Алкилсульфонилзамещенные n-(тиазол-2-ил)бензамиды и их применение для лечения вирусной инфекции гепатита c







Формула / Реферат
1. Соединение формулы
или его соль,
где R1-R5 независимо выбраны из группы, включающей H, OH, (C1-C6)алкилтио, (C1-C6)алкилсульфонил,
-O-CO-(C1-C6)алкил и -O-SO2-(С1-С6)алкил, и
R6 и R9 независимо выбраны из группы, состоящей из Н, (C1-C6)алкилтио, (C1-C6)алкилсульфонила и -O-SO2-(C1-C6)алкила,
при условии, что по меньшей мере один из R1-R6 или R9 должен представлять собой (С1-С6)алкилсульфонил или -O-SO2-(C1-C6)алкил.
2. Соединение по п.1, где указанная соль является фармацевтически приемлемой.
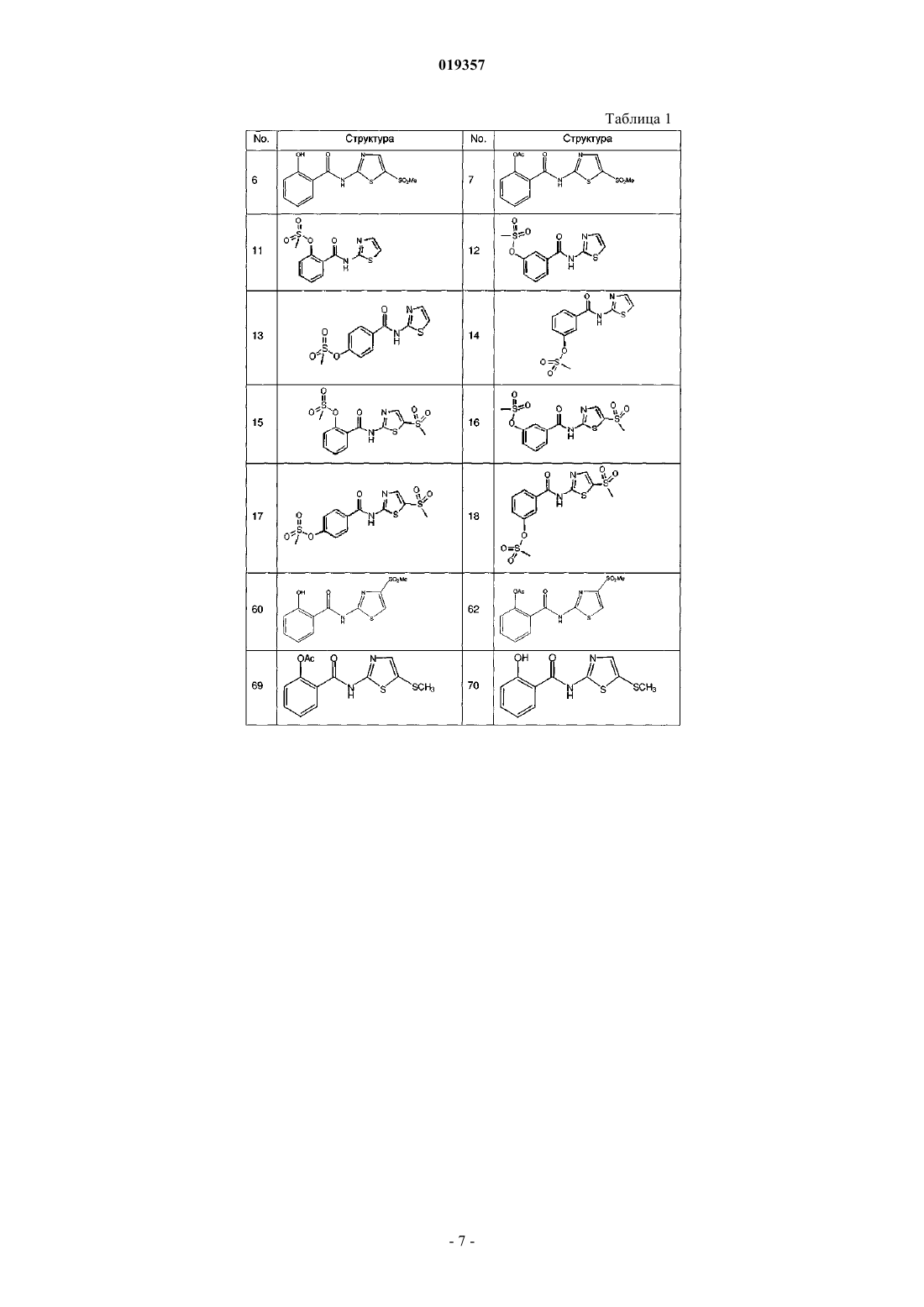
3. Соединение по п.1, выбранное из следующих соединений:
4. Фармацевтическая композиция для лечения вирусной инфекции гепатита С, содержащая эффективное количество соединения формулы (I) по любому из пп.1-3 и фармацевтически приемлемый носитель.
5. Применение соединения формулы
где R1 представляет собой ОН или -O-C(O)-(С1-С6)алкил,
или его фармацевтически приемлемой соли для лечения вирусной инфекции гепатита С.
Текст
(54) АЛКИЛСУЛЬФОНИЛЗАМЕЩННЫЕ N-(ТИАЗОЛ-2-ИЛ)БЕНЗАМИДЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ВИРУСНОЙ ИНФЕКЦИИ ГЕПАТИТА C В изобретении предложены алкилсульфонилзамещенные N-(тиазол-2-ил)бензамиды общей формулы (I) или его соль, где R1-R5 независимо выбраны из группы, включающей H, OH, (C1-C6)алкилтио,(C1-C6)алкилсульфонил, -O-CO-(C1-C6)алкил и -O-SO2-(C1-C6)алкил; и R6 и R9 независимо выбраны из группы, состоящей из Н, (C1-C6)алкилтио, (C1-C6)алкилсульфонила и -O-SO2-(С 1 С 6)алкила, при условии, что по меньшей мере один из R1-R6 или R9 должен представлять собой(C1-С 6)алкилсульфонил или -O-SO2-(С 1-С 6)алкил. Указанные соединения демонстрируют высокую активность в отношении вируса гепатита. Перекрестная ссылка на родственные заявки В заявке на настоящее изобретение испрашивается приоритет на основании предварительной заявки на патент США 60/953758, поданной 3 августа 2007 г., предварительной заявки на патент США 61/046956, поданной 22 апреля 2008 г., и предварительной заявки на патент США 61/056369, поданной 27 мая 2008 г., полное содержание каждой из которых включено в настоящее описание посредством ссылки. Положения относительно финансируемого из федерального бюджета исследования или разработки Изобретение, описанное в настоящем патенте, было создано при поддержке Правительства на основании контракта NIAID NO1-AI-30046 для медицинского центра Университета Джорджтауна (Georgetown University Medical Center). Соответственно, Правительство США может иметь определенные права на настоящее изобретение. Уровень техники В целом, настоящее изобретение относится к области тиазолидных соединений. В частности, изобретение относится к алкилсульфонилзамещенным тиазолидным соединениям. Вирусы гепатита В (HBV) и гепатита С (HCV) являются причиной серьезных проблем в области здравоохранения, поскольку эти вирусы вызывают примерно более 500 миллионов случаев хронических инфекционных заболеваний по всему миру (Chen and Morgan, 2006; Lavanchy, 2004). Оба вируса представляют собой причину серьезного прогрессирующего заболевания печени и являются основными факторами риска практически для всех случаев первичной печеночно-клеточной карциномы (Chen andMorgan, 2006; Lavanchy, 2004; Wong and Lok, 2006). В то время как известные стандарты лечения обеих вирусных инфекций являются эффективными во многих случаях, они являются недостаточными и не приводят к вирусологическому или клиническому "выздоровлению" у большинства индивидуумов(Wong and Lok, 2006). Развитие резистентноети к лекарственным средствам у вируса HBV, включая штаммы, обладающие резистентностью к многочисленным разрешенным к применению агентам, представляет собой все более серьезную клиническую проблему, при этом предполагают, что резистентность к лекарственным средствам будет являться серьезной клинической проблемой при лечении HCV в будущем (Tomei et al., 2005; Tong et al., Yim et al., 2006). Тиазолидные соединения, такие как нитазоксанид (nitazoxanide) (NTZ), являются противоинфекционными и обладают активностью в отношении анаэробных бактерий, простейших и вирусов (Fox et al.,2005; Pankuch and Appelbaum, 2006; Rossignol et al., 2006a; Rossignol and El-Gohary, 2006). ИзначальноNTZ был разработан как средство для лечения кишечных протозойных инфекций, при этом противовирусные свойства NTZ были обнаружены в ходе его разработки для лечения криптоспоридиоза у пациентов с синдромом приобретнного иммунодефицита (СПИДом). NTZ продается в Соединенных Штатах для лечения диареи и энтерита, вызванного криптоспоридией (Cryptosporidium spp) или лямблией кишечной (Giardia lamblia), у взрослых и детей вплоть до 12-месячного возраста (Alinia, Romark Laboratories, Tampa, Florida USA). Клинические исследования продемонстрировали эффективность NTZ при лечении диареи и энтерита, связанного с кишечными протозойными инфекциями, вызванными криптоспоридией (Cryptosporidium spp), лямблией кишечной (G.lamblia), амебой дизентерийной (Entamoebahistolytica) и Blastocystis hominis (Amadi et al., 2002; Oritz et al., 2001; Rossignol et al., 2001, 2005, 2006b). Последние рандомизированные двойные слепые клинические исследования продемонстрировали эффективность NTZ при лечении псевдомембранозного колита, вызванного Clostridium difficile, у взрослых,ротавирусного гастроэнтерита у маленьких детей и норовирусного гастроэнтерита у взрослых (Musher etal., 2006; Rossignol et al., 2006a; Rossignol and El Gohary, 2006). Механизм действия NTZ в отношении анаэробных организмов объясняют нарушением протекания ферментзависимых реакций переноса электронов с участием пируват:ферредоксин оксидоредуктазы (PFOR), необходимых для анаэробного энергетического метаболизма (Hoffman et al., 2006). Механизм противовирусной активности указанного средства не полностью объяснен. После перорального введения 500 мг таблетки NTZ частично абсорбируется из желудочнокишечного тракта и быстро гидролизуется в плазме с образованием активного циркулирующего метаболита, тизоксанида (tizoxanide) (TIZ). NTZ не обнаруживают в плазме. Максимальные концентрации TIZ в сыворотке достигают приблизительно 10 мкг/мл (37 мкМ) (Stockis et al., 2002) после перорального введения одной 500 мг таблетки NTZ (Alinia) с пищей. TIZ находится в глюкурон-конъюгированной форме в печени и выводится с мочой и желчью. Приблизительно две трети пероральной дозы проходят через кишечный тракт и выводятся с калом в виде TIZ (Broekhuysen et al., 2000). Период полувыведения TIZ из плазмы составляет приблизительно 1,5 ч. TIZ не ингибируют ферменты цитохрома Р 450 и, следовательно, не предполагают взаимодействий между лекарственными средствами (Broekhuysen et al., 2000; Stockis et al., 2002). По имеющимся данным,самые частые побочные эффекты в клинических исследованиях включают слабую боль в области живота, головную боль, диарею и тошноту, которая встречается с той же частотой, что и у пациентов, получающих плацебо. В то время как большинство клинических опытов с использованием NTZ включало 3-14 дней лечения, непрерывное применение лекарственного средства в течение 4 лет было оценено как не вызывающее у пациентов с криптоспоридиозом, связанным со СПИДом, каких-либо значительных побочных эффектов, связанных с лекарственным средством (Fox et al., 2005; Rossignol, 2006). В настоящем описании представлены результаты исследований, характеризующие активность NTZ,TIZ и других новых тиазолидов. В частности, продемонстрирована противовирусная активность 2-бензамидо-5-алкилсульфонилтиазолов. Краткое описание изобретения В настоящем изобретении предложены тиазолидные соединения или соли указанных соединений. В некоторых вариантах реализации предложенные соединения отвечают формуле (I) где R1-R5 независимо выбраны из группы, включающей H, OH, (C1-C6)алкилтио,(C1-C6)алкилсульфонил, -O-CO-(C1-C6)алкил и -O-SO2-(C1-C6)алкил; иR6 и R9 независимо выбраны из группы, состоящей из Н, (C1-C6)алкилтио, (C1-C6)алкилсульфонила и -O-SO2-(С 1-С 6)алкила,при условии, что по меньшей мере один из R1-R6 или R9 должен представлять собой(C1-С 6)алкилсульфонил или -O-SO2-(C1-C6)алкил. В некоторых вариантах реализации соединение формулы (I) представляет собой фармацевтически приемлемую соль соединения формулы (I). В одном из вариантов реализации соединение отвечает следующей формуле: Также предложены фармацевтические композиции, содержащие указанные соединения и носитель(например, разбавитель или наполнитель). Фармацевтическая композиция может содержать эффективное количество соединения для лечения инфекции HCV. Также предложено применение соединения согласно изобретению для лечения инфекции HCV у пациента, нуждающегося в таком лечении. Например, у пациента может быть хроническая инфекцияHCV. Краткое описание чертежей На фиг. 1 представлены примеры анализа взаимодействий между интерферонами при комбинированном лечении. Анализ комбинированных видов терапии осуществляли с использованием ПО Calcusyn (Biosoft,Inc., Cambridge, UK). Графики А и В отражают виды лечения HBV; графики C-F отражают виды леченияHCV. Представлены два типа оценки. На панелях А, С и Е представлены графики зависимости Cl-Fa(комбинированный индекс - пораженная фракция (вируса (Belen'kii and Schinazi, 1994). Для указанных графиков комбинированный индекс [Cl] более 1,0 указывает на антагонизм, a Cl менее 1,0 указывает на синергию. Оценка синергии, аддитивности (суммирования) или антагонизма на различных уровнях ингибирования вируса (например, 5% (Fa=0,5) - 99% (Fa=0,99 представлена линиями и точками на графике. Пунктирные 5% (Fa=0,5) - 99% (Fa=0,99 представлены линиями и точками на графике. Пунктирные линии на графике А показывают стандартные отклонения 1,96 (условно не показаны на графике C). На графиках B, D и F представлены консервативные изоболограммы. Для указанных графиков значенияEC50, EC75 и EC90 (50%, 75% и 90% эффективные противовирусные концентрации) для комбинированных видов лечения показаны в виде одной точки. Три линии, исходящие из осей, означают предполагаемые(например, аддитивные) значения EC50, EC75 и EC90 для комбинаций лекарственных средств, рассчитанные исходя из монотерапии. Значения EC50, EC75 и EC90 для комбинаций, обозначенные на графике слева(например, меньше чем) от соответствующих линий, указывают на синергию, а значения, обозначенные на графике справа (например, больше чем) от соответствующих линий, указывают на антагонизм. Фиг. 2 иллюстрирует действие NTZ на уровни нуклеиновых кислот и белков в клетках 2.2.15. Культуры клеток 2.2.15 обрабатывали согласно стандартным процедурам (Korba and Gerin, 1992,Antiviral Res. 19:55). Уровни нуклеиновых кислот HBV определяли путем количественного анализа методом блот-гибридизации (Korba and Gerin, 1992, Antiviral Res. 19:55; 1995, 28:225). Уровни белков HBV определяли путем полуколичественного ферментативного иммуноанализа (EIA) (Korba and Gerin, 1992,Antiviral Res. 19:55; 1995, 28:225). Образцы разбавляли (2-10-кратно), в результате чего приводили уровни в динамические диапазоны чувствительности EIA. Вирионную ДНК HBV, HBsAg и HBeAg анализировали из образцов культуральной среды. РНК HBV, ИР HBV (интермедиаты репликации ДНК HBV) иHBcAg анализировали из внутриклеточных лизатов. Подробное описание предпочтительных вариантов реализации Если не указано иное, единственное число означает "один или несколько". Предложены тиазолидные соединения формулы (I) где R1-R6 имеют значения, приведенные выше. В некоторых вариантах реализации R1, R2, R3, R4 или R5 представляет собой -OS(O2)Rx, где Rx представляет собой алкил. В некоторых вариантах реализации R1, R2, R3, R4 или R5 представляет собой OSO2-CH3. В некоторых вариантах реализации одна из групп R6 или R9 представляет собой Н, а другая представляет собой -SO2-алкил, предпочтительно -SO2CH3, -SO2Et, -SO2iPr или -CH2SO2Me. Предложенные соединения включают 2-бензамидо-5-метилсульфонилтиазолиды, 2-бензамидо-4 метилсульфонилтиазолиды и 2-(тиазол-2-илкарбамоил)фенилметансульфонаты. Согласно некоторым вариантам реализации соединений формулы (I) R1 выбран из H, OH, -O-CO-Rd,SRe и SO2CH3; R2-R5, каждый независимо друг от друга, выбраны из H, OH, -O-CO-Rd; SRe, где Rd и Re,каждый независимо друг от друга, представляют собой (C1-C6)алкил; где R6 представляет собой Н илиSO2CH3; и где в случае, если R1 представляет собой SO2CH3, R6 представляет собой Н; и в случае, если R6 представляет собой SO2CH3, R1 не представляет собой SO2CH3. Согласно другим вариантам реализации соединений формулы (I) указанные соединения отвечают формуле (I), в которой R1-R5, каждый независимо друг от друга, выбраны из H, OH, -O-CO-Rd, SRe; гдеRd и Re, каждый независимо друг от друга, представляют собой (C1-C4)алкил; a R6 представляет собойSO2CH3. Согласно другим вариантам реализации соединений формулы (I) указанные соединения отвечают формуле (I), в которой R1-R5, каждый независимо друг от друга, представляют собой Н. Согласно другим вариантам реализации указанные соединения отвечают формуле (I), в которой R1-R5, каждый независимо друг от друга, представляют собой Н; a R6 представляет собой SO2CH3. Согласно другим вариантам реализации соединений формулы (I) указанные соединения отвечают формуле (I), в которой R1-R5, каждый независимо друг от друга, выбраны из H, -OH, O-CO-Rd, SRe, где Rd и Re, каждый независимо друг от друга, представляют собой (C1-C6)алкил; а R6 представляет собойSO2CH3. Согласно другим вариантам реализации соединений формулы (I) указанные соединения отвечают формуле (I), в которой R1 представляет собой -OS(O2)Rx, где Rx представляет собой алкил; R2-R5, каждый независимо друг от друга, выбраны из H, -OH, -O-CO-Rd, SRe; R6 выбран из H, -OH, SRe, где Rd и Re,каждый независимо друг от друга, представляют собой (C1-С 4)алкил. Согласно другим вариантам реализации соединений формулы (I) указанные соединения отвечают формуле (I), в которой R1 представляет собой -OS(O2)CH3; R2-R5, каждый независимо друг от друга, выбраны из H, OH, O-CO-Rd, SRe, где Rd и Re, каждый независимо друг от друга, представляют собой(C1-C4)алкил; a R6 представляет собой Н. Согласно другим вариантам реализации соединений формулы (I) указанные соединения отвечают формуле (I), в которой R1 представляет собой -OS(O2)CH3; R2-R5, каждый независимо друг от друга,представляют собой Н; а R6 представляет собой Н. Согласно другим вариантам реализации соединений формулы (I) указанные соединения отвечают формуле (I), в которой R1 представляет собой -OS(O2)CH3; R2-R5, каждый независимо друг от друга, выбраны из H, -OH, O-CO-Rd и SRe и где Rd и Re, каждый независимо друг от друга, представляют собой(C1-C6)алкил; a R6 представляет собой Н. Предложенные соединения включают соединения формулы (I), соли и сольваты указанных соединений. Например, в некоторых вариантах реализации соединение согласно настоящему изобретению может представлять собой соль сольвата. Термин "соли" используется в самом широком понимании этого слова. Например, термин "соли" включает водородсодержащие соли и гидроксидсодержащие соли с ионами настоящего соединения. В некоторых вариантах реализации термин "соль" может представлять собой подкласс под названием"фармацевтически приемлемые соли", представляющие собой соли настоящих соединений, обладающие фармакологической активностью и не являющиеся биологически нежелательными или нежелательными в другом отношении. Во всех вариантах реализации соли могут быть образованы с помощью кислот, например, без ограничения, водород, ацетат, адипат, альгинат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, цитрат, камфорат, камфорсульфонат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, фумарат, глюкогептаноат, глицерофосфат, гемисульфат, гептаноат, гексаноат,гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат, лактат, малеат, метансульфонат,2-нафталинсульфонат, никотинат, оксалат, тиоцианат, тозилат и ундеканоат. Во всех вариантах реализации соли могут быть образованы с помощью оснований, например, без ограничения, гидроксид, соли аммония, соли щелочных металлов, такие как соли лития, натрия и калия, соли щелочно-земельных металлов, такие как соли кальция, магния, соли алюминия, соли с органическими основаниями, такими как аммиак, метиламин, диэтиламин, этаноламин, дициклогексиламин, N-метилморфолин, N-метил-Oглюкамин, и соли с аминокислотами, такими как аргинин и лизин. Основные азотсодержащие группы могут быть кватернизированы агентами, включая низшие алкилгалогениды, такие как метил-, этил-, пропил- и бутилхлориды, бромиды и йодиды; диалкилсульфаты, такие как диметил-, диэтил-, дибутил- и диамилсульфаты; длинноцепочечные галогениды, такие как децил-, лаурил-, миристил- и стеарилхлориды, бромиды и йодиды; и аралкилгалогениды, такие как бензил- и фенетилбромиды. В настоящем описании термин "терапевтически приемлемая соль" означает соли или цвиттерионные формы соединений согласно настоящему изобретению, которые являются водо- или жирорастворимыми или диспергируемыми, которые подходят для лечения заболеваний, не вызывая при этом неспецифической токсичности, раздражения и аллергической реакции, которые отвечают разумному соотношению "польза-риск" и которые являются эффективными для их предполагаемого применения. Указанные соли могут быть получены в ходе конечного выделения и очистки соединений или отдельно путем осуществления взаимодействия подходящего соединения в форме свободного основания с подходящей кислотой. Типичные соли присоединения кислот включают ацетат, адипат, альгинат, L-аскорбат,аспартат, бензоат, бензолсульфонат (безилат), бисульфат, бутират, камфорат, камфорсульфонат, цитрат,диглюконат, формиат, фумарат, гентизат, глутарат, глицерофосфат, гликолят, гемисульфат, гептаноат,гексаноат, гиппурат, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат (изетионат), лактат, малеат, малонат, DL-манделат, мезитиленсульфонат, метансульфонат, нафтиленсульфонат, никотинат, 2-нафталинсульфонат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфонат, пикрат,пивалат, пропионат, пироглутамат, сукцинат, сульфонат, тартрат, L-тартрат, трихлорацетат, трифторацетат, фосфат, глутамат, бикарбонат, п-толуолсульфонат (п-тозилат) и ундеканоат. Также основные группы в соединениях согласно настоящему изобретению могут быть кватернизированы метил-, этил-, пропил- и бутилхлоридами, бромидами и иодидами; диметил-, диэтил-, дибутил- и диамилсульфатами; децил-, лаурил-, миристил- и стерилхлоридами, бромидами и иодидами и бензил- и фенетилбромидами. Примеры кислот, которые могут быть использованы для образования терапевтически приемлемых солей присоединения, включают неорганические кислоты, такие как соляная, бромисто-водородная, серная и фосфорная, и органические кислоты, такие как щавелевая, малеиновая, янтарная и лимонная. Соли могут быть также получены путем координации соединений с ионом щелочного или щелочно-земельного металла. Следовательно, настоящее изобретение включает натриевые, калиевые, магниевые и кальциевые соли соединений согласно настоящему изобретению и т.п. Соли присоединения оснований могут быть получены в ходе конечного выделения и очистки соединений путем осуществления взаимодействия карбоксильной, фенольной или аналогичной группы с подходящим основанием, таким как гидроксид, карбонат или бикарбонат катиона металла, или с аммиаком или органическим первичным, вторичным или третичным амином. Катионы терапевтически приемлемых солей включают катионы лития, натрия, калия, кальция, магния и алюминия, а также катионы нетоксичных четвертичных аминов, такие как аммоний, тетраметиламмоний, тетраэтиламмоний,метиламин, диметиламин, триметиламин, триэтиламин, диэтиламин, этиламин, трибутиламин, пиридин,N,N-диметиланилин, N-метилпиперидин, N-метилморфолин, циклогексиламин, прокаин, дибензиламин,N,N-дибензилфенетиламин, 1-фенамин и N,N'-дибензилэтилендиамин. Другие типичные органические амины, подходящие для образования солей присоединения оснований, включают этилендиамин, этаноламин, диэтаноламин, пиперидин и пиперазин. Термин "сольваты" используется в самом широком понимании этого слова. Например, термин"сольваты" включает гидраты, образуемые, когда соединение согласно настоящему изобретению содержит одну или несколько связанных молекул воды. В настоящем описании следующие термины имеют указанные значения. В настоящем описании термин "ацил" отдельно или в комбинации относится к карбонильной группе, присоединенной к алкенильной, алкильной, арильной, циклоалкильной, гетероарильной, гетероциклической или любой другой группе, в случае, когда атом, присоединенный к карбонильной группе, представляет собой атом углерода. Термин "ацетильная" группа относится к группе -C(O)CH3. Примеры ацильных групп включают формильные, алканоильные и ароильные радикалы. Термин "ациламино" включает аминорадикал, содержащий в качестве заместителя ацильную группу. Примером "ациламино" радикала является ацетиламино (CH3C(O)NH-). В настоящем описании термин "алкоксикарбонил" отдельно или в комбинации относится к алкоксигруппе, присоединенной к исходному молекулярному фрагменту посредством карбонильной группы. Примеры таких "алкоксикарбонильных" групп включают метоксикарбонил, этоксикарбонил, пропоксикарбонил, бутоксикарбонил и гексилоксикарбонил. В настоящем описании термин "алкил" отдельно или в комбинации относится к линейному или разветвленному алкильному радикалу, содержащему от 1 до 20 (включительно), предпочтительно от 1 до 10 и более предпочтительно от 1 до 6 атомов углерода. Термин "алкильные группы" используется в самом широком понимании этого слова. Алкильные группы могут содержать заместители, как определено в настоящем описании. Примеры алкильных радикалов включают метил, этил, н-пропил, изопропил, нбутил, изобутил, втор-бутил, трет-бутил, пентил, изоамил, гексил, октил, ноил и т.п. Например,O(C1-C8)алкильные группы включают линейные O(C1-C8)алкильные группы, а также разветвленныеO(C1-C8)алкильные группы. В качестве еще одного примера указанный термин включает циклоалкильные группы, как, например, (C3-C8)алкильные группы включают (C3-C8)циклоалкильные группы. В настоящем описании термин "алкилсульфонил" отдельно или в комбинации относится к алкильной группе, присоединенной к исходному молекулярному фрагменту посредством сульфонильной группы. Примеры алкилсульфонильных групп включают метансульфонил, этансульфонил, третбутансульфонил и т.п. В настоящем описании термин "алкилтио" отдельно или в комбинации относится к алкилтиоэфирному (R-S-) радикалу, при этом термин "алкил" является таким, как определено выше. Примеры подходящих алкилтиоэфирных радикалов включают метилтио, этилтио, н-пропилтио, изопропилтио, нбутилтио, изобутилтио, втор-бутилтио, трет-бутилтио, этоксиэтилтио, метоксипропоксиэтилтио, этоксипентоксиэтоксиэтилтио и т.п. Термины "карбокси" или "карбоксил", используемые либо отдельно, либо с другими терминами,как, например "карбоксиалкил", означают -CO2H. В настоящем описании термины "бензо" или "бенз" отдельно или в комбинации относятся к двухвалентному радикалу C6H4=, полученному из бензола. Примеры включают бензотиофен и бензимидазол. В настоящем описании термин "С-связанный" отдельно или в комбинации относится к любому заместителю, присоединенному к исходному молекулярному фрагменту посредством связи углеродуглерод. В настоящем описании термин "карбонат" отдельно или в комбинации относится к группе-O-C(=O)OR, где R является таким, как определено в настоящем описании. В настоящем описании термин "карбонил" при использовании отдельно включает формил [-C(O)H],а в комбинации представляет собой группу -C(O)-. В настоящем описании термин "карбокси" относится к -C(O)OH или к соответствующему "карбоксилату", такому как производное карбоновой кислоты в виде соли или сложного эфира. Термин"О-карбоксигруппа" относится к группе RC(O)O-, где R является таким, как определено в настоящем описании. Термин "С-карбоксигруппа" относится к группам -C(O)OR, где R является таким, как определено в настоящем описании. В настоящем описании термин "сложный эфир" отдельно или в комбинации относится к карбонилоксигруппе -(C=O)O, соединяющей (в качестве "мостика") два фрагмента, которая связана с атомами углерода. Примеры включают этилбензоат, н-бутилциннамат, фенилацетат и т.п. В настоящем описании термин "эфир" отдельно или в комбинации относится к оксигруппе, соединяющей (в качестве "мостика") два фрагмента, которая связана с атомами углерода. В настоящем описании термин "галогено" или "галоген" отдельно или в комбинации относится к фтору, хлору, брому или йоду. В настоящем описании термин "гидрокси" отдельно или в комбинации относится к -OH. В настоящем описании термин "гидроксиалкил" отдельно или в комбинации относится к линейной или разветвленной алкильной группе, содержащей от одного до примерно десяти атомов углерода, любой из которых может содержать в качестве заместителя один или несколько гидроксильных радикалов. Примеры таких радикалов включают гидроксиметил, гидроксиэтил, гидроксипропил, гидроксибутил и гидроксигексил. В настоящем описании термин "гидроксиалкил" отдельно или в комбинации относится к гидроксигруппе, присоединенной к исходному молекулярному фрагменту посредством алкильной группы. В настоящем описании термин "низший" отдельно или в комбинации означает содержащий от 1 до 6 (включительно) атомов углерода. В настоящем описании термины "сульфонат", "сульфоновая кислота" и "сульфоновый" отдельно или в комбинации относятся к группе -SO3H и аниону указанной группы, так как сульфоновая кислота используется при образовании соли. В настоящем описании термин "сульфанил" отдельно или в комбинации относится к -S и -S-. В настоящем описании термин "сульфонил" отдельно или в комбинации относится к -SO2-. В настоящем описании термины "тиа" и "тио" отдельно или в комбинации относятся к группе -Sили эфиру, в котором кислород замещен на серу. Окисленные производные тиогруппы, а именно сульфинил и сульфонил, включены в определение терминов "тиа" и "тио". В настоящем описании термин "тиоэфир" отдельно или в комбинации относится к тиогруппе, соединяющей (в качестве "мостика") два фрагмента, которая связана с атомами углерода. Термин "алкил" используется в самом широком понимании этого слова. Например, указанный термин относится к разветвленным, неразветвленным и циклическим насыщенным углеводородным цепям,содержащим указанное число атомов углерода. Например, термин "O(C1-C8)алкил" включает линейныйO(C1-C8)алкил, а также разветвленный O(C1-C8)алкил. В качестве еще одного примера термин включает циклоалкил, как, например, "(C1-C8)алкил" включает (C3-C8)циклоалкил. Во всех вариантах реализации указанный термин включает, без ограничения, заместители, такие как метил, этил, пропил, изопропил,бутил, изобутил, втор-бутил, трет-бутил, н-пентил, н-гексил и т.п., если не указано иное. Во всех вариантах соединений формулы (I) группы R2-R5 могут быть одинаковыми, могут быть разными или некоторые из R2-R5 могут быть одинаковыми, в то время как другие являются разными. Возможна любая комбинация. В любом варианте соединений формулы (I) либо R1, либо R6 может представлять собой SO2CH3. Однако если R1 представляет собой SO2CH3, то R6 представляет собой Н. В другом варианте, если R6 представляет собой SO2CH3, то R1 выбран из H, -OH, -O-CO-Rd и SRe. В любом варианте соединений формулы (I), в котором R6 представляет собой SO2CH3, R1 и/или R5 может независимо представлять собой группу или атом, кроме атома водорода. Например, R1 и/или R5 может независимо быть выбран из -OH, -O-CO-Ru. В некоторых вариантах реализации R1 и/или R5 может независимо быть выбран из OH, -O-CO-CH3, О-CO-СН 2 СН 3. Ru представляет собой (C1-C6)алкил. В любом варианте Ru может представлять собой (С 1-С 4)алкил. В любом варианте Ru может представлять собой(C1-C3)алкил. Примеры соединений согласно настоящему изобретению могут включать, без ограничения, следующие соединения, перечисленные в табл. 1. В табл. 2 указаны температуры плавления различных соединений. Таблица 2 Для вышеуказанных соединений, содержащих метилсульфонил (-SO2CH3), авторами настоящего изобретения также предусмотрено, что вместо метилсульфонила может быть использована группа, выбранная из SO2CH2CH3, -SO2CH2CH(CH3)2. Соединение согласно настоящему изобретению, в котором R6 представляет собой SO2CH3, может быть получено путем осуществления взаимодействия ацилгалогенида с амином в подходящих условиях реакции. В некоторых вариантах реализации указанная реакция может быть в общем представлена следующим образом: Соединения согласно настоящему изобретению также могут быть получены в соответствии со следующей схемой реакции: Соединение, представляющее собой 2-(тиазол-2-илкарбамоил)фенилметансульфонат (11), может быть синтезировано способом, описанным в примере 3.0. Другие соединения согласно настоящему изобретению также могут быть получены в соответствии со следующей схемой реакции:(60) и 2-[4-(метилсульфонил)-1,3-тиазол-2-ил]карбамоилфенилацетат (62), могут быть синтезированы способом, описанным в примере 4.0. Другие соединения согласно настоящему изобретению также могут быть получены в соответствии со следующей схемой реакции: Соединения, представляющие собой 4-[4-(метилсульфонил)-1,3-тиазол-2-ил]карбамоилфенилацетат (83),4-гидрокси-N-[4-(метилсульфонил)-1,3-тиазол-2-ил]бензамид (84),3-[4-(метилсульфонил)-1,3-тиазол-2-ил]карбамоилфенилацетат (86) и 3-гидрокси-N-[4-(метилсульфонил)-1,3-тиазол-2-ил]бензамид (87),могут быть синтезированы общими способами, описанными в примере 5.0. Кроме того, соединения согласно настоящему изобретению также могут быть получены в соответствии со следующей схемой реакции: Соединения, представляющие собой 4-[5-(метилсульфонил)-1,3-тиазол-2-ил]карбамоилфенилацетат (78),3-[5-(метилсульфонил)-1,3-тиазол-2-ил]карбамоилфенилацетат (79),4-гидрокси-N-[5-(метилсульфонил)-1,3-тиазол-2-ил]бензамид (80) и 3-гидрокси-N-[5-(метилсульфонил)-1,3-тиазол-2-ил]бензамид (81),могут быть синтезированы общими способами, описанными в примере 6.0. Другие соединения согласно настоящему изобретению также могут быть получены в соответствии со следующей схемой реакции: Соединения, представляющие собой 2-(5-(метилсульфонил)тиазол-2-илкарбамоил)фенилацетат (7) и 2-гидрокси-N-(5-(метилсульфонил)тиазол-2-ил)бензамид (6), могут быть синтезированы способом,описанным в примере 7.0. Настоящее изобретение также включает композицию, содержащую по меньшей мере одно соединение согласно настоящему изобретению в носителе. Термин "носитель" используется в самом широком понимании этого слова. Например, термин "носитель" относится к любым носителям, разбавителям, эксципиентам, смачивающим веществам, буферным веществам, суспендирующим средствам, смазывающим веществам, адъювантам, наполнителям,системам доставки, эмульгаторам, дезинтегрантам, абсорбентам, консервантам, поверхностно-активным веществам, красителям, ароматизаторам и подсластителям. В некоторых вариантах реализации носитель может представлять собой фармацевтически приемлемый носитель, более узкий термин, чем "носитель",- 15019357 так как термин "фармацевтически приемлемый носитель" означает нетоксичный, подходящий для применения в фармацевтической композиции. Настоящее изобретение также относится к фармацевтической композиции, содержащей в фармацевтически приемлемом носителе эффективное количество по меньшей мере одного из соединений согласно настоящему изобретению. Термин "эффективное количество" используется в самом широком понимании этого слова. Указанный термин, например, относится к количеству, необходимому для достижения требуемого эффекта. В некоторых вариантах реализации соединение согласно настоящему изобретению присутствует в фармацевтической композиции в эффективном количестве для лечения инфекции HCV (например, хронической инфекции HCV). Термин "лечение инфекции HCV" может относиться к (i) предупреждению возникновения инфекции HCV у животного, которое может быть предрасположено к инфекции HCV, но у которого она еще не диагностирована; (ii) ингибированию или замедлению инфекции HCV, например приостановлению ее развития; (iii) ослаблению хронической инфекции, например достижению ремиссии;(iv) улучшению симптома у субъекта с хронической инфекцией и/или (v) продлению жизни субъекта с хронической инфекцией. Композиции согласно настоящему изобретению могут быть получены в виде твердых или жидких лекарственных форм или в виде паст или мазей и могут содержать дополнительные активные компоненты. Фармацевтическая композиция согласно настоящему изобретению содержит фармацевтически приемлемый носитель, который, в частности, не ограничен и включает широкий диапазон носителей, известных специалистам в данной области техники, включая смачивающие или диспергирующие вещества(патент США 5578621, включенный в настоящее описание посредством ссылки), производные крахмала(патент США 5578621, включенный в настоящее описание посредством ссылки), наполнители и т.п. Варианты таблеток могут содержать оболочку вещества, которая составляет энтеросолюбильную оболочку,т.е. оболочку, которая является, по существу, нерастворимой в желудочном соке, но, по существу, растворима в кишечных жидкостях. Фармацевтические композиции, содержащие соединения согласно настоящему изобретению, в некоторых вариантах реализации получены для перорального введения и возможно находятся в форме жидкости, например эмульсии или раствора в воде или масле, таком как арахисовое масло, или другой жидкости. Составы неводных мицеллярных растворов могут быть получены согласно способу, описанному в патенте США 5169846, включенному в настоящее описание посредством ссылки. В качестве альтернативы таблетки могут быть получены, например, путем осуществления следующих стадий: влажное гранулирование, сушка и прессование. Нанесение пленочного покрытия может быть в общем случае осуществлено с помощью органических растворителей. Настоящее изобретение представляет собой применение соединения согласно настоящему изобретению для лечения инфекции HCV (например, хронической инфекции HCV). В некоторых вариантах реализации субъект выбран из животных. В некоторых вариантах реализации субъект выбран из млекопитающих. В некоторых вариантах реализации субъект выбран из домашних животных, таких как мыши, собаки, кошки и т.д. В некоторых вариантах реализации субъект выбран из людей. В некоторых вариантах реализации противовирусное лечение или профилактические дозы соединения согласно настоящему изобретению могут зависеть от массы субъекта и могут быть предположены специалистом без проведения излишних исследований исходя из следующих примеров, приведенных в целях иллюстрации и не являющихся ограничивающими. Соединения и композиции согласно настоящему изобретению могут быть введены локально или системно любыми способами, известными специалисту. Например, соединения и композиции согласно настоящему изобретению могут быть введены перорально, парентерально, с помощью спрея для ингаляции, местно, ректально, назально, буккально, вагинально или посредством имплантированного резервуара в дозированных составах, содержащих традиционные нетоксичные фармацевтически приемлемые носители, адъюванты и наполнители. В настоящем описании термин "парентеральный" включает подкожную, внутривенную, внутриартериальную, внутримышечную, интраперитонеальную, интратекальную, внутрижелудочковую, подложечную, интракраниальную или внутрикостную инъекцию и инфузионные способы. Точный протокол введения будет варьироваться в зависимости от различных факторов,включая возраст, массу тела, общее состояние здоровья, пол и режим питания пациента; определение конкретных процедур введения является стандартной процедурой для специалиста. Дозировки порядка от примерно 0,1 до примерно 100 мг/кг активного компонента (соединения) являются подходящими для лечения вышеуказанных состояний (например, 0,1 мг/кг-день). В некоторых вариантах реализации количества находятся в диапазоне от примерно 1 до примерно 10 мг/кг, а в других вариантах реализации количества находятся в диапазоне от примерно 2 до примерно 5 мг/кг. Конкретная дозировка для каждого конкретного пациента будет варьироваться в зависимости от ряда факторов,включая активность и возможную токсичность конкретного используемого соединения; возраст, массу тела, общее состояние здоровья, пол и диету пациента; время введения; скорость выведения из организ- 16019357 ма; комбинацию лекарственных средств; тяжесть конкретного заболевания, подвергаемого лечению; и форму введения. Как правило, результаты исследования эффекта дозы in vitro дают представление о подходящих дозах для введения пациенту. Исследования на моделях заболеваний на животных также являются полезными. Принципы определения подходящих дозировок хорошо известны в данной области техники. Любой режим введения для регулирования времени и последовательности доставки лекарственного средства может быть использован и повторен по мере необходимости для осуществления лечения. Указанный режим может включать многократное применение или предварительное введение и/или совместное введение и/или поствведение с пищей, жидкостью или водой. В некоторых вариантах реализации соединение согласно настоящему изобретению может проявлять селективную противовирусную активность. В настоящем описании термин "селективный противовирусный" означает, что в дозировках, эффективных для предупреждения или лечения вирусного заболевания, активность носит больше противовирусный характер, чем антибактериальный, противогрибковый или антипаразитарный, и кишечная флора субъекта не разрушается до уровней, предполагаемых при использовании широкого спектра антибиотиков. Например, эффективная доза для противовирусного лечения (например, уменьшения вирусной нагрузки по меньшей мере примерно в два раза) может не снижать бактериальный, грибковый или паразитарный уровень в кишечнике (например, более чем примерно в два раза). Другие варианты реализации настоящего изобретения очевидны специалистам в данной области техники исходя из настоящего описания и практического применения изобретения, охарактеризованного в настоящем описании. Описание и примеры приведены для пояснения изобретения, тогда как истинный объем изобретения определяется формулой изобретения. Примеры 1. Материалы и способы. 1.1. Материалы. Ламивудин (Lamivudine) (LMV), адефовир диповоксил (adefovir dipovoxil) (ADV) и 2'-С-метилцитидин приобретали у Moraveck Biochemicals, Inc. (La Brea, CA USA). Рекомбинантный интерферон человека альфа 2b (IFN) приобретали у PBL Biomedical Laboratories (Piscataway, NJ USA). Все остальные тестируемые соединения (фиг. 1) были предоставлены Romark Laboratories, L.C., сыворотку человека (термоинактивированная, смешанная, партия BRH125374) приобретали у Bioreclamation, Inc.(Hicksville, NY). 1.2. Исследования HBV. 1.2.1. Противовирусные анализы. Противовирусные анализы HBV осуществляли, как описано ранее (Korba and Gerin, 1992). Кратко,конфлюэнтные культуры клеток 2.2.15 выдерживали в 96-луночных плоскодонных планшетах для тканевой культуры (конфлюэнтность в данной культуре необходима для активных, высоких уровней репликации HBV, эквивалентных уровням, наблюдаемым у хронически инфицированных индивидуумов (Sells, etal., 1988; Korba and Gerin, 1992). Культуры обрабатывали девятью последовательными ежедневными дозами тестируемых соединений. Уровни ДНК HBV определяли методом количественной блотгибридизации через 24 ч после последнего воздействия. Цитотоксичность определяли по поглощению нейтрального красного красителя через 24 ч после последнего воздействия. 1.2.2. Активность в отношении резистентных к лекарственным средствам генетически модифицированных вариантов HBV. Активность в отношении резистентных к LMV (Allen et al., 1998) и резистентных к ADV (Angus etal., 2003) генетически модифицированных вариантов HBV определяли в ходе 5-дневного анализа с помощью способа временной трансфекции, как описано ранее (Iyer et al., 2004). Противовирусную активность определяли методом количественной Саузерн-блот-гибридизации (Southern blot hybridization) внутриклеточных интермедиатов репликации ДНК HBV (HBV ИР). 1.2.3. Получение белков HBV. Культуры клеток 2.2.15 обрабатывали согласно стандартным процедурам и осуществляли полуколичественный ферментативный иммуноанализ белков HBV, как описано ранее (Korba and Gerin, 1995), за исключением того, что HBeAg анализировали с помощью ETI-EBK Plus (DiaSorin, Inc., Stillwater, MNUSA). Образцы разбавляли (2-10-кратно), в результате чего приводили уровни в динамические диапазоны чувствительности EIA HBsAg и HBeAg анализировали из образцов культуральной среды, а HBeAg анализировали из внутриклеточных лизатов. Внутриклеточную РНК HBV определяли методом количественной Нозерн-блот-гибридизации (Northern blot hybridization) (Korba and Gerin, 1995). 1.3. Исследования HCV. Противовирусную активность тестируемых соединений определяли в ходе 3-дневного анализа с использованием стабильно экспрессирующей репликон HCV линии клеток, AV A5 (субгеномный CONI,генотип Ib) (Blight et al., 2000), поддерживаемой в виде субконфлюэнтных культур в 96-луночных планшетах, как описано ранее (Okuse et al., 2005). Противовирусную активность определяли с помощью анализа методом блот-гибридизации внутриклеточной РНК HCV (приведенного к уровню клеточной РНК В-актина в каждом образце культуры), а цитотоксичность определяли по поглощению нейтрального красного красителя через 3 дня после лечения. Дополнительные исследования проводили с использованием клеток Huh7, содержащих другой репликон HCV, H/FL-Neo, генотип 1, полноразмерная конструкция (Blight et al., 2003). Для исследований с использованием сыворотки человека поддерживали стандартную культуральную среду (содержащую 10% фетальную бычью сыворотку) и условия анализа. 1.4. Оформление результатов. Значения EC50, EC90 и CC50 ( стандартные отклонения [S.D.]) рассчитывали с помощью линейного регрессионного анализа с использованием данных, полученных из всех обработанных культур (Korba andGerin, 1992; Okuse et al., 2005). EC50 и ЕС 90 представляют собой концентрации лекарственных средств,при которых наблюдали 2-кратное или 10-кратное подавление внутриклеточной ДНК HBV или РНКHCV (относительно средних уровней в необработанных культурах) соответственно. CC50 представляет собой концентрацию лекарственного средства, при которой наблюдали в два раза более низкий уровень поглощения красителя нейтрального красного (относительно средних уровней в необработанных культурах). Индекс селективности (S.I.) рассчитывали как соотношение CC50/EC90 для анализов HBV иCC50/EC50 для анализов HCV. Значения EC90 использовали для расчета S.I. в анализах HBV, так как по меньшей мере 3-кратное подавление уровней ДНК HBV, как правило, необходимо для достижения статистической значимости в данном анализе (Korba and Gerin, 1992). Для комбинированных видов лечения ЕС 50, EC90, СС 50 и S.I. представлены для первого указанного соединения. Также указано молярное соотношение соединений в каждой комбинации. 2. Результаты. 2.1. Вирус гепатита В (HBV). 2.1.1. Активности соединений и комбинаций в культурах клеток 2.2.15.NTZ и его активный метаболит, TIZ, демонстрировали селективное ингибирование внутриклеточной репликации HBV и образования внеклеточной формы вируса клетками 2.2.15 (табл. 3). Некоторые другие тиазолиды (см. табл. 3) также являлись эффективными ингибиторами репликации HBV в данном анализе. Комбинации NTZ с одним из двух лекарственных средств, разрешенных для терапии HBV, ламивудином (LMV) и адефовир диповоксилом (ADV), демонстрировали синергетические взаимодействия при использовании для обработки клеток 2.2.15 (табл. 3, фиг. 1 А и 1 В). Анализы анти-HBV осуществляли при конфлюентности, так как это обеспечивает условия для оптимальной репликации HBV (Sells, etal., 1988; Korba and Gerin, 1992). В то время как в условиях противовирусного анализа NTZ демонстрировал минимальную цитотоксичность (100 мкМ, табл. 3), цитотоксичность NTZ в быстроделящихся культурах клеток 2.2.15 была выше (201,3 мкМ). 2.1.2. Активность NTZ и RM4850 в отношении резистентных к лекарственным средствам мутантовNTZ и RM4850 представляли собой эффективные ингибиторы некоторых резистентных к LMV и одной резистентной к ADV конструкции HBV в анализах методом временной трансфекции в клеткахHuh7 (табл. 4). Не наблюдали существенных различий в активности указанных тиазолидов по сравнению с активностью, наблюдаемой для HBV дикого типа для каждого из тестируемых вирусов, резистентных к лекарственным средствам. 2.1.3. Действие NTZ на образование белков HBV. В отличие от большинства вирусов (включая HCV), транскрипция РНК HBV и образование белков эффективно отделены от репликации вирусного генома за счет присутствия долгоживущей популяции ковалентно непрерывных вирусных матричных геномов в ядре клетки-хозяина (ковалентнонепрерывная кольцевая ДНК (сссДНК (см. Locarnini, 2004 для обзора). Внутриклеточная репликация HBV осуществляется в вирусных нуклеокапсидах, расположенных в цитоплазме. В результате большинство соединений, ингибирующих репликацию ДНК HBV (например, аналоги нуклеозидов), как правило, не меняют образование белков HBV, особенно в клеточной культуре. Исходя из предположения о новом механизме действия NTZ в отношении HBV, были проведены исследования для того, чтобы определить, ингибирует ли лекарственное средство образование основных белков HBV. По данным полуколичественного EIA, NTZ понижал уровни внеклеточной поверхностиHBV и е-антигенов (HBsAg, HBeAg), а также уровни внутриклеточного нуклеокапсидного корового антигена HBV (HBcAg) в зависимости от дозы (табл. 5, фиг. 2). Активность NTZ в отношении HBsAg иHBeAg была сходна с активностью, наблюдаемой в отношении образования вириона HBV в том же эксперименте. Относительная активность NTZ в отношении внутриклеточного HBsAg была сходна с активностью, наблюдаемой для ингибирования внутриклеточной репликации ДНК HBV. Количественное нарушение способности EIA обнаруживать белки HBV не наблюдали в образцах контрольных культур, к которым добавляли 10 мкМ NTZ (данные не показаны).NTZ не вызывал уменьшение внутриклеточной РНК HBV по данным нозерн-блот-гибридизации(табл. 5, фиг. 2). В том же эксперименте LMV не влиял на уровни белков HBV или РНК HBV, несмотря на то, что вызывал значительное снижение образования вириона HBV и внутриклеточной репликации ДНК HBV (табл. 5). 2.2. Вирус гепатита C (HCV) 2.2.1. Активность соединения и комбинаций в клеточных культурах репликона HCV.NTZ и TIZ селективно уменьшали внутриклеточную репликацию HCV в клетках AVA5 (табл. 6). Оба соединения обладали большей цитотоксичностью в данной линии клеток, по сравнению с клетками 2.2.15, но указанные соединения были также гораздо более активными в отношении репликации HCV. Комбинации NTZ или TIZ либо с рекомбинантным человеческим интерфероном альфа 2b (IFN), либо с ингибитором NS5B (полимераза HCV), 2'-С-метилцитидином (2'CmeC, Pierra, et al., 2005) проявляли синергические взаимодействия в отношении репликации HCV (табл. 6, фиг. 1 С и 1D). Только два из остальных тиазолидов, RM4832 и RM4863, проявляли активность в отношении HCV(табл. 6). Противовирусная активность NTZ, TIZ и RM4832 в отношении полноразмерного репликона генотипа 1 а (Blight, et al., 2003) была эквивалентна активности, наблюдаемой для клеток AVA5 (генотип 1b) (табл. 6). 2.2.2. Действие предварительного лечения с использованием NTZ перед комбинированными видами лечения. Исходя из наблюдений в ранних клинических исследованиях с использованием NTZ с пегилированным интерфероном был оценен in vitro эффект режима, включающего три дня лечения NTZ с последующим трехдневным лечением комбинацией NTZ с IFN. Предварительное лечение NTZ (монотерапия) приблизительно в 3 раза усиливало эффективность комбинированного лечения NTZ плюс IFN(табл. 7, фиг. 1 Е и 1F). Однако предварительное лечение не влияло на эффективность комбинированного лечения с использованием 2'CmeC (табл. 7). 2.2.3. Действие сыворотки человека на активность в отношении HCV и цитотоксичность TIZ в клетках.NTZ и его циркулирующий метаболит, TIZ, в высокой степени связаны (99%) с белками плазмы в сыворотке человека. Для оценки действия сыворотки человека на активность в отношении HCV и цитотоксичность TIZ сыворотку человека добавляли к культурной среде в различных концентрациях(табл. 8). Значения CC50, EC50 и EC90 TIZ возрастали по мере увеличения концентраций сыворотки человека до 20%. Значения EC50 и EC90 в присутствии 30% сыворотки человека были схожи со значениями в присутствии 20% сыворотки человека (эффект "плато"), что означало достижение максимальной степени связывания с белками. Уровни HCV и РНК В-актина в необработанных культурах были схожи при различных концентрациях сыворотки человека до 30% (данные не показаны). Более высокие концентрации сыворотки человека значительно снижали жизнеспособность клеток (данные не показаны). В нижеприведенных таблицах RM4863 представляет собой то же соединение, что и соединение 6 в табл. 1. Таблица 3 Относительная активность (мкМ) тестируемых соединений в отношении репликации HBV в культурах клеток 2.2.15 ИР HBV, интермедиаты внутриклеточной репликации РНК HBV. Не наблюдали значительной цитотоксической или противовирусной активности в соответствии с указанной концентрацией. Таблица 4 Относительная активность (мкМ) тестируемых соединений в отношении резистентных к лекарственным средствам мутантов HBV в культурах клеток Не наблюдали значительной противовирусной активности в соответствии с указанной концентрацией.HBV Mutant - генетически модифицированный вариант HBV. Таблица 5 Относительная активность (мкМ) NTZ и ламивудина в отношении уровней репликации и белков HBV в культурах клеток 2.2.15. Концентрации для интерферона выражены в IU/мл. Не наблюдали значительной цитотоксической или противовирусной активности в соответствии с указанной концентрацией. Таблица 7 Действие предварительного лечения NTZ (монотерапия) на комбинированную терапию Значения выражены в виде мкМ концентраций лекарственного средства (сначала указано лекарственное средство в случае комбинаций).Concentration of human serum (%) - концентрация сыворотки человека (%). В табл. 9 представлены данные первичного клеточного анализа репликона HCV. Таблица 9 В табл. 10 представлены данные вторичных клеточных анализов репликона HCV с использованием генотипов 1 В и 1 А. Таблица 10 3.0. Синтез 2-(тиазол-2-илкарбамоил)фенилметансульфоната (11). Соединение, представляющее собой 2-(тиазол-2-илкарбамоил)фенилметансульфонат (11), получали согласно следующей схеме синтеза: 3.1. Синтез 2-(1,3-тиазол-2-илкарбамоил)фенилацетата (501). 2-(Ацетилокси)бензойную кислоту (500, 1,80 г, 10,0 ммоль) помещали в 250 мл круглодонную колбу, оснащенную магнитной мешалкой, вакуумным переходником и мембраной. Добавляли эфир (100 мл) и пиридин (1,00 мл, 12,4 ммоль) с получением прозрачного бесцветного раствора, который охлаждали до 0 на ледяной бане перед добавлением по каплям тионилхлорида (875 мкл, 12,0 ммоль) в течение приблизительно 30 с. Густой белый осадок образовывался при добавлении каждой капли. Реакционную смесь перемешивали в течение 90 мин при 0C перед фильтрованием через бумагу и удалением растворителей под вакуумом. Бикарбонат натрия (3,42 г, 40,7 ммоль) и 1,3-тиазол-2-амин (1,00 г, 10,0 ммоль) помещали в 250 мл круглодонную колбу и добавляли воду (30 мл) и этилацетат (30 мл) с образованием бесцветной двухфазной жидкости, которую быстро перемешивали. Неочищенный хлорангидрид суспендировали в этилацетате (10 мл) и по каплям добавляли к быстро перемешиваемой двухфазной жидкости. Полученную двухфазную жидкость накрывали крышкой и быстро перемешивали при комнатной температуре в течение 2 ч. Две фазы разделяли и водную фазу дважды экстрагировали этилацетатом. Смешанные органические фазы промывали солевым раствором, сушили с помощью MgSO4, фильтровали и концентрировали с получением соединения 501 (1,36 г, 52%) в виде бесцветного порошка, который использовали без очистки. Данные для соединения 501: 1 Н-ЯМР (400 МГц, ДМСО-d6)12,58 (br s, 1H), 7,77 (dd, J=7,4, 1,8 Гц, 1H), 7,62 (ddd, J=7,7, 7,7,1,8 Гц, 1H), 7,54 (d, J=3,6 Гц, 1H), 7,40 (ddd, J=7,7, 7,7, 1,4 Гц, 1H), 7,28 (d, J=3,6 Гц, 1H), 7,27 (dd, J=7,7,1,4 Гц) и 2,22 (s, 3 Н) ppm;(40), 101,0 (100) m/z. 3.2. Синтез 2-гидрокси-N-1,3-тиазол-2-илбензамида (502). 2-(1,3-Тиазол-2-илкарбамоил)фенилацетат (501, 663,3 мг, 2,53 ммоль) добавляли в 25 мл круглодонную колбу, оснащенную магнитной мешалкой и водяным холодильником. Воздушное пространство заполняли осушенным азотом и одной порцией добавляли концентрированную соляную кислоту(15,0 мл). Суспензию нагревали до 50C и хорошо перемешивали. Суспендированные твердые вещества растворялись с образованием прозрачного бесцветного раствора перед выпадением твердых веществ в осадок из реакционной смеси, которую затем охлаждали на ледяной бане, а твердые вещества отфильтровывали через фритту средней пористости и промывали большой порцией воды. Осадок на фильтре промывали на фритте метанолом, собирали и растворитель удаляли под вакуумом с получением соединения 502 (481,5 мг, 86%) в виде бесцветного твердого вещества. Данные для соединения 502: 1 Н-ЯМР (400 МГц, ДМСО-d6)12,07 (br s, 2H), 7,99 (dd, J=7,9, 1,5 Гц, 1H), 7,55 (d, J=3,8 Гц, 1H),7,45 (br t, J=7,5 Гц), 7,27 (br d, J=2,7 Гц) и 6,9-7,05 (m, 2H);MC (ИЭР+) m/z (относительная интенсивность): 221,2 (15), 121,1 (50), 101,0 (100). 3.3. Синтез 2-(1,3-тиазол-2-илкарбамоил)фенилметансульфоната (11). 2-Гидрокси-N-1,3-тиазол-2-илбензамид (502, 267,1 мг, 1,21 ммоль) помещали в 10 мл круглодонную колбу, оснащенную магнитной мешалкой и мембраной с входным отверстием для сухого азота. Добавляли дихлорметан (5,0 мл) и триэтиламин (500 мкл, 3,59 ммоль) с образованием светло-розового раствора. Метансульфонилхлорид (100 мкл, 1,3 ммоль) в дихлорметане (приблизительно 1,0 мл) по каплям добавляли к перемешиваемому раствору в течение приблизительно 30 с и реакционную смесь перемешивали при комнатной температуре в течение приблизительно 20 мин, перед тем как погасить насыщеннымNaHCO3, и экстрагировали дихлорметаном. Смешанные органические фазы промывали солевым раствором, сушили над MgSO4, фильтровали и концентрировали под вакуумом. Неочищенный продукт перекристаллизовывали из смеси гексан/этилацетат с получением (284 мг, 78%) соединения 11 в виде бесцветного кристаллического твердого вещества. Пример 11 2-тиазол-2 илкарбамоил)фенилметансульфонат) отвечает эмпирической формуле C11H10N2O4S и имеет молекулярную массу 298,34. Данные для соединения 11: температура плавления=185,5-187,8C; 1 Н-ЯМР (400 МГц, ДМСО-d6)12,63 (br s, 1H), 7,76 (dd, J=7,9, 1,7 Гц, 1H), 7,67 (ddd, J=7,8, 7,8,1,7 Гц, 1H), 7,55 (d, J=3,5 Гц, 1H), 7,48-7,53 (m, 1H), 7,30 (d, J=3,5 Гц, 1H) и 3,35 (s, 3 Н) ppm;(60), получали согласно следующей схеме синтеза:Ahond, A.; Poupat, С.; Potier, P. Synth. Commun. 2002, 32, 1671), суспендировали в этаноле (7,0 мл) и охлаждали на ледяной бане. Добавляли по каплям раствор S-метилбромэтантиоата (1,371 г,5,0582 ммоль, полученный согласно Praveen Rao, P.N.; Amini, M.; Li, H.; Habeeb, A.G.; Knaus, E.E. J. Med.Chem. 2003, 46, 4872-82) в этаноле (7,0 мл) в течение 3 мин. Суспензия становилась гомогенной в конце добавления, баню убирали и реакционную смесь перемешивали при комнатной температуре. Удаляли растворитель и неочищенное вещество разделяли между дихлорметаном и водой. Органические фазы промывали водой и солевым раствором. Смешанные водные фазы снова экстрагировали дихлорметаном,а смешанные органические фазы сушили безводным MgSO4, фильтровали и концентрировали в вакууме с получением стеклообразного вещества оранжевого цвета (с неприятным запахом). Неочищенное вещество адсорбировали на силикагеле (приблизительно 5 г) с использованием этилацетата и пропускали через слой силикагеля, используя гексан (отбрасывали), а затем смесь гексан:этилацетат 9:1. Элюент выпаривали под вакуумом с получением соединения 504 (589 мг, 50%) в виде бесцветного твердого вещества. Данные для соединения 504: 1 Н-ЯМР (400 МГц, CDCl3)8,93 (br s, 1H), 6,40 (s, 1H), 2,45 (s, 3H) и 1,47 (s, 9 Н) ppm; 13 С-ЯМР (100 МГц, CDCl3)160,3, 151,3, 145,4, 105,7, 82,8 (br), 28,2 и 16,2 ppm; триэтиламин (15 мл) с образованием густой суспензии грязно-белого цвета. Раствор ди-третбутилдикарбоната (6,0546 г, 27,742 ммоль) в тетрагидрофуране (24 мл) добавляли к вышеуказанной суспензии и полученную суспензию перемешивали при комнатной температуре в течение 4 ч. Затем реакционную смесь вливали в воду (100 мл) и водный слой экстрагировали этилацетатом. Смешанные органические фазы промывали насыщенным раствором NaHCO3, солевым раствором, сушили над MgSO4,фильтровали и удаляли растворитель под вакуумом. Неочищенный продукт адсорбировали на силикагеле и элюировали через слой силикагеля, используя смесь гексан:этилацетат 9:1. Элюент собирали и выпаривали с получением соединения 506 (5,42 г, 78%) в виде бесцветного кристаллического твердого вещества. Данные для соединения 506: 1 Н-ЯМР (400 МГц, ДМСО-d6)12,75 (br s, 1H), 7,44 (s, 1H) и 1,48 (s, 9 Н) ppm; 13 С-ЯМР (100 МГц, ДМСО-d6)160,1, 152,9 (br), 139,0, 100,5, 81,7 и 27,8 ppm;(100, M79Br-(CH3)2C=CH2+). 4.3 Синтез трет-бутил(4-бром-1,3-тиазол-2-ил)карбамата (507). Тетрагидрофуран (160 мл) и N,N-диизопропиламин (14 мл, 97 ммоль) смешивали в 3-горлой 500 мл круглодонной колбе, оснащенной магнитной мешалкой, мембраной и внутренним термометром. Полученный раствор охлаждали до 0,8C и медленно добавляли н-бутиллитий в гексане (2,50 М, 38 мл,95 ммоль) в течение приблизительно 5 мин с получением светло-желтого раствора (Твнутр макс=10C), который перемешивали и снова оставляли охлаждаться примерно до 0C. Раствор трет-бутил(5-бром-1,3 тиазол-2-ил)карбамата, 506 (8,74 г, 31,3 ммоль) в тетрагидрофуране (30,0 мл) по каплям добавляли к вышеуказанному раствору в течение 16 мин (Твнутр варьировали от 0,9C до максимального значения 7C). Полученную темно-коричневую реакционную смесь перемешивали в течение 15 мин перед тем, как погасить водой (13 мл), а затем перемешивали еще в течение 5 мин. Добавляли насыщенный водный NH4Cl(250 мл) и этилацетат (250 мл) и разделяли фазы. Водную фазу экстрагировали этилацетатом, а смешанные органические фазы промывали солевым раствором, сушили с помощью MgSO4, фильтровали и концентрировали под вакуумом. Неочищенное вещество адсорбировали на силикагеле с использованием этилацетата и элюировали через слой силикагеля, используя 2 л смеси гекс:EtOAc 9:1. Элюент собирали,а растворители удаляли с получением соединения 507 (8,41 г, 96%) в виде бесцветного твердого вещества. Данные для соединения 507: 1 Н-ЯМР (400 МГц, ДМСО-d6)12,75 (br s, 1H), 7,24 (s, 1 Н) и 1,48 (s, 9 Н) ppm; 13 С-ЯМР (400 МГц, CDCl3)160,6, 152,7 (br), 119,8, 110,6, 81,7 и 27,9 ppm; МС (ИЭР+) m/z (относительная интенсивность): 225,1 (100, M81Br-(CH3)2C=CH2+), 223,1 (100,M79Br-(CH3)2C=CH2+). 4.4. Альтернативный синтез трет-бутил(4-метилтио-1,3-тиазол-2-ил)карбамата (504). трет-Бутил(4-бром-1,3-тиазол-2-ил)карбамат (507, 3,9575 г, 14,177 ммоль), иодид меди(I) (2,7718 г,14,554 ммоль) и метилтиолат натрия (5,0242 г, 71,682 ммоль) смешивали в 100 мл колбе, оснащенной магнитной мешалкой и водяным холодильником и мембраной. Воздушное пространство заполняли сухим азотом и добавляли N,N-диметилформамид (26 мл). Реакционная смесь становилась ярко-желтой, а затем бледнела до тусклой серо-розовой суспензии. Реакционную смесь перемешивали при комнатной температуре в течение приблизительно 1 мин перед тем, как поместить на 136C масляную баню, установленную на 140C, и перемешиванием. В течение первых 5-10 мин нагревания цвет тускнел до светложелтого и реакционная смесь становилась гомогенной. Выделение газов/кипение наблюдали при прекращении перемешивания. Реакционную смесь охлаждали до комнатной температуры после 15 ч при 140C, и анализ методом ВЭЖХ показал, что исходное вещество полностью израсходовалось. Реакционную смесь разбавляли этилацетатом (приблизительно 200 мл) и фильтровали через слой целита, элюируя этилацетатом. Смешанные органические фазы промывали смесью 1 М HCl/насыщенный раствор NH4Cl 1:1 (250 мл), в результате чего получали густую эмульсию. Затем всю смесь фильтровали через аморфную целлюлозу и разделяли фазы. Затем органические фазы промывали 0,5 М HCl и насыщенным раствором NaHCO3. Другой очень мелкодисперсный порошок выпадал из раствора при действии основанием. Суспензию снова фильтровали через целит и полученный раствор промывали солевым раствором,сушили с помощью MgSO4, фильтровали и концентрировали под вакуумом с получением зеленого масла(3,17 г). Неочищенный продукт адсорбировали на силикагеле (приблизительно 15 г) EtOAc, сушили в вакууме и элюировали через слой силикагеля (приблизительно 80 г), используя 500 мл гексана (отбрасывали) и 2 л смеси гексан:этилацетат 9:1, который концентрировали в вакууме с получением соединения 504 (2,41 г, 69%) в виде твердого вещества грязно-белого цвета. Данные для соединения 504 приведены выше. 4.5. Синтез 4-(метилтио)-1,3-тиазол-2-амина (508). трет-Бутил[4-(метилтио)-1,3-тиазол-2-ил]карбамат (504, 3,17 г, 12,9 ммоль) растворяли в метиленхлориде (130 мл) и добавляли трифторуксусную кислоту (54 мл) с получением ярко-желтого раствора. Раствор накрывали и перемешивали при комнатной температуре в течение 8 ч, что приводило к завершению реакции. Растворители удаляли под вакуумом и полученное густое масло суспендировали в 0,1 МHCl (50 мл) и удаляли растворитель. Это повторяли один раз и полученные твердые вещества суспендировали в этилацетате (20 мл) и выпаривали с получением мелкодисперсного сыпучего розового твердого вещества (2,0 г). Твердые вещества ресуспендировали в этилацетате (20 мл), обрабатывали ультразвуком и фильтровали через фритту средней пористости, промывая этилацетатом (приблизительно 30 мл). Бледно-лиловые твердые вещества разделяли между насыщенным раствором NaHCO3 (100 мл) и дихлорметаном (100 мл). Фазы разделяли и водную фазу один раз экстрагировали дихлорметаном. Затем смешанные органические фазы промывали солевым раствором, сушили безводным MgSO4, фильтровали и концентрировали под вакуумом с получением соединения 508 (1,33 г, 71%) в виде темного масла, которое затвердевало до кристаллического твердого вещества при охлаждении сухим льдом и отстаивании. Данные для соединения 508: 1 Н-ЯМР (400 МГц, ДМСО-d6)7,06 (br s, 2H), 6,11 (s, 1 Н) и 2,36 (s, 3 Н) ppm; 13 С-ЯМР (100 МГц, ДМСО-d6)168,5, 144,7, 97,37 и 14,7 ppm; МС (ИЭР+) m/z (относительная интенсивность): 147,1 (100, М+Н+), 132,0 (20) и 105,0 (40). 4.6. Синтез 2-[4-(метилтио)-1,3-тиазол-2-ил]карбамоилфенилацетата (509). 4-(Метилтио)-1,3-тиазол-2-амин (508, 672 мг, 4,60 ммоль) растворяли в тетрагидрофуране (10,0 мл) с получением раствора цвета арбуза и охлаждали до 0C. По каплям добавляли раствор ацетилсалицилоилхлорида (0,9915 г) в тетрагидрофуране (1,4 мл) в течение 1 мин, баню убирали и реакционную смесь перемешивали, в то же время позволяя реакционной смеси нагреваться до комнатной температуры в течение приблизительно 40 мин. По каплям добавляли триэтиламин (0,670 мл, 4,81 ммоль) в течение 3 мин с получением темной суспензии, которую перемешивали в течение 15 ч. Твердые вещества удаляли из суспензии путем фильтрования через фритту средней пористости, твердые вещества промывали ТГФ(приблизительно 20 мл) и полученный раствор концентрировали, растворяли в этилацетате, фильтровали через слой магнезола с удалением полярных окрашенных примесей и концентрировали с получением кристаллического твердого вещества оранжевого цвета (1,35 г). Данное неочищенное вещество адсорбировали на силикагеле этилацетатом и очищали путем MPLC (жидкостной хроматографии среднего давления) (элюируя 1 л смеси гекс:EtOAc 6:1, 4:1, 3:1 и 2:1). Фракции объединяли и выпаривали с получением соединения 509 (660,8 мг, 47%) в виде почти бесцветного твердого вещества. Данные для соединения 509: 1 Н-ЯМР (400 МГц, ДМСО-d6)12,69 (br s, 1H), 7,77 (dd, J=7,8, 1,4 Гц, 1H), 7,62 (ddd, J=7,8, 7,8, 1,4 Гц, 1H), 7,40 (ddd, J=7,8, 7,8, 1,4 Гц, 1H), 7,27 (dd, J=8,0, 1,4 Гц, 1H), 6,87 (s, 1H), 2,48 (s, 3 Н) и 2,22 (s, 3H)MC (ИЭР-) m/z (относительная интенсивность): 265,3 (80, M-H-). 4.7. Синтез 2-[4-(метилсульфонил)-1,3-тиазол-2-ил]карбамоилфенилацетата (62). 2-[4-(Метилтио)-1,3-тиазол-2-ил]карбамоилфенилацетат (509, 201,1 мг, 0,6521 ммоль) растворяли в метиленхлориде (21 мл) и одной порцией добавляли м-хлорпербензойную кислоту (317,1 мг,1,378 ммоль). Реакционную смесь накрывали и перемешивали при комнатной температуре в течение 100 мин, пока анализ смеси методом ВЭЖХ не показал полное превращение в целевой сульфон. Реакцию гасили 20% водным Na2S2O3 (20 мл), перемешивали в течение 5 мин и разделяли фазы. Органические фазы промывали насыщенным раствором NaHCO3 и солевым раствором, сушили безводным MgSO4,фильтровали и концентрировали под вакуумом с получением бесцветного белого твердого вещества(211,0 мг). Неочищенный продукт перекристаллизовывали из нагретой в колбе с обратным холодильником смеси этилацетат/гексан (5,0:2,0 мл), фильтровали, промывали гексаном и сушили под вакуумом с получением соединения 62 (133,4 мг, 61%) в виде бесцветного кристаллического твердого вещества. Данные для соединения 62: 1 Н-ЯМР (400 МГц, ДМСО-d6)13,19 (br s, 1H), 8,11 (s, 1H), 7,82 (d, J=7,8 Гц, 1H), 7,65 (dd,J=7,8, 7,8 Гц, 1H), 7,42 (dd, J=7,8, 7,8 Гц, 1H), 7,29 (d, J=7,8 Гц, 1H), 3,23 (s, 3 Н) и 2,23 (s, 3 Н) ppm; 13 С-ЯМР (100 МГц, ДМСО-d6)168,8, 164,8, 160,1, 148,6, 148,3, 133,1, 129,6, 126,0, 125,9, 123,4,120,2, 42,0 и 20,7 ppm; МС (ИЭР+) m/z (относительная интенсивность): 341,1 (10, М+Н+), 299,1 (20), 163,2 (15), 121,1 (100),100,1 (15) и 83,0 (50) m/z; МС (ИЭР-) m/z (относительная интенсивность): 339,2 (10, М-Н-) m/z. 4.8. Синтез 2-гидрокси-N-[4-(метилсульфонил)-1,3-тиазол-2-ил]бензамида (60). 2-[4-(Метилсульфонил)-1,3-тиазол-2-ил]карбамоилфенилацетат (62, 118,2 мг, 0,3473 ммоль) суспендировали в концентрированной соляной кислоте (2,0 мл) и быстро перемешивали. Суспензия мгновенно становилась гомогенной, а затем снова происходило выпадение осадка. Суспензию быстро перемешивали при 50C в течение 16 ч перед охлаждением и фильтрованием через пористую воронку небольшого размера. Твердые вещества промывали водой (приблизительно 5 мл) и сушили под вакуумом с получением соединения 60 в виде бесцветного порошка. Данные для соединения 60: температура плавления=231-235C (запаянная трубка); 1 Н-ЯМР (400 МГц, ДМСО-d6)12,30 (br s, 1H), 11,64 (br s, 1H), 8,12 (s, 1H), 7,96 (dd, J=7,6, 1,6 Гц,1H), 7,65 (ddd, J=7,6, 7,6, 1,6 Гц, 1H), 7,07 (br d, J=7,6 Гц, 1H), 7,01 (br dd, J=7,6, 7,6 Гц, 1H) и 3,25 (s, 3H)MC (ИЭР-) m/z (относительная интенсивность): 297,2 (100, M-H-) m/z. 5.0. Общие процедуры синтеза 4-гидрокси-N-[4-(метилсульфонил)-1,3-тиазол-2-ил]бензамида (84) и 3-гидрокси-N-[4-(метилсульфонил)-1,3-тиазол-2-ил]бензамида (87). Соединения 4-гидрокси-N-[4-(метилсульфонил)-1,3-тиазол-2-ил]бензамид (84) и 3-гидрокси-N-[4(метилсульфонил)-1,3-тиазол-2-ил]бензамид (87) получали согласно следующей общей схеме синтеза: 5.1. Синтез 4-(хлоркарбонил)фенилацетата (510). Тионилхлорид (11,1 мл, 15,3 ммоль) добавляли к 4-ацетоксибензойной кислоте (2,50 г, 13,9 ммоль) и реакционную смесь нагревали в колбе с обратным холодильником. Реакционную смесь охлаждали после нагревания в течение 3,5 ч и концентрировали под вакуумом с получением бесцветного масла. К остатку добавляли толуол и смесь концентрировали под вакуумом с удалением остаточного тионилхлорида. Данную процедуру повторяли еще два раза с получением соединения 510 (2,54 г, 92%) в виде бесцветного масла. Указанное вещество использовали на следующей стадии без дополнительной очистки. Данные для соединения 510: 1 Н-ЯМР (400 МГц, CDCl3)8,18 (d, J=8,9 Гц, 2H), 7,29 (d, J=8,9 Гц, 2H), 2,36 (s, 3 Н) ppm. 5.2. Синтез 3-(хлоркарбонил)фенилацетата (511). С использованием вышеуказанной процедуры в результате реакции тионилхлорида (11,1 мл,15,3 ммоль) и 3-ацетоксибензойной кислоты (2,50 г, 13,9 ммоль) получали соединение 511 (2,72 г, 99%) в виде бесцветного масла. Данное вещество использовали на следующей стадии без дополнительной очистки. Данные для соединения 511: 1 Н-ЯМР (400 МГц, CDCl3)8,03 (ddd, J=8, 2, 1 Гц, 1H), 7,87 (t, J=2 Гц, 1H), 7,52-7,60 (m, 1H), 7,46(ddd, J=8, 2, 1 Гц, 1H), 2,37 (s, 3 Н) ppm. 5.3. Синтез 4-[4-(метилтио)-1,3-тиазол-2-ил]карбамоилфенилацетата (82). К раствору соединения 510 (0,815 г, 4,10 ммоль) и сухого ТГФ (20,0 мл) добавляли раствор триэтиламина (0,572 мл, 4,10 ммоль) и сухого ТГФ (5,00 мл) с последующим добавлением раствора соединения 508 (0,500 г, 3,42 ммоль), растворенного в осушенном ТГФ (15,0 мл). Реакционную смесь перемешивали при комнатной температуре. По окончании перемешивания в течение ночи реакционную смесь концентрировали под вакуумом. Остаток разделяли между насыщенным водным бикарбонатом натрия и дихлорметаном. Фазу дихлорметана промывали второй раз насыщенным раствором бикарбоната натрия, а затем дважды водной 1 М HCl. Фазу дихлорметана сушили над безводным сульфатом магния и концентрировали в вакууме с получением неочищенного соединения 82 (1,15 г, 100%) в виде коричневого твердого вещества. Неочищенный продукт суспендировали в диэтиловом эфире, перемешивали и фильтровали. Фильтрационную пластину несколько раз промывали эфиром и сушили в вакууме с получением соединения 82 (0,692 г, 63%) в виде светло-коричневого твердого вещества. Данные для соединения 82: температура плавления=185,7-188,7C; 1 Н-ЯМР (400 МГц, ДМСО-d6)12,8 (s, 1H), 8,14 (d, J=8,9 Гц, 2H), 7,32 (d, J=8,9 Гц, 2H), 6,89 (s, 1H),2,50 (s, 3H), 2,31 (s, 3 Н) ppm;MC (ИЭР-) m/z (относительная интенсивность): 111,0 (16), 203,2 (31), 307,2 (100). 5.4. Синтез 3-[4-(метилтио)-1,3-тиазол-2-ил]карбамоилфенилацетата (85). К раствору соединения 511 (0,815 г, 4,10 ммоль) и осушенного ТГФ (25,0 мл) добавляли триэтиламин (0,572 мл, 4,10 ммоль) с последующим добавлением раствора соединения 508 (0,500 г,3,42 ммоль), растворенного в осушенном ТГФ (10,0 мл). Реакционную смесь перемешивали при комнатной температуре. По окончании перемешивания в течение ночи реакционную смесь концентрировали под вакуумом. Остаток растворяли в этилацетате и один раз промывали водой, три раза насыщенным водным раствором бикарбоната натрия и один раз солевым раствором. Раствор этилацетата сушили над безводным сульфатом натрия и концентрировали под вакуумом с получением неочищенного соединения 85 (1,28 г, 100%) в виде красной пены. Неочищенный продукт растворяли в дихлорметане. К раствору дихлорметана добавляли силикагель и суспензию концентрировали под вакуумом. Остаток помещали на 90 г картридж с силикагелем и пропускали через колонку, используя для элюирования раствор 20% этилацетата в гексане. Подходящие фракции основного продукта смешивали и концентрировали под вакуумом с получением соединения 85 (0,614 г, 58%) в виде твердого вещества рыжеватого цвета. Данное вещество использовали на следующей стадии без дополнительной очистки. Данные для соединения 85: 1 Н-ЯМР (400 МГц, ДМСО-d6)12,8 (s, 1H), 8,01 (d, J=8 Гц, 1H), 7,87 (t, J=2 Гц, 1H), 7,60 (t, J=8 Гц,1H), 7,42 (ddd, J=8, 2, 1 Гц, 1H), 6,90 (s, 1H), 2,50 (s, 3H), 2,32 (s, 3 Н) ppm. 5.5. Синтез 4-[4-(метилсульфонил)-1,3-тиазол-2-ил]карбамоилфенилацетата (83). Раствор м-хлорпербензойной кислоты (1,0 г, 6,0 ммоль, максимум 77%), растворенной в дихлорметане (30 мл), добавляли к суспензии соединения 82 (0,63 г, 2,0 ммоль) в дихлорметане (60 мл). Реакционная смесь становилась гомогенной, и темно-коричневый раствор становился более светлым с течением времени. По прошествии 1,25 ч к реакционной смеси добавляли насыщенный водный тиосульфат натрия и реакционную смесь энергично перемешивали в течение 20 мин. Желтую смесь разбавляли водой и полученные фазы разделяли. Органическую фазу дважды промывали насыщенным водным бикарбонатом натрия, один раз солевым раствором, сушили безводным сульфатом натрия и концентрировали под вакуумом с получением бежевого твердого вещества. Неочищенный продукт перекристаллизовывали из смеси 50% этилацетат/гексан (220 мл). По прошествии 4 дней суспензию фильтровали, фильтрационную пластину промывали гексаном, а затем диэтиловым эфиром и сушили под вакуумом при 55C в течение ночи с получением соединения 83 (0,20 г, 30%) в виде твердого вещества светло-оранжевого цвета. В результате концентрирования фильтрата в вакууме с последующим осуществлением растирания полученного остатка с диэтиловым эфиром и отфильтровыванием полученного твердого вещества получали дополнительные 45 мг соединения 83 в виде рыжеватого твердого вещества. Данные для соединения 83: температура плавления=242-246C (с разложением (dec; 1 Н-ЯМР (400 МГц, ДМСО-d6)13,2 (s, 1H), 8,18 (d, J=8,7 Гц, 2H), 8,13 (s, 1H), 7,34 (d, J=8,7 Гц, 2H),3,25 (s, 3 Н) и 2,32 (s, 3H) ppm;MC (ИЭР-) m/z (относительная интенсивность): 339,3 (100). 5.6. Синтез 3-[4-(метилсульфонил)-1,3-тиазол-2-ил]карбамоилфенилацетата (86). С использованием процедуры для примера 83, м-хлорпербензойной кислоты (1,0 г, 6,0 ммоль, максимум 77%), растворенной в дихлорметане (30 мл), и соединения 85 (0,61 г, 2,0 ммоль), растворенного в дихлорметане (30 мл), получали неочищенное соединение 86 в виде белого твердого вещества. Неочищенный продукт перемешивали в диэтиловом эфире (30 мл) в течение 30 мин, фильтровали и сушили на воздухе с получением соединения 86 (0,59 г, 87%) в виде белого твердого вещества. Данные для соединения 86: температура плавления=209-212C;MC (ИЭР-) m/z (относительная интенсивность): 339,3 (100). 5.7. Синтез 4-гидрокси-N-[4-(метилсульфонил)-1,3-тиазол-2-ил]бензамида (84). 2 М соляную кислоту (3,0 мл) добавляли к суспензии соединения 83 (0,12 г, 0,35 ммоль) в тетрагидрофуране (3,0 мл) и полученную суспензию нагревали в колбе с обратным холодильником. Реакционная смесь становилась гомогенной при нагревании. По окончании нагревания в колбе с обратным холодильником в течение 1,5 ч реакционную смесь оставляли охлаждаться до комнатной температуры, а затем разделяли между диэтиловым эфиром и водой. Фазу эфира промывали водой, насыщенным водным бикарбонатом натрия и солевым раствором. Фазу эфира сушили безводным сульфатом натрия и концентрировали под вакуумом. Остаток растирали с этилацетатом, растворитель удаляли в потоке азота и полученное твердое вещество сушили под вакуумом при 55C с получением соединения 84 (0,086 г, 82%) в виде светло-желтого твердого вещества. Данные для соединения 84: температура плавления=253-255C (с разложением (dec; 1 Н-ЯМР (400 МГц, ДМСО-d6)12,9 (s, 1H), 10,4 (s, 1H), 8,07 (s, 1H), 8,03 (d, J=8,7 Гц, 2H), 6,89 (d,J=8,7 Гц, 2 Н) и 3,24 (s, 3 Н) ppm;MC (ИЭР-) m/z (относительная интенсивность): 297,3 (100). 5.8. Синтез 3-гидрокси-N-[4-(метилсульфонил)-1,3-тиазол-2-ил]бензамида (87). С использованием вышеуказанной процедуры для примера 84, соединения 86 (0,36 г, 1,0 ммоль),растворенного в тетрагидрофуране (10 мл), и 2 М соляной кислоты (10 мл) получали соединение 87(0,29 г, 91%) в виде белого твердого вещества после концентрирования фазы эфира под вакуумом. Данные для соединения 87: температура плавления=258-259C (с разложением (dec; 1 Н-ЯМР (400 МГц, ДМСО-d6)13,1 (s, 1H), 9,88 (s, 1H), 8,11 (s, 1H), 7,59 (d, J=8 Гц, 1H), 6,47 (s,1H), 7,35 (t, J=8 Гц, 1H), 7,05 (dd, J=8, 2 Гц, 1 Н) и 3,25 (s, 3 Н) ppm;MC (ИЭР-) m/z (относительная интенсивность): 297,3 (100). 6.0. Общие процедуры синтеза 4-гидрокси-N-[5-(метилсульфонил)-1,3-тиазол-2-ил]бензамида (80) и 3-гидрокси-N-[5-(метилсульфонил)-1,3-тиазол-2-ил]бензамида (81). Соединения 4-гидрокси-N-[5-(метилсульфонил)-1,3-тиазол-2-ил]бензамид (80) и 3-гидрокси-N-[5(метилсульфонил)-1,3-тиазол-2-ил]бензамид (81) получали согласно следующей общей схеме синтеза: 6.1. Синтез 5-(метилтио)-1,3-тиазол-2-амина (512). Раствор тиометоксида натрия (1,09 г, 14,8 ммоль), растворенного в метаноле (18,0 мл), добавляли к суспензии 2-амино-5-бромтиазол моногидробромида 505 (2,50 г, 14,0 ммоль) в безводном этаноле(18,0 мл) в течение 5 мин. Реакционная смесь становилась гомогенной. К реакционной смеси добавляли второй раствор тиометоксида натрия (1,09 г, 14,8 ммоль), растворенного в метаноле (12,0 мл). Реакционную смесь нагревали до 45C в течение 40 мин, затем нагревание прекращали и реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи, пока тонкослойная хроматография(EtOAc/гексан 1:1) не показала израсходование большей части исходного вещества наряду с образованием нового продукта. К реакционной смеси добавляли дополнительное количество тиометоксида натрия(0,20 г, 2,85 ммоль) и снова нагревали до 50C. По окончании нагревания в течение 2 ч реакционную смесь охлаждали до комнатной температуры и концентрировали под вакуумом. Остаток растворяли в дихлорметане и три раза промывали водой, один раз солевым раствором, сушили над безводным сульфа- 28019357 том натрия и концентрировали под вакуумом с получением соединения 512 (1,12 г, 55%) в виде оранжевого твердого вещества. Данные для соединения 512: 1 Н-ЯМР (400 МГц, ДМСО-d6)7,20 (s, 2H), 6,97 (s, 1H), 2,29 (s, 3 Н) ppm. 6.2. Синтез 4-[5-(метилсульфанил)-1,3-тиазол-2-ил]карбамоилфенилацетата (76). Согласно процедуре синтеза в примере 82, используя промежуточное соединение 510 (0,815 г,4,10 ммоль), растворенное в осушенном ТГФ (25,0 мл), триэтиламин (0,572 мл, 4,10 ммоль) и соединение 512 (0,500 г, 3,42 ммоль), растворенное в сухом ТГФ (10,0 мл), получали соединение 76 (0,887 г, 84%) в виде рыжеватого твердого вещества. Данные для соединения 76: температура плавления=193,3-195,5C; 1 Н-ЯМР (400 МГц, ДМСО-d6)12,8 (br s, 1H), 8,13 (d, J=8,71 Гц, 2H), 7,58 (s, 1H), 7,32 (d, J=8,71 Гц,2H), 2,46 (s, 3H), 2,31 (s, 3 Н) ppm;MC (ИЭР-) m/z (относительная интенсивность): 292,2 (100), 307,3 (48). 6.3. Синтез 3-[5-(метилсульфанил)-1,3-тиазол-2-ил]карбамоилфенилацетата (77). Согласно процедуре синтеза в примере 85, используя промежуточное соединение 511 (0,815 г,4,10 ммоль), растворенное в осушенном ТГФ (25,0 мл), триэтиламин (0,572 мл, 4,10 ммоль) и соединение 512 (0,500 г, 3,42 ммоль), растворенное в сухом ТГФ (10,0 мл), получали соединение 77 (0,681 г, 65%) в виде рыжеватого твердого вещества. Данные для соединения 77: температура плавления=135,2-136,2C; 1 Н-ЯМР (400 МГц, ДМСО-d6)12,8 (s, 1H), 7,96-8,03 (m, 1H), 7,86 (t, J=2 Гц, 1H), 7,57-7,63 (m, 2H),7,43 (ddd, J=8, 2, 1 Гц, 1H), 2,47 (s, 3H), 2,32 (s, 3 Н) ppm;MC (ИЭР-) m/z (относительная интенсивность): 292,3 (100), 307,3 (49). 6.4. Синтез 4-[5-(метилсульфонил)-1,3-тиазол-2-ил]карбамоилфенилацетата (78). Раствор м-хлорпербензойной кислоты (0,611 г, 2,73 ммоль, максимум 77%), растворенной в дихлорметане (10,0 мл), по каплям добавляли к раствору соединения 76 (0,841 г, 2,73 ммоль), растворенного в дихлорметане (35,0 мл), и реакционную смесь перемешивали при комнатной температуре в течение 1,25 ч. Второй раствор м-хлорпербензойной кислоты (0,611 г, 2,73 ммоль), растворенной в дихлорметане(10,0 мл), добавляли к реакционной смеси в течение 15 мин и полученный раствор перемешивали в течение еще 2,5 ч при комнатной температуре. Реакционную смесь разделяли между дихлорметаном и насыщенным водным тиосульфатом натрия. Органическую фазу промывали насыщенным водным тиосульфатом натрия, дважды насыщенным водным бикарбонатом натрия и один раз солевым раствором. Фазу дихлорметана концентрировали под вакуумом (без высушивания) с получением неочищенного соединения 78 (0,80 г, 86%). Неочищенный продукт перемешивали с хлороформом (250 мл) и фильтровали суспензию. Пластину один раз промывали хлороформом и сушили на воздухе с получением соединения 78(0,559 г, 60%) в виде белого твердого вещества. Данные для соединения 78: температура плавления=279,6-280,6C; 1 Н-ЯМР (400 МГц, ДМСО-d6)13,3 (br s, 1H), 8,20 (s, 1H), 8,18 (d, J=8,71 Гц, 2H), 7,35 (d, J=8,71 Гц,2H), 3,39 (s, 3H), 2,32 (s, 3 Н) ppm;MC (ИЭР-) m/z (относительная интенсивность): 259,3 (17), 339,3 (100). 6.5. Синтез 3-[5-(метилсульфонил)-1,3-тиазол-2-ил]карбамоилфенилацетата (79). Раствор м-хлорпербензойной кислоты (0,458 г, 2,04 ммоль, максимум 77%), растворенной в дихлорметане (8,0 мл), по каплям добавляли к раствору соединения 77 (0,630 г, 2,04 ммоль), растворенного в дихлорметане (25,0 мл), в течение 15 мин. Реакционную смесь перемешивали при комнатной температуре в течение 1,25 ч. Затем второй раствор м-хлорпербензойной кислоты (0,458 г, 2,04 ммоль), растворенной в дихлорметане (8,0 мл), добавляли к реакционной смеси в течение 10 мин и перемешивали в течение еще 4,5 ч при комнатной температуре. Затем реакционную смесь разделяли между дихлорметаном и насыщенным водным тиосульфатом натрия. Органическую фазу снова промывали насыщенным водным тиосульфатом натрия, дважды насыщенным водным бикарбонатом натрия и один раз солевым раствором. Фазу дихлорметана сушили над сульфатом магния и концентрировали под вакуумом с получением неочищенного соединения 79 (0,704 г, 100%), загрязненного следами остаточной 3-хлорбензойной кислоты. Неочищенный продукт снова растворяли в этилацетате и три раза промывали насыщенным водным бикарбонатом натрия, сушили сульфатом магния и концентрировали под вакуумом с получением рыжеватого твердого вещества. Остаток растворяли в ТГФ и светло-коричневый раствор фильтровали через слой магнезола.
МПК / Метки
МПК: C07D 417/12, C07D 277/46, A61K 31/426, A61P 31/14
Метки: алкилсульфонилзамещенные, инфекции, n-(тиазол-2-ил)бензамиды, гепатита, вирусной, лечения, применение
Код ссылки
<a href="https://eas.patents.su/30-19357-alkilsulfonilzameshhennye-n-tiazol-2-ilbenzamidy-i-ih-primenenie-dlya-lecheniya-virusnojj-infekcii-gepatita-c.html" rel="bookmark" title="База патентов Евразийского Союза">Алкилсульфонилзамещенные n-(тиазол-2-ил)бензамиды и их применение для лечения вирусной инфекции гепатита c</a>
Аналог тиофена для лечения вирусной инфекции гепатита с

Номер патента: 16457
Опубликовано: 30.05.2012
Авторы: Чань Чунь Кун Лаваль, Яннопулос Константин Г., Кумар Дас Санджой, Вайанкур Луи, Дени Реал, Фалардо Ги, Пуассон Карл
МПК: A61K 31/4196, A61K 31/4535, A61K 31/381...
Метки: аналог, гепатита, тиофена, лечения, вирусной, инфекции
Формула / Реферат:
1. Соединение, представляющее собойили его фармацевтически приемлемая соль.2. Фармацевтическая композиция, содержащая соединение по п.1 и по меньшей мере один фармацевтически приемлемый носитель или эксципиент.3. Фармацевтическая комбинация, содержащая соединение по п.1 и по меньшей мере один дополнительный агент.4. Фармацевтическая комбинация по п.3, где указанный по меньшей мере один дополнительный агент выбирают из ингибиторов вирусной...
Конъюгаты белок-полимер и способ лечения инфекции вируса гепатита c или гепатита b

Номер патента: 18111
Опубликовано: 30.05.2013
Автор: Линь Ко-Чун
МПК: A61K 47/48
Метки: лечения, белок-полимер, конъюгаты, способ, инфекции, вируса, гепатита
Формула / Реферат:
1. Конъюгат белок-полимер формулы Iв которой каждый из R1, R2, R3, R4 и R5 независимо представляет собой H, C1-5алкил, С2-5алкенил, C2-5алкинил, арил, гетероарил, C3-8циклоалкил или C3-8гетероциклоалкил;каждый из А1 и А2 независимо представляет собой полиэтиленгликоль;каждый из G1, G2 и G3 независимо представляет собой связь или линкерную функциональную группу;Р представляет собой белковый фрагмент;m равно 0 или целому числу 1-10 иn равно...
Применение [d-meala]3-[etval]4-циклоспорина для лечения инфекции гепатита с и фармацевтическая композиция, включающая [d-meala]3-[etval]4-циклоспорин

Номер патента: 12650
Опубликовано: 30.12.2009
Авторы: Вуаньо Грегуар, Моверне Ролан-Ив, Дюмон Жан-Морис, Скальфаро Пьетро
МПК: A61K 38/13, A61P 31/14
Метки: применение, фармацевтическая, композиция, инфекции, лечения, гепатита, d-meala]3-[etval]4-циклоспорина, d-meala]3-[etval]4-циклоспорин, включающая
Формула / Реферат:
1. Применение [D-MeAla]3-[EtVal]4-CsA для изготовления лекарственного средства, предназначенного для лечения инфекции вируса гепатита С (HCV) или для предотвращения рецидива HCV у пациента. 2. Применение по п.1, отличающееся тем, что указанное лекарственное средство дополнительно содержит, по меньшей мере, второй компонент, который является активным в отношении инфекции HCV. 3. Фармацевтическая композиция для лечения инфекции HCV или для...
Производные 6-[2-(фосфонометокси)алкокси]пиримидинов, обладающие антивирусной активностью, способ их получения (варианты) и способ лечения вирусной инфекции

Номер патента: 6020
Опубликовано: 25.08.2005
Авторы: Голи Антонин, Бальзарини Ян М.Р., Де Клерк Эрик Десире Алиса
МПК: C07F 9/6512, A61K 31/675
Метки: получения, инфекции, 6-[2-(фосфонометокси)алкокси]пиримидинов, способ, активностью, лечения, варианты, антивирусной, производные, обладающие, вирусной
Формула / Реферат:
1. Соединение формулы I где R1 означает водород, амино или метилсульфанил, R2 означает водород, метил, галоген, -N(R5)2, гидрокси, защищенную гидроксигруппу или группу формулы Ia R3 независимо означает водород, метил, гидроксиметил, галогенметил или защищенный гидроксиметил, R4 означает водород или галоген, X независимо означает кислород, серу или связь, Z независимо означает гидрокси, сложный эфир или амид, R5 независимо означает водород,...
Производные &beta-d-нуклеозида в качестве лекарственного средства для лечения инфекции вируса гепатита c у хозяина

Номер патента: 7178
Опубликовано: 25.08.2006
Авторы: Соммадосси Жан-Пьер, Лаколла Пауло
МПК: A61K 31/7076, A61P 31/14, A61K 31/7068...
Метки: beta-d-нуклеозида, качестве, лечения, гепатита, средства, производные, инфекции, вируса, лекарственного, хозяина
Формула / Реферат:
1. Применение эффективного против вируса количества производного b-D-нуклеозида структуры или его фармацевтически приемлемой соли или пролекарства, необязательно, в фармацевтически приемлемом носителе или разбавителе при получении лекарственного средства для лечения инфекции вируса гепатита С у хозяина. 2. Применение эффективного против вируса количества производного b-D-нуклеозида структуры или его фармацевтически приемлемой соли или...
Предыдущий патент: Эпихлоргидрин, способ получения и применение
Следующий патент: Аллель- и изотип-специфическая интервенция в ассоциированные с аутоиммунными заболеваниями молекулы класса ii мнс при помощи пептидов
Случайный патент: Новые замещенные производные имидазола.