Органические соединения и их применение для лечения hcv инфекции
Номер патента: 21554
Опубликовано: 30.07.2015
Авторы: Риголлье Паскаль, Раман Пракаш, Брандль Трикси, Зимик Оливер, Сиперсауд Мохиндра
























Формула / Реферат
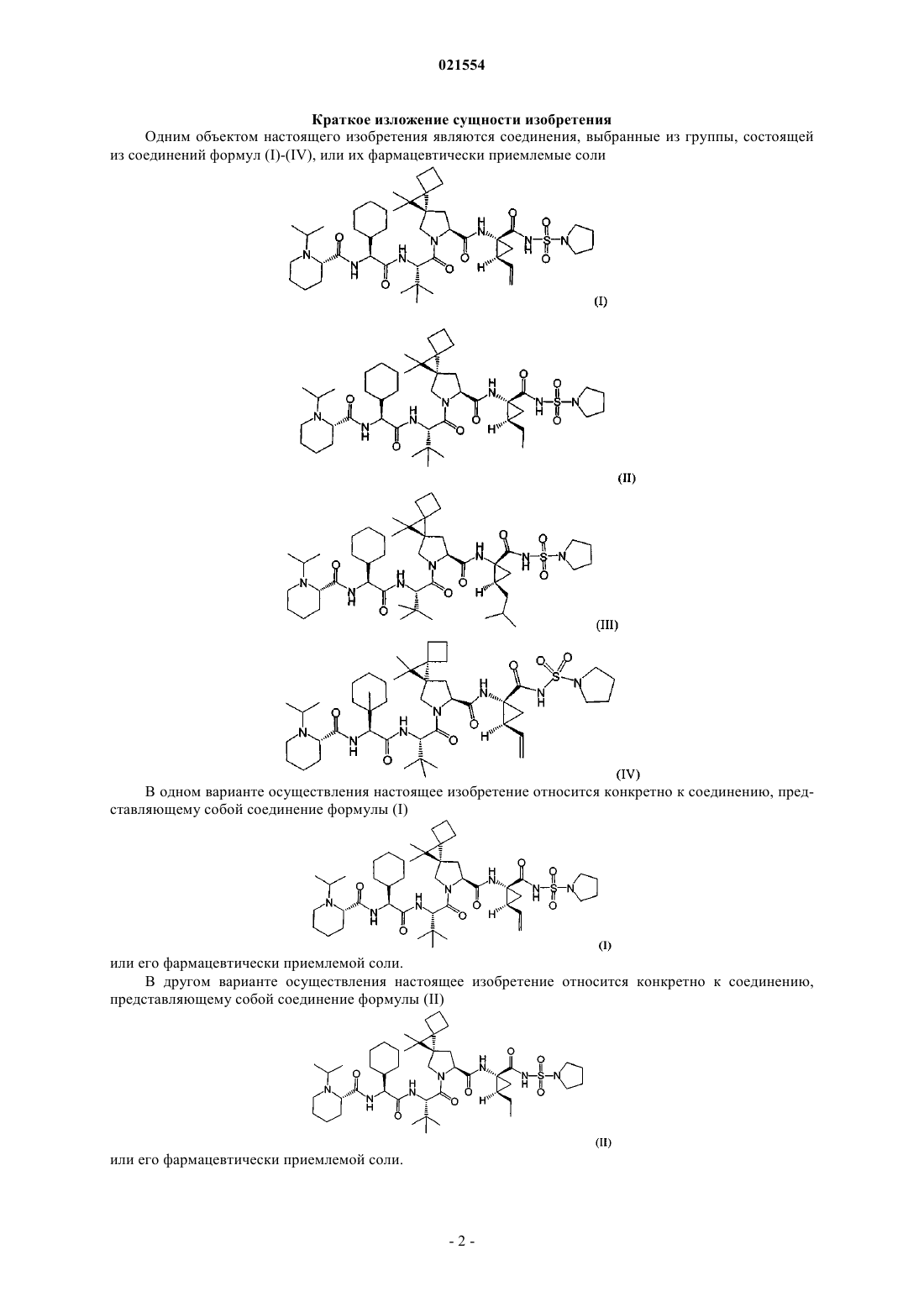
1. Соединение, выбранное из группы, состоящей из соединений формул (I)-(IV), или его фармацевтически приемлемая соль


2. Соединение по п.1, представляющее собой соединение формулы (I)

или его фармацевтически приемлемая соль.
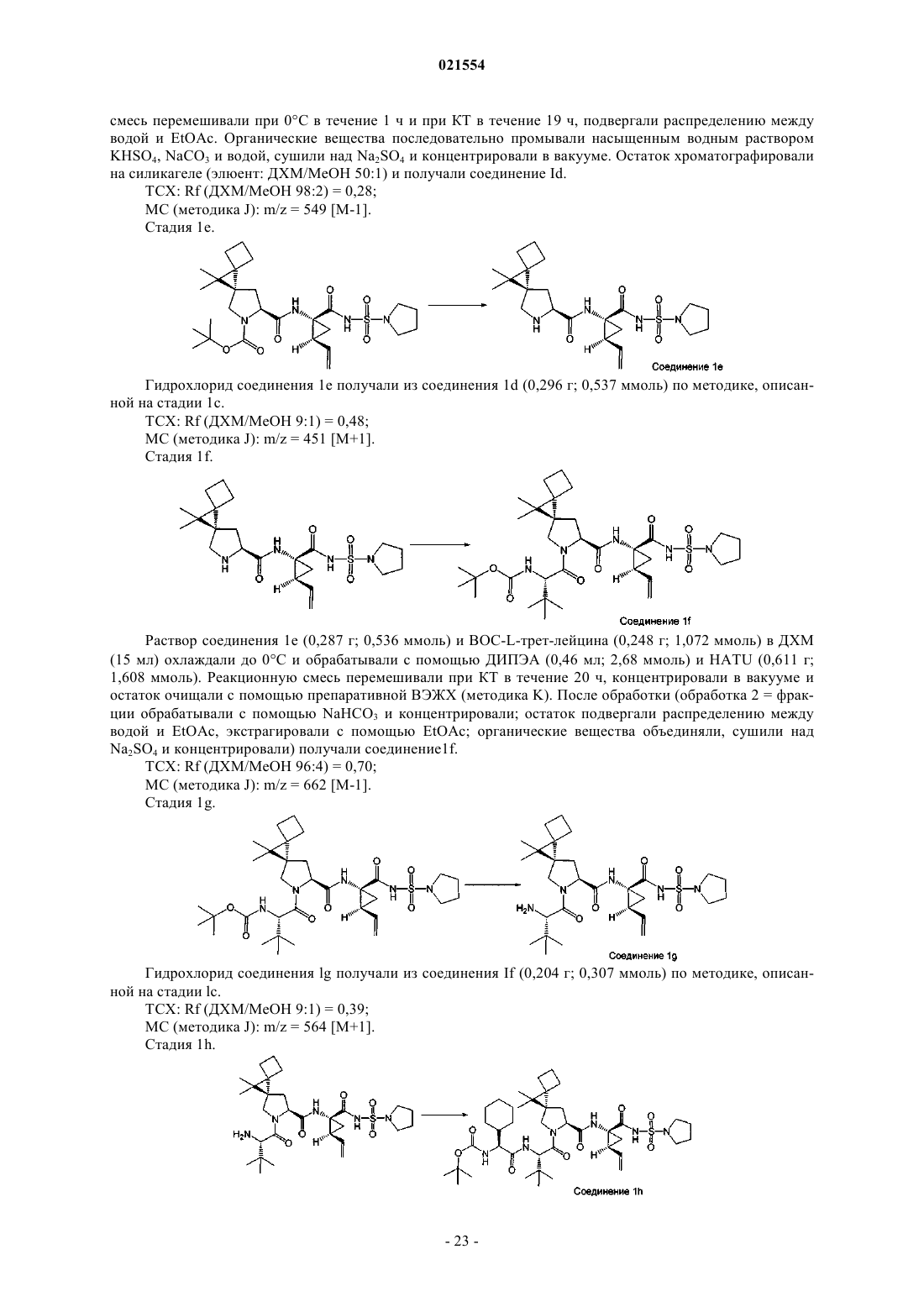
3. Соединение по п.1, представляющее собой соединение формулы (II)

или его фармацевтически приемлемая соль.
4. Соединение по п.1, представляющее собой соединение формулы (III)

или его фармацевтически приемлемая соль.
5. Соединение по п.1, представляющее собой соединение формулы (IV)

или его фармацевтически приемлемая соль.
6. Соединение или его фармацевтически приемлемая соль по любому из пп.1-5, где фармацевтически приемлемая соль представляет собой гидрохлорид.
7. Фармацевтическая композиция, предназначенная для лечения HCV инфекции, включающая соединение по п.1 и фармацевтически приемлемый инертный наполнитель.
8. Фармацевтическая композиция по п.7, включающая соединение формулы (I) и фармацевтически приемлемый наполнитель.
9. Фармацевтическая композиция по п.7, включающая соединение формулы (II) и фармацевтически приемлемый наполнитель.
Текст
ОРГАНИЧЕСКИЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ HCV ИНФЕКЦИИ В изобретении приведены описания органических соединений формул (I)-(IV) или их фармацевтически приемлемых солей, которые применимы для лечения, предупреждения и/или облегчения протекания заболеваний людей, в частности HCV инфекции. Уровень техники Инфицирование вирусом хронического гепатита С (HCV) является большой глобальной проблемой здравоохранения, поскольку во всем мире, по оценкам, им инфицированы 170 млн человек и ежегодно инфицируются еще от 3 до 4 млн (см., например, World Health Organization Fact Sheet No. 164. October 2000). Хотя 25% новых случаев инфицирования являются симптоматическими, у 60-80% пациентов развивается хроническое заболевание печени, и, по оценкам, у 20% из них оно переходит в цирроз, и в год у 1-4% существует опасность развития печеночно-клеточной карциномы (см., например, World HealthEradication. Hepatology, 43:S207-S220). В развитых странах HCV приводит к 50-76% всех случаев рака печени и 2/3 всех случаев трансплантации печени (см., например, World Health Organization Guide onViral Cancers. 2006). И в конечном счете 5-7% инфицированных пациентов умирают от последствий инфицирования с помощью HCV (см., например, World Health Organization Guide on Hepatitis C. 2002). Современным стандартным средством борьбы с инфицированием посредством HCV является пэгилированный интерферон-альфа (IFN-) в комбинации с рибаривином. Однако только до 50% пациентов с генотипом 1 вируса можно успешно лечить этим основанным на интерфероне средством. Кроме того, и интерферон, и рибаривин могут привести к значительным побочным эффектам в диапазоне от симптомов типа гриппа (лихорадка и усталость) до гематологических осложнений (лейкопения, тромбоцитопения), нервно-психиатрических проявлений (депрессия, инсомния, раздражимость), потери массы и аутоиммунных дисфункций (гипотиреоз, диабет) вследствие лечения интерфероном и до выраженной гемолитической анемии вследствие лечения рибаривином. Поэтому все еще необходимы более эффективные и лучше переносимые лекарственные средства.N-концевой серинпротеазный домен, содержащий 180 аминокислот (АК), и С-концевой геликазно/НТФазный домен (АК от 181 до 631). NS3 протеаза считается представителем семейства химотрипсина вследствие сходства белковой последовательности, общей трехмерной структуры и механизма катализа. HCV NS3 серинпротеаза обеспечивает протеолитическое расщепление полипротеина в сочлененияхVirol. 67:3835-3844; Grakoui, A. et al. (1993), J. Virol. 67:2832-2843; Tomei, L. et al. (1993), J. Virol. 67:4017-4026). NS4A, белок, обладающий молекулярной массой, равной около 6 кДа, и содержащий 54 АК, является кофактором активности серинпротеазы NS3 (см., например, Failla, С. et al. (1994), J. Virol. 68:3753-3760; Tanji, Y. et al. (1995), J. Virol. 69:1575-1581). Ауторасщепление сочленения NS3/NS4a серинпротеазой NS3/NS4a протекает внутримолекулярно (т. е. цис), тогда как другие сайты расщепления подвергаются межмолекулярному процессингу (т.е. транс). Показано, что HCV NS3 протеаза важна для репликации вируса и поэтому является привлекательным объектом воздействия для противовирусной химиотерапии. Сохраняется необходимость в новых средствах борьбы с инфицированием HCV, а также лечения связанных с HCV нарушений. Также необходимы соединения, применимые для устранения, предупреждения или ослабления одного или большего количества симптомов HCV, а также необходимы способы устранения, предупреждения или ослабления одного или большего количества симптомов HCV. Кроме того, необходимы новые соединения, способные HCV модулировать активность HCV-серинпротеаз, в особенности NS3/NS4a серинпротеазы, и применение указанных соединений для лечения, предупреждения или ослабления инфицирования посредством HCV. Краткое изложение сущности изобретения Одним объектом настоящего изобретения являются соединения, выбранные из группы, состоящей из соединений формул (I)-(IV), или их фармацевтически приемлемые соли В одном варианте осуществления настоящее изобретение относится конкретно к соединению, представляющему собой соединение формулы (I) или его фармацевтически приемлемой соли. В другом варианте осуществления настоящее изобретение относится конкретно к соединению,представляющему собой соединение формулы (II) или его фармацевтически приемлемой соли. В еще одном варианте осуществления настоящее изобретение относится конкретно к соединению,представляющему собой соединение формулы (III) или его фармацевтически приемлемой соли. Также настоящее изобретение относится конкретно к соединению, представляющему собой соединение формулы (IV) или его фармацевтически приемлемой соли. Соединения, предлагаемые в настоящем изобретении, обладают активностью по отношению к HCV протеазе и могут являться ингибиторами HCV NS3-4A протеазы. Соединения настоящего изобретения могут использоваться для лечения нарушений, связанных сHCV нарушением, выбранных из группы, включающей инфекцию HCV, цирроз печени, хроническое заболевание печени, печеночно-клеточную карциному, криоглобулинемию, неходжкинскую лимфому,фиброз печени и подавленный врожденный внутриклеточный иммунный ответ. При этом HCV выбран из группы, включающей генотипы HCV 1, 2 и/или 3. Настоящее изобретение также относится к фармацевтическим композициям, содержащим соединения формул (I)-(IV), предназначенным для лечения HCV инфекции. Соединения особенно полезны для создания помех клеточному циклу вируса гепатита С для борьбы с инфицированием посредством HCV или ее предупреждения или со связанными с ним физиологическими условиями. Соединения настоящего изобретения могут использоваться в комбинированной терапии для подавления репликации HCV в клетках или для борьбы с инфицированием посредством HCV или ее предупреждения у пациентов с применением соединений, предлагаемых в настоящем изобретении, или фармацевтических композиций, или содержащих их наборов. По данным исследований протеазы HCV NS3-4A и основанного на люциферазе исследования репликонов HCV, описанных ниже в разделе, посвященном примерам, установлено, что соединения, предлагаемые в настоящем изобретении, обладают значениями IC50 для ингибирования HCV, находящимися в диапазоне от 0,1 до более 100 нМ или от 0,5 до 30 нМ, включая, например, от 0,5 до 10 нМ или менее. В некоторых вариантах осуществления соединение, предлагаемое в настоящем изобретении, дополнительно характеризуется как модулятор HCV, включая HCV млекопитающего и предпочтительно включая HCV человека. В предпочтительном варианте осуществления соединение, предлагаемое в настоящем изобретении, является ингибитором HCV. Термины "связанное с HCV патологическое состояние" или "связанное с HCV нарушение" включают нарушения и патологические состояния (например, заболевание), которые связаны с активностьюHCV, например инфицирование посредством HCV у субъекта. Связанные с HCV патологические состояния включают инфицирование посредством HCV, цирроз печени, хроническое заболевание печени, печеночно-клеточную карциному, криоглобулинемию, неходжкинскую лимфому и подавленный врожденный внутриклеточный иммунный ответ. Связанные с HCV патологические состояния часто связаны с NS3 серинпротеазой HCV, которая обеспечивает некоторые стадии процессинга HCV полипротеина в меньшие функциональные белки. NS3 протеаза образует гетеродимерный комплекс с белком NS4A, необходимым кофактором, который усиливает ферментативную активность и предположительно способствует прикреплению HCV к эндоплазматическому ретикулуму. NS3 сначала автокатализирует гидролиз сочленения NS3-NS4A и затем внутримолекулярно расщепляет полипротеин HCV по пересечениям NS4A-NS4B, NS4B-NS5A и NS5A-NS5B. Этот процесс связан с репликацией HCV у субъекта. Ингибирование или модулирование активности одного или большего количества белков NS3, NS4A, NS4B, NS5A и NS5B приводит к ингибированию или модулированию репликации HCV у субъекта и тем самым предупреждает или лечит связанное с HCV патологическое состояние. Связанное с HCV патологическое состояние связано с активностью NS3 протеазы. Также связанное с HCV патологическое состояние связано с активностью гетеродимерного ком-3 021554 плекса NS3-NS4A. Соединения, предлагаемые в настоящем изобретении, представляют собой ингибиторы NS3/NS4A протеазы или ингибиторы NS2/NS3 протеазы. Если не ограничиваться теоретическими соображениями, то можно полагать, что нарушение указанных выше взаимодействий белок-белок соединениями, предлагаемыми в настоящем изобретении,нарушит процессинг вирусного полипротеина NS3 протеазой и тем самым нарушит репликацию вируса. Связанные с HCV нарушения также включают зависимые от HCV заболевания. Зависимые от HCV заболевания включают, например, любое заболевание или нарушение, которое зависит от активности или связано с активностью или неправильной регуляцией по меньшей мере одного штамма HCV. Соединения настоящего изобретения могут быть пригодны в лечении связанных с HCV нарушений,описанных выше, но не ограничивается тем, каким образом соединение осуществляет свою функцию лечения заболевания. Соединения, предлагаемые в настоящем изобретении, могут быть применимы для лечения заболеваний, связанных с ВИЧ, а также с инфицированием посредством ВИЧ и со СПИД (синдром приобретенного иммунодефицита). В некоторых вариантах осуществления настоящее изобретение относится к фармацевтической композиции любого из соединений формул (I)-(IV), предлагаемых в настоящем изобретении. В родственном варианте осуществления настоящее изобретение относится к фармацевтической композиции любого из соединений, предлагаемых в настоящем изобретении, и фармацевтически приемлемого носителя или инертного наполнителя для любого из этих соединений. В некоторых вариантах осуществления настоящее изобретение относится к соединениям как к новым химическим веществам. Соединения настоящего изобретения могут использоваться в составе упаковки средства для лечения нарушения, связанного с HCV. Упаковка средства включает соединение, предлагаемое в настоящем изобретении, упакованное вместе с инструкциями по применению соединения, предлагаемого в настоящем изобретении, в эффективном количестве, для предназначенной цели. Соединения, предлагаемые в настоящем изобретении, применимы в качестве активных средств в фармацевтических композициях, которые эффективны предпочтительно для лечения связанных с HCV нарушений. Фармацевтическая композиция в различных вариантах осуществления содержит активное средство, предлагаемое в настоящем изобретении, в фармацевтически эффективном количестве вместе с другими фармацевтически приемлемыми инертными наполнителями, носителями, наполнителями, разбавителями и т.п. Выражение "фармацевтически эффективное количество" при использовании в настоящем изобретении указывает на количество, необходимое для введения реципиенту или в клетку, ткань или орган реципиента для обеспечения терапевтического результата, предпочтительно анти-HCV эффекта, например, подавления пролиферации вируса HCV или для лечения любого другого связанного с HCV заболевания. Заболевания, которые могут лечиться соединениями, предлагаемыми в настоящем изобретении,включают, например, инфицирование посредством HCV, цирроз печени, хроническое заболевание печени, печеночно-клеточную карциному, криоглобулинемию, неходжкинскую лимфому и подавленный врожденный внутриклеточный иммунный ответ. Также соединения настоящего изобретения могут быть использованы для подавления активностиHCV путем взаимодействия клетки с любым из соединений, предлагаемых в настоящем изобретении, в частности для селективного ингибирования активности одного или большего количества белков NS3,NS4A, NS4B, NS5A и NS5B. Соединение содержится в количестве, эффективном для снижения нагрузки посредством HCV РНК у субъекта. Соединения настоящего изобретения могут быть использованы для приготовления лекарственного средства, предназначенного для борьбы с инфицированием посредством HCV у субъекта. Определения. Термины "лечить", "подвергать лечению", "лечение" включают уменьшение или ослабление по меньшей мере одного симптома, связанного с подвергающимся лечению патологическим состоянием,нарушением или заболеванием или вызванного им. В некоторых вариантах осуществления лечение включает индуцирование ингибированного состояния HCV с последующей активацией модулирующегоHCV соединения, которое, в свою очередь, уменьшит или ослабит по меньшей мере один симптом, связанный с подвергающимся лечению связанным с HCV состоянием, нарушением или заболеванием или вызванный им. Например, лечение может ослабить один или несколько симптомов нарушения или привести к полному устранению нарушения. Термин "субъект" включает организмы, например прокариоты и эукариоты, которые способны страдать от нарушения, связанного HCV или которых оно может поражать. Примеры субъектов включают млекопитающих, например людей, собак, крупный рогатый скот, лошадей, свиней, овец, коз, кошек,кроликов, крыс и трансгенных животных, не являющихся людьми. В некоторых вариантах осуществления субъектом является человек, например человек, который страдает или подвергается опасности страдать или который может страдать от связанного HCV нарушения и заболеваний или патологических состояний, описанных в настоящем изобретении, например от инфицирования посредством HCV. В другом варианте осуществления субъектом является клетка. Выражения "модулирующее HCV соединение", "модулятор HCV" или "ингибитор HCV" означают соединения, которые модулируют, например, ингибируют или иным образом изменяют активность HCV. Аналогичным образом, "ингибитор NS3/NS4A протеазы" или "ингибитор NS2/NS3 протеазы" означает соединение, которое модулирует, например, ингибирует или иным образом изменяет взаимодействие этих протеаз друг с другом. Примеры модулирующих HCV соединений включают соединения формул(I)-(IV), а также соединения, приведенные в примерах 1-19 и табл. А и В (включая их фармацевтически приемлемые соли). При использовании в настоящем изобретении термин "фармацевтически приемлемые соли" означает соли, которые сохраняют биологическую эффективность и свойства соединений, предлагаемых в настоящем изобретении и которые не являются нежелательными в биологическом или ином отношении. Во многих случаях соединения, предлагаемые в настоящем изобретении, могут образовывать соли с кислотой и/или основанием вследствие наличия аминогрупп и/или карбоксигрупп или аналогичных им групп. Фармацевтически приемлемые соли присоединения с кислотами можно образовать с неорганическими кислотами и с органическими кислотами, например ацетат, аспартат, бензоат, безилат, бромид/гидробромид, бикарбонат/карбонат, бисульфат/сульфат, камфорсульфонат, хлорид/гидрохлорид,хлортеофиллонат, цитрат, этандисульфонат, фумарат, глюцептат, глюконат, глюкуронат, гиппурат, гидройодид/йодид, изетиноат, лактат, лактобионат, лаурилсульфат, малат, малеат, малонат, манделат, мезилат, метилсульфат, нафтоат, напсилат, никотинат, нитрат, октадеканоат, олеат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, полигалактуронат, пропионат, стеарат, сукцинат, сульфосалицилат, тартрат, тозилат и трифторацетат. Неорганические кислоты, из которых можно образовать соли, включают, например, хлористоводородную, бромисто-водородную, серную, азотную, фосфорную кислоты и т.п. Органические кислоты, из которых можно образовать соли, включают, например, уксусную, пропионовую, гликолевую, щавелевую, малеиновую, малоновую, янтарную, фумаровую, винную, лимонную, бензойную, миндальную, метансульфоновую, этансульфоновую, толуолсульфоновую, сульфосалициловую кислоты и т.п. Фармацевтически приемлемые соли присоединения с основанием можно образовать с неорганическими и органическими основаниями. Неорганические основания, из которых можно образовать соли, включают, например, основания натрия, калия, аммония, кальция, магния, железа, цинка, меди и т.п.; особенно предпочтительными являются соли аммония, калия, натрия, кальция и магния. Органические основания, из которых можно образовать соли, включают, например, первичные,вторичные и третичные амины, замещенные амины, включая природные замещенные амины, циклические амины, основные ионообменные смолы и т.п., предпочтительно такие как изопропиламин, бензатин,холинат, диэтаноламин, диэтиламин, лизин, меглумин, пиперазин и трометамин. Фармацевтически приемлемые соли, предлагаемые в настоящем изобретении, можно синтезировать из исходного соединения, основного или кислотного фрагмента по обычным химическим методикам. Обычно такие соли можно получить по реакции свободных кислотных форм этих соединений со стехиометрическим количеством соответствующего основания (такого как гидроксид, карбонат, бикарбонат и т.п., Na, Ca, Mg или K) или по реакции свободных основных форм этих соединений со стехиометрическим количеством соответствующей кислоты. Такие реакции обычно проводят в воде или в органическом растворителе или в их смеси. Обычно, если это возможно, предпочтительными являются неводные среды, такие как эфир, этилацетат, этанол, изопропанол или ацетонитрил. Перечни дополнительных подходящих солей приведены, например, в публикации Remington's Pharmaceutical Sciences, 20th ed., MackPublishing Company, Easton, Pa. (1985). При использовании в настоящем изобретении термин "фармацевтически приемлемый наполнитель(носитель)" включает любой и все растворители, диспергирующие среды, покрытия, поверхностноактивные вещества, антиоксиданты, консерванты (например, бактерицидные агенты, фунгицидные агенты), изотонические агенты, агенты, задерживающие всасывание, соли, консерванты, лекарственные средства, стабилизаторы лекарственных средств, связующие, инертные наполнители, разрыхлители, смазывающие агенты, подсластители, ароматизаторы, красители, аналогичные материалы и их комбинации,как это должно быть известно специалисту с общей подготовкой в данной области техники (см., например, публикацию Remington's Pharmaceutical Sciences, 18th Ed. Mack Printing Company, 1990, p. 12891329). За исключением случаев, когда обычный наполнитель (носитель) несовместим с активным ингредиентом, подразумевается его использование в терапевтических или фармацевтических композициях. Термин "терапевтически эффективное количество" соединения, предлагаемого в настоящем изобретении, означает количество соединения, предлагаемого в настоящем изобретении, которое приводит к биологической или медицинской реакции субъекта, или облегчает симптомы, замедляет или задерживает прогрессирование заболевания, или предупреждает заболевание и т.п. В одном неограничивающем варианте осуществления термин "терапевтически эффективное количество" означает количество соединения,предлагаемого в настоящем изобретении, которое при введении субъекту эффективно (1) по меньшей мере, для частичного ослабления, подавления, предупреждения и/или облегчения протекания патологического состояния, или нарушения, или (i) заболевания, опосредуемого активностью NS3/NS4 серинпро-5 021554 теазы; или (2) для уменьшения или ингибирования активности NS3/NS4 серинпротеазы; или (3) для уменьшения или ингибирования репликации по меньшей мере одного вируса, который кодирует NS3 серинпротеазу. В другом неограничивающем варианте осуществления термин "терапевтически эффективное количество" означает количество соединения, предлагаемого в настоящем изобретении, которое при введении в клетку или ткань или в неклеточный биологический материал или среду эффективно, по меньшей мере, для частичного уменьшения или ингибирования вирусной нагрузки и/или репликации вируса. Значение термина "терапевтически эффективное количество", проиллюстрированное в приведенном выше варианте осуществления для NS3 протеазы, является таким же применительно к любым другим относящимся к этому белкам/пептидам/ферментам, таким как NS2 протеаза, NS3 протеаза, NS3 геликаза, белок NS5a и/или NS5b полимераза и т.п. При использовании в настоящем изобретении термин "субъект" означает животное. Предпочтительно, если животным является млекопитающее. Субъект также означает, например, приматов (например, людей), коров, овец, коз, лошадей, собак, кошек, кроликов, крыс, мышей, рыб, птиц и т.п. В предпочтительном варианте осуществления субъектом является примат. В другом предпочтительном варианте осуществления субъектом является человек. При использовании в настоящем изобретении термин "ингибирование" или "подавление" означает ослабление или подавление данного патологического состояния, симптома, или нарушения, или заболевания или значительное ослабление исходной биологической активности или процесса. При использовании в настоящем изобретении термин "лечение" любого заболевания или нарушения в одном варианте осуществления означает улучшение протекания заболевания или нарушения (т.е. замедление или остановку или ослабление развития заболевания или по меньшей мере одного из его клинических симптомов). В другом варианте осуществления "лечение" означает улучшение по меньшей мере одного физического параметра, включая те, которые могут не ощущаться пациентом. В еще одном варианте осуществления "лечение" означает изменение протекания заболевания или нарушения, физическое (например, стабилизацию проявляющегося симптома) или физиологическое (например, стабилизацию физикального параметра), или и то, и другое. В еще одном варианте осуществления "лечение" означает предупреждение или задержку начала или развития, или прогрессирования заболевания или нарушения. При использовании в настоящем изобретении субъект "нуждается в" лечении, если лечение окажет на такой субъект благоприятное биологическое или медицинское воздействие или улучшит качество его жизни. При использовании в настоящем изобретении (в особенности в формуле изобретения) термины, использующиеся в единственном числе, включают множественное число, и наоборот, если в настоящем изобретении не указано иное и если это явно не противоречит контексту. Все методики, описанные в настоящем изобретении, можно проводить в любом подходящем порядке, если в настоящем изобретении не указано иное и если это явно не противоречит контексту. Использование любого и всех примеров или указаний на типичные значения (например, "такой как") в настоящем изобретении предназначено просто для лучшего описания настоящего изобретения и не налагает ограничения на объем настоящего изобретения, определяющийся формулой изобретения. Соединения, предлагаемые в настоящем изобретении, получают в свободной форме или в виде его соли, предпочтительно в форме гидрохлорида. Если в одной молекуле содержатся и основная группа, и кислотная группа, то соединения, предлагаемые в настоящем изобретении, также могут образовать внутренние соли, например цвиттерионные молекулы. Другим объектом настоящего изобретения является фармацевтическая композиция, включающая соединение, предлагаемое в настоящем изобретении, и фармацевтически приемлемый наполнитель. Фармацевтическую композицию можно приготовить для конкретных путей введения, таких как пероральное введение, парентеральное введение и ректальное введение и т.п. Кроме того, фармацевтические композиции, предлагаемые в настоящем изобретении, можно приготовить в твердой форме, включая капсулы, таблетки, пилюли, гранулы, порошки или суппозитории, или в жидкой форме, включая растворы, суспензии или эмульсии. Фармацевтические композиции можно подвергнуть обычной фармацевтической обработке, такой как стерилизация, и/или они могут содержать обычные инертные разбавители,смазывающие агенты или буферные агенты, а также вспомогательные вещества, такие как консерванты,стабилизаторы, смачивающие агенты, эмульгаторы и буферные агенты и т.п. Обычно фармацевтические композиции являются таблетками или капсулами из желатина, включающими активный ингредиент, а также:b) смазывающие агенты, например диоксид кремния, тальк, стеариновую кислоту, ее магниевую или кальциевую соль и/или полиэтиленгликоль; для таблеток такжеc) связующие, например алюмосиликат магния, крахмальную пасту, желатин, трагакантовую камедь, метилцеллюлозу, натриевую соль карбоксиметилцеллюлозы и/или поливинилпирролидон; при необходимостиd) разрыхлители, например крахмалы, агар, альгиновую кислоту или ее натриевую соль, или шипучие смеси; и/илиe) абсорбенты, красители, ароматизаторы и подсластители. На таблетки по методикам, известным в данной области техники, можно нанести пленочное или энтеросолюбильное покрытие. Композиции, подходящие для перорального введения, включают соединение, предлагаемое в настоящем изобретении, в эффективном количестве в виде таблеток, лепешек, водных или масляных суспензий, диспергирующихся порошков или гранул, эмульсий, твердых или мягких капсул или сиропов,или эликсиров. Композиции, предназначенные для перорального применения, получают по любой методике, известной в данной области техники для приготовления фармацевтических композиций, и такие композиции могут содержать один или большее количество агентов, выбранных из группы, включающей подсластители, ароматизаторы, красители и консерванты, чтобы получить фармацевтически привлекательные и обладающие приятным вкусом препараты. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми инертными наполнителями, которые являются подходящими для изготовления таблеток. Этими инертными наполнителями являются, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и разрыхляющие агенты, например кукурузный крахмал или альгиновая кислота; связывающие агенты, например крахмал, желатин или камедь акации; и смазывающие агенты, например стеарат магния, стеариновая кислота или тальк. Таблетки не содержат покрытия или на них по известным методикам наносят покрытие для задержки распада и всасывания в желудочно-кишечном тракте, что обеспечивает непрерывное воздействие в течение более длительного периода времени. Например, можно использовать такое замедляющее вещество, как глицерилмоностеарат или глицерилдистеарат. Препараты для перорального применения также могут представлять собой капсулы из твердого желатина, в которых активный ингредиент смешан с инертным твердым разбавителем, например карбонатом кальция,фосфатом кальция или каолином, или капсулы из мягкого желатина, в которых активный ингредиент смешан с водой или масляной средой, например арахисовым маслом, жидким парафином или оливковым маслом. Некоторые композиции для инъекций представляют собой изотонические водные растворы или суспензии, а суппозитории предпочтительно готовят из эмульсий или суспензий жиров. Указанные композиции можно стерилизовать и/или прибавлять к ним вспомогательные вещества, такие как консервирующие, стабилизирующие, смачивающие или эмульгирующие агенты, стимуляторы растворения, соли для регулирования осмотического давления и/или буферные агенты. Кроме того, они также могут содержать другие терапевтически ценные вещества. Указанные композиции получают по обычным технологиям смешивания, гранулирования или нанесения покрытий, и они содержат примерно 0,1-75% или содержат примерно 1-50% активного ингредиента. Композиции, подходящие для чрескожного введения, включают эффективное количество соединения, предлагаемого в настоящем изобретении, с носителем. Предпочтительные носители включают впитывающиеся фармакологически приемлемые растворители, способствующие проникновению через кожу реципиента. Например, устройства для чрескожного введения представляют собой повязку, включающую защитный слой, резервуар, содержащий соединение необязательно с носителями, необязательно барьер, регулирующий доставку соединения через кожу реципиента с заданной скоростью в течение пролонгированного периода времени, и средства закрепления устройства на коже. Композиции, подходящие для местного введения, например на кожу, или в глаза, или слизистые оболочки, включают водные растворы, суспензии, мази, кремы, гели или распыляемые композиции, например, для подачи в виде аэрозоля и т.п. Такие устройства местного действия являются особенно подходящими для вагинального введения, например для предупреждения инфекции HCV. Они могут содержать солюбилизаторы, стабилизаторы, агенты, регулирующие тоничность, буферные агенты и консерванты. Настоящее изобретение также относится к безводным фармацевтическим композициям и дозированным формам, включающим соединения, предлагаемые в настоящем изобретении, в качестве активных ингредиентов, поскольку вода может облегчать разложение некоторых соединений. Безводные фармацевтические композиции и дозированные формы, предлагаемые в настоящем изобретении, можно получить с использованием безводных или обладающих низкой влажностью ингредиентов в условиях низкой влажности. Безводную фармацевтическую композицию можно готовить и хранить так, чтобы она оставалась безводной. В соответствии с этим безводные композиции предпочтительно упаковывать в материалы, для которых известно, что они защищают от воздействия воды, так чтобы их можно было включить в подходящие наборы препаратов. Примеры подходящих упаковочных средств включают, но не ограничиваются только ими, герметично запаиваемую фольгу, пластмассы, контейнеры для разовых доз (например, флаконы), блистерные упаковки и ленточные упаковки. Фармацевтические композиции могут находиться в дозированных формах, которые включают один или большее количество агентов, которые уменьшают скорость, с которой будет разлагаться соединение,предлагаемое в настоящем изобретении в качестве активного ингредиента. Такие агенты, которые в на-7 021554 стоящем изобретении называют "стабилизаторами", включают, но не ограничиваются только ими, антиоксиданты, такие как аскорбиновая кислота, буферные агенты, регулирующие рН, и солевые буферные агенты и т.п. Фармацевтическая композиция или комбинация, предлагаемая в настоящем изобретении, может содержать в разовой дозе примерно 1-1000 мг активного ингредиента (ингредиентов) для субъекта массой примерно 50-70 кг, примерно 1-500 мг, или примерно 1-250 мг, или примерно 1-150 мг, или примерно 0,5-100 мг, или примерно 1-50 мг активных ингредиентов. Терапевтически эффективная доза соединения, фармацевтической композиции или их комбинаций зависит от вида субъекта, массы тела, возраста и индивидуального состояния, нарушения или заболевания, подвергающегося лечению, и его тяжести. Врач, клиницист или ветеринар с общей подготовкой должен легко определить эффективное количество каждого из активных ингредиентов, необходимое для предупреждения, лечения или подавления прогрессирования нарушения или заболевания. Указанные выше характеристики доз можно определить с помощью проводимых in vitro и in vivo исследований, предпочтительно с использованием млекопитающих, например мышей, крыс, собак, обезьян или изолированных органов, тканей и их препаратов. Соединения, предлагаемые в настоящем изобретении, можно использовать in vitro в виде растворов, например предпочтительно водных растворов, иin vivo энтерально, парентерально, предпочтительно внутривенно, например в виде суспензии или водного раствора. Доза in vitro может находиться в диапазоне примерно от 10-3 до 10-9 М. Терапевтически эффективное количество in vivo в зависимости от пути введения может находиться в диапазоне примерно 0,1-500 или примерно 1-100 мг/кг. Активность соединения, предлагаемого в настоящем изобретении, можно оценить с помощью in vitro и in vivo методик, включая, но не ограничиваясь только ими, методики, приведенные выше. Фармацевтическая композиция может включать соединение формул (I)-(IV) и другое терапевтическое средство (средства). Фармацевтическая композиция необязательно может включать фармацевтически приемлемый инертный наполнитель, описанный выше. Соединения настоящего изобретения могут входить в состав набора, включающего две или большее количество отдельных фармацевтических композиций, по меньшей мере одна из которых содержит соединение формул (I)-(IV. Набор включает приспособление для раздельного хранения указанных композиций, такое как контейнер, разделенный флакон или разделенный пакет из фольги. Примером такого набора является блистерная упаковка, которую обычно используют для упаковки таблеток, капсул и т.п. Набор можно применять для введения различных дозированных форм, например перорально и парентерально, для введения отдельных композиций с разными интервалами или для предохранения отдельных композиций друг от друга. Чтобы содействовать соблюдение пациентом режима лечения, набор,предлагаемый в настоящем изобретении, обычно включает руководства по введению. В комбинированной терапии соединение, предлагаемое в настоящем изобретении, и другое терапевтическое средство могут быть изготовлены и/или приготовлены одним и тем же или разными изготовителями. Кроме того, соединение, предлагаемое в настоящем изобретении, и другое терапевтическое средство могут быть объединены в комбинированном средстве: (i) до передачи комбинированного продукта врачам (например, в случае набора, включающего соединение, предлагаемое в настоящем изобретении, и другое терапевтическое средство); (ii) самими врачами (или под руководством врача) незадолго до введения; (iii) в самом пациенте, например, во время последовательного введения соединения, предлагаемого в настоящем изобретении, и другого терапевтического средства. В соответствии с этим лечения заболевания или патологического состояния, опосредуемого активностью NS3 протеазы, включая, но не ограничиваясь только ими, вирусные инфекции, выбранные из группы, включающей HCV, ВИЧ и т.п., где лекарственное средство приготовлено для введения вместе с другим терапевтическим средством. Соединения формул (I)-(IV могут предназначаться для применения в лечении заболевания или патологического состояния, опосредуемого активностью NS3 протеазы, в котором соединение формул (I)-(IV приготовлено для введения вместе с другим терапевтическим средством. Ссоединения настоящего изобретения могут использоваться для лечения вирусной инфекции, когда пациента ранее (например, за 24 ч) лечили другим терапевтическим средством. Соединение может быть применяться с другим терапевтическим средством для лечения вирусной инфекции, когда пациента ранее (например, за 24 ч) лечили соединением формул (I)-(IV. Соединение, предлагаемое в настоящем изобретении, также можно использовать в комбинации с другими средствами, например с дополнительным модулирующим HCV соединением, которое является или не является соединением формулы (I), для лечения и связанного с HCV нарушения у субъекта. Термин "комбинация" означает фиксированную комбинацию в одной разовой дозированной форме или набор компонентов, предназначенный для комбинированного введения, с помощью которого соединение, предлагаемое в настоящем изобретении, и компонент комбинации можно вводить независимо одновременно или по отдельности через промежутки времени, которые позволяют компонентам комбинации проявить совместный, например синергетический, эффект, или любую их комбинацию. Например, в WO 2005/042020, во всей своей полноте включенной в настоящее изобретение в качестве ссылки, описана комбинация различных ингибиторов HCV с ингибитором цитохрома Р 450 ("CYP"). Любой ингибитор CYP, который улучшает фармакокинетические характеристики соответствующейNS3/4A протеазы, можно применять в комбинации с соединениями, предлагаемыми в настоящем изобретении. Эти ингибиторы CYP включают, но не ограничиваются только ими, ритонавир (WO 94/14436, во всей своей полноте включенная в настоящее изобретение в качестве ссылки), кетоконазол, тролеандомицин, 4-метилпиразол, циклоспорин, NIM811, клометиазол, циметидин, итраконазол, флуконазол, миконазол, флувоксамин, флуоксетин, нефазодон, сертралин, индинавир, нелфинавир, ампренавир, фосампренавир, саквинавир, лопинавир, делавирдин, эритромицин, VX-944 и VX-497. Предпочтительные ингибиторы CYP включают ритонавир, кетоконазол, тролеандомицин, 4-метилпиразол, циклоспорин, NIM811 и клометиазол. Методики определения способности соединения ингибировать активность CYP являются известными (см., например, US 6037157 и Yun, et al. Drug MetabolismDisposition, vol. 21, p. 403-407 (1993); включенный в настоящее изобретение в качестве ссылки). Например, исследуемое соединение можно инкубировать с 0,1, 0,5 и 1,0 мг белка/мл или с использованием другой подходящей концентрации микросом печени человека (например, имеющаяся в продаже охарактеризованная смесь микросом печени) в течение 0, 5, 10, 20 и 30 мин или другого подходящего времени в присутствии системы, генерирующейNADPH. Контрольное инкубирование можно провести при отсутствии микросом печени в течение 0 и 30 мин (с трехкратным повторением). Образцы можно исследовать на наличие соединения. Для последующих исследований, как правило, используют условия инкубации, которые обеспечивают линейную кинетику метаболизма соединения. Для изучения кинетики метаболизма соединения (Km и Vmax) можно использовать методики, известные в данной области техники. Можно определить скорость исчезновения соединения и данные обработать в соответствии с кинетикой Михаэлиса-Ментена по методикам Лайнвивера-Берка, Эди-Ховсти или с помощью нелинейного регрессионного анализа. Можно исследовать ингибирование метаболизма. Например, соединение (одна концентрация, Km) можно инкубировать со смесью микросом печени человека при отсутствии или в присутствии ингибитора CYP (такого как ритонавир) при указанных выше условиях. Понятно, что контрольное инкубирование следует проводить при такой же концентрации органического растворителя, как и при инкубировании с ингибитором CYP. Можно определить концентрации соединения в образцах и скорость исчезновения исходного соединения, скорости представить в виде активности, выраженной в процентах от контрольной. Также известны методики оценки влияния совместного введения субъекту соединения, предлагаемого в настоящем изобретении, и ингибитора CYP (см., например, US2004/0028755; включенный в настоящее изобретение в качестве ссылки). Любые такие методики можно использовать в контексте настоящего изобретения для определения фармакокинетических характеристик комбинации. Затем можно отобрать субъектов, которым будет полезно лечение, предлагаемое в настоящем изобретении. Соответственно, в одном варианте осуществления настоящее изобретение относится к способу введения ингибитора CYP3A4 и соединения, предлагаемого в настоящем изобретении. В другом варианте осуществления настоящее изобретение относится к способу введения ингибитора изофермента 3 А 4("CYP1A2"), изофермента 2 С 9 ("CYP2C9") или изофермента 2 Е 1 ("CYP2E1"). В вариантах осуществления, в которых ингибитором протеазы является VX-950 (или его стереоизомер), ингибитор CYP предпочтительно ингибирует CYP3A4. Также следует учитывать, что активность CYP3A4 у людей наблюдалась во многих случаях. Соответственно, можно ожидать, что варианты осуществления настоящего изобретения, включающие ингибирование изофермента 3 А 4, будут применимы для самых разных пациентов. Соединения, предлагаемые в настоящем изобретении, можно вводить в качестве единственного ингредиента или в комбинации или поочередно с другими противовирусными средствами, предпочтительно средствами, активными по отношению к HCV. При комбинированной терапии эффективные дозы двух или большего количества средств вводят совместно, а при поочередной или последовательной терапии эффективные дозы каждого средства вводят последовательно или одну за другой. Обычно комбинированная терапия предпочтительнее поочередной терапии, поскольку она приводит к нескольким одновременным воздействиям на вирус. Вводимые дозы зависят от скорости всасывания, инактивации и выведения лекарственного средства, а также от других факторов. Следует отметить, что дозы меняются в зависимости от тяжести патологического состояния, которое необходимо облегчить. Также следует понимать, что для каждого конкретного субъекта конкретные режимы дозировки следует регулировать во времени в соответствии с индивидуальной потребностью и решением лица, назначающего или контролирующего назначение композиций. Эффективность соединения для борьбы с вирусной инфекцией можно пролонгировать, дополнить или восстановить путем введения соединения в комбинации или поочередно со вторым и, возможно, третьим противовирусным соединением, которое у резистентного к лекарственному средству вируса приводит к мутации гена, отличающейся от вызываемой основным лекарственным средством. Альтернативно, с помощью такой комбинированной терапии или поочередной терапии можно менять фармакокинетику, биологическое распределение или другие параметры лекарственного средства. Суточные дозы, необходимые для осуществления способа, предлагаемого в настоящем изобретении, меняются в зависимости, например, от использующегося соединения, предлагаемого в настоящем изобретении, реципиента, пути введения, тяжести подвергающегося лечению патологического состояния. Предпочтительный диапазон суточных доз составляет примерно от 1 до 50 мг/кг в сутки в виде разовой дозы или разделенных доз. Для пациентов подходящие суточные дозы составляют примерно от 1 до 20 мг/кг перорально или внутривенно. Для перорального введения подходящая разовая доза составляет примерно от 0,25 до 10 мг/кг активного ингредиента, например, соединения формул (I)-(IV вместе с одним или большим количеством его фармацевтически приемлемых разбавителей или наполнителей(носителей). Количество вспомогательного средства в дозированной форме может меняться значительнее, например от 0,00001 до 1000 мг/кг активного ингредиента. Суточные дозы использующегося вспомогательного средства могут меняться, например, в зависимости от использующегося соединения, реципиента, пути введения и тяжести подвергающегося лечению патологического состояния. Например, ламивудин можно вводить в суточной дозе, равной 100 мг. Пэгилированный интерферон можно вводить парентерально от 1 до 3 раз в неделю, предпочтительно 1 раз в неделю при полной недельной дозе, составляющей от 2 до 10 млн ME, более предпочтительно от 5 до 10 млн ME, наиболее предпочтительно от 8 до 10 млн ME. Поскольку можно применять разные типы вспомогательных средств, их количества могут меняться значительнее, например от 0,0001 до 5000 мг/кг в сутки. Современным стандартом лечения гепатита С является комбинация пэгилированного интерферона альфа с рибавирином, и рекомендованные дозы составляют 1,5 мкг/кг/неделя пэгилированного интерферона альфа-2b или 180 мкг/неделя пэгилированного интерферона альфа-2 а, с прибавлением от 1000 до 1200 мг/сутки рибавирина в течение 48 недель для пациентов с генотипом I или 800 мг/сутки рибавирина в течение 24 недель для пациентов с генотипом 2/3. Соединение, предлагаемое в настоящем изобретении, и вспомогательные средства, предлагаемые в настоящем изобретении, можно вводить любым обычным путем, предпочтительно энтерально, например перорально, например в виде растворов для питья, таблеток или капсул, или парентерально, например, в виде растворов или суспензий для инъекции. Некоторые предпочтительные фармацевтические композиции, например, могут быть основаны на микроэмульсиях, как это описано в UK 2222770 А. Соединение, предлагаемое в настоящем изобретении, вводят вместе с другими лекарственными средствами (вспомогательными средствами), например лекарственным средством, которое обладает противовирусной активностью, предпочтительно активностью против Flaviviridae, наиболее предпочтительно активностью против HCV, например интерфероном, например интерфероном 2 а или интерфероном 2b, например нитроном А, рофероном, авонексом, ребифом или бетафероном, или интерфероном, конъюгированным с растворимым в воде полимером или с альбумином человека, например альбуфероном, противовирусным средством, например рибавирином, ламивудином, соединениями, раскрытыми в патенте US6812219 и WO 2004/002422 А 2 (раскрытия которых во всей своей полноте включены в настоящее изобретение в качестве ссылки), ингибитором HCV или другими факторами, кодирующими вирус Flaviviridae, такими как NS3/4A протеаза, геликаза или РНК полимераза, или пролекарство такого ингибитора, противофиброзным средством, например производным N-фенил-2 пиримидинамина, например иматинибом, иммуномодулирующим средством, например микофеноловой кислотой, ее солью или пролекарством, например микофенолятом натрия или микофенолятом микофенолятом мотефила, или агонистом рецептора S1P, например FTY720 или его аналогом, необязательно фосфорилированным, например, как раскрыто в ЕР 627406 А 1, ЕР 778263 А 1, ЕР 1002792 А 1,WO 02/18395, WO 02/76995, WO 02/06268, JP 2002316985, WO 03/29184, WO 03/29205, WO 03/62252 иWO 03/62248, раскрытия которых во всей своей полноте включены в настоящее изобретение в качестве ссылки. Конъюгаты интерферона с растворимым в воде полимером предпочтительно включают конъюгаты с гомополимерами алкиленоксида, такими как полиэтиленгликоль (ПЭГ) или полипропиленгликоли, полиоксиэтиленированные полиолы, их сополимеры и их блок-сополимеры. В качестве альтернативы полимерам на основе полиалкиленоксидов можно эффективно использовать неантигенные вещества, такие как декстран, поливинилпирролидоны, полиакриламиды, поливиниловые спирты, полимеры на основе углеводов и т.п. Такие конъюгаты интерферон-полимер описаны в патентах US4766106, 4917888, европейской заявке 0236987, европейской заявке 0510356 и международной заявкеWO 95/13090,раскрытия которых во всей своей полноте включены в настоящее изобретение в качестве ссылки. Поскольку модификация полимера значительно снижает антигенный ответ, чужеродный интерферон не должен быть полностью аутологичным. Интерферон, использующийся для приготовления конъюгатов с полимерами, можно приготовить из экстракта интерферона млекопитающего, такого как человек, жвачное животное или крупный рогатый скот, или полученного рекомбинантно. Предпочтительными являются конъюгаты интерферона с полиэтиленгликолем, также известные под названием "пэгилированные интерфероны". Особенно предпочтительными конъюгатами интерферона являются пэгилированные альфаинтерфероны, например пэгилированный интерферон 2a, пэгилированный интерферон 2b; пэгилированный консенсусный интерферон или пэгилированный очищенный интерферон-. Пэгилированный интерферон 2 описан, например, в Европейском патенте 593868 (во всей своей полноте включенном в настоящее изобретение в качестве ссылки) и имеется в продаже, например, под торговым названием ПЕГАСИС (Hoffmann-La Roche). Пэгилированный интерферон 2b описан, например, в Европейском патенте 975369 (во всей своей полноте включенном в настоящее изобретение в качестве ссылки) и имеется в продаже, например, под торговым названием ПЭГ-Интрон-A (Schering Plough). Пэгилированный консенсусный интерферон описан в WO 96/11953 (во всей своей полноте включенной в настоящее изобретение в качестве ссылки). Предпочтительными пэгилированными -интерферонами являются пэгилированный интерферон 2a и пэгилированный интерферон 2b. Также предпочтительным является пэгилированный консенсусный интерферон. Другими предпочтительными вспомогательными средствами являются белки слияния интерферона,например белки слияния интерферона 2a, интерферона 2b; консенсусного интерферона или очищенного интерферона-, каждый из которых слит с другим белком. Некоторые предпочтительные белки слияния включают интерферон (например, интерферон 2b) и альбумин, как описано в патентеUS 6973322 и заявках WO 02/60071, WO 05/003296 и WO 05/077042 (Human Genome Sciences). Предпочтительным интерфероном, конъюгированным с альбумином человека, является альбуферон (Human Genome Sciences). Циклоспорины, которые прочно связываются с циклофилином, но являются неиммуносупрессорными, включают циклоспорины, указанные в патентах US 5767069 и 5981479, и включены в настоящее изобретение в качестве ссылки. [MeIle]4-циклоспорин является предпочтительным неиммуносупрессорным циклоспорином. Некоторые другие производные циклоспорина описаны в WO 2006/039668(Scynexis) и WO 2006/038088 (Debiopharm SA) и включены в настоящее изобретение в качестве ссылки. Циклоспорин считается неиммуносупрессорным, если в реакции смешанной культуры лимфоцитов(РСЛ) он обладает активностью, равной не более 5%, предпочтительно не более 2% от активности циклоспорина А. Реакция смешанной культуры лимфоцитов описана в публикации Т. Meo in "Immunological(0,5106) мышей линии Balb/c (самки, 8-10 недель) в течение 5 дней инкубируют вместе с 0,5106, облученными (2000 рад) или обработанными митомицином С клетками селезенки мышей линии СВА (самки,8-10 недель). Облученные аллогенные клетки индуцируют пролиферативный ответ в клетках селезенкиBalb/c, который можно измерить путем включения в ДНК меченого предшественника. Поскольку стимулирующие клетки облучены (или обработаны митомицином С), они не реагируют на клетки Balb/c с возникновением пролиферации, но сохраняют свою антигенность. Значение IC50, полученное для исследуемого соединения с помощью РСЛ, сопоставляют с полученным для циклоспорина А в параллельном эксперименте. Кроме того, неиммуносупрессорные циклоспорины не способны ингибировать CN и находящийся в прямом направлении путь NF-AT. [MeIle]4-циклоспорин является предпочтительным неиммуносупрессорным связывающим циклофилин циклоспорином для применения в контексте настоящего изобретения. Рибавирин (1D-рибофуранозил-1H-1,2,4-триазол-3-карбоксамид) является синтетическим не индуцирующим интерферон противовирусным аналогом нуклеозида, продающимся под торговым названием виразол (The Merck Index, 11th edition, Editor: Budavar, S., MerckCo., Inc., Rahway, NJ, p. 1304, 1989). В патентах US3798209 и RE29835 (во всей своей полноте включены в настоящее изобретение в качестве ссылки) раскрыт и заявлен рибавирин. Рибавирин структурно сходен с гуанозином и in vitro обладает активностью по отношению к различным ДНК и РНК вирусам, включая Flaviviridae (Gary L. Davis,Gastroenterology, 118:S104-S114, 2000). Рибавирин снижает содержание аминотрансферазы в сыворотке до нормального у 40% пациентов,но не содержание HCV-РНК в сыворотке (Gary L. Davis, Gastroenterology, 118:S104-S114, 2000). Таким образом, по отдельности рибавирин неэффективен для снижения содержания вирусной РНК. Кроме того,рибавирин обладает значительной токсичностью и известно, что он вызывает анемию. Рибавирин не утвержден к применению в монотерапии HCV; он утвержден к применению в комбинации с интерфероном 2 а или интерфероном 2b для борьбы с HCV. Другой предпочтительной комбинацией является комбинация соединения, предлагаемого в настоящем изобретении (например, соединения формулы (I) или любых ее субформул), с неиммуносупрессорным связывающим циклофилин циклоспорином, с микофеноловой кислотой, ее солью или пролекарством и/или агонистом рецептора S1P, например FTY720. Дополнительные примеры соединений, которые можно использовать в комбинированном или поочередном лечении, включают следующие.(f) Сумиферон, очищенная смесь натуральных -интерферонов (Sumitomo, Japan);(k) Консенсусный -интерферон, выпускающийся фирмой Amgen, Inc., Newbury Park, CA. Другие формы интерферона включают интерферон-, ,и , такие как ребиф (интерферон 1 а),выпускающийся фирмой Serono, омниферон (натуральный интерферон), выпускающийся фирмойViragen, РЕБИФ (интерферон 1 а), выпускающийся фирмой Ares-Serono, -интерферон, выпускающийся фирмой BioMedicines; предназначенный для перорального введения интерферон-, выпускающийся фирмой Amarillo Biosciences; интерферон, конъюгированный с растворимым в воде полимером или с альбумином человека, например альбуферон (Human Genome Sciences), противовирусное средство, консенсусный интерферон, интерферон- овец или крупного рогатого скота. Конъюгаты интерферона с растворимым в воде полимером предпочтительно включают конъюгаты с гомополимерами алкиленоксида, такими как полиэтиленгликоль (ПЭГ) или полипропиленгликоли, полиоксиэтиленированные полиолы, их сополимеры и их блок-сополимеры. В качестве альтернативы полимерам на основе полиалкиленоксидов можно эффективно использовать неантигенные вещества, такие как декстран, поливинилпирролидоны, полиакриламиды, поливиниловые спирты, полимеры на основе углеводов и т.п. Поскольку модификация полимера значительно снижает антигенный ответ, чужеродный интерферон не должен быть полностью аутологичным. Интерферон, использующийся для приготовления конъюгатов с полимерами, можно приготовить из экстракта интерферона млекопитающего, такого как человек, жвачное животное или крупный рогатый скот, или полученного рекомбинантно. Предпочтительными являются конъюгаты интерферона с полиэтиленгликолем, также известные под названием "пэгилированные интерфероны".(2) Рибавирин, такой как рибавирин (1D-рибофуранозил-1 Н-1,2,4-триазол-3-карбоксамид), выпускающийся фирмой Valeant Pharmaceuticals, Inc., Costa Mesa, CA); ребетол, выпускающийся фирмойSchering Corporation, Kenilworth, NJ, и копегус, выпускающийся фирмой Hoffmann-La Roche, Nutley,NJ; и новые разрабатывающиеся аналоги рибавирина, такие как левовирин и вирамидин, выпускающиеся фирмой Valeant.(3) Производные тиазолидина, которые при исследовании с помощью ВЭЖХ с обращенной фазой обнаруживают соответствующее ингибирование с белком слияния NS3/4A и субстратом NS5A/5B(Sudo K. et al., Antiviral Research, 1996, 32, 9-18), предпочтительно соединение RD-1-6250, содержащее конденсированный циннамоильный фрагмент, замещенный длинной алкильной цепью, RD4 6205 и RD4 6193.(5) Фенантренхинон, обладающий активностью по отношению к протеазе при исследовании с помощью электрофореза в полиамидном геле с использованием додецилсульфата натрия и при ауторадиографическом исследовании, выделенный из ферментативного культурального бульона Streptomyces sp.,Sch 68631 (Chu M. et al., Tetrahedron Letters, 1996, 37, 7229-7232) и Sch 351633, выделенный из грибовPenicillium griseofulvum, который обнаруживает активность при изучении с помощью сцинтилляционнопроксимального анализа (Chu M. et al., Bioorganic and Medicinal Chemistry Letters, 9, 1949-1952).(6) Ингибиторы протеазы. Изученные примеры включают связанные с субстратом ингибиторы NS3 протеазы (Attwood et al.,Antiviralpeptide derivatives, PCT WO 98/22496, 1998; Attwood et al., Antiviral Chemistry and Chemotherapy,1999, 10, 259-273; Attwood et al., Preparation and use of aminoacid derivatives as anti-viral agents, GermanPCT WO 98/17679), включая альфа-кетоамиды и гидразиномочевины, и ингибиторы, на концах которых расположены электрофилы, такие как бороновая кислота или фосфонат (Llinas-Brunet et al. Hepatitis Cinhibitor peptide analogues, PCT WO 99/07734). Также исследованы несвязанные с субстратом ингибиторы NS3 протеазы, такие как производные 2,4,6-тригидрокси-3-нитробензамида (Sudo K. et al., Biochemical and Biophysical ResearchCommunications, 1997, 238:643-647; Sudo K. et al. Antiviral Chemistry and Chemotherapy, 1998, 9, 186),включая RD3-4082 и RD3-4078, первый замещен по амидной группе цепью, содержащей 14 атомов углерода, и последний подвергает процессингу парафеноксифенильную группу.Sch 68631, фенантренхинон, является ингибитором HCV протеазы (Chu M et al., Tetrahedron Letters,37:7229-7232, 1996). В другом примере тех же авторов Sch 351633, выделенный из грибов Penicilliumgrieofulvum, идентифицирован в качестве ингибитора протеазы (Chu M. et al., Bioorganic and MedicinalChemistry Letters, 9:1949-1952). Активность по отношению к ферменту HCV NS3 протеазе при наномолярной концентрации обеспечена путем выбора селективных ингибиторов, основанных на макромолекуле эглин с. Эглин с, выделенный из пиявок, является активным ингибитором различных серинпротеаз,таких как протеазы А и В S.griseus, -химотрипсин, химаза и субтилизин. Qasim M.A. et al., Biochemistry,36:1598-1607, 1997. Патенты US, в которых раскрыты ингибиторы протеазы, предназначенные для борьбы с HCV,включают, например, патент US6004933, выданный Spruce et al. (во всей своей полноте включенный в настоящее изобретение в качестве ссылки), в котором раскрыт класс ингибиторов цистеинпротеазы для ингибирования HCV эндопептидазы 2; патент US5990276, выданный Zhang et al. (во всей своей полноте включенный в настоящее изобретение в качестве ссылки), в котором раскрыты синтетические ингибиторы NS3 протеазы вируса гепатита С; патент US5538865, выданный Reyes et al. (во всей своей полноте включенный в настоящее изобретение в качестве ссылки). Пептиды в качестве ингибиторов NS3 серинпротеазы HCV раскрыты в WO 02/008251, выданном Corvas International, Inc., и WO 02/08187 иWO 02/008256, выданных Schering Corporation (во всей своей полноте включены в настоящее изобретение в качестве ссылки). Трипептиды-ингибиторы HCV раскрыты в патентах US6534523, 6410531 и 6420380, выданных Boehringer Ingelheim, и WO 02/060926, выданном Bristol Myers Squibb (во всей своей полноте включены в настоящее изобретение в качестве ссылки). Диарилпептиды в качестве ингибиторовNS3 серинпротеазы HCV раскрыты в WO 02/48172, выданном Schering Corporation (включен в настоящее изобретение в качестве ссылки). Имидазолиндиноны, как ингибиторы NS3 серинпротеазы HCV, раскрыты в WO 02/18198, выданном Schering Corporation, и WO 02/48157, выданном Bristol Myers Squibb (во всей своей полноте включены в настоящее изобретение в качестве ссылки). В WO 98/17679, выданномVertex Pharmaceuticals, и WO 02/48116, выданном Bristol Myers Squibb, также раскрыты ингибиторы HCV протеазы (во всей своей полноте включены в настоящее изобретение в качестве ссылки). Ингибиторы NS3-4A серинпротеазы HCV включают BILN 2061, выпускающийся фирмой Boehringer Ingelheim, VX-950, выпускающийся фирмой Vertex, Sch 6/7, выпускающийся фирмой ScheringPlough, и другие соединения, в настоящее время находящиеся на стадии доклинической разработки. Связанные с субстратом ингибиторы NS3 протеазы, включая альфа-кетоамиды и гидразиномочевины, и ингибиторы, на концах которых расположены электрофилы, такие как бороновая кислота или фосфонат; не связанные с субстратом ингибиторы NS3 протеазы, такие как производные 2,4,6-тригидрокси 3-нитробензамида, включают RD3-4082 и RD3-4078, первый замещен по амидной группе цепью, содержащей 14 атомов углерода, и последний подвергает процессингу парафеноксифенильную группу; и Sch 68631, фенантренхинон, ингибитор HCV протеазы.Sch 351633, выделенный из грибов Penicillium grieofulvum, идентифицирован в качестве ингибитора протеазы. Эглин с, выделенный из пиявок, является активным ингибитором различных серинпротеаз,таких как протеазы А и В S.griseus, а-химотрипсин, химаза и субтилизин. В патенте US6004933 (во всей своей полноте включенном в настоящее изобретение в качестве ссылки) раскрыт класс ингибиторов цистеинпротеазы для ингибирования HCV эндопептидазы 2; синтетические ингибиторы NS3 протеазы вируса гепатита С (патент), трипептиды-ингибиторы HCV (патент),диарилпептиды, такие как ингибиторы NS3 серинпротеазы HCV (патент), имидазолиндиноны, как ингибиторы NS3 серинпротеазы HCV (патент). Тиазолидины и бензанилиды (Kakiuchi N. et al. J. FEBS Letters, 421, 217-220; Takeshita N. et al.Analytical Biochemistry, 1997, 247, 242-246). Производные тиазолидина, которые при исследовании с помощью ВЭЖХ с обращенной фазой обнаруживают соответствующее ингибирование с белком слиянияNS3/4A и субстратом NS5A/5B, предпочтительно соединение RD-1-6250, содержащее конденсированный циннамоильный фрагмент, замещенный длинной алкильной цепью, RD4 6205 и RD4 6193. Ингибиторы HCV NS5A, включая BMS-790052, выпускающийся фирмой Bristol-Myers Squibb, и другие соединения, в настоящее время находящиеся в доклинической разработке. Фенантренхинон, обладающий активностью по отношению к протеазе при исследовании с помощью электрофореза в полиамидном геле с использованием додецилсульфата натрия и при ауторадиографическом исследовании, выделенный из ферментативного культурального бульона Streptomyces sp.,Sch 68631 и Sch 351633, выделенный из грибов Penicillium griseofulvum, который обнаруживает активность при изучении с помощью сцинтилляционно-проксимального анализа.(7) Нуклеозидные или ненуклеозидные ингибиторы NS5B РНК-зависимой РНК полимеразы HCV,- 13021554WO 2004/002422 А 2 (во всей своей полноте включенной в настоящее изобретение в качестве ссылки),R803 (Rigel), JTK-003 (Japan Tabacco), HCV-086 (ViroPharma/Wyeth) и соединения, в настоящее время находящиеся на стадии доклинической разработки; глиотоксин (Ferrari R. et al. Journal of Virology, 1999,73, 1649-1654) и натуральный продукт церуленин; 2'-фторнуклеозиды; другие аналоги нуклеозидов, раскрытые в WO 02/057287 А 2, WO 02/057425 А 2, WO 01/90121, WO 01/92282 и патенте US6812219,раскрытие которых во всей своей полноте включено в настоящее изобретение в качестве ссылки. В Idenix Pharmaceuticals описано применение разветвленных нуклеозидов для борьбы с флавивирусами (включая HCV) и пестивирусами, раскрытыми в заявках WO 01/90121 и WO 01/92282 (во всей своей полноте включены в настоящее изобретение в качестве ссылки). Точнее, в Idenix publications описан способ борьбы с инфицированием гепатитом С (и флавивирусами, и пестивирусами) людей и других животных, который включает введение эффективного количества биологически активных 1'-, 2'-, 3'- или 4'-разветвленных B-D или B-L нуклеозидов или их фармацевтически приемлемой соли или пролекарства,по отдельности или в комбинации с другим противовирусным средством, необязательно в фармацевтически приемлемом носителе. Некоторые предпочтительные биологически активные 1'-, 2'-, 3'-, или 4'-разветвленные B-D или B-L нуклеозиды, включая телбивудин, описаны в патентах US6395716 и 6875751, которые включены в настоящее изобретение в качестве ссылки. Другие заявки, в которых раскрыто применение некоторых аналогов нуклеозидов для борьбы с инфицированием вирусом гепатита С, включают РСТ СА 00/01316 (WO 01/32153; поданную 3 ноября 2000 г.) и РСТ/СА 01/00197 (WO 01/60315; поданную 19 февраля 2001 г.), поданную фирмой BioChemCo., Inc., PCT/EP01/09633 (WO 02/18404; опубликованную 21 августа 2001 г.), поданную фирмой Roche,и публикации РСТWO 01/79246 (поданную 13 апреля 2001 г.), WO 02/32920 (поданную 18 октября 2001 г.) и WO 02/48165, выданную Pharmasset, Ltd. (раскрытие которых во всей своей полноте включено в настоящее изобретение в качестве ссылки). В публикации РСТWO 99/43691, выданной Emory University (во всей своей полноте включенной в настоящее изобретение в качестве ссылки), под названием "2'-Fluoronucleosides" раскрыто применение некоторых 2'-фторнуклеозидов для борьбы с HCV. В публикации Eldrup et al. (Oral Session V, Hepatitis С Virus, Flaviviridae; 16th International Conferenceon Antiviral Research (April 27, 2003, Savannah, GA описано соотношение структура-активность для 2'-модифицированных нуклеозидов применительно к ингибированию HCV. В публикации Bhat et al. (Oral Session V, Hepatitis С Virus, Flaviviridae, 2003 (Oral Session V,Hepatitis С Virus, Flaviviridae; 16th International conference on Antiviral Research (April 27, 2003, Savannah,GA); p A75) описаны синтез и фармакокинетические характеристики аналогов нуклеозидов, как возможных ингибиторов репликации HCV РНК. Авторы сообщают, что 2'-модифицированные нуклеозиды проявляют высокую ингибирующую активность при исследовании репликонов с использованием клеток. В публикации Olsen et al. (Oral Session V, Hepatitis С Virus, Flaviviridae; 16th International Conferenceon Antiviral Research (April 27, 2003, Savannah, Gap); A76) также описано влияние 2'-модифицированных нуклеозидов на репликацию HCV РНК.(8) Ингибиторы нуклеотидполимеразы и глиотоксин (Ferrari R. et al. Journal of Virology, 1999, 73,1649-1654) и натуральный продукт церуленин (Lohmann V. et al. Virology, 1998, 249, 108-118).(9) Ингибиторы NS3 геликазы HCV, такие как VP 50406, выпускающийся фирмой ViroPhama, и соединения, выпускающиеся фирмой Vertex. Другие ингибиторы геликазы (Diana G.D. et al., Compounds,compositions and methods for treatment of hepatitis С, патент US5633358 (во всей своей полноте включенный в настоящее изобретение в качестве ссылки); Diana G.D. et al., Piperidine derivatives, pharmaceutical compositions there of and their use in the treatment of hepatitis С, РСТ WO 97/36554).(10) Антисмысловые фосфоротиоатолигодезоксинуклеотиды (S-ODN), комплементарные к последовательности участков в 5'-некодирующем регионе (NCR) вируса (Alt M. et al., Hepatology, 1995, 22,707-717), или нуклеотиды 326-348, включающие 3'-конец NCR, и нуклеотиды 371-388, расположенные в области кодирования ядра HCV РНК (Alt M. et al., Archives of Virology, 1997, 142, 589-599; Galderisi U. etPatent Pub. JP-10101591); такие как ISIS 14803, выпускающийся фирмой Isis Pharm/Elan, ингибитор IRES,выпускающийся фирмой Anadys, ингибиторы IRES, выпускающиеся фирмой Immusol, воздействующие на РНК химические вещества, выпускающиеся фирмой РТС Therapeutics.(12) Рибозимы, такие как стойкие к нуклеазе рибозимы (Maccjak, D.J. et al., Hepatology, 1999, 30,abstract 995) и указанные в патенте US6043077, выданном Barber et al., и в патентах US5869253 и 5610054, выданных Draper et al. (во всей своей полноте включены в настоящее изобретение в качестве(13) si-РНК (малые интерферирующие РНК), воздействующие на геном HCV.(14) Ингибиторы репликации HCV, действующие по любым другим механизмам, такие как выпускающиеся фирмой VP50406ViroPharma/Wyeth, ингибиторы, выпускающиеся фирмой Achillion, Arrow.(15) Ингибиторы других мишеней цикла жизни HCV, включая вхождение вируса, сборку и мейоз.(16) Иммуномодулирующее средство, такое как ингибитор IMPDH, микофеноловая кислота, ее соль или пролекарство, микофенолят натрия или микофенолят мотефила или меримебодиб (VX-497); тимозин альфа-1 (Zadaxin, выпускающийся фирмой SciClone); или агонист рецептора S1P, например FTY720 или его аналог, необязательно фосфорилированный.(17) Противофиброзное средство, такое как производное N-фенил-2-пиримидинамина, иматиниб(Chron-VacC), иммунотерапевтическое средство (терапор), выпускающееся фирмой Avant, средство для терапии Т-клеток, выпускающееся фирмой CellExSys, моноклональные антитела XTL-002, выпускающиеся фирмой STL, ANA 246 и ANA 246, выпускающиеся фирмой Anadys.(19) Различные другие соединения включают 1-аминоалкилциклогексаны (патент US6034134,выданный Gold et al.), алкиллипиды (патент US5922757, выданный Chojkier et al.), витамин Е и другие антиоксиданты (патент US5922757, выданный Chojkier et al.), амантадин, желчные кислоты (патентal.), производные полиадениловой кислоты (патент US5496546, выданный Wang et al.),2',3'-дидезоксиинозин (патент US5026687, выданный Yarchoan et al.), бензимидазолы (патент US5891874, выданный Colacino et al.), экстракты растений (патент US5837257, выданный Tsai et al.,патент US5725859, выданный Omer et al., и патент US6056961) и пиперидины (патент US5830905, выданный Diana et al.); раскрытия которых во всей своей полноте включены в настоящее изобретение в качестве ссылки, а также сквален, телбивудин, N-(фосфоноацетил)-L-аспарагиновая кислота, бензолдикарбоксамиды, производные полиадениловой кислоты, ингибиторы гликозилирования и неспецифические цитопротекторные средства, которые блокируют повреждение клетки, вызванное вирусной инфекцией.(20) Любое другое соединение, в настоящее время находящееся на стадии доклинической или клинической разработки, предназначенное для борьбы с HCV, включая интерлейкин-10 (Schering-Plough),амантадин (симметрел), выпускающийся фирмой Endo Labs Solvay, ингибитор каспазы IDN-6556, выпускающийся фирмой Idun Pharma, HCV/MF59, выпускающийся фирмой Chiron, цивацир (иммуноглобулин гепатита С), выпускающийся фирмой NABI, цеплен (гистаминдихлорид), выпускающийся фирмойMaxim, IDN-6556, выпускающийся фирмой Idun PHARM, T67, ингибитор -тубулина, выпускающийся фирмой Tularik, терапевтическая вакцина для Е 2, выпускающаяся фирмой Innogenetics, FK788, выпускающийся фирмой Fujisawa Helathcare, IdB1016 (силифос, силубинфосфатидилхолинфитосома для перорального введения), ингибитор слияния, выпускающийся фирмой Trimeris, дикатион, выпускающийся фирмой Immtech, средство для очистки крови, выпускающееся фирмой Aethlon Medical, UT 231 В, выпускающийся фирмой United Therapeutics.(21) Пуриннуклеоизидный аналог антагониста T1R7 (toll-подобные рецепторы), разработанные фирмой Anadys, например изоторабин (ANA245) и его пролекарство (ANA975), которые описаны в европейских заявках ЕР 348446 и ЕР 636372, международных заявках WO 03/045968, WO 05/121162 и(22) Другие вспомогательные средства (например, неиммуномодулирующие или иммуномодулирующие соединения), которые можно применять в комбинации с соединением, предлагаемым в настоящем изобретении, включают, но не ограничиваются только ими, указанные в WO 02/18369, которая включена в настоящее изобретение в качестве ссылки. Соединения, предлагаемые в настоящем изобретении, также могут вводиться с другим компонентом, включающим дополнительное средство, выбранное из группы, включающей иммуномодулирующее средство; противовирусное средство; ингибитор протеазы HCV; ингибитор другой мишени цикла жизниHCV; ингибитор CYP или их комбинации. Соответственно, соединения, предлагаемые в настоящем изобретении, могут быть введены с другим противовирусным средством, предпочтительно средством против HCV. Такие противовирусные средства включают, но не ограничиваются только ими, иммуномодулирующие средства, такие как -, -,и -интерфероны, пэгилированные производные интерферона- и тимозин; другие противовирусные средства, такие как рибаривин, амантадин и телбувидин; другие ингибиторы протеаз гепатита С (ингиби- 15021554 торы NS2-NS3 и ингибиторы NS3-NS4A); ингибиторы других мишеней цикла жизни HCV, включая ингибиторы геликазы, полимеразы и металлопротеазы; внутренние ингибиторы входа рибосомы; ингибиторы вирусов широкого спектра действия, такие как ингибиторы IMPDH (например, соединения, приведенные в патентах США 5807876, 6498178, 6344465, 6054472, WO 97/40028, WO 98/40381,WO 00/56331, и микофеноловая кислота и ее производные, включая, но не ограничиваясь только ими,VX-497, VX-148, и/или VX-944); или комбинации любых из приведенных выше соединений. Соединения могут быть введены совместно, например одновременно или последовательно, в терапевтически эффективном количестве вместе со вспомогательным средством, например второе лекарственное средство, определенное выше. Термины "совместное введение" или "комбинированное введение" и т.п. при использовании в настоящем изобретении означают введение выбранных терапевтических средств одному пациенту и включают режимы лечения, при которых средства вводятся необязательно по одному и тому же пути введения или в одно и то же время. Введение фармацевтической комбинации приводит к благоприятному эффекту, например синергетическому терапевтическому эффекту, в отличие от монотерапии с использованием только одного из входящих в нее фармацевтически активных ингредиентов. Каждый компонент комбинации, предлагаемой в настоящем изобретении, можно вводить по отдельности, совместно или в любой их комбинации. Как известно опытным лечащим врачам, дозы интерферона обычно выражают в Международных единицах (ME) (например, от примерно 4 до примерно 12 млн ME). Если дополнительное средство выбрано из числа других ингибиторов CYP, то способ будет включать применение двух или большего количества ингибиторов CYP. Каждый компонент можно вводить в одной или большем количестве дозированных форм. Дозированные формы можно вводить пациенту в любой последовательности. Соединение, предлагаемое в настоящем изобретении, и любое дополнительное средство можно включить в разные дозированные формы. Альтернативно, для уменьшения количества дозированных форм, вводимых пациенту, соединение, предлагаемое в настоящем изобретении, и любое дополнительное средство можно приготовить совместно в любой комбинации. Например, соединение, предлагаемое в настоящем изобретении, являющееся ингибитором, можно включить в одну дозированную форму и дополнительное средство можно включить совместно в другую дозированную форму. Любые отдельные дозированные формы можно вводить в одно и то же время или в разные моменты времени. Альтернативно, композиция, предлагаемая в настоящем изобретении, включает дополнительное средство, описанное в настоящем изобретении. Каждый компонент может содержаться в отдельных композициях, комбинациях композиций или в одной композиции. Применение для связанных с HCV нарушений. Соединения, предлагаемые в настоящем изобретении, обладают ценными фармакологическими характеристиками и применимы для лечения заболеваний. В некоторых вариантах осуществления соединения, предлагаемые в настоящем изобретении, применимы для лечения связанных с HCV нарушений, например, в качестве лекарственных средств для лечения инфицирования посредством HCV. Термин "применение" включает любой один или большее количество следующих вариантов осуществления настоящего изобретения: применение для лечения связанных с HCV нарушений; применение для приготовления фармацевтических композиций, предназначенных для применения для лечения этих заболеваний, например для приготовления лекарственного средства; способы применения соединений,предлагаемых в настоящем изобретении, для лечения этих заболеваний; фармацевтическим препараты,содержащие соединения, предлагаемые в настоящем изобретении, предназначенные для лечения этих заболеваний; и соединения, предлагаемые в настоящем изобретении, предназначенные для применения для лечения этих заболеваний; если это является целесообразным и подходящим, если не указано иное. В частности, подвергающиеся лечению заболевания и, таким образом, являющиеся предпочтительными для применения соединения, предлагаемого в настоящем изобретении, выбраны из числа связанных сHCV нарушений, включая соответствующие инфицированию посредством HCV, а также заболевания,которые зависят от активности одного или большего количества из белков NS3, NS4A, NS4B, NS5A иNS5B или комплекса NS3-NS4A, NS4A-NS4B, NS4B-NS5A или NS5A-NS5B. Термин "применение" также включает варианты осуществления композиций, предлагаемых в настоящем изобретении, которые эффективно связывают белок HCV, так что они могут выступать в качестве маркировки или метки, так что при связывании с флуоресцентной или обычной меткой или при приобретении радиоактивности их можно использовать в качестве реагента для научных исследований или в качестве диагностирующего или визуализирующего реагента. В некоторых вариантах осуществления соединение, предлагаемое в настоящем изобретении, применяют для лечения связанных с HCV заболеваний, и соединение, предлагаемое в настоящем изобретении, применяют в качестве ингибитора любого одного или большего количества HCV. Подразумевается,что применение может представлять собой борьбу с одним или большим количеством штаммов HCV или их подавление. Анализы. Подавление активности HCV можно исследовать с помощью целого ряда анализов, использующихся в данной области техники. Пример такого анализа приведен в публикации Anal Biochem. 1996, 240(1): 60-7; которая во всей своей полноте включена в настоящее изобретение в качестве ссылки. Исследования по определению активности HCV также описаны в представленном ниже экспериментальном разделе. Методика синтеза. Соединения, предлагаемые в настоящем изобретении, получают из общедоступных соединений по методикам, известным специалистам в данной области техники, включая любое одно или большее количество указанных ниже условий, но не ограничиваются только ими. В объеме настоящего описания только легко удаляющаяся группа, которая не является компонентом конкретных конечных искомых соединений, предлагаемых в настоящем изобретении, называется"защитной группой", если из контекста не следует иное. Защита функциональных групп такими защитными группами, сами защитные группы и реакции для их отщепления описаны, например, в стандартных справочниках, таких как, например, Science of Synthesis: Houben-Weyl Methods of MolecularVerlag, Stuttgart 1974. Характеристикой защитных групп является то, что их можно удалить легко (т.е. без протекания нежелательных вторичных реакций), например, путем сольволиза, восстановления, фотолиза или, альтернативно, при физиологических условиях (например, путем ферментативного отщепления). Смеси изомеров, получаемые в контексте настоящего изобретения, по стандартной методике можно разделить на отдельные изомеры; диастереоизомеры можно разделить, например, путем распределения в многофазной смеси растворителей, перекристаллизацией и/или с помощью хроматографического разделения, например, на силикагеле или, например, с помощью жидкостной хроматографии среднего давления на колонке с обращенной фазой; и рацематы можно разделить, например, путем образования солей с оптически чистыми солеобразующими реагентами, и получаемую таким образом смесь диастереоизомеров разделить, например, с помощью фракционной кристаллизации или с помощью хроматографии на колонках с оптически активными веществами. Промежуточные и конечные продукты можно обработать и/или очистить по стандартным методикам, например, с помощью хроматографических методик, методик распределения, (пере)кристаллизации и т.п. Общие условия осуществления способа. Приведенные ниже положения применимы в целом ко всем способам, указанным в настоящем изобретении. Стадии способа синтеза соединений, предлагаемых в настоящем изобретении, можно выполнить при условиях проведения реакций, которые сами по себе известны, предпочтительно при специально указанных, при отсутствии или обычно в присутствии растворителей или разбавителей, включая, например, растворители или разбавители, которые инертны по отношению к используемым реагентам и растворяют их, при отсутствии или в присутствии катализаторов, конденсирующих или нейтрализующих реагентов, например ионообменников, таких как катионообменники, например, в Н+ форме; в зависимости от природы реакции и/или реагентов при пониженной, нормальной или повышенной температуре,например при температуре от примерно -100 до примерно 190 С; включая, например, от примерно -80 до примерно 150 С, например от -80 до -60 С, при комнатной температуре, от -20 до 40 С или при температуре кипения; при атмосферном давлении или в закрытом сосуде, когда это целесообразно, под давлением и/или в инертной атмосфере, например в атмосфере аргона или азота. На всех стадиях реакций образующиеся смеси изомеров можно разделить на отдельные изомеры,например диастереоизомеры или энантиомеры; или на любую необходимую смесь изомеров, например рацематы или смеси диастереоизомеров, например, по методикам, аналогичным описанным в публикации Science of Synthesis: Houben-Weyl Methods of Molecular Transformation. Georg Thieme Verlag,Stuttgart, Germany. 2005. Растворители, из числа которых можно выбрать растворители, которые являются подходящими для любой конкретной реакции, включают указанные специально или, например, воду, сложные эфиры, такие как (низш.) алкил-(низш.) алканоаты, например этилацетат, простые эфиры, такие как алифатические простые эфиры, например диэтиловый эфир, или циклические простые эфиры, например тетрагидрофуран и диоксан, жидкие ароматические углеводороды, такие как бензол и толуол, спирты, такие как мета- 17021554 нол, этанол и 1- или 2-пропанол, нитрилы, такие как ацетонитрил, галогенированные углеводороды, такие как метиленхлорид и хлороформ, амиды кислот, такие как диметилформамид и диметилацетамид,основания, такие как гетероциклические азотистые основания, например пиридин илиN-метилпирролидин-2-он, ангидриды карбоновых кислот, такие как ангидриды (низш.) алкановых кислот, например уксусный ангидрид, циклические, линейные или разветвленные углеводороды, такие как циклогексан, гексан и изопентан, или смеси этих растворителей, например водные растворы, если в описании способов не указано иное. Такие смеси растворителей также можно использовать при обработке,например, с помощью хроматографии или распределения. Соединения, включая их соли, также можно получить в виде гидратов или их кристаллы могут, например, включать растворитель, применяющийся для кристаллизации. Могут содержаться разные кристаллические формы. Настоящее изобретение также относится к таким вариантам осуществления способа, в которых получаемое на любой стадии в качестве промежуточного продукта соединение применяется в качестве исходного вещества, а затем выполняются остальные стадии способа или в которых исходное вещество образуется при условиях проведения реакции или применяется в виде производного, например, в защищенной форме или в форме соли, или соединение, получаемое способом, предлагаемым в настоящем изобретении, получается при условиях проведения реакции и затем обрабатывается in situ. Примеры осуществления настоящего изобретения Настоящее изобретение дополнительно иллюстрируется приведенными ниже примерами, которые не следует рассматривать, в качестве ограничивающих объем настоящего изобретения. Методики анализа, применяющиеся в примерах, являются общепринятыми. Демонстрирование эффективности этих анализов рассматривается в качестве предсказания эффективности для субъектов. Общие методики синтеза. Все исходные вещества, структурные фрагменты, реагенты, кислоты, основания, дегидратирующие реагенты, растворители и катализаторы, использующиеся для синтеза соединений, предлагаемых в настоящем изобретении, имеются в продаже или их можно получить по методикам органического синтеза,известным специалисту с общей подготовкой в данной области техники (Houben-Weyl, 4th Ed. 1952,Methods of Organic Synthesis, Thieme, Volume 21). Кроме того, соединения, предлагаемые в настоящем изобретении, можно получить по методикам органического синтеза, известным специалисту с общей подготовкой в данной области техники, приведенным в представленных ниже примерах. Перечень аббревиатур: Ас - ацетил; АЦН - ацетонитрил;Pd/C - палладий на древесном угле;CH3CN, 4,55-5 мин 20% CH3CN. Методика J: МС. Прибор: Agilent system. Методика: проточно-инжекционная. Детектирование: API-ES, положительный/отрицательный. Методика K: Препаративная ВЭЖХ. Прибор: Gilson system. Колонка: Waters C18 ODB, 5 мкм, 5019 мм. Растворитель: CH3CN (0,1% НСО 2 Н); Н 2 О (0,1% НСО 2 Н). Методика L: Препаративная ВЭЖХ. Прибор: Gilson. Колонка: Sun-Fire prep C18 OBD 5 мкм, колонка 1950 мм (скорость потока 20 мл/мин) или колонка 30100 мм (скорость потока 40 мл/мин). Растворитель: CH3CN (0,1% CF3CO2H) и Н 2 О (0,1% CF3CO2H). Градиентный режим: 0-20 мин: 5-100% CH3CN. Методика М. ЖХСД-МС (ЖХСД - жидкостная хроматография сверхвысокого давления). Прибор: Waters. Колонка: Waters Atlantis, 2,130 мм, скорость потока 0,6 мл/мин. Растворитель: CH3CN (0,1% НСО 2 Н), Н 2 О (0,1% НСО 2 Н). Градиентный режим: 0-2,5 мин: 20-95% CH3CN, 2,5-4,5 мин: 95% CH3CN, 4,5-4,55 мин от 95 до 20%CH3CN, 4,55-5 мин 20% CH3CN. Получение промежуточного продукта I:Winum et al. (Organic Letters, 2001, 3, 2241) в ДХМ (24 мл) обрабатывали пирролидином (0,864 мл; 10,453 ммоль) и перемешивали при КТ в течение 24 ч. Реакционную смесь хроматографировали с помощью ФХ на силикагеле ДХМ (450 мл) обрабатывали с помощью ТФК (120 мл; 1,56 моль) и перемешивали при КТ в течение 7 ч. Реакционную смесь концентрировали в вакууме и оставшееся масло растирали с диизопропиловым эфиром. Полученный порошок промывали диизопропиловым эфиром, сушили в высоком вакууме и получали соединение 1 а. ТСХ: Rf (ДХМ/EtOAc 50:1) = 0,10. Стадия 1b. Раствор (1R,2S)-1-трет-бутоксикарбониламино-2-винилциклопропанкарбоновой кислоты, полученной в соответствии с процедурой, описанной в WO 2000/09558 (8,24 г; 36,3 ммоль), в ТГФ (160 мл) обрабатывали с помощью КДИ (9,09 г; 54,4 ммоль) и кипятили с обратным холодильником в течение 1 ч. Полученную реакционную смесь охлаждали до КТ и обрабатывали соединением 1 а (7,62 г; 50,8 ммоль),затем с помощью ДБУ (8,28 г; 54,4 ммоль). Через 16 ч при КТ реакционную смесь концентрировали, остаток переносили в ДХМ и промывали с помощью насыщенным водным раствором KHSO4 (3). Водные фазы экстрагировали с помощью ДХМ, органические вещества объединяли, сушили над Na2SO4 и концентрировали в вакууме. Остаток хроматографировали на силикагеле (элюент: гексан/EtOAc 4:1) и получали соединение 1b. ЖХМС (методика F) Rt = 3,21 мин; МС (методика J): m/z = 358 [М-1]. Стадия 1 с. Соединение 1b (7,84 г; 21,81 ммоль) обрабатывали с помощью 4 н. HCl в диоксане (84 мл) при КТ. Через 1,5 ч реакционную смесь концентрировали в высоком вакууме и получали соединение 1 с в виде гидрохлорида. ЖХМС (методика Е) Rt = 1,10 мин; МС (методика J): m/z = 260 [М+1]. Стадия Id. 7-трет-Бутиловый эфир (5R,8S)-10,10-диметил-7-азадиспиро[3.0.4.1]декан-7,8-дикарбоновой кислоты получали в соответствии с процедурой, приведенной на стр. 113, строка 12 - стр. 114, строка 7 в находящейся одновременно на рассмотрении заявке на международный патент РСТ/ЕР 08/063460, которая явно включена в настоящее изобретение в качестве ссылки. Раствор гидрохлорида соединения 1 с смесь перемешивали при 0 С в течение 1 ч и при КТ в течение 19 ч, подвергали распределению между водой и EtOAc. Органические вещества последовательно промывали насыщенным водным растворомKHSO4, NaCO3 и водой, сушили над Na2SO4 и концентрировали в вакууме. Остаток хроматографировали на силикагеле (элюент: ДХМ/МеОН 50:1) и получали соединение Id. Гидрохлорид соединения 1 е получали из соединения 1d (0,296 г; 0,537 ммоль) по методике, описанной на стадии 1 с.(15 мл) охлаждали до 0C и обрабатывали с помощью ДИПЭА (0,46 мл; 2,68 ммоль) и HATU (0,611 г; 1,608 ммоль). Реакционную смесь перемешивали при КТ в течение 20 ч, концентрировали в вакууме и остаток очищали с помощью препаративной ВЭЖХ (методика K). После обработки (обработка 2 = фракции обрабатывали с помощью NaHCO3 и концентрировали; остаток подвергали распределению между водой и EtOAc, экстрагировали с помощью EtOAc; органические вещества объединяли, сушили над Гидрохлорид соединения lg получали из соединения If (0,204 г; 0,307 ммоль) по методике, описанной на стадии lc. Раствор соединения lg (1,845 г; 3,07 ммоль) и BOC-L-циклогексилглицина (1,582 г; 6,15 ммоль) в ДХМ (65 мл) охлаждали до 0 С и обрабатывали с помощью ДИПЭА (2,68 мл; 15,37 ммоль), затем с помощью HATU (3,51 г; 9,22 ммоль). Через 16 ч при КТ реакционную смесь подвергали распределению между ДХМ и 1 н. HCl, органические вещества экстрагировали насыщенным водным раствором NaHCO3,сушили над Na2SO4 и концентрировали. Очистка с помощью препаративной ВЭЖХ (методика K) и последующая обработка (обработка 2) давали соединение 1h. ЖХ-МС (методика Е): Rt = 3,18 мин; M/z = 826 [M+Na]. Стадия 1i. Промежуточный гидрохлорид I получали из соединения 1h (1,64 г; 2,042 ммоль) по методике, описанной на стадии 1 с ЖХ-МС (методика Е): Rt = 1,95 мин; M/z = 703 [M+1]. Получение промежуточного продукта II. 7-трет-Бутиловый эфир (5R,8S)-10,10-диметил-7-азадиспиро[3.0.4.1]декан-7,8-дикарбоновой кислоты (32,84 г; 106 ммоль) в ДМФ (1 л) обрабатывали с помощью K2CO3 (22,00 г; 159 ммоль), затем метилйодидом (9,93 мл; 159 ммоль). Реакционную смесь перемешивали при КТ в течение 18 ч, концентрировали в вакууме. Полученный остаток подвергали распределению между водой и EtOAc и экстрагировали с помощью EtOAc. Органические вещества объединяли, промывали рассолом, сушили над Na2SO4 и концентрировали. Остаток хроматографировали на силикагеле (элюент ДХМ/этиловый эфир 120:1) и получали соединение 2 а. ТСХ: Rf (ДХМ/этиловый эфир 120:1) = 0,22; МС (методика J): m/z = 346 [M+Na]. Стадия 2b. Гидрохлорид соединения 2b получали из соединения 2 а (6,3 г; 19,48 ммоль) по методике, описанной на стадии 1 с. МС (методика J): m/z = 224 [М+1]. Соединение 2 с получали из гидрохлорида соединения 2b (7,33 г; 27,93 ммоль) по методике, описанной на стадии 1f, затем очищали с помощью хроматографии на силикагеле (элюент циклогексан/EtOAc 1:1). ТСХ: Rf (гексан/EtOAc 4:1) = 0,37; МС (методика J): m/z = 437 [М+1]. Стадия 2d. Гидрохлорид соединения 2d получали из соединения 2 с (10,55 г; 24,16 ммоль) по методике, описанной на стадии 1c. Соединение 2 е получали из соединения 2d (0,2 г; 0,456 ммоль) по методике, описанной на стадии 1h. ЖХ-МС (методика G): Rt = 2,21 мин; M/z = 598 [M+Na]. Стадия 2f.(6 мл; 2:1:1) перемешивали при КТ 16 ч. Реакционную смесь подвергали распределению между водой иEtOAc. Водную фазу подкисляли с помощью 1 н. HCl и экстрагировали с помощью EtOAc. Органические вещества объединяли, сушили над Na2SO4, концентрировали и получали остаток, который хроматографировали на силикагеле (ДХМ/МеОН 100 до 9:1) и получали соединение 2f.(2 мл) охлаждали до 0 С и обрабатывали с помощью ДИПЭА (0,078 мл; 0,445 ммоль) и HATU (0,102 г; 0,267 ммоль). Реакционную смесь перемешивали при КТ в течение 2 ч, подвергали распределению между ДХМ и 1 н. HCl. Органические вещества промывали насыщенным водным раствором NaHCO3, сушили над Na2SO4, концентрировали в вакууме и получали остаток, который очищали с помощью препаративной ВЭЖХ. После обработки (обработка 2) получали соединение 2g. ЖХ-МС (методика G): Rt = 2,25 мин; M/z = 828 [M+Na]. Стадия 2h. Промежуточный гидрохлорид II получали из соединения 2g (0,02 г; 0,025 ммоль) по методике, описанной на стадии 1 с. ЖХ-МС (методика G): Rt = 1,59 мин; M/z = 706 [М+1]. Получение промежуточного продукта III. 1R,2R)-1-Амино-2-этилциклопропанкарбонил)амида пирролидин-1-сульфоновой кислоты: Раствор метилового эфира (1R,2S)-1-трет-бутоксикарбониламино-2-винилциклопропанкарбоновой кислоты получали в соответствии с процедурой, описанной в WO 20009558, (60,16 г; 249 ммоль) в 2 лEtOH гидрировали при КТ в атмосфере Н 2 при катализе с помощью Rh-Al2O3 (5 г). После завершения катализатор отфильтровывали, раствор концентрировали в высоком вакууме и получали соединение 3 а. МС (методика J): m/z = 266 [M+Na]. Стадия 3b.(440 мл; 2:1:1) перемешивали при КТ в течение 20 ч. Реакционную смесь концентрировали. Полученную водную фазу промывали с помощью EtOAc, охлаждали до 5 С, подкисляли с помощью 6 н. HCl и экстра- 26021554 гировали с помощью EtOAc. Органические вещества объединяли, сушили над Na2SO4, и концентрировали и получали остаток, который кристаллизовали из циклогексана, и получали соединение 3b. ЖХ-МС (методика G): Rt = 1,40 мин; M/z = 252 [M+Na]. Стадия 3 с. Соединение 3 с получали из соединения 3b (5 г; 21,81 ммоль) по методике, описанной на стадии 1b. ЖХ-МС (методика D): Rt = 1,91 мин; M/z = 360 [М-1]. Стадия 3d. 1R,2R)-1-Амино-2-этилциклопропанкарбонил)амидгидрохлорид пирролидин-1-сульфоновой кислоты получали из соединения 3 с (4,602 г; 12,73 ммоль) по методике, описанной на стадии 1 с. ЖХ-МС (методика Е): Rt = 1,06 мин; M/z = 262 [М+1]. Пример 1. Соединение 13 Суспензию (S)-1-изопропилпиперидин-2-карбоновой кислоты (1,39 г; 8,11 ммоль) в ДМФ (150 мл) обрабатывали с помощью HATU (3,86 г; 10,14 ммоль) и ДИПЭА (3,54 мл; 20,29 ммоль) и перемешивали при КТ. Полученный раствор обрабатывали гидрохлоридом промежуточного продукта I (5 г; 6,76 ммоль) и перемешивали при КТ в атмосфере аргона в течение 1 ч. Реакционную смесь переносили в EtOAc,промывали водой. Водную фазу экстрагировали с помощью EtOAc. Органические вещества объединяли,промывали насыщенным водным раствором NaHCO3, сушили над Na2SO4, и концентрировали и получали коричневое масло. Очистка с помощью ФХ на силикагеле (элюент: от циклогексана до смеси циклогексан/ацетон 3:2) давало соединение 1 в виде вспененного вещества. ТСХ: Rf (циклогексан/ацетон 3:2) = 0,23; ЖХ-МС (методика Е): m/z = 856 [М+1]; ВЭЖХ (методика D): Rt = 2,13 мин; 1 Н-ЯМР (500 МГц, ДМСО-d6):(част./млн) = 10,2 (s, 1 Н), 9,6 (bs, 1H), 8,9 (d, 1H), 8,8 (s, 1H), 8,0 (d,1H), 5,5 (dt, 1H), 5,2 (d, 1H), 5,1 (d, 1 Н), 4,55 (d, 1 Н), 4,35 (t, 1 Н), 4,15 (t, 1H), 4,05 (t, 1 Н), 3,4-3,65 (m, 5 Н),3,3 (m, 4 Н), 3,1 (m, 2 Н), 2,85 (bs, 1 Н), 2,5 (t, 1 Н), 2,15 (m, 1 Н), 0,9-2,0 (m, 42 Н), 0,85 (s, 3H), 0,8 (s, 3 Н). Пример 2. Соединение 34 Гидрохлорид соединения 2 получали из промежуточного продукта II (0,28 г; 0,378 ммоль) по методике, описанной для получения соединения 1. ВЭЖХ (методика В): Rt = 3,70 мин; МС (методика J): m/z = 858 [М+1]; 1 Н-ЯМР (400 МГц, метанол-d4):(част./млн) = 8,4 (d, 1H), 4,75 (d, 1H), 4,3 (d, 1H), 4,2 (t, 1 Н), 3,95(bs, 1 Н), 3,4-3,7 (m, 9 Н), 3,0 (m, 1H), 2,15 (m, 1H), 1,05-2,1 (m, 43 Н), 1,05 (s, 9H), 0,9 (s, 3 Н), 0,95 (s, 3 Н). Следующие соединения получали аналогичным образом. Пример 3. Соединение 99(0,61 ммоль) ДИПЭА в 4 мл ДМФ перемешивали при КТ в течение 1 ч. Реакционную смесь разбавляли с помощью ДХМ и промывали 10% водным раствором KHSO4. Водный слой экстрагировали с помощью ДХМ (3) и объединенные органические слои промывали насыщенным водным раствором NaHCO3, сушили над Na2SO4 и концентрировали в вакууме. Неочищенный продукт очищали с помощью препаративной ВЭЖХ и получали искомое соединение. ЖХ-МС (методика Е): Rt = 1,862 мин; Раствор (S)-трет-бутоксикарбониламино(1-метилциклогексил)уксусной кислоты (0,217 г; 0,80 ммоль) (получали в соответствии с Tetrahedron Let. 2007, 48, 6343-6347) и HATU (0,38 г; 1,00 ммоль) в ДХМ (10 мл) перемешивали в течение 20 мин при температуре окружающей среды. После добавления ДИПЭА (0,698 мл; 4,00 ммоль) и [(1R,2S)-1-(пирролидин-1-сульфониламинокарбонил)-2 винилциклопропил]амида(5R,8S)-7-S)-2-амино-3,3-диметилбутирил)-10,10-диметил-7-азадиспиро[3.0.4.1]декан-8-карбоновой кислоты (0,400 г; 0,67 ммоль) в ДХМ (10 мл) реакционную смесь перемешивали в течение 15 ч, растворитель удаляли в вакууме, остаток очищали с помощью препаративной ВЭЖХ Смесь трет-бутилового эфира [(S)-S)-l-(5R,8S)-10,10-диметил-8-[(1R,2S)-1-(пирролидин-1 сульфониламинокарбонил)-2-винилциклопропилкарбамоил]-7-азадиспиро[3.0.4.1]декан-7-карбонил-2,2 диметилпропилкарбамоил)-(1-метилциклогексил)метил]карбаминовой кислоты (0,219 г; 0,27 ммоль) и 2,0 мл HCl (4 M в диоксане) в 2 мл диоксана перемешивали при температуре окружающей среды в течение 2 ч. Смесь концентрировали при пониженном давлении, 2 раза выпаривали вместе с ДХМ и получали искомое соединение, которое использовали на следующей стадии без дополнительной очистки. ЖХ-МС (методика Е): Rt = 1,64 мин; M/z = 717 [М+]; ВЭЖХ (методика A3): Rt = 4,82 мин. Стадия с. Раствор (S)-1-изопропилпиперидин-2-карбоновой кислоты (0,014 г; 0,080 ммоль) и HATU (0,038 г; 0,100 ммоль) в ДМФ (2 мл) перемешивали в течение 30 мин при температуре окружающей среды. После добавления ДИПЭА (0,070 мл; 0,398 ммоль) и [(1R,2S)-1-(пирролидин-1-сульфониламинокарбонил)-2 винилциклопропил]амида 5R,8S)-7-(S)-2-[(S)-2-амино-2-(1-метилциклогексил)ацетиламино]-3,3 диметилбутирил-10,10-диметил-7-азадиспиро[3.0.4.1]декан-8-карбоновой кислоты(0,050 г; 0,066 ммоль) реакционную смесь перемешивали в течение 3 ч, без обработки очищали с помощью препаративной ВЭЖХ (методика L) и получали искомое соединение. ВЭЖХ (методика A3) Rt = 5,40 мин; МС (методика Е): m/z = 870 [М+]; 1 Н-ЯМР (500 МГц, ДМСО-d6): 0,82 (s, 3 Н), 0,83 (s, 3 Н), 0,91 (s, 3 Н), 0,94 (s, 9 Н), 1,15-1,35 (m, 11 Н),1,39-1,55 (m, 6 Н), 1,65-1,85 (m, 12 Н), 2,09-2,17 (m, 1 Н), 2,79-2,86 (m, 1 Н), 3,24-3,36 (m, 6 Н), 3,23-3,36 (m,6 Н), 3,40-3,51 (m, 3 Н), 3,53-3,55 (m, 2 Н), 4,52-4,58 (m, 2 Н), 5,15 (dd, 2 Н), 5,50 (ddd, 1 Н), 8,00 (d, 1H), 8,62(d, 1H), 9,50 (bs, 1H), 10,19 (bs, 1H). Биологическая активность. Пример 5. Исследование NS3-4A протеазы HCV. Ингибирующую активность некоторых соединений, приведенных в табл. А, по отношению кNS3-4A серинпротеазе HCV определяют с помощью однородного исследования с использованием полноразмерного белка NS3-4A (генотип 1 а, штамм HCV-1) и имеющегося в продаже пептидного субстрата с потушенной собственной флуоресценцией, описанного в публикации Taliani, M., et al. 1996, Anal. Biochem. 240:60-67, которая во всей своей полноте включена в настоящее изобретение в качестве ссылки. Пример 6. Основанное на люциферазе исследование репликонов HCV. Противовирусную активность и цитотоксичность некоторых соединений, приведенных в табл. А,исследуют с использованием субгеномного генотипа 1b HCV репликонной линии клеток (Huh-Luc/neoET), содержащих репортерный ген люциферазы, экспрессирование которых происходит под контролем
МПК / Метки
МПК: C07D 401/14, C07D 403/14, A61K 31/403, C07K 5/08, C07D 403/12, C07D 417/14, A61P 31/22, C07D 209/96, C07D 405/14, C07D 401/12
Метки: соединения, применение, инфекции, лечения, органические
Код ссылки
<a href="https://eas.patents.su/30-21554-organicheskie-soedineniya-i-ih-primenenie-dlya-lecheniya-hcv-infekcii.html" rel="bookmark" title="База патентов Евразийского Союза">Органические соединения и их применение для лечения hcv инфекции</a>
Применение йота-каррагинана для лечения риновирусной инфекции и фармацевтическая композиция для лечения риновирусной инфекции, содержащая йота-каррагинан

Номер патента: 15930
Опубликовано: 30.12.2011
Авторы: Претч Александер, Майер Кристиане, Пришл-Грассауэр Ева, Грассауэр Андреас
МПК: A61K 31/731, A61P 31/16, A61K 31/737...
Метки: применение, лечения, инфекции, риновирусной, йота-каррагинан, композиция, содержащая, фармацевтическая, йота-каррагинана
Формула / Реферат:
1. Применение йота-каррагинана в эффективном антивирусном количестве в качестве активного антивирусного ингредиента для изготовления фармацевтической композиции для профилактики или терапевтического лечения риновирусной инфекции.2. Применение по п.1, где указанная риновирусная инфекция является острой или хронической риновирусной инфекцией.3. Применение по п.1 или 2, где композиция пригодна для местного применения или нанесения на слизистую...
Органические соединения железа (iii), их применение и способы получения указанных соединений

Номер патента: 10028
Опубликовано: 30.06.2008
Авторы: Квок Дэвид В.К., Стойнов Николай Минтчев
МПК: A61P 39/04, C01G 49/00, A61K 31/194...
Метки: указанных, способы, органические, применение, получения, соединений, железа, iii, соединения
Формула / Реферат:
1. Способ синтеза формы органического соединения железа (III), который включает: a) получение соли железа трехвалентного; b) прибавление гидроксида щелочного металла к соли железа трехвалентного со скоростью и при температуре, необходимыми для образования однородной коллоидной суспензии полиоксожелеза (polyiron oxo colloidal suspension); c) отделение осадка от суспензии; d) прибавление к осадку органической кислоты; e) образование раствора...
Органические соединения трехвалентного железа фармацевтического качества, их применение и способы их получения

Номер патента: 18245
Опубликовано: 28.06.2013
Авторы: Таун Винстон, Чен Кейт
МПК: A01N 37/00, C07F 15/02
Метки: получения, органические, соединения, трехвалентного, фармацевтического, способы, применение, железа, качества
Формула / Реферат:
1. Применение состава, содержащего цитрат железа (III), для приготовления лекарственного средства для предотвращения или обратного развития кальцификации мягких тканей, причем состав содержит цитрат железа (III), имеющий удельную площадь активной поверхности, определенную методом БЭТ, выше 16,17 м2/г.2. Применение по п.1, в котором цитрат железа имеет истинную скорость растворения в диапазоне от 1,88 до 4,0 мг/см2/мин.3. Применение по п.1, в...
Ингибиторы гиразы и их применение для лечения бактериальной инфекции

Номер патента: 5680
Опубликовано: 28.04.2005
Авторы: Бадиа Майкл, Чарифсон Пол, Стамос Дин, Трюдо Мартэн, Грийо Анн-Лор, Ронкин Стивен
МПК: A61P 31/04, A61K 31/4155, C07D 417/04...
Метки: применение, ингибиторы, бактериальной, лечения, инфекции, гиразы
Формула / Реферат:
1. Способ лечения бактериальной инфекции у млекопитающего, в случае необходимости такого лечения, включающий стадию введения указанному млекопитающему терапевтически эффективного количества соединения формулы или его фармацевтически приемлемой соли, где R1 обозначает необязательно замещеннную группу, выбранную из C1-6 алифатической группы, -C (R4)2(CH2)nNRCOR, -C(R4)2(CH2)nNRCO2(C1-6 алифатической группы), -CO2(C1-6 алифатической группы),...
Алкилсульфонилзамещенные n-(тиазол-2-ил)бензамиды и их применение для лечения вирусной инфекции гепатита c

Номер патента: 19357
Опубликовано: 31.03.2014
Авторы: Семпл Дж.Эдвард, Россиньоль Жан Франсуа
МПК: A61P 31/14, C07D 277/46, A61K 31/426...
Метки: гепатита, лечения, инфекции, n-(тиазол-2-ил)бензамиды, применение, алкилсульфонилзамещенные, вирусной
Формула / Реферат:
1. Соединение формулыили его соль,где R1-R5 независимо выбраны из группы, включающей H, OH, (C1-C6)алкилтио, (C1-C6)алкилсульфонил, -O-CO-(C1-C6)алкил и -O-SO2-(С1-С6)алкил, иR6 и R9 независимо выбраны из группы, состоящей из Н, (C1-C6)алкилтио, (C1-C6)алкилсульфонила и -O-SO2-(C1-C6)алкила,при условии, что по меньшей мере один из R1-R6 или R9 должен представлять собой (С1-С6)алкилсульфонил или -O-SO2-(C1-C6)алкил.2. Соединение по п.1, где...
Предыдущий патент: Коробка передач с двадцатью четырьмя передними передачами
Следующий патент: Легкая головка крепежного средства, обеспечивающая улучшение сборки
Случайный патент: Способ переработки пылевых отходов металло- и ферропроизводства