Производные цефалоспорина и их фармацевтические композиции
Номер патента: 24709
Опубликовано: 31.10.2016
Авторы: Ли Ньян Сук, Ву Сун Хо, Квон Хьюнь Джинь, Парк Тае Кью, Парк Чуль Сун, Чо Юн Лаг, Чае Сан Еунь, Хео Хье Джинь, Ким Йон Зу, Кан Дае Хьюк, Ян Юн Джае, Ох Кьюман, Йоун Джоун Йуль




Формула / Реферат
1. Производное цефалоспорина, представленное химической формулой (1), его сложный эфир или их фармацевтически приемлемая соль

в которой X представляет собой CR, N или С, замещенный Cl (C-Cl);
Y представляет собой C1-C2-алкил, CH(CH3)CO2H или C(CH3)2CO2H;
L представляет собой CH2 или CH=CHCH2;
R1 представляет собой NH2, NHR11 или NH(CH2)mNR11R12;
R2 представляет собой NHR21, NH(CH2)nCOOH, NH(CH2)nNR21R22 или NHC(=O)(CH2)nNR21R22;
R3 представляет собой водород или NH2,
где R представляет собой водород или C1-C3-алкил;
R11 и R21 независимо представляют собой водород, C1-C3-алкил или группу, выбранную из группы, состоящей из

R12 и R22 независимо представляют собой водород или C1-C2-алкил;
m и n независимо представляют собой целое число от 1 до 6.
2. Производное цефалоспорина по п.1, представленное химической формулой (2), его сложный эфир или их фармацевтически приемлемая соль

в которой X представляет собой CR, N или С, замещенный Cl (C-Cl);
Y представляет собой C1-C2-алкил, CH(CH3)CO2H или C(CH3)2CO2H;
L представляет собой CH2 или CH=CHCH2;
R1 представляет собой NH2, NHR11 или NH(CH2)mNR11R12;
R2 представляет собой NHR21, NH(CH2)nNR21R22 или NHC(=O)(CH2)nNR21R22,
где R представляет собой водород или C1-C3-алкил;
R11 и R21 независимо представляют собой водород, C1-C3-алкил или группу, выбранную из группы, состоящей из

R12 и R22 независимо представляют собой водород или C1-C2-алкил;
m и n независимо представляют собой целое число от 1 до 6.
3. Производное цефалоспорина по п.2, его сложный эфир или их фармацевтически приемлемая соль, где
X представляет собой CR, N или С, замещенный Cl (C-Cl);
Y представляет собой CH(CH3)CO2H или C(CH3)2CO2H;
L представляет собой CH2 или CH=CHCH2;
R1 представляет собой NH2 или NH(CH2)mNH2;
R2 представляет собой NHR21, NH(CH2)nNHR21 или NHC(=O)(CH2)nNHR21,
где R представляет собой водород или C1-C3-алкил;
R21 представляет собой группу, выбранную из группы, состоящей из

m и n независимо представляют собой целое число от 1 до 6.
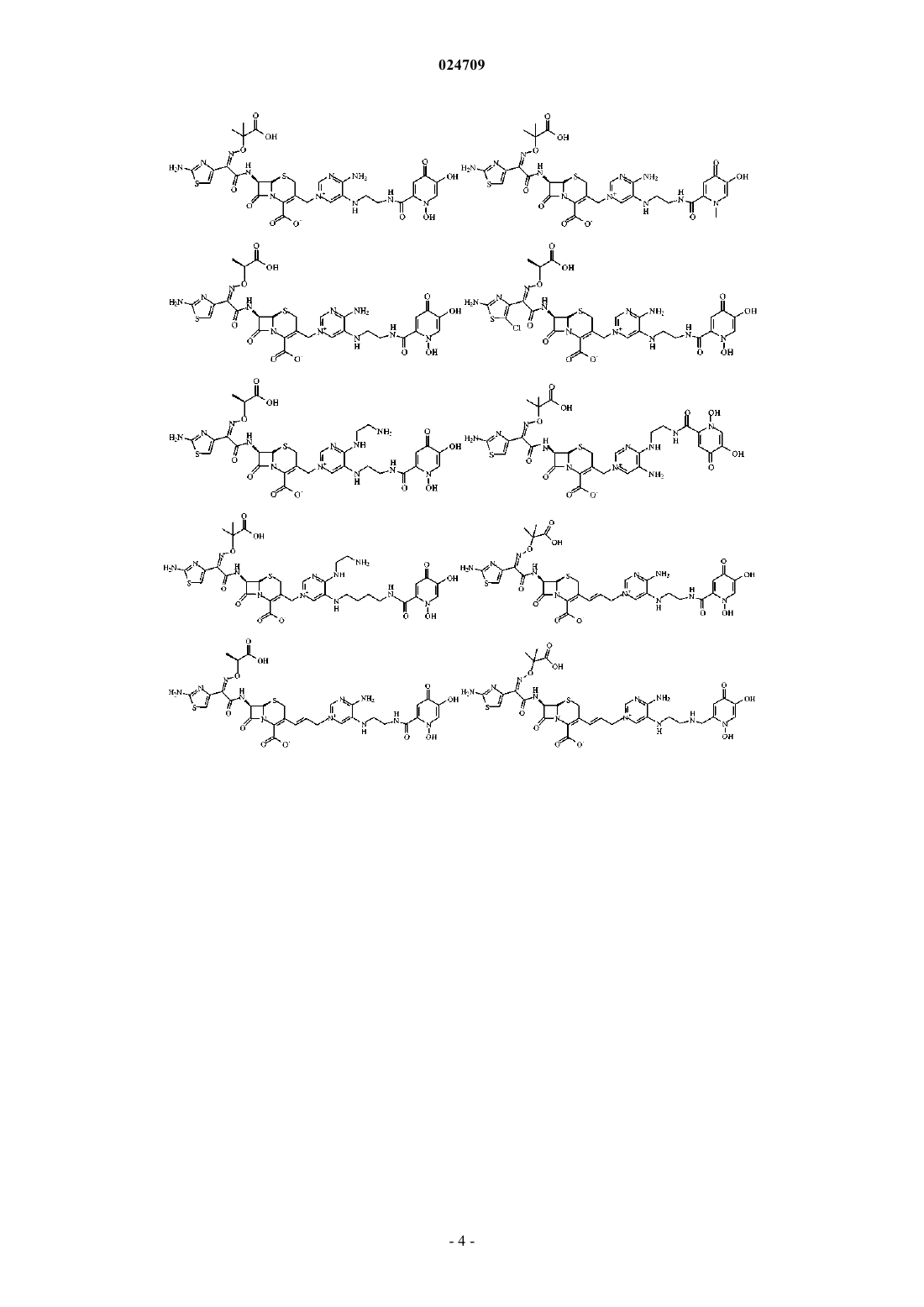
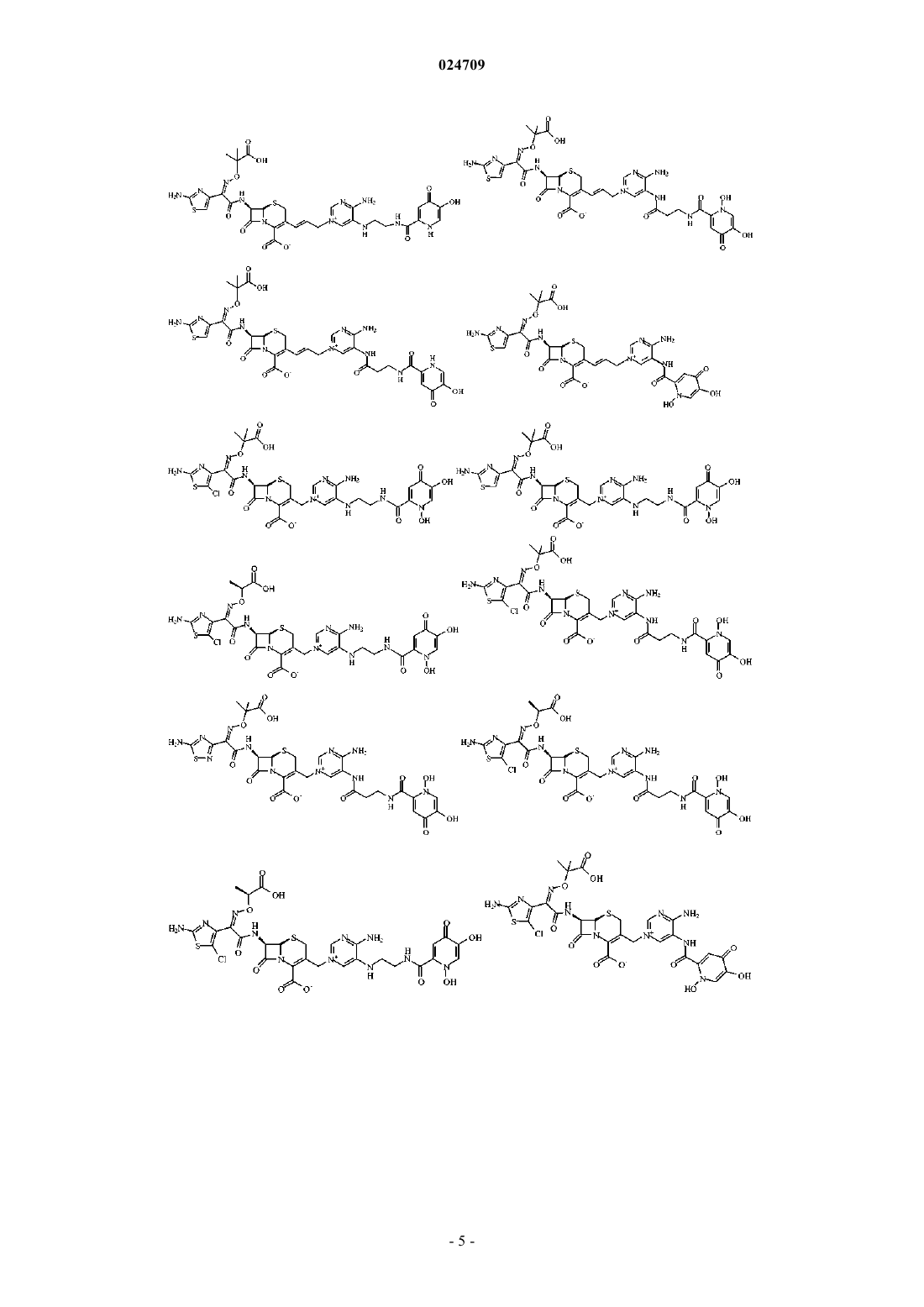
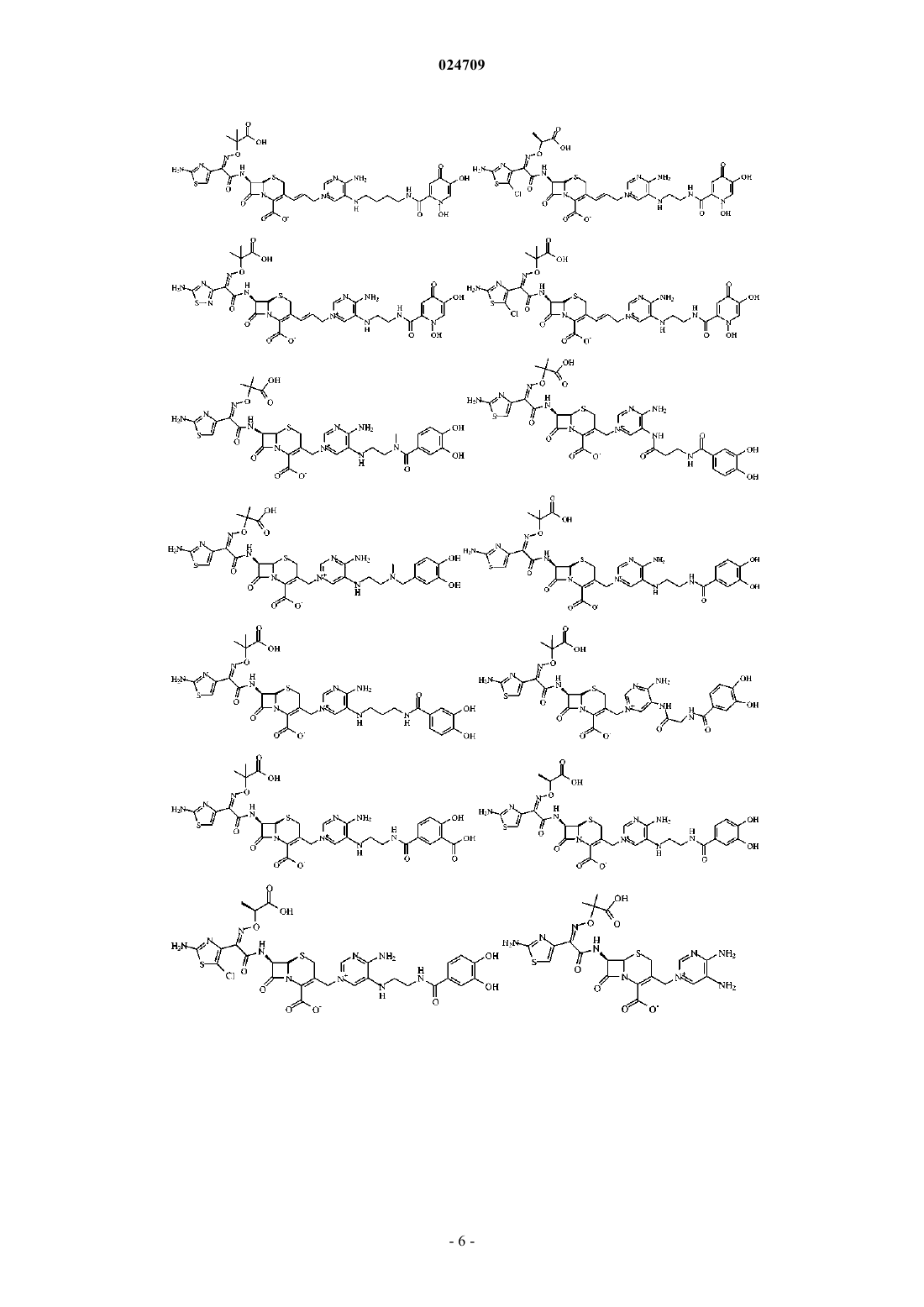
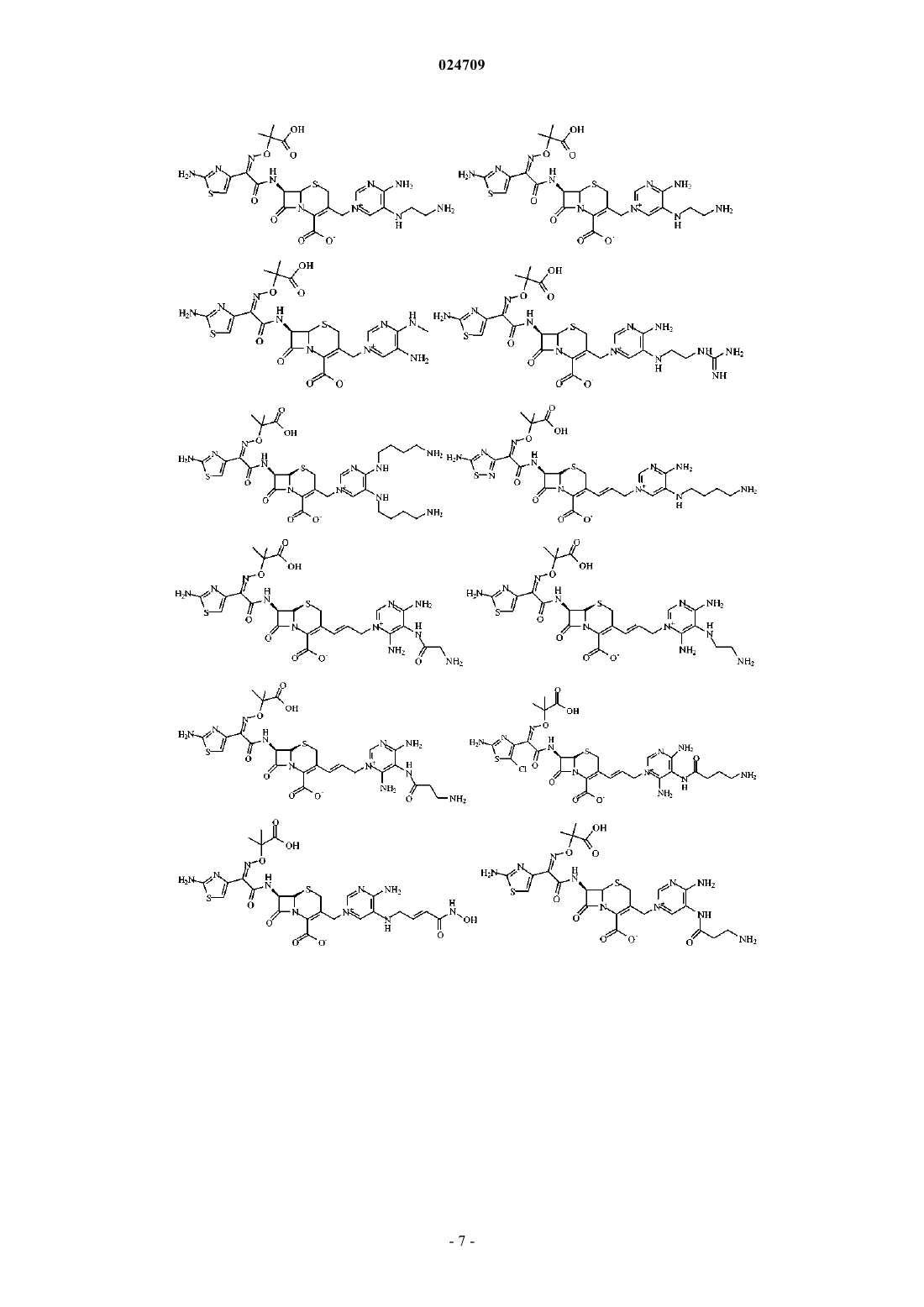
4. Производное цефалоспорина, представленное одной из следующих химических формул, его сложный эфир или их фармацевтически приемлемая соль:






5. Производное цефалоспорина по п.1, представленное одной из следующих химических формул, его сложный эфир или их фармацевтически приемлемая соль:



6. Фармацевтическая композиция, содержащая терапевтически эффективное количество производного цефалоспорина по любому из пп.1-5, его сложного эфира или их фармацевтически приемлемой соли в качестве эффективного ингредиента и фармацевтически приемлемый носитель, разбавитель, адъювант или любую их комбинацию.
7. Способ лечения бактериальной инфекции, включающий использование терапевтически эффективного количества производного цефалоспорина по любому из пп.1-5, его сложного эфира или их фармацевтически приемлемой соли.
8. Способ по п.7, где бактериальная инфекция вызвана грамотрицательной бактерией.
9. Способ по п.8, где грамотрицательная бактерия выбрана из группы, состоящей из Pseudomonas aeruginosa, Acinetobacter baumannii и Klebsiella pneumonia.
Текст
Изобретение относится к новым производным цефалоспорина, представленным химической формулой (1) где X, Y, L, R1, R2 и R3 являются такими, как определено в описании изобретения. Изобретение также относится к фармацевтическим антибиотическим композициям, содержащим новое производное цефалоспорина, представленное химической формулой (1), его пролекарство, его гидрат, его сольват, его изомер или его фармацевтически приемлемую соль в качестве эффективного ингредиента. Согласно изобретению предложены новые производные цефалоспорина, их пролекарство, их гидрат, их сольват, их изомер или их фармацевтически приемлемая соль в качестве ингредиента с низкой токсичностью, эффективного в отношении широкого спектра устойчивых к антибиотикам бактерий, в частности грамотрицательных, который может быть полезен сильной антимикробной активностью. Область техники Настоящее изобретение относится к новым производным цефалоспорина. Настоящее изобретение также относится к фармацевтическим антибиотическим композициям, содержащим новые производные цефалоспорина, их сложные эфиры или их фармацевтически приемлемые соли в качестве эффективного ингредиента. Предшествующий уровень техники Лечение заболеваний, вызванных грамотрицательными бактериями, стало более интенсивным благодаря многочисленным программам развития во время его золотой эры в период с 1960-х вплоть до 1980-х гг. Однако с ростом инфекций, вызванных грамположительными бактериями, такими как MRSA(метициллин-резистентный Staphylococcus aureus), в 1990-е гг. исследования в отношении грамотрицательных бактерий были отодвинуты на второй план. С конца 2000 года в связи с растущей проблемой недостаточности лечения заболеваний, вызванных грамотрицательными бактериями с множественной лекарственной устойчивостью, исследования в отношении грамотрицательных бактерий вновь вызвали интерес. Согласно недавним публикациям Американского общества по инфекционным заболеваниям(IDSA), Европейского центра профилактики и контроля над заболеваниями (ECDC) и Европейского агентства по лекарственным средствам (ЕМЕА) во всем мире существует только 8 эффективных лекарственных средств против грамотрицательных бактерий. В особенности крайне недостаточна разработка новых лекарственных препаратов против грамотрицательных бактерий с множественной лекарственной устойчивостью. В частности, недавно открытая NDM-1 (металло-бета-лактамаза из Нью-Дели) быстро распространяется и стала угрозой для международного сообщества. NDM-1, главным образом, встречается у грамотрицательных бактерий, а единственными двумя эффективными лекарственными средствами в настоящее время являются колистин и тигециклин. Однако эти лекарственные средства используют с осторожностью из-за их токсичности и побочных эффектов. Таким образом, существует крайняя необходимость в замене этих двух лекарственных средств. Быстрое распространение этих патогенных микроорганизмов является проблемой не только отдельных, затронутых ею стран, но все страны должны объединить свои усилия, чтобы контролировать такое распространение. Уже в 2004 году Американское общество по инфекционным заболеваниям (IDSA) опубликовало доклад под названием "Bad Bugs, No Drugs". В этом докладе опубликован список микроорганизмов, у которых в настоящее время возрастает глобальный уровень резистентности. Этот список составлен на основе заболеваемости, смертности от высокопатогенного микроорганизма и отсутствия эффективной лекарственной терапии. Три микроорганизма из этого списка являются грамотрицательными бактериями: изоляты P.aeruginosa, A.baumannii и K.pneumoniae. Это требует государственной поддержки, поскольку они создают серьезные проблемы возникновения эпидемий. В настоящее время существует несколько классов лекарственных средств, применимых в отношении этих бактерий, таких как цефалоспорины, карбапенемы, аминогликозиды и тигециклины. Однако не существует эффективных лекарственных препаратов, применимых против резистентных штаммов, в особенности против акинетобактерий,тигециклины являются единственным эффективным классом лекарственных средств. В 2006 году сообщалось о наличии K.pneumoniae с множественной лекарственной устойчивостью у пациентов с XDR-KP только в восточной части Соединенных Штатов, но в последнее время она распространилась на остальную часть страны. В случае с акинетобактериями инфекция была распространена в масштабах всей страны военнослужащими, которые ранее дислоцировались в странах Ближнего Востока. В качестве основного лечения, главным образом, используют карбапенемы, но число штаммов, устойчивых к карбопенемам, быстро растет, таким образом, остается использовать любое эффективное лечение. Фармацевтические компании проявляют активный интерес в связи с возрастанием потребности в способах лечения заболеваний, вызванных грамотрицательными бактериями, но лишь несколько антибиотиков находятся в разработке. Среди них имеются -лактамные ингибиторы, и некоторые заслуживающие внимания соединения представляют собой CEF-104 и CAZ-104 от компании Novexel, САХ-201 от компании Cubist, и по одному соединению из каждого из следующих классов: полимиксины, тетрациклины и аминогликозиды. Среди эффективных соединений в отношении акинетобактерий находятсяPTK-0796, тетрациклиновая группа, и СВ-182,804, производное полимиксина. Однако эти два соединения широко не используются из-за проблем с токсичностью в их профиле безопасности. В настоящее время цефалоспорины и карбапенемы являются двумя наиболее широко используемыми классами антибиотиков, активных в отношении грамотрицательных микроорганизмов. В классе карбапенемов имипенем и меропенем являются доминирующими на рынке соединениями, но преобладающими соединениями, лидирующими на рынке, являются дженерики. Цефтобипрол был наиболее многообещающим кандидатом в классе цефалоспоринов, но, к сожалению, программа его разработки была прекращена. Поэтому в классе цефалоспоринов дженерики и комбинированная терапия будут основными вариантами лечения. Одной из причин, по которой грамотрицательные бактерии с множественной лекарственной устойчивостью вызывают серьезные проблемы, является то, что большинство штаммов демонстрируют рези-1 024709 стентность к применяемым в настоящее время антибиотикам, которые не оказывают воздействия на многие штаммы. Существует несколько причин повышения резистентности штаммов, но в случае Р.aeruginosa основными причинами резистентности являются мутации на внешней мембране и в пориновых каналах. Из-за этих мутаций многие -лактамные ингибиторы не способны проникнуть в грамотрицательные бактерии. Чтобы преодолеть эту резистентность, вызванную мутациями на внешней мембране и в пориновых каналах, серьезно исследовали антибиотик, переносимый сидерофорами. Ионы железа являются необходимым ингредиентом для роста бактерий. Эти ионы железа обладают высоким сродством к сидерофору,и бактерии продуцируют сидерофор для связывания этих ионов железа и интернализируют их в свою систему. Бактерии обладают распознающими сидерофоры рецепторами клеточных мембран для связывания и интернализации ионов железа. На фиг. 1 представлен механизм связывания бактериями сидерофора и ионов железа при использовании мембранных рецепторов. Следовательно, имитирующая сидерофор группировка может быть присоединена к антибиотику, и рецептор сидерофора бактерии может связаться с антибиотиком. Бактерии затем интернализируют антибиотик. Такая интернализация проходит значительно легче, чем обычная интернализация антибиотика посредством пориновых каналов, а также не подвержена резистентности, вызванной мутацией пориновых каналов. На фиг. 2 представлена интернализация ионов железа путем связывания сидерофора с рецептором бактерии. Хотя проводится много исследований, направленных на преодоление проблемы резистентности с помощью включения сидерофорной группировки, до сих пор немногие были успешными. Одной из причин является то, что для сидерофорной группировки главным образом используют катехол, но он быстро трансформируется под действием катехол-О-метилтрансферазы (COMT) и больше не может связываться с рецептором сидерофора. Много модификаций катехола создано для преодоления этой проблемы, но это часто приводило к низкой эффективности и/или высокой токсичности. Встречались также значительные изменения в расположении сидерофорной группировки в антибиотике. Поэтому существует острая необходимость в разработке лекарственного препарата, обладающего более мощной противомикробной активностью в отношении грамотрицательных бактерий, чем существующие в настоящее время цефалоспорины. В особенности существует крайняя необходимость в разработке цефалоспоринов против резистентных штаммов P.aeruginosa и K.pneumonia. Краткое описание сущности изобретения Техническая задача Авторы настоящего изобретения синтезировали новые производные цефалоспорина, представленные химической формулой (1), в частности новые цефалоспориновые соединения с сидерофорной группой. Соединения по настоящему изобретению обладают более высокой антибактериальной активностью по сравнению с существующими антибиотиками, более эффективны в отношении грамотрицательных бактерий и обладают более сильной антимикробной активностью в отношении основных резистентных штаммов. Таким образом, первая задача настоящего изобретения состоит в том, чтобы предложить новое химическое соединение, представленное химической формулой (1). Вторая задача настоящего изобретения состоит в том, чтобы предложить фармацевтические антибиотические композиции, содержащие новые производные цефалоспорина, их сложные эфиры или их фармацевтически приемлемые соли в качестве эффективного ингредиента. Третья задача настоящего изобретения состоит в том, чтобы предложить способ эффективного антибиотического лечения с помощью предложенных фармацевтических антибиотических композиций и эффективного их количества. Решение задачи Далее будут подробно описаны воплощения настоящего изобретения. Настоящее изобретение относится к новым производным цефалоспорина, представленным химической формулой (1), в частности к новым цефалоспориновым соединениям с сидерофорной группой. Настоящее изобретение также относится к фармацевтическим антибиотическим композициям, содержащим новое производное цефалоспорина, представленное химической формулой (1), его сложный эфир или их фармацевтически приемлемую соль в качестве эффективного ингредиента: Химическая формула (1) где X представляет собой CR, N или Cl-замещенный углерод (С-Cl), R представляет собой водород или C1-C3-алкил;R2 представляет собой NHR21, NH(CH2)nCOOH, NH(CH2)nNR21R22 или NHC(=O)(CH2)nNR21R22; Здесь R11 и R21 независимо представляют собой водород, C1-C3-алкил или выбраны из следующих: каждый из R12 и R22 независимо представляет собой водород или C1-C2-алкил; каждый из m и n независимо представляет собой целое число от 1 до 6;R3 представляет собой водород или NH2. В предпочтительном воплощении производные цефалоспорина, представленные химической формулой (1), представляют собой соединения, имеющие химическую формулу (2), их сложный эфир или их фармацевтически приемлемые соли: Химическая формула (2) где X представляет собой CR, N или Cl-замещенный углерод (С-Cl), R представляет собой водород или C1-C3-алкил;R2 представляет собой NHR21, NH(CH2)nNR21R22 или NHC(=O)(CH2)nNR21R22; Здесь каждый из R11 и R21 независимо представляет собой водород, C1-C3-алкил или выбран из следующих групп: каждый из R12 и R22 независимо представляет собой водород или C1-C2-алкил; каждый из m и n независимо представляет собой целое число от 1 до 6. В более предпочтительном воплощении производные цефалоспорина химической формулы (2) являются соединениями, в которыхX представляет собой CR, N или Cl-замещенный углерод (С-Cl), R представляет собой водород илиR21 выбран из следующих групп:m и n независимо представляют собой целое число от 1 до 6. Настоящее изобретение также относится к производным цефалоспорина, имеющим одну из следующих химических формул, их сложным эфирам или их фармацевтически приемлемым солям: В предпочтительном воплощении производные цефалоспорина по настоящему изобретения представляют собой соединения одной из нижеследующих химических формул, их сложные эфиры или их фармацевтически приемлемые соли: Настоящее изобретение также относится к фармацевтической композиции, содержащей терапевтически эффективное количество производного цефалоспорина по изобретению, его сложного эфира или их фармацевтически приемлемой соли в качестве эффективного ингредиента и фармацевтически приемлемый носитель, разбавитель, адъювант или любую их комбинацию. Настоящее изобретение также относится к способу лечения бактериальной инфекции, включающему использование терапевтически эффективного количества производного цефалоспорина по изобретению, его сложного эфира или их фармацевтически приемлемой соли. В предпочтительном воплощении способа по изобретению бактериальная инфекция вызвана грамотрицательной бактерией. В более предпочтительном воплощении способа по изобретению бактериальная инфекция вызвана грамотрицательной бактерией, выбранной из группы, состоящей из Pseudomonas aeruginosa,Acinetobacter baumannii и Klebsiella pneumonia. Производные цефалоспорина по настоящему изобретению обладают существенной антибактериальной активностью в отношении антибиотикорезистентных грамотрицательных бактерий в низкой концентрации. В частности, соединения по настоящему изобретению демонстрируют более высокую анти- 11024709 микробную активность в отношении P.aeruginosa, A.baumannii и K.pneumonia по сравнению с имеющимися в настоящее время на рынке цефалоспоринами. Когда следующие группы присоединены в положении R11 и R12: эффективность существенно возрастает, и, в частности, следующие гидроксипиридоны демонстрируют очень высокую антибактериальную активность: Использованный здесь термин "алкил" включает структуру прямого или разветвленного типов. Например, (C1-C6)алкил представляет собой метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил,пентил и гексил во всех возможных положениях и все возможные изомеры. Новые производные цефалоспоринов согласно настоящему изобретению могут быть получены в виде их пролекарств или их фармацевтически приемлемых солей для улучшения абсорбции в организме или для увеличения растворимости. Поэтому их пролекарства или их фармацевтически приемлемые соли также входят в объем настоящего изобретения. Использованные здесь термины будут описаны кратко. Термин "фармацевтически приемлемая соль" относится к форме соединения, которая не вызывает значительного раздражения организма, в который его вводят, и не лишает соединение его биологической активности и свойств. Термин "изомер" имеют такое же значение, как указано ниже. Его фармацевтически приемлемая соль может представлять собой нетоксичную соль присоединения кислоты, содержащую фармацевтически приемлемый анион, например могут быть включены соли присоединения кислот, образуемые неорганическими кислотами, такими как соляная кислота, серная кислота, азотная кислота, фосфорная кислота, бромисто-водородная кислота и йодисто-водородная кислота; органическими карбоновыми кислотами, такими как винная кислота, муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота и малеиновая кислота; и сульфоновыми кислотами, такими как метансульфоновая кислота, бензолсульфоновая кислота, пара-толуолсульфоновая кислота и нафталинсульфоновая кислота. Также фармацевтически приемлемые соли карбоновых кислот могут быть получены посредством взаимодействия соединения по настоящему изобретению с основаниями с образованием солей металлов или солей щелочно-земельных металлов, таких как соль лития, соль натрия, соль калия, соль кальция и соль магния; солей с аминокислотами, такими как лизин, аргинин, гуанидин; соли с органическими основаниями, такими как дициклогексиламин, N-метил-D-глутамин, трис-(гидроксиметил)метиламин, диэтаноламин, холин и триэтиламин. Соединение по настоящему изобретению согласно химической формуле (1) может быть превращено в его солевые формы посредством традиционных способов. Термин "изомер" означает соединение по настоящему изобретению или его соль, которая имеет такую же химическую формулуили молекулярную формулу, но оптически или стерически отличается. В число этих изомеров включены структурный изомер, такой как таутомер, изомеры с R- илиS-конфигурацией асимметрического углеродного центра, геометрические изомеры (транс, цис) и все стереоизомеры. Термин "пролекарство" относится к агенту, который превращается в исходное лекарственное средство in vivo. Пролекарства используют часто, поскольку в некоторых ситуациях они могут быть более удобны для введения, чем исходное лекарственное средство. Например, они могут быть биодоступными при пероральном введении, тогда как исходное лекарственное средство таковым не является. Пролекарство также может обладать улучшенной растворимостью в фармацевтических композициях по сравнению с исходным лекарственным средством. Примеры пролекарства включают сложные эфиры соединений по настоящему изобретению и их фармацевтически приемлемые соли, которые могут быть гидролизованы in vivo. Дополнительным примером пролекарства может служить короткий пептид (полиаминокислота), связанный с кислотной группой, где пептид метаболизируется с высвобождением активной группировки. Другие термины, которые включены для описания настоящего изобретения, могут быть интерпретированы с помощью значений, обычно используемых в данной области. Различные виды пролекарственных форм известны в данной области техники. Например, см.: Соединения по настоящему изобретению могут представлять собой полиморфные соединения и полиморфные соединения с антимикробной активностью. Новые производные цефалоспорина согласно настоящему изобретению могут быть получены посредством различных способов в зависимости от типа заместителей. Например, комбинация соединений может быть получена согласно способу, проиллюстрированному ниже. Способы производства по предложенным реакционным схемам приведены только в качестве примеров, и в зависимости от конкретных заместителей реакционные схемы могут быть легко преобразованы специалистами в данной области техники. Таким образом, представленные в качестве примера реакционные схемы способа получения цефалоспориновых соединений согласно настоящему изобретению не ограничивают его, и, если не оговорено особо, обозначения заместителей в реакциях являются такими же, как определено в химической формуле (1). Реакционная схема получения новых производных цефалоспорина согласно химической формуле Как показано на реакционной схеме (1), представляющая собой замещенный пирмидин часть А химической формулы (1) и замещенная защитными группами (P1, P2) часть В взаимодействуют друг с другом, и затем проводят реакцию по удалению защитных групп с помощью кислоты. В реакционной схеме (1), трет-бутил, boc (трет-бутоксикарбонил) или pmb (параметоксибензил) могут быть использованы в качестве защитных групп P1 и Р 2, хотя и не ограничиваются ими, галогены (Cl,Br, I и т.д.) могут быть использованы в качестве углерод-замещенной уходящей группы при взаимодействии с пиримидином. Примеры группы Y в части В реакционной схемы (1) представляют собой диметилацетильную группу, защищенную трет-бутилом, и метилацетильную группу, защищенную дифенилметилом, но не ограничиваются ими. Полярный и апротонный растворитель может быть использован в качестве растворителя в этом взаимодействии. Предпочтительным примером является растворитель, та- 13024709 кой как ДМФА (диметилформамид). Амин в форме основания, такой как TEA (триэтиламин) или DIPEA(N,N-диизопропилэтиламин), может быть использован в качестве оснований при взаимодействии, но в более предпочтительном способе взаимодействия основания не используют вовсе. Вторым взаимодействием в вышеприведенной реакционной схеме (1) является удаление защитной группы при использовании кислот, таких как ТФУК (трифторуксусная кислота) или HCl. В первом взаимодействии по реакционной схеме (1) образуются следующие изомеры (А-2 изомер) в качестве побочного продукта, и для того чтобы уменьшить образование этих побочных продуктов в виде изомеров, может быть проведено следующее взаимодействие, показанное на реакционной схеме (2). Реакционная схема (2) Как показано на реакционной схеме (2), перед взаимодействием с пиримидином части А сначала получают сульфоксидное соединение посредством окисления цефемного соединения с помощью МСРВА (метахлорпербензойная кислота) и затем проводят взаимодействие с частью А. Проводят реакцию восстановления полученного продукта ацетилхлоридом (AcCl) и KI с получением требуемого продукта в качестве основного продукта. В реакционной схеме (2) в качестве растворителя для взаимодействия с МСРВА может быть использован метиленхлорид (МС), но он не ограничивает использования других растворителей. Кроме того, реагент, который может быть использован в реакциях окисления и восстановления, не ограничивается МСРВА и AcCl/KI, а может быть использован окислитель и восстановитель с аналогичными реакционными свойствами. Изобретение также относится к (а) фармацевтическим композициям, содержащим новое производное цефалоспорина, представленное химической формулой (1), его сложный эфир или их фармацевтически приемлемую соль в качестве эффективного ингредиента, и (b) фармацевтическим антибиотическим композициям, содержащим их фармацевтически приемлемый носитель, их разбавитель, их адъювант или любую их комбинацию. Термин "фармацевтическая композиция" означает смесь соединения по настоящему изобретению с другими химическими компонентами, такими как разбавители или носители. Вышеуказанная фармацевтическая композиция облегчает введение соединения в организм. В данной области техники существует множество методик введения соединения, включая пероральное, инъекционное, аэрозольное парентеральное и местное введение, но не ограничиваясь ими. Фармацевтические композиции также могут быть получены посредством взаимодействия соединений с неорганическими или органическими кислотами,такими как соляная кислота, бромисто-водородная кислота, серная кислота, азотная кислота, фосфорная кислота, метансульфоновая кислота, паратолуолсульфоновая кислота, салициловая кислота и тому подобными. Использованный здесь термин "терапевтически эффективное количество" означает количество активного ингредиента, эффективное для облегчения или снятия одного или более чем одного симптома расстройства, которое лечат, или для того, чтобы препятствовать появлению клинических маркеров или возникновению симптомов заболевания, которое необходимо предотвратить. Таким образом, терапевтически эффективное количество означает количество, которое обладает эффектом (1) способствовать снижению темпа прогрессирования заболевания, (2) препятствовать дальнейшему прогрессированию заболевания и/или (3) облегчать (предпочтительно снимать) один или более чем один симптом, связанный с заболеванием. С помощью тестирования соединений в in vivo и in vitro модельных системах можно эмпирически определить терапевтически эффективное количество для лечения заболевания. Термин "носитель" означает химическое соединение, которое способствует внедрению соединения в клетки или ткани. Например, диметилсульфоксид (ДМСО) является обычно используемым носителем,поскольку он способствует проникновению многих органических соединений в клетки или ткани организма. Термин "разбавитель" означает химические соединения, растворенные в воде, которые будут растворять требуемое соединение, а также стабилизировать биологически активную форму этого соединения. Соли, растворенные в буферных растворах, используют в качестве разбавителей в данной области техники. Один широко используемый буферный раствор представляет собой забуференный фосфатом физиологический раствор, поскольку он имитирует солевые условия человеческой крови. Так как буферные соли могут регулировать рН растворов при низких концентрациях, буферные разбавители редко изменяют биологическую активность соединения. Используемые здесь соединения могут быть введены пациенту-человеку в виде соединения как такового или в виде фармацевтической композиции, содержащей соединение с другими активными ингредиентами, в комбинированной терапии или с другими подходящими носителями или эксципиентами. Любая из методик приготовления и введения соединений по настоящему изобретению может быть использована в качестве подходящей и принятой в данной области техники; "Remington's PharmaceuticalSciences," Mack Publishing Co., Easton, PA, 18th edition, 1990. Фармацевтическая композиция по настоящему изобретению может быть приготовлена с помощью известного способа, например посредством традиционных способов смешивания, растворения, гранулирования, дражирования, измельчения в порошок, эмульгирования, инкапсулирования, включения в полимер или лиофилизации. Таким образом, фармацевтические композиции для применения по настоящему изобретению могут быть приготовлены посредством традиционного способа при использовании одного или более чем одного физиологически приемлемого носителя, содержащего эксципиенты и вспомогательные вещества, которые облегчают процесс превращения активных соединений в препараты, которые могут быть использованы в фармацевтике. Подходящая композиция зависит от выбранного пути введения. Любой из широко известных методов, носителей и эксципиентов может быть использован в качестве подходящего и принятого в данной области техники; например в вышеупомянутом Remington's Pharmaceutical Sciences. Для таких целей в настоящем изобретении в соответствии с композицией соединения химической формулы (1) может быть приготовлена инъекционная и пероральная композиция. Для инъекционного введения агенты по настоящему изобретению могут быть приготовлены в виде водных растворов или липидных эмульсий, предпочтительно в физиологически совместимых буферных растворах, таких раствор Хенкса, раствор Рингера или буфер на основе физиологического раствора. Для трансмукозального введения в композиции используют усилители проникновения, соответствующие барьеру, через который необходимо проникать. Такие усилители проникновения широко известны в данной области техники. Для перорального введения соединения могут быть легко приготовлены путем объединения активных соединений с фармацевтически приемлемыми носителями, общеизвестными в данной области техники. Такие носители позволяют приготовить соединения по настоящему изобретению в виде таблеток,пилюль, порошков, гранул, драже, капсул, жидкостей, гелей, сиропов, эмульсий, суспензий и тому подобного для перорального приема пациентом, который подлежит лечению. Предпочтительно композиции находятся в форме капсул, таблеток, пилюль, порошков и гранул, и в особенности более используемыми лекарственными формами являются капсулы и таблетки. Таблетки и пилюли являются предпочтительными для производства в виде кишечно-растворимой композиции. Фармацевтические препараты для перорального применения могут быть получены посредством смешивания одного или более чем одного твердого эксципиента с фармацевтической комбинацией по изобретению, возможно измельчения полученной смеси и обработки смеси гранул, при необходимости после добавления подходящих вспомогательных веществ, для получения таблеток или ядер драже. Подходящими эксципиентами являются, в частности, наполнители, такие как сахара, включая лактозу, сахарозу, маннит или сорбит; препараты целлюлозы, такие как, например кукурузный крахмал, пшеничный крахмал, рисовый крахмал, картофельный крахмал, желатин, трагакантовая камедь, метилцеллюлоза, гидроксипропилметилцеллюлоза,натрий-карбоксиметилцеллюлоза и/или поливинилпирролидон (ПВП). При необходимости могут быть добавлены разрыхлители, такие как поперечно сшитый поливинилпирролидон, агар или альгиновая кислота или ее соль, такая как альгинат натрия, и смазывающие вещества, такие как стеарат магния, и связующие вещества. Фармацевтические препараты, которые могут быть использованы перорально, включают твердые капсулы, состоящие из двух частей, изготовленные из желатина, а также мягкие, запаянные капсулы,изготовленные из желатина и пластификатора, такого как глицерин или сорбит. Твердые капсулы, состоящие из двух частей, могут содержать активные ингредиенты в смеси с наполнителем, таким как лактоза, связующими веществами, такими как крахмалы, и/или смазывающими веществами, такими как тальк или магния стеарат, и, возможно, стабилизаторами. В мягких капсулах активные соединения могут быть растворены или суспендированы в подходящих жидкостях, таких как жирные масла, жидкий парафин или жидкие полиэтиленгликоли. Кроме того, могут быть добавлены стабилизаторы. К тому же композиции по настоящему изобретению могут быть покрыты кишечно-растворимыми полимерами. Все препараты для перорального введения должны находиться в лекарственных формах, подходящих для такого введения. Соединения могут быть приготовлены для парентерального введения посредством инъекции, например посредством болюсной инъекции или непрерывной инфузии. Препараты для инъекций могут быть представлены в стандартной лекарственной форме, например в ампулах или в многодозовых контейнерах, с добавлением консерванта. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных средах, и могут содержать вспомогательные вещества, такие как суспендирующие, стабилизирующие и/или диспергирующие агенты. Кроме того, активный ингредиент может находиться, например, в форме порошка, который может быть перед применением растворен в апирогенной и стерильной воде. Соединения также могут быть приготовлены в виде ректальных композиций, таких как суппозитории или удерживающие клизмы, например, содержащие обычные основы для суппозиториев, такие как масло какао или другие глицериды. Фармацевтические композиции, подходящие для применения по настоящему изобретению, включают композиции, где активные ингредиенты содержатся в количестве, эффективном для достижения назначенной цели. Более конкретно, терапевтически эффективное количество означает количество соединения, эффективное для предотвращения, смягчения или облегчения симптомов заболевания или продления выживаемости субъекта, подвергающегося лечению. Определение терапевтически эффективного количества полностью находится в пределах компетенции специалистов в данной области техники,в особенности в свете представленного здесь подробного описания. При приготовлении в виде единицы дозировки активный ингредиент композиции соединения химической формулы (1) предпочтительно вводят в дозе от 1 до 1500 мг. В зависимости от состояния пациентов, включая возраст, массу тела, пол, путь введения, состояние здоровья и тяжесть заболевания, вводимую дозу соединения по настоящему изобретению определяют согласно указаниям врача или фармацевта. Обычно доза варьируется от примерно 1 до 1500 мг от одного до трех раз в сутки для взрослого. Например, соединения по настоящему изобретению могут быть введены внутримышечно или внутривенно в дозе от 1 до 1500 мг от одного до трех раз в сутки для взрослого. Для некоторых пациентов может быть эффективна более высокая доза. Кроме соединений по настоящему изобретению фармацевтические композиции по настоящему изобретению могут дополнительно содержать, т.е. могут быть приготовлены совместно с одним или более чем одним известным лекарственным средством(ами), выбранным из применимых для клинической практики антибактериальных агентов (например, -лактама, макролида, хинолона или аминогликозида),и противовоспалительным агентом (например, триазолом с антифунгальной активностью или амфотерицином), или могут быть введены в комбинации с одним или более чем одним лекарственным средством(ами). Кроме того, соединения по настоящему изобретению могут быть приготовлены совместно с бактерицидным белком, увеличивающим проницаемость клеточной мембраны, (BPI) или ингибитором эффлюксного насоса или введены в комбинации с ними для повышения активности в отношении грамотрицательных бактерий и антибиотикорезистентных бактерий. Соединения по настоящему изобретению могут быть приготовлены совместно с витамином, например витамином В, таким как витамин В 2, витамин В 6 или витамин В 12, и фолиевой кислотой, или введены в комбинации с ним. Кроме того, соединения по настоящему изобретению могут быть приготовлены в виде препаратов совместно с ингибитором циклооксигеназы (ЦОГ), в частности ингибитором ЦОГ-2,или введены в комбинации с ним. Настоящее изобретение относится к способу антибиотического лечения при использовании фармацевтических антибиотических композиций, содержащих новое производное цефалоспорина, представленное химической формулой (1), его пролекарство, его сложный эфир или их фармацевтически приемлемую соль в качестве эффективного ингредиента. Полезные эффекты изобретения Как описано выше, новое производное цефалоспорина по настоящему изобретению обладает высокой антимикробной активностью в отношении грамотрицательных бактерий, таких как P.aeruginosa,K.pneumonia, A.baumannii, и также в отношении грамотрицательных бактерий с множественной лекарственной устойчивостью, и особенно в отношении создающих наибольшие проблемы грамотрицательных бактерий Pseudomonas aeruginosa с множественной лекарственной устойчивостью. Кроме того, эти соединения демонстрируют высокий потенциал в качестве лекарственных средств на стадии разработки,обладая превосходным фармакокинетическим профилем. Краткое описание графических материалов На фиг. 1 представлено схематическое изображение комплекса железо (Fe)-сидерофор и его рецепторов. На фиг. 2 представлено схематическое изображение процесса переноса иона железа и сидерофора. Описание воплощений Примеры Ниже будет сделана подробная ссылка на различные примеры получения, примеры и тестовые примеры. Поскольку изобретение будет проиллюстрировано одновременно примерами получения, примерами и тестовыми примерами, следует понимать, что настоящее описание не предназначено ограничивать изобретение этими примерами получения, примерами и тестовыми примерами. Следующие примеры получения описывают получение соединений в части А и части В реакционной схемы (1). Пример получения 1. Соединение (A-I).(120 мл), при -78C и добавляли раствор диметилсульфоксида (2,45 мл; 30 ммоль), растворенного в метиленхлориде (20 мл). Полученный раствор перемешивали в течение 10 мин при -78C. Медленно добавляли раствор N-Boc-этаноламина (2 г; 12,4 ммоль), растворенный в метиленхлориде (20 мл), и затем добавляли триэтиламин (8,64 мл; 62 ммоль). Полученный раствор перемешивали в течение 30 мин при-78C и дополнительно 30 мин при комнатной температуре, промывали водой (100 мл) и рассолом(100 мл). Органический слой обезвоживали безводным сульфатом натрия, концентрировали при пониженном давлении и подвергали колоночной хроматографии (n-hex:EA (н-гексан:этилацетат), 3:1-1:1) с получением соединения (I) (270 мг (14%. 1 Н ЯМР (600 МГц, ДМСО-d6)7.83 (s, 1H), 7.49 (s, 1H), 6.88 (d, J=5,4 Гц, 1H), 6.36 (br, 2H), 4.81 (br,1H), 3.13 (m, 4H), 1.39 (s, 9H). 1-2) Получение соединения (A-I). Гидрохлорид 4,5-диаминопиримидина (2,0 г; 18,1 ммоль) и соединение I (3,0 г; 18,8 ммоль) растворяли в метаноле (60 мл) и затем добавляли уксусную кислоту (1,0 г; 18,1 ммоль). Полученный раствор перемешивали в течение 12 ч при комнатной температуре. Добавляли цианоборхлорид натрия (2,2 г; 36,3 ммоль). Полученный раствор перемешивали в течение 3 ч при комнатной температуре, концентрировали при пониженном давлении и подвергали колоночной хроматографии (МС:МеОН, 50:1-20:1) с получением соединения (A-I) (1,09 г (24%. 1 Н ЯМР (600 МГц, хлороформ-d1)8.15 (s, 1H), 7.65 (s, 1H), 5.01 (br, 2H), 3.47 (br, 2 Н), 3.22 (t,J=5,4 Гц, 2 Н), 1.46 (s, 9H). Пример получения 2. Соединение (А-II). 2-1) Получение соединения (II). Койевую кислоту (50 г; 0,35 моль) растворяли в N,N-диметилформамиде (900 мл) и затем последовательно добавляли карбонат калия (58,4 г; 0,42 моль) и 4-метоксибензилхлорид (61,7 г; 0,39 моль). Полученный раствор перемешивали в течение 3 ч при 80C, концентрировали при пониженном давлении и медленно добавляли в воду (800 мл) с получением твердого вещества. Твердое вещество промывали смесью эфир:гексан, 1:1 (800 мл) с получением соединения (II) (90 г (98%. 1 Н ЯМР (600 МГц, хлороформ-d1)7.51 (s, 1 Н), 7.32 (d, J=8.4 Гц, 2 Н), 6.90 (d, J=8.0 Гц, 2 Н), 6.45 (s,1H), 5.00 (s, 2H), 4.45 (s, 2H), 3.81 (s, 3 Н). 2-2) Получение соединения (III). Соединение II (50 г; 0,19 моль) и гидрохлорид гидроксиламина (66,2 г; 0,95 моль) растворяли в пиридине (620 мл). Полученный раствор перемешивали в течение 1 ч при 70C-75C, концентрировали при пониженном давлении, и растворяли в воде (350 мл). 6 н. HCl (рН 1-2) добавляли к полученному раствору при перемешивании при 0C с получением твердого вещества. Твердое вещество промывали эфиром(300 мл) с получением соединения (III) (15 г (30%. 1 Н ЯМР (600 МГц, ДМСО-d6)7.96 (s, 1H), 7.38 (d, J=8,0 Гц, 2 Н), 6.96 (d, J=8,0 Гц, 2 Н), 6.86 (s, 1H),5.54 (br, 1H), 5.03 (s, 2H), 4.45 (s, 2H), 3.74 (s, 3 Н). 2-3) Получение соединения (IV). Соединение (III) (31 г; 0,11 моль) растворяли в N,N-диметилформамиде (350 мл) и затем последовательно добавляли карбонат калия (31 г; 0,22 моль) и 4-метоксибензилхлорид (19,3 г; 0,12 моль). Полученный раствор перемешивали в течение 15 ч при комнатной температуре, концентрировали при пониженном давлении, разбавляли этилацетатом (400 мл) и фильтровали при пониженном давлении. Фильтрат промывали водой (300 мл) и рассолом (300 мл). Органический слой обезвоживали безводным сульфатом натрия. Полученное вещество промывали смесью эфир:гексан, 1:1 (400 мл) с получением соединения (IV) (42 г (95%. 1H ЯМР (600 МГц, хлороформ-d1)7.27-7.21 (m, 5H), 6.99 (s, 1H), 6.90 (d, J=8,0 Гц, 2 Н), 6.86 (d,J=8,0 Гц, 2 Н), 6.49 (s, 1H), 5.03 (s, 2H), 4.93 (s, 2H), 4.50 (s, 2H), 3.82 (s, 3 Н), 3.78 (s, 3 Н). 2-4) Получение соединения (V). Соединение (IV) (20 г; 50,3 ммоль) растворяли в метиленхлориде (580 мл) и затем добавляли дистиллированную воду (50 мл). Полученный раствор перемешивали при 0C. Последовательно добавляли 1 М бромид натрия (30 мл), 1 М бромид тетрабутиламмония (55 мл), TEMPO (тетраметилпиперидинилокси) (2,36 г; 15,1 ммоль), насыщенный раствор гидрокарбоната натрия (110 мл) и раствор гипохлорита натрия (120 мл; 2,01 моль). Полученный раствор перемешивали в течение 1,5 ч при температуре, изменяющейся от 0C до комнатной температуры. Добавляли 1 н. HCl (рН 6-7). Затем добавляли трет-бутанол(380 мл), а затем добавляли 2 М 2-метил-2-бутен, растворенный в тетрагидрофуране (607 мл). После этого добавляли раствор хлорида натрия (45,5 г; 503 ммоль) и моногидрат дигидрофосфата натрия (52 г; 377 ммоль), растворенный в дистиллированной воде (170 мл). Полученный раствор перемешивали в течение 1 ч при комнатной температуре. Полученный раствор выливали в фильтровальную воронку для разделения органического слоя и водного слоя. Органический слой промывали насыщенным раствором дигидрофосфата натрия (800 мл), обезвоживали безводным сульфатом натрия, концентрировали при пониженном давлении и подвергали колоночной хроматографии (МС:МеОН, 50:1-8:1) с получением соединения (V) (40 г (61%. 1H ЯМР (600 МГц, хлороформ-d1)7.35 (d, J=8,4 Гц, 2 Н), 7.25 (d, 8,4 Гц, 2 Н), 6.86 (m, 4H), 6.72 (s,1H), 6.38 (s, 1H), 6.49 (s, 1H), 5.30 (s, 2H), 4.85 (s, 2H), 3.80 (s, 3 Н), 3.79 (s, 3 Н), 3.28 (m, 8 Н), 1.65 (m, 8 Н),1.42 (m, 8 Н), 0.99 (t, J=6,6 Гц, 12 Н). 2-5) Получение соединения (А-II). Соединение (VI) (1,89 г; 10 ммоль) растворяли в N,N-диметилформамиде (50 мл), последовательно добавляли диизопропилэтиламин (7,2 мл; 40 ммоль) и соединение (V) (6,52 г; 10 ммоль) и добавляли гексафторфосфат бензотриазол-1-илокси-трис-пирролидинфосфония (6,24 г; 12 ммоль). Полученный раствор перемешивали в течение 30 мин при комнатной температуре, разбавляли этилацетатом (300 мл),промывали водой (200 мл) и рассолом (150 мл), обезвоживали безводным сульфатом натрия, концентрировали при пониженном давлении и подвергали колоночной хроматографии (МС:МеОН, 40:1-10:1) с получением соединения (А-II) (2,2 г (40%. 1 3-1) Получение соединения (VII). Соединение (II) (1,0 г; 3,81 ммоль) добавляли в 33% метиламин, растворенный в этаноле (19 мл). Полученный раствор перемешивали в течение 20 ч при комнатной температуре, получая белое твердое вещество. Полученный раствор фильтровали при пониженном давлении с получением белого твердого вещества. Белое твердое вещество промывали этанолом (50 мл) и эфиром (20 мл) с получением соединения (VII) (778 мг (75%. 1H ЯМР (600 МГц, ДМСО-d6)7.53 (s, 1H), 7.34 (d, J=9,0 Гц, 2 Н), 6.94 (d, J=9,0 Гц, 2 Н), 6.21 (s, 1H),5.55 (brs, 1H), 4.91 (s, 2H), 4.36 (s, 2H), 3.75 (s, 3 Н), 3.58 (s, 3 Н). 3-2) Получение соединения (VIII). Соединение (VII) (778 мг; 2,83 ммоль) растворяли в диметилсульфоксиде (7 мл) и добавляли триметиламин (1,3 г; 12,7 ммоль), метиленхлорид (7 мл), комплекс триоксида серы (1,35 г; 8,48 ммоль). Полученный раствор перемешивали в течение 2 ч при комнатной температуре, разбавляли хлороформом(150 мл), промывали водой (30 мл), обезвоживали безводным сульфатом натрия, концентрировали при пониженном давлении и подвергали колоночной хроматографии (МС:МеОН, 30:1-10:1) с получением соединения (VIII) (718 мг (93%. 1(d, J=8,4 Гц, 2 Н), 5.18 (s, 2H), 3.86 (s, 3 Н), 3.80 (s, 3 Н). 3-3) Получение соединения (IX). Соединение (VIII) (718 мг; 2,63 ммоль) растворяли в смеси трет-бутаиола (8,5 мл) и тетрагидрофурана (8,5 мл) и затем добавляли 2 М 2-метил-2-бутен (3,3 мл), растворенный в тетрагидрофуране. Полученный раствор перемешивали при комнатной температуре. К полученному раствору добавляли раствор хлорида натрия (1,9 г; 21,0 ммоль) и моногидрат дигидрофосфата натрия (2,1 г; 15,2 ммоль), растворенный в воде (8,5 мл). Полученный раствор перемешивали в течение 1 ч при комнатной температуре, получая белое твердое вещество. Полученный раствор фильтровали при пониженном давлении с получением белого твердого вещества. Белое твердое вещество растворяли в воде (4 мл). Добавляли 1 н. HCl (рН 1-2). Полученное таким образом твердое вещество фильтровали при пониженном давлении и промывали этилацетатом (50 мл) и эфиром (50 мл) с получением соединения (IX) (510 мг (67%. 1H ЯМР (600 МГц, ДМСО-d6)7.79 (s, 1H), 7.37 (d, J=12,6 Гц, 2 Н), 6.96 (d, J=12,6 Гц, 2 Н), 6.71 (s,1H), 4.97 (s, 2H), 3.83 (s, 3 Н), 3.76 (s, 3 Н). 3-4) Получение соединения (А-III). Соединение (А-III) (19 мг (25% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании соединения (VI) (34,5 мг; 0,18 ммоль). 1 Н ЯМР (600 МГц, CD3OD)7.96 (s, 1H), 7.64 (s, 1H), 7.59 (s, 1H), 7.38 (d, J=8,4 Гц, 2 Н), 6.91 (d,J=8,4 Гц, 2 Н), 6.56 (s, 1H), 5.02 (s, 2H), 3.79 (s, 3 Н), 3.77 (s, 3 Н), 3.61 (t, J=6,0 Гц), 3.39 (t, J=6,6 Гц, 2 Н). Пример получения 4. Соединение (XI). 4-1) Получение соединения (X). 2-Аминоэтанол (2,0 г; 32,7 ммоль) растворяли в метиленхлориде (110 мл) и последовательно добавляли бензилоксикарбонилхлорид (5,07 г; 29,8 ммоль) и триэтиламин (4,44 г; 44,6 ммоль). Полученный раствор перемешивали в течение 1 ч при комнатной температуре. К полученному раствору добавляли воду (40 мл). Полученное вещество обезвоживали безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения (X) (4,05 г (70%. 1 Н ЯМР (600 МГц, хлороформ-d1)7.40 (m, 5H), 5.19 (brs, 1H), 5.11 (s, 2H), 3.73 (t, J=4,2 Гц, 2 Н),3.37 (q, J=5,4 Гц, 2 Н), 2.23 (brs, 1H). 4-2) Получение соединения (XI). Соединение (XI) (2,72 г (91% получали посредством способа, аналогичного способу по примеру получения 3-2, при использовании соединения (X) (3 г; 15,3 ммоль). 1 5-1) Получение соединения (XII). 2,4-Дихлор-5-нитропиримидин (3 г; 15,4 ммоль) растворяли в тетрагидрофуране (50 мл) и добавляли изопропилэтиламин (2,0 г; 15,4 ммоль). К полученному раствору медленно добавлялиN-Boc-этилдиамин (2,48 г; 15,4 ммоль), растворенный в тетрагидрофуране (20 мл), при -78C при перемешивании в течение 50 мин, и затем полученный раствор перемешивали в течение 10 мин при комнатной температуре. Полученное вещество концентрировали при пониженном давлении и подвергали колоночной хроматографии (ЕА:Нех, 1:4-1:3) с получением соединения (XII) (3,16 г (64%. 1 Н ЯМР (600 МГц, CDCl3)9.05 (s, 1H), 8.80 (br, 1H), 4.84 (br, 1H), 3.78 (q, J=6 Гц, 2H), 3.48 (q,J=6 Гц, 2 Н), 1.43 (s, 9H). 5-2) Получение соединения (XIII). Соединение (XII) (3,1 г; 9,75 ммоль) растворяли в метаноле (50 мл) и добавляли 10% палладированный уголь (1 г; 0,98 ммоль). Полученный раствор подвергали продувке водородом, перемешивали в течение 40 мин при комнатной температуре, фильтровали через целит и концентрировали при пониженном давлении с получением соединения (XIII) (2,8 г (99%. 1H ЯМР (600 МГц, ДМСО-d6)8.62 (br, 1H), 8.33 (s, 1H), 7.48 (s, 1H), 6.99 (brs, 1H), 5.88 (brs, 2H),3.45 (br, 2H), 3.19 (br, 2H), 1.35 (s, 9H). 5-3) Получение соединения (XIV). Соединение (XIII) (1,02 г; 3,52 ммоль) растворяли в 1,2-дихлорэтане (34 мл) и последовательно добавляли диизопропилэтиламин (455 мг; 3,52 ммоль), соединение (XI) (796 мг; 4,12 ммоль) и триацетоксиборгидрид натрия (1,12 г; 5,28 ммоль). Полученный раствор перемешивали в течение 3 ч при комнатной температуре, разбавляли метиленхлоридом (180 мл), промывали водой (100 мл) и рассолом (100 мл),обезвоживали безводным сульфатом натрия, концентрировали при пониженном давлении и подвергали колоночной хроматографии (МС:МеОН, 60:1-20:1) с получением соединения (XIV) (218 мг (14%. 1H ЯМР (600 МГц, CDCl3)8.22 (s, 1H), 7.60 (s, 1H), 7.34-7.30 (m, 5H), 5.77 (br, 1H), 5.46 (br, 1H),5.18 (br, 1H), 5.11 (s, 2H), 3.58 (br, 2H), 3.54 (br, 2H), 3.38 (br, 2H), 3.20 (br, 2H), 1.39 (s, 9H). 5-4) Получение соединения (XV). Соединение (XV) (150 мг (100% получали посредством способа, аналогичного способу по примеру получения 5-2, при использовании соединения (XIV) (218 мг; 0,51 ммоль) и использовали на следующей стадии без проведения очистки. 5-5) Получение соединения (A-IV). Соединение (A-IV) (198 мг (57% получали посредством способа, аналогичного способу по примеру получения 5-2, при использовании соединения (XV) (150 мг; 0,51 ммоль) и соединения (V) (330 мг; 0,51 ммоль). 1 6-1) Получение соединения (XVI). 4 М HCl, растворенный в 1,4-диоксане, добавляли к соединению (XIII) (90 мг; 0,35 ммоль). Полученный раствор перемешивали в течение 1 ч при комнатной температуре, перегоняли при пониженном давлении и сушили с получением соединения (XVI) (70 мг (100%, которое использовали на следующей стадии без проведения очистки. 6-2) Получение соединения (A-V). Соединение (A-V) (103 мг (59% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании соединения (XVI) (60 мг; 0,31 ммоль) и соединения (V) (206 мг; 0,31 ммоль). 1 Н ЯМР (600 МГц, CDCl3)8.55 (br, 1H), 8.05 (s, 1H), 7.48 (s, 1H), 7.40 (d, J=8,4 Гц, 2H), 7.11 (s, 1H),6.92 (m, 4 Н), 6.69 (br, 1H), 6.60 (d, J=7,8 Гц, 2 Н), 6.33 (s, 1H), 5.35 (s, 2H), 4.48 (s, 2H), 3.80 (s, 3 Н), 3.73 (s,3 Н), 3.44 (br, 2H), 3.34 (br, 2H). Пример получения 7. Соединение (А-VI). 7-1) Получение соединения (XVII). трет-Бутил-4-гидроксибутилкарбамат (1,5 г; 7,93 ммоль) растворяли в метиленхлориде (36 мл) и последовательно добавляли имидазол (1,35 г; 19,8 ммоль) и трет-бутилдиметилсилилхлорид (1,43 г; 9,51 ммоль) при 0C. Полученный раствор разбавляли эфиром (250 мл), промывали водой (240 мл) и рассолом (240 мл), обезвоживали безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения (XVII) (2,4 г (100%. 1 Н ЯМР (600 МГц, CDCl3)4.68 (s, 1H), 3.63 (m, 2H), 3.13 (br, 2H), 1.54 (m, 4H), 1.43 (s, 9H), 0.88 (s,9H), 0.04 (s, 6H). 7-2) Получение соединения (XVIII). Соединение (XVII) (2,5 г; 8,23 ммоль) растворяли в тетрагидрофуране (50 мл) и добавляли 1,6 М н-бутиллитий, растворенный в гексане при 0C. Затем добавляли ди-трет-бутил-дикарбонат (2,15 г; 9,88 ммоль). Полученный раствор перемешивали в течение 2 ч при комнатной температуре, разбавляли эфиром (300 мл), промывали водой (30 мл) и рассолом (30 мл), обезвоживали безводным сульфатом натрия и концентрировали при пониженном давлении. Полученное вещество растворяли в тетрагидрофуране (18 мл) и медленно добавляли 1,0 М бромид тетрабутиламмония (14,8 мл; 14,8 ммоль), растворенный в тетрагидрофуране. Полученный раствор перемешивали в течение 4,5 ч при комнатной температуре, разбавляли эфиром (150 мл), промывали водой (230 мл) и рассолом (40 мл), обезвоживали безводным сульфатом натрия, концентрировали при пониженном давлении и подвергали колоночной хроматографии (ЕА:Нех, 1:4) с получением соединения (XVIII) (1,33 г (56%. 1 Н ЯМР (600 МГц, CDCl3)3.68 (m, 2 Н), 3.61 (t, J=7,2 Гц, 2 Н), 1.68 (m, 2H), 1.59 (m, 2 Н), 1.50 (s,18H). 7-3) Получение соединения (XIX). Соединение (XVIII) (330 мг; 1,14 ммоль) растворяли в диметилсульфоксиде (2,5 мл) и добавляли диизопропилэтиламин (300 мг; 2,30 ммоль), метиленхлорид (2,5 мл) и комплекс триоксида серы (370 мг; 2,28 ммоль) при -20C. Полученный раствор перемешивали в течение 30 мин при комнатной температуре, разбавляли этилацетатом (150 мл), промывали водой (40 мл) и рассолом (40 мл), обезвоживали безводным сульфатом натрия и концентрировали при пониженном давлении с получением соединения 7-4) Получение соединения (XX). Соединение (XX) (220 мг (35% получали посредством способа, аналогичного способу по примеру получения 5-3, при использовании 4,5-диаминопиримидина (182 мг; 1,65 ммоль) и соединения (XIX)H ЯМР (600 МГц, CDCl3)8.09 (s, 1H), 7.63 (s, 1H), 5.84 (br, 2H), 3.65 (t, J=6,6 Гц, 2 Н), 3.15 (t,J=6 Гц, 2 Н), 1.74 (m, 2 Н), 1.68 (m, 2 Н), 1.50 (s, 18H). 7-5) Получение соединения (XXI). Соединение (XXI) (111 мг (89% получали посредством способа, аналогичного способу по примеру получения 6-1, при использовании соединения (XX) (220 мг; 0,58 ммоль). 7-6) Получение соединения (А-VI). Соединение А-VI (153 мг (53% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании соединения (XXI) (110 мг; 0,50 ммоль) и соединения (V) (330 мг; 0,50 ммоль). 1 8-1) Получение соединения (XXII). Соединение (IV) (1,0 г; 2,51 ммоль) растворяли в ацетонитриле (13 мл). Добавляли оксид марганца(IV) (5,5 г; 63,5 ммоль) при 50C. Полученный раствор перемешивали в течение 7 ч, фильтровали через целит, концентрировали при пониженном давлении и подвергали колоночной хроматографии(МС:МеОН, 40:1) с получением соединения (XXII) (0,73 г (24%. 8-2) Получение соединения (XXIII). Соединение (VI) (60 мг; 0,39 ммоль) и соединение (XXII) (232 мг; 0,59 ммоль) растворяли в метаноле (13 мл) и добавляли 10 капель уксусной кислоты. Полученный раствор перемешивали в течение 12 ч при комнатной температуре. Добавляли цианборгидрид натрия (370 мг; 587 ммоль). Полученный раствор перемешивали в течение 3 ч при комнатной температуре, концентрировали при пониженном давлении и подвергали колоночной хроматографии (МС:МеОН, 50:1-20:1) с получением соединения (XXIII) (40 мгH ЯМР (600 МГц, CD3OD)8.05 (s, 1H), 7.50 (s, 1H), 7.20 (d, J=8,4 Гц, 2 Н), 7.16 (d, J=8,4 Гц, 2 Н),7.05 (s, 1H), 6.88 (d, J=8,4 Гц, 2 Н), 6.81 (d, J=8,4 Гц, 2 Н), 6.42 (s, 1H), 5.05 (s, 2H), 4.76 (s, 2H), 3.80 (s, 3H),3.76 (s, 3H), 3.61 (s, 2H), 3.12 (br, 2H), 2.90 (br, 2H). 8-3) Получение соединения (А-VII). Соединение (XXIII) (40 мг; 0,07 ммоль) растворяли в тетрагидрофуране (1 мл) и метаноле (0,5 мл). Добавляли ди-трет-бутил-дикарбонат (18 мг; 0,08 ммоль). Полученный раствор перемешивали при дефлегмации в течение 1,5 ч, концентрировали при пониженном давлении и подвергали колоночной хроматографии (МС:МеОН, 50:1-8:1) с получением соединения (А-VII) (16 мг (34%. 1 9-1) Получение соединения (А-VIII). Соединение (II) (2 г; 7,63 ммоль) растворяли в ацетоне (100 мл) при нагревании и медленно добавляли реактив Джонса (1,88 мл H2SO4; 6 мл дистиллированной воды; 2,14 г CrO3) при 0C. Полученный раствор перемешивали в течение 1 ч при 0C и затем дополнительно перемешивали в течение 1 ч при комнатной температуре. Добавляли метанол (20 мл) и полученный раствор перемешивали в течение 5 мин при комнатной температуре. Полученное твердое вещество удаляли посредством фильтрации при пониженном давлении и фильтрат концентрировали при пониженном давлении. Полученное твердое вещество промывали метанолом с получением соединения (XXV) (560 мг (27%. 1 Н ЯМР (600 МГц, ДМСО-d6)8.34 (s, 1H), 7.37 (d, J=11,4 Гц, 2 Н), 6.97 (d, J=11,4 Гц, 2 Н), 6.92 (s,1H), 4.90 (s, 2H), 3.76 (s, 3 Н). 9-2) Получение соединения (XXVI). Соединение (XXV) (550 мг; 2 ммоль) растворяли в аммиаке (15 мл) и полученный раствор перемешивали в течение 2 ч при дефлегмации. Затем добавляли аммиак (7 мл). Полученный раствор перемешивали в течение 1 ч при дефлегмации, охлаждали до комнатной температуры и концентрировали при пониженном давлении для удаления избытка аммиака. Полученный раствор подкисляли раствором 5 н. HCl и полученное твердое вещество фильтровали при пониженном давлении с получением соединения(XXVI) (500 мг (91%. 1 Н ЯМР (600 МГц, ДМСО-d6)7.88 (br, 1H), 7.38 (d, J=8,4 Гц, 2 Н), 7.19 (br, 1H), 6.91 (d, J=7,8 Гц,2 Н), 5.15 (s, 2H), 3.72 (s, 3 Н). 9-3) Получение соединения (XXVII). Соединение (XXVI) (200 мг; 0,73 ммоль) растворяли в N,N-диметилформамиде (9 мл). Последовательно добавляли карбонат калия (1 г; 7,3 ммоль) и 4-метоксибензилхлорид (570 мг; 3,64 ммоль). Полученный раствор перемешивали в течение 18 ч при 60C, разбавляли этилацетатом (60 мл) и фильтровали при пониженном давлении. Фильтрат промывали водой (330 мл) и рассолом (30 мл). Органический слой обезвоживали безводным сульфатом натрия. Полученное вещество подвергали колоночной хроматографии (SiO2, n-hex:EA, 3:1-1:1) с получением соединения (XXVII) (220 мг (59%. 1H ЯМР (600 МГц, хлороформ-d1)8.22 (s, 1H), 7.70 (s, 1H), 7.39-7.23 (m, 6H), 6.88-6.84 (m, 6 Н),5.31 (s, 2H), 5.15 (s, 2H), 5.12 (s, 2H), 3.79 (s, 3 Н), 3.78 (s, 3 Н), 3.77 (s, 3 Н). 9-4) Получение соединения (XXVIII). Соединение (XXVII) (220 мг; 427 мкмоль) растворяли в тетрагидрофуране (11 мл) и добавляли 2 н. водный раствор гидроксида калия (4,4 мл). Полученный раствор перемешивали в течение 1,5 ч при дефлегмации, охлаждали до комнатной температуры, концентрировали при пониженном давлении для удаления органического растворителя и подкисляли 1 н. раствором HCl. Полученное твердое вещество фильтровали при пониженном давлении с получением соединения (XXVIII) (160 мг (95%. 1(s, 2H), 3.71 (s, 3 Н), 3.70 (s, 3 Н). 9-5) Получение соединения (А-VIII). Соединение (А-VIII) (110 мг (55% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании соединения (VI) (86 мг; 455 мкмоль) и соединения (XXVIII) Гидрохлорид 4,5-диаминопиримидина (100 мг; 0,91 ммоль) и 4-диметиламинопиридин (22 мг; 0,18 ммоль) растворяли в метиленхлориде (3 мл) и добавляли 3-(трет-бутоксикарбонил)пропановую кислоту. Затем медленно добавляли диизопропилкарбодиимид (138 мг; 1,1 ммоль) при 0C. Полученный раствор перемешивали при комнатной температуре, концентрировали при пониженном давлении и подвергали колоночной хроматографии (МС:МеОН, 40:1-10:1) с получением соединения (A-IX) (133 мг 11-1) Получение соединения (XXIX). Соединение (XXIX) (112 мг (100% получали посредством способа, аналогичного способу по примеру получения 6-1, при использовании соединения (А-IX) (146 мг; 0,52 ммоль). 11-2) Получение соединения (А-Х). Соединение (А-Х) (102 мг (61% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании соединения (XXIX) (63 мг; 0,29 ммоль) и соединения (V) (189 мг; 0,29 ммоль). 1 Н ЯМР (600 МГц, CDCl3)8.29 (s, 1H), 8.19 (s, 1H), 7.27 (br, 2H), 7.09 (br, 2H), 6.97 (s, 1H), 6.87 (d,J=8,4 Гц, 2 Н), 6.77 (d, J=7,8 Гц, 2 Н), 6.38 (s, 1H), 5.20 (s, 2H), 4.51 (s, 2H), 3.77 (s, 3H), 3.76 (s, 3H), 3.53 Соединение А-XI (147 мг (86% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании соединения (XXIX) (66,3 мг; 0,30 ммоль) и соединения (XXVIII) Соединение (A-XII) (146 мг (16% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании 4,5-диаминопиримидина (200 мг; 1,81 ммоль) и соединения (V) 14-1) Получение соединения (XXX). Соединение (XXX) (12 г (71% получали посредством способа, аналогичного способу по примеру получения 3-2, при использовании трет-бутил-2-гидроксиэтил(метил)карбамата (17 г; 97 ммоль). 1 Н ЯМР (600 МГц, хлороформ-d1)9.61 (s, 1H), 3.92 (s, 2H), 2.92 (s, 3 Н), 1.46 (s, 9 Н). 14-2) Получение соединения (XXXI). Соединение (XXXI) (180 мг (38% получали посредством способа, аналогичного способу по примеру получения 8-2, при использовании 4,5-диаминопиримидина (200 мг; 1,82 ммоль) и соединенияH ЯМР (600 МГц, хлороформ-d1)8.09 (s, 1H), 7.53 (s, 1H), 6.74 (br, 2H), 3.48-3.32 (m, 4 Н), 2.97 (s,3 Н), 1.46 (s, 9H). 14-3) Получение соединения (XXXII). Соединение (XXXII) получали посредством способа, аналогичного способу по примеру получения 6-1, при использовании соединения (XXXI) (180 мг; 0,67 ммоль) и использовали на следующей стадии без проведения очистки. 14-4) Получение соединения (A-XIII). Соединение (A-XIII) (30 мг (22% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании соединения (XXXII) (70 мг; 0,34 ммоль) и 3-гидрокси-4-(4 метоксибензилокси)бензойной кислоты (103 мг; 0,38 ммоль). 1 Соединение (A-XIV) (35 мг (49% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании соединения (XXIX) (35 мг; 0,16 ммоль) и 3-гидрокси-4-(4 метоксибензилокси)бензойной кислоты (49 мг; 0,18 ммоль). 1 Н ЯМР (600 МГц, ДМСО-d6+D2O)8.22 (s, 1H), 8.16 (s, 1H), 7.41 (d, J=8,4 Гц, 2 Н), 7.29 (d,J=1,8 Гц, 1H), 7.26 (dd, J=1,8 Гц, 9,4 Гц, 1H), 7.04 (d, J=8,4 Гц, 2 Н), 6.95 (d, J=8,4 Гц, 2 Н), 5.09 (s, 2 Н), 3.76 Соединение (A-XV) (221 мг (43% получали посредством способа, аналогичного способу по примеру получения 8-2, при использовании соединения (XXXII) (200 мг; 0,98 ммоль) и 3,4-бис-(4 метоксибензилокси)бензальдегида (408 мг; 1,08 ммоль). 1 Н ЯМР (600 МГц, ДМСО-d6)7.94 (s, 1H), 7.47 (s, 1H), 7.31 (m, 4 Н), 7.08 (br, 1H), 6.99 (br d, 1H),6.90 (m, 5 Н), 5.94 (br, 2H), 4.97 (s, 2H), 4.94 (s, 2H), 3.71 (s, 3H), 3.70 (s, 3 Н), 3.30 (br, 5 Н), 2.85 (br, 2 Н),2.41 (m, 2 Н), 3.02 (d, J=4,2 Гц, 3 Н). Пример получения 17. Соединение (A-XVI). Соединение (A-XVI) (40 мг (20% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании соединения (VI) (95 мг; 500 мкмоль) и 3-гидрокси-4-(4- 25024709 Соединение (А-IX) (450 мг; 1,6 ммоль) растворяли в безводном тетрагидрофуране (12 мл) и медленно добавляли алюмогидрид лития (152 мг; 3,2 ммоль) при 0C. Полученный раствор перемешивали в течение 3 ч при комнатной температуре. Затем добавляли 15% водный раствор гидроксида натрия(200 мкл). Полученный раствор перемешивали в течение 1 ч при комнатной температуре. Полученное таким образом твердое вещество фильтровали при пониженном давлении. Фильтрат концентрировали при пониженном давлении. Полученное вещество подвергали колоночной хроматографии (SiO2,МС:МеОН, 30:1-10:1) с получением соединения (A-XVII) (140 мг (33%. 1 Н ЯМР (600 МГц, хлороформ-d1+CD3OD)8.01 (s, 1H), 7.44 (s, 1H), 3.23-3.12 (m, 4 Н), 1.85 (m, 2 Н),1.48 (s, 9H). Пример получения 19. Соединение (A-XVIII). 19-1) Получение соединения (XXXIII). Соединение (XXXIII) (110 мг (100% получали посредством способа, аналогичного способу по примеру получения 6-1, при использовании соединения (А-XVII) (150 мг; 560 мкмоль). 19-2) Получение соединения (A-XVIII). Соединение (A-XVIII) (35 мг (41% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании соединения (XXXIII) (50 мг; 245 мкмоль) и 3-гидрокси-4-(4 метоксибензилокси)бензойной кислоты (92 мг; 335 мкмоль). 1 Н ЯМР (600 МГц, ДМСО-d6)9.15 (s, 1H), 8.24 (t, J=6 Гц, 1H), 7.78 (s, 1H), 7.38 (s, 1H), 7.37 (d,J=8,4 Гц, 2 Н), 7.26 (d, J=2,4 Гц, 1H), 7.22 (dd, J=8,4 Гц, 2,4 Гц, 1H), 6.99 (d, J=8,4 Гц, 1H), 6.90 (d,J=8,4 Гц), 6.37 (br, 2H), 5.03 (s, 2H), 4.73 (t, J=5,4 Гц, 1H), 3.71 (s, 3 Н), 3.30 (m, 2 Н), 3.04 (q, J=6,6 Гц, 2 Н),1.80 (m, 2H). Пример получения 20. Соединение (A-XIX). 20-1) Получение соединения (XXXIV). Соединение (XXXIV) (854 мг (70% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании 4,5-диаминопиримидина (500 мг; 4,54 ммоль) и 2-(трет-бутоксикарбониламино)уксусной кислоты (876 мг; 4,99 ммоль). 1H ЯМР (600 МГц, хлороформ-d1)8.46 (s, 1H), 8.25 (br, 1H), 8.11 (br, 1H), 5.45 (br d, 3 Н), 3.91 (s,2H), 1.48 (s, 9H). 20-2) Получение соединения (XXXV). Соединение (XXXV) (650 мг (100% получали посредством способа, аналогичного способу по примеру получения 6-1, при использовании соединения (XXXIV) (854 мг; 3,19 ммоль). 20-3) Получение соединения (A-XIX). Соединение (A-XIX) (357 мг (68% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании соединения (XXXV) (250 мг; 1,23 ммоль) и 3-гидрокси-4-(4 метоксибензилокси)бензойной кислоты (337 мг; 1,23 ммоль). 1 21-1) Получение соединения (XXXVI). 4-Бромизофталевую кислоту (1 г; 4,1 ммоль) растворяли в дистиллированной воде (4 мл) и добавляли бикарбонат натрия (1,2 г; 11 ммоль). Полученный раствор перемешивали в течение 1,5 ч при 85C.N1,N1,N2,N2-тетраметилэтан-1,2-диамин (31 мг; 270 мкмоль) и бромид меди (18 мг; 126 мкмоль) растворяли в дистиллированной воде (0,5 мл). Полученный раствор перемешивали в течение 1 ч. Два полученных раствора смешивали и перемешивали в течение 18 ч при 85C, охлаждали до комнатной температуры и подкисляли водным раствором 1 н. HCl. Полученное твердое вещество фильтровали при пониженном давлении, промывали водой и сушили при пониженном давлении с получением соединенияH ЯМР (600 МГц, ДМСО-d6)8.34 (t, J=3,6 Гц, 1H), 8.04 (dt, J=1,2 Гц, 13,2 Гц, 1H), 7.05 (dd,J=3,6 Гц, 12,6 Гц, 1H). 21-2) Получение соединения (XXXVII). Соединение (XXXVI) (720 мг; 3,95 ммоль) растворяли в трифторуксусной кислоте (4,32 мл). Последовательно добавляли ацетон (2 мл) и трифторуксусный ангидрид (TFAA) (1,45 мл). Полученный раствор перемешивали в течение 8 ч при 100C, охлаждали до комнатной температуры и концентрировали при пониженном давлении. Полученное вещество растворяли в этилацетате (100 мл) и промывали 1 н. водным раствором HCl (50 мл). Органический слой обезвоживали безводным сульфатом натрия и концентрировали при пониженном давлении. Полученное таким образом твердое вещество фильтровали при пониженном давлении с получением соединения (XXXVII) (500 мг (57%. 1H ЯМР (600 МГц, хлороформ-d1)8.76 (d, J=2,4 Гц, 1H), 8.30 (dd, J=9,0 Гц, 1,8 Гц, 1H), 7.08 (d,J=8,4 Гц, 1H), 1.78 (s, 6H). 21-3) Получение соединения (А-ХХ). Соединение (А-ХХ) (90 мг (72% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании соединения (VI) (80 мг; 416 мкмоль) и соединения (XXXVII) (78 мг; 350 мкмоль). 1 Н ЯМР (600 МГц, CD3OD)8.42 (d, J=3,6 Гц, 1H), 8.13 (m, 1H), 7.94 (s, 1H), 7.61 (s, 1H), 7.14 (d,12,6 Гц, 1H), 3.66 (t, 9,6 Гц, 2 Гц) 3.40 (t, 9,6 Гц, 2 Н), 1.74 (s, 6H). Пример получения 22. Соединение (A-XXI). 22-1) Получение соединения (XXXVIII). Этил-3,4-дигидроксибензоат (5 г; 28 ммоль) растворяли в N,N-диметилформамиде (50 мл) и добавляли карбонат калия (15 г; 110 ммоль). Полученный раствор перемешивали в течение 2 суток при комнатной температуре, разбавляли этилацетатом (400 мл) и фильтровали при пониженном давлении. Фильтрат промывали водой (3300 мл) и рассолом (300 мл). Органический слой концентрировали при пониженном давлении. К полученному веществу добавляли гексан. Полученное таким образом твердое вещество фильтровали при пониженном давлении с получением соединения (XXXVIII) (11 г (97%. 1H ЯМР (600 МГц, хлороформ-d1)7.64 (d, J=1,8 Гц, 1H), 7.63 (dd, J=7,8 Гц, 2,4 Гц, 1H), 7.38 (m,4 Н), 6.93 (m, 5 Н), 5.13 (s, 2 Н), 5.11 (s, 2 Н), 4.35 (q, J=7,2 Гц, 2 Н), 3.81 (s, 6H), 1.38 (t, J=7,2 Гц, 3 Н). 22-2) Получение соединения (XXXIX). Соединение (XXXVIII) (11 г; 27 ммоль) растворяли в тетрагидрофуране (120 мл) и этаноле (130 мл). Добавляли 2 н. водный раствор гидроксида лития (52 мл). Полученный раствор перемешивали в течение 12 ч при комнатной температуре, концентрировали при пониженном давлении, разбавляли дистиллированной водой (200 мл) и промывали этилацетатом (200 мл). Полученный таким образом слой водного раствора подкисляли 1 н. водным раствором HCl. Полученное таким образом твердое вещество фильтровали при пониженном давлении и сушили под вакуумом с получением соединения (XXXIX) (8,6 г(s, 2H), 5.02 (s, 2H), 3.71 (s, 6H). 22-3) Получение соединения (A-XXI). Соединение (A-XXI) (300 мг (57% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании соединения (VI) (190 мг; 1 ммоль) и соединения (XXXIX) Соединение (A-XXII) (135 мг (59% получали посредством способа, аналогичного способу по примеру получения 8-2, при использовании гидрохлорида 4,5-диаминопиримидина (100 мг; 0,91 ммоль) и трет-бутил 4-оксобутаноата (158 мг; 0,99 ммоль). 1 24-1) Получение соединения (XL). 2,4-Дихлор-5-нитропиримидин (3 г; 15,4 ммоль) растворяли в тетрагидрофуране (52 мл) и медленно добавляли 2 н. метиламин (15,4 мл), растворенный в тетрагидрофуране, при -78C. Полученный раствор перемешивали в течение 10 мин и затем дополнительно перемешивали в течение 50 мин при комнатной температуре. Полученный раствор концентрировали при пониженном давлении, разбавляли этилацетатом (50 мл) и промывали водой (30 мл) и рассолом (30 мл). Полученное вещество обезвоживали безводным сульфатом натрия, концентрировали при пониженном давлении и подвергали колоночной хроматографии (ЕА:Нех, 20:1-5:1) с получением соединения (XL) (925 мг (32%. 1H ЯМР (600 МГц, хлороформ-d1)9.05 (s, 1H), 8.41 (br, 1H), 3.23 (d, J=4,8 Гц, 3 Н). 24-2) Получение соединения (A-XXIII). Соединение (XL) (506 мг; 2,68 ммоль) растворяли в метаноле (5 мл) и добавляли 10% палладированный уголь (285 мг; 0,27 ммоль). Полученный раствор подвергали продувке водородом, перемешивали в течение 2 ч при комнатной температуре, фильтровали через целит и концентрировали при пониженном давлении с получением соединения (A-XXIII) (411 мг (98%. 1 Соединение (VI) (50 мг; 0,32 ммоль) растворяли в метаноле (1 мл). Добавляли триэтиламин (40 мг; 0,40 ммоль) и трет-бутилимино(1 Н-пиразол-1-ил)метилкарбамат (69 мг; 0.32 ммоль). Полученный раствор перемешивали в течение 15 ч при комнатной температуре, концентрировали при пониженном давлении и подвергали колоночной хроматографии (МС:МеОН, 10:1-7:1) с получением соединения Соединение (A-XXV) (189 мг (50% получали посредством способа, аналогичного способу по примеру получения 8-2, при использовании соединения (XLI) (192 мг; 0,68 ммоль) и соединения (XIX) Соединение (A-XXVI) (228 мг (59% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании 4,5-диаминопиримидина (144 мг; 1,31 ммоль) и 4-(трет-бутоксикарбониламино)бутановой кислоты (266 мг; 1,31 ммоль). 1 Н ЯМР (600 МГц, CD3OD)8.30 (s, 1H), 8.24 (s, 1H), 3.18 (t, J=6 Гц, 2 Н), 2.51 (t, J=6,6 Гц, 2 Н), 1.89 28-1) Получение соединения (XLII). Соединение (XLII) (180 мг (100% получали посредством способа, аналогичного способу по примеру получения 6-1, при использовании соединения (А-XXVI) (232 мг; 0,78 ммоль). 28-2) Получение соединения (A-XXVII). Алюмогидрид лития (87 мг; 2,3 ммоль) добавляли в тетрагидрофуран (5 мл) и полученный раствор перемешивали при комнатной температуре. После добавления соединения (XLII) (180 мг; 0,78 ммоль) полученный раствор перемешивали в течение 20 мин при дефлегмации. Добавляли 15% водный раствор гидроксида натрия (0,1 мл). Полученное таким образом твердое вещество фильтровали при пониженном давлении. Фильтрат концентрировали при пониженном давлении и растворяли в метаноле (4 мл). Добавляли ди-трет-бутилдикарбонат (171 мг; 0,78 ммоль). Полученный раствор перемешивали в течение 30 мин при дефлегмации, концентрировали при пониженном давлении и подвергали колоночной хроматографии (МС:МеОН, 45:1-15:1) с получением соединения (A-XXVII) (25,6 мг (11%. 1 Н ЯМР (600 МГц, CDCl3)8.14 (s, 1H), 7.62 (s, 1H), 3.28 (br, 2H), 3.15 (br, 2H), 1.71 (m, 2H), 1.64 Соединение (A-XXVIII) (135 мг (55% получали посредством способа, аналогичного способу по примеру получения 2-5, при использовании сульфата 4,5,6-триаминопиримидина (200 мг; 0,86 ммоль) и 2-(трет-бутоксикарбониламино)уксусной кислоты (166 мг; 0,95 ммоль). 1
МПК / Метки
МПК: C07D 501/34, A61P 31/00, A61K 31/546
Метки: цефалоспорина, производные, композиции, фармацевтические
Код ссылки
<a href="https://eas.patents.su/30-24709-proizvodnye-cefalosporina-i-ih-farmacevticheskie-kompozicii.html" rel="bookmark" title="База патентов Евразийского Союза">Производные цефалоспорина и их фармацевтические композиции</a>
Производные 2,3-бензодиазепина и фармацевтические композиции, содержащие эти производные в качестве активного ингредиента

Номер патента: 5867
Опубликовано: 30.06.2005
Авторы: Гиглер Габор, Леваи Дьердь, Вег Миклош, Мартонне Марко Бернадетт, Сабо Геза, Линг Иштван, Сенаши Габор, Раткаи Зольтан, Шимиг Дьюла, Харшинг Ласло Габор, Баркоци Йожеф, Грефф Зольтан
МПК: C07D 243/02, A61P 25/00, A61K 31/551...
Метки: ингредиента, композиции, содержащие, эти, качестве, производные, фармацевтические, активного, 2,3-бензодиазепина
Формула / Реферат:
1. Производное 2,3-бензодиазепина формулы I где X - водород, хлор или метоксигруппа, Y - водород или галоген, Z - метил или хлор, R - C1-4 алкил или группа формулы -NR1R2, где R1 и R2, независимо, представляют собой водород, C1-4 алкил, C1-4 алкоксил или C3-6 циклоалкил, и его фармацевтически приемлемые соли с кислотами. 2. Производное 2,3-бензодиазепина по п.1, где X - хлор, Y - водород, хлор или бром, R - C1-4 алкил, Z - определен в п.1, и...
Производные 2н-пиридазин-3-она, фармацевтические композиции, содержащие эти производные, и способ получения активного ингредиента

Номер патента: 6246
Опубликовано: 27.10.2005
Авторы: Сенаши Габор, Леваи Дьёрди, Баркоци Йожеф, Эдьед Андраш, Котаи Надь Петер, Вельман Янош, Рацне Байногель Юдит, Гачальи Иштван, Паллаги Каталин, Шимиг Дьюла, Шмидт Эва, Миклошне Ковач Анико
МПК: A61K 31/50, C07D 413/14
Метки: активного, производные, эти, фармацевтические, ингредиента, 2н-пиридазин-3-она, получения, способ, содержащие, композиции
Формула / Реферат:
1. Производное 2H-пиридазин-3-она формулы где R означает атом водорода или C1-4-алкильную группу, X и Y независимо представляют собой атом водорода, атом галогена или группу формулы при условии, что один из X и Y всегда представляет собой группу формулы II, а другой радикал представляет собой атом водорода или атом галогена, где в формуле II n имеет значение 1 или 2, и его фармацевтически приемлемые соли присоединения кислот. 2. Производное...
Фармацевтические композиции, содержащие производные 3-аминоазетидина, новые производные и способ их получения

Номер патента: 4649
Опубликовано: 24.06.2004
Авторы: Бушар Эрве, Ашар Даниель, Букерель Жан, Гризони Серж, Майерс Майкл, Иттэнжер Огюстэн, Филош Брюно
МПК: A61K 31/397, C07D 205/04, A61P 25/00...
Метки: содержащие, фармацевтические, способ, получения, производные, 3-аминоазетидина, новые, композиции
Формула / Реферат:
1. Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение формулы в которой R1 обозначает радикал -NHCOR4 или -N(R5)-Y-R6, Y обозначает CO или SO2, R2 и R3, одинаковые или разные, обозначают либо ароматический радикал, выбранный из фенила, нафтила и инденила, которые могут быть незамещенными или замещенными одним или несколькими заместителями: галоген, алкил, алкокси, формил, гидрокси, трифторметил, трифторметокси,...
Производные гидроксиалкилиндолкарбазола, способ их получения и фармацевтические композиции, которые их содержат

Номер патента: 6201
Опубликовано: 27.10.2005
Авторы: Хикман Джон, Пьер Ален, Марминон Кристель, Моро Паскаль, Пфайфер Брюно, Бизо-Эспьяр Жан-Ги, Ренар Пьер, Прюдом Мишель
МПК: A61K 31/7056, A61P 25/00, C07H 19/23...
Метки: которые, содержат, получения, способ, гидроксиалкилиндолкарбазола, производные, фармацевтические, композиции
Формула / Реферат:
1. Соединения формулы (I) в которой R1 и R2, которые могут быть одинаковыми или разными, каждый, независимо друг от друга, представляет собой группу, выбранную из водорода, линейного или разветвленного (C1-C6)алкила, арил(C1-C6)алкила, в котором алкильная часть может быть линейной или разветвленной, гидрокси, линейного или разветвленного (C1-C6)гидроксиалкила, линейного или разветвленного дигидрокси(C1-C6)алкила, линейного или разветвленного...
Производные бензоксазина в качестве модуляторов 5-нт-6, способ их получения, фармацевтические композиции, содержащие эти производные, и их применение

Номер патента: 9982
Опубликовано: 28.04.2008
Авторы: Кларк Робин Дуглас, Чжао Шухай, Бергер Якоб
МПК: C07D 265/36, A61P 25/18, A61K 31/536...
Метки: производные, композиции, содержащие, качестве, 5-нт-6, бензоксазина, фармацевтические, способ, эти, применение, получения, модуляторов
Формула / Реферат:
1. Соединение формулы его фармацевтически приемлемая соль или пролекарство, где m означает целое число от 0 до 3; n означает 2 или 3; р означает 2; Y означает -S(O2)-; Z1 означает N; R1 и R2 означают водород; R3 означает фенил или нафтил, которые необязательно замещены галогеном, алкилом, алкокси, гидрокси, циано, пиперазинилом, метилсульфониламино, метилсульфонилом, аминокарбонилом, или означает хинолинил, изохинолинил, тиофенил,...
Предыдущий патент: Cпособ получения композиции покрытия с антикоррозионными свойствами (варианты) и способ получения фосфата алюминия
Следующий патент: Электрохимические устройства и способы на основе гидроксидов с использованием окисления металлов
Случайный патент: Способ слежения за продольной осью рельса при скоростной ультразвуковой дефектоскопии и устройство для его осуществления