Ингибиторы гистондеацетилазы, вызывающие дифференцировку клеток, и их применение
Номер патента: 7649
Опубликовано: 29.12.2006
Авторы: Миллер Томас А., Рифкинд Ричард А., Бреслоу Рональд, Бельведер Сандро, Маркс Пол А., Ришон Виктория М., Гершелл Лиланд





Формула / Реферат
1. Соединение, имеющее формулу

где каждый из радикалов R1 и R2 является фенильной, циклогексильной, циклогексиламино-, нафтильной, пиридинамино-, пиперидиновой, 9-пурин-6-амино-, тиазоламино-, гидроксильной, разветвленной или неразветвленной алкильной, алкенильной, алкилокси-, бензилокси-, фенилалкилокси-, пиридинильной, хинолильной или тиеногруппой, в которых алкил представляет собой метил, этил или t-бутил;
где радикал R3 является гидроксиламино-, гидроксильной, амино-, алкиламино- или алкилоксигруппой, в которых алкил представляет собой метил, этил или t-бутил;
где радикал R4 является атомом водорода, галогеном, фенильной группой или циклогексильной группой;
где А могут быть одинаковыми или различными и представлять собой амидную группу или группы -О-, -S-, -NR5- или -СН2-, где радикал R5 является C1-C5алкилом;
в котором n является целым числом от 3 до 10; и где фенильная, нафтильная, хинолильная, алкильная или циклогексильная группы могут быть замещены диметиламиногруппой,
или его фармацевтически приемлемая соль.
2. Соединение по п.1, отличающееся тем, что А представляет собой амидную группу, a R3 представляет собой гидроксиламиногруппу.
3. Соединение по п.1, отличающееся тем, что оно имеет формулу

4. Соединение по п.1, отличающееся тем, что оно имеет формулу

5. Соединение, имеющее формулу

6. Соединение по п.5, отличающееся тем, что n=5.
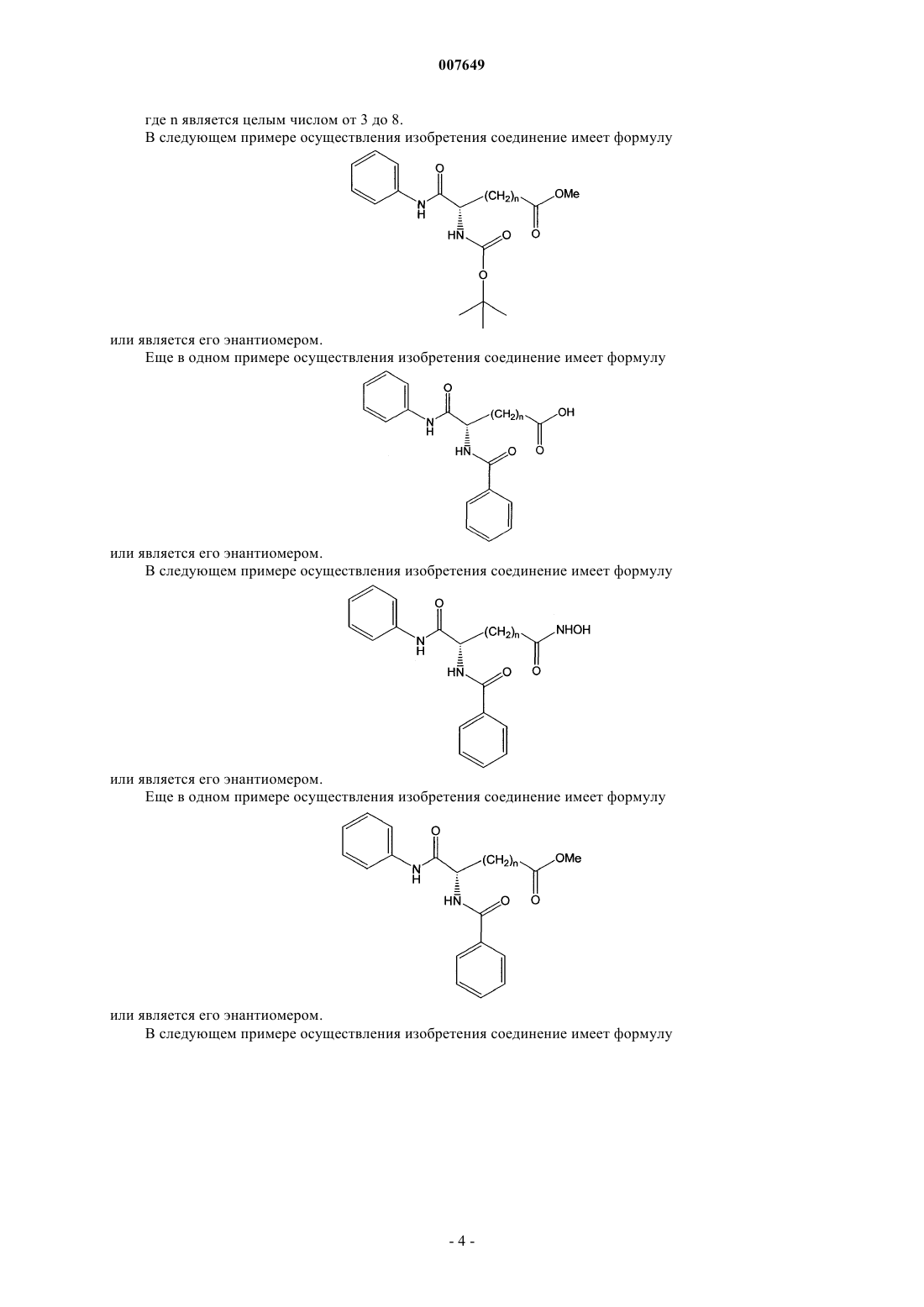
7. Соединение по п.1, отличающееся тем, что оно имеет формулу

или его энантиомер.
8. Соединение по п.7, отличающееся тем, что n=5.
9. Соединение по п.1, отличающееся тем, что оно имеет формулу

или его энантиомер.
10. Соединение по п.9, отличающееся тем, что n=5.
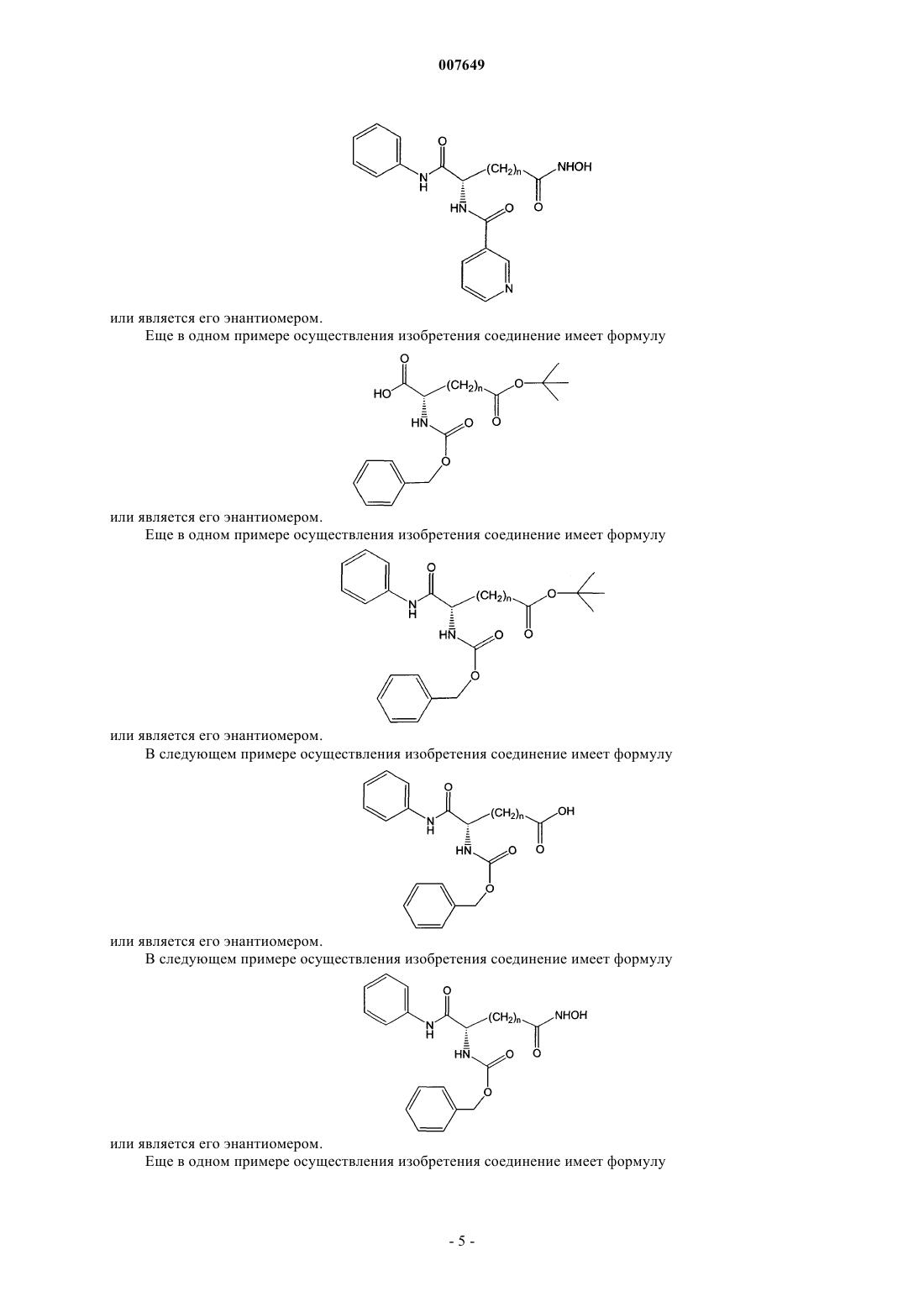
11. Соединение по п.1, отличающееся тем, что оно имеет формулу

или его энантиомер.
12. Соединение по п.11, отличающееся тем, что n=5.
13. Соединение по п.1, отличающееся тем, что оно имеет формулу

14. Соединение по п.1, отличающееся тем, что оно имеет формулу

15. Соединение по п.1, отличающееся тем, что оно имеет формулу

или его энантиомер.
16. Соединение по п.15, отличающееся тем, что n=5.
17. Соединение по п.1, отличающееся тем, что оно имеет формулу

или его энантиомер.
18. Соединение по п.17, отличающееся тем, что n=5.
19. Соединение по п.1, отличающееся тем, что оно имеет формулу

или его энантиомер.
20. Соединение по п.19, отличающееся тем, что n=5.
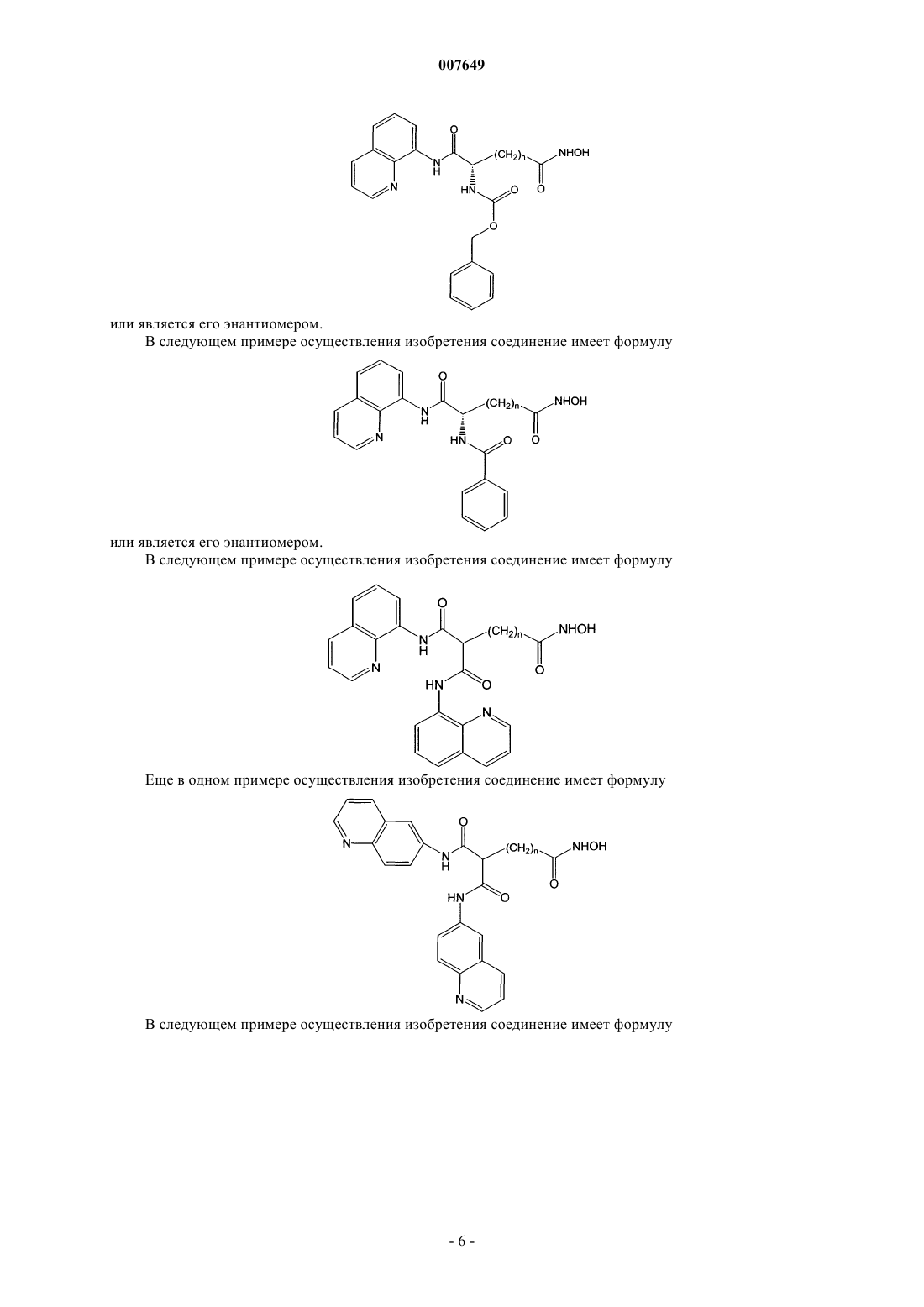
21. Соединение по п.1, отличающееся тем, что оно имеет формулу

или его энантиомер.
22. Соединение по п.21, отличающееся тем, что n=5.
23. Соединение по п.1, отличающееся тем, что оно имеет формулу

или его энантиомер.
24. Соединение по п.23, отличающееся тем, что n=5.
25. Соединение по п.1, отличающееся тем, что оно имеет формулу

или его энантиомер.
26. Соединение по п.25, отличающееся тем, что n=5.
27. Соединение по п.1, отличающееся тем, что оно имеет формулу

или его энантиомер.
28. Соединение по п.27, отличающееся тем, что n=5.
29. Соединение по п.1, отличающееся тем, что оно имеет формулу

где каждый из радикалов R1 и R2 является фенильной, циклогексильной, циклогексиламино-, нафтильной, пиридинамино-, пиперидиновой, алкильной, бензилокси-, фенилалкилокси-, пиридинильной, хинолильной или тиеногруппой, в которых алкил представляет собой метил, этил или t-бутил;
и где фенильная, нафтильная, хинолильная, алкильная или циклогексильная группы могут быть замещены диметиламиногруппой; и
где n является целым числом от 3 до 8.
30. Соединение, имеющее формулу

31. Соединение по п.30, отличающееся тем, что n=5.
32. Соединение, имеющее формулу

33. Соединение по п.32, отличающееся тем, что n=5.
34. Соединение, имеющее формулу

35. Соединение по п.34, отличающееся тем, что n=5.
36. Соединение по п.1, отличающееся тем, что оно имеет формулу

или его энантиомер.
37. Соединение по п.36, отличающееся тем, что n=5.
38. Фармацевтическая композиция, включающая фармацевтически эффективное количество соединения по п.1 и фармацевтически приемлемый носитель.
39. Применение соединения, как оно определено в п.1, в качестве соединения, предназначенного для избирательного индуцирования конечной дифференцировки неопластических клеток и обусловленного этим ингибирования пролиферации этих клеток.
40. Применение соединения, как оно определено в п.1, в качестве соединения, предназначенного для лечения опухоли, характеризующейся пролиферацией неопластических клеток.
41. Применение соединения, как оно определено в п.1, в качестве соединения, предназначенного для дифференцировки опухолевых клеток в опухоли.
42. Применение соединения, как оно определено в п.1, в качестве соединения, предназначенного для ингибирования активности гистондеацетилазы.
Текст
007649 Данная заявка претендует на приоритет и эффект изобретения в соответствии с предварительной заявкой на патент США с регистрационным номером 60/208688, поданной 1 июня 2000 г., и предварительной заявкой на патент США с регистрационным номером 60/152755, поданной 8 сентября 1999 г. На всем протяжении этой заявки ссылки на различные публикации приводятся в виде арабских цифр, заключенных в скобки. Полные ссылки на эти публикации можно найти в конце описания, непосредственно перед формулой изобретения. Содержание этих публикаций полностью включено в данную заявку посредством ссылок для более полного описания состояния области техники, к которой относится изобретение. Предшествующий уровень техники Рак является заболеванием, при котором популяция клеток становится в различной степени невосприимчивой к механизмам регулирования, которые в норме управляют пролиферацией и дифференцировкой. Последним подходом к лечению рака была попытка индукции терминальной дифференцировки неопластических клеток [1]. В модельных культурах клеток была описана дифференцировка при воздействии на клетки различных стимулов, включая циклический АМФ и ретиноевую кислоту [2, 3], акларубицин и другие антрациклины [4]. Существуют многочисленные доказательства того, что неопластическая трансформация не обязательно нарушает способность раковых клеток к дифференцировке [1, 5, 6]. Имеется много примеров опухолевых клеток, которые не реагируют на нормальные регуляторы пролиферации, и, по-видимому, экспрессия программы их дифференцировки блокирована, однако, можно индуцировать их дифференцировку и остановить размножение. Различные вещества, в том числе некоторые относительно простые полярные соединения [5, 7-9], производные витамина D и ретиноевой кислоты [10-12], стероидные гормоны[13], факторы роста [6, 14], протеазы [15, 16], активаторы опухолей [17, 18] и ингибиторы синтеза ДНК или РНК [4, 19-24], могут вызывать появление более дифференцированных характеристик у различных линий трансформированных клеток и первичных культур. В ранних исследованиях некоторых из авторов данного изобретения был идентифицирован ряд полярных соединений, которые эффективно индуцировали дифференцировку во многих линиях трансформированных клеток [8, 9]. Одним из таких эффективных индукторов было гибридное полярно/неполярное соединение N,N'-гексаметилен-бис-ацетамид (НМВА, ГМБА) [9], другим была сибероиланилидгидроксамовая кислота (SAHA, САГК) [39, 50]. Использование этих соединений для индукции эритроидной дифференцировки клеток эритролейкемии мыши (МЭЛ) с подавлением онкогенности обеспечило модель, пригодную для изучения опосредованной индуктором дифференцировки трансформированных клеток [5, 7-9]. Индуцированная ГМБА терминальная эритроидная дифференцировка МЭЛ-клеток является многостадийным процессом. После добавления ГМБА к культуре МЭЛ-клеток (745A-DS19) и до выявления фиксации терминальной дифференцировки имеется латентный период, равный 10-12 ч. Фиксацию определяют как способность клеток проявлять терминальную дифференцировку, несмотря на удаление индуктора [25]. При продолжающемся воздействии ГМБА происходит прогрессивное увеличение численности дифференцирующихся клеток. Авторы данного изобретения описали, что линии МЭЛ-клеток, которым была придана устойчивость к относительно низким концентрациям винкристина, становились заметно более чувствительными к индуцирующему действию ГМБА, и дифференцировку можно было вызвать после короткого латентного периода или вообще без латентного периода [26]. ГМБА способен вызывать фенотипические изменения, соответствующие дифференцировке, в очень разнообразных линиях клеток [5]. Характеристики вызванного лекарственным препаратом эффекта были наиболее широко исследованы на системе эритролейкемических клеток мыши [5, 25, 27, 28]. Индукция дифференцировки МЭЛ-клеток мыши зависит как от времени, так и от концентрации. Минимальная концентрация, необходимая для демонстрации эффекта in vitro в большинстве линий, равна 2-3 мМ; минимальная продолжительность непрерывного воздействия, обычно необходимая для того, чтобы вызвать дифференцировку значительной части (более 20%) популяции без продолжения воздействия лекарственного препарата равна примерно 36 ч. Существует доказательство того, что в путь вызванной индуктором дифференциации включена протеинкиназа С [29]. Исследования in vitro обеспечили основу для оценки потенциала ГМБА как вещества, индуцирующего дифференцировку клеток, при лечении раковых опухолей человека [30]. Было завершено несколько клинических испытаний фазы I с использованием ГМБА [31-36]. Клинические испытания показали, что это соединение может оказывать терапевтическое воздействие на больных раком [35,36]. Однако эти клинические испытания фазы I также продемонстрировали, что потенциальная эффективность ГМБА отчасти ограничена зависимой от дозы токсичностью, которая препятствует достижению оптимальных уровней в крови, и необходимостью внутривенного введения больших количеств вещества в течение длительных периодов времени. Поэтому некоторые из авторов настоящего изобретения перешли к синтезу соединений, являющихся более эффективными и, возможно, менее токсичными, чем ГМБА [37]. Недавно было показано, что определенный класс соединений, вызывающих дифференцировку клеток, ингибирует гистондеацетилазы. Было показано, что несколько экспериментальных противоопухоле-1 007649 вых соединений, таких как трихостатин А (ТСА), трапоксин, сибероиланилидгидроксамовая кислота(SAHA, САГК) и фенилбутират, действуют, по крайней мере частично, через ингибирование гистондеацетилаз [38, 39, 42]. Кроме того, было показано, что диаллилсульфиды и родственные им молекулы [43],оксамфлатин [44], MS-27-275, синтетическое производное бензамида [45], производные бутирата [46],FR901228 [47], депудецин [48] и бисгидроксамид m-карбоксикоричной кислоты [39] ингибируют гистондеацетилазы. In vitro эти соединения могут ингибировать развитие клеток-фибробластов, вызывая остановку клеточного цикла на фазах G1 и G2 [49-52], и могут приводить к терминальной дифференцировке и потере способности к трансформации различных линий трансформированных клеток [49-51]. In vivo фенилбутират эффективен при лечении острой промиелоцитной лейкемии, совместно с ретиноевой кислотой [53]. САГК эффективно предотвращает образование опухолей молочной железы у крыс и опухолей легких у мышей [54, 55]. В патенте США 5369108 [41], выданном некоторым из авторов настоящего изобретения, описаны соединения, пригодные для избирательной индукции терминальной дифференцировки неопластических клеток, причем эти соединения имеют две полярные концевые группы, разделенные гибкой последовательностью метиленовых групп, а одна или обе полярные концевые группы представляют собой крупную гидрофобную группу. Установлено, что такие соединения являются более активными, чем ГМБА и родственные ГМБА соединения. Однако в патенте США 5369108 не указано, что дополнительная крупная гидрофобная группа,находящаяся на том же конце молекулы, что и первая гидрофобная группа, может дополнительно увеличивать активность в отношении дифференцировки примерно в 100 раз при ферментативном анализе и примерно в 50 раз при анализе дифференцировки клеток. Этот новый класс соединений согласно настоящему изобретению может быть пригоден для избирательной индукции терминальной дифференцировки неопластических клеток и вследствие этого может способствовать лечению опухолей у больных. Сущность изобретения Настоящее изобретение предусматривает соединение, имеющее формулу где каждый из радикалов R1 и R3 является фенильной, циклогексильной, циклогексиламино-, нафтильной, пиридинамино-, пиперидиновой, 9-пурин-6-амино-, тиазоламино-, гидроксильной, разветвленной или неразветвленной алкильной, алкенильной, алкилокси-, бензилокси-, фенилалкилокси-, пиридинильной, хинолильной или тиеногруппой, в которых алкил представляет собой метил, этил или t-бутил; где радикал R3 является гидроксамовой кислотой, гидроксиламино-, гидроксильной, амино-, алкиламино- или алкилоксигруппой, в которых алкил представляет собой метил, этил или t-бутил; где радикал R4 является атомом водорода, галогеном, фенильной или циклогексильной группой; где A могут быть одинаковыми или различными и представляют собой амидную группу, -О-, -S-,-NR5- или -СН 2-; где радикал R5 является C1-C5 алкилом; где n является целым числом от 3 до 10; и где фенильная, нафтильная, хинолильная, алкильная или циклогексильная группы могут быть замещены диметиламиногруппой, или фармацевтически приемлемую соль этого соединения. Настоящее изобретение также предусматривает применение указанного соединения для избирательного индуцирования конечной дифференцировки неопластических клеток и обусловленного этим ингибирования пролиферации этих клеток, а также для лечения опухоли, характеризующейся пролиферацией неопластических клеток, для дифференцировки опухолевых клеток в опухоли, и для ингибирования активности гистондеацетилазы. Краткое описание графических материалов Фиг. 1 - влияние соединения 1 согласно настоящему изобретению на дифференцировку МЭЛклеток; фиг. 2 - влияние соединения 1 согласно настоящему изобретению на активность гистондеацетилазы 1; фиг. 3 - влияние соединения 2 согласно настоящему изобретению на дифференцировку МЭЛклеток; фиг. 4 - влияние соединения 3 согласно настоящему изобретению на дифференцировку МЭЛклеток; фиг. 5 - вияние соединения 3 согласно настоящему изобретению на активность гистондеацетилазы 1; фиг. 6 - влияние соединения 4 согласно настоящему изобретению на дифференцировку МЭЛклеток; фиг. 7 - влияние соединения 4 согласно настоящему изобретению на активность гистондеацетилазы 1;-2 007649 фиг. 8 - фотоаффинный маркер (3 Н-498) связывается непосредственно с HDAC 1 (гистондеацетилазой 1); фиг. 9 - САГК вызывает накопление ацетилированных гистонов H3 и H4 в ксенотрансплантате опухоли CWR22 у мышей; фиг. 10. САГК вызывает накопление ацетилированных гистонов H3 и H4 в периферических мононуклеарных клетках крови у больных. САГК вводили посредством внутривенной инфузии 3 раза в день. Пробы отбирали до (пре), сразу после инфузии (пост) и через 2 ч после инфузии; фиг. 11 а-11 е - демонстрируют влияние избранных соединений на очищенную посредством аффинной хроматографии гистондеацетилазу 1 (HDAC1) с меченым эпитопом (Flag) HDAC1. Подробное описание изобретения Настоящее изобретение предусматривает соединение, имеющее формулу каждый из радикалов R1 и R2 является фенильной, циклогексильной, циклогексиламино-, нафтильной,пиридинамино-, пиперидиновой, 9-пурин-6-амино-, тиазоламино-, гидроксилыной, разветвленной или неразветвленной алкильной, алкенильной, алкилокси-, бензилокси-, фенилалкилокси-, пиридинильной,хинолильной или тиеногруппой, в которых алкил представляет собой метил, этил или t-бутил; где радикал R3 является гидроксиламино-, гидроксильной, амино-, алкиламино- или алкилоксигруппой, в которых алкил представляет собой метил, этил или t-бутил; где радикал R4 является атомом водорода, галогеном, фенильной группой или циклогексильной группой; где А могут быть одинаковыми или различными и представлять собой амидную группу или группы-О-, -S-, -NR5- или -СН 2-; где радикал R5 является C1-C5 алкилом; в котором n является целым числом от 3 до 10; и где фенильная, нафтильная, хинолильная, алкильная или циклогексильная группы могут быть замещены диметиламиногруппой, или фармацевтически приемлемую соль этого соединения. В одном примере осуществления А может представлять собой амидную группу, a R3 может представлять собой гидроксиламиногруппу. В другом примере осуществления изобретения соединение имеет формулу Еще в одном примере осуществления изобретения соединение имеет формулу В следующем примере осуществления изобретения соединение имеет формулу где каждый из радикалов R1 и R2 является фенильной, циклогексильной, циклогексиламино-, нафтильной, пиридинамино-, пиперидиновой, алкильной, бензилокси-, фенилалкилокси-, пиридинильной, хинолильной или тиеногруппой, в которых алкил представляет собой метил, этил или t-бутил; и где фенильная, нафтильная, хинолильная, алкильная или циклогексильная группы могут быть замещены диметиламиногруппой; и-3 007649 где n является целым числом от 3 до 8. В следующем примере осуществления изобретения соединение имеет формулу или является его энантиомером. Еще в одном примере осуществления изобретения соединение имеет формулу или является его энантиомером. В следующем примере осуществления изобретения соединение имеет формулу или является его энантиомером. Еще в одном примере осуществления изобретения соединение имеет формулу или является его энантиомером. В следующем примере осуществления изобретения соединение имеет формулу или является его энантиомером. Еще в одном примере осуществления изобретения соединение имеет формулу или является его энантиомером. Еще в одном примере осуществления изобретения соединение имеет формулу или является его энантиомером. В следующем примере осуществления изобретения соединение имеет формулу или является его энантиомером. В следующем примере осуществления изобретения соединение имеет формулу или является его энантиомером. Еще в одном примере осуществления изобретения соединение имеет формулу или является его энантиомером. В следующем примере осуществления изобретения соединение имеет формулу или является его энантиомером. В следующем примере осуществления изобретения соединение имеет формулу Еще в одном примере осуществления изобретения соединение имеет формулу В следующем примере осуществления изобретения соединение имеет формулу В предпочтительном варианте осуществления указанных соединений n = 5. Настоящее изобретение также предполагает охват энантиомеров и солей соединений, перечисленных выше. Любое из описанных соединений может быть включено в фармацевтическую композицию вместе с фармацевтически приемлемым носителем. Любое из соединений может быть также преобразовано в фармацевтически приемлемую соль этого соединения с помощью хорошо известных фармакологических методик. Также могут быть получены предшественники всех соединений с помощью хорошо известных фармакологических методик. Любое из соединений может применяться для индуцирования дифференцировки опухолевых клеток в опухоли, например, путем приведения клеток в контакт с эффективным количеством вещества,вследствие чего происходит дифференцировка опухолевых клеток. Любое из соединений может также применяться для ингибирования активности гистондеацетилазы,например, путем контактирования гистондеацетилазы с эффективным количеством соединения, вследствие чего ингибируется активность гистондеацетилазы. Еще в одном примере осуществления изобретения настоящее изобретение предусматривает фармацевтическую композицию, содержащую фармацевтически эффективное количество любого из вышеуказанных соединений и фармацевтически приемлемый носитель. Еще в одном примере осуществления настоящее изобретение предусматривает применение соединений согласно изобретению для избирательной индукции остановки роста, терминальной дифференцировки или апоптоза неопластических клеток и, вследствие этого, ингибирования пролиферации этих клеток, которое можно осуществить, например, путем контактирования клеток при подходящих условиях с эффективным количеством любого из вышеуказанных соединений. Контактирование следует осуществлять непрерывно в течение длительного периода времени, например, в течение, как минимум, 48 ч, предпочтительно в течение примерно 4-5 дней и более. Такие способы применения соединений согласно изобретению можно осуществлять in vivo или invitro. Если способ применяют in vitro, контактирование можно осуществить посредством инкубации клеток с соединением. Концентрация соединения при контакте с клетками должна быть от примерно 1 нМ до примерно 25 мМ, предпочтительно от примерно 20 нМ до примерно 25 мМ, более предпочтительно от примерно 40 нМ до 100 мкМ и еще более предпочтительно от примерно 40 до примерно 200 нМ. Концентрация зависит от рода каждого отдельного соединения и состояния неопластических клеток. Способ может включать начальную обработку клеток противоопухолевым веществом так, чтобы они приобрели устойчивость к противоопухолевому веществу, и последующий контакт возникающих в результате обработки устойчивых клеток в подходящих условиях с эффективным количеством любого из вышеуказанных соединений, эффективного в отношении индукции терминальной дифференцировки этих клеток. Настоящее изобретение также предусматривает способ лечения больного, имеющего опухоль, характеризующуюся пролиферацией неопластических клеток, который включает введение больному эффективного количества любого из вышеуказанных соединений, эффективного в отношении индукции остановки роста, терминальной дифференцировки и/или апоптоза этих неопластических клеток, и обусловленного этим ингибирования их пролиферации. Применение соединений согласно настоящему изобретению предназначено для лечения больных людей, имеющих опухоли. Однако существует также вероятность того, что это применение может быть эффективным при лечении опухолей у других млекопитающих. Термин опухоль включает любой вид рака, вызванный пролиферацией неопластических клеток, например рак предстательной железы, рак легких, острую лейкемию, множественную миелому, карциному желчного пузыря, карциному почек, карциному молочной железы, колоректальную карциному, нейробластому или меланому. Пути введения соединения согласно настоящему изобретению включают все традиционные и физиологически приемлемые пути, такие как, например, пероральный, легочный, парентеральный (внутри-7 007649 мышечные, внутрибрюшинные, внутривенные (ВВ) или подкожные инъекции), ингаляции (в виде мелкодисперсного порошкообразного состава или в виде мелкодисперсного аэрозоля), трансдермальный,назальный, вагинальный, ректальный или сублингвальный пути введения, и рецептура может быть составлена в виде дозировочных форм, соответствующих каждому пути введения. Настоящее изобретение также предусматривает фармацевтическую композицию, включающую фармацевтически приемлемый носитель, например стерильную воду, не содержащую пирогенов, и терапевтически приемлемое количество любого из вышеуказанных соединений. Предпочтительно эффективное количество есть количество, способное избирательно индуцировать терминальную дифференцировку подходящих неопластических клеток, но меньшее, чем количество, вызывающее токсические эффекты у больного. Настоящее изобретение предусматривает фармацевтическую композицию, описанную выше, в сочетании с противоопухолевым агентом, гормоном, стероидом или ретиноидом. Противоопухолевым агентом может быть один из многочисленных химиотерапевтических агентов,такой как алкилирующий агент, антиметаболит, гормональный агент, антибиотик, колхицин, алкалоид барвинка, L-аспарагиназа, прокарбазин, гидроксимочевина, митотан, нитрозомочевина или имидазолкарбоксамид. Подходящими агентами являются такие агенты, которые способствуют деполяризации тубулина. Предпочтительно противоопухолевый агент является колхицином или алкалоидом барвинка; особо предпочтительны винбластин и винкристин. В тех примерах осуществления, в которых противоопухолевым агентом является винкристин, вводят такое его количество, чтобы клетки стали устойчивыми к винкристину в концентрации, примерно равной 5 мг/мл. Введение агента производят, по существу,так же, как описано выше для введения любых соединений. Предпочтительно введение агента производят в течение, как минимум, 3-5 дней. Введение любого из вышеперечисленных соединений производят так, как описано ранее. Фармацевтическую композицию можно вводить ежедневно в виде 2-6-часовых инфузий в течение периода, равного 3-21 дням, например, ежедневно в виде 4-часовой инфузий в течение 5 дней. Это изобретение можно будет лучше понять на основании подробного описания предпочтительных примеров осуществления, приведенного ниже. Однако специалист в данной области техники легко поймет, что обсуждаемые специфические способы и результаты являются исключительно иллюстративными для настоящего изобретения, которое будет более подробно описано в формуле изобретения, приведенной далее. Описание примеров осуществления изобретения Примеры 1-5 демонстрируют синтез замещенных Lаминопробковых гидроксамовых кислот согласно настоящему изобретению, а примеры 6 и 7 демонстрируют влияние соединений 1-5 на дифференцировку МЭЛ-клеток и на активность гистондеацетилазы. Пример 1. Синтез соединения 1.N-Bocметил-(L)аминосуберат, Boc-Asu(OMe) получали согласно опубликованной ранее процедуре [40]. (Boc - t-бутоксикарбонил; Asu - -аминосуберат (или -аминопробковая кислота.N-Cbzt-бутил-(L)аминосуберата дициклогексиламиновую соль закупали в компании ResearchN-Bocметил-(L)аминосуберат (493 мг, 1,63 ммоль) растворяли в атмосфере Ar (аргона) в 7 мл сухого CH2Cl2. Добавляли EDC (дихлорэтан) (470 мг, 2,45 ммоль), затем - анилин (230 мкл, 2,52 ммоль). Раствор перемешивали при комнатной температуре в течение 2 ч 30 мин, затем промывали разбавленнойHCl (рН 2,4, 25 мл), насыщенным раствором NaHCO3 (10 мл) и Н 2O (210 мл). Продукт очищали посредством колоночной хроматографии (силикагель, гексан:этилацетат 3,5:1). Выход выделенного продукта составил 366 мг (60%). Результаты 1 Н-ЯМР и масс-спектрометрии соответствовали продукту. 90 мг анилида N-Bocметил-(L)аминосуберата (0,238 ммоль) обрабатывали 3,2 мл 25% раствора трифторуксусной кислоты (TFA) в СН 2 Сl2 в течение 30 мин. Растворитель удаляли, а осадок оставляли под глубоким вакуумом на 12 ч. Его растворяли в атмосфере Ar (аргона) в 3 мл сухого CH2Cl2, содержащего бензотриазол-1-ил-окси-трис-пирролидинофосфония гексафторфосфат (РуВОР) (149 мг, 0,286 ммоль), бензойную кислоту (44 мг, 0,357 ммоль) и диизопропилэтиламин (114 мкл, 0,655 ммоль). Раствор перемешивали при комнатной температуре в течение 1 ч. Продукт очищали посредством колоночной хроматографии (силикагель, гексан:этилацетат 3:1-2:1) с получением белого твердого вещества: 75 мг, выход 82%. Результаты 1 Н-ЯМР и масс-спектрометрии соответствовали продукту. Вышеуказанную реакцию сочетания также успешно осуществляли с использованием в качестве реагента 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (EDC). 75 мг (0,196 ммоль) анилида N-бензоилметил-(L)аминосуберата перемешивали в течение 6 ч при 0 С в смеси 1 М NaOH:ТГФ:метилового спирта в соотношении 1:1:1. После полного исчезновения исходного материала раствор нейтрализовали (1 М HCl) и экстрагировали этилацетатом. Органическую фазу собирали и сушили. Удаление растворителя приводило к образованию продукта в виде белого твердого вещества: 67 мг, выход 93%. Результаты 1 Н-ЯМР и масс-спектрометрии соответствовали продукту. К суспензии 26 мг анилида N-бензоилметил-(L)аминосуберата (12) в 1 мл сухого CH2Cl2 добавляли 58 мг H2NOTBDPS (Н 2NО-t-бутилдифенилисилила), а затем - 22 мг EDC. Реакционную смесь перемешивали при комнатной температуре в течение 4 ч. Образовавшуюся в качестве промежуточного продукта защищенную гидроксамовую кислоту очищали посредством колоночной хроматографии (силикагель, СН 2 Сl2:метиловый спирт в соотношении 100:0-98-2). Защиту удаляли посредством обработки 5% трифторуксусной кислотой в CH2Cl2 в течение 1 ч 30 мин. Продукт осаждали из смеси ацетонапентана. 1 Н-ЯМР (d6-ДМСО, 500 МГц) =10,29 (s, 1H), 8,53 (d, 1H), 7,90 (d, 2H), 7,60 (d, 2 Н), 7,53 (m, 1H),7,46 (t, 2H), 7,28 (t, 2H), 7,03 (t, 2H), 4,53 (q, 1H), 1,92 (t, 2H), 1,78 (m, 2 Н), 1,50-1,25 (m, 6 Н). Она была получена из N-Bocметил-Lаминосуберата посредством такой же процедуры, как процедура, использованная для бензоильного аналога. Выходы и поведение при хроматографии были сопоставимыми. 1 Н-ЯМР (d6-ДMCO, 500 МГц) =10,30 (s, 1H), 10,10 (s, 1H), 9,05 (m, 1H), 8,80 (m, 1H), 8,71 (m, 1H),8,24 (m, 1H), 7,60 (m, 2H), 7,30 (m, 2H), 7,04 (m, 1H), 4,56 (m, 1H), 1,93 (t, 2H), 1,79 (m, 2H), 1,55-1,30 (m,6H). Дициклогексиламиновую соль N-Cbz-(L)-Asu(OtBu)-OH (100 мг, 0,178 ммоль) распределяли между 1 М HCl (5 мл) и этилацетатом (10 мл). Органическую фазу удаляли, а водную фазу промывали этилацетатом (33 мл). Органические фракции объединяли, промывали рассолом (12 мл) и сушили (MgSO4). Смесь фильтровали и концентрировали до образования бесцветной пленки (67 мг, 0,176 ммоль, выход 98%). Это соединение сразу же использовали в следующей стадии.N-Cbz-(L)-Asu(OtBu)-OH (67 мг, 0,176 ммоль) растворяли в сухом CH2Cl2 (2,5 мл). Добавляли анилин (17 мкл, 0,187 ммоль), РуВОР (97 мг, 0,187 ммоль) и iPr2Net (46 мкл, 0,266 ммоль) и перемешивали смесь в течение 2 ч. Реакция была завершена по результатам тонкослойной хроматографии (TLC). Смесь разбавляли этилацетатом (5 мл) и водой (5 мл) и разделяли фазы. Водную фазу промывали этилацетатом(33 мл) и объединяли органические фракции. Этот раствор промывали 1 М HCl (12 мл) и рассолом (12 мл), сушили (MgSO4), фильтровали и концентрировали до неочищенного масла. Масло пропускали через слой силикагеля (30% этилацетат/гексан) для удаления основных загрязнений с получением искомого вещества (76 мг, 0,167 ммоль, выход 94%). 1 Н-ЯМР (CDCl3, 400 МГц, без TMS)8,20 (br s, 1H), 7,47 (d, 2H), 7,32 (m, 5H), 7,28 (t, 2 Н), 7,08 (t,1 Н), 5,39 (d, 1 Н), 5,10 (m, 2 Н), 4,26 (m, 1 Н), 2,18 (t, 2 Н), 1,93 (m, 1 Н), 1,67 (m, 1 Н), 1,55 (m, 3 Н), 1,42 (s,9H), 1,36 (m, 3 Н).N-Cbz-(L)-Asu(OtBu)-анилид (76 мг, 0,167 ммоль) растворяли в сухом CH2Cl2 (5 мл) и по каплям добавляли трифторуксусную кислоту (0,5 мл). Через 3 ч по данным тонкослойной хроматографии реакция была завершена. Смесь концентрировали под вакуумом с получением указанного в заголовке соединения (80 мг неочищенного продукта). Это соединение без очистки использовали на следующей стадии. 1 Н ЯМР (ДMCO-d6 400 МГц)11,93 (br s, 1H), 9,99 (br s, 1H), 7,57 (m, 3 Н), 7,34 (m, 5 Н), 7,29 (t, 2 Н),7,03 (t, 1 Н), 5,02 (m, 2 Н), 4,11 (m, 1 Н), 2,17 (t, 2 Н), 1,61 (m, 2 Н), 1,46 (m, 2 Н), 1,27 (m, 4 Н).N-Cbz-(L)-Asu(OH)-анилид (80 мг неочищенного продукта) и O-t-бутилдифенилсилилгидроксиламин (60 мг, 0,221 ммоль) растворяли в CH2Cl2 (4 мл). К смеси добавляли РуВОР (125 мг, 0,241 ммоль) и iPr2NEt (52 мкл, 0,302 ммоль) и перемешивали в течение ночи. Тонкослойная хроматография показала завершение реакции. Смесь концентрировали под вакуумом, а затем пропускали через слой силикагеля (50% этилацетата/гексана) с целью удаления основных загрязнений. Испарение летучих веществ позволило получить 107 мг вещества, которое затем растворили в сухом CH2Cl2 (5 мл) и добавили трифторуксусную кислоту (0,25 мл). Мониторинг посредством тонкослойной хроматографии показал завершение реакции через 1,5 ч. Реакционную смесь концентрировали под вакуумом с целью удаления всех летучих веществ. Осадок переносили в этилацетат (3 мл) и затем медленно добавляли гексан, результатом чего было осаждение белого геля. Супернатант удаляли, а преципитат промывали гексаном(32 мл). Этот материал высушивали при пониженном давлении с получением указанного в заголовке вещества (40 мг, 0,097 ммоль, выход 59%). 1 Н ЯМР (ДМСО-d6, 400 МГц)10,32 (s, 1H), 10,00 (s, 1H), 8,64 (br s, 1H), 7,57 (m, 3 Н), 7, 37 (m, 5 Н),7,30 (t, 2 Н), 7,04 (t, 1 Н), 5,02 (m, 2 Н), 4,12 (m, 1 Н), 1,93 (t, 2H), 1,62 (m, 2 Н) 1,45 (m, 2 Н), 1,29 (m, 4 Н). Получена аналогично соединению 3. Н-ЯМР (ДМСО-d6, 400 МГц)10,45 (s, 1H), 10,31 (s, 1H), 8,85 (dd, 1H), 8,63 (dd, 1 Н), 8,42 (dd, 1 Н),8,13 (dd, 1 Н), 8,68 (m, 2 Н), 7,60 (t, 1 Н), 7,37 (m, 2 Н), 7,28 (m, 2 Н), 5,10 (m, 2 Н), 4,24 (m, 1 Н), 1,93 (t, 2 Н),1,85 (m, 1 Н), 1,70 (m, 1 Н), 1,50 (m, 2 Н), 1,42 (m, 2 Н), 1,30 (m, 2 Н); Образец N-Cbzt-бутил-Lаминосубероил-8-хинолинамида (90 мг, 0,178 ммоль) был получен из предыдущего синтеза. Cbz-группу удаляли посредством гидрирования в метиловом спирте на 5% Pd на С. Образовавшийся свободный амин сочетали с бензойной кислотой с использованием EDC в сухомCH2Cl2 (69% в двух стадиях). После удаления защиты с t-бутилового эфира с помощью трифторуксусной кислоты обычная реакция сочетания с H2NOTBDPS с последующим удалением защиты позволяла получить желаемую гидроксамовую кислоту. 1 Н-ЯМР (d6-ДМСО, 500 МГц)10,55 (s, 1H), 10,30 (s, 1H), 9,03 (m, 1H), 8,78 (m, 1 Н), 8,62 (m, 1 Н),8,40 (m, 1 Н), 7,97 (m, 2 Н), 7,67-7,46 (m, 6 Н), 4,66 (m, 1 Н), 1,94 (t, 2 Н), 1,87 (m, 1 Н), 1,80-1,20 (m, 7 Н).ESI-Macc-спектрометрия: 435 (М+1). Пример 6. Синтез соединения с инвертированной амидной группой. Соединение со следующей формулой: синтезировали посредством обработки малонового эфира из которого удаляли R в реакции с амином и карбодиимидным реагентом с образованием из которого удаляли R' и преобразовывали в гидроксамовую кислоту (NHOH) так же, как в предыдущих примерах.- 12007649 В приведенной выше схеме R может быть t-бутилом, удаляемым трифторуксусной кислотой; R' может быть метилом, удаляемым основанием или Lil; и радикалы R" могут быть одинаковыми или различными, в зависимости от использованного реагента. Пример 7. Влияние соединения 1 (N-бензоил-(L)аминосубероиланилидгидроксамовой кислоты, PhCONH-Asu(NHOH)-NHPh) на дифференцировку МЭЛ-клеток и активность гистондеацетилазы. Дифференцировка клеток эритролейкемии мышей (МЭЛ-клеток) Исследование дифференцировки МЭЛ-клеток было использовано для оценки способности соединения 1 вызывать терминальную дифференцировку. МЭЛ-клетки (логарифмически делящиеся) культивировали с указанными концентрациями соединения 1. После 5-дневного периода культивирования определяли прирост числа клеток с помощью счетчика Coulter, а дифференцировку оценивали микроскопически, используя бензидиновую пробу для определения накопления белка гемоглобина в пересчете на одну клетку. Было обнаружено, как показано на фиг. 1, что соединение 1 (200 нМ) способно вызывать дифференцировку МЭЛ-клеток. Ферментативная активность гистондеацетилазы (HDAC) Влияние соединения 1 на очищенную посредством аффинной хроматографии HDAC1 человека с меченым эпитопом (Flag) оценивали посредством инкубации препарата фермента в отсутствие субстрата на льду в течение 20 мин с указанными количествами соединения 1. Добавляли субстрат (меченый[3 Н]ацетилом гистон, полученный из клеток эритролейкемии мыши), и пробы инкубировали в течение 20 мин при 37 С при общем объеме, равном 30 мкл. Затем реакции останавливали, экстрагировали выделенный ацетат и определяли уровень выделенного радиоактивного изотопа с помощью сцинтилляционного счетчика. Было обнаружено, как показано на фиг. 2, что соединение 1 является сильным ингибитором ферментативной активности HDAC1 (ID50 = 1 нМ). Пример 8. Влияние соединения 2 (N-никотиноил-(L)аминосубероиланилидгидроксамовой кислоты, C5H4NCO-Asu(NHOH)-NHPh) на дифференцировку МЭЛ-клеток. Дифференцировка клеток эритролейкемии мышей (МЭЛ-клеток) Исследование дифференцировки МЭЛ-клеток было использовано для оценки способности соединения 2 вызывать терминальную дифференцировку. МЭЛ-клетки (логарифмически делящиеся) культивировали с указанными концентрациями соединения 2. После 5-дневного периода культивирования микроскопически оценивали дифференцировку, используя бензидиновую пробу для определения накопления белка гемоглобина в пересчете на одну клетку. Было обнаружено, как показано на фиг. 3, что соединение 2 (800 нМ) способно вызывать дифференцировку МЭЛ-клеток. Пример 9. Влияние соединения 3 (N-бензилоксикарбонил-(L)аминосубератанилид-гидроксамовой кислоты, N-Cbz-(L)-Asu(NH-OH)-NHPh) на дифференцировку МЭЛ-клеток и активность гистондеацетилазы. Дифференцировка клеток эритролейкемии мышей (МЭЛ-клеток) Исследование дифференцировки МЭЛ-клеток было использовано для оценки способности соединения 3 вызывать терминальную дифференцировку. МЭЛ-клетки (логарифмически делящиеся) культивировали с указанными концентрациями соединения 3. После 5-дневного периода культивирования микроскопически оценивали дифференцировку, используя бензидиновую пробу для определения накопления белка гемоглобина в пересчете на одну клетку. Было обнаружено, как показано на фиг. 4, что соединение 3 (400 нМ) способно вызывать дифференцировку МЭЛ-клеток. Ферментативная активность гистондеацетилазы (HDAC) Влияние соединения 3 на очищенную посредством аффинной хроматографии HDAC1 человека с меченым эпитопом (Flag) оценивали посредством инкубации препарата фермента в отсутствие субстрата на льду в течение 20 мин с указанными количествами соединения 3. Добавляли субстрат (меченый[3 Н]ацетилом гистон, полученный из клеток эритролейкемии мыши), и пробы инкубировали в течение 20 мин при 37 С при общем объеме, равном 30 мкл. Затем реакции останавливали, экстрагировали выделенный ацетат и определяли уровень выделенного радиоактивного изотопа с помощью сцинтилляционного счетчика. Было обнаружено, как показано на фиг. 5, что соединение 3 является сильным ингибитором ферментативной активности HDAC1 (ID50100 нМ). Пример 10. Влияние соединения 4(N-бензилоксикарбонил-(L)аминоксисубероил-8 хинолинамидгидроксамовой кислоты) на дифференцировку МЭЛ-клеток и активность гистондеацетилазы. Дифференцировка клеток эритролейкемии мышей (МЭЛ-клеток) Исследование дифференцировки МЭЛ-клеток было использовано для оценки способности соединения 4 вызывать терминальную дифференцировку. МЭЛ-клетки (логарифмически делящиеся) культиви- 13007649 ровали с указанными концентрациями соединения 4. После 5-дневного периода культивирования микроскопически оценивали дифференцировку, используя бензидиновую пробу для определения накопления белка гемоглобина в пересчете на одну клетку. Было обнаружено, как показано на фиг. 6, что соединение 4 (40 нМ) способно вызывать дифференцировку МЭЛ-клеток. Ферментативная активность гистондеацетилазы (HDAC) Влияние соединения 4 на очищенную посредством аффинной хроматографии HDAC1 человека с меченым эпитопом (Flag) оценивали посредством инкубации препарата фермента в отсутствие субстрата на льду в течение 20 мин с указанными количествами соединения 4. Добавляли субстрат (меченый[3 Н]ацетилом гистон, полученный из клеток эритролейкемии мыши) и пробы инкубировали в течение 20 мин при 37 С в общем объеме, равном 30 мкл. Затем реакции останавливали, экстрагировали выделенный ацетат и определяли уровень выделенного радиоактивного изотопа с помощью сцинтилляционного счетчика. Было обнаружено, как показано на фиг. 7, что соединение 4 является сильным ингибитором ферментативной активности HDAC1 (ID5010 нМ).SAHA ингибирует активность очищенных посредством аффинной хроматографии HDAC1 (гистондеацетилазы 1) и HDAC3 (гистондеацетилазы 3) [39]. Кристаллографические исследования с SAHA и родственным гистондеацетилазе белком показали, что SAHA ингибирует гистондеацетилазу посредством прямого взаимодействия с каталитическим центром [66]. Дополнительные исследования продемонстрировали, что меченый тритием фотоаффинный аналог SAHA (3 Н-498), содержащий азидную группу[67], непосредственно связывается с гистондеацетилазой 1 (фиг. 8). Эти результаты показывают, что данный класс соединений на основе гидроксамовой кислоты ингибирует активность гистондеацетилазы посредством прямого взаимодействия с белком гистондеацетилазы.SAHA вызывает накопление ацетилированных гистонов Н 3 и Н 4 in vivo. Влияние SAHA было изучено in vivo с использованием ксенотрансплантата опухоли предстательной железы человека CWR22 у мышей [68]. SAHA (50 мг/кг/день) вызывала снижение среднего конечного объема опухоли на 97% по сравнению с контролями при отсутствии выраженной токсичности. Введение SAHA в этой дозе вызывает повышение уровня ацетилированных гистонов Н 3 и Н 4 в ксенотрансплантате опухоли (фиг. 9).SAHA в настоящее время испытывают в клинических испытаниях I фазы на больных с плотными опухолями. SAHA вызывает аккумуляцию ацетилированных гистонов Н 3 и Н 4 в мононуклеарных клетках периферической крови, выделенных от больных, прошедших лечение (фиг. 10). В табл. 1 приведены суммарные результаты примеров 7-10, в которых были исследованы соединения 1-4, а также произведено сравнение результатов с результатами, полученными при использованииSAHA. Таблица 1 Краткое содержание результатов исследования соединений 1-4 и сравнение с результатами, полученными при использовании SAHA Пример 12. Модифицированные ингибиторы HDAC. В дополнительных исследованиях было обнаружено, что соединения 6 и 7, изображенные ниже, являются очень эффективными ингибиторами фермента HDAC. Соединение 6 имело ID50, равную 2,5 нМ, а соединение 7 имело ID50, равную 50 нМ. Это резко отличается от ID50 SAHA, равной 1 мкМ, которая значительно выше. Отметим, что ID50, равная у SAHA 1 мкМ, в целом, того же порядка, что и ее оптимальная для цитодифференцировки МЭЛ-клеток доза, равная 2,5 мкМ, но это близкое сходство не характерно для всех исследованных соединений. В некоторых случаях очень эффективные ингибиторы HDAC были менее эффективны в качестве соединений, вызывающих дифференцировку клеток, возможно из-за того,что вещества метаболизируются во время анализов, проводимых на клетках. Кроме того, не все типы- 14007649 клеток были одинаковыми, и некоторые соединения были значительно более эффективными в отношении опухолевых клеток человека, таких как НТ-29, чем в отношении МЭЛ-клеток. Поэтому ингибирование HDAC является предварительным индикатором. Пример 13. Разработка соединений, не содержащих гидроксамовой кислоты. Для вышеуказанных соединений, представляющих собой гидроксамовые кислоты, было обнаружено, что они подвергаются ферментативному гидролизу значительно быстрее, чем карбоновые кислоты,так что их биологические периоды полураспада коротки. Авторы настоящего изобретения были заинтересованы в получении соединений, которые могли бы быть более стабильными in vivo. Поэтому были разработаны ингибиторы гистондеацетилазы, которые не являются гидроксамовыми кислотами, и которые могут быть использованы в качестве соединений, вызывающих дифференцировку клеток, с более длительными периодами полураспада. Более того, было обнаружено, что вновь полученные соединения обладают большей избирательностью по отношению к гистондеацетилазе, чем, например, SAHA. Были получены соединения, которые имеют двойные связи, наподобие трихостатина A (TSA), чтобы посмотреть, обладают ли результирующие соединения большей эффективностью. Кроме того, цепь вTSA содержит всего 5 атомов углерода, а не 6, как у SAHA. В оксамфлатине содержится цепь из четырех атомов углерода, содержащая двойную связь и этинильный линкер между гидроксамовой кислотой и первым фенильным кольцом, а оксамфлатин был заявлен как эффективный ингибитор HDAC. Авторы включили некоторые из этих характеристик в свои соединения, в том числе в те соединения, которые не являются гидроксамовыми кислотами. Также открыты простые комбинаторные способы скрининга многочисленных соединений такого рода на эффективность и избирательность в отношении ингибирования HDAC. Кроме того, поскольку существует много важных ферментов, содержащих Zn(II), гидроксамовые кислоты, а возможно и некоторые другие группы, координирующие металлы, можно также присоединить к Zn(II) и другим металлам. Так как мишенью для HDAC является боковая цепь гистона, представляющая собой ацетиллизин,были созданы соединения, в которых присутствуют переходные состояния, аналогичные субстрату. На- 15007649 пример, авторы изобретения синтезировали соединения типа SAHA, в которых группа гидроксамовой кислоты -CO-NHOH заменена трифторацетильной группой -CO-CF3. Результирующее соединение (8) легко образует гидрат и поэтому присоединяется к Zn(II) гистондеацетилазы, при этом (9) имитирует переходное состояние (10) для деацетилирования. Это сходно с работой, опубликованной Lipscomb [56],по связыванию с карбоксипептидазой А аналога субстрата (11), содержащего CF3-CO-CH2 группу вместо нормального амида. Гидрат кетона координируется с Zn(II), имитируя переходное состояние для каталитического гидролиза амидного субстрата. Наш синтез отдельного примера 12 из ряда фторкетонов показан на схеме, приведенной ниже: После алкилирования малонового эфира получают альдегид, а затем преобразуют его в трифторметилкарбинол в реакции с реактивом Руппертса [57, 58]. Получают малоновые бис-анилиды, а карбинол окисляют до кетона (12) в реакции с реактивом Десса-Мартина [59]. Другие подходы были испытаны безуспешно. В частности, попытки преобразовать производное карбоновой кислоты непосредственно в трифторметилкетон оказались безуспешными. Соединение 12 было испытано на гистондеацетилазе, и было обнаружено, что оно является ингибитором фермента. Поэтому авторы настоящего изобретения также адаптировали этот синтез для получения аналогов соединения 12 с ненасыщенной цепью и т.п., а также с другими группами на левом конце молекулы. Пример 14. Разработка соединений, в которых группа гидроксамовой кислоты заменена на NHP(O)OH-CH3. Аналог SAHA, в котором CH2-CO-NHOH группа заменена на NH-P(O)OH-CH3, может быть синтезирован по общей схеме, приведенной ниже. Образующееся соединение 13 связывается с Zn(II) гистондеацетилазы таким же образом, как родственная группа связывается с Zn(II) карбоксипептидазы у аналогов, например у аналога, полученного Bartlett [60]. Классическим ингибитором содержащего Zn(II) фермента карбоангидразы является сульфамид,анион которого соединяется с Zn(II) [61]. Поэтому соединение 14, аналог SAHA с сульфамидной группой, синтезируют так, как показано ниже. На последней стадии была проведена реакция бисхлорида карбоновой и сульфокислоты с анилином и аммиаком. Так как хлорид карбоновой кислоты реагирует быстрее, то следует последовательно использовать анилин, а затем аммиак, но последовательность может быть обратной, или можно разделить смесь в том случае, если обе группы имеют сходную реакционную способность. В ходе синтеза соединения 14 был использован тиол (15), который легко можно получить из соответствующей галоидоводородной кислоты. Тиолы также являются ингибиторами ферментов, содержащих Zn(II), таких как карбоксипептидаза А и родственные пептидазы, например фермент, конвертирующий ангиотензин (АСЕ) так, что соединение 15 преобразуется в соединение 16, являющееся ингибитором гистондеацетилазы. Сходный синтез можно использовать для присоединения NH-P(O)OH-CH3 группы к другим соединениям, в частности, к соединениям 6 и 7. Пример 15. Варьирование линкера между Zn(II)-связывающей группой и гидрофобными связывающими группами. На основании результатов, полученных с оксамфлатином, представляется, что фенильное кольцо может быть частью цепи между Zn(II)-связывающей группой и левым участком изображенной молекулы,в частности в тех случаях, когда фенильное кольцо замещено в мета-положении. Поэтому авторы настоящего изобретения предложили синтез для включения таких метазамещенных цепей в другие полученные ими соединения. Были созданы соединения 17 и 18. Для того чтобы вместо гидроксамовой кислоты, присоединенной к фенильному кольцу, получить ариламиды 17 и 18, необходимы простые синтезы, не изображенные в подробностях. Могут быть синтезированы дополнительные соединения, такие как 19 и 20, включающие трифторметилкетоновую группу 12, которая, по имеющимся у авторов сведениям, эффективно связывает Zn(II) гистондеаетилазы. Синтезы включают получение соединений 21 и 22 с последующим добавлением CF3 с образованием карбинола, а затем окисление, как в синтезе 12. Простой синтез включает сочетание по Гекку (Heck) соединений 23 и 24 с этилакрилатом и конверсию эфира в альдегиды 21 и 22 посредством восстановления карбинола и последующего повторного окисления. Все цепи, показанные до сих пор, содержат только атомы углерода, но приемлемыми и даже полезными могут быть тиоэфирные звенья, и они еще более упрощают синтез. Например, сульфамиды, такие как 25 и 26, родственные 19 и 20, могут быть получены соответственно из тиофенола и бромметилсульфамида. Сходный синтез может быть использован для получения соответствующих фосфонамидатов 27- 18007649 и 28, если этот класс соединений окажется пригодным в качестве ингибиторов гистондеацетилазы и веществ, вызывающих дифференцировку клеток. В этом случае N-защищенную m-аминобензойную кислоту используют для ацилирования ариламинов, а затем фосфорилируют анилиновую группу. Пример 16. Варьирование левой части молекулы, несущей гидрофобные группы. Для того чтобы варьировать гидрофобные группы, мы синтезировали соединение 29 в качестве промежуточного продукта, который можно обрабатывать различными аминами с получением соединений 30. После этого удаление защиты с группы гидроксамовой кислоты даст общий класс соединений 31. Синтез показан на схеме, приведенной ниже. В ходе синтеза О-защищенный гидроксиламин ацилируют бромгексановой кислотой, и это соеди- 19007649 нение затем алкилирует бис-пентафторэфир малоновой кислоты. Образующееся соединение 29 затем реагирует с различными аминами, а защитную группу удаляют с помощью кислоты. С использованием этого соединения в качестве исходного материала авторы синтезировали библиотеки родственных соединений, содержащих другие связывающие Zn(II) группы. Например, алкилирование малоната соединением 32 позволяет получить библиотеку фосфонамидатов, а соединение 33 позволяет получить библиотеку CF3-CO. Сходным образом можно получить библиотеку сульфамидов,если в работе, описанной ранее, будет показано, что эта группа перспективна в отношении связыванияZn(II) HDAC. Естественно, что после алкилирования малоната и аминолиза соединение, полученное из 32, будет деметилировано, тогда как соединение, полученное из 33, будет окислено. Это также позволяет расширить структуру соединения 6, производного аминопробковой кислоты. Как было описано выше, оно является одним из наиболее эффективных ингибиторов гистондеацетилазы,исследованных авторами. Авторы получили это соединение, использовав ферментативный гидролиз для достижения оптического разрешения и избирательности между двумя карбометоксигруппами 34, так что авторы могли конвертировать одну из них в амид аминохинолина соединения 6, защищая азот как карбобензоксигруппу. В конце синтеза авторы конвертировали оставшуюся карбометоксигруппу в гидроксамат. Тем не менее, соединение 6 является промежуточным соединением, которое можно использовать для получения других производных. Карбобензоксигруппу из 6 можно удалить, а амин 35 можно ацетилировать различными карбоновыми кислотами с получением библиотеки 36 или хлоридами сульфокислоты с получением соответствующих сульфамидов.- 20007649 Также была синтезирована другая библиотека амидов 37, родственных 6, а затем она была расширена за счет библиотеки других амидов 38 посредством ацилирования аминогруппы после удаления защиты. Авторы также синтезировали группу соединений 39, в которых после удаления карбобензоксигруппы 37 они получили библиотеку сульфамидов, использовав различные сульфонилхлориды. Во всех этих соединениях группа гидроксамовой кислоты может быть защищена. Приведенные выше схемы синтеза можно использовать для создания соединений с большим числом вариаций. Некоторыми группами-заместителями, которые, вероятно, приведут к образованию соединений, имеющих потенциально хорошее сродство к гистондеацетилазе или активных в отношении дифференцировки клеток, являются следующие. Некоторые амины, которые могут быть включены вместо анилина в SAHA или в качестве Х-группы в соединения 37 и 38 Некоторые карбоновые и сульфокислоты, которые могут быть включены в качестве группы Y-CO в соединения 38 или 39 Пример 17. Синтезы с использованием приведенных выше схем. Реактивы и исходные вещества были получены от коммерческих поставщиков и использованы без дополнительной очистки, если не указано иное. Для реакций, чувствительных к влаге, растворители заново перегоняли перед употреблением: тетрагидрофуран перегоняли в атмосфере аргона с металлическим натрием с использованием бензофенона в качестве индикатора; дихлорметан и ацетонитрил перегоняли с порошковым гидридом кальция. Безводный бензол, безводный DIEA и безводный пиридин отбирали с помощью шприца из герметически закрытого флакона, закупленного в компании Aldrich. Третбутанол перед употреблением сушили на молекулярных ситах с диаметром ячеек, равным 4 . Гидрид натрия закупали в виде 60% дисперсии в минеральном масле. Анилин, диизопропиламин, Nметиланилин и бензиловый спирт заново перегоняли перед употреблением. Дейтерированные растворители получали из Изотопных лабораторий Кембриджа (Cambridge Isotope Laboratories). Реакции, чувствительные к воздуху и/или к влаге, проводили в атмосфере сухого аргона в высушенной в термошкафу или с помощью пламени стеклянной посуде, снабженной плотно прилегающей резиновой мембраной. Шприцы и иглы перед употреблением сушили в термошкафу. Реакции при 0 С проводили в ванне, содержавшей лед/воду. Реакции при -78 С проводили в ванне с сухим льдом/ацетоном. Хроматография. Аналитическую тонкослойную хроматографию (TLC) проводили на стеклянных пластинках, предварительно покрытых слоем силикагеля 60 F-254 толщиной 0,25 мм, произведенного компанией ЕМ Science, Германия. Элюированные соединения визуализировали одним или несколькими способами из следующих: коротковолновое ультрафиолетовое излучение, пары I2, окрашивание KMnO4 или окрашиваниеFeCl3. Препаративную тонкослойную хроматографию проводили на листах ватмана с заранее нанесенным покрытием из силикагеля толщиной 500 или 1000 мкм. Испарительную колоночную хроматографию проводили на Kieselgel 60 производства Merck, 230-400 меш. Аппаратура. ЯМР-спектры измеряли на спектрометрах Bruker DPX300 и DRX400; 1 Н наблюдали при 300 и 400 МГц, a 19F - при 376 МГц. Химические смещения приведены как значенияв миллионных долях относительно остаточного пика растворителя. Массовые спектры получали на приборе Nermag R-10-1 для спектров при химической ионизации (CI) или спектров при ударной ионизации электронами и на Jeol JMSLCmate для спектров при ионизации при распылении в электрическом поле (ESI+). CI спектры получали при использовании в качестве ионизируемого газа аммиак (NH3) или метан (СН 4).(35 мл) при 0 С по каплям добавляли ди-t-бутилмалонат (1,20 мл, 5,37 ммоль). Наблюдали выделение газа, раствору давали нагреться до комнатной температуры и перемешивали его в течение 6 ч. Раствор метил-6-бром-2,4-гександиеноата [62] (1,00 г, 4,88 ммоль) в ТГФ (20 мл) готовили в отдельной емкости и перемешивали в водяной бане. К нему из шприца по каплям добавляли смесь, содержащую малонат, и реакцию оставляли для протекания на ночь. Реакцию гасили насыщенным раствором NH4Cl (5 мл), затем- 22007649 добавляли воду (10 мл), и смесь экстрагировали Et2O (315 мл). Органические фракции объединяли и промывали Н 2O (110 мл), затем рассолом, сушили над MgSO4 и фильтровали. После испарения при пониженном давлении с последующей испарительной хроматографией (0-20% этилацетат/гексан) получали соединение 40 в виде прозрачного бесцветного масла (850 мг, 2,49 ммоль, выход 51%). Тонкослойная хроматография Rf 0,66 (20% этилацетат/гексан); 1 Н-ЯМР (CDCl3, 400 МГц)7,26 (dd,1H), 6,26 (dd, 1H), 6,10 (m, 1H), 5,82 (d, 1H), 3,78 (s, 3H), 3,12(t, 1 Н), 2,64 (t, 2H), 1,41s, 18H). К перемешиваемому раствору соединения 40 (200 мг, 0,59 ммоль) в CH2Cl2 (10 мл) добавляли трифторуксусную кислоту (1 мл). Реакцию оставляли для протекания на ночь. Летучие вещества удаляли при пониженном давлении и получали в остатке соединение 41 в виде белого твердого вещества (112 мг, 0,49 ммоль, выход 83%). 1 Н-ЯМР (CD3OD, 400 МГц)7,11 (dd, 1H), 6,33 (dd, IH), 6,16 (m, 1H), 5,81 (d, 1 Н), 3,76 (s, 3H), 3,15 К перемешиваемому раствору оксалилхлорида (2,0 М в CH2Cl2, 11,5 мл, 23,1 ммоль) в CH2Cl2 (100 мл) и ДМФ (1 капля) при 0 С добавляли 4-пентеновую кислоту (2,25 мл, 22,0 ммоль). Смеси предоставляли возможность нагреться до температуры окружающей среды. После прекращения выделения газа температуру смеси снова доводили до 0 С и по каплям добавляли раствор анилина (2,00 мл, 22,0 ммоль) и тетраэтиламмония (6,72 мл, 26,3 ммоль) в CH2Cl2 (5 мл). После нагревания до температуры окружающей среды реакцию оставляли для протекания на 3 ч. Смесь концентрировали при пониженном давлении, а затем распределяли ее между HCl (1N, 10 мл) и этилацетатом (30 мл) и разделяли фазы. Водную фазу экстрагировали этилацетатом (315 мл), органические фазы объединяли, промывали рассолом, сушили над MgSO4 и фильтровали. Концентрирование при пониженном давлении дало желтоватое твердое вещество, которое перекристаллизовывали из толуола с получением соединения 42 в виде белых кристаллов (1,97 г, 11,24 ммоль, выход 51%). Тонкослойная хроматография Rf 0,68 (50% этилацетат/гексан); 1 Н-ЯМР (300 МГц, CDCl3)7,49 (d,2H), 7,29 (t, 2H), 7,08 (t, 1H), 5,88 (m, 1H), 5,10 (dd, 2 Н), 4,42 (br s, 4 Н). К перемешиваемому раствору диизопропиламина (2,06 мл, 14,7 ммоль) в ТГФ (25 мл) при -78 С добавляли n-бутиллитий (2,0 М раствор в гексане, 6,2 мл, 12,4 ммоль) и продолжали перемешивать в течение 20 мин при той же температуре. Затем по каплям добавляли раствор фосфоната 43 а (63) (2,66 г, 11,3 ммоль) в ТГФ (4 мл), что давало насыщенную желтую окраску во время добавления. После пребывания при -78 С в течение 20 мин смесь нагревали до 0 С и по каплям добавляли раствор альдегида 43b (64)(1,78 г, 11,3 ммоль) в ТГФ (4 мл). После добавления раствору позволяли нагреться до температуры окружающей среды и перемешивали его в течение ночи. Раствор разбавляли Et2O (30 мл) и промывали Н 2O(310 мл). Водные растворы, полученные после промывки, объединяли и экстрагировали Et2O (210 мл),органические фазы объединяли, промывали рассолом, сушили над MgSO4 и фильтровали. Испарение при пониженном давлении с последующей испарительной хроматографией (10-20% этилацетат/гексан) дало соединение 43 в виде прозрачного масла (1,54 г, 57%). Тонкослойная хроматография Rf 0,56 (20% этилацетат/гексан); 1 Н-ЯМР (400 МГц, CDCl3)7,22 (dd,1 Н), 6,19 (dd, 1 Н), 6,08 (m, 1 Н), 5,77 (d, 1 Н), 2,42 (m, 2 Н), 2,32 (t,2H), 1,42 (s,9H).(Е,Е)-7-Фенилкарбамоил-гепта-2,4-диеновой кислоты метиловый эфир (44) К перемешиваемому раствору диэфира 43 (1,00 г, 4,61 ммоль) в CH2Cl2 (40 мл) добавляли трифторуксусную кислоту (4,0 мл) и проводили реакцию в течение 6 ч. Смесь концентрировали при пониженном давлении для удаления летучих веществ. Оставалось белое твердое вещество, состоящее из неочищенной кислоты (710 мг, 3,85 ммоль). Эту кислоту (400 мг, 2,17 ммоль) растворяли в CH2Cl2 (20 мл), и к этому раствору при перемешивании добавляли DMAP (13 мг), анилин (218 мкл, 2,39 ммоль) и EDC (500 мг, 2,61 ммоль). Через 1,5 ч смесь разбавляли этилацетатом и промывали Н 2O. Фазы разделяли и водную фазу экстрагировали этилацетатом (315 мл). Органические фазы объединяли и промывали HCl (1N, 15 мл) и рассолом, сушили над MgSO4 и фильтровали. Концентрирование при пониженном давлении давало коричневое твердое вещество. Его растворяли в минимальном количестве CH2Cl2, затем пропускали через слой силикагеля (20-30% этилацетат/гексан, 200 мл) для удаления основных загрязнений. Элюент концентрировали до светло-коричневого масла, которое переносили в небольшое количество CH2Cl2 и из которого осаждали кристаллы добавлением гексана/диэтилового эфира. Исходную жидкость удаляли,кристаллы промывали эфиром, а жидкую фракцию концентрировали, и эту процедуру повторяли несколько раз до получения, в конечном итоге, соединения 44 в виде грязно-белых кристаллов (324 мг, 1,25 ммоль, 58%). Тонкослойная хроматография Rf 0,44 (50% этилацетат/гексан); 1 Н-ЯМР (400 МГц, CDCl3)7,47 (d,1H), 7,30 (t, 2H), 7,24 (m, 1H), 7,09 (t, 1H), 6,24 (dd, 1H), 6,14 (m, 1 Н), 5,81 (d, 1H), 3,72 (s, 3H), 2,60 (m,2H), 2,47 (t, 2H).(Е,Е)-7-(Метилфенилкарбамоил)гепта-2,4-диеновой кислоты метиловый эфир (45) Промежуточное соединение в виде неочищенной кислоты из первой стадии получения 44 (200 мг,1,09 ммоль) и N-метиланилин (130 мкл, 1,19 ммоль) растворяли в CH2Cl2 (10 мл) и перемешивали. Затем добавляли EDC (271 мг, 1,41 ммоль) и DMAP (5 мг), и проводили реакцию в течение ночи. Смесь распределяли между Н 2O и этилацетатом и разделяли фазы. Водную фазу экстрагировали этилацетатом(310 мл), органические фазы объединяли и промывали HCl (1N, 15 мл), затем рассолом, сушили надMgSO4 и фильтровали. После испарения при пониженном давлении осталось чистое соединение 45 в виде коричневого масла (286 мг, 1,05 ммоль, 96%). Тонкослойная хроматография Rf 0,81 (5% метиловый спирт/СН 2 Сl2); 1 Н-ЯМР (300 МГц, CDCl3)7,40 (t, 2H), 7,35 (t, 1H), 7,20 (d, 2H), 7,15 (dd, 1H), 6,20 (m, 2H), 5,76 (d, 1H), 3,70 (s, 3H), 3,24 (s, 3H), 2,42 Эфир соединения 45 (260 мг, 0,95 ммоль) растворяли в метиловом спирте (7,5 мл). Затем добавляли раствор LiOHH2O (200 мг, 4,76 ммоль) в Н 2O (2,5 мл) и перемешивали смесь в течение 6 ч. Реакционную смесь подкисляли HCl (1N) до рН 2, а затем экстрагировали этилацетатом (310 мл). Органические фракции объединяли и промывали Н 2O и рассолом, сушили над MgSO4 и фильтровали. После испарения при пониженном давлении осталось чистое соединение 46 в виде коричневого твердого вещества (200 мг,0,77 ммоль, 81%). Тонкослойная хроматография: Rf 0,13 (40% этилацетат/гексан); 1 Н-ЯМР (300 МГц, CD3OD)7,47 Кислоту 46 (200 мг, 0,77 ммоль) и TBDPSO-NH2 (220 мг, 0,81 ммоль) растворяли в CH2Cl2 (8 мл). К этому раствору во время перемешивания добавляли EDC (178 мг, 0,93 ммоль) и DMAP (5 мг) и реакцию оставляли для протекания на ночь. Смесь концентрировали и затем пропускали через слой силикагеля(этилацетат). После испарения при пониженном давлении осталось светло-коричневое масло (383 мг,0,75 ммоль, выход 97%). Защищенный гидроксамат (270 мг, 0,53 ммоль) растворяли в CH2Cl2 (10 мл) и добавляли трифторуксусную кислоту (0,5 мл). Раствор перемешивали в течение 2 ч и на тонкослойной хроматограмме наблюдали новое пятно, которое окрашивали FeCl3. Раствор концентрировали при пониженном давлении и добавляли диэтиловый эфир, с получением осадка, который прилипал к колбе. Жидкую фазу сливали, осадок растирали с этилацетатом, жидкость удаляли, а испарение всех летучих веществ из осадка дало соединение 47 в виде коричневой смолы (23 мг, 0,084 ммоль, 16%). Тонкослойная хроматография Rf 0,22 (5% метиловый спирт/СН 2 Сl2); 1 Н-ЯМР (400 МГц, CD3OD)7,50 (t, 2H), 7,40 (t, 1H), 2,27 (d, 2H), 7,08 (m, 1H), 6,11 (m, 1H), 5,97 (m, 1H), 5,80 (m, 1H), 3,23 (s, 3 Н),3,39 (m, 2H), 2,21 (t, 2H). Фениламид гидроксамида октандиоевой кислоты (48) Указанное в заголовке соединение 48 получали в виде коричневой смолы (9 мг) через ряд стадий,аналогичных получению соединения 47. Тонкослойная хроматография: Rf 0,20 (5% метиловый спирт/СН 2 Сl2); 1 Н-ЯМР (400 МГц, CD3OD)7,51 (t, 2H), 7,41 (t, 1H), 7,30 (d, 2H), 3,29 (s, 3 Н), 2,11 (m, 4H), 1,58 (m, 4H), 1,22 (m, 4H). Октандиоевой кислоты бензиламид (49) К перемешиваемому раствору субероилхлорида (1,00 мл, 5,55 ммоль) в ТГФ (40 мл) при 0 С по каплям добавляли раствор бензиламина (0,61 мл, 5,55 ммоль) и DIEA (1,45 мл, 8,33 ммоль) в ТГФ (10 мл). Смеси позволяли нагреться до температуры окружающей среды и перемешивали ее в течение 1 ч. Затем добавляли HCl (10 мл, 1N) и перемешивали смесь в течение 0,5 ч. Содержимое разводили этилацетатом(30 мл) и разделяли фазы. Водную фазу экстрагировали этилацетатом (310 мл), органические фазы объединяли, промывали рассолом (5 мл) и сушили над MgSO4. После фильтрования и концентрирования при пониженном давлении оставалось соединение 49 в виде грязно-белого твердого вещества. 1 Н-ЯМР (300 МГц, ДМСО-d6)11,98 (br s, 1H), 9,80 (t, 1H), 7,32 (m, 2H), 7,23 (m, 3 Н), 4,25 (d, 2 Н),2,19 (t, 2 Н), 2,12 (t, 2 Н), 1,50 (m, 4 Н), 1,25 (m, 4 Н). Бензиламида октандиоевой кислоты гидроксиамид (50) Это соединение получали из соединения 49 через его защищенный гидроксамат, как описано для приведенных ранее соединений. Было получено соединение 50 в виде белого твердого вещества. 1 Н-ЯМР (400 МГц, ДMCO-d6)10,30 (s, 1H), 8,27 (t, 1H), 7,28 (m, 2H), 7,23 (m, 3H), 5,65 (d, 2H),2,11 (t, 2H), 1,91 (t, 2H), 1,46 (m, 4 Н), 1,23 (m, 4 Н). Соль N-Cbz-L-2-аминопробковой кислоты 8-t-бутилового эфира дициклогексиламина (100 мг, 0,18 ммоль) растворяли в HCl (5 мл, 1N) и экстрагировали этилацетатом (310 мл). Экстракты объединяли,промывали рассолом и сушили над MgSO4. После испарения оставалась свободная кислота в виде белого твердого вещества (68 мг, 0,179 ммоль). Ее растворяли в CH2Cl2 (2,5 мл), после чего добавляли анилин(17 мкл, 0,19 ммоль), DIEA (46 мкл, 0,27 ммоль) и, наконец, РуВОР (97 мг, 0,19 ммоль). Раствор перемешивали в течение 1 ч, затем концентрировали, а остаток распределяли между Н 2O (5 мл) и этилацетатом (10 мл). Фазы разделяли, и водную фазу экстрагировали этилацетатом (310 мл). Экстракты объединяли, промывали HCl (1N), затем рассолом, сушили над MgSO4 и фильтровали. Концентрирование при пониженном давлении давало твердый остаток, который пропускали через слой силикагеля (30% этилацетат/гексан). Собранный элюент испаряли с получением соединения 51 в виде белого твердого вещества (76 мг, 0,167 ммоль, 94%). Тонкослойная хроматография Rf 0,38 (30% этилацетат/гексан); 1 Н-ЯМР (400 МГц, CDCl3)8,21 (s,1H), 7,48 (d, 2H), 7,32 (m, 5 Н), 7,28 (t, 2H), 7,08 (t, 1H), 5,39 (br d, 1 Н), 5,10 (m, 2 Н), 4,26 (br dd, 1 Н), 2,07 К раствору эфира 51 (76 мг, 0,167 ммоль) в CH2Cl2 (5 мл) добавляли трифторуксусную кислоту (0,5 мл) и реакционный раствор перемешивали в течение 5 ч. Раствор концентрировали при пониженном давлении с получением неочищенного соединения 52 в виде белого твердого вещества (80 мг), которое использовали на следующей стадии без очистки. Тонкослойная хроматография Rf 0,32 (5% метиловый спирт/СН 2 Сl2); 1 Н-ЯМР (400 МГц, ДMCO-d6)11,93 (br s, 1H), 9,99 (s, 1H), 7,58 (d, 2H), 7,55 (d, 1H), 7,35 (m, 4 Н), 7,29 (t, 2H), 7,03 (t, 1H), 5,02 (m, 2H),4,11 (br dd, 1H), 2,17 (t, 2H), 1,59 (m, 2H), 1,48 (m, 2H), 1,22 (m, 4H).(1S)-(6-Гидроксикарбамоил-1-фенилкарбамоилгексил)карбаминовой кислоты бензиловый эфир (53) К раствору неочищенной кислоты 52 (80 мг) и TBDPSO-NH2 (60 мг, 0,221 ммоль) в CH2Cl2 добавляли DIEA (52 мкл, 0,302 ммоль), а затем РуВОР (125 мг, 0,241 ммоль). Раствор перемешивали в течение 3 ч, затем концентрировали при пониженном давлении. Остаток пропускали через слой силикагеля (50% этилацетат/гексан) и испаряли собранный элюент. Получали белую пену (107 мг, 0,164 ммоль, выход 82%), растворяли ее в CH2Cl2 (5 мл), добавляли трифторуксусную кислоту (0,25 мл) и раствор перемешивали в течение 2 ч. В ходе тонкослойной хроматографии обнаруживалось новое пятно, которое окрашивали FeCl3. Смесь концентрировали при пониженном давлении, а остаток растворяли в минимальном количестве этилацетата и осаждали продукт гексаном. Образовавшийся белый гель промывали гексаном- 26007649 и сушили под вакуумом с получением соединения 53 в виде белого твердого вещества (40 мг, 0,097 ммоль, выход из трех стадий 58%). 1 Н-ЯМР (400 МГц, ДМСО-d6)10,31 (s, 1H), 9,99 (s, 1H), 7,59 (d, 2H), 7,56 (d, 1 Н), 7,37 (m, 4 Н), 7,29 Названное соединение получали из дициклогексиламинной соли N-Cbz-L-2-аминопробковой кислоты 8-t-бутилового эфира аналогично получению соединения 51. Испарительная хроматография (0-1% метиловый спирт/СН 2 Сl2) давала соединение 54 в виде светло-коричневого твердого вещества (70 мг,0,138 ммоль, 82%). Тонкослойная хроматография Rf 0,42 (2% метиловый спирт/СН 2 Сl2); 1 Н-ЯМР (400 МГц, CDCl3)10,19 (s, 1H), 8,77 (dd, 1H), 8,71 (dd, 1H), 8,15 (dd, 1H), 7,52 (m, 2H), 7,45 (m, 1 Н), 7,33 (m, 4 Н), 5,50 (br d,1 Н), 5,15 (m, 2 Н), 4,51 (br dd, 1H), 2,17 (t, 2H), 2,00 (m, 1 Н), 1,79 (m, 1 Н), 1,56 (m, 2 Н), 1,45 (m, 2 Н), 1,40 Получена из 54 аналогично 52. Получено соединение 55 в виде коричневого твердого вещества (72 мг, 0,129 ммоль). Тонкослойная хроматография Rf 0,16 (50% этилацетат/гексан); 1 Н-ЯМР(400 МГц, ДMCO-d6)11,92(1S)-[6-Гидроксикарбамоил-1-(хинолин-8-илкарбамоил)гексил]карбаминовой кислоты бензиловый эфир (56) Получен из соединения 55 аналогично получению 53. Получено соединение 56 в виде белого твердого вещества (15 мг, 0,032 ммоль, выход 44%). 1 Н-ЯМР (400 МГц, ДMCO-d6)10,46 (s, 1H), 10,31 (s, 1H), 8,85 (dd, 1H), 8,63 (dd, 1 Н), 8,42 (dd, 1 Н),8,12 (d, 1 Н), 8,66 (m, 2 Н), 7,58 (t, 1 Н), 7,37 (m, 2 Н), 7,28 (m, 2 Н), 7,20-6,90 (1 Н), 5,10 (m, 2 Н), 4,10 (m, 1 Н),1,92 (t,2H), 1,82 (m, 1 Н), 1,68 (m, 1 Н),1,49 (m, 2 Н), 1,40 (m, 2 Н), 1,26 (m, 2 Н). Масс-спектроскопия (ESI+): рассчитано для C25H28N4O5 464, получено 465 [М+Н]+.(7S)-(Циклогексанкарбониламино)-7-фенилкарбамоилгептановой кислоты метиловый эфир (57) К раствору соединения 5 (81 мг, 0,214 ммоль) в CH2Cl2 (10 мл) добавляли трифторуксусную кислоту (0,5 мл) и перемешивали раствор в течение 2 ч. Смесь концентрировали при пониженном давлении. К раствору этого амина (62 мг, 0,223 ммоль) и циклогексанкарбоновой кислоты (31 мкл, 0,245 ммоль) вCH2Cl2 (4 мл) добавляли РуВОР (140 мг, 0,268 ммоль) и DIEA (58 мкл, 0,335 ммоль). Раствор перемешивали в течение 2 ч, концентрировали при пониженном давлении и очищали продукт посредством испарительной хроматографии (40% этилацетат/гексан). После испарения оставалось неочищенное вещество 57 в виде белого твердого вещества (95 мг), содержавшее небольшое количество загрязнения в виде не вступившей в реакцию циклогексановой кислоты. Этот материал использовали на следующей стадии без дополнительной очистки. Тонкослойная хроматография Rf 0,58 (50% этилацетат/гексан); 1 Н-ЯМР (400 МГц, CDCl3)8,58 (s,1H), 7,50 (d, 2H), 7,28 (t, 2H), 7,07 (t, 1H), 6,14 (d, 1H), 4,56 (dt, 1 Н), 3,64 (s, 3 Н), 2,28 (t, 2 Н), 2,13 (tt, 1 Н),1,94 (m, 1 Н), 1,85 (m, 2 Н), 1,76 (m, 2 Н), 1,64 (m, 4 Н), 1,4 (m, 5 Н), 1,22 (m, 4 Н). К раствору эфира 57 (95 мг) в метиловом спирте (2,5 мл) при 0 С добавляли раствор NaOH (1M, 2,5 мл). После добавления образовывался белый осадок, который повторно растворяли путем добавления ТГФ (2,5 мл). Через 3 ч добавляли дополнительное количество NaOH (1 М, 1,0 мл) и поддерживали температуру на уровне 0 С. После полного исчезновения исходного материала по результатам анализа путем тонкослойной хроматографии, реакционную смесь подкисляли HCl (1N) с получением белого осадка. Супернатант удаляли, а из твердого вещества отсасывали жидкость через фильтр. Объединенные жидкости экстрагировали этилацетатом (35 мл), экстракты объединяли, промывали рассолом, сушили над MgSO4 и фильтровали. После концентрирования при пониженном давлении осталось белое твердое вещество, которое объединяли с осадком на фильтре и сушили под вакуумом с получением карбоновой кислоты 58 (75 мг, 0,200 ммоль, 90%). 1 Н-ЯМР (400 МГц, ДМСО-d6)11,95 (s, 1H), 9,98 (s, 1H), 7,90 (d, 1H), 7,58 (d, 1 Н), 7,28 (t, 2H), 7,02CH2Cl2 (4 мл) и добавляли EDC (47 мг, 0,243 ммоль). Раствор перемешивали в течение ночи. После концентрирования при пониженном давлении материал очищали посредством испарительной хроматографии (50% этилацетат/гексан). Испарение объединенных фракций продукта дало белую пену (80 мг, 0,131 ммоль, выход 70%). К раствору этого защищенного гидроксамата в CH2Cl2 (4 мл) и ТГФ (3 мл) добавляли трифторуксусную кислоту (0,25 мл) и перемешивали в течение 1,5 ч. При тонкослойной хроматогра- 28007649 фии наблюдали новое пятно, которое немедленно окрашивали FeCl3. Раствор концентрировали и удаляли все летучие вещества под вакуумом. Остаток растирали с этилацетатом и получали осадок в виде белого геля, который переносили в пластмассовую пробирку с помощью этилацетата (5 мл). Пробирку центрифугировали с получением гранулы осадка, супернатант сливали и добавляли этилацетат (10 мл). Осадок повторно суспендировали путем обработки ультразвуком, затем повторно центрифугировали и осадок сушили под вакуумом. Было получено белое твердое вещество 59 (18 мг, 0,046 ммоль, 35%). 1 Н-ЯМР (400 ЯМР, ДMCO-d6)10,31 (s, 1H), 9,97 (s, 1H), 7,89 (d, 1H), 7,57 (d, 2 Н), 7,28 (t, 2H), 7,02(t, 1H), 4,33 (dt, 1H), 2,22 (t, 2H), 1,91 (t, 2H), 1,61 (m, 6H), 1,68 (m, 2 Н), 1,45 (m, 2 Н), 1,21 (9 Н). Гидроксиамида октандиоевой кислоты хинолин-8-иламид (60) Это соединение было получено из монометилового эфира пробковой кислоты аналогично 48 с использованием 8-аминохинолина. Неочищенный осадок, полученный после удаления защиты с защищенного гидроксамата с помощью трифторуксусной кислоты, переносили в небольшой объем этилацетата и осаждали гексаном с получением соединения 60 в виде белого твердого вещества (18 мг, 0,057 ммоль,21% от карбоновой кислоты). 1 Н-ЯМР (400 МГц, ДMCO-d6)10,31 (s, 1H), 10,02 (s, 1H), 8,92 (dd, 1H), 8,61 (dd, 1H), 8,40 (dd, 1H),7,65 (dd, 1H), 7,63 (dd, 1H), 7,56 (t, 1H), 2,56 (t, 1H), 1,93 (t, 1H), 1,63 (m, 2 Н), 1,49 (m, 2 Н), 1,28 (m, 4 Н). Масс-спектроскопия (ESI+): рассчитано для C17H21N3O3 315, получено 316 [М+Н]+. 2-t-Бутоксикарбонилоктандиоевой кислоты l-t-бутилового эфира 8-этиловый эфир (61) К перемешиваемой суспензии NaH (60% дисперсия, 197 мг, 4,913 ммоль) в ТГФ (25 мл) при 0 С добавляли ди-t-бутилмалонат (1,00 мл, 4,466 ммоль) и смеси давали нагреться до температуры окружающей среды. Через 1 ч выделение газа прекращалось, после чего по каплям добавляли этил-6 бромгексаноат (0,88 мл, 4,913 ммоль). Реакцию проводили при нагревании с обратным холодильником в течение ночи. Реакцию осторожно гасили с помощью Н 2O (10 мл) и разводили этилацетатом. После разделения фаз водную фазу экстрагировали этилацетатом (310 мл). Экстракты объединяли и промывали Н 2O, затем рассолом, сушили над MgSO4 и фильтровали. Концентрирование при пониженном давлении дало желтое масло, которое пропускали через слой силикагеля (10% этилацетат/гексан). После испарения оставался светло-желтый сироп соединения 61 (1,52 г, 4,24 ммоль, выход 95%). Тонкослойная хроматография Rf 0,44 (10% этилацетат/гексан); 1 Н-ЯМР (400 МГц, CDCl3)4,10 (q,2H), 3,08 (t, 1H), 2,26 (t, 2H), 1,76 (m, 2H), 1,60 (m, 2H), 1,43 (s, 18 Н), 1,32 (m, 4H), 1,23 (m, 3 Н). 2-Карбоксиоктандиоевой кислоты 8-этиловый эфир (62) К раствору тройного эфира 61 (500 мг, 1,395 ммоль) в CH2Cl2 (20 мл) добавляли трифторуксусную кислоту (2,0 мл) и реакционную смесь перемешивали в течение ночи. Летучие компоненты испаряли под вакуумом, а остаток повторно растворяли в CH2Cl2 и испаряли с целью удаления всех следовых количеств трифторуксусной кислоты. Получили твердое вещество 62 (327 мг, 1,33 ммоль) и использовали прямо в следующей стадии без дополнительной очистки. 1 Н-ЯМР (400 МГц, ДМСО-d6)12,62 (br s, 2H), 4,03 (q, 2H), 3,16 (t, 1H), 2,25 (t, 2 Н), 1,67 (m, 2 Н),1,49 (m, 2 Н), 1,25 (m, 4 Н), 1,16 (t, 3 Н).- 29007649 7,7-Бис-(хинолин-8-илкарбамоил)гептановой кислоты этиловый эфир (63) Двухосновную кислоту 62 (150 мг, 0,609 ммоль), 8-аминохинолин (211 мг, 1,462 ммоль) и DMAP (5 мг) растворяли в ТГФ (6 мл). К этому раствору добавляли EDC (350 мг, 1,827 ммоль) и реакцию оставляли для протекания на ночь. Смесь концентрировали при пониженном давлении и очищали продукт посредством испарительной хроматографии (40% этилацетат/гексан). После испарения объединенных фракций продукта осталось вещество 63 в виде светло-коричневого твердого вещества (100 мг, 0,201 ммоль, 14%). 1 Н-ЯМР (400 МГц, ДMCO-d6)10,85 (s, 2H), 8,92 (dd, 2H), 8,64 (dd, 2H), 8,40 (dd, 2H), 7,68 (dd, 2H),7,62 (dd, 2H), 7,57 (t, 2H), 4,35 (t, 1H), 3,98 (q, 2H), 2,24 (t, 2H), 2,00 (m, 2 Н), 1,51 (m, 2 Н), 1,37 (m, 4 Н),1,12 (m, 3 Н). 7,7-Бис-(хинолин-8-илкарбамоил)гептановая кислота (64) К раствору эфира 63 (94 мг, 0,212 ммоль) в метиловом спирте (3 мл) и ТГФ (1 мл) добавляли раствор LiOHH2O (44 мг, 1,062 ммоль) в Н 2O (1 мл) и перемешивали смесь в течение 5 ч. После подкисления HCl (1N) до рН, равного 7, добавляли этилацетат (10 мл) и разделяли фазы. Водную фазу экстрагировали этилацетатом (35 мл), экстракты объединяли, промывали насыщенным раствором NH4Cl (3 мл),Н 2O (3 мл), затем рассолом, сушили над MgSO4 и фильтровали. После концентрирования при пониженном давлении осталось соединение 64 в виде белого твердого вещества (94 мг, 0,200 ммоль, выход 94%). Тонкослойная хроматография Rf 0,21 (50% этилацетат/гексан); 1 Н-ЯМР (400 МГц, ДМСО-d6)11,88 (s, 1H), 10,85 (s, 2H), 8,93 (dd, 2H), 8,65 (dd, 2H), 8,40 (dd, 2H), 7,69 (dd, 2H), 7,63 (dd, 2H), 7,58 (t,2H), 4,35 (t, 1H), 2,16 (t, 2H), 2,00 (m, 2H), 1,49 (m, 2 Н), 1,38 (m, 4 Н). 2-(Хинолин-8-илкарбамоил)октандиоевой кислоты 8-гидроксамид 1-хинолин-8-иламид (65)CH2Cl2 (4 мл) и добавляли EDC (57 мг, 0,295 ммоль). Раствор перемешивали в течение ночи, затем концентрировали при пониженном давлении. Очистка посредством испарительной хроматографии (30-50% этилацетат/гексан) и испарение объединенных фракций продукта дало белую пену. К раствору этого защищенного гидроксамата в СН 2 Сl2 (4 мл) добавляли трифторуксусную кислоту (0,2 мл) и перемешивали раствор в течение 4 ч. Тонкослойная хроматография показала полное исчезновение исходного материала и новое пятно, которое окрашивали FeCl3. Раствор концентрировали при пониженном давлении, а остаток разводили минимальным количеством этилацетата. Добавление гексана давало белый осадок, из которого удаляли исходную жидкость. После промывки гексаном осадок сушили под вакуумом с получением соединения 65 в виде белого твердого вещества (30 мг, 0,061 ммоль, 22% от карбоновой кислоты). 1 Н-ЯМР (400 МГц, CDCl3)10,85 (s, 2H), 10,30 (s, 1H), 8,93 (dd, 2H), 8,65 (dd, 2 Н), 8,40 (dd, 2H),7,69 (dd, 2H), 7,63 (dd, 2H), 7,58 (t, 2H), 4,35 (t, 1H), 1,99 (m, 2H), 1,92 (t, 2 Н), 1,48 (m, 2 Н), 1,35 (m, 4 Н). Масс-спектроскопия (ESI+): рассчитано для C27H27N5O4 485, получено 486 [М+Н]+.
МПК / Метки
МПК: C07C 259/00, C07D 215/38, C07D 215/12, A61K 31/27, C07C 229/00, A61K 31/445, C07C 211/00, A61K 31/47, A61K 31/165
Метки: вызывающие, гистондеацетилазы, применение, ингибиторы, дифференцировку, клеток
Код ссылки
<a href="https://eas.patents.su/30-7649-ingibitory-gistondeacetilazy-vyzyvayushhie-differencirovku-kletok-i-ih-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы гистондеацетилазы, вызывающие дифференцировку клеток, и их применение</a>
Новые ингибиторы гистондеацетилазы

Номер патента: 7272
Опубликовано: 25.08.2006
Авторы: Пилатт Изабелль Ноэлль Констанс, Ру Брюно, Анжибо Патрик Рене, Мерпул Ливен, Тен Холтэ Петер, Ван Брандт Свен Францискус Анна, Вердонк Марк Густаф Селин, Дяткин Алексей Борисович
МПК: C07D 207/14, A61P 35/00, A61K 31/4545...
Метки: ингибиторы, новые, гистондеацетилазы
Формула / Реферат:
1. Соединение формулы (I) где n равен 0, 1, 2 или 3 и, когда n равен 0, тогда имеется в виду непосредственная связь; t равен 0, 1, 2, 3 или 4 и, когда t равен 0, тогда имеется в виду непосредственная связь; каждый Q представляет собой азот или каждый X представляет собой азот или каждый Y представляет собой азот или каждый Z представляет собой азот или R1 представляет собой -C(O)NH(OH); R2 представляет собой водород, галоген, гидрокси,...
Способы инактивации клеток-мишеней, агенты и композиции, вызывающие цитолиз, и соединения, используемые для получения этих агентов

Номер патента: 2688
Опубликовано: 29.08.2002
Авторы: Согорд Мортен, Ландо Петер, Абрахмсен Ларс, Калланд Терье, Дохлстен Микаэль, Форсберг Геран
МПК: A61K 39/385, A61K 47/48, A61K 39/085...
Метки: используемые, получения, клеток-мишеней, вызывающие, композиции, способы, соединения, инактивации, агентов, этих, цитолиз, агенты
Формула / Реферат:
1. Способ инактивации клеток-мишеней в присутствии Т-клеток, включающий контактирование клеток мишеней и Т-клеток с суперантигеном в присутствии иммуномодулятора, когда, по меньшей мере, один из элементов - суперантиген или иммуномодулятор - конъюгирован с нацеливающим компонентом, причем нацеливающий компонент выбран из группы, состоящей из антитела, антигенсвязывающего фрагмента антитела, Fab-фрагмента антитела, Fаb2-фрагмента антитела или...
Пиперазинил-, пиперидинил- и морфолинилпроизводные как новые ингибиторы гистондеацетилазы

Номер патента: 7270
Опубликовано: 25.08.2006
Автор: Ван Эмелен Кристоф
МПК: A61K 31/506, A61P 35/00, C07D 401/04...
Метки: морфолинилпроизводные, гистондеацетилазы, пиперазинил, пиперидинил, новые, ингибиторы
Формула / Реферат:
1. Соединение формулы (I) и его фармацевтически приемлемые аддитивные соли где t равен 0 или 1 и, когда t равен 0, тогда имеется в виду непосредственная связь; каждый Q представляет собой азот или ; каждый Х представляет собой азот или ; каждый Y представляет собой азот или ; каждый Z представляет собой -NH-, -О- или -СН2-; R1 представляет собой -C(O)NH(OH); R2 представляет собой водород, гидрокси, C1-6-алкил или арил-С1-6-алкил; -L-...
Ингибиторы адгезии клеток

Номер патента: 3737
Опубликовано: 28.08.2003
Авторы: Олмквист Рональд Г., Ли Вен-Чернг, Лин Ко-Чунг, Хаммонд Чарльз И., Зиммерман Крейг Н., Куэрво Хулио Хернан, Адамс Стивен П., Картер Мэри Бет, Кастро Альфредо К., Энсингер Кэрол Ли
МПК: C07K 14/78, A61P 37/00, A61K 38/04...
Метки: адгезии, клеток, ингибиторы
Формула / Реферат:
1. Соединение, ингибирующее адгезию клеток, формулы (I) Z-(Y1)-(Y2)-(Y3)n-X (I) и его фармацевтически приемлемые производные; отличающееся тем, что Z выбирается из группы, состоящей из алкилсульфонила; аралкилсульфонила; арилсульфонила; циклоалкилсульфонила, необязательно сконденсированного с арилом; гетероциклилсульфонила; гетероциклилалкилсульфонила; алкенилсульфонила, необязательно замещенного арилом; алкинилсульфонила, необязательно...
Ингибиторы адгезии клеток.

Номер патента: 1594
Опубликовано: 25.06.2001
Авторы: Картер Мэри Бет, Куэрво Хулио Хернан, Хаммонд Чарльз И., Кастро Альфредо К., Олмквист Рональд А., Зиммерман Крейг Н., Лин Ко-Чунг, Адамс Стивен П., Энсингер Кэрол Ли, Ли Вен-Чернг
МПК: C07K 14/78, A61K 38/04
Метки: ингибиторы, клеток, адгезии
Формула / Реферат:
1. Соединение, ингибирующее адгезию клеток, формулы (I') Z-N(R1)-С(R2)(A1)-C(O)-N(R1)-C(R2)(А2)-С(О)-{-N(R1)-С(R2)(А3)-С(О)}n-Х которое может быть также описано формулой (I) Z-(Y1)-(Y2)-(Y3)n-X причем Y1=-N(R1)-C(R2)(А1)-С(О)-, Y2=-N(R1)-C(R2)(А2)-С(О)-, и Y3=-N(R1)-C(R2)(А3)-С(О)-, и его фармацевтически приемлемые производные, где Z выбирают из группы, включающей алкил; алифатический ацил, необязательно замещенный группой N-алкил- или...
Предыдущий патент: Жевательная резинка с покрытием, способ ее изготовления и применение для ее покрытия одного или более активных веществ в форме порошка
Следующий патент: Фармацевтическая композиция, содержащая 2,2-дихлор-12 -(4-хлорфенил)додекановую кислоту
Случайный патент: Способ получения спирта