Новые соединения, связывающие фарнезоидный х-рецептор (fxr) (nr1h4) и модулирующие его активность
Номер патента: 24083
Опубликовано: 31.08.2016
Авторы: Кремозер Клаус, Штеенек Кристоф, Кинцель Олаф, Абель Ульрих

























Формула / Реферат
1. Соединение согласно формуле (1), его энантиомер, диастереомер, таутомер, сольват или фармацевтически приемлемая соль:

в которой R выбран из группы, состоящей из COOR6, CONR7R8, тетразолила или Н, где R6 независимо выбран из группы, состоящей из Н или C1-C6-алкила, и R7 и R8 независимо друг от друга выбраны из группы, состоящей из Н, C1-C6-алкила, C1-C6-галоалкила, C1-C6-алкилена-R9, SO2-C1-C6-алкила, R9 выбран из группы, состоящей из COOH, OH или SO3H;
А выбран из группы, состоящей из фенила, пиридила, пиразолила, индолила, тиенила, бензотиенила, индазолила, бензизоксазолила, бензофуранила, бензотриазолила, фуранила, бензотиазолила, тиазолила, каждый из которых возможно замещен одной или двумя группами, независимо выбранными из группы, состоящей из OH, C1-C6-алкила, C3-C6-циклоалкила или галогена;
Q выбран из группы, состоящей из фенила, пиридила, тиазолила, тиофенила, пиримидила, каждый из которых возможно замещен одной или двумя группами, независимо выбранными из группы, состоящей из C1-C6-алкила, галогена или CF3;

где X = СН, N, NO;
R1 выбран из группы, состоящей из водорода, C1-C3-алкила, C3-C6-циклоалкила, C4-C5-алкилциклоалкила, где C1-C3-алкил возможно замещен 1-3 заместителями, независимо выбранными из галогена, гидрокси или C1-C6- алкокси;
R2 и R3 независимо выбраны из группы, состоящей из водорода, C1-C3-алкила, C1-C3-галоалкила, C1-C3-алкокси, C1-C3-галоалкокси и галогена.
2. Соединение по п.1, отличающееся тем, что
R выбран из группы, состоящей из COOR6, CONR7R8, тетразолила или Н, где R6, R7 и R8 независимо выбраны из группы, состоящей из Н, C1-C6-алкила;
А выбран из группы, состоящей из фенила, пиридила, индолила, тиенила, бензотиенила, индазолила, бензизоксазолила, бензофуранила, бензотриазолила, фуранила, бензотиазолила, тиазолила, каждый из которых возможно замещен одной или двумя группами, независимо выбранными из группы, состоящей из OH, C1-C6-алкила, C3-C6-циклоалкила;
Q выбран из группы, состоящей из фенила, пиридила, тиазолила, тиофенила, пиримидила, каждый из которых возможно замещен одной или двумя группами, независимо выбранными из группы, состоящей из C1-C6-алкила, галогена или CF3;

где X = СН, N, NO;
R1 выбран из группы, состоящей из водорода, C1-C3-алкила, C1-C3-галоалкила, C3-C6-циклоалкила, C4-C5-алкилциклоалкила;
R2 и R3 независимо выбраны из группы, состоящей из водорода, C1-C3-алкила, C1-C3-галоалкила, C1-C3-алкокси, C1-C3-галоалкокси и галогена.
3. Соединение по п.1 или 2, имеющее следующую структуру:

где X1 представляет собой СН или N;
R4 и R5 независимо выбраны из группы, состоящей из Н, C1-C6-алкила, галогена или CF3;
R-A выбран из

R1 выбран из группы, состоящей из изопропила и циклопропила;
R2 и R3 независимо выбраны из группы, состоящей из галогена, C1-C3-алкила, метокси и трифторметокси.
4. Соединение по любому из пп.1-3, отличающееся тем, что
А представляет собой фенил;
Q представляет собой возможно замещенный фенил;
X представляет собой СН;
R1 представляет собой циклоалкил;
каждый из R2 и R3 представляет собой галоген.
5. Соединение по п.4, отличающееся тем, что Q представляет собой фенил, замещенный одним заместителем.
6. Соединение по п.5, отличающееся тем, что указанный один заместитель представляет собой галоген.
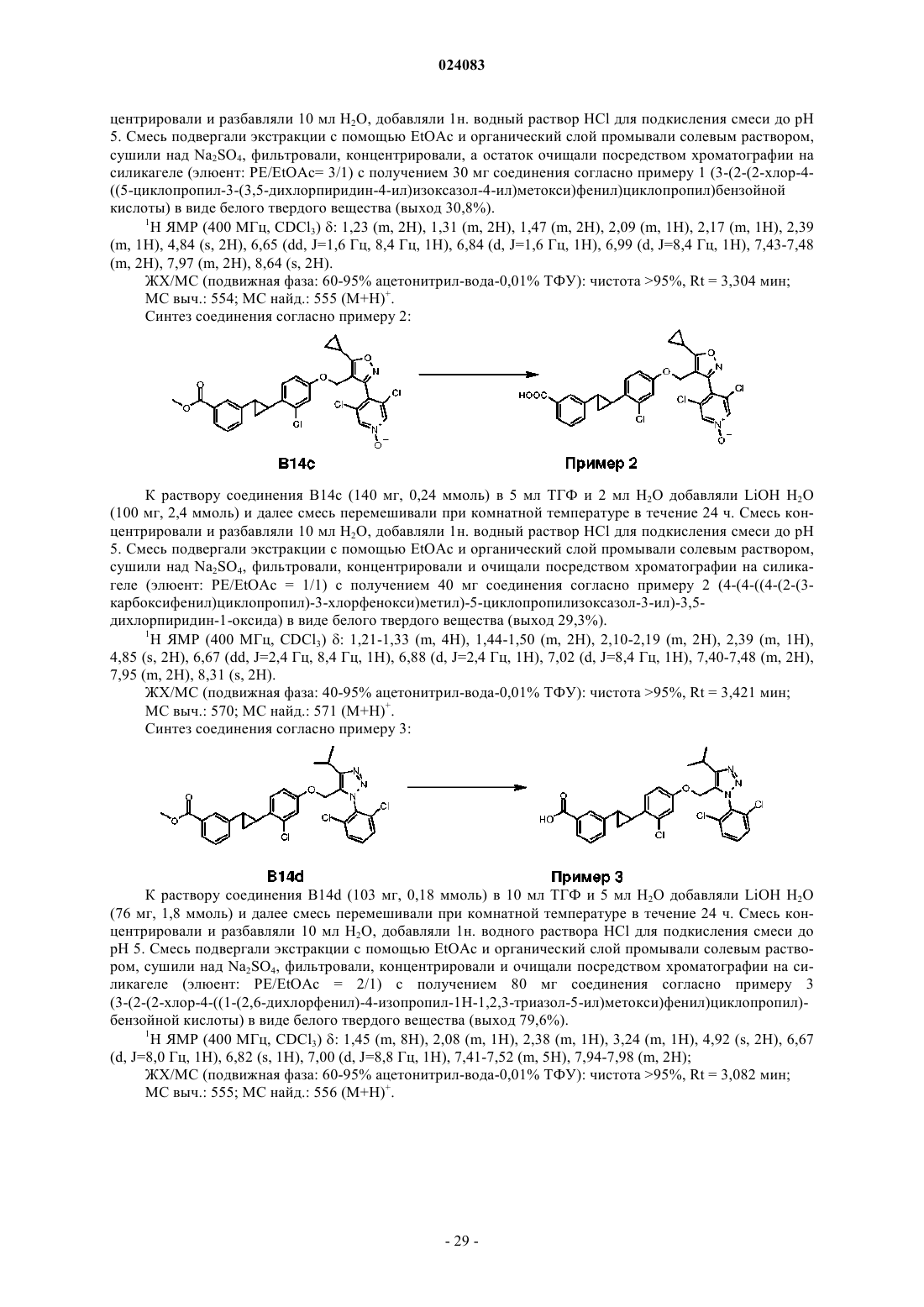
7. Соединение по любому из пп.1-6, выбранное из следующих соединений:
3-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)бензойная кислота;
(-)-3-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)бензойная кислота;
(+)-3-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)бензойная кислота;
3-(2-(2-хлор-4-((3-(2,6-дихлорфенил)-5-изопропилизоксазол-4-ил)метокси)фенил)циклопропил)бензойная кислота;
3-(2-(2-хлор-4-((5-циклопропил-3-(3,5-дихлорпиридин-4-ил)изоксазол-4-ил)метокси)фенил)циклопропил)бензойная кислота;
4-(4-((4-(2-(3-карбоксифенил)циклопропил)-3-хлорфенокси)метил)-5-циклопропилизоксазол-3-ил)-3,5-дихлорпиридин-1-оксид;
3-(2-(2-хлор-4-((1-(2,6-дихлорфенил)-4-изопропил-1H-1,2,3-триазол-5-ил)метокси)фенил)циклопропил)бензойная кислота;
4-((4-(2-(6-(1H-тетразол-5-ил)пиридин-3-ил)циклопропил)-3-хлорфенокси)метил)-5-циклопропил-3-(2,6-дихлорфенил)изоксазол;
5-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)пиколиновая кислота.
8. Соединение по любому из пп.1-6, выбранное из следующих соединений:
3-(2-(6-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)-2-(трифторметил)пиридин-3-ил)циклопропил)бензойная кислота;
4-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)бензойная кислота;
1,3-дигидрокси-2-(гидроксиметил)пропан-2-аминия 4-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)бензоат;
(+)-4-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)бензойная кислота;
(-)-4-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)бензойная кислота;
6-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)-1-метил-1Н-индазол-3-карбоновая кислота;
(+)-6-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)-1-метил-1Н-индазол-3-карбоновая кислота;
(-)-6-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)-1-метил-1Н-индазол-3-карбоновая кислота;
4-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)-N-(метилсульфонил)бензамид;
2-(4-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)бензамидо)этансульфоновая кислота;
4-((4-(2-(4-(1H-тетразол-5-ил)фенил)циклопропил)-3-хлорфенокси)метил)-5-циклопропил-3-(2,6-дихлорфенил)изоксазол;
4-(2-(2-хлор-4-((3-(2,6-дихлорфенил)-5-(2-гидроксипропан-2-ил)изоксазол-4-ил)метокси)фенил)циклопропил)бензойная кислота;
5-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)-1-изопропил-1Н-пиразол-3-карбоновая кислота;
6-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)-1-изопропил-1Н-индазол-3-карбоновая кислота;
4-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)-2,6-диметилбензойная кислота;
4-(2-(2-хлор-4-((5-циклопропил-3-(2-(трифторметокси)фенил)изоксазол-4-ил)метокси)фенил)циклопропил)бензойная кислота;
(+)-2-(4-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)бензамидо)этансульфоновая кислота;
(-)-2-(4-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)бензамидо)этансульфоновая кислота;
2-(4-(2-(2-хлор-4-((5-циклопропил-3-(2,6-дихлорфенил)изоксазол-4-ил)метокси)фенил)циклопропил)бензамидо)уксусная кислота;
4-(2-(2-хлор-4-((4-(2,6-дихлорфенил)-1-изопропил-1H-1,2,3-триазол-5-ил)метокси)фенил)циклопропил)бензойная кислота.
9. Применение соединения по любому из пп.1-8 для получения лекарственного средства для профилактики и/или лечения заболеваний, опосредованных FXR.
10. Применение по п.9
для профилактики и/или лечения хронических внутрипеченочных или некоторых форм внепеченочных холестатических состояний или фиброза печени, возникающего в результате хронических холестатических состояний или острых внутрипеченочных холестатических состояний; и/или
для профилактики и/или лечения стеатоза печени и связанных синдромов, холестатических или фиброзных эффектов, которые связаны с вызванным алкоголем циррозом или с передаваемыми вирусами формами гепатита; и/или
для профилактики и/или лечения печеночной недостаточности или ишемии печени после обширной резекции печени; и/или
для профилактики и/или лечения стеатогепатита, связанного с химиотерапией (CASH); и/или
для профилактики и/или лечения острой печеночной недостаточности.
11. Применение по п.9
для профилактики и/или лечения липидных и липопротеиновых расстройств; и/или
для профилактики и/или лечения диабета II типа и клинических осложнений диабета I и II типов, включая диабетическую нефропатию, диабетическую нейропатию, диабетическую ретинопатию и другие наблюдаемые эффекты клинического проявления длительного диабета; и/или
для профилактики и/или лечения состояний и заболеваний, возникающих в результате хронической жировой и фиброзной дегенерации органов вследствие усиленного накопления липидов и, конкретно, триглицеридов и последующей активации профиброзных путей, таких как неалкогольная жировая болезнь печени (НЖБП) или неалкогольный стеатогепатит (НАСГ); и/или
для профилактики и/или лечения ожирения или метаболического синдрома (общие состояния дислипидемии, диабета и аномально высокого индекса массы тела); и/или
для профилактики и/или лечения острого инсульта или тромбоза, который случается в качестве конечной стадии хронического обструктивного атеросклероза.
12. Применение по п.9 для профилактики и/или лечения гепатоцеллюлярной карциномы и других форм злокачественных неопластических заболеваний печени.
13. Фармацевтическая композиция для применения в профилактике и/или лечении заболеваний, опосредованных FXR, содержащая соединение по любому из пп.1-8 и один или более инертных ингредиентов, представляющих собой фармацевтически приемлемый носитель.
Текст
Изобретение относится к соединениям, которые связываются с рецептором NR1H4 (FRX) и действуют в качестве агонистов рецептора NR1H4 (FRX). Изобретение также относится к применению соединений для получения лекарственного средства для лечения заболеваний и/или состояний за счет связывания указанного ядерного рецептора указанными соединениями, а также к способу синтеза указанных соединений. Настоящее изобретение относится к соединениям, которые связываются с рецептором NR1H4 (фарнезоидный Х-рецептор, FXR) и действуют как агонисты или модуляторы рецептора NR1H4 (FXR). Изобретение также относится к применению указанных соединений для лечения и/или профилактики заболеваний и/или состояний за счет связывания указанного ядерного рецептора указанными соединениями. Многоклеточные организмы находятся в зависимости от сложных механизмов передачи информации между клетками и частями тела. Передаваемая информация может быть весьма сложной, и результатом ее передачи может являться изменение генетических программ, вовлеченных в дифференцировку,пролиферацию или репродукцию клеток. Сигналы, или гормоны, часто представляют собой низкомолекулярные соединения, такие как пептиды, жирные кислоты или производные холестерина. Многие из данных сигналов оказывают свое действие за счет изменения в конечном счете транскрипции конкретных генов. Одной хорошо изученной группой белков, которые опосредуют ответ клетки на множество сигналов, является семейство транскрипционных факторов, известных как ядерные рецепторы, в дальнейшем часто обозначаемые "ЯР". Члены данной группы включают рецепторы стероидных гормонов, витамина D, экдизона, цис- и трансретиноевой кислоты, тиреоидного гормона, желчных кислот, производных холестерина, жирных кислот (и других пролифераторов пероксисом), так же как и так называемые "сиротские" рецепторы, представляющие собой белки, которые структурно сходны с другими членами данной группы, но лиганды для которых неизвестны. Сиротские рецепторы могут указывать на неизвестные сигнальные пути в клетке или могут представлять собой ядерные рецепторы, функционирующие без активации лигандом. Активация транскрипции некоторыми из данных сиротских рецепторов может происходить в отсутствие экзогенного лиганда и/или через пути сигнальной трансдукции,начинающиеся от поверхности клетки (D. Mangelsdorf et al. "The nuclear receptor superfamily: the seconddecade", Cell, 1995, 83(6), 835-839, R. Evans "The nuclear receptor superfamily: a rosetta stone for physiology", Mol. Endocrinol. 2005, 19(6), 1429-1438). В общем случае, в ЯР выделяют три функциональных домена. Аминоконцевой домен, как полагают, обладает некоторой регуляторной функцией. За ним следует ДНК-связывающий домен, в дальнейшем называемый "ДСД", который обычно содержит два цинковых пальца и распознает специфический элемент гормонального ответа, в дальнейшем называемый "ЭГО", в промоторах генов, отвечающих за указанный ответ. Конкретные аминокислотные остатки в "ДСД", как было показано, обеспечивают специфичность связывания с последовательностью ДНК (М. Schena "Mammalian glucocorticoid receptor derivatives enhance transcription in yeast", Science, 1988, 241(4868), 965-967). Лигандсвязывающий домен, в дальнейшем называемый "ЛСД", расположен в карбоксиконцевом участке известных ЯР. В отсутствие гормона ЛСД, по-видимому, препятствует взаимодействию ДСД с ЭГО. Связывание гормона, по-видимому, приводит к конформационному изменению в ЯР и, таким образом, обусловливает это препятствование (A. Brzozowski et al. "Molecular basis of agonism and antagonism in the oestrogenreceptor", Nature, 1997, 389(6652), 753-758). ЯР без ЛСД конститутивно активирует транскрипцию, но на низком уровне. Коактиваторы или транскрипционные активаторы, как полагают, выступают связующим звеном между специфичными к последовательности факторами транскрипции, основным аппаратом транскрипции, и, кроме того, влияют на структуру хроматина целевой клетки. Несколько белков, таких как SRC-1,ACTR и Grip1, взаимодействуют с ЯР способом, при котором взаимодействие усиливается за счет лиганда (D. Heery et al. "A signature motif in transcriptional co-activators mediates binding to nuclear receptors",Nature, 1997, 387(6634), 733-736; T. Heinzel et al. "A complex containing N-CoR, mSin3 and histonecontrol of coregulator recruitment to nuclear receptors", Annu. Rev. Physiol. 2005, 67, 309-333). Модуляторы ядерных рецепторов, такие как стероидные гормоны, влияют на рост и функционирование конкретных клеток за счет связывания с внутриклеточными рецепторами и образования комплексов ядерных рецепторов с лигандами. Комплексы ядерных рецепторов с гормонами затем взаимодействуют с элементом гормонального ответа (ЭГО) в контрольной области конкретных генов и изменяют экспрессию конкретных генов (A. Aranda, A. Pascual "Nuclear hormone receptors and gene expression",Physiol. Rev. 2001, 81(3), 1269-1304). Фарнезоидный Х-рецептор альфа (в дальнейшем также часто называемый NR1H4, когда речь идет о рецепторе человека) представляет собой прототипный ядерный рецептор типа 2, который активирует гены при связывании с промоторным участком целевых генов гетеродимерным образом с ретиноидным Х-рецептором (В. Forman et al. "Identification of a nuclear receptor that is activated by farnesol metabolites",Cell, 1995, 81(5), 687-693). Соответствующими физиологичискими лигандами NR1H4 являются желчные кислоты (D. Parks et al. "Bile acids: natural ligands for an orphan nuclear receptor", Science, 1999, 284(5418),1365-1368; M. Makishima et al. "Identification of a nuclear receptor for bile acids", Science, 1999, 284(5418),1362-1365). Наиболее сильным лигандом является хенодезоксихолевая кислота (ХДХК), которая регулирует экспрессию нескольких генов, участвующих в гомеостазе желчных кислот. Фарнезол и его производные, вместе называемые фарнезоидами, как сообщалось первоначально, активируют крысиный ортолог в высокой концентрации, но они не активируют рецептор человека или мыши. FXR экспрессируется в печени, во всем желудчно-кишечном тракте, включая пищевод, желудок, двенадцатиперстную кишку,-1 024083 тонкий кишечник, толстую кишку, в яичниках, надпочечниках и почках. Помимо контролирования внутриклеточной экспрессии генов, FXR, по-видимому, также вовлечен в паракринную и эндокринную передачу сигналов посредством повышающей регуляции экспрессии цитокина - фактора роста фибробластов 15 (грызуны) или 19 (обезьяны, люди) J. Holt et al. "Definition of a novel growth factor-dependent signal"Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis", Cell Metab. 2005, 2(4), 217-225). Существует одна публикация, в которой высказано предположение о прямом влиянии активацииFXR на выживаемость возбудителей инфекции, таких как бактерии или протозойные паразиты, посредством активирования лизосомального фактора гибели/выживания Тасо-2 в макрофагах (P. Anandet al.macrophages", FEMS Microbiol. Lett. 2005, 250(1), 137-144). Это могло бы проложить путь для дальнейших исследований, направленных на оценку способности FXR выступать в качестве мишени для лекарственного средства для лечения внутриклеточных бактериальных или паразитарных инфекций, таких как туберкулез, проказа, лейшманиоз или трипаносомоз, например болезнь Чагаса. Низкомолекулярные соединения, которые действуют как модуляторы FXR, были предложены в следующих публикациях: WO 2000/037077, WO 2003/015771, WO 2004/048349, WO 2007/076260,WO 2007/092751, WO 2007/140174, WO 2007/140183, WO 2008/051942, WO 2008/157270,WO 2009/005998, WO 2009/012125, WO 2008/025539 и WO 2008/025540. Недавно вышел обзор дополнительных низкомолекулярных модуляторов FXR (R.С. Buijsman et al. "Non-Steroidal Steroid ReceptorModulators", Curr. Med. Chem. 2005, 12, 1017-1075). В публикации WO 2000/037077 предложены соединения, имеющие следующую общую формулуR и R1 независимо представляют собой Н, низший алкил, галоген или CF3;R2 представляет собой низший алкил;R3 и R4 независимо представляют собой Н, низший алкил, галоген, CF3, OH, O-алкил илиO-полигалоалкил. В более предпочтительном варианте реализации в публикации WO 2000/037077 предложено соединение GW4064 которое было также впервые опубликовано в источнике Maloney et al. "Identification of a chemicalacid-based analogs of GW4064", Bioorg. Med. Chem. Lett. 2008, 18(15), 4339-4343 упоминает возможные недостатки данного агониста FXR. Согласно этой публикации GW4064 показывает плохую фармакокинетику с низкой пероральной биодоступностью и низким содержанием вещества в плазме, а также короткий период полувыведения из плазмы. Кроме того, трансстильбеновый фрагмент этой молекулы и родственные агонисты FXR из публи-2 024083rat liver microsomes", Toxicol. Appl. Pharmacol. 2000, 167(1), 46-54. Наконец, авторы Akwabi-Ameyaw et al. упоминают, что трансстильбеновый фрагмент GW4064 ответственен за нестабильность последнего в ультрафиолетовом свете. Фотонестабильность может превратиться в фототоксичность, если соединение при введении его людям подвержено воздействию УФ-света, например, за счет отложения или накопления в коже (см. Colerangle J.B. "Regulatory non-clinical photosafety evaluation - An attempt to merge the FDArisks in patients for a new drug molecule" J. Photochem. Photobiol. B. 2009, 96(1), 57-62). С другой стороны, данный конъюгированный трансстильбен является основой для активностиGW4064, так как авторы упоминают, что простое восстановление двойной связи до этиленовой группы приводит к уменьшению лигандсвязывающих свойств FXR. Соответственно, задачей настоящего изобретения является обеспечение технического решения позволяющего преодолеть недостатки агонистов FXR, содержащих трансстильбеновый фрагмент, предложенных в публикации WO 2000/037077, при сохранении или даже улучшении силы связывания с FXR в противоположность снижению FXR-связывающей активности, наблюдаемому у аналогов GW4064 на основе нафтойной кислоты, описанных в Akwabi-Ameyaw et al. Эта задача была решена путем обеспечения соединений по п.1 формулы изобретения. Предпочтительные варианты реализации предложены в формуле изобретения, а также в приведенном далее описании. Согласно настоящему изобретению применяют превращение трансстильбеновой двойной связи в циклопропильную группу, которое может быть достигнуто путем добавления карбена, с получением соединений, демонстрирующих неожиданное полное устранение вызываемой УФ фотолабильности и в то же время преодоление потенциально токсичных свойств трансстильбена. Получаемые рацемические смеси проявляют свойства агонистов FXR, сходные со значениями, получаемыми для соответствующих молекул, содержащих трансстильбен. Кроме того, хиральное разделение рацемических смесей позволило получить индивидуальные энантиомеры, которые демонстрировали даже лучшие FXR-связывающие и трансактивационные свойства по сравнению с соответствующими молекулами, содержащими трансстильбен, или родственными аналогами на основе нафтойной кислоты, сведения о которых опубликованы в Akwabi-Ameyaw et al. Следовательно, согласно настоящему изобретению предложено более совершенное решение проблемы, связанной с FXR-соединениями, содержащими трансстильбен, в противоположность другим техническим решениям, опубликованным Akwabi-Ameyaw et al. Соединения согласно настоящему изобретению имеют общую химическую структуру в соответствии с формулой (1) где R выбран из группы, состоящей из COOR6, CONR7R8, тетразолила или Н, где R6 независимо выбран из группы, состоящей из Н или низшего алкила, и R7 и R8 независимо друг от друга выбраны из группы, состоящей из Н, низшего алкила, C1-C6-галоалкила, C1-C6-алкилена-R9, SO2-C1-C6-алкила, где R9 выбран из группы, состоящей из COOH, OH или SO3H; А выбран из группы, состоящей из фенила, пиридила, пиразолила, индолила, тиенила, бензотиенила, индазолила, бензизоксазолила, бензофуранила, бензотриазолила, фуранила, бензотиазолила, тиазолила, каждый из которых возможно замещен одной или двумя группами, независимо выбранными из группы, состоящей из OH, низшего алкила, низшего циклоалкила или галогена;Q выбран из группы, состоящей из фенила, пиридила, тиазолила, тиофенила, пиримидила, каждый из которых возможно замещен одной или двумя группами, независимо выбранными из группы, состоящей из низшего алкила, галогена или CF3;R1 выбран из группы, состоящей из водорода, C1-C3-алкила, C3-C6-циклоалкила,C4-C5-алкилциклоалкила, где C1-C3-алкил возможно замещен 1-3 заместителями, независимо выбранными из галогена, гидрокси или C1-C6-алкокси;R2 и R3 независимо выбраны из группы, состоящей из водорода, C1-C3-алкила, C1-C3-галоалкила,C1-C3-алкокси, C1-C3-галоалкокси и галогена. В настоящем изобретении термин "низший алкил" обозначает алкильную группу, которая может быть неразветвленной или разветвленной, предпочтительно неразветвленной, и содержит от 1 до 6,предпочтительно от 1 до 4 атомов углерода. Предпочтительные примеры представляют собой метил и этил, изопропил и т-бутил. Термин "низший циклоалкил" обозначает циклоалкильную группу, содержащую от 3 до 6 атомов углерода. Циклопропил является особенно предпочтительным. Примеры атомов галогена, применяемых в настоящем изобретении в качестве заместителей, как перечислено выше, представляют собой F, Cl и Br, причем Cl является предпочтительным. В предпочтительном варианте реализации в комбинации с любыми выше- и нижеприведенными вариантами реализации соединения согласно настоящему изобретению представлены структурой в соответствии с формулой (1) в которой R выбран из группы, состоящей из COOR6, CONR7R8, тетразолила или Н, где R6, R7 и R8 независимо выбраны из группы, состоящей из Н, низшего алкила; А выбран из группы, состоящей из фенила, пиридила, индолила, тиенила, бензотиенила, индазолила, бензизоксазолила, бензофуранила, бензотриазолила, фуранила, бензотиазолила, тиазолила, каждый из которых возможно замещен одной или двумя группами, независимо выбранными из группы, состоящей из OH, низшего алкила, низшего циклоалкила;Q выбран из группы, состоящей из фенила, пиридила, тиазолила, тиофенила, пиримидила, каждый из которых возможно замещен одной или двумя группами, независимо выбранными из группы, состоящей из низшего алкила, галогена или CF3;R2 и R3 независимо выбраны из группы, состоящей из водорода, C1-C3-алкила, C1-C3-галоалкила,C1-C3-алкокси, C1-C3-галоалкокси и галогена. Предпочтительно R выбран среди COOR6 и CONR7R8, где R6, R7 и R8 имеют значения, определенные выше, более предпочтительно R представляет собой COOR6, где R6 имеет значения, определенные выше, предпочтительно R6 выбран среди Н и низшего алкила. В равной степени в более предпочтительном варианте реализации R представляет собой CONR7R8,где R7 и R8 имеют значения, определенные выше, более предпочтительно R7 и R8 независимо друг от друга выбраны из Н, низшего алкила, C1-C6-алкилена-R9 и SO2-C1-C6-алкила, где R9 выбран из группы,состоящей из COOH и SO3H. А предпочтительно выбран из тех групп, определенных выше, которые не замещены. Более предпочтительно А представляет собой фенил. В альтернативном предпочтительном варианте реализации в комбинации с любыми выше- и нижеприведенными вариантами реализации А предпочтительно выбран из гетероциклических групп пиридила, пиразолила, бензизоксазолила и индазолила, при этом А является незамещенным или замещенным одной или двумя группами, независимо выбранными из OH, низшего алкила, низшего циклоалкила и галогена.Q предпочтительно выбран из тех групп, которые определены выше, замещенных одним заместителем. Предпочтительно заместитель представляет собой галоген, более предпочтительно Cl. В частности,Q представляет собой фенил, замещенный одним атомом галогена, предпочтительно Cl. В альтернативном предпочтительном варианте реализации в комбинации с любыми выше- и нижеприведенными вариантами реализации Q представляет собой пиридильную группу, которая является незамещенной или замещенной одной или двумя группами, предпочтительно одной группой, независимо выбранной из группы, состоящей из низшего алкила, галогена и CF3. В предпочтительном варианте реализации в комбинации с любыми выше- и нижеприведенными вариантами реализации Z выбран из следующих групп: Еще более предпочтительно Z представляет собой следующую группу: Кроме того, Z предпочтительно выбран из структур, определенных выше, где X представляет собой СН. Более предпочтительно Z выбран из структур, определенных выше, где X представляет собой СН иR1 представляет собой циклоалкильную группу, предпочтительно циклопропил. Еще более предпочтительно Z выбран из структур, определенных выше, где X представляет собой СН и R1 представляет собой циклоалкильную группу, предпочтительно циклопропил, и R2 и R3, каждый, представляют собой галоген,наиболее предпочтительно Cl. Наиболее предпочтительным вариантом реализации для Z является определенный выше, где главный гетероциклический скелет представляет собой третью структуру, определенную выше, содержащую 5-членное кольцо с фрагментом O-N. В равной степени предпочтительном варианте реализации в комбинации с любыми выше- и нижеприведенными вариантами реализации Z предпочтительно выбран из структур, определенных выше, гдеX представляет собой N или NO, более предпочтительно X представляет собой N. В предпочтительном варианте реализации в комбинации с любыми выше- и нижеприведенными вариантами реализации R1 выбран из группы, состоящей из водорода, C1-C3-алкила, C3-C6-циклоалкила и С 4-C5-алкилциклоалкила, где C1-C3-алкил не замещен или замещен 1-3 заместителями, предпочтительно 1 или 2 заместителями, независимо выбранными из галогена или гидроксигруппы. Предпочтительные комбинации R, A, Q и Z имеют значения, определенные выше, и настоящее изобретение включает все комбинации предпочтительных вариантов реализации, перечисленных выше. В конкретном предпочтительном варианте реализации R представляет собой COOR6, где R6 имеет значения, определенные выше, предпочтительно R6 выбран из Н и низшего алкила; А представляет собой фенил; Q выбран из тех групп, которые определены выше, замещенных одним заместителем, предпочтительно заместитель представляет собой галоген, более предпочтительно Cl, и, в частности, Q представляет собой фенил, замещенный одним атомом галогена, предпочтительно Cl; и Z выбран среди структур,определенных выше, где X представляет собой СН и R1 представляет собой циклоалкильную группу,предпочтительно циклопропил, и R2 и R3, каждый, представляют собой галоген, наиболее предпочтительно Cl. В более предпочтительном варианте реализации настоящего изобретения соединения имеют общую структуру в соответствии с формулой (2) где X1 выбран из СН и N, предпочтительно СН;R4 и R5 независимо выбраны из группы, состоящей из Н, низшего алкила, галогена или CF3, предпочтительно галогена, более предпочтительно Cl. ще более предпочтительными являются соединения формул (1) или (2), которые имеют структуру,где R-A выбран изR2 и R3 независимо выбраны из группы, состоящей из галогена, C1-C3-алкила, метокси и трифторметокси, предпочтительно представляют собой галоген и наиболее предпочтительно Cl. Наиболее предпочтительные соединения согласно изобретению представляют собой следующие: В равной степени наиболее предпочтительные соединения согласно изобретению представляют собой следующие: Соединения согласно настоящему изобретению могут быть в форме пролекарств. "Пролекарство" означает производное, которое превращается в соединение согласно настоящему изобретению посредством реакции с ферментом, желудочным соком и т.п. при физиологических условиях в живом организме,например посредством реакций окисления, восстановления, гидролиза и т.п., каждая из которых осуществляется ферментативным путем. Примерами пролекарств являются соединения, у которых аминогруппа в соединении согласно настоящему изобретению ацилирована, алкилирована или фосфорилирована с образованием, например, эйкозаноиламиногруппы, аланиламиногруппы, пивалоилоксиметиламиногруппы, или у которых гидроксильная группа ацилирована, алкилирована, фосфорилирована или превращена в борат, например ацетилоксигруппа, пальмитоилоксигруппа, пивалоилоксигруппа, сукцинилоксигруппа, фумарилоксигруппа, аланилоксигруппа, или у которых карбоксильная группа этерифицирована или амидирована. Данные соединения могут быть получены из соединений согласно настоящему изобретению в соответствии с хорошо известными способами. Другие примеры пролекарств представляют собой соединения, где карбоксилат в соединении согласно настоящему изобретению превращен, например, в алкиловый, ариловый, холиновый сложный эфир, сложный эфир, содержащий аминогруппу, ацилоксиметиловый сложный эфир, линоленоиловый сложный эфир. Метаболиты соединений согласно настоящему изобретению также находятся в рамках настоящего изобретения. Там, где возможна таутомерия, такая как, например, кетоенольная таутомерия, соединений согласно настоящему изобретению или их пролекарств, индивидуальные формы, такие как, например, кето- и енольная форма, каждая, находятся в рамках изобретения, так же как и их смеси в любом соотношении. То же справедливо для стереоизомеров, таких как, например, энантиомеры, цис/транс-изомеры, конформеры и т.п. При необходимости изомеры могут быть разделены с помощью способов, хорошо известных в данной области, например с помощью жидкостной хроматографии. Разделение также выполнимо для энантиомеров путем применения, например, хиральных неподвижных фаз. Кроме того, энантиомеры могут быть выделены путем превращения их в диастереомеры, т.е. соединения с энантиомерно чистым вспомогательным соединением, последующим разделением получающихся диастереомеров и отщеплением вспомогательного остатка. Альтернативно, любой энантиомер соединения согласно настоящему изобретению может быть получен в результате стереоселективного синтеза с применением оптически чистых исходных веществ. Другой путь получения чистых энантиомеров из рацемических смесей заключался бы в применении энантиоселективной кристаллизации с хиральными противоионами. Соединения согласно настоящему изобретению могут находиться в форме фармацевтически приемлемой соли или сольвата. Термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований или кислот, включая неорганические основания или кислоты и органические основания или кислоты. В случае, если соединения согласно настоящему изобретению содержат одну или более кислотных или основных групп, изобретение также включает их соответствующие фармацевтически или токсикологически приемлемые соли, в частности их соли, применяемые в фармации. Таким образом, соединения согласно настоящему изобретению, которые содержат кислотные группы, могут быть представлены данными группами и могут быть применены согласно изобретению, например, в виде солей щелочных металлов, солей щелочно-земельных металлов или аммониевых солей. Более точные примеры таких солей включают натриевые соли, калиевые соли,кальциевые соли, магниевые соли или соли с аммиаком или органическими аминами, такими как, например, этиламин, этаноламин, триэтаноламин или аминокислоты. Соединения согласно настоящему изобретению, которые содержат одну или более основных групп, т.е. групп, которые могут быть протонированы, могут быть представлены и могут быть применены согласно изобретению в форме их аддитивных солей с неорганическими или органическими кислотами. Примеры подходящих кислот включают хлороводород, бромоводород, фосфорную кислоту, серную кислоту, азотную кислоту, метансульфоновую кислоту, п-толуолсульфоновую кислоту, нафталиндисульфоновые кислоты, щавелевую кислоту, уксусную кислоту, винную кислоту, молочную кислоту, салициловую кислоту, бензойную кислоту, муравьиную кислоту, пропионовую кислоту, триметилуксусную кислоту, диэтилуксусную кислоту, малоновую кислоту, янтарную кислоту, пимелиновую кислоту, фумаровую кислоту, малеиновую кислоту, яблочную кислоту, сульфаминовую кислоту, фенилпропионовую кислоту, глюконовую кислоту, аскорбиновую кислоту, изоникотиновую кислоту, лимонную кислоту, адипиновую кислоту и другие кислоты, известные специалисту в данной области техники. Если соединения согласно настоящему изобретению одновременно содержат кислотные и основные группы в молекуле, изобретение также включает, наряду с упомянутыми формами солей, внутренние соли или бетаины (цвиттер-ионы). Соответствующие соли могут быть получены с помощью обычных способов, которые известны специалисту в данной области техники, таких как, например, взаимодействие соединений согласно изобретению с органической или неорганической кислотой или основанием в растворителе или диспергирующем агенте или анионный обмен или катионный обмен с другими солями. Настоящее изобретение также включает все соли соединений согласно настоящему изобретению, которые вследствие низкой физиологической совместимости непосредственно не подходят для применения в фармацевтических препаратах, но которые могут быть применены, например, в качестве промежуточных продуктов для химических реакций или для получения фармацевтически приемлемых солей. Кроме того, соединения согласно настоящему изобретению могут присутствовать в форме сольватов, таких как те, которые включают в качестве сольвата воду или фармацевтически приемлемые сольваты, такие как спирты, в частности этанол. К тому же, согласно настоящему изобретению предложены фармацевтические композиции, содержащие по меньшей мере одно соединение согласно настоящему изобретению, или его пролекарство, или его фармацевтически приемлемую соль или сольват в качестве активного ингредиента совместно с фармацевтически приемлемым носителем."Фармацевтическая композиция" означает один или более активных ингредиентов и один или более инертных ингредиентов, которые составляют носитель, также как и любой продукт, который получается,прямо или опосредованно, в результате объединения, комплексообразования или агрегации любых двух или более ингредиентов, или в результате диссоциации одного или более ингредиентов, или в результате других типов реакций или взаимодействий одного или более ингредиентов. Соответственно, фармацевтические композиции согласно настоящему изобретению включают любую композицию, полученную путем смешивания по меньшей мере одного соединения согласно настоящему изобретению и фармацевтически приемлемого носителя. Фармацевтическая композиция согласно настоящему изобретению может дополнительно содержать одно или более других соединений в качестве активных ингредиентов, таких как пролекарство или другие модуляторы ядерных рецепторов. Композиции подходят для перорального, ректального, местного, парентерального (включая подкожное, внутримышечное и внутривенное), офтальмологического (глазного), легочного (назальная или буккальная ингаляция) или назального введения, хотя наиболее подходящий путь введения в любом данном случае будет зависеть от природы и тяжести состояний, подлежащих лечению, и от природы активного ингредиента. Они могут быть удобно представлены в дозированной форме для однократного введения и получены любым из способов, хорошо известных в области фармацевтики. Соединения согласно изобретению могут быть получены с помощью комбинации способов, известных в области техники, включая методики, описанные ниже. Схема I изображает реакцию подходящего соединения формулы (I) с подходящим гетероциклическим соединением формулы II. Схема I Таким образом, подходящее соединение формулы II, в котором R1, R2 и R3 имеют значения, определенные в формуле (1), и Y представляет собой уходящую группу, и подходящее соединение формулы I, в котором R, А и Q имеют значения, определенные в формуле (1), или представляют собой группу, которая приводит к R, как определено в формуле (1), например, путем образования сложного эфира, амида, тетразола или кислоты, подвергают реакции с образованием соединения формулы (1) с подходящими постановками и/или снятиями защитных групп или другими стадиями, известными специалисту в данной области техники или описанными в настоящей заявке. Подходящие уходящие группы хорошо известны в данной области техники и включают галиды, в частности хлорид, бромид и йодид, эфиры сульфокислоты, такие как брозил, тозил, метансульфонил и трифторметансульфонил. Соединение формулы (I) подвергают реакции с соединением формулы (II) в подходящем растворителе, таком как ацетонитрил, диметилформамид, тетрагидрофуран и т.п. в присутствии избытка подходящего основания, такого как гидрид натрия, карбонат калия, карбонат натрия, триэтиламин, диизопропилэтиламин, трет-бутоксид калия и т.д. Такие реакции обычно проводят при температурах от комнатной до кипения выбранного растворителя. Альтернативно, подходящее соединение формулы I, в котором R, А и Q имеют значения, определенные в формуле (1), может быть сконденсировано с подходящим гетероциклическим соединением формулы II, в котором R1, R2 и R3 имеют значения, определенные в формуле (1), и Y представляет собой гидроксил, с применением реакции Мицунобу. Соединения, в которых R представляет собой сложный эфир, можно превратить в соединения формулы (1), в которых R представляет собой кислоту, посредством способов, хорошо известных специалисту в данной области техники. Гидролиз простых алкиловых сложных эфиров можно проводить в подходящих растворителях, таких как ацетонитрил, метанол, этанол, ТГФ и их смесей с водой при температурах примерно 25-100C с подходящими основаниями, такими как гидроксид натрия, гидроксид калия и гидроксид лития. В случае, когда R представляет собой трет-бутиловый сложный эфир, соответствующая кислота может быть получена в кислых условиях, хорошо известных специалистам в данной области техники. Соединения формулы (I) и (II) можно легко получить с помощью способов, которые хорошо известны и установлены в области техники, включая способы и методики, описанные в настоящем документе. Соединения формулы (I) получают из олефиновых предшественников с помощью стандартных реакций циклопропанирования, хорошо известных специалистам в данной области техники (см. H. Lebel,J.-F. Marcoux, C. Molinaro, A.B. Charette, Chem. Rev. 2003, 103, 977-1050). Например, реакция возможно замещенного производного стильбена с реагентом Симмонса-Смита (IZnCH2I) в подходящих растворителях, таких как эфир, ТГФ, гексан, толуол, дихлорметан, при температурах от -20C до примерно кипения выбранного растворителя дает соединения формулы I. Возможно замещенные стильбены могут быть получены путем сочетания Хорнера-Эммонса арилальдегида и арилметиленфосфонового эфира или путем сочетания Хека возможно замещенного стирола с возможно замещенным арилбромидом, йодидом или трифлатом в присутствии палладиевого катализатора. Как изображено на схеме I, циклопропанирование возможно замещенных стильбенов можно проводить после сочетания с возможно замещенным гетероциклом формулы II. Соединения согласно изобретению имеют хиральные центры и могут существовать в оптически активных формах. Если требуется стереоизомер, его можно получить с помощью способов, хорошо известных в данной области техники. Например, рацемическая смесь соединений формулы (1) может быть разделена с помощью хроматографии на хиральной колонке. Альтернативно, рацемическая смесь соединений формулы (1), в которой R представляет собой кислоту, может быть разделена путем кристаллизации с подходящими хиральными аминами, такими как -фенэтиламин, бруцин, цинхонин. Рацемат может также быть химически дериватизирован с другим энантиомерно чистым реагентом, таким как хиральный спирт, который может быть введен в реакцию с группой -COOH с превращением рацемических смесей в смеси диастереомерных эфиров, которые можно разделить с помощью традиционных хроматографических способов или других стандартных способов разделения. Разделение энантиомеров можно также выполнить на уровне рацемических соединений формулы I. Оптически активные соединения формулы (I) могут далее быть подвергнуты реакции с гетероциклическим соединением формулы II, как изложено вкратце на схеме I, с получением оптически активных соединений формулы (1). Перечень фигур чертежей и иных материалов На фиг. 1 а представлена зависимость средней концентрации соединения согласно примеру 12 от времени после перорального введения обезьянам (12 мг/кг; n=3). На фиг. 1b представлена зависимость средней концентрации соединения согласно примеру 4 от времени после перорального введения (12 мг/кг; n=3). На фиг. 1 с представлены уровни исходного соединения в плазме в [нМ] после введения через желудочный зонд одной дозы 5 мг/кг GW4064 и соединения согласно примеру 4 по отдельности мышам С 57BLKS lepr-/- db/db. Данные отображают среднее из 3 индивидуальных измерений в плазме мыши. На фиг. 2 а представлены химическая структура соединения GW4064 и его УФ-спектр. На фиг. 2b представлены химическая структура соединения РХ 20535 и его УФ-спектр. На фиг. 2 с представлены химическая структура соединения согласно примеру 4 и его УФ-спектр. На фиг. 3 а представлен спектр 1 Н ЯМР соединения GW4064 при t=0, до УФ-облучения при 254 нм. На фиг. 3b представлен спектр 1 Н ЯМР соединения РХ 20535 при t=0, до УФ-облучения при 254 нм. На фиг. 3 с представлен спектр 1 Н ЯМР соединения согласно примеру 4 при t=0, до УФ-облучения при 254 нм. На фиг. 3d представлен спектр 1 Н ЯМР соединения GW4064 при времени УФ-облучения при 254 нм t=44. На фиг. 3 е представлен спектр 1 Н ЯМР соединения РХ 20535 при времени УФ-облучения при 254 нм t=44. На фиг. 3f представлен спектр 1 Н ЯМР соединения согласно примеру 4 при времени УФ-облучения при 254 нм t=44. На фиг. 3g представлен спектр 1 Н ЯМР соединения GW4064 при времени УФ-облучения при 254 нм t=154. На фиг. 3h представлен спектр 1 Н ЯМР соединения РХ 20535 при времени УФ-облучения при 254 нм t=154. На фиг. 3i представлен спектр 1 Н ЯМР соединения согласно примеру 4 при времени УФ-облучения при 254 нм t=154. На фиг. 3j представлен спектр 1 Н ЯМР соединения GW4064 при времени УФ-облучения при 254 нмt=704. На фиг. 3k представлен спектр 1 Н ЯМР соединения РХ 20535 при времени УФ-облучения при 254 нм t=704. На фиг. 3l представлен спектр 1 Н ЯМР соединения согласно примеру 4 при времени УФ-облучения при 254 нм t=704. На фиг. 3m приведено сравнение УФ-стабильности, влияния облучения при =366 нм на тонкую пленку с ДМСО. Фиг. 4 иллюстрирует содержание холестерина и триглицеридов в плазме после 8 недель при рационе с высоким содержанием жира (РВСЖ) в присутствии или в отсутствие соединений. Фиг. 5 иллюстрирует содержание холестерина и триглицеридов печени после 8 недель на рационе с высоким содержанием жира (РВСЖ) в присутствии или в отсутствие соединений. Фиг. 6 иллюстрирует индукцию гена-мишени FXR в печени мыши. Способы синтеза Соединения формулы (II) могут быть получены, как изображено на схемах 1 а-1d. Изоксазоловые соединения формулы (II) получают путем реакции возможно замещенных бензальдегидов с гидроксиламином в присутствии подходящего основания, такого как триэтиламин, с последующим хлорированием с подходящим хлорирующим агентом, таким как N-хлорсукцинимид. Полученные хлороксимы подвергают реакции с подходящим -кетоэфиром в основных условиях с подходящим основанием,таким как триэтиламин или метоксид натрия, с получением сложных эфиров изоксазола. Данные сложные эфиры могут быть восстановлены до спиртов формулы (II) с помощью хорошо известных способов,таких как применение литийалюминийгидрида (LAH) или диизобутилалюминийгидрида (DIBAL), и превращены в уходящую группу. 1-Арил-4-алкилтриазоловые соединения формулы (II) могут быть получены путем добавления замещенных пропаргиловых спиртов к замещенным ароматическим азидам и последующего превращения гидроксигруппы в подходящую уходящую группу. 1-Арил-4-алкилтриазоловые соединения формулы (II) могут быть получены путем добавления возможно замещенного азида к ацетиленовому сложному эфиру с последующим восстановлением до спирта и превращением в уходящую группу. Пиразоловые соединения формулы (II) получают путем реакции возможно замещенного фенилгидразина с 1,3-дикетоэфиром с последующим восстановлением и превращением в уходящую группу. Соединения согласно настоящему изобретению синтезировали по одной из схем 2-14, начиная с различных хлорметиларильных (Cl-CH2-Ar) структурных блоков, которые синтезировали в соответствии со схемами 1a-d. Как изображено на схеме 2, в результате реакции одного из хлорметиларильных(Cl-CH2-Ar) структурных блоков А 6 а-е с арилциклопропиларильным структурным блоком В 13 и последующего омыления метилового эфира до свободной кислоты образовались соединения согласно примерам 1-6 (схема 2). Реакция хлорметиларила (Cl-CH2-Ar) А 6 е с (гетеро)арилциклопропиларильным структурным блоком С 6 (схема 3) привела к получению нитрильного предшественника С 7, который или превращали в тетразол (пример 7) путем реакции с NaN3 и NH4Cl, или омыляли до свободной кислоты (пример 8, см. схему 3). Соединение согласно примеру 9 получали в соответствии со схемой 4. Пиридон D2 алкилировали с помощью структурного блока А 6 е до промежуточного продукта D3. За двухстадийной трансформацией сложноэфирной группы в альдегид D5 следовала реакция ХорнераВадсворта-Эммонса (HWE) с получением стильбеноподобного промежуточного продукта D6. Циклопропанирование и гидролиз сложного эфира позволили получить конечное соединение согласно примеру 9. Синтез соединения согласно примеру 10 показан на схеме 5. Фосфонат Е 5 и 3-формилпиридин подвергали реакции с образованием стильбеноподобного промежуточного продукта Е 12, который после снятия защиты и алкилирования с помощью А 6 е подвергали циклопропанированию. Конечный гидролиз сложного эфира позволил получить соединение согласно примеру 10. Соединение согласно примеру 11 синтезировали в соответствии со схемой 6. Бензизоксазолкарбальдегид F5 подвергали реакции с фосфонатом F9 с образованием промежуточного продукта F10, содержащего стильбеновую структуру. Циклопропанирование и снятие защиты позволили получить соединение согласно примеру 11. Соединения согласно примерам 12-14 получали в соответствии со схемой 7, аналогичной схеме 2,но в которой применяли 4-метоксикарбонилфосфонат G1. На схеме 8 показан синтез соединения согласно примеру 15. Стильбеноподобный предшественник Н 6 получают по реакции Хека кросс-сочетания броминдазола Н 4 и олефина Н 5. Циклопропанирование и гидролиз сложного эфира привели к получению конечного соединения согласно примеру 15. Соединение согласно примеру 16 получают в соответствии со схемой 9. Трансстильбен с защитной O-метильной группой сначала циклопропанируют и далее сложноэфирную группу превращают в цианогруппу. O-деметилирование и алкилирование с помощью А 6 е привели к получению промежуточного продукта 17, содержащего цианогруппу, который подвергают реакции с азидом натрия с получением тетразола согласно примеру 16. Синтез соединения согласно примеру 17 показан на схеме 10. А 5 а ацетилировали с последующим бромированием в бензильное положение с применением N-бромсукцинимида (NBS). В результате обработки DBU и K2CO3 образовался промежуточный продукт J3, гидроксигруппа которого была защищена с помощью TBSOTf. За окислением с применением OsO4 и NaIO4 последовало добавление метилмагнийхлорида (реактива Гриньяра) и снятие защиты с гидроксигруппы с помощью тетрабутиламмонийфторида (TBAF) с получением J7. Реакция Мицунобу с 12 а и омыление сложного эфира позволили получить конечное соединение согласно примеру 17. На схеме 11 показан синтез соединения согласно примеру 18. Реакция J6 с диазометаном и последующее снятие защиты TBS позволили получить промежуточный продукт K2. Данный промежуточный продукт превращали в конечное соединение согласно примеру 18 способом, аналогичным тому,который описан для примера 17. На схеме 12 показан синтез соединения согласно примеру 19. Метил-3 оксобутаноат вводили в реакцию с А 3 а с образованием изоксазола L1. Реакция с ДМФА-ДМА с последующей обработкой SiO2 и HCl позволили получить альдегид L3, который восстановили до L4 с помощью NaBH4. За этим последовали защита OH-группы с помощью 3,4-дигидропирана, восстановление сложного эфира с применением DIBAL-H и реакция Мицунобу с 12 а с получением промежуточного продукта L7. Омыление сложного эфира и снятие защиты с гидроксигруппы позволили получить конечное соединение согласно примеру 19. В итоге, настоящее изобретение относится к соединениям в соответствии с общей формулой (I), которые связывают рецептор NR1H4 (FXR) и действуют как агонисты или модуляторы рецептора NR1H4(FXR). Изобретение также относится к применению указанных соединений для лечения и/или профилактики заболеваний и/или состояний за счет связывания указанного ядерного рецептора указанными соединениями. Кроме того, настоящее изобретение относится к применению указанных соединений для получения лекарственного препарата для лечения и/или профилактики заболеваний и/или состояний за счет связывания указанного ядерного рецептора указанными соединениями. Конкретно, настоящее изобретение относится к применению соединений в соответствии с формулой (1) для получения лекарственного препарата для профилактики и/или лечения хронических внутрипеченочных или некоторых форм внепеченочных холестатических состояний, фиброза печени, возникающего в результате хронических холестатических состояний, острых внутрипеченочных холестатических состояний,обструктивных или хронических воспалительных расстройств, которые являются результатом неправильного состава желчи, желудочно-кишечных состояний со сниженным поглощением входящего в рацион жира и жирорастворимых входящих в рацион витаминов, воспалительных заболеваний кишечника, липидных и липопротеиновых расстройств, диабета II типа и клинических осложнений диабета I и II типов, состояний и заболеваний, возникающих в результате хронической жировой и фиброзной дегенерации органов из-за усиленного накопления липидов и конкретно триглицеридов и последующей активации профиброзных путей, ожирения и метаболического синдрома (общие состояния дислипидемии, диабета и аномально высокого индекса массы тела), острого инфаркта миокарда, острого инсульта, тромбоза, который случается в качестве конечной стадии хронического обструктивного атеросклероза, хронических инфекций, вызываемых внутриклеточными бактериями или простейшими паразитами,незлокачественных гиперпролиферативных расстройств,злокачественных гиперпролиферативных расстройств, аденокарциномы толстой кишки и гепатоцеллюлярной карциномы в частности, стеатоза печени и связанных синдромов, печеночной недостаточности или нарушения функции печени как результата хронических заболеваний печени или хирургической резекции печени,инфекции гепатита В, инфекции гепатита С и/или холестатических и фиброзных эффектов, которые связаны с вызванным алкоголем циррозом или с передаваемыми вирусами формами гепатита. Лекарственные средства, упоминаемые в настоящем описании, могут быть получены традиционными способами, включая объединение соединения согласно настоящему изобретению и фармацевтически приемлемого носителя.FXR, как предполагают, является ядерным рецептором желчных кислот. В результате, он модулирует как синтетическую выработку желчных кислот в печени, так и их циркуляцию в кишечнике (путем регулирования белков, связывающих желчные кислоты). Но, помимо участия в физиологии желчных кислот, FXR, по-видимому, вовлечен в регуляцию многих разнообразных физиологических процессов,которые соответствуют в этиологии и для лечения заболеваний, таких разнообразных как холестериновые желчные камни, метаболические расстройства, такие как диабет II типа, дислипидемии или ожирение, хронические воспалительные заболевания, такие как воспалительные заболевания кишечника или хронические внутрипеченочные формы холестаза и многие другие заболевания (Т. Claudel et al. "TheFXR регулирует сложный набор генов ответа в печени и желудочно-кишечном тракте. Продукты генов оказывают влияние на разнообразные физиологические процессы. В ходе функционального анализа FXR первой проанализированной регуляторной сетью была регуляция синтеза желчных кислот. В то время как печеночные Х-рецепторы (LXR) индуцируют ключевой фермент превращения холестерина в желчные кислоты, цитохром Р 450 7 А 1 (Сур 7 А 1), посредством индукции регуляторного ядерного рецептора LRH-1, FXR репрессирует индукцию Сур 7 А 1 посредством активирования синтеза мРНК, кодирующей малый гетеродимерный партнер (SHP), дополнительный ядерный рецептор, который доминантно репрессирует LRH-1. Так как FXR связывает конечные продукты данного пути, первичные желчные кислоты, такие как холевая кислота (ХК) или хенодезоксихолевая кислота (ХДХК), этот процесс можно рассматривать в качестве примера ингибирования по принципу обратной связи на уровне экспрессии генов (В. Goodwin et al. "A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bileacid synthesis by nuclear receptors". Mol. Cell, 2000, 6(3), 507-515). Параллельно репрессии синтеза желчных кислот посредством SHP, FXR индуцирует ряд так называемых АТФ-связывающих кассетных транспортеров (АВС-транспортеров), которые отвечают за экспорт токсичных желчных кислот из цитозоля гепатоцита в канальцы, маленькие ответвления желчного протока, где образуется желчь. Данная гепатопротекторная функция FXR стала впервые очевидной с помощью анализа мышей с выключенным геном FXR (С. Sinai et al. "Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipidhomeostasis". Cell, 2000, 102(6), 731-744), где была показана пониженная или повышенная экспрессия нескольких ABC-транспортеров в печени. В результате дальнейшего детального анализа обнаружили,- 12024083 что главный экскреторный насос солей желчных кислот BSEP или АВСВ 11 (М. Ananthanarayanan et al.transcriptional regulation of the gene encoding the human bile salt export pump". Hepatology, 2002, 35(3), 589596), так же как и ключевой фермент, который опосредует перенос липидов от липопротеинов к фосфолипидам, PLTP (N. Urizar et al. "The farnesoid X-activated receptor mediates bile acid activation ofphospholipid transfer protein gene expression". J. Biol. Chem. 2000, 275(50), 39313-39317) и два ключевых канальцевых мембранных транспортера для фосфолипидов, MRP-2 (ABCC4) (Н. Kast et al. "Regulation ofBiol. Chem. 2003, 278(51), 51085-51090) являются прямыми мишенями для управляемой лигандом активации транскрипции посредством FXR (кратко изложено в М. Miyata "Role of farnesoid X receptor in theImmune Endocr. Metabol. Disord. 2005, 5(3), 289-303.) Тот факт, что FXR, по-видимому, является главным рецептором метаболитов и регулятором синтеза, экспорта и рециркуляции желчных кислот, позволил предложить применение лигандов FXR для индукции выделения желчи и изменения состава желчных кислот в сторону более гидрофильного состава. С разработкой первого синтетического лиганда FXR GW4064 (P. Maloney et al. "Identification of aMed. Res. Rev. 2001, 21(6) 513-522) в качестве модельного соединения и полусинтетического искусственного лиганда на основе желчной кислоты 6-альфа-этил-ХДХК могут быть проанализированы эффекты сверхстимуляции FXR мощными агонистами. Показано, что оба лиганда индуцируют выделение желчи у животных с перевязанными желчными протоками. Более того, кроме желчегонных эффектов, также могут быть показаны гепатопротекторные эффекты (R. Pellicciari et al. "6alpha-ethyl-chenodeoxycholic acidintra- and extrahepatic cholestasis". J. Clin. Invest. 2003, 112(11), 1678-1687). Данный гепатопротекторный эффект был далее сужен до противофиброзного эффекта, который является результатом репрессии тканевых ингибиторов матриксных металлопротеиназ, TIMP-1 и 2, индукции матриксной металлопротеиназы 2 (ММР-2), расщепляющей отложения коллагена в звездчатых клетках печени и последующего снижения уровня мРНК альфа-коллагена и мРНК трансформирующего ростового фактора бета (TGF-beta),которые оба являются профиброзными факторами, агонистами FXR (S. Fiorucci et al. "The nuclear receptorpromotes resolution of liver fibrosis". J. Pharmacol. Exp. Ther. 2005, 314(2), 584-595). Кроме того, противохолестатическая активность была показана в моделях животных с перевязанными желчными протоками,так же как и в моделях животных с холестазом, индуцированным эстрогенами (S. Fiorucci et al.cholestasis". J. Pharmacol. Exp. Ther. 2005, 313(2), 604-612). Генетические исследования показывают, что в наследственных формах холестаза (прогрессивный семейный внутрипеченочный холестаз = ПСВХ, тип I-IV) или понижена ядерная локализация самогоFXR как следствие мутации в гене FIC1 (в ПСВХ I типа, также называемой болезнью Байлера) (F. Chen etreceptor in hereditary cholestasis associated to mutation in ATP8B1". Hum. Mol. Genet. 2004, 13(20), 24512460), или понижены уровни целевого гена FXR, кодирующего экспортный насос фосфолипидов MDR-3(в ПСВХ III типа). В совокупности, существует увеличивающаяся доказательная база, что соединения,связывающие FXR, будут показывать важное клиническое применение в программе лечения хронических холестатических состояний, таких как первичный билиарный цирроз (ПБЦ) или первичный склерозирующий холангит (ПСХ) (рассмотрено в G. Rizzo et al. Curr. Drug Targets Immune Endocr. Metabol.target for cholestatic syndromes" Expert Opin. Ther. Targets 2006, 10(3), 409-421). Глубокое влияние, которое активация FXR имеет на метаболизм желчных кислот и экскрецию, значимо не только для холестатических синдромов, но даже больше для терапии против образования желчных камней. Холестериновые желчные камни образуются вследствие низкой растворимости холестерина, который активно выкачивается из клетки печени в просвет канальцев. Именно относительное про- 13024083 центное содержание трех главных компонентов, желчных кислот, фосфолипидов и свободного холестерина определяет образование смешанных мицелл и, следовательно, условное насыщение раствора свободного холестерина в желчи. Полиморфизмы FXR локализуются как локусы количественных признаков как один фактор, вносящий вклад в возникновение желчекаменной болезни (Н. Wittenburg "FXR andmice", Gastroenterology 2003, 125(3), 868-881). Применяя синтетическое модельное соединение GW4064 для FXR, можно показать, что активация FXR приводит к улучшению индекса насыщения желчи холестерином (= CSI) и прямо - к отсутствию образования желчных камней у мышей C75L, подверженных образованию желчных камней, в то время как продемонстрировано, что лечение лекарственным средством мышей с выключенным геном FXR не оказывает действия на образование желчных камнейMedicine, 2004, 10(12), 1352-1358). Данные результаты определяют FXR как хорошую мишень для разработки низкомолекулярных агонистов, которые можно применить для предотвращения образования холестериновых желчных камней или предотвращения повторного образования желчных камней после хирургического удаления или ударно-волновой литотрипсии (обсуждается в S. Doggrell "New targets in and potential treatments forcholesterol gallstone disease". Curr. Opin. Investig. Drugs 2006, 7(4), 344-348). Таким образом, в одном варианте реализации изобретения соединение согласно формуле (I) и фармацевтические композиции, содержащие указанное соединение, применяют для профилактики и/или лечения обструктивных или хронических воспалительных расстройств, которые возникают вследствие неправильного состава желчи, таких как холелитиаз, также известный как холестериновые желчные камни. Помимо сильного гепатопротекторного и желчегонного, а также противофиброзного эффектов, которые демонстрирует FXR при стимулируемой низкомолекулярными соединениями активации в печени,FXR, вероятно, играет роль в защите кишечника от неопластической трансформации и от развития полипов и их перехода в аденокарциному в кишке (S. Modica et al. "Nuclear bile acid receptor FXR protectsExp. Ther. 2009, 328(2), 469). Подобно ситуации с кишечником, отсутствие FXR приводит к высокому увеличению в образовании гепатоцеллюлярной карциномы (ГЦК), наиболее распространенной формы рака печени (I. Kim et al. "Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice",Carcinogenesis 2007, 28(5), 940 и F. Yang et al." Spontaneous development of liver tumors in the absence of thebile acid receptor farnesoid X receptor". Cancer Res. 2007, 67(3), 863). В то время как функциональный FXR препятствует образованию аденокарциномы толстой кишки и гепатоцеллюлярной карциномы, активацияFXR индуцирует регенерацию печени после гепатэктомии (W. Huang et al. "Nuclear receptor-dependentbile acid signaling is required for normal liver regeneration". Science, 2006, 312 (5771), 233). Объединенные гепатопротекторный эффект, антинеопластический эффект и эффект регенерации печени, связанные с активацией FXR, могут быть терапевтически задействованы для применения агонистов FXR в лечении тяжелых заболеваний печени. В одном варианте реализации соединения согласно изобретению и фармацевтические композиции, содержащие указанные соединения, применяют в лечении заболеваний печени, таких как гепатоцеллюлярный рак (ГЦК), стимулировании возобновления роста печени и уменьшения интенсивности побочных эффектов, связанных с обширной резекцией печени,циррозом печени, независимо от этиологии, и предотвращении или лечении ишемии печени при трансплантации печени или обширном оперативном вмешательстве на печени. Со времени открытия первого синтетического агониста FXR и его введения грызунам стало очевидным, что FXR является ключевым регулятором триглицеридов сыворотки (P. Maloney et al. J. Med.Chem. 2000, 43(16), 2971-2974; T. Willson et al. Med. Res. Rev. 2001, 21(6), 513-522). В течение прошедших шести лет было опубликовано накапливающееся доказательство того, что активация FXR с помощью синтетических агонистов приводит к значительному снижению уровня триглицеридов сыворотки,главным образом в форме сниженных ЛПОНП, но также к сниженному общему холестерину сывороткиhyperlipidemic hamsters". Am. J. Physiol. Endocrinol. Metab. 2006, 290(4), E716-722). Однако снижение количества триглицеридов сыворотки не является единственным эффектом. Лечение мышей db/db или ob/ob синтетическим FXR агонистом GW4064 привело к отмеченному и совместному уменьшению триглицеридов сыворотки, общего холестерина, свободных жирных кислот, кето- 14024083 новых тел, таких как 3-OH бутират. Кроме того, активация FXR запускает внутриклеточный путь сигналинга инсулина в гепатоцитах, что приводит к снижению выхода глюкозы из глюконеогенеза в печени с сопутствующим увеличением гликогена в печени. Лечение FXR положительно влияло как на чувствительность к инсулину, так и на толерантность к глюкозе (K. Stayrook et al. "Regulation of carbohydrate"Potential regulatory role of the farnesoid X receptor in the metabolic syndrome". Biochimie, 2005, 87(1), 9398). Действие на уменьшения массы тела также недавно наблюдали у мышей, которым в избытке давали питание с высоким содержанием липидов. (С. Lihong et al. TXR Agonist, GW4064, Reverses MetabolicDefects in High-Fat Diet Fed Mice". American Diabetes Association (ADA) 66th annual scientific sessions, June 2006, Abstract Number 856-P). Данный эффект потери массы может быть результатом индукции FXR наhuman fibroblast growth factor-19 display increased metabolic rate and decreased adiposity". Endocrinology,2002, 143(5), 1741-1747). В недавних заявках на патент было продемонстрировано действие агонистаFXR на уменьшение массы тела (Stoffel W. et al. "Methods for inhibiting Adipogenesis and for treating Type 2 Diabetes" International Patent Application WO 2004/087076; S. Jones et al. "Methods of using FXR Agonists". International Patent Application WO 2003/080803). При совместном рассмотрении указанные фармакологические эффекты агонистов FXR могут быть задействованы в различных терапевтических путях: компоненты, связывающие FXR, считаются подходящими для лечения диабета II типа благодаря их сенсибилизации инсулина, гликогеногенному и гиполипидемическому эффектам. В одном варианте реализации соединения согласно изобретению и фармацевтические композиции,содержащие указанные соединения, применяют для профилактики и/или лечения диабета II типа, который может быть преодолен за счет FXR-опосредованной повышающей регуляции системной инсулиновой чувствительности и внутриклеточного инсулинового сигналинга в печени, увеличенного периферического потребления глюкозы и ее метаболизма, увеличенного запаса гликогена в печени, уменьшенного выхода глюкозы в сыворотку в ходе печеночного глюконеогенеза. В другом варианте реализации указанные соединения и фармацевтические композиции применяют для профилактики и/или лечения хронических внутрипеченочных состояний, таких как первичный билиарный цирроз печени (ПБЦ), первичный склерозирующий холангит (ПСХ), прогрессирующий семейный холестаз (ПСХ), вызванный алкоголем цирроз и ассоциированный холестаз, и некоторые формы внепеченочных холестатических состояний, таких как холестазы, индуцированные эстрогеном или лекарственными средствами. Настоящее изобретение также относится к соединению согласно формуле (I) или фармацевтической композиции, содержащей указанное соединение, для профилактики и/или лечения желудочно-кишечных заболеваний, характеризуемых уменьшенным поглощением жиров и жирорастворимых витаминов, которые могут преодолены за счет повышения уровней желчных кислот и фосфолипидов в кишечнике. В другом варианте реализации указанные соединения и фармацевтические композиции применяют для предотвращения и/или лечения заболевания выбранного из группы, которая состоит из липидных и липопротеиновых расстройств, таких как гиперхолестеринемия, гипертриглицеридемия и атеросклероз, в качестве клинически проявляющегося состояния, которое может быть улучшено за счет благотворного действия FXR по снижению общего холестерина в плазме, снижению триглицеридов сыворотки, увеличению превращения холестерина печени в желчные кислоты и увеличению чистоты и метаболического превращения ЛПОНП и других липопротеинов в печени. В другом варианте реализации указанные соединения и фармацевтические композиции применяют для профилактики и/или лечения заболеваний, где могут быть совместно использованы гиполипидемические, антихолестатические и антифибротические эффекты медикаментов, нацеленных на FXR, которые могут быть использованы для лечения стеатоза печени и ассоциированных синдромов, таких как неалкогольный стеатогепатит ("НАСГ"), или для лечения холестатических и фибротических эффектов, которые связаны с вызванным алкоголем циррозом, или с передаваемыми вирусами формами гепатита. В сочетании с гиполипидемическими эффектами также было показано, что потеря функциональности FXR ведет к увеличению атеросклероза у мышей с выключенным белком ApoE (Е. Hanniman et al.Lipid Res. 2005, 46(12), 2595-2604). Поэтому агонисты FXR могут иметь клиническое применение как антиатеросклеротические и кардиопротекторные лекарственные средства. Отрицательная регуляция эндотелина-1 в гладкомышечных клетках сосудов может также способствовать благоприятному терапевтическому эффекту (F. He et al. "Downregulation of endothelin-1 by famesoid X receptor in vascular endothelial Изобретение также относится к соединению согласно формуле (I) или фармацевтической композиции, содержащей указанное соединение, для предотвращения и посттравматического лечения сердечнососудистых расстройств, таких как острый инфаркт миокарда, острый инсульт, или тромбоз, который возникает как конечная точка хронического обструктивного атеросклероза. Помимо контролирования образования кишечных полипов, FXR, вероятно экспрессируется в раковых тканях груди и клеточных линиях, а не в здоровых тканях груди и, вероятно, взаимодействует с рецептором эстрогена в клетках РЭ, положительных на рак груди (K. Е. Swales et al. "The famesoid X receptor is expressed in breast cancer and regulates apoptosis and aromatase expression". Cancer Res. 2006, 66(20),10120 и F. Journe et al. "Association between famesoid X receptor expression and cell proliferation in estrogenreceptor-positive luminal-like breast cancer from postmenopausal patients". Breast Cancer Res. Treat. 2009,115(3), 523). Это должно позволить рассматривать FXR также как потенциальную мишень для лечения пролиферативных заболеваний, особенно метастазирующих раковых форм, которые дают метастазы, которые выделяют низкомолекулярную чувствительную форму FXR. В другом варианте реализации указанные соединения и фармацевтические композиции применяют для профилактики и/или лечения злокачественных гиперпролиферативных расстройств, таких как разные формы рака, в частности конкретные формы рака груди, печени или рака толстой кишки, где взаимодействие с лигандом FXR будет иметь благотворное влияние. Наконец, FXR, вероятно, также вовлечен в контроль антибактериальной защиты в кишечникеProc. Natl. Acad. Sci. USA. 2006, 103(10), 3920-3905), хотя точный механизм не установлен. Из этих опубликованных данных, тем не менее, можно заключить, что лечение с помощью агонистов FXR может иметь благотворное влияние при терапии воспалительных расстройств кишечника (ВРК), в частности таких форм, где поражена верхняя (подвздошная) часть кишечника (например, болезнь Крона подвздошной кишки), поскольку это, по-видимому, место действия контроля FXR на бактериальный рост. При ВРК снижение чувствительности адаптивного иммунного ответа каким-то образом ослабляет в кишечнике иммунную систему. Бактериальное разрастание может затем стать причиной инициирования установления хронического воспалительного ответа. Таким образом, подавление бактериального роста механизмами, которые основаны на FXR, может быть ключевым механизмом для предотвращения острых воспалительных случаев. Таким образом, настоящее изобретение также относится к соединению согласно формуле (I) или фармацевтической композиции, содержащей указанное соединение, для предотвращения и/или лечения заболевания, относящегося к воспалительным заболеваниям кишечника, такого как болезнь Крона или язвенный колит. FXR-опосредованное восстановление внешней барьерной функции кишечника или снижение несимбиотической бактериальной нагрузки, как полагают, способствует снижению воздействия бактериальных антигенов на иммунную систему кишечника и может, таким образом, снижать воспалительные реакции. Кроме того, изобретение относится к соединению или фармацевтической композиции для профилактики и/или лечения ожирения и связанных с ним расстройств, таких как метаболический синдром(сочетание условий дислипидемии, диабета и аномально высокого индекса массы тела), которые могут быть преодолены за счет FXR-опосредованного снижения триглицеридов в сыворотке, глюкозы в крови и повышения чувствительности к инсулину и FXR-опосредованной потери массы тела. В одном варианте реализации изобретения, указанное соединение или фармацевтическая композиция предназначена для лечения устойчивых инфекций, вызванных внутриклеточными бактериями или простейшими-паразитами, такими как Mycobacterium spec, (лечение туберкулеза или проказы), Listeriamonocytogenes (лечение листериоза), Leishmania spec, (лейшманиоз), Trypanosoma spec. (болезнь Чагаса,трипаносомоз, сонная болезнь). В другом варианте реализации соединения или фармацевтическая композиция согласно настоящему изобретению подходят для предотвращения и/или лечения клинических осложнений диабета I типа иII типа. Примеры таких осложнений включают диабетическую нефропатию, диабетическую ретинопатию, диабетические нейропатии или периферический облитерирующий эндартериит (ПАОЭ). Другие клинические осложнения сахарного диабета также охвачены настоящим изобретением. Кроме того, состояния и заболевания, возникающие в результате хронической жировой и фиброзной дегенерации органов вследствие усиленного накопления липидов и, в частности, триглицеридов и последующей активации профибротических путей, также могут быть предотвращены и/или подвергнуты лечению с применением соединений или фармацевтической композиции согласно настоящему изобретению. Такие состояния или заболевания включают неалкогольный стеатогепатит (НАСГ) и хронические холестатические состояния в печени, гломерулосклероз и диабетическую нефропатию в почках, вырождения макулы и диабетическую ретинопатию глаз, и нейродегенеративные заболевания, такие как болезнь Альцгеймера в мозге или диабетическую нейропатию в периферической нервной системе. При практическом применении соединения согласно настоящему изобретению могут быть объединены в качестве активного ингредиента в однородную смесь с фармацевтическим носителем согласно традиционным методам приготовления фармацевтических композиций. Носитель может принимать различные формы в зависимости от формы препарата, предназначенного для введения, например перорального или парентерального (в том числе внутривенного). При приготовлении композиций для пероральной дозированной формы могут быть применены любые из обычных фармацевтических сред, такие как,например, вода, гликоли, масла, спирты, ароматизаторы, консерванты, красители и т.п., в случае жидких препаратов для перорального введения, такие как, например, суспензии, эликсиры и растворы, или носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, смазывающие агенты, связующие, разрыхляющие агенты и т.п., и в случае твердых препаратов для перорального введения, таких как, например, порошки, твердые и мягкие капсулы и таблетки, при этом твердые препараты для перорального введения являются предпочтительными по отношению к жидким препаратам. Благодаря легкости введения таблетки и капсулы представляют собой наиболее предпочтительные пероральные единичные дозированные формы, в случае которых, очевидным образом, используют твердые фармацевтические носители. При необходимости на таблетки может быть нанесено покрытие посредством стандартных водных или неводных методов. Такие композиции и препараты должны содержать по меньшей мере 0,1% активного соединения. Процент активного соединения в этих композициях может, конечно, меняться и может соответственно находиться в пределах от примерно 2 до примерно 60% от массы единицы. Количество активного соединения в таких терапевтически подходящих композициях таково, чтобы обеспечить эффективную дозу. Активные соединения можно также вводить интраназально, например в виде жидких капель или спрея. Таблетки, пилюли, капсулы и т.п. могут содержать связующее, такое как трагакант, камедь, кукурузный крахмал или желатин; наполнители, такие как фосфат дикальция; разрыхляющий агент, такой как кукурузный крахмал, картофельный крахмал, альгиновая кислота; смазывающий агент, такой как стеарат магния; и подсластитель, такой как сахароза, лактоза или сахарин. Если единичная дозированная форма представляет собой капсулу, она может содержать, в дополнение к веществам указанного выше типа,жидкий носитель, такой как жирное масло. Различные другие материалы могут присутствовать в качестве покрытия или для изменения физической формы дозированной единицы. Например, таблетки могут быть покрыты шеллаком, сахаром или и шеллаком, и сахаром. Сироп или эликсир может содержать, в дополнение к активному ингредиенту,сахарозу в качестве подсластителя, метил- и пропилпарабены в качестве консервантов, красителей и вкусовую добавку, такую как вишневую или апельсиновую добавку. Поскольку соединения согласно настоящему изобретению в основном представляют собой карбоновые кислоты или аналогичные анионные изостеры и поскольку хорошо известно, что солевые формы ионных лекарственных соединений могут существенно повлиять на биодоступность лекарственных соединений, соединения согласно настоящему изобретению могут также быть применены в качестве солей в различных концентрациях, обеспечивая пероральную доступность состава. Такие фармацевтически приемлемые катионы могут быть выбраны из моно- или двухвалентных ионов, таких как аммоний, ионы щелочных металлов: натрия или калия, или щелочно-земельных металлов: магния или кальция, некоторых фармацевтически приемлемых аминов, таких как трис-(гидроксиметил)аминометан, этилендиамин,диэтиламин, пиперазин или других, или некоторых катионных аминокислот, таких как лизин или аргинин. Соединения согласно настоящему изобретению могут быть также введены парентерально. Растворы или суспензии этих активных соединений могут быть приготовлены в воде, соответствующим образом смешанной с поверхностно-активным веществом, таким как гидроксипропилцеллюлоза. Дисперсии могут быть также приготовлены в глицерине, жидких полиэтиленгликолях и их смесях в маслах. В обычных условиях хранения и применения, полученные препараты содержат консервант для предотвращения роста микроорганизмов. Фармацевтические формы, подходящие для применения для инъекций включают стерильные водные растворы или дисперсии и стерильные порошки для немедленного приготовления стерильных растворов или дисперсий для инъекций. Во всех случаях форма должна быть стерильной и должна быть жидкой в той степени, чтобы ее легко было набрать в шприц. Форма должна быть стабильной в условиях производства и хранения, а также должна быть защищена от загрязняющего действия микроорганизмов,таких как бактерии и грибы. Носитель может представлять собой растворитель или дисперсионную среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), подходящие смеси и растительные масла. Для обеспечения млекопитающих, особенно человека, эффективной дозой соединения согласно настоящему изобретению может быть использован любой подходящий способ введения. Например, могут быть использованы пероральные, ректальные, местные, парентеральные способы введения, введение в глаза, в легкие, в нос и т.п. Дозированные формы включают таблетки, пастилки, дисперсии, суспензии,растворы, капсулы, кремы, мази, аэрозоли и т.п. Предпочтительно соединения согласно настоящему изобретению вводят перорально Эффективная доза применяемого активного ингредиента может меняться в зависимости от конкретного используемого соединения, способа введения, состояния, подлежащего лечению, и тяжести состояния, подлежащего лечению. Такая доза может быть легко установлена специалистом в данной области. При лечении или предотвращении FXR-опосредованных состояний, для которых показаны соединения согласно настоящему изобретению, как правило, достигают удовлетворительных результатов, когда соединения согласно настоящему изобретению вводят в суточной дозе от примерно 0,1 до примерно 100 мг на 1 кг массы тела животного, предпочтительно в виде однократной дневной дозы или разделенных доз на от двух до шести приемов в день или в форме с замедленным высвобождением. Для большинства крупных млекопитающих общая суточная доза составляет от примерно 1,0 до примерно 1000 мг, предпочтительно от примерно 1 до примерно 50 мг. В случае 70 кг взрослого человека, общая суточная доза обычно составляет от примерно 7 до примерно 350 мг. Указанный режим дозирования может быть скорректирован, чтобы обеспечить оптимальный терапевтический эффект. Некоторые аббревиатуры, встречающиеся в настоящем описании:TBS - трет-бутилдиметилсилил. Соединения согласно настоящему изобретению могут быть получены в соответствии с методиками,представленными на следующих схемах и в примерах, с применением соответствующих веществ и далее проиллюстрированы следующими конкретными примерами. Кроме того, используя методики, описанные в в данном документе, в сочетании со знаниями среднего специалиста в данной области могут быть легко получены дополнительные соединения, заявленные в настоящем изобретении. Однако соединения,проиллюстрированные в примерах, не следует рассматривать как единственные соединения, заявленные в изобретении. Примеры дополнительно иллюстрируют подробное описание получения соединений согласно изобретению. Специалисты в данной области могут легко понять, что можно использовать известные изменения условий или процессов следующих препаративных методик для получения указанных соединений. Настоящие соединения, как правило, выделяют в виде их фармацевтически приемлемых солей, таких, как описано выше. Аминные свободные основания, соответствующие выделенным солям, могут быть получены путем нейтрализации с подходящим основанием, таким как водный гидрокарбонат натрия, карбонат натрия,гидроксид натрия и гидроксид калия и экстракцией выделенных аминных свободных оснований в органическом растворителе с последующим испарением. Аминное свободное основание, выделенное таким образом, может быть в дальнейшем превращено в другую фармацевтически приемлемую соль путем растворения в органическом растворителе с последующим добавлением соответствующей кислоты и последующим испарением, осаждением или кристаллизацией. Карбоновые свободные кислоты, соответствующие выделенным солям, могут быть получены путем нейтрализации с подходящей кислотой, такой как водная хлороводородная кислота, гидросульфат натрия, дигидрофосфат натрия, и экстракции выделенной карбоновой свободной кислоты, добычи освобожденных карбоксильных свободных кислот в органическом растворителе с последующим испарением. Карбоновые кислоты, выделенные таким образом,могут быть в дальнейшем превращены в другую фармацевтически приемлемую соль путем растворения в органическом растворителе с последующим добавлением соответствующего основания и последующим испарением, осаждением или кристаллизацией. Иллюстрация получения соединений согласно настоящему изобретению представлена ниже. Если иное не указано на схемах, переменные имеют те же значения, как описано выше. Примеры, представленные ниже, предназначены для иллюстрации конкретных вариантов реализации изобретения. Подходящие исходные вещества, строительные блоки и реагенты, применяемые в синтезе, как описано ниже,коммерчески доступны в Sigma-Aldrich Chemie GmbH, Мюнхен, Германия, Acros Organics, Бельгия или вFisher Scientific GmbH, 58239 Шверте, Германия, например, или могут быть в обычном порядке получены согласно методикам, описанным в литературе, например в "March's Advanced Organic Chemistry: Раствор 2,6-дихлорбензальдегида А 1 а (25 г, 0,14 моль) в 200 мл этанола добавляли к раствору гидрохлорида гидроксиламина (11 г, 0,16 моль) и гидроксида натрия (6,3 г, 0,16 моль) в 100 мл воды. Полученную смесь перемешивали при 90C в течение 24 ч. Объем уменьшали в вакууме примерно на 30 мл,что вызывало осаждение. Далее колбу охлаждали до комнатной температуры, а твердое вещество собирали путем фильтрования и промывали водой (2100 мл). Твердое вещество сушили под вакуумом с получением 25,9 г соединения А 2 а (белое твердое вещество, выход 96%). Синтез соединения А 3 а: В круглодонную колбу вместимостью 500 мл наливали раствор соединения А 2 а (25,9 г, 0,14 моль) в 300 мл ДМФА. Колбу помещали в водяную баню комнатной температуры. Далее в колбу помещали NCS(18,4 г, 0,14 моль). Реакционную смесь перемешивали еще в течение 1 ч, далее содержимое колбы приливали к 400 мл воды и экстрагировали продукт 500 мл Et2O. Органический слой промывали водой(2200 мл) и 100 мл солевого раствора, далее сушили над MgSO4. После фильтрования растворитель удаляли при пониженном давлении с получением 29 г соединения А 3 а в виде желтого масла, которое использовали в следующей реакции без дополнительной очистки. Синтез соединения А 4 а: Перемешанный раствор изобутирилацетата (16 г, 124,8 ммоль) в 120 мл сухого ТГФ обрабатывали раствором метоксида натрия (252 мл, 0,5 М в MeOH), а далее раствором соединения А 3 а (28 г,124,8 ммоль) в 40 мл сухого ТГФ. После перемешивания при комнатной температуре в течение 16 ч растворитель удаляли при пониженном давлении. Остаток распределяли между 800 мл Et2O и 800 мл воды. Органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали посредством хроматографии на силикагеле (элюент: РЕ/EtOAc = 10/1) с получением 24,8 г соединения А 4 а в виде белого твердого вещества (выход 62%). Раствор соединения А 4 а (24,8 г, 79,7 ммоль) в 180 мл сухого ТГФ охлаждали до 0C в атмосфере азота при добавлении по каплям LAH (3,1 г, 79,7 ммоль). Далее реакционной смеси медленно давали нагреться до комнатной температуры в течение 2 ч. Колбу снова охлаждали до 0C и аккуратно добавляли 5 мл MeOH в течение 10 мин. Добавляли насыщенный раствор Na2SO4 и полученное твердое вещество фильтровали через слой целита. Концентрировали с получением 21 г соединения А 5 а в виде желтого твердого вещества (выход 93%). Синтез соединения А 6 а: К раствору бензотриазола (6,77 г, 73,7 ммоль) в 100 мл сухого ДХМ добавляли SOCl2 (8,76 г,73,7 ммоль) при 0C и перемешивали при комнатной температуре в течение 1 ч. Полученную смесь добавляли к раствору соединения А 5 а (21 г, 73,7 ммоль) в 200 мл сухого ДХМ при комнатной температуре и перемешивали в течение 1,5 ч. К смеси добавляли 60 мл воды для того, чтобы погасить реакцию, и подвергали смесь экстракции с помощью ДХМ. Органический слой промывали 1 н. водным растворомNaOH, сушили над Na2SO4, фильтровали, концентрировали и очищали посредством хроматографии на силикагеле (элюент: РЕ/EtOAc = 10/1) с получением 21,7 г соединения А 6 а в виде белого твердого вещества (выход 97,2%). 1 Н ЯМР (400 МГц, CDCl3):1,47 (d, J=6,8 Гц, 6 Н), 3,35 (m, 1H), 4,31 (s, 2H), 7,38-7,48 (m, 3 Н). Синтез соединения А 4 е: К раствору соединения А 3 а (105 г, 0,47 моль) в 400 мл ТЭА добавляли этил-3-циклопропил-3 оксопропаноат (100 г, 0,70 моль). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении и проводили очистку посредством хроматографии на силикагеле (элюент: РЕ/ЕА = 10/1) с получением 87 г соединения А 4 е в виде белого твердого вещества К раствору соединения А 4 е (80 г, 0,26 моль) в 300 мл сухого ТГФ добавляли DIBAL-H (360 мл,0,54 моль) при 0C в атмосфере N2 и перемешивали при комнатной температуре в течение 4 ч. Реакцию гасили MeOH (80 мл) и HCl (1 М), проводили экстракцию с помощью ЕА и органический слой промывали солевым раствором. Проводили концентрирование при пониженном давлении с получением 55 г неочищенного соединения А 5 е, которое использовали в следующей реакции без дополнительной очистки. Синтез соединения А 6 е: К раствору бензотриазола (17,85 г, 194,3 ммоль) в 100 мл сухого ДХМ добавляли SOCl2 (23,09 г,73,7 ммоль) при 0C и перемешивали при комнатной температуре в течение 1 ч. Полученную смесь добавляли к раствору соединения А 5 е (55 г, 194,3 ммоль) в 500 мл сухого ДХМ при комнатной температуре и перемешивали в течение 1,5 ч. К смеси добавляли 120 мл воды, чтобы погасить реакцию, и подвергали смесь экстракции с помощью ДМФА. Органический слой промывали 1 н. водным раствором NaOH, сушили над Na2SO4, фильтровали, концентрировали и очищали посредством хроматографии на силикагеле(элюент: РЕ/ЕА = 10/1) с получением 47,4 г соединения А 6 е в виде белого твердого вещества (выход 81%). К раствору соединения А 1b (5,00 г, 28,4 ммоль) в 40 мл EtOH добавляли смесь NaOH (1,37 г,34,1 ммоль) и NH2OHHCl (2,37 г, 34,1 ммоль) в 15 мл Н 2 О. Полученную смесь перемешивали при 90C в течение 12 ч. Полученный объем концентрировали при пониженном давлении примерно на 10 мл и собирали твердое вещество путем фильтрования. Твердые вещества промывали водой и сушили под вакуумом с получением 5,0 г соединения А 2b в виде белого твердого вещества (выход 92%). Синтез соединения А 3b: В круглодонную колбу вместимостью 100 мл наливали раствор соединения А 2b (5,0 г, 26,2 ммоль) в 50 мл ДМФА. Колбу помещали в водяную баню комнатной температуры. Далее в колбу помещали NCS(4,13 г, 31,4 ммоль). Реакционную смесь перемешивали еще в течение 12 ч, далее содержимое концентрировали при пониженном давлении. Остаток разбавляли Et2O, промывали водой и солевым раствором,сушили над Na2SO4, фильтровали и концентрировали с получением 5,3 г соединения А 3b в виде желтого масла, которое использовали в следующей реакции без дополнительной очистки. Синтез соединения А 4b: Перемешанный раствор этил-3-циклопропил-3-оксопропаноата (3,88 г, 24,5 ммоль) в 40 мл сухого ТГФ обрабатывали 15 мл Et3N, а далее раствором соединения А 3b (5 г, 22,25 ммоль) в 30 мл сухого ТГФ. После перемешивания при комнатной температуре в течение 18 ч растворитель удаляли при пониженном давлении. Полученный остаток распределяли между 100 мл Et2O и 40 мл воды. Органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали посредством хроматографии на силикагеле (элюент: РЕ/EtOAc = 10/1) с получением 3,25 г соединения А 4b в виде белого твердого вещества (выход 45%). Синтез соединения А 5b: К раствору соединения А 4b (2,0 г, 6,10 ммоль) в 30 мл безводного ТГФ по каплям добавляли DIBAL-H (25,6 мл, 25,60 ммоль) при -10C в течение 15 мин в атмосфере N2. Полученную смесь перемешивали при той же температуре в течение 3 ч. Реакцию гасили водой, и проводили экстракцию с помощью ЕА, органический слой промывали солевым раствором, сушили над Na2SO4, Растворитель испаряли, получая неочищенный продукт, который далее очищали посредством флэш-хроматографии на силикагеле(элюент: РЕ/EtOAc = 2/1) с получением 0,44 г соединения А 5b в виде белого твердого вещества (выход 25%). Синтез соединения A6b: К раствору бензотриазола (0,84 г, 9,1 ммоль) в 20 мл сухого ДХМ по каплям добавляли SOCl2(1,08 мг, 9,1 ммоль) при 0C. Полученную смесь перемешивали в течение 30 мин при комнатной температуре в атмосфере N2. Полученный раствор переносили в капельную воронку и по каплям добавляли в течение 10 мин к перемешанному раствору соединения А 5b (2,0 г, 7,0 ммоль) в 20 мл сухого ДХМ. После перемешивания в течение 1 ч, полученную суспензию фильтровали с удалением гидрохлорида бензотриазола. Фильтрат последовательно промывали 30 мл воды дважды, 30 мл 1 н. раствора NaOH и 30 мл солевого раствора, сушили над безводным Na2SO4, фильтровали, концентрировали и очищали посредством хроматографии на силикагеле (элюент: РЕ/EtOAc = 10/1) с получением 0,91 г соединения А 6b в виде белого твердого вещества (выход 42,3%). 1 Н ЯМР (400 МГц, CDCl3) : 1,21-1,35 (m, 4H), 2,14-2,18 (m, 1 Н), 4,38 (s, 2H), 8,67 (s, 2H). Синтез соединения А 6 с: К перемешанному раствору А 6b (0,53 г, 1,75 ммоль) в 10 мл ДХМ добавляли т-СРВА (0,65 мг,3,60 ммоль) при комнатной температуре. После перемешивания в течение 18 ч при комнатной температуре реакцию гасили насыщенным раствором NaHCO3, проводили экстракцию с помощью ДХМ, промывали водой и солевым раствором, сушили над безводным Na2SO4, фильтровали, концентрировали и очищали посредством флэш-хроматографии на силикагеле (элюент: РЕ/EtOAc = 5/1) с получением 318 мг соединения А 6 с в виде белого твердого вещества (выход 57%). 1 Н ЯМР (400 МГц, CDCl3) : 1,23-1,33 (m, 4H), 2,14 (m, 1 Н), 4,40 (s, 2H), 8,31 (s, 2H). Схема 1 с Раствор 2,6-дихлорфенилазида (F, 25 г, 0,13 моль) в толуоле (100 мл) и ацетиленовом спирте (G,52,1 г, 0,53 моль) кипятили с обратным холодильником в атмосфере аргона в течение 35 ч. Толуол удаляли под вакуумом и полученные продукты очищали посредством тщательной колоночной хроматографии и выделяли два триазоловых продукта в виде твердых веществ, соединения A5d (4,5 г, 23%) и соединения A5x (6,5 г, 34%). Получение соединения A6d: К раствору соединения A5d (2,00 г, 7,0 ммоль) в 20 мл ДХМ и 2 мл CCl4 добавляли PPh3 (3,67 г,14,0 ммоль). Далее раствор перемешивали при комнатной температуре в течение 4 ч, данные как ТСХ,так и ЖХ/МС показали, что реакция завершилась. Концентрировали при пониженном давлении и очищали посредством флэш-хроматографии на силикагеле (элюент: РЕ/EtOAc = 10/1) с получением 2,02 г соединения A6d в виде белого твердого вещества (выход 95%). 1 Н ЯМР (400 МГц, CDCl3):1,51-1,49 (d, 6H), 3,24-3,21 (m, 1H), 4,48 (s, 2H), 7,52-7,50 (t, 1 Н), 7,587,54 (d,2H). Схема 2d Раствор 2-бром-1,3-дихлорбензола (13 г, 58 ммоль), 2,2-диметил-2-силабут-3-ин (90 мл, 64 ммоль),CuI (100 мг, 5,5 ммоль), PPh3 (200 мг, 0,75 ммоль) и Pd(PPh3)2Cl2 (235 мг, 0,335 ммоль) в 35 мл NEt3 кипятили в запаянной трубке в атмосфере N2 в течение 24 ч. Данные как ТСХ, так и ЖХ/МС показали, что реакция завершилась. Реакционную смесь концентрировали и добавляли EtOAc. Раствор промывали водой и солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали посредством флэш-хроматографии на силикагеле (элюент: РЕ/ЕА = 100/0) с получением 13 г неочищенного соединения A1f в виде масла (выход 40%, чистота 70%). К раствору неочищенного соединения A1f (13 г, 48,4 ммоль) в 200 мл MeOH добавляли K2CO3 (13 г,94,3 ммоль) и перемешивали смесь при комнатной температуре в течение ночи в атмосфере N2. Реакционную смесь фильтровали и концентрировали при пониженном давлении с получением 4,1 г соединенияA2f в виде белого твердого вещества (выход 56,1%). Синтез соединения A3f: К раствору соединения A2f (2 г, 12 ммоль) в 40 мл сухого ТГФ добавляли n-BuLi (5,6 мл, 2,5 М в гексане, 14 ммоль) при -78C в атмосфере N2 и перемешивали раствор при указанной температуре в течение 30 мин. Далее добавляли раствор этил-хлорацетата (1,65 г, 15 ммоль) в 10 мл сухого ТГФ при-78C. Смесь перемешивали в течение 4 ч. Полученный раствор приливали к насыщенному растворуNH4Cl раствор и добавляли EtOAc для экстракции дважды. Объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали при пониженном давлении и очищали посредством флэшхроматографии на силикагеле (элюент: РЕ/ЕА = 50/1) с получением 1,25 г соединения A3f в виде желтого твердого вещества (выход 44,5%). Синтез соединения A4f: Раствор соединения A3f (3 г, 12,4 ммоль) и 2-азидопропан (20 мл) нагревали при 110C в течение 24 ч в автоклаве. Концентрировали и очищали посредством флэш-хроматографии на силикагеле (элюент: РЕ/ЕА = 4/1) с получением 1 г соединения A4f в виде желтого твердого вещества (выход 34,8%). Синтез соединения A5f: К ледяному холодному раствору соединения A4f (1 г, 1,38 ммоль) в 10 мл безводного ТГФ по каплям добавляли LAH (2 M в ТГФ, 2,76 ммоль). После добавления реакционный раствор перемешивали при указанной температуре в течение 2 ч. Данные ТСХ показали, что реакция завершилась. Медленно добавляли 10 мл MeOH, чтобы погасить реакцию, с последующим добавлением насыщенного раствораNa2SO4. Полученное твердое вещество отфильтровывали, а раствор концентрировали при пониженном давлении и очищали посредством флэш-хроматографии на силикагеле (элюент: РЕ/ЕА = 1/4) с получением 180 мг соединения A5f в виде желтого твердого вещества (выход 45,7%). 1 Н ЯМР (400 МГц, CDCl3) : 7,41-7,43 (m, 2H), 7,30-7,32 (m, 1H), 4,91-4,97 (m, 1H), 4,58-4,59 (d,J=5,6 Гц, 2 Н), 2,70-2,73 (br, 1H), 1,72 (s, 3 Н), 1,70 (s, 3 Н). Синтез соединения A6f: Раствор соединения A5f (200 мг, 0,70 ммоль), CCl4 (1 мл, 10,4 ммоль) и PPh3 (368 мг, 1,40 ммоль) в 5 мл безводного ДХМ перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь кон- 24024083 центрировали при пониженном давлении и очищали посредством флэш-хроматографии на силикагеле(элюент: РЕ/ЕА = 3/1) с получением 100 мг соединения A6f в виде желтого масла (выход 47,1%). 1 Н ЯМР (400 МГц, CDCl3) : 7,45-7,47 (m, 2H), 7,36-7,39 (m, 1H), 4,81-4,85 (m, 1H), 4,51 (s, 1 Н), 1,78SOCl2 (113,3 г, 0,96 моль) медленно добавляли к 700 мл сухого метанола при 0C. После перемешивания в течение 1 ч при комнатной температуре, добавляли соединение В 1 (80 г, 0,48 моль) и перемешивали в течение 1,5 ч. Смесь концентрировали и добавляли водный раствор NaHCO3, чтобы погасить реакцию. Суспензию дважды подвергали экстракции с помощью ДХМ. Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали с получением 85 г соединения В 2, которое использовали в следующей реакции без дополнительной очистки. Синтез соединения В 3: К раствору соединения В 2 (60 г, 0,31 моль) в 500 мл МеОН добавляли раствор NaOH (12,4 г,0,31 моль) в 200 мл МеОН. Смесь перемешивали в течение ночи при комнатной температуре. Смесь концентрировали, а остаток растворяли в 500 мл воды и экстрагировали с помощью Et2O. Водный раствор подкисляли концентрированным раствором HCl до pH 2, образовавшийся белый осадок собирали и сушили под вакуумом с получением 46 г неочищенного соединения В 3 в виде белого твердого вещества. Синтез соединения В 4: К раствору соединения В 3 (46 г, 0,3 моль) в 700 мл сухого ТГФ добавляли раствор ВН 3 в ТГФ (1 М,600 мл, 0,60 моль) при 0C в атмосфере N2 в течение 20 мин. Далее раствор перемешивали в течение ночи при комнатной температуре. Для гашения реакции медленно добавляли 50% водный раствор уксусной кислоты (400 мл). Реакционную смесь концентрировали и далее распределяли между EtOAc и водой. Органическую фазу последовательно промывали 10% водным раствором Na2CO3, H2O и солевым раствором. Полученную смесь сушили над Na2SO4, фильтровали, концентрировали при пониженном давлении и очищали посредством хроматографии на силикагеле (элюент: РЕ/EtOAc = 10/1) с получением 28 г соединения В 4 в виде белого твердого вещества (выход за 3 стадии: 54%). Синтез соединения В 5: К раствору соединения В 4 (28 г, 0,168 моль) в 700 мл Et2O по каплям добавляли PBr3 (17,4 мл,0,185 моль). Раствор перемешивали в течение 2 ч и далее реакционную смесь приливали к 500 мл ледяной воды. Водный слой экстрагировали с помощью Et2O. Объединенные органические слои последовательно промывали насыщенным раствором NaHCO3, водой, и солевым раствором. Органическую фазу сушили над Na2SO4, фильтровали и концентрировали при пониженном давлении с получением 34 г соединения В 5 в виде масла (выход 70%). Синтез соединения В 6: Смесь соединения В 5 (34 г, 148 ммоль) в 34 мл триэтоксифосфина нагревали при 175C в течение 4 ч. Смесь концентрировали при пониженном давлении с получением 40 г соединения В 6, которое использовали в следующей реакции без дополнительной очистки. Синтез соединения В 8: К раствору соединения В 7 (150 г, 1,17 моль), гидроксида кальция (375 г, 5,07 моль) и карбоната натрия (420 г, 3,96 моль) в 1,5 л воды добавляли хлороформ (270 г, 2,29 моль) в течение 80 мин и кипятили реакционную смесь в атмосфере N2 в течение 3 ч. Реакционную смесь охлаждали в ледяной бане. Добавляли 1 л концентрированного водного раствора HCl и 1 л хлороформа. Смесь перемешивали и после разделения фаз удаляли водный слой. Органический слой сушили над Na2SO4, концентрировали и очищали посредством хроматографии на силикагеле (элюент: РЕ/EtOAc = 10/1) с получением 28 г соединения В 8 в виде белого твердого вещества (выход 15,3%). Синтез соединения В 9: К раствору соединения В 8 (11 г, 70,1 ммоль) в 300 мл сухого ДХМ добавляли TBDMSCI (12,7 г,84,2 ммоль), ТЭА (19,6 мл, 140,2 ммоль) и ДМАР (100 мг, кат.). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Смесь концентрировали при пониженном давлении с получением 18,5 г неочищенного соединения В 9, которое использовали в следующей реакции без дополнительной очистки. Синтез соединения В 10: К раствору соединения В 9 (23 г, 80,6 ммоль) в 320 мл сухого ТГФ добавляли гидрид натрия (60% в минеральном масле, 5 г, 123 ммоль) при 0C в течение 30 мин. К полученной смеси добавляли раствор соединения В 6 (18,5 г, 61,5 ммоль) в 160 мл сухого ТГФ при 0C, и перемешивали раствор при комнатной температуре в течение 3 ч. Реакцию гасили насыщенным раствором NH4Cl и проводили экстракцию с помощью EtOAc. Органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали с получением 23 г соединения В 10, которое использовали в следующей реакции без дополнительной очистки. К раствору соединения В 10 (23 г, 80,6 ммоль) в 450 мл сухого ДХМ добавляли TBDMSCI (12,7 г,84,2 ммоль), ТЭА (19,6 мл, 161,2 ммоль) и ДМАР (0,5 г, кат.). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Смесь концентрировали при пониженном давлении и очищали посредством хроматографии на силикагеле (элюент: РЕ/EtOAc = 30/1) с получением 11,8 г соединения В 11 в виде белого твердого вещества (выход за 3 стадии: 41,8%). Синтез соединения В 12: К раствору диэтилцинка (290мл, 1 М в гексане, 290 ммоль) в 600 мл сухого ДХМ добавляли дийодэтан (46 мл, 580 ммоль) при -78C. Раствор перемешивали в течение 30 мин и далее смесь нагревали до-30C. К смеси добавляли ТФУ (38 г, 290 ммоль) и перемешивали в течение 30 мин. К смеси добавляли раствор соединения В 11 (11,8 г, 29 ммоль) в 200 мл ДХМ и смесь перемешивали в течение ночи при комнатной температуре. Реакционную смесь гасили 500 мл 1 н. водного раствора HCl. Органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали досуха и очищали посредством флэш-хроматографии на силикагеле (элюент: РЕ/EtOAc = 20/1) с получением 4,5 г соединения В 12 в виде масла (выход 36,9%). Синтез соединения В 13: Раствор соединения В 12 (4,5 г, 10,8 ммоль) и TBAFH2O (11 г, 42,1 ммоль) в 300 мл сухого ТГФ перемешивали в течение 10 мин при комнатной температуре. Добавляли воду (100 мл) и проводили экстракцию с помощью 500 мл EtOAc. Органический слой промывали солевым раствором, сушили надNa2SO4, концентрировали при пониженном давлении и очищали посредством флэш-хроматографии на силикагеле (элюент: РЕ/EtOAc = 10/1) с получением 2,0 г неочищенного соединения В 13 с чистотой 80% К раствору неочищенного соединения В 13 (200 мг, 0,66 ммоль) и соединения А 6 а (200 мг,0,66 ммоль) в 5 мл ДМФА добавляли K2CO3 (184 мг, 1,33 ммоль). Смесь нагревали в течение ночи при 60C, далее охлаждали до комнатной температуры, разбавляли водой (10 мл) и экстрагировали 30 млEtOAc. Органический слой дважды промывали солевым раствором, сушили над Na2SO4, фильтровали,концентрировали и очищали посредством флэш-хроматографии на силикагеле (элюент: РЕ/EtOAc = 20/1) с получением 220 мг соединения В 14 а в виде белого твердого вещества (выход 59,8%). Синтез соединения В 14b: К раствору неочищенного соединения В 13 (200 мг, 0,66 ммоль) и соединения А 6b (200 мг,0,66 ммоль) в 5 мл ДМФА добавляли K2CO3 (184 мг, 1,33 ммоль). Смесь нагревали в течение ночи при 60C, далее охлаждали до комнатной температуры, разбавляли водой (10 мл) и проводили экстракцию с помощью 30 мл EtOAc. Органический слой дважды промывали солевым раствором, сушили над Na2SO4,фильтровали, концентрировали и очищали посредством хроматографии на силикагеле (элюент: РЕ/EtOAc = 20/1) с получением 100 мг соединения В 14b в виде белого твердого вещества (выход 27,2%). К раствору неочищенного соединения В 13 (200 мг, 0,66 ммоль) и соединения А 6 с (211 мг,0,66 ммоль) в 5 мл ДМФА добавляли K2CO3 (184 мг, 1,33 ммоль). Смесь нагревали в течение ночи при 60C, далее охлаждали до комнатной температуры, разбавляли водой (10 мл) и проводили экстракцию с помощью 30 мл EtOAc. Органический слой дважды промывали солевым раствором, сушили над Na2SO4,фильтровали, концентрировали и очищали посредством флэш-хроматографии на силикагеле (элюент: РЕ/ЕА = 8/1) с получением 140 мг соединения В 14 с в виде белого твердого вещества (выход 38,1%). Синтез соединения B14d: К раствору неочищенного соединения В 13 (200 мг, 0,66 ммоль) и соединения A6d (200 мг,0,66 ммоль) в 5 мл ДМФА добавляли K2CO3 (184 мг, 1,33 ммоль). Смесь нагревали в течение ночи при 60C, далее охлаждали до комнатной температуры, разбавляли водой (10 мл) и проводили экстракцию с помощью 30 мл EtOAc. Органический слой дважды промывали солевым раствором, сушили над Na2SO4,фильтровали, концентрировали и очищали посредством флэш-хроматографии на силикагеле (элюент: РЕ/ЕА = 10/1) с получением 103 мг соединения B14d в виде белого твердого вещества (выход 28%). Синтез соединения В 14 е: К раствору неочищенного соединения В 13 (1 г, 3,3 ммоль, 1,00 экв.) и А 6 е (1 г, 3,3 ммоль, 1 экв.) в 30 мл ДМФА добавляли K2CO3 (1,4 г, 9,9 ммоль, 3,00 экв.). Смесь нагревали в течение ночи при 60C,далее охлаждали до комнатной температуры, разбавляли водой (50 мл) и проводили экстракцию с помощью Et2O (200 мл). Органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали посредством флэш-хроматографии на силикагеле (элюент: РЕ/EtOAc = 20/1) с получением 1,2 г соединения В 14 е, которое использовали в следующей реакции без дополнительной очистки. Синтез соединения согласно примеру 1:(74 мг, 1,8 ммоль) и далее смесь перемешивали при комнатной температуре в течение 24 ч. Смесь кон- 28024083 центрировали и разбавляли 10 мл Н 2 О, добавляли 1 н. водный раствор HCl для подкисления смеси до pH 5. Смесь подвергали экстракции с помощью EtOAc и органический слой промывали солевым раствором,сушили над Na2SO4, фильтровали, концентрировали, а остаток очищали посредством хроматографии на силикагеле (элюент: РЕ/EtOAc= 3/1) с получением 30 мг соединения согласно примеру 1 (3-(2-(2-хлор-45-циклопропил-3-(3,5-дихлорпиридин-4-ил)изоксазол-4-ил)метокси)фенил)циклопропил)бензойной кислоты) в виде белого твердого вещества (выход 30,8%). 1 Н ЯМР (400 МГц, CDCl3) : 1,23 (m, 2H), 1,31 (m, 2H), 1,47 (m, 2H), 2,09 (m, 1H), 2,17 (m, 1 Н), 2,39(m, 2 Н), 7,97 (m, 2 Н), 8,64 (s, 2H). ЖХ/МС (подвижная фаза: 60-95% ацетонитрил-вода-0,01% ТФУ): чистота 95%, Rt = 3,304 мин; МС выч.: 554; МС найд.: 555 (М+Н)+. Синтез соединения согласно примеру 2:(100 мг, 2,4 ммоль) и далее смесь перемешивали при комнатной температуре в течение 24 ч. Смесь концентрировали и разбавляли 10 мл Н 2 О, добавляли 1 н. водный раствор HCl для подкисления смеси до pH 5. Смесь подвергали экстракции с помощью EtOAc и органический слой промывали солевым раствором,сушили над Na2SO4, фильтровали, концентрировали и очищали посредством хроматографии на силикагеле (элюент: РЕ/EtOAc = 1/1) с получением 40 мг соединения согласно примеру 2 (4-(4-4-(2-(3 карбоксифенил)циклопропил)-3-хлорфенокси)метил)-5-циклопропилизоксазол-3-ил)-3,5 дихлорпиридин-1-оксида) в виде белого твердого вещества (выход 29,3%). 1 Н ЯМР (400 МГц, CDCl3) : 1,21-1,33 (m, 4H), 1,44-1,50 (m, 2H), 2,10-2,19 (m, 2H), 2,39 (m, 1 Н),4,85 (s, 2 Н), 6,67 (dd, J=2,4 Гц, 8,4 Гц, 1 Н), 6,88 (d, J=2,4 Гц, 1 Н), 7,02 (d, J=8,4 Гц, 1 Н), 7,40-7,48 (m, 2 Н),7,95 (m, 2 Н), 8,31 (s, 2H). ЖХ/МС (подвижная фаза: 40-95% ацетонитрил-вода-0,01% ТФУ): чистота 95%, Rt = 3,421 мин; МС выч.: 570; МС найд.: 571 (М+Н)+. Синтез соединения согласно примеру 3:(76 мг, 1,8 ммоль) и далее смесь перемешивали при комнатной температуре в течение 24 ч. Смесь концентрировали и разбавляли 10 мл Н 2 О, добавляли 1 н. водного раствора HCl для подкисления смеси доpH 5. Смесь подвергали экстракции с помощью EtOAc и органический слой промывали солевым раствором, сушили над Na2SO4, фильтровали, концентрировали и очищали посредством хроматографии на силикагеле (элюент: РЕ/EtOAc = 2/1) с получением 80 мг соединения согласно примеру 3(3-(2-(2-хлор-4-1-(2,6-дихлорфенил)-4-изопропил-1 Н-1,2,3-триазол-5-ил)метокси)фенил)циклопропил)бензойной кислоты) в виде белого твердого вещества (выход 79,6%). 1 Н ЯМР (400 МГц, CDCl3) : 1,45 (m, 8H), 2,08 (m, 1H), 2,38 (m, 1H), 3,24 (m, 1H), 4,92 (s, 2 Н), 6,67
МПК / Метки
МПК: C07D 261/08, A61K 31/4439, C07D 413/12, C07D 413/04, A61P 1/16, A61K 31/4192, A61K 31/42, A61P 35/00, C07D 249/06, A61P 3/10, A61K 31/443, C07D 413/14
Метки: новые, модулирующие, fxr, связывающие, х-рецептор, фарнезоидный, соединения, nr1h4, активность
Код ссылки
<a href="https://eas.patents.su/30-24083-novye-soedineniya-svyazyvayushhie-farnezoidnyjj-h-receptor-fxr-nr1h4-i-moduliruyushhie-ego-aktivnost.html" rel="bookmark" title="База патентов Евразийского Союза">Новые соединения, связывающие фарнезоидный х-рецептор (fxr) (nr1h4) и модулирующие его активность</a>
Димерные тромбопоэтиновые пептидные миметики, связывающие мpl рецептор и имеющие тромбопоэтичеcкую активность

Номер патента: 3998
Опубликовано: 25.12.2003
Авторы: Фейдже Улрих, Читхэм Дженет, Лиу Чуан-Фа
МПК: C07K 14/52, A61P 7/06, A61K 38/19...
Метки: имеющие, активность, пептидные, рецептор, тромбопоэтичеcкую, связывающие, тромбопоэтиновые, миметики, димерные
Формула / Реферат:
1. Соединение, которое связывается с рецептором mpl, имеющее структуру TMP1(L1)n-TMP2, где TMP1 и TMP2, каждый независимо друг от друга, выбран из группы коровых соединений, имеющих структуру X2 - X3 - X4 - X5 - X6 - X7 - X8 - X9 - X10, причем X2 выбран из группы, включающей Glu, Asp, Lys и Val; X3 выбран из группы, включающей Gly и Ala; X4 представляет собой Pro; X5 выбран из группы, включающей Thr и Ser; X6 выбран из группы, включающей...
Соединения, модулирующие активность ppar&gamma

Номер патента: 4887
Опубликовано: 26.08.2004
Авторы: Хагивара Ацуси, Рубенштейн Стивен М., Хауз Джонатан Б., Фурукава Нобору, Макджи Лоренс Р., Синкаи Хисаси
МПК: C07C 311/21, C07D 213/65, A61K 31/18...
Метки: ppar&gamma, соединения, модулирующие, активность
Формула / Реферат:
1. Соединение формулы где Ar1 представляет собой незамещенный 2-бензотиазолил или замещенный 2-бензотиазолил, имеющий от 1 до 3 заместителей, независимо выбранных из группы, включающей галоген, -OR', -OC(O)R', -NR'R", -SR', -R', -CN, -NO2-, -CO2R', -CONR'R", -C(O)R', -NR"C(O)R', -S(O2)R', -S(O)2NR'R", перфтор(C1-C4)алкокси и перфтор(C1-C4)алкил, где R' и R" являются независимо выбранными из группы, включающей водород,...
Соединения, модулирующие активность ppar, и способы их получения

Номер патента: 9518
Опубликовано: 28.02.2008
Авторы: Браттон Лэрри Дон, Филзен Гари Фредерик, Ауэрбах Брюс Джеффри, Аненгст Пол Чарльз, Триведи Бхарат Калидас, Гейер Эндрю Джордж
МПК: C07C 323/19, C07C 317/22, A61K 31/19...
Метки: активность, получения, модулирующие, ppar, соединения, способы
Формула / Реферат:
1. Соединение, выбранное из [4-(бифенил-4-илметилсульфанил)-5-метокси-2-метилфенокси]уксусной кислоты; [5-метокси-2-метил-4-(4'-трифторметилбифенил-4-илметилсульфанил)фенокси]уксусной кислоты; [4-(2',4'-дихлорбифенил-4-илметилсульфанил)-5-метокси-2-метилфенокси]уксусной кислоты; [5-метокси-2-метил-4-(3'-трифторметилбифенил-4-илметилсульфанил)фенокси]уксусной кислоты; [4-(4'-фторбифенил-4-илметилсульфанил-5-метокси-2-метилфенокси]уксусной...
Соединения, модулирующие активность toll-подобных рецепторов

Номер патента: 19768
Опубликовано: 30.06.2014
Авторы: Хайяши Томоко, Чэнь Майкл, Карсон Деннис А., Коттэм Ховард Б.
МПК: C07D 473/02, A61P 29/00, A61K 31/52...
Метки: модулирующие, активность, рецепторов, toll-подобных, соединения
Формула / Реферат:
1. Соединение, имеющее структуру согласно формуле Iили его фармацевтически приемлемая соль, включая его гидрат,где X представляет собой N или CR2;R представляет собой -OR1, -SR1 или -NRaRb,X1 представляет собой связь или -О-, -S- или -NRc-;Rc представляет собой водород, C1-C10-алкил или замещенный C1-C10-алкил или Rc и R1 вместе с атомом азота могут образовывать гетероциклическое кольцо или замещенное гетероциклическое кольцо;каждый R1...
Соединения, модулирующие активность каннабиноидного рецептора св1, композиции на их основе и их применение

Номер патента: 18900
Опубликовано: 29.11.2013
Авторы: Мюррей Энтони, Альмхольт Дортэ, Хёгберг Томас, Линже Жан-Мишель, Ресевёр Жан-Мари, Ноэрегор Пиа Карина, Бьюрлинг Эмели, Нильсен Петер Одаль
МПК: A61K 31/4155, A61K 31/415, A61P 3/04...
Метки: композиции, соединения, основе, применение, св1, модулирующие, рецептора, активность, каннабиноидного
Формула / Реферат:
1. Соединение формулы (I) или его соль, гидрат, сольват или N-окисьгде X означает связь или -СН2-;Z обозначает радикал, выбранный из группы, состоящей из следующих радикалов формул (1)-(26):где R обозначает (C1-С6)алкил, фенил или бензил, возможно замещенный R5 в кольце; илимоноциклическое неароматическое карбоциклическое кольцо, содержащее 3-6 атомов в кольце;R3 обозначает водород, (C1-С3)алкил;R4 обозначает радикал формулы...
Предыдущий патент: Спортивное покрытие, использующее структурные модули
Следующий патент: Вводимые перорально кортикостероидные композиции
Случайный патент: Синтетические молекулярные конструкции