Противораковая комбинация
Номер патента: 20589
Опубликовано: 30.12.2014
Авторы: Лебовиц Питер, Дамбл Мелисса, Лакер Сильви, Кумар Ракеш




























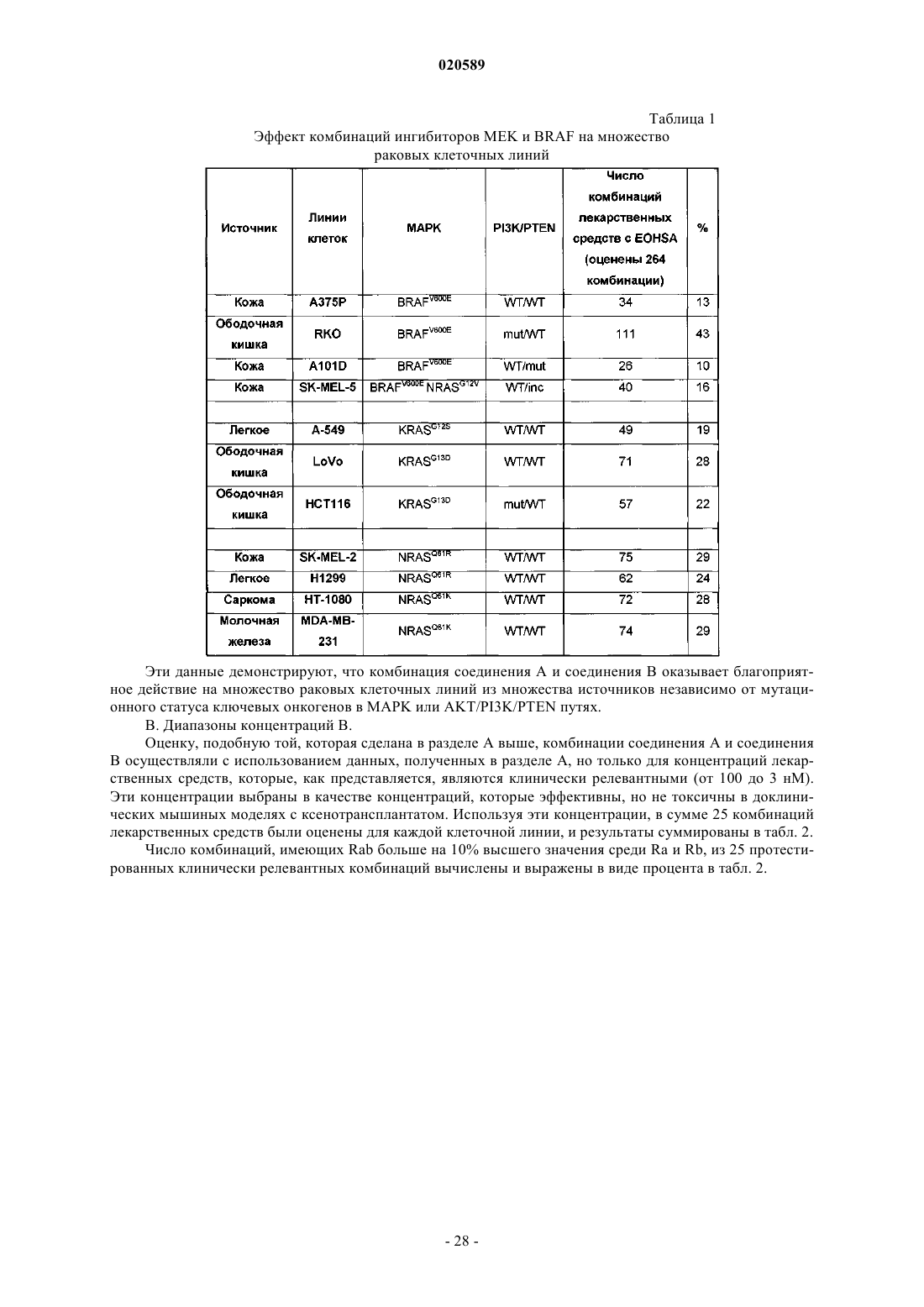
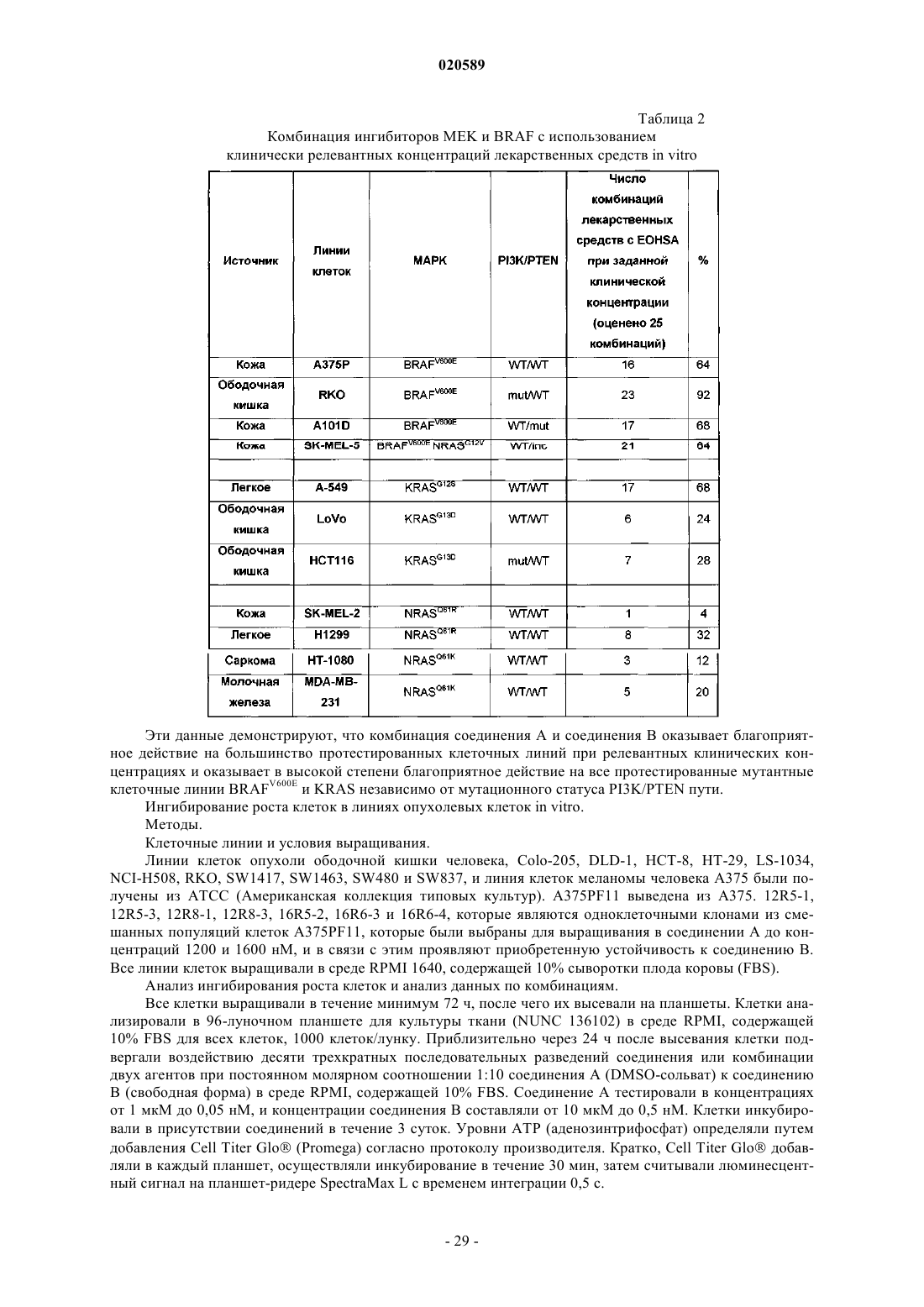
Формула / Реферат
1. Комбинация, содержащая:
(1) соединение формулы (I)

или его фармацевтически приемлемую соль или фармацевтически приемлемый сольват;
(2) соединение формулы (II)

или его фармацевтически приемлемую соль,
для применения в лечении расстройств, при которых ингибирование активности MEK и/или B-Raf оказывает благоприятное действие.
2. Комбинация по п.1, где соединение (1) находится в форме диметилсульфоксидного сольвата, а соединение (2) находится в форме метансульфонатной соли.
3. Комбинационный набор, содержащий комбинацию по любому из пп.1, 2 вместе с фармацевтически приемлемым(и) носителем или носителями, для применения в лечении расстройств, при которых ингибирование активности MEK и/или B-Raf оказывает благоприятное действие.
4. Применение комбинации по любому из пп.1, 2 в изготовлении лекарственного средства для лечения рака.
5. Комбинация по любому из пп.1, 2 для применения в лечении рака.
6. Фармацевтическая композиция, содержащая комбинацию по любому из пп.1, 2 вместе с фармацевтически приемлемым разбавителем или носителем, для применения в лечении расстройств, при которых ингибирование активности MEK и/или B-Raf оказывает благоприятное действие.
7. Способ лечения рака у человека, нуждающегося в таком лечении, включающий введение терапевтически эффективного количества:
(1) соединения формулы (I)

или его фармацевтически приемлемой соли или фармацевтически приемлемого сольвата;
(2) соединения формулы (II)

или его фармацевтически приемлемой соли.
8. Способ по п.7, где рак выбран из рака молочной железы, рака легкого, рака ободочной кишки, меланомы и саркомы.
9. Способ по п.7 или 8, где рак представляет собой меланому.
10. Способ по любому из пп.7-9, где соединение (1) находится в форме диметилсульфоксидного сольвата, а соединение (2) находится в форме метансульфонатной соли.
Текст
Новая комбинация, содержащая ингибитор MEK N-3-[3-циклопропил-5-(2-фтор-4 йодфениламино)-6,8-диметил-2,4,7-триоксо-3,4,6,7-тетрагидро-2 Н-пиридо[4,3-d]пиримидин-1 ил]фенилацетамид или его фармацевтически приемлемую соль или фармацевтически приемлемый сольват с ингибитором В-Raf, в частности N-3-[5-(2-амино-4-пиримидинил)-2(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил-2,6-дифторбензолсульфонамидом или его фармацевтически приемлемой солью, фармацевтические композиции, содержащие указанные соединения, и способы применения таких комбинаций и композиций в лечении состояний, при которых благобриятным является ингибирование MEK и/или B-Raf, например рака. Область изобретения Настоящее изобретение относится к способу лечения рака у млекопитающего и к комбинациям, полезным в таком лечении. B частности, способ относится к новой комбинации, содержащей ингибиторMEK (митоген-активируемая регулируемая внеклеточными сигналами киназа) N-3-[3-циклопропил-5(2-фтор-4-йод-фениламино)-6,8-диметил-2,4,7-триоксо-3,4,6,7-тетрагидро-2 Н-пиридо[4,3-d]пиримидин 1-ил]фенилацетамид или его фармацевтически приемлемую соль или сольват с ингибитором B-Raf, в частностиN-3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил-2,6 дифторбензолсульфонамидом или его фармацевтически приемлемой солью, к фармацевтическим композициям, содержащим указанные ингибиторы, и к способам применения таких комбинаций и композиций в лечении состояний, при которых полезным является ингибирование MEK и/или B-Raf, например рака. Уровень техники Эффективное лечение гиперпролиферативных расстройств, включая рак, является остающейся актуальной целью в области онкологии. Как правило, рак является результатом нарушения регуляции нормальных процессов, контролирующих деление клеток, дифференцировку клеток и апоптическую гибель клеток, и характеризуется пролиферацией злокачественных клеток, которые обладают потенциалом неограниченного роста, локального распространения и системного метастазирования. Нарушение регуляции нормальных процессов включает аномалии в путях сигнальной трансдукции и ответную реакцию на факторы в отличие от тех, которые имеют место в нормальных клетках. Важным крупным семейством ферментов является семейство ферментов протеинкиназ. B настоящее время известно о существовании примерно 500 разных протеинкиназ. Протеинкиназы катализируют фосфорилирование аминокислотной боковой цепи в различных белках путем переноса -фосфата комплекса ATP-Mg2+ на указанную аминокислотную боковую цепь. Эти ферменты контролируют большинство процессов передачи сигнала внутри клеток и, тем самым, регулируют функцию, рост, дифференцировку и деструкцию (апоптоз) клеток посредством обратимого фосфорилирования гидроксильных групп сериновых, треониновых и тирозиновых остатков в белках. Исследования показали, что протеинкиназы являются ключевыми регуляторами многих клеточных функций, включая сигнальную трансдукцию, регуляцию транскрипции, подвижность клеток и деление клеток. Было также показано, что несколько онкогенов кодируют протеинкиназы, и это свидетельствует о том, что киназы играют роль в онкогенезе. Эти процессы жестко регулируются, часто сложными перекрестными путями, где каждая киназа сама регулируется одной или более киназами. Следовательно, аберрантная или несоответствующая активность протеинкиназ может способствовать возникновению болезненных состояний, ассоциированных с такой аберрантной активностью киназ, включая доброкачественные и злокачественные пролиферативные расстройства, а также заболевания, являющиеся результатом несоответствующей активации иммунной и нервной систем. Ввиду их физиологической значимости, разнообразия и убиквитарности протеинкиназы стали одним из самых важных и всесторонне исследуемых семейств ферментов в биохимических и медицинских исследованиях. Семейство протеинкиназ подразделяется, как правило, на два основных подсемейства: протеинтирозинкиназы и протеинсерин/треонинкиназы в зависимости от аминокислотного остатка, который они фосфорилируют. Протеинсерин/треонинкиназы (PSTK) включают циклический AMP (аденозинмонофосфат)- и циклический GMP (гуанозинмонофосфат)-зависимые протеинкиназы, кальций- и фосфолипидзависимую протеинкиназу, кальций- и кальмодулинзависимые протеинкиназы, казеинкиназы, протеинкиназы цикла деления клеток и др. Эти киназы обычно являются цитоплазматическими или ассоциированными с фракциями частиц клеток, возможно посредством якорных белков. Аберрантная активность протеинсерин/треонинкиназ предполагается или вовлечена во многие различные патологии, такие как ревматоидный артрит, псориаз, септический шок, потеря кости, многие виды рака и другие пролиферативные заболевания. Соответственно, серин/треонинкиназы и пути сигнальной трансдукции, частью которых они являются, представляют собой важные мишени для разработки лекарственных средств. Тирозинкиназы фосфорилируют тирозиновые остатки. Тирозинкиназы играют равноценно важную роль в клеточной регуляции. Эти киназы включают несколько рецепторов для молекул, таких как факторы роста и гормоны, включая рецептор эпидермального фактора роста, инсулиновый рецептор, рецептор тромбоцитарного фактора роста и др. Исследования показали, что многие тирозинкиназы представляют собой трансмембранные белки с их рецепторными доменами, расположенными на внешней поверхности клетки и их киназными доменами на внутренней поверхности. Большая работа проводится также с целью идентификации модуляторов тирозинкиназ. Рецепторные тирозинкиназы (RTK) катализируют фосфорилирование некоторых тирозильных аминокислотных остатков в различных белках, включая их самих, которые регулируют рост, пролиферацию и дифференцировку клеток. Ниже по ходу нескольких RTK лежат несколько сигнальных путей, среди них Ras-Raf-MEK-ERK киназный путь. B настоящее время известно, что активация Ras GTP-азных белков в ответ на факторы роста, гормоны, цитокины и т.д. стимулирует фосфорилирование и активацию Raf киназ. Эти киназы затем фосфорилируют и активируют внутриклеточные протеинкиназы MEK1 и MEK2, которые, в свою очередь, фосфорилируют и активируют другие протеинкиназы, ERK1 и 2. Этот путь передачи сигнала,-1 020589 также известный как путь митоген-активированной протеинкиназы (MAPK) или цитоплазматический каскад, опосредует клеточные ответы на ростовые сигналы. Основная его функция заключается в том,чтобы связать активность рецепторов на клеточной мембране с модификацией цитоплазматических или ядерных мишеней, которые регулируют пролиферацию, дифференцировку и выживание клеток. Конститутивная активация этого пути достаточна для индуцирования клеточной трансформации. Дисрегуляционная активация МАР-киназного пути вследствие аберрантной активации рецепторных тирозинкиназ, мутаций Ras или мутаций Raf часто обнаруживается при раке у человека и представляет собой основной фактор, определяющий аномальный контроль роста. При злокачественностях у человека мутации Ras являются обычными и идентифицированы примерно в 30% случаев рака. Семейство RasGTP-азных белков (белки, которые превращают гуанозинтрифосфат в гуанозиндифосфат) передают сигналы от активированных рецепторов факторов роста к нижележащим внутриклеточным партнерам. Особое место среди мишеней, рекрутируемых активной мембраносвязанной Ras, занимает семейство Raf серин/треонинпротеинкиназ. Семейство Raf состоит из трех родственных киназ (А-, В- и C-Raf), которые действуют ниже по ходу как эффекторы Ras. Ras-опосредованная активация Raf, в свою очередь, запускает активацию MEK1 и MEK2 (MAP/ERK киназы 1 и 2), которые, в свою очередь, фосфорилируютERK1 и ERK2 (внеклеточные сигнал-регулируемые киназы 1 и 2) на тирозине-185 и треонине-183. Активированные ERK1 и ERK2 транслоцируются и аккумулируются в ядрах, где они могут фосфорилировать различные субстраты, включая факторы транскрипции, которые контролируют рост и выживание клеток. Учитывая важность пути Ras/Raf/MEK/ERK в развитии рака у человека, киназные компоненты сигнального каскада объединяются как потенциально важные мишени для модулирования прогрессирования заболевания при раке и других пролиферативных заболеваниях.MEK1 и MEK2 являются членами большего семейства киназ с двойственной специфичностью(MEK1-7), которые фосфорилируют треониновые и тирозиновые остатки различных МАР-киназ. MEK1 и MEK2 кодируются разными генами, но они имеют высокую гомологию (80%) как в С-концевых каталитических киназных доменах, так и в большей части N-концевой регуляторной области. Онкогенезные формы MEK1 и MEK2 обнаружены при раке у человека, но конститутивная активация MEK приводит,как было показано, к клеточной трансформации. B дополнение к Raf, MEK также может быть активирована другими онкогенами. До настоящего времени известными субстратами MEK1 и MEK2 являются только ERK1 и ERK2. Эта необычная субстратная специфичность в дополнение к уникальной способности фосфорилировать как тирозиновые, так и треониновые остатки позволяет считать MEK1 и MEK2 критическими в каскаде сигнальной трансдукции, который дает возможность интегрировать многие внеклеточные сигналы в MAPK-путь. Соответственно, было установлено, что ингибитор белка MAPK-киназного пути (например, MEK) должен иметь значение как антипролиферативный, проапоптический и антиинвазивный агент для применения в сдерживании распространения и/или лечении пролиферативного или инвазивного заболевания. Более того, известно также, что соединение, обладающее ингибиторной активностью в отношенииMEK, эффективно индуцирует ингибирование активности ERK1/2 и супрессию пролиферации клеток(The Journal of Biological Chemistry, vol. 276, No. 4, p. 2686-2692, 2001), и, как предполагается, такое соединение эффективно воздействует на заболевания, вызываемые нежелательной клеточной пролиферацией, такие как заболевания опухолевого генеза и/или рак. Были идентифицированы мутации в различных Ras GTP-азах и в B-Raf киназе, которые могут приводить к длительной и конститутивной активации MAPK-пути, в конечном счете приводящей к повышению деления и выживания клеток. Как следствие, эти мутации строго связаны с образованием, развитием и прогрессированием целого ряда различных видов рака у человека. Биологическая роль Raf киназ, и конкретно B-Raf, в сигнальной трансдукции описана в Davies, H., et al., Nature (2002), 9:1-6; Garnett,M.J.Marais, R., Cancer Cell (2004), 6:313-319; Zebisch, A.Troppmair, J., Cell. Mol. Life Sci. (2006),63:1314-1330; Midgley, R.S.Kerr, D.J., Crit. Rev. Onc/Hematol. (2002), 44:109-120; Smith, R.A., et al.,Curr. Top. Med. Chem. (2006), 6:1071-1089 и Downward, J., Nat. Rev. Cancer (2003), 3:11-22. Естественные мутации киназы B-Raf, которые активируют передачу сигнала MAPK-путем, обнаружены в большом проценте меланом человека (Davies (2002), см. выше) и раковых опухолей щитовидной железы (Cohen et al., J. Nat. Cancer Inst. (2003), 95(8):625-627 и Kimura et al., Cancer Res. (2003),63(7):1454-1457), а также менее часто, но все же в значительной доле встречаются при следующих видах рака: аденокарцинома Баррета (Garnett et al., Cancer Cell. (2004), 6, 313-319, и Sommerer et al., Oncogene(2004), 23(2):554-558),карциномы желчных протоков (Zebisch et al., Cell. Mol. Life Sci. (2006), 63:1314-1330), рак молочной железы (Davies (2002), см. выше),рак шейки матки (Moreno-Bueno et al., Clin. Cancer Res. (2006), 12(12):3865-3866),холангиокарцинома (Tannapfel et al., Gut. (2003), 52(5):706-712),опухоли центральной нервной системы, включая первичные опухоли ЦНС, такие как глиобластомы, астроцитомы и эпендимомы (Knobbe et al., Acta Neuropathol. (Berl.) (2004), 108(6):467-470; Davies(2002), см. выше, и Garnett et al., Cancer Cell. (2004), см. выше), и вторичные опухоли ЦНС (т.е. метастазы в центральную нервную систему опухолей, имеющих происхождение за пределами центральной нервной системы),колоректальный рак, включая карциному ободочной кишки толстого кишечника (Yuen et al., CancerRes. (2002), 62(22):6451-6455, Davies (2002), см. выше, и Zebisch et al., Cell Mol. Life Sci (2006),рак желудка (Lee et al., Oncogene (2003), 22(44):6942-6945),карцинома в области головы и шеи, включая плоскоклеточную карциному в области головы и шеи(Cohen et al., J. Nat. Cancer Inst. (2003), 95(8):625-627 и Weber et al., Oncogene (2003), 22(30):4757-4759),гематологические виды рака, включая лейкозы (Garnett et al., Cancer Cell. (2004), см. выше), в частности острый лимфобластный лейкоз (Garnett et al., Cancer Cell. (2004), см. выше и Gustafsson et al.,Leukemia (2005), 19(2):310-312),острый миелогенный лейкоз (AML (ангиомиолипомы (Lee et al., Leukemia (2004), 18(1):170-172 иChristiansen et al., Leukemia (2005), 19(12):2232-2240),миелодиспластические синдромы (Christiansen et al., Leukemia (2005), см. выше) и хронический миелогенный лейкоз (Mizuchi et al., Biochem Biophys Res Commun. (2005), 326(3):645-651),ходжкинская лимфома (Figl et al., Arch Dermatol. (2007), 143(4):495-499),неходжкинская лимфома (Lee et al., Br. J. Cancer. (2003), 89(10):1958-1960),мегакариобластный лейкоз (Eychene et al., Oncogene (1995), 10(6):1159-1165) и множественная миелома (Ng et al., Br. J. Haematol. (2003), 123(4):637-645),печеночно-клеточная карцинома (Garnett et al., Cancer Cell (2004), рак легкого (Brose et al., CancerRes. (2002), 62(23):6997-7000, Cohen et al., J. Nat. Cancer Inst. (2003), см. выше и Davies (2002), см. выше),включая мелкоклеточный рак легкого (Pardo et al., EMBO J. (2006), 25(13):3078-3088) и немелкоклеточный рак легкого (Davies (2002), см. выше),рак яичника (RussellMcCluggage J. Pathol. (2004), 203(2):617-619 и Davies (2002), см. выше),рак эндометрия (Garnett et al., Cancer Cell. (2004), см. выше, и Moreno-Bueno et al., Clin. Cancer Res.(Cell Mol Life Sci (2006 и эритролейкозом (Zebisch et al., Cell. Mol. Life Sci. (2006. С учетом роли, которую играют киназы семейства Raf в этих видах рака, и на основании экспериментальных исследований с использованием целого ряда преклинических и терапевтических агентов,включая агент, избирательно нацеленный на ингибирование активности киназы B-Raf (King A.J., et al.,(2006), Cancer Res. 66:11100-11105), общепризнано, что ингибиторы одной или более киназ семействаRaf полезны для лечения таких видов рака или другого состояния, ассоциированного с Raf киназой. Мутация B-Raf также вовлечена в другие состояния, включая кардиофациальный кожный синдром(Rodriguez-Viciana et al., Science (2006), 311(5765):1287-1290) и поликистозное заболевание почек (Nagaoet al., Kidney Int. (2003), 63(2):427-437). Несмотря на то что имеются недавние достижения в лечении рака соединениями, такими как ингибиторы MEK и B-Raf, все еще остается потребность в более эффективном и/или усиленном лечении индивидуумов, страдающих раковыми заболеваниями. Краткое изложение сущности изобретения Авторы настоящего изобретения идентифицировали комбинацию химиотерапевтических агентов,которая обеспечивает повышенную активность по сравнению с монотерапией. B частности, описана комбинация лекарственных средств, содержащая ингибитор MEK N-3-[3-циклопропил-5-(2-фтор-4-йодфениламино)-6,8-диметил-2,4,7-триоксо-3,4,6,7-тетрагидро-2 Н-пиридо[4,3-d]пиримидин-1 ил]фенилацетамид или его фармацевтически приемлемую соль или фармацевтически приемлемый сольват в комбинации с ингибитором B-Raf N-3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3 тиазол-4-ил]-2-фторфенил-2,6-дифторбензолсульфонамидом или его фармацевтически приемлемой солью. Ингибитор MEK по изобретению представляет собой соединение, имеющее структурную формулу (I) или его фармацевтически приемлемую соль или фармацевтически приемлемый сольват (в данном описании "соединение А"). Ингибитор B-Raf по изобретению представляет собой соединение, имеющее структурную формулу (II) или его фармацевтически приемлемую соль (в данном описании "соединение В"). В первом аспекте настоящего изобретения предложена комбинация, содержащая: или его фармацевтически приемлемую соль или фармацевтически приемлемый сольват; или его фармацевтически приемлемую соль. В другом аспекте настоящего изобретения предложена комбинация, содержащаяN-3-[3-циклопропил-5-(2-фтор-4-йод-фениламино)-6,8-диметил-2,4,7-триоксо-3,4,6,7-тетрагидро 2 Н-пиридо[4,3-d]пиримидин-1-ил]фенилацетамид диметилсульфоксид (сольват) и N-3-[5-(2-амино-4 пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил-2,6-дифторбензолсульфонамида метансульфонат. В другом аспекте настоящего изобретения предложена комбинация, содержащая: или его фармацевтически приемлемую соль или фармацевтически приемлемый сольват; или его фармацевтически приемлемую соль,для применения в терапии. В другом аспекте настоящего изобретения предложена комбинация, содержащая: или его фармацевтически приемлемую соль или фармацевтически приемлемый сольват; или его фармацевтически приемлемую соль,для применения в лечении рака. В другом аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая: или его фармацевтически приемлемую соль или фармацевтически приемлемый сольват; или его фармацевтически приемлемую соль вместе с фармацевтически приемлемым разбавителем или носителем. В еще одном аспекте предложено применение комбинации, содержащей: или его фармацевтически приемлемую соль или фармацевтически приемлемый сольват; или его фармацевтически приемлемую соль,в изготовлении лекарственного средства для лечения рака. В еще одном аспекте предложен способ лечения рака у млекопитающего, включающий введение указанному млекопитающему:(1) терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли или фармацевтически приемлемого сольвата; или его фармацевтически приемлемой соли. В еще одном аспекте предложен способ лечения рака у человека, нуждающегося в таком лечении,включающий введение терапевтически эффективного количества комбинации N-3-[3-циклопропил-5-(2 фтор-4-йод-фениламино)-6,8-диметил-2,4,7-триоксо-3,4,6,7-тетрагидро-2 Н-пиридо[4,3-d]пиримидин-1 ил]фенилацетамида или его фармацевтически приемлемой соли или фармацевтически приемлемого сольвата и N-3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил-2,6 дифторбензолсульфонамида или его фармацевтически приемлемой соли. В еще одном аспекте предложен способ лечения рака у человека, нуждающегося в таком лечении,включающий введение терапевтически эффективного количества комбинации N-3-[3-циклопропил-5-(2 фтор-4-йод-фениламино)-6,8-диметил-2,4,7-триоксо-3,4,6,7-тетрагидро-2 Н-пиридо[4,3-d]пиримидин-1 ил]фенилацетамида диметилсульфоксидного сольвата и N-3-[5-(2-амино-4-пиримидинил)-2-(1,1 диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил-2,6-дифторбензолсульфонамида метансульфоната. В еще одном аспекте данного изобретения предложен способ лечения рака у млекопитающего, нуждающегося в таком лечении, включающий введение терапевтически эффективного количества комбинации по изобретению, причем комбинацию вводят в пределах установленного периода времени и в течение некоторой продолжительности времени. Краткое описание графических материалов Фиг. 1 представляет собой график, демонстрирующий ингибирование роста опухоли в результате введения ингибитора MEK, представляющего собой соединение А, ингибитора B-Raf, представляющего собой соединение B, и их комбинации. Фиг. 2 представляет собой график, демонстрирующий ингибирование роста опухоли в результате введения ингибитора MEK, представляющего собой соединение А, ингибитора B-Raf, представляющего собой соединение B, и их комбинации. Подробное описание изобретения В данном описании ингибитор MEK N-3-[3-циклопропил-5-(2-фтор-4-йод-фениламино)-6,8 диметил-2,4,7-триоксо-3,4,6,7-тетрагидро-2 Н-пиридо[4,3-d]пиримидин-1-ил]фенилацетамид или его фармацевтически приемлемая соль или фармацевтически приемлемый сольват представляет собой соединение формулы (I) или фармацевтически приемлемую соль или фармацевтически приемлемый сольват. Для удобства данная группа из возможных соединения и солей или сольватов собирательно обозначена как соединение А, что означает, что ссылка на соединение A относится к указанному соединению или к любой его фармацевтически приемлемой соли или любому его фармацевтически приемлемому сольвату в альтернативе. В зависимости от соглашения по присвоению названий соединение формулы (I) может быть надлежащим образом упоминаться как N-3-[3-циклопропил-5-[(2-фтор-4-йодфенил)амино]-6,8-диметил-2,4,7 триоксо-3,4,6,7-тетрагидропиридо[4,3-d]пиримидин-1(2H)-ил]фенилацетамид. Используемый в данном описании ингибитор B-Raf N-3-[5-(2-амино-4-пиримидинил)-2-(1,1 диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил-2,6-дифторбензолсульфонамид или его фармацевтически приемлемая соль представляет собой соединение формулы (II) или его фармацевтически приемлемую соль. Для удобства данная группа из возможных соединения и солей собирательно обозначена как соединение B, что означает, что ссылка на соединение B относится к указанному соединению или к его фармацевтически приемлемой соли в альтернативе. Используемый в данном документе термин "комбинация по изобретению" относится к комбинации,содержащей соединение A и соединение В. Используемый в данном документе термин "неоплазия" относится к аномальному росту клеток или ткани и охватывает доброкачественные, т.е. нераковые новообразования, и злокачественные, т.е. раковые новообразования. Термин "неопластический" означает или относится к новообразованию. Используемый в данном документе термин "агент" означает вещество, которое оказывает желаемый эффект в ткани, системе, животном, млекопитающем, человеке или другом субъекте. Соответственно,термин "антинеопластический агент" означает вещество, оказывающее антинеопластический эффект в ткани, системе, животном, млекопитающем, человеке или другом субъекте. Следует также иметь в виду,что "агент" может представлять собой единственное соединение или комбинацию или композицию двух или более соединений. Под термином "лечение" и производными от него терминами в данном документе подразумевается терапевтическая терапия. При ссылке на конкретное состояние лечение означает:(1) облегчение состояния или одного или более биологических проявлений данного состояния;(2) препятствование (а) одной или более точкам в биологическом каскаде, приводящем к данному состоянию или ответственному за данное состояние, или (б) одному или более биологическим проявлениям данного состояния;(3) ослабление одного или более симптомов, следствий или побочных эффектов, ассоциированных с данным состоянием, или одного или более симптомов, следствий или побочных эффектов, ассоциированных с данным состоянием или его лечением; или(4) замедление прогрессирования состояния или одного или более биологических проявлений данного состояния. В данном документе "предупреждение" относится к профилактическому введению лекарственного средства, по существу, для минимизации вероятности или тяжести состояния или его биологического проявления либо для препятствования началу такого состояния или его биологического проявления. Специалист в данной области поймет, что "предупреждение" не является абсолютным термином. Профилактическая терапия целесообразна, например, тогда, когда считается, что субъект имеет высокий риск развития рака, например когда субъект имеет серьезную семейную историю рака или когда субъект подвергался воздействию канцерогена. Используемый в данном документе термин "эффективное количество" означает, что количество лекарственного средства или фармацевтического агента, которое будет вызывать биологическую или медицинскую ответную реакцию ткани, системы, животного или человека, которую стремится достичь,например, исследователь или клиницист. Кроме того, термин "терапевтически эффективное количество" означает любое количество, которое по сравнению с соответствующим субъектом, не получавшим такое количество, приводит к повышению качества лечения, излечению, предупреждению или ослаблению заболевания, расстройства, или к снижению побочного эффекта, или к снижению скорости прогрессирования заболевания или расстройства. Этот термин также охватывает количества, эффективные в отношении усиления нормальной физиологической функции. Соединения A и/или B могут содержать один или более хиральных атомов или же могут существовать в виде энантиомеров. Соответственно, соединения по данному изобретению включают смеси энан-7 020589 тиомеров, а также чистые энантиомеры или энантиомерно обогащенные смеси. Понятно также, что все таутомеры и смеси таутомеров охвачены соединением A и соединением В. Очевидно также, что соединения A и B могут присутствовать, по отдельности или оба, в виде сольватов. Используемый в данном документе термин "сольват" относится к комплексу различной стехиометрии, образованному растворенным веществом (в данном изобретении соединения формулы (I) или(II) или их соли) и растворителем. Такие растворители в целях данного изобретения не могут затрагивать биологическую активность растворенного вещества. Примеры подходящих растворителей включают, без ограничения, воду, метанол, диметилсульфоксид, этанол и уксусную кислоту. B одном воплощении используемым растворителем является фармацевтически приемлемый растворитель. Примеры подходящих фармацевтически приемлемых растворителей включают, без ограничения, воду, этанол и уксусную кислоту. B другом воплощении используемым растворителем является вода. Соединения A и B могут обладать способностью кристаллизоваться более чем в одной форме, т.е. могут обладать свойством, известным как полиморфизм, и подразумевается, что такие полиморфные формы ("полиморфы") охвачены соединениями A и В. Полиморфизм обычно может возникать в ответ на изменения температуры или давления или и того, и другого и может также возникать в результате вариаций в процессе кристаллизации. Полиморфы можно различать по различным характеристикам, известным в данной области, таким как картины дифракции рентгеновских лучей, растворимость и точка плавления. Соединение А, наряду с его фармацевтически приемлемыми солями, а также в виде их сольватов,раскрыто и заявлено как полезное в качестве ингибитора активности MEK, в частности в лечении рака, вWO 2005/121142. Соединение A представляет собой соединение примера 4-1, приведенного в указанной заявке. Соединение A может быть получено, как описано в WO 2005/121142. Предпочтительно соединение A находится в форме диметилсульфоксидного сольвата. Предпочтительно соединение A находится в форме натриевой соли. Предпочтительно соединение A находится в форме сольвата, выбранного из следующих: гидрат, сольват с уксусной кислой, этанолом, нитрометаном,хлорбензолом, 1-пентанолом, изопропиловым спиртом, этиленгликолем и 3-метил-1-бутанолом. Эти сольваты и солевые формы могут быть получены специалистом в данной области, руководствуясь описанием в WO 2005/121142. Соединение B, наряду с его фармацевтически приемлемыми солями, раскрыто и заявлено как полезное в качестве ингибитора активности B-Raf, в частности в лечении рака, в международной патентной заявке PCT/US09/42682. Получение соединения B описано в примерах 58 а-58 е указанной заявки. Заявка РСТ опубликована 12 ноября 2009 г. в виде публикации WO 2009/137391, и она включена в данное описание посредством ссылки. Более подробно, соединение B может быть получено описанными ниже способами. Способ 1. Соединение B (первая кристаллическая форма) - N-3-[5-(2-амино-4-пиримидинил)-2-(1,1 диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил-2,6-дифторбензолсульфонамид Суспензию N-3-[5-(2-хлор-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил 2,6-дифторбензолсульфонамида (196 мг, 0,364 ммоль) и аммиака в метаноле 7 М (8 мл, 56,0 ммоль) нагревали в герметично закрытой пробирке до 90C в течение 24 ч. Реакционную смесь разбавляли DCM(дихлорметаном), добавляли силикагель и концентрировали. Неочищенный продукт подвергали хроматографии на силикагеле, элюируя с градиентом от 100% DCM до 1:1 [DCM:(9:1 EtOAc:MeOH)]. Чистые фракции концентрировали с получением сырого продукта. Этот сырой продукт снова очищали обращенно-фазовой ВЭЖХ (высокоэффективная жидкостная хроматография) (градиент смеси ацетонитрил:вода с 0,1%TFA в обоих). Объединенные чистые фракции концентрировали, затем распределяли между DCM и насыщенным NaHCO3. DCM-слой отделяли и сушили над Na2SO4. Было получено указанное в заголовке соединение, N-3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил-2,6 дифторбензолсульфонамид (94 мг, выход 47%). 1 Н ЯМР (400 МГц, DMSO (диметилсульфоксид)-d6)м.д. (миллионные доли): 10.83 (s, 1 Н), 7.93 (d,J=5.2 Гц, 1 Н), 7.55-7.70 (m, 1 Н), 7.35-7.43 (m, 1 Н), 7.31 (t, J=6.3 Гц, 1 Н), 7.14-7.27 (m, 3 Н), 6.70 (s, 2 Н),5.79 (d, J=5.13 Гц, 1 Н), 1.35 (s, 9 Н). МС (ЭРИ) (масс-спектрометрия с электрораспылительной ионизацией): 519,9 [М+Н]+. Способ 2. Соединение B (альтернативная кристаллическая форма) - N-3-[5-(2-амино-4-пиримидинил)-2-(1,1 диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил-2,6-дифторбензолсульфонамид. 19,6 мг N-3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил-2,6 дифторбензолсульфонамида (может быть получен в соответствии с примером 58 а) объединяли с 500 мкл этилацетата в сосуде емкостью 2 мл при комнатной температуре. Суспензию подвергали воздействию температурного цикла между 0-40C в течение 48 ч. Полученную суспензию оставляли охлаждаться до комнатной температуры и твердое вещество собирали вакуумной фильтрацией. Твердое вещество анализировали методами спектроскопии комбинационного рассеяния, ДРЛП (дифракция рентгеновских лучей на порошке), ДСК/ТГА (дифференциальная сканирующая калориметрия/термогравиметрический анализ), которые показали, что кристаллическая форма отличается от кристаллической формы, полученной способом, описанным в примере 58 а, выше. Способ 3. Соединение B (альтернативная кристаллическая форма, большая партия) - N-3-[5-(2-амино-4 пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил-2,6-дифторбензолсульфонамид(250 мл, 5 об.). Содержимое перемешивали, охлаждали до 15C и добавляли пиридин (26,2 мл, 1,1 экв.). После добавления пиридина температуру содержимого реактора доводили до 15C и через капельную воронку начинали добавление 2,6-дифторбензолсульфонилхлорида (39,7 мл, 1,0 экв.). Температуру во время добавления поддерживали 25C. После окончания добавления содержимое реактора нагревали до 20-25C и выдерживали в течение ночи. Добавляли этилацетат (150 мл) и дихлорметан удаляли дистилляцией. Сразу после окончания дистилляции реакционную смесь разбавляли еще раз этилацетатом (5 об.) и концентрировали. Реакционную смесь разбавляли этилацетатом (10 об.) и водой (4 об.) и содержимое нагревали до 50-55C при перемешивании до тех пор, пока все твердое вещество не растворилось. Слои расслаивали и разделяли. Органический слой разбавляли водой (4 об.) и содержимое нагревали до 50-55 в течение 20-30 мин. Слои расслаивали и затем разделяли и этилацетатный слой упаривали при пониженном давлении до 3 объемов. Добавляли этилацетат (5 об.) и снова выпаривали при пониженном давлении до 3 объемов. Затем в реактор добавляли циклогексан (9 об.) и содержимое нагревали до температуры дефлегмации в течение 30 мин, затем охлаждали до 0C. Твердое вещество отфильтровывали и промывали циклогексаном (2100 мл). Твердое вещество сушили на воздухе в течение ночи с получением метил-3-[(2,6-дифторфенил)сульфонил]амино-2-фторбензоата (94,1 г, 91%). Стадия В. Метил-3-[(2,6-дифторфенил)сульфонил]амино-2-фторбензоат (490 г, 1 экв.), полученный в соответствии со стадией A выше, растворяли в THF (2,45 л, 5 об.) и перемешивали и охлаждали до 0-3C. 1 М раствор лития бис-(триметилсилил)амида в THF (5,25 л, 3,7 экв.) загружали в реакционную смесь, затем добавляли 2-хлор-4-метилпиримидин (238 г, 1,3 экв.) в THF (2,45 л, 5 об.). Реакционную смесь затем перемешивали в течение 1 ч. Реакционную смесь гасили добавлением 4,5 М HCl (3,92 л, 8 об.). Водный слой (нижний слой) удаляли и выбрасывали. Органический слой концентрировали при пониженном давлении до 2 л. IPAC (изопропилацетат) (2,45 л) добавляли в реакционную смесь, которую затем концентрировали до 2 л. Добавляли IPAC (0,5 л) и МТВЕ (метил-трет-бутиловый эфир) (2,45 л) и осуществляли перемешивание в течение ночи в атмосфере N2. Твердое вещество отфильтровывали. Твердое вещество и маточный фильтрат вместе добавляли обратно и перемешивали в течение нескольких часов. Твердое вещество отфильтровывали и промывали МТВЕ (5 об.). Твердое вещество помещали в вакуумный шкаф при 50C в течение ночи. Твердое вещество сушили в вакуумном шкафу при 30C в течение выходных дней с получением N-3-[(2-хлор-4-пиримидинил)ацетил]-2-фторфенил-2,6-дифторбензолсульфонамида В реакционный сосуд загружали N-3-[(2-хлор-4-пиримидинил)ацетил]-2-фторфенил-2,6 дифторбензолсульфонамид (30 г, 1 экв.), затем добавляли дихлорметан (300 мл). Реакционную суспензию охлаждали до 10C и добавляли N-бромсукцинимид ("NBS") (12,09 г, 1 экв.) приблизительно равными порциями при перемешивании в течение 10-15 мин между каждым добавлением. После последнего добавления NBS реакционную смесь нагревали до 20C и перемешивали в течение 45 мин. Затем в реакционный сосуд добавляли воду (5 об.), смесь перемешивали и затем слои разделяли. К дихлорметановому слою снова добавляли воду (5 об.), смесь перемешивали и слои разделяли. Дихлорметановые слои концентрировали до 120 мл. B реакционную смесь добавляли этилацетат (7 об.) и смесь концентрировали до 120 мл. Затем в реакционную смесь добавляли диметилацетамид (270 мл) и охлаждали смесь до 10C. 2,2-диметилпропантиоамид (1,3 г, 0,5 экв.) двумя равными порциями добавляли к содержимому реактора при перемешивании в течение 5 мин между добавлениями. Реакционную смесь нагревали до 20-25C. Через 45 мин содержимое сосуда нагревали до 75C и выдерживали в течение 1,75 ч. Реакционную смесь затем охлаждали до 5C и медленно загружали воду (270 мл), поддерживая температуру ниже 30C. Затем загружали этилацетат (4 об.), смесь перемешивали и слои разделяли. B водный слой снова загружали этилацетат (7 об.) и содержимое перемешивали и разделяли. Снова в водный слой загружали этилацетат (7 об.) и содержимое перемешивали и разделяли. Органические слои объединяли и промывали водой (4 об.) 4 раза и перемешивали в течение ночи при 20-25C. Органические слои затем концентрировали при нагревании и в вакууме до 120 мл. Содержимое сосуда затем нагревали до 50C и медленно добавляли гептаны (120 мл). После добавления гептанов содержимое сосуда нагревали до температуры дефлегмации, затем охлаждали до 0C и выдерживали в течение 2 ч. Твердое вещество отфильтровывали и промывали гептанами (22 об.). Твердый продукт затем сушили под вакуумом при 30C с получениемN-3-[5-(2-Амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил-2,6 дифторбензолсульфонамид. В реакторе для работы под давлением на 1 галлон (3,78 л) смесь N-3-[5-(2-хлор-4-пиримидинил)-2(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил-2,6-дифторбензолсульфонамида (120 г), полученный в соответствии со стадией С выше, и гидроксида аммония (28-30%, 2,4 л, 20 об.) нагревали в герметично закрытом реакторе для работы под давлением до 98-103C и перемешивали при этой температуре в течение 2 ч. Реакционную смесь охлаждали медленно до комнатной температуры (20C) и перемешивали в течение ночи. Твердое вещество отфильтровывали и промывали минимальным количеством маточной жидкости и сушили под вакуумом. Твердое вещество добавляли к смеси EtOAc (15 об.)/вода (2 об.), нагревали до полного растворения при 60-70C и водный слой удаляли и выбрасывали. B EtOAc-слой загружали воду (1 об.), нейтрализовали водн. HCl до pH 5,4-5,5 и добавляли воду (1 об.). Водный слой удаляли и выбрасывали при 60-70C. Органический слой промывали водой (1 об.) при 60-70C и водный слой удаляли и выбрасывали. Органический слой фильтровали при 60C и концентрировали до 3 объемов. B смесь загружали EtOAc (6 об.) и смесь нагревали и перемешивали при 72C в течение 10 мин, затем охлаждали 20C и перемешивали в течение ночи. EtOAc удаляли вакуумной дистилляцией, концентрируя реакционную смесь до 3 объемов. Реакционную смесь выдерживали при 65-70C в течение 30 мин. Загружали кристаллы продукта, имеющего ту же самую кристаллическую форму, что и продукт, полученный в примере 58b (и получаемый по методике примера 58b) выше, в гептановой суспензии. Гептан (9 об.) медленно добавляли при 65-70C. Суспензию перемешивали при 65-70C в течение 2-3 ч и затем медленно охлаждали до 0-5C. Продукт фильтровали, промывали смесью EtOAc/гептан К раствору N-3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил 2,6-дифторбензолсульфонамида (204 мг, 0,393 ммоль) в изопропаноле (2 мл) добавляли метансульфоновую кислоту (0,131 мл, 0,393 ммоль) и раствор перемешивали при комнатной температуре в течение 3 ч. Образовался белый осадок и суспензию фильтровали и промывали диэтиловым эфиром с получением указанного в заголовке продукта в виде белого кристаллического твердого вещества (210 мг, выход 83%). 1 Н ЯМР (400 МГц, DMSO-d6)м.д. 10.85 (s, 1 Н), 7.92-8.05 (m, 1 Н), 7.56-7.72 (m, 1 Н), 6.91-7.50 (m,7 Н), 5.83-5.98 (m, 1 Н), 2.18-2.32 (m, 3 Н), 1.36 (s, 9 Н). МС (ЭРИ): 520,0 [М+Н]+. Способ 5. Соединение B (альтернативное воплощение мезилатной соли) - N-3-[5-(2-амино-4-пиримидинил)2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил-2,6-дифторбензолсульфонамида метансульфонат.N-3-[5-(2-Амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2-фторфенил-2,6 дифторбензолсульфонамид (который может быть получен согласно примеру 58 а) (2,37 г, 4,56 ммоль) объединяли с предварительно профильтрованным ацетонитрилом (5,25 об., 12,4 мл). Добавляли предварительно профильтрованный раствор метансульфоновой кислоты (1,1 экв., 5,02 ммоль, 0,48 г) в H2O(0,75 экв., 1,78 мл) при 20C. Температуру полученной смеси повышали до 50-60C, одновременно перемешивая на низкой скорости. Сразу после того, как температура смеси достигла 50-60C, добавляли затравочную суспензиюN-3-[5-(2-амино-4-пиримидинил)-2-(1,1-диметилэтил)-1,3-тиазол-4-ил]-2 фторфенил-2,6-дифторбензолсульфонамида метансульфоната (1,0% мас./мас., суспендированный в 0,2 объемах предварительно профильтрованного ацетонитрила) и смесь перемешивали при скорости,достаточно быстрой для того, чтобы твердое вещество не оседало, при 50-60C в течение 2 ч. Смесь затем охлаждали до 0-5C со скоростью 0,25C/мин и выдерживали при 0-5C в течение 6 ч. Смесь фильтровали и влажный осадок на фильтре промывали дважды предварительно профильтрованным ацетонитрилом. Первую промывку выполняли 14,2 мл (6 об.) предварительно профильтрованного ацетонитрила, а вторую промывку выполняли 9,5 мл (4 об.) предварительно профильтрованного ацетонитрила. Влажное твердое вещество сушили при 50C под вакуумом с получением 2,39 г (выход 85,1%) продукта. В типичных случаях соли по настоящему изобретению представляют собой фармацевтически приемлемый соли. Соли, охваченные термином "фармацевтически приемлемые соли", относятся к нетоксичным солям соединения по данному изобретению. Соли соединений по настоящему изобретению могут включать соли присоединения кислоты по азоту на заместителе в соединении по настоящему изобретению. Репрезентативные соли включают следующие соли: ацетат, бензолсульфонат, бензоат, бикарбонат,бисульфат, битартрат, борат, бромид, кальций-эдетат, камзилат, карбонат, хлорид, клавуланат, цитрат,дигидрохлорид, эдетат, эдисилат, эстолат, эзидат, фумарат, глуцептат, глюконат, глутамат, гликоллиларсанилат, гексилрезорцинат, соль с гидрабамином, гидробромид, гидрохлорид, гидроксинафтоат, йодид,изетионат, лактат, лактобионат, лаурат, малат, малеат, манделат, мезилат, метилбромид, метилнитрат,метилсульфат, монокалималеат, мукат, напсилат, нитрат, соль с N-метилглюкамином, оксалат, памоат(эмбонат), пальмитат, пантотенат, фосфат/дифосфат, полигалактуронат, калиевую соль, салицилат, натриевую соль, стеарат, субацетат, сукцинат, таннат, тартрат, теоклат, тозилат, триэтиодид, триметиламмониевую соль и валерат. Другие соли, которые не являются фармацевтически приемлемыми, могут быть полезными в получении соединения по данному изобретению, и эти соли составляют еще один ас- 11020589 пект изобретения. Соли легко могут быть получены специалистом в данной области техники. Хотя существует возможность того, что для применения в терапии можно вводить соединения A иB в виде химических веществ, возможно присутствие активного ингредиента в виде фармацевтической композиции. Соответственно, согласно данному изобретению предложены также фармацевтические композиции, содержащие соединение A и/или соединение B и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов. Соединения A и B такие, как описано выше. Носитель(и), разбавитель(и) или эксципиент(ы) должен(жны) быть приемлемым(и) в смысле совместимости с другими ингредиентами композиции, обеспечивающими приготовление фармацевтической композиции, и не должен(жны) оказывать вредного воздействия на реципиента. B соответствии с другим аспектом данного изобретения предложен также способ приготовления фармацевтической композиции, включающий смешивание соединения A и/или соединения B с одним или более фармацевтически приемлемыми носителями, разбавителями или эксципиентами. Такие используемые составляющие фармацевтических композиций могут присутствовать в отдельных фармацевтических комбинациях или могут быть приготовлены вместе в виде одной фармацевтической композиции. Соответственно, согласно данному изобретению предложена комбинация фармацевтических композиций, одна из которых содержит соединение A и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов, а другая фармацевтическая композиция содержит соединение B и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов соединение A и соединение B такие, как описано выше, и могут быть использованы в любых композициях, описанных выше. Фармацевтические композиции могут быть представлены в стандартных лекарственных формах,содержащих предопределенное количество активного ингредиента на стандартную дозу. Как известно специалистам в данной области, количество активного ингредиента на дозу будет зависеть от состояния,которое лечат, от пути введения и от возраста, массы тела и состояния пациента. Предпочтительными композициями стандартных лекарственных форм являются композиции, содержащие суточную дозу, или субдозу, или ее соответствующую долю активного ингредиента. Кроме того, такие фармацевтические композиции могут быть приготовлены любым способом, известным в области фармации. Соединения A и B можно вводить любым подходящим путем. Подходящие пути включают пероральный, ректальный, назальный, местный (включая трансбуккальный и сублингвальный), вагинальный и парентеральный (включая подкожный, внутримышечный, внутривенный, интрадермальный, интратекальный и эпидуральный) пути. Следует иметь в виду, что предпочтительный путь может варьировать в зависимости, например, от состояния реципиента, комбинации и вида рака, подлежащего лечению. Следует также иметь в виду, что каждый вводимый агент может быть введен одним и тем же путем или разными путями и что соединения A и B могут быть объединены вместе в фармацевтической композиции. Фармацевтические композиции, адаптированные для перорального введения, могут быть представлены в виде дискретных единиц, таких как капсулы или таблетки; в виде порошков или гранул; в виде растворов или суспензий в водных или неводных жидкостях; в виде съедобных пенок или муссов; в виде жидких эмульсий типа масло-в-воде или жидких эмульсий типа вода-в-масле. Например, для перорального введения в форме таблетки или капсулы активный лекарственный компонент может быть объединен с пероральным нетоксичным фармацевтически приемлемым инертным носителем, таким как этанол, глицерин, вода и т.п. Порошки получают путем измельчения соединения до подходящего тонкого размера частиц и смешивания с измельченным подобным образом фармацевтическим носителем, таким как пищевой углевод, такой как, например, крахмал или маннит. Корригент, консервант, диспергирующий агент и окрашивающий агент также могут присутствовать. Капсулы изготавливают путем приготовления порошковой смеси, как описано выше, и заполнения ею сформованных желатиновых оболочек. Глиданты и смазывающие вещества, такие как коллоидный диоксид кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль, могут быть добавлены в порошковую смесь перед операцией заполнения. Разрыхлитель или солюбилизирующий агент, такой как агар-агар, карбонат кальция или карбонат натрия, также может быть добавлен для улучшения доступности лекарственного средства при проглатывании капсулы. Кроме того, при желании или необходимости, в смесь также могут быть введены подходящие связывающие вещества, смазывающие вещества, разрыхляющие агенты и красители, порошковая смесь может быть пропущена через таблеточную машину, в результате могут быть получены неидеально сформованные агломераты, раздробленные на гранулы. Гранулы могут быть смазаны и введены в смесь. Подходящие связывающие вещества включают крахмал, желатин, природные сахара, такие как глюкоза или бета-лактоза, зерновые подсластители, природные и синтетические камеди, такие как аравийская камедь,трагакант или альгинат натрия, карбоксиметилцеллюлозу, полиэтиленгликоль, воски и т.п. Смазывающие вещества, используемые в этих лекарственных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и т.п. Разрыхлители включают, без ограничения, крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и т.п. Таблетки формуют, например,путем приготовления порошковой смеси, гранулирования или агрегирования, добавления смазывающего вещества и разрыхлителя и прессования в таблетки. Порошковую смесь готовят смешиванием соедине- 12020589 ния, соответственно измельченного, с разбавителем или основой, как описано выше, и возможно со связывающим веществом, таким как карбоксиметилцеллюлоза, альгинат, желатин или поливинилпирролидон, замедлителем растворения, таким как парафин, ускорителем резорбции, таким как четвертичная соль, и/или агентом абсорбции, таким как бентонит, каолин или дикальция фосфат. Гранулирование порошковой смеси может быть осуществлено путем увлажнения связывающим веществом, таким как сироп, крахмальная паста, акадийский растительный клей или растворы целлюлозных или полимерных материалов, и продавливания через сито. B качестве альтернативы для предотвращения прилипания к таблеточным формовочным матрицам гранулы могут быть смазаны добавлением стеариновой кислоты,стеаратной соли, талька или минерального масла. Смазанную смесь затем прессуют в таблетки. Соединения по настоящему изобретению могут быть объединены также со свободно текучим инертным носителем и прямо спрессованы в таблетки, минуя стадии гранулирования или агломерации. Может быть осуществлено покрытие таблеток прозрачной или непрозрачной защитной оболочкой, состоящей из изолирующего слоя шеллака, покрытия из сахара или полимерного материала и полировочного покрытия из воска. Для распознавания разных дозировок в покрытия могут быть добавлены красители. Пероральные жидкости, такие как растворы, сиропы и эликсиры, могут быть приготовлены в стандартной лекарственной форме, в которой содержится предопределенное количество соединения. Сиропы могут быть приготовлены путем растворения соединения в подходящем образом корригированном водном растворе, а эликсиры готовят с использованием нетоксичного спиртового носителя. Суспензии могут быть приготовлены путем диспергирования соединения в нетоксичном носителе. Могут быть также добавлены солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и эфиры полиоксиэтиленсорбита, консерванты, корригирующая добавка, такая как масло перечной мяты, или натуральные подсластители или сахарин, или другие искусственные подсластители и т.п. Если это целесообразно, стандартные лекарственные препараты для перорального введения могут быть микроинкапсулированными. Может быть также приготовлена композиция для пролонгированного или длительного высвобождения, например, путем покрытия оболочкой или заключения дисперсного материала в полимеры, воск или подобный материал. Агенты для применения согласно настоящему изобретению можно вводить также в форме липосомных систем доставки, таких как небольшие однослойные везикулы, крупные однослойные везикулы и многослойные везикулы. Липосомы могут быть образованы из различных фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины. Агенты для применения согласно настоящему изобретению можно доставлять также с использованием моноклональных антител в качестве индивидуальных носителей, с которыми сопряжены молекулы соединения. Соединения могут быть также связаны с растворимыми полимерами в качестве носителей направленной доставки лекарственных средств. Такие полимеры могут включать поливинилпирролидон,пирановый сополимер, полигидроксипропилметакриламид-фенол, полигидроксиэтиласпартамид-фенол или полиэтиленоксидеполилизин, замещенный пальмитоильными остатками. Кроме того, соединения могут быть сопряжены с полимерами класса биоразлагаемых полимеров, используемыми для достижения контролируемого высвобождения лекарственного средства, например с полимолочной кислотой,полиэпсилон-капролактоном, полигидроксимасляной кислотой, полиортоэфирами, полиацеталями, полидигидропиранами, полицианоакрилатами и поперечно сшитыми или амфипатическими блоксополимерами гидрогелей. Фармацевтические композиции, адаптированные для трансдермального введения, могут быть представлены в виде отдельных пластырей, которые должны оставаться в тесном контакте с эпидермисом реципиента в течение длительного периода времени. Например, активный ингредиент может доставляться из пластыря посредством ионтофореза, как в общем описано в Pharmaceutical Research, 3(6), 318(1986). Фармацевтические композиции, адаптированные для местного введения, могут быть приготовлены в виде мазей, кремов, суспензий, лосьонов, порошков, растворов, пластырей, гелей, спреев, аэрозолей или масел. Для лечения глаза или других внешних тканей, например ротовой полости и кожи, композиции предпочтительно наносят в виде мази или крема для местного применения. При приготовлении мази активный ингредиент может быть использован в парафиновой или смешивающейся с водой мазевой основе. Альтернативно, активный ингредиент может быть включен в состав крема с основой масло-в-воде или вода-в-масле. Фармацевтические композиции, адаптированные для местного введения в глаз, включают глазные капли, в которых активный ингредиент растворен или суспендирован в подходящем носителе, в частности в водном растворителе. Фармацевтические композиции, адаптированные для местного введения в ротовую полость, включают лепешки, пастилки и полоскания. Фармацевтические композиции, адаптированные для ректального введения, могут быть представлены в виде суппозиториев или клизм. Фармацевтические композиции, адаптированные для назального введения, где носитель представляет собой твердое вещество, включают крупнозернистый порошок, имеющий размер частиц, например,20-500 мкм. Порошок вводят таким путем, при котором его вдыхают через нос, т.е. путем быстрой ингаляции через носовые проходы из контейнера с порошком, который держат близко к носу. Подходящие композиции, в которых носитель представляет собой жидкость для введения в виде назального спрея или в виде назальных капель, включают водные или масляные растворы активного ингредиента. Фармацевтические композиции, адаптированные для введения путем ингаляции, включают тонкодисперсные пыли или туманы, которые могут быть генерированы различными типами дозирующих аэрозольных баллончиков, небулайзеров или инсуффляторов. Фармацевтические композиции, адаптированные для вагинального введения, могут быть представлены в виде пессариев, тампонов, кремов, гелей, паст, пен или спреев. Фармацевтические композиции, адаптированные для парентерального введения, включают водные и неводные стерильные инъецируемые растворы, которые могут содержать антиоксиданты, буферные агенты, бактериостатические агенты и растворенные вещества, которые делают препарат изотоническим с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут содержать суспендирующие агенты и загустители. Композиции могут быть представлены в однодозовых или многодозовых контейнерах, например в запаянных ампулах и флаконах, и могут храниться в высушенном сублимацией (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед использованием. Растворы и суспензии для немедленной инъекции могут быть приготовлены из стерильных порошков, гранул и таблеток. Следует иметь в виду, что в дополнение к ингредиентам, в частности к ингредиентам, упомянутым выше, композиции могут содержать другие агенты, традиционные в области, имеющей отношение к рассматриваемому типу препарата. Например, препараты, подходящие для перорального введения, могут содержать корригенты. Если не дано иного определения, во всех описанных здесь протоколах введения доз режим введения соединений необязательно должен начинаться вместе с началом лечения и заканчиваться вместе с окончанием лечения, требуется только, чтобы число следующих друг за другом суток, в которые вводят оба соединения, и возможное число следующих друг за другом суток, в которые вводят только одно из соединений-компонентов, или указанный протокол введения доз, включая количество введенного соединения, имели место в некоторой точке в ходе лечения. Соединения A и B могут быть использованы в комбинации в соответствии с изобретением путем введения одновременно в единой фармацевтической композиции, содержащей оба соединения. Альтернативно, комбинацию можно вводить по отдельности в отдельных фармацевтических композициях, каждая из которых содержит одно из соединений A и B, последовательно, где, например, соединение A или соединение B вводят первым, а другое соединение вводят вторым. Такое последовательное введение может быть близким по времени (например, одновременно) или удаленным по времени. Кроме того, не имеет значения, вводят ли соединения в одной и той же лекарственной форме. Например, одно соединение можно вводить местно, а другое соединение можно вводить перорально. Предпочтительно оба соединения вводят перорально. Таким образом, в одном из воплощений одну или более доз соединения A вводят одновременно или по отдельности с одной или более дозами соединения В. Если не дано иного определения, во всех описанных здесь протоколах введения доз режим введения соединений необязательно должен начинаться вместе с началом лечения и заканчиваться вместе с окончанием лечения, требуется только, чтобы число следующих друг за другом суток, в которые вводят оба соединения, и возможное число следующих друг за другом суток, в которые вводят только одно из соединений-компонентов, или указанный протокол введения доз, включая количество введенного соединения, имели место в некоторой точке в ходе лечения. В одном из воплощений многократные дозы соединения A вводят одновременно или по отдельности с многократными дозами соединения В. В одном из воплощений многократные дозы соединения A вводят одновременно или по отдельности с одной дозой соединения В. В одном из воплощений одну дозу соединения A вводят одновременно или по отдельности с многократными дозами соединения В. В одном из воплощений одну дозу соединения A вводят одновременно или по отдельности с одной дозой соединения В. Во всех вышеуказанных воплощениях соединение A можно вводить первым, или соединение B можно вводить первым. Комбинации могут быть представлены в виде комбинационного набора. Под использованным в данном документе термином "комбинационный набор" или "набор компонентов" подразумевается фармацевтическая(ие) композиция или композиции, которую(ые) используют для введения соединения A и соединения B по изобретению. Когда оба соединения вводят одновременно, тогда комбинационный на- 14020589 бор может содержать соединение A и соединение В в единой фармацевтической композиции, такой как таблетка, или в отдельных фармацевтических композициях. Когда соединения A и B вводят не одновременно, тогда комбинационный набор будет содержать соединение A и соединение B в отдельных фармацевтических композициях либо в одной упаковке, либо соединение A и соединение B в отдельных фармацевтических композициях в отдельных упаковках. В одном аспекте предложен набор компонентов, содержащий следующие компоненты: соединение A совместно с фармацевтически приемлемыми эксципиентами, разбавителями или носителями и соединение B совместно с фармацевтически приемлемыми эксципиентами, разбавителями или носителями. В одном из воплощений данного изобретения предложен набор компонентов, содержащий следующие компоненты: соединение A совместно с фармацевтически приемлемыми эксципиентами, разбавителями или носителями и соединение B совместно с фармацевтически приемлемыми эксципиентами, разбавителями или носителями,где компоненты находятся в форме, которая пригодна для последовательного, раздельного и/или одновременного введения. В одном из воплощений набор компонентов включающий в себя первый контейнер, содержащий соединение A совместно с фармацевтически приемлемым эксципиентом, разбавителем или носителем; и второй контейнер, содержащий соединение B совместно с фармацевтически приемлемым эксципиентом, разбавителем или носителем, и контейнерное средство для вмещения указанных первого и второго контейнеров. Комбинационный набор может быть также снабжен инструкцией, такой как инструкции по дозировкам и введению. Такие инструкции по дозировкам и введению могут быть инструкциями, предназначенными для врача, например этикетка с указанием лекарственного продукта, или они могут быть инструкциями, предоставляемые врачом, такие как инструкции для пациента. Используемый в данном документе термин "нагрузочная доза" следует понимать как означающий однократную дозу или кратковременный режим введения соединения A или соединения B, имеющего дозировку выше, чем поддерживающая доза, вводимая субъекту, например, для быстрого увеличения уровня концентрации лекарственного средства в крови. Предпочтительно кратковременный режим для применения здесь будет составлять от 1 до 14 суток; предпочтительно от 1 до 7 суток; предпочтительно от 1 до 3 суток; предпочтительно в течение 3 суток; предпочтительно в течение 2 суток; предпочтительно в течение 1 суток. B некоторых воплощениях "нагрузочная доза" может увеличивать концентрацию лекарственного средства в крови до терапевтически эффективного уровня. B некоторых воплощениях"нагрузочная доза" может увеличивать концентрацию лекарственного средства в крови до терапевтически эффективного уровня в сочетании с поддерживающей дозой лекарственного средства. "Нагрузочную дозу" можно вводить один раз в сутки или более чем один раз в сутки (например, до 4 раз в сутки). Предпочтительно вводить "нагрузочную дозу" один раз в сутки. Предпочтительно нагрузочная доза будет в 2-100 раз выше поддерживающей дозы; предпочтительно в 2-10 раз; предпочтительно в 2-5 раз; предпочтительно в 2 раза; предпочтительно в 3 раза; предпочтительно в 4 раза; предпочтительно в 5 раз. Предпочтительно нагрузочную дозу будут вводить в течение 1-7 суток; предпочтительно 1-5 суток; предпочтительно 1-3 суток; предпочтительно в течение 1 суток; предпочтительно в течение 2 суток; предпочтительно в течение 3 суток с последующим протоколом введения поддерживающей дозы. Используемый в данном документе термин "поддерживающая доза" означает дозу, которую последовательно вводят (например, по меньшей мере двукратно) и которая предназначена либо для медленного подъема уровней концентрации соединения в крови до терапевтически эффективного уровня, либо для поддержания такого терапевтически эффективного уровня. Поддерживающую дозу обычно вводят один раз в сутки, и суточная доза поддерживающей дозы ниже, чем общая суточная доза нагрузочной дозы. Предпочтительно комбинации по данному изобретению вводят в пределах "установленного периода времени". Под используемым в данном документе термином "установленный период времени" и производными от него терминами подразумевается интервал времени между введением одного из соединения A и соединения B и другого из соединения A и соединения В. Если не дано иного определения, то установленный период времени может включать одновременное введение. Когда оба соединения по изобретению вводят один раз в сутки, тогда установленный период времени относится к введению соединения A и соединения B в течение одних суток. Когда одно или оба соединения по изобретению вводят чаще чем один раз в сутки, тогда установленный период времени рассчитывают по первому введению каждого соединения в конкретные сутки. Все введения соединения по изобретению, которые следуют за первым введением в течение конкретных суток, не учитываются при расчете установленного периода времени. Соответственно, если соединения вводят в "установленный период времени" и вводят не одновременно, то их оба вводят одно за другим не позднее чем через 24 ч, в этом случае установленный период времени будет составлять примерно 24 ч; соответственно их оба будут вводить одно за другим не позднее чем через 12 ч, в этом случае установленный период времени будет составлять примерно 12 ч; соответственно их оба будут вводить одно за другим не позднее чем через 11 ч, в этом случае установленный период времени будет составлять примерно 11 ч; соответственно их оба будут вводить одно за другим не позднее чем через 10 ч, в этом случае установленный период времени будет составлять примерно 10 ч; соответственно их оба будут вводить одно за другим не позднее чем через 9 ч, в этом случае установленный период времени будет составлять примерно 9 ч; соответственно их оба будут вводить одно за другим не позднее чем через 8 ч, в этом случае установленный период времени будет составлять примерно 8 ч; соответственно их оба будут вводить одно за другим не позднее чем через 7 ч, в этом случае установленный период времени будет составлять примерно 7 ч; соответственно их оба будут вводить одно за другим не позднее чем через 6 ч, в этом случае установленный период времени будет составлять примерно 6 ч; соответственно их оба будут вводить одно за другим не позднее чем через 5 ч, в этом случае установленный период времени будет составлять примерно 5 ч; соответственно их оба будут вводить одно за другим не позднее чем через 4 ч, в этом случае установленный период времени будет составлять примерно 4 ч; соответственно их оба будут вводить одно за другим не позднее чем через 3 ч, в этом случае установленный период времени будет составлять примерно 3 ч; соответственно их оба будут вводить одно за другим не позднее чем через 2 ч, в этом случае установленный период времени будет составлять примерно 2 ч; соответственно их оба будут вводить одно за другим не позднее чем через 1 ч, в этом случае установленный период времени будет составлять примерно 1 ч. B данном документе введение соединения A и соединения B менее чем примерно через 45 мин считается одновременным введением. Соответственно, когда комбинацию по изобретению вводят в пределах "установленного периода времени", соединения вводят совместно в течение некоторой "продолжительности времени". Под используемым в данном документе термином "продолжительность времени" и производными от него терминами подразумевается, что оба соединения по изобретению вводят в течение указанного количества следующих друг за другом суток. Относительно введения в пределах "установленного периода времени". Соответственно, оба соединения будут вводить в пределах установленного периода времени в течение по меньшей мере одних суток, в этом случае продолжительность времени будет составлять по меньшей мере одни сутки; соответственно во время курса лечения оба соединения будут вводить в пределах установленного периода времени в течение по меньшей мере 3 следующих друг за другом суток, в этом случае продолжительность времени будет составлять по меньшей мере 3 суток; соответственно во время курса лечения оба соединения будут вводить в пределах установленного периода времени в течение по меньшей мере 5 следующих друг за другом суток, в этом случае продолжительность времени будет составлять по меньшей мере 5 суток; соответственно во время курса лечения оба соединения будут вводить в пределах установленного периода времени в течение по меньшей мере 7 следующих друг за другом суток, в этом случае продолжительность времени будет составлять по меньшей мере 7 суток; соответственно во время курса лечения оба соединения будут вводить в пределах установленного периода времени в течение по меньшей мере 14 следующих друг за другом суток, в этом случае продолжительность времени будет составлять по меньшей мере 14 суток; соответственно во время курса лечения оба соединения будут вводить в пределах установленного периода времени в течение по меньшей мере 30 следующих друг за другом суток, в этом случае продолжительность времени будет составлять по меньшей мере 30 суток. Дополнительно относительно введения в пределах "установленного периода времени". Соответственно, во время курса лечения соединение A и соединение B будут вводить в пределах установленного периода времени в течение 1-4 суток за период 7 суток и на протяжении остальных суток этого 7-суточного периода будут вводить только соединение А. Соответственно, этот 7-суточный протокол повторяют 2 циклами или в течение 14 суток; соответственно 4 циклами или в течение 28 суток; соответственно при непрерывном введении. Соответственно, во время курса лечения соединение A и соединение B будут вводить в пределах установленного периода времени в течение 1-4 суток за период 7 суток и на протяжении остальных суток 7-суточного периода будут вводить только соединение В. Соответственно, этот 7-суточный протокол повторяют 2 циклами или в течение 14 суток; соответственно 4 циклами или в течение 28 суток; соответственно для непрерывного введения. Подходящим является введение соединения B в течение следующих друг за другом суток на протяжении периода времени 7 суток. Подходящим является введение соединения B по схеме через день на протяжении каждого 7-суточного периода. Соответственно, во время курса лечения соединение A и соединение B будут вводить в пределах установленного периода времени в течение 3 суток за период 7 суток и на протяжении остальных суток этого 7-дневного периода будут вводить только соединение В. Соответственно, этот 7-суточный протокол повторяют 2 циклами или в течение 14 суток; соответственно 4 циклами или в течение 28 суток; соответственно для непрерывного введения. Соответственно, соединение A будут вводить 3 следующих друг за другом суток на протяжении периода 7 суток. Соответственно, во время курса лечения соединение A и соединение B будут вводить в пределах установленного периода времени в течение 2 суток за период 7 суток и на протяжении остальных суток этого 7-дневного периода будут вводить только соединение В. Соответственно, этот 7-суточный протокол повторяют 2 циклами или в течение 14 суток; соответственно 4 циклами или в течение 28 суток; соответственно для непрерывного введения. Соответственно, соединение A будут вводить 2 следующих друг за другом суток на протяжении периода 7 суток. Соответственно, во время курса лечения соединение A и соединение B будут вводить в пределах установленного периода времени в течение 1 суток за период 7 суток, и на протяжении остальных суток этого 7-суточного периода будут вводить только соединение В. Соответственно, этот 7-суточный протокол повторяют 2 циклами или в течение 14 суток; соответственно4 циклами или в течение 28 суток; соответственно для непрерывного введения. Соответственно, если соединения не вводят во время "установленного периода", то их вводят последовательно. Под термином "последовательное введение" и производными от него терминами, используемыми в данном документе, подразумевается, что одно из соединения A и соединения B вводят на протяжении двух или более следующих друг за другом суток, а другое из соединения A и соединения B последовательно вводят на протяжении двух или более следующих друг за другом суток. Предусмотрены также перерывы во введении между последовательным введением одного из соединения A и соединения B и другого из соединения A и соединения В. Перерыв во введении представляет собой период времени в сутках после последовательного введения одного из соединения A и соединения B и до введения другого из соединения A и соединения B, когда не вводят ни соединение А, ни соединение В. Подходящим перерывом во введении будет период времени в сутках, выбранный из следующих: 1, 2, 3, 4, 5,6, 7, 8, 9, 10, 11, 12, 13 и 14 суток. Относительно последовательного введения. Соответственно, одно из соединения A и соединения B вводят на протяжении 1-30 следующих друг за другом суток, затем следует возможный перерыв во введении, затем следует введение другого из соединения A и соединения B на протяжении 1-30 следующих друг за другом суток. Соответственно, одно из соединения A и соединения B вводят на протяжении 2-21 следующих друг за другом суток, затем следует возможный перерыв во введении, затем следует введение другого из соединения A и соединения B на протяжении 2-21 следующих друг за другом суток. Соответственно, одно из соединения A и соединения B вводят на протяжении 2-14 следующих друг за другом суток, затем следует перерыв во введении 1-14 суток, затем следует введение другого из соединения A и соединения B на протяжении 2-14 следующих друг за другом суток. Соответственно, одно из соединения A и соединения B вводят на протяжении 3-7 следующих друг за другом суток, затем следует перерыв во введении 3-10 суток, затем следует введение другого из соединения A и соединения B на протяжении 3-7 следующих друг за другом суток. Соответственно, соединение B будут вводить первым в последовательности, затем следует возможный перерыв во введении, затем следует введение соединения А. Соответственно, соединение B вводят на протяжении 1-21 следующих друг за другом суток, затем следует возможный перерыв во введении,затем следует введение соединения A на протяжении 1-21 следующих друг за другом суток. Соответственно, соединение B вводят на протяжении 3-21 следующих друг за другом суток, затем следует перерыв во введении 1-14 суток, затем следует введение соединения A на протяжении 3-21 следующих друг за другом суток. Соответственно, соединение B вводят на протяжении 3-21 следующих друг за другом суток, затем следует перерыв во введении 3-14 суток, затем следует введение соединения A на протяжении 3-21 следующих друг за другом суток. Соответственно, соединение B вводят на протяжении 21 следующих друг за другом суток, затем следует возможный перерыв во введении, затем следует введение соединения A на протяжении 14 следующих друг за другом суток. Соответственно, соединение B вводят на протяжении 14 следующих друг за другом суток, затем следует перерыв во введении 1-14 суток, затем следует введение соединения A на протяжении 14 следующих друг за другом суток. Соответственно,соединение B вводят на протяжении 7 следующих друг за другом суток, затем следует перерыв во введении 3-10 суток, затем следует введение соединения A на протяжении 7 следующих друг за другом суток. Соответственно, соединение B вводят на протяжении 3 следующих друг за другом суток, затем следует перерыв во введении 3-14 суток, затем следует введение соединения A на протяжении 7 следующих друг за другом суток. Соответственно, соединение B вводят на протяжении 3 следующих друг за другом суток, затем следует перерыв во введении 3-10 суток, затем следует введение соединения A на протяжении 3 следующих друг за другом суток. Соответственно, соединение A будут вводить первым в последовательности, затем следует возможный перерыв во введении, затем следует введение соединения В. Соответственно, соединение A вводят на протяжении 1-21 следующих друг за другом суток, затем следует возможный перерыв во введении,затем следует введение соединения B на протяжении 1-21 следующих друг за другом суток. Соответственно, соединение A вводят на протяжении 3-21 следующих друг за другом суток, затем следует перерыв во введении 1-14 суток, затем следует введение соединения B на протяжении 3-21 следующих друг за другом суток. Соответственно, соединение A вводят на протяжении 3-21 следующих друг за другом су- 17020589 ток, затем следует перерыв во введении 3-14 суток, затем следует введение соединения B на протяжении 3-21 следующих друг за другом суток. Соответственно, соединение A вводят в течение 21 следующих друг за другом суток, затем следует возможный перерыв во введении, затем следует введение соединения B на протяжении 14 следующих друг за другом суток. Соответственно, соединение A вводят на протяжении 14 следующих друг за другом суток, затем следует перерыв во введении 1-14 суток, затем следует введение соединения B на протяжении 14 следующих друг за другом суток. Соответственно, соединение A вводят на протяжении 7 следующих друг за другом суток, затем следует перерыв во введении 310 суток, затем следует введение соединения B на протяжении 7 следующих друг за другом суток. Соответственно, соединение A вводят на протяжении 3 следующих друг за другом суток, затем следует перерыв во введении 3-14 суток, затем следует введение соединения B на протяжении 7 следующих друг за другом суток. Соответственно, соединение A вводят на протяжении 3 следующих друг за другом суток,затем следует перерыв во введении 3-10 суток, затем следует введение соединения B на протяжении 3 следующих друг за другом суток. Следует иметь в виду, что после введения в пределах "установленного периода времени" и введения на протяжении "следующих друг за другом суток" может следовать повторное введение доз или может следовать альтернативный протокол введения доз, и перерыв во введении может предшествовать повторному введению дозы или альтернативному протоколу введения доз. Подходящее количество соединения A (в пересчете на несолевое/несольватированное количество),вводимое как часть комбинации по настоящему изобретению, будет представлять собой количество, выбранное из примерно 0,125-10 мг; подходящее количество будет выбрано из примерно 0,25-9 мг; подходящее количество будет выбрано из примерно 0,25-8 мг; подходящее количество будет выбрано из примерно 0,5-8 мг; подходящее количество будет выбрано из примерно 0,5-7 мг; подходящее количество будет выбрано из примерно 1-7 мг; подходящее количество будет составлять примерно 5 мг. Подходящее количество соединения А, вводимое как часть комбинации по настоящему изобретению, будет представлять собой количество, выбранное из примерно 0,125-10 мг. Например, количество соединения А,вводимое как часть комбинации по настоящему изобретению, может составлять 0,125, 0,25, 0,5, 0,75, 1,1,5, 2, 2,5, 3, 3,5, 4, 4,5, 5, 5,5, 6, 6,5, 7, 7,5, 8, 8,5, 9, 9,5, 10 мг. Подходящее количество соединения B (в пересчете на несолевое/несольватированное количество),вводимое как часть комбинации по настоящему изобретению, будет представлять собой количество, выбранное из примерно 10-600 мг. Подходящее количество будет выбрано из примерно 30-300 мг; подходящее количество будет выбрано из примерно 30-280 мг; подходящее количество будет выбрано из примерно 40-260 мг; подходящее количество будет выбрано из примерно 60-240 мг; подходящее количество будет выбрано из примерно 80-220 мг; подходящее количество будет выбрано из примерно 90-210 мг; подходящее количество будет выбрано из примерно 100-200 мг, подходящее количество будет выбрано из примерно 110-190 мг, подходящее количество будет выбрано из примерно 120-180 мг, подходящее количество будет выбрано из примерно 130-170 мг, подходящее количество будет выбрано из примерно 140-160 мг, подходящее количество будет составлять 150 мг. Соответственно, количество соединения B,вводимое как часть комбинации по настоящему изобретению, будет представлять собой количество, выбранное из примерно 10-300 мг. Например, количество соединения B, вводимое как часть комбинации по настоящему изобретению, будет составлять количество, выбранное из 10, 20, 30, 40, 50, 60, 70, 80, 85, 90,95, 100, 105, 110, 115, 120, 125, 130, 135, 140, 145, 150, 155, 160, 165, 170, 175, 180, 185, 190, 195, 200, 205,210, 215, 220, 225, 230, 235, 240, 245, 250, 255, 260, 265, 270, 275, 280, 285, 290, 295 и 300 мг. Выбранное подходящее количество соединения B вводят от 1 до 4 раз в сутки. Выбранное подходящее количество соединения B вводят два раза в сутки. Соответственно, соединение B вводят в количестве 150 мг два раза в сутки. Выбранное подходящее количество соединения B вводят один раз в сутки. В данном описании все количества, конкретно указанные для соединения A и соединения B, указаны как количество свободного или несолевого соединения. Способ лечения Комбинации по изобретению могут быть полезны при расстройствах, при которых ингибированиеMEK и/или B-Raf оказывает благоприятное действие. Таким образом, согласно настоящему изобретению предложена также комбинация по изобретению для применения в терапии, в частности в лечении расстройств, при которых ингибирование активностиMEK и/или B-Raf оказывает благоприятное действие, в частности рака. В еще одном аспекте изобретения предложен способ лечения расстройства, при котором ингибирование MEK и/или B-Raf оказывает благоприятное действие, включающий введение комбинации по изобретению. В еще одном аспекте изобретения предложено применение комбинации по изобретению в изготовлении лекарственного средства для лечения расстройства, при котором ингибирование MEK и/или B-Raf оказывает благоприятное действие. Типично, расстройство представляет собой рак, при котором ингибирование MEK и/или B-Raf оказывает благоприятное действие. Примеры видов рака, подходящих для лечения комбинацией по изобретению, включают, но не ограничены ими, как первичные, так и вторичные метастатические формы рака в области головы и шеи, молочной железы, легкого, ободочной кишки, яичника и предстательной железы. Подходящим является рак, выбранный из следующих: рак в области головы и шеи, рак молочной железы, рак легкого, рак ободочной кишки, рак яичника, рак предстательной железы, глиомы, глиобластомы,астроцитомы, мультиформная глиобластома, синдром Баннаян-Зонана (Bannayan-Zonana), болезнь Коудена (Cowden), болезнь Лермитт-Дуклос (Lhermitte-Duclos), воспалительный рак молочной железы,опухоль Вильмса (Wilms), саркома Юинга (Ewing), рабдомиосаркома, эпендимома, медуллобластома,рак ободочной кишки, рак в области головы и шеи, рак почки, рак легкого, рак печени, меланома, рак яичника, рак поджелудочной железы, рак предстательной железы, саркома, остеосаркома, гигантоклеточная опухоль кости, рак щитовидной железы, лимфобластный Т-клеточный лейкоз, хронический миелогенный лейкоз, хронический лимфоцитарный лейкоз, волосковоклеточный лейкоз, острый лимфобластный лейкоз, острый миелогенный лейкоз, AML (ангиомиолипомы), хронический нейтрофильный лейкоз, острый лимфобластный Т-клеточный лейкоз, плазмацитома, иммунобластный крупноклеточный лейкоз, мантийноклеточный лейкоз, множественная миелома, мегакариобластный лейкоз, множественная миелома, острый мегакариоцитарный лейкоз, промиелоцитарный лейкоз, эритролейкоз, злокачественная лимфома, ходжкинская лимфома, неходжкинская лимфома, лимфобластная Т-клеточная лимфома, лимфома Беркитта (Burkitt), фоликулярная лимфома, нейробластома, рак мочевого пузыря, уротелиальный рак, рак легкого, рак вульвы, рак шейки матки, рак эндометрия, почечно-клеточный рак, мезотелиома, эзофагеальный рак, рак слюнной железы, печеночно-клеточный рак, рак желудка, рак носоглотки,рак щеки, рак ротовой полости, GIST (гастроинтестинальная стромальная опухоль) и рак яичка. Дополнительно, примеры рака, подлежащего лечению, включают аденокарциному Баррета; карциному желчных протоков; рак молочной железы; рак шейки матки; холангиокарциному; опухоли центральной нервной системы, включая первичные опухоли ЦНС, такие как глиобластомы, астроцитомы(например, мультиформную глиобластому) и эпендимомы, и вторичные опухоли ЦНС (т.е. метастазы в центральную нервную систему опухолей, имеющих происхождение вне центральной нервной системы); колоректальный рак, включая карциному ободочной кишки толстого кишечника; рак желудка; карциному в области головы и шеи, включая плоскоклеточную карциному в области головы и шеи; гематологические виды рака, включая лейкозы и лимфомы, такие как острый лимфобластный лейкоз, острый миелогенный лейкоз (AML), миелодиспластические синдромы, хронический миелогенный лейкоз, ходжкинскую лимфому, неходжкинскую лимфому, мегакариобластный лейкоз, множественную миелому и эритролейкоз; печеночно-клеточную карциному; рак легкого, включая мелкоклеточный рак легкого и немелкоклеточный рак легкого; рак яичника, рак эндометрия; рак поджелудочной железы; аденому гипофиза; рак предстательной железы; почечно-клеточный рак, саркому; рак кожи, включая меланомы; и рак щитовидной железы. Соответственно, настоящее изобретение относится к способу лечения или уменьшения тяжести рака, выбранного из следующих: глиобластомы головного мозга (глиомы), синдром Баннаян-Зонана, болезнь Коудена, болезнь Лермитт-Дуклос, рак молочной железы, рак ободочной кишки, рак в области головы и шеи, рак почки, рак легкого, рак печени, меланома, рак яичника, рак поджелудочной железы, рак предстательной железы, саркома и рак щитовидной железы. Соответственно, настоящее изобретение относится к способу лечения или уменьшения тяжести рака, выбранного из следующих: рак яичника, рак молочной железы, рак поджелудочной железы и рак предстательной железы. Комбинация по изобретению может быть использована сама по себе или в комбинации с одним или более другими терапевтическими агентами. Таким образом, согласно изобретению в еще одном аспекте предложена комбинация, содержащая комбинацию по изобретению с дополнительным терапевтическим агентом или агентами, композициями и лекарственными средствами, содержащими комбинацию, и применение дополнительной комбинации, композиций и лекарственных средств в терапии, в частности в лечении заболеваний, реагирующих на ингибирование MEK и/или киназы В. В одном из воплощений комбинация по изобретению может быть использована в сочетании с другими терапевтическими способами лечения рака. B частности, в антинеопластической терапии предусмотрена комбинированная терапия с другими химиотерапевтическими, гормональными, антительными агентами, а также с хирургическими и/или радиационными методами лечения, иными, чем упомянутые выше. Комбинированные терапии согласно настоящему изобретению включают введение соединения A и соединения B, а также возможное применение других терапевтических агентов, включая другие антинеопластические агенты. Такую комбинацию агентов можно вводить вместе или по отдельности, и при введении по отдельности введение можно выполнять одновременно или последовательно в любом порядке, как близко, так и удаленно по времени. B одном из воплощений фармацевтическая комбинация содержит соединение A и соединение B и возможно по меньшей мере один дополнительный антинеопластический агент. Терапевтически эффективные количества соединения A и соединения B являются такими, как указано выше. Терапевтически эффективное количество дополнительных терапевтических агентов по настоящему изобретению будет зависеть от ряда факторов, включая, например, возраст и массу тела млекопитающего, точное состояние, требующее лечения, тяжесть состояния, характер композиции и путь введения. B конечном счете, терапевтически эффективное количество будет находиться на усмотрении лечащего врача или ветеринара. Относительное распределение введения по времени будет выбрано с целью достижения желаемого совместного терапевтического эффекта. В одном из воплощений дополнительная противораковая терапия представляет собой хирургическое вмешательство и/или радиотерапию. В одном из воплощений дополнительная противораковая терапия представляет собой по меньшей мере один дополнительный антинеопластический агент. Любой антинеопластический агент, который обладает активностью против восприимчивой опухоли, которую лечат, может быть использован в данной комбинации. Типичные полезные антинеопластические агенты включают, но ими не ограничены, антимикротрубочковые агенты, такие как дитерпеноиды и алкалоиды барвинка; платиновые координационные комплексы; алкилирующие агенты, такие как азотистые аналоги иприта, оксазафосфорины, алкилсульфонаты, нитрозомочевины и триазены; антибиотики, такие как антрациклины, актиномицины и блеомицины; ингибиторы топоизомеразы II, такие как эпиподофиллотоксины; антиметаболиты, такие как аналоги пурина и пиримидина и антифолатные соединения, ингибиторы топоизомеразы I, такие как камптотецины, гормоны и аналоги гормонов, ингибиторы путей сигнальной трансдукции, ингибиторы ангиогенеза, опосредованного нерецепторными тирозинкиназами, иммунотерапевтические агенты, проапоптические агенты и ингибиторы передачи сигнала в клеточном цикле. Антимикротрубочковые агенты или антимитотические агенты. Антимикротрубочковые агенты или антимитотические агенты представляют собой фазаспецифические агенты, активные в отношении микротрубочек опухолевых клеток во время М или митозной фазы клеточного цикла. Примеры антимикротрубочковых агентов включают, без ограничения,дитерпеноиды и алкалоиды барвинка. Дитерпеноиды из природных источников являются фаза-специфическими противораковыми агентами, которые действуют в G2/M фазах клеточного цикла. Считается, что дитерпеноиды стабилизируют-тубулиновую субъединицу микротрубочек за счет связывания с этим белком. Разборка данного белка,как представляется, затем ингибируется с остановкой митоза и последующей гибелью клетки. Примеры дитерпеноидов включают, без ограничения, паклитаксел и его аналог доцетаксел. Паклитаксел,5,20-эпокси-1,2,4,7,10,13-гекса-гидрокситакс-11-ен-9-он-4,10-диацетат-2 бензоат-13-эфир с (2R,3S)-N-бензоил-3-фенилизосерином, является природным дитерпеновым продуктом, выделенным из тихоокеанского тиса коротколистного Taxus brevifolia, и коммерчески доступен в форме раствора для инъекций TAXOL. Он является членом таксанового семейства терпенов. Паклитаксел был разрешен для клинического применения в лечении резистентного рака яичника в США(Markman et al., Yale Journal of Biology and Medicine, 64:583, 1991, McGuire et al., Ann. Intem., Med.,111:273, 1989) для лечения рака молочной железы (Holmes et al., J. Nat. Cancer Inst., 83:1797, 1991). Он является потенциальным кандидатом для лечения новообразований кожи (Einzig et al., Proc. Am. Soc.Clin. Oncol., 20:46) и карцином в области головы и шеи (Forastire et al., Sem. Oncol., 20:56, 1990) Данное соединение также демонстрирует потенциал для лечения поликистозного заболевания почек (Woo et al.,Nature, 368:750, 1994), рака легкого и малярии. Лечение пациентов таклитакселом приводит к подавлению костного мозга (множественные клеточные линии дифференцировки, Ignoff, R.J. et al., CancerChemotherapy Pocket Guide, 1998), связанному с длительностью введения доз выше пороговой концентрации (50 нМ) (Kearns, С.М. et al., Seminars in Oncology, 3(6):16-23, 1995). Доцетаксел, (2R,3S)-N-карбокси-3-фенилизосерин, N-трет-бутиловый эфир, 13-эфир с 5-20-эпокси-1,2,4,7,10,13-гексагидрокситакс-1-1-ен-9-он-4-ацетат-2-бензоатом, тригидратом, коммерчески доступен в форме раствора для инъекций TAXOTERE. Доцетаксел показан для лечения рака молочной железы. Доцетаксел является полусинтетическим производным паклитаксела (см. выше), полученным с использованием природного предшественника, 10-деацетил-баккатина III, экстрагированного из хвои европейского тиса. Алкалоиды барвинка представляют собой фаза-специфические антиненопластические агенты, получаемые из растения барвинок. Алкалоиды барвинка действуют в М фазе (митоз) клеточного цикла путем специфического связывания с тубулином. B результате, связанная молекула тубулина неспособна полимеризоваться в микротрубочки. Митоз, как полагают, останавливается в метафазе с последующей гибелью клетки. Примеры алкалоидов барвинка включают, но ими не ограничены, винбластин, винкристин и винорелбин. Винбластин, винкалейкобластина сульфат, коммерчески доступен в форме VELBAN в виде раствора для инъекций. Хотя он имеет возможное показание в качестве терапии второй линии различных солидных опухолей, он в основном показан для лечения рака яичка и различных лимфом, включая болезнь Ходжкина и лимфоцитарные и гистиоцитарные лимфомы. Миелосупрессия является лимитирующим дозу побочным эффектом винбластина. Винкристин, винкалейкобластин, 22-оксо-, сульфат, коммерчески доступен в форме ONCOVIN в виде раствора для инъекций. Винкристин показан для лечения острых лейкозов, а также нашел примене- 20020589 ние в схемах лечения ходжкинских и неходжкинских злокачественных лимфом. Алопеция и неврологические эффекты являются самыми распространенными побочными эффектами винкристина, и реже имеют место миелосупрессия и эффекты в отношении слизистой оболочки желудочно-кишечного тракта. Винорелбин,3',4'дидегидро-4'-дезокси-С'-норвинкалейкобластин[R-(R,R)-2,3 дигидроксибутандиоат (1:2) (соль)], коммерчески доступен в форме инъекционного раствора винорелбина тартрата (NAVELBINE) и является полусинтетическим алкалоидом барвинка. Винорелбин показан для применения в качестве единственного агента или в комбинации с другими химиотерапевтическими агентами, такими как цисплатин, в лечении различных солидных опухолей, в частности немелкоклеточного рака легкого, запущенного рака молочной железы и гормон-резистентного рака предстательной железы. Миелосупрессия является самым распространенным лимитирующим дозу побочным эффектом винорелбина. Платиновые координационные комплексы. Платиновые координационные комплексы представляют собой неспецифические в отношении фазы противораковые агенты, которые взаимодействуют с ДНК. Платиновые комплексы входят в опухолевые клетки, подвергаются гидратации и образуют внутри- и межцепочечные поперечные связи с ДНК и, в результате, оказывают пагубные биологические эффекты в отношении данной опухоли. Примеры платиновых координационных комплексов включают, но ими не ограничены, оксалиплатин, цисплатин и карбоплатин. Цисплатин, цис-диаминдихлороплатина, коммерчески доступен в форме PLATINOL в виде раствора для инъекций. Цисплатин показан главным образом для лечения метастатического рака яичка и рака яичника, а также прогрессирующего рака мочевого пузыря. Карбоплатин, диамин[1,1-циклобутан-дикарбоксилат(2-)-О,О']платина, коммерчески доступен в форме PARAPLATIN в виде раствора для инъекций. Карбоплатин показан главным образом для лечения первой и второй линий прогрессирующего рака яичника. Алкилирующие агенты. Алкилирующие агенты представляют собой неспецифические в отношении фазы противораковые агенты и сильные электрофилы. Обычно алкилирующие агенты образуют ковалентные связи, в результате алкилирования с ДНК через нуклеофильные группировки молекулы ДНК, такие как фосфатная, амино, сульфгидрильная, гидроксильная, карбоксильная и имидазольная группы. Такое алкилирование нарушает функцию нуклеиновой кислоты, приводя к гибели клетки. Примеры алкилирующих агентов включают, но ими не ограничены, азотистые аналоги иприта, такие как циклофосфамид, мелфалан и хлорамбуцил; алкилсульфонаты, такие как бусульфан; нитрозомочевины, такие как кармустин; и триазены, такие как дакарбазин. Циклофосфамид, 2-[бис-(2-хлорэтил)амино]тетрагидро-2 Н-1,3,2-оксазафосфорин 2-оксида моногидрат, коммерчески доступен в форме раствора для инъекций или таблеток, такой как CYTOXAN. Циклофосфамид показан в качестве единственного агента или в комбинации с другими химиотерапевтическими агентами для лечения злокачественных лимфом, множественной миеломы и лейкозов. Мелфалан, 4-[бис-(2-хлорэтил)амино]-L-фениламин, коммерчески доступен в форме раствора для инъекций или таблеток ALKERAN. Мелфалан показан для паллиативного лечения множественной миеломы и неоперабельной эпителиальной карциномы яичника. Супрессия костного мозга является самым распространенным лимитирующим дозу побочным эффектом мелфалана. Хлорамбуцил, 4-[бис-(2-хлорэтил)амино]бензолбутановая кислота, коммерчески доступен в форме таблеток LEUKERAN. Хлорамбуцил показан для паллиативного лечения хронического лимфолейкоза и злокачественных лимфом, таких как лимфосаркома, гигантоклеточная фолликулярная лимфома и болезнь Ходжкина. Бусульфан, 1,4-бутандиола диметансульфонат, коммерчески доступен в форме таблетокMYLERAN. Бусульфан показан для паллиативного лечения хронического миелогенного лейкоза. Кармустин, 1,3-[бис-(2-хлорэтил)-1-нитрозомочевина, коммерчески доступен как BiCNU в форме отдельных ампул с лиофилизированным веществом. Кармустин показан для паллиативного лечения в качестве единственного агента или в комбинации с другими агентами против опухолей головного мозга,множественной миеломы, болезни Ходжкина и неходжкинских лимфом. Дакарбазин, 5-(3,3-диметил-1-триазено)имидазол-4-карбоксамид, коммерчески доступен какDTIC-Dome в форме отдельных ампул с веществом. Дакарбазин показан для лечения метастатической злокачественной меланомы и в комбинации с другими агентами для терапии второй линии болезни Ходжкина. Антибиотические антинеопластические агенты. Антибиотические антинеопластические агенты представляют собой неспецифические в отношении фазы агенты, которые связываются с ДНК или встраиваются в ДНК. Обычно такое действие приводит к стабильным комплексам ДНК или к разрыву цепи и, в результате, к нарушению обычной функции нуклеиновых кислот и к гибели клеток. Примеры антибиотических антинеопластических агентов включают,но ими не ограничены, актиномицины, такие как дактиномицин; антроциклины, такие как даунорубицин и доксорубицин; и блеомицины. Дактиномицин, известный также как актиномицин D, коммерчески доступен в инъецируемой форме как COSMEGEN. Дактиномицин показан для лечения опухоли Вильмса и рабдомиосаркомы. Даунорубицин,(8S-цис-)-8-ацетил-10-[(3-амино-2,3,6-тридезоксиL-ликсогексопиранозил]7,8,9,10-тетрагидро-6,8,11-тригидрокси-1-метокси-5,12-нафтацендиона гидрохлорид, коммерчески доступен в липосомной инъецируемой форме как DAUNOXOME или в инъецируемой форме какCERUBIDINE. Даунорубицин показан для вызывания ремиссии в процессе лечения острого нелимфоцитарного лейкоза и прогрессирующей саркомы Капоши, ассоциированной с ВИЧ. Доксорубицин,(8S,10S)-10-[(3-амино-2,3,6-тридезоксиL-ликсогексопиранозил)окси]-8 гликолоил, 7,8,9,10-тетрагидро-6,8,11-тригидрокси-1-метокси-5,12-нафтаценедиона гидрохлорид, коммерчески доступен в инъецируемой форме как RUBEX или ADRIAMYCIN RDF. Доксорубицин показан главным образом для лечения острого лимфобластного лейкоза и острого миелобластного лейкоза,но он также является полезным компонентом в лечении некоторых солидных опухолей и лимфом. Блеомицин, смесь цитотоксических гликопептидных антибиотиков, выделенных из штаммаStreptomyces verticillus, коммерчески доступен как BLENOXANE. Блеомицин показан для паллиативного лечения, в качестве единственного агента или в комбинации с другими агентами, плоскоклеточного рака, лимфом и рака яичка. Ингибиторы топоизомеразы II. Ингибиторы топоизомеразы II включают, без ограничения, эпиподофиллотоксины. Эпиподофиллотоксины являются фаза-специфическими антинеопластическими агентами, получаемыми из растения мандрагора. Эпиподофиллотоксины обычно воздействуют на клетки в S и G2 фазах клеточного цикла, образуя тройной комплекс с топоизомеразой II и ДНК, что вызывает разрывы цепи ДНК. Разрывы цепи аккумулируются, и клетка гибнет. Примеры эпиподофиллотоксинов включают, но ими не ограничены, этопозид и тенипозид. Этопозид, 4'-деметил-эпиподофиллотоксин 9[4,6-O-(R)-этилиденD-глюкопиранозид], коммерчески доступен в форме инъецируемого раствора или капсул как VePESID и общеизвестен как VP-16. Этопозид показан в качестве единственного агента или в комбинации с другими химиотерапевтическими агентами для лечения рака яичка и немелкоклеточного рака легкого. Тенипозид, 4'-деметил-эпиподофиллотоксин 9[4,6-O-(R)-тенилиденD-глюкопиранозид], коммерчески доступен в форме раствора для инъекций как VUMON и общеизвестен как VM-26. Тенипозид показан в качестве единственного агента или в комбинации с другими химиотерапевтическими агентами для лечения острого лейкоза у детей. Антиметаболитные антинеопластические агенты. Антиметаболитные неопластические агенты представляют собой фаза-специфические антинеопластические агенты, которые действуют в S-фазе (синтез ДНК) клеточного цикла, ингибируя синтез ДНК или ингибируя синтез пуриновых или пиримидиновых оснований, и за счет этого ограничивает синтез ДНК. B результате, S-фаза не продолжается и происходит гибель клеток. Примеры антиметаболитных антинеопластических агентов включают, но ими не ограничены, фторурацил, метотрексат, цитарабин,меркаптопурин, тиогуанин и гемцитабин. 5-Фторурацил, 5-фтор-2,4-(1 Н,3 Н)-пиримидиндион, коммерчески доступен как фторурацил. Введение 5-фторурацила приводит к ингибированию синтеза тимидилата, и он также встраивается в РНК и ДНК. Обычным результатом является гибель клеток. 5-Фторурацил показан в качестве единственного агента или в комбинации с другими химиотерапевтическими агентами для лечения рака молочной железы, рака ободочной кишки, рака прямой кишки, рака желудка и рака поджелудочной железы. Другие фторпиримидиновые аналоги включают 5-фтор-дезоксиуридин (флоксуридин) и 5-фтордезоксиуридина монофосфат. Цитарабин, 4-амино-1D-арабинофуранозил-2(1 Н)-пиримидинон, коммерчески доступен какCYTOSAR-U и общеизвестен как Ara-С. Считается, что цитарабин проявляет специфичность в отношении клеточной фазы в S-фазе, ингибируя элонгацию цепи ДНК посредством терминального встраивания цитарабина в растущую цепь ДНК. Цитарабин показан в качестве единственного агента или в комбинации с другими химиотерапевтическими агентами для лечения острого лейкоза. Другие цитидиновые аналоги включают 5-азацитидин и 2',2'-дифтордезоксицитидин (гемцитабин). Меркаптопурин, 1,7-дигидро-6 Н-пурин-6-тиона моногидрат, коммерчески доступен какPURINETHOL .Меркаптопурин проявляет специфичность в отношении клеточной фазы в S-фазе, ингибируя синтез ДНК по еще не установленному механизму. Меркаптопурин показан в качестве единственного агента или в комбинации с другими химиотерапевтическими агентами для лечения острого лейкоза. Полезным меркаптопуриновым аналогом является азатиоприн. Тиогуанин, 2-амино-1,7-дигидро-6 Н-пурин-6-тион, коммерчески доступен как TABLOID. Тиогуанин проявляет специфичность в отношении клеточной фазы в S-фазе, ингибируя синтез ДНК по еще не установленному механизму. Тиогуанин показан в качестве единственного агента или в комбинации с другими химиотерапевтическими агентами для лечения острого лейкоза. Другие пуриновые аналоги включают пентостатин, эритрогидроксинониладенин, флударабина фосфат и кладрибин. Гемцитабин, 2'-дезокси-2',2'-дифторцитидина моногидрохлорид (-изомер), коммерчески доступен как GEMZAR. Гемцитабин проявляет специфичность в отношении клеточной фазы в S-фазе, блокируя прохождение клеток через G1/S барьер. Гемцитабин показан в комбинации с цисплатином для лечения локально прогрессирущего немелкоклеточного рака легкого и отдельно для лечения локально прогрессирующего рака поджелудочной железы. Метотрексат, N-[4-(2,4 диамино-6-птеридинил)метил]метиламино]бензоил]-L-глутаминовая кислота, коммерчески доступен как натрий-метотрексат. Метотрексат проявляет эффекты в отношении клеточной фазы, в частности в S-фазе, ингибируя синтез, репарацию и/или репликацию ДНК через ингибирование редуктазы дигидрофолиевой кислоты, которая требуется для синтеза пуриновых нуклеотидов и тимидилата. Метотрексат показан в качестве единственного агента или в комбинации с другими химиотерапевтическим агентами для лечения хориокарциномы, менингеального лейкоза, неходжкинской лимфомы и карцином молочной железы, головы, шеи, яичника и мочевого пузыря. Ингибиторы топоизомеразы I. Камптотецины, включая камптотецин и производные камптотецина, доступны или находятся в разработке в качестве ингибиторов топоизомеразы I. Считается, что цитотоксическая активность камптотецинов связана с их ингибиторной активностью в отношении топоизомеразы I. Примеры камптотецинов включают, но ими не ограничены, иринотекан и топотекан и различные оптические формы 7-(4-метилпиперазино-метилен)-10,11-этилендиокси-20-камптотецина, описанные ниже. Иринотекан HCl, (4S)-4,11-диэтил-4-гидрокси-9-[(4-пиперидинопиперидино)карбонилокси]-1 Нпирано[3',4',6,7]индолизино[1,2-b]хинолин-3,14(4 Н,12 Н)-диона гидрохлорид, коммерчески доступен в форме раствора для инъекций CAMPTOSAR. Иринотекан представляет собой производное камптотецина, которое связывается, вместе со своим активным метаболитом SN-38, с комплексом топоизомеразаI-ДНК. Считается, что его цитотоксичность проявляется в результате непоправимых разрывов двойной цепи, вызываемых взаимодействием тройного комплекса топоизомераза I:ДНК:иринотекан или SN-38 с ферментами репликации. Иринотекан показан для лечения метастатического рака ободочной кишки или прямой кишки. ТопотеканHCl,(S)-10-[(диметиламино)метил]-4-этил-4,9-дигидрокси-1 Нпирано[3',4',6,7]индолизино[1,2-b]хинолин-3,14-(4 Н,12 Н)-диона моногидрохлорид, коммерчески доступен в форме раствора для инъекций HYCAMTIN. Топотекан представляет собой производное камптотецина, которое связывается с комплексом топоизомераза 1-ДНК и предотвращает религирование единичных разрывов цепи, вызванных топоизомеразой I в ответ на торсионную деформацию молекулы ДНК. Топотекан показан для лечения второй линии метастатического рака яичника и мелкоклеточного рака легкого. Гормоны и аналоги гормонов. Гормоны и аналоги гормонов являются соединениями, полезными для лечения видов рака, при которых существует связь между гормоном(ами) и ростом и/или отсутствием роста раковой опухоли. Примеры гормонов и аналогов гормонов, полезных в лечении рака, включают, но ими не ограничены, адренокортикостероиды, такие как преднизон и преднизолон, которые полезны в лечении злокачественной лимфомы и острого лейкоза у детей; аминоглютетимид и другие ингибиторы ароматазы, такие как анастрозол, летразол, форазол и экземестан, полезные в лечении адренокортикальной карциномы и гормон-зависимой карциномы молочной железы, содержащих рецепторы эстрогенов; прогестрины, такие как мегестрола ацетат, полезные в лечении гормон-зависимого рака молочной железы и карциномы эндометрия; эстрогены, андрогены и антиандрогены, такие как флутамид, нилутамид, бикалутамид, ципротерона ацетат и 5-редуктазы, такие как финастерид и дутастерид, полезные в лечении карциномы предстательной железы и доброкачественной гипертрофии предстательной железы; антиэстрогены, такие как тамоксифен, торемифен, ралоксифен, дролоксифен, йодоксифен, а также избирательные модуляторы рецепторов эстрогенов (SERMS), такие как те, которые описаны в патентах США 5681835, 5877219 и 6207716, полезные в лечении гормонзависимой карциномы молочной железы и других чувствительных видов рака; и гонадотропин-высвобождающий гормон (GnRH) и его аналоги, которые стимулируют высвобождение лютеинизирующего гормона (LH) и/или фолликулостимулирующего гормона (FSH), для лечения карциномы предстательной железы, например агонисты и антагонисты LHRH, такие как гозерелина ацетат и лупролид. Ингибиторы пути сигнальной трансдукции. Ингибиторами пути сигнальной трансдукции являются те ингибиторы, которые блокируют или ингибируют химический процесс, индуцирующий внутриклеточное изменение. B данном описании это изменение представляет собой клеточную пролиферацию или дифференцировку. Ингибиторы сигнальной трансдукции, полезные в настоящем изобретении, включают ингибиторы рецепторных тирозинкиназ,нерецепторных тирозинкиназ, блокаторы SH2/SH3 домена, ингибиторы серин/треониновых киназ, фосфотидилинозитол-3-киназ, миоинозитольной передачи сигнала и Ras онкогены. Несколько протеинтирозинкиназ катализируют фосфорилирование специфических тирозильных остатков в различных белках, вовлеченных в регуляцию клеточного роста. Такие протеинтирозинкиназы могут быть в широком смысле классифицированы как рецепторные или нерецепторные киназы. Рецепторные тирозинкиназы являются трансмембранными белками, имеющими внеклеточный лигандсвязывающий домен, трансмембранный домен и тирозинкиназный домен. Рецепторные тирозинкиназы вовлечены в регуляцию клеточного роста и обычно называются рецепторами факторов роста. Было показано, что несоответствующая или неконтролируемая активация многих этих киназ, т.е. аберрантная киназная рецепторная активность в отношении факторов роста, например в результате сверхэкспрессии или мутации, приводит к неконтролирующему клеточному росту. Соответственно, аберрантную активность таких киназ связывают с ростом злокачественной ткани. Следовательно, ингибиторы таких киназ могут обеспечивать лечение рака. Рецепторы факторов роста включают, например рецептор эпидермального фактора роста (EGFr), рецептор тромбоцитарного фактора роста (PDGFr), erbB2, erbB4, ret, рецептор васкулярного эндотелиального фактора роста (VEGFr), тирозинкиназу с доменами, гомологичными иммуноглобулиноподобному и эпидермальному фактору роста (TIE-2), рецептор инсулинового фактора роста-I (IGF-I), макрофагальный колониестимулирующий фактор (cfms), ВТК, ckit, cmet, рецепторы факторов роста фибробластов (FGF), Trk-рецепторы (TrkA, TrkB и TrkC), эфриновые (eph) рецепторы иRET-протоонкоген. Несколько ингибиторов ростовых рецепторов находятся в стадии разработки и включают антагонисты лигандов, антитела, ингибиторы тирозинкиназ и антисмысловые олигонуклеотиды. Рецепторы факторов роста и агенты, которые ингибируют функцию рецепторов факторов роста, описаны, например, в Kath, John С., Exp. Opin. Ther. Patents (2000), 10(6):803-818; Shawver et al., DDT. Vol 2,No. 2, February 1997 и Lofts, F.J. et al., "Growth factor receptors as targets", New Molecular Targets for CancerChemotherapy, ed. Workman, Paul and Kerr, David, CRC press 1994, London. Тирозинкиназы, которые не являются киназами рецепторов факторов роста, называются нерецепторными тирозинкиназами. Нерецепторные тирозинкиназы, полезные в настоящем изобретении, которые являются мишенями или потенциальными мишенями противораковых лекарственных средств, включаютcSrc, Lck, Fyn, Yes, Jak, cAbl, FAK (киназа фокальной адгезии), тирозинкиназу Брутона и Bcr-Abl. Такие нерецепторные киназы и агенты, которые ингибируют функцию нерецепторных тирозинкиназ, описаны в Sinh, S. и Corey, S.J. (1999), Journal of Hematotherapy and Stem Cell Research, 8(5):465-80 и Bolen, J.B.,Brugge, J.S. (1997), Annual review of Immunology. 15:371-404. Блокаторы доменов SH2/SH3 являются агентами, которые разрывают связывание с доменами SH2 или SH3 у ряда ферментов или адапторных белков, включая, PI3-K р 85 субъединицу, киназы семействаSrc, адапторные молекулы (She, Crk, Nek, Grb2) и Ras-GAP. SH2/SH3-домены в качестве мишеней для противораковых лекарственных средств обсуждаются в Smithgall, Т.Е. (1995), Journal of Pharmacologicaland Toxicological Methods. 34(3):125-32. Ингибиторы серин/треонинкиназ, включая блокаторы МАР-киназного каскада, которые включают блокаторы Raf киназ (rafk), митоген-активируемых или внеклеточных регулируемых киназ (MEK) и внеклеточных регулируемых киназ (ERK); и блокаторы членов С-семейства протеинкиназ, в том числе блокаторы PKC (альфа, бета, гамма, эпсилон, мю, лямбда, йота, дзета), членов семейства киназ IkB (IKKa,IKKb), семейства киназ PKB, семейства киназ akt и TGF-бета рецепторных киназ. Такие серин/треонинкиназы и их ингибиторы описаны в Yamamoto, Т., Taya, S., Kaibuchi, K. (1999), Journal of(1995), Cancer Treatment and Research. 78:3-27, Lackey, K. et al., Bioorganic and Medicinal Chemistry Letters,(10), 2000, 223-226; патент США 6268391 и Martinez-lacaci, L., et al., Int. J. Cancer (2000), 88(1):44-52. Ингибиторы членов семейства фосфотидилинозитол-3-киназ, включая блокаторы PI3-киназы, ATM,DNA-PK и Ku, также полезны в настоящем изобретении. Такие киназы рассматриваются в Abraham, R.T.(1996), Current Opinion in Immunology. 8(3): 412-8; Canman, C.E., Lim, D.S. (1998), Oncogene. 17(25):33013308; Jackson, S.P. (1997), International Journal of Biochemistry и Cell Biology, 29(7):935-8 и Zhong, H. et al.,Cancer res, (2000), 60(6):1541-1545. Также полезными в настоящем изобретении являются ингибиторы миоинозитольной передачи сигналов, такие как блокаторы фосфолипазы С и миоинозитольные аналоги. Такие сигнальные ингибиторы описаны в Powis, G., and Kozikowski A. (1994), New Molecular Targets for Cancer Chemotherapy ed., PaulWorkman and David Kerr, CRC press 1994, London. К другой группе ингибиторов путей сигнальной трансдукции относятся ингибиторы Ras онкогена. Такие ингибиторы включают ингибиторы фарнезилтрансферазы, геранил-геранил-трансферазы и СААХ-протеаз, а также антисмысловые олигонуклеотиды, рибозимы и иммунотерапевтические средства. Было показано, что такие ингибиторы блокируют активацию ras в клетках, содержащих мутантныйras дикого типа, тем самым действуя как антипролиферативные агенты. Ингибирование ras онкогена рассматривается в Scharovsky, O.G., Rozados, V.R., Gervasoni, S.I. Matar, P. (2000), Journal of Biomedical Как упомянуто выше, антитела против лигандсвязывающей рецепторной киназы также могут служить в качестве ингибиторов сигнальной трансдукции. Эта группа ингибиторов путей сигнальной трансдукции включает применение гуманизированных антител против внеклеточного лигандсвязывающего домена рецепторных тирозинкиназ, например Imclone C225 EGFR-специфическое антитело (см. Green,M.C. et al., Monoclonal Antibody Therapy for Solid Tumors, Cancer Treat. Rev. (2000), 26(4), 269-286);Growth in Mice, Cancer Res. (2000), 60, 5117-5124). Антиангиогенные агенты. Антиангиогенные агенты, включая ингибиторы ангиогенеза, опосредованного нерецепторной MEK,также могут быть полезны. Антиангиогенные агенты - такие как те, которые ингибируют эффекты сосудистого эндотелиального фактора роста (например, антитело против фактора роста эндотелиальных клеток сосудов бевацизумаб [Avastin], и соединения, действующие по другим механизмам (например, линомид, ингибиторы функции интегринов v3 и ангиостатин. Иммунотерапевтические агенты. Агенты, применяемые в иммунотерапевтических схемах лечения, также могут быть полезны в комбинации с соединениями формулы (I). Иммунотерапевтические подходы, включая, например ex vivo и invivo подходы, направленные на повышение иммуногенности опухолевых клеток пациента, такие как трансфекция цитокинами, например, интерлейкином-2, интерлейкином-4 или гранулоцитарным макрофагальным колониестимулирующим фактором, подходы, направленные на снижение Т-клеточной анергии, подходы с использованием трансфицированных иммунных клеток, таких как трансфицированные цитокинами дендритные клетки, подходы с использованием трансфицированных цитокинами опухолевых клеточных линий и подходы с использованием антиидиотипических антител. Протоапоптические агенты. Агенты, применяемые в протоапоптических схемах лечения (например, bcl-2 антисмысловые олигонуклеотиды), также могут быть использованы в комбинации по настоящему изобретению. Ингибиторы сигнальных путей, задействованных в клеточном цикле. Ингибиторы сигнальных путей, задействованных в клеточном цикле, ингибируют молекулы, вовлеченные в контроль клеточного цикла. Семейство протеинкиназ, названное циклинзависимыми киназами(CDK), и их взаимодействие с семейством белков, названных циклинами, контролируют ход эукариотического клеточного цикла. Согласованная активация и инактивация разных циклин/CDK-комплексов необходима для нормального хода клеточного цикла. Несколько ингибиторов сигнальных путей, задействованных в клеточном цикле, находятся в стадии разработки. Примеры циклинзависимых киназ, включая CDK2, CDK4 и CDK6, и ингибиторов для них описаны, например, в Rosania et al., Exp. Opin. Ther.Patents (2000), 10(2):215-230. В одном из воплощений комбинация по настоящему изобретению содержит соединение формулы I или его соль или сольват и по меньшей мере один антинеопластический агент, выбранный из антимикротрубочковых агентов, платиновых координированных комплексов, алкилирующих агентов, антибиотиков, ингибиторов топоизомеразы II, антиметаболитов, ингибиторов топоизомеразы I, гормонов и аналогов гормонов, ингибиторов путей сигнальной трансдукции, ингибиторов ангиогенеза, опосредованного нерецепторной тирозин-специфичной MEK-киназой, иммунотерапевтических агентов, проапоптических агентов и ингибиторов сигнальных путей, задействованных в клеточном цикле. В одном из воплощений комбинация по настоящему изобретению содержит соединение формулы I или его соль или сольват и по меньшей мере один антинеопластический агент, который представляет собой антимикротрубочковый агент, выбранный из дитерпеноидов и алкалоидов барвинка. В еще одном воплощении по меньшей мере один антинеопластический агент представляет собой дитерпеноид. В еще одном воплощении по меньшей мере один антинеопластический агент представляет собой алкалоид барвинка. В одном из воплощений комбинация по настоящему изобретению содержит соединение формулы I или его соль или сольват и по меньшей мере один антинеопластический агент, который представляет собой платиновый координационный комплекс. В еще одном воплощении по меньшей мере один антинеопластический агент представляет собой паклитаксел, карбоплатин или винорелбин. В еще одном воплощении по меньшей мере один антинеопластический агент представляет собой карбоплатин. В еще одном воплощении по меньшей мере один антинеопластический агент представляет собой винорелбин. В еще одном воплощении по меньшей мере один антинеопластический агент представляет собой паклитаксел. В одном из воплощений комбинация по настоящему изобретению содержит соединение формулы I или его соль или сольват и по меньшей мере один антинеопластический агент, который представляет собой ингибитор пути сигнальной трансдукции. В еще одном воплощении ингибитор пути сигнальной трансдукции представляет собой ингибитор киназы рецептора фактора роста VEGFR2, TIE2, PDGFR, BTK, erbB2, EGFr, IGFR-1, TrkA, TrkB, TrkC или c-fms. В еще одном воплощении ингибитор пути сигнальной трансдукции представляет собой ингибитор серин/треонинкиназы rafk, akt или PKC-дзета. В еще одном воплощении ингибитор пути сигнальной трансдукции представляет собой ингибитор нерецепторной тирозинкиназы, выбранной из киназ семейства src. В еще одном воплощении ингибитор пути сигнальной трансдукции представляет собой ингибиторc-src. В еще одном воплощении ингибитор пути сигнальной трансдукции представляет собой ингибиторRas онкогена, выбранный из ингибиторов фарнезилтрансферазы и геранил-геранил-трансферазы. В еще одном воплощении ингибитор пути сигнальной трансдукции представляет собой ингибитор серин/треонинкиназы, выбранный из группы, состоящей из PI3K. В еще одном воплощении ингибитор пути сигнальной трансдукции представляет собой двойственный ингибитор В одном из воплощений комбинация по настоящему изобретению содержит соединение формулы I или его соль или сольват и по меньшей мере один антинеопластический агент, который представляет собой ингибитор передачи сигналов клеточного цикла. В еще одном воплощении ингибитор передачи сигналов клеточного цикла представляет собой ингибитор CDK2, CDK4 или CDK6. В одном из воплощений млекопитающим в способах и применениях по настоящему изобретению является человек. Соответственно, настоящее изобретение относится к способу лечения или уменьшения тяжести рака, в который вовлечены Raf, Ras, MEK и PI3K/Pten, каждый из которых может быть либо дикого типа,либо мутантным. Это включает, но не ограничивается пациентами с раковыми заболеваниями, которые имеют мутантный RAF, RAS дикого типа, MEK дикого типа и PI3K/PTEN дикого типа; мутантный RAF,мутантными RAS, MEK дикого типа и PI3K/PTEN дикого типа; мутантный RAF, мутантный RAS, мутантный MEK и PI3K/PTEN дикого типа; и мутантный RAF, RAS дикого типа, мутантный MEK иPI3K/PTEN дикого типа. Термин "дикого типа", как известно в данной области, относится к полипептидной или полинуклеотидной последовательности, которая имеется у нативной популяции, без генетической модификации. Как также известно в данной области, "мутантый" включает полипептидную или полинуклеотидную последовательность, имеющую по меньшей мере одну модификацию аминокислоты или нуклеиновой кислоты по сравнению с соответствующей аминокислотой или нуклеиновой кислотой, находящейся соответственно в полипептиде или полинуклеотиде дикого типа. Термин "мутантный" охватывает однонуклеотидный полиморфизм (SNP), когда последовательность цепи нуклеиновой кислоты отличается от сравниваемой самой распространенной обнаруженной (дикого типа) цепью нуклеиновой кислоты одной парой оснований. Виды рака, которые являются видами рака либо с дикого типа, либо с мутантными Raf, Ras, MEK или мутантными PI3K/Pten, идентифицируют известными методами. Например, опухолевые клетки дикого типа или мутантные опухолевые клетки могут быть идентифицированы методами амплификации и секвенирования ДНК, методами детекции ДНК и РНК, включая, без ограничения, нозерн-блоттинг и саузерн-блоттинг соответственно, и/или различными методами с использованием биочиповых и матричных технологий. Полипептиды дикого типа и мутантные полипептиды могут быть детектированы различными методами, включая, без ограничения, иммунодиагностические методы, такие как ELISA (твердофазный иммуноферментный анализ), вестерн-блоттинг или иммуноцитохимические методы. Соответственно, могут быть использованы методы пирофосфоролиз-активированной полимеризации (РАР) и/или PCR(полимеразная цепная реакция) (Liu, Q et al., Human Mutation, 23:426-436 (2004). Приведенные ниже примеры предназначены только для иллюстрации и никоим образом не ограничивают объем изобретения. Примеры Пример 1. Композиция капсулы. Пероральную лекарственную форму для введения комбинации по настоящему изобретению изготавливают путем заполнения стандартной, состоящей из двух частей твердой желатиновой капсулы ингредиентами в соотношениях, указанных в табл. А. Таблица А Несмотря на то что предпочтительные воплощения данного изобретения проиллюстрированы примером, приведенным выше, следует иметь в виду, что данное изобретение не ограничено раскрытыми здесь точными инструкциями, и что права на все модификации, подпадающие под объем прилагаемой формулы изобретения, зарезервированы. Анализы.In vitro исследования комбинаций ингибиторов BRAF и MEK на линиях раковых клеток из множества источников с разными мутациями. А. Диапазоны концентраций А. Эксперименты с лекарственными комбинациями проводили в 384-луночных планшетах. Клетки вносили в 384-луночные планшеты в количестве 500 клеток на лунку в культуральной среде, подходящей для каждого типа клеток, дополненной 10% FBS (сыворотка плода коровы) и 1% пенициллина/стрептомицина, и инкубировали в течение ночи при 37C, 5% СО 2. Шестнадцать концентраций 2-кратных разведений каждого лекарственного средства тестировали в матрице для ингибирования клеточного роста. Тестированные концентрации для соединения A (свободная форма) составляли от 1 мкМ до 0,03 нМ и для соединения B (DMSO-сольват) составляли от 10 мкМ до 0,3 нМ. Клетки обрабатывали комбинацией соединений и инкубировали при 37C в течение 72 ч. Рост клеток измеряли с использованием реагента CellTiter-Glo в соответствии с протоколом производителя, и сигналы считывали на планшет-ридере PerkinElmer EnVision в режиме люминесценции с 0,5-секундным считыванием. Результаты выражали в виде процента ингибирования по сравнению с клетками, обработанными DMSO, и делали коррекцию с учетом фона путем вычитания значений, полученных для лунок, не содержащих клетки. Ответную реакцию (процент ингибирования по сравнению с необработанными образцами и приведенный только к среде) на соединение "А" в концентрации "a" (Ra) и ответную реакцию на соединение"В" в концентрации "b" (Rb) сравнивали с ответной реакцией на смесь соединений "А" и "В" в концентрациях "а" и "b" соответственно (Rab). Используя эти значения, рассчитывали превышение относительно наивысшего значения, показанного одним агентом (Excess Over Highest Single Agent (EOHSA, для каждой концентрации каждой из протестированных клеточных линий:Rab10% высшего значения среди Ra и Rb = более чем аддитивная;Rab-10% высшего значения среди Ra и Rb = антагонизм. Используя эту формулу, если Rab больше не менее чем на 10%, чем наивысшее значение среди Ra иRb, то комбинация лекарственных средств считается более чем аддитивной. Если Rab меньше не менее чем на 10%, чем наивысшее значение среди Ra и Rb, то комбинация лекарственных средств является антагонистической. Для каждой из протестированных клеточных линий было подсчитано число комбинаций в матрице 1616 с более чем аддитивным ответом (те, где Rab больше не менее чем на 10%, чем высшее значение среди Ra и Rb) перечислено. Число более чем аддитивных комбинаций (из 264 протестированных) указано в табл. 1. Из этой таблицы видно, что комбинации концентраций оказывают на указанные клеточные линии особенно благоприятное влияние (выделены серым цветом), если более чем 20% (51 комбинация из 256 протестированных) протестированных комбинаций демонстрируют EOHSA 10%. Таблица 1 Эффект комбинаций ингибиторов MEK и BRAF на множество раковых клеточных линий Эти данные демонстрируют, что комбинация соединения A и соединения B оказывает благоприятное действие на множество раковых клеточных линий из множества источников независимо от мутационного статуса ключевых онкогенов в MAPK или AKT/PI3K/PTEN путях. В. Диапазоны концентраций В. Оценку, подобную той, которая сделана в разделе A выше, комбинации соединения A и соединенияB осуществляли с использованием данных, полученных в разделе А, но только для концентраций лекарственных средств, которые, как представляется, являются клинически релевантными (от 100 до 3 нМ). Эти концентрации выбраны в качестве концентраций, которые эффективны, но не токсичны в доклинических мышиных моделях с ксенотрансплантатом. Используя эти концентрации, в сумме 25 комбинаций лекарственных средств были оценены для каждой клеточной линии, и результаты суммированы в табл. 2. Число комбинаций, имеющих Rab больше на 10% высшего значения среди Ra и Rb, из 25 протестированных клинически релевантных комбинаций вычислены и выражены в виде процента в табл. 2. Таблица 2 Комбинация ингибиторов MEK и BRAF с использованием клинически релевантных концентраций лекарственных средств in vitro Эти данные демонстрируют, что комбинация соединения A и соединения B оказывает благоприятное действие на большинство протестированных клеточных линий при релевантных клинических концентрациях и оказывает в высокой степени благоприятное действие на все протестированные мутантные клеточные линии BRAFV600E и KRAS независимо от мутационного статуса PI3K/PTEN пути. Ингибирование роста клеток в линиях опухолевых клеток in vitro. Методы. Клеточные линии и условия выращивания. Линии клеток опухоли ободочной кишки человека, Colo-205, DLD-1, HCT-8, HT-29, LS-1034,NCI-H508, RKO, SW1417, SW1463, SW480 и SW837, и линия клеток меланомы человека А 375 были получены из АТСС (Американская коллекция типовых культур). A375PF11 выведена из А 375. 12R5-1,12R5-3, 12R8-1, 12R8-3, 16R5-2, 16R6-3 и 16R6-4, которые являются одноклеточными клонами из смешанных популяций клеток A375PF11, которые были выбраны для выращивания в соединении A до концентраций 1200 и 1600 нМ, и в связи с этим проявляют приобретенную устойчивость к соединению В. Все линии клеток выращивали в среде RPMI 1640, содержащей 10% сыворотки плода коровы (FBS). Анализ ингибирования роста клеток и анализ данных по комбинациям. Все клетки выращивали в течение минимум 72 ч, после чего их высевали на планшеты. Клетки анализировали в 96-луночном планшете для культуры ткани (NUNC 136102) в среде RPMI, содержащей 10% FBS для всех клеток, 1000 клеток/лунку. Приблизительно через 24 ч после высевания клетки подвергали воздействию десяти трехкратных последовательных разведений соединения или комбинации двух агентов при постоянном молярном соотношении 1:10 соединения A (DMSO-сольват) к соединениюB (свободная форма) в среде RPMI, содержащей 10% FBS. Соединение A тестировали в концентрациях от 1 мкМ до 0,05 нМ, и концентрации соединения B составляли от 10 мкМ до 0,5 нМ. Клетки инкубировали в присутствии соединений в течение 3 суток. Уровни АТР (аденозинтрифосфат) определяли путем добавления Cell Titer Glo (Promega) согласно протоколу производителя. Кратко, Cell Titer Glo добавляли в каждый планшет, осуществляли инкубирование в течение 30 мин, затем считывали люминесцентный сигнал на планшет-ридере SpectraMax L с временем интеграции 0,5 с.
МПК / Метки
МПК: A01N 43/90, A61K 31/519
Метки: комбинация, противораковая
Код ссылки
<a href="https://eas.patents.su/30-20589-protivorakovaya-kombinaciya.html" rel="bookmark" title="База патентов Евразийского Союза">Противораковая комбинация</a>
Противораковая комбинация, содержащая производные замещённого акрилоилдистамицина и антитела, ингибирующие факторы роста или их рецепторы

Номер патента: 11574
Опубликовано: 28.04.2009
Авторы: Джерони Мария Кристина, Чиомеи Марина, Песенти Энрико
МПК: A61P 35/00, A61K 39/395, A61K 31/40...
Метки: антитела, содержащая, роста, акрилоилдистамицина, факторы, ингибирующие, производные, противораковая, замещённого, рецепторы, комбинация
Формула / Реферат:
1. Комбинация, содержащая производное акрилоилдистамицина формулы (I) где R1 является атомом хлора или брома; R2 является дистамицином или подобным дистамицину каркасом или фармацевтически приемлемой его солью; и антитело, ингибирующее фактор роста или его рецептор. 2. Комбинация по п.1, где антитело, ингибирующее фактор роста или его рецептор, выбрано из группы, состоящей из бевацизумаба, цетуксимаба, панитумумаба, матузумаба, нимотузумаба,...
Композиция опухолеассоциированных пептидов и относящаяся к ним противораковая вакцина

Номер патента: 18457
Опубликовано: 30.08.2013
Авторы: Шор Оливер, Вайншенк Тони, Сингх Харпрет, Вальтер Штеффен, Левандровски Петер, Хильф Норберт, Траутвайн Клаудиа
МПК: A61K 38/16, A61K 39/00, A61K 38/08...
Метки: относящаяся, композиция, пептидов, вакцина, ним, противораковая, опухолеассоциированных
Формула / Реферат:
1. Фармацевтическая композиция, включающая по крайней мере два пептида, содержащих аминокислотную последовательность SEQ ID № 1 и SEQ ID № 2, и/или содержащих вариантную аминокислотную последовательность, которая по крайней мере на 80% идентична последовательностям с SEQ ID № 1 и SEQ ID № 2, и/или полинуклеотид, содержащий нуклеиновую кислоту, кодирующую SEQ ID № 1 и SEQ ID № 2, или вариантную аминокислотную последовательность, и фармацевтически...
Опухолеассоциированные пептиды, связывающиеся с молекулами человеческого лейкоцитарного антигена (hla) i или ii класса, и относящаяся к ним противораковая вакцина

Номер патента: 13466
Опубликовано: 30.04.2010
Авторы: Вайншенк Тони, Вальтер Штеффен, Сингх Харпреет, Эммерлих Нильс
МПК: C07K 14/47, A61K 39/00, A61K 38/00...
Метки: пептиды, класса, лейкоцитарного, молекулами, относящаяся, опухолеассоциированные, человеческого, связывающиеся, ним, вакцина, противораковая, hla, антигена
Формула / Реферат:
1. Опухолеассоциированный пептид, который выбран из группы пептидов, включающих по меньшей мере одну последовательность в соответствии с SEQ ID № 1 или SEQ ID № 2 или их вариант, при условии, что пептид не является интактным человеческим опухолеассоциированным полипептидом, причем указанный пептид обладает общей длиной между 9 и 30 аминокислотами.2. Опухолеассоциированный пептид по п.1, причем пептид состоит или преимущественно состоит из...
Фармацевтическая комбинация

Номер патента: 20046
Опубликовано: 29.08.2014
Авторы: Кайзер Рольф, Хильберг Франк, Шапиро Дейвид, Штефаник Мартин Фридрих
МПК: A61P 1/16, A61K 31/496, A61K 31/519...
Метки: комбинация, фармацевтическая
Формула / Реферат:
1. Фармацевтическая комбинация, включающая 3-Z-[1-(4-(N-((4-метилпиперазин-1-ил)метилкарбонил)-N-метиламино)анилино)-1-фенилметилен]-6-метоксикарбонил-2-индолинон или его фармацевтически приемлемую соль и N-[4-[2-(2-амино-4,7-дигидро-4-оксо-1Н-пирроло[2,3-d]пиримидин-5-ил)этил]бензоил]-L-глутаминовую кислоту или ее фармацевтически приемлемую соль.2. Фармацевтическая комбинация по п.1, в которой фармацевтически приемлемой солью...
Комбинация на основе производного камптотецина и комбретастатина

Номер патента: 5400
Опубликовано: 24.02.2005
Автор: Биссери Мари-Кристин
МПК: A61P 35/00, A61K 31/4745
Метки: основе, комбинация, камптотецина, комбретастатина, производного
Формула / Реферат:
1. Фармацевтическая комбинация, включающая эффективное количество 7-этил-10-[4-(1-пиперидино)-1-пиперидино]карбонилоксикамптотецина (CPT-11) в комбинации с эффективным количеством комбретастатина для лечения солидных опухолей, где указанный комбретастатин имеет следующую формулу: 2. Комбинация по п.1, где указанная солидная опухоль является аденокарциномой ободочной кишки. ...
Предыдущий патент: Тиено[2,3-b]пиридиндионовые активаторы амфк и их терапевтическое применение
Следующий патент: Способы улучшения жизнеспособности растений
Случайный патент: Статор электрической машины