Гидроксиэтиламиносульфонамиды аминокислот в качестве ингибиторов протеаз ретровирусов.
Номер патента: 470
Опубликовано: 26.08.1999
Авторы: Девадас Балекудру, Гетмен Даниел П., Фрескос Джон Н., Васкез Майкл Л., Макдонэлд Джозеф Дж., Браун Дейвид Л., Декрессенцо Гэри А., Нагараян Сринивазан, Сикорски Джеймс А.





























Формула / Реферат
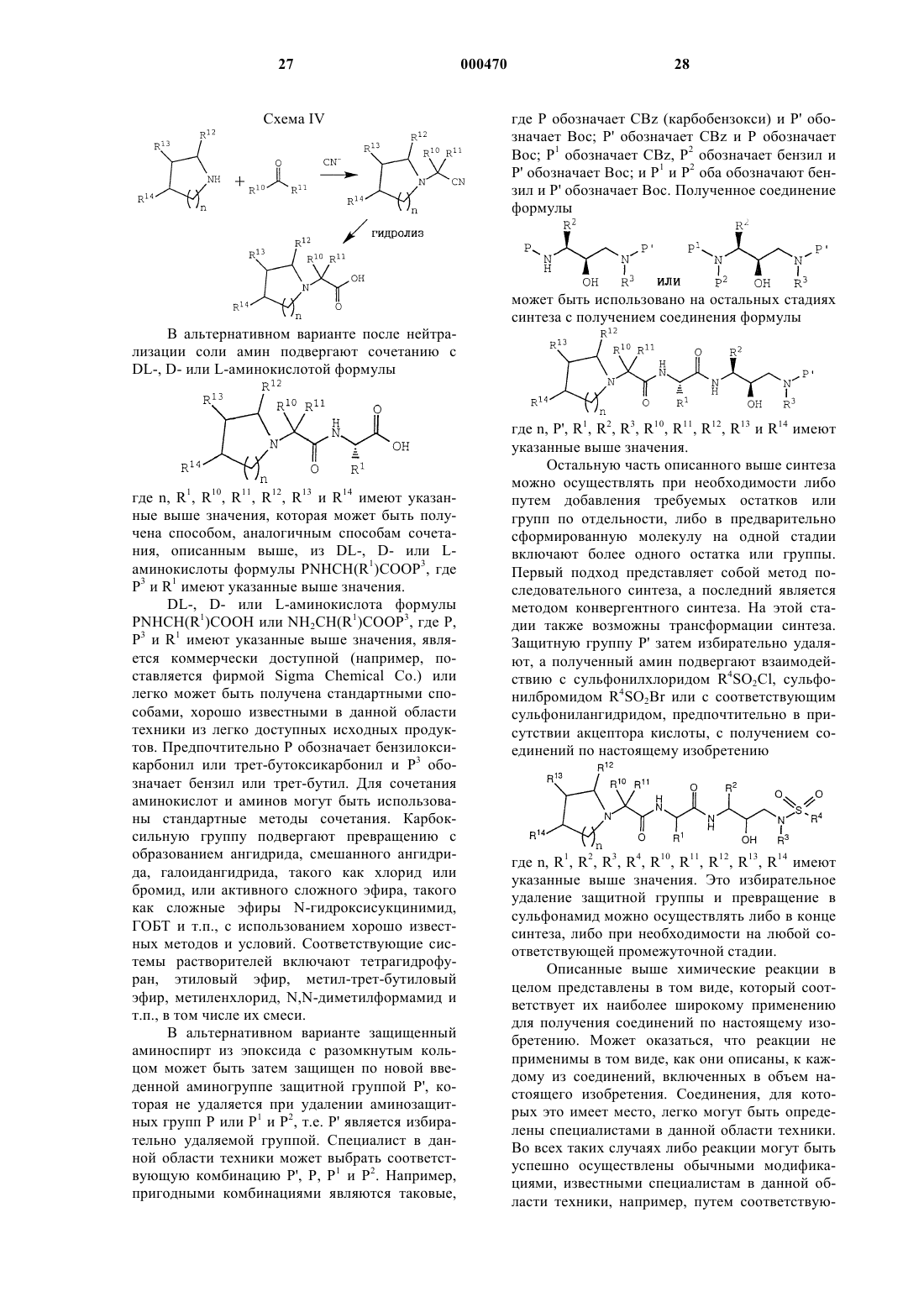
1. Соединение формулы

где n равно 1 или 2;
R1 обозначает алкил с 1-5 атомами углерода, алкенил с 2-5 атомами углерода, алкинил с 2-5 атомами углерода, гидроксиалкил с 1-3 атомами углерода, алкоксиалкил, состоящий из алкила с 1-3 атомами и алкокси с 1-3 атомами углерода, цианалкил, включающий алкил с 1-3 атомами углерода, имидазолилметил, -CH2CONH2, -CH2CH2CONH2, -CH2S(O)2NH2, -СН2SСН2, -СН2S(О)СН3, -СН2S(O)2СН3, -С(СН3)2SСН3, -C(CH3)2S(O)CH3 или -C(CH3)2S(O)2CH3;
R2 обозначает алкил с 1-5 атомами углерода, аралкил, включающий алкил с 1-3 атомами углерода, алкилтиоалкил, включающий алкил с 1-3 атомами углерода, арилтиоалкил, включающий алкил с 1-3 атомами углерода, или циклоалкилалкил, состоящий из алкила с 1-3 атомами углерода и 3-6-членного углеводородного кольца;
R3 обозначает алкил с 1-5 атомами углерода, циклоалкил с 5-8-членным кольцом или циклоалкилметил с 3-6-членным кольцом;
R4 обозначает арил, сконденсированный с бензольным ядром 5-6-членный гетероарил или сконденсированный с бензольным ядром 5-6-членный гетероцикл; или группу формулы

где А и В, каждый независимо друг от друга, обозначает О, S, SO или SO2;
R6 обозначает дейтерий, алкил с 1-5 атомами углерода, фтор или хлор;
R7 обозначает водород, дейтерий, метил или фтор или хлор;
или группу формулы

где Z обозначает О, S или NH и
R9 обозначает группу формулы

где Y обозначает О, S или NH;
X обозначает связь, О или NR21;
R20 обозначает водород, алкил с 1-5 атомами углерода, алкенил с 2-5 атомами углерода, алкинил с 2-5 атомами углерода, аралкил, включающий алкил с 1-5 атомами углерода, гетероаралкил, состоящий из 5-6-членного кольца и алкила с 1-5 атомами углерода, гетероциклоалкил, состоящий из 5-6-членного кольца и алкила с 1-5 атомами углерода, аминоалкил с 2-5 атомами углерода, N-монозамещенный или N,N-дизамещенный аминоалкил, включающий алкил с 2-5 атомами углерода, где указанные заместители представляют собой алкил с 1-3 атомами углерода, аралкил с 1-3 атомами углерода, карбоксиалкил с 1-5 атомами углерода, алкоксикарбонилалкил, включающий алкил с 1-5 атомами углерода, цианалкил с 1-5 атомами углерода или гидроксиалкил с 2-5 атомами углерода;
R21 обозначает водород или алкил с 1-3 атомами углерода;
или группа формулы NR20R21 обозначает 5-6-членный гетероцикл; и
R22 обозначает алкил с 1-3 атомами углерода или группу R20R21N-алкил, где алкил имеет 1-3 атома углерода;
R10 обозначает водород, алкил, гидроксиалкил или алкоксиалкил, где алкил имеет 1-3 атома углерода;
R11 обозначает водород, алкил с 1-5 атомами углерода, гидроксиалкил с 1-4 атомами углерода, алкоксиалкил с 1-3 атомами углерода, бензил, имидазолилметил, -CH2CH2CONH2, -CH2CONH2, -СН2СН2SСН3 или -CH2SCH3 или их сульфоновые или сульфоксидные производные;
R12 обозначает водород, гидроксиалкил или алкоксиалкил, где алкил имеет 1-3 атома углерода; и
R13 и R14 каждый независимо друг от друга обозначает водород, гидрокси, алкокси, 2-гидроксиэтокси, гидроксиалкил или алкоксиалкил; где алкил имеет 1-3 атома углерода; или
R12 и R13 или R13 и R14 вместе с атомами углерода, к которым они присоединены, обозначают 5-6-членный гетероарил или бензо, каждый из которых необязательно замещен, по крайней мере, одной гидрокси- или алкоксигруппой с 1-3 атомами углерода;
или его фармацевтически приемлемая соль, пролекарство или сложный эфир.
2. Соединение по п.1, где R1 обозначает алкил с 1-4 атомами углерода, алкенил с 2-3 атомами углерода, алкинил с 3-4 атомами углерода, цианметил, имидазолилметил, -CH2CONH2, -CH2CH2CONH2, -CH2S(O)2NH2, -СН2SСН3, -СН2S(O)СН3, -СН2S(O)2СН3, -С(СН3)2SСН3, -C(CH3)2S(O)CH3 или -С(СН3)2S(O)2СН3; и
R2 обозначает алкил с 3-5 атомами углерода, арилметил, алкилтиоалкил, включающий алкил с 1-3 атомами углерода, арилтиометил или циклоалкилметил с 5-6-членным углеводородным кольцом;
R3 обозначает алкил с 1-5 атомами углерода, циклоалкилметил с 3-6-членным кольцом, циклогексил или циклогептил;
R4 обозначает фенил, 2-нафтил, 4-метоксифенил, 4-гидроксифенил, 3,4-диметоксифенил, 3-аминофенил, 4-аминофенил, 2-аминобензотиазол-5-ил, 2-аминобензотиазол-6-ил, бензотиазол-5-ил, бензотиазол-6-ил, бензоксазол-5-ил, 2,3-дигидроксибензофуран-5-ил, бензофуран-5-ил, 1,3-бензодиоксол-5-ил или 1,4-бензодиоксан-6-ил; или группу формулы

где А и В каждый обозначает О;
R6 обозначает дейтерий, метил, этил, пропил, изопропил или фтор; и
R7 обозначает водород, дейтерий, метил или фтор;
или группу формулы

где Z обозначает О, S или NH и
R9 обозначает группу формулы

где Y обозначает О, S или NH;
Х обозначает связь, О или NR21;
R20 обозначает водород, алкил с 1-5 атомами углерода, фенилалкил, включающий алкил с 1-3 атомами углерода, гетероциклоалкил, состоящий из 5-6-членного кольца и алкила с 1-3 атомами углерода, или N-монозамещенный или N,N-дизамещенный аминоалкил с 2-3 атомами углерода, где указанные заместители представляют собой алкильные радикалы с 1-3 атомами углерода; и
R21 обозначает водород или метил;
или группа формулы -NR20R21 обозначает пирролидинил, пиперидинил, пиперазинил, 4-метилпиперазинил, 4-бензилпиперазинил, морфолинил или тиаморфолинил; и
R22 обозначает алкил с 1-3 атомами углерода,
или его фармацевтически приемлемая соль, пролекарство или сложный эфир.
3. Соединение по п.2, где R1 обозначает изопропил, втор-бутил, трет-бутил, 3-пропинил, имидазолилметил, -CH2CONH2, -СН2SСН3, -СН2S(О)СН3, -СН2S(O)2СН3, -С(СН3)2SСН3, -С(СН3)2S(O)СН3 или -С(СН3)2S(O)2СН3;
R2 обозначает изобутил, н-бутил, CH3SCH2CH2-, фенилтиометил, (2-нафтилтио) метил, бензил, 4-метоксифенилметил, 4-гидроксифенилметил, 4-фторфенилметил или циклогексилметил;
R3 обозначает пропил, изоамил, изобутил, бутил, циклогексил, циклогептил, циклопентилметил или циклогексилметил;
R4 обозначает фенил, 2-нафтил, 4-метоксифенил, 4-гидроксифенил, бензотиазол-5-ил, бензотиазол-6-ил, бензоксазол-5-ил, 2,3-дигидробензофуран-5-ил, бензофуран-5-ил, 1,3-бензодиоксол-5-ил, 2-метил-1,3-бензодиоксол-5-ил, 2,2-диметил-1,3-бензодиоксол-5-ил, 2,2-дидейтерий-1,3-бензодиоксол-5-ил, 2,2-дифтор-1,3-бензодиоксол-5-ил или 1,4-бензодиоксан-6-ил; или группу формулы

где Z обозначает О, S или NH и
R9 обозначает группу формулы

где Y обозначает О, S или NH;
Х обозначает связь, О или NR21;
R20 обозначает водород, метил, этил, пропил, изопропил, изобутил, бензил, 2-(1-пирролидинил)этил, 2-(1-пиперидинил)этил, 2-(1-пиперазинил)этил, 2-(4-метилпиперазин-1-ил)этил, 2-(1-морфолинил)этил, 2-(1-тиаморфолинил)этил или 2-(N,N-диметиламино) этил;
R21 обозначает атом водорода; и
R22 обозначает метил;
R10 и R12 каждый обозначает атом водорода;
R11 обозначает водород, метил, изопропил, бутил, втор-бутил, изобутил, гидроксиметил или гидроксиэтил; и
R13 и R14 каждый независимо друг от друга обозначает водород, гидрокси, метокси или этокси; или
R12 и R13 или R13 и R14 вместе с атомами углерода, к которым они присоединены, обозначают бензил, который необязательно замещен, по крайней мере, одной гидрокси- или метоксигруппой,
или его фармацевтически приемлемая соль, пролекарство или сложный эфир.
4. Соединения по п.3, где n равно 1;
R1 обозначает втор-бутил, трет-бутил, изопропил, 3-пропинил или -С(СН3)2S(O)2СН3;
R2 обозначает бензил, 4-фторфенилметил или циклогексилметил;
R4 обозначает фенил, 4-метоксифенил, 4-гидроксифенил, бензотиазол-5-ил, бензотиазол-6-ил, 2,3-дигидробензофуран-5-ил, бензофуран-5-ил, 1,2-бензодиоксол-5-ил, 2-метил-1,3-бензодиоксол-5-ил, 2,2-диметил-1,3-бензодиоксол-5-ил, 2,2-дидейтерий-1,3-бензодиоксол-5-ил, 2,2-дифтор-1,3-бензодиоксол-5-ил, 1,4-бензодиоксан-6-ил, 2-(метоксикарбониламино)бензотиазол-6-ил или 2-(метоксикарбониламино)бензимидазол-5-ил;
R11 обозначает атом водорода; и
R13 и R14 каждый независимо друг от друга обозначает водород, гидрокси, метокси или этокси,
или его фармацевтически приемлемая соль, пролекарство или сложный эфир.
5. Соединение по п.1, где фармацевтически приемлемая соль представляет собой соль соляной кислоты, соль серной кислоты, соль фосфорной кислоты, соль щавелевой кислоты, соль малеиновой кислоты, соль янтарной кислоты, соль лимонной кислоты или соль метансульфокислоты.
6. Соединение по п.5, где фармацевтически приемлемая соль представляет собой соль соляной кислоты, соль щавелевой кислоты, соль лимонной кислоты или соль метансульфокислоты.
7. Соединение по п.1, выбранное из группы, включающей:
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-3,3-диметилбутанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-3-метилбутанамид;
2S-[[(пирролидин-1-ил)ацетил] амино]-N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил) амино] -1S-(фенилметил) пропил] - 3S - метилпентанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(1,3-бензодиоксол-5-ил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил)пропил]-4-пентинамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[фенилсульфонил] (2-метилпропил) амино]-1S- (фенилметил) пропил]-3,3-диметилбутанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[фенилсульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-3-метилбутанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[фенилсульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил] -3S-метилпентанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[фенилсульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-4-пентинамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-3,3-диметилбутанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-3-метилбутанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2-метилпропил) амино]-1S-(фeнилмeтил)пpoпил]-3S-метилпентанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(4-метоксифенил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил) пропил]-4-пентинамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(2,3-дигидробензофуран-5-ил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-3,3-диметилбутанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(2,3-дигидробензофуран-5-ил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-3-метилбутанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(2,3-дигидробензофуран-5-ил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-3S-метилпентанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(2,3-дигидробензофуран-5-ил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]4-пентинамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(бензотиазол-6-ил)сульфонил] (2-метилпропил) амино]-1S- (фенилметил) пропил]-3,3-диметилбутанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(бензотиазол-6-ил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-3-метилбутанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(бензотиазол-6-ил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-3S-метилпентанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(бензотиазол-6-ил)сульфонил] (2-метилпропил) амино]-1S-(фeнилмeтил)пропил]-4-пентинамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(2-нафтил) сульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-3,3-диметилбутанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(2-нафтил) сульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-3-метилбутанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(2-нафтил) сульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-3S-метилпентанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(2-нафтил) сульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-4-пентинамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(1,4-бензодиоксан-6-ил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил) пропил]-3,3-диметилбутанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(1,4-бензодиоксан-6-ил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил) пропил]-3-метилбутанамид;
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(1,4-бензодиоксан-6-ил)сульфонил] (2-метилпропил)амино]-1S-(фенилметил)пропил]-3S-метилпентанамид; и
2S-[[(пирролидин-1-ил) ацетил] амино]-N-[2R-гидрокси-3-[[(1,4-бензодиоксан-6-ил)сульфонил] (2-метилпропил) амино]-1S-(фенилметил)пропил]-4-пентинамид.
8. Фармацевтическая композиция, включающая соединение по п.1 и фармацевтически приемлемый носитель.
9. Применение соединения по п.1 для приготовления лекарства, предназначенного для ингибирования протеазы ретровируса.
10. Применение соединения по п.8 для приготовления лекарства, предназначенного для лечения ретровирусной инфекции.
11. Применение соединения по п.1 для приготовления лекарства, предназначенного для предотвращения репликации ретровируса.
12. Способ предотвращения репликации ретровируса in vitro, включающий применение эффективного количества соединения по п.1.
13. Применение композиции по п.8 для приготовления лекарства, предназначенного для лечения СПИДа.
Текст
1 Заявка является частичным продолжением заявки на патент США 08/402287, поданной на имя настоящего заявителя 10 марта 1995 г., которая полностью включена в настоящее описание в качестве ссылки. Предпосылки создания изобретения Настоящее изобретение относится к ингибиторам протеаз ретровирусов, в частности к новым соединениям, к композиции и способу ингибирования ретровирусных протеаз, таких как протеаза вируса иммунодефицита человека(ВИЧ). Настоящее изобретение, в частности,относится к соединениям из класса гидроксиэтиламинсульфонамидов аминокислот, обладающих способностью ингибировать протеазы ретровирусов, к композиции и к способу ингибирования протеаз ретровирусов, профилактическому предотвращению ретровирусной инфекции или распространения ретровируса и к лечению ретровирусной инфекции, например ВИЧ-инфекции. Изобретение также относится к способу получения таких соединений, а также к промежуточным продуктам, используемым в этих способах. Во время цикла репликации ретровирусов продукты транскрипции гена gag и gag-pol транслируются в виде протеинов. Эти протеины далее подвергаются воздействию (процессингу) кодируемой вирусом протеазы (или протеиназы), что приводит к образованию ферментов вируса и структурных протеинов ядра вируса. В основном протеины-предшественники гена gag процессируются в протеины ядра, а протеиныпредшественники гена роl процессируются в ферменты вируса, например в обратную транскриптазу и протеазу ретровируса. Было установлено, что правильный процессинг протеинов-предшественников протеазой ретровируса необходим для сборки инфекционных вирионов. Например, было подтверждено, что мутации"сдвига рамки" в протеазной области гена pol ВИЧ препятствуют процессингу протеинапредшественника гена gag. Также было подтверждено, что сайтнаправленный мутагенез остатка аспарагиновой кислоты в активном сайте протеазы ВИЧ предотвращает процессинг протеина-предшественника гена gag. Таким образом, были предприняты попытки ингибировать репликацию вируса путем ингибирования активности протеаз ретровирусов. Ингибирование протеазы ретровируса обычно включает использование переходной миметической фазы, при этом протеаза ретровируса подвергается действию миметического соединения, которое конкурентно по отношению к протеинам gag и gag-pol связывается(обычно обратимым образом) с ферментом, тем самым ингибируя специфический процессинг структурных протеинов и высвобождение самой протеазы ретровируса. Таким образом, может быть осуществлено эффективное ингибирование репликационных протеаз ретровируса. 2 Специально для ингибирования протеаз, в частности для ингибирования протеазы ВИЧ,было предложено несколько классов соединений. Такие соединения включают изостеры гидроксиэтиламина и изостеры восстановленного амида. См., например, ЕР 0346847; ЕР 0342541;HIV-1 Protease", Science, 249, 527 (1990). Например, в патенте США 5157041, международных заявках WO 94/04491, WO 94/04492, WO 94/04493, WO 94/05639, WO 92/08701 и в заявке на патент США 08/294468, поданной 23 августа 1994 г. (каждый документ полностью включен в настоящее описание в качестве ссылки) описаны ингибиторы протеазы ретровируса, включающие изостеры гидроксиэтиламина, гидроксиэтилмочевины или гидроксиэтилсульфонамида. Известны несколько классов соединений,которые используются в качестве ингибиторов протеолитического фермента ренина. См., например, патент США 4599198; патент Великобритании 2184730; патент Великобритании 2209752; европейскую заявку ЕР 0264795; патент Великобритании 2200115 и U.S. Sir Н 725. Среди этих публикаций в патенте Великобритании 2200115, патенте Великобритании 2209752,европейской заявке ЕР 0264795, U.S. Sir H725 и в патенте США 4599198 описаны содержащие мочевину ингибиторы ренина из класса гидроксиэтиламинов. В европейской заявке ЕР 0468641 описаны ингибиторы ренина и промежуточные продукты для получения ингибиторов, которые включают сульфонамидсодержащие соединения из класса гидроксиэтиламинов,такие, как 3-(трет-бутоксикарбонил) аминоциклогексил-1-(фенилсульфонил)амино-2(5)бутанол. В патенте Великобритании 2200115 описаны сульфамоилсодержащие ингибиторы ренина из класса гидроксиэтиламинов, а в ЕР 0264795 описаны некоторые сульфонамидсодержащие ингибиторы ренина из класса гидроксиэтиламинов. Однако известно, что хотя протеазы ренина и ВИЧ обе классифицируются как аспартильные протеазы, обычно нельзя предсказать, что соединения, эффективные в качестве ингибиторов ренина, будут эффективными ингибиторами и для протеазы ВИЧ. Краткое описание изобретения Настоящее изобретение относится к определенным соединениям, являющимся ингибиторами протеазы ретровируса, их аналогам и фармацевтически приемлемым солям, сложным эфирам и пролекарствам. Соединения, являющиеся предметом изобретения, характеризуются как соединения-ингибиторы, представляющие собой гидроксиэтиламиносульфонамиды аминокислот. Соединения по изобретению преимущественно ингибируют протеазы ретровирусов, 3 такие как протеаза вируса иммунодефицита человека (ВИЧ). Таким образом, настоящее изобретение также включает фармацевтические композиции, способы ингибирования протеаз ретровирусов и способы лечения или профилактики ретровирусной инфекции, такой как ВИЧинфекция. Изобретение также включает способ получения таких соединений, а также промежуточные продукты, используемые в таких способах. Подробное описание изобретения Настоящее изобретение относится к ингибирующему протеазу ретровируса соединению формулы или к его фармацевтически приемлемой соли,пролекарству или сложному эфиру, гдеR2 обозначает алкил, аралкил, алкилтиоалкил, арилтиоалкил или циклоалкилалкил; предпочтительно R2 обозначает алкил с 1-5 атомами углерода, аралкил, включающий алкил с 1-3 атомами углерода, алкилтиоалкил, включающий алкил с 1-3 атомами углерода, арилтиоалкил,включающий алкил с 1-3 атомами углерода,циклоалкилалкил, состоящий из алкила с 1-3 атомами углерода и 3-6-членного углеводородного кольца; более предпочтительно R2 обозначает алкил с 3-5 атомами углерода, арилметил,алкилтиоалкил, включающий алкил с 1-3 ато 000470 4 мами углерода, арилтиометил или циклоалкилметил, включающий 5-6-членное углеводородное кольцо; еще более предпочтительно R2 обозначает изобутил, н-бутил, CH3SCH2CH2-, бензил, фенилтиометил, (2-нафтилтио) метил, 4 метоксифенилметил, 4-гидроксифенилметил, 4 фторфенилметил или циклогексилметил; и еще более предпочтительно R2 обозначает бензил, 4 фторфенилметил или циклогексилметил; наиболее предпочтительно R2 обозначает бензил;R3 обозначает алкил, циклоалкил или циклоалкилалкил; предпочтительно R3 обозначает алкил с 1-5 атомами углерода, циклоалкил,включающий 5-8-членное кольцо, или циклоалкилметил, включающий 3-6-членное кольцо; более предпочтительно R3 обозначает пропил,изоамил, изобутил, бутил, циклопентилметил,циклогексилметил, циклогексил или циклогептил; наиболее предпочтительно R3 обозначает изобутил или циклопентилметил;R4 обозначает арил, гетероарил или гетероциклил; предпочтительно R4 обозначает арил,сконденсированный с бензольным ядром 5-6 членный гетероарил или сконденсированный с бензольным ядром 5-6-членный гетероцикл; или где А и В каждый независимо друг от друга обозначает O, S, SO или SO2; предпочтительно А и В каждый обозначает О;R6 и R7 каждый независимо друг от друга обозначает фтор или хлор; и предпочтительноN,N-дизамещенный аминоалкил, где указанные заместители представляют собой алкил или аралкил, карбоксиалкил, алкоксикарбонилалкил,цианалкил или гидроксиалкил; предпочтительноR20 обозначает водород, алкил с 1-5 атомами углерода, алкенил с 2-5 атомами углерода, алкинил с 2-5 атомами углерода, аралкил, включающий алкил с 1-5 атомами углерода, гетероаралкил, состоящий из 5-6-членного кольца и алкила с 1-5 атомами углерода, гетероциклоалкил, состоящий из 5-6-членного кольца и алкила с 1-5 атомами углерода, аминоалкил с 2-5 атомами углерода, N-монозамещенный или N,Nдизамещенный аминоалкил, включающий алкил с 2-5 атомами углерода, где указанные заместители представляют собой алкил с 1-3 атомами углерода, аралкил, включающий алкил с 1-3 атомами углерода, карбоксиалкил с 1-5 атомами углерода, алкоксикарбонилалкил, включающий алкил с 1-5 атомами углерода, цианалкил с 1-5 атомами углерода или гидроксиалкил с 2-5 атомами углерода; более предпочтительно R20 обозначает водород, алкил с 1-5 атомами углерода,фенилалкил, включающий алкил с 1-3 атомами углерода, гетероциклоалкил, состоящий из 5-6 членного кольца и алкила с 1-3 атомами углерода, или N-монозамещенный или N,Nдизамещенный аминоалкил с 2-3 атомами углерода, где указанные заместители представляют собой алкил с 1-3 атомами углерода; и наиболее предпочтительно R20 обозначает водород, метил, этил, пропил, изопропил, изобутил, бензил,2-(1-пирролидинил)этил,2-(1-пиперидинил) этил, 2-(1-пиперазинил)этил, 2-(4-метилпиперазин-1-ил)этил, 2-(1-морфолинил)этил, 2-(1 тиаморфолинил)этил или 2-(N,N-диметиламино) этил;R21 обозначает водород или алкил; предпочтительно R21 обозначает атом водорода или алкил с 1-3 атомами углерода; более предпочтительно R21 обозначает водород или метил; и наиболее предпочтительно R21 обозначает атом водорода; или группа формулы NR20R21 обозначает гетероцикл; предпочтительно группа формулы NR20R21 обозначает 5-6-членный гетероцикл; более предпочтительно группа формулыR22 обозначает алкил или R20R21N-aлкил; предпочтительно R22 обозначает алкил илиR20R21N- алкил, где алкил имеет 1-3 атома углерода; и более предпочтительно R22 обозначает алкил с 1-3 атомами углерода; иR10 обозначает водород, алкил, гидроксиалкил или алкоксиалкил, где алкил имеет 1-3 атома углерода; предпочтительно R10 обозначает атом водорода;R11 обозначает алкил с 1-5 атомами углерода, предпочтительно метил, изопропил, бутил,втор-бутил или изобутил; гидроксиалкил с 1-4 атомами углерода, предпочтительно гидроксиметил или гидроксиэтил; алкоксиалкил с 1-4 атомами углерода, предпочтительно метоксиметил или метоксиэтил; или водород, бензил, имидазолилметил, -CH2CH2CONH2, -CH2CONH2,-CH2CH2SCH3 или -CH2SCH3 или их сульфоновые или сульфоксидные производные; более предпочтительно R11 обозначает атом водорода;R12 обозначает водород, гидроксиалкил или алкоксиалкил; предпочтительно R12 обозначает водород, гидроксиалкил или алкоксиалкил,где алкил имеет 1-3 атома углерода; предпочтительно R12 обозначает атом водорода; и R13 и R14 каждый независимо друг от друга обозначает водород, гидрокси, алкокси, гидроксиалкокси,гидроксиалкил или алкоксиалкил; предпочтительно R13 и R14 каждый независимо друг от друга обозначает водород, гидрокси, алкокси, 2 гидроксиэтокси, гидроксиалкил или алкоксиал 7 кил; где алкил имеет 1-3 атома углерода; более предпочтительно R13 и R14 каждый независимо друг от друга обозначает водород, гидрокси,метокси или этокси; илиR12 и R13 или R13 и R14 вместе с атомами углерода, к которым они присоединены, обозначают 5-6 членный гетероарил или бензо, каждый из которых необязательно замещен, по крайней мере, одной гидрокси- или алкоксигруппой с 1-3 атомами углерода; предпочтительно R12 и R13 или R13 и R14 вместе с атомами углерода, к которым они присоединены, обозначают бензо, который необязательно замещен,по крайней мере, одной гидрокси- или метоксигруппой. Абсолютная стереохимия атома углерода-СН(ОН)-группы предпочтительно является (R)формой. Абсолютная стереохимия атома углерода -CH(R1)-группы предпочтительно является(S)-формой. Абсолютная стереохимия атома углерода -CH(R2)- группы предпочтительно является (S)-формой. Семейством соединений, представляющих особенный интерес среди соединений формулы или их фармацевтически приемлемые соли,пролекарства или сложные эфиры, где n, R1, R2,R3 и R4 имеют значения, указанные выше. Семейством соединений, представляющих дополнительный интерес среди соединений формулы II, являются соединения, которые соответствуют формуле или их фармацевтически приемлемые соли,пролекарства или сложные эфиры, где R1, R2,R3, R4 и n имеют значения, указанные выше. Наиболее предпочтительное семейство соединений формулы III состоит из соединений или их фармацевтически приемлемых солей,пролекарств или сложных эфиров, гдеR4 имеет значения, указанные выше. Представляющие интерес соединения включают следующие: 2S-(пирролидин-1-ил)ацетил]амино]-N[2R-гидрокси-3-(1,3-бензодиоксол-5-ил)суль 000470(2-метилпропил)амино]-1S-(фeнилмeтил)пропил]-4-пентинамид; 2S-(пирролидин-1-ил)ацетил]амино]-N[2R-гидрокси-3-(2-нафтил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил]-3,3 диметилбутанамид; 2S-(пирролидин-1-ил)ацетил]амино]-N[2R-гидрокси-3-(2-нафтил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил]-3 метилбутанамид; 2S-(пирролидин-1-ил)ацетил]амино]-N[2R-гидрокси-3-(2-нафтил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропил]-3Sметилпентанамид; 2S-(пирролидин-1-ил)ацетил]амино]-N[2R-гидрокси-3-(2-нафтил)сульфонил](2-метилпропил)амино]-1S-(фeнилмeтил)пропил]-4 пентинамид; 2S-(пирролидин-1-ил)ацетил]амино]-N[2R-гидрокси-3-(1,4-бензодиоксан-6-ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил) пропил]-3,3-диметилбутанамид; 2S-(пирролидин-1-ил)ацетил]амино]-N[2R-гидрокси-3-(1,4-бензодиоксан-6-ил)сульфонил](2-метилпропил)амино]-1S-(фeнилмeтил) пропил]-3-метилбутанамид; 2S-(пирролидин-1-ил)ацетил]амино]-N[2R-гидрокси-3-(1,4-бензодиоксан-6-ил)сульфонил](2-метилпропил)амино]-1S-(фeнилмeтил) пропил]-3S-метилпентанамид; и 2S-(пирролидин-1-ил)ацетил]амино]-N[2R-гидрокси-3-(1,4-бензодиоксан-6-ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил) пропил]-4-пентинамид. В контексте данного описания понятие"алкил" индивидуально или в комбинации обозначает алкильный радикал с прямой или разветвленной цепью, содержащий предпочтительно от 1 до 8 атомов углерода, более предпочтительно от 1 до 5 атомов углерода, наиболее предпочтительно от 1 до 3 атомов углерода. Примеры таких радикалов включают метил,этил, н-пропил, изопропил, н-бутил, изобутил,втор-бутил, трет-бутил, пентил, изоамил, гексил, октил и т.п. Понятие "алкенил" индивидуально или в комбинации обозначает углеводородный радикал с прямой или разветвленной цепью, имеющий одну или более двойных свя 000470 10 зей и содержащий предпочтительно от 2 до 10 атомов углерода, более предпочтительно от 2 до 8 атомов углерода, наиболее предпочтительно от 2 до 5 атомов углерода. Примеры соответствующих алкенильных радикалов включают этенил,пропенил,2-метилпропенил,1,4 бутадиенил и т.п. Понятие "алкинил" индивидуально или в комбинации обозначает углеводородный радикал с прямой или разветвленной цепью, имеющий одну или более тройных связей и содержащий предпочтительно от 2 до 10 атомов углерода, более предпочтительно от 2 до 5 атомов углерода. Примеры алкинильных радикалов включают этинил, пропинил (пропаргил), бутинил и т.п. Понятие "алкокси" индивидуально или в комбинации обозначает алкилэфирный радикал, где понятие "алкил" имеет значения, указанные выше. Примеры соответствующих алкилэфирных радикалов включают метокси, этокси, н-пропокси, изопропокси, нбутокси, изобутокси, втор-бутокси, третбутокси и т.п. Понятие "циклоалкил" индивидуально или в комбинации обозначает насыщенный или частично насыщенный моно-, би- или трициклический алкильный радикал, где каждый циклический фрагмент предпочтительно содержит в кольце от 3 до 8 атомов углерода,более предпочтительно от 3 до 7 атомов углерода, наиболее предпочтительно от 5 до 6 атомов углерода, и который необязательно может представлять собой конденсированную с бензогруппой кольцевую систему, которая необязательно замещена, как это определено в настоящем описании по отношению к арилу. Примеры таких циклоалкильных радикалов включают циклопропил, циклобутил, циклопентил, циклогексил,циклогептил, октагидронафтил, 2,3-дигидро-1Hинденил, адамантил и т.п. Предполагается, что понятия "бициклический" и "трициклический",используемые в данном описании, включают как конденсированные кольцевые системы, такие как нафтил и -карболинил, так и замещенные кольцевые системы, такие как бифенил,фенилпиридил, нафтил и дифенилпиперазинил. Понятие "циклоалкилалкил" обозначает алкильный радикал, как определено выше, который замещен циклоалкильным радикалом, как определено выше. Примеры таких циклоалкилалкильных радикалов включают циклопропилметил, циклобутилметил, циклопентилметил, циклогексилметил, 1-циклопентилэтил, 1-циклогексилэтил,2-циклопентилэтил,2-циклогексилэтил, циклобутилпропил, циклопентилпропил, циклогексилбутил и т.п. Понятие "бензо" индивидуально или в комбинации обозначает двухвалентный радикал C6H4=, производный от бензола. Понятие "арил" индивидуально или в комбинации обозначает фенильный или нафтильный радикал, необязательно замещенный одним или несколькими заместителями, выбранными из алкила, алкокси, галогена, гидрокси, амино, нитро, циано, галоидалкила, карбок 11 си, алкоксикарбонила, циклоалкила, гетероцикло, алканоиламино, амидо, амидино, алкоксикарбониламино, N-алкиламидино, алкиламино,диалкиламино, N-алкиламидо, N,N-диалкиламидо, аралкоксикарбониламино, алкилтио, алкилсульфинила, алкилсульфонила и т.п. Примерами арильных радикалов являются фенил, паратолил, 4-метоксифенил, 4-(трет-бутокси) фенил, 3-метил-4-метоксифенил, 4-фторфенил, 4 хлорфенил, 3-нитрофенил, 3-аминофенил, 3 ацетамидофенил, 4-ацетамидофенил, 2-метил-3 ацетамидофенил,4-СF3-фенил,2-метил-3 аминофенил, 3-метил-4-аминофенил, 2-амино-3 метилфенил, 2,4-диметил-3-аминофенил, 4 гидроксифенил, 3-метил-4-гидроксифенил, 1 нафтил, 2-нафтил, 3-амино-1-нафтил, 2-метил-3 амино-1-нафтил,6-амино-2-нафтил,4,6 диметокси-2-нафтил, пиперазинилфенил и т.п. Понятия "аралкил" и "аралкокси" индивидуально или в комбинации обозначают алкильный или алкоксильный радикалы, как определено выше, в которых, по крайней мере, один атом водорода замещен арильным радикалом, как определено выше, например бензил, бензилокси, 2-фенилэтил, дибензилметил, гидроксифенилметил, метилфенилметил, дифенилметил,дифенилметокси, 4-метоксифенилметокси и т.п. Понятие "аралкоксикарбонил" индивидуально или в комбинации обозначает группу формулы аралкил-О-С(О)-, в которой понятие "аралкил" имеет указанное выше значение. Примерами аралкоксикарбонильного радикала являются бензилоксикарбонил и 4-метоксифенилметоксикарбонил. Понятие "арилокси" обозначает группу формулы арил-O-, в которой понятие "арил" имеет указанное выше значение. Понятие "алканоил" индивидуально или в комбинации обозначает ацильный радикал, производный от алканкарбоновой кислоты, примеры которого включают ацетил, пропионил, бутирил, валерил,4-метилвалерил и т.п. Понятие "циклоалкилкарбонил" обозначает ацильный радикал формулы циклоалкил-С(О)-, в котором понятие "циклоалкил" имеет указанное выше значение, такой как циклопропилкарбонил, циклогексилкарбонил,адамантилкарбонил, 1,2,3,4-тетрагидро-2-нафтоил, 2-ацетамидо-1,2,3,4-тетрагидро-2-нафтоил,1-гидрокси-1,2,3,4-тетрагидро-6-нафтоил и т.п. Понятие "аралканоил" обозначает ацильный радикал, производный от замещенной арилом алканкарбоновой кислоты, такой как фенилацетил, 3-фенилпропионил (гидроциннамоил), 4 фенилбутирил, (2-нафтил) ацетил, 4-хлоргидроциннамоил, 4-аминогидроциннамоил, 4 метоксигидроциннамоил и т.п. Понятие "ароил" обозначает ацильный радикал, производный от арилкарбоновой кислоты, где "арил" имеет указанные выше значения. Примеры таких ароильных радикалов включают замещенный и незамещенный бензоил или нафтоил, например бензоил, 4-хлорбензоил, 4-карбоксибензоил, 4(бензилоксикарбонил) бензоил, 1-нафтоил, 2 000470 12 нафтоил, 6-карбокси-2-нафтоил, 6-(бензилоксикарбонил)-2-нафтоил, 3-бензилокси-2-нафтоил,3-гидрокси-2-нафтоил,3-(бензилоксиформамидо)-2-нафтоил и т.п. Понятие "гетероцикл" индивидуально или в комбинации обозначает насыщенный или частично ненасыщенный моноциклический, бициклический или трициклический гетероцикл, содержащий в кольце, по крайней мере, один, предпочтительно 1-4, более предпочтительно 1-2 атома азота, кислорода или серы и имеющий предпочтительно 3-8 членов в каждом кольце, более предпочтительно 3-7 и наиболее более предпочтильно 5-6 членов в каждом кольце. Подразумевается, что понятие"гетероцикл" включает сульфоны, сульфоксиды,N-оксиды третичного азота в кольце и кольцевые системы, сконденсированные с карбоциклическим радикалом и бензогруппой. В таких гетероциклических радикалах, по крайней мере,один, предпочтительно 1-4, более предпочтительно 1-2 атома углерода, необязательно могут быть замещены галогеном, алкилом, алкокси-,гидрокси-, оксогруппой, арилом, аралкилом,гетероарилом, гетероаралкилом, амидино-, Nалкиламидино-, алкоксикарбониламино-, алкилсульфониламиногруппой и т.п., и/или вторичный атом азота (т.е. -NH-) может быть замещен гидроксигруппой, алкилом, аралкоксикарбонилом, алканоилом, гетероаралкилом, фенилом или фенилалкилом, и/или третичный атом азота(т.е. =N-) может быть замещен оксидогруппой."Гетероциклоалкил" обозначает алкильный радикал, как определено выше, в котором, по крайней мере, один атом водорода замещен гетероциклом, как определено выше, например пирродинилметил, тетрагидротиенилметил, пиридилметил и т.п. Понятие "гетероарил" индивидуально или в комбинации обозначает ароматический гетероцикл, как определено выше,который необязательно замещен, как определено выше по отношению к определениям арила и гетероцикла. Примерами таких гетероциклических и гетероарильных групп являются пирролидинил, пиперидинил, пиперазинил, морфолинил, тиаморфолинил, пирролил, имидазолил 13 5-бензимидазолил,метилендиоксифен-4-ил,метилендиоксифен-5-ил,этилендиоксифенил,бензотиазолил, бензопиранил, бензофурил, 2,3 дигидробензофурил, бензоксазолил, тиофенил и т.п. Понятие "циклоалкилалкоксикарбонил" обозначает ацильную группу, производную от циклоалкилалкоксикарбоновой кислоты формулы циклоалкилалкил-O-СООН, где циклоалкилалкил имеет указанное выше значение. Понятие "арилоксиалканоил" обозначает ацильную группу формулы арил-O-алканоил, где арил и алканоил имеют указанные выше значения. Понятие "гетероциклоалкоксикарбонил" обозначает ацильную группу, производную от гетероциклоалкил-O-СООН, где гетероциклоалкил имеет указанное выше значение. Понятие "гетероциклоалканоил" обозначает ацильную группу,производную от гетероциклоалкилкарбоновой кислоты, где гетероцикл имеет указанные выше значения. Понятие "гетероциклоалкоксикарбонил" обозначает ацильную группу, производную от гетероциклоалкил-О-СООН, где гетероцикл имеет указанные выше значения. Понятие "гетероарилоксикарбонил" обозначает ацильную группу, производную от карбоновой кислоты и представленную формулой гетероарил-OСООН, где гетероарил имеет указанные выше значения. Понятие "аминокарбонил" индивидуально или в комбинации обозначает замещенную аминогруппой карбонильную (карбамоильную) группу, где аминогруппа может представлять собой первичную, вторичную или третичную аминогруппу, содержащую заместители,выбранные из группы, включающей алкил,арил, аралкил, циклоалкил, циклоалкилалкил и т.п. Понятие "аминоалканоил" обозначает ацильную группу, производную от алкилкарбоновой кислоты и замещенную аминогруппой,где аминогруппа может представлять собой первичную, вторичную или третичную аминогруппу, содержащую заместители, выбранные из группы, включающей алкил, арил, аралкил,циклоалкил, циклоалкилалкил и т.п. Понятие"галоген" обозначает фтор, хлор, бром или йод. Понятие "галоидалкил" обозначает алкил,имеющий указанные выше значения, где один или более атомов водорода замещены галогеном. Примеры таких галоидалкильных радикалов включают хлорметил, 1-бромэтил, фторметил,дифторметил,трифторметил,1,1,1 трифторэтил и т.п. Понятие "уходящая группа"(L или W) обычно обозначает группы, легко замещаемые нуклеофильной группой, такой как амин, тиол или спиртовая нуклеофильная группа. Такие уходящие группы хорошо известны в данной области техники. Примеры таких уходящих групп включают, но не ограничены ими,N-гидроксисукцинимид, N-гидроксибензотриазол, галогениды, трифлаты, тозилаты и т.п. Предпочтительные уходящие группы при необходимости указаны в данном описании. 14 Способы получения соединений формулыI описаны ниже. Следует отметить, что общая методика относится к получению соединений,имеющих определенную стереохимическую конфигурацию, например, в которых абсолютная стереохимическая конфигурация относительно гидроксильной группы обозначена как(R), однако такая методика в целом применима и к соединениям противоположной конфигурации, например, у которых стереохимическая конфигурация относительно гидроксильной группы обозначена как (S). Кроме того, соединения, имеющие (R)-стереохимическую конфигурацию, могут использоваться для получения соединений, имеющих (S)-стереохимическую конфигурацию. Например, соединение, имеющее (R)-стереохимическую конфигурацию, может быть преобразовано в таковое, имеющее (S)-стереохимическую конфигурацию, хорошо известными способами. Получение соединений формулы I Соединения по настоящему изобретению,представленные выше формулой I, могут быть получены с использованием следующих общих способов, схематически показанных на схемах I и II.N-защищенное хлоркетоновое производное аминокислоты формулы где Р обозначает аминозащитную группу и R2 имеет указанные выше значения, восстанавливают до соответствующего спирта, используя соответствующий восстановитель. Пригодные аминозащитные группы хорошо известны в данной области техники и включают карбобензокси, трет-бутоксикарбонил и т.п. Предпочтительной аминозащитной группой является карбобензокси. Предпочтительным N-защищенным хлоркетоном является N-бензилоксикарбонилL-фенилаланинхлорметилкетон. Предпочтительным восстановителем является борогидрид натрия. Реакцию восстановления проводят при температуре от -10 С до приблизительно 25 С,предпочтительно при приблизительно 0 С, в пригодной системе растворителей, такой как,например, тетрагидрофуран, и т.п. Nзащищенные хлоркетоны являются коммерчески доступными, например, поставляются фирмой Bachem, Inc. Torrance, California. В альтернативном варианте хлоркетоны могут быть получены способом, описанным у S.J. Fittkau, J.Prakt. Chem., 315, 1037 (1973), с последующим получением N-защищенных производных с использованием методов, хорошо известных в данной области техники. Галоидспирт может применяться непосредственно, как описано ниже, или же предпочтительно его подвергают взаимодействию предпочтительно при комнатной температуре с пригодным основанием в соответствующей сис 000470 16 теме растворителей с получением защищенного аминоэпоксида формулы:N где Р и R2 имеют указанные выше значения. Пригодные системы растворителей для получения аминоэпоксида включают этанол, метанол,изопропанол, тетрагидрофуран, диоксан и т.п., в том числе их смеси. Пригодные основания для получения эпоксида из восстановленного хлоркетона включают гидроксид калия, гидроксид натрия, трет-бутоксид калия, DBU и т.п. Предпочтительным основанием является гидроксид калия. В альтернативном варианте защищенный аминоэпоксид может быть получен согласно находящимся в процессе одновременного рассмотрения международным заявкам на имя настоящего заявителя PCT/US 93/04804 (WO 93/23388) и PCT/US 94/12201 и заявке на патент США, находящейся у патентного поверенного с регистрационным номеромС-2860 (каждая из этих публикаций полностью включена в настоящее описание в качестве ссылки), в которых представлены методы получения хирального эпоксида, хирального циангидрина, хирального амина и других хиральных промежуточных продуктов, которые могут использоваться для получения ингибиторов протеазы ретровируса,при этом процесс начинают с DL-, D- или Lаминокислот, которые подвергают взаимодействию с соответствующей аминозащитной группой в пригодном растворителе, получая аминозащищенный эфир аминокислоты. Например,для получения ингибиторов по настоящему изобретению может быть использована защищенная L-аминокислота следующей формулы где Р 3 обозначает карбоксизащитную группу,например, метил, этил, бензил, трет-бутил, 4 метоксифенилметил и т.п.; R2 имеет указанные выше значения; и Р 1 и Р 2 и/или Р' независимо друг от друга выбирают из аминозащитных групп, которые включают, но не ограничены ими, аралкил, замещенный аралкил, циклоалкенилалкил и замещенный циклоалкенилалкил, аллил, замещенный аллил, ацил, алкоксикарбонил, аралкоксикарбонил и силил. Примеры аралкила включают, но не ограничены ими, бензил, ортометилбензил, тритил и бензгидрил, который необязательно может быть замещен галогеном,С 1-С 8 алкилом, алкокси-, гидрокси-, нитрогруппой, алкиленом, амино-, алкиламино-, ациламиногруппой и ацилом, или их соли, такие как соли фосфония и аммония. Примеры арильных 17 групп включают фенил, нафталинил, инданил,антраценил, дуренил, 9-(9-фенилфлуоренил) и фенантренил, циклоалкенилалкил или замещенный циклоалкенилалкил, включающий C6-С 10 циклоалкилы. Пригодные ацильные группы включают карбобензокси, трет-бутоксикарбонил, изобутоксикарбонил, бензоил, замещенный бензоил, бутирил, ацетил, трифторацетил, трихлорацетил, фталоил и т.п. Предпочтительно Р 1 и Р 2 независимо друг от друга выбирают из аралкила и замещенного аралкила. Более предпочтительно Р 1 и Р 2 каждый обозначает бензил. Кроме того, защитные группы Р 1 и/или Р 2 и/или Р' могут образовывать гетероциклическое кольцо с азотом, к которому они присоединены,например, 1,2-бис (метилен) бензол, фталимидил, сукцинимидил, малеимидил и т.п., и эти гетероциклические группы дополнительно могут включать примыкающие арильные и циклоалкильные кольца. Кроме того, гетероциклические группы могут быть моно-, ди- или тризамещенными, например, нитрофталимидил. Понятие "силил" относится к атому кремния, необязательно замещенному одной или несколькими алкильной, арильной или аралкильной группами. Пригодные силильные защитные группы включают, но не ограничены ими, триметилсилил, триэтилсилил, триизопропилсилил, третбутилдиметилсилил, диметилфенилсилил, 1,2 бис(диметилсилил)бензол, 1,2-бис(диметилсилил)этан и дифенилметилсилил. Путем силилирования аминных функций с получением моноили бисдисилиламина можно получать производные аминоспирта, аминокислоты, эфиров аминокислоты и амида аминокислоты. В случае аминокислот, эфиров аминокислот и амидов аминокислот восстановление карбонильной функции позволяет получить требуемый моноили биссилиловый аминоспирт. Силилирование аминоспирта может привести к получениюN,N,O-трисилильного производного. Удаление силильной функции из функции силилового эфира легко осуществляют обработкой, например, таким реагентом, как гидроксид металла или фторид аммония, или на отдельной стадии реакции, или in situ в процессе получения аминоальдегидного реагента. Пригодными силилирующими агентами являются, например, триметилсилилхлорид,трет-бутилдиметилсилилхлорид, фенилдиметилсилилхлорид, дифенилметилсилилхлорид или продукты их сочетания с имидазолом или ДМФ. Способы силилирования аминов и удаления силильных защитных групп хорошо известны специалистам в данной области техники. Способы получения этих аминовых производных из соответствующих аминокислот,амидов аминокислот или эфиров аминокислот хорошо известны специалистам в области органической химии, включающей химию аминокислот/эфиров аминокислот или аминоспиртов.Lаминокислоты восстанавливают до соответствующего спирта. Например, аминозащищенный эфир L-аминокислоты может быть восстановлен гидридом диизобутилалюминия при -78 С в пригодном растворителе, таком, как толуол. Предпочтительные восстановители включают алюмогидрид лития, борогидрид лития, борогидрид натрия, боран, три- трет-бутоксиалюмогидрид лития, комплекс боран/ТГФ. Наиболее предпочтительным восстановителем является гидрид диизобутилалюминия (ДИБАЛ-Г) в толуоле. Образовавшийся спирт затем превращают, например, путем окисления по Сверну в соответствующий альдегид формулы где Р 1, Р 2 и R2 имеют указанные выше значения. Например, раствор спирта в дихлорметане добавляют к охлажденному (от -75 С до -68 С) раствору оксалилхлорида в дихлорметане и ДМСО в дихлорметане и перемешивают в течение 35 мин. Пригодные окислители включают, например, комплекс триоксид серы-пиридин и ДМСО,оксалилхлорид и ДМСО, ацетилхлорид или ангидрид и ДМСО, трифторацетилхлорид или ангидрид и ДМСО, метансульфонилхлорид и ДМСО или тетрагидротиафен-S-оксид, толуолсульфонилбромид и ДМСО, трифторметансульфониловый ангидрид (трифлиновый ангидрид) и ДМСО, пентахлорид фосфора и ДМСО, диметилфосфорилхлорид и ДМСО и изобутилхлорформиат и ДМСО. Условия окисления описаны у Reetz и др., [Angew. Chem., 99, стр. 1186,(1987), Angew. Chem., Int. Ed. EngL, 26, стр. 1141, (1987)], где применяли оксалилхлорид и ДМСО при -78 С. Предпочтительный способ окисления,описанный в настоящем изобретении, основан на использовании комплекса триоксид серыпиридин, триэтиламина и ДМСО при комнатной температуре. Эта система позволяет получить очень высокий выход требуемого хирального защищенного аминоальдегида, который не нуждается в очистке для дальнейшего применения,т.е. не требует очистки килограммовых количеств промежуточных продуктов с помощью хроматографии, что делает менее опасными крупномасштабные процессы. Проведение реакции при комнатной температуре также устраняет необходимость использования низкотемпературного реактора, что делает процесс более пригодным для коммерческого производства. Реакция может быть проведена в атмосфере инертного газа, такого как азот или аргон,или нормального либо сухого воздуха при атмосферном давлении или в закрытом реакционном сосуде при избыточном давлении. Предпочтительной является атмосфера азота. Альтерна 19 тивные аминные основания включают, например, трибутиламин, триизопропиламин, Nметилпиперидин, N-метилморфолин, азабициклононан,диизопропилэтиламин,2,2,6,6 тетраметилпиперидин, N,N-диметиламинопиридин или смеси этих оснований. Предпочтительным основанием является триэтиламин. Альтернативами чистому ДМСО в качестве растворителя являются смеси ДМСО с непротонными или галогенированными растворителями, такими как тетрагидрофуран, этилацетат, толуол,ксилол, дихлорметан, этилендихлорид и т.п. Биполярные апротонные сорастворители включают ацетонитрил, диметилформамид, диметилацетамид, ацетамид, тетраметилмочевину и ее циклический аналог,N-метилпирролидон,сульфолан и т.п. Кроме N,N-дибензилфенилаланинола в качестве предшественника альдегидов для получения соответствующих Nмонозамещенного [либо Р 1, либо Р 2 обозначает Н] или N,N-дизамещенного альдегида могут применяться указанные выше производные фенилаланинола. Кроме того, для получения альдегидов может быть осуществлено гидридное восстановление амида или эфирного производного соответствующего защищенного по атому азота с помощью бензильной (или другой пригодной) защитной группы фенилаланина, замещенного фенилаланина или циклоалкильного аналога производного фенилаланина. Перенос гидрида представляет собой дополнительный метод синтеза альдегидов в условиях, при которых исключены конденсации альдегида, ср. с окислением по Оппенауэру. В этом процессе альдегиды также могут быть получены способами восстановления защищенного фенилаланина и аналогов фенилаланина или их амидных или эфирных производных, например, с помощью амальгамы натрия с НСl в этаноле или лития, натрия, калия либо кальция в аммиаке. Температура реакции может составлять от приблизительно -20 С до приблизительно 45 С и предпочтительно от приблизительно 5 С до приблизительно 25 С. Два дополнительных способа получения защищенного по атому азота альдегида включают окисление соответствующего спирта хлорной известью в присутствии каталитического количества свободного радикала 2,2,6,6-тетраметил-1-пиридилокси. Во втором способе окисление спирта до альдегида осуществляют с помощью каталитического количества перрутената тетрапропиламмония в присутствии N-метилморфолин-Nоксида. В альтернативном варианте хлорангидридное производное защищенного фенилаланина или производного фенилаланина, как описано выше, может быть восстановлено с помощью водорода и катализатора, такого как Pd на карбонате бария или сульфате бария, с или без добавления смягчающего катализатор агента, та 000470 20 кого как сера или тиол (восстановление по Роземунду). Полученный в результате окисления по Сверну альдегид затем подвергают взаимодействию с таким реагентом, как галоидметиллитий, который получают in situ при взаимодействии алкиллитиевого или ариллитиевого соединения с дигалоидметаном, представленным формулой Х 1 СН 2 Х 2, где Х 1 и Х 2 независимо друг от друга обозначают I, Вr или Сl. Например, раствор альдегида и хлорйодметана в ТГФ охлаждают до -78 С и добавляют раствор нбутиллития в гексане. Образовавшийся продукт представляет собой смесь диастереомеров соответствующих аминозащищенных эпоксидов формул Диастереомеры могут быть разделены, например, хроматографией или в альтернативном варианте диастереомерные продукты могут быть разделены после взаимодействия на последующих стадиях. Вместо L-аминокислоты может быть использована D-аминокислота для получения соединений,имеющих(S)стереохимическую конфигурацию относительно атома углерода, связанного с R2. Присоединение хлорметиллития или бромметиллития к хиральному аминоальдегиду является высоко диастереоселективным. Предпочтительно хлорметиллитий или бромметиллитий получают in situ при взаимодействии дигалоидметана и н-бутиллития. Пригодные метиленирующие галоидметаны включают хлорйодметан, бромхлорметан, дибромметан, дийодметан, бромфторметан и т.п. Сложный эфир сульфокислоты и продукта присоединения, например, бромистого водорода к формальдегиду также представляет собой метиленирующий агент. Тетрагидрофуран является предпочтительным растворителем, однако могут использоваться другие растворители, такие как толуол,диметоксиэтан, этилендихлорид, метиленхлорид, как в виде чистых растворителей, так и в виде их смесей. Биполярные апротонные растворители, такие как ацетонитрил, ДМФ, Nметилпирролидон, пригодны в качестве растворителей или в качестве части смеси растворителей. Реакция может быть проведена в атмосфере инертного газа, такого как азот или аргон. нБутиллитий может быть заменен другими металлорганическими реагентами, такими как метиллитий, трет-бутиллитий, втор-бутиллитий,фениллитий, фенилнатрий и т.п. Реакцию можно осуществлять при температурах от приблизительно -80 до 0 С, но предпочтительно от приблизительно -80 до -20 С. Наиболее предпочтительные температуры реакции составляют от -40 до -15 С. Реагенты можно добавлять по отдельности, однако в определенных условиях 21 предпочтительны совместные добавления. Предпочтительным давлением при проведении реакции является атмосферное, однако в определенных условиях, таких как повышенная влажность окружающей среды, целесообразно повышенное давление. Альтернативные способы превращения в эпоксиды по настоящему изобретению включают замену на другие виды заряженных предшественников агентов метиленирования с последующей их обработкой основанием с получением аналогичного аниона. Примеры этих видов включают тозилат или трифталат триметилсульфоксония, галогенид тетраметиламмония,галогенид метилдифенилсульфоксония, где галогенид представляет собой хлорид, бромид или йодид. Превращение альдегидов по настоящему изобретению в их эпоксидное производное также можно осуществлять в несколько стадий. Например, добавление аниона тиоанизола, полученного, например, из бутил- или ариллитиевого реагента, к защищенному аминоальдегиду,и окисление полученного защищенного аминосульфидного спирта хорошо известными окислителями, такими как перекись водорода, третбутилгипохлорит, хлорная известь или перйодат натрия, приводит к получению сульфоксида. Алкилирование сульфоксида, например, метилйодидом или бромидом, метилтозилатом,метилмезилатом, метилтрифлатом, этилбромидом, изопропилбромидом, бензилхлоридом или т.п. можно проводить в присутствии органического или неорганического основания. В альтернативном варианте защищенный аминосульфидный спирт может быть алкилирован, например, с помощью указанных выше алкилирующих агентов с получением солей сульфония,которые затем превращают в требуемые эпоксиды с помощью трет-амина или неорганических оснований. Требуемые эпоксиды образуются (с использованием наиболее предпочтительных условий) избирательно по отношению к диастереомерам в соотношении, по крайней мере, приблизительно 85:15 (соотношение S:R). Продукт может быть очищен хроматографией с получением диастереомерно- и энантиомерно чистого продукта, но обычно он может быть использован непосредственно без очистки для получения ингибиторов протеазы ретровируса. Описанный выше способ применим как для смесей оптических изомеров, так и для разделенных соединений. Если требуется конкретный оптический изомер, то он может быть выделен с помощью выбора исходных продуктов, например Lфенилаланина,D-фенилаланина,L-фенилаланинола, D-фенилаланинола, D-гексагидрофенилаланинола и т.п., или же разделение можно осуществлять на промежуточных или конечной стадиях. Для образования солей, сложных эфиров или амидов соединений по настоящему 22 изобретению могут быть использованы хиральные вспомогательные вещества, такие как один или два эквивалента камфорсульфокислоты,лимонной кислоты, камфарной кислоты, 2 метоксифенилуксусной кислоты и т.п. Эти соединения или производные могут быть подвергнуты кристаллизации или выделены хроматографическим путем с использованием либо хиральной, либо нехиральной колонки, как это известно специалистам в данной области техники. Затем аминоэпоксиды подвергают взаимодействию в пригодной системе растворителей с равным количеством или предпочтительно с избытком соответствующего амина формулыR3NH2, где R3 обозначает водород или имеет указанные выше значения. Реакцию можно проводить в широком диапазоне температур, например, от приблизительно 10 С до приблизительно 100 С, но предпочтительно, хотя это не является необходимым, проводить ее при температуре, при которой начинается кипение с обратным холодильником растворителя. Пригодные системы растворителей включают протонные, апротонные и биполярные органические растворители, как, например, такие, в которых растворителем является спирт, такой как метанол, этанол, изопропанол и т.п., простые эфиры, такие как тетрагидрофуран, диоксан и т.п., и толуол, N,N-диметилформамид, диметилсульфоксид и их смеси. Предпочтительным растворителем является изопропанол. Полученный продукт является производным 3-(N-защищенный амин)-3-(R2)-1-(NНR3)-пропан-2-ола (в настоящем описании названный аминоспиртом),которое может быть представлено формулами где Р, Р 1, Р 2, R2 и R3 имеют указанные выше значения. В альтернативном варианте вместо аминоэпоксида может быть использован галоидспирт. Затем указанный выше спирт подвергают взаимодействию в соответствующем растворителе с сульфонилхлоридом R4SO2Cl, сульфонилбромидом R4SО 2 Вr или с соответствующим сульфонилангидридом, предпочтительно в присутствии акцептора кислоты. Пригодные для осуществления реакции растворители включают метиленхлорид, тетрагидрофуран и т.д. Пригодные акцепторы кислоты включают триэтиламин,пиридин и т.п. Образовавшееся сульфонамидное производное может быть представлено, в зависимости от используемого эпоксида, формулами 23 где Р, Р 1, Р 2, R2, R3 и R4 имеют указанные выше значения. Эти промежуточные продукты пригодны для получения соединений-ингибиторов по настоящему изобретению. Сульфонилгалогениды формулы R4SO2X могут быть получены взаимодействием соответствующего арила, гетероарила и конденсированного с бензогруппой гетероциклического реагента Гриньяра или литиевого реагента с сульфурилхлоридом или диоксидом серы с последующим окислением галогеном, предпочтительно хлором. Арил, гетероарил и конденсированный с бензогруппой гетероциклический реагент Гриньяра или литиевый реагент могут быть получены из соответствующих им галогенидов(таких как хлор- или бромсодержащие соединения), которые являются коммерчески доступными или легко могут быть получены из коммерчески доступных исходных продуктов с использованием методов, известных в данной области техники. Кроме того, тиолы могут быть окислены до сульфонилхлоридов с использованием хлора в присутствии воды в тщательно контролируемых условиях. Кроме того, сульфоновые кислоты, такие,как арилсульфоновые кислоты, могут быть превращены в сульфонилгалогениды с использованием таких реагентов, как РСl5, SOCl2,ClC(O)C(O)Cl и т.п., а также в ангидриды с использованием дегидратирующих реагентов. Сульфоновые кислоты могут в свою очередь быть получены по методам, хорошо известным в данной области техники. Некоторые сульфоновые кислоты являются коммерчески доступными. Вместо сульфонилгалогенидов для получения соединений, в которых фрагмент -SO2 замещен фрагментом -SO- или -S-, соответственно могут использоваться сульфинилгалогениды (R4SOX) или сульфенилгалогенды (R4SX). Арилсульфоновые кислоты, конденсированные с бензогруппой гетероциклические сульфоновые кислоты или гетероарилсульфоновые кислоты могут быть получены сульфированием ароматического кольца по методам, хорошо известным в данной области техники, такими, например, как взаимодействие с серной кислотой,SO3, с комплексами на основе SO3, такими как ДМФ(SО 3), пиридин(SО 3), N,N-диметилацетамид(SO3) и т.п. Предпочтительно арилсульфонилгалогениды получают из ароматических соединений взаимодействием с ДМФ(SO3) иSOCl2 или СlС(O)С(O)Сl. Реакции можно осуществлять постадийно или в одном сосуде. Арилсульфоновые кислоты, конденсированные с бензогруппой гетероциклосульфоновые кислоты, гетероарилсульфоновые кислоты,арилмеркаптаны, конденсированные с бензогруппой гетероциклические меркаптаны, гетероарилмеркаптаны, арилгалогениды, конденсированные с бензогруппой гетероциклические галогениды, гетероарилгалогениды и т.п. являются коммерчески доступными или легко могут 24 быть получены из коммерчески доступных исходных продуктов с использованием стандартных методов, хорошо известных в данной области техники. Например, ряд сульфоновых кислот (R4SO3H), представленных формулами где А, В, Z, R6, R7 и R9 имеют указанные выше значения, можно получить из 1,2-бензодитиола,2-меркаптофенола, 1,2-бензодиола, 2-аминобензотиазола, бензотиазола, 2-аминобензимидазола,бензимидазола и т.п., которые являются коммерчески доступными, патент США 4595407 на имя Carter; патент США 4634465 на имя Ehrenfreund и др.; Yoder и др., J. Heterocycl. Chem. 4: 166-167 (1967); Cole и др., Aust. J. Chem. 33: 675-680 (1980); Cabiddu и др., Synthesis 797-798Ansink и Cerfontain, Rec. Trav. Chim. Pays-Bas 108: 395-403 (1989); и Kajihara и Tsuchiya, EP 638564 А 1 (каждый документ полностью включен в данное описание в качестве ссылки). Например, 1,2-бензодитиол, 2-меркаптофенол или 1,2-бензодиол могут быть подвергнуты взаимодействию с R6R7C(L')2, где L' имеет указанные выше значения и предпочтительно обозначает Вr или I, в присутствии основания, такого как гидроксид, или с R6R7C=O в присутствии кислоты, такой как толуолсульфоновая кислота, или с Р 2 О 5 с получением замещенного конденсированного с бензогруппой гетероцикла формулы который затем может быть сульфонилирован до указанной выше сульфоновой кислоты. Например, CF2Br2 или CD2Br2 может быть подвергнут взаимодействию с 1,2-бензолдитиолом, 2 меркаптофенолом или 1,2-бензолдиолом в присутствии основания с получением соответственно соединений где А и В обозначают О или S и D обозначают атом дейтерия. Кроме того, когда А и/или В обозначают S, сера может быть окислена с использованием способов, описанных ниже применительно к сульфоновым или сульфоксидным производным. После получения сульфонамидного производного аминозащитную группу Р или аминозащитные группы Р 1 и Р 2 удаляют в условиях,которые не оказывают воздействия на оставшуюся часть молекулы. Эти способы хорошо известны в данной области техники и включают кислотный гидролиз, гидрогенолиз и т.п. В предпочтительном способе используется удаление защитной группы, например удаление кар 25 бобензоксигруппы, путем гидрогенолиза в присутствии палладия на угле в пригодной системе растворителей, такой как спирт, уксусная кислота и т.п. или их смеси. Когда защитная группа представляет собой трет-бутоксикарбонильную группу, она может быть удалена с использованием неорганической или органической кислоты, например, НСl или трифторуксусной кислоты, в пригодной системе растворителей,например, в диоксане или метиленхлориде. Полученный продукт представляет собой производное аминовой соли. После нейтрализации соли амин подвергают сочетанию с DL-, D- или L-аминокислотой,соответствующей формуле PNHCH(R1)СООН,где Р и R1 имеют указанные выше значения, с последующим удалением защитных групп амина, как описано выше, и сочетанию с соединением формулы где R10 и R11 имеют указанные выше значения,W обозначает уходящую группу, такую как мезилат, бром или хлор, и L обозначает уходящую группу, такую как галогенид, ангидрид, активный сложный эфир и т.п. Например, когда R10 иR11 оба обозначают атом водорода, то может быть использован бромацетилгалогенид, хлорацетилгалогенид или соответствующий ангидрид. В заключение путем взаимодействия указанного выше промежуточного продукта с циклическим амином формулы можно получить антивирусное соединение по настоящему изобретению, имеющее формулу где n, R1, R2, R3, R4, R10, R11, R12, R13 и R14 имеют указанные выше значения. Амины формулы 26 или могут быть легко получены из коммерчески доступных исходных продуктов с использованием стандартных способов, хорошо известных в данной области техники, например, таких продуктов, как 4-гидроксипролин, 3-гидроксипиперидин, 4-гидроксипиперидин, 3-пирролин,изонипекотиновая кислота, 5-гидроксииндол, 5 гидроксииндол-3-уксусная кислота, 5,6-диметоксииндол, изохинолин, хинолин, 5-гидроксиизохинолин,8-гидрокси-5-нитрохинолин,6,7 диметокси-1,2,3,4-тетрагидроизохинолин и т.п. В альтернативном варианте после нейтрализации соли амин подвергают сочетанию сDL-, D- или L-аминокислотой, соответствующей формуле PNHCH(R1)COOH, где Р и R1 имеют указанные выше значения, с последующим удалением защитных групп амина, как описано выше, и сочетанием амина с удаленными защитными группами с циклической аминокислотой формулы где n, R10, R11, R12, R13 и R14 имеют указанные выше значения, например, такой как пирролидин-1-илуксусная кислота,пиперидин-1 илуксусная кислота и т.п., с получением антивирусных соединений по настоящему изобретению. Циклические аминокислоты являются коммерчески доступными или легко могут быть получены из защищенной карбоновой кислоты с уходящей группой W (как определено выше) путем взаимодействия с циклическим амином,как показано на схеме III, или в альтернативном варианте путем взаимодействия циклического амина с соответствующим образом замещенным кетоном в присутствии аниона цианида с последующим гидролизом цианогруппы с получением соответствующей карбоновой кислоты, как показано на схеме IV, где n, Р 3, R10, R11, R12, R13 и R14 имеют указанные выше значения. В альтернативном варианте после нейтрализации соли амин подвергают сочетанию с где n, R1, R10, R11, R12, R13 и R14 имеют указанные выше значения, которая может быть получена способом, аналогичным способам сочетания, описанным выше, из DL-, D- или Lаминокислоты формулы PNHCH(R1)СООР 3, где Р 3 и R1 имеют указанные выше значения.PNHCH(R1)СООН или NH2CH(R1)COOP3, где Р,Р 3 и R1 имеют указанные выше значения, является коммерчески доступной (например, поставляется фирмой Sigma Chemical Co.) или легко может быть получена стандартными способами, хорошо известными в данной области техники из легко доступных исходных продуктов. Предпочтительно Р обозначает бензилоксикарбонил или трет-бутоксикарбонил и Р 3 обозначает бензил или трет-бутил. Для сочетания аминокислот и аминов могут быть использованы стандартные методы сочетания. Карбоксильную группу подвергают превращению с образованием ангидрида, смешанного ангидрида, галоидангидрида, такого как хлорид или бромид, или активного сложного эфира, такого как сложные эфиры N-гидроксисукцинимид,ГОБТ и т.п., с использованием хорошо известных методов и условий. Соответствующие системы растворителей включают тетрагидрофуран, этиловый эфир, метил-трет-бутиловый эфир, метиленхлорид, N,N-диметилформамид и т.п., в том числе их смеси. В альтернативном варианте защищенный аминоспирт из эпоксида с разомкнутым кольцом может быть затем защищен по новой введенной аминогруппе защитной группой Р', которая не удаляется при удалении аминозащитных групп Р или Р 1 и Р 2, т.е. Р' является избирательно удаляемой группой. Специалист в данной области техники может выбрать соответствующую комбинацию Р', Р, Р 1 и Р 2. Например,пригодными комбинациями являются таковые, 000470 28 где Р обозначает CBz (карбобензокси) и Р' обозначает Воc; Р' обозначает CBz и Р обозначает Воc; Р 1 обозначает CBz, Р 2 обозначает бензил и Р' обозначает Воc; и Р 1 и Р 2 оба обозначают бензил и Р' обозначает Воc. Полученное соединение формулы может быть использовано на остальных стадиях синтеза с получением соединения формулы где n, Р', R1, R2, R3, R10, R11, R12, R13 и R14 имеют указанные выше значения. Остальную часть описанного выше синтеза можно осуществлять при необходимости либо путем добавления требуемых остатков или групп по отдельности, либо в предварительно сформированную молекулу на одной стадии включают более одного остатка или группы. Первый подход представляет собой метод последовательного синтеза, а последний является методом конвергентного синтеза. На этой стадии также возможны трансформации синтеза. Защитную группу Р' затем избирательно удаляют, а полученный амин подвергают взаимодействию с сульфонилхлоридом R4SO2Cl, сульфонилбромидом R4SO2Br или с соответствующим сульфонилангидридом, предпочтительно в присутствии акцептора кислоты, с получением соединений по настоящему изобретению где n, R1, R2, R3, R4, R10, R11, R12, R13, R14 имеют указанные выше значения. Это избирательное удаление защитной группы и превращение в сульфонамид можно осуществлять либо в конце синтеза, либо при необходимости на любой соответствующей промежуточной стадии. Описанные выше химические реакции в целом представлены в том виде, который соответствует их наиболее широкому применению для получения соединений по настоящему изобретению. Может оказаться, что реакции не применимы в том виде, как они описаны, к каждому из соединений, включенных в объем настоящего изобретения. Соединения, для которых это имеет место, легко могут быть определены специалистами в данной области техники. Во всех таких случаях либо реакции могут быть успешно осуществлены обычными модификациями, известными специалистам в данной области техники, например, путем соответствую 29 щей защиты взаимодействующих групп, путем использования альтернативных обычных реагентов, путем обычной модификации условий реакции и т.п., либо для получения соответствующих соединений по настоящему изобретению могут быть использованы другие реакции,представленные в данном описании или иные обычные реакции. Во всех способах получения все исходные продукты являются известными или могут быть легко получены из известных исходных продуктов. Предполагается, что специалист в данной области техники может без дальнейших исследований, используя предшествующее описание,применить настоящее изобретение в его наиболее полном объеме. Поэтому представленные ниже предпочтительные конкретные варианты осуществления должны рассматриваться исключительно как иллюстративные, а не как ограничивающие каким-либо образом объем изобретения. Все реагенты использовали в том виде, как они были получены, без очистки. Все протонные и углеродные ЯМР-спектры получали с использованием спектрометра ядерного магнитного резонанса либо типа Varian VXR-300, либо типа VXR-400. Следующие примеры иллюстрируют получение соединений-ингибиторов по настоящему изобретению и промежуточных продуктов, используемых для получения соединений-ингибиторов по настоящему изобретению. Пример 1. Получение 2S-[бис(фенилметил)амино] бензопропанола. Способ 1. Получение 2S-[бис(фенилметил) амино]бензопропанола восстановлением фенилметилового эфира N,N-бис(фенилметил)-Lфенилаланина с помощью ДИБАЛ. Стадия 1. Раствор, содержащий Lфенилаланин (50,0 г, 0,302 моля), гидроксид натрия (24,2 г, 0,605 моля) и карбонат калия(83,6 г, 0,605 моля) в воде (500 мл), нагревали до 97 С. Затем медленно добавляли (время добавления 25 мин) бензилбромид (108,5 мл, 0,605 моля). Смесь перемешивали при 97 С в течение 30 мин в атмосфере азота. Раствор охлаждали до комнатной температуры и экстрагировали толуолом (2 х 250 мл). Объединенные органические слои промывали водой и соляным раствором,сушили над сульфатом магния, фильтровали и концентрировали, получая масло. Идентичность продукта подтверждали следующим образом. Аналитическая ТСХ (смесь 10% этилацетата/гексан, силикагель) показала, что основной компонент со значением Rf 0,32 является тре 000470 30 буемым трибензилированным соединением, т.е. фенилметиловым эфиром N,N-бис(фенилметил)-L-фенилаланина. Это соединение может быть очищено с помощью колоночной хроматографии (силикагель, смесь 15% этилацетата/гексан). Обычно продукт является достаточно чистым для его использования непосредственно на следующей стадии без дополнительной очистки. 1 Н-ЯМР-спектр соответствовал таковому,известному по опубликованным литературным данным. 1 Н-ЯМР (CDCl3) , 3,00 и 3,14 (АВХсистема, 2 Н, JAB=14,1 Гц, JAX=7,3 Гц и JBX=5,9 Гц), 3,54 и 3,92 (АВ-система, 4 Н, JAB=13,9 Гц),3,71 (t, 1 Н, J=7,6 Гц), 5,11 и 5,23 (АВ-система,2 Н, JAB=12,3 Гц), и 7,18 (m, 20 Н). MC-EI: m/z 434 (М-1). Стадия 2. Фенилметиловый эфир бензилированного фенилаланина (0,302 моля), полученный с помощью предыдущей реакции, растворяли в толуоле (750 мл) и охлаждали до -55 С. Добавляли 1,5 М раствор ДИБАЛ в толуоле(443,9 мл, 0,666 моля) с такой скоростью, чтобы поддерживать температуру на уровне от -55 до 50 С (время добавления 1 ч). Смесь перемешивали в течение 20 мин в атмосфере азота и затем реакцию прекращали при -55 С путем медленного добавления метанола (37 мл). Затем холодный раствор сливали на холодный (5 С) 1,5 н. раствор НСl (1,8 л). Выпавший в осадок твердый продукт (приблизительно 138 г) отфильтровывали и промывали толуолом. Твердый продукт суспендировали в смеси толуола (400 мл) и воды (100 мл). Смесь охлаждали до 5 С и обрабатывали 2,5 н. NaOH (186 мл) и затем перемешивали при комнатной температуре до растворения твердого продукта. Толуоловый слой отделяли от водной фазы и промывали водой и соляным раствором, сушили над сульфатом магния, фильтровали и концентрировали до объема 75 мл (89 г). К остатку добавляли этилацетат (25 мл) и гексан (25 мл), после чего начинал кристаллизоваться требуемый спиртовой продукт. Через 30 мин для усиления дальнейшей кристаллизации добавляли еще 50 мл гексана. Твердый продукт отфильтровывали и промывали 50 мл гексана, получая 34,9 г продукта в первом выходе. Второй выход продукта (5,6 г) выделяли повторной фильтрацией маточного раствора. Оба выхода объединяли и перекристаллизовывали из этилацетата (20 мл) и гексана(30 мл), получая 40 г S-2-[бис(фенилметил) амино]бензопропанола, 40%-ный выход из Lфенилаланина. Дополнительные 7 г (7%) продукта могут быть получены в результате перекристаллизации концентрированного маточного раствора. ТСХ продукта с Rf = 0,23 (смесь 10% этилацетата/гексан, силикагель); 1 Н-ЯМР (CDCl3)2,44 (m, 1H), 3,09 (m,2H), 3,33 (m, 1H), 3,48 и 3,92 (АВ-система, 4 Н, 31+42,4 (с 1,45, СH2 Сl2); ДСК 77,67 С; Анализ: рассчитано для С 23H25 ОN: С 83,34; Н 7,60; N 4,23; обнаружено С 83,43; Н 7,59; N 4,22; ЖХВР на хиральной стационарной фазе: колонка Cyclobond I SP (250 х 4,6 мм внутренний диаметр (В.Д., подвижная фаза: метанол/триэтиламмонийацетатный буфер с рН 4,2(58:42, объем/объем), скорость потока 0,5 мл/мин, определение детектором при 230 нм и температуре 0 С. Время удержания: 11,25 мин,время удержания требуемого энантиомера: 12,5 мин. Способ 2. Получение S-2-[бис(фенилметил)амино]бензопропанола путемL-Фенилаланинол (176,6 г, 1,168 моля) добавляли к перемешиваемому раствору карбоната калия (484,6 г, 3,506 моля) в 710 мл воды. Смесь нагревали до 65 С в атмосфере азота. Добавляли раствор бензилбромида (400 г, 2,339 моля) в 3 А этаноле (305 мл) с такой скоростью,чтобы поддерживать температуру на уровне между 60 и 68 С. Двухфазный раствор перемешивали при 65 С в течение 55 мин и затем давали охладиться до 10 С при интенсивном перемешивании. Маслянистый продукт отвердевал в виде небольших гранул. Продукт разбавляли 2,0 л водопроводной воды и перемешивали в течение 5 мин до растворения неорганических побочных продуктов. Продукт выделяли фильтрацией при пониженном давлении и промывали водой до получения значения рН 7. Полученный неочищенный продукт сушили на воздухе в течение ночи, получая полусухой твердый продукт (407 г), который перекристаллизовывали из 1,1 л этилацетата/гептана (1:10 по объему). Продукт выделяли фильтрацией (при -8 С),промывали 1,6 л холодной смеси (-10 С) этилацетата/гептана (1:10 по объему) и сушили на воздухе, получая 339 г (выход 88%) S-2[бис(фенилметил)амино]бензопропанола, tпл = 71,5 -73,0 С. При необходимости дополнительное количество продукта может быть получено из маточного раствора. Другие аналитические характеристики идентичны таковым для соединения, полученного согласно способу 1. Пример 2. Получение 2S-[бис(фенилметил)амино] бензопропанальдегида. Способ 1. 2S-[бис(фенилметил)амино] бензопропанол (200 г, 0,604 моля) растворяли в триэтиламине (300 мл, 2,15 моля). Смесь охлаждали до 12 С и добавляли раствор комплекса триоксид серы/пиридин (380 г, 2,39 моля) в 32 ДМСО (1,6 л) с такой скоростью, чтобы поддерживать температуру на уровне от 8 до 17 С(время добавления 1,0 ч). Раствор перемешивали при температуре окружающей среды в атмосфере азота в течение 1,5 ч и к этому моменту по данным ТСХ-анализа (смесь 33% этилацетата/гексан, силикагель) реакция была завершена. Реакционную смесь охлаждали ледяной водой и реакцию прекращали добавлением 1,6 л холодной воды (10-15 С) в течение 45 мин. Образовавшийся раствор экстрагировали этилацетатомMgSO4 (280 г) и фильтровали. Растворитель удаляли с помощью роторного испарителя при 35-40 С и затем сушили в вакууме, получая 198,8 г 2S-[бис(фенилметил)амино] бензопропанальдегида в виде масла светло-желтого цвета(99,9%). Полученный неочищенный продукт был достаточно чистым для его использования непосредственно на следующей стадии без дополнительной очистки. Аналитические характеристики соединения соответствовали таковым,известным по опубликованным литературным данным.C23H24NO 330,450, обнаружено: 330,1836. Анализ: рассчитано для C23H23ON: С 83,86; Н 7,04; N 4,25; обнаружено С 83,64; Н 7,42; N 4,19. ЖХВР на хиральной стационарной фазе(S,S): колонка Pirkle-Whelk-О 1 (250 х 4,6 мм внутренний диаметр (В.Д., подвижная фаза: гексан/изопропанол (99,5:0,5 по объему), скорость потока 1,5 мл/мин, определение УФдетектором при 210 нм. Время удержания требуемого S-изомера: 8,75 мин, время удержанияR-энантиомера: 10,62 мин. Способ 2. Раствор оксалилхлорида (8,4 мл,0,096 моля) в дихлорметане (240 мл) охлаждали до -74 С. Затем медленно добавляли раствор ДМСО (12,0 мл, 0,155 моля) в дихлорметане (50 мл) с такой скоростью, чтобы поддерживать температуру на уровне -74 С (время добавления 1,25 ч). Смесь перемешивали в течение 5 мин,затем добавляли раствор S-2-[бис(фенилметил) амино] бензопропанола (0,074 моля) в 100 мл дихлорметана (время добавления 20 мин, температура от -75 до -68 С). Раствор перемешивали при -78 С в течение 35 мин в атмосфере азота. Затем в течение 10 мин (температура от -78 до -68 С) добавляли триэтиламин (41,2 мл, 0,295 моля), после чего осаждалась аммониевая соль. Холодный раствор перемешивали в течение 30 мин и затем добавляли воду (225 мл). Дихлорметановый слой отделяли от водной фазы и 33 промывали водой, соляным раствором, сушили над сульфатом магния, фильтровали и концентрировали. Остаток разбавляли этилацетатом и гексаном и затем фильтровали для последующего удаления аммониевой соли. Фильтрат концентрировали, получая S-[бис (фенилметил) амино] бензопропанальдегид. Альдегид использовали на последующей стадии без очистки. Способ 3. К смеси, содержащей 1,0 г (3,0 ммоля) S-2-[бис(фенилметил)амино] бензопропанола, 0,531 г (4,53 ммоля) Nметилморфолина, 2,27 г молекулярных сит (4) и 9,1 мл ацетонитрила, добавляли 53 мг (0,15 ммоля) тетрапропиламмонийперрутената(ТПАП). Смесь перемешивали в течение 40 мин при комнатной температуре и концентрировали при пониженном давлении. Остаток суспендировали в 15 мл этилацетата, фильтровали через подушку из силикагеля. Фильтрат концентрировали при пониженном давлении, получая продукт, содержащий приблизительно 50% S-2-[бис(фенилметил)амино] бензопропанальдегида в виде масла светложелтого цвета. Способ 4. К раствору, содержащему 1,0 г(0,03 ммоля) 2,2,6,6-тетраметил-1-пиперидинилокси в качестве свободного радикала (ТЕМПО), 0,32 г (3,11 ммоля) бромида натрия, 9,0 мл этилацетата и 1,5 мл воды. Смесь охлаждали до 0 С и медленно добавляли в течение 25 мин водный раствор, содержащий 2,87 мл 5%-ной бытовой хлорной извести, содержащей 0,735 г(8,75 ммоля) бикарбоната натрия и 8,53 мл воды. Смесь перемешивали при 0 С в течение 60 мин. При перемешивании в течение 10 мин добавляли еще две порции (по 1,44 мл каждая) хлорной извести. Двухфазной смеси давали разделиться. Водный слой экстрагировали дважды 20 мл этилацетата. Объединенный органический слой промывали 4,0 мл раствора, содержащего 25 мг йодида калия и воду (4,0 мл), 20 мл 10%ного водного раствора тиосульфата натрия и затем соляным раствором. Органический раствор сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая 1,34 г неочищенного масла, содержащего небольшое количество требуемого альдегида, т.е. S-[бис(фенилметил) амино]бензопропанальдегида. Способ 5. По методике, аналогичной описанной в способе 1 данного примера, за исключением того, что использовали 3,0 эквивалента комплекса триоксид серы/пиридин, выделяли(191,7 г, 0,58 моля) и хлорйодметан (56,4 мл,0,77 моля) в тетрагидрофуране (1,8 л), охлаждали до температуры от -30 до -35 С в реакторе из нержавеющей стали в атмосфере азота (можно также успешно работать и при более низкой температуре, такой как -70 С, однако более высокие температуры легче получать при крупномасштабных процессах). Затем добавляли раствор н-бутиллития в гексане (1,6 М, 365 мл, 0,58 моля) с такой скоростью, чтобы поддерживать температуру ниже -25 С. После добавления смесь перемешивали при температуре от -30 до-35 С в течение 10 мин. Дополнительные добавления реагентов осуществляли следующим образом. (1) Добавляли дополнительную порцию хлорйодметана (17 мл), а затем н-бутиллитий(110 мл) при температуре не выше -25 С. После добавления смесь перемешивали при температуре от -30 до -35 С в течение 10 мин. Эту процедуру повторяли еще один раз. (2) Добавляли дополнительную порцию хлорйодметана (8,5 мл, 0,11 моля), а затем н-бутиллитий (55 мл,0,088 моля) при температуре не выше -25 С. После добавления смесь перемешивали при температуре от -30 до -35 С в течение 10 мин. Эту процедуру повторяли пять раз. (3) Добавляли дополнительную порцию хлорйодметана (8,5 мл, 0,11 моля), а затем н-бутиллитий (37 мл,0,059 моля) при температуре не выше -25 С. После добавления смесь перемешивали при температуре от -30 до -35 С в течение 10 мин. Эту процедуру повторяли еще один раз. Прекращали внешнее охлаждение и смесь нагревали до температуры окружающей среды в течение 4-16 ч, после чего ТСХ (силикагель, смесь 20% этилацетата/гексан) показала, что реакция завершена. Реакционную смесь охлаждали до 10 С и реакцию прекращали добавлением 1452 г 16%-ного раствора хлорида аммония (полученного растворением 232 г хлорида аммония в 1220 мл воды), поддерживая температуру ниже 23 С. Смесь перемешивали в течение 10 мин и разделяли органический и водный слои. Водную фазу экстрагировали этилацетатом (2 х 500 мл). Этилацетатный слой объединяли с тетрагидрофурановым слоем. Объединенный раствор сушили над сульфатом магния (220 г), фильтровали и концентрировали на роторном испарителе при 65 С. Остаток в виде коричневого масла сушили при 70 С в вакууме (0,8 бар) в течение 1 ч, получая 222,8 г неочищенного продукта. 35 100%. Вследствие относительной нестабильности продукта на силикагеле неочищенный продукт обычно использовали непосредственно на следующей стадии без дополнительной очистки). Соотношение диастереомеров неочищенной смеси определяли протонным ЯМР:(2S)/(2R): 86:14. Характеристики минорного и основного эпоксидных диастереомеров в этой смеси определяли с помощью ТСХ-анализа (силикагель, смесь 10% этилацетата/гексан),Rf=0,29 и 0,32 сответственно. Аналитический образец каждого из этих диастереомеров получали очисткой с помощью хроматографии на силикагеле (смесь 3% этилацетата/гексан), получая следующие характеристики:H-ЯМР (300 МГц, СDСl3)2,20 (m, 1H),2,59 (m, 1H), 2,75 (m, 2H), 2,97 (m, 1H), 3,14 (m,1H), 3,85 (АВ-система, 4 Н), 7,25 (m, 15H). ЖХВР на хиральной стационарной фазе: колонка Pirkle-Whelk-О 1 (250 х 4,6 мм В.Д.), подвижная фаза: гексан/изопропанол (99,5:0,5 по объему), скорость потока 1,5 мл/мин, определение УФ-детектором при 210 нм. Время удержания продукта (8): 9,38 мин, время удержания энантиомера (4): 13,75 мин. Способ 2. Раствор, содержащий 0,704 моля неочищенного альдегида и хлорйодметан (7,0 мл, 0,096 моля) в тетрагидрофуране (285 мл) охлаждали до -78 С в атмосфере азота. Затем добавляли 1,6 М раствор н-бутиллития в гексане(25 мл, 0,040 моля) с такой скоростью, чтобы поддерживать температуру на уровне -75 С(время добавления 15 мин). После первого добавления вновь добавляли хлорйодметан (1,6 мл, 0,022 моля), а затем н-бутиллитий (23 мл,0,037 моля), поддерживая температуру на уровне -75 С. Смесь перемешивали в течение 15 мин. Каждый из реагентов, хлорйодметан (0,70 мл, 0,010 моля) и н-бутиллитий (5 мл, 0,008 моля), добавляли еще четыре раза в течение 45 мин при -75 С. Охлаждающую баню затем удаляли и раствор нагревали до 22 С в течение 1,5 ч. Смесь сливали на 300 мл насыщенного водного раствора хлорида аммония. Отделяли тетрагидрофурановый слой. Водную фазу экстрагировали этилацетатом (1 х 300 мл). Объединенные органические слои промывали соляным раство 000470 36 ром, сушили над сульфатом магния, фильтровали и концентрировали, получая масло коричневого цвета (27,4 г). Продукт может использоваться на следующей стадии без дальнейшей очистки. Требуемый диастереомер может быть очищен перекристаллизацией на последующей стадии. Продукт также может быть очищен хроматографией. Способ 3. Раствор, содержащий S[бис(фенилметил)амино]бензопропанальдегид(178,84 г, 0,54 моля) и бромхлорметан (46 мл,0,71 моля) в тетрагидрофуране (1,8 л), охлаждали до температуры от -30 до -35 С в реакторе из нержавеющей стали в атмосфере азота (можно также успешно работать при более низкой температуре, такой как -70 С, однако более высокие температуры легче получать при крупномасштабных процессах). Раствор н-бутиллития в гексане (1,6 М, 340 мл, 0,54 моля) добавляли с такой скоростью, чтобы поддерживать температуру ниже -25 С. После добавления смесь перемешивали при температуре от -30 до -35 С в течение 10 мин. Дополнительные добавления реагентов осуществляли следующим образом.(1) Добавляли дополнительную порцию бромхлорметана (14 мл), а затем н-бутиллитий (102 мл) при температуре не выше -25 С. После добавления смесь перемешивали при температуре от -30 до -35 С в течение 10 мин. Эту процедуру повторяли еще один раз. (2) Добавляли дополнительную порцию бромхлорметана (7 мл, 0,11 моля), а затем н-бутиллитий (51 мл, 0,082 моля) при температуре не выше -25 С. После добавления смесь перемешивали при температуре от -30 до -35 С в течение 10 мин. Эту процедуру повторяли пять раз. (3) Добавляли дополнительную порцию бромхлорметана (7 мл, 0,11 моля),а затем н-бутиллитий (51 мл, 0,082 моля) при температуре не выше -25 С. После добавления смесь перемешивали при температуре от -30 до-35 С в течение 10 мин. Эту процедуру повторяли еще один раз. Прекращали внешнее охлаждение и смесь нагревали до температуры окружающей среды в течение 4-16 ч, после чего ТСХ (силикагель, смесь 20% этилацетата/гексан) показала, что реакция завершена. Реакционную смесь охлаждали до 10 С и реакцию прекращали добавлением 1452 г 16%-ного раствора хлорида аммония (полученного растворением 232 г хлорида аммония в 1220 мл воды),поддерживая температуру ниже 23 С. Смесь перемешивали в течение 10 мин и разделяли органический и водный слои. Водную фазу экстрагировали этилацетатом (2 х 500 мл). Этилацетатный слой объединяли с тетрагидрофурановым слоем. Объединенный раствор сушили над сульфатом магния (220 г), фильтровали и концентрировали на роторном испарителе при 65 С. Остаток в виде коричневого масла сушили при 70 С в вакууме (0,8 бар) в течение 1 ч, получая 222,8 г неочищенного продукта. 37 Способ 4. Использовали методику, описанную в способе 3 данного примера, за исключением того, что температура реакции составляла -20 С. Полученный N,N,S-трис (фенилметил)-2S-оксиранметанамин представлял собой смесь диастереомеров меньшей чистоты, чем таковая, полученная по способу 3. Способ 5. Использовали методику, описанную в способе 3 данного примера, за исключением того, что температура реакции составляла от -70 до -78 С. Полученный N,N,Sтрис(фенилметил)-2S-оксиранметанамин представлял собой смесь диастереомеров, которую непосредственно применяли на следующей стадии без дополнительной очистки. Способ 6. Использовали методику, описанную в способе 3 данного примера, за исключением того, что непрерывное добавление бромхлорметана и н-бутиллития осуществляли при температуре от -30 до -35 С. После осуществления реакции и соответствующих обработок по методике, описанной в способе 3 этого примера, выделяли требуемый N,N,S-трис (фенилметил)-2S-оксиранметанамин с сопоставимым выходом и чистотой. Способ 7. Использовали методику, описанную в способе 2 данного примера, за исключением того, что вместо хлорйодметана применяли дибромметан. После осуществления реакции и соответствующих обработок по методике,описанной в способе 2 этого примера, выделяли требуемый Получение N-[3(S)-[N,N-бис(фенилметил) амино]-2-(R)-гидрокси-4-фенилбутил]-N-изобутиламина. К раствору, содержащему N,N-дибензил 3(S)-амино-1,2(S)-эпокси-4-фенилбутан (388,5 г,1,13 моля) в изопропаноле (2,7 л) (или в этилацетате), в течение 2 мин добавляли изобутиламин (1,7 кг, 23,1 моля). Температура повышалась от 25 до 30 С. Раствор нагревали до 82 С и перемешивали при этой температуре в течение 1,5 ч. Теплый раствор концентрировали при пониженном давлении при 65 С. Остаток в виде масла коричневого цвета переносили в колбу объемом 3 л и сушили в вакууме (0,8 мм рт.ст.) в течение 16 ч, получая 450 г 3(S)-[N,Nбис(фенилметил)амино]-4-фенилбутан-2R-ола в виде неочищенного масла. Аналитический образец основного диастереомерного продукта получали очисткой небольшого количества образца неочищенного продукта с помощью хроматографии на силикагеле (смесь 40% этилацетата/гексан). ТСХ-анализ: силикагель, смесь 40% 38 этилацетата/гексан; Rf = 0,28; ЖХВР-анализ: ультрасферная колонка типа ODS, 25% триэтиламина/фосфатный буфер рН 3 - ацетонитрил,скорость потока 1 мл/мин, УФ-детектор; время удержания 7,49 мин; МСВР рассчитано для:(М+1) 417,616,обнаружено: 417,2887. Аналитический образец минорного диастереомерного продукта, т.е. 3(S)-[N,Nбис(фенилметил)амино]-4-фенилбутан-2S-ола,получали очисткой небольшого количества образца неочищенного продукта с помощью хроматографии на силикагеле (смесь 40% этилацетата/гексан). Пример 5.-N-изобутиламина. К раствору щавелевой кислоты (8,08 г,89,72 ммоля) в метаноле (76 мл) в течение 15 мин добавляли раствор неочищенного 3(S)[N,N-бис(фенилметил)амино]-1-(2-метилпропил)амино-4-фенилбутан-2R-ола 39,68 г, который содержал приблизительно 25,44 г (61,06 ммоля) 3(S),2(R)-изомера и приблизительно 4,49 г (10,78 ммоля) 3(S),2(S) -изомера в этилацетате (90 мл). Смесь перемешивали при комнатной температуре в течение приблизительно 2 ч. Твердый продукт выделяли фильтрацией, промывали этилацетатом (2 х 20 мл) и сушили в вакууме в течение приблизительно 1 ч, получая 21,86 г (выход изомера 70,7%) соли 97%-ной диастереометрической чистоты (по данным о площади пиков при ЖХВР-анализе). ЖХВРанализ: колонка типа Vydec-пептид/протеин С 18, УФ-детектор, 254 нм, скорость потока 2 мл/мин, градиент А = 0,05%-ная трифторуксусная кислота в воде, Б = 0,05%-ная трифторуксусная кислота в ацетонитриле, 0 мин 75% А/25% Б, 30 мин 10% А/90% Б, 35 мин 10% А/90% Б, 37 мин 75% А/25% Б; время удержания 10,68 мин для 3(S),2(R)-изомера и 9,73 мин для 3(S),2(S)-изомера). tпл 174,99 С; микроанализ: рассчитано: С 71,05%, Н 7,50%, N 5,53%; обнаружено: С 71,71%, Н 7,75%, N 5,39%. В альтернативном варианте дигидрат щавелевой кислоты (119 г, 0,94 моля) добавляли в круглодонную колбу объемом 5000 мл, снабженную механической мешалкой и капельной воронкой. Добавляли метанол (1000 мл) и смесь перемешивали до полного растворения. В течение 20 мин добавляли раствор неочищенного 3(S)-[N,N-бис(фенилметил)амино]-1-(2-метилпропил)амино-4-фенилбутан-2(R)-ола в этилацетате (1800 мл, 0,212 г изомеров аминоспирта/мл, 0,9160 моля). Смесь перемешивали в те 39 чение 18 ч и твердый продукт выделяли центрифугированием при 400xg в виде шести порций. Каждую порцию промывали 125 мл этилацетата. Затем соль собирали и сушили в течение ночи при давлении 1 торр, получая 336,3 г продукта (71% по отношению к общему содержанию аминоспирта). Данные ЖХВР/МС (электроспрей) соответствовали требуемому продукту (m/z 417 [М+Н]+). В альтернативном варианте неочищенный 3(S)-[N,N-бис(фенилметил)амино]-1-(2-метилпропил)амино-4-фенилбутан-2(R)-ол (5 г) растворяли в метил-трет-бутиловом эфире (МТБЭ)(10 мл) и добавляли щавелевую кислоту (1 г) в метаноле (4 мл). Смесь перемешивали в течение приблизительно 2 ч. Образовавшийся твердый продукт фильтровали, промывали холодным МТБЭ и сушили, получая 2,1 г белого твердого продукта 98,9%-ной диастереометрической чистоты (по данным о площади пиков при ЖХВРанализе). Пример 6. Получение ацетата N-[3(S)[N,N-бис (фенилметил)амино]-2-(R)-гидрокси-4 фенилбутил]-N-изобутиламина. К раствору неочищенного 3(S)-[N,Nбис(фенилметил)амино]-1-(2-метилпропил)амино-4-фенилбутан-2(R)-ол в метил-трет-бутиловом эфире (МТБЭ) (45 мл, 1,1 г изомеров аминоспирта/мл) по каплям добавляли уксусную кислоту (6,9 мл). Смесь перемешивали при комнатной температуре в течение приблизительно 1 ч. Растворитель удаляли в вакууме, получая масло коричневого цвета в виде продукта приблизительно 85%-ной диастереометрической чистоты (по данным о площади пиков при ЖХВР-анализе). Коричневое масло кристаллизовали следующим образом: 0,2 г масла растворяли при нагревании в первом растворителе,получая прозрачный раствор, добавляли второй растворитель до тех пор, пока раствор не становился мутным, смесь вновь нагревали до получения прозрачного раствора, вносили затравку в виде продукта приблизительно 99%-ной диастереометрической чистоты, охлаждали до комнатной температуры и затем выдерживали в холодильнике в течение ночи. Кристаллы фильтровали, промывали вторым растворителем и сушили. Диастереомерную чистоту кристаллов оценивали по площади пиков при ЖХВРанализе. Результаты приведены в таблице 1. Первый растворитель МТБЭ МТБЭ Метанол Толуол Толуол Гептан Гексан Вода Гептан Гексан Таблица 1 Выход по Диастереомассе (г) мерная чистота В альтернативном варианте неочищенный 3(S)-[N,N-бис(фенилметил)амино]-1-(2-метил 40 пропил)амино-4-фенилбутан-2(R)-ол (50,0 г,содержащий приблизительно 30,06 г (76,95 ммоля) 3(S),2(R)-изомера и приблизительно 5,66 г (13,58 ммоля) 3(S),2(S)-изомера) растворяли в метил-трет-бутиловом эфире (45,0 мл). К этому раствору в течение приблизительно 10 мин добавляли уксусную кислоту (6,90 мл, 120,6 ммоля). Смесь перемешивали при комнатной температуре в течение приблизительно 1 ч и концентрировали при пониженном давлении. Маслянистый остаток очищали перекристаллизацией из метил-трет-бутилового эфира (32 мл) и гептана (320 мл). Твердый продукт выделяли фильтрацией, промывали холодным гептаном и сушили в вакууме в течение приблизительно 1 ч, получая 21,34 г (выход изомера 58,2%) моноацетатной соли 96%-ной диастереометрической чистоты (по данным о площади пиков при ЖХВР-анализе). tпл 105-106 С; микроанализ: рассчитано: С 75,53%, Н 8,39%, N 5,87%; обнаружено С 75,05%, Н 8,75%, N 5,71%. Пример 7. Получение L-тартрата N-[3(S)[N,N-бис (фенилметил)амино]-2-(R)-гидрокси-4 фенилбутил]-N-изобутиламина. Неочищенный 3(S)-[N,N-бис(фенилметил) амино]-1-(2-метилпропил)амино-4-фенилбутан 2R-ол (10,48 г, содержащий приблизительно 6,72 г (16,13 ммоля) 3 (S), 2 (R)- изомера и приблизительно 1,19 г (2,85 ммоля) 3(S),2(S)изомера) растворяли в тетрагидрофуране (10,0 мл). К этому раствору в течение приблизительно 5 мин добавляли раствор L-винной кислоты(2,85 г, 19 ммолей) в метаноле (5,0 мл). Смесь перемешивали при комнатной температуре в течение приблизительно 10 мин и концентрировали при пониженном давлении. К маслянистому остатку добавляли метил-трет-бутиловый эфир (20,0 мл) и смесь перемешивали при комнатной температуре в течение приблизительно 1 ч. Твердый продукт выделяли фильтрацией,получая 7,50 г неочищенной соли. Неочищенную соль очищали перекристаллизацией из этилацетата и гептана при комнатной температуре, получая 4,13 г (выход изомера 45,2%) солиL-винной кислоты 95%-ной диастереометрической чистоты (по данным о площади пиков при ЖХВР-анализе). Микроанализ: рассчитано: С 67,76%, Н 7,41%, N 4,94%; обнаружено: С 70,06%, Н 7,47%, N 5,07%. Пример 8. Получение дигидрохлорида N[3(S)-[N,N-бис(фенилметил)амино]-2(R)-гидрокси-4-фенилбутил]-N-изобутиламина. Неочищенный 3(S)-[N,N-бис(фенилметил) амино]-1-(2-метилпропил)амино-4-фенилбутан 2(R)-ол (10,0 г, содержащий приблизительно 6,41 г (15,39 ммоля) 3(S),2(R)-изомера и приблизительно 1,13 г (2,72 ммоля) 3(S),2 (S)изомера) растворяли в тетрагидрофуране (20,0 мл). К этому раствору в течение приблизительно 5 мин добавляли соляную кислоту (20 мл,6,0 н.). 41 Смесь перемешивали при комнатной температуре в течение приблизительно 1 ч и концентрировали при пониженном давлении. Остаток перекристаллизовывали из этанола при 0 С,получая 3,20 г (выход изомера 42,7%) дихлориданой соли 98%-ной диастереометрической чистоты (по данным о площади пиков при ЖХВРанализе). Микроанализ: рассчитано: С 68,64%,Н 7,76%, N 5,72%; обнаружено С 68,79%, Н 8,07%, N 5,55%. Пример 9. Получение толуолсульфонатаN-[3(S)-[N,N-бис(фенилметил)амино]-2(R)-гидрокси-4-фенилбутил]-N-изобутиламина. Неочищенный 3(S)-[N,N-бис(фенилметил) амино]-1-(2-метилпропил)амино-4-фенилбутан 2(R)-ол (5,0 г, содержащий приблизительно 3,18 г (7,63 ммоля) 3(S),2(R)-изомера и приблизительно 0,56 г (1,35 ммоля) 3(S),2(S)-изомера) растворяли в метил-трет-бутиловом эфире (10,0 мл). К этому раствору в течение приблизительно 5 мин добавляли раствор толуолсульфокислоты (2,28 г, 12 ммолей) в метил-третбутиловом эфире (2,0 мл) и метанол (2,0 мл). Смесь перемешивали при комнатной температуре в течение приблизительно 2 ч и концентрировали при пониженном давлении. Остаток перекристаллизовывали из метил-трет-бутилового эфира и гептана при 0 С, фильтровали, промывали холодным гептаном и сушили в вакууме,получая 1,85 г (выход изомера 40,0%) соли монотолуолсульфокислоты 97%-ной диастереометрической чистоты (по данным о площади пиков при ЖХВР-анализе). Пример 10. Получение метансульфонатаN-[3(S)-[N,N-бис(фенилметил)амино]-2(R)-гидрокси-4-фенилбутил]-N-изобутиламина. Неочищенный 3(S)-[N,N-бис(фенилметил) амино]-1-(2-метилпропил)амино-4-фенилбутан 2(R)-ол (10,68 г, содержащий приблизительно 6,85 г (16,44 ммоля) 3(S),2(R)-изомера и приблизительно 1,21 г (2,90 ммоля) 3(S),2(S)изомера) растворяли в тетрагидрофуране (10,0 мл). К этому раствору добавляли метансульфокислоту (1,25 мл, 19,26 ммолей). Смесь перемешивали при комнатной температуре в течение приблизительно 2 ч и концентрировали при пониженном давлении. Маслянистый остаток перекристаллизовывали из метанола и воды при 0 С, фильтровали, промывали холодным метанолом/водой (1:4) и сушили в вакууме, получая 2,40 г (выход изомера 28,5%) соли монометансульфокислоты 98%-ной диастереометрической чистоты (по данным о площади пиков при ЖХВР-анализе). Пример 11. Получение N-бензил-L-фенилаланинола. Способ 1. L-Фенилаланинол (89,51 г, 0,592 моля) растворяли в 375 мл метанола в атмосфере инертного газа, добавляли 35,52 г (0,592 моля) ледяной уксусной кислоты и 50 мл метанола,а затем добавляли раствор, содержащий 62,83 г 42 Смесь охлаждали до приблизительно 15 С и приблизительно в течение 40 мин добавляли раствор, содержащий 134,6 г (2,14 моля) цианборгидрида натрия в 700 мл метанола, поддерживая температуру между 15 С и 25 С. Смесь перемешивали при комнатной температуре в течение 18 ч. Смесь концентрировали при пониженном давлении и распределяли между 1 л 2 М раствора гидроксида аммония и 2 л простого эфира. Эфирный слой промывали 1 л 1 М раствора гидроксида аммония, дважды 500 мл воды, 500 мл соляного раствора и сушили над сульфатом магния в течение 1 ч. Эфирный слой фильтровали, концентрировали при пониженном давлении и неочищенный твердый продукт перекристаллизовывали из 110 мл этилацетата и 1,3 л гексана, получая 115 г (выход 81%) Nбензил-L-фенилаланинола в виде белого твердого вещества. Способ 2. L-Фенилаланинол (5 г, 33 ммоля) и 3,59 г (33,83 ммоля) бензальдегида растворяли в 55 мл 3 А этанола в атмосфере инертного газа в вибраторе Парра и смесь нагревали до 60 С в течение 2,7 ч. Смесь охлаждали до приблизительно 25 С, добавляли 0,99 г 5%-ной платины на угле и смесь гидрировали при давлении водорода 60 фунтов/кв.дюйм и 40C в течение 10 ч. Катализатор отфильтровывали, продукт концентрировали при пониженном давлении и неочищенный твердый продукт перекристаллизовывали из 150 мл гептана, получая 3,83 г (выход 48%) N-бензил-L-фенилаланинола в виде белого твердого вещества. Пример 12. ПолучениеN-бензил-L-фенилаланинол (2,9 г, 12 ммолей) растворяли в 3 мл триэтиламина и 27 мл метанола и добавляли 5,25 г (24,1 ммоля) дитрет-бутилдикарбоната. Смесь нагревали до 60 С в течение 35 мин и концентрировали при пониженном давлении. Остаток растворяли в 150 мл этилацетата и дважды промывали 10 мл холодной (0-5 С) разбавленной соляной кислоты (рН от 2,5 до 3),15 мл воды, 10 мл соляного раствора, сушили над сульфатом магния,фильтровали и концентрировали при пониженном давлении. Неочищенный продукт в виде масла очищали с помощью хроматографии на силикагеле (этилацетат:гексан в качестве раствора для элюирования, 12:3), получая 3,98 гN-(трет-бутоксикарбонил)-Nбензил-L-фенилаланинола в 2,8 мл толуола, добавляли 2,4 мг (0,015 ммоля) 2,2,6,6-тетраметил 1-пиперидинилокси (ТЕМПО) в качестве свободного радикала, 0,1 г (0,97 ммоля) бромида натрия, 2,8 мл этилацетата и 0,34 мл воды. Смесь охлаждали до 0 С и в течение 30 мин медленно добавляли 4,2 мл водного раствора 43 5%-ной бытовой хлорной извести, содержащего 0,23 г (3,0 мл, 2,738 ммоля) бикарбоната натрия. Смесь перемешивали при 0 С в течение 10 мин. Добавляли еще три порции (по 0,4 мл каждая) хлорной извести с последующим перемешиванием в течение 10 мин после каждого добавления для поглощения всего исходного продукта. Двухфазной смеси давали разделиться. Водный слой дважды экстрагировали 8 мл толуола. Объединенный органический слой промывали 1,25 мл раствора, содержащего 0,075 г йодида калия,бисульфат натрия (0,125 г) и воду (1,1 мл), 1,25 мл 10%-ного водного раствора тиосульфата натрия, 1,25 мл фосфатного буфера с рН 7 и 1,5 мл соляного раствора. Органический раствор сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении,получая 0,32 г (выход 100%) N-(трет-бутоксикарбонил) - N -бензил- L-фенилаланиналя. Способ 2. К раствору, содержащему 2,38 гN-(трет-бутоксикарбонил)-Nбензил-L-фенилаланинола в 3,8 мл (27,2 ммоля) триэтиламина, при 10 С добавляли раствор, содержащий 4,33 г (27,2 ммоля) комплекса триоксид серы/пиридин в 17 мл диметилсульфоксида. Смесь нагревали до комнатной температуры и перемешивали в течение 1 ч. Добавляли воду(16 мл) и смесь экстрагировали 20 мл этилацетата. Органический слой промывали 20 мл 5%ной лимонной кислоты, 20 мл воды, 20 мл соляного раствора, сушили над сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении, получая 2,37 г (выход 100%) N-(трет-бутоксикарбонил)-N-бензил-Lфенилаланиналя. Пример 14. Получение 3(S)-[N-(трет-бутоксикарбонил)-N-бензиламино]-1,2-(S)-эпокси-4-фенилбутана. Способ 1. Раствор, содержащий 2,5 г (7,37 ммоля) N-(трет-бутоксикарбонил)-N-бензил-Lфенилаланинала и 0,72 мл хлорйодметана в 35 мл ТГФ, охлаждали до -78 С. Медленно добавляли, поддерживая температуру ниже -70 С,4,64 мл раствора н-бутиллития (1,6 М в гексане,7,42 ммоля). Смесь перемешивали в течение 10 мин при температуре на уровне между -70 и-75 С. Последовательно добавляли еще две порции по 0,22 мл хлорйодметана и 1,4 мл нбутиллития и после каждого добавления смесь перемешивали в течение 10 мин при температуре между -70 и -75 С. Последовательно добавляли еще четыре порции по 0,11 мл хлорйодметана и 0,7 мл н-бутиллития и после каждого добавления смесь перемешивали в течение 10 мин 44 при температуре между -70 и -75 С. Смесь нагревали до комнатной температуры в течение 3,5 ч. Реакцию прекращали при температуре ниже 5 С добавлением 24 мл ледяной воды. Двухфазные слои разделяли и водный слой дважды экстрагировали 30 мл этилацетата. Объединенные органические слои трижды промывали 10 мл воды, затем 10 мл соляного раствора,сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении,получая 2,8 г неочищенного масла желтого цвета. Это неочищенное масло (выход 100%) представлял собой смесь диастереомерных эпоксидовN,S-бис(фенилметил)-N-(трет-бутоксикарбонил)-2R-оксиранметанамина. Неочищенную смесь использовали непосредственно на следующей стадии без очистки. Способ 2. К суспензии, содержащей 2,92 г(13,28 ммоля) йодида триметилсульфоксония в 45 мл ацетонитрила, добавляли 1,49 г (13,28 ммоля) трет-бутоксида калия. Добавляли раствор, содержащий 3,0 г (8,85 ммоля) N-(третбутоксикарбонил)-N-бензил-L-фенилаланиналя в 18 мл ацетонитрила и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь разбавляли 150 мл воды и экстрагировли дважды 200 мл этилацетата. Органические слои объединяли и промывали 100 мл воды, 50 мл соляного раствора, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении, получая 3,0 г неочищенного масла желтого цвета. Неочищенный продукт очищали с помощью хроматографии на силикагеле (этилацетат:гексан в качестве раствора для элюирования, 1:8), получая 1,02 г (выход 32,7%) смеси двух диастереомеров N,S-бис(фенилметил)-N-(трет-бутоксикарбонил)-2S-оксиранметанамина и N,S-бис(фенилметил)-N-(третбутоксикарбонил)-2R-оксиранметанамина. Способ 3. К суспензии, содержащей 0,90 г(4,42 ммоля) йодида триметилсульфония в 18 мл ацетонитрила, добавляли 0,495 г (4,42 ммоля) трет-бутоксида калия. Добавляли раствор, содержащий 1,0 г (2,95 ммоля) N-(третбутоксикарбонил)-N-бензил-L-фенилаланиналя в 7 мл ацетонитрила, и смесь перемешивали при комнатной температуре в течение 1 ч. Смесь разбавляли 80 мл воды и экстрагировали дважды 80 мл этилацетата. Органические слои объединяли и промывали 100 мл воды, 30 мл соляного раствора, сушили над сульфатом натрия,фильтровали и концентрировали при пониженном давлении, получая 1,04 г неочищенного масла желтого цвета. Неочищенный продукт представлял собой смесь двух диастереомеровN,S-бис(фенилметил)-N-(трет-бутоксикарбонил)-2R-оксиранметанамина) в 0,98 мл изопропанола, добавляли 0,71 мл (7,14 ммоля) изобутиламина. Смесь кипятили с обратным холодильником при 85-90 С в течение 1,5 ч. Смесь концентрировали при пониженном давлении и продукт в виде масла очищали с помощью хроматографии на силикагеле (хлороформ:метанол в качестве раствора для элюирования, 100:6),получая 330 мг 3S-[N-(трет-бутоксикарбонил)N-(фенилметил)амино]-1-(2-метилпропил)амино-4-фенилбутан-2R-ола в виде бесцветного масла (выход 54,5%). Также выделяли 3S-[N(трет-бутоксикарбонил)-N-(фенилметил)амино]1-(2-метилпропил)амино-4-фенилбутан-2S-ол. Когда в качестве исходного материала использовали очищенный N,S-бис(фенилметил)-N(трет-бутоксикарбонил)-2S-оксиранметанамин,то после очистки с помощью хроматографии выделяли 3S-[N-(трет-бутоксикарбонил)-N(фенилметил)амино]-1-(2-метилпропил)амино 4-фенилбутан-2R-ол с выходом 86%. Пример 16.(жидкий, 1,0 М в ТГФ), поддерживая температуру ниже 5 С. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 ч. Смесь охлаждали до 0 С и медленно добавляли 20 мл воды с целью разложить избыток РН 3 и прекратить образование продукта в смеси, поддерживая температуру ниже 12 С. После прекращения реакции смесь перемешивали в течение 20 мин и концентрировали при по 000470 46 ниженном давлении. Смесь, содержащую продукт, экстрагировали трижды 60 мл этилацетата. Органические слои объединяли и промывали 20 мл воды, 25 мл насыщенного раствора хлорида натрия и концентрировали при пониженном давлении, получая 1,1 г неочищенного масла. Неочищенный продукт очищали с помощью хроматографии на силикагеле(хлороформ:метанол в качестве раствора для элюирования, 10:6), получая 900 мг (выход 94,4%) 3S(N-трет-бутоксикарбонил) амино-4-фенилбутан 1,2R-диола в виде белого твердого вещества. Пример 17. Получение 3S-(N-трет-бутоксикарбонил) амино-2R-гидрокси-4-фенилбут-1-илтолуолсульфоната. К раствору, содержащему 744,8 мг (2,65 ммоля) 3S-(N-трет-бутоксикарбонил)амино-4 фенилбутан-1,2R-диола в 13 мл пиридина, при 0 С в виде одной порции добавляли 914 мг толуолсульфонилхлорида. Смесь перемешивали при температуре от 0 до 5 С в течение 5 ч. К реакционной смеси добавляли смесь, содержащую 6,5 мл этилацетата и 15 мл 5%-ного водного раствора бикарбоната натрия, и перемешивали в течение 5 мин. Смесь, содержащую продукт, экстрагировали трижды 50 мл этилацетата. Органические слои объединяли и промывали 15 мл воды, 10 мл насыщенного раствора хлорида натрия и концентрировали при пониженном давлении, получая приблизительно 1,1 г плотного твердого вещества желтого цвета. Неочищенный продукт очищали с помощью хроматографии на силикагеле (этилацетат:гексан в качестве раствора для элюирования, 1:3), получая 850 мг (выход 74%) 3S-(N-трет-бутоксикарбонил)амино-2R-гидрокси-4-фенилбут-1-илтолуолсульфоната в виде белого твердого вещества. Пример 18. Получение 3S-[N-(трет-бутоксикарбонил) амино]-1-(2-метилпропил)амино-4-фенилбутан 2R-ола. К раствору, содержащему 90 мг (0,207 ммоля) 3S-(N-трет-бутоксикарбонил)амино-2Rгидрокси-4-фенилбут-1-илтолуолсульфоната в 0,143 мл изопропанола и 0,5 мл толуола, добавляли 0,103 мл (1,034 ммоля) изобутиламина. Смесь нагревали до 80-85 С и перемешивали в течение 1,5 ч. Смесь, содержащую продукт,концентрировали при пониженном давлении 47 при 40-50 С и очищали с помощью хроматографии на силикагеле (хлороформ:метанол в качестве раствора для элюирования, 10:1), получая 54,9 мг(выход 76,8%) 3S-[N-(третбутоксикарбонил)амино]-1-(2-метилпропил) амино-4-фенилбутан-2R-ола в виде белого твердого вещества. Пример 19.N-бензилоксикарбонил-Lфенилаланинхлорметилкетона в смеси с 807 мл метанола и 807 мл тетрагидрофурана, при -2 С в течение 100 мин добавляли 13,17 г (0,348 моля,1,54 экв.) твердого борогидрида натрия. Растворители удаляли при пониженном давлении при 40 С и остаток растворяли в этилацетате (приблизительно 1 л). Раствор промывали последовательно 1 М бисульфатом калия, насыщенным раствором бикарбоната натрия и затем насыщенным раствором хлорида натрия. После высушивания над безводным сульфатом магния и фильтрации раствор отгоняли при пониженном давлении. К образовавшемуся маслу добавляли гексан (приблизительно 1 л) и смесь нагревали при перемешивании до 60 С. После охлаждения до комнатной температуры собирали твердый продукт и промывали 2 л гексана. Образовавшийся твердый продукт перекристаллизовывали из горячего этилацетата и гексана, получая 32,3 г (выход 43%) N-бензилоксикарбонил-3(S)амино-1-хлор-4-фенил-2(S)-бутанола, tпл 150151 С и M+Li+ = 340. Раздел Б. К раствору, содержащему 6,52 г(0,116 моля, 1,2 экв.) гидроксида калия в 968 мл абсолютного этанола, при комнатной температуре добавляли 32,3 г (0,097 моля) N-СВz-3(S)амино-1-хлор-4-фенил-2(S)-бутанола. После перемешивания в течение 15 мин растворитель удаляли при пониженном давлении и твердый продукт растворяли в метиленхлориде. После промывки водой, сушки над сульфатом магния,фильтрации и упаривания получали 27,9 г белого твердого вещества. После перекристаллизации из горячего этилацетата и гексана получали 22,3 г (выход 77%) N-бензилоксикарбонил-3(S)амино-1,2(S)-эпокси-4-фенилбутана, tпл 102103C и МН+ 298. Раздел В. Раствор N-бензилоксикарбонил 3(S)-амино-1,2(S)-эпокси-4-фенилбутана (1,00 г,3,36 ммоля) и изобутиламина (4,90 г, 67,2 ммоля, 20 экв.) в 10 мл изопропилового спирта нагревали с обратным холодильником в течение 1,5 ч. Раствор охлаждали до комнатной темпе 000470 48 ратуры, концентрировали в вакууме и затем сливали при перемешивании в 100 мл гексана,после чего продукт кристаллизовался из раствора. Продукт выделяли фильтрацией и сушили на воздухе, получая 1,18 г (выход 95%) N-3(S)фенилметилкарбамоил)]амино-2(R)-гидрокси-4 фенилбутил]-N-[(2-метилпропил)]амина,C22H30N2O3, tпл 108,0-109,5 С, МН+ m/z = 371. Пример 20.(483 мкл, 3,8 ммоля) получали фенилметил [2Rгидрокси-3-[(3-метилбутил)(фенилсульфонил) амино]-1S-(фенилметил)пропил]карбамат. После хроматографии на силикагеле, элюируя хлороформом, содержащим 1% этанола, получали чистый продукт. Анализ: рассчитано для Получение 2R-гидрокси-3-(4-аминофенил)сульфонил](2-метилпропил)амино]-1S(фенилметил)пропиламина. Раздел А. Получение фенилметилового эфира 2R-гидрокси-3-(4-нитрофенил)сульфонил](2-метилпропил)амино]-1S-(фенилметил) пропилкарбаминовой кислоты. К раствору, содержащему 4,0 г (10,8 ммоля)(3,27 г, 32,4 ммоля) триэтиламина. Раствор охлаждали до 0 С и добавляли 2,63 г (11,9 ммоля) 4-нитробензолсульфонилхлорида, перемешивали в течение 30 мин при 0 С, а затем в течение 1 ч при комнатной температуре. Добавляли этилацетат, промывали 5%-ной лимонной кислотой,насыщенным бикарбонатом натрия, соляным раствором, сушили и концентрировали, получая 5,9 г неочищенного продукта. Этот продукт перекристаллизовывали из этилацетата/гексана,получая 4,7 г чистого фенилметилового эфира 2R-гидрокси-3-(4-нитрофенил)сульфонил] (2 49 метилпропил)амино]-1S-(фенилметил) пропилкарбаминовой кислоты, m/е = 556 (М+Н). Раздел Б. Получение 2R-гидрокси-3-(4 аминофенил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропиламина. Раствор, содержащий 3,0 г (5,4 ммоля) фенилметилового эфира 2R-гидрокси-3-(4 нитрофенил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропилкарбаминовой кислоты в 20 мл этилацетата, гидрировали в течение 3,5 ч с использованием в качестве катализатора 1,5 г 10%-ного палладия на угле при избыточном давлении водорода 35 фунтов/кв.дюйм. Катализатор удаляли фильтрацией и раствор концентрировали,получая 2,05 г требуемого 2R-гидрокси-3-(4 аминофенил)сульфонил](2-метилпропил) амино]-1S-(фенилметил) пропиламина, m/е = 392 (М+Н). Пример 22. Получение 2R-гидрокси-3-(3-аминофенил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропиламина. Раздел А. Получение фенилметилового эфира 2R-гидрокси-3-(3-нитрофенил)сульфонил](2-метилпропил)амино]-1S-(фенилметил) пропилкарбаминовой кислоты. К раствору, содержащему 1,1 г (3,0 ммоля)(0,94 г, 9,3 ммоля) триэтиламина. Раствор охлаждали до 0 С и добавляли 0,67 г (3,0 ммоля) 3 нитробензолсульфонилхлорида, перемешивали в течение 30 мин при 0 С, затем в течение 1 ч при комнатной температуре. Добавляли этилацетат, промывали 5%-ной лимонной кислотой,насыщенным бикарбонатом натрия, соляным раствором, сушили и концентрировали, получая 1,74 г неочищенного продукта. Этот продукт перекристаллизовывали из этилацетата/гексана,получая 1,40 г чистого фенилметилового эфира 2R-гидрокси-3-(3-нитрофенил)сульфонил](2 метилпропил)амино]-1S-(фенилметил)пропилкарбаминовой кислоты, m/е = 562 (M+Li). Раздел Б. Получение 2R-гидрокси-3-(3 аминофенил)сульфонил] (2-метилпропил) амино] -1S- (фенилметил)пропиламина. Раствор, содержащий 1,33 г (2,5 ммоля) фенилметилового эфира 2R-гидрокси-3-(3 нитрофенил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропилкарбаминовой кислоты в 40 мл смеси (1:1) метанол: тетрагидрофуран, гидрировали в течение 1,5 ч с использо 000470 50 ванием в качестве катализатора 0,70 г 10%-ного палладия на угле при избыточном давлении водорода 40 фунтов/кв.дюйм. Катализатор удаляли фильтрацией и раствор концентрировали,получая 0,87 г требуемого 2R-гидрокси-3-(3 аминофенил)сульфонил](2-метилпропил)амино] Получение 2S-[(пирролидин-1-ил)ацетил] амино]-N-[2R-гидрокси-3-[N-(2-метилпропил)N1-(2,3-дигидробензофуран-5-илсульфонил) амино]-1S-(фенилметил)пропил]-3S-метилпентанамида. Раздел А. Получение гидрохлорида пирролидинуксусной кислоты. Раствор, содержащий 19,6 г (101 ммоль) трет-бутилбромацетата в 150 мл ТГФ, охлаждали в ледяной бане и по каплям обрабатывали в течение приблизительно 0,5 ч раствором, содержащим 14,4 г (202 ммоля) пирролидина в 75 мл ТГФ, получая осадок белого цвета. Баню удаляли и образовавшуюся в результате реакции суспензию перемешивали в течение двух ч. Твердый продукт удаляли фильтрацией и фильтрат концентрировали при пониженном давлении, получая прозрачную жидкость над окрашенным в оранжевый цвет твердым продуктом. Жидкость охлаждали в ледяной бане,затем обрабатывали 40 мл (80 ммолей) 4 н. НСl в диоксане и перемешивали в течение 15 ч. Растворители удаляли в вакууме и остаток растирали с диэтиловым эфиром, а затем фильтровали,получая 12,9 г требуемой кислоты в виде беловатого твердого вещества. Раздел Б. Получение 5-(2,3-дигидробензофуранил)сульфонилхлорида. К раствору, содержащему 3,35 г безводного N,N-диметилформамида, при 0 С в атмосфере азота добавляли 6,18 г сульфурилхлорида,после чего образовывался твердый продукт. После перемешивания в течение 15 мин добавляли 4,69 г 2,3-дигидробензофурана и смесь выдерживали при 100 С в течение 2 ч. Реакционную смесь охлаждали, сливали на ледяную воду, экстрагировали метиленхлоридом, сушили над сульфатом магния, фильтровали и концентрировали неочищенный продукт. Этот продукт перекристаллизовывали из этилацетата, получая 2,45 г 5-(2,3-дигидробензофуранил)сульфонилхлорида. Раздел В. Получение фенилметилового эфира 2R-гидрокси-3-(2,3-дигидробензофуран 5-ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропилкарбаминовой кислоты. К раствору, содержащему 1,11 г (3,0 ммоля)(0,94 г, 9,3 ммоля) триэтиламина. Раствор охлаждали до 0 С и добавляли 0,66 г 5-(2,3 дигидробензофуранил)сульфонилхлорида, перемешивали в течение 15 мин при 0 С, а затем в течение 2 ч при комнатной температуре. Добавляли этилацетат, промывали 5%-ной лимонной кислотой, насыщенным бикарбонатом натрия,соляным раствором, сушили и концентрировали, получая 1,62 г неочищенного продукта. Этот продукт перекристаллизовывали из диэтилового эфира, получая 1,17 г чистого фенилметилового эфира 2R-гидрокси-3-(2,3-дигидробензофуран 5-ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропилкарбаминовой кислоты. Раздел Г. Получение 2R-гидрокси-3-(2,3 дигидробензофуран-5-ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропиламина. Раствор, содержащий 2,86 г фенилметилового эфира 2R-гидрокси-3-(2,3-дигидробензофуран-5-ил)сульфонил](2-метилпропил)амино]1S-(фенилметил)пропилкарбаминовой кислоты в 30 мл тетрагидрофурана, гидрировали в течение 16 ч с использованием 0,99 г 10%-ного палладия на угле при избыточном давлении водорода 50 фунтов/кв.дюйм. Катализатор удаляли фильтрацией и фильтрат концентрировали, получая 1,99 г требуемого 2R-гидрокси-3-(2,3 дигидробензофуран-5-ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропиламина. Раздел Д. Получение 2S-[(карбобензилокси)амино]-N-[2R-гидрокси-3-[(3-метилпропил)(2,3-дигидробензофуран-5-илсульфонил) амино]-1S-(фенилметил)пропил]-3S-метилпентанамида. Раствор, содержащий 5,8 г (22,0 ммоля) NCBz-L-изолейцина в 45 мл безводного N,Nдиметилформамида (ДМФ), охлаждали до 0 С и к нему добавляли 3,9 г (28,7 ммоля) Nгидроксибензотриазола (ГОБТ) и 4,2 г (22,0 ммоля) гидрохлорида 1-(3-диметиламинопропил)-3-этилкарбодиимида (ЭДК). Через 20 мин ледяную баню удаляли и перемешивание продолжали еще в течение 40 мин. К реакционному раствору затем добавляли раствор, содержащий 8,0 г (19,1 ммоля) 2R-гидрокси-3-(2,3 дигидробензофуран-5-ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропиламина и 2,2 г (22,0 ммоля) 4-метилморфолина в 25 мл безводного ДМФ, и перемешивали в течение 15 ч. Растворители удаляли в вакууме и остаток распределяли между 300 мл этилацетата и 120 мл 5%-ного раствора бисульфата калия. Слои разделяли и органический слой промывали, используя по 120 мл насыщенного раствора бикарбоната натрия, воды и соляного раствора, а затем сушили над безводным сульфатом магния,фильтровали и концентрировали в вакууме, получая 16,7 г неочищенного продукта. Неочищенный продукт кристаллизовали из этанола,получая 12,0 г (94%) 2S-[(карбобензилокси)-1S-(фенилметил)пропил]-3S-метилпентанамида: m/е = 762 (M+Li). Раздел Е. Получение 2S-амино-N-[2Rгидрокси-3-[(3-метилпропил)(2,3-дигидробензофуран-5-илсульфонил)амино]-1S-(фенилметил)пропил]-3S-метилпентанамида. В сосуд Фишера-Портера, снабженный магнитной мешалкой, загружали 11,9 г (17,9 ммоля) 2S-[(карбобензилокси)амино]-N-[2Rгидрокси-3-[(3-метилпропил)(2,3-дигидробензофуран-5-илсульфонил)амино]-1S-(фенилметил) пропил]-3S-метилпентанамида и 75 мл тетрагидрофурана (ТГФ). Раствор гидрировали в течение 4 ч при комнатной температуре в присутствии в качестве катализатора 5 г 10%-ного палладия на угле (50 мас.% воды) при избыточном давлении водорода 50 фунтов/кв.дюйм. Катализатор удаляли фильтрацией и растворитель удаляли в вакууме. Остаток растворяли в 300 мл этилацетата и промывали, используя по 120 мл насыщенного раствора бикарбоната натрия и соляного раствора, а затем сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении, получая 8,8 г требуемого продукта, m/е = 532 (М+Н). Раздел Ж. Получение 2S-[(пирролидин-1 ил)ацетил]амино]-N-[2R-гидpoкcи-3-[N1-(2-мeтилпpoпил)-N-(2,3-дигидpoбeнзoфypaн-5-илсульфонил)амино]-1S-(фенилметил)пропил]-3Sметилпентанамида. Раствор, содержащий 3,7 г (22,1 ммоля) гидрохлорида пирролидинуксусной кислоты в 45 мл безводного ДМФ, охлаждали до 0 С и к нему добавляли 3,4 г (24,7 ммоля) ГОБТ и 3,6 г(19,0 ммолей) ЭДК. Через 20 мин ледяную баню удаляли и перемешивание продолжали еще в течение 40 мин. Затем к реакционному раствору добавляли раствор, содержащий 8,8 г (16,5 ммоля) 2S-амино-N-[2R-гидрокси-3-[(3-метилпропил)(2,3-дигидробензофуран-5-илсульфонил) амино]-1S-(фенилметил)пропил]-3S-метилпентанамида и 4,5 г (44,1 ммоля) 4-метилморфолина в 25 мл безводного ДМФ, и перемешивали в течение 16 ч. Растворители удаляли в вакууме и остаток распределяли между 300 мл этилацетата и 120 мл 5%-ного раствора бисульфата калия. Слои разделяли и органический слой промывали, используя по 120 мл насыщенного раствора бикарбоната натрия, воды и соляного раствора, а затем сушили над безводным сульфатом магния, фильтровали и концентрировали в вакууме, получая 9,9 г неочищенного продукта. Вновь проводили реакцию сочетания,используя 2,5 г (15,1 ммоля) гидрохлорида пирролидинуксусной кислоты, 2,3 г (17,0 ммолей) ГОБТ, 2,45 г (12,8 ммоля) ЭДК, 3,0 г (30,0 ммолей) 4-метилморфолина и 9,9 г неочищенного продукта вместо амина, используемого в разделе Б. Повторяли обработку реакционной смеси и получали 10,2 г неочищенного продукта. Очи 53 стку осуществляли на хроматографе типа Prep 2000 на силикагеле с использованием смеси 70100% (5% метанола/95% этилацетата)/гексан,получая требуемый продукт в виде белого твердого вещества, m/e = 649 (M+Li). Пример 24. Получение N-[(1,1-диметилэтокси) карбонил]-N-[2-метилпропил]-3S-[N-(фенилметоксикарбонил)амино]-2R-гидрокси-4-фенилбутиламина. К раствору, содержащему 7,51 г (20,3 ммоля) N-[3S-[(фенилметоксикарбонил)амино]-2Rгидрокси-4-фенилбутил]-2-метилпропиламина в 67 мл безводного тетрагидрофуран, добавляли 2,25 г (22,3 ммоля) триэтиламина. После охлаждения до 0 С добавляли 4,4 г (20,3 ммоля) дитрет-бутилдикарбоната и перемешивание продолжали при комнатной температуре в течение 21 ч. Летучие компоненты удаляли в вакууме,добавляли этилацетат, затем промывали 5%-ной лимонной кислотой, насыщенным бикарбонатом натрия, соляным раствором, сушили над сульфатом магния, фильтровали и концентрировали, получая 9,6 г неочищенного продукта. После хроматографии на силикагеле с использованием смеси 30% этилацетат/гексан получали 8,2 г чистого N-3S-(фенилметилкарбамоил)амино]-2R-гидрокси-4-фенил]-1-[(2-метилпропил)амино-2-(1,1-диметилэтоксил) карбонил]бутана, масс-спектр m/е = 477 (M+Li). Пример 25. Получение 2S-бромацетил]амино]-N-[2Rгидрокси-3-[N1-(3-метилбутил)-N1-(фенилсульфонил)амино]-1S-(фенилметил)пропил]-3,3 диметилбутанамида. Раздел А. К раствору, содержащему NCBz-L-изолейцин (450 мг, 1,7 ммоля) и Nгидроксибензотриазол (260 мг, 1,7 ммоля) в ДМФ (10 мл), добавляли ЭДК (307 мг, 1,6 ммоля). Раствор перемешивали при комнатной температуре в течение 60 мин и затем добавляли 2R-гидрокси-3-[N-(3-метилбутил)-N-(фенилсульфонил)амино]-1S-(фенилметил)пропиламин(585 мг, 1,5 ммоля) в ДМФ (2 мл). Реакционную смесь перемешивали в течение 16 ч при комнатной температуре, а затем сливали на 50%-ный насыщенный раствор бикарбоната натрия (200 мл). Водную смесь трижды экстрагировали этилацетатом (50 мл). Объединенные этилацетатные слои промывали водой (50 мл) и насы 000470 54 щенным раствором NaCl (50 мл), а затем сушили над сульфатом магния. После фильтрации и концентрирования получали масло, которое хроматографировали на силикагеле (50 г),элюируя 20%-ным этилацетатом в гексане. Таким путем получали фенилметил [1S-2Rгидрокси-3-[(3-метилбутил)(фенилсульфонил) амино]-1S-(фенилметил)пропил]амино]карбонил]-2,2-диметилпропил]карбамат в виде твердого вещества. Анализ: рассчитано дляC35H47N3O6S: С 65,91; Н 7,47; N 6,59; обнаружено: С 65,42; Н 7,24; N 6,55. Раздел Б. Раствор фенилметил [1S-2Rгидрокси-3-[(3-метилбутил)(фенилсульфонил) амино]-1S-(фенилметил)пропил]амино]карбонил]-2,2-диметилпропил]карбамата (200 мг, 0,31 ммоля) в метаноле (15 мл) гидрировали в присутствии 10%-ного палладия на угле в течение 2 ч. Реакционную смесь фильтровали через диатомовую землю и концентрировали до масла. Раздел В. Полученный в разделе Б свободный амин (150 мг, 0,3 ммоля) смешивали с диизопропилэтиламином (114 мкл, 0,33 ммоля) в дихлорметане (5 мл). К этой смеси по каплям добавляли бромацетилхлорид (27 мкл, 0,33 ммоля). Реакционную смесь перемешивали в течение 30 мин при комнатной температуре,после чего разбавляли дихлорметаном (30 мл) и экстрагировали 1 н. HCl, водой и затем насыщенным раствором NaCl (по 25 мл каждого). Органический слой сушили над MgSO4 и концентрировали до твердого вещества. 2Sбромацетил]амино]-N-[2R-гидрокси-3-[(3-метилбутил)(фенилсульфонил)амино]-1S-(фенилметил)пропил]-3,3-диметилбутанамид был достаточно чистым для использования на следующей стадии. Этот продукт также может быть получен при замене бромацетилхлорида на бромуксусный ангидрид или же может быть использован хлорацетилхлорид либо хлоруксусный ангидрид. Пример 26.Nгидроксисукцинамида (2,79 г, 7,1 ммоля) и реакционную смесь перемешивали в атмосфере азота в течение 16 ч. Содержимое реакционной смеси концентрировали в вакууме и остаток 55 растворяли в этилацетате, промывали бисульфатом калия и насыщенным хлоридом натрия. Органический слой сушили над сульфатом магния,фильтровали и концентрировали, получая 4,3 г неочищенного продукта, который хроматографировали с использованием смеси этилацетат/гексан (3:1), получая 3,05 г (выход 72%) 2S(1,1-диметилэтокси)карбонил]амино]-N-[2Rгидрокси-3-[(3-метилбутил)фенилсульфонил) амино]-1S-(фенилметил)пропил]-3-метилпентанамида. Раздел Б. Полученный в разделе А продукт (3,05 г, 5,0 ммолей) растворяли в 20 мл 4 н. НСl в диоксане и перемешивали в атмосфере азота в течение 1,5 ч. Содержимое концентрировали в вакууме и обрабатывали диэтиловым эфиром. Неочищенную гидрохлоридную соль отсасывали при давлении 1 мм рт.ст. до высушивания, получая 2,54 г продукта в виде его гидрохлорида. Раздел В. Гидрохлорид амина (2,54 г, 5,0 ммолей) растворяли в 50 мл тетрагидрофурана и к этой смеси добавляли 4-метилморфолин (1,01 г, 10 ммолей), что приводило к образованию осадка. К этой суспензии добавляли хлоруксусный ангидрид (0,865 г, 5,0 ммолей) и перемешивали в течение 40 мин. Содержимое концентрировали в вакууме и остаток распределяли между этилацетатом (200 мл) и 5%-ным KHSO4. Органический слой промывали насыщенным бикарбонатом натрия и насыщенным хлоридом натрия, сушили над сульфатом магния, фильтровали и концентрировали, получая неочищенный продукт. После очистки с помощью хроматографии на силикагеле с использованием в качестве элюента смеси этилацетат/гексан (1:1) получали 1,89 г чистого хлорацетамида. Пример 27.N,Nдиметилформамида, добавляли 1,91 г (10 ммолей) ЭДК и перемешивали при 0 С в течение 1 ч. К этой смеси добавляли раствор, содержащий 4,0 г (10,0 молей) 2R-гидрокси-3-[(2-метилпропил)(4-метоксифенилсульфонил)амино]-1S(фенилметил)пропиламина в 6 мл N,N-диметилформамида, и раствор перемешивали в течение 16 ч. Растворитель удаляли с помощью роторного испарителя, заменяли его этилацетатом и промывали насыщенным бикарбонатом натрия, 000470 56 5%-ной лимонной кислотой и соляным раствором. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали, получая 6,1 г неочищенного продукта, который хроматографировали на силикагеле с использованием в качестве элюента смеси этилацетат/гексан (1:1), получая 5,12 г (выход 83%) 2S[(трет-бутоксикарбонил)амино]-N-[2R-гидрокси-3-[(3-метилпропил)(4-метоксифенилсульфонил)амино]-1S-(фенилметил)пропил]-3Sметилпентанамида. Раздел Б. 5,00 г (8,0 ммолей) продукта, полученного в разделе А, растворяли в 20 мл 4 н. НСl в диоксане и перемешивали в течение 20 мин. Осажденный продукт упаривали дважды из диэтилового эфира, получая гидрохлорид 2S(амино)-N-[2R-гидрокси-3-[(3-метилпропил)(4 метоксифенилсульфонил)амино]-1S-(фенилметил)пропил]-3S-метилпентанамида,который использовали в разделе В без дополнительной очистки. Раздел В. Гидрохлорид амина, полученный в разделе Б, растворяли в 45 мл метиленхлорида и добавляли 3,0 г N,N-диизопропилэтиламина, а затем 1,22 г (7,11 ммоля) хлоруксусного ангидрида. Раствор перемешивали при комнатной температуре в течение 30 мин. Содержимое концентрировали в вакууме и остаток распределяли между этилацетатом и водой. Органический слой промывали 5%-ной лимонной кислотой, насыщенным бикарбонатом натрия и соляным раствором. Органическую фазу сушили над сульфатом магния, фильтровали и концентрировали в вакууме, получая неочищенный продукт 2S-[(хлорацетил)амино]-N-[2R-гидрокси-3-[(3 метилпропил)(4-метоксифенилсульфонил) амино]-1S-(фенилметил)пропил]-3S-метилпентанамид в виде пены белого цвета, который использовали в разделе Г без дополнительной очистки. Раздел Г. 4,8 г (8,0 ммолей) 2S[(хлорацетил)амино]-N-[2R-гидрокси-3-[(3-метилпропил)(4-метоксифенилсульфонил)амино]1S-(фенилметил)пропил]-3S-метилпентанамида растворяли в 15 мл тетрагидрофурана и к этой смеси добавляли воду, а затем 2,8 мл (40 ммолей) пирролидина и реакционную смесь перемешивали в течение 1,5 ч. Растворители удаляли в вакууме и остаток растворяли в этилацетате. Смесь последовательно промывали насыщенным бикарбонатом натрия и соляным раствором, сушили над сульфатом магния, фильтровали и концентрировали в вакууме, получая 5,6 г неочищенного продукта. После очистки с помощью быстрой хроматографии на силикагеле с использованием в качестве элюента 1-3%-ного метанола в дихлорметане получали 3,2 г 2S[(пирролидин-1-ил)ацетиламино]-N-[2R-гидрокси-3-[N-(2-метилпропил)-N1-(4-метоксифенилсульфонил)амино]-1S-(фенилметил)пропил]-3Sметилпентанамида в виде белого твердого вещества. Получение фенилметилового эфира 2Rгидрокси-3-(2-аминобензотиазол-6-ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил) пропилкарбаминовой кислоты. Фенилметиловый эфир 2R-гидрокси-3-(4 аминофенил)сульфонил](2-метилпропил)амино]-1S-(фенилметил)пропилкарбаминовой кислоты (0,30 г, 0,571 ммоля) добавляли к тщательно перемешанному порошку безводного сульфата меди (1,20 г) и тиоцианата калия (1,50 г), а затем добавляли безводный метанол (6 мл) и образовавшуюся суспензию темно-коричневого цвета кипятили с обратным холодильником в течение 2 ч. Реакционную смесь фильтровали, фильтрат разбавляли водой (5 мл) и кипятили с обратным холодильником. К реакционной смеси добавляли этанол, охлаждали и фильтровали. После концентрирования фильтрата получали остаток, который хроматографировали (этилацетат/гексан, 80:20), получая 0,26 г (78%) требуемого соединения в виде твердого вещества. Пример 29. Получение фенилметилового эфира 2Rгидрокси-3-(бензотиазол-6-ил)сульфонил](2 метилпропил)амино]-1S-(фенилметил)пропилкарбаминовой кислоты. Способ 1. Фенилметиловый эфир 2Rгидрокси-3-(2-аминобензотиазол-6-ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил) пропилкарбаминовой кислоты (0,25 г, 0,429 ммоля) добавляли к раствору изоамилнитрита(0,116 мл, 0,858 ммоля) в диоксане (5 мл) и смесь выдерживали при 85 С. После прекращения выделения азота реакционную смесь концентрировали и остаток очищали с помощью хроматографии (гексан:этилацетат, 5:3), получая 0,130 г (53%) требуемого продукта в виде твердого вещества. Способ 2. Неочищенный бензотиазол-6 сульфонилхлорид в этилацетате (100 мл) добавляли к N-[3S-бензилоксикарбониламино-2Rгидрокси-4-фенил]-N-изобутиламину (1,03 г,2,78 ммоля),а затем добавлялиNметилморфолин (4 мл). После перемешивания при комнатной температуре в течение 18 ч реакционную смесь разбавляли этилацетатом (100 58 мл), промывали лимонной кислотой (5%, 100 мл), бикарбонатом натрия (насыщенный, 100 мл) и соляным раствором (100 мл), сушили Получение фенилметилового эфира 2Rгидрокси-3-(2-аминобензотиазол-5-ил)сульфонил](2-метилпропил)амино]-1S-(фенилметил) пропилкарбаминовой кислоты и фенилметилового эфира 2R-гидрокси-3-(2-аминобензотиазол-7-ил)сульфонил](2-метилпропил)амино]-1S(фенилметил)пропилкарбаминовой кислоты. Фенилметиловый эфир 2R-гидрокси-3-[(3 аминофенилсульфонил)(2-метилпропил)амино]1S-(фенилметил)пропилкарбаминовой кислоты(0,36 г, 0,685 ммоля) добавляли к тщательно перемешанному порошку безводного сульфата меди (1,44 г) и тиоцианата калия (1,80 г), а затем добавляли безводный метанол (10 мл) и образовавшуюся суспензию темно-коричневого цвета кипятили с обратным холодильником в течение 2 ч. Реакционную смесь фильтровали и фильтрат разбавляли водой (5 мл) и кипятили с обратным холодильником. К реакционной смеси добавляли этанол, охлаждали и фильтровали. После концентрирования фильтрата получали остаток, который хроматографировали (этилацетат:гексан, 1:1), получая 0,18 г (45%) 7-изомера в виде твердого вещества. После дополнительной очистки на колонках (этилацетат:гексан,3:2) получали 0,80 г (20%) 5-изомера в виде твердого вещества. Пример 31. 59 К раствору хлорметилкетона N-бензилоксикарбонил-L-фенилаланина (75 г, 0,2 моля) в смеси с 800 мл метанола и 800 мл тетрагидрофурана в течение 100 мин добавляли борогидрид натрия (13,17 г, 0,348 моля, 1,54 экв.). Раствор перемешивали при комнатной температуре в течение 2 ч и затем концентрировали в вакууме. Остаток растворяли в 1000 мл этилацетата и промывали 1 н. KHSO4, насыщенным воднымNaHCO3, насыщенным водным NaCl, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме, получая масло. Неочищенный продукт растворяли в 1000 мл гексана при 60 С и давали охладиться до комнатной температуры, после чего образовывались кристаллы, которые выделяли фильтрацией и промывали избыточным количеством гексана. Затем этот твердый продукт перекристаллизовывали из горячего этилацетата и гексана, получая 32,3 г (выход 43%) N-бензилоксикарбонил-3(S)амино-1-хлор-4-фенил-2(S)-бутанола, tпл 150151 С, МС (FAB): МLi+ = 340. Раздел Б. 3(S)-[N-(бензилоксикарбонил) амино]-1,2(S)-эпокси-4-фенилбутан. Раствор гидроксида калия (6,25 г, 0,116 моля, 1,2 экв.) в 970 мл абсолютного этанола обрабатывалиN-бензилоксикарбонил-3(S)амино-1-хлор-4-фенил-2(S)-бутанолом (32,3 г,0,097 моля). Этот раствор перемешивали при комнатной температуре в течение 15 мин и затем концентрировали в вакууме, получая белое твердое вещество. Это твердое вещество растворяли в дихлорметане и промывали водой,сушили над безводным MgSO4, фильтровали и концентрировали в вакууме, получая белое твердое веществ. Твердое вещество перекристаллизовывали из гексана и этилацетата, получая 22,3 г (выход 77%) 3(S)-[N-(бензилоксикарбонил)амино]-1,2(S)-эпокси-4-фенилбутана,tпл 102-103 С, МС (FAB): МН+ 298. Раздел В. N-[3(S)-бензилоксикарбониламино-2(R)-гидрокси-4-фенил]-N-изобутиламин. РастворN-бензилкарбонил-3(S)-амино 1,2(S)-эпокси-4-фенилбутана (50,0 г, 0,168 моля) и изобутиламина (246 г, 3,24 моля, 20 экв.) в 650 мл изопропилового спирта кипятили с обратным холодильником в течение 1,25 ч. Раствор охлаждали до комнатной температуры, концентрировали в вакууме и затем сливали при перемешивании в 1 л гексана, после чего продукт кристаллизовался из раствора. Продукт выделяли фильтрацией и сушили на воздухе, получая 57,56 г(461 мг, 2,61 ммоля). Раствор перемешивали при комнатной температуре в течение 16 ч и затем концентрировали в вакууме. Остаток растворяли в этилацетате и этот раствор промывали 1 н.KHSO4, насыщенным водным NaHCO3, соляным раствором, сушили над безводным MgSO4,фильтровали и концентрировали, получая 1,234 г прозрачного масла. Масло кристаллизовали из смеси простого эфира и гексана, получая 729,3 мг требуемого вещества (выход 56,5%), tпл 9599 С, МС (FAB): MH+ = 511. Раздел Д. 3S-амино-1-[N-(2-метилпропил)N-(4-метоксифенилсульфонил)амино]-4-фенил 2R-бутанол. Раствор фенилметил [2(R)-гидрокси-3-[N(2-метилпропил)-N-(4-метоксифенилсульфонил) амино]-1S-(фенилметил)пропил]карбамата(671,1 мг, 1,31 ммоля), полученного в разделе Г,в 10 мл метанола гидрировали при избыточном давлении 40 фунтов/кв.дюйм при комнатной температуре в течение 15 ч в присутствии 50 мг 10%-ного палладия на угле. Катализатор удаляли фильтрацией через диатомовую землю и фильтрат концентрировали, получая пену белого цвета, 474 мг (выход 96%), МС (FAB): MH+= 377. Пример 32. Получение 1,3-бензодиоксол-5-сульфонилхлорида. Способ 1. К раствору, содержащему 4,25 г безводного N,N-диметилформамида, при 0 С в атмосфере азота добавляли 7,84 г сульфурилхлорида, после чего образовывался твердый продукт. После перемешивания в течение 15 мин добавляли 6,45 г 1,3-бензодиоксола и смесь выдерживали при 100 С в течение 2 ч. Реакционную смесь охлаждали, сливали на ледяную воду, экстрагировали метиленхлоридом, сушили над сульфатом магния, фильтровали и концентрировали, получая 7,32 г неочищенного продукта в виде масла черного цвета. Этот продукт хроматографировали на силикагеле, используя смесь 20% метиленхлорида/гексан, получая 1,9 г (1,3-бензодиоксол-5-ил)сульфонилхлорида. Способ 2. В круглодонную колбу объемом 22 л, снабженную механической мешалкой, охлаждающим конденсатором, кожухом с подогревом, капельной воронкой с уравновешенным давлением, добавляли комплекс триоксид серы/ДМФ (2778 г, 18,1 моля). Затем добавляли дихлорэтан (4 л) и начинали перемешивание. После этого через капельную воронку в течение 5 мин добавляли 1,3-бензодиоксол (1905 г, 15,6 моля). Далее температуру повышали до 75 С и поддерживали на этом уровне в течение 22 ч(ЯМР-анализ показал, что реакция завершена через 9 ч). Реакционную смесь охлаждали до
МПК / Метки
МПК: C07K 5/062, A61K 38/55
Метки: ретровирусов, ингибиторов, аминокислот, качестве, протеаз, гидроксиэтиламиносульфонамиды
Код ссылки
<a href="https://eas.patents.su/30-470-gidroksietilaminosulfonamidy-aminokislot-v-kachestve-ingibitorov-proteaz-retrovirusov.html" rel="bookmark" title="База патентов Евразийского Союза">Гидроксиэтиламиносульфонамиды аминокислот в качестве ингибиторов протеаз ретровирусов.</a>
Производные индола в качестве антагонистов возбуждающих аминокислот

Номер патента: 308
Опубликовано: 29.04.1999
Авторы: Де Маджистрис Элизабетта, Конти Надия, Ферьяни Альдо, Ди Фабио Романо
МПК: C07D 209/42, A61K 31/40
Метки: аминокислот, индола, возбуждающих, производные, качестве, антагонистов
Формула / Реферат:
1. Соединение формулы (I) или его соль, или метаболически лабильный сложный эфир, где m равно 2, a R представляет собой хлор в положении 4 и 6, А представляет собой незамещенную этенильную группу в трансконфигурации; R1 представляет собой водород, С1-4алкил, возможно замещенный карбоксилом, С3-6циклоалкил, фенил, возможно замещенный метоксилом, 3-пиридил, 4-тетрагидропиранил, R2 представляет собой водород или метил, R3 представляет...
Замещенные 2-фенилпиридины в качестве гербицидов

Номер патента: 83
Опубликовано: 25.06.1998
Авторы: Клинтц Ральф, Хампрехт Герхард, Мисслитц Ульф, Кениг Хартманн, Хайстрахер Элизабет, Шэфер Петер, Вальтер Хельмут, Вестфален Карл-Отто
МПК: C07D 213/61, A01N 43/40
Метки: 2-фенилпиридины, гербицидов, замещенные, качестве
Формула / Реферат:
1. Замещенные 2-фенилпиридины общей формулы 1 в которой переменные имеют следующее значение: n обозначает 0 или 1; R1, R3 и R4 независимо друг от друга обозначают водород, галоген, С1-C4алкил, C1-C4галогеналкил, гидрокси, C1-C4алкокси, С1-C4галогеналкокси, нитро, амино, С1-C4алкиламино, ди-(С1-C4 алкил)амино, меркапто, C1-C4алкилтио, С1-C4галогеналкилтио, циано, карбоксил, аминокарбонил, (С1-C4 алкиламино)карбонил или...
Замещенные 2-фенилпиридины в качестве гербицидов

Номер патента: 85
Опубликовано: 25.06.1998
Авторы: Вальтер Хельмут, Мисслитц Ульф, Кениг Хартманн, Хампрехт Герхард, Хайстрахер Элизабет, Клинтц Ральф, Шэфер Петер, Вестфален Карл-Отто
МПК: C07D 213/61, A01N 43/40
Метки: замещенные, качестве, 2-фенилпиридины, гербицидов
Формула / Реферат:
1. Замещенные 2-фенилпиридины общей формулы I в которой переменные имеют следующее значение: n обозначает 0 или 1; R1, R3 и R4 независимо друг от друга обозначают водород, галоген, C1-C4алкил, C1-C4 галогеналкил, гидрокси, C1-C4алкокси, C1-C4галогеналкокси, нитро, амино, C1-C4 алкиламино, ди-(C1-C4алкил)амино, меркапто, C1-C4алкилтио, C1-C4галогеналкилтио, циано, гидроксикарбонил, (C1-C4алкокси)карбонил, аминокарбонил,...
Замещенные 2-фенилпиридины в качестве гербицидов

Номер патента: 84
Опубликовано: 25.06.1998
Авторы: Гетц Норберт, Харреус Альбрехт, Шэфер Петер, Кениг Хартманн, Вестфален Карл-Отто, Мисслитц Ульф, Хайстрахер Элизабет, Вальтер Хельмут, Хампрехт Герхард, Клинтц Ральф
МПК: C07D 213/61, A01N 43/40
Метки: 2-фенилпиридины, качестве, гербицидов, замещенные
Формула / Реферат:
1. Замещенные 2-фенилпиридины общей формулы I в которой переменные имеют следующие значения: n обозначает 0 или 1; R1, R3 и R4 независимо друг от друга обозначают водород, галоген, C1-C4алкил, C1-C4 галогеналкил, гидрокси, C1-C4алкокси, C1-C4галогеналкокси, нитро, амино, C1-C4 алкиламино, ди-(C1-C4алкил)амино, меркапто, C1-C4алкилтио, C1-C4галогеналкилтио, циано, карбоксил, (C1-C4алкокси)карбонил, аминокарбонил, C1-C4алкиламинокарбонил...
Бис-(2-галоидэтил)аминофенилзамещенные производные дистамицина и их применение в качестве противоопухолевого и противовирусного средства

Номер патента: 33
Опубликовано: 26.02.1998
Авторы: Берия Итало, Францетти Кристина, Коззи Паоло, Кополонго Лаура
МПК: C07D 207/34, A61K 31/40
Метки: противоопухолевого, бис-(2-галоидэтил)аминофенилзамещенные, средства, применение, качестве, противовирусного, производные, дистамицина
Формула / Реферат:
1.Бис-(2-галоидэтил) аминофенилзамещенные производные дистамицина формулы (I) в которой n обозначает 2, 3 или 4; один из R и R1 обозначает водород, С1-С4 -алкил, СF3 или С1-С4 -алкокси, а другой независимо обозначает СF3, С1-С4 -алкил или С1-С4 -алкокси; и Х обозначает галоген; и его фармацевтически приемлемые соли. 2. Соединение формулы (I) по п.1, в котором n обозначает 3; Х обозначает хлор; один из R и R1 обозначает водород или С1-С4...
Предыдущий патент: Получение производных бициклогексана
Следующий патент: Производные 2,3-бензодиазепина
Случайный патент: Устройство для ввода кабелей питания и волокна оптоволоконной сети населенного района