Гетероарильные соединения в качестве лигандов 5-ht4 рецептора
Номер патента: 22374
Опубликовано: 30.12.2015
Авторы: Нироджи Рамакришна, Камбхампати Рамасастри, Равула Йотхсна, Мохаммед Абдул Рашид, Роаяпалли Правин Кумар, Схинде Анил Карбхари, Ярлагадда Суреш, Равелла Сриниваса Рао, Патнала Срирамахандра Мурти, Джаяраджан Прадип, Бхирапунени Гопинадх, Джасти Венкатесварлу






















Формула / Реферат
1. Соединение общей формулы (I)
 в которой
в которой представляет собой
представляет собой или
или ;
; представляет собой
представляет собой или
или
![]() представляет собой точку присоединения;
представляет собой точку присоединения;

R2 представляет собой (C3-6)циклоалкил или гетероциклил, где гетероциклил представляет собой неароматическое моноциклическое кольцо из 2-6 атомов углерода, структура которого включает в себя 1 или 2 гетероатома, и необязательно замещен водородом, (C1-3)алкилом или -CO-OR3;
R3 представляет собой (C1-3)алкил;
Y представляет собой C или O;
m представляет собой целое число от 0 до 1, при условии что, когда m представляет 0, R1 представляет собой (C3-6)циклоалкил или гетероциклил, определенный выше;
n представляет собой целое число от 0 до 2;
p представляет собой целое число от 0 до 1,
или его фармацевтически приемлемая соль.
2. Соединение по п.1, которое выбрано из группы, состоящей из
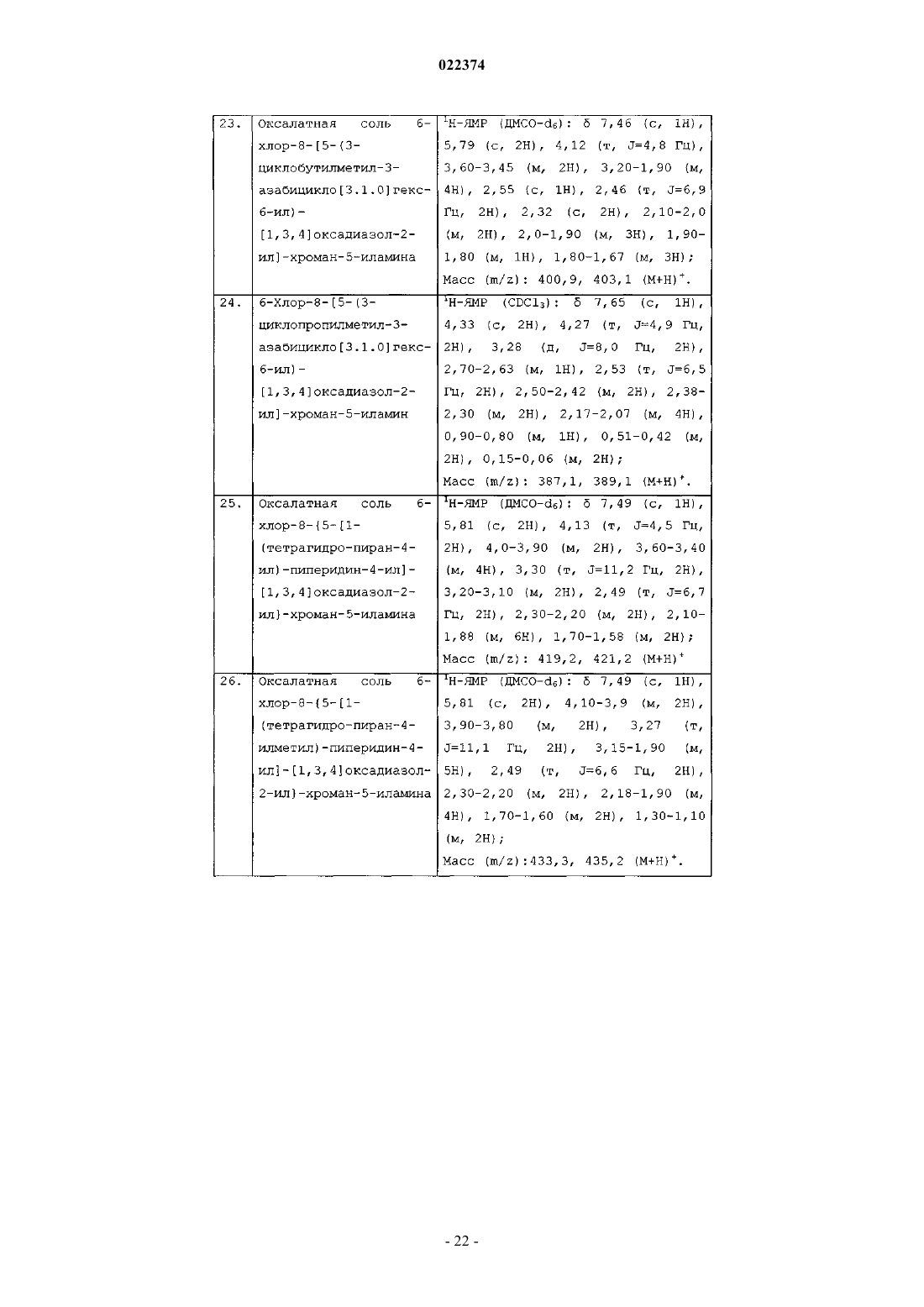
гемифумарата 6-хлор-8-[5-(1-циклопропилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина;
L(+)-тартратной соли 6-хлор-8-[5-(1-циклобутилпиперидин-4-илметил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина;
6-хлор-8-[5-(1-циклобутилпиперидин-4-илметил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина;
оксалатной соли 1-изопропил-3-{5-[1-(3-метоксипропил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил}-1Н-индазола;
L(+)-тартратной соли 3-[5-(1-циклобутилпиперидин-4-илметил)-[1,3,4]сксадиазол-2-ил]-1Н-индазол-2-ил]-1-изопропил-1Н-индазола;
оксалатной соли 6-хлор-8-[5-(3-циклобутил-3-азабицикло[3.1.0]гекс-6-ил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина;
оксалатной соли этилового эфира 4-[5-(8-амино-7-хлор-2,3-дигидробензо[1,4]диоксан-5-ил)-[1,3,4]оксадиазол-2-ил]-[1,4']бипиперидинил-1'-карбоновой кислоты;
оксалатной соли 5-хлор-7-{5-[1-(тетрагидропиран-4-ил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил}-2,3-дигидробензофуран-4-иламина;
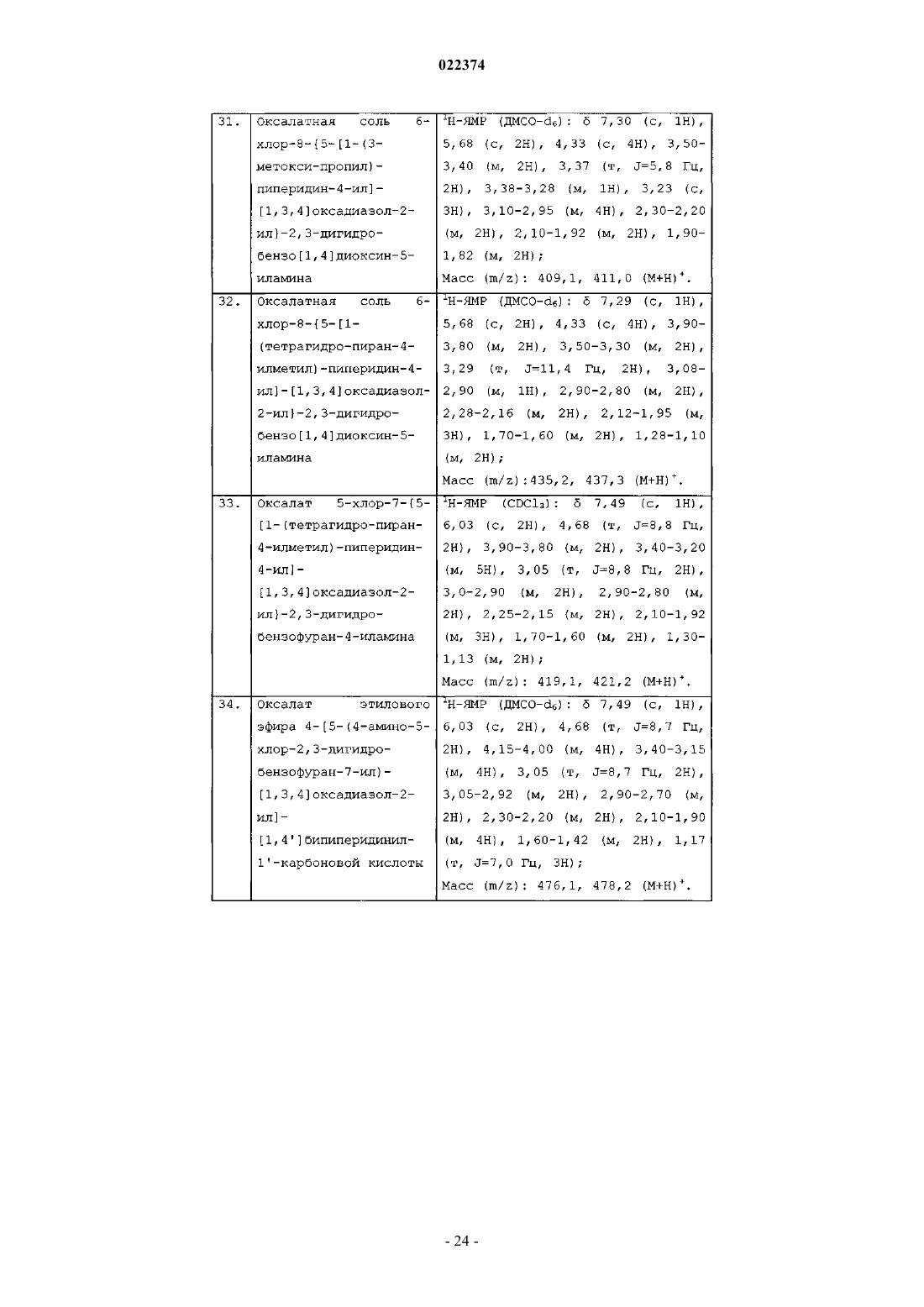
6-хлор-8-{5-[1-(2-метоксиэтил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил}хроман-5-иламина;
6-хлор-8-{5-[1-(3-метилбутил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил}хроман-5-иламина;
6-хлор-8-[5-(1-циклобутилметилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина;
6-хлор-8-[5-(1-циклопропилметилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина;
6-хлор-8-[5-(1-изопропилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина;
6-хлор-8-{5-[1-(3-метоксипропил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил}хроман-5-иламина;
6-хлор-8-[5-(1-циклобутилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина;
6-хлор-8-[5-(1-циклобутилметилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]-2,3-дигидробензо[1,4]диоксин-5-иламина;
6-хлор-8-[5-(1-циклобутилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]-2,3-дигидробензо[1,4]диоксин-5-иламина;
6-хлор-8-[5-(1-циклопентилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина;
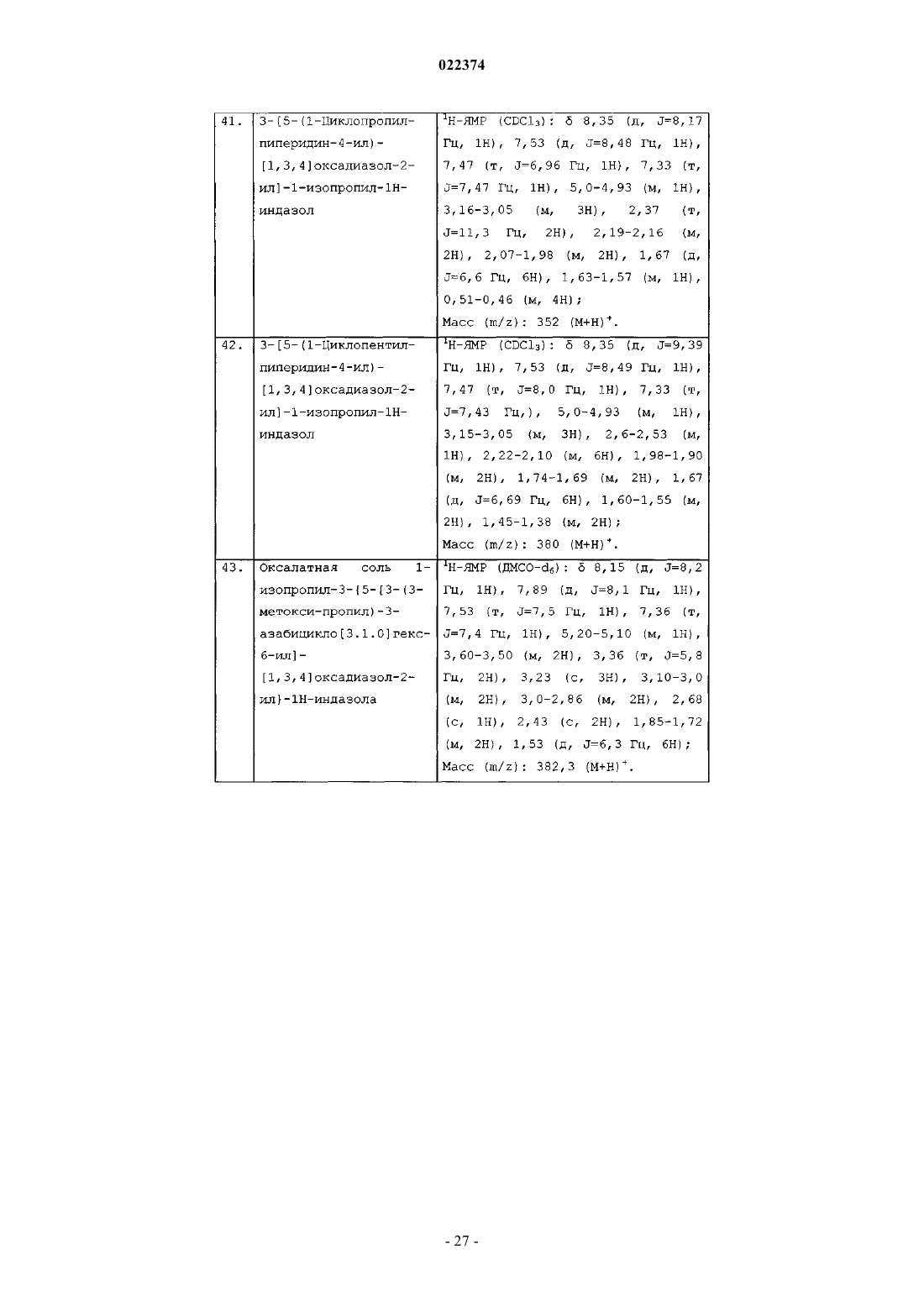
6-хлор-8-[5-(2-пиперидин-1-илэтил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина;
этилового эфира 4-[5-(5-амино-6-хлор-хроман-8-ил)-[1,3,4]оксадиазол-2-ил]-[1,4']бипиперидинил-1'-карбоновой кислоты;
6-хлор-8-[5-(3-пиперидин-1-илпропил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина;
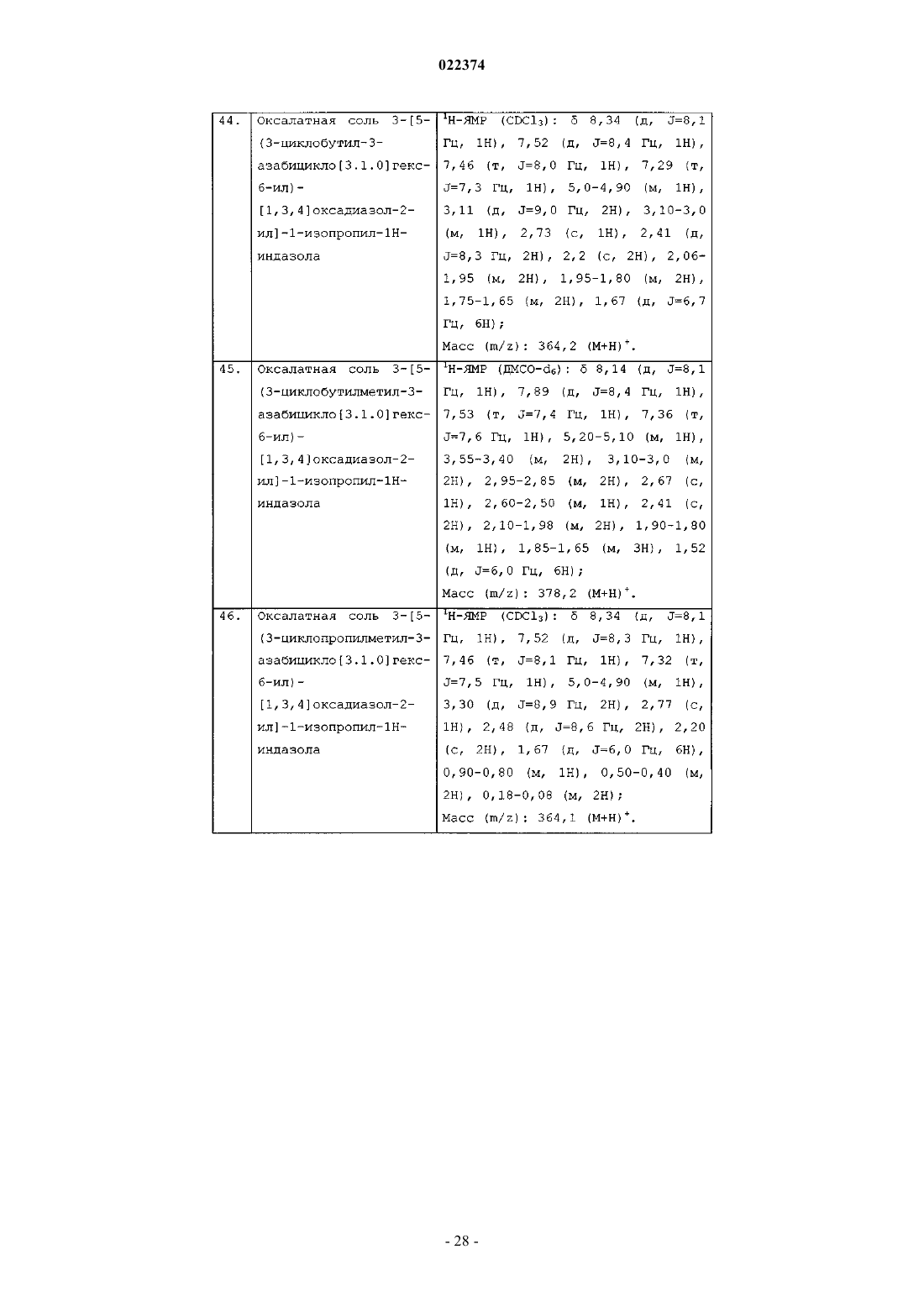
оксалатной соли 6-хлор-8-[5-(1-циклопентилпиперидин-4-илметил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина;
оксалатной соли 6-хлор-8-[5-(3-изопропил-3-азабицикло[3.1.0]гекс-6-ил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина;
оксалатной соли 6-хлор-8-[5-(3-циклобутилметил-3-азабицикло[3.1.0]гекс-6-ил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина;
6-хлор-8-[5-(3-циклопропилметил-3-азабицикло[3.1.0]гекс-6-ил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина;
оксалатной соли 6-хлор-8-{5-[1-(тетрагидропиран-4-ил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил}хроман-5-иламина;
оксалатной соли 6-хлор-8-{5-[1-(тетрагидропиран-4-илметил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил}хроман-5-иламина;
оксалатной соли 5-хлор-7-[5-(1-циклопропилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]-2,3-дигидробензофуран-4-иламина;
оксалатной соли 5-хлор-7-[5-(1-циклобутилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]-2,3-дигидробензофуран-4-иламина;
оксалатной соли 6-хлор-8-[5-(1-циклопропилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]-2,3-дигидробензо[1,4]диоксин-5-иламина;
оксалатной соли 6-хлор-8-{5-[1-(тетрагидропиран-4-ил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил}-2,3-дигидробензо[1,4]диоксин-5-иламина;
оксалатной соли 6-хлор-8-{5-[1-(3-метоксипропил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил}-2,3-дигидробензо[1,4]диоксин-5-иламина;
оксалатной соли 6-хлор-8-{5-[1-(тетрагидропиран-4-илметил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил}-2,3-дигидробензо[1,4]диоксин-5-иламина;
оксалата 5-хлор-7-{5-[1-(тетрагидропиран-4-илметил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил}-2,3-дигидробензофуран-4-иламина;
оксалата этилового эфира 4-[5-(4-амино-5-хлор-2,3-дигидробензофуран-7-ил)-[1,3,4]оксадиазол-2-ил]-[1,4']бипиперидинил-1'-карбоновой кислоты;
3-[5-(1-циклобутилметилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]-1-изопропил-1Н-индазола;
1-изопропил-3-{5-[1-(2-метоксиэтил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил}-1Н-индазола;
3-[5-(1-циклобутилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]-1-изопропил-1Н-индазола;
1-изопропил-3-[5-(1-изопропилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]-1Н-индазола;
3-[5-(1-циклопропилметилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]-1-изопропил-1Н-индазола;
1-изопропил-3-{5-[1-(3-метилбутил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил}-1Н-индазола;
3-[5-(1-циклопропилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]-1-изопропил-1Н-индазола;
3-[5-(1-циклопентилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]-1-изопропил-1Н-индазола;
оксалатной соли 1-изопропил-3-{5-[3-(3-метоксипропил)-3-азабицикло[3.1.0]гекс-6-ил]-[1,3,4]оксадиазол-2-ил}-1Н-индазола;
оксалатной соли 3-[5-(3-циклобутил-3-азабицикло[3.1.0]гекс-6-ил)-[1,3,4]оксадиазол-2-ил]-1-изопропил-1Н-индазола;
оксалатной соли 3-[5-(3-циклобутилметил-3-азабицикло[3.1.0]гекс-6-ил)-[1,3,4]оксадиазол-2-ил]-1-изопропил-1Н-индазола;
оксалатной соли 3-[5-(3-циклопропилметил-3-азабицикло[3.1.0]гекс-6-ил)-[1,3,4]оксадиазол-2-ил]-1-изопропил-1H-индазола;
оксалатной соли 1-изопропил-3-{5-[1-(тетрагидропиран-4-илметил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил}-1Н-индазола;
оксалатной соли 1-изопропил-3-[5-(1-(тетрагидропиран-4-ил)пиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]-1Н-индазола;
оксалатной соли 1-изопропил-3-[5-(2-пиперидин-1-илэтил)-[1,3,4]оксадиазол-2-ил]-1Н-индазола
или их фармацевтически приемлемых солей.
3. Способ получения соединения формулы (I) по п.1, включающий:
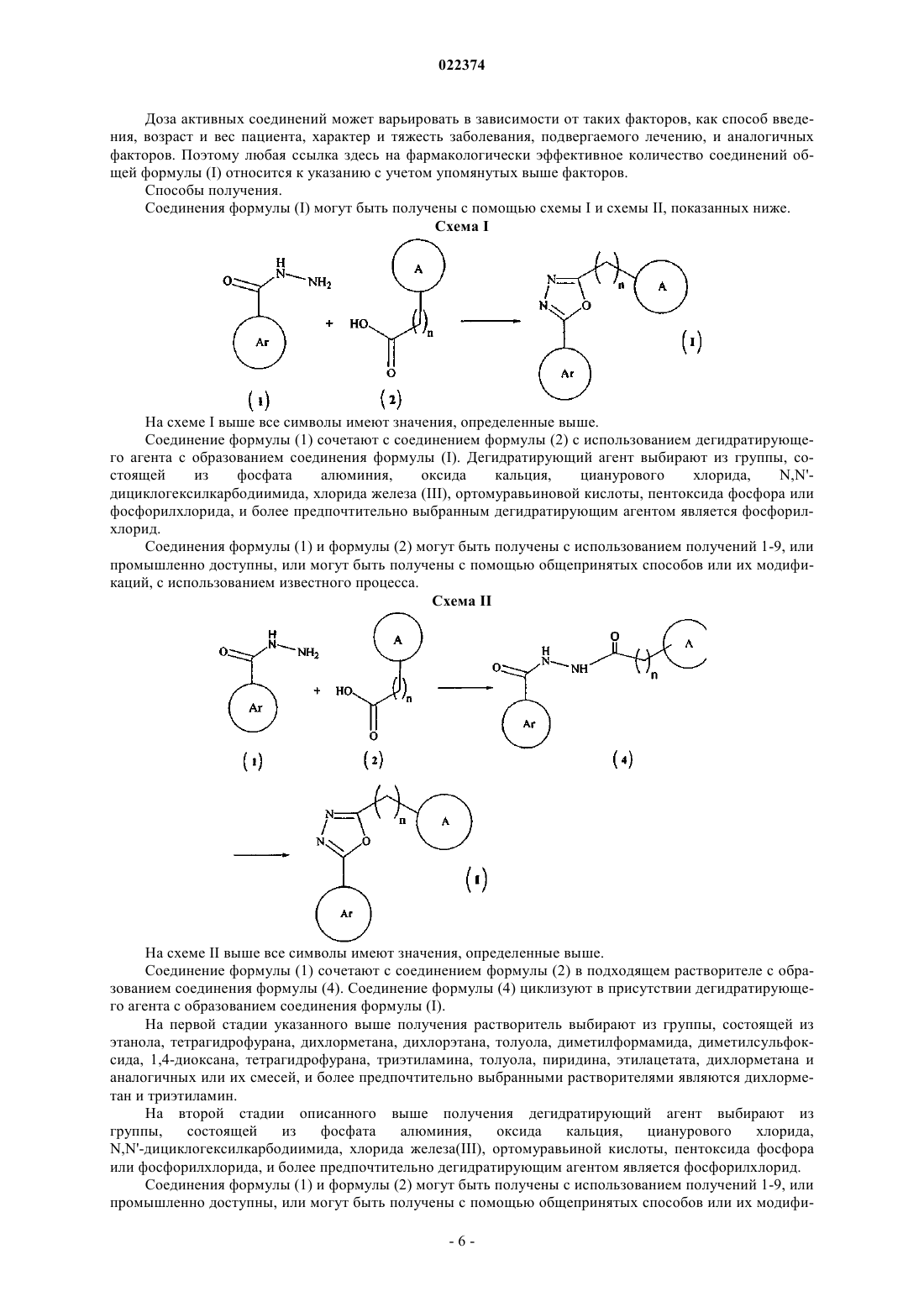
(а) сочетание соединения формулы (1) с соединением формулы (2)

в присутствии дегидратирующего агента с образованием соединения формулы (I), в которой все заместители имеют значения, определенные в п.1;
(b) необязательно, превращение соединения формулы (I) в его фармацевтически приемлемые соли.
4. Способ получения соединения формулы (I) по п.1, включающий:
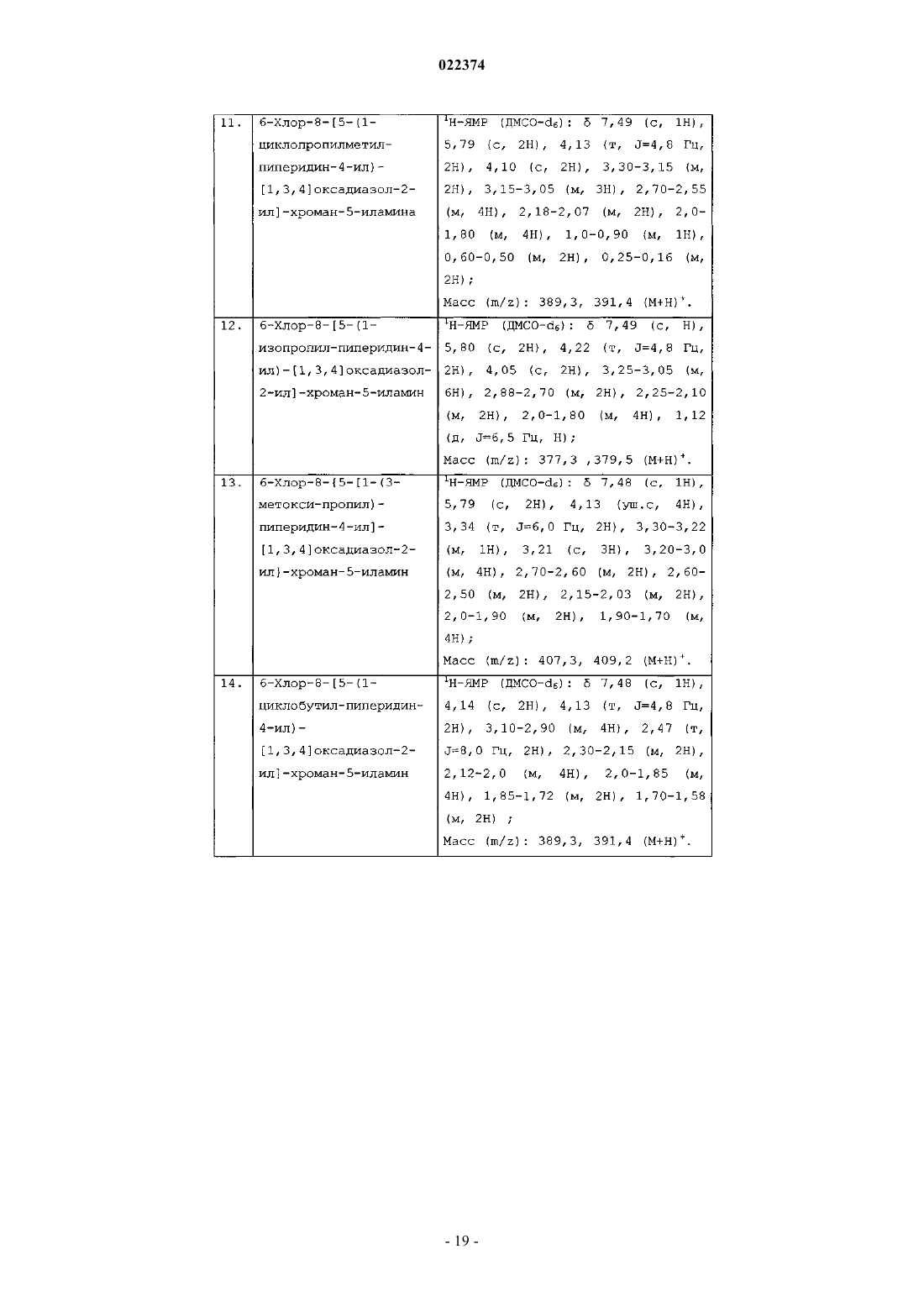
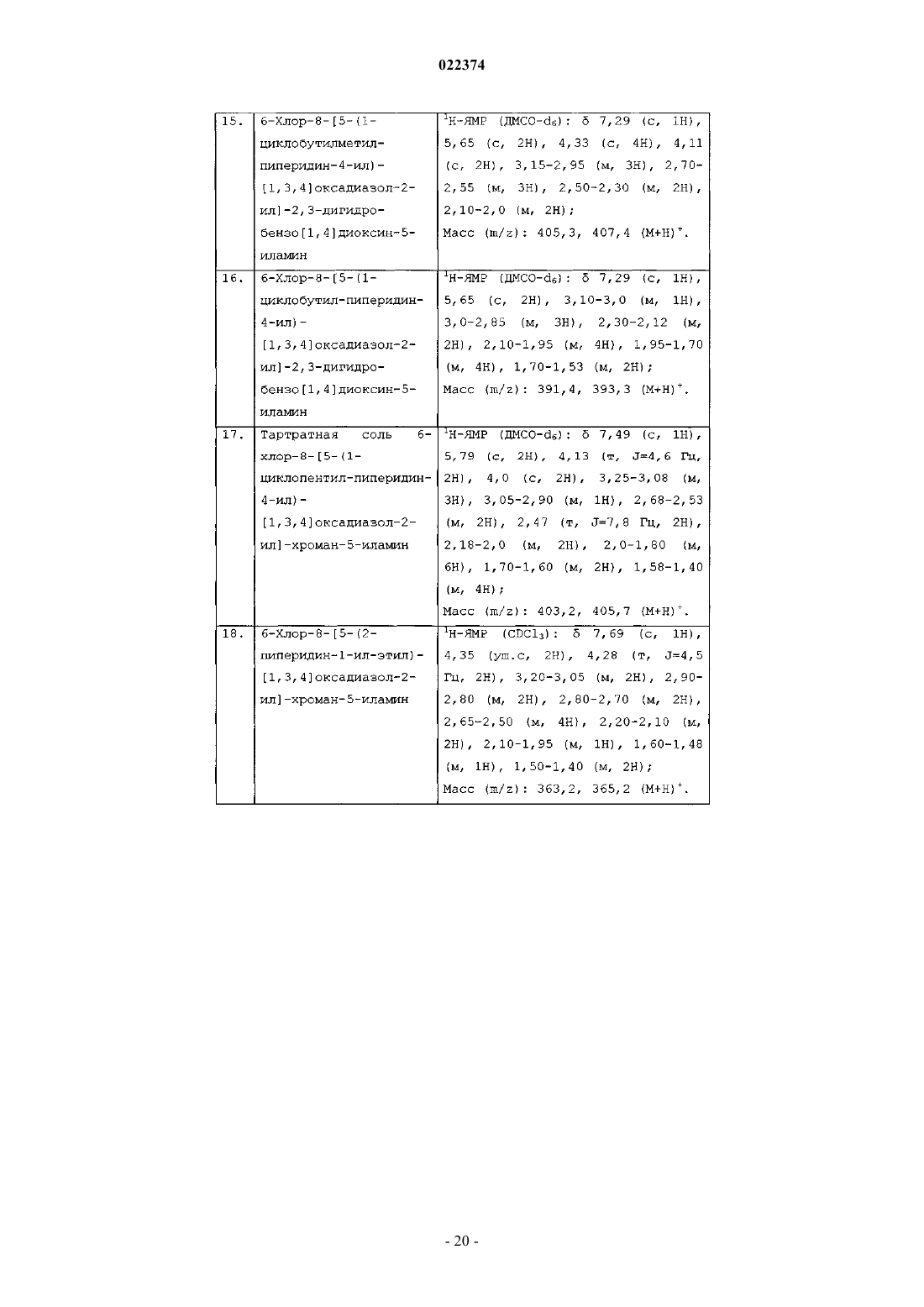
(а) сочетание соединения формулы (1) с соединением формулы (2)

в присутствии подходящего растворителя с образованием соединения формулы (4)

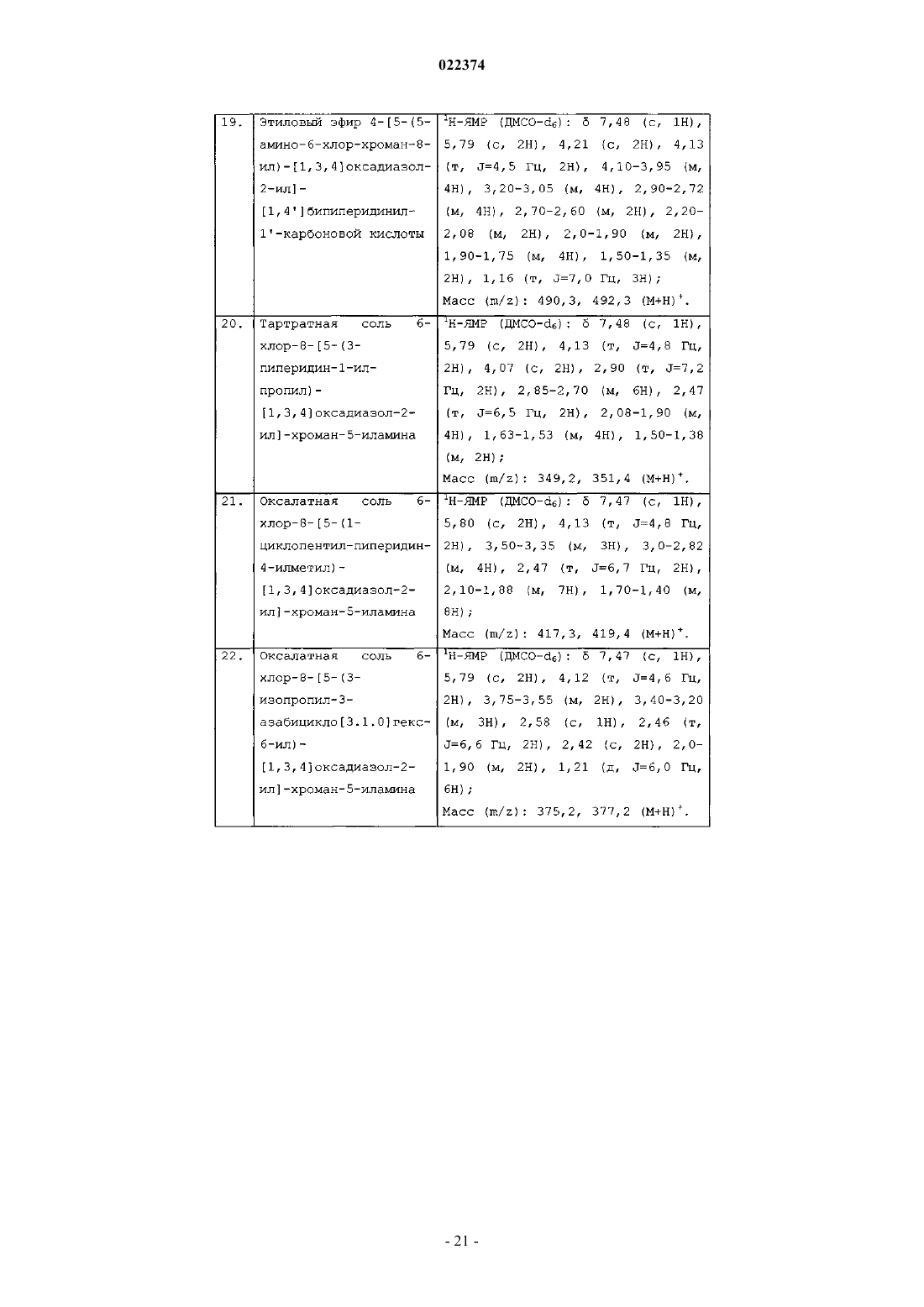
(b) циклизацию соединения формулы (4) с образованием соединения формулы (I), в которой все заместители имеют значения, определенные в п.1;
(c) необязательно, превращение соединения формулы (I) в его фармацевтически приемлемые соли.
5. Фармацевтическая композиция для лечения клинических состояний, опосредованных 5-HT4 рецепторами, включающая соединение по любому из пп.1, 2 и фармацевтически приемлемые эксципиенты.
6. Фармацевтическая композиция по п.5 для лечения таких состояний, как синдром гиперактивности с дефицитом внимания, болезнь Альцгеймера, когнитивные расстройства, деменция или шизофрения.
7. Способ лечения когнитивных расстройств, деменции, синдрома гиперактивности с дефицитом внимания, шизофрении и боли, включающий введение пациенту, нуждающемуся в этом, эффективного количества соединения или его фармацевтически приемлемой соли по любому из пп.1, 2.
8. Применение соединения по любому из пп.1, 2 для получения лекарственного средства для лечения заболеваний, опосредованных 5-HT4 рецепторами.
9. Применение соединения по п.8 для лечения клинических состояний, таких как синдром гиперактивности с дефицитом внимания, болезнь Альцгеймера, когнитивные расстройства, деменция или шизофрения.
Текст
Изобретение относится к новым соединениям формулы (I) и их фармацевтически приемлемым солям и к композициям, содержащим их: Кроме того, изобретение относится к способу получения указанных выше соединений и их фармацевтически приемлемых солей. Соединения формулы (I) полезны при лечении различных расстройств, которые связаны с 5-HT4 рецепторами.(71)(73) Заявитель и патентовладелец: СУВЕН ЛАЙФ САЙЕНСИЗ ЛИМИТЕД (IN) Область изобретения Настоящее изобретение относится к новым соединениям формулы (I) и их фармацевтически приемлемым солям и композициям, содержащим их, для лечения различных расстройств, которые связаны с 5-HT4 рецепторами. Предпосылки изобретения 5-HT4 официально признан (Humphrey et al., 1993) и идентифицирован во множестве тканей многих видов (см. обзор FordClarke, 1993). Найдено, что модуляторы 5-HT4 рецептора (например, агонисты и антагонисты) являются полезными для лечения множества заболеваний, таких как гастроэзофагеальный рефлюкс, желудочно-кишечное заболевание, расстройство перистальтики желудка, неязвенная диспепсия, функциональная диспепсия, синдром раздраженного кишечника, констипация, диспепсия, эзофагит,гастроэзофагеальное заболевание, тошнота, заболевания центральной нервной системы, когнитивные расстройства, деменция, синдром гиперактивности с дефицитом внимания, шизофрения и сердечнососудистые расстройства, такие как сердечная недостаточность и сердечная аритмия (Corsi. M. et al.,Pharmacological analysis of 5-hydroxytryptamine effects on electrically stimulated human isolated urinaryUS 20080269211 раскрыты некоторые 5-HT4 рецепторные соединения. Хотя описаны некоторые лиганды 5-NT4 рецепторов, все еще существует потребность и простор в обнаружении новых лекарств с новыми химическими структурами для лечения расстройств, опосредованных 5-HT4 рецептором. Краткое содержание изобретения Настоящее изобретение относится к новым 5-HT4 лигандным соединениям формулы (I) представляет собой представляет собой представляет собой точку присоединения;R2 представляет собой циклоалкил или гетероциклил и необязательно замещен водородом, алкилом или -CO-OR3;m представляет целое собой число от 0 до 1; при условии что, когда m представляет 0, R1 представ-1 022374 ляет собой циклоалкил или гетероциклил;n представляет собой целое число от 0 до 2;p представляет собой целое число от 0 до 1; или их фармацевтически приемлемым солям. Настоящее изобретение относится к применению терапевтически эффективного количества соединения формулы (I), к получению лекарственного средства для лечения различных расстройств, которые связаны с 5-HT4 рецепторами. В частности, соединения данного изобретения полезны в лечении разнообразных расстройств, таких как синдром гиперактивности с дефицитом внимания, болезнь Альцгеймера, когнитивные расстройства, деменция или шизофрения. В еще одном аспекте изобретение относится к фармацевтическим композициям, содержащим терапевтически эффективное количество по крайней мере одного соединения формулы (I), и его фармацевтически приемлемых солей, в смеси с фармацевтически приемлемым эксципиентом. В еще одном аспекте изобретение относится к способу лечения с применением соединений формулы (I). В еще одном аспекте изобретение дополнительно относится к способу получения соединений формулы (I) и их фармацевтически приемлемых солей. Характерные соединения настоящего изобретения включают соединения, указанные ниже, и их фармацевтически приемлемые соли. Настоящее изобретение не следует рассматривать как ограниченное ими. Подробное описание изобретения Если не указано иное, следующие термины, используемые в описании и в формуле изобретения,имеют значения, данные ниже. Термин "алкил" обозначает углеводородный радикал с прямой или разветвленной цепью, состоящий единственно из атомов углерода и водорода, не содержащий ненасыщенности, имеющий от одного до трех атомов углерода, и который присоединен к остатку молекулы с помощью одинарной связи. Примерные "алкильные" группы включают в их число метил, этил, н-пропил, изопропил и аналогичные. Термин "циклоалкил" обозначает неароматическое моноциклическое кольцо из 3-8 атомов углерода. Примерные "циклоальные" группы включают циклопропил, циклобутил, циклопентил и аналогичные. Термин "гетероциклил" обозначает неароматическое моноциклическое кольцо из 2-7 атомов углерода, структура которого включает в себя 1-3 гетероатома, данные дополнительные атомы могут повторяться в кольце более одного раза. Примерные "гетероциклильные" группы включают в себя пирролидинил, пиперидинил, пиперазинил, морфолинил и аналогичные. Фраза "фармацевтически приемлемые соли" указывает, что вещество или композиция должны быть химически и/или токсикологически совместимыми с другими ингредиентами, включая рецептурные формы, для млекопитающих, подвергаемых лечению ими. Фраза "терапевтически эффективное количество" определяется как количество соединения настоящего изобретения, которое (i) лечит конкретную болезнь, состояние или расстройство, (ii) устраняет один или более симптомов конкретного заболевания, состояния или расстройства, (iii) задерживает наступление одного или более симптомов описываемого здесь заболевания, состояния или расстройства. Промышленные реагенты использовались без дополнительной очистки. Комнатная температура относится к 25-40C. Если не указано иное, все масс спектры снимались с использованием ESI условий. 1HЯМР спектры регистрировались при 400 МГц с помощью инструмента Bruker. В качестве растворителя использовали дейтерированный хлороформ, метанол или диметилсульфоксид. TMS использовали в качестве внутреннего ссылочного стандарта. Величины химического сдвига выражены в частях на миллион(5). Для обозначения мультиплетности ЯМР сигналов использованы следующие сокращения: s=синглет,bs=широкий синглет, d=дублет, t=триплет, q=квартет, qui=квинтет, h=гептет, dd=двойной дублет,dt=двойной триплет, tt=триплет триплетов, m=мультиплет. Хроматография относится к колоночной хроматографии, выполняемой с использованием силикагеля 100-200 меш, и осуществляли ее под давлением азота в условиях флэш-хроматографии. Фармацевтические композиции. Для того чтобы использовать соединения формулы (I) в терапии, их обычно формулируют в виде фармацевтической композиции в соответствии со стандартной фармацевтической практикой. Фармацевтические композиции настоящего изобретения могут формулироваться общепринятым образом с использованием одного или более фармацевтически приемлемых эксципиентов. Фармацевтически приемлемым эксципиентом является носитель или разбавитель. Так, активные соединения изобретения можно формулировать для перорального, внутриназального или парентерального введения (например, внутривенно, внутримышечно или подкожно). Такие фармацевтические композиции и процессы их получения хорошо известны в технике (The Science and Practice of Pharmacy,D.B. Troy, 21st Edition, WilliamsWilkinc, 2006). Доза активных соединений может варьировать в зависимости от таких факторов, как способ введения, возраст и вес пациента, характер и тяжесть заболевания, подвергаемого лечению, и аналогичных факторов. Поэтому любая ссылка здесь на фармакологически эффективное количество соединений общей формулы (I) относится к указанию с учетом упомянутых выше факторов. Способы получения. Соединения формулы (I) могут быть получены с помощью схемы I и схемы II, показанных ниже. Схема I На схеме I выше все символы имеют значения, определенные выше. Соединение формулы (1) сочетают с соединением формулы (2) с использованием дегидратирующего агента с образованием соединения формулы (I). Дегидратирующий агент выбирают из группы, состоящей из фосфата алюминия,оксида кальция,цианурового хлорида,N,N'дициклогексилкарбодиимида, хлорида железа (III), ортомуравьиновой кислоты, пентоксида фосфора или фосфорилхлорида, и более предпочтительно выбранным дегидратирующим агентом является фосфорилхлорид. Соединения формулы (1) и формулы (2) могут быть получены с использованием получений 1-9, или промышленно доступны, или могут быть получены с помощью общепринятых способов или их модификаций, с использованием известного процесса. Схема II На схеме II выше все символы имеют значения, определенные выше. Соединение формулы (1) сочетают с соединением формулы (2) в подходящем растворителе с образованием соединения формулы (4). Соединение формулы (4) циклизуют в присутствии дегидратирующего агента с образованием соединения формулы (I). На первой стадии указанного выше получения растворитель выбирают из группы, состоящей из этанола, тетрагидрофурана, дихлорметана, дихлорэтана, толуола, диметилформамида, диметилсульфоксида, 1,4-диоксана, тетрагидрофурана, триэтиламина, толуола, пиридина, этилацетата, дихлорметана и аналогичных или их смесей, и более предпочтительно выбранными растворителями являются дихлорметан и триэтиламин. На второй стадии описанного выше получения дегидратирующий агент выбирают из группы,состоящей из фосфата алюминия,оксида кальция,цианурового хлорида,N,N'-дициклогексилкарбодиимида, хлорида железа(III), ортомуравьиной кислоты, пентоксида фосфора или фосфорилхлорида, и более предпочтительно дегидратирующим агентом является фосфорилхлорид. Соединения формулы (1) и формулы (2) могут быть получены с использованием получений 1-9, или промышленно доступны, или могут быть получены с помощью общепринятых способов или их модифи-6 022374 каций, с использованием известного процесса. Если необходимо, фармацевтически приемлемые соли соединений формулы (I) могут быть получены удобно по реакции с соответствующей кислотой или производным кислоты. Подходящие фармацевтически приемлемые соли понятны специалистам в данной области и включают в себя соли, описанные в J. Pharm. Sci., 1977, 66, 1-19, такие как кислотно-аддитивные соли, образуемые с неорганическими кислотами, например соляной, бромисто-водородной, серной, азотной или фосфорной кислотой, и органическими кислотами, например янтарной, малеиновой, уксусной, фумаровой, лимонной, яблочной, винной, бензойной, п-толуиловой, п-толуолсульфоновой, бензолсульфоновой,метансульфоновой или нафталинсульфоновой кислотой. Наиболее предпочтительными солями соединений формулы (I) являются оксалат, тартрат, фумарат, метансульфонат, гидрохлорид и сульфат. На основе клинической разработки соединения выбирают солевую форму соединения и эффективную дозу. Оксалатная соль является наиболее предпочитаемой солью для соединения в форме свободного основания примеров 3 и 4. Фумаратная соль является наиболее предпочитаемой солью для соединения в форме свободного основания примера 1. На основе клинической разработки соединений из соединений в виде свободного основания примеров 1-74 специалист в данной области может легко получить все предпочтительные соли данного изобретения. Примеры Новые соединения настоящего изобретения были получены в соответствии со следующими экспериментальными процедурами с использованием соответствующих материалов и условий. Получение 1. Получение гидразида 5-амино-6-хлорхроман-8-карбоновой кислоты. Стадия (i). Получение метил 4-амино-2-гидроксибензоата. К перемешиваемому раствору 4-аминосалициловой кислоты (50 г, 326,7 ммоль) в метаноле (375 мл) при 0C добавляли концентрированную серную кислоту (99,7 мл, 1,87 ммоль) при подержании температуры реакции ниже 20C. Реакционную смесь постепенно нагревали до кипения с обратным холодильником, и после завершения реакции спустя 6 ч ее охлаждали до температуры ледяной бани и подщелачивали водным раствором гидроксида натрия (10 н., 214,5 мл). Белый осадок, который образовывался, отфильтровывали, промывали водой, эфиром и сушили в вакууме, получая метил-4-амино-2 гидроксибензоат (50,70 г). Выход: 93%. 1H-ЯМР (ДМСО-d6):10,76 (уш.с, 1H), 7,43 (д, J=8,6 Гц, 1H), 6,13 (уш.с, 2H), 6,10 (дд, J=8,6, 2,0 Гц,1H), 5,99 (д, J=2,0 Гц, 1H), 3,79 (с, 3H); Масс (m/z): 168 (М+H)+. Стадия (ii). Получение метил-4-ацетиламино-2-гидроксибензоата. Раствор метил-4-амино-2-гидроксибензоата (50,7 г, 303,6 ммоль, полученный на вышеуказанной стадии) в этилацетате (750 мл) добавляли к перемешиваемому раствору воды (250 мл) и бикарбоната натрия (34,9 г, 415,5 ммоль), охлаждаемому при 0C, с последующим ацетилхлоридом (29,7 мл,415,5 ммоль) на протяжении периода 15 мин. Реакционную смесь постепенно подогревали до комнатной температуры и перемешивали в течение 2 ч. Два слоя разделяли, органический слой промывали солевым раствором, сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении,получая 4-ацетиламино-2-гидроксибензоат (63,5 г). Выход: 99%. 1H-ЯМР (CDCl3):10,86 (уш.с, 1H), 7,78 (д, J=8, 6 Гц, 1H), 7,23 (с, 1H), 7,16 (уш.с, 1H), 7,10 (д,J=8,6 Гц, 1H), 6,13 (уш.с, 1H), 3,92 (с, 3H), 2,19 (с, 3H); Масс (m/z): 208 (M-H)+. Стадия (iii). Получение метил-4-ацетиламино-5-хлор-2-гидроксибензоата. К перемешиваемому раствору метил-4-ацетиламино-2-гидроксибензоата (61,4 г, 294,0 ммоль, полученному на указанной выше стадии) в дихлорэтане (1,2 л) добавляли N-хлорсукцинимид (58,8 г,441 ммоль) и реакционную смесь нагревали с обратным холодильником в течение 3 ч. Летучие вещества удаляли при пониженном давлении; выпавшее таким образом в осадок твердое соединение разбавляли водой (1,0 л) и фильтровали. Сырой продукт разбавляли 1:9 смесью (метанола и дихлорметана) и промывали солевым раствором. Органический слой сушили над безводным сульфатом натрия и летучие вещества удаляли при пониженном давлении, получая метил-4-ацетиламино-5-хлор-2-гидроксибензоатH-ЯМР (ДМСО-d6):10,49 (уш.с, 1H), 9,47 (с, 1H), 7,75 (с, 1H), 7,72 (с, 1H), 3,85 (с, 3H), 2,16 (с,3H); Масс (m/z): 244, 246 (M+H)+. Стадия (iv). Получение метил-4-ацетиламино-5-хлор-2-(проп-2-инилокси)бензоата. К перемешиваемому раствору 4-ацетиламино-5-хлор-2-гидроксибензоата (30 г, 123,2 полученного на предыдущей стадии) в диметилформамиде (246 мл) добавляли карбонат калия (42,5 г, 308 ммоль). Реакционную смесь охлаждали до 0C и добавляли пропаргилбромид (22,3 мл, 150,3 ммоль) на протяже-7 022374 нии периода 15 мин. Реакционную смесь подогревали до комнатной температуры и перемешивали в течение 5 ч, прежде чем помещали в холодную воду. Выпавшие в осадок твердые вещества отфильтровывали и неочищенный продукт растворяли в 1:9 смеси (метанол:дихлорметан) и промывали солевым раствором. Органический слой сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении, получая указанное в заголовке соединение (25,2 г). Выход: 73%. 1H-ЯМР (ДМСО-d6):9,60 (с, 1H), 7,91 (с, 1H), 7,76 (с, 1H), 4,82 (с, 2H), 3,77 (с, 3H), 3,61 (с, 1H),2,15 (с, 3H); Масс (m/z): 282, 284 (M+H)+. Стадия (v). Получение метил-5-ацетиламино-6-хлор-2H-хромен-8-карбоксилата. Перемешиваемый раствор метил-4-ацетиламино-5-хлор-2-(проп-2-инилокси)бензоата (25 г,88,8 ммоль, полученного на предыдущей стадии) в даутерме А (127 мл) нагревали до 220C в течение 3 ч. Реакционную смесь охлаждали до 60-70C и выливали в гексан. Выпавшие в осадок твердые вещества отфильтровывали и промывали гексаном, получая метил-5-ацетиламино-6-хлор-2H-хромен-8 карбоксилат (16,2 г). Выход: 64,8%. 1(с, 3H), 2,06 (с, 3H); Масс (m/z): 282, 284 (M+H)+. Стадия (vi). Получение метил-5-ацетиламино-6-хлорхроман-8-карбоксилата. К раствору метил-5-ацетиламино-6-хлор-2H-хромен-8-карбоксилата (20,5 г, 72,9 ммоль, полученного на указанной выше стадии) в этаноле (300 мл) добавляли Pd/C (10% мас./мас., 8,6 г). С использованием давления аэростата применяли давление водорода. Реакционную смесь перемешивали при комнатной температуре в течение 5 ч и фильтровали через подушку из целита. Фильтрат концентрировали досуха,получая метил-5-ацетиламино-6-хлорхроман-8-карбоксилат (18,88 г). Выход: 91,3%. 1H-ЯМР (ДМСО-d6):9,65 (с, 1H), 7,55 (с, 1H), 4,16 (т, J=4,5 Гц, 2H), 3,76 (с, 3H), 2,58 (т, J=6,3 Гц,2H), 2,05 (с, 3H), 1,87 (м, 2H); Масс (m/z): 284, 286 (M+H)+. Стадия (vii). Получение 5-амино-6-хлорхроман-8-карбоновой кислоты. К метил-5-ацетиламино-6-хлорхроман-8-карбоксилату (18,88 г, 66,6 ммоль, полученному на указанной выше стадии) добавляли водный раствор гидроксида натрия (1,4 н, 475 мл) и реакционную смесь нагревали с обратным холодильником в течение 6 ч. Реакционную смесь подкисляли 2 н. гидрохлоридом при 0C и выпавший в осадок продукт отфильтровывали и сушили в вакууме, получая 5-амино-6-хлорхроман-8-карбоновую кислоту (14,07 г). Выход: 92,9%. 1H-ЯМР (ДМСО-d6):11,8 (уш.с, 1H), 7,48 (с, 1H), 5,74 (уш.с, 2H), 4,09 (т, J=4,6 Гц, 2H), 2,43 (т,J=6,4 Гц, 2H), 1,91 (м, 2H); Масс (m/z): 228, 230 (M+H)+. Стадия (viii). Получение метил-5-амино-6-хлорхроман-8-карбоксилата. К перемешиваемому раствору 5-амино-6-хлорхроман-8-карбоновой кислоты (13,5 г, 59,34 ммоль,полученной на указанной выше стадии) в метаноле (68 мл), охлаждаемому при 0C, добавляли по каплям концентрированную серную кислоту (18,10 мл). Реакционную смесь постепенно подогревали до комнатной температуры и перемешивали в течение 4 ч. Реакционную смесь охлаждали до 0C, разбавляли водой (202 мл) и подщелачивали гидроокисью натрия (10 М, 57,9 мл). Продукт, который выпадал в осадок,отфильтровывали и сушили в вакууме, получая метил-5-амино-6-хлорхроман-8-карбоксилат (10,5 г). Выход: 7 0,2%. 1H-ЯМР (CDCl3):7,75 (с, 1H), 4,37 (уш.с, 2H), 4,24 (т, J=5,0 Гц, 2H), 3,83 (с, 3H), 2,49 (т, J=6,6 Гц,2H), 2,10 (м, 2H); Масс (m/z): 242, 244 (M+H)+. Стадия (ix). Получение гидразида 5-амино-6-хлорхроман-8-карбоновой кислоты. К перемешиваемому раствору метил-5-амино-6-хлорхроман-8-карбоксилата (10,0 г, 41,4 ммоль, полученному на вышеуказанной стадии) в этаноле (82 мл), добавляли гидразингидрат (31,05 мл). Температуру реакции постепенно поднимали до кипения с обратным холодильником и реакционную смесь перемешивали при данной температуре в течение 5 ч. Летучие вещества удаляли при пониженном давлении,сырую массу растворяли в 10% метаноле в дихлорметане и промывали водой, солевым раствором, сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении, получая гидразид 5-амино-6-хлорхроман-8-карбоновой кислоты (9,3 г). Выход: 93%. 1-8 022374 Масс (m/z): 242, 244 (M+H)+. Получение 2. Получение метил-1-изопропил-1H-индазол-3-илкарбоксилата. Стадия (i). Получение метил-1H-индазол-3-илкарбоксилата. К перемешиваемому раствору индазол-3-карбоновой кислоты (80,5 г, 0,497 ммоль, полученной на вышеуказанной стадии) в метаноле (2 л), охлаждаемому при 0C, добавляли тионилхлорид (120 мл,1,59 ммоль) на протяжении периода 1 ч. Температуру реакции постепенно повышали и реакционную смесь нагревали с обратным холодильником в течение 5 ч. Летучие вещества удаляли и сырую массу разбавляли дихлорметаном, промывали водным бикарбонатом натрия, сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении, получая указанное в заголовке соединениеH-ЯМР (CDCl3):13,2 (уш.с, 1H), 8,23 (д, J=8,2 Гц, 1H), 7,86 (д, J=8,4 Гц, 1H), 7,48 (т, J=7,4 Гц,1H), 7,35 (т, J=7,6 Гц, 1H), 4,09 (с, 3H); Масс (m/z): 177 (M+H)+. Стадия (ii). Получение метил-1-изопропил-1H-индазол-3-илкарбоксилата. К перемешиваемому раствору метил-1H-индазол-3-илкарбоксилата (80,0 г, 0,454 ммоль, полученного на вышеуказанной стадии) в сухом диметилформамиде (500 мл) при 0C добавляли порциями на протяжении 30 мин гидрид натрия (60% в минеральном масле, 23,7 г, 0,592 ммоль). Реакционную смесь постепенно подогревали до комнатной температуры и перемешивали в течение 45 мин перед ее охлаждением снова до 0C. К реакционной смеси добавляли изопропилиодид (55 мл, 0,54 5 ммоль) и перемешивали при комнатной температуре в течение 4 ч. Реакционную смесь выливали в раздробленный лед,перемешивали в течение 10 мин и экстрагировали этилацетатом (2250 мл). Объединенный органический слой промывали водой (2500 мл), солевым раствором, сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении, получая сырую массу, которую очищали на колонке с силикагелем, получая 1-изопропил-1H-имидазол-3-илкарбоксилат (40,0 г). Выход: 40%. 1H-ЯМР (CDCl3):8,24 (д, J=8,1 Гц, 1H), 7,52 (д, J=8,4 Гц, 1H), 7,43 (т, J=7,2 Гц, 1H), 7,31 (т,J=7,6 Гц, 1H), 4,96 (м, 1H), 4,04 (с, 3H), 1,66 (д, J=6,7 Гц, 6H); Масс (m/z): 219 (M+H)+. Стадия (iii). Получение гидразида 1-изопропил-1H-индазол-3-илкарбоновой кислоты. К перемешиваемому раствору метил-1-изопропил-1H-индазол-3-илкарбоксилата (40,0 г,183,5 ммоль, полученного на вышеуказанной стадии) в этаноле при комнатной температуре добавляли гидразингидрат (130 мл, 2,56 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 2 ч. Летучие вещества удаляли при пониженном давлении и сырую массу разбавляли дихлорметаном, промывали водой, солевым раствором, сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении, получая указанное в заголовке соединение (37,52 г). Выход: 93%. 1(Chem. Pharm. Bull. 1998, 46(1), 42-52; 3,93 г, 18,4 ммоль) в метаноле (36,8 мл), охлаждаемому при 0C,добавляли тионилхлорид (6,0 мл). Реакционную смесь постепенно подогревали до комнатной температуры и нагревали с обратным холодильником в течение 2 ч. Летучие вещества удаляли при пониженном давлении; сырую массу разбавляли водным раствором бикарбоната натрия и экстрагировали этилацетатом. Объединенный органический слой сушили над безводным сульфатом натрия и растворитель удаляли в вакууме, получая 4-амино-5-хлор-2,3-дигидробензофуран-7-карбоксилат (3,89 г). Выход: 92,9%. 1H-ЯМР (ДМСО-d6):7,43 (с, 1H), 6,06 (уш.с, 2H), 4,60 (т, J=8,8 Гц, 2H), 3,68 (с, 3H), 2,97 (т,J=8,8 Гц, 2H); Масс (m/z): 228,0, 230,1 (M+H)+. Стадия (ii). Получение гидразида 4-амино-5-хлор-2,3-дигидробензофуран-7-карбоновой кислоты. К перемешиваемому раствору 4-амино-5-хлор-2,3-дигидробензофуран-7-карбоксилата (3,88 г,17,07 ммоль, полученного на вышеуказанной стадии) в этаноле (34,1 мл) добавляли гидразингидрат(11,5 мл, 236,2). Температуру реакции постепенно увеличивали до кипения с обратным холодильником и реакционную смесь перемешивали при данной температуре в течение 5 ч. Летучие вещества удаляли при пониженном давлении, сырую массу растирали с избытком эфира и пентана, получая гидразид 4-амино 5-хлор-2,3-дигидробензофуран-7-карбоновой кислоты (3,76 г).(Journal of Medicinal Chemistry,. 1993, 36, 4121; 2,2 г, 9,58 ммоль) в метаноле (38,3 мл), охлаждаемому при 0C, добавляли тионилхлорид (2,78 мл). Реакционную смесь постепенно подогревали до комнатной температуры, а затем нагревали с обратным холодильником в течение 3 ч. Летучие вещества удаляли при пониженном давлении; сырую массу разбавляли водным раствором бикарбоната натрия и экстрагировали этилацетатом. Объединенный органический слой сушили над безводным сульфатом натрия и растворитель удаляли в вакууме, получая указанное в заголовке соединение (2,12 г). Выход: 90,9%. 1(2,1 г, 8,6 ммоль, полученного на вышеуказанной стадии) в этаноле (34,4 мл) добавляли гидразингидрат(6,2 мл, 129,3 ммоль). Температуру реакции постепенно увеличивали до кипения с обратным холодильником и реакционную смесь перемешивали при комнатной температуре в течение 3 ч. Летучие вещества удаляли при пониженном давлении; сырую массу растирали с избытком эфира и пентана, получая гидразид 8-амино-7-хлор-2,3-дигидробензо[1,4]диоксан-5-карбоновой кислоты (2,1 г). Выход: 100%. 1H-ЯМР (ДМСО-d6):8,80 (уш.с, 1H), 7,27 (с, 1H), 5,40 (уш.с, 2H), 4,46 (уш.с, 2H), 4,40-4,25 (м, 4H); Масс (m/z): 244,1, 246,1 (M+H)+. Получение 5. Получение 1-циклопропилпиперидин-4-карбонилхлорида. Стадия (i). Получение 1-циклопропилпиперидин-4-карбонитрила. К перемешиваемому раствору 1-циклопропил-4-пиперидона (Alfa Aesar, 3,0 г, 21,5 ммоль) в смеси 1,2-диметоксиэтана (72 мл) и этанола (2,2 мл), охлаждаемому при 0C, добавляли п-толуолсульфонилметилизоцианид (5,45 г, 27,95 ммоль). На протяжении периода 1 ч добавляли твердый третичный бутоксид калия (5,54 г, 49,45 ммоль). Реакционную смесь перемешивали при данной температуре в течение дополнительного 1 ч и постепенно подогревали до комнатной температуры. После перемешивания в течение 2 ч при данной температуре ее охлаждали до 0C, разбавляли солевым раствором и этилацетатом. Органический слой отделяли, сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении, получая сырой продукт, который очищали на колонке с силикагелем, получая 1-циклопропилпиперидин-4-карбонитрил (1,32 г). Выход: 41,2%. 1H-ЯМР (CDCl3):2,82 (м, 2H), 2,63 (м, 1H), 2,49 (м, 2H), 1,98-1,78 (м, 4H), 1,70-1,58 (м, 1H), 0,500,40 (м, 2H), 0,40-0,35 (м, 2H); Масс (m/z): 151 (M+H)+. Стадия (ii). Получение 1-циклопропилпиперидин-4-карбоновой кислоты. Смесь 1-циклопропилпиперидин-4-карбонитрила (1,32 г, 8,8 ммоль, полученного на вышеуказанной стадии) и соляной кислоты (6 н., 35,2 мл) нагревали с обратным холодильником в течение 3 ч. Летучие вещества удаляли при пониженном давлении; следы воды удаляли с помощью совместной перегонки с толуолом. Полученный таким образом сырой продукт растирали несколько раз с эфиром и сушили в вакууме, получая 1-циклопропилпиперидин-4-карбоновую кислоту (2,02 г). Выход: 100%. 1(м, 1H), 3,55-3,45 (м, 1H), 2,10-1,85 (м, 4H), 1,20-1,10 (м, 2H), 0,80-0,70 (м, 2H); Масс (m/z): 170 (M+H)+. Стадия (iii). Получение 1-циклопропилпиперидин-4-карбонилхлорида. К перемешиваемой смеси 1-циклопропилпиперидин-4-карбоновой кислоты (10,0 г, 48,6 ммоль, полученной на предыдущей стадии) в дихлорметане (198 мл), охлаждаемой при 0C, добавляли сухой диметилформамид (2 мл) с последующим добавлением по каплям оксалилхлорида (12,5 мл, 145,8 ммоль). Реакционную смесь постепенно подогревали до комнатной температуры и перемешивали в течение 1 ч. Летучие вещества удаляли при пониженном давлении и получали сырой 1-циклопропилпиперидин-4 карбонилхлорид (11,0 г). Сырой продукт использовали в следующей реакции без очистки. Выход: 100%. 1H-ЯМР (ДМСО-d6):10,66 (уш.с, 1H), 3,50-3,42 (м, 2H), 3,40-3,30 (м, 1H), 3,15-3,0 (м, 2H), 2,802,65 (м, 1H), 2,10-1,80 (м, 4H), 1,15-1,08 (м, 2H), 0,80-0,70 (м, 2H); Масс (m/z): 184 (M+H)+. Получение 6. Получение (1-циклобутилпиперидин-4-ил)уксусной кислоты. Стадия (i). Получение трет-бутил-4-этоксикарбонилметилен-пиперидин-1-карбоксилата. К перемешиваемому раствору 1-Boc-4-пиперидона (2,0 г, 10,03 ммоль) в бензоле (40 мл) при комнатной температуре добавляли реагент Wittig (5,23 г, 15 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 10 ч и летучие вещества удаляли при пониженном давлении, получая сырую массу, которую очищали с помощью хроматографии на колонке с силикагелем, получая трет-бутил 4-этоксикарбонилметиленпиперидин-1-карбоксилат (2,05 г). Выход: 76%. 1(2,05 г, 7,62 ммоль, полученного на вышеуказанной стадии) в этаноле (30 мл) при комнатной температуре добавляли Pd/C (10 мас.%, 600 мг). Для реакции в течение 5 ч применяли аэростатное давление водорода. Реакционную смесь фильтровали через подушку из целита и летучие вещества удаляли при пониженном давлении, получая трет-бутил-4-этоксикарбонилметилпиперидин-1-карбоксилат (1,98 г). Выход: 95,8%. 1H-ЯМР (CDCl3):4,20-4,0 (м, 4H), 2,83-2,65 (м, 2H), 2,23 (д, J=6,8 Гц, 2H), 2,0-1,88 (м, 1H), 1,751,68 (м, 2H), 1,45 (с, 9H), 1,26 (т, J=7,0 Гц, 3H), 1,25-1,05 (м, 2H); Масс (m/z): 272 (M+H)+. Стадия (iii). Получение этилового эфира пиперидин-4-илуксусной кислоты. К перемешиваемому раствору трет-бутил-4-этоксикарбонилметилпиперидин-1-карбоксилата(1,98 г, 7,3 ммоль, полученного на вышеуказанной стадии) в изопропиловом спирте (5 мл), охлаждаемому при 0C, добавляли раствор сухого изопропанольного гидрохлорида (3 н, 15 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Летучие вещества удаляли при пониженном давлении и сырой продукт растирали с эфиром несколько раз, и сушили в вакууме, получая этиловый эфир пиперидин-4-илуксусной кислоты (1,57 г). Выход: 100%. 1H-ЯМР (ДМСО-d6):4,03 (кв., 2H), 3,23-3,15 (м, 2H), 2,86-2,78 (м, 2H), 2,24 (д, J=6,8 Гц, 2H), 2,01,85 (м, 1H), 1,81-1,72 (м, 2H), 1,40-1,25 (м, 2H), 1,14 (т, J=6,9 Гц, 3H); Масс (m/z): 172 (M+H)+. Стадия (iv). Получение этилового эфира (1-циклобутилпиперидин-4-ил)уксусной кислоты. Смесь циклобутанона (0,3 мл, 3,94 ммоль) в уксусной кислоте (0,19 мл, 3,28 ммоль) добавляли к перемешиваемому раствору этилового эфира пиперидин-4-илуксусной кислоты (562 мг, 3,28 ммоль, полученного на вышеуказанной стадии) в дихлорметане, охлаждаемому при 0C. Добавляли порциями твердый триацетоксиборгидрид натрия (1,39 г, 7,2 ммоль) на протяжении периода 15 мин. Реакционную смесь постепенно подогревали до комнатной температуры и перемешивали в течение 16 ч. Реакционную смесь охлаждали до 0C и подщелачивали насыщенным раствором бикарбоната натрия (pH 7,5). Два слоя разделяли, органический слой промывали солевым раствором, сушили над безводным сульфатом натрия и летучие вещества удаляли при пониженном давлении, получая этиловый эфир(м, 2H), 1,95-1,80 (м, 2H), 1,80-1,60 (м, 7H), 1,35-1,20 (м, 2H), 1,27 (т, J=7,1 Гц, 3H); Масс (m/z): 226 (M+H)+. Стадия (v). Получение (1-циклобутилпиперидин-4-ил)уксусной кислоты. К перемешиваемой смеси этилового эфира (1-циклобутилпиперидин-4-ил)уксусной кислоты(652,9 мг, 2,90 ммоль, полученного на вышеуказанной стадии), тетрагидрофурана (6 мл) и воды (6,0 мл),охлаждаемой при 0C, добавляли моногидрат гидроксида лития (133 мг, 3,19 ммоль) одной партией. Реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Реакционную смесь снова охлаждали до 0C и подкисляли 2 н. соляной кислотой до pH 2-3. Летучие вещества удаляли при пониженном давлении и следы воды удаляли с помощью азеотропной перегонки с толуолом, получая- 11022374 Масс (m/z): 198 (M+H)+. Получение 7. Получение 1-(3-метоксипропил)пиперидин-4-карбоновой кислоты. Стадия (i). Получение этил-1-(3-метоксипропил)пиперидин-4-карбоксилата. К перемешиваемому раствору этилизонипекотината (22,0 г, 140 ммоль) в ацетонитриле (250 мл) при комнатной температуре добавляли карбонат цезия (97 г, 298 ммоль) с последующим 1-бром-3-метоксипропаном (20 мл, 154 ммоль) и реакционную смесь нагревали до кипения с обратным холодильником в течение 4 ч. Реакционную смесь охлаждали до комнатной температуры и фильтровали через небольшую подушку из целита. Летучие вещества удаляли при пониженном давлении, получая этил-1-(3-метоксипропил)пиперидин-4-карбоксилат (31,0 г). Выход: 99%. 1H-ЯМР (CDCl3):4,12 (кв., 2H), 3,41 (т, J=6,4 Гц, 2H), 2,90-2,85 (м, 2H), 2,38 (т, J=7,4 Гц, 2H), 2,342,20 (м, 1H), 2,05-1,93 (м, 2H), 1,92-1,85 (м, 2H), 1,80-1,70 (м, 4H), 1,23 (т, J=7,1 Гц, 3H); Масс (m/z): 230 (M+H)+. Стадия (ii). Получение 1-(3-метоксипропил)пиперидин-4-карбоновой кислоты. К перемешиваемой смеси этил-1-(3-метоксипропил)пиперидин-4-карбоксилата (33,0 г, 144,1 ммоль,полученного на вышеуказанной стадии), тетрагидрофурана (200 мл) и воды (200 мл) добавляли моногидрат гидроксида лития (6,1 г, 144,1 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч перед разбавлением этилацетатом. Два слоя разделяли и водный слой подкисляли до pH 3-4 концентрированной соляной кислотой, а летучие вещества удаляли при пониженном давлении, получая 1-(3-метоксипропил)пиперидин-4-карбоновую кислоту (35,0 г). Выход: 100%. 1(SYNLETT, 1996, 1097; 5,0 г, 18,3 ммоль) в тетрагидрофуране (74 мл), охлаждаемому при 0C, добавляли ВНз-ДМС (2 н. раствор в тетрагидрофуране 36 мл, 73,2 ммоль) на протяжении периода 30 мин. Реакционную температуру постепенно повышали до кипения с обратным холодильником в течение 6 ч. После охлаждения реакционной смеси до 0C ее гасили добавлением водного раствора хлорида аммония и экстрагировали этилацетатом. Объединенный органический слой сушили над безводным сульфатом натрия,и растворитель удаляли при пониженном давлении. Сырой продукт очищали с помощью хроматографии на колонке с силикагелем, получая этил-3-бензил-3-азабицикло[3.1.0]гексан-6-карбоксилат (2,8 г). Выход: 62,5%. 1H-ЯМР (CDCl3):7,40-7,20 (м, 5H), 4,14 (кв., 2H), 3,61 (с, 2H), 3,05 (д, J=9,0 Гц, 2H), 2,44 (д,J=8,7 Гц, 2H), 2,14 (т, J=2,6 Гц, 1H), 1,97 (с, 2H), 1,28 (т, J=7,1 Гц, 3H). Масс (m/z): 246,2 (M+H)+. Стадия (ii). Получение этил-3-азабицикло[3.1.0]гексан-6-карбоксилата. К перемешиваемому раствору этил-3-бензил-3-азабицикло[3.1.0]гексан-6-карбоксилата (2,0 г,8,1 ммоль, полученного на указанной выше стадии) в метаноле (20 мл) добавляли гидроксид палладия(468 мг). С использованием водородного баллона (аэростата) к реакционной смеси применяли давление водорода. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и фильтровали через небольшую подушку из целита. Летучие вещества удаляли при пониженном давлении, получая этил-3-азабицикло[3.1.0]гексан-6-карбоксилат (1,22 г). Выход: 96%. 1H-ЯМР (CDCl3):4,11 (кв., 2H), 3,11 (д, J=11,6 Гц, 2H), 2,98 (д, J=11,7 Гц, 2H), 2,02 (с, 2H), 1,49 (т,J=3,0 Гц, 1H), 1,24 (т, J=4,2 Гц, 3H); Масс (m/z): 156,1 (M+H)+. Стадия (iii). Получение этил-3-циклобутил-3-азабицикло[3.1.0]гексан-6-карбоксилата. Смесь циклобутанона (157 мг, 2,19 ммоль) в уксусной кислоте (0,11 мл, 1,56 ммоль) добавляли к перемешиваемому раствору этил-3-азабицикло[3.1.0]гексан-6-карбоксилата (243 мг, 1,56 ммоль, полученного на предыдущей стадии) в дихлорметане, охлаждаемому при 0C. На протяжении периода 15 мин порциями добавляли твердый триацетоксиборгидрид натрия (727 мг, 3,43 ммоль). Реакционную смесь постепенно подогревали до комнатной температуры и перемешивали в течение 16 ч. Реакционную смесь охлаждали до 0C и подщелачивали насыщенным раствором бикарбоната натрия (pH 7,5). Два слоя разделяли, органический слой промывали солевым раствором, сушили над безводным сульфатом натрия и летучие вещества удаляли при пониженном давлении,получая этил-3-циклобутил-3 азабицикло[3.1.0]гексан-6-карбоксилат (219 мг).H-ЯМР (CDCl3):4,11 (кв., 2H), 3,10-2,90 (м, 3H), 2,34 (д, J=8,8 Гц, 2H), 2,04 (с, 1H), 1,93 (с, 2H),2,0-1,80 (м, 3H), 1,80-1,55 (м, 3H), 1,25 (т, J=7,1 Гц, 3H); Масс (m/z): 210,2 (M+H)+. Стадия (iv). Получение 3-циклобутил-3-азабицикло[3.1.0]гексан-6-карбоновой кислоты. К перемешиваемой смеси этил-3-циклобутил-3-азабицикло[3.1.0]гексан-6-карбоксилата (218 мг,1,04 ммоль, полученного на вышеуказанной стадии), тетрагидрофурана (2 мл) и воды (2,0 мл), охлаждаемой при 0C, одной порцией добавляли моногидрат гидроксида лития (133 мг, 3,19 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 24 ч. Реакционную смесь снова охлаждали до 0C и подкисляли 2 н. соляной кислотой до pH 2-3. Летучие вещества удаляли при пониженном давлении и следы воды удаляли с помощью азеотропной перегонки с толуолом, получая 3 циклобутил-3-азабицикло[3.1.0]гексан-6-карбоновую кислоту (180 мг). Выход: 92%. 1H-ЯМР (ДМСО-d6):2,98-2,86 (м, 1H), 2,78 (д, J=8,5 Гц, 2H), 2,20 (д, J=8,1 Гц, 2H), 1,90-1,80 (м,2H), 1,82-1,68 (м, 2H), 1,65-1,55 (м, 2H), 1,49 (с, 2H), 1,42 (с, 1H); Масс (m/z): 182,3 (M+H)+. Получение 9. Получение 1'-этилового эфира [1,4']-бипиперидинил-4,1'-дикарбоновой кислоты. Стадия (i). Получение этил-4-оксопиперидин-1-карбоксилата. К перемешиваемому раствору гидрохлорида пиперидин-4-она (2,0 г, 14,7 ммоль) в DCM (60 мл),охлаждаемому при 0C, добавляли триэтиламин (5,15 мл, 36,75 ммоль) и этилхлорформиат (1,59 мл,16,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч перед разбавлением водой. Два слоя разделяли, органический слой сушили над безводным сульфатом натрия и летучие вещества удаляли при пониженном давлении, получая этил-4-оксопиперидин-1-карбоксилат (3,14 г). Выход: 98%. 1H-ЯМР (CDCl3):4,22 (кв., 2H), 3,79 (т, J=6,0 Гц, 2H), 2,48 (т, J=6,0 Гц, 2H), 1,31 (т, J=7,1 Гц, 3H); Масс (m/z): 172,1 (M+H)+. Стадия (ii). Получение диэтилового эфира [1,4']бипиперидинил-4,1'-дикарбоновой кислоты. Смесь этил-4-оксопиперидин-1-карбоксилата (3,14 г, 18,3 ммоль, полученного на вышеуказанной стадии) в уксусной кислоте (1,05 мл, 18,3 ммоль) добавляли к перемешиваемому раствору этилизонипекотината (2,87 мл, 18,3 ммоль) в дихлорметане (10 мл), охлаждаемому при 0C. На протяжении 15 мин добавляли порциями твердый триацетоксиборгидрид натрия (11,6 г, 54,9 ммоль). Реакционную смесь постепенно подогревали до комнатной температуры и перемешивали в течение 16 ч. Реакционную смесь охлаждали до 0C и подщелачивали насыщенным раствором бикарбоната натрия (pH 7,5). Два слоя разделяли, органический слой промывали солевым раствором, сушили над безводным сульфатом натрия и летучие вещества удаляли при пониженном давлении. Сырой продукт очищали с помощью хроматографии на колонке с силикагелем, получая диэтиловый эфир [1,4']бипиперидинил-4,1'-дикарбоновой кислоты (5,51 г). Выход: 96,3%. 1(м, 4H), 1,60-1,43 (м, 2H), 1,32-1,22 (м, 6H); Масс (m/z): 313,2 (M+H)+. Стадия (iii). Получение 1'-этилового эфира [1,4']бипиперидинил-4,1'-дикарбоновой кислоты. К перемешиваемой смеси диэтилового эфира [1,4']бипиперидинил-4,1'-дикарбоновой кислоты(5,51 г, 17,67 ммоль), тетрагидрофурана (34 мл) и воды (34 мл), охлаждаемой при 0C, добавляли моногидрат гидроксида лития (742,0 мг, 17,67 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, разбавляли этилацетатом. Два слоя разделяли, водный слой подкисляли 2 н. соляной кислотой до pH 3-4 и летучие вещества удаляли при пониженном давлении, получая 1'-этиловый эфир [1,4']бипиперидинил-4,1'-дикарбоновой кислоты (5,0 г). Выход: 94%. 1(м, 2H), 2,90-2,65 (м, 2H), 2,60-2,50 (м, 1H), 2,18-2,08 (м, 2H), 2,05-1,94 (м, 4H), 1,60-1,50 (м, 2H), 1,16 (т,J=7,0 Гц, 3H); Масс (m/z): 285,1 (M+H)+. Пример 1. Получение гемифумарата 6-хлор-8-[5-(1-циклопропилпиперидин-4-ил)[1,3,4]оксадиазол-2-ил]хроман-5-иламина. Стадия (i). Получение N-(1-циклопропилпиперидин-4-карбонил)-N'-(5-амино-6-хлорхроман-8 карбонил)гидразина. К перемешиваемому раствору гидразида 5-амино-6-хлорхроман-8-карбоновой кислоты (8,0 г,33,1 ммоль, полученного в Получении 1) в дихлорметане (200 мл), охлаждаемому при 0C, добавляли триэтиламин (13,9 мл, 99,9 ммоль) и раствор 1-циклопропилпиперидин-4-карбонилхлорида (11,0 г) в ди 1 хлорметане (200 мл). Реакционную смесь подогревали до комнатной температуры и перемешивали в течение 16 ч перед разбавлением ее водой (160 мл). Два слоя разделяли, органический слой сушили над безводным сульфатом натрия и летучие вещества удаляли при пониженном давлении, получая указанное в заголовке соединение (10,5 г). Выход: 81%. 1(м, 2H), 1,70-1,60 (м, 2H), 1,60-1,42 (м, 3H), 0,42-0,35 (м, 2H), 0,30-0,22 (м, 2H); Масс (m/z): 393, 395 (M+H)+. Стадия (ii). Получение 6-хлор-8-[5-(1-циклопропилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]хроман 5-иламина. К N-(1-циклопилпиперидин-4-карбонил)-N'-(5-амино-6-хлорхроман-8-карбонил)гидразину (10,5 г,26,7 ммоль, полученному на вышеуказанной стадии) добавляли фосфорилхлорид (53,5 мл). Температуру реакции постепенно повышали до 120C. Реакционную смесь перемешивали при данной температуре в течение 1 ч, охлаждали до комнатной температуры и растирали с гексанами (3100 мл). Сырую реакционную смесь разбавляли 10% водным раствором бикарбоната натрия и экстрагировали 1:9 смесью метанола в дихлорметане. Органический слой сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении и сырой продукт очищали на колонке с силикагелем, получая 6-хлор-8[5-(1-циклопропилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина (8,8 г). Выход: 87,9%. 1(14 г, 37,3 ммоль, полученного на вышеуказанной стадии) в этаноле (280 мл) нагревали до кипения с обратным холодильником до тех пор, пока не получался прозрачный раствор. Смесь охлаждали до комнатной температуры и добавляли фумаровую кислоту (4,32 г, 37,3 ммоль). Реакционную смесь нагревали до кипения с обратным холодильником в течение 1 ч. Летучие вещества удаляли при пониженном давлении и полученную таким образом фумаратную соль перекристаллизовывали из изопропанола, получая гемифумарат 6-хлор-8-[5-(1-циклопропилпиперидин-4-ил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина (14,0 г). Выход: 92,8%. 1H-ЯМР (ДМСО-d6):7,48 (с, 1H), 6,60 (с, 1H), 5,75 (с, 2H), 4,13 (т, J=4,8 Гц, 2H), 3,0-2,90 (м, 3H),2,52-2,42 (м, 2H), 2,40-2,30 (м, 2H), 2,01-1,90 (м, 4H), 1,75-1,62 (м, 3H), 0,48-0,40 (м, 2H), 0,35-0,28 (м,2H); Масс (m/z): 375, 377 (M+H)+. Пример 2. Получение L(+)-тартратной соли 6-хлор-8-[5-(1-циклобутилпиперидин-4-илметил)[1,3,4]оксадиазол-2-ил]хроман-5-иламина. Стадия (i). Получение 6-хлор-8-[5-(1-циклобутилпиперидин-4-илметил)-[1,3,4]оксадиазол-2 ил]хроман-5-иламина. К (1-циклобутилпиперидин-4-ил)уксусной кислоте (725 мг, 3,52 ммоль, полученной в получении 4) добавляли фосфорилхлорид (4 мл). Смесь перемешивали в течение 15 мин и добавляли гидразид 5-амино-6-хлорхроман-8-карбоновой кислоты (500 мг, 2,0 ммоль). Реакционную смесь постепенно нагревали до кипения с обратным холодильником в течение 30 мин. Реакционную смесь охлаждали до комнатной температуры, растирали с гексанами (220 мл) и сырую массу подщелачивали водным раствором бикарбоната натрия. Подщелоченную смесь экстрагировали 10% метанолом в дихлорметане. Органический слой сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении и очищали на колонке с силикагелем, получая 6-хлор-8-[5-(1-циклобутилпиперидин-4-илметил)[1,3,4]оксадиазол-2-ил]хроман-5-иламина (250 мг). Выход: 30%. 1H-ЯМР (CDCl3):7,68 (с, 1H), 4,35 (уш.с, 2H), 4,28 (т, J=5,0 Гц, 2H), 2,93-2,88 (м, 2H), 2,83 (д,J=6,9 Гц, 2H), 2,73-2,62 (м, 1H), 2,54 (т, J=6,6 Гц, 2H), 2,20-2,10 (м, 2H), 2,08-2,0 (м, 2H), 1,95-1,65 (м, 9H),1,48-1,35 (м, 2H); Масс (m/z): 403, 405 (M+H)+. Стадия (ii). Получение L(+)-тартратной соли 6-хлор-8-[5-(1-циклобутилпиперидин-4-илметил)[1,3,4]оксадиазол-2-ил]хроман-5-иламина. К перемешиваемому раствору 6-хлор-8-[5-(1-циклобутилпиперидин-4-илметил)-[1,3,4]оксадиазол 2-ил]хроман-5-иламина (175,7 мг, 0,436 ммоль, полученного на вышеуказанной стадии) в метаноле (2 мл) добавляли L(+)-винную кислоту (65,4 мг, 0,436 ммоль). Реакционную смесь перемешивали в течение 1 ч при комнатной температуре и летучие вещества удаляли при пониженном давлении, получая сырую массу, которую растирали несколько раз с эфирным растворителем, получая L(+)-тартрат 6-хлор-8-[5-(1 циклобутилпиперидин-4-илметил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина (206,2 мг). Выход: 85,5%. 1(м, 1H), 3,10-3,0 (м, 2H), 2,84 (д, J=6,7 Гц, 2H), 2,48 (т, J=7,6 Гц, 2H), 2,33-2,15 (м, 2H), 2,10-2,0 (м, 2H),2,0-1,85 (, 5H), 1,85-1,72 (м, 2H), 1,70-1,58 (м, 2H), 1,45-1,30 (м, 2H); Масс (m/z): 403, 405 (M+H)+. Пример 3. Получение оксалатной соли 1-изопропил-3-5-[1-(3-метоксипропил)пиперидин-4-ил][1,3,4]оксадиазол-2-ил-1H-индазола. Стадия (i). Получение 1-изопропил-3-5-[1-(3-метоксипропил)пиперидин-4-ил]-[1,3,4]оксадиазол-2 ил-1H-индазола. К смеси гидразида 1-изопропил-1H-индазол-3-карбоновой кислоты (15,0 г, 68,8 ммоль) и гидрохлорида 1-(3-метоксипропил)пиперидин-4-карбоновой кислоты (20,9 г, 88,2 ммоль, полученного в Получении 7), охлаждаемой при 0C, добавляли фосфорилхлорид (130 мл). Температуру реакции постепенно повышали до 100C и перемешивали 2 ч. После завершения реакции реакционную смесь охлаждали до 0C и растирали с гексанами (3250 мл). Сырой продукт подщелачивали водным раствором гидроксида натрия и экстрагировали 5% метанолом в дихлорметане. Объединенный органический слой сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Сырой продукт очищали с помощью хроматографии на колонке с силикагелем, получая 1-изопропил-3-5-[1-(3 метоксипропил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил-1 Н-индазол (15,78 г). Выход: 59%. 1H-ЯМР (CDCl3):8,35 (д, J=8,1 Гц, 1H), 7,53 (д, J=8,5 Гц, 1H), 7,47 (т, J=7,0 Гц, 1H), 7,33 (т,J=7,4 Гц, 1H), 5,05-4,90 (м, 1H), 3,44 (т, J=6,4 Гц, 2H), 3,35 (с, 3H), 3,15-2,97 (м, 3H), 2,48 (т, J=7,3 Гц, 2H),2,26-2,02 (м, 6H), 1,88-1,75 (м, 2H), 1,67 (д, J=6,7 Гц, 6H); Масс (m/z): 384,5 (M+H)+. Стадия (ii). Получение оксалатной соли 1-изопропил-3-5-[1-(3-метоксипропил)пиперидин-4-ил][1,3,4]оксадиазол-2-ил-1 Н-индазола. К перемешиваемому раствору 1-изопропил-3-5-[1-(3-метоксипропил)пиперидин-4-ил][1,3,4]оксадиазол-2-ил-1H-индазола (12,55 г, 32,7 ммоль, полученного на вышеуказанной стадии) в 2-пропаноле (200 мл) добавляли щавелевую кислоту (4,12 г, 32,7 ммоль). После перемешивания при комнатной температуре в течение 1 ч реакционную смесь дополнительно разбавляли 2-пропанолом и нагревали с обратным холодильником в течение 2 ч. Кристаллический продукт, который выпадал в осадок после охлаждения реакционной смеси до комнатной температуры, отфильтровывали, сушили в вакууме,получая оксалатную соль 1-изопропил-3-5-[1-(3-метоксипропил)пиперидин-4-ил][1,3,4]оксадиазол-2-ил-1H-индазола (16,4 г). Выход: 88%. 1(i). Получение 3-[5-(1-циклобутилпиперидин-4-илметил)-[1,3,4]оксадиазол-2-ил]-1 изопропил-1H-индазола. К смеси гидразида 1-изопропил-1H-индазол-3-карбоновой кислоты (120 мг, 0,55 ммоль) и гидрохлорида (1-циклобутилпиперидин-4-ил)уксусной кислоты (147 мг, 0,74 ммоль, полученной в Получении 6), охлаждаемой при 0C, добавляли фосфорилхлорид (1,5 мл). Температуру реакции постепенно повышали до 100C и перемешивали 2 ч. После завершения реакции реакционную смесь охлаждали до 0C и растирали с гексанами (325 мл). Сырой продукт охлаждали до 0C, подщелачивали водным раствором гидроксида натрия и экстрагировали 5% метанолом в дихлорметане. Объединенный органический слой сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Сырой продукт очищали с помощью хроматографии на колонке с силикагелем, получая 3-[5-(1-циклобутилпиперидин-4-илметил)-[1,3,4]оксадиазол-2-ил]-1-изопропил-1H-индазол (62 мг). Выход: 30%. 1 Стадия (ii). Получение L(+)-тартратной соли 3-[5-(1-циклобутилпиперидин-4-илметил)[1,3,4]оксадиазол-2-ил]-1-изопропил-1H-индазола. К перемешиваемому раствору 3-[5-(1-циклобутилпиперидин-4-илметил)-[1,3,4]оксадиазол-2-ил]-1 изопропил-1H-индазола (62 мг, 0,16 ммоль, полученного на вышеуказанной стадии) в 2-пропаноле(5,0 мл) добавляли L(+)-винную кислоту (26 мг, 0,16 ммоль). После перемешивания при комнатной температуре в течение 1 ч летучие вещества удаляли при пониженном давлении и сырой продукт растирали несколько раз с эфиром, получая L(+)-тартратную соль 3-[5-(1-циклобутилпиперидин-4-илметил)[1,3,4]оксадиазол-2-ил]-1-изопропил-1H-индазола (81 мг). Выход: 94%. 1H-ЯМР (ДМСО-d6):8,18 (д, J=8,1 Гц, 1H), 7,90 (д, J=8,5 Гц, 1H), 7,54 (т, J=7,4 Гц, 1H), 7,38 (т,J=7,6 Гц, 1H), 5,22-5,10 (м, 1H), 4,11 (с, 2H), 3,30-3,20 (м, 2H), 3,20-3,05 (м, 2H), 3,0 (д, J=6,8 Гц, 2H), 2,452,30 (м, 1H), 2,10-1,90 (м, 4H), 1,90-1,80 (м, 2H), 1,78-1,65 (м, 2H), 1,50-1,40 (м, 2H); Масс (m/z): 380,2 (M+H)+. Пример 5. Получение 6-хлор-8-[5-(3-циклобутил-3-азабицикло[3.1.0]гекс-6-ил)-[1,3,4]оксадиазол-2 ил]хроман-5-иламина щавелевой кислоты. Стадия (i). Получение 6-хлор-8-[5-(3-циклобутил-3-азабицикло[3.1.0]гекс-6-ил)-[1,3,4]оксадиазол-2 ил]хроман-5-иламина щавелевой кислоты. К 3-циклобутил-3-азабицикло[3.1.0]гексан-6-карбоновой кислоте (74 мг, 0,40 ммоль, полученной в Получении 8) добавляли фосфорилхлорид (1 мл). Смесь перемешивали в течение 15 мин и добавляли гидразид 5-амино-6-хлорхроман-8-карбоновой кислоты (80 мг, 0,33 ммоль). Реакционную смесь постепенно нагревали до кипения с обратным холодильником в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры, растирали с гексанами (220 мл) и сырую массу подщелачивали водным раствором бикарбоната натрия. Подщелоченную смесь экстрагировали 10% метанолом в дихлорметане. Органический слой сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении и очищали на колонке с силикагелем, получая 6-хлор-8-[5-(3-циклобутил-3-азабицикло-[3.1.0]гекс 6-ил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина (18 мг). Выход: 14,0%. 1(м, 2H), 1,30-1,20 (м, 1H). Масс (m/z): 387,1, 389,2 (M+H)+. Стадия (ii). Получение оксалатной соли 6-хлор-8-[5-(3-циклобутил-3-азабицикло[3.1.0]гекс-6-ил)[1,3,4]оксадиазол-2-ил]хроман-5-иламина. К перемешиваемому раствору 6-хлор-8-[5-(3-циклобутил-3-азабицикло[3.1.0]гекс-6-ил)[1,3,4]оксадиазол-2-ил]хроман-5-иламина (18 мг, 0,05 ммоль, полученного на вышеуказанной стадии) в 2-пропаноле (3 мл), добавляли щавелевую кислоту (6,0 мг, 0,05 ммоль). После перемешивания при комнатной температуре в течение 1 ч реакционную смесь дополнительно разбавляли 2-пропанолом и нагревали с обратным холодильником в течение 2 ч. Летучие вещества удаляли при пониженном давлении и сырой продукт, который получался, растирали с эфиром, сушили в вакууме, получая оксалатную соль 6-хлор-8-[5-(3-циклобутил-3-азабицикло[3.1.0]гекс-6-ил)-[1,3,4]оксадиазол-2-ил]хроман-5-иламина(м, 1H); Масс (m/z): 387,1, 389,2 (M+H)+. Пример 6. Получение оксалатной соли этилового эфира 4-[5-(8-амино-7-хлор-2,3 дигидробензо[1,4]диоксан-5-ил)-[1,3,4]оксадиазол-2-ил]-[1,4']бипиперидинил-1'-карбоновой кислоты. Стадия (i). Получение этилового эфира 4-[5-(8-амино-7-хлор-2,3-дигидробензо[1,4]диоксан-5-ил)[1,3,4]оксадиазол-2-ил]-[1,4']бипиперидинил-1'-карбоновой кислоты. К 1'-этиловому эфиру [1,4']бипиперидинил-4,1'-дикарбоновой кислоты (372 мг, 1,02 ммоль, полученному в Получении 9) добавляли фосфорилхлорид (3,2 мл). Смесь перемешивали в течение 15 мин и добавляли гидразид 8-амино-7-хлор-2,3-дигидробензо[1,4]диоксан-5-карбоновой кислоты (200 мг,0,82 ммоль). Реакционную смесь постепенно нагревали до кипения с обратным холодильником в течение 1 ч. Реакционную смесь охлаждали до комнатной температуры, растирали с гексанами (250 мл) и сырую массу подщелачивали водным раствором бикарбоната натрия. Подщелоченную смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении и очищали на колонке с силикагелем, получая этиловый эфир 4-[5-(8-амино-7-хлор-2,3-дигидробензо[1,4]диоксан-5-ил)-[1,3,4]оксадиазол-2-ил]-[1,4']бипиперидинил 1'-карбоновой кислоты (110 мг). Выход: 27,5%.H-ЯМР (CDCl3):7,42 (с, 1H), 4,50-4,36 (м, 6H), 4,33-4,20 (м, 2H), 4,12 (кв., 2H), 3,03-2,92 (м, 3H),2,83-2,70 (м, 2H), 2,55-2,42 (м, 1H), 2,42-2,30 (м, 2H), 2,15-2,06 (м, 2H), 2,04-1,90 (м, 2H), 1,86-1,78 (м,2H), 1,55-1,40 (м, 2H), 1,26 (т, J=7,1 Гц, 3H); Масс (m/z): 492,1, 494,3 (M+H)+. Стадия (ii). Получение оксалатной соли этилового эфира 4-[5-(8-амино-7-хлор-2,3 дигидробензо[1,4]диоксин-5-ил)-[1,3,4]оксадиазол-2-ил]-[1,4']бипиперидинил-1'-карбоновой кислоты. К перемешиваемому раствору этилового эфира 4-[5-(8-амино-7-хлор-2,3-дигидробензо[1,4]диоксин 5-ил)-[1,3,4]оксадиазол-2-ил]-[1,4']бипиперидинил-1'-карбоновой кислоты (100 мг, 0,20 ммоль, полученного на предыдущей стадии) в этаноле (3 мл) добавляли щавелевую кислоту (23 мг, 0,18 ммоль). После перемешивания при комнатной температуре в течение 1 ч реакционную смесь дополнительно разбавляли 2-пропанолом и нагревали с обратным холодильником в течение 2 ч. Летучие вещества удаляли при пониженном давлении и полученный сырой продукт растирали с эфиром и сушили в вакууме, получая оксалатную соль этилового эфира 4-[5-(8-амино-7-хлор-2,3-дигидробензо[1,4]диоксин-5-ил)[1,3,4]оксадиазол-2-ил]-[1,4']бипиперидинил-1'-карбоновой кислоты (115 мг). Выход: 97,4%. 1H-ЯМР (ДМСО-d6):7,29 (с, 1H), 5,66 (уш.с, 2H), 4,33 (с, 4H), 4,15-4,05 (м, 2H), 4,03 (кв., 2H),3,40-3,15 (м, 4H), 3,10-2,90 (м, 2H), 2,90-2,70 (м, 2H), 2,26-2,18 (м, 2H), 2,08-1,90 (м, 4H), 1,58-1,42 (м,2H), 1,17 (т, J=7,0 Гц, 3H); Масс (m/z): 492,1, 494,3 (M+H)+. Пример 7. Получение оксалатной соли 5-хлор-7-5-[1-(тетрагидропиран-4-ил)пиперидин-4-ил][1,3,4]оксадиазол-2-ил-2,3-дигидробензофуран-4-иламина. Стадия (i). Получение 5-хлор-7-5-[1-(тетрагидропиран-4-ил)пиперидин-4-ил]-[1,3,4]оксадиазол-2 ил-2,3-дигидробензофуран-4-иламина. К 1-(тетрагидропиран-4-ил)пиперидин-4-карбоновой кислоте (168,2 мг, 0,58 ммоль) добавляли фосфорилхлорид (1,76 мл). Смесь перемешивали в течение 15 мин и добавляли гидразид 4-амино-5-хлор 2,3-дигидробензофуран-7-карбоновой кислоты (101,2 мг, 0,44 ммоль, полученный в Получении 3). Реакционную смесь постепенно нагревали до температуры кипения с обратным холодильником в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, растирали с гексанами (220 мл) и сырую массу подщелачивали водным раствором бикарбоната натрия. Подщелоченную смесь экстрагировали 10% метанолом в дихлорметане. Органический слой сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Остаточную массу очищали на колонке с силикагелем, получая 5-хлор-7-5-[1-(тетрагидропиран-4-ил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил-2,3 дигидробензофуран-4-иламина (23,5 мг). Выход: 13,1%. 1H-ЯМР (CDCl3):7,64 (с, 1H), 4,84 (т, J=8,7 Гц, 2H), 4,31 (уш.с, 2H), 4,10-4,0 (м, 2H), 3,39 (т,J=11,4 Гц, 2H), 3,10 (т, J=8,7 Гц, 2H), 3,10-2,95 (м, 3H), 2,62-2,50 (м, 1H), 2,45-2,25 (м, 2H), 2,20-1,95 (м,4H), 1,88-1,75 (м, 2H), 1,75-1,60 (м, 2H); Масс (m/z): 405,2, 407,4 (M+H)+. Стадия (ii). Получение оксалатной соли 5-хлор-7-5-[1-(тетрагидропиран-4-ил)пиперидин-4-ил][1,3,4]оксадиазол-2-ил-2,3-дигидробензофуран-4-иламина. К перемешиваемому раствору 5-хлор-7-5-[1-(тетрагидропиран-4-ил)пиперидин-4-ил][1,3,4]оксадиазол-2-ил-2,3-дигидробензофуран-4-иламина (20,4 мг, 0,05 ммоль) в этаноле (2 мл) добавляли щавелевую кислоту (6,0 мг, 0,05 ммоль). После перемешивания при комнатной температуре в течение 1 ч реакционную смесь дополнительно разбавляли 2-пропанолом и нагревали с обратным холодильником в течение 2 ч. Летучие вещества удаляли при пониженном давлении и полученный сырой продукт растирали с эфиром, сушили в вакууме, получая оксалатную соль 5-хлор-7-5-[1-(тетрагидропиран-4 ил)пиперидин-4-ил]-[1,3,4]оксадиазол-2-ил-2,3-дигидробензофуран-4-иламина (22,5 мг). Выход: 90,3%. 1 Биологические анализы. Пример 50. Определение величин EC50 5-HT4 рецептора. Для анализа на основе клеток использовали систему стабильной линии CHO клеток, экспрессирующих человеческий 5-HT4 рецептор, и pCRE-Luc репортер. В данном анализе предлагается подход на нерадиоактивной основе определения связывания соединения с GPCR. В данном конкретном анализе измеряют уровень внутриклеточного циклического AMP, который модулируется за счет активации или ингибирования рецептора. Рекомбинантные клетки преследуют репортерный ген люциферазы под контролем сАМР ответного элемента. Указанные выше клетки выращивали на 96 луночных белых планшетах с прозрачным дном в средеHams F12, содержащей 10% фетальную бычью сыворотку (FBS). Перед добавлением соединений или стандартного агониста клетки хранили в сыворотке на протяжении ночи. К клеткам добавляли увеличивающиеся концентрации испытуемых соединений в среде OptiMEM. Инкубирование продолжали при 37C в CO2 в течение 4 ч. Среду удаляли и клетки промывали солевым раствором с фосфатным буфером. Клетки лизировали и измеряли активность люциферазы в люминометре. Единицы люминесценции наносили на график против концентрации соединений с использованием программного обеспеченияGraphpad. Величины EC50 соединений определяли как концентрацию, требуемую при стимуляции активности люциферазы на 50%.
МПК / Метки
МПК: A61K 31/445, A61K 31/4245, A61P 25/00, C07D 413/14
Метки: лигандов, гетероарильные, 5-ht4, качестве, рецептора, соединения
Код ссылки
<a href="https://eas.patents.su/30-22374-geteroarilnye-soedineniya-v-kachestve-ligandov-5-ht4-receptora.html" rel="bookmark" title="База патентов Евразийского Союза">Гетероарильные соединения в качестве лигандов 5-ht4 рецептора</a>
1,2-дигидро-2-оксохинолиновые соединения в качестве лигандов рецептора 5-нт4

Номер патента: 20227
Опубликовано: 30.09.2014
Авторы: Кандикере Нагарадж Вишвоттам, Нироджи Рамакришна, Равула Йотхсна, Ахмад Иштияк, Камбхампати Рама Састри, Схинде Анил Карбхари, Бхирапунени Геопинадх, Патнала Срирамахандра Мурти, Джаяраджан Прадип, Джасти Венкатесварлу, Мохаммед Абдул Рашид
МПК: A61K 31/4704, A61P 25/00, C07D 401/12...
Метки: рецептора, качестве, 1,2-дигидро-2-оксохинолиновые, 5-нт4, соединения, лигандов
Формула / Реферат:
1. Соединение общей формулы (I)или его фармацевтически приемлемые соли;где R1 представляет собой водород, гидрокси, галоген, нитро, амин или метокси;R2 представляет собой водород, (С1-С4)алкил или -СН2-фенил;R3 представляет собойR4 представляет собой водород, гидрокси или фенил; R5 представляет собой водород, (С1-С4)алкил или пирролидин;R6 представляет собой фенил или пиридин;R7 и R8 вместе с атомом N образуют морфолин или пирролидин.2....
Сульфоновые соединения в качестве лигандов 5-ht6 рецептора

Номер патента: 22043
Опубликовано: 30.10.2015
Авторы: Ребалли Веена, Баданже Раджеш Кумар, Нироджи Рамакришна, Ахмад Иштияк, Абрахам Ренни, Джасти Венкатесварлу, Камбхампати Рама Састри, Схинде Анил Карбхари, Намала Рамбабу, Чиндхе Анил Кашинатх, Мулла Мохамад Садик Абдулхамид
МПК: A61K 31/4468, A61K 31/4709, A61K 31/4725...
Метки: рецептора, 5-ht6, лигандов, соединения, качестве, сульфоновые
Формула / Реферат:
1. Соединение общей формулы (I)в котором представляет собойпри условии, что указанная связь между кольцоми группой SO2 не является сульфонамидной связью;в каждом случае R1 представляет собой водород, хлор, бром, фтор, метил или метокси;R2 представляет собой водород;R3 представляет собой водород или метил;n равно 2,или его фармацевтически приемлемые соли.2. Соединение по п.1, которое выбрано из группы, состоящей...
Соединения хинуклидина в качестве лигандов никотинового ацетилхолинового рецептора α7

Номер патента: 17628
Опубликовано: 30.01.2013
Авторы: Ван Нэнхуэй, Олсон Ричард Е., Ивуагву Кристиана И., Макдональд Ивар М., Кинг Далтон, Зуси Ф.Кристофер, Кук II Джеймс Х., Мэкор Джон Е.
МПК: C07D 498/20, A61K 31/439, A61P 25/28...
Метки: ацетилхолинового, хинуклидина, соединения, качестве, alpha;7, никотинового, рецептора, лигандов
Формула / Реферат:
1. Соединение формулы I или его стереоизомергде R1 выбирают из группы, содержащей изоксазолил, пиразолил, оксазолил, тиазолил, имидазолил, оксадиазолил, тиадиазолил, триазолил, пиридинил, пиразинил, пиридазинил, пиримидинил, триазинил, хинолинил, изохинолинил, хиноксалинил, хиназолинил, нафтиридинил, индазолил, индолил, 2-индолонил, бензизоксазолил, бензоизотиазолил, бензоксазолил, бензотиазолил, бензимидазолил, фуропиридинил, тиенопиридинил,...
Бициклические соединения в качестве лигандов a4b2 никотинового ацетилхолинового рецептора

Номер патента: 20506
Опубликовано: 28.11.2014
Авторы: Нироджи Рамакришна, Мохаммед Абдул Рашид, Джасти Венкатесварлу, Камбхампти Рама Састри, Джаяраджан Прадип, Ахмад Иштияк, Бхирапунени Гопинадх, Вирамалла Сринивас, Равелла Сриниваса Рао, Схинде Анил Карбхари
МПК: A61P 25/00, A61K 31/435, A61K 31/403...
Метки: бициклические, качестве, никотинового, соединения, рецептора, ацетилхолинового, лигандов
Формула / Реферат:
1. Соединение общей формулы (I)где R представляет собой пиридинил;R1 представляет собой водород или С1-8алкил;R2 представляет собой водород;R3 представляет собой водород или С1-8алкил;m представляет собой 1;n представляет собой 1 или 2;р представляет собой 1,или его стереоизомеры или его фармацевтически приемлемые соли.2. Соединение по п.1, выбранное из группы, содержащейгидрохлорид...
Гетероарильные соединения в качестве бетамиметиков для лечения заболеваний дыхательных путей

Номер патента: 12727
Опубликовано: 30.12.2009
Авторы: Череда Энцо, Буйсу Тьерри, Хёнке Кристоф, Конетцки Инго, Шнапп Андреас, Лустенбергер Филипп
МПК: A61P 11/00, A61K 31/538, C07D 401/12...
Метки: соединения, заболеваний, дыхательных, качестве, лечения, бетамиметиков, путей, гетероарильные
Формула / Реферат:
1. Соединения формулы 1 в которой n обозначает 2, 3, m обозначает 1, 2 или 3, X обозначает NR2 или О, А обозначает СО, В обозначает группу с двумя связями, выбранную из О, CR3R4-О, NR5, CR3R4-NR5, CH=CH или СН2-СН2, R1 обозначает Н, C1-С6алкил, C1-С6галоалкил, С3-С6циклоалкил, галоген, ОН, CN, NO2, О-С1-С6алкил, СООН или СОО-С1-С4алкил, R2 обозначает Н, С1-С4алкил, С1-2алкилен-С3-С6-циклоалкил, фенилэтил или бензил, R3 обозначает Н или...
Предыдущий патент: Раздаточная головка для покапельной раздачи жидкости
Следующий патент: Производные пиразолоспирокетонов для применения в качестве ингибиторов ацетил-коа-карбоксилаз
Случайный патент: Пирролопиримидины как ингибиторы протеинкиназы