Замещенные соединения [(5h-пирроло[2,1-c][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2-диметилпропановой кислоты в качестве обратных агонистов h1/антагонистов 5-ht2a двойного действия
Номер патента: 22708
Опубликовано: 29.02.2016
Авторы: Сандерсон Адам Ян, Камп Анна Мари, Коатес Дэвид Эндрю, Галлахер Питер Таддеус, Ледгард Эндрю Джеймс























Формула / Реферат
1. Соединение формулы

где R1 представляет собой хлор или метил;
R2 представляет собой метил, этил, изопропил, хлор, бром, трифторметил или метилтио; и
R3 представляет собой водород или метокси;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, отличающееся тем, что R3 представляет собой водород, или его фармацевтически приемлемая соль.
3. Соединение по п.1 или 2, отличающееся тем, что R1 представляет собой хлор, или его фармацевтически приемлемая соль.
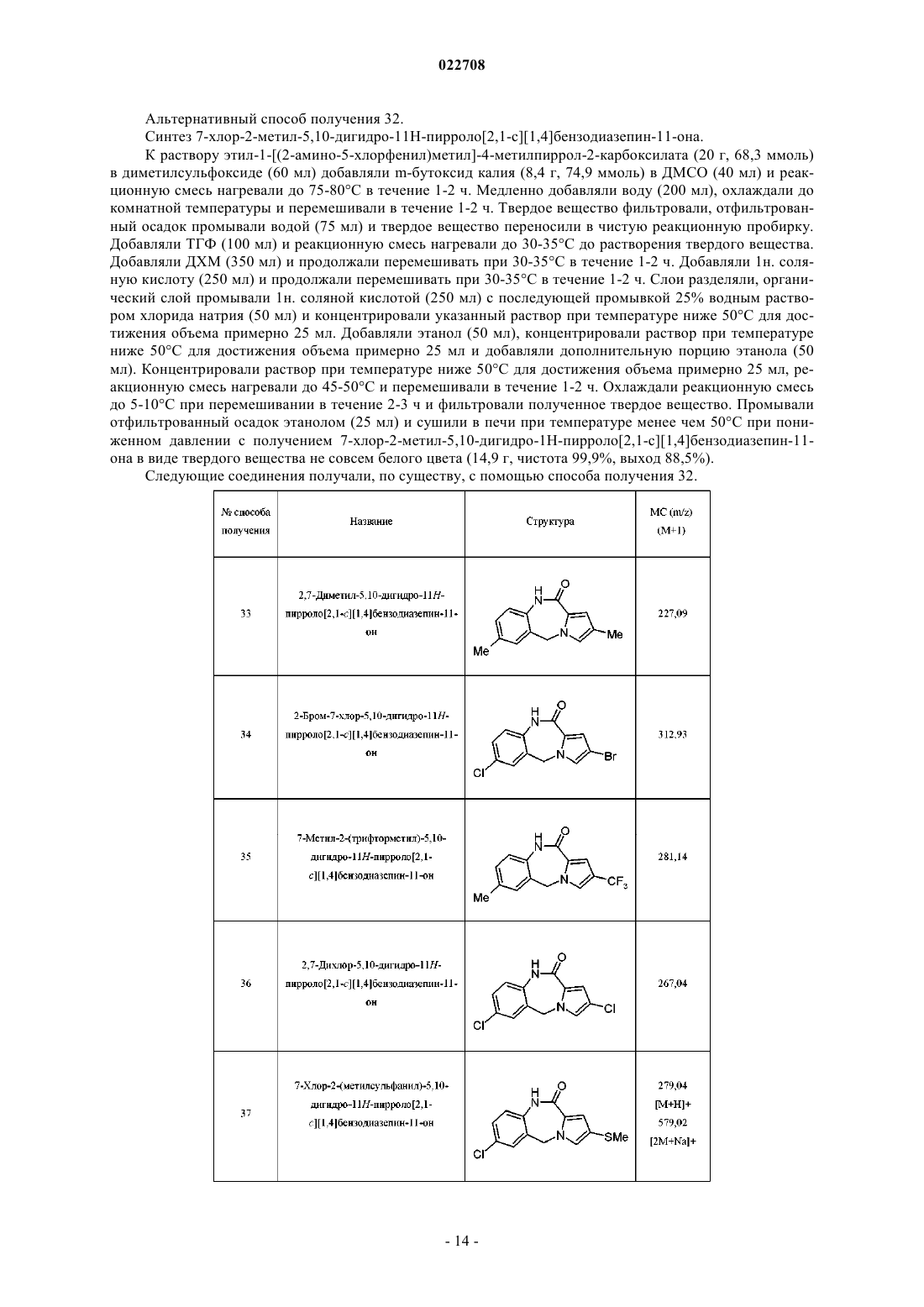
4. Соединение по п.1, представляющее собой 3-[4-(7-хлор-2-метил-5H-пирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2-диметилпропановую кислоту или ее фармацевтически приемлемую соль.
5. Соединение по п.1, представляющее собой дигидрохлорид 3-[4-(7-хлор-2-метил-5H-пирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2-диметилпропановой кислоты.
6. Фармацевтическая композиция для лечения расстройств сна, содержащая соединение по любому из пп.1-5 или его фармацевтически приемлемую соль в комбинации по меньшей мере с одним фармацевтически приемлемым носителем, разбавителем или наполнителем.
7. Применение соединения по любому из пп.1-5 или его фармацевтически приемлемой соли для лечения бессонницы.
8. Применение соединения по любому из пп.1-5 или его фармацевтически приемлемой соли для лечения бессонницы, где указанная бессонница характеризуется затруднениями при засыпании или поддержании сна или затруднениями как при засыпании, так и при поддержании сна.
9. Применение по любому из пп.7 или 8 для лечения бессонницы у человека.
10. Фармацевтическая композиция для лечения расстройств сна, содержащая соединение по любому из пп.1-5 или его фармацевтически приемлемую соль в комбинации по меньшей мере с одним фармацевтически приемлемым носителем, разбавителем или наполнителем и селективный ингибитор обратного захвата серотонина.
Текст
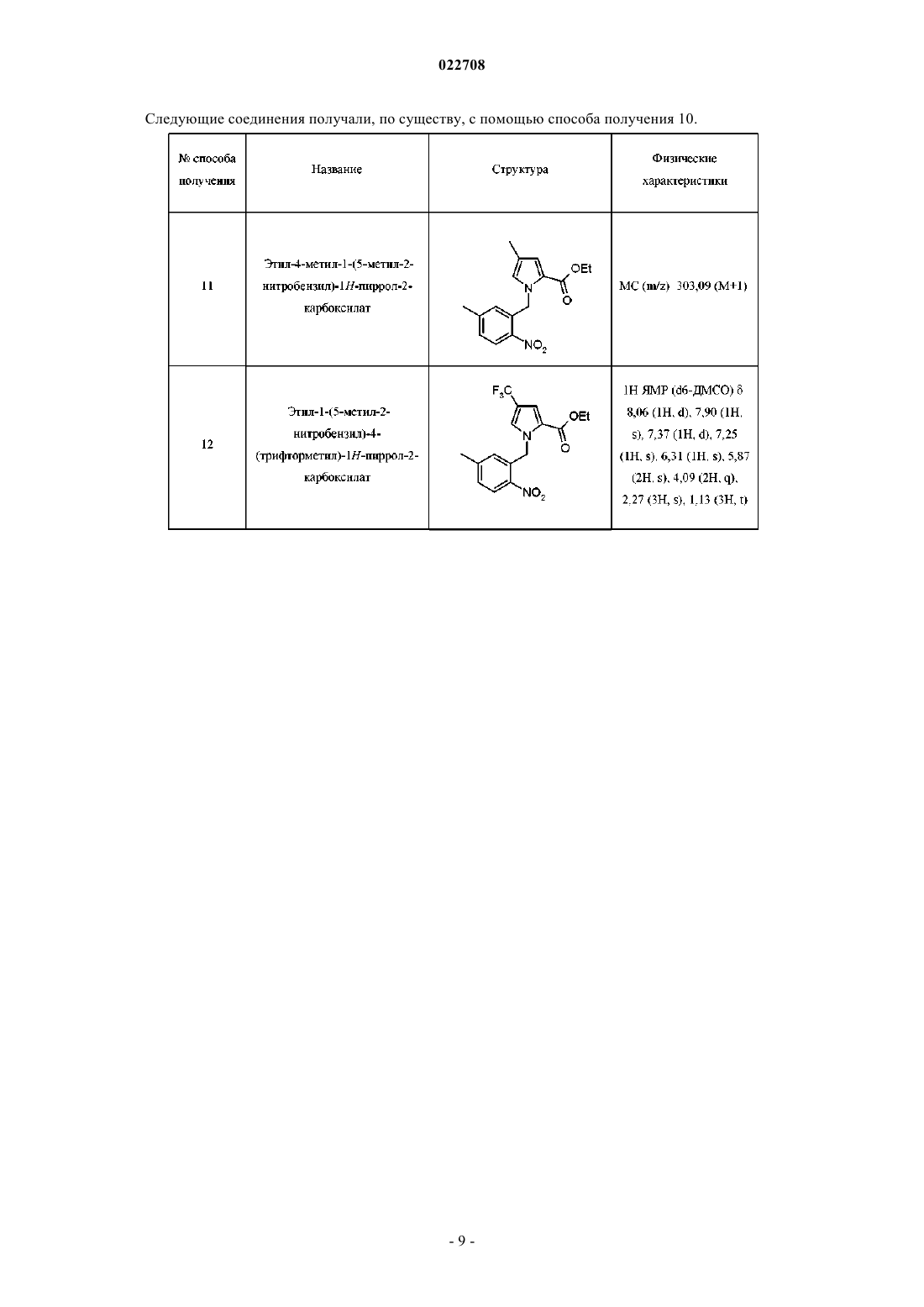
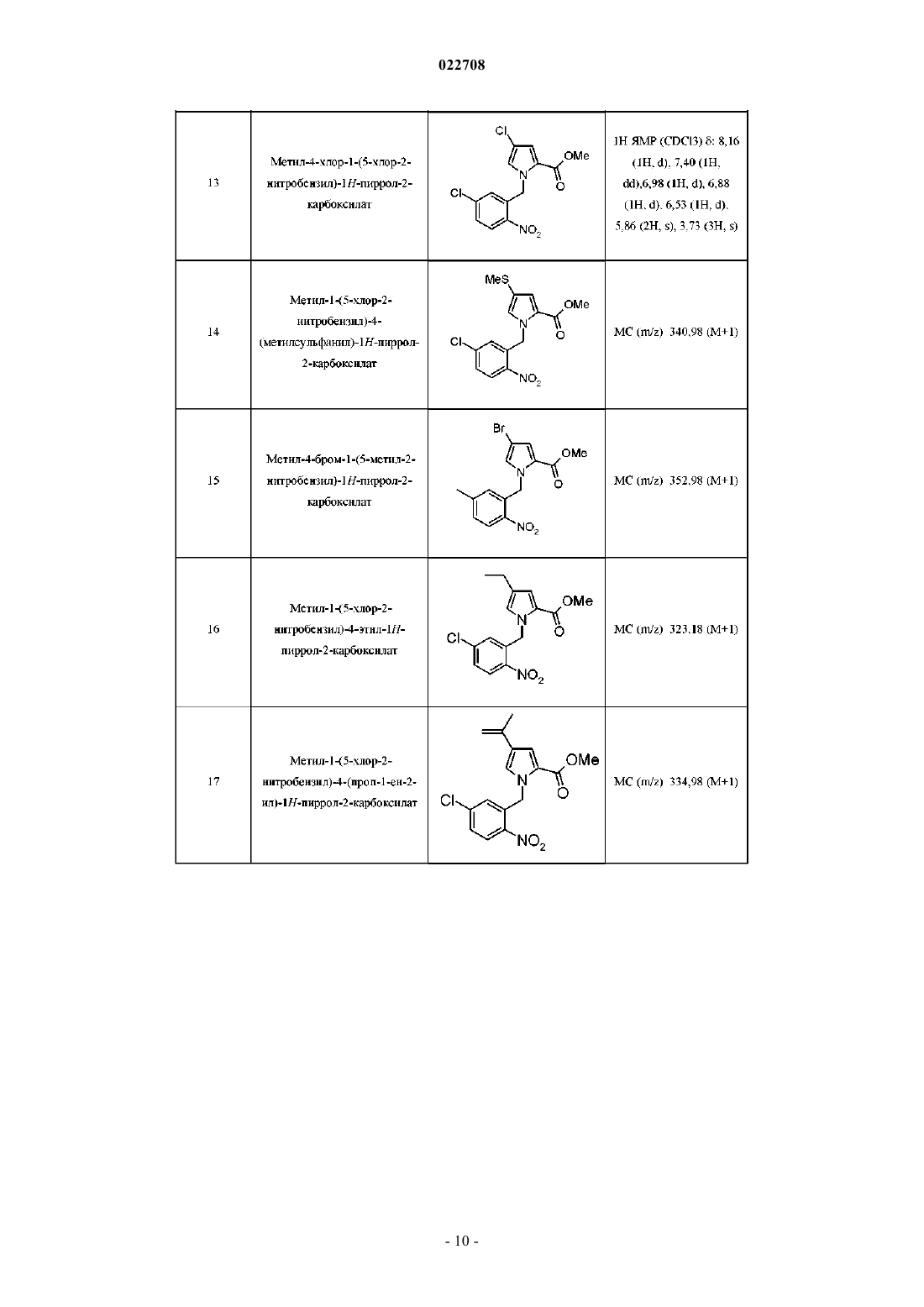
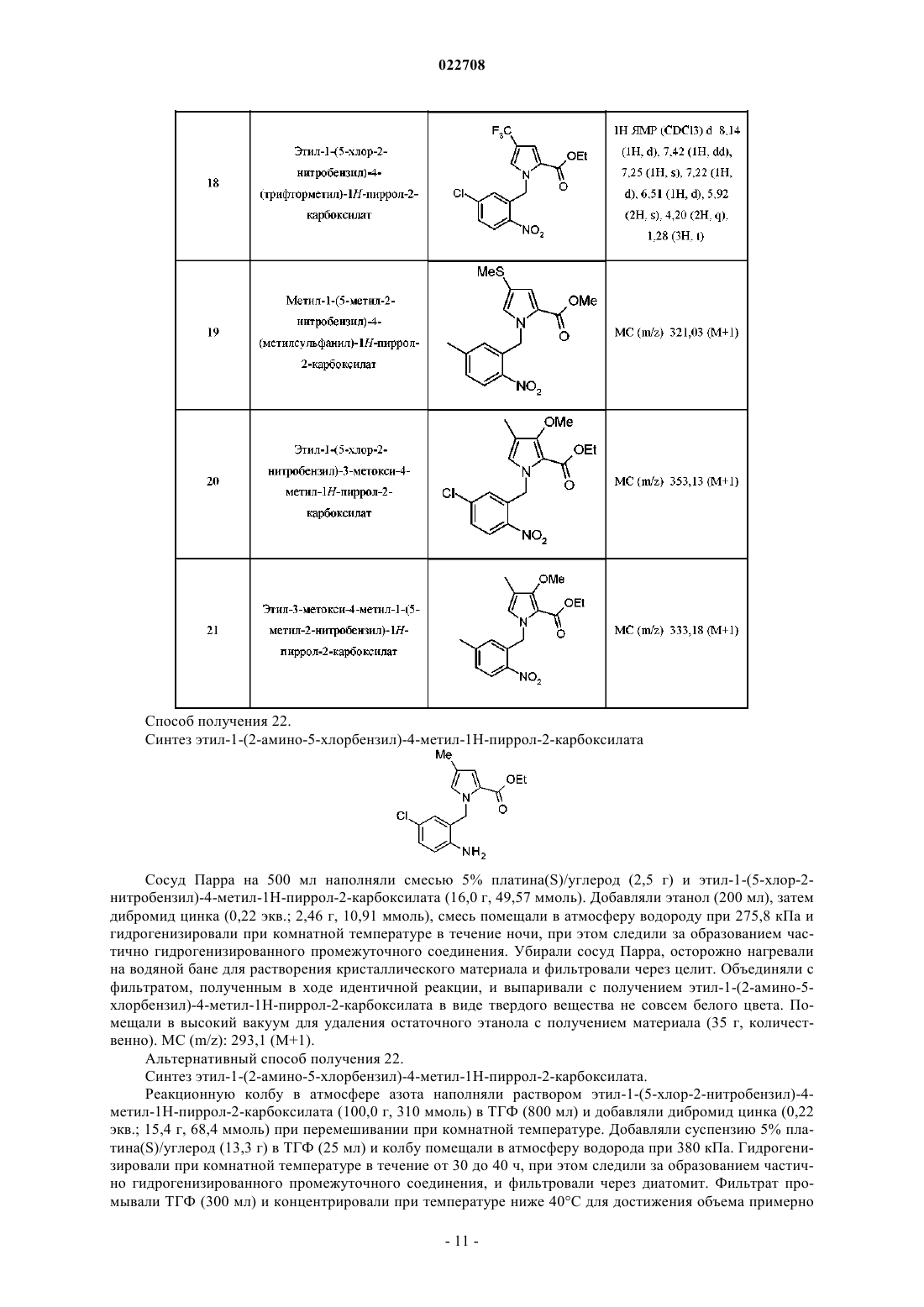
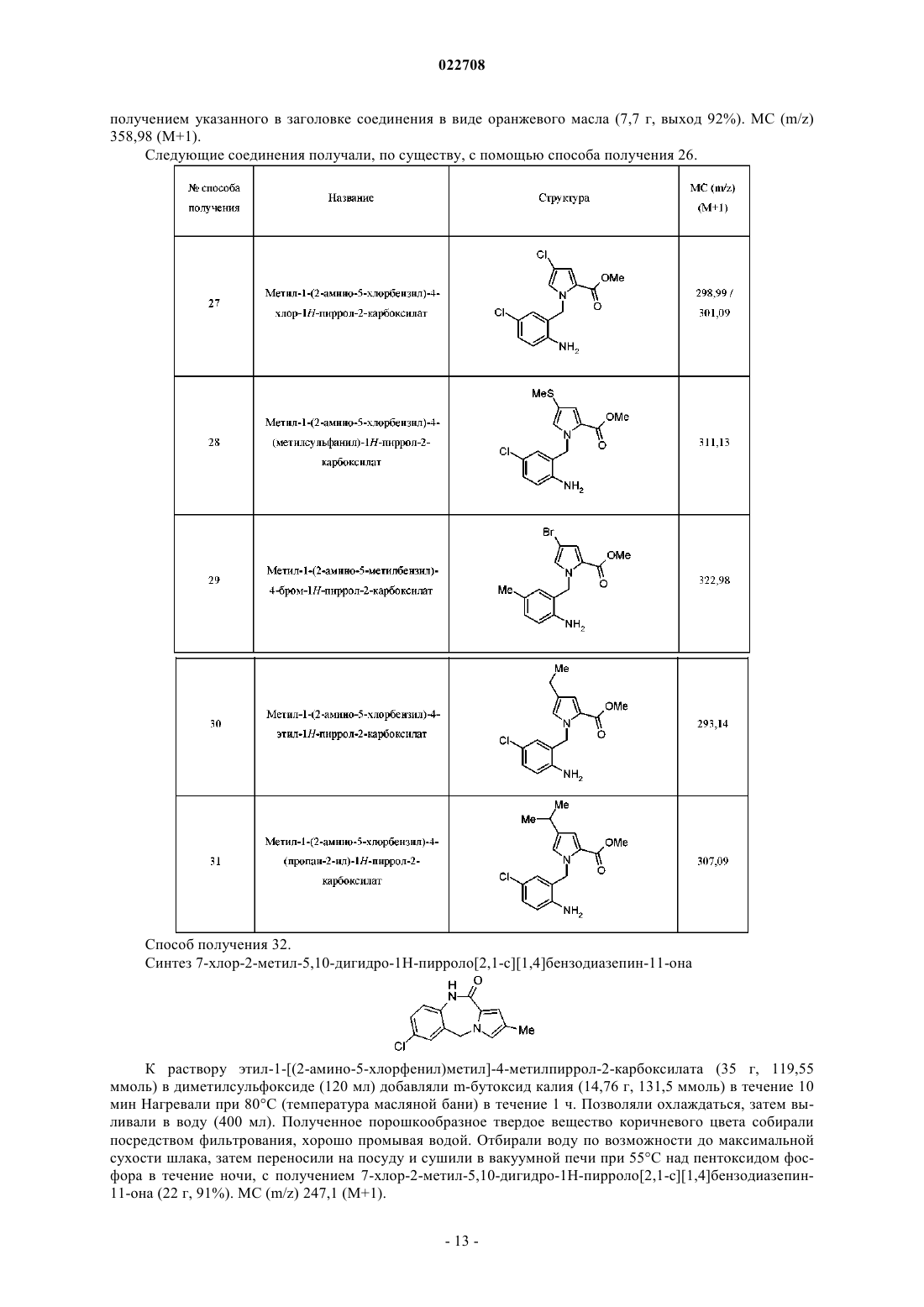
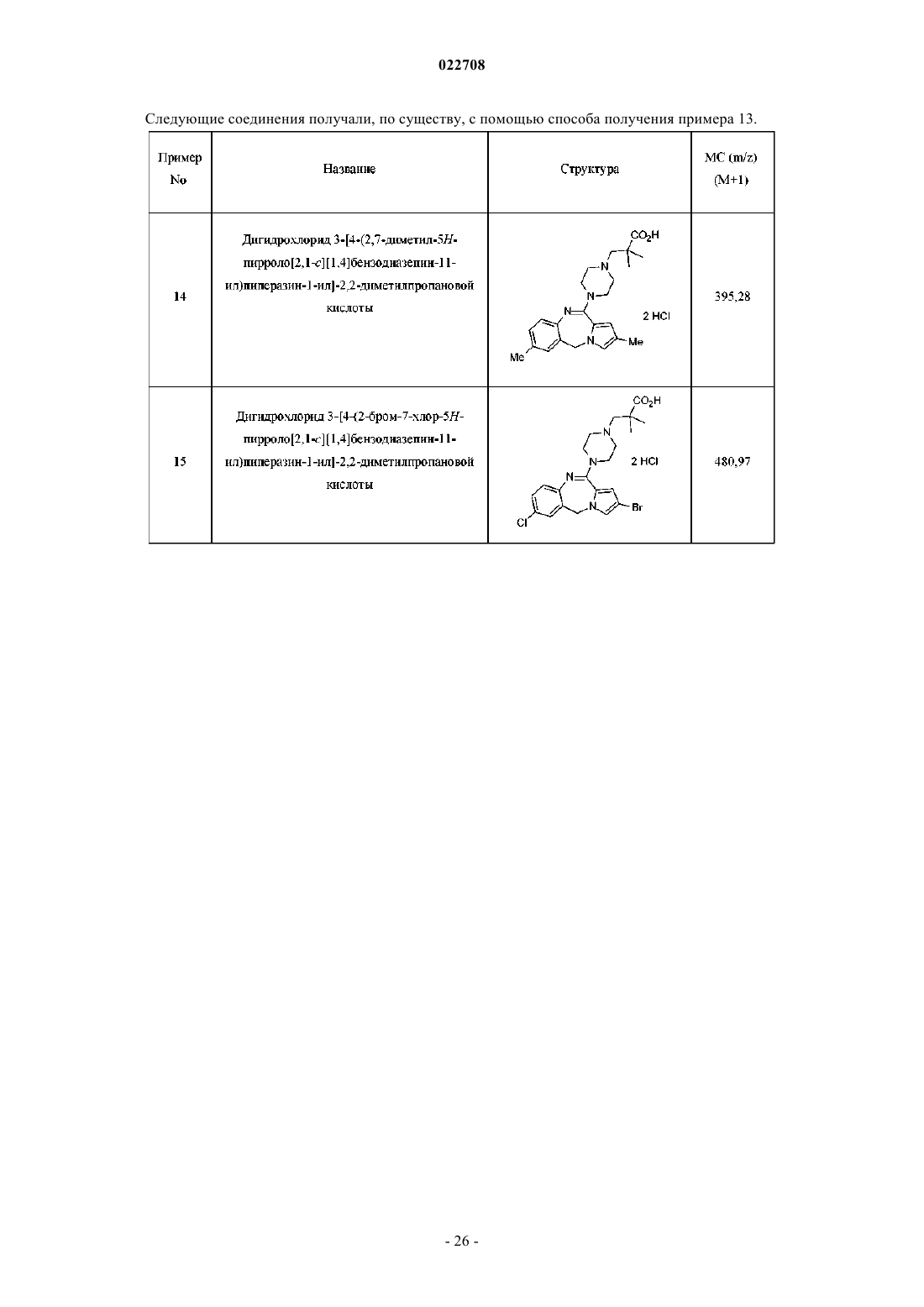
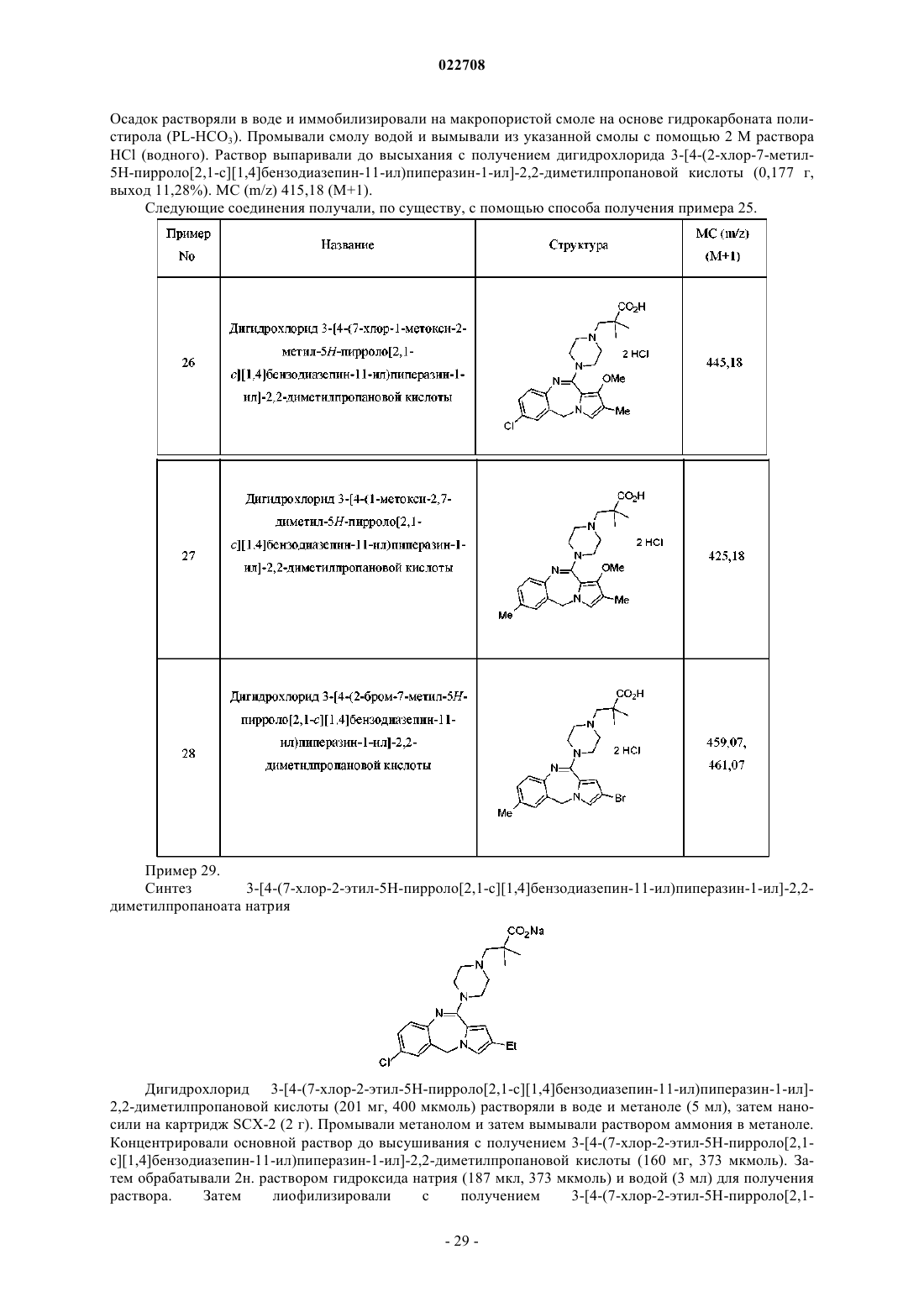
ЗАМЕЩЕННЫЕ СОЕДИНЕНИЯ [(5H-ПИРРОЛО[2,1-c][1,4]БЕНЗОДИАЗЕПИН-11 ИЛ)ПИПЕРАЗИН-1-ИЛ]-2,2-ДИМЕТИЛПРОПАНОВОЙ КИСЛОТЫ В КАЧЕСТВЕ ОБРАТНЫХ АГОНИСТОВ H1/АНТАГОНИСТОВ 5-HT2A ДВОЙНОГО ДЕЙСТВИЯ Предложен двойной антагонист рецепторов Н 1/5-НТ 2 А, имеющий формулу(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) Гистамин играет важную роль в различных физиологических процессах благодаря его взаимодействию по меньшей мере с четырьмя различными рецепторами, сопряженными с G-белком, - рецепторами Н 1-Н 4. В ЦНС рецепторы H1 играют ключевую роль в регуляции цикла сна, и антагонисты/обратные агонисты H1, как известно, вызывают сонливость. Подобным образом, серотонин играет важную роль в различных физиологических процессах благодаря его взаимодействию по меньшей мере с четырнадцатью различными рецепторами, сопряженными с G-белком. Модулирование рецепторов 5-НТ 2 А в ЦНС играет ключевую роль в регуляции цикла сна, и антагонисты 5-НТ 2 А, как было показано, обеспечивают улучшение медленного сна и поддержание сна у пациентов, страдающих бессонницей. Соединения, обладающие активностью обратного агониста или антагониста H1 или 5-НТ 2 А, использовались в лечении бессонницы (например, доксепин и тразодон соответственно) и проявляли значительное фармакологическое действие в исследованиях сна на животных. Однако на сегодняшний день не имеется коммерчески доступных селективных обладающих двойным действием обратных агонистов/антагонистов Н 1/5-HT2A. В патенте США 4192803 описаны конкретные замещенные 4-(5H-пирроло[2,1 с][1,4]бензодиазепин-11-ил)пиперазинильные соединения для применения в качестве антипсихотических или нейролептических средств. Согласно настоящему изобретению предложено семейство замещенных соединений [(5Hпирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2-диметилпропановой кислоты, обладающих высокой активностью обратных агонистов в отношении рецептора H1 и высокой активностью антагонистов в отношении рецептора 5-НТ 2 А. Конкретные соединения согласно настоящему изобретению также являются селективными в отношении рецепторов H1 и 5-НТ 2 А, в частности, по сравнению с другими гистаминовыми рецепторами, серотониновыми рецепторами и другими физиологически значимыми рецепторами, в частности, по сравнению с рецептором 5-НТ 2 С, рецептором GABAA, мускариновыми рецепторами, допаминергическими рецепторами, адренергическими рецепторами и hERG-каналом. Также было показано на животных моделях, что некоторые соединения могут подходить для лечения расстройств сна, характеризующихся плохим поддержанием сна. Полагают, что соединения согласно изобретению как таковые подходят для лечения расстройств сна, характеризующихся плохой латентностью сна или плохим поддержанием сна, или как плохой латентностью сна, так и плохим поддержанием сна, например, для лечения бессонницы, такой как хроническая или временная первичная бессонница, или хроническая или временная вторичная бессонница, или как временная, так и хроническая вторичная бессонница. Примеры вторичной бессонницы включают, но не ограничиваются указанными, бессонницу, связанную с депрессивными расстройствами (например, большим депрессивным расстройством, дистимией и/или циклотимией), бессонницу, связанную с тревожными расстройствами (например, генерализованным тревожным расстройством и/или социофобией), бессонницу, связанную с болью (например, фибромиалгией, хронической костной или суставной болью, например, связанную с воспалительным артритом или остеоартритом или диабетической невропатической болью), бессонницу, связанную с аллергическими реакциями (например, аллергической астмой, кожным зудом, ринитом, застоем и т.д.), бессонницу,связанную с легочными расстройствами или расстройствами дыхательных путей (например, синдромом обструктивного апноэ во сне, реактивным заболеванием дыхательных путей и т.д.), бессонницу, связанную с психическими расстройствами, деменцией и/или нейтродегенеративными заболеваниями, и/или бессонницу, связанную с расстройствами сна в результате нарушения циркадных ритмов (например, расстройство сна при сменной работе, расстройство, представляющее собой синдром смены часовых поясов,расстройство с отсроченным наступлением фазы сна, расстройство с ускоренным наступлением фазы сна и синдром не 24-часового цикла "сон-бодрствование" и т.д.). Кроме того, некоторые соединения согласно настоящему изобретению при совместном введении с селективными ингибиторами обратного захвата серотонина демонстрируют усиление их действия на медленную фазу сна (NREM-сон) и поддержание сна. Согласно настоящему изобретению предложены соединения формулы I где R1 представляет собой хлор или метил;R3 представляет собой водород или метокси; или их фармацевтически приемлемая соль. Согласно другому аспекту изобретения предложена фармацевтическая композиция, содержащая соединение формулы I или его фармацевтически приемлемую соль в комбинации по меньшей мере с одним фармацевтически приемлемым носителем, разбавителем или наполнителем. Кроме того, согласно указанному аспекту изобретения предложена фармацевтическая композиция, приготовленная с возможностью обеспечения лечения бессонницы, например бессонницы, которая характеризуется удлиненным латентным периодом сна или плохим поддержанием сна или как удлиненным латентным периодом сна,так и плохим поддержанием сна, такой как, например, первичная бессонница, синдром смены часовых поясов, расстройство сна при сменной работе, расстройство с отсроченным наступлением фазы сна, расстройство с ускоренным наступлением фазы сна и/или расстройства с не 24-часовым циклом "сонбодрствование", содержащая соединение формулы I или его фармацевтически приемлемую соль в комбинации с одним или более фармацевтически приемлемыми наполнителями, носителями или разбавителями. Согласно другому варианту реализации указанного аспекта изобретения предложена фармацевтическая композиция, содержащая соединение согласно формуле I или его фармацевтически приемлемую соль в комбинации по меньшей мере с одним фармацевтически приемлемым носителем, наполнителем или разбавителем и, необязательно, другими терапевтическими ингредиентами. Согласно другому варианту реализации указанного аспекта изобретения фармацевтическая композиция также содержит второй терапевтический агент, который представляет собой ингибитор обратного захвата серотонина, например,циталопрам, пароксетин, флуоксетин и/или флувоксетин. Согласно настоящему изобретению также предложен способ лечения бессонницы, например бессонницы, которая характеризуется удлиненным латентным периодом сна или плохим поддержанием сна или как удлиненным латентным периодом сна, так и плохим поддержанием сна, как, например, первичной бессонницы, синдрома смены часовых поясов, расстройства сна при сменной работе, расстройства с отсроченным наступлением фазы сна, расстройства с ускоренным наступлением фазы сна и/или расстройств с не 24-часовым циклом "сон-бодрствование", у млекопитающего, включающий введение млекопитающему, нуждающемуся в указанном лечении, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Согласно другому варианту реализации указанного аспекта изобретения, способ также включает одновременное, совместное или последовательное введение в комбинации второго терапевтического агента, который представляет собой ингибитор обратного захвата серотонина, такой как, например, циталопрам, пароксетин, флуоксетин и/или флувоксетин. Согласно одному конкретному варианту реализации указанных способов лечения, млекопитающее представляет собой человека. Согласно настоящему изобретению также предложено соединение формулы I или его фармацевтически приемлемая соль для применения в терапии. В пределах указанного аспекта согласно настоящему изобретению предложено соединение формулы I или его фармацевтически приемлемая соль для применения для лечения бессонницы. Согласно другим вариантам реализации изобретения бессонница характеризуется удлиненным латентным периодом сна или плохим поддержанием сна или как удлиненным латентным периодом сна, так и плохим поддержанием сна, такая как, например, первичная бессонница,синдром смены часовых поясов, расстройство сна при сменной работе, расстройство с отсроченным наступлением фазы сна, расстройство с ускоренным наступлением фазы сна, и/или расстройств с не 24 часовым циклом "сон-бодрствование". Согласно другому варианту реализации указанного аспекта изобретения предложено соединение согласно формуле I или его фармацевтически приемлемая соль, для применения в одновременном, раздельном или последовательном введении в комбинации с ингибитором обратного захвата серотонина, например циталопрамом, пароксетином, флуоксетином и/или флувоксетином, для лечения бессонницы. Согласно одному конкретному варианту реализации указанных аспектов изобретения применение представляет собой применение у млекопитающих, в частности у людей. Согласно другому аспекту настоящего изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли для получения лекарственного средства для лечения бессонницы, например первичной бессонницы, которая характеризуется удлиненным латентным периодом сна или плохим поддержанием сна или как удлиненным латентным периодом сна, так и плохим поддержанием сна, например, первичной бессонницы, синдрома смены часовых поясов, расстройства сна при сменной работе, расстройства с отсроченным наступлением фазы сна, расстройства с ускоренным наступлением фазы сна и/или расстройств с не 24-часовым циклом "сон-бодрствование". Согласно другому варианту реализации указанного аспекта изобретения предложено применение соединения формулы I или его фармацевтически приемлемой соли и второго терапевтического агента, который представляет собой ингибитор обратного захвата серотонина, например циталопрам, пароксетин, флуоксетин и/или флувоксетин, для получения лекарственного средства для лечения бессонницы, например бессонницы, для которой характерен удлиненный период латентного сна и/или плохое поддержание сна, например, первичной бессонницы, синдрома смены часовых поясов, расстройства сна при сменной работе, расстройства с отсроченным наступлением фазы сна, расстройства с ускоренным наступлением фазы сна и/или расстройств с не 24-часовым циклом "сон-бодрствование". Для ясности, в настоящем изобретении используется следующая нумерация в структуре трициклического кольца: Соединения согласно настоящему изобретению содержат основные и кислотные фрагменты и соответственно, реагируют с рядом органических и неорганических кислот и оснований с образованием фармацевтически приемлемых солей. Предполагается, что фармацевтически приемлемые соли каждого из соединений согласно настоящему изобретению включены в объем настоящего изобретения. Термин"фармацевтически приемлемая соль" при использовании в настоящем изобретении относится к любой соли соединения согласно изобретению, которая является, по существу, не токсичной для живых организмов. Такие соли включают соли, перечисленные в источнике Journal of Pharmaceutical Science, 66, 219 (1977), хорошо известные специалистам в данной области техники. Предпочтительные классы соединений согласно настоящему изобретению представляют собой соединения, в которых: 1) R3 представляет собой водород; 2) R3 представляет собой метокси; 3) R1 представляет собой хлор; 4) R1 представляет собой метил; 5) R1 представляет собой хлор и R3 представляет собой водород; 6) R1 представляет собой метил и R3 представляет собой водород; 7) R2 представляет собой метил, этил или изопропил; 8) R2 представляет собой метил; 9) R2 представляет собой хлор или бром; 10) R2 представляет собой хлор; 11) R2 представляет собой трифторметил и 12) R2 представляет собой метилтио. Необходимо понимать, что другие предпочтительные соединения представляют собой соединения,представляющие собой комбинацию указанных выше предпочтительных вариантов выбора конкретного заместителя или заместителей с предпочтительными вариантами выбора других заместителей. Примеры указанных комбинаций включают, но не ограничиваются указанными, следующие предпочтительные классы соединений: 13) предпочтительные соединения любого из предпочтительных классов 7-12 (которые представляют собой предпочтительные варианты R2), где R3 представляет собой водород (предпочтительный класс 1); 14) предпочтительные соединения любого из предпочтительных классов 7-12 (которые представляют собой предпочтительные варианты R2), где R1 представляет собой хлор и R3 представляет собой водород (предпочтительный класс 5); 15) предпочтительные соединения предпочтительного класса 8, где R3 представляет собой метокси(предпочтительный класс 2). Конкретные предпочтительные соединения представляют собой соединения, описанные в примерах, включая их свободные основания и фармацевтически приемлемые соли. Одно конкретное предпочтительное соединение представляет собой 3-[4-(7-хлор-2-метил-5Hпирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2-диметилпропановую кислоту или ее фармацевтически приемлемую соль (т.е. соединение примера 1 и его фармацевтически приемлемые соли). Аббревиатуры, используемые в настоящем изобретении, имеют следующие определения:"БСА" означает бычий сывороточный альбумин."DMEM" означает модифицированную по способу Дульбекко среду Игла."DPBS" означает забуференный фосфатом Дульбекко физиологический раствор."ДСК" означает дифференциальную сканирующую калориметрию."HBSS" означает сбалансированный солевой раствор Хэнкса."ВЭЖХ" означает высокоэффективную жидкостную хроматографию."ч" означает час или часы."IC50" означает концентрацию, при которой достигается 50% максимального подавления."i.v." означает внутривенный или внутривенно."mFST" означает тест принудительного плавания на мышах; животная модель для оценки антидепрессантной активности."ЯМР" означает ядерный магнитный резонанс."ТГФ" означает тетрагидрофуран. Общая химия. Соединения согласно настоящему изобретению можно получать согласно следующим схемам синтеза с помощью общих способов, хорошо известных и признанных в данной области техники. Подходящие условия реакции для этапов указанных схем хорошо известны в данной области, и подходящие замены растворителей и кореагентов находятся в пределах компетенции специалистов в данной области. Подобным образом, специалистам в данной области техники очевидно, что синтетические промежуточные соединения при необходимости или при желании могут быть выделены и/или очищены с помощью различных хорошо известных методов и что зачастую возможно непосредственно использовать различные промежуточные соединения в последующих этапах синтеза с незначительной очисткой или без очистки. Кроме того, специалистам в данной области техники очевидно, что в некоторых случаях порядок введения фрагментов не является критичным. Конкретный порядок этапов, необходимых для получения соединений согласно настоящему изобретению, зависит от конкретного синтезируемого соединения,исходного соединения и относительной подвижности замещенных фрагментов, что очевидно специалистам в области химии. Все заместители, если иное не указано, являются такими, как указано выше, и все реагенты хорошо известны и являются общепринятыми в данной области техники. В целом, соединение формулы I может быть получено из соединения формулы II, где Pg представляет собой подходящую группу для защиты карбоксильной группы (схема 1). Более конкретно, соединение формулы II, где Pg представляет собой С 1-С 3-алкильную группу, подвергают реакции с подходящим агентом для удаления защитной группы, таким как водный раствор гидроксида натрия в органическом сорастворителе, таком как изопропиловый спирт, с получением соединения формулы I после нейтрализации с помощью кислоты. Схема 1 В целом, соединение формулы II может быть получено из соединения формулы III или соединения формулы IV (схема 2). Более конкретно, соединение формулы III подвергают реакции с фосфорилхлоридом в подходящем растворителе, таком как метоксибензол или дихлорметан, с получением промежуточного соединения, представляющего собой иминохлорид. Указанный иминохлорид может быть выделен или непосредственно подвергнут реакции с Pg-2,2-диметил-3-пиперазин-1-илпропаноатом в присутствии подходящего основания, такого как карбонат калия, с получением соединения формулы II. Реакцию осуществляют в подходящем растворителе, таком как ацетонитрил. Альтернативно, иминохлорид можно подвергать реакции с пиперазином в присутствии подходящего основания, такого как карбонат цезия, с получением соединения формулы IV. Реакцию осуществляют в подходящем растворителе, таком как ацетонитрил. Соединение формулы IV алкилируют с помощью Pg-2,2-диметил-3-оксопропаноата в присутствии подходящего восстанавливающего агента, такого как триацетоксиборгидрид натрия, с получением соединения формулы II. Реакцию, как правило, осуществляют в растворителе, таком как дихлорметан. Схема 2 Соединение формулы III может быть получено из соединения формулы V, где R4 представляет собой метил или этил (схема 3). Более конкретно, соединение формулы V подвергают реакции с агентом,подходящим для восстановления арильной нитрогруппы до соответствующего анилина. Подходящие восстанавливающие агенты включают водород в присутствии катализатора на основе переходного металла, такого как платина, железо в уксусной кислоте и дихлорид олова в соляной кислоте. Соответствующий анилин может быть сначала выделен или непосредственно подвергаться реакции в условиях циклизации с получением соединения формулы III. Циклизацию осуществляют в присутствии кислоты,такой как соляная кислота, или основания, такого как m-бутоксид калия. Соединение формулы V, где R4 представляет собой метил или этил, может быть получено, как описано в способах получения, или согласно методикам, известным в области химии для получения структурных аналогов указанных соединений. Схема 3 В следующих иллюстративных способах получения и примерах реагенты получали из различных коммерческих источников. Растворители в целом удаляли при пониженном давлении (выпаривали). В некоторых способах указанный выход обозначает выход неочищенных продуктов, которые выделяли путем выпаривания или фильтрования и использовали непосредственно без дальнейшей очистки. Способ получения 1. Синтез метил-2,2-диметил-3-оксопропаноата. К периодинану Десса-Мартина (106 г, 250 ммоль), суспендированному в ДХМ (1000 мл), добавляли метил-3-гидрокси-2,2-диметилпропаноат (33 г, 250 ммоль) при 0C и перемешивали при комнатной температуре в течение 18 ч. Реакционную смесь фильтровали через слой целита и фильтрат концентрировали. Концентрированный фильтрат промывали пентаном (2200 мл). Слой пентана отделяли и концентрировали в вакууме с получением метил-2,2-диметил-3-оксопропаноата (31,93 г, количественный выход). 1 Раствор метил-2,2-диметил-3-оксопропаноата (30,88 г, 237,31 ммоль) и трет-бутил пиперазин-1 карбоксилата (34,00 г, 182,55 ммоль) в ДХМ (500 мл) перемешивали при комнатной температуре в течение 20 мин. Добавляли уксусную кислоту (2 экв.; 20,92 мл, 365,09 ммоль) с последующим добавлением триацетоксиборгидрида натрия (1,4 экв.; 54,17 г, 255,56 ммоль) в течение 0,5 ч и перемешивали полученную смесь при комнатной температуре в течение ночи. Осторожно гасили водой (250 мл) и переносили смесь в делительную воронку с ДХМ (300 мл). Полученный органический слой промывали солевым раствором. Сушили над MgSO4, фильтровали и выпаривали с получением трет-бутил-4-(3-метокси-2,2 диметил-3-оксопропил)пиперазин-1-карбоксилата (58 г, 100%). МС (m/z): 301,2 (М+1). Способ получения 3. Синтез дигидрохлорида метил-2,2-диметил-3-(пиперазин-1-ил)пропаноата К раствору трет-бутил-4-(3-метокси-2,2-диметил-3-оксопропил)пиперазин-1-карбоксилата (58,0 г,193,08 ммоль) в изопропиловом спирте (150 мл) добавляли 4 М раствор соляной кислоты в диоксане (4 экв.; 193,08 мл, 772,31 ммоль) в течение 15 мин, при этом наблюдали выделение газа и образование мелкодисперсного осадка. Нагревали при 55C в течение 3 ч с получением белого осадка. Охлаждали до 10C и собирали твердое вещество белого цвета посредством фильтрования, промывали дополнительной порцией изопропилового спирта (30 мл), затем EtOAc. Сушили в вакуумной печи при 45C в течение 1 ч с получением дигидрохлорида метил-2,2-диметил-3-(пиперазин-1-ил)пропаноата (31 г, выход 59%). МС Дигидрохлорид метил-2,2-диметил-3-(пиперазин-1-ил)пропаноата (112,0 г, 409,95 ммоль) растворяли в воде (250 мл). Добавляли твердый бикарбонат натрия с получением водного слоя, имеющего pH 4, и полученную смесь экстрагировали в растворе 10% ДХМ в диэтиловом эфире (300 мл) для удаления небольшого количества темного твердого материала и окраски. Удаляли органический слой. Ощелачивали водную фазу до pH 8 с помощью 2 М водного раствора гидроксида натрия, затем насыщали твердым хлоридом натрия. Экстрагировали в 10% растворе изопропанола в хлороформе. Добавляли дополнительную порцию 2 М водного раствора гидроксид натрия для поддержания pH 8. Снова экстрагировали 10% раствором изопропанола в хлороформе. Процесс экстрагирования продолжали до достижения восстановления 85% ожидаемого материала. Промывали раствор изопропанола в хлороформе солевым раствором,сушили над Na2SO4, фильтровали и выпаривали с получением гидрохлорида метил-2,2-диметил-3(пиперазин-1-ил)пропаноата (82,1 г, выход %). МС (m/z): 201,1 (М+1). Способ получения 5. Синтез метил-4-этенил-1H-пиррол-2-карбоксилата(1,3 экв.; 3,1 г, 12,7 ммоль) и карбоната калия (3 экв.; 4,1 г, 29,4 ммоль) в смеси 1,4-диоксана (20 мл) и воды (10 мл) дегазировали путем пропускания азота в течение 10 мин. Добавляли трис(дибензилиденацетинил)бис-палладий (Pd2(dba)3) (0,01 экв.; 0,0925 г, 98,0 мкмоль) и 1,1'-бис-(ди-третбутилфосфино)ферроцен (dtbpf) (0,03 экв.; 0,0853 г, 294,1 мкмоль) и нагревали при 95C в течение 3 ч. Охлаждали до комнатной температуры, затем распределяли между водой (50 мл) и смесью гексаны/диэтиловый эфир, 1:1 (100 мл). Органический слой промывали водой (250 мл), затем солевым раствором. Сушили над Na2SO4, фильтровали и выпаривали с получением метил-4-этенил-1H-пиррол-2-6 022708 карбоксилат в виде бледно-желтого масла (1,5 г). Использовали без дополнительной очистки. 1 Н-ЯМР (CDCl3) : 8,90 (1H, br. s), 7,02 (1H, m), 6,95 (1H, m), 6,56 (1H, q) 5,48 (1H, dd), 5,05 (1H, dd),3,86 (3H, s). Способ получения 6. Синтез метил-4-этил-1H-пиррол-2-карбоксилата Смесь 5% палладия на углероде (0,2 г) в этаноле (25 мл) выливали в сосуд Парра на 500 мл и добавляли метил-4-этенил-1H-пиррол-2-карбоксилат (1,4 г, 9,3 ммоль). Гидрогенизировали при 206,8 кПа в течение 3 ч с достижением полного превращения по результатам ЖХ/МС. Фильтровали через целит и выпаривали с получением масла. Пропускали через слой силикагеля с использованием смеси изогексан/этилацетат (100:0-75:25) с получением указанного в заголовке соединения в виде бледно-желтого масла (1,12 г). 1(0,3 г, 0,06 ммоль), трис-(дибензилиденацетонил)бис-палладия (Pd2(dba)3) (0,3 г, 0,06 ммоль), трифосфат калия (2,54 г, 11,95 ммоль) и метанол (12 мл). Полученную смесь нагревали в герметизированной пробирке в микроволновой печи при 140C в течение 30 мин. Реакционную смесь охлаждали, фильтровали и шлак промывали метанолом, выпаривали фильтрат при пониженном давлении и хроматографировали на силикагеле при элюировании смесью изогексан/дихлорметан (градиентное элюирование, 100:0-0:100). Фракции, содержащие продукт, выпаривали с получением метил-4-(проп-1-ен-2-ил)-1H-пиррол-2 карбоксилата (0,679 г, выход 68,79%). МС (m/z): 166,1 (М+1). Способ получения 8. Синтез этил-3-метокси-4-метил-1H-пиррол-2-карбоксилата К смеси этилового эфира 3-гидрокси-4-метил-1H-пиррол-2-карбоновой кислоты (3,5 г, 20,69 ммоль) и 2 М гидроксида натрия (75 мл, 1500 ммоль) добавляли диметилсульфат (3 мл, 3,99 г, 31,63 ммоль) и реакционную смесь интенсивно перемешивали в течение 30 мин при комнатной температуре (применяли охлаждающую водяную баню). Собирали полученный осадок и промывали водой. Добавляли дополнительную порцию диметилсульфата (3 мл, 3,99 г, 31,63 ммоль) к фильтрату и перемешивали в течение 30 мин. Собирали полученный осадок и промывали водой. Добавляли дополнительную порцию диметилсульфата (3 мл, 3,99 г, 31,63 ммоль) к фильтрату и перемешивали в течение 1 ч. Объединяли выделенные осадки и сушили в вакуумной печи при 50C в течение 30 мин с получением этил-3-метокси-4-метил-1Hпиррол-2-карбоксилата (3,221 г, выход 84,98%). МС (m/z): 184,09 (М+1). Способ получения 9. Синтез этил-1-(5-хлор-2-нитробензил)-4-метил-1H-пиррол-2-карбоксилата Интенсивно перемешивали раствор 2-(бромметил)-4-хлор-1-нитробензола (25,02 г, 99,88 ммоль) в ДХМ (200 мл) и к указанному раствору добавляли раствор этил-4-метил-1H-пиррол-2-карбоксилата (15 г,97,92 ммоль) в ДХМ (200 мл). Затем добавляли 30-гидратированный гидроксид тетра-н-бутиламмония(0,508 г) и охлаждали перемешанную смесь под азотом до 5C с использованием внешней ледяной бани. Добавляли по каплям 25% водный раствор гидроксида натрия (200 мл) в течение 15 мин, наблюдая повышение внутренней температуры до 10C. Перемешивали и позволяли нагреваться до комнатной температуры. Перемешивали при комнатной температуре в течение 3,5 ч. Добавляли дополнительную порцию 50% водного раствора гидроксида натрия (20 мл) и перемешивали при комнатной температуре в течение дополнительного 1 ч. Прекращали перемешивать и позволяли слоям разделяться. Переносили в делительную воронку и промывали органический слой 1 н. соляной кислотой (200 мл) и с использованием индикаторной бумаги определяли, что pH указанного слоя имел кислое значение. Затем промывали органический слой водой (600 мл) и затем солевым раствором (600 мл) и в конечном итоге сушили надNa2SO4. Фильтровали смесь и удаляли растворитель в вакууме при 40C с получением оранжевого масла. Очищали путем пропускания через слой силикагеля с использованием смеси изогексан/этилацетат (градиентное элюирование от 10 до 20%) с получением этил-1-(5-хлор-2-нитробензил)-4-метил-1H-пиррол-2 карбоксилата в виде желтого масла (33 г, количественный выход). МС (m/z): 322,98 (М+1). Альтернативный способ получения 9. Синтез этил-1-(5-хлор-2-нитробензил)-4-метил-1H-пиррол-2-карбоксилата. К раствору этил-4-метил-1H-пиррол-2-карбоксилата (30 г, 196 ммоль) в ДМФА (240 мл) в атмосфере азота добавляли карбонат цезия (96 г, 295 ммоль) при перемешивании и реакционную смесь нагревали до температуру от 40 до 45C в течение 1 ч. Охлаждали реакционную смесь до комнатной температуры и добавляли по каплям раствор 2-(бромметил)-4-хлор-1-нитробензола (54 г, 216 ммоль) в ДМФА (120 мл). Перемешивали полученную суспензию в течение от 1,5 до 2,0 ч, твердое вещество фильтровали и промывали отфильтрованный осадок ДМФА (45 мл). Переносили твердое вещество в чистую реакционную пробирку, добавляли воду (200 мл) и охлаждали суспензию до температуры от 10 до 15C при перемешивании в течение от 1 до 2 ч. Фильтровали суспензию, промывали водой (75 мл) и переносили твердое вещество в другую реакционную пробирку. Добавляли этанол (125 мл) и перемешивали суспензию при температуре от 5 до 10C в течение от 1 до 2 ч. Твердое вещество фильтровали, промывали отфильтрованный осадок этанолом (20 мл) и сушили твердое вещество в печи при температуре менее 50C при пониженном давлении с получением этил-1-(5-хлор-2-нитробензил)-4-метил-1H-пиррол-2-карбоксилата в виде твердого вещества желтого цвета (54 г, чистота 94,1%, выход 73,3%). Способ получения 10. Синтез этил-4-бром-1-(5-хлор-2-нитробензил)-1H-пиррол-2-карбоксилата Гидрат натрия, суспендированный в 60% минеральном масле, (1,2 экв., 1,1 г, 27,52 ммоль) промывали малым количеством изогексана (2). Суспендировали в N,N-диметилацетамиде (15 мл) и охлаждали на ледяной бане. Добавляли этил-4-бром-1H-пиррол-2-карбоксилат (5,0 г, 22,93 ммоль) порциями в течение 15 мин, позволяя снижаться интенсивности выделения газа между добавлениями. Перемешивали при комнатной температуре в течение 15 мин с получением коричневого раствора. Добавляли порциями 2(бромметил)-4-хлор-1-нитробензол (1,15 экв., 6,61 г, 26,37 ммоль) в течение 15 мин и перемешивали полученный пурпурный раствор при комнатной температуре в течение 2,5 ч. Охлаждали на ледяной бане и гасили водой (10 мл). Распределяли между 1 М соляной кислотой (100 мл) и EtOAc (300 мл). Органический слой промывали водой (2100 мл), затем солевым раствором. Сушили над Na2SO4 и обесцвечивали с помощью угля. Фильтровали через целит и выпаривали с получением этил-4-бром-1-(5-хлор-2 нитробензил)-1H-пиррол-2-карбоксилата в виде оранжевого масла (9,1 г, количественный выход). 1 Следующие соединения получали, по существу, с помощью способа получения 10. Сосуд Парра на 500 мл наполняли смесью 5% платина(S)/углерод (2,5 г) и этил-1-(5-хлор-2 нитробензил)-4-метил-1H-пиррол-2-карбоксилата (16,0 г, 49,57 ммоль). Добавляли этанол (200 мл), затем дибромид цинка (0,22 экв.; 2,46 г, 10,91 ммоль), смесь помещали в атмосферу водороду при 275,8 кПа и гидрогенизировали при комнатной температуре в течение ночи, при этом следили за образованием частично гидрогенизированного промежуточного соединения. Убирали сосуд Парра, осторожно нагревали на водяной бане для растворения кристаллического материала и фильтровали через целит. Объединяли с фильтратом, полученным в ходе идентичной реакции, и выпаривали с получением этил-1-(2-амино-5 хлорбензил)-4-метил-1H-пиррол-2-карбоксилата в виде твердого вещества не совсем белого цвета. Помещали в высокий вакуум для удаления остаточного этанола с получением материала (35 г, количественно). МС (m/z): 293,1 (М+1). Альтернативный способ получения 22. Синтез этил-1-(2-амино-5-хлорбензил)-4-метил-1H-пиррол-2-карбоксилата. Реакционную колбу в атмосфере азота наполняли раствором этил-1-(5-хлор-2-нитробензил)-4 метил-1H-пиррол-2-карбоксилата (100,0 г, 310 ммоль) в ТГФ (800 мл) и добавляли дибромид цинка (0,22 экв.; 15,4 г, 68,4 ммоль) при перемешивании при комнатной температуре. Добавляли суспензию 5% платина(S)/углерод (13,3 г) в ТГФ (25 мл) и колбу помещали в атмосферу водорода при 380 кПа. Гидрогенизировали при комнатной температуре в течение от 30 до 40 ч, при этом следили за образованием частично гидрогенизированного промежуточного соединения, и фильтровали через диатомит. Фильтрат промывали ТГФ (300 мл) и концентрировали при температуре ниже 40C для достижения объема примерно 200 мл. Добавляли ДХМ (250 мл), концентрировали раствор при температуре ниже 40C для достижения объема примерно 200 мл и добавляли дополнительную порцию ДХМ (600 мл). Указанный процесс при необходимости можно повторять для удаления нежелательного уровня ТГФ из реакционной смеси. Добавляли воду (500 мл), разделяли слои и органический слой промывали водой (300 мл) с последующей промывкой 25% водным раствором хлорида натрия (250 мл). Концентрировали раствор при 40C для достижения объема примерно 200 мл, добавляли гептан (400 мл) и концентрировали раствор при температуре ниже 40C для достижения объема примерно 200 мл. Добавляли гептан (400 мл) и реакционную смесь нагревали до 40-45C при перемешивании в течение 2-3 ч. Охлаждали реакционную смесь до 510C и продолжали перемешивать при указанной температуре в течение 1-2 ч. Фильтровали полученное твердое вещество и сушили в печи при менее чем 50C при пониженном давлении с получением этил-1(2-амино-5-хлорбензил)-4-метил-1H-пиррол-2-карбоксилата (81,4 г, чистота 95,8%, выход 85,9%) в виде твердого вещества желтого цвета Следующие соединения получали, по существу, с помощью способа получения 22. К хорошо перемешанному раствору этил-4-бром-1-(5-хлор-2-нитробензил)-1H-пиррол-2 карбоксилата (9,1 г, 23,48 ммоль) в уксусной кислоте (60,00 мл) при 70C порциями добавляли железо (5 экв., 6,56 г, 117,38 ммоль) в течение 0,5 ч. Между добавлениями наблюдали экзотермический эффект с поддержанием температуры 85C при удаленной масляной бане. Реакционная смесь становится более густой по мере угасания экзотермического эффекта и можно добавлять оставшуюся часть железа. Перемешивали при 85C в течение 0,5 ч. Охлаждали до комнатной температуры и выливали в воду (200 мл). Экстрагировали хлороформом (2200 мл). Объединяли органические слои и промывали водой (2100 мл), затем насыщенным водным раствором NHCO3. Сушили над Na2SO4, фильтровали и выпаривали с получением указанного в заголовке соединения в виде оранжевого масла (7,7 г, выход 92%). МС (m/z) 358,98 (М+1). Следующие соединения получали, по существу, с помощью способа получения 26. К раствору этил-1-[(2-амино-5-хлорфенил)метил]-4-метилпиррол-2-карбоксилата (35 г, 119,55 ммоль) в диметилсульфоксиде (120 мл) добавляли m-бутоксид калия (14,76 г, 131,5 ммоль) в течение 10 мин Нагревали при 80C (температура масляной бани) в течение 1 ч. Позволяли охлаждаться, затем выливали в воду (400 мл). Полученное порошкообразное твердое вещество коричневого цвета собирали посредством фильтрования, хорошо промывая водой. Отбирали воду по возможности до максимальной сухости шлака, затем переносили на посуду и сушили в вакуумной печи при 55C над пентоксидом фосфора в течение ночи, с получением 7-хлор-2-метил-5,10-дигидро-1H-пирроло[2,1-с][1,4]бензодиазепин 11-она (22 г, 91%). МС (m/z) 247,1 (М+1). Альтернативный способ получения 32. Синтез 7-хлор-2-метил-5,10-дигидро-11H-пирроло[2,1-с][1,4]бензодиазепин-11-она. К раствору этил-1-[(2-амино-5-хлорфенил)метил]-4-метилпиррол-2-карбоксилата (20 г, 68,3 ммоль) в диметилсульфоксиде (60 мл) добавляли m-бутоксид калия (8,4 г, 74,9 ммоль) в ДМСО (40 мл) и реакционную смесь нагревали до 75-80C в течение 1-2 ч. Медленно добавляли воду (200 мл), охлаждали до комнатной температуры и перемешивали в течение 1-2 ч. Твердое вещество фильтровали, отфильтрованный осадок промывали водой (75 мл) и твердое вещество переносили в чистую реакционную пробирку. Добавляли ТГФ (100 мл) и реакционную смесь нагревали до 30-35C до растворения твердого вещества. Добавляли ДХМ (350 мл) и продолжали перемешивать при 30-35C в течение 1-2 ч. Добавляли 1 н. соляную кислоту (250 мл) и продолжали перемешивать при 30-35C в течение 1-2 ч. Слои разделяли, органический слой промывали 1 н. соляной кислотой (250 мл) с последующей промывкой 25% водным раствором хлорида натрия (50 мл) и концентрировали указанный раствор при температуре ниже 50C для достижения объема примерно 25 мл. Добавляли этанол (50 мл), концентрировали раствор при температуре ниже 50C для достижения объема примерно 25 мл и добавляли дополнительную порцию этанола (50 мл). Концентрировали раствор при температуре ниже 50C для достижения объема примерно 25 мл, реакционную смесь нагревали до 45-50C и перемешивали в течение 1-2 ч. Охлаждали реакционную смесь до 5-10C при перемешивании в течение 2-3 ч и фильтровали полученное твердое вещество. Промывали отфильтрованный осадок этанолом (25 мл) и сушили в печи при температуре менее чем 50C при пониженном давлении с получением 7-хлор-2-метил-5,10-дигидро-1H-пирроло[2,1-с][1,4]бензодиазепин-11 она в виде твердого вещества не совсем белого цвета (14,9 г, чистота 99,9%, выход 88,5%). Следующие соединения получали, по существу, с помощью способа получения 32. 7-хлор-2-(трифторметил)-5,10-дигидро-1H-пирроло[2,1 К смеси этил-1-(5-хлор-2-нитробензил)-4-(трифторметил)-1H-пиррол-2-карбоксилата (1 экв., 1,98 г,5,26 ммоль) и этанола (100 мл) при 50C добавляли дихлорид олова (3 экв., 3,02 г, 15,77 ммоль) в 5 М соляной кислоте (20 мл, 100 ммоль). Нагревали в течение ночи и затем в течение дополнительных 26 ч,удаляли большую часть этанола в вакууме и разбавляли полученный раствор водой и собирали полученный осадок посредством фильтрования, хорошо промывали водой и сушили в вакууме при 40C. Абсорбировали твердое вещество на силикагеле, хроматографировали при элюировании смесью ДХМ/метанол(5 95) с получением нециклизованного аминоэфира (0,260 г) и 7-хлор-2-(трифторметил)-5,10-дигидро 1H-пирроло[2,1-с][1,4]бензодиазепин-11-она (0,613 г). Нагревали нециклизованный эфир в смеси 5 М соляной кислоты (10 мл, 50 ммоль) в течение 80 ч. Удаляли этанол в вакууме и собирали полученное осажденное твердое вещество посредством фильтрования и промывали водой с получением 7-хлор-2(трифторметил)-5,10-дигидро-1H-пирроло[2,1-с][1,4]бензодиазепин-11-она (0,17 г). Объединяли с полученным ранее 7-хлор-2-(трифторметил)-5,10-дигидро-1H-пирроло[2,1-с][1,4]бензодиазепин-11-оном с получением 0,794 г (выход 50%). МС (m/z) 300,99 (М+1). Следующее соединение получали, по существу, с помощью способа получения 43. К суспензии 7-хлор-2-метил-5,10-дигидро-1H-пирроло[2,1-с][1,4]бензодиазепин-11-она (40,6 г,164,58 ммоль) в метоксибензоле (250 мл) при 70C добавляли одной порцией N,N-диметиланилин (2,8 экв.; 58,59 мл, 460,8 ммоль) с последующим добавлением фосфорилхлорида (2,3 экв. (молярных); 35,18 мл, 378,52 ммоль) в течение 20 мин, при этом по мере добавления контролировали экзотермическую реакцию. Нагревали полученный темный раствор при 90C в течение 1,5 ч. Добавляли дополнительную порцию фосфорилхлорида (0,33 экв.; 5,05 мл, 54,31 ммоль) и нагревали смесь при 90C в течение дополнительного 1 ч для достижения полного превращения. Выпаривали почти до высыхания и распределяли осадок между водой (500 мл) и этилацетатом (2500 мл). Объединяли органические слои и промывали водой (500 мл), затем солевым раствором. Сушили над Na2SO4, фильтровали и выпаривали на силикагеле. Пропускали через слой силикагеля с использованием смеси изогексан/этилацетат (градиентное элюирование от 5 до 25%). Объединяли фракции продукта и выпаривали до получения малого количества растворителя. Добавляли изогексан (150 мл) и собирали полученный желтый порошок посредством фильтрования с получением 7,11-дихлор-2-метил-5H-пирроло[2,1-с][1,4]бензодиазепина (34,0 г, выход 78%). МС (m/z): 265,1 (М+1). Альтернативный способ синтеза. В атмосфере N2 к смеси 7-хлор-2-метил-5,10-дигидро-1H-пирроло[2,1-с][1,4]бензодиазепин-11-она(100 г, 405 ммоль), N,N-диметиланилина (138 г, 1,14 моль) и анизола (550 мл) добавляли фосфорилхлорид (124 г, 819 ммоль). Нагревали до 80-85C и перемешивали в течение от 4 до 6 ч. Концентрировали до 1,5-2,5 объемов в общем при поддержании температуры ниже 75C. Охлаждали до 15-25C. Добавляли по каплям дихлорметан (600 мл) и перемешивали в течение 1 ч. Добавляли полученную смесь к воде (600 мл) при поддержании температуры между 10 и 35C. Перемешивали в течение 1 ч и затем слои разделяли. Экстрагировали водный слой дихлорметаном (600 мл). Промывали объединенные органические слои 1,0 н. HCl (600 г) с последующей промывкой 7% NHCO3 (600 г). Фильтровали органический слой через диатомит (от 20 до 30 г) и силикагель (от 60 до 100 г) Концентрировали полученный фильтрат до 1,5-2,5 объемов в общем при поддержании температуры ниже 45C. Добавляли гептан (от 270 г до 410 г) и концентрировали полученный фильтрат до 1,5-2,5 объемов в общем при поддержании температуры ниже 45C. Добавляли гептан (150-210 г), охлаждали до температуры от 5 до 10C и перемешивали в течение от 2 до 3 ч. Фильтровали, промывали отфильтрованный осадок гептаном и сушили в вакууме при температуре ниже 45C с получением указанного в заголовке соединения в виде твердого вещества бледножелтого цвета (выход от 90 до 95%) Следующие соединения получали, по существу, с помощью способа получения 45. К 7-хлор-2-метил-5,10-дигидро-1H-пирроло[2,1-с][1,4]бензодиазепин-11-ону (1,117 г, 4,53 ммоль) в ДХМ (50 мл) добавляли фосфорилхлорид (1,47 мл, 2,43 г, 15,85 ммоль) и перемешивали при комнатной температуре в течение выходных. К реакционной смеси добавляли ледяную воду, затем добавляли ДХМ,отделяли водный слой и промывали слой ДХМ водой и раствором гидрокарбоната натрия. Сушили раствор ДХМ раствор MgSO4, фильтровали и выпаривали растворитель в вакууме с получением 7,11 дихлор-2-метил-5H-пирроло[2,1-с][1,4]бензодиазепина (1,167 г). Дигидрохлорид метил-2,2-диметил-3-(пиперазин-1-ил)пропаноата (1,61 г, 5,89 ммоль) растворяли в воде и наносили на два картриджа, заполненных SCX-2 (10 г). Указанные картриджи промывали метанолом и элюировали 2 М аммиаком в метаноле. Концентрировали в вакууме, затем растворяли полученное масло в ацетонитриле (40 мл). Добавляли 7,11-дихлор-2-метил-5H-пирроло[2,1-с][1,4]бензодиазепин(1,167 г, 4,4 ммоль) к раствору ацетонитрила. Раствор ацетонитрила разделяли и помещали в 2 пробирки для микроволновой печи, в каждую указанную пробирку для микроволновой печи добавляли карбонат калия (0,89 г, 6,79 ммоль), нагревали и перемешивали при 140C в микроволновой печи в течение 3,5 ч. Реакционной смеси позволяли охлаждаться, фильтровали, промывали твердое вещество, собравшееся в виде шлака, ацетонитрилом, затем фильтрат концентрировали в вакууме, добавляли метанол и затем выпаривали до высыхания. Обрабатывали дополнительной порцией метанола и собирали полученный осадок, промывали указанный осадок метанолом с получением метил-3-[4-(7-хлор-2-метил-5H-пирроло[2,1 с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2-диметилпропаноата в виде кристаллического твердого вещества (1,1 г). МС (m/z): 429,18 (М+1). Альтернативный способ синтеза метил-3-[4-(7-хлор-2-метил-5H-пирроло[2,1-с][1,4]бензодиазепин- 17022708 11-ил)пиперазин-1-ил]-2,2-диметилпропаноата. К суспензии 7,11-дихлор-2-метил-5H-пирроло[2,1-с][1,4]бензодиазепина (34,0 г, 128,23 ммоль) в ацетонитриле (300 мл) добавляли гидрохлорид метил-2,2-диметил-3-(пиперазин-1-ил)пропионата (2,1 экв.; 63,75 г, 269,29 ммоль) и карбонат калия (4 экв.; 70,89 г, 512,93 ммоль). Смесь нагревали при температуре обратной конденсации в течение ночи. Выпаривали до высыхания. Осадок распределяли между водой (500 мл) и EtOAc (2500 мл). Объединяли органические слои и промывали водой (500 мл), затем солевым раствором. Сушили над сульфатом натрия, фильтровали и выпаривали до получения коричневого масла. Добавляли изогексан (150 мл) и зародыш кристалла из способа получения 50. Позволяли кристаллизоваться в течение 2 ч, затем переносили колбу в холодильник и позволяли реакционной смеси отстаиваться. Собирали полученное тяжелое кристаллическое твердое вещество посредством фильтрования, промывая холодным изогексаном. Сушили в вакуумной печи при 40C в течение 1 ч с получением метил-3-[4-(7-хлор-2-метил-5H-пирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2 диметилпропаноата (53,6 г, 97% выход). МС (m/z): 429,18 (М+1). Второй альтернативный способ синтеза метил-3-[4-(7-хлор-2-метил-5H-пирроло[2,1 с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2-диметилпропаноата. Смесь 7,11-дихлор-2-метил-5H-пирроло[2,1-с][1,4]бензодиазепина (10,0 г, 37,7 ммоль), дигидрохлорида метил-2,2-диметил-3-(пиперазин-1-ил)пропионата (19,4 г, 71,0 ммоль), диизопропиламина (22,9 г, 226 ммоль) и ацетонитрила (80 г) нагревали при температуре от 80 до 85C в течение от 22 до 26 ч. Охлаждали до температуры от 30 до 40C и добавляли этилацетат (80 г). Добавляли по каплям воду (80 г). Перемешивали в течение от 40 до 60 мин и разделяли слои. Водный слой экстрагировали этилацетатом (60-80 г). Объединенные органические слои промывали 25% водным раствором хлоридом натрия(240 г). Органический слой концентрировали до 1,5 объемов и 3,5 объемов в общем при поддержании температуры ниже 50C. Добавляли гептан (41-55 г) при температуре от 40 до 50C и перемешивали в течение от 2 до 3 ч. Концентрировали до 1,5-3,0 объемов в общем при поддержании температуры ниже 50C. Охлаждали до температуры от 0 до 10C и перемешивали в течение от 2 до 3 ч. Фильтровали, промывали отфильтрованный осадок гептаном (от 3,0 до 10,0 г) и сушили в вакууме при температуре ниже 60C с получением указанного в заголовке соединения (16,0 г, 94,7%, мас.%, выход 94%) в виде светлого желтого твердого вещества. Способ получения 51. Синтез метил-3-[4-(2,7-дихлор-5H-пирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2 диметилпропаноата К раствору 2,7-дихлор-5,10-дигидро-1H-пирроло[2,1-с][1,4]бензодиазепин-11-она (1 экв.; 2,505 г,9,38 ммоль) в хлороформе (80 мл) добавляли фосфорилхлорид (5 экв.; 4,36 мл, 7,19 г, 46,89 ммоль), нагревали и перемешивали при 50C в течение ночи. Реакционный раствор декантировали и выпаривали в вакууме до получения масла. Указанное масло растворяли в ДХМ, промывали насыщенным водным раствором гидрокарбоната натрия. Раствор ДХМ сушили над Na2SO4, фильтровали и концентрировали до высыхания с получением 2,7,11-трихлор-5H-пирроло[2,1-с][1,4]бензодиазепина в виде окрашенного кремообразного твердого вещества. Одновременно обессоливали гидрохлорид метил-2,2-диметил-3(пиперазин-1-ил)пропаноата (0,837 г, 3,54 ммоль) путем растворения в метаноле, наносили раствор метанола на колонку, заполненную SCX-2 (10 г), промывали метанолом и затем элюировали 2,5 М аммиаком в растворе метанола, выпаривали аммиачную фракцию метанола с получением метил-2,2-диметил-3(пиперазин-1-ил)пропаноата (0,665 г) в виде масла. Указанное масло смешивали с 2,7,11-трихлор-5Hпирроло[2,1-с][1,4]бензодиазепином (0,505 г, 1,77 ммоль), карбонатом калия (0,733 г, 5,31 ммоль) и ацетонитрилом (20 мл) и нагревали при температуре обратной конденсации в течение ночи. Охлаждали реакционную смесь до комнатной температуры и фильтровали, промывая шлак EtOAc, затем выпаривали фильтрат в вакууме до образования твердого вещества. Хроматографировали на силикагеле при элюировании смесью метанол/ДХМ (градиентное элюирование 2,98-8,92). Выпаривали фракции, содержащие продукт, с получением метил-3-[4-(2,7-дихлор-5H-пирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1 ил]-2,2-диметилпропаноата (0,69 г). MC (m/z) 449,13/451,07 (М+1). Следующие соединения получали, по существу, с помощью способа получения 51. Метил-1-(5-метил-2-нитробензил)-4-(метилсульфанил)-1H-пиррол-2-карбоксилат (3,49 ммоль; 1,12 г) обрабатывали уксусной кислотой (15 мл), затем обрабатывали железным порошком (10,5 ммоль; 585 мг), затем медленно нагревали на масляной бане при 80C. Через несколько часов повышали температуру масляной бани до 90C. Затем реакционную смесь оставляли при 60C в течение 2 дней. Затем кон- 20022708 центрировали до высыхания для удаления уксусной кислоты, затем обрабатывали EtOAc и промывали через слой силикагеля дополнительной порцией EtOAc до тех пор, пока оранжевый цвет не переставал выходить. Концентрировали элюент красного цвета до высыхания, затем обрабатывали метанолом, затем заново концентрировали, растворяли в ДХМ (20 мл) и обрабатывали фосфорилхлоридом (1,0 мл). Реакционную смесь нагревали на масляной бане при 50C в течение ночи. Затем реакционную смесь охлаждали до комнатной температуры (RT) и обрабатывали льдом, затем дважды промывали водой и затемNHCO3 (водным раствором). Органический слой сушили над MgSO4, фильтровали и концентрировали до получения смолы. Между тем дигидрохлорид метил-2,2-диметил-3-(пиперазин-1-ил)пропаноата (1,60 г,5,86 ммоль) растворяли в воде, затем наносили на картридж, заполненный SCX-2 (210 г), и промывали метанолом и затем элюировали аммиаком в метаноле. Концентрировали основной раствор до высыхания до получения масла. Затем указанное масло растворяли в ацетонитриле (50 мл) и добавляли к смеси смолы и карбоната калия (1,0 г), затем нагревали до обратной конденсации. Через 2 ч реакционную смесь нагревали в микроволновой печи до 140C в течение 2 ч. Затем указанную реакционную смесь фильтровали и экстрагировали ацетонитрилом и ацетоном. Объединяли и концентрировали исходные растворы до высыхания на силикагеле, затем очищали с помощью флэш-хроматографии на силикагеле (40 г) (1050% EtOAc в гексане). Отбирали фракции, которые содержали чистый продукт, объединяли и концентрировали с получением метил-2,2-диметил-3-[4-(7-метил-2-(метилсульфанил)-5H-пирроло[2,1 с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]пропаноата (418 мг; 27% выход). МС (m/z): 441,18 (М+1). Способ получения 61. Синтез 7-хлор-2-(метилсульфанил)-11-(пиперазин-1-ил)-5H-пирроло[2,1-с][1,4]бензодиазепина(1,38 г, 4,75 ммоль) в хлороформе (20 мл) добавляли фосфорилхлорид (5 экв.; 2,21 мл, 3,64 г, 23,76 ммоль) и нагревали и перемешивали при 45C в течение 2 ч. Повышали температуру реакции до 60C и перемешивали в течение дополнительных 2 ч. Удаляли хлороформ в вакууме и растворяли осадок в ДХМ(100 мл) и промывали насыщенным раствором бикарбоната натрия (100 мл). Фильтровали слой ДХМ через фрит для разделения фаз и выпаривали растворитель в вакууме с получением вязкого масла. Растворяли масло в ацетонитриле (10 мл) и добавляли карбонат цезия (3 экв.; 4,65 г, 14,26 ммоль). Добавляли к указанной смеси пиперазин (10 экв.; 4,09 г, 47,52 ммоль) в сухом ацетонитриле (10 мл), нагревали и перемешивали смесь при температуре обратной конденсации в течение ночи. Охлаждали реакционную смесь, добавляли насыщенный раствор хлорида аммония (100 мл) и экстрагировали EtOAc (250 мл). Объединяли органические слои и промывали водой (3100 мл), сушили над MgSO4, фильтровали и выпаривали растворитель в вакууме с получением 7-хлор-2-(метилсульфанил)-11-(пиперазин-1-ил)-5Hпирроло[2,1-с][1,4]бензодиазепина (1,0 г, 60%). МС (m/z): 347,13 (М+1). Способ получения 62. Синтез метил-3-[4-(7-хлор-2-(метилсульфанил)-5H-пирроло[2,1-с][1,4]бензодиазепин-11 ил)пиперазин-1-ил]-2,2-диметилпропаноата(5 мл) и перемешивали в атмосфере азота в течение 30 мин при комнатной температуре. К реакционной смеси добавляли триацетоксиборгидрид натрия (3 экв.; 1,89 г, 8,56 ммоль) и перемешивали в атмосфере азота в течение ночи при комнатной температуре. Добавляли метанол и наносили на колонку, заполненную SCX-2, промывали метанолом и элюировали 2 М аммиаком в метаноле. Выпаривали раствор метанола с получением метил-3-[4-(7-хлор-2-(метилсульфанил)-5H-пирроло[2,1-с][1,4]бензодиазепин-11 ил)пиперазин-1-ил]-2,2-диметилпропаноата (1,23 г, 84%). MC (m/z): 461,12 (М+1). Смешивали метил-3-[4-(2-бром-7-метил-5H-пирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1 ил]-2,2-диметилпропаноат (1,00 экв.; 600,00 мг, 1,27 ммоль), 4,4,5,5-тетраметил-2-(проп-1-ен-2-ил)-1,3,2 диоксаборолан (3,00 экв.; 638,93 мг, 3,80 ммоль) в метаноле (15 мл). Добавляли предварительно перемешанную смесь 1,09 мас.% трис-(дибензилиденацетонил)биспалладия (Pd2(dba)3), 1,16 мас.% 1,1'-бис-(дитрет-бутилфосфино)ферроцена (dtbpf) и 97,75 мас.% трифосфата калия (1,06 г) и нагревали при 140C в течение 25 мин в микроволновой печи. Фильтровали реакционную смесь через целит и разбавляли до объема 25 мл метанолом. Гидрогенизировали раствор метанола путем пропускания со скоростью 1 мл/мин через проточный гидрогенизатор H-Cube с использованием каталитического картриджа 10%Pd/C, при 50C. Выпаривали растворитель в вакууме с получением метил-2,2-диметил-3-4-[7-метил 2-(пропан-2-ил)-5H-пирроло[2,1-с][1,4]бензодиазепин-11-ил]пиперазин-1-илпропаноата. МС (m/z): 437,28 (М+1). Способ получения 64. Синтез метил-3-[4-(2-этил-7-метил-5H-пирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]2,2-диметилпропаноата Смешивали метил-3-[4-(2-бром-7-метил-5H-пирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1 ил]-2,2-диметилпропаноат (1,00 экв.; 620,00 мг, 1,31 ммоль), триэтенилбороксинпиридин (1,5 экв.; 472,79 мг, 1,96 ммоль;) в метаноле (15 мл). Добавляли предварительно перемешанную смесь 1,09 мас.% трис(дибензилиденацетонил)биспалладия (Pd2(dba)3), 1,16 мас.% 1,1'-бис-(ди-трет-бутилфосфино)ферроцена(dtbpf) и 97,75 мас.% трифосфата калия (1,10 г) и нагревали при 140C в течение 25 мин в микроволновой печи. Фильтровали реакционную смесь через целит и разбавляли до объема 25 мл метанолом. Гидрогенизировали раствор метанола путем пропускания со скоростью 1 мл/мин через проточный гидрогенизатор H-Cube с использованием каталитического картриджа 10%Pd/C при 50C. Выпаривали растворитель в вакууме с получением метил-3-[4-(2-этил-7-метил-5H-пирроло[2,1-с][1,4]бензодиазепин-11 ил)пиперазин-1-ил]-2,2-диметилпропаноата. МС (m/z): 423,18 (М+1). Пример 1. Синтез 3-[4-(7-хлор-2-метил-5H-пирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2 диметилпропановой кислоты К суспензии метил-3-[4-(7-хлор-2-метил-5H-пирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1 ил]-2,2-диметилпропаноата (28,0 г, 65,27 ммоль) в смеси изопропилового спирта (150 мл) и воды (150 мл) добавляли гидроксид натрия (3 экв., 7,83 г, 195,82 ммоль) и нагревали при 70C в течение 4 ч с получением прозрачного раствора. Нагревали при 70C в течение дополнительного 1 ч, затем немного охлаждали. Добавляли 5 М соляную кислоту с получением pH 8 и преципитировали твердое вещество белого цвета. Добавляли дополнительную порцию 5 М соляной кислоты с получением pH между 6,5 и 7. Напо- 22022708 ловину уменьшали объем растворителя и затем охлаждали колбу в холодильнике в течение 0,5 ч. Собирали полученное твердое вещество белого цвета посредством фильтрования и сушили в течение ночи в вакуумной печи при 40C над пентоксидом фосфора с получением 3-[4-(7-хлор-2-метил-5H-пирроло[2,1 с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2-диметилпропановой кислоты (26,5 г, выход 98%). МС(m/z) 415,3 (М+1). Температура плавления = 246,5C (ДСК, начальный момент). Следующие соединения получали, по существу, с помощью способа получения примера 1. К суспензии 3-[4-(7-хлор-2-метил-5H-пирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2 диметилпропановой кислоты (24,25 г, 58,44 ммоль) в изопропиловом спирте (250 мл) при 60C добавляли 4 М раствор соляной кислоты в диоксане (2,4 экв., 35,07 мл, 140,26 ммоль) в течение 10 мин с получением чистого раствора. Позволяли немного охлаждаться, затем выпаривали до получения твердого вещества не совсем белого цвета. Растирали с малым количеством диэтилового эфира и собирали порошковидное кремообразное твердое вещество посредством фильтрования. Сушили в вакуумной печи при 40C в течение ночи. Измельчали до получения тонкодисперсного порошка и сушили в вакуумной печи при 60C в течение 6 ч. За уровнем остаточного изопропилового спирта следили с помощью 1H ЯМР. Растворяли в теплом этаноле (350 мл) и выпаривали до высыхания. Растирали с этанолом (50 мл) и снова выпаривали до высыхания. Растирали с сухим диэтиловым эфиром (200 мл) и собирали полученное твердое вещество посредством фильтрования. Сушили в течение 6 ч в вакуумной печи при 50C с получением дигидрохлорида 3-[4-(7-хлор-2-метил-5H-пирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2 диметилпропановой кислоты (26,9 г, 94% выход). МС (m/z) 415,2 (М+1). Следующие соединения получали, по существу, с помощью способа получения примера 13. Дигидрохлорид метил-2,2-диметил-3-(пиперазин-1-ил)пропаноата (1,49 г, 5,47 ммоль) растворяли в воде и абсорбировали на колонке, заполненной SCX2. Колонку промывали метанолом и элюировали метил-2,2-диметил-3-пиперазин-1-илпропаноатом с 2 М аммиаком в метаноле. Удаляли метанол в вакууме и добавляли метил-2,2-диметил-3-(пиперазин-1-ил)пропаноат к ацетонитрилу (12 мл). К раствору ацетонитрила добавляли 2,11-дихлор-7-метил-5H-пирроло[2,1-с][1,4]бензодиазепин (0,85 г, 3,22 ммоль) и бикарбонат натрия (0,405 г, 4,83 ммоль). Нагревали и перемешивали в микроволновой печи в течение 30 мин при 140C. Охлаждали до комнатной температуры, реакционную смесь абсорбировали на силикагеле и очищали с помощью хроматографии (градиентное элюирование смесью EtOAc/изогексан 0 100%100 0%). Собирали фракции, содержащие продукт, выпаривали растворитель в вакууме и осадок растворяли в метаноле (10 мл) и добавляли гидроксид лития (0,235 г, 9,65 ммоль). Нагревали и перемешивали раствор метанола в микроволновой печи в течение 12,5 мин при 140C. Охлаждали до комнатной температуры и окисляли с помощью уксусной кислоты, затем растворитель выпаривали при пониженном давлении. Осадок растворяли в избытке 2 М раствора HCl (водного) и затем выпаривали до высыхания. Осадок растворяли в воде и иммобилизировали на макропористой смоле на основе гидрокарбоната полистирола (PL-HCO3). Промывали смолу водой и вымывали из указанной смолы с помощью 2 М раствораHCl (водного). Раствор выпаривали до высыхания с получением дигидрохлорида 3-[4-(2-хлор-7-метил 5H-пирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2-диметилпропановой кислоты (0,177 г,выход 11,28%). МС (m/z) 415,18 (М+1). Следующие соединения получали, по существу, с помощью способа получения примера 25. Дигидрохлорид 3-[4-(7-хлор-2-этил-5H-пирроло[2,1-с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]2,2-диметилпропановой кислоты (201 мг, 400 мкмоль) растворяли в воде и метаноле (5 мл), затем наносили на картридж SCX-2 (2 г). Промывали метанолом и затем вымывали раствором аммония в метаноле. Концентрировали основной раствор до высушивания с получением 3-[4-(7-хлор-2-этил-5 Н-пирроло[2,1 с][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2-диметилпропановой кислоты (160 мг, 373 мкмоль). Затем обрабатывали 2 н. раствором гидроксида натрия (187 мкл, 373 мкмоль) и водой (3 мл) для получения раствора. Затем лиофилизировали с получением 3-[4-(7-хлор-2-этил-5H-пирроло[2,1- 29
МПК / Метки
МПК: C07D 487/04
Метки: двойного, обратных, соединения, 5h-пирроло[2,1-c][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2-диметилпропановой, замещенные, действия, качестве, 5-ht2а, агонистов, кислоты
Код ссылки
<a href="https://eas.patents.su/30-22708-zameshhennye-soedineniya-5h-pirrolo21-c14benzodiazepin-11-ilpiperazin-1-il-22-dimetilpropanovojj-kisloty-v-kachestve-obratnyh-agonistov-h1-antagonistov-5-ht2a-dvojjnogo-dejjstviya.html" rel="bookmark" title="База патентов Евразийского Союза">Замещенные соединения [(5h-пирроло[2,1-c][1,4]бензодиазепин-11-ил)пиперазин-1-ил]-2,2-диметилпропановой кислоты в качестве обратных агонистов h1/антагонистов 5-ht2a двойного действия</a>
Соединения пиперазинилпиразинов в качестве агонистов или антагонистов серотонин 5-ht2 рецептора

Номер патента: 6552
Опубликовано: 24.02.2006
Автор: Нильссон Бьерн М.
МПК: A61K 31/445, A61K 31/4427, A61K 31/497...
Метки: пиперазинилпиразинов, соединения, качестве, серотонин, рецептора, агонистов, антагонистов, 5-ht2
Формула / Реферат:
1. Соединение общей формулы (I) в которой: (i) X и Y оба являются азотом и Z является CH, образуя производное пиразина, или (ii) X и Z оба являются CH и Y является азотом, образуя производное пиридина, или (iii) X является C-CF3, Z является CH и Y является азотом, образуя производное 4-трифторметилпиридина, или (iv) Y и Z оба являются азотом и X является CH, образуя производное пиримидина, и где R1 и R2, каждый независимо, выбирают из группы A,...
Производные фениламида 4-(бензил)пиперазин-1-карбоновой кислоты и родственные соединения в качестве модуляторов амида жирной кислоты гидролазы для лечения страхов, боли и других состояний

Номер патента: 12589
Опубликовано: 30.10.2009
Авторы: Сейерстад Марк, Аподака Ричард, Брайтенбухер Дж.Гай, Сяо Вэй, Паттабираман Канака
МПК: A61P 25/28, A61P 25/04, A61P 25/22...
Метки: жирной, страхов, лечения, кислоты, состояний, соединения, других, качестве, модуляторов, амида, производные, 4-(бензил)пиперазин-1-карбоновой, боли, фениламида, гидролазы, родственные
Формула / Реферат:
1. Соединение формулы (I) в которой Z означает -N- или >СН; R1 представляет собой -Н или -C1-4алкил; Ar1 означает 2-тиазолил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, каждый из которых является незамещенным или замещенным по атому углерода кольца одной или двумя Ra группами; где каждый остаток Ra независимо выбран из группы, включающей -C1-4алкил, -C2-4алкенил, -ОН, -OC1-4алкил, атом галогена, -CF3,...
Нафтилсульфоновые кислоты и родственные соединения в качестве агонистов поглощения глюкозы

Номер патента: 6704
Опубликовано: 24.02.2006
Авторы: Спивак Уэйн Р., Сонюйянь Ши, Лум Роберт Т., Парк Джеонг Веонг, Прасад В.В.С.В.Манчем, Скоу Стивен Р., Козловски Майкл Р., Робинсон Луиз
МПК: C07C 237/42, A61K 31/16, A61K 31/18...
Метки: соединения, нафтилсульфоновые, кислоты, родственные, поглощения, агонистов, глюкозы, качестве
Формула / Реферат:
1. Соединение формулы где R1 означает -C(O)R45 -SO2R4 или -C(O)NHR4; R3, каждый независимо, означает OH, -SO2OH, -SO2NH2, -C(O)OH или тетразолил; R4 выбирают из группы, включающей C1-C6алкил [необязательно замещенный одним или двумя карбонильными аминогруппами, фенилами (необязательно замещенными одним галогеном, C1-C6алкилом или C1-C6алкокси), фенилокси (необязательно замещенным одним галогеном, C1-C6алкилом или C1-C6алкокси), нафтил или...
(фенил)амиды 7-(пиперазин-1-илметил)-1h-индол-2-карбоновой кислоты и родственные соединения в качестве ингибиторов мар-киназы p38 для лечения заболеваний дыхательных путей

Номер патента: 21053
Опубликовано: 31.03.2015
Авторы: Гёггель Рольф, Химмельсбах Франк, Даманн Георг, Юнг Биргит, Вагнер Хольгер
МПК: A61P 11/00, C07D 209/42, A61K 31/404...
Метки: 7-(пиперазин-1-илметил)-1h-индол-2-карбоновой, качестве, дыхательных, кислоты, фенил)амиды, лечения, родственные, мар-киназы, путей, соединения, заболеваний, ингибиторов
Формула / Реферат:
1. Соединения общей формулы (I)в которой Ar обозначает заместитель формулы (II)R3 обозначает C1-C6-алкил, C3-C6-циклоалкил, C1-С4-алкоксигруппу, С1-C2-перфторалкил, 3-метилоксетан-3-ил, C1-C2-перфторалкоксигруппу, морфолинил, при этом циклоалкильный остаток необязательно может быть замещен C1-C3-алкилом;R4 обозначает Н, C1-С4-алкил, C1-С4-алкоксигруппу;R5 обозначает Н, C1-C5-алкилсульфониламиногруппу, C3-C6-циклоалкилсульфониламиногруппу,...
Замещенные бициклогексанкарбоновые кислоты и их производные в качестве антагонистов рецептора возбуждающих аминокислот, способ их получения и применение.

Номер патента: 894
Опубликовано: 26.06.2000
Авторы: Монн Джеймс Э., Мэсси Стивен М., Домингес-Фернандес Кармен, Хелтон Дэвид Р.
МПК: A61K 31/19, C07C 211/38, C07D 233/58...
Метки: производные, антагонистов, рецептора, применение, кислоты, замещенные, аминокислот, возбуждающих, получения, бициклогексанкарбоновые, качестве, способ
Формула / Реферат:
1. Соединение формулы где Х представляет собой связь, S, О или NRa; R представляет собой группу (1-4С)алкил или группу фенил(1-4С)алкил, либо дифенил(1-4С)алкил, в которой фенильное кольцо является незамещенным или замещенным одним, двумя или тремя заместителями, выбранными независимо из галогена, (1-4С)алкила, (1-4С)алкокси, (1-4С)фторалкила, (1-4С)фторалкокси, фенила, фенокси, 3-трифторметилфенокси и 4-хлорфенокси; Ra представляет водород...
Предыдущий патент: Санитарный агент
Следующий патент: Система автоматического распознавания конфигурации ветви привода и/или количества активных подвижных элементов рабочего органа рабочей машины
Случайный патент: Детектор землетрясения