S-фторметиловый эфир 6.aльфа., 9.альфа.-дифтор-17.aльфа. -[(2-фуранилкарбонил)окси]-11.бета.-гидрокси-16.aльфа.-метил-3-оксо-андроста-1,4-диен-17.бета.- карботиокислоты в качестве противовоспалительного агента
Номер патента: 5992
Опубликовано: 25.08.2005
Авторы: Биггадик Кейт, Кут Стивен Джон, Найс Розалин Кей







Формула / Реферат
1. Соединение формулы (I)

и его сольваты.
2. Соединение формулы (I) по п.1 в несольватированной форме.
3. Соединение формулы (I) в несольватированной форме по п.2 в полиморфной модификации формы 1, отличающейся тем, что она имеет профиль XRPD (дифракции рентгеновских лучей на порошке), имеющий пик около 18,9° 2q .
4. Соединение формулы (I) в несольватированной форме по п.2 в полиморфной модификации формы 2, отличающейся тем, что она имеет профиль XRPD, имеющий пики около 18,4 и 21,5° 2q .
5. Соединение формулы (I) в несольватированной форме по п.2 в полиморфной модификации формы 3, отличающейся тем, что она имеет профиль XRPD, имеющий пики около 18,6 и 19,2° 2q .
6. Соединение формулы (I) по п.1 в виде кристаллического твердого вещества в форме, по существу, стехиометрического сольвата с ацетоном.
7. Соединение формулы (I) по п.1 в виде кристаллического твердого вещества в форме, по существу, стехиометрического сольвата с тетрагидрофураном.
8. Соединение формулы (I) по п.1 в виде кристаллического твердого вещества в форме, по существу, стехиометрического сольвата с изопропанолом.
9. Соединение формулы (I) по п.1 в виде кристаллического твердого вещества в форме, по существу, стехиометрического сольвата с метилэтилкетоном.
10. Соединение формулы (I) по п.1 в виде кристаллического твердого вещества в форме, по существу, стехиометрического сольвата с диметилформамидом.
11. Применение соединения формулы (I) или его физиологически приемлемого сольвата по любому из пп.1-5 в качестве противовоспалительного или противоаллергического агента в ветеринарии или медицине человека.
12. Применение соединения формулы (I) или его физиологически приемлемого сольвата по любому из пп.1-5 для производства лекарственного средства для лечения воспалительных и/или аллергических состояний.
13. Фармацевтическая композиция, содержащая соединение формулы (I) или его физиологически приемлемый сольват по любому из пп.1-5 в смеси с одним или более чем одним физиологически приемлемым разбавителем или носителем.
14. Фармацевтическая композиция по п.13, которая не находится под давлением и пригодна для местного введения в легкое через полость рта в виде сухого порошка.
15. Фармацевтическая композиция по п.13 или 14, которая содержит лактозу или крахмал в качестве разбавителя или носителя.
16. Фармацевтическая композиция по п.13, которая не находится под давлением и пригодна для местного введения в носовую полость.
17. Фармацевтическая композиция по п.16, которая содержит воду в качестве разбавителя или носителя.
18. Фармацевтическая аэрозольная композиция, содержащая соединение формулы (I) или его физиологически приемлемый сольват по любому из пп.1-5 и фторуглерод или водородсодержащий хлорфторуглерод в качестве пропеллента, возможно, в комбинации с поверхностно-активным веществом или сорастворителем.
19. Фармацевтическая аэрозольная композиция по п.18, отличающаяся тем, что лекарственное средство является полностью растворенном в этой композиции.
20. Фармацевтическая аэрозольная композиция по п.18, отличающаяся тем, что лекарственное средство находится в виде частиц, при этом указанная композиция не содержит стабилизатор в форме добавки воды (то есть воды, добавленной в дополнение к появляющейся в композиции воде) или аминокислоты, ее производного либо их смеси.
21. Фармацевтическая аэрозольная композиция по любому из пп.18-20, которая содержит соединение формулы (I) или его физиологически приемлемый сольват по любому из пп.1-3, и фторуглерод или водородсодержащий хлорфторуглерод в качестве пропеллента, и суспендирующий агент, который растворим в пропелленте.
22. Фармацевтическая аэрозольная композиция по п.21, где суспендирующий агент представляет собой олигомолочную кислоту или ее производное.
23. Фармацевтическая аэрозольная композиция по любому из пп.18-22, где пропеллент выбран из 1,1,1,2-тетрафторэтана, 1,1,1,2,3,3,3-гептафтор-н-пропана и их смесей.
24. Фармацевтическая аэрозольная композиция по любому из пп.18-20, которая состоит, по существу, из соединения формулы (I) или его физиологически приемлемого сольвата по любому из пп.1-3, возможно, в комбинации с другим терапевтически активным агентом и пропеллентом, выбранным из 1,1,1,2-тетрафторэтана, 1,1,1,2,3,3,3-гептафтор-н-пропана и их смесей.
25. Фармацевтическая композиция по любому из пп.13-23, которая дополнительно содержит другой терапевтически активный агент.
26. Фармацевтическая композиция по п.25, в которой указанный другой терапевтически активный агент представляет собой агонист b2-адренорецепторов.
27. Фармацевтическая композиция, содержащая комбинацию соединения формулы (I) или его физиологически приемлемого сольвата по любому из пп.1-5 вместе с ингибитором фосфодиэстеразы 4 совместно с физиологически приемлемым разбавителем или носителем.
28. Способ лечения человека или животного, имеющего воспалительное и/или аллергическое состояние, при котором указанному человеку или животному вводят эффективное количество соединения формулы (I) или его физиологически приемлемого сольвата по любому из пп.1-5.
29. Способ получения соединения формулы (I) по п.1 или его сольвата, при котором осуществляют алкилирование по 17b-карботиокислотной группировке соединения формулы (II)

или его соли.
30. Способ по п.29, где алкилирование осуществляют путем взаимодействия соединения формулы (II) или его соли с фторметилгалогенидом.
31. Способ получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 1 по п.3, при котором осуществляют кристаллизацию соединения формулы (I) в присутствии несольватирующего растворителя.
32. Способ получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 1 по п.3, при котором осуществляют десольватацию соединения формулы (I) в сольватированной форме.
33. Способ получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 1 по п.3, при котором соединение формулы (I) растворяют в метилизобутилкетоне, этилацетате или метилацетате и получают соединение формулы (I) в виде несольватированной формы 1 путем добавления несольватирующего антирастворителя.
34. Соединение формулы (II)

или его соль.
35. Соединение формулы (II) по п.34 в форме твердой кристаллической соли.
36. Соединение формулы (II) по п.35 в форме соли с диизопропилэтиламином.
37. Способ получения соединения формулы (II) по п.34, при котором
(а) соединение формулы (III)

подвергают взаимодействию с активированным производным 2-фуранкарбоновой кислоты, например, в количестве по меньшей мере 2 моль активированного производного на моль соединения формулы (III) с получением соединения формулы (IIA)

и
(б) удаляют присоединенную через атом серы 2-фуроильную группировку из соединения формулы (IIA) посредством взаимодействия продукта стадии (а) с органическим первичным или вторичным аминным основанием, способным к образованию водорастворимого 2-фуроиламида.
38. Способ получения соединения формулы (II) по п.37, при котором, если продукт стадии (б) растворяют в органическом растворителе, по существу не смешивающемся с водой, дополнительно осуществляют стадию, на которой соединение формулы (II) очищают путем вымывания амидного побочного продукта со стадии (б) посредством водной промывки.
39. Способ получеэшя соединения формулы (II) по п.37, при котором, если продукт стадии (б) растворяют в смешивающемся с водой растворителе, дополнительно осуществляют стадию, на которой соединение формулы (II) очищают путем обработки продукта стадии (б) водной средой таким образом, чтобы осадить чистое соединение формулы (II) или его соль.
40. Способ получения соединения формулы (II) по п.34, при котором
(а) соединение формулы (III)

подвергают взаимодействию с активированным производным 2-фуранкарбоновой кислоты в количестве по меньшей мере 2 моль активированного производного на моль соединения формулы (III) с получением соединения формулы (IIA), как оно определено в п.37, и
(б) удаляют присоединенную через атом серы 2-фуроильную группировку из соединения формулы (IIA) посредством взаимодействия продукта стадии (а) с дополнительным молем соединения формулы (III) с получением 2 моль соединения формулы (II).
41. Соединение формулы (IIA)

42. Соединение формулы (VI)

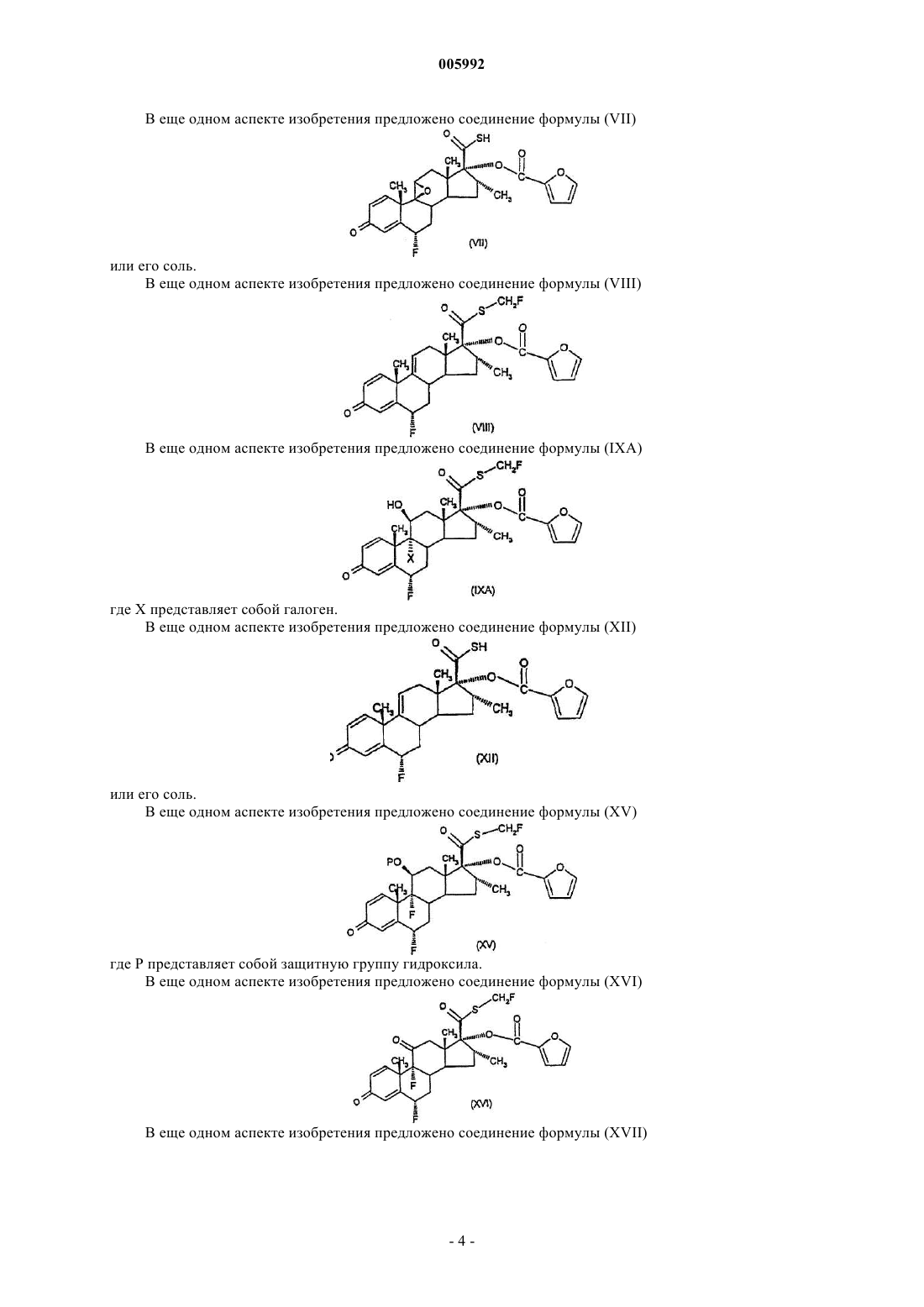
43. Соединение формулы (VII)

или его соль.
44. Соединение формулы (VIII)

45. Соединение формулы (IXA)

где X представляет собой галоген.
46. Соединение формулы (XII)

или его соль.
47. Соединение формулы (XV)

где P представляет собой защитную группу гидроксила.
48. Соединение формулы (XVI)

49. Соединение формулы (XVII)

или его соль, где P представляет собой защитную группу гидроксила.
50. Соединение формулы (XX)

или его соль либо производное, где 11-карбонильная группа замаскирована.
51. Соединение формулы (XXIII)

где L представляет собой уходящую группу, отличную от фтора.
52. Способ получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 2 по п.4, при котором соединение формулы (I) в несольватированной форме растворяют в метаноле или безводном дихлорметане и осуществляют перекристаллизацию соединения формулы (I) в виде несольватированной полиморфной модификации формы 2.
53. Способ получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 3 по п.5, при котором соединение формулы (I) или его сольват растворяют в дихлорметане в присутствии воды и осуществляют перекристаллизацию соединения формулы (I) в виде несольватированной полиморфной модификации формы 3.
54. Способ получения соединения формулы (I) по п.1 или его сольвата, при котором соединение формулы (VI)

подвергают взаимодействию с источником фтора.
55. Способ получения соединения формулы (I) по п.1 или его сольвата, при котором удаляют защиту или демаскируют соединение формулы (I), в котором 11-b-гидроксигруппа защищена или замаскирована.
56. Способ по п.55, где 11-b-гидроксигруппа защищена, при котором удаляют защиту с соединения формулы (XV)

где P представляет собой защитную группу гидроксила.
57. Способ по п.55, где 11-b-гидроксигруппа замаскирована, при котором восстанавливают соединение формулы (XVI)

или его производное, где 11-карбонильная группа замаскирована.
58. Способ получения соединения формулы (I) по п.1 или его сольвата, при котором соединение формулы (XXIII)

где L представляет собой уходящую группу, подвергают взаимодействию с источником фтора.
59. Соединение формулы (X)

60. Способ получения соединения формулы (I) по п.1 или его сольвата, при котором удаляют защиту или демаскируют производное соединения формулы (I), в котором 3-карбонильная группа защищена или замаскирована.
61. Способ получения соединения формулы (II), как оно определено в п.34, при котором соединение формулы (X), как оно определено в п.59, обрабатывают реагентом, подходящим для превращения карбоновой кислоты в карботиокислоту.
Текст
005992 Настоящее изобретение относится к новому противовоспалительному и противоаллергическому соединению андростанового ряда и способам его получения. Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим данное соединение, и к их терапевтическим применениям, в частности, для лечения воспалительных и аллергических состояний. Для лечения воспалительных расстройств или заболеваний, таких как астма и ринит, известны и широко применяются глюкокортикоиды, обладающие противовоспалительными свойствами. Например,в патенте США 4335121 раскрыты S-фторметиловый эфир 6,9-дифтор-17-(1-оксопропокси)-11 гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты (известный под международным непатентованным наименованием как пропионат флутиказона) и его производные. Как правило, применение глюкокортикоидов, в особенности у детей, в некоторых странах ограничено вследствие весьма вероятных побочных действий. Побочные действия, которых опасаются при приеме глюкокортикоидов,включают в себя подавление системы гипоталамус-гипофиз-надпочечники (НРА), воздействия на рост костей у детей и плотность костей у пожилых, офтальмологические осложнения (формирование катаракты и глаукома) и кожную атрофию. Кроме того, некоторые глюкокортикоидные соединения имеют сложные пути метаболизма, при этом образование активных метаболитов может сделать фармакодинамику и фармакокинетику таких соединений трудными для понимания. Несмотря на то, что современные стероиды гораздо безопаснее первоначально введенных, создание новых молекул, которые имеют превосходные противовоспалительные свойства с предсказуемыми фармакокинетическими и фармакодинамическими свойствами, с приемлемым профилем побочных действий и с удобной схемой лечения, остается задачей исследования. Авторами изобретения в настоящее время идентифицировано новое глюкокортикоидное соединение, которое, по существу, удовлетворяет этим требованиям. Таким образом, согласно одному аспекту данного изобретения предложено соединение формулы (I) и его сольваты. Химическим названием соединения формулы (I) является S-фторметиловый эфир 6,9-дифтор 17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты. Ссылки ниже на соединение по данному изобретению включают в себя как соединение формулы(I), так и его сольваты, в частности фармацевтически приемлемые сольваты. Предпочтительным является соединение формулы (I) в несольватированной форме. Предпочтительно соединение формулы (I) в несольватированной форме представляет собой полиморфную модификацию формы 1, формы 2 или формы 3. В одном аспекте изобретения предложено соединение формулы (I) в несольватированной форме в полиморфной модификации формы 1, характеризующейся тем, что она имеет профиль дифракции рентгеновских лучей на порошке (XRPD), имеющий пик около 18,9 2. В другом аспекте изобретения предложено соединение формулы (I) в несольватированной форме в полиморфной модификации формы 2, характеризующейся тем, что она имеет профиль XRPD, имеющий пики около 18,4 и 21,5 2. В еще одном аспекте изобретения предложено соединение формулы (I) в несольватированной форме в полиморфной модификации формы 3, характеризующейся тем, что она имеет профиль XRPD,имеющий пики около 18,6 и 19,2 2. В предпочтительном аспекте предложено соединение формулы (I) в виде кристаллического твердого вещества в форме, по существу, стехиометрического сольвата с ацетоном. В другом предпочтительном аспекте предложено соединение формулы (I) в виде кристаллического твердого вещества в форме, по существу, стехиометрического сольвата с тетрагидрофураном. В еще одном предпочтительном аспекте предложено соединение формулы (I) в виде кристаллического твердого вещества в форме, по существу, стехиометрического сольвата с изопропанолом. В еще одном предпочтительном аспекте предложено соединение формулы (I) в виде кристаллического твердого вещества в форме, по существу, стехиометрического сольвата с метилэтилкетоном. В еще одном предпочтительном аспекте предложено соединение формулы (I) в виде кристаллического твердого вещества в форме, по существу, стехиометрического сольвата с диметилформамидом. Согласно одному аспекту изобретения предложено применение соединения формулы (I) или его физиологически приемлемого сольвата, как они определены выше, в качестве противовоспалительного или противоаллергического агента в ветеринарии или медицине человека.-1 005992 Согласно другому аспекту изобретения предложено применение соединения формулы (I) или его физиологически приемлемого сольвата, как они определены выше, для производства лекарственного средства для лечения воспалительных и/или аллергических состояний. В следующем аспекте изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его физиологически приемлемый сольват, как они определены выше, в смеси с одним или более чем одним физиологически приемлемым разбавителем или носителем, которая предпочтительно не находится под давлением и пригодна для местного введения в легкое через полость рта в виде сухого порошка и которая предпочтительно содержит лактозу или крахмал в качестве разбавителя или носителя. В другом аспекте изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его физиологически приемлемый сольват, как они определены выше, в смеси с одним или более чем одним физиологически приемлемым разбавителем или носителем, которая предпочтительно не находится под давлением и пригодна для местного введения в носовую полость и которая предпочтительно содержит воду в качестве разбавителя или носителя. В еще одном аспекте изобретения предложена фармацевтическая аэрозольная композиция, содержащая соединение формулы (I) или его физиологически приемлемый сольват, как они определены выше,и фторуглерод или водородсодержащий хлорфторуглерод в качестве пропеллента, возможно, в комбинации с поверхностно-активным веществом или сорастворителем; где лекарственное средство предпочтительно является полностью растворенным в этой композиции, более предпочтительно указанное лекарственное средство находится в виде частиц, при этом указанная композиция не содержит стабилизатор в форме добавки воды (т.е. воды, добавленной в дополнение к появляющейся в композиции воде) или аминокислоты, ее производного либо их смеси. В предпочтительном аспекте изобретения фармацевтическая аэрозольная композиция по изобретению содержит соединение формулы (I) или его физиологически приемлемый сольват, как определено для полиморфной модификации формы 1, и фторуглерод или водородсодержащий хлорфторуглерод в качестве пропеллента, и суспендирующий агент, который растворим в пропелленте; где суспендирующий агент предпочтительно представляет собой олигомолочную кислоту или ее производное; и где пропеллент предпочтительно выбран из 1,1,1,2-тетрафторэтана, 1,1,1,2,3,3,3-гептафтор-н-пропана и их смесей. В другом аспекте изобретения предложена фармацевтическая аэрозольная композиция, как она определена выше, которая состоит, по существу, из соединения формулы (I) или его физиологически приемлемого сольвата, как они определены выше, возможно, в комбинации с другим терапевтически активным агентом и пропеллентом, выбранным из 1,1,1,2-тетрафторэтана, 1,1,1,2,3,3,3-гептафтор-н-пропана и их смесей. В еще одном аспекте изобретения предложена фармацевтическая композиция по изобретению, которая дополнительно содержит другой терапевтически активный агент, где указанный другой терапевтически активный агент предпочтительно представляет собой агонист 2-адренорецепторов. В еще одном аспекте изобретения предложена фармацевтическая композиция, содержащая комбинацию соединения формулы (I) или его физиологически приемлемого сольвата, как они определены выше, вместе с ингибитором фосфодиэстеразы 4 совместно с физиологически приемлемым разбавителем или носителем. В следующем аспекте изобретения предложен способ лечения человека или животного, имеющего воспалительное и/или аллергическое состояние, при котором указанному человеку или животному вводят эффективное количество соединения формулы (I) или его физиологически приемлемого сольвата, как они определены выше. В еще одном аспекте изобретения предложен способ получения соединения формулы (I) по изобретению или его сольвата, при котором осуществляют алкилирование по 17-карботиокислотной группировке соединения формулы (II) или его соли. В предпочтительном способе по изобретению алкилирование осуществляют путем взаимодействия соединения формулы (II) или его соли с фторметилгалогенидом. В особом аспекте изобретения предложен способ получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 1, как она определена выше, при котором осуществляют кристаллизацию соединения формулы (I) в присутствии несольватирующего растворителя.-2 005992 В еще одном аспекте изобретения предложен способ получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 1, как она определена выше, при котором осуществляют десольватацию соединения формулы (I) в сольватированной форме. В еще одном аспекте изобретения предложен способ получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 1, как она определена выше, при котором соединение формулы (I) растворяют в метилизобутилкетоне, этилацетате или метилацетате и получают соединение формулы (I) в виде несольватированной формы 1 путем добавления несольватирующего антирастворителя. В следующем аспекте изобретения предложено соединение формулы (II), как оно определено выше,или его соль, более предпочтительно - в форме твердой кристаллической соли, наиболее предпочтительно - в форме соли с диизопропилэтиламином. В еще одном аспекте изобретения предложен способ получения соединения формулы (II), при котором подвергают взаимодействию с активированным производным 2-фуранкарбоновой кислоты, например в количестве по меньшей мере 2 моль активированного производного на моль соединения формулы (III), с получением соединения формулы (IIА)(б) удаляют присоединенную через атом серы 2-фуроильную группировку из соединения формулы(IIА) посредством взаимодействия продукта стадии (а) с органическим первичным или вторичным аминным основанием, способным к образованию водорастворимого 2-фуроиламида; причем если продукт стадии (б) растворяют в органическом растворителе, по существу не смешивающемся с водой, то дополнительно осуществляют стадию, на которой соединение формулы (II) очищают путем вымывания амидного побочного продукта со стадии (б) посредством водной промывки; или если продукт стадии (б) растворяют в смешивающемся с водой растворителе, то дополнительно осуществляют стадию, на которой соединение формулы (II) очищают путем обработки продукта стадии(б) водной средой таким образом, чтобы осадить чистое соединение формулы (II) или его соль. В другом аспекте изобретения предложен способ получения соединения формулы (II), при котором(а) соединение формулы (III), как оно определено выше, подвергают взаимодействию с активированным производным 2-фуранкарбоновой кислоты в количестве по меньшей мере 2 моль активированного производного на моль соединения формулы (III) с получением соединения формулы (IIА), как оно определено выше, и(б) удаляют присоединенную через атом серы 2-фуроильную группировку из соединения формулы(IIА) посредством взаимодействия продукта стадии (а) с дополнительным молем соединения формулы(III) с получением 2 моль соединения формулы (II). В одном аспекте изобретения предложено соединение формулы (IIА), как оно определено выше. В другом аспекте изобретения предложено соединение формулы (VI)-3 005992 В еще одном аспекте изобретения предложено соединение формулы (VII) или его соль. В еще одном аспекте изобретения предложено соединение формулы (VIII) В еще одном аспекте изобретения предложено соединение формулы (IXA) где X представляет собой галоген. В еще одном аспекте изобретения предложено соединение формулы (XII) или его соль. В еще одном аспекте изобретения предложено соединение формулы (XV) где Р представляет собой защитную группу гидроксила. В еще одном аспекте изобретения предложено соединение формулы (XVI) В еще одном аспекте изобретения предложено соединение формулы (XVII) или его соль, где Р представляет собой защитную группу гидроксила. В еще одном аспекте изобретения предложено соединение формулы (XX) или его соль либо производное, где 11-карбонильная группа замаскирована. В еще одном аспекте изобретения предложено соединение формулы (XXIII) где L представляет собой уходящую группу, отличную от фтора. В следующем аспекте изобретения предложен способ получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 2, как она определена выше, при котором соединение формулы (I) в несольватированной форме растворяют в метаноле или безводном дихлорметане и осуществляют перекристаллизацию соединения формулы (I) в виде несольватированной полиморфной модификации формы 2. В еще одном аспекте изобретения предложен способ получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 3, как она определена выше, при котором соединение формулы (I) или его сольват растворяют в дихлорметане в присутствии воды и осуществляют перекристаллизацию соединения формулы (I) в виде несольватированной полиморфной модификации формы 3. В еще одном аспекте изобретения предложен способ получения соединения формулы (I) по изобретению или его сольвата, при котором соединение формулы (VI), как оно определено выше, подвергают взаимодействию с источником фтора. В еще одном аспекте изобретения предложен способ получения соединения формулы (I) по изобретению или его сольвата, при котором удаляют защиту или демаскируют соединение формулы (I), в котором 11 гидроксигруппа защищена или замаскирована. В одном предпочтительном способе по изобретению удаляют защиту с соединения формулы (XV), как оно определено выше. В другом предпочтительном способе по изобретению восстанавливают соединение формулы (XVI), как оно определено выше, или его производное, где 11-карбонильная группа замаскирована. В еще одном аспекте изобретения предложен способ получения соединения формулы (I) по изобретению или его сольвата, при котором соединение формулы (XXIII), как оно определено выше, подвергают взаимодействию с источником фтора. В еще одном аспекте изобретения предложено соединение формулы (X) В еще одном аспекте изобретения предложен способ получения соединения формулы (I) по изобретению или его сольвата, при котором удаляют защиту или демаскируют производное соединения формулы (I), в котором 3-карбонильная группа защищена или замаскирована.-5 005992 В еще одном аспекте изобретения предложен способ получения соединения формулы (II), как оно определено выше, при котором соединение формулы (X), как оно определено выше, обрабатывают реагентом, подходящим для превращения карбоновой кислоты в карботиокислоту. Соединение формулы (I) оказывает потенциально полезные противовоспалительные или противоаллергические действия, в частности при местном введении, продемонстрированные, например, его способностью связываться с глюкокортикоидным рецептором и препятствовать осуществлению ответа через этот рецептор. Следовательно, соединение формулы (I) полезно в лечении воспалительных и/или аллергических расстройств. Соединение (I) подвергается высокоэффективному печеночному метаболизму в крысиных и человеческих системах in vitro с образованием 17-карбоновой кислоты (X) как единственного главного метаболита. Этот метаболит синтезировали, и было показано, что он более чем в 1000 раз менее активен,чем родоначальное соединение в исследованиях функциональной активности глюкокортикоидов in vitro. Такой эффективный печеночный метаболизм отражен в данных in vivo на крысах, которые демонстрируют плазменный клиренс со скоростью, почти равной печеночному кровотоку, и пероральную биодоступность 1%, что согласуется с экстенсивным метаболизмом первого прохождения. Исследования in vitro метаболизма в гепатоцитах человека показали, что соединение (I) метаболизируется идентичным пропионату флутиказона образом, но превращение соединения (I) в неактивный кислый метаболит происходит приблизительно в 5 раз быстрее, чем в случае с пропионатом флутиказона. Полагают, что такая очень эффективная инактивация в печени будет минимизировать системное воздействие на человека, приводя к улучшенному профилю безопасности. Кроме того, вдыхаемые стероиды всасываются через легкое, и такой способ всасывания вносит значительный вклад в системное воздействие. Следовательно, сниженное легочное всасывание могло бы обеспечить улучшенный профиль безопасности. Исследования, проведенные с соединением формулы (I),показали значительно более слабое системное действие соединения формулы (I) после доставки сухого порошка в легкие анестезированных свиней, чем в случае с пропионатом флутиказона. Полагают, что улучшенный профиль безопасности дает возможность продемонстрировать желаемые противовоспалительные эффекты соединения формулы (I) при введении один раз в день. Дозирование один раз в день считается значительно более удобным для пациентов, чем схема дозирования два раза в день, которая обыкновенно используется для пропионата флутиказона. Примеры болезненных состояний, при которых применимо соединение по данному изобретению,включают в себя кожные заболевания, такие как экзема, псориаз, аллергический дерматит, нейродермит,зуд и аллергические реакции; воспалительные состояния носа, горла или легких, такие как астма (включая индуцированные аллергеном астматические реакции), ринит (включая сенную лихорадку), носовые полипы, хроническое обструктивное заболевание легких, коллагеновую болезнь легких и фиброз; воспалительные состояния кишечника, такие как неспецифический язвенный колит и болезнь Крона; и аутоиммунные заболевания, такие как ревматоидный артрит. Кроме того, соединение по данному изобретению можно использовать в лечении конъюнктивы и конъюнктивита. Специалистам в данной области техники будет ясно, что ссылка здесь на лечение распространяется как на профилактику, так и на лечение установленных состояний. Как упомянуто выше, соединение формулы (I) полезно в медицине человека или ветеринарии, в частности в лечении пациентов с воспалительными и/или аллергическими состояниями, в особенности для лечения один раз в день. Соединение по данному изобретению может быть приготовлено в виде препарата для введения любым удобным способом, и поэтому в пределах объема данного изобретения также находятся фармацевтические композиции, содержащие соединение формулы (I) или его физиологически приемлемый сольват совместно, при желании, в смеси с одним или более чем одним физиологически приемлемым разбавителем или носителем. Фармацевтические композиции, подходящие для введения один раз в день,представляют особый интерес. Кроме того, предложен способ приготовления таких фармацевтических композиций, при котором смешивают указанные ингредиенты. Соединение по изобретению может быть, например, приготовлено в виде препарата для перорального, трансбуккального, сублингвального, парентерального, местного или ректального введения, в особенности для местного введения. Термин местное введение, как он использован здесь, включает в себя введение путем инсуффляции и ингаляции. Примеры различных типов препарата для местного введения включают в себя мази,лосьоны, кремы, гели, пены, препараты для доставки с помощью трансдермальных пластырей, порошки,спреи, аэрозоли, капсулы или картриджи для применения в ингаляторе или инсуффляторе либо капли(например глазные капли или капли в нос), растворы/суспензии для распыления, суппозитории, пессарии, удерживающие клизмы и жевательные либо сосательные таблетки или шарики (например, для лечения афтозных язв), либо липосомные или микроинкапсулированные препараты.-6 005992 Композиции для местного введения в легкое преимущественно включают в себя сухие порошковые композиции и композиции в виде спрея. Сухие порошковые композиции для местной доставки в легкое могут быть представлены, например, в капсулах и картриджах для применения в ингаляторе или инсуффляторе, например, из желатина. Обычно композиции содержат порошковую смесь для ингаляции соединения по изобретению и подходящую порошковую основу, такую как лактоза или крахмал. Обычно каждая капсула и картридж могут содержать от 20 мкг до 10 мг соединения формулы (I). Альтернативно, соединение по изобретению может быть представлено без эксципиентов. Упаковка такой композиции может быть пригодной для доставки одной дозы или множества доз. В случае доставки множества доз количество композиции может быть отмерено предварительно (как, например, в Diskus, см. GB 2242134, или Diskhaler, см. GB 2178965,2129691 и 2169265) или отмерено при применении (как, например, в Turbuhaler, см. ЕР 69715). Примером устройства для одной дозы является Rotahaler (см. GB 2064336). Ингаляционное устройство Diskus включает в себя вытянутую полоску, образованную из подложки, имеющей большое количество углублений, расположенных по всей ее длине, и покрывающего листа, прикрепленного к подложке герметично, но с возможностью отслаивания, с целью разграничения большого количества контейнеров, причем каждый контейнер содержит вдыхаемый препарат, содержащий соединение формулы (I), предпочтительно в комбинации с лактозой. Предпочтительно, чтобы данная полоска была достаточно гибкой для того, чтобы быть смотанной в рулон. Покрывающий лист и подложка предпочтительно будут иметь находящиеся впереди концевые участки, которые не прикреплены друг к другу, и по меньшей мере один из указанных находящихся впереди концевых участков сконструирован с целью присоединения к наматывающему устройству. Кроме того, предпочтительно, чтобы герметичное крепление между подложкой и покрывающим листом простиралось на всю их ширину. Покрывающий лист может предпочтительно отслаиваться от подложки в продольном направлении от первого конца указанной подложки. Фармацевтические композиции, которые не находятся под давлением и пригодны для местного введения в легкое через полость рта в виде сухого порошка (в особенности таковые без эксципиента или приготовленные в виде препарата с разбавителем или носителем, таким как лактоза или крахмал, наиболее предпочтительно лактоза), представляют особый интерес. Например, композиции в виде спрея могут быть приготовлены в виде водных растворов или суспензий либо аэрозолей, доставляемых из находящихся под давлением упаковок, таких как дозирующий ингалятор, с применением подходящего сжиженного пропеллента. Аэрозольные композиции, подходящие для ингаляции, могут представлять собой либо суспензию, либо раствор и обычно содержат соединение формулы (I) и подходящий пропеллент, такой как фторуглерод или водородсодержащий хлорфторуглерод либо их смеси, в частности гидрофторалканы, в особенности 1,1,1,2-тетрафторэтан,1,1,1,2,3,3,3-гептафтор-н-пропан или их смесь. Аэрозольная композиция может возможно содержать дополнительные эксципиенты, используемые при приготовлении лекарственных средств, хорошо известные в данной области техники в качестве поверхностно-активных веществ, например олеиновую кислоту или лецитин, и сорастворителей, например этанол. Одна композиция, взятая в качестве примера, не содержит эксципиентов и состоит, по существу, из (например, состоит из) соединения формулы (I) (предпочтительно в несольватированной форме, например в виде формы 1) (возможно в комбинации с другим терапевтически активным ингредиентом) и пропеллента, выбранного из 1,1,1,2-тетрафторэтана,1,1,1,2,3,3,3-гептафтор-н-пропана и их смеси. Другая композиция, взятая в качестве примера, содержит соединение формулы (I) в виде частиц, пропеллент, выбранный из 1,1,1,2-тетрафторэтана, 1,1,1,2,3,3,3 гептафтор-н-пропана и их смеси, и суспендирующий агент, который растворим в пропелленте, например олигомолочную кислоту или ее производное, как описано в WO 94/21229. Предпочтительным пропеллентом является 1,1,1,2-тетрафторэтан. Как отмечено в другом месте этого описания, соединение формулы (I), по-видимому, не образует сольват с 1,1,1,2-тетрафторэтаном. Как правило, находящиеся под давлением композиции будут содержаться в небольших жестяных банках (например, в алюминиевых банках), закрывающихся с помощью клапана (например, дозирующего клапана) и помещенных в привод,снабженный раструбом. Желательно, чтобы лекарственные средства для введенияпосредством ингаляции имели контролируемый размер частиц. Оптимальный размер частиц для ингаляции в бронхиальную систему обычно составляет 1-10 мкм, предпочтительно 2-5 мкм. Обычно частицы, имеющие размер более 20 мкм, являются слишком большими, чтобы при ингаляции достичь малых дыхательных путей. Для достижения таких размеров частиц частицы соединения формулы (I) при производстве могут быть уменьшены в размере традиционными способами, например посредством микронизации. Желаемая фракция может быть отделена посредством воздушной сепарации или просеивания. Предпочтительно частицы будут кристаллическими, полученными, например, способом, при котором осуществляют смешивание в камере с непрерывным потоком в присутствии ультразвукового излучения текущего раствора соединения формулы (I) в качестве лекарственного средства в жидком растворителе с текущим жидким антирастворителем для указанного лекарственного средства (например, как описано в международной патентной заявке PCT/GB 99/04368), или же способом, при котором пропускают струю раствора вещества в жидком растворителе и по касательной к ней струю жидкого антирастворителя для указанного вещества в цилиндрическую сме-7 005992 сительную камеру, имеющую аксиальное выходное отверстие, так что указанные струи таким образом очень хорошо перемешиваются посредством образования завихрения, что таким образом приводит к осаждению кристаллических частиц вещества (например, как описано в международной патентной заявке PCT/GB 00/04327). Обычно, когда используется такой эксципиент, как лактоза, размер частиц эксципиента будет значительно больше размера частиц вдыхаемого лекарственного средства по настоящему изобретению. Когда эксципиент представляет собой лактозу, как правило, он будет представлен в виде измельченной лактозы, причем не более чем у 85% частиц лактозы MMD (средний массовый диаметр,mass median diameter) будет составлять 60-90 мкм и не менее чем у 15% MMD будет составлять менее 15 мкм. Композиции для местного введения в нос (например, для лечения ринита) включают в себя находящиеся под давлением аэрозольные композиции и водные композиции, вводимые в нос с помощью нагнетательного насоса. Композиции, которые не находятся под давлением и пригодны для местного введения в носовую полость, представляют особый интерес. Для этой цели такая композиция предпочтительно содержит воду в качестве разбавителя или носителя. Водные композиции для введения в легкое или нос могут быть снабжены такими традиционными эксципиентами, как забуферивающие агенты, изменяющие тонус агенты и им подобное. Кроме того, водные композиции могут быть введены в нос посредством распыления. Другие возможные формы представления включают в себя следующее. Мази, кремы и гели могут быть приготовлены, например, с водной или масляной основой с добавлением подходящих загустителя и/или гелеобразующего агента и/или растворителей. Так, например, такие основы могут включать в себя воду и/или масло, такое как вазелиновое масло или растительное масло, такое как арахисовое масло или касторовое масло, либо растворитель, такой как полиэтиленгликоль. Загустители и гелеобразующие агенты, которые могут быть использованы в соответствии с природой данной основы, включают в себя мягкий парафин, стеарат алюминия, цетостеариловый спирт, полиэтиленгликоли, природный липид, обнаруживаемый на поверхности шерсти (woolfat), пчелиный воск, производные карбоксиполиметилена и целлюлозы и/или глицерилмоностеарат, и/или неионные эмульгирующие агенты. Лосьоны могут быть приготовлены с водной или масляной основой и, как правило, также будут содержать один или более чем один эмульгирующий агент, стабилизатор, диспергирующий агент, суспендирующий агент или загуститель. Порошки для наружного применения могут быть приготовлены с помощью подходящей порошковой основы, например талька, лактозы или крахмала. Капли могут быть приготовлены с водной или неводной основой, содержащими также один или более чем один диспергирующий агент, солюбилизирующий агент, суспендирующий агент или консервант. При желании композиции по данному изобретению могут быть забуферены путем добавления подходящих забуферивающих агентов. Доля активного соединения формулы (I) в композициях для местного применения по данному изобретению зависит от определенного типа препарата, который следует приготовить, но, как правило, будет лежать в пределах от 0,001 до 10 мас.%. Однако, как правило, для большинства типов препаратов применяемая доля преимущественно будет лежать в пределах от 0,005 до 1% и предпочтительно от 0,01 до 0,5%. Однако в порошках для ингаляции или инсуффляции применяемая доля обычно будет лежать в пределах от 0,1 до 5%. Аэрозольные композиции предпочтительно приготавливают таким образом, чтобы каждая отмеренная доза или пшик аэрозоля содержал 1-2000 мкг, например 20-2000 мкг, предпочтительно приблизительно 20-500 мкг соединения формулы (I). Введение может быть осуществлено один раз в день или несколько раз в день, например 2, 3, 4 или 8 раз, каждый раз с введением 1, 2 или 3 доз. Предпочтительно соединение формулы (I) доставляют один или два раза в день, более предпочтительно один раз в день. Суммарная суточная доза аэрозоля обычно будет лежать в интервале от 10 мкг до 10 мг, например от 100 мкг до 10 мг, предпочтительно 200-2000 мкг. Композиции для местного применения могут быть введены посредством одной или более чем одной аппликации в день на пораженную область; преимущественно могут быть использованы окклюзионные повязки, превышающие по площади области кожи. С помощью адгезивной резервуарной системы можно добиться непрерывной или пролонгированной доставки. Для приема внутрь соединение по изобретению может быть, например, приготовлено в виде препарата традиционным образом для перорального, парентерального или ректального введения. Препараты для перорального введения включают в себя сиропы, эликсиры, порошки, гранулы, таблетки и капсулы,которые обычно содержат традиционные эксципиенты, такие как связывающие агенты, наполнители,смазывающие вещества, разрыхлители, увлажняющие агенты, суспендирующие агенты, эмульгирующие агенты, консерванты, буферные соли, корригенты, красители и/или подсластители, как целесообразно. Однако предпочтительны стандартные лекарственные формы, которые описаны ниже.-8 005992 Предпочтительные формы препарата для приема внутрь представляют собой стандартные лекарственные формы, т.е. таблетки и капсулы. Такие стандартные лекарственные формы содержат от 0,1 до 20 мг, предпочтительно от 2,5 до 10 мг соединения по изобретению. Соединение согласно данному изобретению, как правило, может быть введено путем приема внутрь в случаях, когда показана системная адренокортикальная терапия. В общих чертах, препараты для приема внутрь могут содержать от 0,05 до 10% активного ингредиента в зависимости от типа рассматриваемого препарата. Суточная доза может варьировать от 0,1 до 60 мг, например 5-30 мг, в зависимости от состояния, которое лечат, и требуемой продолжительности лечения. Препараты с замедленным высвобождением или препараты с энтеросолюбильным покрытием могут быть полезны, в частности, для лечения воспалительных расстройств кишечника. Фармацевтические композиции по данному изобретению, кроме того, могут быть использованы в комбинации с другим терапевтически активным агентом, например агонистом 2-адренорецепторов, антигистаминным или противоаллергическим средством. Таким образом, согласно данному изобретению предложена в еще одном аспекте комбинация, содержащая соединение формулы (I) или его физиологически приемлемый сольват совместно с другим терапевтически активным агентом, например агонистом 2-адренорецепторов, антигистаминным или противоаллергическим средством. Примеры агонистов 2-адренорецепторов включают в себя сальметерол (например, в виде рацемата или единственного энантиомера, такого как R-энантиомер), сальбутамол, формотерол, сальмефамол(salmefamol), фенотерол или тербуталин и их соли, например соль ксинафоат сальметерола, соль сульфат или свободное основание сальбутамола или соль фумарат формотерола. Примеры антигистаминных препаратов включают в себя метапирилен или лоратадин. Другие подходящие комбинации включают в себя, например, другие противовоспалительные агенты, к примеру NSAID (нестероидные противовоспалительные средства) (например, кромогликат натрия,недокромил натрия, ингибиторы фосфодиэстеразы 4, антагонисты лейкотриена, ингибиторы индуцибельной изоформы синтазы оксида азота (iNOS), ингибиторы триптазы и эластазы, антагонисты -2 интегрина и агонисты аденозина 2 а), или антимикробные агенты (например, антибиотики, противовирусные средства). Особый интерес представляет применение соединения формулы (I) в комбинации с ингибитором фосфодиэстеразы 4 (PDE4). РDЕ 4-специфичный ингибитор, полезный в этом аспекте данного изобретения, может быть любым соединением, про которое известно, что оно ингибирует фермент PDE4, или которое описано как действующее в качестве ингибитора PDE4, и которое является ингибитором толькоPDE4, не соединением, которое ингибирует другие члены PDE-семейства так же, как и PDE4. Как правило, предпочтительно использование ингибитора PDE4, который имеет соотношение значений IС 50, равное приблизительно 0,1 или более, а именно IC50 для каталитической формы PDE4, которая связывает ролипрам с высокой аффинностью, деленное на IC50 для такой формы, которая связывает ролипрам с низкой аффинностью. В целях данного описания цАМФ-каталитический сайт (циклический аденозинмонофосфат), который связывает R- и S-ролипрам с низкой аффинностью, именуют низкоаффинным сайтом связывания (LPDE4), а другую форму этого каталитического сайта, который связывает ролипрам с высокой аффинностью, именуют высокоаффинным сайтом связывания (HPDE4). Этот терминHPDE4 не следует путать с термином hPDE4, который используется для обозначения PDE4 человека. Были проведены начальные эксперименты по оценке и обоснованию анализа на связывание [3 Н]ролипрама. Детальное описание этой работы дается в разделе Анализы на связывание, подробно описанном ниже. Предпочтительными ингибиторами PDE4 для использования в этом изобретении будут такие соединения, которые имеют полезный терапевтический индекс, т.е. соединения, которые предпочтительно ингибируют цАМФ-каталитическую активность, когда данный фермент находится в форме, которая связывает ролипрам с низкой аффинностью, тем самым снижая побочные действия, которые несомненно связаны с ингибированием формы, которая связывает ролипрам с высокой аффинностью. Другой способ установления этого заключается в том, что предпочтительные соединения будут иметь соотношение значений IC50, равное приблизительно 0,1 или более, а именно IC50 для каталитической формы PDE4, которая связывает ролипрам с высокой аффинностью, деленное на IC50 для такой формы, которая связывает ролипрам с низкой аффинностью. Дальнейшим улучшением этого стандартного показателя является такой показатель, когда ингибитор PDE4 имеет соотношение значений IС 50, равное приблизительно 0,1 или более; причем указанное отношение представляет собой отношение значения IC50 для случая конкурирования со связыванием 1 нМ [3 Н]R-ролипрама с формой PDE4, которая связывает ролипрам с высокой аффинностью, к значениюIC50 для случая ингибирования каталитической активности формы PDE4, которая связывает ролипрам с низкой аффинностью, при использовании в качестве субстрата 1 мкМ [3 Н]-цАМФ. Примерами полезных ингибиторов PDE4 являются(S)-(-)-этил-[4-(3-циклопентилокси-4-метоксифенил)пирролидин-2-илиден]ацетат. Наиболее предпочтительными являются такие ингибиторы PDE4, которые имеют соотношение значений IC50 больше 0,5, и в особенности такие соединения, которые имеют это соотношение больше 1,0. Предпочтительными соединениями являются цис-4-циано-4-(3-циклопентилокси-4-метоксифенил) циклогексан-1-карбоновая кислота, 2-карбометокси-4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-он и цис-[4-циано-4-(3-циклопропилметокси-4-дифторметоксифенил)циклогексан-1-ол]; они являются примерами соединений, которые связываются предпочтительно с низкоаффинным сайтом связывания и которые имеют соотношение значений IC50 0,1 или более. Другие представляющие интерес соединения включают в себя соединения, раскрытые в патенте США 5552438, выданном 3 сентября 1996 г.; этот патент и соединения, которые в нем раскрыты, включены сюда в полном объеме посредством ссылки. Соединением, представляющим особый интерес, которое раскрыто в патенте США 5552438, является цис-4-циано-4-[3-(циклопентилокси)-4-метоксифенил]циклогексан-1-карбоновая кислота (также известная как циломаласт (cilomalast и ее соли, сложные эфиры, пролекарства или физические формы; AWD-12-281 от Astra (Hofgen, N. et al. 15th EFMC Int.(ссылка по CAS162401-32-3) и пталазинон (pthalazinone) (WO 9947505) от Byk-Gulden; или соединение, идентифицированное как Т-440 (Tanabe Seiyaku; Fuji, K. et al. J. Pharmacol. Exp. Then, 1998,284(1): 162). Анализы на связывание фосфодиэстеразы и ролипрама Метод анализа 1 А. Определили, что выделенная человеческая моноцитарная PDE4 и hrPDE (человеческая рекомбинантная PDE4) существуют главным образом в низкоаффинной форме. Тогда активность тестируемых соединений относительно низкоаффинной формы PDE4 может быть оценена с использованием стандартных анализов каталитической активности PDE4 с применением в качестве субстрата 1 мкМ [3 Н]цАМФ (Torphy et al. J. of Biol. Chem., Vol. 267,3, pp. 1798-1804, 1992). Супернатанты мозга крыс, полученные в результате высокоскоростного центрифугирования, использовали в качестве источника белка, и оба энантиомера [3 Н]-ролипрама готовили с удельной активностью, равной 25,6 Ки/ммоль. Исходя из опубликованной методики модифицировали условия стандартного анализа, чтобы они были идентичны условиям анализа PDE за исключением конечных условий для цАМФ: 50 мМ Трис-HCl (рН 7,5), 5 мМ MgCl2, 50 мкМ 5'-АМФ и 1 нМ [3 Н]-ролипрам (Torphy et al. J. ofBiol. Chem., Vol. 267,3, pp. 1798-1804, 1992). Анализ проводили в течение 1 ч при 30 С. Реакцию останавливали и связанный лиганд отделяли от свободного лиганда, используя харвестер клеток Брандела(Brandel). Конкурирование за высокоаффинный сайт связывания оценивали в условиях, которые идентичны таковым, используемым для измерения активности низкоаффинной PDE, за исключением того,что [3 Н]-цАМФ не присутствовал. Метод анализа 1 Б. Измерение фосфодиэстеразной активности. Активность PDE анализировали, используя [3 Н]-цАМФ-SРА- или [3 Н]-цГМФ (циклический гуанозин-монофосфат)-SРА-ферментный анализ, как описано поставщиком (Amercham Life Science). Реакции проводили в 96-луночных планшетах при комнатной температуре в 0,1 мл реакционного буфера, содержащего (конечные концентрации): 50 мМ Трис-HCl, рН 7,5, 8,3 мМ MgCl2, 1,7 мМ EGTA (этиленгликольтетрауксусная кислота), [3 Н]-цАМФ или [3 Н]-цГМФ (приблизительно 2000 распадов в минуту/пмоль), фермент и различные концентрации ингибиторов. Анализ осуществляли в течение 1 ч и останавливали посредством добавления 50 мкл SPA-иттрийсиликатных шариков в присутствии сульфата цинка. Планшеты встряхивали и оставляли стоять при комнатной температуре в течение 20 мин. Образование меченного радиоактивным изотопом продукта оценивали с помощью стинцилляционной спектрометрии. Анализ на связывание [3 Н]R-ролипрама 3 Анализ на связывание [ Н]R-ролипрама проводили путем модификации метода Шнайдера (Schneider) и соавторов, см. Nicholson et al., Trends Pharmacol. Sci., Vol. 12, pp. 19-27 (1991) и McHale et al., Mol.al., Mol. Pharmacol. Vol. 39, 376-384 (1991). В результате конкурирование за связывание [3 Н]R-ролипрама- 10005992 дает независимое подтверждение эффективности ингибирования PDE4 немечеными конкурентами. Анализ осуществляли при 30 С в течение 1 ч в 0,5 мкл буфера, содержащего (конечные концентрации): 50 мМ Трис-HCl, рН 7,5, 5 мМ МgСl2, 0,05%-ный бычий сывороточный альбумин, 2 нМ [3 Н]R-ролипрам(5,7104 распадов в минуту/пмоль) и различные концентрации не меченных радиоактивным изотопом ингибиторов. Реакцию останавливали путем добавления 2,5 мл ледяного реакционного буфера (без [3 Н]R-ролипрама) и осуществления быстрой вакуумной фильтрации (харвестер клеток Брандела) через фильтры Ватман GF/B, которые были замочены в 0,3%-ном полиэтиленимине. Фильтры промывали дополнительными 7,5 мл холодного буфера, сушили, и радиоактивность подсчитывали с помощью жидкостной стинцилляционной спектрометрии. Согласно данному изобретению фармацевтическая комбинация, содержащая соединение формулы(I) или его физиологически приемлемый сольват совместно с ингибитором PDE4, может быть удобно представлена для применения в форме фармацевтического препарата совместно с физиологически приемлемым разбавителем или носителем. Отдельные соединения таких комбинаций могут быть введены либо последовательно, либо одновременно в отдельных или комбинированных фармацевтических препаратах. Подходящие дозы известных терапевтических агентов будут без труда определены специалистами в данной области техники. Неожиданно было обнаружено, что соединение формулы (I) имеет существенную склонность к образованию сольватов с обычно используемыми органическими растворителями. Такие сольваты являются, по существу, стехиометрическими, например отношение соединения формулы (I) к растворителю близко к 1:1, например, согласно анализу заявителя, было определено, что оно находится в интервале 0,95-1,05:1. Например, авторами изобретения получены сольваты с растворителями, такими как ацетон,диметилформамид (DMF), диметилацетамид (DMAc), тетрагидрофуран (THF), N-метил-2-пирролидон,изопропанол и метилэтилкетон. Однако сольватация соединения формулы (I) непредсказуема, поскольку авторами изобретения обнаружено, что хотя оно образует сольват с изопропанолом, но, по-видимому,оно не образует сольват с этанолом или метанолом. Более того, по-видимому, оно не образует сольват с 1,1,1,2-тетрафторэтаном, этилацетатом, метилацетатом, толуолом, метилизобутилкетоном (MIBK) или водой. Однако вследствие токсичности многих органических растворителей необходимо было разработать специальные условия конечной стадии обработки (обсужденные позже) для того, чтобы сделать возможным получение соединения формулы (I) в несольватированной форме. Авторами изобретения неожиданно было обнаружено, что соединение формулы (I) в несольватированной форме может существовать в ряде полиморфных форм. Конкретно авторы изобретения идентифицировали полиморфные формы, которые можно различить с помощью дифракции рентгеновских лучей на порошке (XRPD) и которые названы авторами изобретения как форма 1, форма 2 и форма 3. Повидимому, форма 3 является нестабильной второстепенной полиморфной модификацией формы 2. В общих чертах данные формы характеризуются своими XRPD-профилями следующим образом: форма 1: пик около 18,9 2; форма 2: пики около 18,4 и 21,5 2; форма 3: пики около 18,6 и 19,2 2. В пределах интервала 21-23 2 форма 3 показывает единичный пик, в то время как форма 2 показывает пару пиков. Во всех случаях присутствует пик при 7 2, однако, интенсивность этого пика намного выше в случае форм 2 и 3, чем в случае формы 1. Картины XRPD полиморфных модификаций показаны чертежом на фиг. 1. Превращение формы 2 в форму 1 во времени в водной суспензии при температуре окружающей среды показано на фиг. 2. При превращении формы 2 в форму 1 особенно заметны потеря пика, характерного для формы 2 (отмеченного В) около 18,4 2, явное уменьшение интенсивности у пика около 7 2 (отмеченного А) и появление пика, характерного для формы 1 (отмеченного С) около 18,9 2. Температурная зависимость формы 3 показана на фиг. 4. Температуру изменяли согласно профилю,показанному на фиг. 5. Из фиг. 4 можно видеть, что форма 3 сначала превращается в форму 2 в температурном интервале 30-170 С и затем превращается в форму 1 в температурном интервале 170-230 С. При превращении формы 3 в форму 2 особенно заметны разделение одного пика в интервале 21-23 2 на два пика в пределах того же самого интервала и сдвиг влево пика около 18,6 2 до приблизительно 18,4 2. При превращении формы 2 в форму 1 могут наблюдаться изменения, похожие на изменения, отмеченные в предыдущем параграфе. Профили дифференциальной сканирующей калориметрии (DSC) и термогравиметрического анализа (TGA) формы 1 показаны на фиг. 3. Эти профили характеризуются переходом около 280-300 С (обычно близко к 298 С), соответствующим эндотермическому событию при DSC и химической деградации при TGA. В условиях проведенных экспериментов профили DSC форм 2 и 3 принципиально не отличались, и таким образом DSC не является подходящей методикой для различения данных 3 форм. Продемонстрированное на фиг. 3 отсутствие активности в профилях TGA и DSC ниже приблизительно 298 С предполагает, что вещество демонстрирует хорошую физическую и химическую стабильность при обычных рабочих температурах.- 11005992 Как показано в примерах, энтальпию растворения форм 1 и 3 определяли в определенных органических растворителях и, соответственно, энтальпия перехода из формы 3 в форму 1 оценена как 5,1-6,7 кДж/моль. Таким образом, авторы изобретения предпочитают соединение формулы (I) в несольватированной форме 1, поскольку эта форма оказывается термодинамически наиболее стабильной при температуре окружающей среды и, кроме того, оказывается наименее чувствительной к нежелательной сорбции влаги(см. результаты в разделе "Примеры"). Тем не менее, в других условиях предпочтительной может быть форма 2 (или форма 3). Несмотря на то, что применение соединения формулы (I) в сольватированной форме не является предпочтительным, тем не менее авторами изобретения неожиданно обнаружено, что некоторые сольватные формы имеют особо привлекательные физико-химические свойства, которые делают их полезными в качестве промежуточных соединений в получении соединения формулы (I) в несольватированной форме (например, посредством удаления растворителя в качестве конечной стадии). Например, авторами изобретения обнаружено, что некоторые стехиометрические сольваты могут быть выделены в виде твердых веществ в строго кристаллической форме, включая соединение формулы (I) в виде сольвата с метилэтилкетоном; соединение формулы (I) в виде сольвата с изопропанолом; соединение формулы (I) в виде сольвата с тетрагидрофураном; соединение формулы (I) в виде сольвата с ацетоном. Еще одним особым преимуществом этих сольватов является тот факт, что десольватация сольвата(например, путем нагревания) приводит к образованию несольватированной формы, как предпочтительной формы 1. Вышеупомянутые сольваты обладают относительно низкой токсичностью и подходят для использования в производстве в промышленном масштабе. Соединение формулы (I) в виде сольвата сDMF, который также может быть выделен в виде твердого вещества в кристаллической форме, также представляет интерес для использования в прогрессивной обработке до несольватированной формы 1. Соединение формулы (I) и его сольваты могут быть получены по методике, описанной здесь далее. Способ по данному изобретению для получения соединения формулы (I) или его сольвата включает в себя алкилирование по 17-карботиокислотной группировке тиокислоты формулы (II), как она определена выше, или ее соли, причем соединение формулы (II) можно подвергать взаимодействию с соединением формулы FCH2L, где L представляет собой уходящую группу (например, атом галогена, мезильную или тозильную группу или им подобное), например, с соответствующим фторметилгалогенидом в стандартных условиях. Фторметилгалогенидный реагент предпочтительно является бромфторметаном. Как будет отмечено позже, соединение формулы (II) предпочтительно используют в виде соли, в частности соли с диизопропилэтиламином. В предпочтительном способе получения соединения формулы (I) соединение формулы (II) или его соль обрабатывают бромфторметаном, возможно, в присутствии межфазного катализатора. Предпочтительным растворителем является метилацетат или более предпочтительным этилацетат, возможно, в присутствии воды. Присутствие воды улучшает растворимость как исходного вещества, так и продукта, а применение межфазного катализатора приводит к увеличению скорости реакции. Примеры межфазных катализаторов, которые могут быть использованы, включают в себя (но не ограничиваются ими) бромид тетрабутиламмония, хлорид тетрабутиламмония, бромид бензилтрибутиламмония, хлорид бензилтрибутиламмония, бромид бензилтриэтиламмония, хлорид метилтрибутиламмония и хлорид метилтриоктиламмония. THF также может быть с успехом применен в качестве растворителя для реакции, в которой присутствие межфазного катализатора вновь обеспечивает существенно более быструю скорость реакции. Продукт, находящийся в органической фазе, предпочтительно промывают сначала водным раствором кислоты, например разбавленной HCl, для того, чтобы удалить аминные соединения, такие как триэтиламин и диизопропилэтиламин, и затем водным раствором основания, например бикарбонатом натрия, для того, чтобы удалить любой непрореагировавший предшественник соединение формулы (II). Как будет отмечено позже, если соединение формулы (I), полученное таким образом в растворе этилацетата, подвергнуть дистилляции и добавить толуол, то выкристаллизуется несольватированная форма 1. Соединения формулы (II) могут быть получены из соответствующего 17-гидроксильного производного формулы (III), как оно определено выше, с использованием, например, методики, описаннойG.H. Phillipps et al., (1994) Journal of Medicinal Chemistry, 37, 3717-3729. Например, данная стадия обычно включает в себя добавление реагента, подходящего для проведения этерификации, например активированного производного 2-фуранкарбоновой кислоты, такого как активированный сложный эфир или предпочтительно 2-фуроилгалогенид, например 2-фуроилхлорид (используемый по меньшей мере в 2 кратном молярном количестве по отношению к соединению формулы (III, в присутствии органического основания, например триэтиламина. Второй моль 2-фуроилхлорида взаимодействует с тиокислотной группировкой в соединении формулы (III) и его необходимо удалить, например, путем взаимодействия с амином, таким как диэтиламин.- 12005992 Однако у этого способа имеются недостатки, заключающиеся в том, что получающееся соединение формулы (II) трудно очистить от загрязнения побочным продуктом 2-фуроилдиэтиламидом. Поэтому авторами изобретения были созданы несколько улучшенных способов для осуществления этого превращения. В первом таком улучшенном способе авторами изобретения было открыто, что при использовании более полярного амина, такого как диэтаноламин, получается более водорастворимый побочный продукт(в данном случае 2-фуроилдиэтаноламид), который позволяет получить соединение формулы (II) или его соль с высокой чистотой, поскольку этот побочный продукт эффективно может быть удален путем промывания водой. В способе получения соединения формулы (II) по изобретению на стадии (а) активированное производное 2-фуранкарбоновой кислоты предпочтительно может быть активированным эфиром 2 фуранкарбоновой кислоты, но более предпочтительно 2-фуроилгалогенидом, в особенности 2 фуроилхлоридом. Подходящим растворителем для этой реакции является этилацетат или метилацетат(предпочтительно метилацетат) (когда следует стадия с участием органического растворителя, по существу не смешивающегося с водой) либо ацетон (когда следует стадия с участием смешивающегося с водой растворителя). В норме будет присутствовать органическое основание, например триэтиламин. На стадии (б) предпочтительным органическим основанием является диэтаноламин. Основание может быть подходящим образом растворено в растворителе, например метаноле. Как правило, стадии (а) и (б) будут осуществлены при пониженной температуре, например между 0 и 5 С. В случае использования органического растворителя, не смешивающегося с водой, водной промывкой может служить вода, однако,применение соляного раствора приводит к более высоким выходам и поэтому является предпочтительным. В случае использования смешивающегося с водой растворителя, водная среда представляет собой,например, разбавленный водный раствор кислоты, такой как разбавленная HCl. В альтернативном способе получения соединения формулы (II) по изобретению на стадии (а) активированное производное 2-фуранкарбоновой кислоты предпочтительно может быть активированным эфиром 2-фуранкарбоновой кислоты, но более предпочтительно 2-фуроилгалогенидом, в особенности 2 фуроилхлоридом. Подходящим растворителем для этой стадии является ацетон. В норме будет присутствовать органическое основание, например триэтиламин. На стадии (б) подходящим растворителем является DMF или диметилацетамид. В норме будет присутствовать органическое основание, например триэтиламин. Как правило, стадии (а) и (б) будут осуществлены при пониженной температуре, например между 0 и 5 С. Продукт может быть выделен путем обработки кислотой и путем промывания водой. Вышеупомянутый способ является очень эффективным вследствие того, что при его осуществлении не образуется какой-либо фуроиламидный побочный продукт (таким образом, предоставляет, в частности, преимущества для окружающей среды), поскольку избыточный моль фуроильной группировки вступает в реакцию с дополнительным молем соединения формулы (II) с образованием дополнительного моля соединения формулы (II). Дополнительные общие условия превращения соединения формулы (III) в соединение формулы (II) в двух описанных способах будут хорошо известны специалистам в данной области техники. Однако в соответствии с предпочтительным набором условий авторами изобретения обнаружено,что соединение формулы (II) преимущественно может быть выделено в форме твердой кристаллической соли. Предпочтительной солью является соль, образованная с основанием, таким как триэтиламин, 2,4,6 триметилпиридин, диизопропилэтиламин или N-этилпиперидин. Такие солевые формы соединения формулы (II) более стабильны, легче фильтруются и сушатся и могут быть выделены с более высокой чистотой, чем свободная тиокислота. Наиболее предпочтительной солью является соль, образованная с диизопропилэтиламином. Соль с триэтиламином также представляет интерес. Соединения формулы (III) могут быть получены в соответствии с методиками, описанными в GB 2088877 В. Соединения формулы (III) также могут быть получены с помощью способов, содержащих следующие стадии: Стадия (а) включает в себя окисление раствора, содержащего соединение формулы (V). Стадия (а) предпочтительно будет проведена в присутствии растворителя, содержащего метанол,воду, тетрагидрофуран, диоксан или диметиловый эфир диэтиленгликоля. Для увеличения выхода и производительности предпочтительными растворителями являются метанол, вода или тетрагидрофуран и более предпочтительными вода или тетрагидрофуран, в особенности вода и тетрагидрофуран в качестве растворителя. Диоксан или диметиловый эфир диэтиленгликоля также являются предпочтительными- 13005992 растворителями, которые могут быть возможно (и предпочтительно) использованы вместе с водой. Растворитель предпочтительно будет присутствовать в количестве от 3 до 10 объемов относительно количества исходного вещества (1 ед. массы), более предпочтительно от 4 до 6 объемов, в особенности 5 объемов. Окислитель предпочтительно присутствует в количестве 1-9 молярных эквивалентов относительно количества исходного вещества. Например, если используют 50 мас.%-ный водный раствор йодной кислоты, то окислитель может присутствовать в количестве от 1,1 до 10 ед. массы относительно количества исходного вещества (1 ед. массы), более предпочтительно от 1,1 до 3 ед. массы, в особенности 1,3 ед. массы. Стадия окисления предпочтительно будет включать в себя применение химического окислителя. Окислителем более предпочтительно будет йодная или йодноватая кислота либо их соль. Наиболее предпочтительно окислителем будет йодная кислота или периодат натрия, в особенности йодная кислота. Альтернативно (или в дополнение к этому), также будет понятно, что стадия окисления может включать в себя любую подходящую реакцию окисления, например реакцию, в которой используется воздух и/или кислород. Если в реакции окисления используется воздух и/или кислород, то используемый в указанной реакции растворитель предпочтительно будет метанолом. Стадия (а) предпочтительно будет включать в себя инкубирование реагентов при комнатной или несколько повышенной температуре, скажем, приблизительно 25 С, например, в течение 2 ч. Соединение формулы (IV) может быть выделено посредством перекристаллизации из реакционной смеси путем добавления антирастворителя. Подходящим антирастворителем для соединения формулы (IV) является вода. Неожиданно авторами изобретения было обнаружено, что весьма желательно контролировать условия, при которых происходит осаждение соединения формулы (IV) в результате добавления антирастворителя, например воды. Когда перекристаллизацию осуществляют с использованием охлажденной воды (например, смеси вода/лед при температуре 0-5 С), хотя можно рассчитывать и на лучшие свойства антирастворителя, авторами изобретения было обнаружено, что полученный кристаллический продукт является очень объемистым, походит на мягкий гель, и его трудно фильтровать. Не ограничиваясь теорией, авторы изобретения предполагают,что этот продукт с низкой плотностью содержит большое количество сольватированного растворителя внутри кристаллической решетки. В противоположность этому, если используют условия приблизительно 10 С или выше (например, приблизительно температура окружающей среды), то получают гранулированный продукт похожей на песок консистенции, который очень легко фильтруется. Кристаллизация в этих условиях обычно начинается, спустя приблизительно 1 ч, и обычно завершается в пределах нескольких часов (например, 2 ч). Не ограничиваясь теорией, авторы изобретения полагают, что этот гранулированный продукт содержит мало или вообще не содержит сольватированного растворителя внутри кристаллической решетки. Стадия (б) обычно включает в себя добавление реагента, подходящего для превращения карбоновой кислоты в карботиокислоту, например, с использованием газа сероводорода совместно с подходящим агентом сочетания, например карбонилдиимидазолом (CDI), в присутствии подходящего растворителя,например диметилформамида. В альтернативном способе получения соединения формулы (II), в котором соединение формулы (X) обрабатывают реагентом, подходящим для превращения карбоновой кислоты в карботиокислоту, предпочтительно используют газ сероводород совместно с подходящим агентом сочетания, таким как CDI, в присутствии подходящего растворителя, например DMF. Соединения формулы (X) могут быть получены по методике, аналогичной описанной здесь. Примеры подходящих источников фтора в альтернативном способе получения соединения формулы (I) или его сольвата с участием соединения формулы (VI), включают в себя фторид (например, фторид натрия) или более предпочтительно HF. Предпочтительным реагентом является водный раствор HF. Может быть использован такой растворитель, как THF или DMF. Соединение формулы (VI) может быть получено способом, при котором(а) осуществляют алкилирование соединения формулы (VII), как оно определено выше, или его соли;(в) осуществляют этерификацию соединения формулы (IX) В способе (а) могут быть использованы условия, аналогичные описанным выше для превращения соединения формулы (II) в соединение формулы (I). Обычно соединение формулы (VII) будут подвергать взаимодействию с соединением формулы FCH2L, где L представляет собой уходящую группу (например, атом галогена, мезильную или тозильную группу или им подобное), например, с соответствующим фторметилгалогенидом, в стандартных условиях. Фторметилгалогенидный реагент предпочтительно представляет собой бромфторметан. Способ (б) предпочтительно осуществляют в две стадии (1) образование галогенгидрина, в особенности бромгидрина (например, в результате взаимодействия с бромоданом (bromodan) или эквивалентным реагентом) с последующей (2) обработкой основанием, таким как гидроксид натрия, с тем, чтобы осуществить замыкание цикла. Продуктом стадии (1) является соединение формулы (IXA), где X представляет собой галоген, в особенности Вr, которое является новым промежуточным соединением и при желании может быть выделено. В способе (в) подходящим реагентом может быть активированное производное 2-фуранкарбоновой кислоты, такое как активированный сложный эфир или предпочтительно 2-фуроилгалогенид, например 2-фуроилхлорид, в присутствии органического основания, например триэтиламина. Эту реакцию можно проводить при повышенной температуре, например при приблизительно 60 С, или при температуре окружающей среды, в присутствии катализатора ацилирования, например диметиламинопиридина(DMAP). Соединения формулы (VII) могут быть получены способом, при котором осуществляют этерификацию соединения формулы (XI) Могут быть использованы условия, аналогичные описанным выше для превращения соединения формулы (III) в соединение формулы (II). Например, подходящим реагентом может быть активированное производное 2-фуранкарбоновой кислоты, такое как активированный сложный эфир или предпочтительно 2-фуроилгалогенид, например 2-фуроилхлорид, в присутствии органического основания, например триэтиламина. Соединение формулы (XI) известно (J. Labelled Compd Radiopharm. (1997) 39(7), 567-584). Соединение формулы (VIII) может быть получено способом, при котором(а) осуществляют алкилирование соединения формулы (XII), как оно определено выше, или его соли; либо(б) осуществляют этерификацию соединения формулы (XIII) В способе (а) могут быть использованы условия, аналогичные описанным выше для превращения соединения формулы (II) в соединение формулы (I). Обычно соединение формулы (XII) будут подвергать взаимодействию с соединением формулы FCH2L, где L представляет собой уходящую группу (например, атом галогена, мезильную или тозильную группу или им подобное), например, с соответствующим фторметилгалогенидом в стандартных условиях. Фторметилгалогенидный реагент предпочтительно представляет собой бромфторметан.- 15005992 В способе (б) могут быть использованы условия, аналогичные использованным выше для превращения соединения формулы (IX) в соединение формулы (VI). Например, подходящим реагентом может быть активированное производное 2-фуранкарбоновой кислоты, такое как активированный сложный эфир или предпочтительно 2-фуроилгалогенид, например 2-фуроилхлорид, в присутствии органического основания, например триэтиламина. Соединения формулы (IX) и (XIII) могут быть получены путем алкилирования соответствующих тиокислот (XI) и (XIV) (определенных ниже) с использованием методики, аналогичной уже описанной(например, путем взаимодействия с соединением формулы FCH2L, где L представляет собой уходящую группу (например, атом галогена, мезильную или тозильную группу или им подобное), например, с использованием соответствующего фторметилгалогенида, в стандартных условиях. Фторметилгалогенидный реагент предпочтительно представляет собой бромфторметан. Тиокислота формулы (XI) является известным соединением (J. Labelled Compd Radiopharm. (1997) 39(7), 567-584). Соединение формулы (XII) может быть получено способом, при котором осуществляют этерификацию соединения формулы (XIV) или его соли. Этот способ можно осуществлять с использованием методики, аналогичной уже описанной. Например, подходящим реагентом может быть активированное производное 2-фуранкарбоновой кислоты,такое как активированный сложный эфир или предпочтительно 2-фуроилгалогенид, например 2 фуроилхлорид, в присутствии органического основания, например триэтиламина. Соединение формулы (XIV) может быть получено из соответствующей карбоновой кислоты, например, способом, аналогичным описанному выше для превращения соединения формулы (IV) в соединение формулы (III). Вышеуказанная соответствующая карбоновая кислота известна (Upjohn, WO 90/15816). Еще один альтернативный способ получения соединения формулы (I) или его сольвата включает в себя удаление защиты с соединения формулы (I), в котором 11 гидроксигруппа защищена или замаскирована, или демаскировку этого соединения. Первый такой способ включает в себя удаление защиты с соединения формулы (XV), где Р представляет собой защитную группу гидроксила. Примеры защитных групп гидроксила Р описаны в Protective Groups in Organic Chemistry Ed JFWand Sons, 1991). Примеры подходящих защитных групп гидроксила Р включают в себя группы, выбранные из карбоната, алкила (например, трет-бутила или метоксиметила), аралкила (например, бензила, пнитробензила, дифенилметила или трифенилметила), гетероциклических групп, таких как тетрагидропиранил, ацила (например, ацетила или бензила) и силильных групп, таких как триалкилсилил (например,трет-бутилдиметилсилил). Защитные группы гидроксила могут быть удалены с использованием традиционных методик. Так, например, карбонат может быть удален путем обработки основанием, а алкильные, силильные, ацильные и гетероциклические группы могут быть удалены путем сольволиза, например гидролиза в кислотных или основных условиях. Аралкильные группы, такие как трифенилметил, могут аналогичным образом быть удалены путем сольволиза, например гидролиза в кислотных условиях. Аралкильные группы, такие как бензил или п-нитробензил, могут быть отщеплены путем гидрогенолиза в присутствии катализатора на основе благородных металлов, такого как палладий на угле. пНитробензил также может быть отщеплен с помощью фотолиза. 11 Гидроксигруппа может быть замаскирована в виде карбонильной группы. Таким образом, второй такой способ включает в себя восстановление соединения формулы (XVI), как оно определено выше. Восстановление до соединения формулы (I) может быть достигнуто, например, путем обработки гидридным восстановителем, таким как борогидрид, например борогидрид натрия. 11-Кетон (XVI) также может быть замаскирован. Примеры замаскированных производных соединения формулы (XVI) включают в себя (1) кетальные производные, например кетали, образованные в результате обработки соединения формулы (XVI) спиртом, например метанолом, этанолом или этан-1,2 диолом, (2) дитиокетальные производные, например дитиокетали, образованные в результате обработки соединения формулы (XVI) тиолом, например метантиолом, этантиолом или этан-1,2-дитиолом, (3) монотиокетальные производные, например монотиокетали, образованные в результате обработки соединения формулы (XVI), например, 1-гидроксиэтан-2-тиолом, (4) производные, образованные в результате обработки соединения формулы (XVI) аминоспиртом, например эфедрином, (5) имины, образованные в- 16005992 результате обработки соединения формулы (XVI) аминами, (6) оксимы, образованные в результате обработки соединений формулы (XVI) гидроксиламинами. Такие производные соединения формулы (XVI) заявлены авторами изобретения в качестве аспекта данного изобретения. Такие замаскированные производные могут быть превращены обратно в кетон традиционными способами, например кетали, имины и оксимы превращают в карбонил путем обработки разбавленной кислотой, а дитиокетали превращают в кетон с помощью ряда способов, как описано Р.С. Bulman Page etal. (1989), Tetrahedron, 45, 7643-7677, и в ссылках, имеющихся в указанном документе. Соединения формулы (XV) могут быть получены способом, при котором(а) осуществляют алкилирование соединения формулы (XVII), как оно определено выше, или его соли, где Р представляет собой защитную группу гидроксила; либо(б) осуществляют этерификацию соединения формулы (XVIII) На стадии (а) могут быть использованы условия, аналогичные описанным выше для превращения соединения формулы (II) в соединение формулы (I). Обычно соединение формулы (XVII) будут подвергать взаимодействию с соединением формулы FCH2L, где L представляет собой уходящую группу (например, атом галогена, мезильную или тозильную группу или им подобное), например, с соответствующим фторметилгалогенидом, в стандартных условиях. Фторметилгалогенидный реагент предпочтительно представляет собой бромфторметан. На стадии (б) могут быть использованы условия, аналогичные использованным выше для превращения соединения формулы (IX) в соединение формулы (VI). Например, подходящим реагентом может быть активированное производное 2-фуранкарбоновой кислоты, такое как активированный сложный эфир или предпочтительно 2-фуроилгалогенид, например 2-фуроилхлорид, в присутствии органического основания, например триэтиламина. Соединение формулы (XVIII) может быть получено путем алкилирования соответствующей тиокислоты с использованием методики, аналогичной уже описанной (например, путем взаимодействия с соединением формулы FCH2L, где L представляет собой уходящую группу (например, атом галогена,мезильную или тозильную группу или им подобное), например, с использованием соответствующего фторметилгалогенида, в стандартных условиях. Фторметилгалогенидный реагент предпочтительно представляет собой бромфторметан. Соответствующие тиокислоты является известными соединениями или могут быть получены по стандартной методике. Соединение формулы (XVIII) может быть альтернативно получено посредством защиты соответствующего гидроксипроизводного. Соединение формулы (XVII) может быть получено способом, при котором осуществляют этерификацию соединения формулы (XIX) или его соли, где Р представляет собой защитную группу гидроксила. Этот способ можно осуществлять с использованием методики, аналогичной уже описанной для превращения соединений формулы (III) в (II). Например, подходящим реагентом может быть активированное производное 2-фуранкарбоновой кислоты, такое как активированный сложный эфир или предпочтительно 2-фуроилгалогенид, например 2-фуроилхлорид, в присутствии органического основания,например триэтиламина. Соединения формулы (XIX) могут быть получены путем защиты соответствующего гидроксипроизводного (III), в котором сначала была защищена тиокислота, с которой затем удаляют защиту. Соединения формулы (XVI) могут быть получены способом, при котором(а) осуществляют алкилирование соединения формулы (XX), как оно определено выше, или его соли либо производного, где 11-карбонильная группа замаскирована; либо(б) осуществляют этерификацию соединения формулы (XXI) или его производного, где 11-карбонильная группа замаскирована. На стадии (а) могут быть использованы условия, аналогичные описанным выше для превращения соединения формулы (III) в соединение формулы (II). Обычно соединение формулы (XX) будут подвергать взаимодействию с соединением формулы FCH2L, где L представляет собой уходящую группу (например, атом галогена, мезильную или тозильную группу или им подобное), например, с соответствующим фторметилгалогенидом, в стандартных условиях. Фторметилгалогенидный реагент предпочтительно представляет собой бромфторметан. На стадии (б) могут быть использованы условия, аналогичные использованным выше для превращения соединения формулы (IX) в соединение формулы (VI). Например, подходящим реагентом может быть активированное производное 2-фуранкарбоновой кислоты, такое как активированный сложный эфир или предпочтительно 2-фуроилгалогенид, например 2-фуроилхлорид, в присутствии органического основания, например триэтиламина. Соединение формулы (XXI) или его производное, где 11-кетонная группа замаскирована, может быть получено путем алкилирования соответствующей тиокислоты с использованием методики, аналогичной уже описанной (например, путем взаимодействия с соединением формулы FCH2L, где L представляет собой уходящую группу (например, атом галогена, мезильную или тозильную группу или им подобное), например, с использованием соответствующего фторметилгалогенида, в стандартных условиях. Фторметилгалогенидный реагент предпочтительно представляет собой бромфторметан. Соответствующие тиокислоты является известными соединениями или могут быть получены из соответствующих карбоновых кислот способами, аналогичными описанным выше. Соединение формулы (XX) можно получить способом, при котором осуществляют этерификацию соединения формулы (XXII) или его производного, где 11-кетонная группа замаскирована. Этот способ можно осуществить с использованием методики, аналогичной уже описанной. Например, подходящим реагентом может быть активированное производное 2-фуранкарбоновой кислоты, такое как активированный сложный эфир или предпочтительно 2-фуроилгалогенид, например 2-фуроилхлорид, в присутствии органического основания,например триэтиламина. Соединения формулы (XXII) и их производные, где 11-кетон замаскирован, могут быть получены путем окисления соответствующего гидроксипроизводного (IV) с последующей маскировкой кетона и затем превращения группы карбоновой кислоты в тиокислоту (см., например, превращение соединений формулы (IV) в (III. В альтернативном способе получения соединений формулы (I) или их сольватов, группа L в соединениях формулы (XXIII) представляет собой уходящую группу (например, галогенид, отличный от фторида, такой как хлорид, йодид, или эфир сульфокислоты, такой как мезилат, тозилат, трифлат). Источником фтора в этом способе по изобретению предпочтительно является фторид-ион, например KF. Дополнительные подробности этого превращения могут быть получены из документа G.H. Phillipps et al., (1994) Journal of Medicinal Chemistry, 37, 3717-3729 или J. Labelled Compd Radiopharm.(1997)39(7), 567-584. Соединения формулы (XXIII) могут быть получены способами, аналогичными описанным здесь. Соответствующие новые промежуточные соединения формулы (VI), (VIII), (IX), (IXA), (XV) и (XVI), где группировка -CH2F заменена группировкой -CH2L (где L представляет собой уходящую группу, отличную от фтора) заявлены в качестве аспекта данного изобретения. В альтернативном способе получения соединений формулы (I) или их сольватов с удалением защиты или демаскировки производного соединения формулы (I), 3-карбонильная может быть замаскирована способом, аналогичным описанному выше в отношении маскировки карбонильной группы в положении- 18005992 11. Так, 3-карбонил может быть замаскирован, например, в виде кеталя, монотиокеталя, дитиокеталя,производного с аминоспиртом, оксима или имина. Данная карбонильная группа может быть возвращена в исходное состояние с помощью традиционных способов, например, кетали превращают в карбонил путем обработки разбавленной кислотой, а дитиокетали превращают в кетон с помощью ряда методов,как описано Р.С. Bulman Page et al., (1989), Tetrahedron, 45, 7643-7677 и ссылках в этом документе. Некоторые промежуточные соединения являются новыми, и они предложены авторами изобретения совместно, где целесообразно, с их солями и сольватами в качестве аспекта данного изобретения. Как особо отмечено выше, в способе получения соединения формулы (I) в несольватированной форме несольватирующий растворитель на стадии кристаллизации (а) представляет собой этанол, метанол, воду, этилацетат, толуол, метилизобутилкетон или их смесь, или на стадии (б) осуществляют десольватацию соединения формулы (I) в сольватированной форме (например, в форме сольвата с ацетоном, изопропанолом, метилэтилкетоном, DMF или тетрагидрофураном), например, путем нагревания. Как правило, десольватацию на стадии (б) будут осуществлять при температуре, превышающей 50 С, предпочтительно при температуре, превышающей 100 С. Как правило, нагревание будут осуществлять под вакуумом. Предпочтительное соединение формулы (I) в несольватированной форме получено вышеупомянутым способом. В способе получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 1 несольватирующий антирастворитель представляет собой как изооктан или толуол. Согласно первому предпочтительному воплощению этого способа соединение формулы (I) можно растворить в этилацетате и соединение формулы (I) в виде несольватированной полиморфной модификации формы 1 можно получить путем добавления толуола в качестве антирастворителя. Для повышения выхода раствор этилацетата предпочтительно используют горячим и после того, как добавлен толуол,смесь подвергают дистилляции для понижения содержания этилацетата. Согласно второму предпочтительному воплощению этого способа соединение формулы (I) можно растворить в метилизобутилкетоне и соединение формулы (I) в виде несольватированной полиморфной модификации формы 1 можно получить путем добавления изооктана в качестве антирастворителя. В способе получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 2, соединение формулы (I) обычно будут растворять в горячем метаноле или безводном дихлорметане и оставлять для охлаждения. В способе получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 3, соединение формулы (I) или его сольват (в частности, сольват с ацетоном) растворяют в дихлорметане в присутствии воды, которая составляет обычно 1-3% воды по объему. Способ получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 3 включает в себя растворение соединения формулы (I) или его сольвата (в частности, как сольвата с ацетоном) в дихлорметане в присутствии воды (обычно 1-3% воды по объему) и перекристаллизацию соединения формулы (I) в виде несольватированной полиморфной модификации формы 3. Преимущества соединения формулы (I) и/или его сольватов либо полиморфных модификаций могут включать в себя тот факт, что данное вещество демонстрирует превосходные противовоспалительные свойства с предсказуемыми фармакокинетическим и фармакодинамическим действием, с приемлемым профилем побочных действий и сочетается с удобной схемой лечения людей. Дополнительные преимущества могут включать в себя тот факт, что данное вещество обладает желаемыми физическими и химическими свойствами, которые обеспечивают производство препаратов, готовых к применению, и хранение. Краткое описание графических материалов Фиг. 1 - четеж профилей XRPD полиморфных модификаций формы 1, формы 2 и формы 3 несольватированного соединения формулы (I); фиг. 2 - чертеж профилей XRPD полиморфных модификаций формы 1, формы 2 и смеси (50:50) полиморфных модификаций формы 1 и формы 2 несольватированного соединения формулы (I) вместе с временной зависимостью для профиля смеси (50:50) формы 1 и формы 2; фиг. 3 - профили DSC и TGA полиморфной модификации формы 1 несольватированного соединения формулы (I); фиг. 4 - температурная зависимость профиля XRPD несольватированной формы 3 соединения формулы (I), полученная для 5 моментов времени; фиг. 5 - температурный и временной профиль для экспериментов XRPD на фиг. 4. Следующие не ограничивающие примеры иллюстрируют данное изобретение. Примеры Общие методы. 1 Н ЯМР-спектры регистрировали при 400 МГц и химические сдвиги выражают как число частей на миллион относительно тетраметилсилана. Для описания многообразия сигналов используют следующие сокращения: s (синглет), d (дублет), t (триплет), q (квартет), m (мультиплет), dd (дублет дублетов), ddd- 19005992 упакованным картриджам с силикагелем, содержащим КР-Sil на основе 12i флэш-хроматографического модуля. LCMS (сочетание жидкостной хроматографии и масс-спектрометрии) проводили на колонке Supelcosil LCABZ+PLUS (3,3 см 4,6 мм внутренний диаметр) с элюированием 0,1%-ной НСО 2 Н и 0,01 М ацетатом аммония в воде (растворитель А) и 0,05%-ной НСО 2 Н, 5%-ной водой в ацетонитриле (растворитель Б), при скорости потока 3 мл/мин, используя следующий градиент элюирования: 0-0,7 мин 0% Б,0,7-4,2 мин 100% Б, 4,2-5,3 мин 0% Б, 5,3-5,5 мин 0% Б. Масс-спектры регистрировали на спектрометреFisons VG Platform, используя электрораспыление в режиме положительной и отрицательной регистрации ионов (ES+ve и ES-ve). Профили DSC и TGA получали, используя синхронный температурный анализатор NetzschSTA449C, используя негерметичный сосуд с потоком газообразного азота и температурный градиент 10 С/мин. Характеристики сорбции влаги получали, используя микровесы сорбции воды Hiden Igasorb. Программа предусматривает пошаговое увеличение относительной влажности (ОВ) от 0 до 90% ОВ и затем обратное снижение до 0% ОВ шагами по 10% ОВ. Анализ XRPD, результаты которого показаны на фиг. 1 и 2, проводили на порошковом дифрактометре Phillips X'pert MPD, серийный номер DY667. Способ заключается в изменении 2 от 2 до 45 с величиной шага 2, равной 0,02, и временем сбора, равным 1 с для каждого шага. На фиг. 4 показаны результаты данного анализа XRPD с применением этого же прибора с термоприставкой Anton Parr TTK с использованием способа изменения 2 от 2 до 35 с величиной шага 2,равной 0,04, и временем сбора, равным 1 с. Промежуточные соединения. Промежуточное соединение 1. 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16 метил-3-оксоандроста-1,4-диен-17-карботиокислота. Раствор 6,9-дифтор-11,17-дигидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты (полученной в соответствии с методикой, описанной в GB 2088877 В) (18 г, 43,64 ммоль) в безводном дихлорметане (200 мл) и триэтиламине (15,94 мл, 114 ммоль) обрабатывали при температуре 5 С раствором 2-фуроилхлорида (11,24 мл, 114 ммоль) в безводном дихлорметане (100 мл) в течение приблизительно 40 мин. Раствор перемешивали при температуре 5 С в течение 30 мин. Полученное твердое вещество собирали путем фильтрования, последовательно промывали 3,5%-ным водным раствором гидрокарбоната натрия, водой, 1 М соляной кислотой и водой и сушили в вакууме при 60 С, получая твердое вещество кремового цвета. Дихлорметановый фильтрат последовательно промывали 3,5%-ным раствором гидрокарбоната натрия, водой, 1 М соляной кислотой, водой, сушили (Na2SO4) и упаривали, получая твердое вещество кремового цвета, которое объединяли с полученным ранее. Объединенные фракции твердого вещества (26,9 г) суспендировали в ацетоне (450 мл) и перемешивали. Добавляли диэтиламин (16,8 мл, 162 ммоль) и смесь перемешивали при комнатной температуре в течение 4,5 ч. Смесь концентрировали, осадок собирали путем фильтрования и промывали небольшим количеством ацетона. Промывки и фильтрат объединяли, концентрировали и наносили на колонку с силикагелем Biotage, которую элюировали смесью хлороформ/метанол в отношение 24:1. Фракции, содержащие более полярный компонент, объединяли и упаривали, получая твердое вещество кремового цвета. Его объединяли с выделенным ранее твердым веществом и сушили в вакууме, получая твердое вещество светло-бежевого цвета (19,7 г). Его растворяли в теплой воде, рН доводили до значения 2 концентрированной соляной кислотой и смесь экстрагировали этилацетатом. Органический экстракт сушили (Na2SO4) и упаривали,получая после сушки при 50 С указанное в заголовке соединение в виде твердого вещества кремового цвета (18,081 г, 82%); время удерживания для LCMS 3,88 мин; m/z 507 МН+; ЯМР(CDCl3) включает в себя 7.61 (1 Н, m), 7.18-7.12 (2 Н, m), 6.52 (1 Н, dd, J=4, 2 Гц), 6.46 (1 Н, s), 6.41 (1 Н, dd, J=10, 2 Гц), 5.47 и 5.35 (1 Н, 2m), 4.47 (1 Н, bd, J=9 Гц), 3.37 (1 Н, m), 1.55 (3 Н, s), 1.21 (3 Н, s), 1.06 (3 Н, d, J=7 Гц). Промежуточное соединение 1. 6,9-Дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16 метил-3-оксоандроста-1,4-диен-17-карботиокислота (первый альтернативный способ). Перемешиваемую суспензию 6,9-дифтор-11,17-дигидрокси-16-метил-3-оксоандроста-1,4 диен-17-карботиокислоты (полученной в соответствии с методикой, описанной в GB 2088877 В) (1 ед. массы, 49,5 г) в ацетоне (10 об.) охлаждают до 0-5 С, обрабатывают триэтиламином (0,51 ед. массы, 2,1 экв.), поддерживая температуру ниже 5 С, и перемешивают в течение 5 мин при 0-5 С. Затем в течение как минимум 20 мин добавляют 2-фуроилхлорид (0,65 ед. массы, 2,05 экв.), поддерживая температуру реакционной смеси 0-5 С. Реакционную смесь перемешивают в течение 30 мин при 0-5 С, затем отбирают образец для анализа посредством высокоэффективной жидкостной хроматографии (ВЭЖХ). В течение приблизительно 15 мин добавляют раствор диэтаноламина (1,02 ед. массы, 4 экв.) в метаноле (0,8 об.) с последующей промывкой метанолом (0,2 об.) и реакционную смесь перемешивают при 0-5 С в течение 1 ч. Еще раз отбирают образец для анализа посредством ВЭЖХ, после чего реакционную смесь нагревают приблизительно до 20 С и обрабатывают водой (1,1 ед. массы). Далее реакционную смесь в течение приблизительно 20 мин обрабатывают раствором HCl (SG1.18 (11,5 М), 1 об.) в воде (10 об.),поддерживая температуру реакционной смеси ниже 25 С. Суспензию перемешивают при 20-23 С в тече- 20005992 ние по меньшей мере 30 мин, затем фильтруют. Осадок на фильтре промывают водой (32 об.). Продукт сушат в вакууме при температуре приблизительно 60 С в течение ночи, получая указанное в заголовке соединение в виде твердого вещества белого цвета (58,7 г, 96,5%). Промежуточное соединение 1. 6,9-Дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16 метил-3-оксоандроста-1,4-диен-17-карботиокислота (второй альтернативный способ). Перемешиваемую суспензию 6,9-дифтор-11,17-дигидрокси-16-метил-3-оксоандроста-1,4 диен-17-карботиокислоты (полученной в соответствии с методикой, описанной в GB 2088877B) (1 ед. массы, 49,5 г) в ацетоне (10 об.) охлаждают до 0-5 С, обрабатывают триэтиламином (0,51 ед. массы, 2,1 экв), поддерживая температуру ниже 5 С, и перемешивают в течение 5 мин при 0-5 С. Затем в течение как минимум 20 мин добавляют 2-фуроилхлорид (0,65 ед. массы, 2,05 экв.), поддерживая температуру реакционной смеси 0-5 С. Реакционную смесь перемешивают в течение по меньшей мере 30 мин и затем разбавляют водой (10 об.), поддерживая температуру реакционной смеси в интервале 0-5 С. Полученный осадок собирают с помощью фильтрования и последовательно промывают смесью ацетон/вода (50/50 2 об.) и водой (22 об.). Продукт сушат под вакуумом при температуре приблизительно 55 С в течение ночи, получая 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста 1,4-диен-17-ил-S-(2-фуранилкарбонил)тиоангидрид в виде твердого вещества белого цвета (70,8 г,98,2%). (ЯМР(CD3CN) 0.99 (3 Н, d) (J = 7.3 Гц), 1.24 (3 Н, s), 1.38 (1 Н, m) (J = 3,9 Гц), 1.54 (3 Н, s), 1.67(1 Н, m), 1.89 (1 Н, широкий d) (J = 15, 2 Гц), 1.9-2.0 (1 Н, m), 2.29-2.45 (3 Н, m), 3.39 (1 Н, m), 4.33 (1 Н, m),4.93 (1 Н, широкий s), 5.53 (1 Н, ddd) (J = 6.9, 1,9 Гц; JHF = 50,9 Гц), 6.24 (1 Н, m), 6.29 (1 Н, dd) (J = 10,3, 2,0 Гц), 6.63 (2 Н, m), 7.24-7.31 (3 Н, m), 7.79 (1 Н, dd) (J1 Гц), 7.86 (1 Н, dd) (J1 Гц. Часть этого продукта (0,56 г) смешивают с 6,9-дифтор-11,17-дигидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислотой (0,41 г) в молярном отношении 1:1 в DMF (10 об., принимая во внимание общую стероидную загрузку). Реакционную смесь обрабатывают триэтиламином (приблизительно 2,1 экв.) и перемешивают приблизительно при 20 С в течение приблизительно 6 ч. К реакционной смеси добавляют воду (50 об.), содержащую дополнительное количество конц. HCl (0,5 об.), и полученный в результате этого осадок собирают с помощью фильтрования. Слой промывают водой (25 об.) и сушат в вакууме приблизительно при 55 С в течение ночи, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,99 г, 102%). Промежуточное соединение 1 А. Соль 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси 16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты с диизопропилэтиламином. Перемешиваемую суспензию 6,9-дифтор-11,17-дигидрокси-16-метил-3-оксоандроста-1,4 диен-17-карботиокислоты (полученной в соответствии с методикой, описанной в GB 2088877 В) (49,5 г) в метилацетате (500 мл) обрабатывают триэтиламином (35 мл), поддерживая температуру реакционной смеси в интервале 0-5 С. Добавляют 2-фуроилхлорид (25 мл) и смесь перемешивают при температуре 05 С в течение 1 ч. Добавляют раствор диэтаноламина (52,8 г) в метаноле (50 мл) и смесь перемешивают при температуре 0-5 С в течение по меньшей мере 2 ч. Добавляют разбавленную соляную кислоту (приблизительно 1 М, 550 мл), поддерживая температуру реакционной смеси ниже 15 С, и смесь перемешивают при 15 С. Отделяют органическую фазу и водную фазу экстрагируют обратно метилацетатом(2250 мл). Все органические фазы объединяют, последовательно промывают соляным раствором (5250 мл) и обрабатывают диизопропилэтиламином (30 мл). Реакционную смесь концентрируют посредством дистилляции при атмосферном давлении приблизительно до объема 250 мл и охлаждают до температуры 25-30 С (кристаллизация требуемого продукта обычно происходит во время стадии дистилляция/(последующее охлаждение). Добавляют трет-бутилметиловый эфир (ТВМЕ) (500 мл), суспензию дополнительно охлаждают и выдерживают при 0-5 С в течение по меньшей мере 10 мин. Продукт отфильтровывают, промывают охлажденным ТВМЕ (2200 мл) и сушат под вакуумом приблизительно при 40-50 С (75,3 г, 98,7%). ЯМР (CDCl3) : 7.54-7.46 (1 Н, m), 7.20-7.12 (1 Н, dd), 7.07-6.99 (1 Н, dd), 6.48-6.41(2 Н, m), 6.41-6.32 (1 Н, dd), 5.51-5.28 (1 Н, dddd 2JH-F =50 Гц), 4.45-4.33 (1 Н, bd), 3.92-3.73 (3 Н, bm), 3.273.14 (2 Н, q), 2.64-2.12 (5 Н, m), 1.88-1.71 (2 Н, m), 1.58-1.15 (3 Н, s), 1.50-1.38 (15 Н, m), 1.32-1.23 (1 Н, m),1.23-1.15 (3 Н, s), 1.09-0.99 (3 Н, d). Промежуточное соединение 1 Б. Соль 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси 16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты с триэтиламином. Перемешиваемую суспензию промежуточного соединения 1 (30 г) в этилацетате (900 мл) обрабатывают триэтиламином (1,05 мол. экв., 8,6 мл) и смесь перемешивают при температуре приблизительно 20 С в течение 1,5 ч. Осадок отфильтровывают, промывают этилацетатом (22 об.) и сушат в вакууме при 45 С в течение 18 ч, получая указанное в заголовке соединение в виде твердого вещества белого цвета (28,8 г, 80%). ЯМР (CDCl3) : 7.59-7.47 (1 Н, m), 7.23-7.13 (1 Н, dd), 7.08-6.99 (1 Н, d), 6.54-6.42 (2 Н,m), 6.42-6.32 (1 Н, dd), 5.55-5.26 (1 Н, dddd 2JH-F =50 Гц), 4.47-4.33 (1 Н, bd), 3.88-3.70 (1 Н, bm), 3.31-3.09- 21005992 Примеры Пример 1. Несольватированная форма 1 S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты. Суспензию промежуточного соединения 1 (2,5 г, 4,94 ммоль) растворяли в безводном N,Nдиметилформамиде (25 мл) и добавляли гидрокарбонат натрия (465 мг, 5,53 ммоль). Смесь перемешивали при -20 С, добавляли бромфторметан (0,77 мл, 6,37 ммоль) и смесь перемешивали при -20 С в течение 2 ч. Добавляли диэтиламин (2,57 мл, 24,7 ммоль) и смесь перемешивали при -20 С в течение 30 мин. Смесь добавляли к 2 М соляной кислоте (93 мл) и перемешивали в течение 30 мин. Добавляли воду (300 мл), осадок собирали путем фильтрования, промывали водой и сушили в вакууме при 50 С, получая твердое вещество белого цвета, которое подвергали перекристаллизации из смеси ацетон/вода (получая сольват S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16 метил-3-оксоандроста-1,4-диен-17-карботиокислоты с ацетоном) и сушили в вакууме при 50 С, получая указанное в заголовке соединение (2,351 г, 88%), время удерживания для LCMS 3,66 мин; m/z 539 МН+; ЯМР(CDCl3) включает в себя 7.60 (1 Н, m), 7.18-7.11 (2 Н, m), 6.52 (1 Н, dd, J4.2 Гц), 6.46 (1 Н, s), 6.41(3 Н, s), 1.16 (3 Н, s), 1.06 (3 Н, d, J=7 Гц). Фармакологическая активность Фармакологическая активность in vitro. Фармакологическую активность оценивали в функциональном анализе in vitro активности агониста глюкокортикоидов, которая обычно может служить основой для предсказания противовоспалительной или противоаллергической активности in vivo. В экспериментах этого раздела соединение формулы (I) использовали в в виде несольватированной формы 1. Функциональный анализ основан на анализе, описанном К.Р. Ray et al., Biochem. J. (1997), 328, 707715. Клетки А 549, стабильно трансфицированные репортерным геном, содержащим чувствительные элементы NF-В промотора гена ELAM, соединенного с sPAP (секретируемой щелочной фосфатазой),обрабатывали тестируемыми соединениями в соответствующих дозах в течение 1 ч при 37 С. Затем клетки в течение 16 ч стимулировали фактором некроза опухоли (TNF, 10 нг/мл), в течение которых с помощью стандартного колориметрического анализа измеряют количество продуцируемой щелочной фосфатазы. Строили кривые доза-ответ, из которых оценивали величины ЕС 50 (концентрация тестируемого соединения, при которой наблюдается эффект, равный 50% от максимального). В этом исследовании соединение из примера 1 продемонстрировало значение ЕС 50 1 нМ. Глюкокортикоидный рецептор (ГР) может функционировать по меньшей мере по двум различным механизмам, посредством позитивной регуляции экспрессии генов через прямое связывание ГР со специфическими последовательностями в промоторах генов и негативной регуляции экспрессии генов,управляемой другими транскрипционными факторами (такими как NFB или АР-1) через их прямое взаимодействие с ГР. В варианте описанного выше способа для контроля этих функций были созданы две репортерные плазмиды и их, посредством трансфекции, по отдельности ввели в эпителиальные клетки легких человека А 549. Первая клеточная линия содержит репортерный ген люциферазы светляков под контролем синтетического промотора, который специфически отвечает на активацию транскрипционного фактораNFB при стимуляции с помощью TNF. Вторая клеточная линия содержит репортерный ген люциферазы renilla под контролем синтетического промотора, который содержит 3 копии консенсусного глюкокортикоидного чувствительного элемента и который отвечает на прямую стимуляцию глюкокортикоидами. Одновременное измерение трансактивации и трансрепрессии проводили путем смешивания этих двух клеточных линий в отношении 1:1 в 96-луночном планшете (40000 клеток на лунку) и выращивания в течение ночи при 37 С. Тестируемые соединения растворяли в диметилсульфоксиде (DMSO) и добавляли к клеткам в конечной концентрации DMSO, равной 0,7%. После инкубации в течение 1 ч добавляли 0,5 нг/мл TNF (RD Systems) и через следующие 15 ч при 37 С измеряли уровни люциферазы светляков и люциферазы renilla, используя набор Packard Firelite и следуя инструкциям производителя. Строили кривые доза-ответ, из которых определяли величины EC50. Трансактивация (ГР) Трансрепрессия(NFB) Фармакологическая активность in vivo. Фармакологическую активность in vivo оценивали с использованием модели эозинофилии сенсибилизированных овальбумином коричневых норвежских (Brown Norway) крыс. Эта модель предназначена- 22005992 для имитации индуцированной аллергеном эозинофилии легких, главного компонента воспаления легких при астме. В экспериментах этого раздела соединение формулы (I) использовали в виде несольватированной формы 1. Соединение (I) вызывало на этой модели дозозависимое ингибирование эозинофилии легких после введения в виде внутритрахеальной суспензии (ВТ) в физиологическом растворе за 30 мин до введения овальбумина. Значительное ингибирование достигается после введения разовой дозы 30 мкг соединения(I), и ответ оказался значительно (р=0,016) сильнее ответа, который наблюдался с эквивалентной дозой пропионата флутиказона в этом же исследовании (69% ингибирование с соединением (I) в сравнении с 41% ингибированием с пропионатом флутиказона). В крысиной модели дегенерации тимуса 3 дневные ВТ дозы по 100 мкг соединения (I) индуцировали значительно меньшие снижения массы тимуса (р=0,004) по сравнению с эквивалентной дозой пропионата флутиказона в этом же исследовании (67%-ное снижение массы тимуса с соединением (I) в сравнении с 78%-ным снижением с пропионатом флутиказона). Взятые вместе эти результаты указывают на превосходный терапевтический индекс соединения (I) в сравнении с пропионатом флутиказона. Метаболизм in vitro в гепатоцитах крысы и человека. Инкубация соединения (I) с гепатоцитами крысы или человека показывает, что данное соединение и пропионат флутиказона метаболизируются одинаково, при этом единственным существенным продуцируемым метаболитом является 17 карбоновая кислота (X). Исследование скорости появления этого метаболита при инкубации соединения (I) с гепатоцитами человека (37 С, 10 мкМ концентрация лекарственного средства, гепатоциты от 3 субъектов, 0,2 и 0,7 миллионов клеток/мл) показывает, что соединение (I) метаболизируется приблизительно в 5 раз быстрее, чем пропионат флутиказона: Среднее продуцирование метаболита составляет 102-118 пмоль/ч для соединения (I) и 18,8-23,0 пмоль/ч для пропионата флутиказона. Фармакокинетика после внутривенного (ВВ) и перорального введения у крыс. Соединение (I) вводили перорально (0,1 мг/кг) и ВВ (0,1 мг/кг) самцам крыс Wister Han и определяли фармакокинетические параметры. Соединение (I) продемонстрировало незначительную биодоступность при пероральном введении (0,9%) и плазменный клиренс 47,3 мл/мин/кг, приближающийся к скорости печеночного кровотока (плазменный клиренс пропионата флутиказона = 45,2 мл/мин/кг). Фармакокинетика после внутритрахеального введения сухого порошка у свиней. Анестезированным свиньям (2) внутритрахеально вводили дозы гомогенной смеси соединения (I) (1 мг) и пропионата флутиказона (1 мг) в виде сухого порошка, смешанного с лактозой (10 мас.%). В течение времени вплоть до 8 ч после введения доз отбирали образцы крови. После экстрагирования и проведения анализа с помощью LC-MS/MS-методики определяли уровни в плазме соединения (I) и пропионата флутиказона, нижние пределы количественного анализа этих методов составили 10 и 20 пг/мл для соединения (I) и пропионата флутиказона соответственно. С использованием этих методов уровень соединения (I) можно было количественно измерить вплоть до 2 ч после введения, а уровень пропионата флутиказона можно было количественно измерить вплоть до 8 ч после введения. Максимальные концентрации в плазме для обоих соединений наблюдали в течение 15 мин после введения. Данные по периоду полувыведения из плазмы при ВВ введении (0,1 мг/кг) использовали для расчета величин AUC (площади под кривой) (0-inf) для соединения (I). Это компенсирует плазменный профиль соединения (I) при определении только вплоть до 2 ч после ВТ введения дозы и удаляет любой наклон, обусловленный наличием ограниченного числа данных между соединением (I) и пропионатом флутиказона. Величины Сmах и AUC (0-inf) демонстрируют заметно сниженную системную продолжительность действия соединения (I) по сравнению с пропионатом флутиказона Фармакокинетические параметры как для соединения (I), так и для пропионата флутиказона были одинаковыми у анестезированной свиньи после внутривенного введения смеси двух этих соединений в дозе 0,1 мг/кг. Клиренс этих двух глюкокортикоидов аналогичен в этой экспериментальной модели на свиньях. Пример 1. Несольватированная форма 1 S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты (первый альтернативный способ). Маловязкую суспензию промежуточного соединения 1 А (12,61 г, 19,8 ммоль, эквивалент 10 г промежуточного соединения 1) в этилацетате (230 мл) и воде (50 мл) обрабатывают межфазным катализатором (хлоридом бензилтрибутиламмония, 10 мол.%), охлаждают до 3 С и обрабатывают бромфторметаном (1,10 мл, 19,5 ммоль, 0,98 экв.), промывая в предварительно охлажденном (0 С) этилацетате (ЕtOАс)(20 мл). Суспензию перемешивают в течение ночи, позволяя нагреться до 17 С. Водный слой отделяют и органическую фазу последовательно промывают 1 М HCl (50 мл), 1%-ным (м/о) раствором NaHCO3(350 мл) и водой (250 мл). Этилацетатный раствор подвергают дистилляции при атмосферном давлении до тех пор, пока температура дистиллята не достигнет приблизительно 73 С, в этот момент добавляют толуол (150 мл). Дистилляцию продолжают при атмосферном давлении до тех пор, пока не будет удален весь оставшийся ЕtOАс (температура дистиллята приблизительно 103 С). Полученную суспензию охлаждают, выдерживают при температуре 10 С и отфильтровывают. Слой промывают толуолом(230 мл) и продукт сушат в сушильном шкафу под вакуумом при 60 С до постоянной массы, получая указанное в заголовке соединение (8,77 г, 82%). Пример 1. Несольватированная форма 1 S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты (второй альтернативный способ). Суспензию сольвата с ацетоном S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты (полученного,например, в соответствии с примером 11) (50,0 г) в ацетоне (1500 мл) и воде (75 мл) нагревали до температуры дефлегмации. Полученную смесь осветляли с помощью фильтрования в горячем состоянии(фильтровальная бумага Ватман 54 (Whatman 54, во время которого в фильтрате кристаллизовалось некоторое количество твердого вещества. К фильтрату добавляли дополнительное количество ацетона(200 мл), получая в флегме прозрачный раствор. Раствор подвергали дистилляции при атмосферном давлении до тех пор, пока в флегме не появилась мутность (собрали приблизительно 750 мл растворителя). К горячему раствору добавляли толуол (1000 мл) и дистилляцию при атмосферном давлении продолжали, что привело к кристаллизации при температуре приблизительно 98 С. Дистилляцию растворителя продолжали, пока температура реакционной смеси не достигла 105 С (собрали приблизительно 945 мл растворителя). Смесь охлаждали до температуры окружающей среды, затем охлаждали и выдерживали при температуре 10 С в течение 10 мин. Продукт отфильтровывали, промывали толуолом (150 мл) и имеющуюся в нем влагу удаляли. Продукт сушили при температуре приблизительно 60 С под вакуумом в течение 16 ч, получая указанное в заголовке соединение в виде плотного твердого вещества белого цвета (37,8 г, 83,7%). Картина XRPD продукта из примера 1 показана на фиг. 1. Профили DSC и TGA показаны на фиг. 3. Пример 2. Несольватированная форма 2 S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты. Суспензию S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси 16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты (полученного, например, в соответствии с примером 1, первым способом) (6,0 г) в дихлорметане (180 мл) нагревали до температуры дефлегмации,получая прозрачный раствор. Данный раствор осветляли с помощью фильтрования в горячем состоянии(фильтровальная бумага Ватман 54) и подвергали дистилляции при атмосферном давлении (собрали приблизительно 100 мл растворителя), что привело к кристаллизации при температуре дефлегмации. Смесь выдерживали при температуре дефлегмации в течение приблизительно 30 мин и медленно охлаждали до температуры окружающей среды. Смесь дополнительно охлаждали и выдерживали при 10-20 С в течение 2 ч. Суспензию охлаждали до температуры ниже 10 С и продукт отфильтровывали, имеющуюся в нем влагу удаляли и сушили при температуре приблизительно 60 С под вакуумом в течение ночи,получая твердое вещество белого цвета (4,34 г, 71%). Более чистый образец несольватированной формы 2 S-фторметилового эфира 6,9-дифтор-17[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты получали путем кристаллизации охлаждением S-фторметилового эфира 6,9-дифтор-17-[(2 фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты (полученного, например, в соответствии с примером 1, первым способом) в метаноле (60 об., подвергнутых дистилляции при атмосферном давлении до приблизительно 37,5 об.). Продукт выделяли путем фильтрования и сушили в сушильном шкафу при 60 С под вакуумом в течение 16 ч, получая электростатическое твердое вещество белого цвета (4,34 г, 71%). Картина XRPD продукта из примера 2 показана на фиг. 1. Пример 3. Несольватированная форма 3 S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты. Суспензию сольвата с ацетоном S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты (полученного,например, в соответствии с примером 11) (20,0 г) в дихлорметане (800 мл, 40 об.) и воде (10 мл, 0,5 об.) нагревали до температуры дефлегмации, получая прозрачный раствор. Данный раствор осветляли с помощью фильтрования в горячем состоянии (фильтровальная бумага Ватман 54), во время которого в фильтрате кристаллизовалось некоторое количество твердого вещества, которое полностью растворялось при нагревании до температуры дефлегмации. Раствор подвергали дистилляции при атмосферном давлении (собрали приблизительно 400 мл растворителя) и оставляли охлаждаться до температуры окружающей среды. Смесь дополнительно охлаждали и выдерживали при температуре 10 С в течение 10 мин. Продукт отфильтровывали, имеющуюся в нем влагу удаляли и сушили при температуре приблизительно 60 С под вакуумом в течение ночи, получая твердое вещество белого цвета (12,7 г, 70%). Картина XRPD продукта из примера 3 показана на фиг. 1 и 4. Пример 4. Взаимопревращение форм 1. 2 и 3 несольватированного S-фторметилового эфира 6,9 дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты. Суспендирование смеси формы 1 и формы 2 в воде при температуре окружающей среды показало,что компоненты полностью превращаются со временем в форму 1. Результаты XRPD показаны на фиг. 2. Похожие результаты получены после суспендирования смеси формы 1 и формы 2 в этаноле при температуре окружающей среды. На основании этих результатов можно сделать вывод, что форма 1 является термодинамически более стабильной полиморфной формой из этих двух форм. Температурные XRPD-исследования формы 3 проводили как показано на фиг. 4. Температурный и временной профиль показаны на фиг. 5, а 5 записей самописца, показанные на фиг. 4, были получены в точках равновесия, показанных на фиг. 5. Результаты указывают на то, что форма 3 превращается сначала в форму 2, а затем в форму 1 по мере повышения температуры. Пример 5. Сорбция влаги формами 1, 2 и 3 несольватированного S-фторметилового эфира 6,9 дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты. Характеристики сорбции влаги данных трех форм определяли путем наблюдения за изменением массы твердого вещества в условиях пошагового увеличения, а затем снижения влажности. Были получены следующие результаты: форма 1: поглощение 0,18% мас./мас. влаги в интервале относительной влажности 0-90% при 25 С,форма 2: поглощение 1,1-2,4% мас./мас. влаги в интервале относительной влажности 0-90% при 25 С,форма 3: поглощение 1,2-2,5% мас./мас. влаги в интервале относительной влажности 0-90% при 25 С. Пример 6. Энтальпия растворения форм 1 и 3 несольватированного S-фторметилового эфира 6,9 дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты. Энтальпии растворения в диметилсульфоксиде (DMSO) и ацетонитриле определяли при 25 С. Были получены следующие результаты: Форма 1 Форма 3 Ацетонитрил(результаты в кДж/моль) Из этих результатов можно определить, что энтальпия фазового превращения формы 3 в форму 1 составляет приблизительно 5,1-6,7 кДж/моль. Исходя из предположения, что энтропия фазового превращения мала, поскольку обе формы являются несольватированными, энтальпия фазового превращения может быть приравнена к свободной энергии перехода. Таким образом, эти результаты означают, что форма 1 является термодинамически наиболее стабильной формой при 25 С.- 25005992 Пример 7. Сольват S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11 гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты с метилэтилкетоном. Суспензию S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси 16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты (полученного, например, в соответствии с примером 1) (400 мг) в метилэтилкетоне (3,2 мл) нагревают до температуры дефлегмации, получая прозрачный раствор. Часть растворителя отгоняют посредством дистилляции при атмосферном давлении(приблизительно 1 мл) и смесь охлаждают до температуры приблизительно 20 С. Выкристаллизовавшийся продукт отфильтровывают, сушат при температуре приблизительно 20 С под вакуумом, получая указанное в заголовке соединение в виде твердого вещества белого цвета (310 мг, 68%). ЯМР(CDCl3) включает в себя пики, описанные в примере 1 для родоначального соединения, и следующие дополнительные пики растворителя: 2.45 (2 Н, q), 2.14 (3 Н, s), 1.06 (3 Н, t). Пример 8. Сольват S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11 гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты с изопропанолом. Раствор S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16 метил-3-оксоандроста-1,4-диен-17-карботиокислоты (полученного, например, в соответствии с примером 1) (150 мг) в изопропаноле (15 мл) оставляют для медленной кристаллизации в течение периода приблизительно 8 недель. Полученные короткие и толстые кристаллы выделяют путем фильтрования, получая указанное в заголовке соединение в виде твердого вещества белого цвета. ЯМР(CDCl3) включает в себя пики, описанные в примере 1 для родоначального соединения, и следующие дополнительные пики растворителя: 4.03 (1 Н, m), 1.20 (6 Н, d). Пример 9. Сольват S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11 гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты с тетрагидрофураном. Суспензию S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси 16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты (полученного, например, в соответствии с примером 1) (150 мг) в TGF (20 об.) нагревают, получая прозрачный раствор. Растворитель оставляют медленно испаряться в течение периода 6 дней, получая указанное в заголовке соединение в виде твердого вещества белого цвета. Альтернативно, раствор в TGF по каплям добавляют к раствору бикарбоната калия (2% мас./мас.) в воде (50 об.) и выпавший в осадок продукт собирают путем фильтрования, получая указанное в заголовке соединение в виде твердого вещества белого цвета. ЯМР(CDCl3) включает в себя пики, описанные в примере 1 для родоначального соединения, и следующие дополнительные пики растворителя: 3.74 (4 Н, m), 1.85 (4 Н, m). Пример 9, Сольват S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11 гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты с тетрагидрофураном (альтернативный способ). Маловязкую суспензию соли 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16 метил-3-оксоандроста-1,4-диен-17-карботиокислоты с триэтиламином (полученной, например, в соответствии с получением промежуточного соединения 1 Б) (1,2 г) в TGF (10 мл) обрабатывают межфазным катализатором (бромидом тетрабутиламмония, обычно между 8 и 14 мол.%), охлаждают до температуры приблизительно 3 С и обрабатывают бромфторметаном (0,98 экв.). Суспензию перемешивают в течение от 2 до 5 ч, оставляют нагреваться до 17 С. Реакционную смесь выливают в воду (30 об.), перемешивают при температуре приблизительно 10 С в течение 30 мин и отфильтровывают. Собранное твердое вещество промывают водой (43 об.) и продукт сушат в сушильном шкафу под вакуумом при 60 С в течение ночи, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,85 г, 87%). Пример 10. Сольват S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11 гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты с DMF. Смесь промежуточного соединения 1 (4,5 г, 8,88 ммоль) в DMF (31 мл) обрабатывают бикарбонатом калия (0,89 г, 8,88 ммоль) и смесь охлаждают до -20 С. Добавляют раствор бромфторметана (0,95 г,8,50 ммоль, 0,98 экв.) в DMF (4,8 мл) при 0 С и смесь перемешивают при -20 С в течение 4 ч. Затем смесь перемешивают при -20 С в течение дополнительных 30 мин, добавляют к 2 М соляной кислоте(100 мл) и перемешивают в течение дополнительных 30 мин при 0-5 С. Осадок собирают с помощью вакуумного фильтрования, промывают водой и сушат при температуре 50 С, получая указанное в заголовке соединение (4,47 г, 82%). ЯМР(CD3OD) включает в себя пики, описанные в примере 1 для родоначального соединения, и следующие дополнительные пики растворителя: 7.98 (1 Н, bs), 2.99 (3 Н, s), 2.86(3 Н, s). Пример 11. Сольват S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11 гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты с ацетоном. Раствор промежуточного соединения 1 (530,1 г, 1 ед. массы) в диметилформамиде (DMF) (8 об.) обрабатывают гидрокарбонатом калия (0,202 ед. массы, 1,02 экв.) и смесь охлаждают до -173 С при перемешивании. Затем добавляют бромфторметан (BFM) (0,22 ед. массы, 0,99 экв.) и реакционную смесь перемешивают при -173 С в течение по меньшей мере 2 ч. Далее реакционную смесь в течение приблизи- 26005992 тельно 10 мин добавляют к воде (17 об.) при температуре 53 С с последующей промывкой водой (1 об.). Суспензию перемешивают при 5-10 С в течение по меньшей мере 30 мин и затем фильтруют. Осадок на фильтре (сольват S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11 гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты с DMF) промывают водой (44 об.) и продукт сушат, размазывая на фильтре. Влажный осадок возвращают в сосуд, добавляют ацетон (5,75 об.) и кипятят с обратным холодильником в течение 2 ч. Смесь охлаждают до температуры 523 С, добавляют воду (5,75 об.), поддерживая температуру 523 С. Затем смесь охлаждают до температуры 203 С, фильтруют и сушат в вакууме при 605 С в течение ночи, получая указанное в заголовке соединение в виде твердого вещества белого цвета (556,5 г, 89%). ЯМР(CDCl3) включает в себя пики, описанные в примере 1 для родоначального соединения, и следующие дополнительные пики растворителя: 2.17 (6H,s). Пример 12. Сухая порошковая композиция, содержащая несольватированную форму 1 S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты. Сухую порошковую композицию готовили следующим образом: несольватированная форма 1 S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты (полученная в соответствии с примером 1, первым альтернативным способом и измельченная до MMD 3 мкм) - 0,20 мг,измельченная лактоза (в которой не более 85% частиц имеют MMD 60-90 мкм и не менее 15% частиц имеют MMD менее 15 мкм - 12 мг. Готовят легко снимающуюся блистерную полоску, содержащую 60 блистеров, каждый из которых заполнен только что описанной композицией. Пример 13. Аэрозольная композиция, содержащая несольватированную 1 S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-113-гидрокси-16-метил-3-оксоандроста-1,4-диен-17 карботиокислоты. Алюминиевый контейнер заполняли композицией следующим образом: несольватированная форма 1 S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17-карботиокислоты (полученная в соответствии с примером 1 первым альтернативным способом и измельченная до MMD 3 мкм) - 250 мкг,1,1,1,2-тетрафторэтан - до 50 мкл (количества на одно действие),в общем количестве подходит для 120 действий, и контейнер соединяют с дозирующим клапаном,выполненным с возможностью высвобождения 50 мкл за одно действие. Пример 14. Назальная композиция, содержащая несольватированную 1 S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11-гидрокси-16-метил-3-оксоандроста-1,4-диен-17 карботиокислоты. Композицию для доставки в носовую полость готовили следующим образом. Несольватированная форма 1 S-фторметилового эфира 6,9-дифтор-17-[(2-фуранилкарбонил)окси]-11 гидрокси-16-метил-3-оксоандроста-1,4-диен-17 карботиокислоты (полученная в соответствии с примером 1 первым альтернативным способом, измельченная) 10 мг Полисорбат 20 0,8 мг Сорбитанмонолаурат 0,09 мг Дигидрат дигидрофосфата натрия 94 мг Безводный гидрофосфат натрия 17,5 мг Хлорид натрия 48 мг Деминерализованная вода До 10 мл Композицией заполняли распылительный насос, способный доставлять большое количество отмеренных доз (Валуа (Valois. По всему описанию и следующей далее формуле изобретения, за исключением контекста, требующего другого толкования, будет понятно, что слово содержать и такие варианты как содержит и содержащий подразумевают включение сформулированного целого или стадии либо группы целых, но не исключение любого другого целого или стадии либо группы целых или стадий. Патенты и патентные заявки, описанные в этой заявке, включены в этот документ посредством ссылки. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение формулы (I) и его сольваты. 2. Соединение формулы (I) по п.1 в несольватированной форме. 3. Соединение формулы (I) в несольватированной форме по п.2 в полиморфной модификации формы 1, отличающейся тем, что она имеет профиль XRPD (дифракции рентгеновских лучей на порошке),имеющий пик около 18,9 2. 4. Соединение формулы (I) в несольватированной форме по п.2 в полиморфной модификации формы 2, отличающейся тем, что она имеет профиль XRPD, имеющий пики около 18,4 и 21,5 2. 5. Соединение формулы (I) в несольватированной форме по п.2 в полиморфной модификации формы 3, отличающейся тем, что она имеет профиль XRPD, имеющий пики около 18,6 и 19,2 2. 6. Соединение формулы (I) по п.1 в виде кристаллического твердого вещества в форме, по существу, стехиометрического сольвата с ацетоном. 7. Соединение формулы (I) по п.1 в виде кристаллического твердого вещества в форме, по существу, стехиометрического сольвата с тетрагидрофураном. 8. Соединение формулы (I) по п.1 в виде кристаллического твердого вещества в форме, по существу, стехиометрического сольвата с изопропанолом. 9. Соединение формулы (I) по п.1 в виде кристаллического твердого вещества в форме, по существу, стехиометрического сольвата с метилэтилкетоном. 10. Соединение формулы (I) по п.1 в виде кристаллического твердого вещества в форме, по существу, стехиометрического сольвата с диметилформамидом. 11. Применение соединения формулы (I) или его физиологически приемлемого сольвата по любому из пп.1-5 в качестве противовоспалительного или противоаллергического агента в ветеринарии или медицине человека. 12. Применение соединения формулы (I) или его физиологически приемлемого сольвата по любому из пп.1-5 для производства лекарственного средства для лечения воспалительных и/или аллергических состояний. 13. Фармацевтическая композиция, содержащая соединение формулы (I) или его физиологически приемлемый сольват по любому из пп.1-5 в смеси с одним или более чем одним физиологически приемлемым разбавителем или носителем. 14. Фармацевтическая композиция по п.13, которая не находится под давлением и пригодна для местного введения в легкое через полость рта в виде сухого порошка. 15. Фармацевтическая композиция по п.13 или 14, которая содержит лактозу или крахмал в качестве разбавителя или носителя. 16. Фармацевтическая композиция по п.13, которая не находится под давлением и пригодна для местного введения в носовую полость. 17. Фармацевтическая композиция по п.16, которая содержит воду в качестве разбавителя или носителя. 18. Фармацевтическая аэрозольная композиция, содержащая соединение формулы (I) или его физиологически приемлемый сольват по любому из пп.1-5 и фторуглерод или водородсодержащий хлорфторуглерод в качестве пропеллента, возможно, в комбинации с поверхностно-активным веществом или сорастворителем. 19. Фармацевтическая аэрозольная композиция по п.18, отличающаяся тем, что лекарственное средство является полностью растворенном в этой композиции. 20. Фармацевтическая аэрозольная композиция по п.18, отличающаяся тем, что лекарственное средство находится в виде частиц, при этом указанная композиция не содержит стабилизатор в форме добавки воды (то есть воды, добавленной в дополнение к появляющейся в композиции воде) или аминокислоты, ее производного либо их смеси. 21. Фармацевтическая аэрозольная композиция по любому из пп.18-20, которая содержит соединение формулы (I) или его физиологически приемлемый сольват по любому из пп.1-3, и фторуглерод или водородсодержащий хлорфторуглерод в качестве пропеллента, и суспендирующий агент, который растворим в пропелленте. 22. Фармацевтическая аэрозольная композиция по п.21, где суспендирующий агент представляет собой олигомолочную кислоту или ее производное.- 28005992 23. Фармацевтическая аэрозольная композиция по любому из пп.18-22, где пропеллент выбран из 1,1,1,2-тетрафторэтана, 1,1,1,2,3,3,3-гептафтор-н-пропана и их смесей. 24. Фармацевтическая аэрозольная композиция по любому из пп.18-20, которая состоит, по существу, из соединения формулы (I) или его физиологически приемлемого сольвата по любому из пп.1-3, возможно, в комбинации с другим терапевтически активным агентом и пропеллентом, выбранным из 1,1,1,2-тетрафторэтана, 1,1,1,2,3,3,3-гептафтор-н-пропана и их смесей. 25. Фармацевтическая композиция по любому из пп.13-23, которая дополнительно содержит другой терапевтически активный агент. 26. Фармацевтическая композиция по п.25, в которой указанный другой терапевтически активный агент представляет собой агонист 2-адренорецепторов. 27. Фармацевтическая композиция, содержащая комбинацию соединения формулы (I) или его физиологически приемлемого сольвата по любому из пп.1-5 вместе с ингибитором фосфодиэстеразы 4 совместно с физиологически приемлемым разбавителем или носителем. 28. Способ лечения человека или животного, имеющего воспалительное и/или аллергическое состояние, при котором указанному человеку или животному вводят эффективное количество соединения формулы (I) или его физиологически приемлемого сольвата по любому из пп.1-5. 29. Способ получения соединения формулы (I) по п.1 или его сольвата, при котором осуществляют алкилирование по 17-карботиокислотной группировке соединения формулы (II) или его соли. 30. Способ по п.29, где алкилирование осуществляют путем взаимодействия соединения формулы(II) или его соли с фторметилгалогенидом. 31. Способ получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 1 по п.3, при котором осуществляют кристаллизацию соединения формулы (I) в присутствии несольватирующего растворителя. 32. Способ получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 1 по п.3, при котором осуществляют десольватацию соединения формулы (I) в сольватированной форме. 33. Способ получения соединения формулы (I) в виде несольватированной полиморфной модификации формы 1 по п.3, при котором соединение формулы (I) растворяют в метилизобутилкетоне, этилацетате или метилацетате и получают соединение формулы (I) в виде несольватированной формы 1 путем добавления несольватирующего антирастворителя. 34. Соединение формулы (II) или его соль. 35. Соединение формулы (II) по п.34 в форме твердой кристаллической соли. 36. Соединение формулы (II) по п.35 в форме соли с диизопропилэтиламином. 37. Способ получения соединения формулы (II) по п.34, при котором- 29005992 подвергают взаимодействию с активированным производным 2-фуранкарбоновой кислоты, например, в количестве по меньшей мере 2 моль активированного производного на моль соединения формулы (III) с получением соединения формулы (IIА)(б) удаляют присоединенную через атом серы 2-фуроильную группировку из соединения формулы(IIА) посредством взаимодействия продукта стадии (а) с органическим первичным или вторичным аминным основанием, способным к образованию водорастворимого 2-фуроиламида. 38. Способ получения соединения формулы (II) по п.37, при котором, если продукт стадии (б) растворяют в органическом растворителе, по существу не смешивающемся с водой, дополнительно осуществляют стадию, на которой соединение формулы (II) очищают путем вымывания амидного побочного продукта со стадии (б) посредством водной промывки. 39. Способ получения соединения формулы (II) по п.37, при котором, если продукт стадии (б) растворяют в смешивающемся с водой растворителе, дополнительно осуществляют стадию, на которой соединение формулы (II) очищают путем обработки продукта стадии (б) водной средой таким образом,чтобы осадить чистое соединение формулы (II) или его соль. 40. Способ получения соединения формулы (II) по п.34, при котором подвергают взаимодействию с активированным производным 2-фуранкарбоновой кислоты в количестве по меньшей мере 2 моль активированного производного на моль соединения формулы (III) с получением соединения формулы (IIА), как оно определено в п.37, и(б) удаляют присоединенную через атом серы 2-фуроильную группировку из соединения формулы(IIА) посредством взаимодействия продукта стадии (а) с дополнительным молем соединения формулы(III) с получением 2 моль соединения формулы (II). 41. Соединение формулы (IIА)
МПК / Метки
МПК: C07J 31/00, A61K 31/58, A61P 5/44
Метки: качестве, агента, противовоспалительного, 2-фуранилкарбонил)окси]-11.бета.-гидрокси-16.aльфа.-метил-3-оксо-андроста-1,4-диен-17.бета, эфир, 9.альфа.-дифтор-17.aльфа, карботиокислоты, 6.aльфа, s-фторметиловый
Код ссылки
<a href="https://eas.patents.su/30-5992-s-ftormetilovyjj-efir-6alfa-9alfa-diftor-17alfa-2-furanilkarboniloksi-11beta-gidroksi-16alfa-metil-3-okso-androsta-14-dien-17beta-karbotiokisloty-v-kachestve-protivovospalitelnogo.html" rel="bookmark" title="База патентов Евразийского Союза">S-фторметиловый эфир 6.aльфа., 9.альфа.-дифтор-17.aльфа. -[(2-фуранилкарбонил)окси]-11.бета.-гидрокси-16.aльфа.-метил-3-оксо-андроста-1,4-диен-17.бета.- карботиокислоты в качестве противовоспалительного агента</a>
Комплексы лимфотоксина альфа/бета и антител против рецептора лимфотоксина-бета в качестве противоопухолевых агентов

Номер патента: 96
Опубликовано: 27.08.1998
Авторы: Броунинг Джеффри Л., Бенджамин Кристофер, Мейер Вернер
МПК: A61K 38/19, G01N 33/53
Метки: лимфотоксина, против, противоопухолевых, антител, качестве, лимфотоксина-бета, агентов, рецептора, комплексы
Формула / Реферат:
1. Способ лечения или уменьшения прогрессирования, тяжести или эффектов неоплазии, включающий введение терапевтически эффективного количества гетеромерного комплекса лимфотоксина a/b в присутствии терапевтически эффективного количества антитела к рецептору лимфотоксина b и/или g-интерферона. 2. Способ лечения или уменьшения прогрессирования, тяжести или эффектов неоплазии, включающий введение терапевтически эффективного количества антитела к...
Способ получения дроспиренона (6&beta, 7β 15&beta, 16&beta – диметилен -3 – оксо -17α-прегн- 4 -ен-21,17-карболактона, drsp) и 6&beta,7β15&beta,16&beta – диметилен -5&beta-гидрокси -3- оксо – 17&alpha – андростан – 21,17 – карболактон (zk 90965) в качестве промежуточного продукта

Номер патента: 1947
Опубликовано: 22.10.2001
Авторы: Мор Йорг-Торштен, Никкиш Клаус
МПК: C07J 53/00
Метки: оксо, промежуточного, дроспиренона, диметилен, 6&beta, способ, качестве, 5&beta-гидрокси, ен-21,17-карболактона, 90965, 17α-прегн, продукта, 6&beta,7β15&beta,16&beta, 16&beta, андростан, drsp, 21,17, получения, карболактон, 17&alpha, 15&beta, 7&beta
Формула / Реферат:
1. Способ получения дроспиренона (6b,7b;15b,16b-диметилен-3-оксо-17a-прегн-4-ен-21,17-карболактон DRSP) осуществляемый каталитическим гидрированием 17a-(3-гидрокси-1-пропинил)-6b,7b;15b,16b-диметилен-5b-андростан-3b,5,17b-триола (ZK 34506) с получением 17a-(3-гидрокси-1-пропил)-6b,7b;15b,16b-диметилен-5b-андростан-3b,5,17b-триола (ZK 92836) окислением в присутствии соли рутения до...
Дигидрат (2r,3s)-3-трет-бутоксикарбониламино-2-гидрокси-3-фенилпропионат 4,10beta-диацетокси-2alpha-бензоилокси-5beta, 20-эпокси-1-гидрокси-9-оксо-19-нор-циклопропа[g]такс-ii-ен-13alpha-ила и способ его получения

Номер патента: 668
Опубликовано: 28.02.2000
Авторы: Дидье Эрик, Отелен Жан-Рене, Лавинь Мишель
МПК: A61K 31/335, C07D 305/14
Метки: способ, 2r,3s)-3-трет-бутоксикарбониламино-2-гидрокси-3-фенилпропионат, дигидрат, 4,10beta-диацетокси-2alpha-бензоилокси-5beta, 20-эпокси-1-гидрокси-9-оксо-19-нор-циклопропа[g]такс-ii-ен-13alpha-ила, получения
Формула / Реферат:
1. Дигидрат (2R,3S)-3-трет-бутоксикарбониламино-2-гидрокси-3-фенилпропионата 4,10b-диацетокси-2a-бензоилокси-5b,20-эпокси-1-гидрокси-9-оксо-19-нор-циклопропа[g]такс-11-ен-13a-ила. 2. Способ получения дигидрата (2R,3S)-3-трет-бутоксикарбониламино-2-гидрокси-3-фенилпропионата 4,10b-диацетокси-2a-бензоилокси-5b-20-эпокси-1-гидрокси-9-оксо-19-нор-циклопропа[g]такс-11-ен-13a-ила, заключающийся в том, что кристаллизуют...
Применение ( – ) – цис – 5,7 – дигидрокси – 2 – (2 – хлорфенил) – 8 – [4 - (3 - гидрокси - 1 - метил) пиперидинил] – 4 – бензопиран – 4 – она в качестве ингибитора пролиферации клеток гладких мышц

Номер патента: 4786
Опубликовано: 26.08.2004
Авторы: Дюмон Дженнифер А., Паттерсон Уинстон Кемпбелл
МПК: A61P 9/10, A61K 31/445
Метки: дигидрокси, пиперидинил, применение, клеток, мышц, цис, пролиферации, гладких, метил, бензопиран, качестве, ингибитора, гидрокси, хлорфенил
Формула / Реферат:
1. Способ ингибирования пролиферации клеток гладких мышц, включающий введение пациенту эффективного количества соединения (-)-цис-5,7-дигидрокси-2-(2-хлорфенил)-8-[4-(3-гидрокси-1-метил)пиперидинил]-4H-бензопиран-4-она (флавопиридола), причем указанное эффективное количество составляет менее 70%, предпочтительно менее 60% или, в частности, менее 50% дозировки, которая была бы необходимой для подавления роста опухоли. 2. Применение...
Полиморфная форма 2-(r) – (1- (r) – (3,5-бис (трифторметил) фенил) этокси) -3- (s) – (4-фтор) фенил – 4 – (3 – (5-оксо-1h, 4h-1, 2, 4 – триазоло) метил)морфолина в качестве антагониста рецептора тахикинина

Номер патента: 2405
Опубликовано: 25.04.2002
Авторы: Макколи Джеймс, Крокер Луис
МПК: A61K 31/5375, A61P 25/28, C07D 265/32...
Метки: этокси, тахикинина, 5-оксо-1h, рецептора, форма, полиморфная, 4h-1, 4-фтор, триазоло, качестве, антагониста, 2-(r, трифторметил, 3,5-бис, фенил, метил)морфолина
Формула / Реферат:
1. Полиморфная форма соединения 2-(R)-(1-(R)-(3,5-бис(трифторметил)фенил)этокси)-3-(S)-(4-фтор)фенил-4-(3-(5-оксо-1Н,4Н-1,2,4-триазоло)метил)морфолина, обозначенная как форма I, по существу, отличающаяся рентгеновской порошковой дифрактограммой с характерными отражениями приблизительно 12,0, 15,3, 16,6, 17,0, 17,6, 19,4, 20,0, 21,9, 23,6, 23,8 и 24,8ш (2 тэта). 2. Фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и...
Предыдущий патент: Имидазольные соединения, арил или гетероарил конденсированные, в качестве противовоспалительных и анальгетических средств
Следующий патент: Улучшенный способ получения меламина высокой чистоты с высоким выходом
Случайный патент: Способ получения 2-этокси-1-((2'-((гидроксиамино)иминометил)дифенил-4-ил)метил)-1h-бензо[d]имидазол-7-карбоновой кислоты и ее сложных эфиров