-d6)1,3-2,3 (м, СН), 2,5 (ушир.с, 1 Н), 3,5 (ушир.с, 1 Н), 3,95 (с, 4 Н, СН 2 СН 2). Этиленкеталь 6-гидроксииминоадамантан-2-ила (2,4 г) растворяли 7 М NH3/MeOH (100 мл), добавляли никель Ренея (1 г) и смесь гидрировали при 14 С. Смесь фильтровали и выпаривали для получения 2,0 г промежуточного продукта (4). ЯМР (ДМСО-d6)1,3-2,3 (м, СН), 3,23 (ушир.с, 2 Н, NH2), 3,95 (с, 4 Н, СН 2 СН 2). Пример A3. Раствор 3-метокси-5-метилбензолацетонитрила (0,016 моль) в ТГФ (20 мл) охлаждали до -40 С и затем добавляли NaГМДС (0,0355 моль) по каплям и смесь перемешивали в течение 1 ч при -30 С. Смесь йодметана (0,0355 моль) в ТГФ (q.s.) добавляли по каплям при -30 С и реакционную смесь перемешивали в течение 1 ч при -40 С, затем смеси позволяли достичь комнатной температуры и перемешивали в течение ночи. Полученную смесь обрабатывали 1 Н HCl и слои разделяли. Сырой продукт экстрагировали и обрабатывали CH2Cl2/гексан (3/2) для выделения желаемого продукта, получая 2,5 г (83 %) промежуточного продукта (5). Гидрохлорид калия 6 Н в воде (20 мл) добавляли к раствору промежуточного продукта (5) (0,013 моль) в этаноле (40 мл) и затем реакционную смесь перемешивали в течение 4 ч в микроволновых условиях при 160 С. Смесь разводили водой и экстрагировали ДИПЭ. Водный слой подкисляли конц. HCl до рН 1 и экстрагировали дихлорметаном. Органические экстракты промывали водой и солевым раствором, затем сушили и растворитель выпаривали. Полученный остаток растирали в гексане и желаемый продукт собирали, получая 1,69 г (61,5 %) промежуточного продукта (6). Раствор промежуточного продукта (6) (0,005 моль) в дихлорметане (20 мл) охлаждали до -78 С и затем по каплям добавляли трибромборан (1 М) в дихлорметане (10,5 мл). Реакционной смеси позволяли достичь комнатной температуры и перемешивали в течение ночи при комнатной температуре. Добавляли воду (50 мл), затем 6 Н KOH (10 мл) и смесь перемешивали в течение 30 мин. Водный слой отделяли и экстрагировали дихлорметаном, затем подкисляли конц. HCl до рН 1 и экстрагировали дихлорметаном(3 х 40 мл). Органические экстракты промывали водой и солевым раствором, сушили и растворитель выпаривали, получая 0,620 г промежуточного продукта (7). Хлор(1,1-диметилэтил)диметилсилан (0,0048 моль), 1 Н-имидазол (0,0048 моль) и N,N-диметил-4 пиридинамин (0,020 г) добавляли к раствору промежуточного продукта (7) (0,0032 моль) в дихлорметане(30 мл) и затем реакционную смесь перемешивали в течение ночи при комнатной температуре. Полученный осадок фильтровали и фильтрат выпаривали. Остаток (1,6 г) растирали в ДИПЭ и затем желаемый продукт собирали, получая 0,85 г промежуточного продукта (8). Пример А 4. Раствор цианида калия (0,09 моль) в воде (20 мл) добавляли к раствору 1-бром-3-(бромметил)-5 метилбензола [51719-69-8] (0,085 моль) в этаноле (100 мл) и реакционную смесь перемешивали в течение ночи при комнатной температуре, затем смесь (18 г) очищали колоночной хроматографией на силикагеле(элюент: CH2Cl2/гептан 2/1). Фракции продукта собирали и растворитель выпаривали, получая 7,5 г- 12009710 Раствор промежуточного продукта (9) (0,036 моль) в ТГФ (150 мл) охлаждали до -40 С в атмосфере азота, добавляли по каплям NaГМДС (2 М) в ТГФ (0,080 моль) при -25 С и реакционную смесь перемешивали в течение 1 ч при -30 С. Смесь йодметана (0,080 мл) в ТГФ (20 мл) добавляли по каплям при 30 С и полученной смеси позволяли достичь комнатной температуры, затем перемешивали в течение ночи. Добавляли HCl (1 Н, 100 мл) и слои разделяли. Водный слой экстрагировали 2 раза EtOAc, затем органические слои комбинировали, промывали 5% раствором NaHCO3, водой, солевым раствором и сушили. Наконец, растворитель выпаривали, получая 8,2 г промежуточного продукта (10). Смесь гидроксида калия (10 г) в воде (60 мл) добавляли к раствору промежуточного продукта (10)(0,034 моль) в этаноле (160 мл) и затем реакционную смесь перемешивали и нагревали в колбе с обратным холодильником в течение выходных дней. Смесь разводили ледяной водой и экстрагировали дихлорметаном для получения экстракта (I) и водного слоя (I). Водный слой (I) подкисляли HCl и экстрагировали дихлорметаном. Экстракт промывали солевым раствором, сушили и растворитель выпаривали,получая 8 г остатка (ЖХМС: 90% Ч). Остаток растирали в гексане и две фракции продукта собирали,получая фракцию 1: 2,8 г промежуточного продукта (11).N,N-Диэтилэтанамин (0,005 моль), 2-акриловую кислоту, фенилметиловый эфир (0,002 моль),трис(4-метилфенил)фосфин (0,0006 моль) и затем Pd2 (дибензилиденацетон)3 комплекс (0,0002 моль) добавляли к раствору промежуточного продукта (11) (0,001 моль) в ДМФ (6 мл). Реакционную смесь нагревали до 90 С и встряхивали в течение 4 ч при 90 С. Смесь разводили EtOAc и ДИПЭ, затем полученный осадок фильтровали и фильтрат промывали 3 раза водой. Водный слой подкисляли 1 Н HCl и экстрагировали EtOAc. Органический слой промывали солевым раствором, сушили, фильтровали и растворитель выпаривали, получая 0,340 г промежуточного продукта (12). Пример А 5.[2495-35-4] (0,002 моль). Добавляли трис(4-метилфенил)фосфин (0,0006 моль) и Pd2(дибензилиденацетон)3 комплекс (0,0002 моль) и затем реакционную смесь встряхивали в течение 4 ч при 90 С. Смесь разводили EtOAc и промывали водой. Водные слои собирали, подкисляли 1 Н HCl до рН 1-2 и экстрагировали EtOAc. Экстракты собирали, промывали водой и солевым раствором, сушили, фильтровали и растворитель выпаривали (вак.), получая 0,340 г промежуточного продукта (13). Пример А 6. 3-Бром-,-диметилбензолуксусную кислоту [81606-47-5] (0,001 моль) растворяли в N,N-диэтилэтанамине (q.s.), и раствор дегазировали, и затем добавляли N,N-диэтилэтанамин (0,005 моль), 4-(1-оксо 2-пропенил)морфолин [5117-12-4] (0,002 моль), трис(4-метилфенил)фосфин (0,0005 моль) и Pd2 (дибензилиденацетон)3 комплекс (0,00015 моль). Реакционную смесь встряхивали в течение ночи при 90 С и разводили EtOAc. Катализатор фильтровали через дикалит и промывали EtOAc, затем добавляли воду и органический слой отделяли. Водный слой экстрагировали EtOAc, подкисляли HCl до рН 1 и снова экстрагировали EtOAc. Экстракты промывали водой и солевым раствором, фильтровали и растворитель выпаривали, получая 0,291 г промежуточного продукта (14). Пример А 7. Смесь этилового эфира 3-бром-,-диметилбензолуксусной кислоты [81606-46-4] (0,0018 моль), 2 акрилонитрила (1 г), уксусной кислоты, соли палладия (2+) (0,0006 моль), DPPP [6737-42-4] (0,0012 моль) и уксусной кислоты, калиевой соли (1 г) в этаноле (150 мл) подвергали реакции в течение 16 ч при 100 С и затем растворитель выпаривали. Остаток растворяли в дихлорметане и полученный раствор промывали. Сырой продукт очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/гептан 3/2). Фракции продукта собирали и растворитель выпаривали, получая 0,750 г промежуточного продукта (15). Промежуточный продукт (15) (0,0031 моль) восстанавливали палладием на активированном угле(кат. кол-во) и затем никелем Ренея (кат. кол-во). После поглощения водорода (3 экв.) катализаторы фильтровали и фильтрат выпаривали, получая 0,7 г промежуточного продукта (16). 1,1'-Окси-бис-[2-хлорэтан] [111-44-4] (0,0025 моль) добавляли к раствору промежуточного продукта (16) (0,0012 моль) и карбоната калия (0,006 моль) в ДМФ (15 мл) и затем реакционную смесь перемешивали в течение 22 ч при 100 С. Смесь фильтровали и осадок на фильтре разводили EtOAc, затем промывали водой и сушили. Наконец, растворитель выпаривали, получая 0,6 г промежуточного продукта (17). Гидроксид калия (6 мл) добавляли к раствору промежуточного продукта (17) (0,0012 моль) в этаноле (12 мл) и затем реакционную смесь перемешивали и нагревали в колбе с обратным холодильником в течение 1 ч. Смесь охлаждали, разводили водой и экстрагировали. Водный слой подкисляли конц. HCl и экстрагировали дихлорметаном. Органический слой промывали водой и солевым раствором и затем растворитель выпаривали. Водный слой концентрировали (вак.) и полученный концентрат промывали метанолом. Наконец, растворитель выпаривали, получая 0,400 г промежуточного продукта (18). Пример А 8.[22972-63-0] (0,011 моль) в этаноле (40 мл) и реакционную смесь перемешивали и нагревали в колбе с обратным холодильником в течение 5 дней, затем смесь разводили водой и экстрагировали дихлорметаном. Водный слой подкисляли HCl и экстрагировали дихлорметаном. Экстракты промывали водой и солевым раствором, затем сушили и растворитель выпаривали, получая 0,190 г промежуточного продукта (19). Пример А 9.N,N-Диэтилэтанамин (0,005 моль), 4-(1-оксо-2-пропенил)морфолин [5117-12-4] (0,002 моль), трис(4 метилфенил)фосфин [1038-95-5] (0,0006 моль) и затем Pd2 (дибензилиденацетон)3 комплекс (0,00016 моль) добавляли к раствору промежуточного продукта (11) (0,001 моль) в ДМФ (10 мл), затем реакционную смесь перемешивали в течение ночи при 90 С и разводили EtOAc. Полученную смесь промывали водой и затем водный слой подкисляли 1 Н HCl до рН 1 и экстрагировали EtOAc. Экстракт сушили и растворитель выпаривали, получая 0,500 г остатка (ЖХМС: 69%). Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH 99/1). Фракции продукта собирали и растворитель выпаривали,получая 0,196 г (62%) промежуточного продукта (20). Пример А 10. 2-Феноксибензолацетонитрил [25562-98-5] (0,010 мл) растворяли в ТГФ (40 мл) в атмосфере азота и смесь охлаждали до -40 С, затем добавляли по каплям NaГМДС (2 М) в ТГФ (0,025 моль) и смесь перемешивали в течение 30 мин. Смесь йодометана (0,030 моль) в ТГФ, а.п. (10 мл) добавляли по каплям и после достижения комнатной температуры реакционную смесь перемешивали в течение 2 ч. Смесь фильтровали через дикалит, затем осадок на фильтре промывали EtOAc и к фильтрату добавляли 0,1 М HCl (60 мл). Водный слой отделяли и экстрагировали 2 раза EtOAc. Органические слои комбинировали, промывали водой и солевым раствором, затем сушили, фильтровали и растворитель выпаривали, получая 2,7 г промежуточного продукта (21). Раствор промежуточного продукта (21) (0,002 моль) в гидроксиде калия (6 М) в воде (10 мл) и этаноле (20 мл) помещали в тефлоновый сосуд микроволновой лабораторной установки (Milestone Inc.) и- 14009710 раствор перемешивали в закрытом сосуде в течение 6 ч при 170 С. Полученную смесь затем охлаждали и промывали EtOAc. Водный слой отделяли и подкисляли HCl. Наконец, полученный осадок фильтровали,получая промежуточный продукт (22). Пример A11. 3,5-Дифторбензолацетонитрил [122376-76-5] (0,013 моль) растворяли ТГФ, а.п. (60 мл) в атмосфере азота и смесь охлаждали до -30 С, затем по каплям добавляли NaГМДС (2 М) в ТГФ (0,029 моль) и смесь перемешивали в течение 1 ч. Смесь йодметана (0,030 моль) в ТГФ, а.п. (10 мл) добавляли по каплям и при достижении комнатной температуры реакционную смесь перемешивали в течение 6 ч. Смесь фильтровали через дикалит, затем осадок на фильтре промывали EtOAc и фильтрат обрабатывали 1 Н HCl. Органический слой отделяли, промывали водой и солевым раствором, затем сушили, фильтровали и растворитель выпаривали, получая 2,4 г промежуточного продукта (23). Раствор промежуточного продукта (23) (0,013 моль) в гидроксиде калия (6 М) в воде (20 мл) и этаноле (40 мл) перемешивали и нагревали в колбе с обратным холодильником в течение 24 ч, после охлаждения реакционную смесь промывали EtOAc. Водный слой подкисляли HCl и полученный осадок фильтровали, получая 1,5 г (60%) промежуточного продукта (24). Пример А 12. 2,6-Дифторбензолацетонитрил [654-01-3] (0,013 моль) растворяли в ТГФ (25 мл) в атмосфере азота и смесь охлаждали до -40 С, затем по каплям добавляли NaГМДС (2 М) в ТГФ (0,028 моль) и смесь перемешивали в течение 30 мин. Йодметан (0,028 моль) добавляли по каплям и при достижении комнатной температуры реакционную смесь перемешивали в течение 6 ч. Смесь фильтровали через дикалит, затем осадок на фильтре промывали EtOAc и фильтрат обрабатывали 1 Н HCl. Органический слой отделяли,промывали водой и солевым раствором, затем сушили, фильтровали и растворитель выпаривали. Остаток (2,2 г) очищали колоночной хроматографией на силикагеле (элюент: дихлорметан). Фракции продукта собирали и растворитель выпаривали, получая 1,4 г промежуточного продукта (25). Соляную кислоту (40 мл) добавляли к раствору промежуточного продукта (25) (0,006 моль) в ледяной уксусной кислоте (20 мл) и затем реакционную смесь перемешивали и нагревали в колбе с обратным холодильником в течение 24 ч. Растворитель выпаривали, затем остаток растворяли в дихлорметане и промывали карбонатом натрия (1 М). Водный слой подкисляли конц. HCl и экстрагировали дихлорметаном. Органические экстракты собирали, сушили и растворитель выпаривали, получая 0,6 г (72 %) промежуточного продукта (26). Пример А 13. Хлорид олова(II) (0,068 моль) добавляли к 3,4-дигидро-4-[(триметилсилил)окси]-2 Н-1-бензопиран 4-карбонитрилу [74187-63-6] (0,017 моль) в атмосфере азота, затем добавляли уксусную кислоту (20 мл) и соляную кислоту (20 мл) и реакционную смесь перемешивали и нагревали в колбе с обратным холодильником в течение ночи в атмосфере азота. Смесь охлаждали, выливали на лед и экстрагировали дихлорметаном. Органический слой промывали, сушили, фильтровали и растворитель выпаривали, получая 1,4 г остатка (54% Ч). Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/СН 3 ОН 98/2). Фракции продукта собирали и растворитель выпаривали, получая 1 г промежуточного продукта (27). Пример А 14.(0,125 г) в трихлорметане (5 л) перемешивали на льду в атмосфере азота. Триметилсиланкарбонитрил[7677-24-9] (0,067 моль) добавляли по каплям и реакционную смесь перемешивали в течение ночи. Добавляли дихлорметан (50 мл) и смесь промывали 2 раза раствором карбоната натрия. Органический слой сушили, фильтровали и растворитель выпаривали, получая 4 г промежуточного продукта (28). Смесь промежуточного продукта (28) (0,0072 моль) в уксусной кислоте (15 мл) и соляной кислоте(15 мл) перемешивали и нагревали в колбе с обратным холодильником в течение ночи в атмосфере азота и затем реакционную смесь охлаждали. Смесь выливали в воду и экстрагировали дихлорметаном. Органический слой экстрагировали разведенным раствором гидроксида натрия, затем водный слой подкисляли соляной кислотой и экстрагировали дихлорметаном. Органический слой отделяли, сушили (MgSO4),фильтровали и растворитель выпаривали, получая 1 г остатка (56% Ч). Остаточную фракцию очищалиFlash-40 колоночной хроматографией Biotage (элюент: CH2Cl2/СН 3 ОН 99/1). Фракции продукта собирали и растворитель выпаривали, получая 0,39 г (28%) промежуточного продукта (29). Пример А 15.(0,021 моль) в уксусной кислоте (40 мл) и соляной кислоте (40 мл) перемешивали и нагревали в колбе с обратным холодильником в течение ночи при установках Dean-Starck. Реакционную смесь охлаждали и экстрагировали дихлорметаном. Органический слой промывали раствором Na2CO3, затем водный слой подкисляли HCl до рН 2 и экстрагировали дихлорметаном. Органический слой отделяли, промывали,сушили (MgSO4), фильтровали и растворитель выпаривали, получая 0,7 г промежуточного продукта (30). Пример А 16. Смесь 3,4-дигидро-5,7-диметил-1(2 Н)-нафталенона [13621-25-5] (0,02 моль) и йодида цинка (0,125 г) в трихлорметане (5 мл) перемешивали на льду и добавляли триметилсиланкарбонитрил [7677-24-9] (0,075 моль). Реакционную смесь перемешивали в течение ночи и промывали 2 раза раствором NaHCO3. Органический слой сушили, фильтровали и растворитель выпаривали, получая 5,7 г промежуточного продукта (31). Смесь промежуточного продукта (31) (0,02 моль) в уксусной кислоте (40 мл) и соляной кислоте (40 мл) перемешивали и нагревали в колбе с обратным холодильником в течение 3 дней в атмосфере азота. Реакционную смесь охлаждали и экстрагировали дихлорметаном. Органический слой экстрагировали раствором Na2CO3, затем водный слой подкисляли соляной кислотой и экстрагировали дихлорметаном. Органический слой отделяли, промывали, сушили (MgSO4), фильтровали и растворитель выпаривали, получая 1,2 г (29%) промежуточного продукта (32). Пример А 17.[2627-86-3] (0,01 моль) в этаноле (20 мл) перемешивали и нагревали в колбе с обратным холодильником в течение выходных дней и затем растворитель выпаривали (вакуум), получая 2,8 г промежуточного продукта (33). Промежуточный продукт (33) (0,001 моль) забирали в ТГФ (безводный) (5 мл) и смесь охлаждали до 0 С в атмосфере азота, затем добавляли тетрагидроборат натрия (0,00115 моль) и трифторуксусную кислоту (0,00344 моль) и реакционную смесь перемешивали при 0 С. Добавляли дихлорметан (10 мл) и- 16009710 насыщенный раствор NaHCO3. Органический слой отделяли, промывали NaHCO3, сушили и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/EtOAc 95/5). Две фракции продукта собирали и растворитель выпаривали, получая 0,130 г промежуточного продукта (34) и 0,090 г промежуточного продукта (35). Пример А 18.N,N-диэтилэтанамина (0,2 г) в 2-пропаноне (10 мл) и воде (10 мл) перемешивали и затем добавляли бис-(1,1 диметилэтил)овый эфир дикарбоновой кислоты [24424-99-5] (0,0022 моль). Реакционную смесь перемешивали в течение выходных дней, затем вливали в дихлорметан и промывали водой. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали, получая 0,38 г промежуточного продукта (36). В. Получение соединений. Пример В 1. 2,2-Диметил-(4-хлорфенил)уксусную кислоту [6258-30-6] (2,0, 10 ммоль) и гидрохлорид 2-аминоадамантана [13074-39-0] (1,9 г, 10 ммоль) растворяли в дихлорметане (50 мл), добавляли HOBt (2,7 г, 20 моль),N,N-диэтилэтанамин (2,1 г, 20 ммоль) и EDCI (2,1 г, 11 ммоль) и смесь перемешивали в течение ночи. Реакционную смесь промывали 15% лимонной кислотой, насыщ. NaHCO3 и солевым раствором, сушили над MgSO4 и выпаривали в вакууме. Остаток перекристаллизовывали из изопропанола, получая 2,0 Соединение 1 (1,7 г, 5 ммоль) растворяли в метаноле (100 л), добавляли 0,5 г палладия на активированном угле (10%) и СаО (1 г) и смесь гидрогенезировали при 50 С. После поглощения 1 экв. водорода реакционную смесь фильтровали, выпаривали досуха. Остаток растворяли в дихлорметане, промывали насыщ. NaHCO3, сушили и выпаривали. Остаток перекристаллизовывали из диизопропилового эфира,получая 0,65 г (60%) соединения 2. ЯМР: (ДМСО-d6)1,4-1,8 (м, СН), 1,49 (с, 6 Н, (СН 3)2), 3,79 (д, 1 Н, СН), 6,21 (д, 1 Н, NH), 7,25-7,37 2,2-Диметилфенилуксусную кислоту [826-55-1] растворяли в сухом дихлорметане, добавляли оксалилхлорид и 1 каплю ДМФ. После перемешивания в течение 2 ч растворитель выпаривали досуха, снова растворяли в 10 мл дихлорметана и добавляли к раствору 2-аминоадамантана [13074-39-0] и триэтиламина в дихлорметане. Смесь перемешивали в течение ночи, экстрагировали 15% лимонной кислотой, насыщ. NaHCO3 и солевым раствором, сушили над MgSO4 и выпаривали в вакууме. Остаток перекристаллизовывали из изопропилового эфира. ЯМР: (CDCl3)1,3-1,8 (м, СН), 1,55 (с, 6 Н, (СН 3)2), 2,31 (с, 6 Н, 2 хСН 3), 3,96 (д, 1 Н, СН), 5,50 (д, 1 Н,NH), 6,91 (с, 1 Н, Ar-H), 6,99 (с, 2 Н, Ar-H). Пример В 4.- 17009710 2-Метил-2-(3-метоксифенил)пропионовую кислоту (2,0 г, 10 моль) и гидрохлорид 2-аминоадамантана [13074-39-0] (1,9 г, 10 ммоль) растворяли в дихлорметане (50 мл), добавляли HOBt (2,7 г, 20 моль),N,N-диэтилэтанамин (2,1 г, 20 ммоль) и EDCI (2,1 г, 11 ммоль) и смесь перемешивали в течение ночи. Реакционную смесь промывали 15% лимонной кислотой, насыщ. NaHCO3 и солевым раствором, сушили над MgSO4 и выпаривали в вакууме. Остаток перекристаллизовывали из изопропанола, получая 2,0 г Соединение 4 растворяли в сухом дихлорметане, охлаждали до -78 С и добавляли трибромид бора. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, вливали в водный аммиак и экстрагировали дихлорметаном. Органические слои промывали солевым раствором, сушили и выпаривали. Твердый остаток перекристаллизовывали из этилацетата, получая соединение 5. ЯМР: (ДМСО-d6)1,4-1,8 (м, СН), 1,44 (с, 6 Н, (СН 3)2), 3,79 (д, 1 Н, СН), 6,18 (д, 1 Н, NH), 6,65-7,16 Соединение 4 растворяли ДМФ и добавляли этилбромацетат вместе с карбонатом калия. Смесь перемешивали при 60 С в течение ночи, выливали на лед и экстрагировали дихлорметаном. Органический слой промывали 1 М NaHCO3 и солевым раствором и выпаривали. Остаток растворяли в этаноле, добавляли гидроксид калия 1 М и смесь перемешивали в течение 2 ч. Раствор подкисляли 1 М HCl, экстрагировали EtOAc, органический слой сушили и выпаривали. Остаток перекристаллизовывали из этилацетата,получая соединение 6. ЯМР: (ДМСО-d6)1,4-1,8 (м, СН), 1,47 (с, 6 Н, (СН 3)2), 3,78 (д, 1 Н, СН), 4,67 (с, 2 Н, СН 2 СООН),6,23 (д, 1 Н, NH), 6,77-7,3 (м, 4 Н, Ar-H). Пример В 5. Соединение 4 растворяли в ДМФ и добавляли гидрохлорид хлорида диметиламиноэтила, затемK2CO3. Смесь перемешивали при 60 С в течение ночи, выливали на лед и экстрагировали дихлорметаном. Органический слой промывали 1 М NaHCO3 и солевым раствором и выпаривали. Остаток растворяли в иPrOH при нагревании, добавляли щавелевую кислоту и кристаллы амина фильтровали, получая соединение 7. ЯМР: (ДМСО-d6)1,4-1,8 (м, СН), 1,49 (с, 6 Н, (СН 3)2), 2,78 (с, 6 Н, N(CH3)2), 3,43 (т, 2 Н, СН 2), 3,79NaHCO3 и солевым раствором, сушили над MgSO4 и выпаривали в вакууме. Остаток хроматографировали на силикагеле (элюенты 3-5% МеОН в дихлорметане), получая 1,8 г соединения 8: ЯМР: (CDCl3)1,2-1,85 (м, СН), 1,59 (с, 6 Н, (СН 3)2), 1,95-2,00 (м, 2 Н, СН), 3,91 (дт, 1 Н, СН), 5,32 (д,1 Н, NH), 7,25-7,47 (м, 5 Н, Ar-H); и 1,8 г соединения 9: ЯМР: (CDCl3)1,2-1,7 (м, СН), 1,56 (с, 6 Н, (СН 3)2), 2,05-2,10 (м, 2 Н, СН), 3,83 (дт, 1 Н, СН), 5,32 (д,1 Н, NH), 7,25-7, 50 (м, 5 Н, Ar-H). Пример В 7. Соединение 8 (80 мг) растворяли в дихлорметане (2 мл) и охлаждали до -78 С в атмосфере азота. Добавляли DAST (0,1 мл) и смесь перемешивали и нагревали до комнатной температуры. Добавляли насыщенный NaHCO3 и слои разделяли. Органический слой промывали солевым раствором, сушили(MgSO4) и выпаривали. Остаток перекристаллизовывали из диизопропилового эфира для получения 40 мг Соединение 8 (100 мг, 0,3 ммоль) растворяли в дихлорметане (2 мл), охлаждали до -78 С и добавляли трибромид бора (0,15 мл, 1,5 ммоль). Реакционную смесь согревали до комнатной температуры, разводили дихлорметаном и выливали на смесь льда и аммиака. Слои разделяли, органический слой промывали солевым раствором, сушили (MgSO4) и выпаривали. Остаток перекристаллизовывали из этилацетата, получая соединение 11. ЖХ-МС: М+1 393,34, 395,34. ЯМР: (CDCl3)1,25-1,52 (м, СН), 1,57 (с, 6 Н, (СН 3)2), 1,90-2,42 (м, СН), 3,97 (дт, 1 Н, СН), 5,37 (д,1 Н, NH), 6,28-7,30 (м, 4 Н, Ar-H). Пример В 9. 2,2-Диметилфенилуксусную кислоту [826-55-1] (0,5 г, 2,7 моль) растворяли в сухом дихлорметане,добавляли оксалилхлорид (0,4 г) и 1 каплю ДМФ. После перемешивания в течение 2 ч раствор выпаривали досуха, снова растворяли в 10 мл дихлорметана и добавляли к раствору этиленкеталя 6-оксоадамантан-2-иламина (0,6 г, 2,7 ммоль) и N,N-диэтилэтанамина (0,5 мл) в дихлорметане. Смесь перемешивали в течение ночи, экстрагировали 15% лимонной кислотой, насыщ. NaHCO3 и солевым раствором, сушили над MgSO4 и выпаривали в вакууме. Остаток очищали на силикагеле (элюент: 5% МеОН в дихлорметане) и соединение 12 перекристаллизовывали из изопропилового эфира, получая 600 мг (50%). ЯМР: (CDCl3)1,52-2,05 (м, СН), 1,60 (с, 6 Н, (СН 3)2), 3,85 (дт, 1 Н, СН), 3,85-3,90 (м, 4 Н, СН 2 СН 2),5,45 (д, 1 Н, NH), 7,23-7,42 (м, 5 Н, Ar-H). Пример В 10. Кеталь из примера В 9 (450 мг) растворяли в ацетоне (10 мл), добавляли 1 М HCl (5 мл) и смесь перемешивали в течение 3 ч при 45 С. Реакционную смесь концентрировали и экстрагировали дихлорметаном. Органические слои промывали насыщ. NaHCO3 и солевым раствором и выпаривали. Остаток перекристаллизовывали из этанола, получая 300 мг соединения 13. ЯМР: (CDCl3)1,52-1,75 (м, СН), 1,60 (с, 6 Н, (СН 3)2), 1,95-2,15 (м, 2 Н, СН), 2,30 (д, 2 Н, СН), 2,50 (с,2 Н, СН), 4,12 (дт, 1 Н, СН), 5,45 (д, 1 Н, NH), 7,27-7,47 (м, 5 Н, Ar-H). Соединение 13 (50 мг) растворяли в МеОН и добавляли NaBH4 (50 мг). Смесь перемешивали при комнатной температуре в течение 6 ч. Добавляли 1 М HCl и смесь экстрагировали дихлорметаном. Органическую фазу промывали солевым раствором, сушили и выпаривали. Хроматография на силикагеле 1-Фенилциклопропанкарбоновую кислоту (0,00028 моль) добавляли к смеси N-циклогексилкарбодиимида (0,0004 моль) на полимерной подложке в дихлорметане (5 мл). Смесь перемешивали в течение 15 мин. Добавляли 2-метил-2-пропанамин (0,0002 моль) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Смолу отфильтровывали и фильтрат выпаривали. Остаток очищали на предварительно укомплектованной силикагелевой колонке для жидкостной хроматографии (14 мл; элюент: дихлорметан). Фракции продукта собирали и растворитель выпаривали, получая соединение 17. Пример В 13. Карбодиимид на полимерной подложке (0,0004 моль) суспендировали в дихлорметане (5 мл). Затем добавляли 1-фенилциклопропанкарбоновую кислоту (0,00028 моль) и N,N-диметил-4-пиридинаминамин(0,0002 моль; 6 величин) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Смесь фильтровали. Осадок на фильтре промывали дихлорметаном и растворитель фильтрата выпаривали. Остаток очищали хроматографией с испарительной колонкой на TRIKONEX FlashTube (элюент: гексан/EtOAc 9/2). Фракции продукта собирали и затем экстрагировали и экстракты выпаривали, получая 0,037 соединения 31. Пример В 14. Смесь m,-диметилгидратроповой кислоты (0,001 моль), 1-гидрокси-1 Н-бензотриазола (0,0011 моль) и моногидрохлорида N'-(этилкарбонимидоил)-N,N-диметил-1,3-пропандиамина (0,00105 моль) в дихлорметане (5 мл) перемешивали до полного растворения (20 мин) при комнатной температуре. Добавляли смесь гидрохлорида 2-адамантанамина (0,0013 моль) в дихлорметане (2 мл), триэтиламин (1 мл) и ДМФ (0,5 мл) и полученную смесь перемешивали в течение ночи при комнатной температуре. Добавляли воду (2 мл) и смесь перемешивали в течение 10 мин. Смесь фильтровали через Extrelut и растворитель фильтрата выпаривали. Остаток очищали флэш-хроматографией на TRIKONEX FlashTube (элюент: CH2Cl2/EtOAc 95/5). Фракции продукта собирали и очищали ВЭЖХ. Фракции продукта собирали и растворитель выпаривали, получая соединение (89). Суспензию соединения (89) (0,005 моль), 1-бром-2,5-пирролидиндиона (0,0055 моль) и 2,2'-азо-бис(2-метилпропионитрила [cas: 78-67-1] (0,030 г) в тетрахлорметане (50 мл) перемешивали и нагревали в колбе с обратным холодильником в течение 1 ч, затем осадок фильтровали и растворитель выпаривали. Остаток растворяли в дихлорметане и раствор промывали 2% раствором NaHCO3, водой и солевым раствором. Смесь сушили и растворитель выпаривали, получая 2 г продукта. Часть этого остатка (0,100 г) очищали высокоэффективной жидкостной хроматографией. Фракции продукта собирали и растворитель выпаривали, получая соединение (270). Суспензию соединения (270) (0,0013 моль), цианида калия (0,0065 моль) и йодида калия (0,00013 моль) в ацетонитриле (10 мл) перемешивали в течение ночи при комнатной температуре и затем растворитель выпаривали. Остаток растворяли в дихлорметане и раствор экстрагировали Н 2 О. Смесь фильтровали через Extrelut и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: гексан/EtOAc 2/1). Фракции продукта собирали и растворитель выпаривали, получая 0,12 г (96%) соединения (161). Смесь соединения (161) (0,0009 моль) в смеси аммиака в метаноле (50 мл) гидрировали при 14 С с никелем Ренея (кат. кол-во) в качестве катализатора. После поглощения водорода (2 экв.) катализатор фильтровали и фильтрат выпаривали, получая 0,270 г (88%) соединения (170).(8 мл) перемешивали в течение 15 мин и добавляли по каплям смесь 1-хлор-2-(хлорметокси)этана(0,00066 моль) в N,N-диметилформамиде (q.s.), затем реакционную смесь перемешивали в течение выходных дней при комнатной температуре. Смесь нагревали в течение ночи до 65 С и добавляли экстра 1-хлор-2(хлорметокси)этан (0,030 г). Полученную смесь перемешивали в течение 3 ч при 65 С, затем вливали в воду и экстрагировали дихлорметаном. Продукт очищали высокоэффективной жидкостной хроматографией. Фракции продукта собирали и остаток встряхивали с активированным углем, получая 0,021 г (8,5%) соединения (191). Пример В 15. Смесь йодметана (0,001 моль) в N,N-диметилформамиде (1 мл) добавляли по каплям к суспензии соединения (170) (0,0003 моль) и карбоната калия (0,001 моль) в N,N-диметилформамиде (3 мл) и реакционную смесь перемешивали в течение ночи при комнатной температуре, затем смесь вливали в воду и промывали дихлорметаном. Полученную смесь фильтровали через Extrelut и растворитель выпаривали,получая продукт (ЯМР: CTS, ЖХМС: 100% MW 382). Осадок растирали с ДИПЭ; полученный осадок фильтровали и сушили, получая 0,075 г (65%) соединения (178). Пример В 16.(0,0006 моль) и 1-гидрокси-1 Н-бензотриазола (0,0008 моль) в дихлорметане (5 мл), ДМФ (1 мл) и N,Nдиэтилэтанамине (3 мл) перемешивали, затем добавляли моногидрохлорид N'-(этилкарбонимидоил)-N,Nдиметил-1,3-пропандиамина (0,00045 моль) и реакционную смесь перемешивали в течение ночи. Добавляли воду (2 мл), смесь перемешивали в течение 10 мин и фильтровали через Extrelut. Растворитель выпаривали и остаток очищали колоночной флэш-хроматографией на TRIKONEX FlashTube (элюент:CH2Cl2/EtOAc 98/2). Фракции продукта собирали и растворитель выпаривали. Остаток далее очищали высокоэффективной жидкостной хроматографией. Фракции продукта собирали и растворитель выпаривали. Остаток растворяли в дихлорметане и промывали раствором Na2CO3. Смесь фильтровали через(100 мл) подвергали реакции при 125 С в течение 16 ч и затем растворитель выпаривали. Остаток (0,5 г) очищали колоночной хроматографией на силикагеле (элюент: дихлорметан). Собирали две фракции продукта и растворитель выпаривали, получая 0,120 г (97%) соединения (180). Смесь соединения (180) (0,0003 моль) в ТГФ (40 мл) гидрировали с палладием на активированном угле (10%) (0,03 г) в качестве катализатора. После поглощения водорода (1 экв.) катализатор отфильтро- 21009710 вывали и фильтрат выпаривали. Остаток растворяли в дихлорметане и остаток очищали колоночной хроматографией на силикагеле (элюент: дихлорметан). Две фракции продукта собирали и растворитель выпаривали, получая 0,045 г соединения (193). Смесь соединения (193) (0,00015 моль) и 1,4-диоксана (0,5 мл) в соляной кислоте (2 мл) перемешивали в течение 1 ч при 70 С и затем растворитель выпаривали. Остаток растворяли в дихлорметане и фильтровали через слой силикагеля (дихлорметан). Фильтрат выпаривали и полученный остаток сушили,получая 0,025 г (45%) соединения (196). Пример В 17.(0,082 г) в ДМА (50 мл) подвергали реакции в микроволновой печи при 150 С в течение 45 мин. Затем реакционную смесь вливали в воду и экстрагировали EtOAc/ДИПЭ. Экстракты промывали водой и фильтровали через Extrelut, затем растворитель выпаривали. Водную фазу экстрагировали дихлорметаном и фильтровали через Extrelut. Растворитель выпаривали и остаток очищали высокоэффективной жидкостной хроматографией с обращенной фазой. Фракции продукта собирали и растворитель выпаривали, получая соединение (159). Пример В 18. Бутиллитий (0,0011 моль) добавляли по каплям в атмосфере N2 при -78 С к раствору соединения(271) (0,0005 моль) в ТГФ (5 мл) и смесь перемешивали в течение 30 мин. Затем по каплям добавляли смесь йодпропана (0,0006 моль) в ТГФ (5 мл) и реакционную смесь перемешивали в течение 1 ч при -78 С. Смеси позволяли нагреться в течение ночи и затем добавляли насыщенный раствор NH4Cl (5 мл). Органический слой отделяли, промывали, фильтровали через Extrelut и растворитель выпаривали. Остаток(0,170 г) очищали на предварительно укомплектованной силикагелевой колонке для жидкостной хроматографии (5 г) (элюент: гексан/EtOAc 10/1). Фракции продукта собирали и растворитель выпаривали. Остаток очищали высокоэффективной жидкостной хроматографией с обращенной фазой. Фракции продукта собирали и растворитель выпаривали, получая соединение (166). Пример В 19. Смесь соединения (271) (0,00013 моль), Pd2(дибензилиденацетон)3 комплекса (0,026 г), 1,1'-бис-(дифенилфосфино)ферроцена (0,033 г) и Zn/Zn(CN)2 (0,012 г/0,105 г) в ДМА (50 мл) подвергали реакции в микроволновой печи при 150 С в течение 15 мин, затем реакционную смесь вливали в воду и экстрагировалиEtOAc/ДИПЭ. Экстракты промывали водой и растворитель выпаривали. Остаток очищали твердофазной экстракцией на предварительно укомплектованной силикагелевой колонке для жидкостной хроматографии(элюент: CH2Cl2). Фракции продукта собирали и растворитель выпаривали, получая 0,055 г соединения (272). Смесь соединения (272) (0,001 моль) в растворе аммиака в метаноле (50 мл) гидрировали при 14 С с никелем Ренея (кат. кол-во) в качестве катализатора. После поглощения водорода (2 экв.) катализатор фильтровали и фильтрат выпаривали. Остаток растворяли в дихлорметане, раствор фильтровали и фильтрат выпаривали, получая 0,270 г соединения (273).(q.s.) перемешивали в течение 15 мин при комнатной температуре. Затем по каплям добавляли смесь 4 хлорбутаноилхлорида [4635-59-0] (0,000165 моль) в дихлорметане (2,5 мл) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Смесь промывали HCl (1 Н), 5% растворомNaHCO3 и водой. Полученную смесь фильтровали через Extrelut и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/СН 3 ОН 99/1). Фракции продукта собирали и растворитель выпаривали, получая 0,072 г соединения (274) (бесцветное масло). Хлорид N,N,N-триэтилбензолметанаминия (0,00015 моль) и гидроксид натрия (50%) (0,5 мл) добавляли к раствору соединения (274) (0,00014 моль) в дихлорметане (5 мл) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Смесь промывали 2 раза HCl (1 Н), 5% растворомNaHCO3 и водой. Полученную смесь фильтровали через Extrelut и растворитель выпаривали, получая 0,050 г бесцветного масла. Остаток очищали твердофазной экстракцией на предварительно упакованной силикагелевой колонке для жидкостной хроматографии (элюент: CH2Cl2/CH3OH 90/10). Фракции продукта собирали и растворитель выпаривали, получая 0,024 г соединения (160). Пример В 20. Смесь промежуточного продукта (29) (0,0019 моль) в N,N-диэтилэтанамине (2 мл) и дихлорметане(15 мл) перемешивали и добавляли 1-гидрокси-1 Н-бензотриазол (0,002 моль). Затем добавляли N'-(этилкарбоимидоил)-N,N-диметил-1,3-пропандиамин (0,002 моль) и смесь перемешивали в течение 10 мин. Добавляли гидрохлорид 2-адамантанамина (0,0022 моль) и реакционную смесь перемешивали в течение ночи. Добавляли раствор лимонной кислоты (2 мл) и полученную смесь фильтровали через Extrelut. Фильтрат выпаривали и остаток очищали колоночной флэш-хроматографией на TRIKONEX FlashTube(элюент: CH2Cl2/EtOAc 90/10). Фракции продукта собирали и растворитель выпаривали. Такую оставшуюся фракцию очищали высокоэффективной жидкостной хроматографией, затем фракции продукта собирали и растворитель выпаривали, получая 0,155 г (25%) соединения (171). Смесь соединения (171) (0,00044 моль) в метаноле (50 мл) гидрировали в течение ночи с палладием на активированном угле (0,1 г) в качестве катализатора. После поглощения водорода (1 экв.) катализатор отфильтровывали и фильтрат выпаривали, затем остаток сушили (вак.), получая 0,12 г соединения (172). Пример В 21. Смесь соединения (170) (0,0006 моль) и формальдегида (0,2 г) в метаноле (40 мл) гидрировали при 50 С с палладием на активированном угле (0,05 г) в качестве катализатора в присутствии раствора тиофена (0,1 мл). После поглощения водорода (2 экв.) катализатор отфильтровывали и фильтрат выпаривали. Остаток растворяли в дихлорметане, промывали HCl (1 Н), 5% раствором NaHCO3 и солевым раствором. Смесь фильтровали через Extrelut и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/(CH3OH/NH3 (1% 90/10). Фракции продукта собирали и растворитель выпаривали, получая соединение (192). Пример В 22.(дибензилиденацетон)3 комплекса (0,0001 моль), трис(2-метилфенил)фосфина [6163-58-2] (0,00025 моль) и N,Nдибутил-1-бутанамина (0,0025 моль) в ДМФ (5 мл) перемешивали в течение ночи при 90 С и затем реакционную смесь охлаждали. Добавляли воду (3 мл) и смесь экстрагировали EtOAc. Органический слой отделяли, сушили(MgSO4), фильтровали и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле(элюент: дихлорметан). Фракции продукта собирали и растворитель выпаривали, получая 0,167 г соединения (198). Смесь соединения (198) (0,0003 моль) в уксусной кислоте (4 мл) и соляной кислоте (2 мл) перемешивали в течение ночи при 60 С, затем реакционную смесь охлаждали и экстрагировали дихлорметаном. Органический слой отделяли, промывали, сушили, фильтровали и растворитель выпаривали. Остаток очищали высокоэффективной жидкостной хроматографией. Фракции продукта собирали и растворитель выпаривали, получая 0,045 г соединения (202). Смесь промежуточного продукта (36) (0,0013 моль) в дихлорметане (10 мл) и N,N-диэтилэтанамина(3 мл) перемешивали и добавляли 1-гидрокси-1 Н-бензотриазол (0,002 моль). Затем добавляли моногидрохлорид N'-(этилкарбонимидоил)-N,N-диметил-1,3-пропандиамина (0,002 моль) и смесь перемешивали в течение 10 мин. После добавления ДМФ (2 мл) добавляли гидрохлорид 2-адамантанамина (0,0016 моль) и реакционную смесь перемешивали в течение ночи. Смесь промывали водой (2 мл), раствором гидроксида калия и снова водой. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH 98/2). Фракции продукта собирали и растворитель выпаривали, получая 0,176 г соединения (204). Смесь соединения (204) (0,00036 моль) в растворе ТФУ в дихлорметане (28%) (3 мл) перемешивали в течение 3 ч и затем растворитель выпаривали. Остаток растворяли в дихлорметане и раствор промывали раствором Na2CO3. Органический слой отделяли, фильтровали через Extrelut и растворитель выпаривали, получая 0,116 г соединения (208). Пример В 24.(0,0028 моль) и гидрохлорида 2-адамантанамин (0,0028 моль) в N,N-диэтилэтанамине (2 мл) и смеси дихлорметана, ТГФ и ДМФ (1/1/0,5) (50 мл) перемешивали в течение выходных дней, затем реакционную смесь вливали в воду и экстрагировали дихлорметаном. Экстракты промывали раствором лимонной кислоты (15%) и органический слой сушили, затем фильтровали. Растворитель выпаривали и остаток очищали колоночной флэш-хроматографией на TRIKONEX FlashTube (элюент: CH2Cl2/EtOAc 90/10). Фракции продукта собирали и растворитель выпаривали, получая 0,18 г соединения (252). Пример В 25. 2-Изоцианатотрицикло[3,3,1,13,7]декан [71189-14-5] (0,0053 моль) добавляли к раствору 1,2,3,4-тетрагидрохинолина (0,00586 моль) в EtOAc (10 мл) и реакционную смесь перемешивали в течение ночи. Растворитель выпаривали и остаток перекристаллизовывали из 2-пропанола. Наконец, желаемый продукт собирали, получая 0,500 г соединения (200); т.п. 163-165 С. Пример В 26. 1-[(3,4-Дигидро-1(2 Н)-хинолинил)карбонил]-3-метил-1 Н-имидазол, йодид [213134-25-9] (0,01 моль) добавляли к раствору 4-аминотрицикло[3,3,1,13,7]декан-1-ола [75375-89-2] (0,01 моль) и N,N-диэтилэтанамина (0,01 моль) в смеси дихлорметана, ТГФ и ДМФ (1/1/0,2) (100 мл) и реакционную смесь перемешивали в течение ночи. Смесь промывали 1 Н HCl, 2 Н гидроксидом калия и хлоридом натрия, затем сушили и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: гексан/EtOAc 3/11/1). Две фракции продукта собирали и растворитель выпаривали, получая 1,5 г- 24009710 1-Метилпиперазин (0,0015 моль) добавляли в 1 части к раствору соединения (270) (0,0003 моль) в дихлорметане (5 мл) и затем смесь перемешивали в течение ночи при комнатной температуре. Добавляли гидроксид натрия (1 Н) (1 мл) и реакционную смесь перемешивали тщательно в течение 30 мин. Слои разделяли и водный слой экстрагировали. Органический слой сушили, фильтровали и растворитель выпаривали, получая соединение (231). Пример В 28. Морфолин (0,0012 моль) добавляли к раствору соединения (270) (0,00044 моль) в дихлорметане (10 мл) и затем смесь перемешивали в течение ночи при комнатной температуре. Добавляли гидроксид натрия(1 Н) (1 мл) и реакционную смесь перемешивали тщательно в течение 15 мин. Водный слой отделяли и затем органический слой промывали водой и фильтровали через Extrelut. Фильтрат выпаривали и остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/CH3OH 99/1). Фракции продукта собирали и растворитель выпаривали, получая соединение (232). Пример В 29. Смесь 1-изохинолинкарбоновой кислоты (0,0056 моль) в ДМФ (50 мл) перемешивали и добавляли 1-гидрокси-1 Н-бензотриазол (0,0067 моль). Затем добавляли моногидрохлорид N'-(этилкарбонимидоил)-N,N-диметил-1,3-пропандиамина (0,00067 моль) и смесь перемешивали в течение 20 мин. Добавляли 1-адамантанамин[768-94-5] (0,0067 моль) и реакционную смесь перемешивали в течение 3 ч. Полученную смесь вливали в воду и затем экстрагировали EtOAc. Отделенный органический слой промывали, сушили (MgSO4), фильтровали и растворитель выпаривали. Оставшуюся фракцию очищали колоночной хроматографией на силикагеле (элюент: дихлорметан). Фракции продукта собирали и растворитель выпаривали, получая 1,5 г соединения (265). Смесь соединения (265) (0,004 моль) и соляной кислоты (12 Н) (1 мл) в метаноле (50 мл) гидрировали в течение ночи с платиной на активированном угле (1 г) в качестве катализатора. После поглощения водорода(2 экв.) катализатор фильтровали и фильтрат выпаривали. Остаток растворяли в дихлорметане и промывали раствором карбоната натрия. Органический слой отделяли, сушили (MgSO4), фильтровали и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/СН 3 ОН 99/195/5). Две фракции продуктов собирали и растворитель выпаривали, получая 0,8 г соединения (267). Пример В 30. Соединение 238 (0,0036 моль) растворяли в CH2Cl2 (50 мл) и раствор охлаждали до -70 С. Затем по каплям добавляли DAST (0,0015 моль) и реакционную смесь перемешивали в течение 30 мин при -70 С. После удаления холодной бани смеси позволяли достичь комнатной температуры в течение 1 ч и затем по частям добавляли насыщ. раств. NaHCO3. Отделенный органический слой промывали водой и солевым раствором, затем сушили, фильтровали и растворитель выпаривали. Остаток очищали колоночной хроматографией на силикагеле (элюент: CH2Cl2/СН 3 ОН 98/2). Фракции продукта собирали и растворитель выпаривали, получая 1 г соединения (278) (ЖХМС: 94% Ч). Смесь соединения (278) (0,002 моль) в ТГФ (50 мл) гидрировали с Pd/C 10% (0,2 г) в качестве катализатора. После поглощения водорода (2 экв.) катализатор фильтровали и фильтрат выпаривали (вак). Остаток растирали в ДИПЭ и после собирания сырой продукт очищали колоночной хроматографией на силикагеле (элюент:CH2Cl2/СН 3 ОН 99/1). Фракции продукта собирали и их растворитель выпаривали, получая соединение (279). Пример В 31. Суспензию промежуточного продукта 12 (0,0192 моль), N'-(этилкарбонимидоил)-N,N-диметил-1,3 пропандиамина (0,021 моль) и HOBt (0,021 моль) в ДМФ (10 мл) перемешивали в течение 30 мин при- 25009710 комнатной температуре, затем добавляли гидрохлорид 2-аминоадамантана [62058-03-1] (0,0231 моль) в ДМФ (q.s.) и реакционную смесь перемешивали в течение ночи. Полученный сырой продукт растирали с ДИПЭ и желаемый продукт собирали, получая 7,8 г соединения (280) (83%). Смесь соединения (280) (0,0065 моль) в ТГФ (150 мл) гидрировали с В (1 г) в качестве катализатора. После поглощения водорода (2 экв.) катализатор фильтровали и фильтрат выпаривали (вак.), получая 2,6 г соединения (281) (100%).(0,000275 моль) и HOBt (0,000275 моль) в ДМФ (10 мл) перемешивали в течение 30 мин при комнатной температуре и затем добавляли В (0,000325 моль). Реакционную смесь перемешивали в течение ночи при комнатной температуре, промывали водой и 5% раств. NaHCO3 и затем фильтровали через Extrelut. Растворитель выпаривали и остаток (0,200 г) очищали колоночной хроматографией на силикагеле (2 г)(элюент: CH2Cl2/CH3OH 95/5). Фракции чистого продукта собирали и растворитель выпаривали. Наконец, желаемый продукт сушили (вак.), получая 0,106 г соединения (277). В табл. 1, 2 и 3 перечислены соединения по настоящему изобретению, полученные в соответствии с одним из вышеуказанных примеров. Таблица 1
МПК / Метки
МПК: C07C 237/22, C07C 271/22, C07C 235/36, C07C 233/23, C07C 233/41, C07C 233/22, C07C 233/58, C07C 233/14, C07C 237/20, C07C 233/32, C07C 233/11
Метки: адамантилацетамиды, 11-бета, гидроксистероиддегидрогеназы, ингибиторы
Код ссылки
<a href="https://eas.patents.su/30-9710-adamantilacetamidy-kak-ingibitory-11-beta-gidroksisteroiddegidrogenazy.html" rel="bookmark" title="База патентов Евразийского Союза">Адамантилацетамиды как ингибиторы 11-бета гидроксистероиддегидрогеназы</a>
Ингибиторы 11-бета-гидроксистероиддегидрогеназы типа 1

Номер патента: 5274
Опубликовано: 30.12.2004
Авторы: Барф Тьерд, Эмонд Рикард, Валлгорда Йерк, Нильссон Марианне, Курц Гвидо
МПК: A61P 3/00, A61K 31/426, C07D 277/52...
Метки: 11-бета-гидроксистероиддегидрогеназы, ингибиторы, типа
Формула / Реферат:
1. Соединение формулы (II) где T представляет фенил, нафтил, гетероарил, означающий моноциклическую, би- или трициклическую ароматическую кольцевую систему, в которой только одно кольцо является ароматическим, имеющую от 5 до 14 атомов в кольце, в которой одно или несколько атомов кольца являются иными, чем углерод, такими как азот, сера, кислород и селен; фенил-C2-алкенил или нафтил-C2-алкенил, необязательно независимо замещенные [R]n, где n...
Бета-аминотетрагидроимидазо – (1, 2 – а) – пиразины и тетрагидротриазоло – (4, 3 – а ) – пиразины как ингибиторы дипептидилпептидазы для лечения или предотвращения диабета

Номер патента: 6845
Опубликовано: 28.04.2006
Авторы: Ксу Дзинйоу, Фишер Майкл Г., Парми Эмма Р., Ким Доосеоп, Вебер Энн Э., Маккосс Малкольм, Эдмондсон Скотт Д.
МПК: C07D 487/04, A61P 3/10, A61K 31/4985...
Метки: ингибиторы, лечения, бета-аминотетрагидроимидазо, предотвращения, дипептидилпептидазы, пиразины, тетрагидротриазоло, диабета
Формула / Реферат:
1. Соединение, выбранное из группы, состоящей из и их фармацевтически приемлемых солей. 2. Соединение по п.1, выбранное из группы, состоящей из или его фармацевтически приемлемая соль. 3. Соединение по п.2, которое представляет или его фармацевтически приемлемая соль. 4. Соединение по п.2, которое представляет или его фармацевтически приемлемая соль. 5. Соединение по п.2, которое представляет или его фармацевтически приемлемая соль....
S-фторметиловый эфир 6.aльфа., 9.альфа.-дифтор-17.aльфа. -[(2-фуранилкарбонил)окси]-11.бета.-гидрокси-16.aльфа.-метил-3-оксо-андроста-1,4-диен-17.бета.- карботиокислоты в качестве противовоспалительного агента

Номер патента: 5992
Опубликовано: 25.08.2005
Авторы: Кут Стивен Джон, Найс Розалин Кей, Биггадик Кейт
МПК: A61K 31/58, C07J 31/00, A61P 5/44...
Метки: карботиокислоты, s-фторметиловый, 6.aльфа, агента, 9.альфа.-дифтор-17.aльфа, эфир, 2-фуранилкарбонил)окси]-11.бета.-гидрокси-16.aльфа.-метил-3-оксо-андроста-1,4-диен-17.бета, противовоспалительного, качестве
Формула / Реферат:
1. Соединение формулы (I) и его сольваты. 2. Соединение формулы (I) по п.1 в несольватированной форме. 3. Соединение формулы (I) в несольватированной форме по п.2 в полиморфной модификации формы 1, отличающейся тем, что она имеет профиль XRPD (дифракции рентгеновских лучей на порошке), имеющий пик около 18,9° 2q . 4. Соединение формулы (I) в несольватированной форме по п.2 в полиморфной модификации формы 2, отличающейся тем, что она...
Комплексы лимфотоксина альфа/бета и антител против рецептора лимфотоксина-бета в качестве противоопухолевых агентов

Номер патента: 96
Опубликовано: 27.08.1998
Авторы: Бенджамин Кристофер, Броунинг Джеффри Л., Мейер Вернер
МПК: G01N 33/53, A61K 38/19
Метки: качестве, лимфотоксина, противоопухолевых, рецептора, против, антител, комплексы, лимфотоксина-бета, агентов
Формула / Реферат:
1. Способ лечения или уменьшения прогрессирования, тяжести или эффектов неоплазии, включающий введение терапевтически эффективного количества гетеромерного комплекса лимфотоксина a/b в присутствии терапевтически эффективного количества антитела к рецептору лимфотоксина b и/или g-интерферона. 2. Способ лечения или уменьшения прогрессирования, тяжести или эффектов неоплазии, включающий введение терапевтически эффективного количества антитела к...
Ненуклеозидные ингибиторы обратной транскриптазы

Номер патента: 5598
Опубликовано: 28.04.2005
Автор: Симоно Брюно
МПК: C07D 471/14, A61K 31/55
Метки: ненуклеозидные, ингибиторы, транскриптазы, обратной
Формула / Реферат:
1. Соединение общей формулы I где R2 выбирают из группы, включающей H, F, Cl, C1-C4алкил, C3-C4циклоалкил и CF3; R4 обозначает H или Me; R5 обозначает H, Me или Et при условии, что R4 и R5 оба не обозначают Me, и если R4 обозначает Me, то R5 не обозначает Et; R11 обозначает Me, Et, циклопропил, пропил, изопропил или циклобутил и Q выбирают из группы, включающей или его фармацевтически приемлемая соль. 2. Соединение по п.1, где R11 обозначает...
Предыдущий патент: Конструкция теплицы с рельсовой системой
Следующий патент: Способ лечения липодистрофии у млекопитающего
Случайный патент: Многослойное стекло, содержащее встроенный усилительный элемент