Ингибиторы jak2 и их применение для лечения миелопролиферативных заболеваний и злокачественной опухоли
Номер патента: 22488
Опубликовано: 29.01.2016
Авторы: Ван Хунхе, Ингрим Дженнифер, Гребински Джеймс В., Харт Эми, Пьюрандэер Ашок В., Шрёдер Гретчен


























Формула / Реферат
1. Соединение формулы (I)

или его фармацевтически приемлемая соль,
где X представляет собой С1-6алкил;
Y представляет собой С1-4алкил;
R представляет собой

каждый из которых необязательно конденсирован с 5- или 6-членным углеродным кольцом, которое является насыщенным, частично ненасыщенным или полностью ненасыщенным или гетероциклом, причем указанный гетероцикл содержит один гетероатом, выбранный из NR3 или S, указанные конденсированные углеродное кольцо или гетероцикл необязательно замещены 0-3 R1;
R1 представляет собой Н, галоген, CN, C1-6алкил, замещенный 0-3 Rc, CF3, CONRaRa, NRaRa, COORb, SO2-С1-4алкил, C(O)Rd, С3-6циклоалкил, замещенный 0-3 Re, фуранил, тетрагидропиранил или пиридинил;
R2 представляет собой Н, C1-6алкил, замещенный 0-3 Rc, С(О)О-С1-4алкил, SO2-С1-4алкил, С3-6циклоалкил, замещенный 0-3 Re, тетрагидропиранил;
R3 представляет собой Н, С(О)О-С1-4алкил;
Ra представляет собой Н, С1-6алкил, замещенный 0-3 Re, С3-6циклоалкил, замещенный 0-3 Re, тетрагидропиранил или диоксотетрагидротиофенил;
Rb представляет собой Н или С1-6алкил;
Rc представляет собой Н, галоген, CN, ОН, О-С1-4алкил, О-С1-4алкил-О-С1-4алкил, NH2, N(C1-4алкил)2, C(O)N(C1-4алкил)2, SO2-С1-6алкил или морфолинил или пиперазинил, каждый из которых необязательно замещен 0-1 С1-4алкилом;
Rd представляет собой С1-6алкил или азеридинил, азетидинил, пирролидинил, пиперидинил, морфолинил, пиперазинил, диоксидотиоморфолинил или тетрагидропиранил, каждый из которых замещен 0-2 Re; и
Re представляет собой Н, галоген, CN, С1-4алкил, ОН, О-С1-4алкил, SO2-C1-4алкил, NHC(O)-C1-4алкил, морфолинил, ОС(О)-С1-4алкил, C(O)N(C1-4алкил)2 или О-С1-4алкил-О-С1-4алкил.
2. Соединение по п.1, где
X представляет собой этил;
Y представляет собой метил и
R представляет собой

каждый из которых необязательно замещен 0-3 R1;
или его фармацевтически приемлемая соль.
3. Соединение по п.2, где R представляет собой

4. Соединение по п.2, где R представляет собой

каждый из которых необязательно замещен 0-2 R1.
5. Соединение по п.2, где
R представляет собой
R1 представляет собой Н, галоген, CN, С1-6алкил, замещенный 0-3 Rc, CF3, CONRaRa, COORb, SO2-С1-4алкил, C(O)Rd, С3-6циклоалкил, замещенный 0-3 Re, или пиридинил;
Ra представляет собой Н, С1-6алкил, замещенный 0-3 Re, С3-6циклоалкил, замещенный 0-3 Re, тетрагидропиранил или диоксотетрагидротиофенил;
Rb представляет собой Н или С1-6алкил;
Rc представляет собой Н, галоген, ОН, О-С1-4алкил, SO2-С1-4алкил или морфолинил;
Rd представляет собой С1-6алкил или азетидинил, пирролидинил, морфолинил, пиперазинил или диоксидотиоморфолинил, каждый из которых замещен 0-2 Re; и
Re представляет собой Н, галоген, CN, ОН, О-С1-4алкил, SO2-C1-4алкил, NHC(O)-С1-4алкил или морфолинил;
или его фармацевтически приемлемая соль.
6. Соединение по п.2, где
R представляет собой
R1 представляет собой Н, галоген, С1-6алкил, замещенный 0-3 Rc, CF3, CONRaRa, COORb, C(O)Rd, С3-6циклоалкил, замещенный 0-3 Re, или фуранил;
R2 представляет собой Н, C1-6алкил, замещенный 0-3 Rc, SO2-С1-4алкил, С3-6циклоалкил, замещенный 0-3 Re, или тетрагидропиранил;
Ra представляет собой Н или С1-6алкил, замещенный 0-3 Re;
Rb представляет собой Н или C1-6алкил;
Rc представляет собой Н, галоген, CN, ОН, О-С1-6алкил, О-С1-4алкил-О-С1-4алкил, NH2, N(С1-4алкил)2, С(О)N(С1-4алкил)2, SO2-С1-4алкил или морфолинил или пиперазинил, каждый из которых необязательно замещен 0-1 С1-4алкилом;
Rd представляет собой С1-6алкил или морфолинил, пиперазинил или диоксидотиоморфолинил, каждый из которых замещен 0-2 Re;
Re представляет собой Н, С1-4алкил, CN, ОН, NHC(O)-С1-4алкил или морфолинил;
или его фармацевтически приемлемая соль.
7. Соединение по п.6, где
R1 представляет собой С1-6алкил, замещенный 0-3 Rc; и R2 представляет собой С1-6алкил.
8. Соединение по п.1, которое представляет собой N,N-дициклопропил-4-(1,5-диметил-1Н-пиразол-3-иламино)-6-этил-1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7-карбоксамид

или его фармацевтически приемлемую соль.
9. Фармацевтическая композиция для лечения пролиферативных заболеваний и рака, содержащая одно или несколько соединений по любому из пп.1-8 или их фармацевтически приемлемые соли и фармацевтически приемлемый носитель.
10. Применение соединения по любому из пп.1-8 или его фармацевтически приемлемых солей в терапии пролиферативных заболеваний и рака.
11. Применение соединения по любому из пп.1-8 или его фармацевтически приемлемых солей для производства лекарственного препарата для лечения миелопролиферативных заболеваний, лейкоза или лимфомы.
12. Применение по п.11, причем лейкоз выбран из острого миелоидного лейкоза, включая рефрактерный острый миелоидный лейкоз, или острого лимфоидного лейкоза.
13. Применение по п.11, причем миелопролиферативное заболевание представляет собой истинную полицитемию, эссенциальную тромбоцитемию или первичный миелофиброз.
14. Применение соединения по любому из пп.1-8 или его фармацевтически приемлемых солей в качестве активного средства для лечения миелопролиферативных заболеваний или лейкоза или лимфомы у млекопитающих.
15. Применение по п.14, причем лейкоз выбран из острого миелоидного лейкоза, включая рефрактерный острый миелоидный лейкоз, или острого лимфоидного лейкоза.
16. Применение по п.14, причем миелопролиферативное заболевание представляет собой истинную полицитемию, эссенциальную тромбоцитемию или первичный миелофиброз.
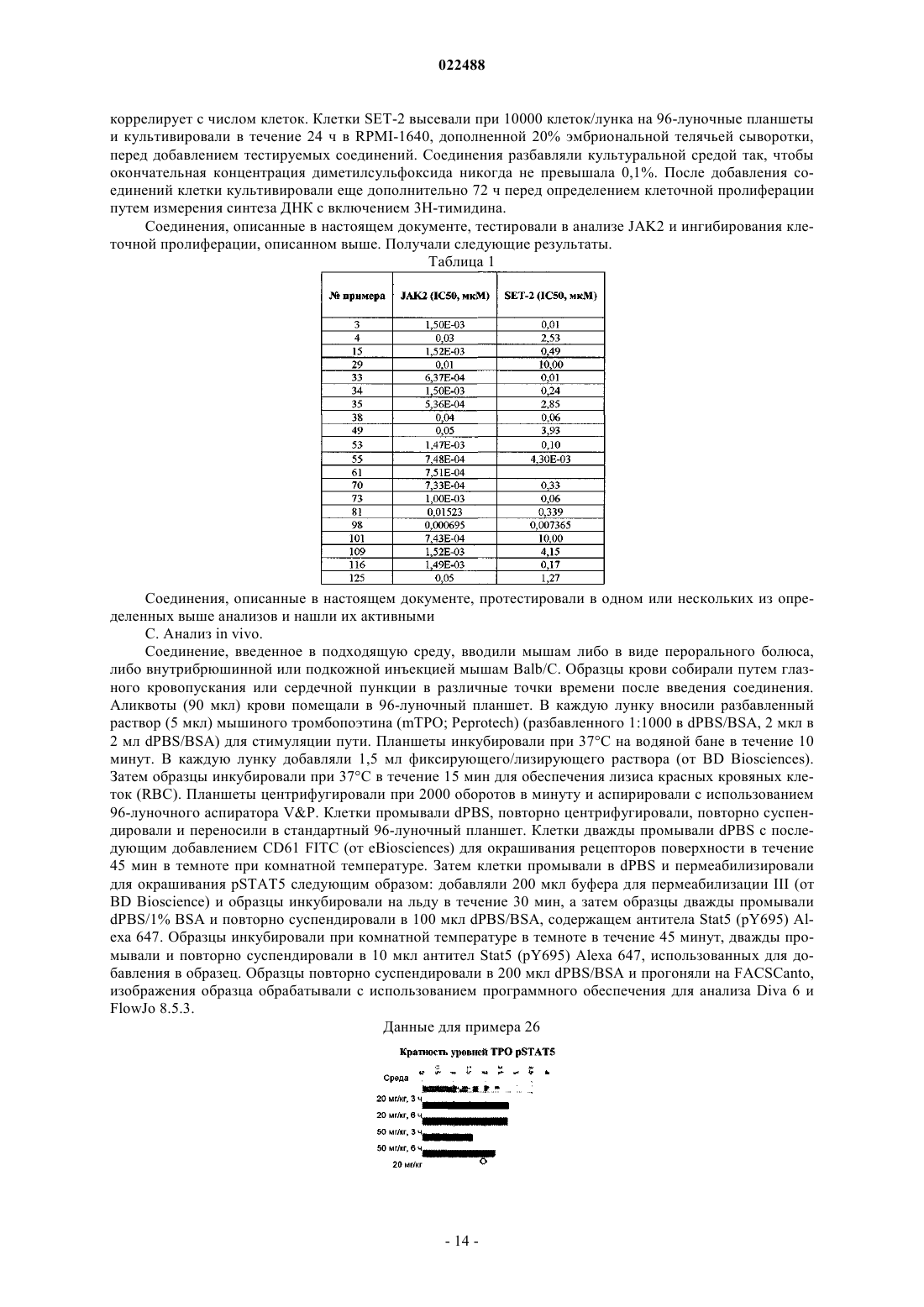
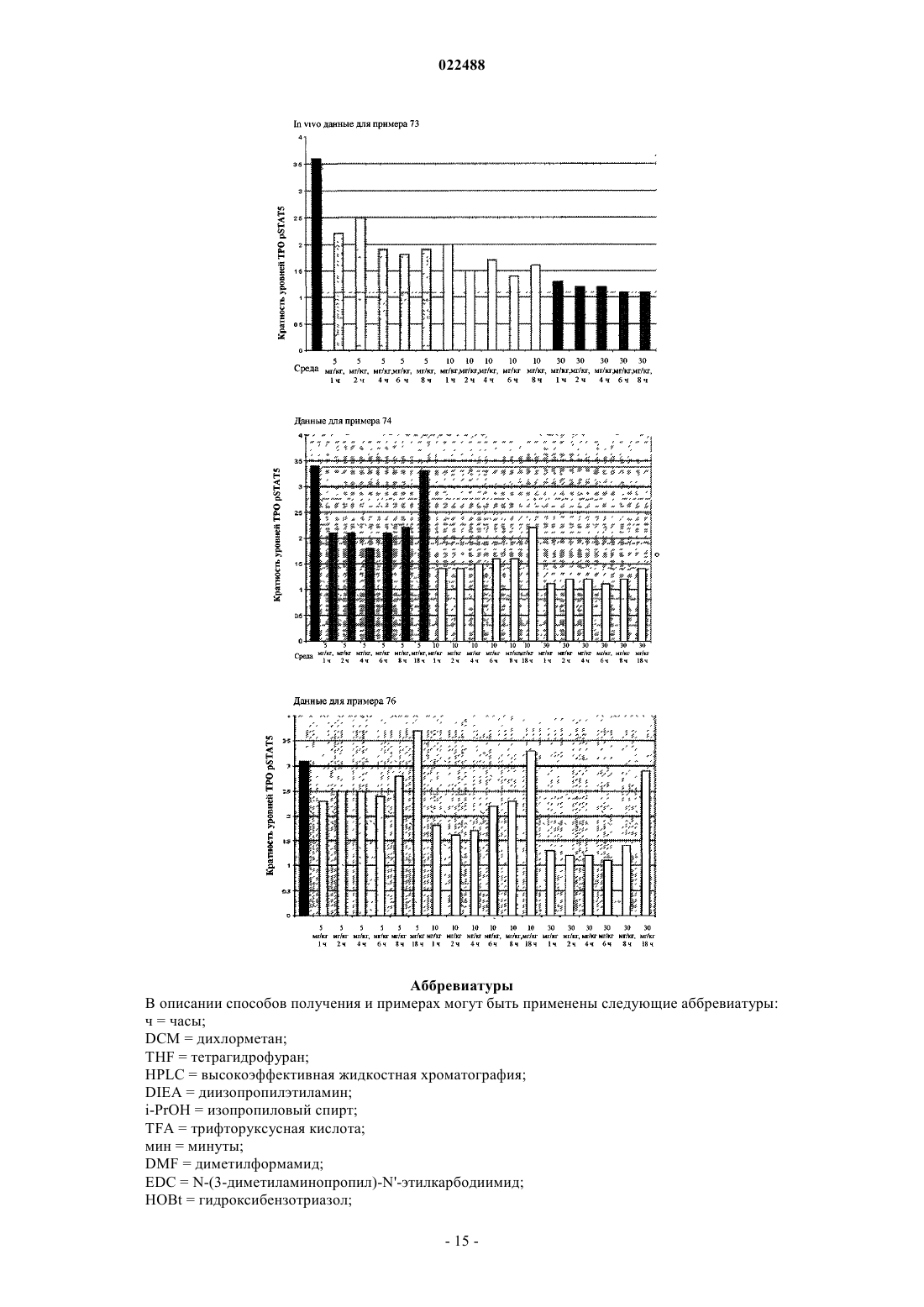
Текст
ИНГИБИТОРЫ JAK2 И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ МИЕЛОПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ И ЗЛОКАЧЕСТВЕННОЙ ОПУХОЛИ и их фармацевтически приемлемым солям. Соединения формулы I ингибируют тирозинкиназную активность JAK2, что делает их применимыми в качестве антипролиферативных средств для лечения злокачественной опухули и других заболевания. Область техники Изобретение относится к новым соединениям, которые применимы в качестве противораковых/антипролиферативных средств. Настоящее изобретение также относится к способу применения соединений в лечении пролиферативных заболеваний, таких как злокачественная опухоль, и к фармацевтическим композициям, содержащим эти соединения. Уровень техники Попытки определить новые терапевтические подходы к лечению Ph(-) миелопролиферативного заболевания были подкреплены наблюдениями конститутивной активации сигнального пути JAK-Stat у больных MPD. В частности, однонаправленная мутация валина на фенилаланин по остатку 617 (JAK2V617F) в JAK2 наблюдалась у большинства больных PV (95%), ЕТ (50-60%) и PMF (50-60%) (табл. 2,Kralovics et al., 2005; Baxter et al., 2005; Tefferi et al., 2005). Мутация V617F находится на участке генаJAK2, кодирующего псевдокиназный домен, который, как полагают, функционирует как автоингибиторный домен с регулированием тирозинкиназной активности JAK2. Мутации в экзоне 12 JAK2, которые также приводят к конститутивной киназной активности JAK2, также наблюдались с меньшей частотойal., 2007). JAK2 является членом семейства нерецепторных тирозинкиназ, которое также включает JAK1,Тук 2 и JAK3, и функционирует как медиатор передачи сигнала с рецептора цитокина (для обзора см.Murray, 2007). При связывании цитокина с его собственным рецептором связанные с рецептором членыJAK семейства активируются и фосфорилируют STAT, латентный фактор транскрипции, который при опосредованном JAK фосфорилировании подвергается димеризации и транслокации в ядро для регулирования генной экспрессии. Генетические и биохимические исследования установили отдельные комбинации взаимодействия члена JAK семейства с отдельным цитокиновым рецептором. Например, взаимодействие рецепторов эритропоэтина (ЕРО), тромбопоэтина (ТРО) и гранулоцитарного колониестимулирующего фактора (GM-CSF) приводит к преобладающей активации JAK2 для опосредованной нисходящей передачи сигнала. В соответствии с патофизиологией MPD, связанного с мутацией JAK2-V617F, эти цитокины способствуют дифференцировке и размножению клеточных типов, лежащих в основе развитияPV, ЕТ и PMF, соответственно. В отличие от других генных событий активации экспрессия JAK2-V617F не является достаточной для обеспечения трансформации в клеточных модельных системах, и была показана необходимость коэкспрессии цитокиновых рецепторов I типа, что подчеркивает важную функциональную зависимость взаимодействия JAK-цитокиновый рецептор (Lu et al., 2005). Интересно, что активирующие мутации в рецепторе ТРО (MPL, замена триптофана на лейцин по остатку 515), которые приводят к конститутивной активации JAK2-Stat, были выявлены у больных MPD, страдающих JAK2V617F-негативными PMF и ЕТ (5% и 1%, соответственно) (Pikman et al., 2006). Эти наблюдения показывают, что передача сигнала JAK-Stat может быть активирована при MPD посредством мутации в нескольких точках пути взаимоисключающим образом, и предполагают возможность существования дополнительных мутаций пути при JAK2-V617F и MPL-W515L-негативной MPD. Важное подтверждение передачи сигнала JAK2 как ведущего фактора Ph(-) MPD получили на моделях грызунов, у которых передача сигнала при мутации JAK2-V617F была восстановлена в компартменте стволовой кроветворной клетки. В некоторых лабораториях было продемонстрировано, что вирусная трансдукция JAK2-V617F в костный мозг мыши и последующая реимплантация реципиентным мышам воссоздавали некоторые аспекты MPD человека (Wernig et al. 2006, Lacout et al., 2006, Bumm etal., 2006, Zaeleskas et al., 2006). Эти признаки включали повышенный гематокрит, спленомегалию от экстрамедуллярного гемопоэза, гранулоцитоз и фиброз костного мозга, каждый из которых также проявляется при истинной полицитемии. Интересно, что в отличие от состояния человека в этих мышиных моделях тромбоцитоз не наблюдался, и, как предполагается, был вызван вторичными генными событиями,которые участвуют в размножении тромбоцитов (Wernig et al., 2006). Подобное воссоздание мутации рецептора ТРО (MPL-W515L) в костном мозге грызунов приводило к развитию миелопролиферативного заболевания с более быстрым началом, чем у животных с JAK2-V617F, что напоминало первичный миелофиброз, включающий спленомегалию, гепатомегалию и ретикулиновый фиброз костного мозга (Pikman et al., 2006). Также в отличие от модели JAK2-V617F у мышей, экспрессирующих MPL-W515L, наблюдался сильный тромбоцитоз, возможно, указывающий на доминантную функцию рецепторной активации в размножении этой линии по сравнению с JAK2-V617. Тем не менее, в совокупности эти наблюдения подчеркивают роль как MPL-W515L, так и JAK2-V617F как ведущих мутаций, лежащих в основе развития MPD у человека. Ключевым вопросом генетической основы MPD является роль дополнительных генных событий,которые участвуют в развитии заболевания, помимо JAK2 и MPL. Несколько доказательств свидетельствую о дополнительных генных изменениях в развитии MPD. Действительно, у больных MPD часто встречается митотическая рекомбинация с образованием двух аллелей JAK2-V617F, что указывает на отбор по клеточным клонам, гомозиготным по мутантной киназе (Levine et al. 2005). В связи с этим важно получить животных с условным включением JAK2-V617F и определить фенотипические последствия гомозиготной нагрузки JAK2-V617F по сравнению с гетерозиготной. Кроме того, имеются данные о наследуемом через гаметы аллеле, который предшествует и предрасполагает пациентов к приобретениюJAK2-V617F (Goerttler et al., 2005; Levine et al., 2006), а также о потере хромосомного участка 20q у некоторых больных MPD. Хотя конверсия MPD в AML клинически наблюдается на умеренных уровнях, а активирующие хромосомные транслокации JAK наблюдаются при лейкозе, эпидемиологические данные свидетельствуют о сомнительности того, что JAK2-V617F является ведущим генным фактором в этом случае, что позволяет предположить, что для полной лейкозной трансформации необходимы дополнительные генные изменения (Theocharides et al., 2007). Несмотря на эти замечания ингибирование JAK2 низкомолекулярными ингибиторами является достаточным для модулирования развития заболевания на доклинических животных моделях, что позволяет предположить, что активации JAK2 достаточно для поддержания MPD (Paradani et al., 2007). Будет важно идентифицировать эти дополнительные генные изменения и выяснить, как эти генные изменения участвуют в развитии заболевания PV, ЕТ и PMF в связи с JAK2-V617F и MPL-W515L. Это также будет иметь важное значение для реализации подходов к выяснению, являются ли мутантными другие компоненты пути JAK2 у больных MPD, не связанных с приобретением мутаций JAK2-V617F, экзона 12 JAK2 или MPL-W515L. В патентной публикация WO 2006/122137 раскрываются соединения, которые применимы в качестве IKK ингибиторов. В примереА 171 раскрывается соединение формулы которое, как было выявлено, обладает слабой активностью в отношении к JAK2 в анализе, описанном в настоящем документе ниже. Подробное описание изобретения Изобретение относится к соединениям формулы I, фармацевтическим композициям, содержащим такие соединения, и к способам применения таких соединений. В соответствии с настоящим изобретением раскрыты соединения формулы I или фармацевтически приемлемая соль, где X представляет собой С 1-4 алкил; Y представляет собой С 1-4 алкил; R представляет собой каждый из которых необязательно конденсирован с 5- или 6-членным углеродным кольцом, которое является насыщенным, частично ненасыщенным или полностью ненасыщенным или гетероциклом,причем указанный гетероцикл содержит один гетероатом, выбранный из NR3 или S, указанные конденсированные углеродное кольцо или гетероцикл необязательно замещены 0-3 R1;Rc представляет собой Н, галоген, CN, ОН, О-С 1-4 алкил, О-С 1-4 алкил-О-С 1-4 алкил, NH2, N(C1-4 алкил)2, C(O)N(C1-4 алкил)2, SO2-С 1-4 алкил или морфолинил или пиперазинил, каждый из которых необязательно замещен 0-1 С 1-4 алкилом;Rd представляет собой С 1-6 алкил или азеридинил, азетидинил, пирролидинил, пиперидинил, морфолинил, пиперазинил, диоксидотиоморфолинил или тетрагидропиранил, каждый из которых замещен 0-2Re представляет собой Н, галоген, CN, С 1-4 алкил, ОН, О-С 1-4 алкил, SO2-C1-4 алкил, NHC(O)-С 1-4 алкил, морфолинил, ОС(О)-С 1-4 алкил, С(О)N(С 1-4 алкил)2 или O-C1-4 алкил-О-С 1-4 алкил; при этом "гетероцикл" является насыщенным, частично ненасыщенным или полностью ненасыщенным, и содержит 1, 2,3 или 4 гетероатомов, независимо выбранных из N, О и S. В другом варианте осуществления представлены соединения формулы (I), где каждый из которых необязательно замещен 0-3 R1. В другом варианте осуществления представлены соединения формулы (I), гдеY представляет собой метил иX представляет собой этил. В другом варианте осуществления представлены соединения формулы (I), где В другом варианте осуществления представлены соединения формулы (I), где R представляет собой каждый из которых необязательно замещен 0-2 R1. В другом варианте осуществления представлены соединения формулы (I), гдеRd представляет собой C1-6 алкил или азетидинил, пирролидинил, морфолинил, пиперазинил или диоксидотиоморфолинил, каждый из которых замещен 0-2 Re;Re представляет собой Н, галоген, CN, ОН, О-С 1-6 алкил. SO2-C1-4 алкил, NHC(O)-С 1-6 алкил или морфолинил. В другом варианте осуществления представлены соединения формулы (I), гдеRc представляет собой Н, галоген, CN, ОН, О-С 1-4 алкил, О-С 1-4 алкил-О-С 1-4 алкил, NH2, N(C1-4 алкил)2, С(О)N(С 1-4 алкил)2, SO2-С 1-4 алкил или морфолинил или пиперазинил, каждый из которых необязательно замещен 0-1 С 1-6 алкилом;Rd представлет собой С 1-6 алкил или морфолинил, пиперазинил или диоксидотиоморфолинил, каждый из которых замещен 0-2 Re; иRe представляет собой Н, С 1-4 алкил, CN, ОН, NHC(O)-C1-4 алкил или морфолинил. В другом варианте осуществления представлены соединения формулы (I), гдеR2 представляет собой С 1-6 алкил. В другом варианте осуществления представлены соединения формулы (I), где соединение формулы(I) выбрано из представленных в качестве примера соединений. В другом варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение по настоящему изобретению или его фармацевтически приемлемую соль. В другом варианте осуществления настоящее изобретение относится к фармацевтической композиции, содержащей соединение формулы I или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель. В другом варианте осуществления настоящее изобретение относится к способу лечения миелопролиферативного заболевания, такого как истинная полицитемия, эссенциальная тромбоцитемия и первичный миелофиброз, включая множественную миелому, нейробластому, глиобластому, системный мастоцитоз и гемобластозы, такие как острый миелоидный лейкоз (включая рефрактерный острый миелоидный лейкоз) и острый лимфоидный лейкоз. В другом варианте осуществления настоящее изобретение также относится к применению соединения формулы I по настоящему изобретению для производства лекарственного препарата для лечения пролиферативного расстройства и/или злокачественной опухоли. Настоящее изобретение может быть осуществлено в других конкретных формах без отклонения от его сущности или основных признаков. Настоящее изобретение охватывает все отмеченные в настоящем документе комбинации предпочтительных аспектов и/или вариантов осуществления настоящего изобретения. Подразумевается, что возможные варианты осуществления по настоящему изобретению могут быть взяты в сочетании с любым другим вариантом осуществления или вариантами осуществления для описания дополнительных более предпочтительных вариантов осуществления. Также подразумевается,что каждый отдельный элемент предпочтительных вариантов осуществления представляет собой свой собственный независимый предпочтительный вариант осуществления. Кроме того, подразумевается, что для описания дополнительного варианта осуществления любой элемент варианта осуществления объединен со всевозможными другими элементами из любого варианта осуществления. Определения В настоящем описании могут быть использованы следующие определения терминов. Если не указано иное, первоначальное определение, предусмотренное в документе для группы или термина, подходит к такой группе или термину по всему настоящему описанию отдельно или в составе другой группы. Соединения по настоящему изобретению могут содержать один или несколько асимметричных центров. Если не указано иное, в настоящее изобретение включены все хиральные (энантиомерные и диастереоизомерные) и рацемические формы соединений по настоящему изобретению. В соединениях также может находиться много геометрических изомеров олефинов, двойных связей C=N и т.п., и все такие стабильные изомеры предусмотрены настоящим изобретением. Описаны цис- и трансгеометрические изомеры соединений по настоящему изобретению, и они могут быть выделены в виде смеси изомеров или в виде отдельных изомерных форм. Соединения по настоящему изобретению могут быть выделены в оптически активных или рацемических формах. Из предшествующего уровня техники хорошо известно, как получать оптически активные формы, например, путем повторного растворения рацемических форм или путем синтеза из оптически активных исходных веществ. Подразумеваются все хиральные (энантиомерные и диастереомерные) и рацемические формы и все геометрические изомерные формы структурной формулы, если конкретно не обозначена специфическая стереохимия или изомерная форма. Используемый в настоящем документе термин "замещенный" означает, что каждый один или несколько атомов водорода на обозначенном атоме замещены путем выбора из обозначенной группы при условии, что нормальная валентность атома не превышена, и что замещение приводит к стабильному соединению. Если заместитель представляет собой кетогруппу (т.е. =0), 2 атома водорода на атоме замещены. Кетоновые заместители не присутствуют в ароматических фрагментах. Используемые двойные связи в кольце представляют собой двойные связи, которые образованы между двумя смежными атомами в кольце (например, С=С, C=N или N=N). Когда любая переменная (например, R3) встречается более одного раза в любом компоненте или формуле соединения, ее определение в каждом случае не зависит от ее определения при каждых других появлениях. Таким образом, например, если группа представлена как замещенная 0-2 R3, тогда указанная группа может необязательно быть замещена R3 группами числом до двух, и R3 при каждом случае выбран независимо от определения R3. Также комбинации заместителей и/или переменных являются допустимыми, только если такие комбинации приводят к стабильным соединениям. Если связь с заместителем представлена как перекрестная связь, связывающая два атома в кольце,тогда такой заместитель может быть связан с любым атомом на кольце. Если заместитель представлен без указания атома, через который такой заместитель связан с оставшимся соединением настоящей формулы, тогда такой заместитель может быть связан через любой атом в таком заместителе. Комбинации заместителей и/или переменных являются допустимыми, только если такие комбинации приводят к стабильным соединениям В случаях, где в соединениях по настоящему изобретению присутствуют атомы азота (например,амины), они могут быть превращены в N-оксиды путем обработки окислителем (например, МСРВА и/или пероксидами водорода) для получения других соединений по настоящему изобретению. Таким образом, считается, что все представленные и заявленные атомы азота охватывают как представленный атом азота, так и его производное N-оксид (N О). Подразумевается, что используемые в настоящем документе термины "алкил" или "алкилен" включают как разветвленные, так и неразветвленные насыщенные алифатические углеводородные группы,содержащие точно определенное число атомов углерода. Например, подразумевается, что "C1-10 алкил"(или алкилен) включает С 1, С 2, С 3, С 4, С 5, С 6, С 7, C8, C9 и С 10 алкильные группы. Кроме того, например,"C1-С 6 алкил" обозначает алкил, содержащий 1-6 атомов углерода. Алкильные группы могут быть незамещенными или замещенными, таким образом один или несколько его атомов водорода замещены другой химической группой. Пример алкильных групп включает без ограничения метил (Me), этил (Et),пропил (например, н-пропил и изопропил), бутил (например, н-бутил, изобутил, трет-бутил), пентил (например, н-пентил, изопентил, неопентил) и т.п. Подразумевается, что "алкенил" или "алкенилен" включают углеводородные цепи неразветвленной или разветвленной конфигурации и содержат одну или несколько двойных связей углерод-углерод, которые могут возникать в любом устойчивом положении вдоль цепи. Например, подразумевается, что"С 2-6 алкенил" (или алкенилен) включает С 2, С 3, С 4, С 5 и С 6 алкенильные группы. Примеры алкенила включают без ограничения этенил, 1-пропенил, 2-пропенил, 2-бутенил, 3-бутенил, 2-пентенил, 3-пентенил, 4-пентенил, 2-гексенил, 3-гексенил, 4-гексенил, 5-гексенил, 2-метил-2-пропенил, 4-метил-3-пентенил и т.п. Подразумевается, что "алкинил" или "алкинилен" включает углеводородные цепи неразветвленной или разветвленной конфигурации и содержат одну или несколько тройных связей углерод-углерод, которые могут возникать в любом устойчивом положении вдоль цепи. Например, подразумевается, что "С 2-6 алкинил" (или алкинилен) включает С 2, С 3, С 4, C5 и С 6 алкинильные группы, такие как этинил, пропинил,бутинил, пентинил, гексинил и т.п."Гало" или "галоген" включают в себя фтор, хлор, бром и йод. Термин "циклоалкил" относится к циклическим алкильным группам, включая моно-, би- или полициклические кольцевые системы. Подразумевается, что С 3-10 циклоалкил включает С 3, С 4, С 5, С 6 и С 7 циклоалкильные группы. Пример циклоалкильных групп включает без ограничения циклопропил, циклобутил, циклопентил, циклогексил, норборнил и т.п. Подразумевается, что используемый в настоящем документе термин "углеродное кольцо" или "карбоциклический остаток" означает любое стабильное 3-,4-, 5-, 6- или 7-членное моноциклическое или бициклическое или 7-, 8-, 9-, 10-, 11-, 12- или 13-членное бициклическое или трициклическое кольцо, каждое из которых может быть насыщенным, частично ненасыщенным, ненасыщенным или ароматическим. Примеры таких углеродных циклов включают без ограничения циклопропил, циклобутил, циклобутенил, циклопентил, циклопентенил, циклогексил, циклогептенил, циклогептил, циклогептенил, адамантил, циклооктил. циклооктенил, циклооктадиенил,[3.3.0]бициклооктан, [4.3.0]бициклононан, [4.4.0]бициклодекан, [2.2.2]бициклооктан, фторенил, фенил,нафтил, инданил, адамантил, антраценил и тетрагидронафтил (тетралин). Указанные выше соединенные мостиковой связью кольца также включены в определение углеродного цикла (например,[2.2.2]бициклооктан). Если не указано иное, предпочтительные углеродные циклы представляют собой циклопропил, циклобутил, циклопентил, циклогексил, фенил и инданил. Подразумевается, что при использовании термина "углеродное кольцо" он включает в себя "арил". Если один или несколько атомов углерода связаны с двумя несмежными атомами углерода возникает соединенное мостиковой связью кольцо. Предпочтительные мостики представляют собой один или два атома углерода. Отметим, что мостик всегда превращает моноциклическое кольцо в трициклическое кольцо. Если кольцо соединено мос-5 022488 тиковой связью, в мостике также могут находиться перечисленные для кольца заместители. Термин "арил" относится к моноциклическим или бициклическим ароматическим углеводородным группам, содержащим 6-12 атомов углерода в части кольца, таким как фенильная, нафтильная, бифенильная и дифенильная группы, каждая из которых может быть замещена. Термин "замещенный арил" относится к арильной группе, замещенной, например, одним-четырьмя заместителями, такими как алкил, замещенный алкил, алкенил, замещенный алкенил, алкинил, замещенный алкинил, арил, замещенный арил, арилалкил, галоген, трифторметокси, трифторметил, гидрокси,алкокси, алканоил, алканоилокси, арилокси, арилалкилокси, амино, алкиламино, ариламино, арилалкиламино, диалкиламино, алканоиламино, тиол, алкилтио, уреидо, нитро, циано, карбокси, карбоксиалкил,карбамид, алкоксикарбонил, алкилтионо, арилтионо, арилсульфониламин, сульфоновая кислота, алкилсульфонил, сульфонамидо, арилокси и т.п. Заместитель может быть дополнительно замещен гидрокси,галогеном, алкилом, алкокси, алкенилом, алкинилом, арилом или арилалкилом. Термин "гетероарил" относится к необязательно замещенной ароматической группе, например, которая представляет собой 4-7-членную моноциклическую, 7-11-членную бициклическую или 10-15 членную трициклическую кольцевую систему, которая содержит по меньшей мере один гетероатом и по меньшей мере одно содержащее атом углерода кольцо, например пиридин, тетразол, индазол. Подразумевается, что используемые в настоящем документе термины "гетероцикл", "гетероциклил", "гетероциклическая система" или "гетероциклическая группа" означают стабильное 5-, 6- или 7 членное моноциклическое или бициклическое или 7-, 8-, 9-, 10-, 11-, 12-, 13- или 14-членное бициклическое гетероциклическое кольцо, которое является насыщенным, частично ненасыщенным или полностью ненасыщенным, и которое состоит из атомов углерода и 1, 2, 3 или 4 гетероатомов, независимо выбранных из N, О и S; и содержит любую бициклическую группу, в которой любое из вышеопределенных гетероциклических колец конденсировано с бензольным кольцом. Гетероатомы азота и серы необязательно могут быть окислены (т.е., NO и S(O)p). Атом азота может быть замещенным или незамещенным (т.е.,N или NR, где R представляет собой Н или другой заместитель, если определено). Гетероциклическое кольцо может быть присоединено к своей боковой группе через любой гетероатом или атом углерода,что приводит к стабильной структуре. Описанные в настоящем документе гетероциклические кольца могут быть замещенными на атоме углерода или азота, если получающееся соединение стабильно. Атом азота в гетероцикле необязательно может быть кватернизированным. Предпочтительно, если общее число атомов S и О в гетероцикле превышает 1, тогда эти гетероатомы не смежны друг с другом. Предпочтительно, чтобы общее число атомов S и О в гетероцикле было не более 1. Подразумевается, что при использовании термина "гетероцикл" он включает в себя гетероарил. Примеры гетероциклов включают без ограничения акрилинил, азоцинил, бензимидазолил, бензофуранил, бензотиофуранил, бензоксазолил, бензоксазолинил, бензтиазолил, бензтриазолил, бензтетразолил, бензизоксазолил, бензизотиазолил, бензимидазолинил, карбазолил, 4aH-карбазолил, карболинил,хроманил, хроменил, циннолинил, декагидрохинолинил, 2H,6H-1,5,2-дитиазинил, дигидрофуро[2.3-b] тетрагидрофуран, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индоленил, индолинил, индолизинил, индолил, 3 Н-индолил, изатионил, изобензофуранил, изохроманил,изоиндазолил, изоиндолинил, изоиндолил, изохинолинил, изотиазолил, изотиазолопиридинил, изоксазолил, изоксазолопиридинил, метилендиоксифенил, морфолинил, нафтиридинил, октагидроизохинолинил,оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, охиндолил, пиримидинил, фенантридинил, фенантролинил, феназинил, фенотиазинил,феноксатинил, феноксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонил, 4-пиперидонил,пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридооксазол, пиридоимидазол, пиридотиазол, пиридинил, пиримидинил, пирролидинил, пирролинил, 2-пирролидонил, 2 Н-пирролил, пирролил, хиназолинил, хинолинил, 4H-хинолизинил, хинохалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил,6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, тиенил, тиенотиазолил, тиенооксазолил, тиеноимидазолил, тиофенил, триазинил, 1,2,3 триазолил, 1,2,4-триазолил, 1,2,5-триазолил, 1,3,4-триазолил и ксантенил. Также включены конденсированное кольцо и спиросоединения, содержащие, например, вышеуказанные гетероциклы. Предпочтительные 5-10-членные гетероциклы включают без ограничения пиридинил, фуранил, тиенил, пирролил, пиразолил, пиразинил, пиперазинил, пиперидинил, имидазолил, имидазолидинил, индолил, тетразолил, изоксазолил, морфолинил, оксазолил, оксадиазолил, оксазолидинил, тетрагидрофуранил, тиадиазинил, тиадиазолил, тиазолил, триазинил, триазолил, бензимидазолил, 1H-индазолил, бензофуранил, бензотиофуранил, бензтетразолил, бензотриазолил, бензизоксазолил, бензоксазолил, охиндолил, бензоксазолинил, бензтиазолил, бензизотиазолил, изатионил, изохинолинил, октагидроизохинолинил, тетрагидроизохинолинил, тетрагидрохинолинил, изоксазолопиридинил, хиназолинил, хинолинил,изотиазолопиридинил, тиазолопиридинил, оксазолопиридинил, имидазолопиридинил и пиразолопиридинил. Предпочтительные 5-6-членные гетероциклы включают без ограничения пиридинил, фуранил, тиенил, пирролил, пиразолил, пиразинил, пиперазинил, пиперидинил, имидазолил, имидазолидинил, индо-6 022488 лил, тетразолил, изоксазолил, морфолинил, оксазолил, оксадиазолил, оксазолидинил, тетрагидрофуранил, тиадиазинил, тиадиазолил, тиазолил, триазинил и триазолил. Также включены конденсированное кольцо и спиросоединения, содержащие, например, вышеуказанные гетероциклы. В другом варианте осуществления гетероциклы включают без ограничения пиридил, пиридинил,изоксазил, изохинолинил, тиенил, пиразолил. фуранил, пирролил, тиазолил, имидазолил, пиразинил,тиадиазолил, пиримидинил, пиридазинил, оксазолил, изотиазолил, оксадиазолил, инданонил, пиперазинил, пиранил или пирролил. Также включены меньшие гетероциклы, такие как эпоксиды и азиридины. Подразумевается, что используемые в настоящем документе термины "ароматическая гетероциклическая группа" или "гетероарил" означают стабильные моноциклические и полициклические ароматические углеводороды, которые включают по меньшей мере один гетероатом в гетероцикле, такой как атом серы, кислорода или азота. Гетероарильные группы включают без ограничения пиридил, пиримидинил,пиразинил, пиридазинил, триазинил, фурил, хинолил, изохинолил, тиенил, имидазолил, тиазолил, индолил, пирроил, оксазолил, бензофурил, бензотиенил, бензтиазолил, изоксазолил, пиразолил, триазолил,тетразолил, индазолил, 1,2,4-тиадиазолил, изотиазолил, пуринил, карбазолил, бензимидазолил, индолинил, бензодиоксоланил, бензодиоксан и т.п.Гетероарильные группы могут быть замещены или незамещены. Атом азота может быть замещен или незамещен (т.е., N или NR, где R представляет собой Н или другой заместитель, если определено). Гетероатомы азота и серы могут быть необязательно окислены(т.е, NО и S(O)p). Было отмечено, что общее число атомов S и О в ароматическом гетероцикле не более 1. Соединенные мостиковой связью кольца также включены в определение гетероцикла. Если один или несколько атомов (т.е., С, О, N или S) связаны с двумя несмежными атомами углерода или азота, то возникают соединенные мостиковой связью кольца. Предпочтительные мостики включают без ограничения один атом углерода, два атома углерода, один атом азота, два атома азота и группу углерод-азот. Отметили, что мостик всегда превращает моноциклическое кольцо в трициклическое кольцо. Если кольцо соединено мостиковой связью, в мостике также могут находиться перечисленные для кольца заместители. Термины "карбоциклическое кольцо" или "углеродное кольцо" относятся к стабильным, насыщенным, частично насыщенным или ненасыщенным, моно- или бициклическим углеводородным кольцам,содержащим 3-12 атомов. В частности, они включают моноциклическое кольцо, содержащее 5 или 6 атомов, или бициклическое кольцо, содержащее 9 или 10 атомов. Подходящие значения включают циклопропан, циклобутил, циклопентил, циклогексил, циклогептил, дигидроинденил и тетрагидронафтил. Термин "необязательно замещенный", так как он относится к "карбоциклическому кольцу" или "углеродному циклу", в настоящем документе означает, что карбоциклическое кольцо может быть замещено в одном или нескольких взаимозаменяемых положениях кольца одной или несколькими группами, независимо выбранными из алкила (предпочтительно, низшего алкила), алкокси (предпочтительно, низшего алкокси), нитро, моноалкиламино (предпочтительно, низшего алкиламино), диалкиламино (предпочтительно, ди[низший алкил]амино), циано, галогена, галоалкила (предпочтительно, трифторметила), алканоила, аминокарбонила, моноалкиламинокарбонила, диалкиламинокарбонила, алкиламидо (предпочтительно низшего алкиламидо), алкоксиалкила (предпочтительно, низшего алкокси[низшего алкила]), алкоксикарбонила (предпочтительно, низшего алкоксикарбонила), алкилкарбонилокси (предпочтительно,низшего алкилкарбонилокси) и арила (предпочтительно, фенила), причем указанный арил был необязательно замещен галогеном, низшим алкилом и низшими алкоксигруппами. Термин "гетероатомы" включает кислород, серу и азот. Соединения формулы I могут существовать в свободной форме (без ионизации) или могут образовывать соли, которые также подпадают под объем настоящего изобретения. Предпочтительными являются фармацевтически приемлемые соли (т.е. нетоксичные, физиологически приемлемые), хотя применимы также другие соли, например, при выделении или очистке соединений по настоящему изобретению. Соединения формулы I могут образовывать соли с щелочными металлами, такими как натрий, калий и литий, с щелочно-земельными металлами, такими как кальций и магний, с органическими основаниями, такими как дициклогексиламин, трибутиламин, пиридин и аминокислотами, такими как аргинин,лизин и т.п.Специалистам настоящей области техники известно образование таких солей. Соединения формулы I могут образовывать соли с разными органическими и неорганическими кислотами. Такие соли включают соли, образованные с хлоридом водорода, бромидом водорода, метансульфоновой кислотой, серной кислотой, уксусной кислотой, трифторуксусной кислотой, оксолиновой кислотой, малеиновой кислотой, бензолсульфоновой кислотой, толуолсульфоновой кислотой и т.д. (например, нитраты, фосфаты, бораты, тартраты, цитраты, сукцинаты, бензоаты, аскорбаты, салицилаты и т.п.). Специалистам настоящей области техники известно образование таких солей. Кроме того, могут быть образованы цвиттерионы ("внутренние соли"). Предусмотрены все стереоизомеры соединений по настоящему изобретению или в смеси, или в чистой или по существу чистой форме. Определение соединений по настоящему изобретению охватывает все возможные стереоизомеры и их смеси. В особенности оно охватывает рацемические формы и выде-7 022488 ленные оптические изомеры, характеризующиеся точно определенной активностью. Рацемические формы могут быть повторно растворены при помощи физических способов, таких как, например, фракционная кристаллизация, разделение или кристаллизация диастереомерных производных или разделение путем хиральной колоночной хроматографии. Отдельные оптические изомеры могут быть получены из рацематов традиционными способами, такими как, например, образование соли с оптически активной кислотой с последующей кристаллизацией. Подразумевается, что настоящее изобретение включает все изотопы атомов, которые встречаются в настоящих соединениях. Изотопы включают такие атомы, которые характеризуются одинаковым атомным числом, но разными массовыми числами. В качестве общего примера и без ограничения изотопы водорода включают дейтерий и тритий. Изотопы углерода включают 13 С и 14 С. Как правило, с применением соответствующего изотопно меченного реагента вместо использованного в ином случае немеченого реагента известными специалистам настоящей области техники традиционными методиками или способами, аналогичными описанным в настоящем документе, могут быть получены изотопно меченные соединения настоящего изобретения. Соединения формулы I также могут находиться в формах пролекарства. Поскольку известно, что пролекарства усиливают многочисленные желательные качества фармацевтических препаратов (например, растворимость, биологическую доступность, технологические свойства и т.д.), соединения по настоящему изобретению могут быть доставлены в форме пролекарства. Таким образом, подразумевается,что настоящее изобретение охватывает пролекарства заявленных в настоящем документе соединений,способы их доставки и содержащие их композиции. Подразумевается, что "пролекарство" включают любые ковалентно связанные носители, которые высвобождают активное исходное лекарственное средство по настоящему изобретению in vivo после введения такого пролекарства млекопитающему и т.д. Пролекарства по настоящему изобретению получают путем модификации присутствующих в соединении функциональных групп, в том смысле, что модификации расщепляются до исходного соединения или путем рутинной манипуляции, или in vivo. Пролекарства включают соединения по настоящему изобретению, где гидрокси-, амино- или сульфгидрильная группа связаны с любой группой, которая при условии введения пациенту пролекарства по настоящему изобретению расщепляется с образованием свободной гидроксильной, свободной аминной или свободной сульфгидрильной группы, соответственно. Примеры пролекарств включают без ограничения ацетатные, формиатные и бензоатные производные спирта и амина. Из предшествующего уровня техники хорошо известны различные формы пролекарств. Для примеров таких производных пролекарств см.:c) Н. Bundgaard, Advanced Drug Delivery Reviews, 8, 1-38 (1992). Кроме того, подразумевается, что в объем настоящего изобретения также включены сольваты (например, гидраты) соединения формулы I. Способы сольватации являются общеизвестными из предшествующего уровня техники. Подразумевается, что "стабильное соединение" и "стабильная структура" обозначают соединение,достаточно стойкое для того, чтобы смогло выдержать выделение из реакционной смеси с достаточной степенью чистоты, и технологию превращения в эффективный лекарственный препарат. Предпочтительно, чтобы перечисленные здесь соединения не содержали N-галоген, S(O)2H или S(O)H группу. Используемые в настоящем документе термины "лечить" или "лечение" охватывает лечение болезненного состояния у млекопитающих, в частности у людей, и включают: (а) профилактику возникновения болезненного состояния у млекопитающих, в частности, когда млекопитающие предрасположены к болезненному состоянию, но его наличие еще не диагостировано; (b) ингибирование болезненного состояния, т.е., подавление его развития; и/или (с) облегчение болезненного состояния, т е., вызывание регрессии болезненного состояния. Подразумевается, что "'терапевтически эффективное количество" включает количество соединения по настоящему изобретению, которое эффективно при условии введения отдельно или в комбинации. Также подразумевается, что "терапевтически эффективное количество" также включает количество комбинации эффективных для лечения заболевания соединений. Кроме того, настоящее изобретение включает композиции, содержащие одно или несколько соединений по настоящему изобретению и фармацевтически приемлемый носитель."Фармацевтически приемлемый носитель" относится к общеизвестным из предшествующего уровня техники средам для доставки биологически активных средств животным, в частности, млекопитающим. Фармацевтически приемлемые носители сформулированы на основании ряда факторов в пределах компетенции специалистов в данной области техники. Они включают без ограничения, тип и природу сформулированного активного средства; субъект, которому вводили композицию, содержащую средство; предполагаемый путь введения композиции; и намеченное терапевтическое показание. Фармацевтически приемлемые носители включают как водную, так и не водную жидкую среду, а также разные твердые и полутвердые лекарственные формы. В дополнение к активному средству такие носители могут включать несколько разных ингредиентов и добавок, такие дополнительные ингредиенты были включены в состав по разным причинам, хорошо известным специалистам в данной области техники, например, для стабилизации активного средства, связующих и т.д. Описания подходящих фармацевтических приемлемых носителей и факторы их выбора были обнаружены в разных легкодоступных источниках, таких как, например, Remington's Pharmaceutical Sciences, 17th ed, Mack Publishing Company, Easton, PA, 1985, которые включены в настоящий документ посредством ссылки во всей своей полноте. Применимость Изобретение основывается на открытии, что определенные соединения являются ингибиторами протеинкиназ. Более конкретно, соединения, такие как описанные в настоящем изобретении, ингибируют протеинтирозинкиназную активность членов семейства рецепторов JAK. Эти ингибиторы найдут применение в лечении пролиферативных заболеваний, зависящих от передачи сигнала одним или несколькими из этих рецепторов. Такие заболевания включают миелопролиферативные заболевания, солидные опухоли поджелудочной железы, предстательной железы, легкого, головы и шеи, молочной железы, толстой кишки, яичника, а также другие типы опухолей, включающие множественную миелому,меланому, нейробластому, глиобластому и гематологические злокачественные новообразования, такие как острый миелогенный лейкоз. Настоящее изобретение также относится к фармацевтической композиции соединения формулы I или его фармацевтически приемлемой соли и фармацевтически приемлемого носителя в лечении гиперпролиферативного нарушения у млекопитающего. В частности, указанная фармацевтическая композиция, как предполагается, ингибирует рост и/или метастазирование тех первичных и рецидивных солидных опухолей, которые связаны с Flt-3 (Fms-подобной киназой-3), JAK2, JAK3 и JAK1, особенно тех опухолей, которые в значительной степени зависимы от JAK2, в отношении их роста и распространения,включая, например, злокачественные опухоли системы крови, щитовидной железы, молочной железы,толстой кишки, поджелудочной железы, или опухолей разнообразных типов, включая множественную миелому, меланому, нейробластому и глиобластому. В дополнение к активности JAK2 ингибиторная активность по отношению к JAK3/JAK1 или TYK2 может быть применима в лечении определенных злокачественных опухолей, характеризующихся воспалительным компонентом. Таким образом, согласно следующему аспекту настоящее изобретение относится к применению соединения формулы I или его фармацевтически приемлемой соли в производстве лекарственного препарата для применения в достижении антипролиферативного эффекта у теплокровного животного, такого как человек. Согласно следующему признаку настоящее изобретение относится к способу достижения антипролиферативного эффекта у теплокровного животного, такого как человек, нуждающийся в таком лечении,который предусматривает введение указанному животному эффективного количества соединения формулы I или его фармацевтически приемлемой соли, как определено в настоящем документе выше. В силу способности ингибировать киназы Flt-3, JAK2 и JAK3 соединения по настоящему изобретению могут быть использованы для лечения пролиферативных заболеваний, включая злокачественную опухоль. Таким образом, настоящее изобретение относится к способам лечения разнообразных злокачественных опухолей, включающих без ограничения следующие: карциному, включающую карциному мочевого пузыря (включая прогрессирующую и метастатическую злокачественную опухоль мочевого пузыря), молочной железы, толстой кишки (включая колоректальную злокачественную опухоль), почки, печени, легкого (включая мелко- и немелкоклеточную злокачественную опухоль легкого и аденокарциному легкого), яичника, предстательной железы, яичек, мочеполового тракта, лимфатической системы, прямой кишки, гортани, поджелудочной железы (включая экзокринную карциному поджелудочной железы), пищевода, желудка, желчного пузыря, шейки матки,щитовидной железы и кожи (включая плоскоклеточную карциному); опухоли кроветворной системы лимфоидного происхождения, включающие лейкоз, острый лимфоцитарный лейкоз, острый лимфобластный лейкоз, В-клеточную лимфому, Т-клеточную лимфому, лимфому Ходжкина, неходжкинскую лимфому, волосатоклеточную лимфому, гистиоцитарную лимфому и лимфому Беркитта; опухоли кроветворной системы миелоидного происхождения, включающие острый и хронический миелогенный лейкоз, миелодиспластический синдром, миелоидный лейкоз и промиелоцитарный лейкоз; опухоли центральной и периферической нервной системы, включающие астроцитому, нейробластому, глиому и невриному; опухоли мезенхимного происхождения, включающие фибросаркому, рабдомиосаркому и остеосаркому; и другие опухоли, включающие меланому, пигментную ксеродерму, кератоакантому, семиному, фолликулярную злокачественную опухоль щитовидной железы и тератокарциному. Изобретение относится к способам лечения лейкоза, миелопролиферативных заболеваний, таких как истинная полицитемия, эссениальная тромбоцитопения и миелофиброз, множественная миелома,злокачественная опухоль толстой кишки, злокачественная опухоль молочной железы и злокачественная опухоль желудка. Антипролиферативное лечение, определенное в настоящем документе выше, может быть применено в качестве монотерапии или может включать в дополнение к соединению по настоящему изобретению одно или несколько других веществ и/или типов лечения. Такое лечение может быть осуществлено путем одновременного, последовательного или раздельного введения отдельных компонентов лечения. Соединения по настоящему изобретению также могут быть применимыми в комбинации с известными противораковыми и цитотоксическими средствами и типами лечения, включая облучение. При формулировании в виде фиксированной дозы в таких комбинированных продуктах используются соединения по настоящему изобретению в диапазоне дозировки, описанном ниже, и другое фармацевтически активное средство в одобренном диапазоне его дозировки. Соединения формулы I могут быть применены последовательно с известными противораковыми или цитотоксическими средствами и лечением, включая облучение, когда комбинированный состав является неприемлемым. Термин "противораковое" средство включает любое известное средство, которое является применимым для лечения злокачественной опухоли, включая следующие: 17-этинилэстрадиол, диэтилстилбестрол, тестостерон, преднизон, флуоксиместерон, дромостанолон пропионат, тестолактон, мегестролацетат, метилпреднизолон, метилтестостерон, преднизолон, триамцинолон, хлоротрианисен, гидроксипрогестерон, аминоглутетимид, эстрамустин, медроксипрогестеронацетат, лейпролид, флутамид, торемифен, золадекс; ингибиторы матричной металлопротеиназы; ингибиторы VEGF, такие как антитела кEGFR, включая гефитиниб, эрлотиниб, ABX-EGF, EMD72000, 11F8 и цетуксимаб; ингибиторы Eg5, такие как SB-715992, SB-743921 и MKI-833; ингибиторы pan Her, такие как канертиниб, EKB-569, CI-1033,АЕЕ-788, XL-647, mAb 2C4 и GW-572016; ингибиторы киназы, например, Gleevec и дазатиниб (Sprycel); Casodex (бикалутамид, Astra Zeneca), тамоксифен; ингибиторы киназы MEK-1, ингибиторы киназы МАРК, ингибиторы киназы PI3; ингибиторы PDGF, такие как иматиниб; антиангиогенные и антиваскулярные средства, которые путем нарушения тока крови к солидным опухолям приводят злокачественные клетки в состояние покоя, лишая их питания; кастрацию, которая делает зависимые от андрогена карциномы непролиферативными; ингибиторы нерецепторных и рецепторных тирозинкиназ; ингибиторы интегриновой передачи сигнала; действующие на тубулин средства, такие как винбластин, винкристин, винорелбин, винфлунин, паклитаксел, доцетаксел, 7-0-метилтиометилпаклитаксел, 4-дезацетил-4 метилкарбонатпаклитаксел, 3' -трет-бутил-3'-N-трет-бутилоксикарбонил-4-дезацетил-3'-дефенил-3'-Nдебензоил-4-О-метоксикарбонилпаклитаксел, С-4-метилкарбонатпаклитаксел, эпотилон А, эпотилон В,эпотилон С, эпотилон D, дезоксиэпотилон А, дезоксиэпотилон В, [1S-[1R,3R(E),7R,10S,11R,12R,16S-7,11-дигидрокси-8,8,10,12,16-пентаметил-3-[1-метил-2-(2-метил-4-тиазолил)этенил]-4-аза 17-оксабицикло[14.1.0]гептадекан-5,9-дион (иксабепилон), [1S-[1R,3R(E),7R,10S,11R,12R,16S3-[2-[2-(аминометил)-4-тиазолил]-1-метилэтенил]-7,11-дигидрокси-8,8,10,12,16-пентаметил-4-17-диоксабицикло[14.1.0]-гептадекан-5,9-дион и их производные; ингибиторы CDK, антипролиферативные ингибиторы клеточного цикла, эпидофиллотоксин, этопозид, VM-26; противоопухолевые ферменты, например, ингибиторы топоизомеразы I, камптотецин, топотекан, SN-38; прокарбазин; митоксантрон; платиновые координационные комплексы, такие как цисплатин, карбоплатин и оксалиплатин; модификаторы биологического ответа; ингибиторы роста; антигормональные терапевтические средства; лейковорин; тегафур; антиметаболиты, такие как антагонисты пурина (например, 6-тиогуанин и 6-меркаптопурин; антагонисты глутамина, например, DON (АТ-125; d-оксонорлейцин); ингибиторы рибонуклеотидредуктазы; ингибиторы mTOR и гемопоэтические факторы роста. Дополнительные цитотоксические средства включают циклофосфамид, доксорубицин, даунорубицин, митоксантрон, мелфалан, гексаметилмеламин, тиотепу, цитарабин, идатрексат, триметрексат, дакарбазин, L-аспарагиназу, бикалутамид, лейпролид, пиридобензоиндольные производные, интерфероны и интрелейкины. В области медицинской онкологии традиционной практикой является применение комбинации различных форм лечения для каждого больного злокачественной опухолью. В медицинской онкологии другим компонентом(ами) такого лечения в дополнение к антипролиферативному лечению, определенному в настоящем документе выше, может быть хирургия, лучевая терапия или химиотерапия. Такая химиотерапия может охватывать три основные категории терапевтических средств:(i) антиангиогенные средства, которые действуют путем различных механизмов из тех, что определены в настоящем документе выше (например, линомид, ингибиторы функции интегрина av3, ангиостатин, разоксан);(ii) цитостатические средства, такие как антиэстрогены (например, тамоксифен, торемифен, ралоксифен, дролоксифен, йодоксифен), прогестогены (например, мегестрол ацетат), ингибиторы ароматазы(например, флутамид, нилутамид, бикалутамид, ципротерон ацетат), агонисты и антагонисты LHRH (например, госерелин ацетат, лейпролид), ингибиторы тестостерон-5 а-дигидроредуктазы (например, финастерид), ингибиторы фарнезилтрансферазы, противоинвазионные средства (например, ингибиторы металлопротеиназы, такие как маримастат, и ингибиторы функции рецептора активатора плазминогена урокиназного типа) и ингибиторы функции фактор роста, (такие факторы роста включают, например,EGF, FGF, тромбоцитарный фактор роста и фактор роста гепатоцитов, такие ингибиторы включают антитела к фактору роста, антитела к рецептору фактора роста, такие как Avastin (бевацизумаб) и Erbitux (цетуксимаб); ингибиторы тирозинкиназы и ингибиторы серин/треонин киназы); и(iii) антипролиферативные/противоопухолевые лекарственные средства и их комбинации, используемые в медицинской онкологии, такие как антиметаболиты (например, антифолаты, такие как метотрексат, фторпиримидины, такие как аналоги 5-фторурацила, пурина и аденозина, цитозинарабинозид); интеркалирующие противораковые антибиотики (например, антрациклины, такие как доксорубицин,дауномицин, эпирубицин и идарубицин, митомицин-С, дактиномицин, метрамицин); производные платины (например, цисплатин, карбоплатин); алкилирующие средства (например, мустраген, мелфалан,хлорамбуцил, бусулфан, циклофосфамид, ифосфамид, нитрозомочевины, тиотепа; антимитотические средства (например, подобные алкалоидам барвинка винкристин, винорелбин, винбластин и винфлунин) и таксоиды, такие как Taxol (паклитаксел), Taxotere (доцетаксел), и новейшие направленные на микротрубочки средства, такие как аналоги эпотилона (иксабепилон), аналоги дискодермолида и аналоги элеутеробина; ингибиторы топоизомеразы (например, эпиподофиллотоксины, такие как этопозид и тенипозид, амсакрин, топотекан, иринотекан); ингибиторы клеточного цикла (например, флавопиридолы); модификаторы биологических ответов и протеасомные ингибиторы, такие как Velcade (бортезомиб). Как указано выше, соединения формулы I по настоящему изобретению представляют интерес, благодаря их антипролиферативным эффектам. Такие соединения по настоящему изобретению, как ожидается, будут применимыми для лечения широкого диапазона болезненных состояний, включающих злокачественную опухоль, псориаз и ревматоидный артрит. Более конкретно, соединения формулы I применимы в лечении разнообразных злокачественных опухолей, включающих без ограничения следующие: карциному, включая карциному предстательной железы, панкреатическую протоковую аденокарциному, молочной железы, толстой кишки, легкого, яичника, поджелудочной железы и щитовидной железы; опухоли центральной и периферической нервной системы, включая нейробластому, глиобластому и медуллобластому; гематологические злокачественные опухоли, такие как острый миелогенный лейкоз (AML), и другие опухоли, включая меланому и множественную миелому. В связи с ключевой ролью киназ в регуляции клеточной пролиферации в целом ингибиторы могут выступать в качестве обратимых цитостатических средств, которые могут быть применимыми в лечении любого патологического процесса, который характеризуется аномальной клеточной пролиферацией, например, доброкачественная гиперплазия предстательной железы, семейный аденоматоз толстой кишки,нейрофиброматоз, легочный фиброз, артрит, псориаз, гломерулонефрит, рестеноз после ангиопластики или сосудистой хирургии, образование гипертрофического рубца и воспалительное заболевание кишечника. Соединения формулы I особенно применимы в лечении опухолей, имеющих высокую встречаемость тирозинкиназной активности, таких как опухоли предстательной железы, толстой кишки, головного мозга, щитовидной железы и поджелудочной железы. Благодаря введению композиции (или комбинации) соединений по настоящему изобретению, развитие опухолей у млекопитающего хозяина снижается. Соединения формулы I также могут быть применимыми в лечении других злокачественных заболеваний (таких как острый миелогенный лейкоз). Фармацевтические композиции по настоящему изобретению, содержащие активный ингредиент,могут иметь форму, подходящую для перорального применения, например, такую как таблетки, пастилки, лепешки, водные или масляные суспензии, диспергируемые порошки или гранулы, эмульсии, твердые или мягкие капсулы или сиропы или эликсиры. При введении соединения в соответствии с настоящим изобретением человеку суточная дозировка,как правило, будет определяться назначением врача, при этом дозировка обычно варьирует в зависимости от возраста, массы тела, пола и реакции отдельного пациента, а также от тяжести симптомов у пациента. При формулировании в виде фиксированной дозы в таких комбинированных продуктах используются соединения по настоящему изобретению в диапазоне дозировки, описанном выше, и другое фармацевтически активное средство или лечение в одобренном диапазоне его дозировки. Соединения формулыI также могут быть введены последовательно с известными противораковыми или цитотоксическими средствами, когда комбинированный состав является неприемлемым. Настоящее изобретение не ограничивается последовательностью введения; соединения формулы I могут быть введены или до, или после введения известного противоракового или цитотоксического средства(средств). Соединения могут быть введены в диапазоне дозировки от около 0,05 до 200 мг/кг/сутки, предпочтительно менее чем 100 мг/кг/сутки, в одной дозе или в 2-4 дробных дозах. Дозировка и состав Соединения по настоящему изобретению могут быть введены в составе таких пероральных лекарственных форм, как таблетки, капсулы (каждая из которых включает составы замедленного высвобождения или регулируемого по времени высвобождения), пилюли, порошки, гранулы, эликсиры, настойки,суспензии, сиропы и эмульсии. Они также могут быть введены во внутривенной (болюсной или инфузионной), внутрибрюшинной, подкожной или внутримышечной форме, причем все используемые лекарственные формы хорошо известны среднему специалисту в области фармации. Они могут быть введены отдельно, но обычно будут вводиться с фармацевтическим носителем, выбранным на основе выбранного пути введения и стандартной фармацевтической практики. Режим дозировки для соединений по настоящему изобретению будет, конечно, варьировать в зависимости от известных факторов, таких как фармакодинамические характеристики конкретного средства,способ и путь его введения; вид, возраст, пол, состояние здоровья, медицинское состояние и масса тела реципиента; природа и степень симптомов; вид сопутствующего лечения; частота лечения; путь введения, почечная и печеночная функция пациента и желаемый эффект. Врач или ветеринар может определить и назначить эффективное количество лекарственного средства, необходимого для лечения злокачественной опухоли. В качестве общей рекомендации суточная пероральная доза каждого активного ингредиента при использовании для указанных эффектов будет находиться в диапазоне от около 0,001 до 1000 мг/кг массы тела, предпочтительно от около 0,001 до 100 мг/кг массы тела в сутки и наиболее предпочтительно от около 0,001 до 20 мг/кг/сутки. При внутривенном введении наиболее предпочтительные дозы будут находиться в диапазоне от около 0,1 до около 10 мг/кг. Соединения по настоящему изобретению могут быть введены в одной суточной дозе, или общая суточная доза может быть введена дробными дозами за два, три или четыре раза в сутки. Соединения по настоящему изобретению могут быть введены в интраназальной форме путем местного применения подходящих интраназальных сред или чрескожными путями с использованием чрескожных пластырей. При введении в форме чрескожной системы доставки введение дозировки будет,конечно, непрерывным, а не периодическим, в течение всего режима введения. Соединения обычно вводятся в смеси с подходящими фармацевтическими разбавителями, наполнителями или носителями (совместно названными в настоящем документе фармацевтическими носителями), соответственно выбранными по отношению к предполагаемой форме введения, а именно, пероральным таблеткам, капсулам, эликсирам, сиропам и т.п., и в соответствии с традиционной фармацевтической практикой. Например, для перорального введения в форме таблетки или капсулы активный лекарственный компонент может быть скомбинирован с пероральным, нетоксичным, фармацевтически приемлемым инертным носителем, таким как лактоза, крахмал, сахароза, глюкоза, метилцеллюлоза, стеарат магния,фосфат дикальция, сульфат кальция, маннит, сорбит и т.п.; для перорального введения в жидкой форме пероральные лекарственные компоненты могут быть скомбинированы с любым пероральным, нетоксичным, фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и т.п.Более того, если желательно или необходимо, в смесь также могут быть включены подходящие связывающие средства, смазывающие вещества, разрыхлители и красители. Подходящие связывающие средства включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, кукурузные подсластители, натуральные и синтетические камеди, такие как гуммиарабик, трагакант или альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль, воск и т.п. Смазывающие средства, применяемые в этих лекарственных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п. Разрыхлители включают без ограничения крахмал, метилцеллюлозу, агар,бентонит, ксантановую камедь и т.п. Соединения по настоящему изобретению также могут быть введены в форме липосомных систем доставки, таких как небольшие моноламеллярные везикулы, большие моноламеллярные везикулы и мультиламеллярные везикулы. Липосомы могут быть сформированы из разнообразных фосфолипидов,таких как холестерин, стеариламин или фосфатидилхолины. Соединения по настоящему изобретению также могут быть соединены с растворимыми полимерами в качестве нацеливающих лекарственное средство носителей. Такие полимеры могут включать поливинилпирролидон, пирановый сополимер, полигидроксипропилметакриламидфенол,полигидроксиэтиласпартамидфенол или полиэтиленоксидполилизин, замещенный пальмитоиловыми остатками. Кроме того, соединения по настоящему изобретению могут быть соединены с классом биоразлагаемых полимеров, применимых для достижения регулированного высвобождения лекарственного средства, например, полимолочная кислота, полигликолевая кислота, сополимеры полимолочной и полигликолевой кислоты, полиэпсилон-капролактон, полигидроксимасляная кислота, сложные полиортоэфиры, полиацетали, полидигидропираны, полицианоацилаты и сшитые или амфипатичесукие блок- 12022488 сополимеры гидрогелей. Лекарственные формы (фармацевтические композиции), подходящие для введения, могут содержать от около 1 мг до около 1000 мг активного ингредиента на единицу дозировки. В таких фармацевтических композициях активный ингредиент, как правило, будет присутствовать в количестве около 0,1-95 мас.% от общей массы композиции. Желатиновые капсулы могут содержать активный ингредиент и порошкообразные носители, такие как лактоза, крахмал, производные целлюлозы, стеарат магния, стеариновая кислота и т.п. Подобные разбавители могут быть использованы для создания прессованных таблеток. И таблетки, и капсулы могут быть изготовлены как продукты замедленного высвобождения для обеспечения непрерывного высвобождения лекарственного препарата в течение часов. Прессованные таблетки могут быть покрыты сахаром или покрыты пленкой для маскировки какого-либо неприятного вкуса и для защиты таблетки от атмосферного воздействия, или покрыты энтеросолюбильным покрытием для селективной дезинтеграции в желудочно-кишечном тракте. Жидкие лекарственные формы для перорального введения могут содержать краситель и вкусовую добавку с целью увеличения приемлемости для пациента. В основном, подходящими носителями для парентеральных растворов являются вода, подходящее масло, солевой раствор, водная декстроза (глюкоза) и растворы родственных сахаров, и гликоли, такие как пропиленгликоль или полиэтиленгликоли. Растворы для парентерального введения предпочтительно содержат растворимую в воде соль активного ингредиента, подходящие стабилизирующие средства и,если необходимо, буферные вещества. Подходящими стабилизирующими средствами являются противоокислительные средства, такие как бисульфит натрия, сульфит натрия или аскорбиновая кислота, или отдельно, или в комбинации. Также используются лимонная кислота и ее соли и EDTA натрия. К тому же парентеральные растворы могут содержать консерванты, такие как бензалкония хлорид, метил- или пропилпарабен и хлорбутанол. Подходящие фармацевтические носители описаны в Remington's Pharmaceutical Sciences, Mack Publishing Company, стандартный текст ссылки в этой области. Биологические анализы Анализ тирозинкиназы JAK2 Анализы выполняли в 384-луночных планшетах с V-образным дном. Окончательный аналитический объем составлял 30 мкл, полученный путем 15 мкл добавлений фермента и субстратов (меченный флуоресцеином пептид и АТФ) и тестируемых соединений в аналитический буфер (100 мМ HEPES, рН 7,4, 10 мМ MgCl2, 25 мМ бета-глицерофосфата, 0,015% Brij35 и 4 мМ DTT). Реакцию инициировали комбинацией JAK2 с субстратами и тестируемыми соединениями. Реакционную смесь инкубировали при комнатной температуре в течение 60 мин и реакцию прекращали добавлением 45 мкл 35 мМ EDTA к каждому образцу. Реакционную смесь анализировали на Caliper LabChip 3000 с помощью электрофоретического разделения флуоресцентного субстрата и фосфорилированного продукта. Данные ингибирования рассчитывали путем сравнения с реакциями в контроле без фермента для 100% ингибирования и с реакциями только в среде для 0% ингибирования. Окончательная концентрация реагентов в анализах составляла для АТФ 30 мкМ; для флуоресцентного пептида JAK2 1,5 мкМ; для JAK2 1 нМ и для DMSO 1,6%. Строили кривые зависимости от дозы для определения концентрации, необходимой для ингибирования 50% киназной активности (IC50) Соединения растворяли при 10 мМ в диметилсульфоксиде(DMSO) и оценивали в одиннадцати концентрациях, каждую в двух повторностях. Значения IC50 получали с помощью нелинейного регрессионного анализа. Анализ тирозинкиназы JAK3 Анализы выполняли в 384-луночных планшетах с V-образным дном. Окончательный аналитический объем составлял 30 мкл, полученный путем 15 мкл добавлений фермента и субстратов (меченный флуоресцином пептид и АТФ) и тестируемых соединений в аналитический буфер (100 мМ HEPES, рН 7,4, 10 мМ MgCl2, 25 мМ бета-глицерофосфата, 0,015% Brij35 и 4 мМ DTT). Реакцию инициировали комбинацией JAK3 с субстратами и тестируемыми соединениями. Реакционную смесь инкубировали при комнатной температуре в течение 60 мин и реакцию прекращали добавлением 45 мкл 35 мМ EDTA к каждому образцу. Реакционную смесь анализировали на Caliper LabChip 3000 с помощью электрофоретического разделения флуоресцентного субстрата и фосфорилированного продукта. Данные ингибирования рассчитывали путем сравнения с реакциями в контроле без фермента для 100% ингибирования и реакциями только в среде для 0% ингибирования. Окончательная концентрация реагентов в анализах составляла для АТФ 8 мкМ; для флуоресцентного пептида JAK3 1,5 мкМ; для JAK3 2,5 нМ и для DMSO 1,6%. Строили кривые зависимости от дозы для определения концентрации, необходимой для ингибирования 50% киназной активности (IC50). Соединения растворяли при 10 мМ в диметилсульфоксиде(DMSO) и оценивали в одиннадцати концентрациях, каждую в двух повторностях. Значения IC50 получали с помощью нелинейного регрессионного анализа. Анализ ингибирования клеточной пролиферации Соединения оценивали по их способности ингибировать клеточную пролиферацию с использованием анализа, с помощью которого измеряют включение тимидина в ДНК делящейся клетки, что напрямую коррелирует с числом клеток. Клетки SET-2 высевали при 10000 клеток/лунка на 96-луночные планшеты и культивировали в течение 24 ч в RPMI-1640, дополненной 20% эмбриональной телячьей сыворотки,перед добавлением тестируемых соединений. Соединения разбавляли культуральной средой так, чтобы окончательная концентрация диметилсульфоксида никогда не превышала 0,1%. После добавления соединений клетки культивировали еще дополнительно 72 ч перед определением клеточной пролиферации путем измерения синтеза ДНК с включением 3 Н-тимидина. Соединения, описанные в настоящем документе, тестировали в анализе JAK2 и ингибирования клеточной пролиферации, описанном выше. Получали следующие результаты. Таблица 1 Соединения, описанные в настоящем документе, протестировали в одном или нескольких из определенных выше анализов и нашли их активными С. Анализ in vivo. Соединение, введенное в подходящую среду, вводили мышам либо в виде перорального болюса,либо внутрибрюшинной или подкожной инъекцией мышам Balb/C. Образцы крови собирали путем глазного кровопускания или сердечной пункции в различные точки времени после введения соединения. Аликвоты (90 мкл) крови помещали в 96-луночный планшет. В каждую лунку вносили разбавленный раствор (5 мкл) мышиного тромбопоэтина (mTPO; Peprotech) (разбавленного 1:1000 в dPBS/BSA, 2 мкл в 2 мл dPBS/BSA) для стимуляции пути. Планшеты инкубировали при 37 С на водяной бане в течение 10 минут. В каждую лунку добавляли 1,5 мл фиксирующего/лизирующего раствора (от BD Biosciences). Затем образцы инкубировали при 37 С в течение 15 мин для обеспечения лизиса красных кровяных клеток (RBC). Планшеты центрифугировали при 2000 оборотов в минуту и аспирировали с использованием 96-луночного аспиратора VP. Клетки промывали dPBS, повторно центрифугировали, повторно суспендировали и переносили в стандартный 96-луночный планшет. Клетки дважды промывали dPBS с последующим добавлением CD61 FITC (от eBiosciences) для окрашивания рецепторов поверхности в течение 45 мин в темноте при комнатной температуре. Затем клетки промывали в dPBS и пермеабилизировали для окрашивания pSTAT5 следующим образом: добавляли 200 мкл буфера для пермеабилизации III (отBD Bioscience) и образцы инкубировали на льду в течение 30 мин, а затем образцы дважды промывалиdPBS/1% BSA и повторно суспендировали в 100 мкл dPBS/BSA, содержащем антитела Stat5 (pY695) Alexa 647. Образцы инкубировали при комнатной температуре в темноте в течение 45 минут, дважды промывали и повторно суспендировали в 10 мкл антител Stat5 (pY695) Alexa 647, использованных для добавления в образец. Образцы повторно суспендировали в 200 мкл dPBS/BSA и прогоняли на FACSCanto,изображения образца обрабатывали с использованием программного обеспечения для анализа Diva 6 иFlowJo 8.5.3. Данные для примера 26 Аббревиатуры В описании способов получения и примерах могут быть применены следующие аббревиатуры: ч = часы;EtOAc = этилацетат; АсОН = уксусная кислота; Реагент ВОР = гексафторфосфат бензотриазол-1-илокси-трис-(диметиламино)фосфония; Насыщенный солевой раствор = насыщенный водный раствор хлорида натрия;NIS = N-йодсукцинамид. Способы получения С применением показанного на схеме 1 общего способа могут быть получены соединения общей формулы I, в которой группа R представляет собой тиазол (как в Ia1), а группы R1 и R2 представляют собой CF3 или алкил или циклоалкил, или они сцеплены с образованием насыщенного карбоциклического или гетероциклического кольца, или где группа R2 представляет собой COORb. Дихлорзамещенное промежуточное соединение II (полученное с использованием опубликованной в WO 200612237 методики) может быть объединено с 2,4-диметоксибензилом, и полученный вторичный амин кэпировали подходящей защитной группой (Boc) (III). Второй атом хлора может быть превращен в соответствующий амин (IV) посредством промежуточного соединения бензофенонимина. Аминосоединение может быть галогенировано до промежуточного соединения V. Соединение V могут подвергать опосредованному переходным металлом образованию индольного кольца и полученный азот индола кэпировали этилйодидом с получением соединения VI. Путем гидролиза сложного эфира с последующим образованием амидной связи и отщепления защитных групп с кислотной обработкой будет получен амин VII. Амин VII может быть превращен в тиомочевину VIII первым сцеплением с бензоилизотиоцианатом с последующей обработкой водным основанием. Образование тиазола может быть осуществлено путем конденсации с производным а-бромкетона (R1CHBrCOR2). Схема 1 С применением схемы 2 могут быть получены соединения общей формулы Ia2, в которой группа R1 представляет собой CONRaRa. Промежуточное соединение тиомочевины (VIII) может быть соединено сEtO2CCHBrCOR1 с получением сложного эфира тиазола (IX). Сложный эфир может быть гидролизирован, и кислота может быть сцеплена с амином с получением производного тиазоламида (Ia). Схема 2 Подобным образом с применением показанного на схеме 3 общего протокола могут быть получены соединения общей формулы Ia3, в которой группа R1 представляет собой CONRaRa. Схема 3 Путем конденсации а,а'-дигалогенкетона могут быть получены соединения общей формулы Ia, в которой R1 представляет собой галоген (Cl, Br или I), как показано на схеме 4. Схема 4 Альтернативно, производное тиомочевины VIII при комнатной температуре может быть превращено в С-5 незамещенный тиазол XI и затем сразу галогенировано с применением электрофильного источника галогена или путем металлирования с последующим гашением электрофильным галогенирующим средством (схема 5). Схема 5 С применением показанной на схеме 6 обычной методики синтеза могут быть синтезированы соединения общей формулы 1 а 5, в которой R1 представляет собой SO2Rb Схема 6 С применением схемы 7 могут быть получены соединения общей формулы Ia, в которой R1 и R2 объединены с образованием ароматического или гетероароматического кольца. Схема 7 Альтернативно, эти соединения могут быть получены путем первого сцепления анилина или гетероанилина (XVI) с изотиоцианатом (XV) с последующим окислительным образованием кольца (схема 8). С применением показанной на схеме 9 обычной методики синтеза могут быть получены соединения общей формулы Ib1. Анилин VII может быть объединен с соединением XVII у-дитиометилкетона (полученным при комнатной температуре с применением методики, описанной у Synlett, p 2331 (2008 при основных условиях для получения XVIII. Путем постадийной конденсации Вос-защищенного производного гидразина может быть получен необходимый пиразол Ib1. Схема 9 Соединения общей формулы Ib1 или If1 и 1f также могут быть получены путем сцепления производного галогена С-4 (XIX) с соответствующим образом замещенным производным 2-аминопиразола(XX) путем осуществления взаимодействия, катализированного переходным металлом (схема 10). Схема 10 С применением схемы 11 могут быть синтезированы соединения общей формулы Ib2, в которой R2 группа представляет собой CONRaRa. Анилин VII может быть объединен с производным удитиометилкетона XXII, (полученный с применением методики из Tetrahedron, p 2631 (2003 с получением промежуточного соединения XXIII. Путем постадийной конденсации Вос-защищенного производного гидразина может быть получен необходимый альдегид пиразола XXIV. Альдегид может быть окислен с применением оксона или гипохлорита натрия с получением карбоновой кислоты XXV. Пиразоламид Ib2 может быть получен путем соединения кислоты XXV с амином. Схема 11 С применением показанного в схеме 12 общего протокола могут быть получены соединения общей формулы Icl. Анилин VII может быть соединен с хлорацетилхлоридом и полученный амид может быть обработан тиоамидом (R2CSNH2) с получением тиазола Ic1. Схема 12 В соответствии со схемой 13 могут быть получены соединения общей формулы Id1. При условиях реакции дегидратации ранее описанное производное изотиоцианата XV может быть объединено с амидином XXV с получением 1,2,4-тиадиазола (Id1). Схема 13 С применением показанной на схеме 14 методики синтеза могут быть получены соединения общей формулы Ie1. Изотиоцианат XV может быть объединен с азидом XXVI в присутствии фосфина с получением 1,3-оксазола Ie1. Схема 14 С применением показанной на схеме 15 методики синтеза могут быть получены соединения общей формулы Ig1. Амин VII может быть объединен с ацилизотиоцианатом XXVII. Ацилтиоуреидо может быть конденсирован с производным гидразина с получением производного 1,2,4-триазола Ig1. Схема 15 В 200 мл круглодонной колбе растворяли 4,6-дихлор-1-метил-1H-имидазо[4,5-с]пиридин (15 г, 74,2 ммоль) (полученный согласно WO 2006122137 в примере А 1.5) в 2,4-диметоксибензиламине (25 мл, 166 ммоль). На протяжении 3 ч реакционную смесь нагревали до 110 С и охлаждали до комнатной температуры. Добавляли воду и путем осаждения отделяли твердое вещество коричневого цвета. Путем вакуумной фильтрации получали твердое вещество коричневого цвета. Твердое вещество промывали этилаце- 19022488 татом и гексаном с получением 6-хлор-N-2,4-диметоксибензил)-1-метил-1H-имидазо[4,5-с]пиридин-4 амина (23,459 г, выход 95%) в виде твердого вещества кремового цвета.NaHMDS (1,0 М THF, 2,366 мл, 2,366 ммоль). После перемешивания при -78 С в течение 30 мин добавляли ВОС 2 О (0,483 мл, 2,082 ммоль). Холодную баню удаляли и реакцию оставляли нагреваться до к.т. в течение ночи. При помощи HPLC наблюдали 50% преобразования. Раствор повторно охлаждали до-78 С. Добавляли NaHMDS (1,0 М THF, 2,366 мл, 2,366 ммоль) и смесь перемешивали 30 мин перед добавлением ВОС 2 О (0,483 мл, 2,082 ммоль). Холодную баню удаляли и реакционную среду нагревали до комнатной температуры 5 ч. При пониженном давлении растворитель удаляли и остаток делили между этилацетатом и водой. Органический слой дважды промывали водой, сушили над безводным сульфатом магния, отфильтровывали и концентрировали в вакууме. Неочищенный остаток очищали при помощи флэш-хроматографии с применением колонки ISCO 40g и с элюированием 50-100% этилацетатом/гексаном с получением трет-бутил-6-хлор-1-метил-1H-имидазо[4,5-с]пиридин-4-ил-(2,4-диметоксибензил)карбамата (0,638 г, выход 78%) в виде белого пенистого твердого вещества.(0,672 г, 2,063 ммоль) добавляли безводный диоксан (4 мл). Реакционную смесь в течение ночи нагревали до 90 С. После охлаждения до к т. растворитель удаляли в вакууме. Остаток делили между этилацетатом и водой. Органический слой дважды промывали водой, сушили над безводным сульфатом магния,отфильтровывали и концентрировали в вакууме. Неочищенный остаток очищали при помощи флэшхроматографии с применением колонки ISCO 40g и с элюированием 50-100% этилацетатом/гексаном с получением трет-бутил-2,4-диметоксибензил(6-(дифенилметиленамино)-1-метил-1H-имидазо[4,5-с]пиридин-4-ил)карбамата (0,757 г, выход 89%) в виде светло-желтого твердого вещества. В 1000 мл круглодонной колбе перемешивали трет-бутил-2,4-диметоксибензил(6-(дифенилметиленамино)-1-метил-1H-имидазо[4,5-с]пиридин-4-ил)карбамат (пример 1 С, 9,2 г, 15,93 ммоль) в THF (50 мл). Добавляли 1 н. HCl (33 мл). Через 2 мин реакцию гасили 1 н. NaOH (65 мл) и этилацетатом (100 мл). Органический слой отделяли, сушили над безводным сульфатом натрия, отфильтровывали и концентрировали в вакууме. Остаток сушили в вакууме 1 ч и растирали с простым эфиром (4). Трет-бутил-6-амино- 20022488 1-метил-1H-имидазо[4,5-с]пиридин-4-ил-(2,4-диметоксибензил)карбамат (6,587 г, 15,93 ммоль, выход 100%) выделяли в виде твердого вещества кремового цвета. Вещество переносили на следующую стадию без дополнительной очистки. В круглодонной колбе растворяли трет-бутил-6-амино-1-метил-1 Н-имидазо[4,5-с]пиридин-4-ил(2,4-диметоксибензил)карбамат (пример 1D, 7,887 г, 19,08 ммоль) в MeCN (200 мл) и охлаждали до 0 С. В оставшемся MeCN (50 мл) растворяли N-йодсукцинимид (4,51 г, 20,03 ммоль) и в течение 40 мин по каплям добавляли в реакционную смесь. Реакционную смесь перемешивали при 0 С в течение дополнительных 10 мин и гасили 2 М гидросульфитом натрия (125 мл). Перемешивание и температуру поддерживали в течение 50 мин. Смесь переносили в делительную воронку. Водный слой экстрагировалиCH2Cl2 (3100 мл). Объединенные органические слои промывали водой и насыщенным солевым раствором, сушили над Na2SO4, отфильтровывали и концентрировали в вакууме. Неочищенный остаток очищали при помощи флэш-хроматографии с применением колонки ISCO 120g и с элюированием 20-100% этилацетатом/CH2Cl2 с получением трет-бутил-6-амино-7-йод-1-метил-1 Н-имидазо[4,5-с]пиридин-4-ил(2,4-диметоксибензил)карбамата (8,436 г, выход 82%) в виде бледно-желтого твердого вещества. В сосуд с трет-бутил-6-амино-7-йод-1-метил-1H-имидазо[4,5-с]пиридин-4-ил-(2,4-диметоксибензил)карбаматом (пример IE, 3,49 г, 6,47 ммоль) в DMA (43,1 мл) добавляли Pd2(dba)3 (0,474 г, 0,518 ммоль), этилпируват (7,22 мл, 64,7 ммоль) и N-метилдициклогексиламин (2,77 мл, 12,94 ммоль). Реакционную смесь барботировали аргоном в течение 10 мин и нагревали при 60 С 6 ч. Реакцию завершали при помощи LCMS. После охлаждения до комнатной температуры, реакционную смесь разбавляли этилацетатом и насыщенным водным NaHCO3. Полученную эмульсию фильтровали через вакуумный фильтр через слой целита и переносили в делительную воронку. Водный слой экстрагировали этилацетатом. Объединенные органические слои промывали 10% водным раствором LiCl (2). Органическую фазу сушили над MgSO4, отфильтровывали и концентрировали в вакууме. Остаток коричневого цвета очищали при помощи флэш-хроматографии с применением колонки ISCO 120g и с элюированием 40-80% этилацетатом/гексаном с получением этил-4-(трет-бутоксикарбонил-(2,4-диметоксибензил)амино)-1-метил 1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7-карбоксилата (2,830 г, выход 86%). К раствору этил-4-(трет-бутоксикарбонил-(2,4-диметоксибензил)амино)-1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7-карбоксилата (пример 1F, 5,046 г, 9,90 ммоль) в DMF (49,5 мл) добавляли этилйодид (1,601 мл, 19,81 ммоль) и карбонат цезия (6,45 г, 19,81 ммоль). Реакционную смесь нагревали при 60 С 60 мин и охлаждали до комнатной температуры. Реакционную смесь разбавляли этилацетатом и фильтровали через вакуумный фильтр с удалением карбоната цезия. Добавляли насыщен- 21022488 ный водный бикарбонат натрия и водный слой экстрагировали этилацетатом (2). Объединенные органические слои промывали 10% хлоридом лития (3), сушили над безводным сульфатом магния, отфильтровывали и концентрировали в вакууме. Неочищенный продукт переносили на следующую стадию. К раствору этил-4-(трет-бутоксикарбонил-(2,4-диметоксибензил)амино)-6-этил-1-метил-1,6-дигидроимидазо[4.5-dпирроло[2.3-b]пиридин-7-карбоксилата (пример 1G, 5,32 г, 9,9 ммоль) в этаноле (49,5 мл) добавляли водный раствор 1 н. NaOH (49,5 мл). Реакционную смесь нагревали при 60 С 3 ч и охлаждали до к.т. Этанол удаляли путем концентрирования в вакууме. Остаток подкисляли 1 н. HCl и продукт собирали путем вакуумной фильтрации. 4-(трерт-Бутоксикарбонил-(2,4-диметоксибензил)амино)-6-этил 1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7-карбоновую кислоту (4,3 г, 8,44 ммоль, выход 85% за две стадии) выделяли в виде белого твердого вещества после сушки в вакууме. В смесь 4-(трет-бутоксикарбонил-(2,4-диметоксибензил)амино)-6-этил-1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7-карбоновой кислоты (пример 1 Н, 0,89 г, 1,747 ммоль) в ацетонитриле (17,47 мл) добавляли гидрохлорид дициклопропиламина (0,303 г, 2,271 ммоль), N-метилморфолин(0,499 мл, 4,54 ммоль), DMAP (0,021 г, 0.175 ммоль) и НАТО (0,797 г, 2,096 ммоль). Реакционную смесь перемешивали при к.т. 5 ч и нагревали до 50 С в течение 45 мин. Реакционную смесь охлаждали до комнатной температуры. Ацетонитрил удаляли в вакууме и остаток делили между насыщенным водным бикарбонатом натрия и дихлорметаном. Водный слой экстрагировали дихлорметаном. Объединенные органические слои сушили над безводным сульфатом натрия, отфильтровывали и концентрировали в вакууме. Неочищенное твердое вещество очищали при помощи флэш-хроматографии с применением колонки К раствору трет-бутил-7-(дициклопропилкарбамоил)-6-этил-1-метил-1,6-дигидроимидазо[4.5-d] пирроло[2.3-b]пиридин-4-ил-(2,4-диметоксибензил)карбамата (пример 1I, 2,66 г, 4,51 ммоль) в дихлорметане (33,8 мл) добавляли TFA (11,28 мл). Реакционную смесь перемешивали при к.т. в течение 45 мин и концентрировали в вакууме. Неочищенный остаток делили между этилацетатом и насыщенным водным бикарбонатом натрия. Водный слой экстрагировали этилацетатом (2) и объединенные органически вещества сушили над безводным сульфатом натрия, отфильтровывали и концентрировали в вакууме. Неочищенное твердое вещество очищали при помощи флэш-хроматографии с применением колонки(12,18 мг, 0,013 ммоль), BINAP (24,84 мг, 0,040 ммоль) и трет-бутоксида натрия (13,63 мг, 0,142 ммоль) добавляли PhMe (887 мкл). Смесь барботировали газообразным аргоном в течение 5 мин и нагревали до 85 С в течение 5,5 ч. Реакционную смесь отфильтровывали через слой целита и концентрировали. Неочищенный остаток очищали при помощи препаративной HPLC с получением N,N-дициклопропил-6 этил-1-метил-4-(тиазол-2-иламино)-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7-карбоксамида (4 мг, выход 10,49%) вместе с некоторым количеством восстановленного исходного вещества. Раствор 4-амино-N,N-дициклопропил-6-этил-1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b] пиридин-7-карбоксамида (пример 1J, 234 мг, 0,69 ммоль) и бензоилизотиоцианата (0,1 мл, 0,83 ммоль) в ацетоне (3 мл) перемешивали при комнатной температуре в течение 3 ч. Реакционную смесь охлаждали до 0 С и добавляли воду. Полученное твердое вещество коричневого цвета собирали путем фильтрации и сушили на воздухе с получением 4-(3-бензоилтиоуреидо)-N,N-дициклопропил-6-этил-1-метил-1,6 дигидроимидазо[4.5-d]пирроло[2 3-b]пиридин-7-карбоксамида (306 мг, выход 88%).[2.3-b]пиридин-7-карбоксамид (пример 2 А, 306 мг, 0,61 ммоль) и 1 н. NaOH (10 мл, 10,00 ммоль) нагревали при 60 С в течение 1 ч в 10 мл EtOH. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Оставшийся водный раствор экстрагировали (3) этилацетатом. Органическую фазу сушили над Na2SO4, отфильтровывали и концентрировали. Очисткой при помощи хроматографии на силикагеле (дихлорметан/метанол 0-4%) получали N,N-дициклопропил-6-этил-1-метил-4-тиоуреидо-1,6 дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7-карбоксамид в виде желтого твердого вещества (199 мг,выход 82%).N,N-дициклопропил-6-этил-1-метил-4-тиоуреидо-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7-карбоксамид (пример 2 В, 40 мг, 0,10 ммоль) и хлорацетон (62,4 мг, 0,40 ммоль) нагревали при 80 С в течение 20 мин в EtOH (2 мл). Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли в вакууме. Добавляли насыщенный раствор NaHCO3 и водный слой экстрагировали(3) дихлорметаном. Органические слои объединяли, сушили над Na2SO4, отфильтровывали и концентрировали. Неочищенное вещество очищали при помощи хроматографии на силикагеле (дихлорметан/метанол 0-7%) с получением N,N-дициклопропил-6-этил-1-метил-4-(4-метилтиазол-2-иламино)-1,6 дигадроимидазо[4.5-d]пирроло[2.3-b]пиридин-7-карбоксамида (9 мг, выход 12%) в виде белого твердого вещества.N,N-Дициклопропил-6-этил-4-(4-(фторметил)тиазол-2-иламино)-1-метил-1,6-дигидроимидазо[4.5-d] пирроло[2.3-b]пиридин-7-карбоксаиид. 3 А. Получение 1-бром-3-фторпропан-2-она К раствору 1-фторпропан-2-она (0,51 мл, 7,02 ммоль) в тетрахлориде углерода (25 мл) по каплям добавляли бром (0,36 мл, 7,02 ммоль). Раствор нагревали при 45 С в течение 3 ч. Реакционную смесь охлаждали до комнатной температуры и гасили водой. После экстрагирования дихлорметаном (3) объединенные органические слои сушили над Na2SO4, отфильтровывали и концентрировали. Очисткой при помощи хроматографии на силикагеле (0-20% EtOAc/гексаны) получали 1-бром-3-фторпропан-2-он в виде желтого масла (118 мг, 65%). 1 Н ЯМР (400 МГц, хлороформ-d)ppm 5,14 (s, 1 Н) 5,02 (s, 1 Н) 4,10 (d, J=2,86 Гц, 2 Н). 3. Получение N,N-дициклопропил-6-этил-4-(4-(фторметил)тиазол-2-иламино)-1-метил-1,6-дигидроимидазо[4.5-d1 пирроло[2.3-b]пиридин-7-карбоксамида. Получали из N,N-дициклопропил-6-этил-1-метил-4-тиоуреидо-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7-карбоксамида (пример 2 В) и 1-бром-3-фторпропан-2-она (пример 3 А) с применением методики согласно примеру 2. К раствору 1-(изопропилсульфонил)пропан-2-она (418 мг, 2,55 ммоль) в 1:1 AcOH/DCM (16 мл) при 0 С добавляли по каплям сульфурилхлорид (0,248 мл, 3,05 ммоль). Реакционную смесь перемешивали при к.т. в течение 4 ч. Растворитель удаляли в вакууме, затем разбавляли дихлорметаном. Органический слой промывали (2) насыщенным раствором NaHCO3, сушили над Na2SO4, отфильтровывали и концентрировали с получением 800 мг бесцветного масла. Вещество использовали без дополнительной очистки. 1 Н ЯМР (500 МГц, хлороформ-d)ppm 5,19 (s, 1 Н) 3,65 (dt, J=13,66, 6,90 Гц, 1 Н) 2,56 (s, 3 Н) 1,45[2.3-b]пиридин-7-карбоксамида (пример 2 В) и 1-хлор-1-(изопропилсульфонил)пропан-2-она (пример 5 а) с применением методики согласно примеру 2.[4.5-d]пирроло[2.3-b]пиридин-7-карбоксамид. Раствор 4-амино-N,N-дициклопропил-6-этил-1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7-карбоксамида (пример 1J, 70 мг, 0,21 ммоль) и бензоилизотиоцианата (36 мкл, 0,27 ммоль) в ацетоне (1 мл) перемешивали при комнатной температуре в течение 1,5 ч. Растворитель удаляли в вакууме, остаток переносили в EtOH (1,5 мл) и добавляли K2CO3 (40 мг, 0,290 ммоль). Реакционную смесь нагревали при 60 С в течение 3 ч. После охлаждения до комнатной температуры добавляли 2-бром-1(пиридин-2-ил)этанон, гидробромид (139,4 мг, 0,50 ммоль) и реакционную смесь нагревали при 80 С в течение 24 ч. Реакционную смесь охлаждали до комнатной температуры и концентрировали. Остаток очищали при помощи хроматографии на силикагеле (0-7% МеОН/дихлорметан). Повторной очисткой препаративной HPLC получали 10,2 мг (выход 10%) N,N-дициклопропил-6-этил-1-метил-4-(4-(пиридин 2-ил)тиазол-2-иламино)-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7-карбоксамид в виде твердого вещества коричневого цвета. Получение N,N-дициклопропил-6-этил-1-метил-4-(4-(трифторметил)тиазол-2-иламино)-1,6-дигидроимидазол[4.5-d]пирроло[2.3-b]пиридин-7-карбоксамид. Это соединение получали в соответствии с примером 2 с применением 3-бром-1,1,1-трифторацетона с получением N,N-дициклопропил-6-этил-1-метил-4-(4-(трифторметил)тиазол-2-иламино)-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7-карбоксамида.(37,8 мкл, 0,281 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель удаляли в вакууме. Остаток переносили в этанол (1500 мкл) и добавляли K2CO3 (49,3 мг,0,357 ммоль). Реакционную смесь нагревали до 60 С в течение 3 ч. Добавляли 2-бромпропаналь (45,4 мг,0,332 ммоль). Через 3 ч добавляли дополнительное количество 2-бромпропаналя (-100 мг). Перемешивали при 60 С в течение 16 ч. Добавляли дополнительный 2-бромпропаналь и реакционную смесь перемешивали при 60 С 3 ч. Этанол удаляли путем концентрирования в вакууме. Неочищенный остаток очищали при помощи флэш-хроматографии с применением колонки ISCO 12g и с элюированием 2-10%MeOH/CH2Cl2). Дополнительную очистку завершали при помощи препаративной HPLC. N,Nдициклопропил-6-этил-1-метил-4-(5-метилтиазол-2-иламино)-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b] пиридин-7-карбоксамид (20 мг, выход 17,47%) выделяли в виде белого твердого веществ MS (ESI) масса/заряд 436,0 (М+Н). 1 Н ЯМР (500 МГц, DMSO-d6)ppm 10,56 (br s, 1H), 8,12 (s, 1H), 7,23 (s, 1H), 7,12 (s, 1H), 4,62 (q,2H, J= 6,78 Гц), 4,04 (s, 3H), 2,87-2,99 (m, 2H), 2,39 (s, 3H), 1,35-1,46 (m, 3H), 0,72-0,81 (m, 4H), 0,59-0,71[4.5-d]пирроло[2.3-b]пиридин-7-карбоксамида (пример 1J) с применением методики согласно примеру 8. 4-Амино-N,N-дициклопропил-6-этил-1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7 карбоксамид (1J, 300 мг, 0,89 ммоль) и бензоилизотиоцианат (0,16 мл, 1,15 ммоль) в ацетоне (4 мл) перемешивали при к.т. в течение 1,5 ч. Растворитель удаляли в вакууме и остаток переносили в EtOH (6 мл). Добавляли K2CO3 (172 мг, 1,241 ммоль) и реакционную смесь нагревали при 60 С в течение 3 ч. Реакционную смесь охлаждали до к.т. и добавляли этил-3-бром-2-оксопропаноат (692 мг, 3,55 ммоль). Реакционную смесь нагревали при 70 С в течение ночи. После завершения стадии кристаллизации в реакционную смесь добавляли 1 н. NaOH (6,3 мл, 6,30 ммоль) и нагревали при 60 С в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли в вакууме. Оставшийся водный раствор подкисляли до рН 1 и осадок удаляли вакуумной фильтрацией, промывали водой, EtOAc и затем дихлорметаном с получением 2-(7-(дициклопропилкарбамоил)-6-этил-1-метил-1,6-дигидроимидазо[4.5d]пирроло[2.3-b]пиридин-4-иламино)тиазол-4-карбоновой кислоты (325 мг, выход 79%) в виде твердого вещества коричневого цвета. К 2-(7-(дициклопропилкарбамоил)-6-этил-1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-4-иламино)тиазол-4-карбоновой кислоте (пример 12 А, 44 мг, 0,095 ммоль) в DMF (2 мл) добавляли НАТО (108 мг, 0,284 ммоль), DIPEA (0,066 мл, 0,378 ммоль) и диметиламин (0,013 мл, 0,189 ммоль). Реакционную смесь перемешивали при к.т. в течение 3 ч, затем растворитель удаляли в вакууме. Остаток очищали при помощи препаративной HPLC с получением 2-(7-(дициклопропилкарбамоил)-6-этил-1 метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-4-иламино)-N,N-диметилтиазол-4-карбоксамида(10 мг, выход 21%) в виде коричневого твердого вещества. Этил-2-(7-(дициклопропилкарбамоил)-6-этил-1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-4-иламино)тиазол-4-карбоксилат. Это соединение получали в соответствии с примером 2 с применением этилбромпирувата и N,Nдициклопропил-6-этил-1-метил-4-тиоуреидо-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7 карбоксамида (пример 2 В) с получением этил-2-(7-(дициклопропилкарбамоил)-6-этил-1-метил-1,6 дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-4-иламино)тиазол-4-карбоксилата в виде желтого твердого вещества. Этил-2-(7-(дициклопропилкарбамоил)-6-этил-1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b] пиридин-4-иламино)тиазол-5-карбоксилат. Это соединение получали в соответствии с примером 2 с применением этил-2-хлор-2-формилацетата и N,N-дициклопропил-6-этил-1-метил-4-тиоуреидо-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b] пиридин-7-карбоксамида (пример 2 В) с получением этил-2-(7-(дициклопропилкарбамоил)-6-этил-1 метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-4-иламино)тиазол-5-карбоксилата (18,5 мг, выход 17%) в виде бледно-желтого твердого вещества. К раствору этил-2-(7-(дициклопропилкарбамоил)-6-этил-1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-4-иламино)тиазол-5-карбоксилата (пример 19, 14,6 мг, 0,030 ммоль) в EtOH (296 мкл) добавляли водный раствор 1 н. NaOH (296 мкл). Реакционную смесь перемешивали при к.т. 3 ч и перемешивали при 50 С 18 ч. После охлаждения до комнатной температуры этанол удаляли путем концентрирования в вакууме. Остаток подкисляли 1 н. HCl и твердый продукт собирали путем фильтрация. 2-(7(Дициклопропилкарбамоил)-6-этил-1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-4-иламино)тиазол-5-карбоновую кислоту выделяли в виде бледно-желтого твердого вещества, которую использовали в неочищенном виде в следующей реакции.MS (ESI) масса/заряд 465,9 (М+Н). 20. Получение N-циклопропил-2-(7-(дициклопропилкарбамоил)-6-этил-1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-4-иламино)тиазол-5-карбоксамида. В смесь 2-(7-(дициклопропилкарбамоил)-6-этил-1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b] пиридин-4-иламино)тиазол-5-карбоновой кислоты (пример 20 А, 13,97 мг, 0,030 ммоль) в DMF (300 мкл) добавляли циклопропиламин (4,16 мкл, 0,060 ммоль), HATU (14,83 мг, 0,039 ммоль) и основание Хюнинга (15,72 мкл, 0,090 ммоль). Реакционную смесь перемешивали при к.т. 1 ч и концентрировали в вакууме. Остаток переносили в МеОН и очищали при помощи препаративной HPLC. N-Циклопропил-2-(7(дициклопропилкарбамоил)-6-этил-1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-4-иламино)тиазол-5-карбоксамид (3 мг. выход 19,82%) выделяли в виде белого твердого вещества.N,N-Дициклопропил-6-этил-1-метил-4-(5-(морфолин-4-карбонил)тиазол-2-иламино)-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7-карбоксамид. Это соединение получали в соответствии с примером 20 с применением морфолина и 2-(7-(дициклопропилкарбамоил)-6-этил-1-метил-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-4-иламино) тиазол-5-карбоновой кислоты (пример 20 А) с получением N,N-дициклопропил-6-этил-1-метил-4-(5(морфолин-4-карбонил)тиазол-2-иламино)-1,6-дигидроимидазо[4.5-d]пирроло[2.3-b]пиридин-7-карбоксамида в виде белого твердого вещества.
МПК / Метки
МПК: A61K 31/437, A61P 35/00, C07D 471/14
Метки: ингибиторы, злокачественной, заболеваний, применение, лечения, миелопролиферативных, опухоли
Код ссылки
<a href="https://eas.patents.su/30-22488-ingibitory-jak2-i-ih-primenenie-dlya-lecheniya-mieloproliferativnyh-zabolevanijj-i-zlokachestvennojj-opuholi.html" rel="bookmark" title="База патентов Евразийского Союза">Ингибиторы jak2 и их применение для лечения миелопролиферативных заболеваний и злокачественной опухоли</a>
Замещенные производные бензимидазола и их применение для лечения злокачественной опухоли

Номер патента: 6802
Опубликовано: 28.04.2006
Авторы: Клерк Франсуа, Реснер Манфред, Карре Шанталь, Депати Изабелль, Депре Стефани, Ангуйан-Бонифас Одиль, Ами Франсуа
МПК: A61P 35/00, C07D 235/30, A61K 31/4184...
Метки: применение, замещенные, производные, опухоли, лечения, бензимидазола, злокачественной
Формула / Реферат:
1. Соединения формулы (I) где А представляет собой фенил, где R1 выбран из одной или нескольких аналогичных групп, выбранных из алкила, возможно, замещенного алкокси, гетероалкилом, арилом, ацилом, группой ацильного производного, галогеном; алкокси, возможно, замещенного алкилом, гетероалкилом, арилом, гетероарилом, алкоксиалкилом, гидроксиалкиламидом или перфторалкоксигруппой или алкилтио, возможно, замещенного амидом или перфторалкилтио;...
Композиции для доставки лекарственных средств с высокой водорастворимостью (варианты) и их применение в лечении злокачественной опухоли

Номер патента: 12522
Опубликовано: 30.10.2009
Автор: Чэнь Эндрю Сянь
МПК: A61K 36/24, A61K 31/475, A61K 9/00...
Метки: средств, злокачественной, лечении, доставки, варианты, лекарственных, опухоли, композиции, водорастворимостью, применение, высокой
Формула / Реферат:
1. Композиция, содержащая триглицеридное масло, эмульгатор, стабилизатор, воду и алкалоид барвинка или его фармацевтически приемлемую соль, где: (a) композиция представляет собой эмульсию, содержащую масляную и водную фазу, и (b) не менее 80% алкалоида барвинка или его фармацевтически приемлемой соли присутствует в масляной фазе. 2. Композиция по п.1, где алкалоид барвинка представляет собой винкристин, винбластин или винорелбин. 3. Композиция...
Фармацевтическая композиция и комбинированное лекарственное средство, содержащее ингибиторы hmg-coa-редуктазы и фосфодиэстеразы 4, и их применение для лечения воспалительных заболеваний легких

Номер патента: 21461
Опубликовано: 30.06.2015
Авторы: Маркс Дегенхард, Браун Клеменс, Вользен Андреа, Воллин Штефан-Лутц
МПК: A61K 31/343, A61K 31/22, A61K 31/277...
Метки: фармацевтическая, легких, композиция, воспалительных, средство, комбинированное, лекарственное, фосфодиэстеразы, ингибиторы, лечения, заболеваний, hmg-coa-редуктазы, применение, содержащее
Формула / Реферат:
1. Фармацевтическая композиция для профилактики или лечения хронических воспалительных заболеваний легких, которая содержит компонент (А) ингибитор фосфодиэстеразы 4 типа (ингибитор PDE4) и компонент (Б) ингибитор редуктазы 3-гидрокси-3-метилглутарил кофермента А (ингибитор HMG-CoA-редуктазы) и по меньшей мере одно фармацевтически приемлемое вспомогательное вещество, где ингибитором PDE4 является...
Применение производного антрациклина для лечения опухоли печени

Номер патента: 4570
Опубликовано: 24.06.2004
Авторы: Валота Ольга, Керр Дэвид, Паччарини Мария Аделе
МПК: A61K 31/70, A61P 35/00
Метки: опухоли, печени, антрациклина, лечения, применение, производного
Формула / Реферат:
1. Применение метоксиморфолино доксорубицина (MMDX) формулы для изготовления лекарственного средства для внутрипеченочного введения через печеночную артерию при лечении опухоли печени у человека. 2. Применение по п.1, где опухоль печени представляет собой опухоль, первично ограниченную печенью. 3. Применение по п.2, где опухоль, первично ограниченная печенью, представляет собой печеночно-клеточный рак (HCC) или холангиокарциному. 4....
2-(1н-индазол-6-иламино)бензамидные соединения как ингибиторы протеинкиназ, полезные для лечения офтальмологических заболеваний

Номер патента: 8501
Опубликовано: 29.06.2007
Авторы: Кэйниа Роберт Стивен, Палмер Синтия Луиз, Борчардт Аллен Джон
МПК: A61P 27/02, A61K 31/416, A61P 35/00...
Метки: лечения, 2-(1н-индазол-6-иламино)бензамидные, полезные, офтальмологических, ингибиторы, заболеваний, соединения, протеинкиназ
Формула / Реферат:
1. Соединение 2-{3-[(Е)-2-(4,6-диметилпиридин-2-ил)винил]-1H-индазол-6-иламино}-N-(4-гидрокси-бут-2-инил)бензамид, представленное формулой или его фармацевтически приемлемая соль. 2. Способ лечения возрастной дегенерации желтого пятна у млекопитающего, включающий введение указанному млекопитающему терапевтически эффективного количества соединения или его фармацевтически приемлемой соли по п.1. 3. Способ лечения хориоидальной неоваскуляризации у...
Предыдущий патент: Птеридины, полезные в качестве агрохимикатов и продуктов для здоровья животных
Следующий патент: Новые пептиды и способы их получения и применения
Случайный патент: Фармацевтическая композиция для генерации противовирусного иммунного ответа у человека и животного