Хиназолиновые соединения
Номер патента: 21439
Опубликовано: 30.06.2015
Авторы: Филп Джоанн, Калландер Лара С., Лоухорн Брайан Гриффин




























Формула / Реферат
1. Соединение согласно формуле I

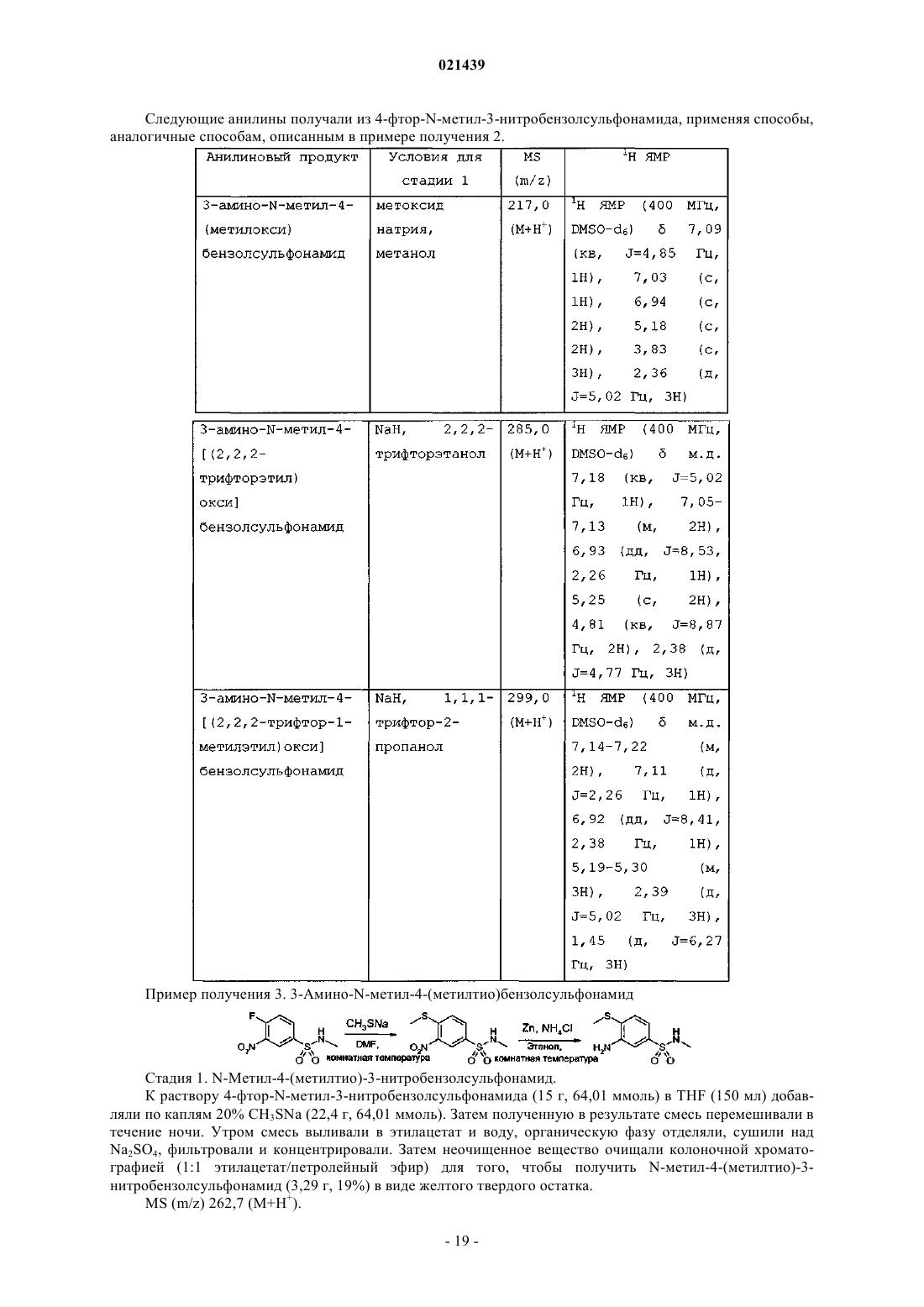
в которой R1 представляет собой C1-C4-алкил;
R2 представляет собой Н или галоген;
R3 представляет собой Н, галоген, C1-C8-алкил, C1-C8-галогеналкил, гидроксил, гидрокси-C1-C8-алкил-, C1-C8-алкокси, C3-C8-циклоалкилокси, C1-C8-галогеналкокси, C1-C8-алкилтио-, C1-C8-галогеналкилтио-, C3-C8-циклоалкилтио-, C1-С8-алкилсульфонил, C1-C8-галогеналкилсульфонил, С3-С8-циклоалкилсульфонил, фенил, 5-членный гетероарил или -N(Ra)(Rb), где указанный 5-членный гетероарил содержит один кольцевой гетероатом азота, кислорода или серы и необязательно содержит 1, 2 или 3 дополнительных кольцевых атома азота и указанный упомянутый фенил или 5-членный гетероарил необязательно замещен от одного до трех раз, независимо, галогеном, C1-C6-алкилом, C1-C4-галогеналкилом, C1-C6-алкокси, C1-C4-галогеналкокси, гидрокси-C1-C4-алкилом или -N(Ra)(Rb);
каждый Ra независимо представляет собой C1-C4-алкил, в котором упомянутый C1-C4-алкил необязательно замещен от одного до трех раз, независимо, галогеном, гидроксилом, C1-C6-алкокси, амино, C1-C6-алкиламино, ди-C1-C6-алкиламино, -СО2Н, -CO2-C1-C6-алкилом, -CONH2, -CONH-C1-C6-алкилом или -CON-(C1-C6-алкил)(C1-C6-алкилом);
Rb представляет собой C1-C4-алкил или
Ra и Rb, взятые вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членное гетероциклическое кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из азота, кислорода и серы, где упомянутое кольцо необязательно замещено от одного до двух раз, независимо, галогеном, C1-C4-алкилом, C1-C4-галогеналкилом, амино, C1-C4-алкиламино, ди-C1-C4-алкиламино, гидроксилом, гидрокси-C1-C4-алкилом, оксо, C1-C4-алкокси, C1-C4-галогеналкокси или C1-C4-алкокси-C1-C4-алкилом;
R4 представляет собой Н;
каждый R5, R6 и R7 независимо представляет собой Н, галоген, нитро, амино, C1-C4-алкиламино, ди-C1-C4-алкиламино, гидроксил или ORc;
каждый Rc независимо представляет собой C1-C8-алкил или 5-6-членный гетероциклоалкил, где упомянутый C1-C8-алкил необязательно замещен от одного до трех раз, независимо, галогеном, гидроксилом, C1-C4-алкокси, амино, C1-C4-алкиламино или -N(Ra)(Rb) и указанный 5-6-членный гетероциклоалкил выбирают из группы, состоящей из пирролидинила, тетрагидрофуранила, тетрагидротиенила, дигидрофурила, оксазолинила, тиазолинила, пиразолинила, пиперидила (или пиперидинила), пиперазинила, морфолинила, тетрагидропиранила, дигидропиранила, 1,3-диоксанила, тетрагидро-4Н-1,4-тиазинила, 1,4-диоксанила, 1,3-оксатианила и 1,3-дитианила; или
R6 и R7, взятые вместе, представляют собой -O-C1-C2-алкил-О-;
или его фармацевтически приемлемая соль.
2. Соединение или фармацевтически приемлемая соль по п.1, в котором R3 представляет собой Н, галоген, C1-C6-алкил, C1-C6-галогеналкил, C1-C6-алкокси, C5-C6-циклоалкилокси, С1-С6-галогеналкокси, C1-C6-алкилтио-, C5-C6-циклоалкилтио, C1-C6-галогеналкилтио, C1-C6-алкилсульфонил, C1-С6-галогеналкилсульфонил, C5-C6-циклоалкилсульфонил, фенил, 5-членный гетероарил или -N(Ra)(Rb), где упомянутый гетероарил содержит один гетероатом, выбранный из N, О и S, или содержит один атом азота и один другой гетероатом, выбранный из N, О и S, или содержит два атома азота и один другой гетероатом, выбранный из N, О и S; и упомянутый фенил или гетероарил необязательно замещен от одного до трех раз, независимо, галогеном, C1-C6-алкилом, C1-C4-галогеналкилом или -N(Ra)(Rb).
3. Соединение или фармацевтически приемлемая соль по п.1, в котором R3 представляет собой Н, галоген, C1-C8-алкил, C1-C8-галогеналкил, гидроксил, гидрокси-С1-С8-алкил-, C1-C8-алкокси, C3-C8-циклоалкилокси, C1-C8-галогеналкокси, C1-C8-алкилтио-, C1-C8-галогеналкилтио-, C3-C8-циклоалкилтио-, фенил, 5-членный гетероарил или -N(Ra)(Rb), где упомянутый фенил или гетероарил необязательно замещен от одного до трех раз, независимо, галогеном, C1-C6-алкилом, C1-C4-галогеналкилом, C1-C6-алкокси, C1-C4-галогеналкокси, гидрокси-C1-C4-алкилом или -N(Ra)(Rb).
4. Соединение или фармацевтически приемлемая соль по п.1, в котором R3 представляет собой Н, галоген, C1-C6-алкил, C1-C6-галогеналкил, гидрокси-C1-C6-алкил-, C1-C6-алкокси, C3-C6-циклоалкилокси, C1-C6-галогеналкокси, C1-C6-алкилтио-, C3-C6-циклоалкилтио-, C1-C6-галогеналкилтио-, фенил, 5-членный гетероарил или -N(Ra)(Rb), в которых упомянутый гетероарил содержит один гетероатом, выбранный из N, О и S, или содержит один атом азота и один другой гетероатом, выбранный из N, О и S, или содержит два атома азота и один другой гетероатом, выбранный из N, О и S; и упомянутый фенил или гетероарил необязательно замещен от одного до трех раз, независимо, галогеном, C1-C6-алкилом, C1-C4-галогеналкилом или -N(Ra)(Rb).
5. Соединение или фармацевтически приемлемая соль по п.1, в котором R3 представляет собой Н, галоген, C1-C4-алкокси, C5-C6-циклоалкилокси, C1-C4-галогеналкокси, C1-C4-алкилтио-, C1-C4-галогеналкилтио-, C5-C6-циклоалкилтио-, C1-C4-алкилсульфонил, C1-C4-галогеналкилсульфонил, C5-C6-циклоалкилсульфонил или N(Ra)(Rb).
6. Соединение или фармацевтически приемлемая соль по любому одному из пп.1-5, в котором
каждый Ra независимо представляет собой -СН3, -СН2СН3 или -CH2CF3;
Rb представляет собой -СН3 или
Ra и Rb, взятые вместе с азотом, к которому они присоединены, представляют собой пирролидин-1-ил, пиперидин-1-ил или морфолин-4-ил, где упомянутые пирролидин-1-ил, пиперидин-1-ил или морфолин-4-ил необязательно замещены от одного до двух раз, независимо, F, -СН3 или -CF3.
7. Соединение или фармацевтически приемлемая соль по любому одному из пп.1-6, в котором
каждый R5, R6 и R7 независимо представляет собой Н, галоген, нитро, амино, C1-C4-алкиламино, ди-C1-C4-алкиламино, гидроксил или ORc;
каждый Rc независимо представляет собой C1-C4-алкил, пирролидинил, тетрагидрофуранил, тетрагидротиенил, пиперидинил, пиперазинил, морфолинил, тетрагидропиранил, тетрагидро-4Н-1,4-тиазинил или 1,4-диоксанил, где упомянутый C1-C4-алкил необязательно замещен галогеном, гидроксилом, трифторметилом, C1-C4-алкокси, амино, C1-C4-алкиламино, ди(С1-С4-алкил)амино, пирролидин-1-илом, пиперидин-1-илом, пиперазин-1-илом, морфолин-4-илом или тетрагидро-4Н-1,4-тиазин-1-илом; или
R6 и R7, взятые вместе, представляют собой -O-C1-C2-алкил-О-.
8. Соединение или фармацевтически приемлемая соль по любому одному из пп.1-7, в котором R6 представляет собой Н.
9. Соединение или фармацевтически приемлемая соль по любому одному из пп.1-7, в котором
каждый R6 и R7 представляет собой ORc;
каждый Rc независимо представляет собой C1-C8-алкил, необязательно замещенный от одного до трех раз, независимо, галогеном, гидроксилом, C1-C4-алкокси, амино, C1-C4-алкиламино или ди-C1-C4-алкиламино.
10. Соединение или фармацевтически приемлемая соль по п.1, в котором
R1 представляет собой C1-C4-алкил;
R2 представляет собой Н или галоген;
R3 представляет собой Н, галоген, C1-C4-алкокси, C5-C6-циклоалкилокси, C1-C4-галогеналкокси, C1-C4-алкилтио-, C1-C4-галогеналкилтио-, C5-C6-циклоалкилтио-, C1-C4-алкилсульфонил, C1-C4-галогеналкилсульфонил, C5-C6-циклоалкилсульфонил или -N(Ra)(Rb);
каждый Ra независимо представляет собой C1-C4-алкил, в котором упомянутый C1-C4-алкил необязательно замещен от одного до трех раз, независимо, галогеном, гидроксилом, C1-C4-алкокси, амино, C1-C4-алкиламино, ди-C1-C4-алкиламино, -СО2Н, -СО2-C1-C4-алкилом, -CONH2, -CONH-C1-C4-алкилом или -CON-(C1-C4-алкил)(C1-C4-алкилом); и
Rb представляет собой C1-C4-алкил или
Ra и Rb, взятые вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членное гетероциклическое кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из азота, кислорода и серы, где упомянутое кольцо необязательно замещено от одного до двух раз, независимо, галогеном, C1-C4-алкилом, C1-C4-галогеналкилом, амино, C1-C4-алкиламино, ди(C1-C4-алкил)амино, гидроксилом, гидрокси-C1-C4-алкилом, оксо, C1-C4-алкокси, C1-C4-галогеналкокси или C1-C4-алкокси-C1-C4-алкилом;
R4 представляет собой Н;
каждый R5, R6 и R7 независимо представляет собой Н, галоген, нитро, амино, C1-C4-алкиламино, ди-C1-C4-алкиламино, гидроксил или ORc;
каждый Rc представляет собой C1-C4-алкил, который необязательно замещен галогеном, гидроксилом, трифторметилом, C1-C4-алкокси, амино, C1-C4-алкиламино, ди-C1-C4-алкиламино, пирролидин-1-илом, пиперидин-1-илом, пиперазин-1-илом, морфолин-4-илом, тетрагидро-4Н-1,4-тиазин-4-илом; или
R6 и R7, взятые вместе, представляют собой -O-C1-C2-алкил-О-.
11. Соединение или фармацевтически приемлемая соль по п.1, в котором
R1 представляет собой C1-C3-алкил;
R2 представляет собой Н;
R3 представляет собой Н, галоген, C1-C6-алкил, C1-C6-галогеналкил, гидрокси-C1-C6-алкил-, C1-C6-алкокси, С3-С6-циклоалкилокси, C1-C6-галогеналкокси, C1-C6-алкилтио-, С3-С6-циклоалкилтио-, C1-C6-галогеналкилтио-, фенил, 5-членный гетероарил или -N(Ra)(Rb), где упомянутый гетероарил содержит один гетероатом, выбранный из N, О и S, или содержит один атом азота и необязательно один дополнительный гетероатом, выбранный из N, О и S, или содержит два атома азота и необязательно один дополнительный гетероатом, выбранный из N, О и S; и упомянутый фенил или гетероарил необязательно замещен от одного до трех раз, независимо, галогеном, C1-C6-алкилом, C1-C4-галогеналкилом или -N(Ra)(Rb);
каждый Ra независимо представляет собой C1-C4-алкил, в котором упомянутый C1-C4-алкил необязательно замещен гидроксилом, C1-C6-алкокси, амино, C1-C6-алкиламино или ди(C1-C6-алкил)амино; и
Rb представляет собой C1-C4-алкил или
Ra и Rb, взятые вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членное гетероциклическое кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из азота, кислорода и серы, в котором упомянутое кольцо необязательно замещено от одного до двух раз, независимо, C1-C4-алкилом, C1-C4-галогеналкилом, гидрокси-C1-C4-алкилом, оксо или С1-С4-алкокси-С1-С4-алкилом;
R4 представляет собой Н;
R5 представляет собой Н;
каждый R6 и R7 представляет собой ORc;
каждый Rc независимо представляет собой C1-C4-алкил, необязательно замещенный от одного до трех раз, независимо, галогеном.
12. Соединение, выбранное из






или его фармацевтически приемлемая соль.
13. Фармацевтическая композиция для лечения заболевания, опосредованного сердечным тропонином I (TNNI3K), содержащая соединение или фармацевтически приемлемую соль по любому одному из пп.1-12 и одно или более фармацевтически приемлемых вспомогательных веществ.
14. Способ ингибирования TNNI3K, включающий контакт TNNI3K с соединением или фармацевтически приемлемой солью по любому одному из пп.1-12.
15. Способ лечения застойной сердечной недостаточности, включающий введение нуждающемуся пациенту терапевтически эффективного количества соединения или фармацевтически приемлемой соли по любому одному из пп.1-12.
16. Способ лечения застойной сердечной недостаточности, включающий введение нуждающемуся пациенту терапевтически эффективного количества фармацевтической композиции по п.13.
Текст
в которой R1, R2, R3, R4, R5, R6 и R7 являются такими, как определено в настоящем описании, и способам их получения и применения. Область техники, к которой относится изобретение Настоящее изобретение относится к соединениям, которые ингибируют TNNI3K, и способам их получения и применения. Более конкретно, настоящее изобретение относится к хиназолинам в качествеTNNI3K ингибиторов. Уровень техники Киназа, взаимодействующая с сердечным тропонином I (TNNI3K), также известная как CARK (для кардиальной панкирин-повторяющей киназы), представляет собой белковую киназу, которая обладает высокоселективной экспрессией для сердечных тканей, и показано, что она взаимодействует с компонентами саркомера, включая тропонин I (Zhao, Y. et al., J. Mol. Med., 2003, 81, 297-304; Feng, Y. et al., Gen.Physiol. Biophys., 2007, 26, 104-109; Wang, H. et al., J. Cell. Mol. Med., 2008, 12, 304-315). Хотя в настоящее время субстраты для TNNI3K не обнаружены, недавние сообщения предполагают, что данный белок играет роль в развитии кардиомиоцитной гипертрофии, вызванной давлением, и сократительной дисфункции (Wheeler, F.С. et al., Mamm. Genome, 2005, 16, 414-423; Wang, X. et al. "TNNI3K, a cardiacspecific kinase, promotes cardiac hypertrophy in vivo", Poster presentation at the 2006 Scientific Sessions of theAmerican Heart Association, Chicago, IL, Wheeler, F.C. et al., PLos Genet, 2009, 5(9), e1000647 и Pu, W.T.,PLos Genet, 2009, 5(9), e1000643). Ингибирование киназной активности TNNI3K может нарушать данные сигнальные пути и обеспечивать облегчением и/или купированием гипертрофии сердца, наблюдаемой у пациентов с постепенно ухудшающейся сердечной недостаточностью. В ответ на механические, нейрогормональные и генетические стимулы сердце будет подвергаться гипертрофии или мышечному росту и ремоделированию для того, чтобы поддерживать достаточный сердечный выброс для удовлетворения потребностей ткани в кислороде. Тогда как данные морфологические изменения первоначально выглядят как компенсирующие, продолжительная дисрегуляция гипертрофической сигнализации может приводить к сердечной недостаточности, патофизиологическому состоянию, при котором сердце не может более адекватно функционировать в качестве насоса (Mudd, J.О.and Kass, D.A., Nature, 2008, 451, 919-928). Предотвращение или купирование патологической гипертрофии сердца обладает потенциалом для задержки или предотвращения развития застойной сердечной недостаточности (McKinsey, Т.A. and Kass, D.A., Nat. Rev. Drug Discov., 2007, 6, 617-635; Kaye, D.M. andKrum, H., Nat. Rev. Drug Discov., 2007, 6, 127-139). Сердечная недостаточность является ответственной за сниженное качество жизни и преждевременную смерть значительной части страдающих ею пациентов, и она характеризуется нарушенным функционированием сердца или из-за пониженной насосной функции (систолическая дисфункция), или из-за пониженного наполнения (диастолическая дисфункция). Застойная сердечная недостаточность (CHF) характеризуется нарушенной функцией левого желудочка, повышенным сопротивлением периферических и легочных сосудов и пониженной переносимостью физических нагрузок и одышкой. Предполагается, что распространение сердечной недостаточности будет увеличиваться среди пожилых людей, что делает необходимым поиск новых и улучшенных способов лечения сердечной недостаточности. Сущность изобретения Настоящее изобретение относится к новым хиназолинам. Конкретно, настоящее изобретение относится к соединению согласно формуле IR3 представляет собой Н, галоген, C1-C8-алкил, C1-C8-галогеналкил, гидроксил, гидрокси-C1-C8 алкил, C1-C8-алкокси, C3-C8-циклоалкилокси, C1-C8-галогеналкокси, C1-C8-алкилтио-, C1-C8-галогеналкилтио-, C3-C8-циклоалкилтио-, C1-C8-алкилсульфонил, C1-C8-галогеналкилсульфонил, C3-C8-циклоалкилсульфонил, фенил, 5-членный гетероарил или N(Ra)(Rb), где упомянутый фенил или гетероарил необязательно замещен от одного до трех раз, независимо, галогеном, C1-C6-алкилом, C1-C4-галогеналкилом, C1-C6-алкокси, С 1-С 4-галогеналкокси, гидрокси-C1-C4-алкилом или -N(Ra)(Rb); каждый Ra независимо представляет собой C3-C4-алкил, в котором упомянутый C1-C4-алкил необязательно замещен от одного до трех раз, независимо, галогеном, гидроксилом, C1-C6-алкокси, амино, C1-C6-алкиламино, (C1-C6-алкил)(C1-C6-алкил)амино, -СО 2 Н, -CO2-C1-C6-алкилом, -CONH2,-CONH-C1-C6-алкилом или -CON-(C1-C6-алкил)(C1-C6-алкилом); иRa и Rb, взятые вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членное гетероциклическое кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из азота, кислорода и серы, в котором упомянутое кольцо необязательно замещено от одного до двух раз,независимо,галогеном,C1-C4-алкилом,C1-C4-галогеналкилом,амино,C1-C4-алкиламино,(C1-C4-алкил)(C1-C4-алкил)амино, гидроксилом, гидрокси-C1-C4-алкилом, оксо, C1-C4-алкокси,C1-C4-галогеналкокси или C1-C4-алкокси-С 1-С 4-алкилом;R4 представляет собой Н; каждый R5, R6 и R7 независимо представляет собой Н, галоген, нитро, амино, C1-C4-алкиламино,(C1-C4-алкил)(C1-C4-алкил)амино, гидроксил или ORc; каждый Rc независимо представляет собой C1-C8-алкил или 5-6-членный гетероциклоалкил, в котором упомянутый C1-C8-алкил необязательно замещен от одного до трех раз, независимо, галогеном, гидроксилом, C1-C4-алкокси, амино, C1-C4-алкиламино или -N(Ra)(Rb); илиR6 и R7, взятые вместе, представляют собой -O-C1-C2-алкил-О-; или его соли, в частности фармацевтически приемлемой соли. Соединения настоящего изобретения представляют собой ингибиторы TNNI3K и могут быть пригодны для лечения болезней и расстройств сердца, в частности сердечной недостаточности. Соответственно, кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим соединение настоящего изобретения. Кроме того, настоящее изобретение относится к способам ингибирования TNNI3K и лечению состояний, связанных с ней, применяя соединение настоящего изобретения или фармацевтическую композицию, содержащую соединение настоящего изобретения. Подробное описание изобретения Как применяют в настоящем изобретении, термин "алкил" относится к насыщенному, нормальному или разветвленному углеводородному фрагменту, который может быть незамещенным или замещенным одним или более заместителями, определенными в настоящем изобретении. Примеры алкилов включают,но не ограничиваются, метил (Me), этил (Et), пропил, изопропил, бутил, изобутил, трет-бутил и пентил. Термин "C1-C4" относится к алкилу, содержащему от 1 до 4 атомов углерода. Когда термин "алкил" применяют в комбинации с другими группами заместителей, такой как "галогеналкил", или "гидроксиалкил", или "арилалкил", предполагается, что термин "алкил" включает двухвалентный углеводородный радикал с нормальной или разветвленной цепью. Например, предполагается,что "арилалкил" относится к радикалу - алкиларилу, в котором его алкильный фрагмент представляет собой двухвалентный углеводородный радикал с нормальной или разветвленной цепью и его арильный фрагмент представляет собой, как определено в настоящем изобретении, и он представлен соединяющей группировкой, показанной в бензильной группе (-СН 2-фенил). Как применяют в настоящем изобретении, термин "циклоалкил" относится к неароматическому, насыщенному, циклическому углеводородному кольцу. Термин "C3-C8-циклоалкил" относится к неароматическому циклическому углеводородному кольцу, содержащему от 3 до 8 кольцевых атомов углерода. Примеры "C3-C8-циклоалкильных" групп, пригодных в настоящем изобретении, включают циклопропил,циклобутил, циклопентил, циклогексил, циклогептил и циклооктил."Алкокси" относится к группе, содержащей алкильный радикал, присоединенный через соединяющий атом кислорода. Термин "C1-C4-алкокси" относится к углеводородному радикалу с нормальной или разветвленной цепью, содержащему по меньшей мере 1 и вплоть до 4 атомов углерода, присоединенному через соединяющий атом кислорода. Примеры "C1-C4-алкокси" групп, пригодных в настоящем изобретении, включают, но не ограничиваются, метокси, этокси, н-пропокси, изопропокси, н-бутокси, вторбутокси и трет-бутокси."Алкилтио-" относится к группе, содержащей алкильный радикал, присоединенный через соединяющий атом серы. Термин "C1-C4-алкилтио-" относится к углеводородному радикалу с нормальной или разветвленной цепью, содержащему по меньшей мере 1 и вплоть до 4 атомов углерода, присоединенному через соединяющий атом серы. Примеры "C1-C4-алкилтио-" групп, пригодных в настоящем изобретении,включают, но не ограничиваются, метилтио-, этилтио-, н-пропилтио-, изопропилтио-, н-бутилтио-, вторбутилтио- и трет-бутилтио-."Алкилсульфонил" относится к группе, содержащей алкильный радикал, присоединенный через сульфонильный радикал (т.е. -S(=O)2-). Термин "C1-C4-алкилсульфонил" относится к углеводородному радикалу с нормальной или разветвленной цепью, содержащему по меньшей мере 1 и вплоть до 4 атомов углерода, присоединенному через сульфонильный радикал. Примеры "C1-C4-алкилсульфонильной" группы, пригодной в настоящем изобретении, включают, но не ограничиваются, метансульфонил, этансульфонил, пропансульфонил и бутансульфонил."Циклоалкилокси" и "циклоалкилтио" относятся к группе, содержащей атомы насыщенного карбоциклического кольца, присоединенные через соединяющий атом кислорода или серы соответственно. Примеры "циклоалкилокси" групп включают, но не ограничиваются, циклопропокси, циклобутокси,циклопентилокси, циклогексилокси и подобные."Арил" представляет собой группу или фрагмент, содержащий ароматический, одновалентный моноциклический или бициклический углеводородный радикал, содержащий от 6 до 10 кольцевых атомов углерода, который может быть незамещенным или замещенным одним или более заместителями, определенными в настоящем изобретении, и с которым может быть конденсировано одно или более циклоал-2 021439 кильных колец, которые могут быть незамещенными или замещенными одним или более заместителями,определенными в настоящем изобретении. Обычно в соединениях настоящего изобретения арил представляет собой фенил. Гетероциклические группы могут представлять собой гетероарильные или гетероциклоалкильные группы."Гетероциклоалкил" представляет собой группу или фрагмент, содержащий неароматический, одновалентный моноциклический или бициклический радикал, который является насыщенным или частично ненасыщенным, содержащим 3-10 кольцевых атомов, включая 1-4 гетероатома, выбранных из азота,кислорода и серы, и который может быть незамещенным или замещенным одним или более заместителями, определенными в настоящем изобретении. Типичные примеры гетероциклоалкилов включают, но не ограничиваются, азетидинил, пирролидил (или пирролидинил), пиперидинил, пиперазинил, морфолинил, тетрагидро-4 Н-1,4-тиазинил, тетрагидрофуран (или тетрагидрофуранил), дигидрофуран, оксазолинил, триазолинил, пиразолинил, тетрагидропиран, дигидропиран, 1,3-диоксоланил, 1,3-диоксанил,1,4-диоксанил, 1,3-оксатиоланил, 1,3-оксатианил, 1,3-дитианил, азабицикло[3,2,1]октил, азабицикло[3,3,1]нонил, азабицикло[4,3,0]нонил, оксабицикло[2,2,1]гептил и 1,5,9-триазациклододецил. Обычно в соединениях настоящего изобретения гетероциклоалкильные группы представляют собой 5- и/или 6-членные гетероциклоалкильные группы, такие как пирролидил (или пирролидинил), тетрагидрофуран (или тетрагидрофуранил), тетрагидротиенил, дигидрофуран, оксазолинил, триазолинил, пиразолинил, пиперидил (или пиперидинил), пиперазинил, морфолинил, тетрагидропиран, дигидропиран,1,3-диоксанил, тетрагидро-4 Н-1,4-тиазинил, 1,4-диоксанил, 1,3-оксатианил и 1,3-дитианил."Гетероарил" представляет собой группу или фрагмент, содержащий ароматический одновалентный моноциклический или бициклический радикал, содержащий 5-10 кольцевых атомов, включая 1-4 гетероатома, выбранных из азота, кислорода и серы, который может быть незамещенным или замещенным одним или более заместителями, определенными в настоящем изобретении. Данный термин также включает бициклические гетероциклические арильные соединения, содержащие фрагмент арильного кольца, конденсированный с фрагментом гетероциклоалкильного кольца, содержащие 5-10 кольцевых атомов, включая 1-4 гетероатома, выбранных из азота, кислорода и серы, которые могут быть незамещенным или замещенным одним или более заместителями, определенными в настоящем изобретении. Типичные примеры гетероарилов включают, но не ограничиваются, тиенил, пирролил, имидазолил, пиразолил, фуран (или фуранил), изотиазолил, изоксазолил, оксазолил, оксадиазолил, тиазолил, пиридил(или пиридинил), пиразинил, пиримидинил, пиридазинил, триазинил, тетразинил, триазолил, тетразолил,бензо[b]тиенил, изобензофурил, 2,3-дигидробензофурил, хроменил, хроманил, индолизинил, изоиндолил, индолил, индазолил, пуринил, изохинолил, хинолил, фталазинил, нафтиридинил, хиназолинил, бензотиазолил, бензимидазолил, тетрагидрохинолинил, циннолинил, птеридинил и изотиазолил. Обычно гетероарильные группы, присутствующие в соединениях настоящего изобретения, представляют собой 5- и/или 6-членные моноциклические гетероарильные группы. Выбранные 5-членные гетероарильные группы содержат один кольцевой гетероатом азота, кислорода или серы и необязательно содержат 1, 2 или 3 дополнительных кольцевых атома азота. Выбранные 6-членные гетероарильные группы содержат 1, 2, 3 или 4 кольцевых гетероатома азота. Выбранные 5- или 6-членные гетероарильные группы включают тиенил, пирролил, имидазолил, пиразолил, фурил, изотиазолил, изоксазолил,оксазолил, оксадиазолил, тиазолил, триазолил, тетразолил, пиридил, пиразинил, пиримидинил, пиридазинил и триазинил."Оксо" представляет собой кислородный фрагмент с двойной связью; например, при присоединении непосредственно к атому углерода он образует карбонильный фрагмент (С=O). Термины "галоген" и "гало" представляют собой заместитель, являющийся хлором, фтором, бромом или йодом. Предполагается, что "гидрокси" или "гидроксил" обозначают радикал -OH. Как применяют в настоящем изобретении, термин "соединение (соединения) настоящего изобретения" относится к соединению формулы I (как определено выше) в любой форме, т.е. любой солевой или несолевой форме (например, в виде свободной кислоты или свободного основания или в виде их фармацевтически приемлемой соли) и любой его физической форме (например, включая нетвердые формы(например, жидкие или полутвердые формы) и твердой форме (например, аморфные или кристаллические формы, специфические полиморфные формы, сольваты, включая гидраты (например, моно-, ди- и гемигидраты и смеси различных форм. Как применяют в настоящем изобретении, термин "необязательно замещенный" обозначает, что группы могут быть или незамещенными, или замещенными одним или более конкретными заместителями. Предполагается, что альтернативные определения для различных групп и групп, являющихся заместителями, формулы I, приводимые во всем описании, конкретно описывают каждый фрагмент соединения, описанный в настоящем изобретении, индивидуально, а также группы одного или более фрагментов соединения. Объем настоящего изобретения включает любую комбинацию значений данных групп и групп, являющихся заместителями. Кроме того, настоящее изобретение относится к соединению согласно формуле I, в которой:R3 представляет собой Н, галоген, C1-C4-алкокси, C5-C6-циклоалкилокси, C1-C4-галогеналкокси,C1-C4-алкилтио-,C1-C4-галогеналкилтио-,C5-C6-циклоалкилтио-,С 1-С 4-алкилсульфонил,C1-C4-галогеналкилсульфонил, С 5-С 6-циклоалкилсульфонил или -N(Ra)(Rb); каждый Ra независимо представляет собой C1-C4-алкил, в котором упомянутый C1-C4-алкил необязательно замещен от одного до трех раз, независимо, галогеном, гидроксилом, C1-C4-алкокси, амино,C1-C4-алкиламино,(C1-C4-алкил)(C1-C4-алкил)амино,-СО 2 Н,-CO2-C1-C4-алкилом,-CONH2,-CONH-C1-C4-алкилом или -CON(C1-C4-алкил)(C1-C4-алкилом); иRa и Rb, взятые вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членное гетероциклическое кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из азота, кислорода и серы, в котором упомянутое кольцо необязательно замещено от одного до двух раз,независимо,галогеном,C1-C4-алкилом,C1-C4-галогеналкилом,амино,C1-C4-алкиламино,(C1-C4-алкил)(C1-C4-алкил)амино, гидроксилом, гидрокси-C1-C4-алкилом, оксо, C1-C4-алкокси,C1-C4-галогеналкокси или C1-C4-алкокси-C1-С 4-алкилом;R4 представляет собой Н; каждый R5, R6 и R7 независимо представляет собой Н, галоген, нитро, амино, C1-C4-алкиламино,(C1-C4-алкил)(C1-C4-алкил)амино, гидроксил или ORc; и каждый Rc независимо представляет собой C1-C4-алкил, пирролидинил, тетрагидрофуранил, тетрагидротиенил, пиперидинил, пиперазинил, морфолинил, тетрагидропиран, тетрагидро-4 Н-1,4-тиазинил или 1,4-диоксанил, в которых упомянутый C1-C4-алкил необязательно замещен галогеном, гидроксилом,трифторметилом, C1-C4-алкокси, амино, C1-C4-алкиламино, (C1-C4-алкил)(C1-C4-алкил)амино, пирролидинилом, тетрагидрофуранилом, тетрагидротиенилом, пиперидинилом, пиперазинилом, морфолинилом,тетрагидропиранилом, тетрагидро-4 Н-1,4-тиазинилом или 1,4-диоксанилом; илиR6 и R7, взятые вместе, представляют собой -O-C1-C2-алкил-О-; или его соли, в частности фармацевтически приемлемой соли. В другом варианте осуществления настоящего изобретения:R3 представляет собой Н, галоген, C1-C8-алкил, C1-C8-галогеналкил, гидроксил, гидрокси-C1-C8 алкил-, C1-С 8-алкокси, C3-C8-циклоалкилокси, C1-C8-галогеналкокси, C1-C8-алкилтио-, C1-C8-галогеналкилтио-, C3-C8-циклоалкилтио-, фенил, 5-членный гетероарил или -N(Ra)(Rb), в котором упомянутый фенил или гетероарил необязательно замещен от одного до трех раз, независимо, галогеном,C1-C6-алкилом, C1-C4-галогеналкилом, C1-C6-алкокси, C1-С 4-галогеналкокси, гидрокси-C1-C4-алкилом или -N(Ra)(Rb); каждый Ra независимо представляет собой C1-C4-алкил, в котором упомянутый C1-C4-алкил необязательно замещен от одного до трех раз, независимо, галогеном, гидроксилом, C1-C6-алкокси, амино,C1-C6-алкиламино,(C1-C6-алкил)(C1-C6-алкил)амино,-СО 2 Н,-CO2-C1-C6-алкилом,-CONH2,-CONH-C1-C6-алкилом или -CON-(C1-C6-алкил)(C1-C6-алкилом); иRa и Rb, взятые вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членное гетероциклическое кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из азота, кислорода и серы, в котором упомянутое кольцо необязательно замещено от одного до двух раз,независимо,галогеном,C1-C4-алкилом,C1-C4-галогеналкилом,амино,C1-C4-алкиламино,(C1-C4-алкил)(C1-C4-алкил)амино, гидроксилом, гидрокси-C1-C4-алкилом, оксо, C1-C4-алкокси,C1-C4-галогеналкокси или C1-C4-алкокси-C1-С 4-алкилом;R5 представляет собой Н; каждый R6 и R7 представляет собой ORc; каждый Rc независимо представляет собой C1-C8-алкил, необязательно замещенный от одного до трех раз, независимо, галогеном, гидроксилом, C1-C4-алкокси, амино, C1-C4-алкиламино или(C1-C4-алкил)(C1-C4-алкил)амино; или его соль, в частности фармацевтически приемлемую соль. В другом варианте осуществления настоящего изобретения:R3 представляет собой Н, галоген, C1-C6-алкил, C1-C6-галогеналкил, гидрокси-C1-C6-алкил-,C1-C6-алкокси, С 3-С 6-циклоалкилокси, C1-C6-галогеналкокси, C1-C6-алкилтио-, C3-C6-циклоалкилтио-,C1-C6-галогеналкилтио-, фенил, 5-членный гетероарил или -N(Ra)(Rb),в которых упомянутый гетероарил содержит один гетероатом, выбранный из N, О и S, или содер-4 021439 жит один атом азота и необязательно содержит один дополнительный гетероатом, выбранный из N, О иS, или содержит два атома азота и необязательно содержит один дополнительный гетероатом, выбранный из N, О и S; и упомянутый фенил или гетероарил необязательно замещен от одного до трех раз, независимо, галогеном, C1-C6-алкилом, C1-C4-галогеналкилом или -N(Ra)(Rb); каждый Ra независимо представляет собой C1-C4-алкил, в котором упомянутый C1-C4-алкил необязательно замещен гидроксилом, трифторметилом, C1-C6-алкокси, амино, C1-C6-алкиламино илиRa и Rb, взятые вместе с атомом азота, к которому они присоединены, образуют 5- или 6-членное гетероциклическое кольцо, необязательно содержащее один дополнительный гетероатом, выбранный из азота, кислорода и серы, в которых упомянутое кольцо необязательно замещено от одного до двух раз, независимо, C1-C4-алкилом, C1-С 4-галогеналкилом, гидрокси-C1-C4-алкилом, оксо илиR5 представляет собой Н; каждый R6 и R7 представляет собой ORc; каждый Rc независимо представляет собой C1-C4-алкил, необязательно замещенный от одного до трех раз, независимо, галогеном; или его соль, в частности фармацевтически приемлемая соль. В конкретном варианте осуществления настоящего изобретения R1 представляет собой -СН 3. В другом конкретном варианте осуществления настоящего изобретения R2 представляет собой Н или F. В еще другом конкретном варианте осуществления настоящего изобретения R2 представляет собой Н. В другом варианте осуществления настоящего изобретения R3 представляет собой Н, галоген,C1-C6-алкил, С 1-С 6-галогеналкил, C1-C6-алкокси, C5-C6-циклоалкилокси, C1-C6-галогеналкокси,C1-C6-алкилтио-,C5-C6-циклоалкилтио-,C1-C6-галогеналкилтио-,C1-C6-алкилсульфонил,C1-С 6-галогеналкилсульфонил, C5-C6-циклоалкилсульфонил, фенил, 5-членный гетероарил или-N(Ra)(Rb), в которых упомянутый гетероарил содержит один гетероатом, выбранный из N, О и S, или содержит один атом азота и необязательно содержит один дополнительный гетероатом, выбранный из N,О и S, или содержит два атома азота и необязательно содержит один дополнительный гетероатом, выбранный из N, О и S; и упомянутый фенил или гетероарил необязательно замещен от одного до трех раз,независимо, галогеном, C1-C6-алкилом, C1-C4-галогеналкилом или -N(Ra)(Rb). В следующем варианте осуществления настоящего изобретения R3 представляет собой Н,галоген, C1-C8-алкил, C1-C8-галогеналкил, гидроксил, гидрокси-C1-C8-алкил-, C1-С 8-алкокси,C3-C8-циклоалкилокси,C1-C8-галогеналкокси,C1-C8-алкилтио-,C1-C8-галогеналкилтио-,C3-C8-циклоалкилтио-, фенил, 5-членный гетероарил или -N(Ra)(Rb), в которых упомянутый фенил или гетероарил необязательно замещен от одного до трех раз, независимо, галогеном, C1-C6-алкилом,С 1-C4-галогеналкилом, C1-C6-алкокси, C1-C4-галогеналкокси, гидрокси-C1-C4-алкилом или -N(Ra)(Rb). В еще следующем варианте осуществления настоящего изобретения R3 представляет собой Н, галоген, C1-C6-алкил, C1-C6-галогеналкил, гидрокси-C1-C6-алкил-, C1-C6-алкокси, C3-C6-циклоалкилокси,C1-C6-галогеналкокси, C1-C6-алкилтио-, C3-C6-циклоалкилтио-, C1-C6-галогеналкилтио-, фенил,5-членный гетероарил или -N(Ra)(Rb), в которых упомянутый гетероарил содержит один гетероатом, выбранный из N, О и S, или содержит один атом азота и необязательно содержит один дополнительный гетероатом, выбранный из N, О и S, или содержит два атома азота и необязательно содержит один дополнительный гетероатом, выбранный из N, О и S; и упомянутый фенил или гетероарил необязательно замещен от одного до трех раз, независимо, галогеном, C1-C6-алкилом, C1-C4-галогеналкилом или N(Ra)(Rb). В еще следующем варианте осуществления настоящего изобретения R3 представляет собой Н,галоген,C1-C4-алкокси,C5-C6-циклоалкилокси,C1-C4-галогеналкокси,C1-C4-алкилтио-,C1-C4-галогеналкилтио-, C5-C6-циклоалкилтио-, C1-С 4-алкилсульфонил, C1-C4-галогеналкилсульфонил,С 5-С 6-циклоалкилсульфонил или -N(Ra)(Rb). В конкретном варианте осуществления настоящего изобретения каждый Ra независимо представляет собой -СН 3, -СН 2 СН 3 или -CH2CF3, Rb представляет собой -СН 3 или Ra и Rb, взятые вместе с азотом, к которому они присоединены, представляют собой пирролидин-1-ил, пиперидин-1-ил или морфолин-4-ил,в которых упомянутые пирролидин-1-ил, пиперидин-1-ил или морфолин-4-ил необязательно замещены от одного до двух раз, независимо F, -СН 3 или -CF3. В другом конкретном варианте осуществления настоящего изобретения R3 представляет собой Н, F,Cl, -OCH3, -OCH(СН 3)2, -OCF3, -OCH2CF3, -OCH(CH3)CF3, -SCH3, -SCH2CF3, -SO2CH3, -N(CH3)2,-N(CH3)CH2CH3, -N(CH3)CH2CF3, 2-(трифторметил)пирролидин-1-ил, 2,5-(диметил)пирролидин-1-ил,пиперидин-1-ил, 4,4-дифторпиперидин-1-ил или морфолин-4-ил.-5 021439 В следующем варианте осуществления настоящего изобретения каждый R5, R6 и R7 независимо представляет собой Н, галоген, нитро, амино, C1-C4-алкиламино, (C1-C4-алкил)(C1-С 4-алкил)амино, гидроксил или ORc и каждый Rc независимо представляет собой C1-C4-алкил, пирролидинил, тетрагидрофуранил, тетрагидротиенил, пиперидинил, пиперазинил, морфолинил, тетрагидропиран, тетрагидро-4 Н-1,4 тиазинил или 1,4-диоксанил, в которых упомянутый C1-C4-алкил необязательно замещен галогеном, гидроксилом, трифторметилом, C1-C4-алкокси, амино, C1-C4-алкиламино, (C1-C4-алкил)(C1-C4-алкил)амино,пирролидинилом, тетрагидрофуранилом, тетрагидротиенилом, пиперидинилом, пиперазинилом, морфолинилом, тетрагидропиранилом, тетрагидро-4 Н-1,4-тиазинилом или 1,4-диоксанилом; или R6 и R7, взятые вместе, представляют собой -О-C1-C2-алкил-О-. В конкретном варианте осуществления настоящего изобретения R5 представляет собой H, Cl,-OCH3, -OCH2CH3, -OCH(CH3)2, -O(CH2)2OCH3 или -О-(тетрагидропиран-4-ил). В другом конкретном варианте осуществления настоящего изобретения R5 представляет собой Н. В другом варианте осуществления настоящего изобретения каждый R6 и R7 представляет собойOR , где и каждый Rc независимо представляет собой C1-C8-алкил, необязательно замещенный от одного до трех раз, независимо, галогеном, гидроксилом, C1-C4-алкокси, амино, C1-C4-алкиламино или(C1-C4-алкил)(C1-C4-алкил)амино. В следующем варианте осуществления настоящего изобретения каждый R6 и R7 представляет собойOR , где каждый Rc независимо представляет собой C1-C4-алкил, необязательно замещенный от одного до трех раз, независимо, галогеном. В еще следующем варианте осуществления настоящего изобретения каждый R6 и R7 представляет собой ORc, где каждый Rc независимо представляет собой C1-C4-алкил, необязательно замещенныйC1-C4-алкокси. В конкретном варианте осуществления настоящего изобретения R6 представляет собой H, Cl, I,-OCH3, -OCH2CH3, -OCH(СН 3)2, -О(СН 2)2OCH3, -O(СН 2)3-морфолин-4-ил, -NH2, -NHCH3 или -N(CH3)2. В другом конкретном варианте осуществления настоящего изобретения R6 представляет собой Н. В другом конкретном варианте осуществления настоящего изобретения R7 представляет собой Н,-OH, -OCH3, -OCH2CH3, -OCH(CH3)2, -O(CH2)3Cl, -O(CH2)2OCH3, -O(CH2)3OCH3, -O(CH2)3-морфолин-4 ил, -NH2, -NHCH3 или -NO2. В другом конкретном варианте осуществления настоящего изобретения R6 и R7, взятые вместе,представляют собой O(CH2)2O-. Конкретными соединениями настоящего изобретения являются: и их соли, в частности фармацевтически приемлемые соли. Типичные соединения настоящего изобретения включают соединения примеров 1-88. Соединения согласно формуле I могут содержать один или более асимметрических центров (также называемых хиральными центрами) и могут, следовательно, существовать в виде отдельных энантиомеров, диастереомеров или других стереоизомерных форм или в виде их смеси. Хиральные центры, такие как хиральные атомы углерода, могут также присутствовать в заместителе, таком как алкильная группа. Когда стереохимия хирального центра, присутствующего в формуле I или в любой химической структуре, показанной в настоящем изобретении, не указана, предполагается, что структура включает все отдельные стереоизомеры и все их смеси. Таким образом, соединения согласно формуле I, содержащие один или более хиральных центров, можно применять в виде рацемических смесей, энантиомерно обогащенных смесей или в виде энантиомерно чистых отдельных стереоизомеров. Отдельные стереоизомеры соединения согласно формуле I, которые содержат один или более асимметрических центров, можно разделять способами, известными специалистам в данной области техники. Например, данное разделение можно осуществлять (1) получением диастереомерных солей, комплексов или других производных; (2) избирательной реакцией со специфическим по отношению к стереоизомеру реагентом, например ферментативным окислением или восстановлением; или (3) газожидкостной или жидкостной хроматографией в хиральном окружении, например на хиральной подложке, такой как оксид кремния со связанным хиральным лигандом или в присутствии хирального растворителя. Специалисту в данной области техники ясно, что, когда требуемый стереоизомер превращают в другую химическую молекулу одним из способов разделения, описанным выше, требуется дополнительная стадия для высвобождения требуемой формы. Альтернативно, конкретные стереоизомеры можно получить асимметрическим синтезом, применяя оптически активные реагенты, субстраты, катализаторы или растворители, или превращением одного энантиомера в другой асимметрическим превращением. Когда описанному соединению или его соли дают название или изображают структурой, должно быть ясно, что соединение или соль, включая их сольваты (в частности, гидраты), могут существовать в кристаллических формах, некристаллических формах или их смеси. Соединение или соль или их сольваты (в частности, гидраты) могут также обладать полиморфизмом (т.е. способностью находиться в различных кристаллических формах). Данные различные кристаллические формы обычно известны как"полиморфы." Должно быть ясно, что, когда соединению дается название или оно изображается структурой, описанное соединение или его сольваты (в частности, гидраты) также включают все их полиморфы. Полиморфы имеют одинаковый химический состав, но отличаются укладкой, размещением в пространстве и другими описательными свойствами кристаллического твердого состояния. Полиморфы, следовательно, могут обладать различными физическими свойствами, такими как геометрия, плотность, твердость, способность к деформации, стабильность и растворимость. Полиморфы обычно обладают различными температурами плавления, ИК-спектрами и порошковыми рентгеновскими дифрактограммами,которые можно применять для идентификации. Специалисту в данной области техники ясно, что различные полиморфы можно получить, например, изменением или подбором условий, применяемых для кристаллизации/перекристаллизации соединения. Что касается сольватов соединений настоящего изобретения или их солей, которые могут находиться в кристаллической форме, специалистам в данной области техники ясно, что можно получить фармацевтически приемлемые сольваты, в которых молекулы растворителя встраиваются в кристаллическую решетку в процессе кристаллизации. Сольваты могут включать неводные растворители, такие как эта- 11021439 нол, изопропанол, DMSO, уксусную кислоту, этаноламин и этилацетат или они могут включать воду в качестве растворителя, которая встраивается в кристаллическую решетку. Сольваты, в которых вода является растворителем, который встраивается в кристаллическую решетку, обычно называют "гидратами". Гидраты включают стехеометрические гидраты, а также составы, содержащие различное количество воды. Настоящее изобретение включает все данные сольваты. Из-за их потенциального применения в медицине соли соединений формулы I являются предпочтительно фармацевтически приемлемыми. Соединения настоящего изобретения являются основаниями, в которых требуемую солевую форму можно получить любым подходящим способом, известным в данной области техники, включая обработку свободного основания неорганической кислотой, такой как хлороводородная кислота, бромоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и подобной, или органической кислотой, такой как уксусная, трифторуксусная, малеиновая, янтарная,миндальная, фумаровая, малоновая, пировиноградная, щавелевая, гликолевая, салициловая, пиранозидильная, такая как глюкуроновая или галактуроновая, альфа-гидрокси кислота, такая как лимонная кислота или винная кислота, аминокислота, такая как аспарагиновая кислота или глютаминовая кислота,ароматическая кислота, такая как бензойная кислота или коричная кислота, сульфоновая килсота, такая как п-толуолсульфокислота, метансульфокислота, этансульфокислота или подобной. Примеры фармацевтически приемлемых солей включают сульфаты, пиросульфаты, бисульфаты, сульфиты, бисульфиты,фосфаты, хлориды, бромиды, йодиды, ацетаты, пропионаты, деканоаты, каприлаты, акрилаты, формиаты, изобутираты, капроаты, гептаноаты, пропиолаты, оксалаты, малонаты, сукцинаты, субераты, себакаты, фумараты, малеаты, бутен-1,4-диоаты, гексин-1,6-диоаты, бензоаты, хлорбензоаты, метилбензоаты,динитробензоаты, гидроксибензоаты, метоксибензоаты, фталаты, фенилацетаты, фенилпропионаты, фенилбутираты, цитраты, лактаты, -гидроксибутираты, гликоллаты, тартраты, манделаты и сульфонаты,такие как ксилолсульфонаты, метансульфонаты, пропансульфонаты, нафталин-1-сульфонаты и нафталин-2-сульфонаты. Если основное соединение настоящего изобретения выделяют в виде соли, соответствующую форму свободного основания данного соединения можно получить подходящим способом, известным в данной области техники, включая обработку соли неорганическим или органическим основанием, подходящим неорганическим или органическим основанием, имеющим большее pKa, чем форма свободного основания данного соединения. Общие способы получения Соединения формулы I можно получить, применяя синтетические способы, показанные на схемах ниже или разработанные на основе знаний специалиста в области органической химии. Способ получения, приведенный на данных схемах, является применимым для получения соединений настоящего изобретения, имеющих ряд различных R1 и R2 групп, применяя подходящие исходные соединения, которые можно подходящим образом защитить при необходимости, для достижения совместимости с реакциями,приведенными в настоящем изобретении. Последующее деблокирование, при необходимости, дает соединения обычно описанных свойств. Когда схемы показаны с соединениями только формулы I, они являются иллюстративными для способов, которые можно применять для получения соединений настоящего изобретения. Названия соединений получают, применяя программное обеспечение для получения названия соединений ACD/Name Pro V6,02, полученной у Advanced Chemistry Development, Inc., 110 Yonge Street,14th Floor, Toronto, Ontario, Canada, M5C 1T4 (http://www.acdlabs.com/). Как показано на схеме 1, соединения формулы I можно получить при ряде условий реакцией ариламина (например, Ar-NH-R2, конкретно, Ar-NH2) с активированным хиназолином. Схема 1 а) изопропанол, w (6-15 бар), 150 С, 15 мин; b) изопропанол, w (1-2 бар), 100 С, 30 мин. Как показано на схемах 2-5, соединения формулы I можно получить с помощью стандартных превращений функциональных групп из заранее полученных промежуточных веществ. а) Н 2, Pd/C, MeOH, 25 С, 2,5 ч; b) H2CO, NaBH(OAc)3, AcOH, CH2Cl2, 25 С, 2 ч. Настоящее изобретение также включает различные дейтерированные формы соединений формулыI. Любой имеющийся атом водорода, соединенный с атомом углерода, можно независимо заместить атомом дейтерия. Специалист в данной области техники знает, как получить дейтерированные формы соединений формулы(например,N-(дейтерометил)амины или Ra/Rb алкилы) можно получить общепринятыми способами (см., например,метил-d3-амин, имеющийся в продаже у Aldrich Chemical Co., Milwaukee, WI, Cat. No. 489689-2). Применение данных соединений согласно схеме 1 позволит получить соединения формулы I, в которых различные атомы водорода N-метильной или пиримидинильной групп замещены атомом дейтерия. Способы применения. Настоящее изобретение относится к способу ингибирования TNNI3K, который включает контакт киназы с соединением формулы I или его солью, в частности его фармацевтически приемлемой солью. Настоящее изобретение также относится к способу лечения TNNI3K-опосредованного заболевания или расстройства, включающему введение эффективного количества соединения формулы I или его соли, в частности его фармацевтически приемлемой соли, пациенту, конкретно, нуждающемуся в нем человеку. Как применяют в настоящем изобретении, "пациент" относится к человеку или другому млекопитающему. Конкретно, настоящее изобретение относится к способу ингибирования TNNI3K активности, включающему контакт киназы с эффективным количеством соединения формулы I или его фармацевтически приемлемой солью. Например, TNNI3K активность можно ингибировать в ткани сердца млекопитающего введением нуждающемуся пациенту эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Соединения настоящего изобретения могут быть, в частности, пригодными для лечения TNNI3Kопосредованных заболеваний или расстройств, в частности, ингибированием TNNI3K активности, где данные заболевания или расстройства выбраны из сердечной недостаточности, в частности застойной сердечной недостаточности; гипертрофии сердца и сердечной недостаточности или застойной сердечной недостаточности в результате гипертрофии сердца. Соединения настоящего изобретения могут также быть пригодными для лечения сердечной недостаточности или застойной сердечной недостаточности в результате ишемии миокарда или инфаркта миокарда. Предполагается, что терапевтически "эффективное количество" относится к количеству соединения, которое при введении пациенту, нуждающемуся в данном лечении, является достаточным для осуществления лечения, как определено в настоящем изобретении. Таким образом, например, терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемой соли является количеством агента настоящего изобретения, которое при введении нуждающемуся пациенту является достаточным для модулирования или ингибирования активности TNNI3K так, что заболевание, которое опосредуется данной активностью, ослабляется, облегчается или предотвращается. Количество указанного соединения, которое будет соответствовать данному количеству, будет изменяться в зависимости от факторов, таких как конкретное соединение (например, активность (pXC50), эффективность (ЕС 50) и биологический период полувыведения конкретного соединения), заболевания и его тяжести, параметров(например, возраст, размер и вес) нуждающегося в лечении пациента, но, тем не менее, может быть обычно определено специалистом в данной области техники. Кроме того, продолжительность лечения и период времени введения (период времени между дозами и время дозирования, например, перед/вместе с/после еды) соединением будет изменяться в зависимости от параметров нуждающегося в лечении млекопитающего (например, веса), конкретного соединения и его свойств (например, фармацевтических характеристик), заболевания или состояния и его тяжести и конкретного состава и способа, которые применяют, но, тем не менее, она может быть определена специалистом в данной области техники. Предполагается, что "лечение" обозначает, по меньшей мере, ослабление заболевания у пациента,где заболевание вызвано или опосредовано TNNI3K. Способы лечения для ослабления заболевания включают применение соединений настоящего изобретения любым общепринятым приемлемым способом, например, для предотвращения, замедления, профилактики, терапии или вылечивания заболевания. Соединения формулы I настоящего изобретения могут быть пригодными для лечения сердечной недостаточности, в частности застойной сердечной недостаточности. Соединения формулы I настоящего изобретения могут быть пригодны для лечения гипертрофии сердца и сердечной недостаточности или застойной сердечной недостаточности в результате гипертрофии сердца, ишемии миокарда или инфаркта миокарда. Соединения настоящего изобретения можно вводить любым подходящим способом введения,включая как системное введение, так и местное введение. Системное введение включает пероральное введение, парентеральное введение, трансдермальное введение, ректальное введение и введение ингаляцией. Парентеральное введение относится к способам введения, отличным от энтерального и трансдермального введения, а также введения ингаляцией, и обычно осуществляется инъекцией или вливанием. Парентеральное введение включает внутривенную, внутримышечную и подкожную инъекцию или вливание. Ингаляция относится к введению в легкие пациента, ингаляции через рот или через носовые проходы. Местное введение включает нанесение на кожу. Соединения настоящего изобретения можно вводить один раз или согласно режиму дозирования, в котором ряд доз вводится с разной периодичностью времени в течение указанного периода времени. Например, дозы можно вводить один, два, три или четыре раза в день. Дозы можно вводить до достижения требуемого терапевтического эффекта или неопределенный период времени для поддержания требуемого терапевтического эффекта. Подходящие режимы дозирования для соединения настоящего изобретения зависят от фармакокинетических свойств данного соединения, таких как абсорбция, распределение и период полувыведения, которые может определить специалист в данной области техники. Кроме того,подходящие режимы дозирования, включая продолжительность осуществления данных режимов, для соединения настоящего изобретения зависят от состояния, подвергающегося лечению, тяжести состояния, подвергающегося лечению, возраста и физического состояния пациента, подвергающегося лечению,истории болезни пациента, подвергающегося лечению, природы сопутствующей терапии, требуемого терапевтического эффекта и подобных факторов, в пределах знаний и компетенции специалиста в данной области техники. Кроме того, специалистам в данной области техники должно быть ясно, что подходящие режимы дозирования могут требовать приспосабливания, принимая во внимание ответную реакцию пациента на режим дозирования, или с течением времени, поскольку пациенту требуются изменения. Лечение TNNI3K-опосредованных заболеваний можно осуществлять, применяя соединения настоящего изобретения, в качестве монотерапии или в дуальной или многокомпонентной терапии, такой как в комбинации с другими сердечно-сосудистыми средствами, например в комбинации с одним или более из следующих агентов: бета-блокатор, АСЕ ингибитор, блокатор рецепторов ангиотензина (ARB),блокатор кальциевых каналов, диуретик, ингибитор ренина, центрально действующее антигипертензивное средство, двойной ACE/NEP ингибитор, ингибитор альдостеронсинтазы и антагонист рецепторов альдостерона, которые вводят в эффективных количествах, как это известно в данной области техники. Примеры подходящих бета-блокаторов включают тимолол (такой как BLOCARDEN), картеололTENORMIN), пиндолол (такой как VISKEN), бисопролол, буциндолол, эсмолол, ацебутолол, лабеталол, небиволол, целипролол, зоталол и окспренолол. Примеры подходящих АСЕ ингибиторов включают алацеприл, беназеприл, беназаприлат, каптоприл, церонаприл, цилазаприл, делаприл, еналаприл, еналаприлат, фозиноприл, лизиноприл, моексипирил, мовелтоприл, периндоприл, квинаприл, квинаприлат,рамиприл, рамиприлат, спираприл, темокаприл, трандолаприл и зофеноприл. Предпочтительными АСЕ ингибиторами являются беназеприл, еналприл, лизиноприл и рамиприл. Примеры подходящих блокаторов рецептора ангиотензина включают кандесартан, эпросартан, ирбесартан, лосартан, олмесартан, тазосартан, телмисартан и валсартан. Примеры подходящих блокаторов кальциевых каналов включают дигидропиридины (DHP) и соединения, не являющиеся DHP. Подходящие DHP включают амлодипин, фелодипин, риозидин, израдипин, лацидипин, никардипин, нифедипин, нигульпидин, нилудипин, нимодифин, низолдипин, нитрендипин и нивалдипин и их фармацевтически приемлемые соли. Подходящие блокаторы кальциевых каналов, не являющиеся DHP, представляют собой флунаризин, прениламин, дилтиазем, фендилин, галлопамил, мибефрадил, анипамил, тиапамил и верампимил и их фармацевтически приемлемые соли. Подходящий диуретик представляет собой тиазидное производное, выбранное из амилорида, хлортиазида, гидрохлортиазида, метилхлортиазида и хлорталидона. Подходящим ингибитором ренина является алискирен. Примеры подходящих центрально действующих антигипертензивных средств включают клонидин, гуанабенз, гуанфацин и метилдопу. Примеры подходящих двойныхACE/NEP ингибиторов включают омапатрилат, фасидотрил и фасидотрилат. Примеры подходящих ингибиторов альдостеронсинтазы включают анастрозол, фадрозол и экземестан. Примеры подходящих антагонистов рецептора альдостерона включают спиронолактон и эплеренон. Кроме того, настоящее изобретение включает применение соединений настоящего изобретения в качестве активного терапевтического вещества, в частности, для лечения заболевания, опосредованногоTNNI3K. Конкретно, настоящее изобретение включает применение соединений настоящего изобретения для лечения сердечной недостаточности, в частности застойной сердечной недостаточности; гипертрофии сердца; сердечной недостаточности или застойной сердечной недостаточности в результате гипертрофии сердца и сердечной недостаточности или застойной сердечной недостаточности в результате ишемии миокарда или инфаркта миокарда. В другом аспекте настоящее изобретение включает применение соединений настоящего изобретения для получения лекарственного средства для применения в лечении вышеуказанных заболеваний. Композиции. Соединения настоящего изобретения будут обычно, но необязательно, формулироваться в фармацевтическую композицию перед введением пациенту. Соответственно, в другом аспекте настоящее изобретение относится к фармацевтическим композициям, содержащим соединение настоящего изобретения и фармацевтически приемлемое вспомогательное вещество. Фармацевтические композиции настоящего изобретения можно получить и упаковать в виде нерасфасованной формы, из которой можно извлечь эффективное количество соединения настоящего изобретения, и затем дать пациенту, такой как порошки, сиропы и растворы для инъекции. Альтернативно,фармацевтические композиции настоящего изобретения можно получить и упаковать в виде единичных лекарственных форм. Для перорального применения, например, можно вводить одну или более таблеток или капсул. Доза фармацевтической композиции содержит, по меньшей мере, терапевтически эффективное количество соединения настоящего изобретения (т.е. соединение формулы I или соль, в частности его фармацевтически приемлемые соли). При получении в виде единичной лекарственной формы фармацевтические композиции могут содержать от 1 до 1000 мг соединения настоящего изобретения. Фармацевтические композиции настоящего изобретения обычно содержат одно соединение настоящего изобретения. Однако в определенных вариантах осуществления фармацевтические композиции настоящего изобретения содержат более одного соединения настоящего изобретения. Кроме того, фармацевтические композиции настоящего изобретения могут необязательно дополнительно содержать одно или более дополнительных фармацевтически активных соединений. Как применяют в настоящем изобретении, "фармацевтически приемлемое вспомогательное вещество" обозначает вещество, композицию или среду, участвующую в придании формы или консистенции композиции. Каждое вспомогательное вещество должно быть совместимо с другими ингредиентами фармацевтической композиции при смешении так, чтобы избежать взаимодействий, которые могли бы заметно снижать эффективность соединения настоящего изобретения, при введении пациенту, и взаимодействий, которые могли бы приводить в результате к фармацевтическим композициям, которые были бы фармацевтически неприемлемыми. Кроме того, каждое вспомогательное вещество должно быть, конечно, достаточно высокой чистоты, чтобы сделать его фармацевтически приемлемым. Соединения настоящего изобретения и фармацевтически приемлемое вспомогательное вещество или вспомогательные вещества будут обычно формулировать в лекарственную форму, подобранную для введения пациенту требуемым способом введения. Общепринятые лекарственные формы включают формы, подобранные для (1) перорального введения, такие как таблетки, капсулы, каплеты, пилюли, пас- 15021439(2) парентерального введения, такие как стерильные растворы, суспензии и порошки для растворения;(3) трансдермального введения, такие как трансдермальные пластыри; (4) ректального введения, такие как суппозитории; (5) ингаляции, такие как аэрозоли и растворы; и (6) местного введения, такие как крема, мази, лосьоны, растворы, пасты, спреи, пены и гели. Подходящие фармацевтически приемлемые вспомогательные вещества будут изменяться в зависимости от конкретной выбранной лекарственной формы. Кроме того, подходящие фармацевтически приемлемые вспомогательные вещества можно выбрать для конкретной функции, которую они будут выполнять в композиции. Например, определенные фармацевтически приемлемые вспомогательные вещества можно выбрать в связи с их способностью облегчать получение однородных лекарственных форм. Определенные фармацевтически приемлемые вспомогательные вещества можно выбрать в связи с их способностью облегчать получение стабильных лекарственных форм. Определенные фармацевтически приемлемые вспомогательные вещества можно выбрать в связи с их способностью облегчать перенос или транспортировку соединения или соединений настоящего изобретения при введении пациенту из одного органа в другой орган или из одной части тела в другую часть тела. Определенные фармацевтически приемлемые вспомогательные вещества можно выбрать в связи с их способностью улучшать соблюдение больным режима и схемы лечения. Подходящие фармацевтически приемлемые вспомогательные вещества включают следующие типы вспомогательных веществ: разбавители, наполнители, связующие, разрыхлители, смазывающие вещества, глиданты, агенты для гранулирования, покрывающие агенты, смачивающие агенты, растворители,сорастворители, суспендирующие агенты, эмульгаторы, подсластители, ароматизаторы, агенты, корригирующие запах, красители, агенты, предотвращающие слипание, увлажнители, хелатирующие агенты,пластификаторы, агенты, увеличивающие вязкость, антиоксиданты, консерванты, стабилизаторы, поверхностно-активные вещества и буферные агенты. Специалистам в данной области техники ясно, что определенные фармацевтически приемлемые вспомогательные вещества могут выполнять более чем одну функцию и могут выполнять альтернативные функции в зависимости от того, какое количество вспомогательного вещества присутствует в составе и какие другие ингредиенты присутствуют в составе. Специалисты в данной области техники могут выбрать подходящие фармацевтически приемлемые вспомогательные вещества в подходящих количествах для применения в настоящем изобретении. Кроме того, существует ряд источников, которые являются доступными специалистам в данной области техники, которые описывают фармацевтически приемлемые вспомогательные вещества и могут быть пригодны при выборе подходящих фармацевтически приемлемых вспомогательных веществ. Примеры включают Remington's Pharmaceutical Sciences (Mack Publishing Company), The Handbook of PharmaceuticalPharmaceutical Association and the Pharmaceutical Press). Фармацевтические композиции настоящего изобретения получают, применяя методики и способы,известные специалистам в данной области техники. Некоторые из способов, обычно применяемых в данной области техники, описывают в Remington's Pharmaceutical Sciences (Mack Publishing Company). В одном аспекте настоящее изобретение относится к твердой пероральной лекарственной форме,такой как таблетка или капсула, содержащей эффективное количество соединения настоящего изобретения и разбавитель или наполнитель. Подходящие разбавители и наполнители включают лактозу, сахарозу, декстрозу, маннит, сорбит, крахмал (например, кукурузный крахмал, картофельный крахмал и предварительно клейстеризованный крахмал), целлюлозу и ее производные (например, микрокристаллическую целлюлозу), сульфат кальция и диосновный фосфат кальция. Пероральная твердая лекарственная форма может дополнительно содержать связующее. Подходящие связующие включают крахмал (например, кукурузный крахмал, картофельный крахмал и предварительно клейстеризованный крахмал), желатин, камедь, альгинат натрия, альгиновую кислоту, трагакант, гуаровую камедь, повидон, целлюлозу и ее производные (например, микрокристаллическую целлюлозу). Пероральная твердая лекарственная форма может дополнительно содержать разрыхлитель. Подходящие разрыхлители включают кросповидон, крахмал гликолят натрий, кроскармеллозу, альгиновую кислоту и карбоксиметилцеллюлозу натрия. Пероральная твердая лекарственная форма может дополнительно содержать смазывающее вещество. Подходящие смазывающие вещества включают стеариновую кислоту, стеарат магния, стеарат кальция и тальк. Примеры Следующие примеры иллюстрируют настоящее изобретение. Не предполагается, что данные примеры ограничивают объем настоящего изобретения, но скорее дают руководство специалисту в данной области техники по получению и применению соединений, композиций и способов настоящего изобретения. При описании конкретных вариантов осуществления настоящего изобретения специалисту в данной области техники ясно, что различные изменения и модификации можно осуществлять, не выходя за пределы сущности и объема настоящего изобретения. В следующем экспериментальном описании можно применять следующие сокращения: Стадия 1. 4-Фтор-3-нитробензолсульфонилхлорид. 1-Фтор-2-нитробензол (50,0 г, 0,354 моль) добавляли к хлорсульфокислоте (91 г, 0,778 моль) при 65 С. Затем полученную в результате смесь грели при 100 С в течение 18 ч. Смесь охлаждали до комнатной температуры, выливали на лед и экстрагировали хлористым метиленом. Затем объединенные органические слои промывали NaHCO3, затем соляным раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме для того, чтобы получить 4-фтор-3-нитробензолсульфонилхлорид (55,3 г, 65% выход) в виде коричневого масла. Стадия 2. 4-Фтор-N-метил-3-нитробензолсульфонамид. К раствору 4-фтор-3-нитробензолсульфонилхлорида (43 г, 179,5 ммоль) в THF (500 мл) добавляли триэтиламин (150 мл, 1,08 моль). Смесь охлаждали до -35 С и добавляли по каплям гидрохлорид метиламина (14,5 г, 215,4 ммоль) в воде. Через 1 ч смесь нагревали до комнатной температуры и разбавляли смесью 1:1 вода/этилацетат. Органический слой отделяли и промывали насыщенным водным бикарбонатом натрия, затем соляным раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме. Неочищенный остаток очищали колоночной хроматографией (20% этилацетат/петролейный эфир) для получения 4-фтор-N-метил-3-нитробензолсульфонамида (38 г, 90% выход) в виде желтого твердого остатка. Пример получения 2. 3-Амино-N-метил-4-[(1-метилэтил)окси]бензолсульфонамидNaH (0,440 г, 11 ммоль) добавляли к 20 мл изопропанола и полученную в результате смесь перемешивали при комнатной температуре. Через 30 мин добавляли 4-фтор-N-метил-3 нитробензолсульфонамид (2,34 г, 10 ммоль). Затем реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь выливали в EtOAc и воду. Органическую фазу отделяли, сушили надNa2SO4 и концентрировали в вакууме для получения неочищенного продукта. Очистка колоночной хроматографиейN-метил-4-[(1-метилэтил)окси]-3 нитробензолсульфонамид (1,6 г, 58% выход) в виде желтого твердого остатка.(20 мл) в атмосфере азота добавляли Pd/C (0,160 г). Затем колбу вакуумировали и заполняли водородом три раза. Полученную в результате смесь перемешивали в атмосфере водорода в течение ночи при комнатной температуре. Затем смесь фильтровали и концентрировали для того, чтобы получить 3-амино-N-метил-4-[(1-метилэтил)окси]бензолсульфонамид (1,1 г, 77%) в виде белого твердого остатка. 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 7,01-7,10 (м, 2 Н), 6,87-6,98 (м, 2 Н), 5,08 (уш.с, 2 Н), 4,63 (дт,J=5,93, 11,98 Гц, 1 Н), 2,34-2,41 (м, 3 Н), 1,29 (д, J=6,02 Гц, 6 Н); Следующие анилины получали из 4-фтор-N-метил-3-нитробензолсульфонамида, применяя способы,аналогичные способам, описанным в примере получения 2. Стадия 1. N-Метил-4-(метилтио)-3-нитробензолсульфонамид. К раствору 4-фтор-N-метил-3-нитробензолсульфонамида (15 г, 64,01 ммоль) в THF (150 мл) добавляли по каплям 20% CH3SNa (22,4 г, 64,01 ммоль). Затем полученную в результате смесь перемешивали в течение ночи. Утром смесь выливали в этилацетат и воду, органическую фазу отделяли, сушили надNa2SO4, фильтровали и концентрировали. Затем неочищенное вещество очищали колоночной хроматографией (1:1 этилацетат/петролейный эфир) для того, чтобы получить N-метил-4-(метилтио)-3 нитробензолсульфонамид (3,29 г, 19%) в виде желтого твердого остатка. Стадия 2. 3-Амино-N-метил-4-(метилтио) бензолсульфонамид. К раствору N-метил-4-(метилтио)-3-нитробензолсульфонамида (1,0 г, 3,81 ммоль) в 10 мл этанола и 10 мл NH4Cl добавляли цинковую пыль (2,5 г, 3,81 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем смесь фильтровали и разбавляли этилацетатом и водой. Органическую фазу отделяли, промывали водой и соляным раствором, сушили над MgSO4, фильтровали и концентрировали для того, чтобы получить 3-амино-N-метил-4-(метилтио)бензолсульфонамид (0,500 г, 56%) в виде белого твердого остатка. 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 7,06 (д, J=8,03 Гц, 1 Н), 6,86 (с, 1 Н), 6,67-6,76 (м, 1 Н), 5,28 (уш.с,2 Н), 2,17 (с, 3 Н), 2,21 (с, 3 Н);MS (m/z) 232,7 (М+Н+). Следующие анилины получали из 4-фтор-N-метил-3-нитробензолсульфонамида, применяя способы,аналогичные способам, описанным в примере получения 3. Стадия 1. N-Метил-4-(4-морфолинил)-3-нитробензолсульфонамид. К раствору 4-фтор-N-метил-3-нитробензолсульфонамида (2,00 г, 8,54 ммоль) и морфолина (0,744 г,8,54 ммоль) в THF (100 мл) добавляли N,N-диизопропилэтиламин (2,21 г, 17,08 ммоль). Полученный в результате раствор перемешивали при 50 С в течение ночи. Утром реакционную смесь охлаждали до комнатной температуры и концентрировали досуха в вакууме. Остаток растворяли в этилацетате и промывали водой и соляным раствором, сушили над MgSO4, фильтровали и концентрировали в вакууме для получения N-метил-4-(4-морфолинил)-3-нитробензолсульфонамида (2,5 г, 97%) в виде красного масла.MS (m/z) 302,0 (М+Н+). Стадия 2. 3-Амино-N-метил-4-(4-морфолинил)бензолсульфонамид. К смеси N-метил-4-(4-морфолинил)-3-нитробензолсульфонамида (2,5 г, 8,30 ммоль) в THF (100 мл) в атмосфере азота добавляли Pd/C (0,8 г). Затем колбу вакуумировали и заполняли водородом три раза. Полученную в результате смесь перемешивали в атмосфере водорода при 50 С в течение ночи. Затем смесь фильтровали и концентрировали для того, чтобы получить 3-амино-N-метил-4-(4 морфолинил)бензолсульфонамид (1,98 г, 88%). 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 7,07-7,17 (м, 2 Н), 7,01 (д, J=8,28 Гц, 1 Н), 6,94 (дд, J=1,88,8,16 Гц, 1 Н), 5,20 (с, 2 Н), 3,72-3,81 (м, 4 Н), 2,80-2,89 (м, 4 Н), 2,38 (д, J=4,77 Гц, 3 Н); Следующие анилины получали из 4-фтор-N-метил-3-нитробензолсульфонамида и указанного амина, применяя способы, описанные в примере получения 4. К смеси 4-фтор-N-метил-3-нитробензолсульфонамида (1,6 г, 6,83 ммоль) в THF (50 мл) в атмосфере азота добавляли Pd/C (0,600 г). Затем колбу вакуумировали и заполняли водородом. Полученную в результате смесь перемешивали в атмосфере водорода в течение ночи при 50 С. Затем смесь фильтровали и концентрировали для того, чтобы получить 3-амино-4-фтор-N-метилбензолсульфонамид (1,25 г, 89%) в виде грязно-белого твердого остатка. 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 7,26 (кв, J=4,85 Гц, 1 Н), 7,13-7,22 (м, 2 Н), 6,90 (ддд, J=2,38,4,27, 8,41 Гц, 1 Н), 5,63 (с, 2 Н), 2,40 (д, J=5,02 Гц, 3 Н);-40 С перед обработкой раствором гидрохлорида метиламина (2,64 г, 39,1 ммоль) в 10 мл воды с последующей обработкой TEA (5,44 мл, 39,1 ммоль). Реакционную смесь перемешивали и нагревали до комнатной температуры в течение 1 ч перед распределением между 350 мл EtOAc и 30 мл соляного раствора. Органический слой промывали дважды соляным раствором, сушили над MgSO4 и подвергали флэшхроматографии (330 г силикагеля, 0-40% EtOAc/гексан) для того, чтобы получить 4-хлор-N-метил-3 нитробензолсульфонамид (6,38 г, 65%) в виде светло-желтого твердого остатка.(50,0 мл) обрабатывали железом (14,15 г, 253 ммоль) и NH4Cl (13,55 г, 253 ммоль), грели при 90 С в течение 4 ч перед охлаждением и фильтровали через целит. Остаток на фильтре промывали EtOAc и объединенные фильтраты фильтровали снова для удаления выпавшего NH4Cl перед концентрированием. Полученное в результате неочищенное вещество распределяли между 350 мл EtOAc и 50 мл насыщенного водного NaHCO3. Органический слой промывали соляным раствором, сушили над MgSO4, концентрировали и подвергали колоночной флэш-хроматографии (330 г силикагеля, 0-15% EtOAc/DCM) для того,чтобы получить 3-амино-4-хлор-N-метилбензолсульфонамид (5,60 г, 100%) в виде светло-желтого кристаллического твердого остатка. 1 Н ЯМР (400 МГц, MeOD)м.д. 7,39 (д, J=8,28 Гц, 1 Н), 7,27 (д, J=2,26 Гц, 1 Н), 7,03 (дд, J=8,28, 2,26 Гц, 1 Н), 2,54 (с, 3 Н); Стадия 1. Фенилметил [(4-фтор-3-нитрофенил)сульфонил]метилкарбамат. Раствор 4-фтор-N-метил-3-нитробензолсульфонамида (2 г, 8,54 ммоль) в THF (20 мл) обрабатывали триэтиламином (2,380 мл, 17,08 ммоль) с последующим добавлением по каплям бензилхлорформиата(3,75 мл, 11,10 ммоль). Смесь перемешивали при 25 С в течение 5 ч перед концентрированием. Остаток обрабатывали водой и экстрагировали CH2Cl2. Органические экстракты промывали (соляным раствором),сушили (сульфат натрия), концентрировали и подвергали флэш-хроматографии (25-50% EtOAc-гексан) для получения желтого твердого остатка, который суспендировали в смеси EtOAc-гексан, собирали фильтрацией и промывали гексаном для получения фенилметил[(4-фтор-3 нитрофенил)сульфонил]метилкарбамата (1 г, 32%) в виде белого твердого остатка.(10 мл) при 25 С обрабатывали 2,2,2-трифторэтиламином (0,592 г, 5,97 ммоль) и перемешивали в течение 20 ч перед концентрированием для получения желтого масла, которое растворяли в смесиEtOAc/гексан. Образовывался желтый осадок, который собирали фильтрацией и промывали гексаном для получения фенилметилметил(3-нитро-4-[(2,2,2-трифторэтил)амино]фенилсульфонил)карбамата(1,07 г, 88%) в виде желтого твердого остатка.MS (m/z) 448,1 (М+Н+). Стадия 3. Фенилметилметил(4-[метил(2,2,2-трифторэтил)амино]-3-нитрофенилсульфонил)карбамат. Раствор фенилметилметил(3-нитро-4-[(2,2,2-трифторэтил)амино]фенилсульфонил)карбамата (1 г,2,24 ммоль) в DMF (1 мл) при 25 С обрабатывали гидридом натрия (0,179 г, 4,47 ммоль) и перемешивали в течение 2 мин перед обработкой йодометаном (0,42 мл, 6,71 ммоль). Через 1 ч смесь разбавляли водой и экстрагировали EtOAc. Органический экстракт промывали (соляной раствор), сушили (сульфат натрия), концентрировали и подвергали флэш-хроматографии (10-35% EtOAc-гексан) для получения фенилметилметил(4-[метил(2,2,2-трифторэтил)амино]-3-нитрофенилсульфонил)карбамата (539 мг, 52%) в виде желтого масла.MS (m/z) 462,1 (М+Н+). Стадия 4. 3-Амино-N-метил-4-[метил(2,2,2-трифторэтил)амино]бензолсульфонамид. Раствор фенилметилметил(4-[метил(2,2,2-трифторэтил)амино]-3-нитрофенилсульфонил)карбамата (539 мг, 1,17 ммоль) в MeOH (10 мл) при 25 С обрабатывали 10% палладием на угле (124 мг,0,117 ммоль) и перемешивали в атмосфере водорода (баллон) в течение ночи перед фильтрованием через целит. Фильтрат снова фильтровали через 0,45-микронный шприцевой фильтр и концентрировали для получения 3-амино-N-метил-4-[метил(2,2,2-трифторэтил)амино]бензолсульфонамида (320 мг, 92%) в виде коричневого масла. 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 7,14-7,20 (м, 2 Н), 7,12 (д, J=2,26 Гц, 1 Н), 6,95 (дд, J=8,28,2,26 Гц, 1 Н), 5,23 (с, 2 Н), 3,82 (кв, J=9,87 Гц, 2 Н), 2,83 (с, 3 Н), 2,39 (д, J=5,02 Гц, 3 Н); Стадия 1. 2-Фтор-5-нитробензолсульфонилхлорид. Смесь 1-фтор-4-нитробензола (3,0 г, 21,3 ммоль) в хлорсульфокислоте (5,5 мл, 84 ммоль) перемешивали при 90-100 С в течение 8 ч перед охлаждением до комнатной температуры и медленно выливали в ледяную воду и экстрагировали EtOAc. Органический экстракт промывали насыщенным водным(сульфат натрия) и концентрировали для получения 2-фтор-5-нитробензолсульфонилхлорида (3,2 г, 63%) в виде бесцветного масла, которое непосредственно применяли на следующей стадии. Стадия 2. 2-Фтор-N-метил-5-нитробензолсульфонамид. Раствор 2-фтор-5-нитробензолсульфонилхлорида (3,2 г, 12,6 ммоль) в THF (30 мл) при -45 С обрабатывали гидрохлоридом метиламина (1,0 г, 15,1 ммоль) и триэтиламином (2,1 мл, 15,1 ммоль) и перемешивали в течение 30 мин. Затем смесь обрабатывали 6 М водной HCl для доведения рН до 3 и нагревали до комнатной температуры перед разбавлением водой и экстрагированием EtOAc. Органический экстракт сушили (сульфат натрия), концентрировали и подвергали флэш-хроматографии (5-20%EtOAc-петролейный эфир) для получения 2-фтор-N-метил-5-нитробензолсульфонамида в виде желтого твердого остатка (3,0 г, 93%).MS (m/z) 235,1 (M+H+). Стадия 3. 5-Амино-2-фтор-N-метилбензолсульфонамид. Раствор 2-фтор-N-метил-5-нитробензолсульфонамида (3,0 г, 12,8 ммоль) в MeOH (40 мл) обрабатывали 10% палладием на угле (300 мг, 0,28 ммоль) и перемешивали в атмосфере водорода (40 psi) в течение 8 ч перед фильтрованием через целит и концентрированием для получения 5-амино-2-фтор-N-метилбензолсульфонамида (2,5 г, 96%) в виде грязно-белого твердого остатка. 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 7,40-7,49 (м, 1 Н), 7,01-7,09 (м, 1 Н), 6,94 (дд, J=5,95, 2,87 Гц,1 Н), 6,71-6,77 (м, 1 Н), 5,49 (уш.с, 2 Н), 2,45 (д, J=4,85 Гц, 3 Н);MS (m/z) 205,1 (М+Н+). Следующие анилины получали из указанных нитробензолов, применяя способы, аналогичные способам, описанным в примере получения 8. Стадия 1. 2,4-Дифтор-5-нитробензолсульфонилхлорид. Смесь 2,4-дифтор-1-нитробензола (20 г, 126 ммоль) в хлорсульфокислоте (44 г, 378 ммоль) перемешивали при 100 С в течение 48 ч перед тем, как вылить ее в ледяную воду и экстрагировать EtOAc. Органический экстракт сушили (Na2SO4), концентрировали и остаток растирали со смесью 10%EtOAc-петролейный эфир для получения 2,4-дифтор-5-нитробензолсульфонилхлорида в виде коричневого масла (21 г, 81%), который применяли непосредственно на следующей стадии. Стадия 2. 2,4-Дифтор-N-метил-5-нитробензолсульфонамид. Раствор 2,4-дифтор-5-нитробензолсульфонилхлорида (21 г, 81 ммоль) в THF (400 мл) при -60 С обрабатывали гидрохлоридом метиламина (6,6 г, 97 ммоль) и затем обрабатывали по каплям триэтиламином (22,6 мл, 162 ммоль). После перемешивания в течение 6 ч при -(60-40)С смесь доводили до рН 3 добавлением 15% водной HCl, разбавляли водой и экстрагировали EtOAc. Органические экстракты сушили (Na2SO4), концентрировали и подвергали флэш-хроматографии (17% EtOAc-петролейный эфир) для получения 2,4-дифтор-N-метил-5-нитробензолсульфонамида (8 г, 38%) в виде коричневого твердого остатка. 1 Н ЯМР (400 МГц, CDC13)м.д. 8,66-8,74 (м, 1 Н), 7,20-7,25 (м, 1 Н), 4,81-4,91 (м, 1 Н), 2,78-2,81 (м,3 Н). Стадия 3. 4-(Диметиламино)-2-фтор-N-метил-5-нитробензолсульфонамид. Раствор 2,4-дифтор-N-метил-5-нитробензолсульфонамида (8,0 г, 31,6 ммоль) в CH2Cl2 (200 мл) при-20 С обрабатывали гидрохлоридом диметиламина (2,56 г, 31,6 ммоль). Полученную в результате смесь обрабатывали по каплям триэтиламином и перемешивали в течение 1 ч перед обработкой 15% воднойHCl для доведения рН до 3, разбавляли водой и экстрагировали EtOAc. Органический экстракт сушили(сульфат натрия), концентрировали и подвергали флэш-хроматографии (20-50% EtOAc-петролейный эфир) для получения 4-(диметиламино)-2-фтор-N-метил-5-нитробензолсульфонамида (4,0 г, 46%) в виде желтого твердого остатка.THF (100 мл) при 25 С обрабатывали Cs2CO3 (20,5 г, 62,9 ммоль) и перемешивали в течение 8 ч перед разбавлением, добавлением воды и экстрагированием EtOAc. Органический экстракт сушили (Na2SO4),концентрировали и подвергали флэш-хроматографии (3% EtOAc-петролейный эфир) для получения 4-фтор-1-нитро-2-[(2,2,2-трифторэтил)окси]бензола (10 г, 67%) в виде желтого твердого остатка.(82 мл, 125,5 ммоль) перемешивали при 50 С в течение 8 ч перед тем, как ее выливали на лед и экстрагировали EtOAc. Органические экстракты сушили (Na2SO4) и концентрировали для получения 2-фтор-5 нитро-4-[(2,2,2-трифторэтил)окси]бензолсульфонилхлорида (15 г, неочищенный) в виде коричневого масла, которое применяли непосредственно на следующей стадии. Стадия 3. 2-Фтор-N-метил-5-нитро-4-[(2,2,2-трифторэтил)окси]бензолсульфонамид. Смесь 2-фтор-5-нитро-4-[(2,2,2-трифторэтил)окси]бензолсульфонилхлорида (15 г, неочищенный) вTHF (150 мл) при -45 С обрабатывали гидрохлоридом метиламина (5,96 г, 89 ммоль) и затем обрабатывали по каплям триэтиламином (12,4 мл, 89 ммоль). После перемешивания в течение 1 ч при -45 С смесь доводили до рН 3 добавлением водной 3 М HCl, нагревали до комнатной температуры, разбавляли водой и экстрагировали EtOAc. Органический экстракт сушили (Na2SO4), концентрировали и подвергали флэшхроматографии (9-17% EtOAc-петролейный эфир) для получения 2-фтор-N-метил-5-нитро-4-[(2,2,2 трифторэтил)окси]бензолсульфонамида (10 г, 72% для двух стадий) в виде желтого твердого остатка.MS (m/z) 333,0 (М+Н+). Стадия 4. 5-Амино-2-фтор-N-метил-4-[(2,2,2-трифторэтил)окси]бензолсульфонамид. Смесь 2-фтор-N-метил-5-нитро-4-[(2,2,2-трифторэтил)окси]бензолсульфонамида (10 г, 30,1 ммоль) в MeOH (150 мл) обрабатывали 10% Pd/C (1 г) и перемешивали в атмосфере Н 2 (45 psi) при 45 С в течение 10 ч перед фильтрованием. Фильтрат концентрировали для получения 5-амино-2-фтор-N-метил-4[(2,2,2-трифторэтил)окси]бензолсульфонамида (8 г, 88%) в виде белого твердого остатка. 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 7,40 (кв, J=5,07 Гц, 1 Н), 7,10 (д, J=11,69 Гц, 1 Н), 7,05 (д,J=7,28 Гц, 1H), 5,04 (с, 2H), 4,83 (кв, J=8,82 Гц, 2 Н), 2,42 (д, J=4,41 Гц, 3 Н); Стадия 1. N-Метил-4-(метилсульфонил)-3-нитробензолсульфонамид. Смесь N-метил-4-(метилтио)-3-нитробензолсульфонамида (1,0 г, 3,81 ммоль) в AcOH (15 мл) обрабатывали по каплям 30% водной Н 2 О 2 (5 мл, 44 ммоль) и перемешивали при 117 С в течение 2 ч перед охлаждением до комнатной температуры и разбавлением водой (100 мл). Образовавшийся осадок собирали фильтрацией и сушили для получения N-метил-4-(метилсульфонил)-3-нитробензолсульфонамида(1,5 г, неочищенный) в виде желтого твердого остатка, который применяли непосредственно на следующей стадии. Стадия 2. 3-Амино-N-метил-4-(метилсульфонил)бензолсульфонамид. Раствор N-метил-4-(метилсульфонил)-3-нитробензолсульфонамида (1,5 г, неочищенный) в EtOH(20 мл) и насыщенном водном NH4Cl (20 мл) обрабатывали Zn пылью (3,3 г, 51 ммоль) и перемешивали при 50 С в течение ночи перед фильтрованием (EtOAc промывка). Фильтрат разбавляли водой и экстрагировали EtOAc. Органический экстракт промывали водой и соляным раствором, сушили (MgSO4), кон- 25021439 центрировали и подвергали флэш-хроматографии (33% EtOAc-петролейный эфир) для получения 3-амино-N-метил-4-(метилсульфонил)бензолсульфонамида (950 мг, 94% 2 стадии) в виде желтого твердого остатка. 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 7,73 (д, J=8,28 Гц, 1 Н), 7,58 (кв, J=4,94 Гц, 1 Н), 7,34 (д,J=1,76 Гц, 1 Н), 7,01 (дд, J=8,53, 1,76 Гц, 1 Н), 6,48 (с, 2 Н), 3,18 (с, 3 Н), 2,47 (д, J=5,02 Гц, 3 Н);J. Med. Chem. (1999), 42, 3860) в POCl3 (2 мл) грели при 80-90 С в течение 8 ч перед концентрированием. Остаток обрабатывали насыщенным водным NaHCO3 и экстрагировали EtOAc. Органический экстракт сушили (Na2SO4) и концентрировали для получения 4,5-дихлор-6,7-бис-(метилокси)хиназолина (70 мг,65%) в виде желтого твердого остатка. 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 8,94 (с, 1 Н), 7,56 (с, 1 Н), 4,05 (с, 3 Н), 3,87 (с, 3 Н);MS (m/z) 258,9 (М+Н+). Следующие 4-хлорхиназолины получали из указанных имеющихся в продаже или известных хиназолинонов, применяя способы, аналогичные способам, описанным в примере получения 12.MeOH (1 мл) при 25 С обрабатывали Cs2CO3 (1,7 г, 5,2 ммоль). После перемешивания смеси в течение 30 мин добавляли йодоэтан (0,42 мл, 5,2 ммоль) и смесь перемешивали при 60 С в течение 40 ч перед охлаждением до комнатной температуры. Твердый остаток собирали фильтрацией и промывали MeOH для получения 6-(этилокси)-7-(метилокси)-4(1 Н)-хиназолинона (560 мг, 49%).MS (m/z) 221,1 (М+Н+). Стадия 2. 4-Хлор-6-(этилокси)-7-(метилокси)хиназолин. Смесь 6-(этилокси)-7-(метилокси)-4(1 Н)-хиназолинона (560 мг, 2,54 ммоль) в POCl3 (5 мл,53,6 ммоль) обрабатывали 1 каплей DMF и грели при 100 С в течение 2 ч перед концентрированием. Остаток обрабатывали насыщенным водным NaHCO3 и экстрагировали CH2Cl2. Органические экстракты сушили(сульфат натрия) и концентрировали для получения 4-хлор-6-(этилокси)-7(метилокси)хиназолина (416 мг, 1,74 ммоль, 69%) в виде грязно-белого твердого остатка.MS (m/z) 239,0 (М+Н+). Следующие хиназолины получали, применяя способы, аналогичные способам, описанным в примере получения 13, заменой йодоэтана на стадии 1 указанным алкилгалогенидом. Стадия 1. 5-(Этилокси)-7-(метилокси)-4(1 Н)-хиназолинон. Смесь [5-гидрокси-7-(метилокси)-4-оксо-3(4 Н)-хиназолинил]метил-2,2-диметилпропаноата (1 г,3,26 ммоль, полученный согласно J. Med. Chem. (2006), 49 6465), трифенилфосфина (1,370 г, 5,22 ммоль) и этанола (0,210 мл, 3,59 ммоль) в DCM (10 мл) при 0 С обрабатывали по каплям в течение 2 мин 40% раствором DEAD в толуоле (2,379 мл, 5,22 ммоль). После добавления смесь нагревали до комнатной температуры и перемешивали в течение 3 ч перед концентрированием. Остаток растворяли в 7 М аммиаке в метаноле (10 мл, 70,0 ммоль) и смесь перемешивали в течение 3 дней. Твердый остаток собирали фильтрацией и промывали MeOH и CH2Cl2 для получения 5-(этилокси)-7-(метилокси)-4(1 Н)хиназолинона (286 мг, 40%) в виде белого твердого остатка.MS (m/z) 221,1 (М+Н+). Стадия 2. 4-Хлор-5-(этилокси)-7-(метилокси)хиназолин. Смесь 5-(этилокси)-7-(метилокси)-4(1 Н)-хиназолинона (285 мг, 1,29 ммоль) в POCl3 (10 мл,107 ммоль) обрабатывали 1 каплей DMF и перемешивали при 100 С в течение 1 ч перед концентрированием. Остаток обрабатывали насыщенным водным NaHCO3 и экстрагировали CH2Cl2. Органические экстракты сушили (сульфат натрия), концентрировали и подвергали флэш-хроматографии (1-3%MeOH-CH2Cl2) для получения 4-хлор-5-(этилокси)-7-(метилокси)хиназолина (260 мг, 84%) в виде белого твердого остатка. 1MS (m/z) 239,0 (М+Н+). Следующие хиназолины получали, применяя способы, аналогичные способам, описанным в примере получения 14 заменой этанола на стадии 1 указанным спиртом.HCl в Et2O (40,9 мл, 82 ммоль) и полученный в результате светло-серый твердый остаток собирали и промывали эфиром для получения HCl соли анилина (11,5 г), которую обрабатывали оксалилхлоридом(16,72 мл, 191 ммоль), и смесь грели при 170 С в течение 1,5 ч перед охлаждением до комнатной температуры и обработки MeOH. Полученную в результате смесь нагревали до кипения и фильтровали в горячем состоянии для получения красного твердого остатка, который промывали MeOH и сушили на воздухе для получения 4,5,6-трис-(метилокси)-1H-индол-2,3-диона (10,5 г, 81%) в виде красно-оранжевого твердого остатка.(70,5 мл, 582 ммоль) при 100 С обрабатывали 30% водным пероксидом водорода (12,9 мл, 126 ммоль) по каплям в течение 30 мин при такой скорости, чтобы предотвратить выливание из колбы вспенивающейся смеси. После завершения добавления смесь перемешивали в течение 10 мин перед охлаждением и разбавлением с добавлением льда. Смесь доводили до рН 8 добавлением концентрированной HCl (70 мл) и затем подкисляли до рН 3 добавлением ледяной уксусной кислоты. Образовавшийся твердый остаток собирали фильтрацией, промывали водой и сушили в вакууме для получения 6-амино-2,3,4-трис(метилокси)бензойной кислоты (2,7 г, 28%) в виде коричневого твердого остатка.MeOH (20 мл) при 25 С обрабатывали раствором 2 М TMS-диазометана в Et2O (14,85 мл, 29,7 ммоль) и перемешивали в течение 10 мин перед концентрированием для получения метил 6-амино-2,3,4-трис(метилокси)бензоата (2,87 г, 99%) в виде коричневого твердого остатка.(3,72 г, 35,7 ммоль) в 2-метоксиэтаноле (25 мл) перемешивали при 125 С в течение 2 ч перед концентрированием. Остаток суспендировали в воде и твердый остаток собирали фильтрацией, промывали водой и сушили в вакууме (45 С) для получения 5,6,7-трис-(метилокси)-4(1 Н)-хиназолинона (1 г, 34%) в виде светло-коричневого твердого остатка.MS (m/z) 237,1 (М+Н+). Стадия 5. 4-Хлор-5,6,7-трис-(метилокси)хиназолин. 5,6,7-трис-(Метилокси)-4(1 Н)-хиназолинон (0,8 г, 3,39 ммоль) обрабатывали POCl3 (10 мл,107 ммоль) и одной каплей DMF и перемешивали при 100 С в течение 1 ч перед концентрированием. Остаток обрабатывали насыщенным водным NaHCO3 и экстрагировали CH2Cl2. Органические экстракты сушили (сульфат натрия) и концентрировали для получения 4-хлор-5,6,7-трис-(метилокси)хиназолина(770 мг, 89%) в виде коричневого твердого остатка. 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 8,88 (с, 1 Н), 7,37 (с, 1 Н), 4,04 (с, 3 Н), 3,94 (с, 3 Н), 3,91 (с, 3 Н); Раствор 3-амино-N-метил-4-[(2,2,2-трифторэтил)окси]бензолсульфонамида (3,0 г, 10,7 ммоль) в изо-PrOH (100 мл) обрабатывали 4-хлор-6-(метилокси)-7-[(фенилметил)окси]хиназолином (3,2 г,10,7 ммоль, полученный согласно J. Med. Chem. (1999), 42, 5369), и перемешивали при 80 С в течение 2 ч перед концентрированием. Остаток промывали петролейным эфиром и сушили для полученияN-метил-3-(6-(метилокси)-7-[(фенилметил)окси]-4-хиназолиниламино)-4-[(2,2,2-трифторэтил)окси]бензолсульфонамида (4,2 г, 72%) в виде коричневого твердого остатка. 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 11,42 (уш.с, 1 Н), 8,75 (с, 1 Н), 8,20 (с, 1 Н), 7,80-7,84 (м, 2 Н),7,49-7,57 (м, 4 Н), 7,35-7,49 (м, 4 Н), 5,34 (с, 2 Н), 4,91 (кв, J=8,82 Гц, 2 Н), 3,99 (с, 3 Н), 2,45 (д, J=5,07 Гц,3 Н);MS (m/z) 549,1 (М+Н+). Следующие соединения получали с помощью способов, аналогичных способам, описанным в примере получения 16, применяя в качестве исходных веществ указанные хиназолин и анилин.
МПК / Метки
МПК: A61K 31/517, A01N 43/54
Метки: хиназолиновые, соединения
Код ссылки
<a href="https://eas.patents.su/30-21439-hinazolinovye-soedineniya.html" rel="bookmark" title="База патентов Евразийского Союза">Хиназолиновые соединения</a>
Хиназолиновые соединения

Номер патента: 20330
Опубликовано: 30.10.2014
Авторы: Мюррей Питер Джон, Кинг-Андервуд Джон, Брукфилд Фредерик Артур, Харди Джордж, Браун Кристофер Джон, Ито Кадзухиро
МПК: A61K 31/519, A61P 11/00, A61P 11/06...
Метки: хиназолиновые, соединения
Формула / Реферат:
1. Соединение формулы (I)в которой R1 выбран из группы, состоящей из-H;разветвленной или неразветвленной C1-10 алкильной цепи, в которой по меньшей мере 1 атом углерода заменен кислородом;-CH2CH2CH2OH;-CH2OCH2CH2OCH2CH2OH;-CH2NHCONH2;-C1-3 алкиленСО2Н;-CH2CH2CH2CH2CO2H;-CH2CH2CH2C(O)NHCH(CH3)2;-CH2CH2CH2C(O)(CH3)2;-CH2CH2CH2NHCH2CH2OCH3;-CH2CH2CH2N(CH2CH2OCH3)2;-CH2CH2CH2C(O)N(CH2CH2OCH3)2;-CH2NHC(O)CH2NHC(O)CH3;-CH2CH2CH2C(O)NHCH2CH2OCH3;X...
Замещенные хиназолиновые соединения

Номер патента: 19110
Опубликовано: 30.01.2014
Авторы: Лериш Каролине, Оклер Эрик, Ле-Ру Жак, Миддлмисс Дейвид Н.
МПК: A61K 31/502, C07D 401/14, A61P 35/00...
Метки: замещенные, соединения, хиназолиновые
Формула / Реферат:
1. Соединение, имеющее структуру (I), или его фармацевтически приемлемая сольгде R1 представляет собой фенил, необязательно замещенный фрагментами R9 и R10, иR2 представляет собой водород или метил,R3 представляет собой водород, С1-С4алкил, циклоалкил, гетероциклоалкил, арил, гетероарил,-CN, -CF3, -OR4, -OCOR4, -COR4, -NR4R5, -NR4COR5, -NR4COOR5, -(С1-С4алкил)OR4, -(C1-C4алкил)COR4, -(С1-С4алкил)NR4R5, -(С1-C4алкил)NR4COR5,...
Хинолиновые и хиназолиновые соединения, применяемые в терапии, особенно при лечении доброкачественной гиперплазии простаты

Номер патента: 2851
Опубликовано: 31.10.2002
Автор: Фокс Дэвид Натан Абрахам
МПК: C07D 401/14, A61K 31/4709, A61P 13/08...
Метки: лечении, применяемые, доброкачественной, гиперплазии, хинолиновые, хиназолиновые, соединения, простаты, особенно, терапии
Формула / Реферат:
1. Соединение формулы I где R1 представляет собой C1-4 алкокси, необязательно замещенную одним или несколькими атомами фтора; R2 представляет собой Н или C1-6 алкокси, необязательно замещенную одним или несколькими атомами фтора; R3 представляет собой 5- или 6-членное гетероциклическое кольцо, содержащее, по меньшей мере, один гетероатом, выбранный из N, О и S, причем этот цикл необязательно замещен одной или несколькими группами, выбранными из...
Новые 4-(тетразол-5-ил) хиназолиновые производные в качестве противораковых средств

Номер патента: 18716
Опубликовано: 30.10.2013
Авторы: Пулла Редди Муддасани, Адибхатла Кали Сатиа Бхуджанга Рао, Венкайах Човдари Наннапанени, Анантханени Лакшми, Пула Субба Рао, Конаканчи Дурга Прасад
МПК: C07D 239/72, A61P 35/00, A61K 31/498...
Метки: новые, хиназолиновые, производные, 4-(тетразол-5-ил, качестве, противораковых, средств
Формула / Реферат:
1. 4-(Тетразол-5-ил)хиназолиновое производное формулы Iв которой каждый R1 представляет собой С1-С6алкил;R2 представляет собой водород или его выбирают из группы, состоящей из замещенного бензила, пиридин-2-илметила и 1-метила-1Н-имидазол-2-илметила, где заместители для бензила независимо выбирают из группы, состоящей из нитро и амино;или его фармацевтически приемлемая соль.2. 4-(Тетразол-5-ил)хиназолиновое производное формулы I или его...
6,7-диалкокси хиназолиновые производные, эффективные для лечения нарушений, связанных с раком

Номер патента: 18514
Опубликовано: 30.08.2013
Авторы: Венкайя Чоудари Наннапанени, Джиотхи Прасад Раманадхам, Адибхатла Кали Сатьа Бхуджанга Рао, Нагешвара Рао Боллепалли
МПК: A61K 31/517, A61P 35/00, C07D 239/94...
Метки: связанных, эффективные, хиназолиновые, нарушений, лечения, производные, 6,7-диалкокси, раком
Формула / Реферат:
1. Применение хиназолинового производного формулыили его фармацевтически приемлемой соли для получения лекарственного средства для лечения рака легких, рака молочной железы.2. Применение по п.1, отличающееся тем, что3. Применение по п.2, отличающееся тем, что фармацевтически приемлемая соль представляет собой моногидрохлорид (NRC-2694A).4. Применение по п.2, отличающееся тем, что фармацевтически приемлемая соль представляет собой дигидрохлорид...
Предыдущий патент: Способ и система для управления роумингом мобильного оборудования
Следующий патент: Способ производства фармацевтически активного вещества
Случайный патент: Способ производства кисломолочного продукта "наринэ"