Производные аминоиндазола, активные в качестве ингибиторов киназ, способ их получения и содержащая их фармацевтическая композиция
Номер патента: 7430
Опубликовано: 27.10.2006
Авторы: Салом Барбара, Д'анелло Маттео, Мартина Катия, Вульпетти Анна, Амичи Раффаэлла
























Формула / Реферат
1. Способ лечения заболеваний, обусловленных и/или связанных с измененной (нарушенной) активностью протеинкиназ, включающий введение нуждающемуся в этом млекопитающему эффективного количества производного аминоиндазола, представленного формулой (I)

где R выбирают из группы, состоящей из -NHCOR', -NHCONHR', -NHCONR'R", -NHSO2R' или -NHCOOR', где R' и R" каждый независимо выбран из прямого или разветвленного C1-С6алкила, С2-С6 алкенила или -алкинила, С3-С6циклоалкила или циклоалкилС1-С6алкила, арила, гетероарила, арилС1-С6 алкила, 5- или 6-членного гетероцикла или гетероциклилС1-С6алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода или серы, причем R, R' и R" необязательно замещены в любом из свободных положений одной или более группами, например от 1 до 6 групп, выбранных из галогена, нитро-, оксогрупп (=O), карбокси, циано, алкила, перфторалкила, алкенила, алкинила, циклоалкила, арила, гетероциклила, аминогрупп и их производных, таких как, например, алкиламино, диалкиламино, ариламино, диариламино, уреидо, алкилуреидо или арилуреидо; карбониламиногрупп и их производных, таких как формиламино, алкилкарбониламино, алкенилкарбониламино, арилкарбониламино, алкоксикарбониламино; гидроксигрупп и их производных, таких как алкокси, арилокси, алкилкарбонилокси, арилкарбонилокси, циклоалкенилокси или алкилиденаминоокси; карбонильных групп и их производных, таких как алкилкарбонил, арилкарбонил, алкоксикарбонил, арилоксикарбонил, циклоалкилоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил; сульфированных производных, таких как алкилтио, арилтио, алкилсульфонил, арилсульфонил, алкилсульфинил, арилсульфинил, арилсульфонилокси, аминосульфонил, алкиламиносульфонил или диалкиламиносульфонил; или R является фталимидогруппой формулы (II), приведенной ниже

любой R1, если присутствует, занимает положение 5 или 6 индазольного кольца и представляет собой группу, необязательно дополнительно замещенную, как указано выше для R' или R";
m равно 0 или 1;
или ее фармацевтически премлемой соли.
2. Способ по п.1, где заболевание, обусловленное и/или связанное с измененной активностью протеинкиназ, является нарушением клеточной пролиферации, выбранным из группы, включающей рак, болезнь Альцгеймера, вирусные инфекции, аутоиммунные заболевания и нейродегенеративные нарушения.
3. Способ по п.2, где рак выбран из карциномы, сквамозной клеточной карциномы, гематопоэтических опухолей лимфоидной или миелоидной линии, опухоли мезенхимного происхождения, опухоли центральной и периферической нервной системы, меланомы, семиномы, тератокарциномы, остеосаркомы, пигментной ксеродермы, кератоксантомы, фолликулярной опухоли щитовидной железы и саркомы Капоши.
4. Способ по п.1, в котором нарушение клеточной пролиферации выбрано из доброкачественной гиперплазии простаты, семейного аденоматоза, полипоза, нейрофиброматоза, псориаза, пролиферации гладкомышечных клеток, связанной с атеросклерозом, легочного фиброза, артроидного гломерулонефрита и постоперационного стеноза и рестеноза.
5. Способ по п.1, который обеспечивает ингибирование опухолевого ангиогенеза и метастазирования.
6. Способ по п.1, дополнительно включающий назначение млекопитающему, нуждающемуся в этом, радиационной терапии или химиотерапии по схеме в комбинации по крайней мере с одним цитостатическим или цитотоксическим агентом.
7. Способ по п.1, в котором нуждающимся в лечении млекопитающим является человек.
8. Способ ингибирования активности протеинкиназ, который включает контактирование указанной киназы с эффективным количеством соединения формулы (I), как определено в п.1.
9. Производное аминоиндазола, представленное формулой (I)

R выбирают из группы, состоящей из -NHCOR', -NHCONHR', -NHCONR'R", -NHSO2R' или -NHCOOR', где R' и R" каждый независимо выбран из прямого или разветвленного C1-С6алкила, С2-С6 алкенила или -алкинила, С3-С6циклоалкила или циклоалкилС1-С6алкила, арила, гетероарила, арилС1-С6 алкила, 5- или 6-членного гетероцикла или гетероциклилС1-С6алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода или серы, причем R, R' и R" необязательно замещены в любом из свободных положений одной или более группами, например, от 1 до 6 групп, выбранных из галогена, нитро-, оксогрупп (=O), карбокси, циано, алкила, перфторалкила, алкенила, алкинила, циклоалкила, арила, гетероциклила, аминогрупп и их производных, таких как, например, алкиламино, диалкиламино, ариламино, диариламино, уреидо, алкилуреидо или арилуреидо; карбониламиногрупп и их производных, таких как формиламино, алкилкарбониламино, алкенилкарбониламино, арилкарбониламино, алкоксикарбониламино; гидроксигрупп и их производных, таких как алкокси, арилокси, алкилкарбонилокси, арилкарбонилокси, циклоалкенилокси или алкилиденаминоокси; карбонильных групп и их производных, таких как алкилкарбонил, арилкарбонил, алкоксикарбонил, арилоксикарбонил, циклоалкилоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил; сульфированных производных, таких как алкилтио, арилтио, алкилсульфонил, арилсульфонил, алкилсульфинил, арилсульфинил, арилсульфонилокси, аминосульфонил, алкиламиносульфонил или диалкиламиносульфонил; или R является фталимидогруппой формулы (II), приведенной ниже

любой R1, если присутствует, занимает положение 5 или 6 индазольного кольца и представляет собой группу, необязательно дополнительно замещенную, как указано выше для R' или R";
m равно 0 или 1;
или ее фармацевтически приемлемую соль; при условии что:
a) когда R является -NHCOR' и m равно 0, тогда R' является отличным от метила, н-пропила, бензила, 2,2-дифенилэтила, 3,5-диметилизоксазол-4-ила, 2-(морфолин-4-ил)этила или фенила, необязательно замещенного хлором, гидрокси, метилом, нитро или амино;
b) когда индазол замещен в положении 5 или 6 метоксигруппой, тогда R является отличным от 3-(N,N-диэтиламино)пропиламино, 3-[(3-метил)морфолин-4-ил]пропиламино или 1-гидрокси-2-метил-2-пропиламино;
c) соединение 3-фталимидоиндазол исключается.
10. Соединение формулы (I) по п.9, где R является группой -NHCOR' и R', R1 и m являются такими, как определено в п.9.
11. Соединение формулы (I) по п.10, где m равно 1, и R1 и R1 выбирают, каждый независимо, из C1-С6алкила, С3-С6циклоалкила или циклоалкилС1-С6алкила, арила, гетероарила, арилС1-С6алкила, 5- или 7-членного гетероциклила или гетероциклилС1-С6алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода или серы.
12. Соединение формулы (I) по п.9, где R является группой -NHCONHR' или -NHCONR'R", и R', R", R1 и m являются такими, как определено в п.9.
13. Соединение формулы (I) по п.12, где m равно 1 и R1, R' и R" выбирают, каждый независимо, из C1-С6алкила, С3-С6циклоалкила или циклоалкилС1-С6алкила, арила, гетероарила, арилС1-С6алкила, 5- или 7-членного гетероциклила или гетероциклилС1-С6алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода или серы.
14. Соединение формулы (I) по п.9, где R является -NHSO2R' группой и R', R1 и m являются такими, как определено в п.9.
15. Соединение формулы (I) по п.14, где m равно 1, и R1 и R' выбирают, каждый независимо, из C1-С6алкиыр, С3-С6циклоалкила или циклоалкилС1-С6алкила, арила, гетероарила, арилС1-С6алкила, 5- или 7-членного гетероциклила или гетероциклилС1-С6алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода или серы.
16. Соединение формулы (I) по п.9, где R является группой -NHCOOR' и R', R1 и m являются такими, как определено в п.9.
17. Соединение формулы (I) по п.16, где m равно 1 и R1 и R' выбирают, каждый независимо, из C1-С6алкила, С3-С6циклоалкила или циклоалкилС1-С6алкила, арила, гетероарила, арилС1-С6алкила, 5- или 7-членного гетероциклила или гетероциклилС1-С6алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода или серы.
18. Соединение формулы (I) по п.9, где R является фталимидогруппой формулы (II), и R1 и m являются такими, как определено в п.9.
19. Соединение формулы (I) по п.18, где m равно 1, и R1 выбирают из С2-С6алкенила, С3-С6алкинила, арила, гетероарила, арилС1-С6алкила, 5- или 7-членного гетероциклила или гетероциклилС1-С6алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода или серы.
20. Соединение формулы (I), как определено в п.9, необязательно в форме фармацевтически приемлемой соли, выбранное из группы, включающей следующее:
1) метил 2-({3-[(анилинокарбонил)амино]-1Н-индазол-5-ил}окси)бутаноат;
2) N-бензил-N'-[5-(2-пирролидин-1-илэтокси)-1Н-индазол-3-ил]мочевина;
3) метил 2-[(3-{[(бензиламино)карбонил]амино}-1Н-индазол-5-ил)окси]бутаноат;
4) N-изопропил-N'-{5-[(2-оксо-1-фенилпирролидин-3-ил)окси]-1Н-индазол-3-ил}мочевина;
5) 2-[(3-{[(изопропиламино)карбонил]амино}-1Н-индазол-5-ил)окси]-N-фенилпропанамид,
6) метил 2-[(3-{[(изопропиламино)карбонил]амино}-1Н-индазол-5-ил)окси]бутаноат;
7) N-изопропил-N'-{5-[2-(4-метил-1,3-тиазол-5-ил)этокси]-1Н-индазол-3-ил}мочевина;
8) N-[5-(бут-3-инилокси)-1Н-индазол -3-ил]-N'-изопропилмочевина;
9) метил 2-({3-[(3-фенилпропаноил)амино]-1Н-индазол-5-ил}окси)бутаноат;
10) N-{5-[(2-оксо-1-фенилпирролидин-3-ил)окси]-1Н-индазол-3-ил}циклопропанкарбоксамид;
11) метил 2-({3-[(циклопропилкарбонил)амино]-1Н-индазол-5-ил}окси)бутаноат;
12) 2-(4-трет-бутилфенокси)-N-[5-(2-пирролидин-1-ил-этокси)-1Н-индазол-3-ил]ацетамид;
13) 2-(4-метоксифенил)-N-[5-(2-пирролидин-1-илэтокси)-1Н-индазол-3-ил]ацетамид;
14) метил 2-[(3-{[(4-метоксифенил)ацетил]амино}-1Н-индазол-5-ил)окси]бутаноат;
15) N-изопропил-N'-{6-[(2-оксо-1-фенилпирролидин-3-ил)окси]-1Н-индазол-3-ил}мочевина;
16) N-{6-[(2-метилбензил)окси]-1Н-индазол-3-ил}циклопропанкарбоксамид;
17) N-{6-[(2-оксо-1-фенилпирролидин-3-ил)окси]-1Н-индазол-3-ил}циклопропанкарбоксамид;
18) метил 2-({3-[(циклопропилкарбонил)амино]-1Н-индазол-6-ил}окси)бутаноат;
19) метил 2-({3-[(3-хлорбензоил)амино]-1Н-индазол-6-ил}окси)бутаноат;
20) N-бензил-N'-(5-гидрокси-2Н-индазол-3-ил)мочевина;
21) N-(5-гидрокси-2Н-индазол-3-ил)-N'-изопропилмочевина;
22) 2-(4-трет-бутилфенокси)-N-(5-гидрокси-2Н-индазол-3-ил)ацетамид;
23) N-(5-гидрокси-2Н-индазол-3-ил)-2-(4-метоксифенил)ацетамид;
24) N-(6-гидрокси-2Н-индазол-3-ил)-N'-фенилмочевина;
25) N-(6-гидрокси-2Н-индазол-3-ил)-3-фенилпропанамид;
26) N-(6-гидрокси-2Н-индазол-3-ил)циклопропанкарбоксамид.
21. Способ получения соединений формулы (I) и их фармацевтически приемлемых солей, как определено в п.9, где R является таким, как определено в п.9, но отличным от фталимидогруппы формулы (II), который включает:
а) взаимодействие в кислых условиях производного 2-аминобензонитрила формулы (III)

где m имеет значения, определенные выше в п.9, если присутствует, R''' является метильной или бензильной группой; с нитритом натрия в присутствии хлорида олова, с получением соединения формулы (IV)

b) взаимодействие соединения формулы (IV) с фталевым ангидридом, с получением соединения формулы (V)

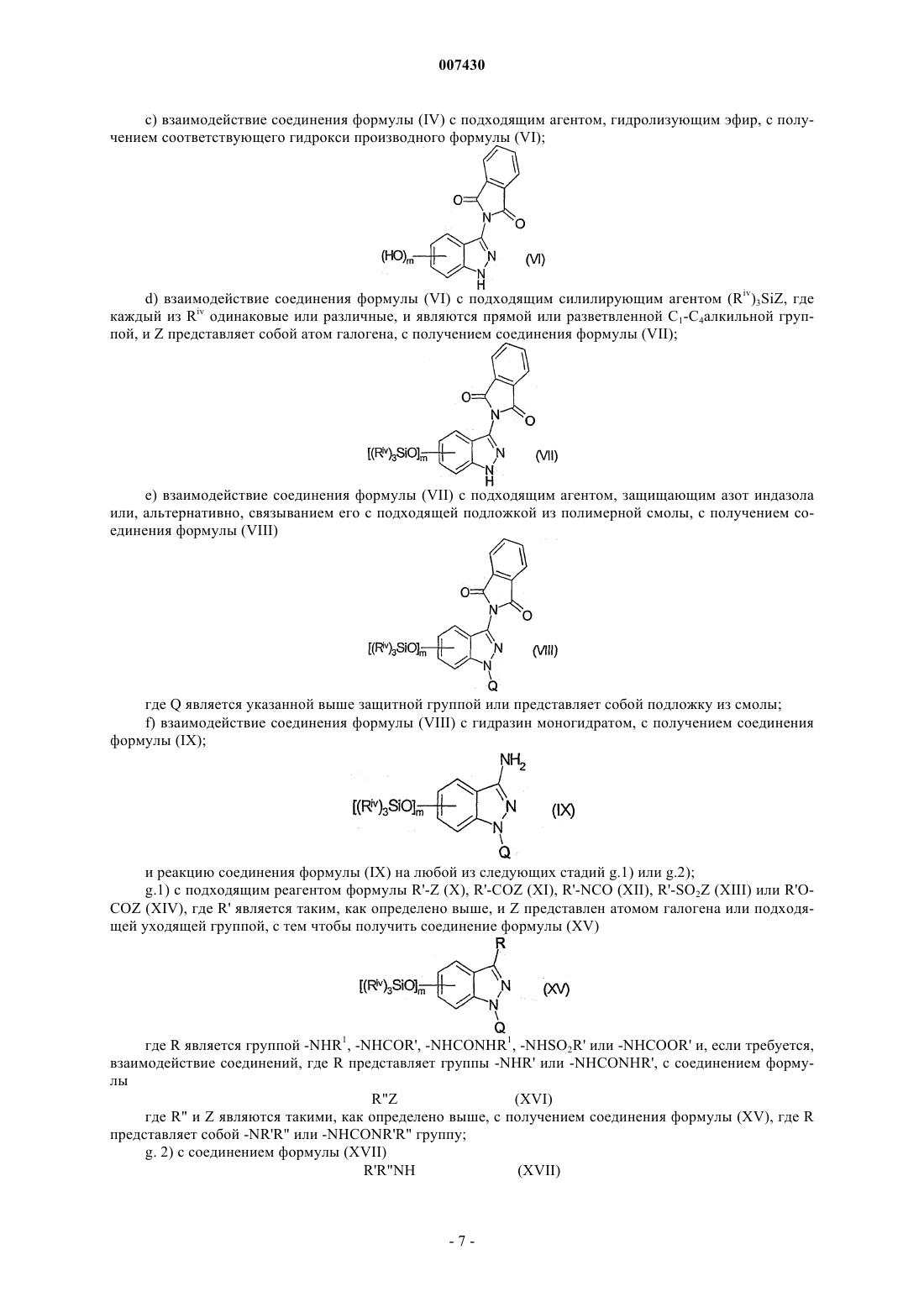
с) взаимодействие соединения формулы (V) с подходящим агентом, расщепляющим эфирную связь, с получением соответствующего гидроксипроизводного формулы (VI)

d) взаимодействие соединения формулы (VI) с подходящим силилирующим агентом (Riv)3SiZ, где каждый Riv является одинаковым или отличным, прямым или разветвленным С1-С4алкилом, и Z является атомом галогена, с получением соединения формулы (VII)

е) взаимодействие соединения формулы (VII) с подходящим защитным агентом атома азота индазола или, альтернативно, связывание его с подходящей полимерной смолой, для получения соединения формулы (VIII)

где Q является указанной выше защитной группой или представляет смолу подложки;
f) взаимодействие соединения формулы (VIII) с гидразин моногидратом, с получением соединения формулы (IX)

и взаимодействие соединения формулы (IX) по любой из следующих стадий g.l) или g. 2);
g.1) с подходящим реагентом формулы R'-Z (X), R'-COZ (XI), R'-NCO (XII), R'-SO2Z (XIII) или R'OCOZ (XIV), где R' имеет значения, определенные в п.9, и Z представляет собой атом галогена или подходящую уходящую группу, с получением соответствующего соединения формулы (XV)

где R является -NHCOR', -NHCONHR', -NHSO2R' или -NHCOOR' группой и, если требуется, взаимодействие соединений, имеющих в качестве R -NHCONHR' группу, с соединением формулы
R"Z (XVI)
где R" имеет значения, определенные в п.9, и Z определено выше, с получением соединений формулы (XV), где R является -NHCONR'R" группой;
g.2) с соединением формулы (XVII)
R'R"NH (XVII)
где R' и R" являются такими, как определено выше, в присутствии 4-нитрофенилхлорформиата, для получения соответствующего соединения формулы (XV), где R является -NHCONR'R" группой;
h) взаимодействие любого из указанных выше соединений формулы (XV) с тетрабутиламмонийфторидом для получения соединения формулы (XVIII)

i) взаимодействие соединения формулы (XVIII) с производным формулы
R1-Z (XIX)
где R1 является таким, как определено в п.9, и Z является атомом галогена, подходящей уходящей группой или гидроксигруппой, для получения соединения формулы (XX)

j) снятие защиты с соединения формулы (XX) или, альтернативно, отщепление полимерной смолы для того, чтобы получить желаемое соединение формулы (I) и, если требуется, превращение его в другое соединение формулы (I) и/или в его фармацевтически приемлемую соль.
22. Способ получения по п.21, где соединение формулы (III) является 2-амино-4-метоксибензонитрилом или 2-амино-5-бензилоксибензонитрилом.
23. Способ получения по п.21, где стадию а) проводят в присутствии хлористо-водородной кислоты.
24. Способ получения по п.21, где на стадии d) силилирующим агентом является трет-бутилдиметилсилилхлорид.
25. Способ получения по п.21, где на стадии е) производное индазола формулы (VII) присоединено к хлортритилхлоридной полимерной смоле.
26. Способ получения соединений формулы (I) и их фармацевтически приемлемых солей, как определено в п.9, гдх R является фталимидогруппой формулы (II), приведенной ниже

способ получения которых включает взаимодействие соединений формулы (VIII)

где Q является защитной группой или представляет смолу подложки; с тетрабутиламмонийфторидом для получения соединения формулы (XVIII)

где R является фталимидной группой; и взаимодействие соединения формулы (XVIII) с производным формулы
R1-Z (XIX)
где R1 является таким, как определено в п.9, и Z является атомом галогена, подходящей уходящей группой или гидроксигруппой, для получения соединения формулы (XX)

и снятие защиты с соединения формулы (XX) или, альтернативно, отщепление полимерной смолы для того, чтобы получить желаемое соединение формулы (I) и, если требуется, превращение его в другое соединение формулы (I) и/или в его фармацевтически приемлемую соль.
27. Соединение формулы (I), как определено в п.9, и его фармацевтически приемлемые соли, которые получают, например, посредством методики комбинаторной химии, как в способе получения по п.21, первоначальным взаимодействием соединения формулы (IХа)

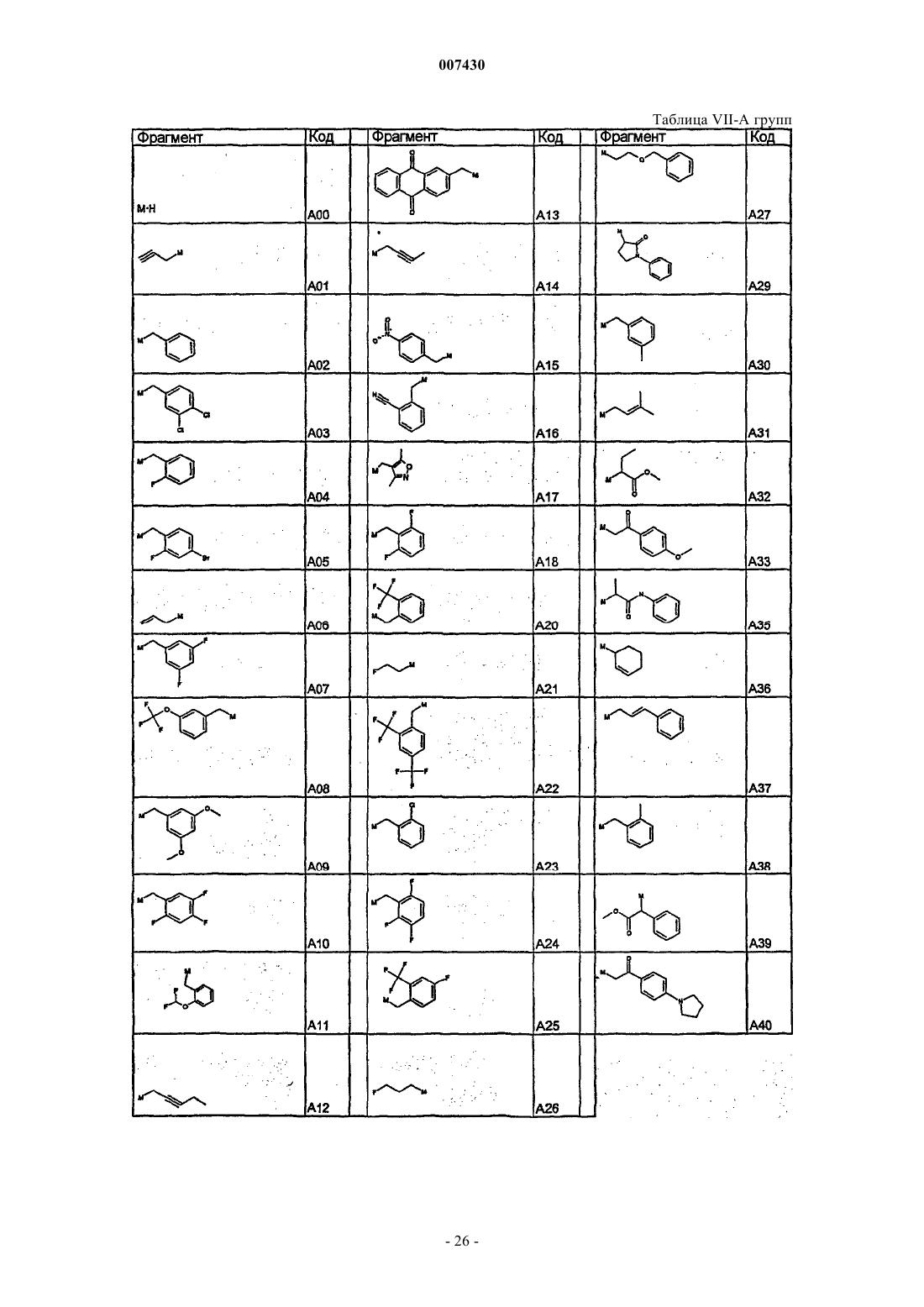
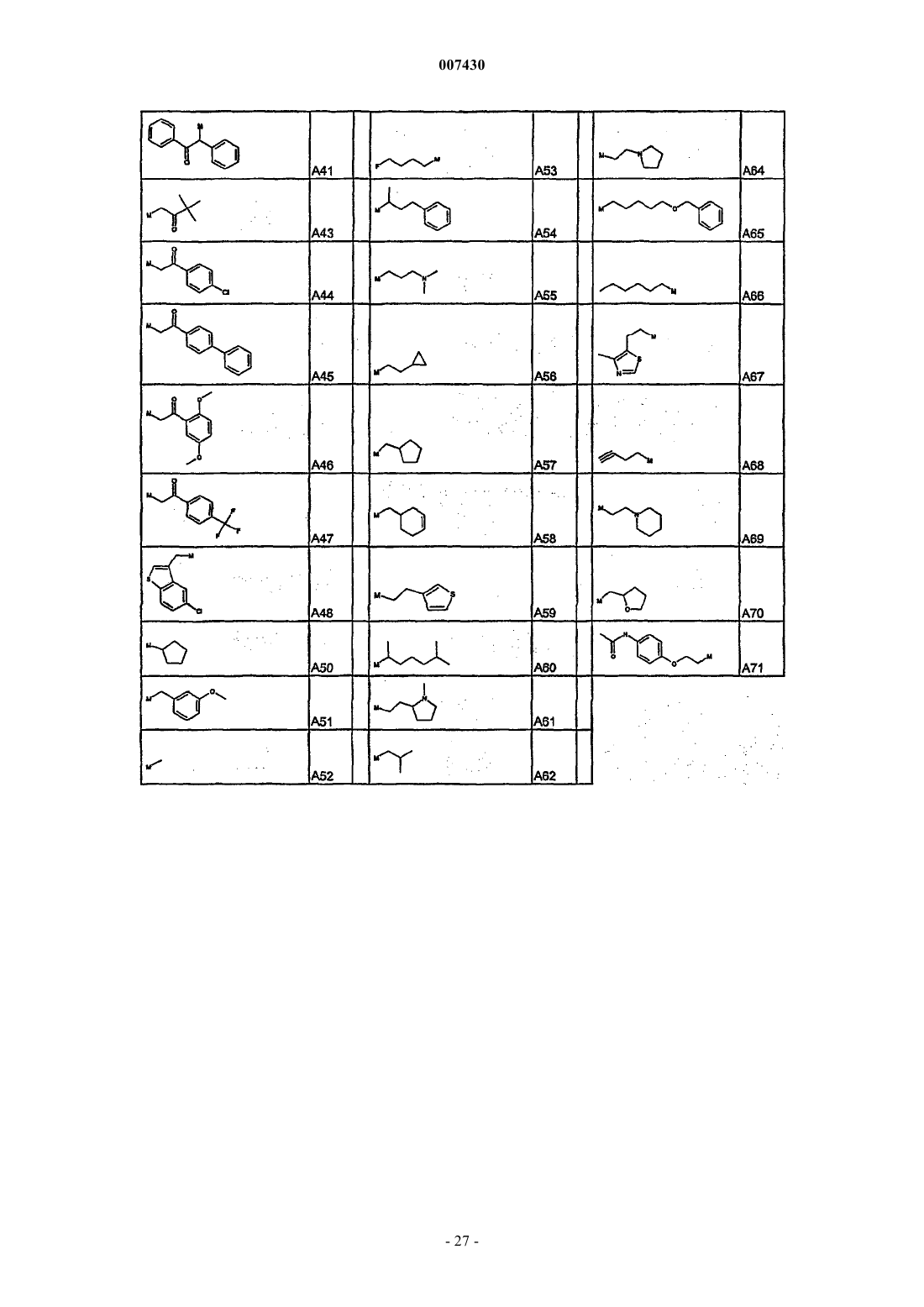
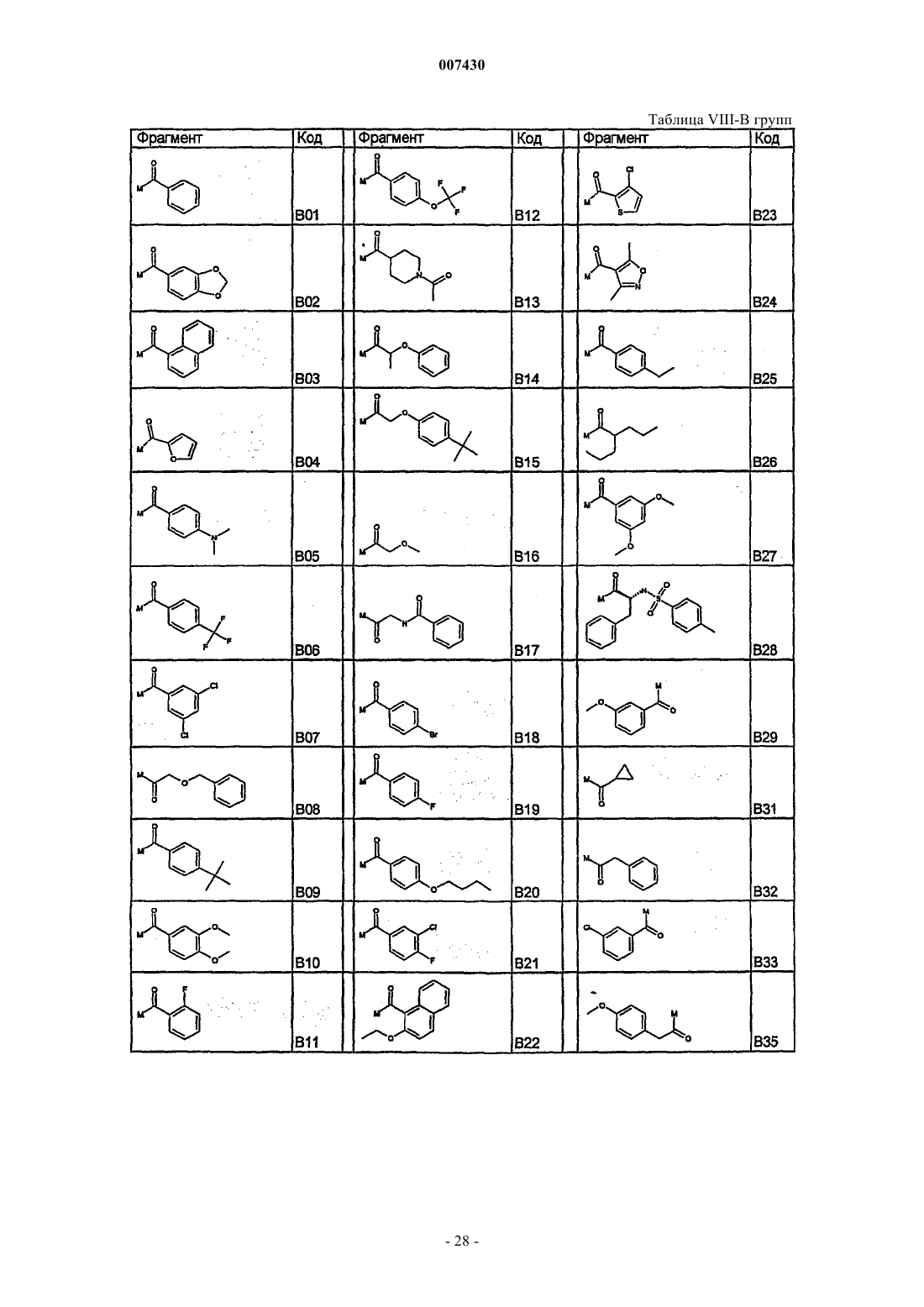
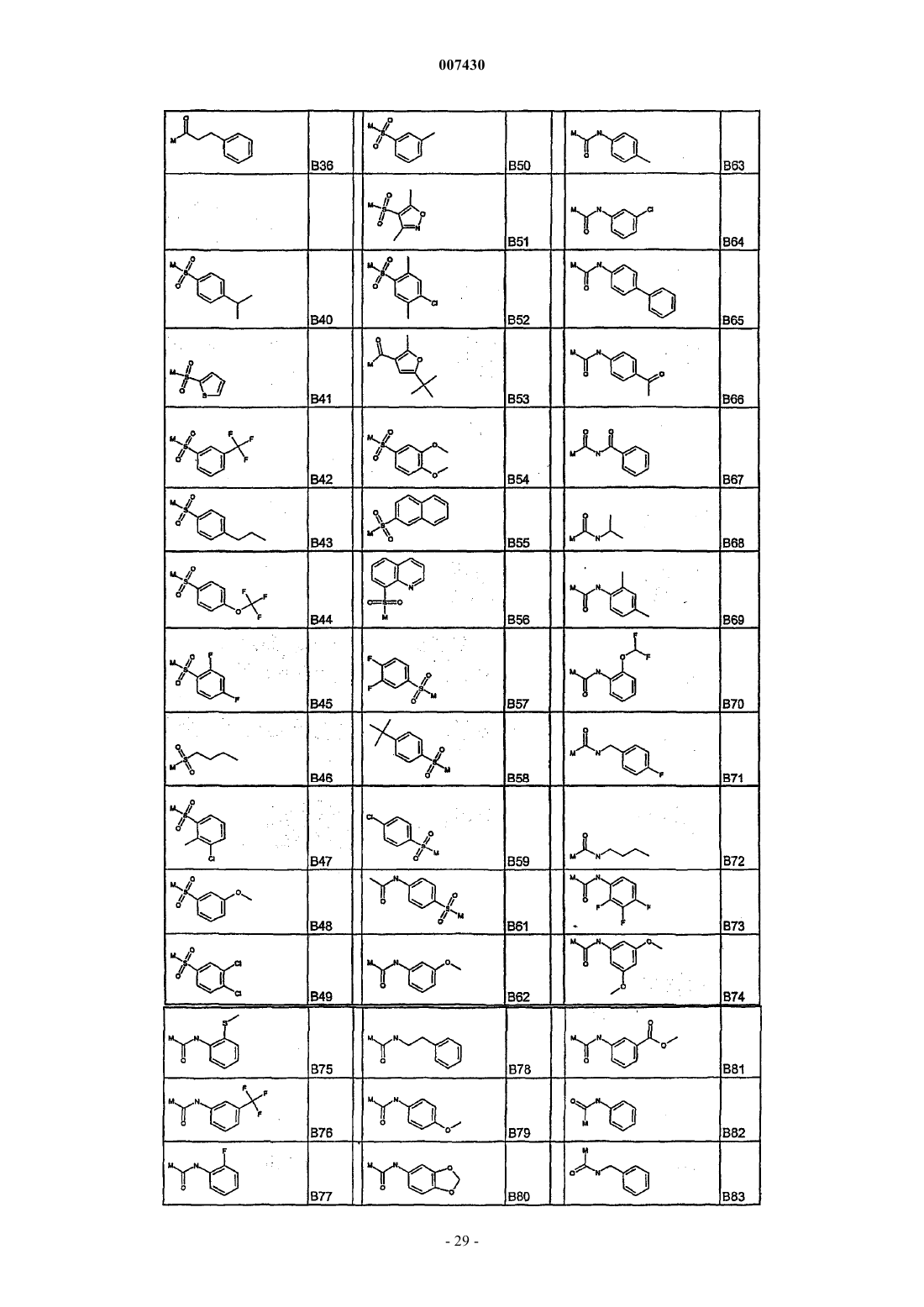
с каждым из соединений формулы (X), как установлено в табл. I:
Таблица I. Соединения формулы R'-Z (X)


для получения множества соединений формулы (XVa)

с последующим взаимодействием каждого из производных формулы (XVa) с тетрабутиламмонийфторидом, и затем с одним из производных формулы (XIX), как указано в табл. II:
Таблица II. Соединения формулы R1-Z (XIX), где Z является бромом



или в табл. III:
Таблица III. Соединения формулы R1-Z (XIX), где Z является гидроксигруппой


и последующим снятием защиты с полученного соединения или, альтернативно, отщеплением полимерной смолы для того, чтобы получить желаемое соединение формулы (I) и, если требуется, превращением его в другое соединение формулы (I) и/или в его фармацевтически приемлемую соль.
28. Соединение формулы (I), как определено в п.9, и его фармацевтически приемлемые соли, которые получают, например, посредством методики комбинаторной химии, как в способе получения по п.21, первоначальным взаимодействием соединения формулы (IXb)

с каждым из соединений формулы (X), как установлено в табл. I:
Таблица I. Соединения формулы R'-Z (X)


для получения множества соединений формулы (XVb)

затем путем взаимодействия каждого из производных формулы (XVb) с тетрабутиламмонийфторидом, и затем с одним из производных формулы (XIX), как указано в табл. II:
Таблица II. Соединения формулы R1-Z (XIX), где Z является бромом



или в табл. III:
Таблица III. Соединения формулы R1-Z (XIX), где Z является гидроксигруппой


и последующим снятием защиты с полученного соединения или, альтернативно, отщеплением полимерной смолы для того, чтобы получить желаемое соединение формулы (I) и, если требуется, превращением его в другое соединение формулы (I) и/или в его фармацевтически приемлемую соль.
29. Соединение формулы (I), как определено в п.9, и его фармацевтически приемлемые соли, которые получают, например, посредством методики комбинаторной химии, как в способе получения по п.21, первоначальным взаимодействием соединения формулы (IХа)

с каждым из соединений формулы (XI), как указано в табл. IV:
Таблица IV. Соединения формулы R'COZ (XI)


для получения множества соединений формулы (XVc)

затем путем взаимодействия каждого из производных формулы (XVc) с тетрабутиламмонийфторидом, и затем с одним из производных формулы (XIX), как указано в табл. II:
Таблица II. Соединения формулы R1-Z (XIX), где Z является бромом


или в табл. III:
Таблица III. Соединения формулы R1-Z (XIX), где Z является гидроксигруппой


и последующим снятием защиты с полученного соединения или, альтернативно, отщеплением полимерной смолы для того, чтобы получить желаемое соединение формулы (I) и, если требуется, превращением его в другое соединение формулы (I) и/или в его фармацевтически приемлемую соль.
30. Соединение формулы (I), как определено в п.9, и его фармацевтически приемлемые соли, которые получают, например, посредством методики комбинаторной химии, как в способе получения по п.21, первоначальным взаимодействием соединения формулы (IXb)

с каждым из соединений формулы (XI), как указано в табл. IV:
Таблица IV. Соединения формулы R'COZ (XI)



для получения множества соединений формулы (XVd)

затем путем взаимодействия каждого из производных формулы (XVd) с тетрабутиламмонийфторидом, и затем с одним из производных формулы (XIX), как указано в табл. II.
Таблица II. Соединения формулы R1-Z (XIX), где Z является бромом



или в табл. III:
Таблица III. Соединения формулы R1-Z (XIX), где Z является гидроксигруппой


и последующим снятием защиты с полученного соединения или, альтернативно, отщеплением полимерной смолы для того, чтобы получшть желаемое соединение формулы (I) и, если требуется, превращением его в другое соединение формулы (I) и/или в его фармацевтически приемлемую соль.
31. Соединение формулы (I), как определено в п.9, и его фармацевтически приемлемые соли, которые получают, например, посредством методики комбинаторной химии, как в способе получения по п.21, первоначальным взаимодействием соединения формулы (IХа)

с каждым из соединений формулы (XII), как указано в табл. V:
Таблица V. Соединения формулы R'-NCO (XII)


для получения множества соединений формулы (XVe)

затем путем взаимодействия каждого из производных формулы (XVe) с тетрабутиламмонийфторидом и затем с одним из производных формулы (XIX), как указано в табл. II:
Таблица II. Соединения формулы R1-Z (XIX), где Z является бромом



или в табл. III:
Таблица III. Соединения формулы R1-Z (XIX), где Z является гидроксигруппой


и последующим снятием защиты с полученного соединения или, альтернативно, отщеплением полимерной смолы для того, чтобы получить желаемое соединение формулы (I) и, если требуется, превращением его в другое соединение формулы (I) и/или в его фармацевтически приемлемую соль.
32. Соединение формулы (I), как определено в п.9, и его фармацевтически приемлемые соли, которые получают, например, посредством методики комбинаторной химии, как в способе получения по п.21, первоначальным взаимодействием соединения формулы (IXb)

с каждым из соединений формулы (XII), как указано в табл. V:
Таблица V. Соединения формулы R'-NCO (XII)


для получения множества соединений формулы (XVf)

затем путем взаимодействия каждого из производных формулы (XVf) с тетрабутиламмонийфторидом и затем с одним из производных формулы (XIX), как указано выше в табл. II:
Таблица II. Соединения формулы R1-Z (XIX), где Z является бромом



или в табл. III:
Таблица III. Соединения формулы R1-Z (XIX), где Z является гидроксигруппой


и последующим снятием защиты с полученного соединения или, альтернативно, отщеплением полимерной смолы для того, чтобы получить желаемое соединение формулы (I) и, если требуется, превращением его в другое соединение формулы (I) и/или его фармацевтически приемлемую соль.
33. Соединение формулы (I), как определено в п.9, и его фармацевтически приемлемые соли, которые получают, например, посредством методики комбинаторной химии, как в способе получения по п.21, первоначальным взаимодействием соединения формулы (IХа)

с каждым из соединений формулы (XIII), как указано в табл. VI:
Таблица VI. Соединения формулы R'-SO2Z (XIII)


для получения множества соединений формулы (XVg)

затем путем взаимодействия каждого из производных формулы (XVg) с тетрабутиламмонийфторидом, и затем с одним из производных формулы (XIX), как указано в табл. II:
Таблица II. Соединения формулы R1-Z (XIX), где Z является бромом


или в табл. III:
Таблица III. Соединения формулы R1-Z (XIX), где Z является гидроксигруппой

и последующим снятием защиты с полученного соединения или, альтернативно, отщеплением полимерной смолы для того, чтобы получить желаемое соединение формулы (I) и, если требуется, превращением его в другое соединение формулы (I) и/или в его фармацевтически приемлемую соль.
34. Соединение формулы (I), как определено в п.9, и его фармацевтически приемлемые соли, которые получают, например, посредством методики комбинаторной химии, как в способе получения по п.21, первоначальным взаимодействием соединения формулы (IXb)

с каждым из соединений формулы (XIII), как указано в табл. VI:
Таблица VI. Соединения формулы R'-SO2Z (XIII)


для получения множества соединений формулы (XVh)

затем путем взаимодействия каждого из производных формулы (XVh) с тетрабутиламмонийфторидом, и затем с одним из производных формулы (XIX), как указано в табл. II:
Таблица II. Соединения формулы R1-Z (XIX), где Z является бромом


или в табл. III:
Таблица III. Соединения формулы R1-Z (XIX), где Z является гидроксигруппой


и последующим снятием защиты с полученного соединения или, альтернативно, отщеплением полимерной смолы для того, чтобы получить желаемое соединение формулы (I) и, если требуется, превращением его в другое соединение формулы (I) и/или в его фармацевтически приемлемую соль.
35. Библиотека двух или более производных аминоиндазола, представленных формулой (I)

где R выбирают из группы, состоящей из -NHCOR', -NHCONHR', -NHCONR'R", -NHSO2R' или -NHCOOR', где R' и R" являются, каждая независимо, группой, выбранной из прямого или разветвленного C1-С6алкила, С2-С6алкенила или -алкинила, С3-С6циклоалкила или циклоалкилС1-С6алкила, арила, гетероарила, арилС1-С6алкила, 5- или 6-членного гетероцикла или гетероциклилС1-С6алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода или серы, причем R, R' и R" необязательно замещены в любом из свободных положений одной или более группами, например от 1 до 6 групп, выбранных из галогена, нитро-, оксогрупп (=O), карбокси, циано, алкила, перфторалкила, алкенила, алкинила, циклоалкиыр, арила, гетероциклила, аминогрупп и их производных, таких как, например, алкиламино, диалкиламино, ариламино, диариламино, уреидо, алкилуреидо или арилуреидо; карбониламиногрупп и их производных, таких как формиламино, алкилкарбониламино, алкенилкарбониламино, арилкарбониламино, алкоксикарбониламино; гидроксигрупп и их производных, таких как, алкокси, арилокси, алкилкарбонилокси, арилкарбонилокси, циклоалкенилокси или алкилиденаминоокси; карбонильных групп и их производных, таких как алкилкарбонил, арилкарбонил, алкоксикарбонил, арилоксикарбонил, циклоалкилоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил; сульфированных производных, таких как алкилтио, арилтио, алкилсульфонил, арилсульфонил, алкилсульфинил, арилсульфинил, арилсульфонилокси, аминосульфонил, алкиламиносульфонил или диалкиламиносульфонил; или R является фталимидогруппой формулы (II), приведенной ниже

любой R1, если присутствует, занимает положение 5 или 6 индазольного кольца и представляет собой группу, необязательно дополнительно замещенную, как указано выше для R' или R";
m равно 0 или 1;
или их фармацевтически приемлемых солей.
36. Соединение формулы (I), необязательно в форме его фармацевтически приемлемой соли, как указано в одной из таблиц с IX по XVI:
Таблица IX


Таблица X

Таблица XI









Таблица XII






Таблица XIII




Таблица XIV


Таблица XV



Таблица XVI


37. Фармацевтическая композиция, включающая эффективное количество аминоиндазола формулы (I), как определено в п.9 и по крайней мере один фармацевтически приемлемый эксципиент, носитель или разбавитель.
38. Фармацевтическая композиция по п.37, дополнительно включающая один или более химиотерапевтический агент, в виде комбинированного состава для совместного, раздельного или последовательного применения в противораковой терапии.
39. Продукт или набор, включающий соединение п.9 или его фармацевтическую композицию, как определено в п.37, и один или несколько химиотерапевтических агентов, в виде комбинированного состава для совместного, раздельного или последовательного применения в противораковой терапии.
40. Соединение формулы (I) или его фармацевтически приемлемая соль, как определено в п.9, для использования в качестве медикамента.
41. Применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено в п.9, в производстве медикамента для лечения заболеваний, обусловленных и/или связанных с измененной активностью протеинкиназ.
42. Применение по п.41 для лечения опухолей.
Текст
007430 Данное изобретение касается производных аминоиндазола, активных в качестве ингибиторов киназ и, в особенности, оно касается производных 3-аминоиндазола, способа их получения, содержащих их фармацевтических композиций и применения в качестве терапевтических агентов, особенно в лечении заболеваний, связанных с нарушением метаболизма протеинкиназ. Неудовлетворительная работа протеинкиназ (РК) является отличительным признаком множества заболеваний. Огромная часть онкогенов и протоонкогенов, участвующих в развитии рака у человека,кодируется РК. Повышенная активность РК также вовлечена в развитие доброкачественных заболеваний,таких как доброкачественная гиперплазия простаты, семейный (наследственный) аденоматоз, полипоз,нейрофиброматоз, псориаз, пролиферация гладкомышечных клеток, связанная с атеросклерозом, легочный фиброз, артроидный гломерулонефрит и постоперационный стеноз и рестеноз. РК также вовлечены в воспалительные состояния и в размножение вирусов и паразитов. РК могут также играть главную роль в патогенезе и развитии нейродегенеративных нарушений. В качестве общей ссылки в отношении неудовлетворительной работы или нарушения регуляции РК смотри, например, Current Opinion in Chemical Biology 1999, 3, 459-465. Главным объектом данного изобретения является разработка соединений, применимых в терапии в качестве агентов для борьбы с множеством заболеваний, обусловленных и/или связанных с нарушением протеинкиназной активности. Другим объектом является создание соединений, которые наделены свойствами ингибирования активности множества протеинкиназ. Авторы изобретения обнаружили, что некоторые производные 3-аминоиндазола, здесь и далее вкратце называемые производными индазола или индазолами, обладают активностью ингибирования множества протеинкиназ и, следовательно, применимы в терапии лечения заболеваний, связанных с нарушением регуляции протеинкиназ. Более точно, индазолы этого изобретения применимы в лечении ряда опухолей, включая, но не ограничиваясь ими: карциному таких органов как мочевой пузырь, молочная железа, толстая кишка, почки, печень, легкие (включая мелкоклеточный рак легкого), пищевод, желчный пузырь, яичники, поджелудочная железа,желудок, шейка матки, щитовидная железа, простата и кожа, включая карциному сквамозных клеток; гематопоэтические опухоли лимфоидной линии, включая лейкемию, острую лимфоцитную лейкемию, острую лимфобластическую лейкемию, В-клеточную лимфому, Т-клеточную лимфому, лимфому Ходжкина, лимфому ворсистых клеток и лимфому Беркитта; гематопоэтические опухоли миелоидной линии, включая острую и хроническую миелогенную лейкемии, миелодиспластический синдром и промиелоцитарную лейкемию; опухоли мезенхимного происхождения, включая фибросаркому и рабдомиосаркому; опухоли центральной и периферической нервной системы, включая астроцитому, нейробластому,глиому и шванному; другие опухоли, включая меланому, семиному, тератокарциному, остеосаркому, пигментную ксеродерму, кератоксантому, фолликулярную опухоль щитовидной железы и саркому Капоши. Благодаря ключевой роли РК в регуляции клеточной пролиферации, эти индазолы также применимы в лечении ряда нарушений клеточной пролиферации, таких как, например, доброкачественная гиперплазия простаты, семейный аденоматоз, полипоз, нейрофиброматоз, псориаз, пролиферация гладкомышечных клеток, связанная с атеросклерозом, легочный фиброз, артроидный гломерулонефрит и постоперационный стеноз и рестеноз. Соединения данного изобретения могут применяться в лечении болезни Альцгеймера, предположительно благодаря факту участия cdk5 в фосфорилировании тау-белка (J. Biochem., 117, 741-749, 1995). Соединения данного изобретения, являясь модуляторами апоптоза, могут также применяться в лечении рака, вирусных инфекций, для предотвращения развития СПИДа у HIV-инфицированных индивидов, аутоимунных заболеваний и нейродегенеративных нарушений. Соединения данного изобретения могут применяться для ингибирования опухолевого ангиогенеза и метастазирования. Соединения данного изобретения могут применяться в качестве ингибиторов циклинзависимых киназ (cdk) и также в качестве ингибиторов других протеинкиназ, таких как протеинкиназа С в различных изоформах: Met, PAK-4, PAK-5, ZC-1, STLK-2, DDR-2, Aurora 1, Aurora 2, Bub-1, PLK, Chkl, Chk2, HER2,raf1, MEK1, МАРК, EGF-R, PDGF-R, FGF-R, IGF-R, VEGF-R, PI3K, weel kinase, Src, Аbl, Akt, ILK, MK-2,IKK-2, Cdc7, Nek, и, таким образом, быть эффективными в лечении заболеваний, связанных с другими протеинкиназами. Некоторые индазолы и аминоиндазолы известны в уровне техники как синтетические или химические промежуточные продукты, как полимерные стабилизаторы, как терапевтические агенты и даже как ингибиторы протеинкиназ. В качестве примера, ряд алкиламиноиндазолов рассмотрен в US 28939 (переизданный патент US 3133081) Smithkline Co., как имеющие активность миорелаксантов и аналгетиков; среди них 3 метиламино-5-трифторметилиндазол и 3-диэтиламино-5-трифторметилиндазол.Bioorg. Med. Chem. Lett. (1998), 8(7), 715-720 как ингибиторы HIV-протеаз. Производные диарилмочевины, применимые в лечении заболеваний, отличных от рака, раскрыты как ингибиторы р 38-киназы в WO 99/32111 принадлежащей Bayer Co.; среди конкретно представленных соединений N-[4-[(пиридил-4-ил)окси]фенил]-N'-6-[хлор-(индазол-3-ил)]мочевина. Производные имидазопиридина, дополнительно замещенные арильными группами, например, индазолиламинокарбонилфенилом, рассмотрены как антагонисты фактора, активирующего тромбоциты,(PAF) в WO 91/17162, Pfizer Ltd. Соединения индазола, дополнительно замещенные в положении 3 группами, отличными от амино,или их производные описаны в WO 01/02369, принадлежащем Agouron Pharmaceuticals Inc., как обладающие активностью ингибиторов протеинкиназ. Меркаптоцианоакрилоиламино- или алкилтиоцианоакрилоиламино-гетероциклы раскрыты в качестве соединений, применимых в лечении нарушений, связанных с усиленным клеточным ростом, в US 5714514, Hoechst. Производные 1-ациламино-3-(N-арилсульфонил-N-алкоксиамино)-2-гидроксипропана,где арильный остаток также содержит индазольные группы, описаны в качестве ингибиторов HIVаспартилпротеазы в WO 99/65870 Vertex Pharmaceuticals Inc. Ряд других производных индазола известны как терапевтические агенты, в частности, 3-[3(морфолин-4-ил)пропиониламино]индазол, 3-(N,N-диэтиламино)пропиламино-5-метоксииндазол, 3-[(3 метил)морфолин-4-ил]пропиламино-5-метоксииндазол, 3-(N,N-диэтиламино)пропиламино-5-метилиндазол и 3-[(3-метил)морфолин-4-ил]пропиламино-5-метилиндазол рассмотрены как обладающие анальгезирующей и противовоспалительной активностью [смотри US 4751302 и JP-A-60061569, Asahi ChemicalIndustry]; 3-[(2-гидроксифенил)карбониламино]индазол раскрыт как антимикробный агент [смотриPharmazie (1990), 45 (6), 441-2]. Некоторые другие индазолы, известные в уровне техники, в основном описаны как химические промежуточные продукты или как соединения для целей, отличных от терапевтических, например, как стабилизаторы полимеров, отбеливающие агенты, красители и подобные вещества. Среди них: 3-(этоксикарбониламино)индазол [смотри Chemical Abstracts 92 (1980):215400]; 3 ацетиламиноиндазол и 3-бензоиламиноиндазол [смотри J. Org. Chem. (1996), 61 (24), 8397-8401]; 3 бутириламиноиндазол, 3-[(4-хлорфенил)карбониламино]индазол, 3-[(4-метилфенил)карбониламино] индазол и 3-[(3,3-дифенил)пропиониламино]индазол [смотри Acta Chim. Hung, (1990), 127 (6), 795-802]; 3[(3,5-диметилизоксазол-4-ил)карбониламино]индазол [смотри J. Heterocyl. Chem. (1974), 11(4), 623-6]; 3[(4-нитрофенил)карбониламино]индазол и 3-(фенилацетиламино) индазол [смотри J. Chem. Soc., Perkin(1973): 136158]; 3-[(1-гидрокси-2-метил)-2-пропил]амино-6,7-диметоксииндазол [смотри US 4864032,принадлежащий Ortho Pharmaceutical Co.]. Кроме того, 3-фталимидоиндазол и 4-хлор-3-фталимидоиндазол как синтетические промежуточные продукты в получении фармацевтических препаратов, имеющие анальгезирующую и противовоспалительную активность, рассмотрены в US 4751302, принадлежащем Asahi Chemical Industry Co. Сульфониламиноиндазолы и, в большей степени, длинно-цепочечные алкилоксифенилсульфониламиноиндазолы в качестве соединений, образующих циановые красители, рассмотрены в JP-A-08022109,Heisei. Широкие классы соединений пиразола, применимых в качестве ингибиторов протеинкиназ, также раскрыты Vertex Pharmaceuticals Inc. в ряде патентных заявок, таких как WO 02/62789, WO 02/59112, WO 02/59111, WO 02/57259, WO 02/50066, WO 02/50065, WO 02/22608, WO 02/22607, WO 02/22606, WO 02/22605, WO 02/22604, WO 02/22603 и WO 02/22601. Таким образом, настоящее изобретение предлагает метод лечения заболеваний, обусловленных и/или связанных с измененной активностью протеинкиназ, путем введения нуждающемуся в этом млекопитающему эффективного количества аминоиндазола, представленного формулой (I)-NHCOOR', где R' и R" каждый независимо выбран из прямого или разветвленного C1-С 6 алкила, С 2-С 6 алкенила или -алкинила, С 3-С 6 циклоалкила или циклоалкилС 1-С 6 алкила, арила, гетероарила, арилС 1-С 6 алкила, 5- или 6-членного гетероцикла или гетероциклилС 1-С 6 алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода или серы; причем R, R' , R необязательно замещены в любом из свободных положений одной или более группами, например, от 1 до 6 групп, выбранных из галогена, нитро-, оксогрупп (=O), карбокси, циано, алкила, перфторалкила, алкенила, алкинила, циклоалкила, арила, гетероциклила, аминогрупп и их производных, таких как, например, алкиламино, диалкиламино, ариламино, диариламино, уреидо, алкилуреидо или арилуреидо; карбониламиногрупп и их производных, таких как формиламино, алкилкарбониламино, алкенилкарбониламино, арилкарбониламино, алкоксикарбониламино; гидроксигрупп и их производных, таких как, алкокси, арилокси, алкилкарбонилокси, арилкарбонилокси, циклоалкенилокси или алкилиденаминоокси; карбонильных групп и их производных, таких как алкилкарбонил, арилкарбонил, алкоксикарбонил, арилоксикарбонил, циклоалкилоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил; сульфированных производных, таких как алкилтио, арилтио, алкилсульфонил, арилсульфонил, алкилсульфинил, арилсульфинил, арилсульфонилокси, аминосульфонил, алкиламиносульфонил или диалкиламиносульфонил; или R является фталимидогруппой формулы (II), приведенной ниже и где любой R1, если присутствует, занимает положение 5 или 6 индазольного кольца и представляет группу, необязательно дополнительно замещенную, как указано выше для R' или R";m равно 0 или 1; или его фармацевтически приемлемой соли. В предпочтительном воплощении способа, описанного выше, заболевание, обусловленное и/или связанное с измененной активностью протеинкиназ, выбирают из группы, состоящей из рака, нарушений клеточной пролиферации, болезни Альцгеймера, вирусных инфекций, аутоимунных заболеваний и нейродегенеративных нарушений. Специфические типы рака, которые поддаются лечению, включают карциному, сквамозную клеточную карциному, гематопоэтические опухоли миелоидной или лимфоидной линии, опухоли мезенхимного происхождения, опухоли центральной и периферической нервной системы, меланому, семиному, тератокарциному, остеосаркому, пигментную ксеродерму, кератоксантому, тиреоидный фолликулярный рак и саркому Капоши. В другом предпочтительном воплощении способа, описанного выше, нарушение клеточной пролиферации выбирают из группы, состоящей из доброкачественной гиперплазии простаты, семейного аденоматоза, полипоза, нейрофиброматоза, псориаза, пролиферации гладкомышечных клеток, связанной с атеросклерозом, легочного фиброза, артроидного гломерулонефрита и постхирургического стеноза и рестеноза. Кроме того, объект метода данного изобретения также обеспечивает ингибирование ангиогенеза и метастазирования опухоли. Настоящее изобретение далее предлагает производное аминоиндазола, представленное формулой-NHCOOR', где R' и R" каждый независимо выбран из прямого или разветвленного C1-С 6 алкила, С 2-С 6 алкенила или -алкинила, С 3-С 6 циклоалкила или циклоалкилС 1-С 6 алкила, арила, гетероарила, арилС 1 С 6 алкила, 5- или 6-членного гетероцикла или гетероциклилС 1-С 6 алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода или серы; причем R, R', R необязательно замещены в любом из свободных положений одной или более группами, например, от 1 до 6 групп, выбранных из галогена, нитро-,оксогрупп (=O), карбокси, циано, алкила, перфторалкила, алкенила, алкинила, циклоалкила, арила, гетероциклила, аминогрупп и их производных, таких как, например, алкиламино, диалкиламино, ариламино,диариламино, уреидо, алкилуреидо или арилуреидо; карбониламиногрупп и их производных, таких как формиламино, алкилкарбониламино, алкенилкарбониламино, арилкарбониламино, алкоксикарбониламино; гидроксигрупп и их производных, таких как, алкокси, арилокси, алкилкарбонилокси, арилкарбонилокси, циклоалкенилокси или алкилиденаминоокси; карбонильных групп и их производных, таких как алкилкарбонил, арилкарбонил, алкоксикарбонил, арилоксикарбонил, циклоалкилоксикарбонил, амино-3 007430 карбонил, алкиламинокарбонил, диалкиламинокарбонил; сульфированных производных, таких как алкилтио, арилтио, алкилсульфонил, арилсульфонил, алкилсульфинил, арилсульфинил, арилсульфонилокси, аминосульфонил, алкиламиносульфонил или диалкиламиносульфонил; или R является фталимидогруппой формулы (II), приведенной ниже любой R1, если присутствует, занимает положение 5 или 6 индазольного кольца и представляет группу, необязательно дополнительно замещенную, как указано выше для R' или R";m равно 0 или 1; или его фармацевтически приемлемую соль; при условии чтоb) когда индазол замещен в позиции 5 или 6 метокси группой, тогда R отличен от 3-(N,Nдиэтиламино)пропиламина, 3-[(3-метил)морфолин-4-ил]пропиламина или 1-гидрокси-2-метил-2 пропиламина;c) 3-фталимидоиндазол исключается. Соединения формулы (I), как объект данного изобретения, могут иметь ассиметрические атомы углерода и могут, таким образом, существовать или в виде рацемической смеси, или как индивидуальные оптические изомеры. Таким образом, все возможные изомеры и их смеси, и также метаболиты и фармацевтически приемлемые биопредшественники (иначе называемые как пролекарства) соединений формулы (I), также как любой терапевтический метод лечения, их включающий, также входят в объем данного изобретения. В данном изобретении, если не указано иначе, под термином прямой или разветвленный C1-С 6 алкил понимается такая группа, как, например, метил, этил, н-пропил, изопропил, н-бутил, изобутил, вторбутил, трет-бутил, н-пентил, н-гексил и тому подобные группы. Под термином прямой или разветвленный С 2-С 6 алкенил или -алкинил понимается ненасыщенная углеводородная цепь, имеющая двойную или тройную связь, такая как, например, винил, этинил, 1 пропенил, аллил, 1- или 2-пропинил, 1-, 2- или 3-бутенил, 1-, 2- или 3-бутинил, пентенил, пентинил, гексенил, гексинил и тому подобные группы. Под термином С 3-С 6 циклоалкил подразумевается группа, такая как, например, циклопропил, циклобутил, циклопентил, или циклогексил. Под термином арил подразумевается моно-, би- или поли-карбоциклический либо гетероциклический углеводород с от 1 до 4 кольцевых групп, или конденсированных друг с другом, или присоединенных друг к другу одинарными связями, в котором по крайней мере одно из карбоциклических или гетероциклических колец является ароматическим. Не ограничивающими примерами арильных групп являются, например, группы фенила, инданила,бифенила, - или -нафтила, флуоренила, 9,10-дигидроантраценила, пиридила, пиразинила, пиримидинила, пиридазинила, индолила, имидазолила, имидазопиридила, 1,2-метилендиоксифенила, тиазолила,изотиазолила, пирролила, пирролилфенила, фурила, фенилфурила, бензотетрагидрофуранила, оксазолила, изоксазолила, пиразолила, хроменила, тиенила, бензотиенила, изоиндолинила, бензоимидазолила,тетразолила, тетразолилфенила, пирролидинилтетразолила, изоиндолинилфенила, хинолинила, изохинолинила, 2,6-дифенилпиридила, хиноксалинила, пиразинила, фенилхинолинила, бензофуразанила, 1,2,3 триазолила, 1-фенил-1,2,3-триазолила, и тому подобные группы. Под термином 5- или 6-членный гетероцикл, таким образом включая ароматические гетероциклические группы, также называемые арильными группами, далее подразумевается насыщенный или частично насыщенный 5- или 6-членный карбоцикл, в котором один или более атомов углерода заменены от 1 до 3 гетероатомами, такими как азот, кислород или сера. Примерами 5- или 6-членных гетероциклических групп, необязательно бензоконденсированных или дополнительно замещенных, являются 1,3-диоксолан, пиран, пирролидин, пирролин, имидазолидин,пиразолидин, пиразолин, пиперидин, пиперазин, морфолин, тетрагидрофуран и тому подобные группы. В соответствии с вышеуказанными значениями, определенными для R1, R' и R", любые из вышеприведенных групп могут быть дополнительно необязательно замещены в любом из свободных положений одной или более группами, например, от 1 до 6 групп, выбранных из галогена, нитро-, оксогрупп(=O), карбокси, циано, алкила, перфторалкила, алкенила, алкинила, циклоалкила, арила, гетероарила,гетероциклила, аминогрупп и их производных, таких как, например, алкиламино, диалкиламино, ариламино, диариламино, уреидо, алкилуреидо или арилуреидо; карбониламиногрупп и их производных, та-4 007430 ких как, например, формиламино, алкилкарбониламино, алкенилкарбониламино, арилкарбониламино,алкоксикарбониламино; гидроксигрупп и их производных, таких как, например, алкокси, арилокси, алкилкарбонилокси, арилкарбонилокси, циклоалкенилокси или алкилиденаминоокси; карбонильных групп и их производных, таких как, например, алкилкарбонил, арилкарбонил, алкоксикарбонил, арилоксикарбонил, циклоалкилоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил; сульфированных производных, таких как, например, алкилтио, арилтио, алкилсульфонил, арилсульфонил,алкилсульфинил, арилсульфинил, арилсульфонилокси, аминосульфонил, алкиламиносульфонил или диалкиламиносульфонил. В свою очередь, когда целесообразно, каждая из вышеуказанных групп может быть далее замещена одной или более из вышеупомянутых групп. Среди этих последних групп и если не указано противоположное в данном изобретении, под термином атом галогена подразумевается атом фтора, хлора, брома или иода. Под термином перфторалкил подразумевается прямая или разветвленная C1-С 6 алкильная группа,как определено выше, в которой более чем один атом водорода замещен атомами фтора. Примерами перфорированных алкильных групп являются, например, трифторметил, 2,2,2 трифторэтил, 1,2-дифторэтил, 1,1,1,3,3,3-гексафторпропил-2-ил и тому подобные. Из всего вышеперечисленного квалифицированному специалисту должно быть понятно, что любая группа, название которой было указано как составное название, такое как, например, циклоалкилалкил,арилалкил, гетероциклилалкил, алкокси, алкилтио, арилокси, арилалкокси, гетероциклилокси, гетероциклилалкокси, алкилкарбонилокси и тому подобные, должна пониматься обычно как составленная из частей, из которых она происходит. В качестве примера, термин гетероциклилалкил характеризуется алкильной группой, которая дополнительно замещена гетероциклильной группой, как определено выше. Фармацевтически приемлемые соли соединений формулы (I) представляют собой соли присоединения неорганической или органической кислоты, например азотной, хлористоводородной, бромистоводородной, серной, хлорной, фосфорной, уксусной, трифторуксусной, пропионовой, гликолевой, молочной,щавелевой, малоновой, яблочной, малеиновой, виннокаменной, лимонной, бензойной, коричной, миндальной, метансульфоновой, изатиновой и салициловой кислот, также как и соли с неорганическими или органическими основаниями, например, щелочных или щелочно-земельных металлов, особенно гидроксиды, карбонаты или бикарбонаты натрия, кальция или магния, ациклические или циклические амины,предпочтительнее метиламин, этиламин, диэтиламин, триэтиламин или пиперидин. Из всего вышесказанного квалифицированному специалисту должно быть ясно, что в соединениях формулы (I), где m равно 0, -OR1 группы отсутствуют и, следовательно, не существует R1 групп, связанных с индазольным скелетом через атом кислорода. В таком случае, следовательно, положения 5 или 6 в соответствии с описанной ниже системой нумерации являются незамещенными (или замещенными водородом). С другой стороны, когда m равно 1, одна -OR1 группа (т.е. R1) присутствует в любой из позиций 5 или 6 индазольного кольца. Более предпочтительными, в пределах этого класса, являются соединения, где m равно 1 и R1 занимает любое из положений 5 или 6 индазольного кольца. Еще более предпочтительными являются соединения, где R1, R' и R" выбраны, каждый независимо,из С 2-С 6 алкенила, С 3-С 6 алкинила, арила, гетероарила, арилС 1-С 6 алкила, 5- или 7-членного гетероциклила или гетероциклилС 1-С 6 алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода или серы. Другой класс предпочтительных соединений данного изобретения представлен соединениями формулы (I), где R является -NHCOR' группой и R', R1 и m являются такими, как определено выше. Более предпочтительными, в пределах этого класса, являются соединения, где m равно 1 и R1 занимает любое из положений 5 или 6 индазольного кольца. Еще более предпочтительными являются соединения, где R1 и R' выбраны, каждый независимо, изC1-С 6 алкила, С 3-С 6 циклоалкила или циклоалкилС 1-С 6 алкила, арила, гетероарила, арилС 1-С 6 алкила, 5- или 7-членного гетероциклила или гетероциклилС 1-С 6 алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода, серы. Другой класс предпочтительных соединений данного изобретения представлен соединениями формулы (I), где R является -NHCONHR1 или -NHCONR'R" группой, и R', R", R1 и m являются такими, как определено выше. Более предпочтительными, в пределах этого класса, являются соединения, где m равно 1 и R1 занимает любое из положений 5 или 6 индазольного кольца.-5 007430 Еще более предпочтительными являются соединения, где R1, R' и R" выбраны, каждый независимо,из C1-С 6 алкила, С 3-С 6 циклоалкила или циклоалкилС 1-С 6 алкила, арила, гетероарила, арилС 1-С 6 алкила, 5 или 7-членного гетероциклила или гетероциклилС 1-С 6 алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода и серы. Другой класс предпочтительных соединений данного изобретения представлен соединениями формулы (I), где R является -NHSO2R' группой, и R', R1 и m являются таковыми, как определено выше. Более предпочтительными, в пределах этого класса, являются соединения, где m равно 1 и R1 занимает любое из положений 5 или 6 индазольного кольца. Еще более предпочтительными являются соединения, где R1 и R' выбраны, каждый независимо, изC1-С 6 алкила, С 3 С 6 циклоалкила или циклоалкилС 1-С 6 алкила, арила, гетероарила, арилС 1-С 6 алкила, 5- или 7-членного гетероциклила или гетероциклилС 1-С 6 алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода и серы. Другой класс предпочтительных соединений данного изобретения представлен соединениями формулы (I), где R является -NHCOOR' группой, и R', R1 и m являются такими, как определено выше. Более предпочтительными в пределах этого класса, являются соединения, где m равно 1 и R1 занимает любое из положений 5 или 6 индазольного кольца. Еще более предпочтительными являются соединения, где R1 и R' выбраны, каждый независимо, изC1-С 6 алкила, С 3-С 6 циклоалкила или циклоалкилС 1-С 6 алкила, арила, гетероарила, арилС 1-С 6 алкила, 5- или 7-членного гетероциклила или гетероциклилС 1-С 6 алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода и серы. Другой класс предпочтительных соединений данного изобретения представлен соединениями формулы (I), где R является фталимидогруппой формулы (II), и R1 и m являются таковыми, как определено выше. Более предпочтительными, в пределах этого класса, являются соединения, где m равно 1 и R1 занимает любое из положений 5 или 6 индазольного кольца. Еще более предпочтительными являются соединения, где R1 выбран из С 2-С 6 алкенила, С 3-С 6 алкинила, арила, гетероарила, арилС 1-С 6 алкила, 5- или 7-членного гетероциклила или гетероциклилС 1-С 6 алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода или серы. Конкретные примеры соединений формулы (I), необязательно в форме фармацевтически приемлемых солей, описаны в экспериментальной части. Как указано выше, следующим объектом настоящего изобретения является способ получения производных аминоиндазола формулы (I). Следовательно, соединения формулы (I) и их фармацевтически приемлемые соли, где R является таким, как определено выше, но отличным от фталимидогруппы формулы (II), могут быть получены способом, включающим: а) взаимодействие в кислых условиях производного 2-аминобензонитрила формулы (III) где m является таким, как определено выше и, если присутствует, R представляет собой метильную или бензильную группу, с нитритом натрия в присутствии хлорида олова, с получением соединения формулы (IV);b) взаимодействие соединения формулы (IV) с фталевым ангидридом с получением соединения формулы (V);-6 007430 с) взаимодействие соединения формулы (IV) с подходящим агентом, гидролизующим эфир, с получением соответствующего гидрокси производного формулы (VI);d) взаимодействие соединения формулы (VI) с подходящим силилирующим агентом (Riv)3SiZ, где каждый из Riv одинаковые или различные, и являются прямой или разветвленной С 1-С 4 алкильной группой, и Z представляет собой атом галогена, с получением соединения формулы (VII); е) взаимодействие соединения формулы (VII) с подходящим агентом, защищающим азот индазола или, альтернативно, связыванием его с подходящей подложкой из полимерной смолы, с получением соединения формулы (VIII) где Q является указанной выше защитной группой или представляет собой подложку из смолы;f) взаимодействие соединения формулы (VIII) с гидразин моногидратом, с получением соединения формулы (IX); и реакцию соединения формулы (IX) на любой из следующих стадий g.1) или g.2);g.1) с подходящим реагентом формулы R'-Z (X), R'-COZ (XI), R'-NCO (XII), R'-SO2Z (XIII) или R'OCOZ (XIV), где R' является таким, как определено выше, и Z представлен атомом галогена или подходящей уходящей группой, с тем чтобы получить соединение формулы (XV)(XVI) где R" и Z являются такими, как определено выше, с получением соединения формулы (XV), где R представляет собой -NR'R" или -NHCONR'R" группу;-7 007430 где R' и R" также являются таковыми, как определено выше, в присутствии 4 нитрофенилхлорформиата, с получением соответствующего соединения формулы (XV), где R являетсяh) взаимодействие любых из вышеуказанных соединений формулы (XV) с тетрабутиламмонийфторидом, с получением соединения формулы (XVIII) взаимодействие соединения формулы (XVIII) с производным формулыR1-Z где R1 является таким, как определено выше, и Z является атомом галогена, подходящей уходящей группой или гидрокси группой, с получением соединения формулы (XX);j) удаление защитной группы в соединении формулы (XX) или, альтернативно, отщепление полимерной смолы, с тем чтобы получить желаемое соединение формулы (I) и, если требуется, превращением его в другое соединение формулы (I) и/или в его фармацевтически приемлемую соль. Из всего вышесказанного квалифицированному специалисту в этой области должно быть понятно,что если соединение формулы (I), полученное согласно выше описанному процессу, получено в виде смеси изомеров, их разделение на отдельные изомеры формулы (I), проведенное в соответствии со стандартными методиками, также находится в рамках настоящего изобретения. Аналогично, превращение в свободное соединение (I) его соответствующей соли, в соответствии с хорошо известными методами в данной области, входит в объем настоящего изобретения. Что касается стадии а) способа получения, соединение формулы (III) предпочтительно подвергать взаимодействию с нитритом натрия 2-амино-4-метоксибензонитрилом или 2-амино-5-бензилоксибензонитрилом. Соль диазония восстанавливают в присутствии хлорида олова в кислых условиях, например, в хлористоводородной или серной кислоте. Реакция может быть проведена в смеси воды и подходящего растворителя, такого как, например,метанол, этанол, и тому подобного, при значении температуры в интервале от около 0 С до около 10 С. Реакция может быть осуществлена добавлением нитрита натрия к раствору соединения формулы(III) в концентрированной хлористо-водородной кислоте, в то время как перемешивание продолжают от 1 до 3 ч. Далее суспензия может быть по каплям перенесена в раствор хлорида олова в концентрированной хлористоводородной кислоте и охлаждена при примерно 0 С, в то время как перемешивание продолжают в течение подходящего периода времени, например, от примерно 4 до примерно 6 ч. Что касается стадии b) способа получения, соединение формулы (IV) подвергают взаимодействию с фталевым ангидридом в соответствии со стандартными методами получения фталимидопроизводных. Реакция может быть проведена во множестве растворителей, включая хлороформ, ацетонитрил, диоксан,тетрагидрофуран, диметилформамид, диметилацетамид и тому подобные; предпочтительно в ацетонитриле. Таким образом, фталевый ангидрид добавляется к раствору соединения формулы (V). Температура затем доводится до нужного значения, например, от примерно 70 С до примерно 100 С, предпочтительно до 80 С. Перемешивание продолжают в течение требуемого периода времени, который варьирует от примерно 1 до примерно 4 ч. Что касается стадии с) способа получения, соединение формулы (V) превращают в соответствующее гидроксипроизводное посредством взаимодействия с подходящим агентом, гидролизующим эфир,таким как, например, соль гидрохлорида пиридиния, иодотриметилсилан или трибромид бора. Реакция может быть проведена в свободном от примесей хлориде пиридиния или, с другими реагентами, в дихлорметане или хлороформе. Предпочтительно использовать свободный от примесей хлорид пиридиния. Соответственно, смесь хлорида пиридиния и соединения формулы (V) доводят до приемлемой температуры в интервале значений от примерно 180 С до примерно 200 С, в то время как перемешивание проводят в течение периода времени от примерно 1 до примерно 3 ч. Что касается стадии d) способа получения, соединение формулы (VI) подвергают взаимодействию с силильным производным, предпочтительно с трет-бутилдиметилсилилхлоридом (TBDMSCl), с тем что-8 007430 бы получить соответствующее производное силилового эфира. Реакция может быть проведена в присутствии подходящего основания, такого как, например, 1,5-диазабицикло[4.3.0]нон-5-ен(DBN) или, более предпочтительно, 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU). В связи с этим, трет-бутилдиметилсилилхлорид (TBDMSCl) добавляют к раствору соединения формулы (VI). Реакция может быть проведена во множестве растворителей, таких как дихлорметан, ацетонитрил, диметилформамид, и тому подобное; дихлорметан предпочтителен. Температура может варьировать от примерно 20 С до примерно 40 С в то время как продолжают перемешивание в течение времени от примерно 1 до 4 ч. Что касается стадии е) способа получения, индазольное производное формулы (VII), получаемое таким образом, или защищено по индазольному атому азота, или, альтернативно, связано с подходящей полимерной смолой. Реакция введения защитной группы может быть проведена в соответствии со стандартными методиками, хорошо известными в данной области, например, с использованием подходящих защитных групп азота, таких как, например, трет-бутилоксикарбонильная группа (ВОС). В этом же положении, в качестве альтернативы, индазол формулы (VII) может быть обычным методом сшит с инертным полимерным носителем, таким как, например, 2-хлортритилхлоридная смола,тритилхлоридная смола, паранитрофенилкарбонатная смола Wang или бром-(4-метоксифенил)полистирол, которые все обычно используются в данной области. Понятно, что этот же путь особенно удобен преимущественно для получения соединений формулы(I) в условиях твердофазного синтеза (SPS), которые типично адаптированы для составления библиотек соединений, относящихся к методикам комбинаторной химии, например, как описано ниже. Реакцию со смолой проводят в присутствии незначительного избытка подходящего основания, например амина, например, диизопропилэтиламина (DIPEA), триэтиламина (TEA), 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU), или 2-трет-бутиламино-2-диэтиламино-1,3-диметилпергидро-1,3,2-диазафосфорина, в подходящем растворителе, например, дихлорметане, хлороформе, тетрагидрофуране, диметилформамиде, диметилацетамиде и тому подобном. Предпочтительно, реакцию проводят в дихлорметане при температуре около 20 С. Реакция может быть осуществлена путем добавления к суспензии смолы, основания и соединения формулы (VII), и путем перемешивания при температуре около 20 С в течение необходимого времени,например, до 24 ч. Что касается стадии f) способа получения, производное формулы (VIII) обрабатывают моногидратом гидразина, с тем чтобы отщепить фталимидогруппу. Реакцию предпочтительно проводят с использованием большого избытка, например, более чем с 10 эквивалентами гидразин гидрата или моногидрата, в присутствии подходящих растворителей, таких как,например, галогенированные углеводороды, низшие спирты и их смеси. Предпочитительными растворителями являются дихлорметан, этанол и их смеси. Реакция может быть проведена путем добавления гидразина к раствору соединения формулы (VIII) и перемешивания в течение необходимого времени при температуре, варьирующей от 20 С до примерно 45 С. Предпочтительно продолжать перемешивание реакционной смеси при температуре около 40 С примерно 16 ч. Что касается любой из стадий g.l) или g. 2) способа получения, аминопроизводное формулы (IX) подвергают взаимодействию с подходящим реагентом формул с (X) по (XIV), или с соединением формулы (XVII), в соответствии с хорошо известными методами. Обычно, соединение формулы (IX) может быть подвергнуто взаимодействию: с соединением формулы (XI), чтобы получить соответствующее -NHCOR' ацильное производное; соединением формулы(XII), чтобы получить соответствующее -NHCONHR' уреидопроизводное; соединением формулы (XIII),чтобы получить соответствующее -NHSO2R' производное; соединением формулы (XIV), чтобы получить соответствующее -NHCOOR' производное. Альтернативно, соединение формулы (IX) может быть подвергнуто взаимодействию с соединением формулы R'R"NH (XVII) в присутствии 4-нитрофенилхлорформиата, чтобы получить соответствующее уреидо -NHCONR'R" производное. Любую из вышеперечисленных реакций проводят в соответствии со стандартными методами,обычно используемыми при получении функционализированных аминопроизводных, начиная с соответствующего амина. Предпочтительно, в рамках определения соединений формулы (X), Z представляет собой подходящую уходящую группу, например, йодной, бромной или бороновой кислоты, в рамках определения соединений формулы (XI), (XIII) или (XIV), Z представляет собой атом галогена и, даже более предпочтительно, атом хлора. Помимо указанного выше, квалифицированному специалисту должно быть ясно, что, если требуется, любое из вышеуказанных соединений формулы (XV), полученных таким образом, и где R представляет собой группу -NHCONHR', может быть далее превращено в соответствующее производное, имеющее в качестве R группу -NHCONR'R", соответственно.-9 007430 Эти реакции также проводятся в соответствии с обычными методами путем взаимодействия соответствующего промежуточного соединения формулы (XV) с подходящим производным формулы (XVI). В этом случае, соединение формулы (IX) растворяют в подходящем растворителе, таком как дихлорметан, диметилформамид, тетрагидрофуран, диоксан и тому подобном, и добавляется подходящее основание, такое как триэтиламин, диизопропилэтиламин, карбонат натрия и тому подобное. Затем добавляют соединение общей формулы (XI), (XIII) или (XIV), и смесь перемешивается в течение примерно от 2 до 15 ч при температуре, варьирующей от примерно 20 С до примерно 80 С. При использовании изоцианата общей формулы (XII) условия реакции такие, как указано выше, за исключением того, что добавление основания может быть необязательным. Во всех указанных реакциях может быть необязательно использован подходящий катализатор, такой как диметиламинопиридин. По-существу, аналогичные методики могут быть применены, когда соединение формулы (XII) подвергают взаимодействию с соединением формулы (X) с получением соответствующего функционального аминопроизводного формулы (XIV), в соответствии с хорошо известными методами. В качестве примера, соединение формулы (IX), действуя в соответствии с обычными методами,может быть подвергнуто взаимодействию с производным формулы (X), где Z является атомом галогена,например, иода или брома, и R' является арилалкильной группой, такой как, например, бензил. Альтернативно, соединение формулы (IX) может быть подвергнуто взаимодействию с производным формулы (X), где Z является атомом брома и R' является арильной группой, в присутствии палладиевого катализатора, такого как, например, трис(дибензилиденацетон)дипалладий, ацетат палладия или 1,1'бис(дифенилфосфино)ферроцендихлорпалладий, путем добавления подходящего основания, например,трет-бутоксида калия, карбоната цезия и тому подобного, и палладиевого лиганда, такого как 2,2'-бис(дифенилфосфино)-1,1'-бинафтил, три-о-толилфосфин, три-н-бутилфосфин, три-трет-бутилфосфин и тому подобного, для получения соответствующего производного формулы (XV). В этом случае, соединение формулы (IX) суспендируют в подходящем безводном растворителе, таком как толуол, N-метил-2-пирролидон, диметоксиэтан, диоксан и тому подобном, и добавляют соединение формулы (X), катализатор, основание и лиганд. Суспензию затем помещают в подходящие температурные условия примерно от 50 С до примерно 100 С, в то время как перемешивание продолжают в течение времени от 8 до 5 ч. Реакцию проводят в инертной атмосфере. В соответствии со стадией h) получения, соединение формулы (XV) подвергают взаимодействию с тетрабутиламмонийфторидом для того, чтобы получить соответствующее гидроксипроизводное формулы (XVIII). Соединение формулы (XV) может быть таким образом суспендировано в безводном растворителе, таком как диоксан, тетрагидрофуран или подобном, и добавляют раствор тетрабутиаммоний фторида в подходящем растворителе. Раствор перемешивают примерно от 2 ч до примерно 16 ч при температуре, варьирующей от примерно 20 С до примерно 50 С. Продукт формулы (XVIII), полученный таким образом, может быть далее подвергнут взаимодействию в соответствии со стадией i) получения, с подходящим производным формулы (XIX). В частности, реакцию с соединением формулы (XIX), где Z является атомом галогена, таким как бром или хлор, или подходящей уходящей группой, проводят в присутствии основания, такого как, например, гидроксид натрия, гидрид натрия, 2-трет-бутиламино-2-диэтиламино-1,3-диметилпергидро 1,3,2-диазафосфорин или, более предпочтительно, карбонат цезия, для получения соответствующего эфирного производного формулы (XX). В этом случае, соединение формулы (XVIII) суспендируют в подходящем растворителе, таком как диметилацетамид, тетрагидрофуран, диоксан, или более предпочтительно диметилформамид, и добавляют основание. Смесь перемешивают от примерно 5 ч до примерно 36 ч при температуре, варьирующей от примерно 20 С до примерно 80 С. Альтернативно, эти же соединения формулы (XX) могут быть получены взаимодействием производного формулы (XVIII) с соединением формулы (XIX), где Z является гидроксилом, в оперативных условиях Митсунобу (Mitsunobu), например, в присутствии трифенилфосфина и диизопропилазодикарбоксилата. В этом случае, трифенилфосфин, диизопропилазодикарбоксилат и соединение общей формулы(XIX) растворяют в подходящем растворителе, таком как тетрагидрофуран, диоксан и подобном, и раствор переносят в смесь соединения формулы (XVIII), растворенного в подходящем растворителе, таком как тетрагидрофуран, диоксан и подобном, в присутствии подходящего основания, такого как триэтиламин или диизопропилэтиламин. Смесь перемешивают в течение времени, варьрующего от примерно 2 ч до примерно 15 ч при температуре, варьирующей от 0 до 20 С. В конечном итоге, в соответствии со стадией j) получения, с соединения формулы (XX) снимается защита индазольного атома азота обработкой, в соответствии с обычным методом, в кислых условиях. Соединение формулы (XX) суспендируют в подходящем растворителе, таком как метиловый спирт, этиловый спирт и подобном, и добавляют концентрированный раствор хлористо-водородной кислоты. Смесь перемешивают в течение необходимого времени от примерно 5 ч до примерно 15 ч при темпера- 10007430 туре, варьирующей от примерно 20 С до примерно 40 С, предпочтительно при около 20 С. Альтернативно, это же промежуточное соединение формулы (XX) отщепляется от смолы, с которой оно связано. Отщепление смолы может быть проведено, например, в присутствии трифторуксусной кислоты,чтобы получить желаемое соединение формулы (I). Смолу суспендируют в растворе смеси 5-95% трифторуксусной кислоты в дихлорметане, и смесь перемешивают при примерно 20 С в течение времени,варьирующего от примерно 5 мин до примерно 3 ч. Из всего указанного выше, квалифицированному специалисту должно быть ясно, что соединения формулы (I), где R1 и m являются такими, как определено выше, и R представляет собой фталимидогруппу формулы (II), и их фармацевтически приемлемые соли могут быть получены в соответствии с аналогичным способом путем взаимодействия соединения формулы (VIII) по стадиям h) , i) и j) способа получения, чтобы получить желаемое производное формулы (I), несущее фталимидогруппу (II) вместо R группы. Предпочтительно, при получении соединений формулы (I), где R является сульфонамидогруппой(-NHSO2R') , приведенный выше синтетический путь может быть удобным образом модифицирован изменением порядка стадий снятия защиты. В частности, соединения формулы (I), где R является -NHSO2R' группой, могут быть предпочтительно получены взаимодействием промежуточных производных формулы (VIII), полученных в соответствии со стадией (е) способа получения, с тетрабутиламмонийфторидом как указано на стадии h) способа получения, с получением соединения формулы (XVIII), где R является фталимидогруппой. Таким образом полученные соединения формулы (XVIII) затем подвергаются взаимодействию с производным формулы (XIX) в соответствии со стадией (i) способа получения, с получением соединения формулы (XX), где R является фталимидогруппой. Указанные выше соединения формулы (XX) затем подвергаются взаимодействию с гидразин моногидратом, в соответствии со стадией (f) способа получения, с получением соединения формулы (XX), гдеR является -NH2. В конечном итоге, указанные выше соединения формулы (XX) затем подвергаются взаимодействию с подходящим производным формулы (XIII) в соответствии со стадией (g.1) способа получения, с получением соответствующих сульфонамидопроизводных формулы (XX), где R представляет собой заданную NHSO2R' группу, которые далее подвергают снятию защиты или отщепляют от смолы в соответствии со стадией (j) способа получения. При получении соединений формулы (I) в соответствии с любым вариантом способа получения, которые все входят в объем данного изобретения, необязательные функциональные группы обоих исходных веществ, как реагента, так и его промежуточного соединения, которые могут дать начало нежелательным побочным реакциям, должны быть соответствующим образом защищены в соответствии с обычными методиками. Более того, превращение этих последних соединений в соединения со снятой защитой может быть проведено в соответствии с известными методиками. Фармацевтически приемлемые соли соединений формулы (I) или, альтернативно, свободные от солей соединения, могут быть получены в соответствии с обычными методами. Соединения формулы (III) известны или просто получаются в соответствии с известными методами. В качестве примера, 2-амино-4-метоксибензонитрил может быть получен, следуя описанному в ЕР-А 257583, на имя ShionogiCo; 2-амино-5-бензилоксибензонитрил может быть получен, как описано в J.Heterocycl. Chem. (1972), 9(4), 759-73. Если это соединение коммерчески доступно само по себе, все соединения формулы (X), (XI), (XII),(XIII), (XIV), (XVI), (XVII) и (XIX) известны или легко получаются в соответствии с хорошо известными методами. Аналогично, любой реагент настоящего способа получения, включая силильное производное(Riv)3SiZ, а также полимерную смолу, являются коммерчески доступными или легко получаемыми из коммерчески доступных материалов. Как указано выше, соединения формулы (I) данного изобретения обычно получают в соответствии с методиками комбинаторной химии, широко известными в данной области, путем серийного проведения упомянутых выше реакций между несколькими промежуточными продуктами и работе в SPS условиях(условиях твердофазного синтеза). Все предпочтительные соединения изобретения, когда это удобно в форме их фармацевтически приемлемых солей, указаны здесь и определены как продукты, полученные определенным способом, то есть продукты формулы (I), которые могут быть получены, например, указанным способом. Таким образом, предлагаются новые соединения изобретения и их фармацевтически приемлемые соли, которые могут быть получены, например, с использованием методики приведенного выше способа получения, путем первоначального взаимодействия соединения формулы (IХа) с каждым из соединений формулы (X), как указано выше в таблице I, с получением множества соединений формулы (XVa) с последующим взаимодействием каждого из производных формулы (XVa) с тетрабутиламмонийфторидом, как указано на стадии h) способа получения, и затем с каждым из производных формулы (XIX),как указано в таблицах II или III, и проведением последующих операций в соответствии со стадией j) способа получения. Также предлагаются новые соединения данного изобретения и их фармацевтически приемлемые соли, которые можно получить, например, методом комбинаторной химии, как в вышеприведенном способе, путем первоначального взаимодействия соединения формулы (IXb) с каждым из соединений формулы (X), как указано выше в таблице I, с получением множества соединений формулы (XVb) с последующим взаимодействием каждого из производных формулы (XVb) с тетрабутиламмонийфторидом, как указано на стадии h) способа получения, и затем с каждым из производных формулы (XIX),как указано в таблицах II или III, и проведением последующих операций как указано на стадии j) способа получения. Также предлагаются новые соединения данного изобретения и их фармацевтически приемлемые соли, которые можно получить, например, методом комбинаторной химии, как в вышеприведенном способе, путем первоначального взаимодействия соединения формулы (IХа) с каждым из соединений формулы (XI), как указано выше в таблице IV, с получением множества соединений формулы (XVc) с последующим взаимодействием каждого из производных формулы (XVc) с тетрабутиламмонийфторидом, как указано на стадии h) способа получения, и затем с каждым из производных формулы (XIX),как указано в таблицах II или III, и проведением последующих операций, как указано на стадии j) способа получения. Также предлагаются новые соединения данного изобретения и их фармацевтически приемлемые соли, которые можно получить, например, методом комбинаторной химии, как в вышеприведенном способе, путем первоначального взаимодействия соединения формулы (IXb) с каждым из соединений формулы (XI), как указано выше в таблице IV, с получением множества соединений формулы (XVd) с последующим взаимодействием каждого из производных формулы (XVd) с тетрабутиламмонийфторидом, как указано на стадии h) способа получения, и затем с каждым из производных формулы (XIX),как указано в таблицах II или III, и проведением последующих операций, как указано на стадии j) способа получения. Также предлагаются новые соединения данного изобретения и их фармацевтически приемлемые соли, которые можно получить, например, методом комбинаторной химии, как в выше приведенном способе, путем первоначального взаимодействия соединения формулы (IХа) с каждым из соединений формулы (XII), как указано выше в таблице V, с получением множества соединений формулы (XVe) с последующим взаимодействием каждого из производных формулы (XVe) с тетрабутиламмонийфторидом, как указано на стадии h) способа получения, и затем с каждым из производных формулы (XIX),как указано в таблицах II или III, и проведением последующих операций, как указано на стадии j) способа получения. Также предлагаются новые соединения данного изобретения и их фармацевтически приемлемые соли, которые можно получить, например, методом комбинаторной химии, как в выше приведенном способе, путем первоначального взаимодействия соединения формулы (IXb) с каждым из соединений формулы (XII), как указано выше в таблице V, с получением множества соединений формулы (XVf) с последующим взаимодействием каждого из производных формулы (XVf) с тетрабутиламмонийфторидом, как указано на стадии h) способа получения, и затем с каждым из производных формулы (XIX),как указано в таблицах II или III, и проведением последующих операций, как указано на стадии j) способа получения.- 13007430 Также предлагаются новые соединения данного изобретения и их фармацевтически приемлемые соли, которые можно получить, например, методом комбинаторной химии, как в выше приведенном способе, путем первоначального взаимодействия соединения формулы (IХа) с каждым из соединений формулы (XIII), как указано выше в таблице VI, с получением множества соединений формулы (XVg) с последующим взаимодействием каждого из производных формулы (XVg) с тетрабутиламмонийфторидом, как указано на стадии h) способа получения, и затем с каждым из производных формулы (XIX),как указано в таблицах II или III, и проведением последующих операций, как указано на стадии j) способа получения. Также предлагаются новые соединения данного изобретения и их фармацевтически приемлемые соли, которые можно получить, например, методом комбинаторной химии, как в выше приведенном способе, путем первоначального взаимодействия соединения формулы (IXb) С каждым из соединений формулы (XIII), как указано выше в таблице VI, с получением множества соединений формулы (XVh) с последующим взаимодействием каждого из производных формулы (XVg) с тетрабутиламмонийфторидом, как указано на стадии h) способа получения, и затем с каждым из производных формулы (XIX),как указано в таблицах II или III, и проведением последующих операций, как указано на стадии j) способа получения. Таблица I. Соединения формулы R'-Z (X)- 18007430 Таким образом, еще одним объектом данного изобретения является библиотека двух или более производных аминоиндазола, представленных формулой (I)-NHCOOR', где R' и R" каждый независимо выбран из прямого или разветвленного C1-С 6 алкила, С 2-С 6 алкенила или -алкинила, С 3-С 6 циклоалкила или циклоалкилС 1-С 6 алкила, арила, гетероарила, арилС 1-С 6 алкила, 5- или 6-членного гетероцикла или гетероциклилС 1-С 6 алкила с от 1 до 3 гетероатомами, выбранными из азота, кислорода или серы; причем R, R', R необязательно замещены в любом из свободных положений одной или более группами, например, от 1 до 6 групп, выбранных из галогена, нитро-, оксогрупп (=O), карбокси, циано, алкила, перфторалкила, алкенила, алкинила, циклоалкила, арила, гетероциклила, аминогрупп и их производных, таких как, например, алкиламино, диалкиламино, ариламино, диариламино, уреидо, алкилуреидо или арилуреидо; карбониламиногрупп и их производных, таких как формиламино, алкилкарбониламино, алкенилкарбониламино, арилкарбониламино, алкоксикарбониламино; гидроксигрупп и их производных, таких как, алкокси, арилокси, алкилкарбонилокси, арилкарбонилокси, циклоалкенилокси или алкилиденаминоокси; карбонильных групп и их производных, таких как алкилкарбонил, арилкарбонил, алкоксикарбонил, арилоксикарбонил, циклоалкилоксикарбонил, аминокарбонил, алкиламинокарбонил, диалкиламинокарбонил; сульфированных производных, таких как алкилтио, арилтио, алкилсульфонил, арилсульфонил, алкилсульфинил, арилсульфинил, арилсульфонилокси, аминосульфонил, алкиламиносульфонил или диалкиламиносульфонил; или R является фталимидогруппой (II), приведенной ниже где любой R1, если присутствует, занимает положение 5 или 6 индазольного кольца и представляет группу, необязательно дополнительно замещенную, как указано выше для R' или R";m равно 0 или 1; или его фармацевтически приемлемой соли. Из всего выше сказанного специалисту должно быть понятно, что если таким образом получена библиотека производных индазола, например, состоящая из нескольких тысяч соединений формулы (I),она может быть использована с большими преимуществами для скрининга в отношении имеющихся киназ, как было описано ранее. Смотри в качестве общей ссылки: библиотеки соединений и их использование в качестве инструментов для скрининга биологических активностей, J. Med. Chem. 1999, 42, 2373-2382; и Bioorg. Med.Chem. Lett. 10 (2000), 223-226. Фармакология Соединения формулы (I) активны в качестве ингибиторов протеинкиназ и, таким образом, применимы, например, для предотвращения неконтролируемой пролиферации опухолевых клеток. В терапии они могут использоваться в лечении различных опухолей, таких как, например, карцином, например карциномы молочных желез, легочной карциномы, карциномы мочевого пузыря, карциномы прямой кишки, опухолей яичников и эндометрия, сарком, например, сарком мягкой и костной ткани, и гематологических злокачественных заболеваний, например, лейкозов. Дополнительно, соединения формулы (I) также применимы при лечении других клеточных пролиферативных нарушений, таких как псориаз, пролиферация гладкомышечных клеток, связанная с атеросклерозом и постхирургическим стенозом и рестенозом и при лечении болезни Альцгеймера. Ингибирующая активность предполагаемых cdk/циклин ингибиторов и активность выбранных соединений была определена методом анализа, основанным на использовании SPA технологии (AmershamPharmacia Biotech). Данный анализ включает в себя перенос киназой радиоактивно меченного фосфатного остатка на биотинилированный субстрат. Результирующему 33 Р-меченному биотинилированному продукту позволяют связаться с SPA-гранулами, покрытыми стрептавидином (емкость биотина 130 пмоль/мг), и световое излучение измеряют сцинтилляционным счетчиком.- 19007430 Анализ ингибирующей активности cdk2/Cyclin A Киназная активность: 4 мкМ в домашнем субстрате биотинилированного гистона H1 (SigmaН 5505), 10 мкМ АТФ (0,1 микроКи Р 33-АТФ), 4,2 нг комплекса Cyclin A/CDK2, ингибитора в конечном объеме 30 мкл буфера (Трис НСl 10 мМ рН 7,5, MgCl2 10 мМ, ДТТ 7,5 мМ + 0,2 мг/мл БСА) были добавлены в каждую лунку 96-луночного планшета с U-образным дном. После 30 мин инкубации при комнатной температуре реакция была остановлена смесью 100 мкл PBS + 32 мМ ЭДТА + 0,1% Триton Х-100 + 500 мкл АТФ, содержащей 1 мг SPA-гранул. Затем объем, равный 110 мкл, переносят в планшете Optiplate. После 20 мин инкубации для поглощения субстрата, добавляют 100 мкл 5 М CsCl для того, чтобы позволить гранулам образовать слой на поверхности планшета, и оставляют на 4 ч до того, как будет посчитана радиоактивность в считывающем устройстве. Определение IC50: ингибиторы были протестированы при различных концентрациях, варьирующих от 0,0015 до 10 мкМ. Экспериментальные данные были проанализированы с помощью компьютерной программы GraphPad Prizm с использованием уравнения логистики с четырьмя параметрами: у = нижняя точка+(верхняя точка)/(1+10logIc50-x)угол наклона кривой,где х - логарифм концентрации ингибитора, а у - ответ; у начинается с нижней точки и идет вверх сигмовидной кривой. Подсчет Ki: Экспериментальный метод: реакцию проводили в буфере (10 мМ Трис, рН 7,5, 10 мМ MgCl2, 0,2 мг/мл БСА, 7,5 мМ ДТТ), содержащем 3,7 нМ фермента, гистона и АТФ (постоянный уровень охлажденного/меченного АТФ 1/3000). Реакция была остановлена с помощью ЭДТА, и данный субстрат был адсорбирован на фосфомембране (Multiscreen 96-луночные планшеты от Millipore). После интенсивной промывки планшеты multiscreen считываются на поверхностном счетчике. Контроль (нулевое время) был измерен для каждой концентрации АТФ и гистона. Схема эксперимента: скорости реакции были измерены при четырех различных концентрациях АТФ, субстрата (гистона) и ингибитора. 80-точечная матрица концентраций была разработана в области значений соответственно Km АТФ и субстрата, и значений IC50 для ингибитора (0,3, 1, 3, 9-кратные значения данной Km или IC50). Предварительный эксперимент в отсутствие ингибитора и при различных концентрациях АТФ и субстрата позволяет определить единую конечную временную точку (10 мин) в линейном ряду реакции для эксперимента определения Ki. Оценки кинетических параметров: кинетические параметры были оценены методом одновременной нелинейной регрессии наименьших квадратов с использованием [Уравн.1] (конкурирующий ингибитор относительно АТФ, случайный механизм) с использованием полного набора данных (80 точек): где А=[АТФ], В=[субстрат], I=[ингибитор], Vm= максимальная скорость, Ка, Kb, Ki - константы диссоциации АТФ, субстрата и ингибитора, соответственно,и- фактор кооперативности между субстратом и связыванием АТФ и субстратом и связыванием ингибитора, соответственно. Дополнительно, выбранные соединения были охарактеризованы выборкой ser/threo киназ, жестко взаимосвязанных с клеточным циклом (cdk2/cyclin E, cdkl/cyclin B1, cdk5/p25, cdk4/cyclin D1), и также в отношении специфичности МАРК, РКА, EGFR, IGF1-R, и Aurora-2. Анализ ингибирующей активности cdk2/Cyclin E Киназная реакция: 10 мкМ в домашнем субстрате биотинилированного гистона H1 (SigmaН 5505), 30 мкМ АТФ (0,3 микроКи Р 33-АТФ), 4 нг GST-Cyclin E/CDK2 комплекса, ингибитор в конечном объеме 30 мкл буфера (ТРИС HCl 10 мМ, рН 7,5, MgCl2 10 мМ, ДТТ 7,5 мМ + 0,2 мг/мл БСА) были добавлены в каждую лунку 96-луночного планшета с U-образным дном. После 60 мин инкубации при комнатной температуре, реакция была остановлена с помощью 100 мкл PBS + 32 мМ ЭДТА + 0,1% Триton Х-100 + 500 мкМ АТФ, содержащего 1 мг SPA-гранул. Затем объем, равный 110 мкл, переносят в планшет Optiplate. После 20 мин инкубации для поглощения субстрата, было добавлено 100 мкл 5 М CsCl, чтобы позволить гранулам образовать слой на поверхности планшета, и выдерживали 4 ч до подсчета радиоактивности в Top-Count считывающем устройстве. Определение IC50: смотри выше. Анализ ингибирующей активности cdkl/Cyclin Bl Киназная реакция: 4 мкМ в домашнем субстрате биотинилированного гистона H1 (SigmaН-5505),20 мкМ АТФ (0,2 микроKи Р 33-АТФ), 3 нг Cyclin B/CDK1 комплекса, ингибитор в конечном объеме 30 мкл буфера (ТРИС HCl 10 мМ, рН 7,5, MgCl2 10 мМ, ДТТ 7,5 мМ + 0,2 мг/мл БСА) были добавлены в каждую лунку 96-луночного планшета с U-образным дном. После 20 мин инкубации при комнатной температуре, реакция была остановлена с помощью 100 мкл PBS + 32 мМ ЭДТА + 0,1% Триton Х-100 + 500 мкМ АТФ, содержащего 1 мг SPA-гранул. Затем объем, равный 110 мкл, переносят в планшет Optiplate.- 20007430 После 20 мин инкубации для поглощения субстрата, было добавлено 100 мкл 5 М CsCl для того,чтобы позволить гранулам образовать слой на поверхности планшета Optiplate и выдерживали 4 ч до подсчета радиоактивности в Top-Count считывающем устройстве. Определение IC50: смотри выше. Анализ ингибирующей активности cdk5/p25 Анализ ингибирующей активности cdk5/p25 был проведен в соответствии со следующим протоколом. Киназная реакция: 10 мкМ биотинилированного субстрата гистона H1 (SigmaН-5505), 30 мкМ АТФ (0,3 микроКи Р 33-АТФ), 15 нг CDK5/p25 комплекса, ингибитор в конечном объеме 30 мкл буфера(ТРИС HCl 10 мМ, рН 7,5, MgCl2 10 мМ, ДТТ 7,5 мМ + 0,2 мг/мл БСА) были добавлены в каждую лунку 96-луночного планшета с U-образным дном. После 30 мин инкубации при комнатной температуре, реакция была остановлена с помощью 100 мкл PBS + 32 мМ ЭДТА + 0,1% Триton Х-100 + 500 мкМ АТФ,содержащего 1 мг SPA-гранул. Затем объем, равный 110 мкл, переносят в планшет Optiplate. После 20 мин инкубации для поглощения субстрата, было добавлено 100 мкл 5 М CsCl для того,чтобы позволить гранулам образовать слой на поверхности планшета Optiplate и выдерживали 4 ч до подсчета радиоактивности в Top-Count считывающем устройстве. Определение IC50: смотри выше. Анализ ингибирующей активности cdk4/Cyclin Dl Киназная реакция: 0,4 мкМ мышиного субстрата GST-Rb (769-921) ( sc-4112 от Santa Cruz), 10 мкМ АТФ (0,5 микроКи Р 33-АТФ), 100 нг бакуловируса, экспрессирующего GST-cdk4/GST-Cyclin D1,подходящие концентрации ингибитора в конечном объеме 50 мкл буфера (ТРИС HCl 10 мМ, рН 7,5,MgCl2 10 мМ, ДТТ 7,5 мМ + 0,2 мг/мл БСА) были добавлены в каждую лунку 96-луночного планшета сU-образным дном. После 40 мин инкубации при 37 С, реакция была остановлена с помощью 20 мкл 120 мМ ЭДТА. Поглощение: 60 мкл из каждой лунки были перенесены на планшет Multiscreen, чтобы позволить субстрату связаться с фосфоцеллюлозным фильтром. Планшеты были затем промыты 3 раза растворомPBS, свободным от Са/Mg по 150 мкл/на лунку и профильтрованы фильтрационной системой Multiscreen. Детектирование: фильтры были оставлены для высыхания при 37 С, затем было добавлено по 100 мкл/на лунку сцинцилятора и 33 Р-меченный Rb фрагмент был детектирован при подсчете радиоактивности в Top-Count считывающем устройстве. Определение IC50: смотри выше. Анализ ингибирующей активности МАРК Киназная реакция: 10 мкМ в домашнем, биотинилированном субстрате MBP (SigmaМ-1891) , 15 мкМ АТФ (0,15 микроКи Р 33-АТФ), 30 нг GST-МАРК (Upstate Biothecnology14-173), ингибитор в конечном объеме 30 мкл буфера (ТРИС HCl 10 мМ, рН 7,5, MgCl2 10 мМ, ДТТ 7,5 мМ + 0,2 мг/мл БСА) были добавлены в каждую лунку 96-луночного планшета с U-образным дном. После 30 мин инкубации при комнатной температуре, реакция была остановлена с помощью 100 мкл PBS + 32 мМ ЭДТА + 0,1% Триton X-100 + 500 мкМ АТФ, содержащего 1 мг SPA-гранул. Затем объем, равный 110 мкл, переносят в планшет Optiplate. После 20 мин инкубации для поглощения субстрата, было добавлено 100 мкл 5 М CsCl для того,чтобы позволить гранулам образовать слой на поверхности планшета Optiplate и выдерживали 4 ч до подсчета радиоактивности в Top-Count считывающем устройстве. Определение IC50: смотри выше. Анализ ингибирующей активности РКА Киназная реакция: 10 мкМ в домашнем субстрате биотинилированного гистона H1 (SigmaН 5505), 10 мкМ АТФ (0,2 микроМ Р 33-АТФ), 0,45 U РКА, (Sigma2645) ингибитор в конечном объеме 30 мкл буфера (ТРИС HCl 10 мМ, рН 7,5, MgCl2 10 мМ, ДТТ 7,5 мМ + 0,2 мг/мл БСА) были добавлены в каждую лунку 96-луночного планшета с U-образным дном. После 90 мин инкубации при комнатной температуре, реакция была остановлена с помощью 100 мкл PBS + 32 мМ ЭДТА + 0,1% Триton Х-100 + 500 мкМ АТФ, содержащего 1 мг SPA-гранул. Затем объем, равный 110 мкл, переносят в планшет Optiplate. После 20 мин инкубации для поглощения субстрата, было добавлено 100 мкл 5 М CsCl для того,чтобы позволить гранулам образовать слой на поверхности планшета Optiplate и выдерживали 4 ч до подсчета радиоактивности в Top-Count считывающем устройстве. Определение IC50: смотри выше. Анализ ингибирующей активности EGFR Киназная реакция: 10 мкМ в домашнем субстрате MBP (SigmaМ-1891), 2 мкМ АТФ (0,04 микроКи Р 33-АТФ), 36 нг GST-EGFR экспрессируемого клетками насекомых, ингибитор в конечном объеме 30 мкл буфера (HEPES 50 мМ, рН 7,5, MgCl2 3 мМ, MnCl2 3 мМ, DTT 1 мМ, NaVO3 3 мкМ + 0,2 мг/мл БСА) были добавлены в каждую лунку 96-луночного планшета с U-образным дном. После 20 мин инкубации при комнатной температуре, реакция была остановлена с помощью 100 мкл PBS + 32 мМ ЭДТА +- 21007430 0,1% Триton X-100 + 500 мкМ АТФ, содержащего 1 мг SPA-гранул. Затем объем, равный 110 мкл, переносят в планшет Optiplate. После 20 мин инкубации для поглощения субстрата, было добавлено 100 мкл 5 М CsCl для того,чтобы позволить гранулам образовать слой на поверхности планшета Optiplate, и выдержано 4 ч до подсчета радиоактивности в Top-Count. Определение IC50: смотри выше. Анализ ингибирующей активности IGF1-R Анализ ингибирующей активности IGF1-R был проведен в соответствии со следующим протоколом. Киназная реакция: 10 мкМ субстрата биотинилизированного MBP (Sigma cat.М-1891), 0-20 мкм ингибитора, 6 мкМ АТФ, 1 микроКи Р 33-АТФ и 22,5 нг GST-IGF1-R (преинкубированного в течение 30 мин при комнатной температуре с охлажденным 60 мкМ ледяным АТФ) в конечном объеме 30 мкл буфера (50 мМ HEPES, рН 7,9, 3 мМ MnCl2, 1 мМ ДТТ, 3 мкМ NaVO3) были добавлены в каждую лунку 96 луночного планшета с U-образным дном. После 35 мин инкубации при комнатной температуре, реакция была остановлена с помощью 100 мкл PBS содержащего 32 мМ ЭДТА, 500 мкМ охлажденного АТФ,0,1% Триton X-100 и 10 мг/мл стрептавидина, покрывающего SPA-гранулы. После 20 мин инкубации было отобрано 110 мкл суспензии и перенесено в 96-луночные планшеты Optiplate, содержащие 100 мкл 5 М CsCl. После 4 ч, планшеты считывали в течение 2 мин в счетчике радиоактивности Packard TopCount. Анализ ингибирующей активности Aurora-2 Киназная реакция: 8 мкМ биотинилированного пептида (4 повтора LRRWSLG) , 10 мкМ АТФ (0,5 мкКиР 33g-АТФ), 15 нг Aurora2, ингибитор в конечном объеме 30 мкл буфера (HEPES 50 мМ, рН 7,0,MgCl2 10 мМ, 1 мМ ДТТ, 0,2 мг/мл БСА, 3 мкМ ортованадата) были добавлены в каждую лунку 96 луночного планшета с U-образным дном. После 30 мин инкубации при комнатной температуре, реакция была остановлена и биотинилированный пептид был поглощен добавлением 100 мкл суспензии гранул. Расслаивание: 100 мкл 5 М CsCl2 были добавлены в каждую лунку и выдерживались в течение 4 ч перед подсчетом радиоактивности в счетчике радиоактивности Top-Count. Определение IC50: смотри выше. Анализ ингибирующей активности Cdc7/dbf4 Анализ ингибирующей активности Cdc7/dbf4 был проведен в соответствии со следующим протоколом.Biotin-MCM2 субстрат является транс-фосфорилированным Cdc7/Dbf4 комплексом в присутствии АТФ, обнаруживаемого с помощью 33-АТФ. Фосфорилированный субстрат Biotin-MCM2 затем поглощается SPA-гранулами, покрытыми стрептавидином и степень фосфорилирования оценивается подсчетом. Анализ ингибирующей активности Cdc7/dbf4 был проведен в 96-луночном планшете в соответствии со следующим протоколом. В каждую лунку планшета были добавлены:- 10 мкл тестируемого соединения (12 возрастающих концентраций в интервале от нМ до мкМ для создания кривой доза-ответ);- 10 мкл смеси охлажденного АТФ (10 мкМ конечная концентрация) и радиоактивного АТФ (1/2500 молярное соотношение с охлажденным АТФ) были затем использованы для начала реакции, которую проводили при 37 С. Субстрат, фермент и АТФ были разведены в 50 мМ HEPES, рН 7,9, содержащем 15 мМ MgCl2, 2 мМ ДТТ, 3 мкМ NaVO3, 2 мМ глицерофосфата и 0,2 мг/мл БСА. Растворитель для тестируемых соединений также содержал 10% ДМСО. После 20 мин инкубации при комнатной температуре, реакция была остановлена путем добавления в каждую лунку 100 мкл PBS, рН 7,4, содержащего 50 мМ ЭДТА, 1 мМ охлажденного АТФ, 0,1% Триton Х-100 и 10 мг/мл SPA - гранул, покрытых стрептавидином. После 15 мин инкубации при комнатной температуре для того, чтобы провзаимодействовали биотинилированный МСМ 2-стрептавидин и SPA-гранулы, гранулы были помещены в 96-луночный фильтрующий планшет (UnifilterR GF/B) с использованием Packard Cell Harvester (Filtermate) , промыты дистиллированной водой и затем обсчитаны с использованием Top Count (Packard). Из полученных значений вычли контрольные значения, и затем экспериментальные данные (каждая точка в виде 3-х значений) были проанализированы для определения IC50 с использованием метода нелинейной регрессии (Sigma Plot). Соединения формулы (I) данного изобретения подходят для введения их млекопитающим, например, людям, они могут быть введены обычными способами, и причем дозировка зависит от возраста, веса, особенностей пациента и способа введения.- 22007430 Например, подходящая дозировка соединений формулы (I), предназначенная для перорального способа введения, может варьировать от примерно 10 до примерно 500 мг на дозу, от 1 до 5 раз в день. Соединения данного изобретения могут быть введены в различных лекарственных формах, например, перорально, в форме таблеток, капсул, таблеток покрытых сахаром или пленкой, жидких растворов или суспензий; ректально в форме суппозиториев; парентерально, например внутримышечно, или с помощью внутривенной и/или внутриоболочечной и/или интраспинальной инъекции или инфузии. Кроме того, соединения данного изобретения могут быть введены либо как индивидуальные агенты, либо, альтернативно, в комбинации с известными методами противораковой терапии, такими как радиационная терапия или химиотерапия по схеме в комбинации с цитостатическими или цитотоксическими агентами, агентами типа антибиотиков, алкилирующими агентами, антиметаболитными агентами,гормональными агентами, иммунологическими агентами, агентами типа интерферона, ингибиторами циклооксигеназ (например, СОХ-2 ингибиторами), ингибиторами матриксных металлопротеаз, ингибиторами теломераз, ингибиторами тирозинкиназ, агентами рецепторов антиростового фактора, анти-HER агентами, анти-EGFR агентами, агентами антиангиогенеза, ингибиторами фарнезилтрансфераз, ингибиторами ras-raf-сигнальной трансдукции, ингибиторами клеточного цикла, другими ингибиторами cdk,агентами связывания тубулина, ингибиторами топоизомеразы I, ингибиторами топоизомеразы II, и тому подобными. В качестве примера, соединения данного изобретения могут быть введены в комбинации с одним или несколькими химиотерапевтическими агентами, такими как, например, экземестан, форместан, анастрозол, летрозол, фадрозол, таксан, производные таксана, инкапсулированные таксаны, СРТ-11, производные каптотецина, антрациклиновые гликозиды, например, доксорубицин, идарубицин, эпирубицин,этопозид, навелбин, винбластин, карбоплатин, цисплатин, эстрамустин, целекоксиб, тамоксифен, ралоксифен, Sugen SU-5416, Sugen SU 6668, герцептин, и тому подобные, необязательно в виде их липосомальных препаратов. При приготовлении препаратов с фиксированной дозой такие комбинированные продукты используют соединения данного изобретения в интервале доз, описанном выше, и другие фармацевтически активные агенты в пределах принятого интервала доз. Соединения формулы (I) могут быть использованы последовательно с противораковыми агентами,когда комбинированный состав является неприемлемым. Настоящее изобретение также включает фармацевтические композиции, содержащие соединение формулы (I) или его фармацевтически приемлемую соль в сочетании с фармацевтически приемлемым эксципиентом (который может быть носителем или разбавителем). Данные фармацевтические композиции, содержащие соединения данного изобретения, обычно приготовляются следующими традиционными способами и вводятся в фармацевтически приемлемой форме. Например, твердые формы для перорального применения могут содержать, совместно с активным соединением, разбавители, например лактозу, декстрозу, сахарозу, сукрозу, целлюлозу, кукурузный крахмал или картофельный крахмал; смазывающие вещества, например, окись кремния, тальк, стеариновую кислоту, стеарат магния или кальция, и/или полиэтиленгликоли; связывающие агенты, например,крахмалы, аравийскую камедь, желатин, метилцеллюлозу, карбоксиметилцеллюлозу или поливинилпирролидон; дезагрегирующие агенты, например крахмал, альгиновую кислоту, альгинаты или натрийкрахмалгликолят/ вспенивающиеся композиции; красители; подсластители; увлажняющие агенты, такие как лецитин, полисорбаты, лаурилсульфаты; и, в основном, нетоксичные и фармакологически неактивные субстанции, используемые в фармацевтических препаратах. Описанные препараты могут быть получены по известной схеме, например, посредством процессов перемешивания, гранулирования, таблетирования,покрытия сахаром или покрытия пленкой. Жидкие дисперсионные системы для перорального применения могут быть, например, сиропами,эмульсиями и суспензиями. Сиропы могут содержать в качестве носителя, например, сахарозу или сахарозу с глицерином,и/или маннитом, и/или сорбитом. Суспензии и эмульсии могут содержать в качестве носителя, например, натуральную смолу, агар,альгинат натрия, пектин, метилцеллюлозу, карбоксиметилцеллюлозу или поливиниловый спирт. Суспензия или растворы для внутримышечных инъекций могут содержать, совместно с активным компонентом, фармацевтически приемлемый носитель, например, стерильную воду, оливковое масло,этилолеат, гликоли, например, пропиленгликоль, и, если требуется, подходящее количество лидокаина гидрохлорида. Растворы для внутривенных инъекций или инфузий могут содержать в качестве носителя,например, стерильную воду или предпочтительно они могут быть в форме стерильных, водных, изотонических солевых растворов или они могут содержать в качестве носителя пропиленгликоль. Суппозитории могут содержать совместно с активным компонентом фармацевтически приемлемый носитель, например масло какао, полиэтиленгликоль, ПАВ на основе сложного эфира жирной кислоты и полиоксиэтилен сорбитана или лецитин.- 23007430 Следующие примеры, представленные здесь, предназначены для лучшей иллюстрации настоящего изобретения, но не имеют целью каким-либо образом его ограничить. Основные методы Флэш-хроматографию проводят на силикагеле (Merck grade 9395, 60 А). Времена удерживания жидкостной хроматографии высокого давления (ВЭЖХ: значения RT) были определены следующим образом: Способ 1. Инструментарий: ВЭЖХ система Waters 2790, оборудованная 996 Waters PDA детектором и Micromass mod. ZQ масс-спектрометром с одинарной квадрупольной линзой, оборудованный источником электрораспыления ионов (ESI). Условия хроматографии: колонка RP18 Waters X Terra (4, 6 х 50 мм, 3,5 мкм) ; подвижной фазой А был 5 мМ буфер, содержащий ацетат аммония (рН 5,5, со смесью уксусная кислота/ацетонитрил 95:5), а подвижной фазой В была смесь Н 2 О/ацетонитрил (5:95). Градиент от 10 до 90% насыщения раствором В- 8 мин, удержание 90% В - 2 минуты. УФ детектирование при 220 нм или 254 нм. Скорость потока 1 мл/мин. Вводимый объем - 10 мкл. Полное сканирование, диапазон масс от 100 до 800 а.м.е. Капиллярное напряжение 2,5 KV; температура источника 120 С; cone (конус) 10 V. Времена удерживания (ВЭЖХ r.t.) даны в минутах при 220 нм или при 254 нм. Значения масс даны в виде соотношения m/z. Способ 2. Инструментарий: Waters 2790 Alliance система, оборудованная термостатируемым автоколлектором; УФ детектором с двойной длиной волны 2487; Satin Interface; двуходовой кран LabPro, массспектрометром Waters с одинарной квадрупольной линзой, с ESI-интефейсом; хемилюминесцентным азотным детектором Antek (CLND) 8060. Условия хроматографии: колонка Zorbax SB C8 (4,6 х 50 мм; 5 мкм); подвижной фазой А был раствор, содержащий 0,01% муравьиной кислоты в ацетонитриле, и подвижной фазой В был раствор, содержащий 0,01% муравьиной кислоты в метаноле. Градиент от 0 до 95% насыщения раствором В - 10 мин, удержание 95% В - 2 минуты. УФ детектирование при 220 нм. Скорость потока 1 мл/мин. Вводимый объем - 10 мкл. Полное сканирование, диапазон масс от 120 до 1000 а.м.е. Капиллярное напряжение 2,8 KV; температура источника была 115 С; cone (конус) 32 V. Времена удерживания (ВЭЖХ r.t.) даны в минутах при 220 нм или при 254 нм. Значения масс даны в виде соотношения m/z. Способ 3. Инструментарий: НР 1100 ВЭЖХ бинарный насос; автоколлектор Gilson 215; УФ-детектор с одной длиной волны НР 1100; испарительный детектор светового рассеяния (ELS) Sedex 75 с (Sedere, France); масс-спектрометр PE/Sciex API-2000. Условия хроматографии: колонки YMC ODS-AQ 4,6 х 50 мм, 5 мкм S5; с подвижными фазами ВЭЖХ, состоящими из раствора 0,5% муравьиной кислоты в воде, для ВЭЖХ (А) из раствора 0,5% муравьиной кислоты в растворе ацетонитрила (В) марки для ВЭЖХ. Градиент ВЭЖХ, показанный в таблице, был получен при инжекции по 5 мкл для каждого образца. УФ-детектирование при 220 нм. Источник турбоионного рассеивания был использован при напряжении ионного рассеивания, равном 5 кВ, при температуре, равной 475 С, и с напряжением на выходе и кольцевом напряжении, равном 10V и 250V, соответственно. Положительные ионы были сканированы в Q1 в интервале значений а.м.е. от 160 до 800. При необходимости, соединения были очищены препаративной ВЭЖХ на колонке Waters SymmetryC18 (19 х 50 мм, 5 мкм) с использованием waters системы препаративной ВЭЖХ 600, оборудованной 996 Н-ЯМР-спектрометрия была проведена на Mercury VX 400, работающем при частоте, равной 400,45 МГц, снабженным 5 мм двойным резонансным зондом [1 Н (15N-31P) ID-PFG Varian]. Как прежде было показано, несколько соединений формулы (I) синтезировались параллельно, в соответствии с методиками комбинаторной химии. Поэтому, ряд соединений, получаемых таким образом, были обычным образом и однозначно идентифицированы, как указано в кодирующей системе таблиц с IX по XVI, и с помощью времени удерживания в ВЭЖХ (способы с 1 по 3) и масс. Каждый код, который идентифицирует одно конкретное соединение формулы (I), состоит из трех частей А-М-В. А представляет собой любой заместитель R1- [смотри формулу (I)] и присоединен к остатку индазольной структуры через атом кислорода, с тем чтобы получить производные индазола, которые замещены в положении 5 (А-М 1-В) или в положении 6 (А-М 2-В); каждый А радикал (заместитель) представлен в следующей таблице VII. Совместно с -NH-группой в положении 3 индазольной структуры, к которой она присоединена, BNH- представляет собой R-группу формулы (I); каждый В радикал (заместитель) представлен в следующей таблице VIII. М относится к центральному ядру двухвалентной 3-аминоиндазольной структуры, имеющей -Огруппу в положении 5 или 6 и замещена группами А и В. В частности, М может варьировать между M1 или М 2 как в формуле, приведенной ниже, и каждая служит для идентификации соединения, будучи замещенной А-O- группами в положении 5 (Ml) или в положении 6 (М 2) Для простоты изложения, каждая А или В группы таблиц VII и VIII были идентифицированы соответствующей химической формулой, также показывающей точку присоединения к остатку молекулы М. Так, в качестве примера, соединение А 21-М 1-В 10 таблицы XI (смотри пример 11, номер ввода 429),представляет собой индазол Ml, замещенный в положении 5 (через атом кислорода) группой А 21 и в положении 3 (через -NH-группу) группой В 10; аналогично, соединение А 10-М 2-В 70 таблицы XII (смотри пример 12, номер ввода 281), представляет собой индазол М 2, замещенный в положении 6 (через атом кислорода) группой А 10 и в положении 3 (через -NH-группу) группой В 70:- 29007430 Пример 1. 6-Метокси-1 Н-индазол-3-амин. К ледяной суспезии, содержащей 66,35 г (0,448 моль) 2-амино-4-метоксибензонитрила в 530 мл концентрированной HCl, добавляют по каплям раствор, содержащий 37,07 г (0,537 моль) нитрита натрия в 55 мл воды. После 1,5 ч холодную суспензию добавляют по каплям при 5 С к предварительно приготовленному раствору, содержащему 679,25 г (3,58 моль) хлорида олова в 530 мл концентрированной хлористоводородной кислоты. После 3 ч холодную суспензию фильтруют, и влажную твердую массу обрабатывают 1,7 л кипящей воды в течение 30 мин. Горячий хлопьевидный раствор очищают фильтрацией через тканевой фильтр. Ледяные жидкости обрабатывают по каплям 0,8 л 17% NaOH. Твердую фазу фильтруют и сушат в вакууме при 50 С: 67,2 г продукта получают в виде твердого вещества светлокоричневого цвета. Выход = 91,9%. Т.пл. =195-197 С (разлож.) ВЭЖХ время удержания (в.у.) 1,9 [М+Н]+ = 164. Н 1 ЯМР (ДMCO-d6), диагностические сигналы (м.д.): 3,74 (с, 3H), 5,17 (широкий с, 2 Н), 6,5 (дд, 1 Н),6,6 (д, 1 Н), 7,5 (д, 2 Н), 11,07 (с, 1 Н). Пример 2. 2(6-Метокси-1 Н-индазол-3-ил)-1 Н-изоиндол-1,3(2 Н)-дион. 20 г (0,122 моль) 6-метокси-1 Н-индазол-3-амина, 20 г (0,135 моль) фталевого ангидрида и 140 мг(1,22 ммоль) 4-диметиламинопиридина кипятят с обратным холодильником в 0,4 л ацетонитрила в течение 2,5 ч. Смесь охлаждают до 5 С и фильтруют с получением первой порции продукта (24,2 г) . Маточные растворы концентрируют в вакууме и обрабатывают 70 мл трет-бутилметилового эфира (ТБМЭ): вторую продукта (5,8 г) порцию получают фильтрацией. Затем получают всю массу продукта, равную 30 г, в виде твердого вещества желтого цвета. Выход = 83,6%. Т.пл = 193-195 С. ВЭЖХ в.у. 4,7 [М+Н]+ = 294 [2 М+Н]+ = 587 [3 М+Н]+ = 880. Н 1 ЯМР (ДМСО-d6), диагностические сигналы (м.д.): 3,84 (с, 3H), 6,78 (дд, 1 Н), 6,96 (д, 1 Н), 7,55(дд, 1 Н), 7,91-8,1 (м, 4 Н), 13,14 (с, 1 Н). Пример 3. 2(6-Гидрокси-1 Н-индазол-3-ил)-1 Н-изоиндол-1,3(2 Н)-дион. Смесь, состоящую из 24,2 г (82,5 ммоль) 2(6-Метокси-1 Н-индазол-3-ил)-1 Н-изондол-1,3(2 Н)диона и 73,4 г (0,635 моль) гидрохлорида пиридина нагревают при 200 С в течение 4 ч. Полученный коричневый раствор охлаждают до 140 С и медленно переливают в хорошо перемешиваемую смесь из 250 мл 0,2 н. HCl и 350 мл этилацетата. Органический слой отделяют, и водный слой подсаливают (45 г NaCl) и экстрагируют дважды 350 мл этилацетата. Органические экстракты высушивают над сульфатом натрия и концентрируют в вакууме до небольшого объема. Осадок фильтруют и высушивают: получают 15,89 г продукта в виде твердого вещества желтого цвета. Выход = 68,9%. Т.пл = 265-270 С (разл.) ВЭЖХ в.у. 3,7 [М+Н]+ = 280 [2 М+Н]+ = 559 [3 М+Н]+ = 838. Н 1 ЯМР (ДМСО-d6), диагностические сигналы (м.д.): 6,65 (дд, 1 Н), 6,8 (с, 1 Н), 7,44 (д, 1 Н), 7,97 (м,4 Н), 9,73 (широкий с, 1 Н), 12,86 (с, 1 Н). Пример 4. 2-(6[трет-бутил(диметил)силил]окси-1 Н-индазол-3-ил)-1 Н-изоиндол-1,3(2 Н)-дион. К суспензии 15,03 г (53,82 ммоль) 2(6-гидрокси-1 Н-индазол-3-ил)-1 Н-изондол-1,3(2 Н)-диона в 150 мл дихлорметана добавляют раствор, содержащий 20,19 г (0,134 моль) TBDMS хлорида в 75 мл дихлорметана. Полученную смесь обрабатывают, добавляя по каплям 12,06 мл (80,73 ммоль) 1,8 диазабицикло[5,4,0]ундец-7-ена (DBU) при комнатной температуре, получая прозрачный раствор. После 3 ч реакционную смесь выливают в 250 мл 0,5 н. HCl. Водную фазу отделяют и экстрагируют 120 мл дихлорметана. Органические экстракты высушивают над сульфатом натрия и растворитель выпаривают в вакууме. Влажный неочищенный продукт перемешивают в 50 мл этилацетата при 50 С. Затем примерно половину растворителя выпаривают в вакууме и смесь обрабатывают, добавляя по каплям 100 мл циклогексана. Продукт выделяют отсасыванием, получая его в виде твердого вещества светло-желтого цвета(д, 1 Н) , 7,54 (д, 1 Н) , 7,93 (м, 2 Н), 8,1 (м, 2 Н). Пример 5. 5-Бензилокси-1 Н-индазол-3-амин. К ледяной суспезии, состоящей из 63,27 г (0,282 моль) 2-амино-5-(бензилокси)бензонитрила в 500 мл концентрированной HCl, добавляют по каплям раствор, содержащий 23,32 г (0,338 моль) нитрита натрия в 75 мл воды. После 2 ч холодную суспензию добавляют по каплям при 2 С к предварительно приготовленному раствору, содержащему 509,25 г (2,36 моль) хлорида олова в 380 мл концентрированной хлористоводородной кислоты. После 3 ч холодную суспензию фильтруют и влажную твердую массу обрабатывают 1,8 л кипящей воды и 300 мл этанола 95 в течение 30 мин. Горячий хлопьевидный раствор очищают фильтрацией через тканевый фильтр. Жидкости концентрируют для удаления этанола и обрабатывают, добавляя по каплям 0,35 л 35% NaOH при 4 С. Твердую фазу отфильтровывают и высушивают в вакууме при 50 С: 73,82 г продукта были получены в виде твердого вещества светло-коричневого цвета. Т.пл. =193-195 С ВЭЖХ время удержания (в.у.) 4.7[М]+ = 240 [2 М+Н]+ = 479. Н 1 ЯМР (ДМСО-d6) , диагностические сигналы (м.д.): 5,03 (с, 2 Н), 5,16 (широкий с, 1 Н), 6,96 (д,1 Н), 7,13 (д, 1 Н), 7,26 (д, 1 Н), 7,27-7,49 (м, 5 Н).
МПК / Метки
МПК: A61K 31/416, C07D 403/04, A61P 35/00, C07D 231/56
Метки: содержащая, активные, способ, качестве, ингибиторов, аминоиндазола, производные, композиция, получения, киназ, фармацевтическая
Код ссылки
<a href="https://eas.patents.su/30-7430-proizvodnye-aminoindazola-aktivnye-v-kachestve-ingibitorov-kinaz-sposob-ih-polucheniya-i-soderzhashhaya-ih-farmacevticheskaya-kompoziciya.html" rel="bookmark" title="База патентов Евразийского Союза">Производные аминоиндазола, активные в качестве ингибиторов киназ, способ их получения и содержащая их фармацевтическая композиция</a>
Сульфонамидные производные в качестве ингибиторов рассасывания костной ткани и ингибиторов адгезии клеток,способ их получения, применение и фармацевтическая композиция

Номер патента: 3102
Опубликовано: 26.12.2002
Авторы: Карниато Дени, Шойнеманн Карлхайнц, Катбертсон Роберт Эндрю, Макдауэлл Роберт, Кнолле Йохен, Пейман Ануширван, Гурвест Жан-Франсуа, Гадек Томас, Бодари Сара Кэтрин, Вилл Дэвид Вильям
МПК: A61P 19/10, C07D 239/42, A61K 31/505...
Метки: рассасывания, ингибиторов, получения, костной, композиция, производные, качестве, адгезии, применение, клеток,способ, сульфонамидные, ткани, фармацевтическая
Формула / Реферат:
1. Сульфонамидные производные общей формулы I где R1 и R2 вместе образуют двухвалентный (С2-С3)алкиленовый радикал; R4 является Н или (С1-С6)алкилом; R5 является (С1-С10)алкилом, (С6-С14)арилом, (C5-C14)гетероарилом или (С6-С14)арил(С1-С10)алкильной группой, где арил, гетероарил или алкил возможно замещены R3, или 2-оксобицикло[2.2.1]гепт-1-илметильной группой; R3 является (C1-C4)алкилом, (C1-C4)алкилокси, галогеном, трифторметилом, циано,...
Производные аминофталазинона, активные как ингибиторы киназ, способ их получения и содержащие их фармацевтические композиции

Номер патента: 6645
Опубликовано: 24.02.2006
Автор: Пуличи Маурицио
МПК: A61P 35/00, C07D 237/34, C07D 237/32...
Метки: аминофталазинона, производные, киназ, фармацевтические, получения, ингибиторы, способ, композиции, содержащие, активные
Формула / Реферат:
1. Способ лечения заболеваний, вызванных и/или ассоциированных с измененной активностью протеинкиназы, который включает введение млекопитающему, нуждающемуся в данном лечении, эффективного количества аминофталазинонового производного, представленного формулой (I) где Ra и Rb, каждый независимо, представляют собой атом водорода или группу, необязательно далее замещенную, выбранную из прямого или разветвленного C1-C6алкила, C3-C6циклоалкила или...
Новые производные арилглицинамида, способ их получения и фармацевтическая композиция , содержащая эти соединения

Номер патента: 2201
Опубликовано: 28.02.2002
Авторы: Шпек Георг, Юнг Биргит, Шромм Курт, Шнорренберг Герд, Эссер Франц, Доллингер Хорст
МПК: A61P 9/00, C07D 295/14, A61K 31/395...
Метки: фармацевтическая, способ, получения, арилглицинамида, эти, соединения, композиция, производные, содержащая, новые
Формула / Реферат:
1. Производные арилглицинамида общей формулы (I) или их фармацевтически приемлемые соли, где Ar означает незамещенный или 1-5-кратнозамещенный фенил или незамещенный или 1-2-кратнозамещенный нафтил, причем заместители фенила и нафтила независимо друг от друга являются галогеном (фтором, хлором, бромом, йодом), алкилом с 1-4 атомами углерода, O-(С1-С4)алкилом, трифторметилом, трифторметокси или NR12R13, где R12 и R13 независимо друг от друга...
Пирролидинил-, пиперидинил- или гомопиперидинилзамещенные производные, способ их получения, их применение в медицине, содержащая их фармацевтическая композиция и способ ее получения

Номер патента: 4648
Опубликовано: 24.06.2004
Авторы: Де Брюин Марсель Франс Леопольд, Вигеринк Пит Том Берт Поль, Ван Эмелен Кристоф, Вирсхюэрен Вим Гастон
МПК: A61K 31/505, A61P 9/00, C07D 405/14...
Метки: применение, способ, производные, гомопиперидинилзамещенные, композиция, пирролидинил, медицине, содержащая, получения, фармацевтическая, пиперидинил
Формула / Реферат:
1. Соединение формулы (I) его стереохимически изомерная форма или его фармацевтически приемлемая кислотно-аддитивная соль, в которых R1, R2 и R3 каждый представляет водород; -Z1-Z2- представляет двухвалентный радикал формулы -O-CH(R4)-CR2-CH2- (a-4), R4 представляет водород; Alk представляет C1-6алкандиил, необязательно замещенный гидроксигруппой; представляет двухвалентный радикал формулы в которой m равно 0 или 1; R6...
Фармацевтическая композиция, содержащая в качестве активного начала уратоксидазу, и лиофилизат для ее получения

Номер патента: 24
Опубликовано: 30.12.1997
Авторы: Брель Тьерри, Байоль Ален, Дюпэн Патрис, Алеман Клод
МПК: A61K 38/44
Метки: содержащая, уратоксидазу, композиция, лиофилизат, фармацевтическая, начала, активного, получения, качестве
Формула / Реферат:
1. Фармацевтическая композиция, содержащая в качестве активного начала ураток-сидазу, отличающаяся тем, что она содержит эффективное количество уратоксидазы и 0,1-10 мг/мл блоксополимера этиленоксида и пропиленоксида общей формулы: НО(СН2СН2О)75[СН(СН3)СН2O]30(CH2CH2O)75H в среде буферного раствора. 2. Композиция по п.1, отличающаяся тем, что она содержит 0,5-5 мг/мл указанного блоксополимера. 3. Композиция по п.1, отличающаяся тем, что...
Предыдущий патент: Азабициклические замещённые конденсированные гетероарильные соединения
Следующий патент: Андрогенная фармацевтическая композиция и способ для лечения депрессии
Случайный патент: Ингибиторы циклинзависимых киназ