Производные 2-арил-4-метилпентановой кислоты в качестве модуляторов γ-секретазы



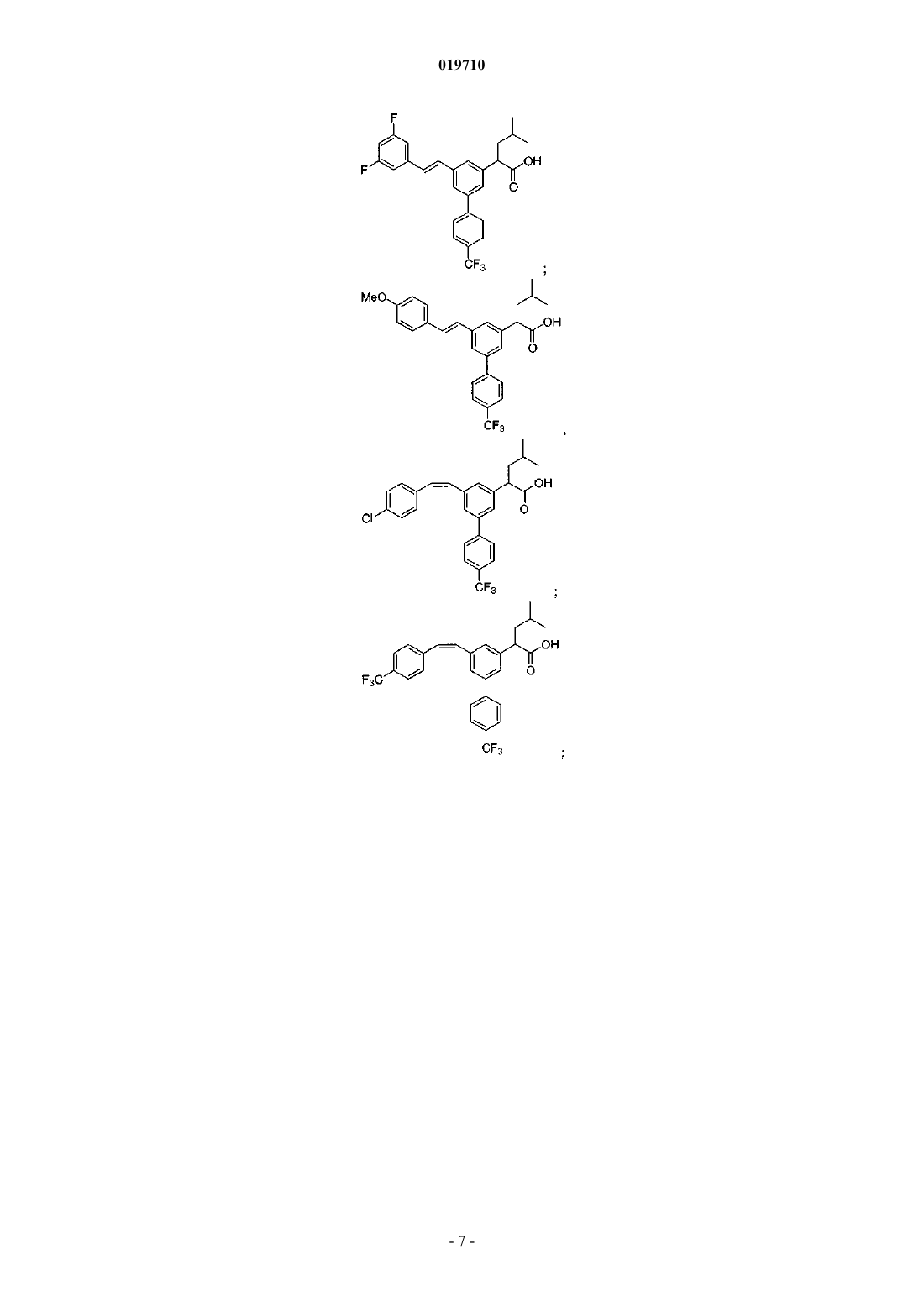
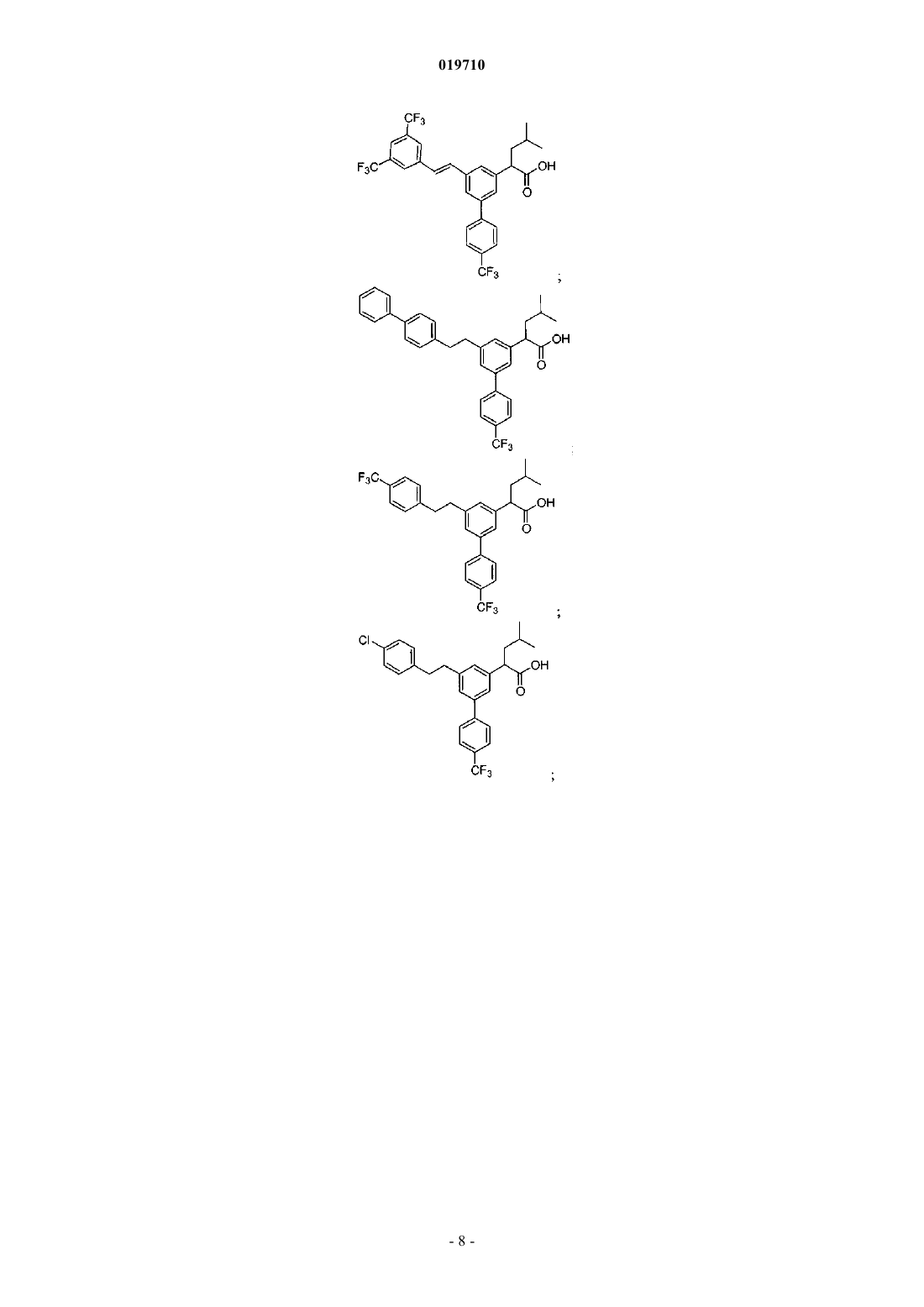



















Формула / Реферат
1. Соединение с общей формулой (I)

где А представляет собой фенил, пиридил или бифенил;
X представляет собой СН2, СН2СН2, С(O), СН=СН, СºС или СНОН;
R1 представляет собой изо-С4Н9;
R3 представляет собой Н, CF3, F, Cl, ОСН3, С(1-4)алкил или CN;
R4 представляет собой Н, F, Cl или CF3;
R5 и R6 независимо выбирают из группы, включающей CF3, H, F, Cl;
R7 и R8 представляют собой Н;
R9 представляет собой Н,
и его фармацевтически приемлемые соли.
2. Соединение по п.1, где
R1 представляет собой изо-С4Н9;
R4 выбирают из группы, включающей CF3, H, F, Cl;
R5 и R6 независимо выбирают из группы, включающей CF3, H, F, Cl;
R7 и R8 представляют собой Н;
R9 представляет собой Н,
и его фармацевтически приемлемые соли.
3. Соединение по п.2, где
R3 представляет собой Н, CF3, F, Cl, OCH3, С(1-4)алкил или CN;
R4, R5, R6 представляют собой Н, CF3, Cl и F,
и его фармацевтически приемлемые соли.
4. Соединение по п.3, где R9 представляет собой Н, и его фармацевтически приемлемые соли.
5. Соединение по п.4, где
R1 представляет собой СН2СН(СН3)2;
R4 и R5 представляют собой CF3, Cl, F или Н;
R6 представляет собой CF3;
R7 и R8 представляют собой Н,
и его фармацевтически приемлемые соли.
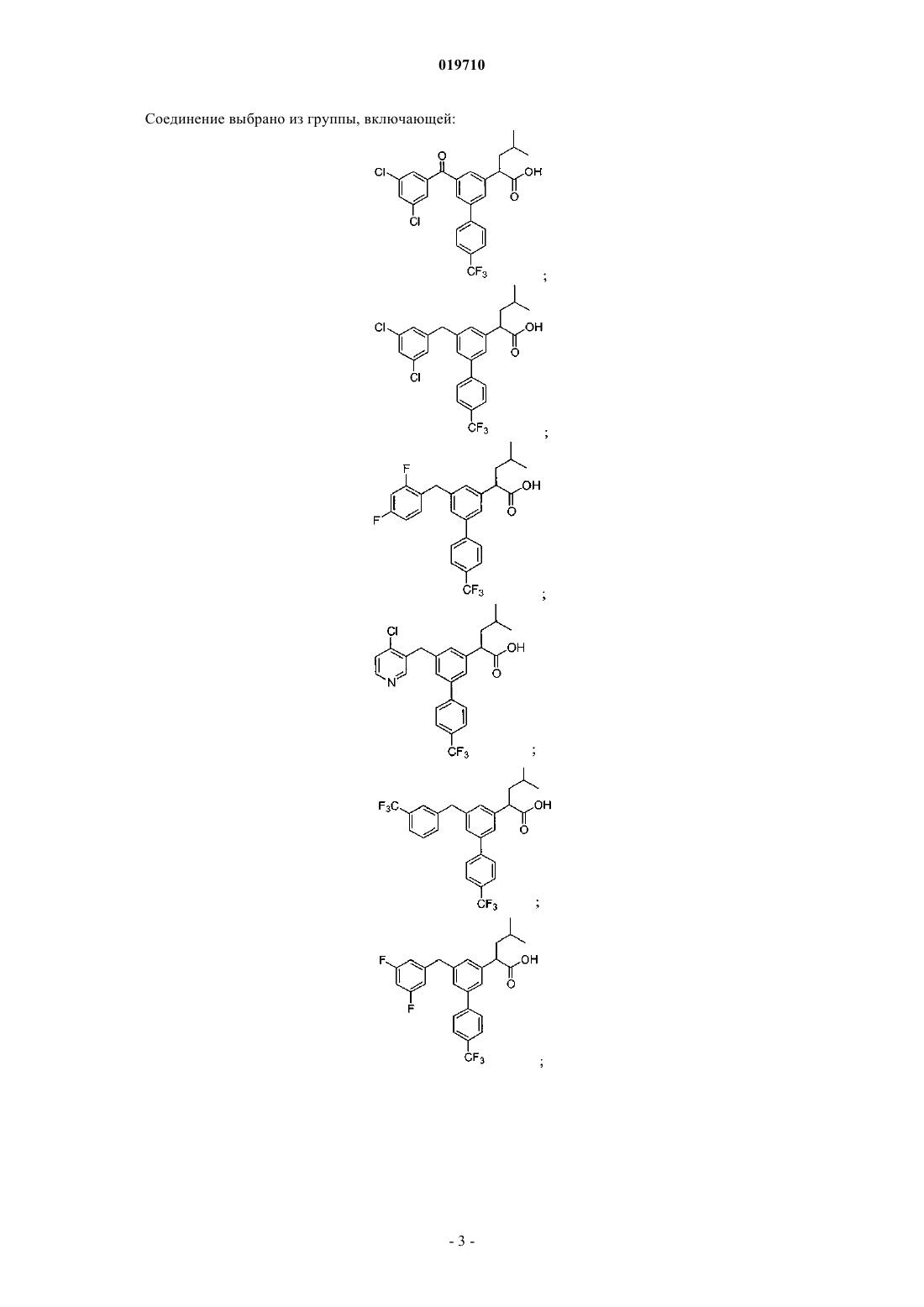
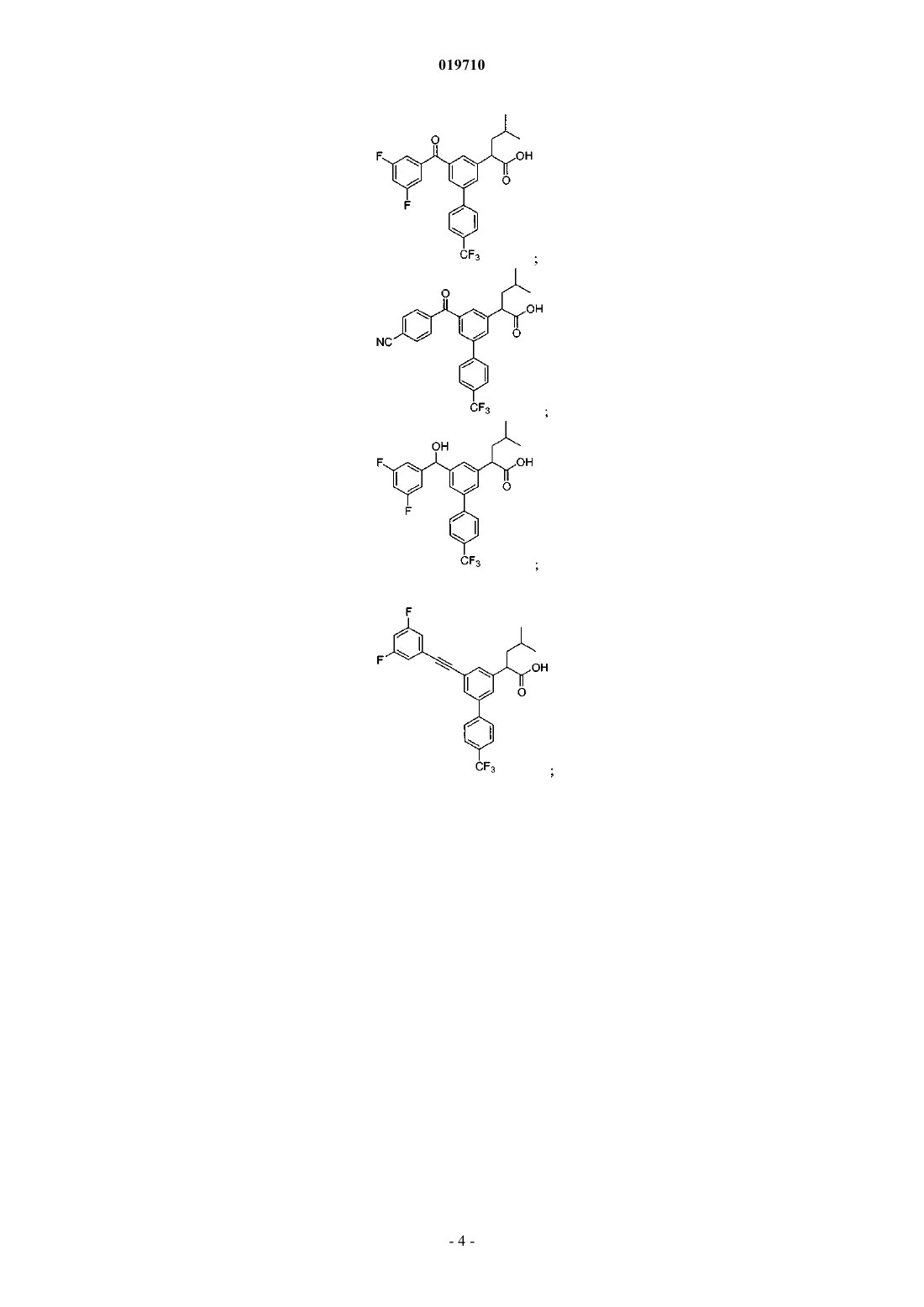
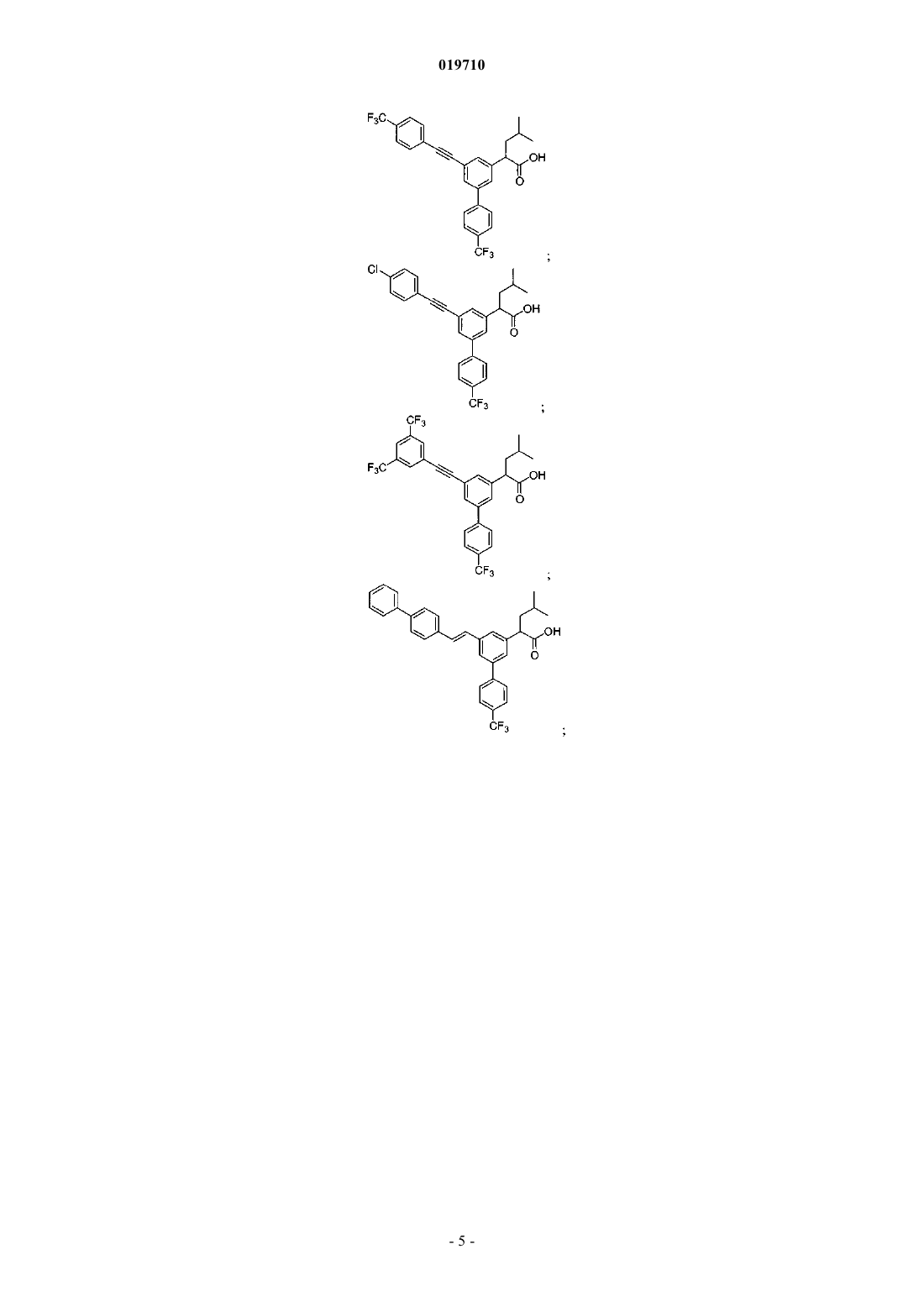
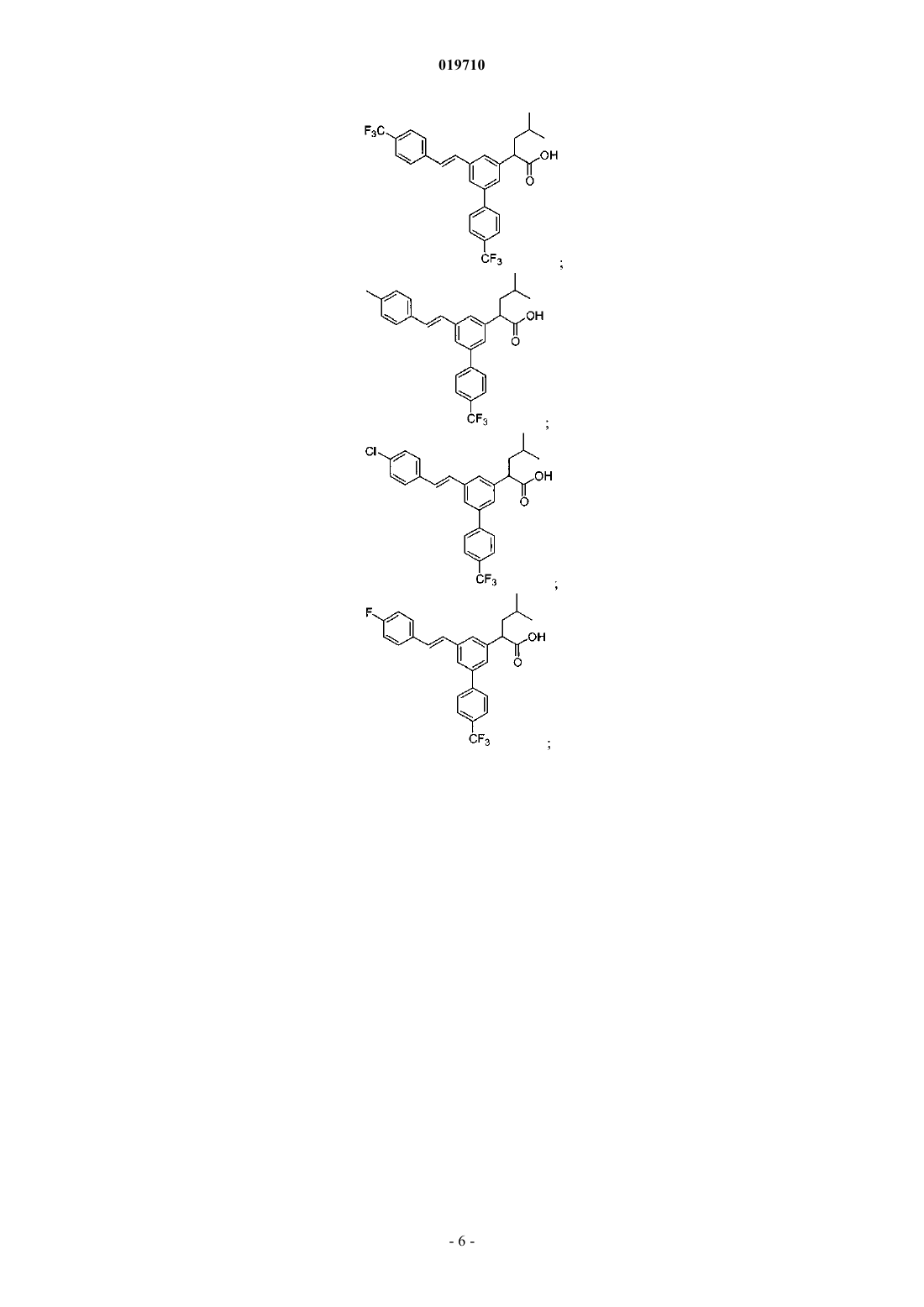
6. Соединение, выбранное из группы, включающей:








и его фармацевтически приемлемые соли.
7. Фармацевтическая композиция для лечения млекопитающего, обладающая активностью модулятора γ-секретазы, содержащая соединение по любому из пп.1-6 в форме смеси с инертным носителем.
8. Способ лечения млекопитающего путем модуляции γ-секретазы, включающий введение указанному млекопитающему терапевтически эффективного количества соединения по любому из пп.1-6.
9. Способ лечения заболевания, связанного с повышенным уровнем образования Аβ42 у млекопитающего, включающий введение указанному млекопитающему терапевтически эффективного количества соединения по любому из пп.1-6.
10. Способ по п.9, в котором указанное заболевание представляет собой болезнь Альцгеймера.
Текст
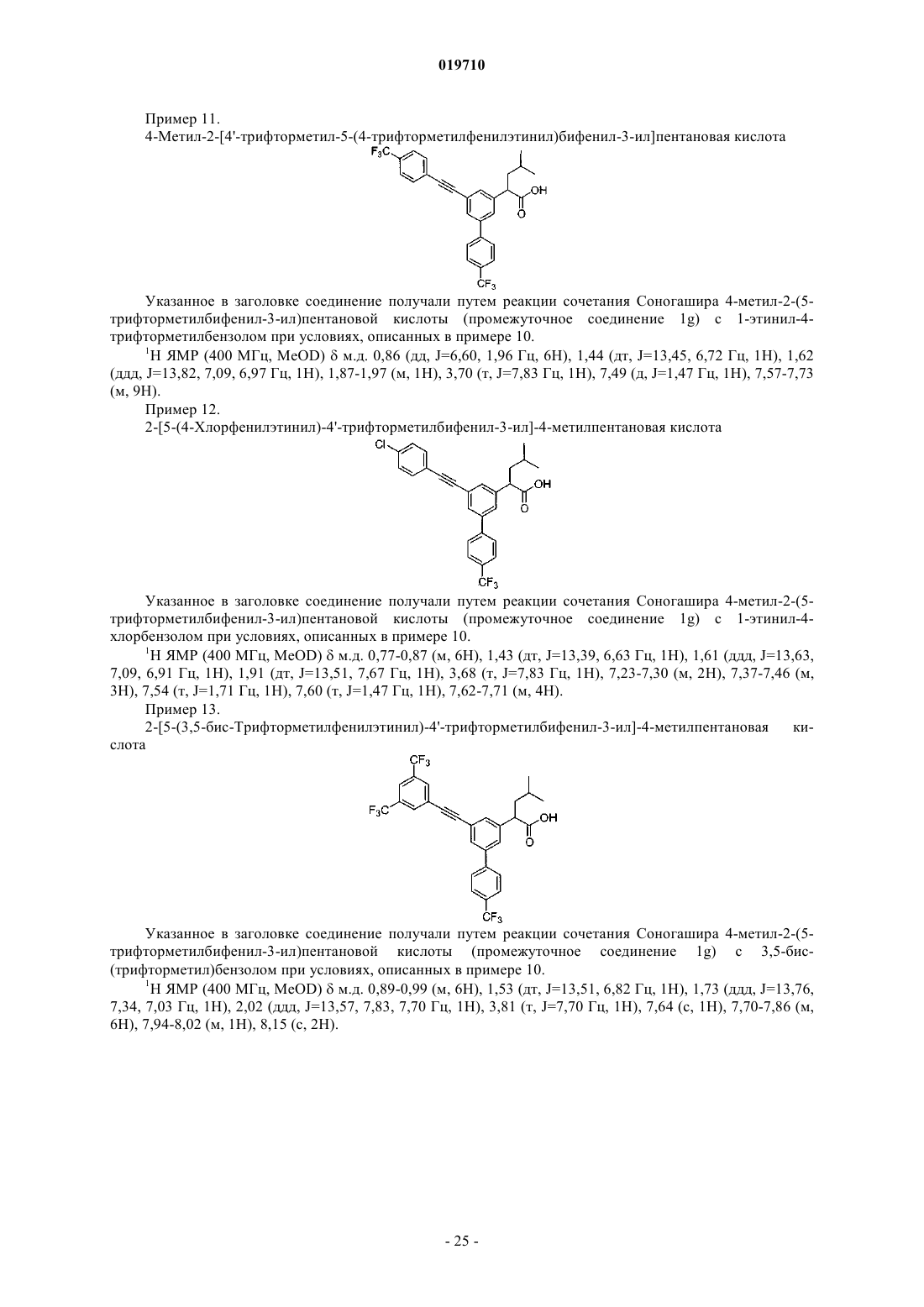
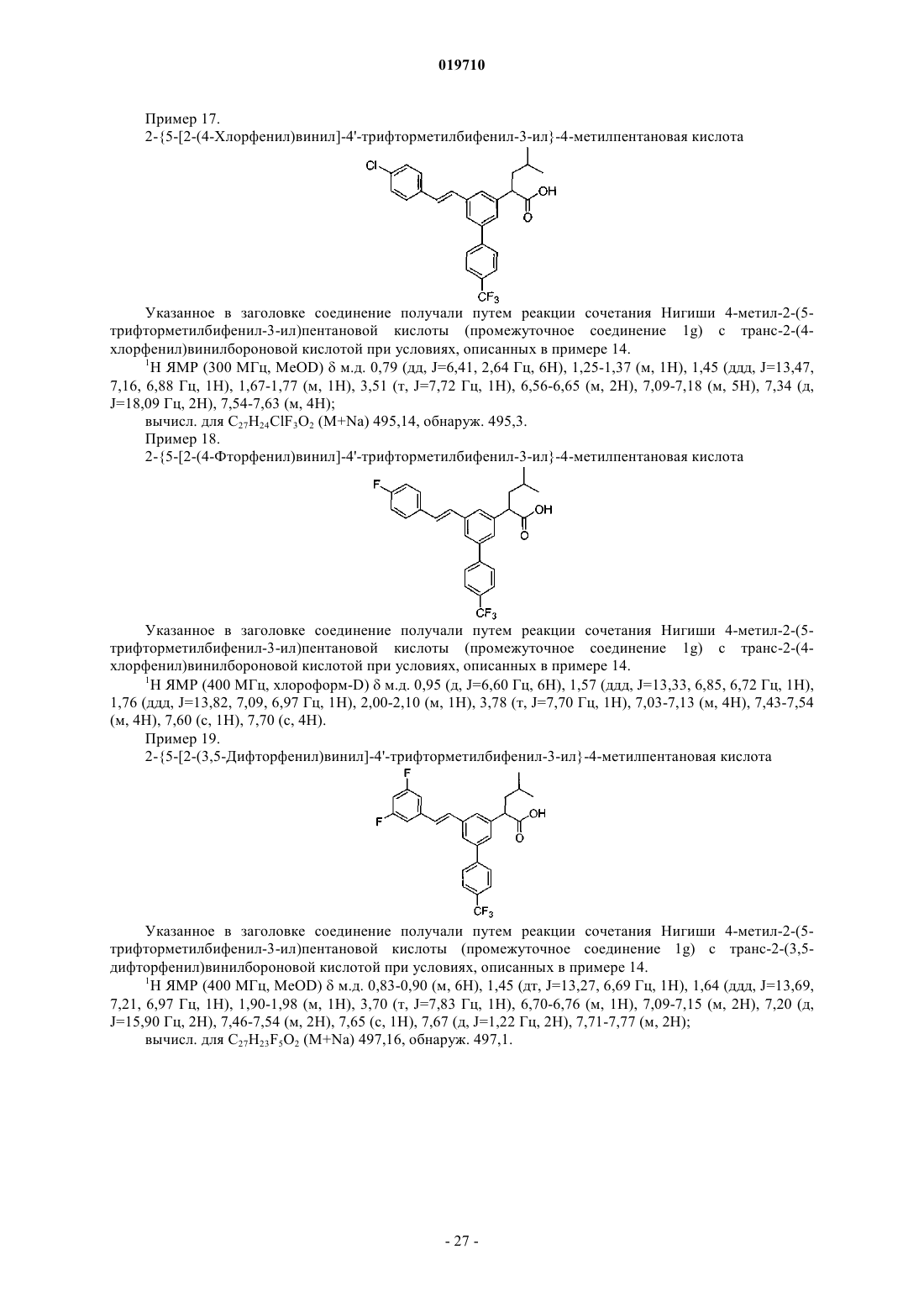
(I) могут применяться для лечения заболеваний, связанных с активностью -секретазы, включая болезнь Альцгеймера Хо Чих Юнг (US) Медведев В.Н. (RU) Перекрестные ссылки на смежные изобретения Данное заявление является притязанием на приоритет при заполнении предварительной заявки сер.60/981209, США, зарегистрированной 19 октября 2007 г. Полная информация соответствующей вышеупомянутой патентной заявки приводится здесь в качестве ссылки для любых целей. Область изобретения Настоящее изобретение связано с использованием соединений, имеющих общую формулу I, определения А, X, R1, R3, R4, R5, R6, R7, R8 и R9 приведены в настоящей спецификации. Соединения с формулой I могут применяться для лечения заболеваний, связанных с активностью -секретазы, включая болезнь Альцгеймера. Вводная информация об изобретении Болезнь Альцгеймера (БА) - прогрессирующее нейродегенеративное нарушение, характеризующееся потерей памяти, познавательной способности и устойчивости поведения. БА поражает 6-10% населения в возрасте старше 65 лет и до 50% в возрасте старше 85 лет. Это самая распространенная причина деменции и третья по частоте причина смерти, после сердечно-сосудистых и раковых заболеваний. В настоящее время эффективного лечения БА не существует. Общая сумма расходов, связанных с БА в США, превышает 100 млрд USD ежегодно. Этиология БА сложна, однако известны определенные факторы риска ее развития, включающие (1) возраст, (2) семейный анамнез и (3) травмы головы; прочие факторы включают токсины окружающей среды и низкий уровень образования. Специфические нейропатологические повреждения лимбической коры и коры больших полушарий включают внутриклеточные нейрофибриллярные клубки, состоящие из гиперфосфорилированного тау-белка и внеклеточные отложения фибриллярных агрегатов амилоидных бета-пептидов (амилоидные бляшки). Важным компонентом амилоидных бляшек являются бета-амилоидные пептиды (А-бета, Абета или A) различной длины. Считается, что основной причиной формирования бляшек является один из вариантов означенных пептидов A1-42 (Абета-42). Другой вариант - пептид A1-40 (Абета-40). Бета-амилоид представляет собой продукт протеолиза бета-амилоидного белка-предшественника (бета-АРР или АРР). Наследственные рано возникающие аутосомно-доминантные формы БА связаны с миссенс-мутациями гаммаамилоидного белка-предшественника (гамма-АРР или АРР) и в белках пресенилинах 1 и 2. У некоторых больных поздно возникающие формы БА коррелировались со специфическим аллелем гена аполипопротеина Е (АпоЕ) и, как обнаружилось совсем недавно, с мутацией гена альфа 2-макроглобулина, которая имеет место по крайней мере у 30% больных БА. Несмотря на неоднородность, все формы БА характеризуются сходными патологическими изменениями. Генетический анализ дает самые лучшие возможности логичного подхода к терапевтическому лечению БА. Все известные к настоящему времени мутации количественно или качественно затрагивают производство амилоидогенных пептидов, известных как Абета-пептиды (A), в частности A42, и с высокой вероятностью подтверждают "гипотезу амилоидного каскада" (Tanzi and Bertram, 2005, Cell 120, 545). Вероятная связь между образованием A пептида и патологией БА подчеркивает необходимость лучшего понимания механизмов образования A и подтверждает обоснованность терапевтического подхода, направленного на модуляцию содержания A. Высвобождение A пептидов модулируется по крайней мере двумя протеолитическими агентами,известными как - и -секретаза и отщепляющими соответственно фрагменты с N-конца (связь Met-ASP) и С-конца (остатки 37-42) пептида А. В секреторном пути имеется подтверждение того, что -секретаза отщепляет первый из указанных, результатом чего является секреция s-APP (s) и удержание карбоксиконцевого остатка (CTF) с молекулярной массой 11 кДа. Считается, что последний дает начало Апептидам после расщепления -секретазой. У больных с определенными мутациями особого белка (пресинилина) избирательно повышается содержание более длинной изоформы, A42, и такие мутации соотносятся с возникновением семейной болезни Альцгеймера в более раннем возрасте. Таким образом, по мнению многих исследователей, A42 является основным виновником патогенеза болезни Альцгеймера. К настоящему времени стало очевидно, что активность -секретазы нельзя приписать одному конкретному белку; фактически, она обусловлена совокупностью нескольких белков.-секретазная активность присуща белковому комплексу, состоящему по крайней мере из четырех компонентов: гетеродимер пресенилина (ПС), никастрин, aph-1 и pen-2. Гетеродимер ПС состоит из фрагментов ПС с амино- и карбоксильными концами, образовавшихся в результате эндопротеолиза белка-предшественника. Две аспартатные группы каталитического участка расположены на поверхности этого гетеродимера. Недавно было высказано предположение о том, что никастрин служит в качестве рецептора субстрата -секретазы. Функции прочих членов семейства -секретаз неизвестны, но все они необходимы для активности (Steiner, 2004. Curr. Alzheimer Research 1(3): 175-181). Таким образом, несмотря на то что молекулярный механизм второй стадии расщепления остается неясным до настоящего времени, -секретазный комплекс стал одной из основных мишеней в поиске соединений для лечения болезни Альцгеймера. Были предложены различные стратегии воздействия на -секретазу при болезни Альцгеймера, от непосредственного влияния на каталитический центр до разработки субстрат-специфичных ингибиторов и модуляторов -секретазной активности (Marjaux et al., 2004. Drug Discovery Today: Therapeutic Strategies, Vol. 1, 1-6). Соответственно, был описан ряд соединений, мишенями которых являются секретазыPatents 14, 1403-1420). Действительно, этот результат был недавно подтвержден биохимическими исследованиями, показавшими воздействие определенных НСПВП на -секретазу (Weggen et al. (2001), Nature 414, 6860, 212 иWO 01/78721 и US 2002/0128319; Morihara et al. (2002), J. Neurochem. 83, 1009; Eriksen (2003), J. Clin. Invest. 112, 440). Потенциальными ограничениями применения НСПВП для лечения БА является их ингибирующее действие на циклооксигеназы, способное привести к нежелательным побочным явлениям, и низкое проникновение в ЦНС (Peretto et al., 2005, J. Med. Chem. 48, 5705-5720). Таким образом, существует сильная потребность в новых соединениях, способных модулировать активность -секретазы, тем самым открывая новые пути в лечении болезни Альцгеймера. Цель настоящего изобретения - создание таких соединений. Краткое описание изобретения Изобретение включает соединения, имеющие общую формулу (I)X представляет собой СН 2, СН 2 СН 2, С(О), СН=СН, СС или СНОН;R5 и R6 независимо выбирают из группы, включающей CF3, H, F, Cl;R9 представляет собой Н,и его фармацевтически приемлемые соли. Подробное описание изобретения Изобретение включает соединения, имеющие общую формулу (I)X представляет собой СН 2, СН 2 СН 2, С(O), СН=СН, СС или СНОН;R5 и R6 независимо выбирают из группы, включающей CF3, H, F, Cl;R9 представляет собой Н,и его фармацевтически приемлемые соли. Соединение выбрано из группы, включающей: и его фармацевтически приемлемые соли. В другом осуществлении изобретение относится к способу лечения заболевания, сопровождающегося повышенным уровнем образования А 42, у млекопитающих, включающему введение млекопитающим терапевтически эффективного количества соединения формулы I. Специалист в этой области может видеть, что соединения формулы I могут иметь один или более асимметричных углеродных атомов в своей структуре. Это предполагает, что настоящее изобретение включает формы соединений, состоящие из одного энантиомера, рацемические смеси и смеси с избытком какого-либо энантиомера. Некоторые из соединений изобретения и/или их солей существуют в форме разных стереоизомеров. Все эти формы являются предметом данного изобретения. Ниже описаны примеры солей соединений, соответствующих настоящему изобретению. Приведенный ниже список различных солей не может рассматриваться как полный и не ограничивается перечисленным. Соединения, относящиеся к изобретению и содержащие одну или более кислотных групп, могут использоваться соответственно, как и их соли щелочных металлов, соли щелочно-земельных металлов или аммония. Более точные примеры таких солей включают соли натрия, калия, кальция, магния, а также соли аммония или органических аминов, например этиламин, этаноламин, триэтаноламин или аминокислоты. Термин "фармацевтически приемлемый" означает одобренный контрольными органами, например ЕМЕА (Европа), и/или FDA (США), и/или другими национальными контрольными органами, для использования в лечении живых существ, предпочтительнее людей. Соответствующие соли компонентов в соответствии с изобретением можно получить традиционными методами, известными специалистам, например смешиванием с органическими или неорганическими основаниями в растворителе или дисперсной среде либо путем катионного обмена с другими солями. Кроме того, изобретение включает все соли соединений, относящихся к изобретению, которые из-за низкой физиологической совместимости не подходят для непосредственного применения в качестве фармацевтических препаратов, но которые можно использовать, например, в качестве промежуточных компонентов в химических реакциях или для приготовления фармацевтически приемлемых солей либо для изучения способности к модулированию -секретазной активности соединения, относящегося к изобретению, любым подходящим способом, например in vitro. Изобретение также связано с теми соединениями, которые предназначаются для использования в качестве медикаментов. Соединения соответствуют определению выше, кроме того, относительно медикаментов, осуществления, описанные ниже, относительно применения изобретения, т.е. состав, применение и сочетания, также относятся к этому аспекту изобретения. В частности, соединения, относящиеся к изобретению, подходят для лечения болезни Альцгеймера. Более подробная информация об использовании приведена ниже. Соединения могут использоваться для модуляции активности -секретазы. Использующийся в данном документе термин "модуляция -секретазной активности" относится к воздействию на превращение АРР -секретазного комплекса. Это предпочтительно относится к эффекту,при котором общая скорость превращения АРР остается в основном такой же, как без применения указанных соединений, но относительное количество продуктов изменяется, предпочтительнее сопровождаясь снижением образования A42-пептида. Например, могут образовываться разные формы Абета (Абета-38 или другие пептидные соединения с более короткой аминокислотной цепью вместо Абета-42) или разное относительное количество продукта (например, другое соотношение Абета-40 и Абета-42, предпочтительнее повышенное).-секретазную активность можно измерить путем оценки превращения АРР, например, определив количество образовавшихся Абета пептидов, прежде всего Абета-42 (см. раздел "Примеры" ниже). Ранее было показано, что -секретазный комплекс также участвует в превращениях белка Notch. Это сигнальный белок, играющий решающую роль в процессах развития (например, обзор в SchweisguthF. (2004), Curr. Biol. 14, R129). Что касается применения указанных соединений для модуляции -секретазной активности, особенно благоприятным представляется отсутствие влияния на активность -секретазы в отношении Notchбелка во избежание возможных нежелательных побочных явлений. Таким образом, предпочтительными являются соединения, не влияющие на активность секретазного комплекса в отношении Notch-белка. В контексте изобретения "влияние на активность в отношении Notch-белка" включает как ингибирование, так и активацию этой активности с определенным коэффициентом. Соединение определяется как не оказывающее влияния на активность в отношении Notch-белка, если этот коэффициент менее 20, предпочтительнее менее 10, еще предпочтительнее менее 5 и предпочтительнее всего менее 2, при определении соответствующим способом, как описано Shimizu et al. (2000),Mol. Cell. Biol, 20: 6913 при концентрации 30 мкмоль. Такая модуляция -секретазной активности возможна, в частности, у животных, таких как млекопитающие. В качестве примера можно привести таких млекопитающих, как мыши, крысы, морские свинки,обезьяны, собаки и кошки. Модуляция возможна также у человека. В конкретном осуществлении изобретения указанная модуляция производится in vitro или в культуре клеток. Как известно специалистам в этой области, имеется несколько способов in vitro с культурами клеток. В качестве примера можно привести способ определения С-конечных фрагментов АРР в клеточных линиях трансгенных животных путем вестерн-блоттинга, включая, среди прочих, описанные Yan et al.,1999, Nature 402, 533-537. Пример способа определения -секретазы in vitro описан в WO 03/008635. В этом способе соответствующий пептидный субстрат добавляется к препарату -секретазы и измеряется способность к расщеплению субстрата. Концентрацию разных продуктов расщепления -секретазой (A-пептиды) можно определить разными способами, известными специалистам в этой области. Примеры таких способов включают определение пептидов способом масс-спектрометрии или обнаружение с помощью антител. Примеры способов анализа состава растворимых A-пептидов в среде для культур клеток и биологических жидкостях включают, среди прочих, описанные в работе Wang et al., 1996, J. Biol. Chem. 271,31894-31902. В этом способе используется сочетание иммунопреципитации Абета-пептидов специфическими антителами и обнаружение и количественное определение пептидных молекул методом времяпролетной масс-спектрометрии с ионизацией лазерной десорбцией с использованием матрицы. Примеры ELISA-методов измерения образования Абета-40- и Абета-42-пептидов включают, среди прочих, описанные в работе Vassar et al., 1999, Science 286, 735-741. Дальнейшая информация описана,например, в работе N. Ida et al. (1996), J. Biol. Chem. 271, 22908, and M. Jensen et al. (2000), Mol. Med. 6,291. Подходящие антитела можно приобрести, например, в компании Genetics Company, Inc., Швейцария. Тест-системы на основе антител выпускает также компания Innogenetics, Бельгия. Для этих методов можно использовать клетки, экспрессирующие -секретазный комплекс, а также клетки после трансфекции, временно или постоянно экспрессирующие некоторые или все интеракторы-секретазного комплекса. Для таких исследований подходят многочисленные имеющиеся клеточные линии, известные специалистам. Особенно хорошо подходят клетки и клеточные линии нейронного или глиального происхождения. Кроме того, могут использоваться клетки и ткани мозга, а также их гомогенаты и препараты мембран (Xia et al., 1998, Biochemistry 37, 16465-16471). Такие исследования можно проводить, например, для изучения влияния соединений, относящихся к изобретению, в разных экспериментальных условиях и конфигурациях. Кроме того, такие анализы могут проводиться в рамках функциональных исследований секретазного комплекса. Например, один или более интеракторов (дикого типа или несущих определенные мутации и/или модификации) -секретазного комплекса животного, предпочтительно млекопитающего, или, предпочтительнее всего, человека, могут экспрессироваться в определенных клеточных линиях, что дает возможность изучения эффекта соединений, относящихся к изобретению. Мутантные формы взаимодействующих компонентов могут быть либо мутантными формами, описанными у определенных животных, предпочтительнее млекопитающих и наиболее предпочтительно людей, или мутантными формами, не описанными ранее. Модификации интеракторов -секретазного комплекса включают физиологическую модификацию этих интеракторов и другие модификации, описанные как модификации белков в биологической системе. Примеры таких модификаций включают, среди прочих, гликозилирование, фосфорилирование, фенилирование, миристилирование и фарнезилирование. Кроме того, соединения, относящиеся к настоящему изобретению, могут использоваться для производства медикаментов для модуляции -секретазной активности. Активность -секретазы можно регулировать разными способами, что будет сопровождаться изменением профиля различных A-пептидов. Соответствующие дозировки, пути введения, формы и др. описаны ниже. Далее, изобретение относится к применению компонентов формулы I для лечения заболевания, связанного с повышенным уровнем образования А 42. Заболевания, сопровождающиеся повышением образования Абета-пептида и его отложением в тканях головного мозга, включают болезнь Альцгеймера(БА), церебральную амилоидную ангиопатию, мультиинфарктную деменцию, деменцию боксеров или синдром Дауна; предпочтительно БА. Использующийся в данном документе термин "лечение" относится ко всем процессам, направленным на замедление, прерывание, приостановку или прекращение прогресса болезни, но не обязательно означает полное устранение всех симптомов. Использующийся в настоящем документе термин "повышенный уровень образования А 42" относится к состоянию, при котором образование A42-пептида возрастает в результате общего повышения превращения АРР или предпочтительнее в результате изменения характера превращения АРР по сравнению с АРР дикого типа и непатологической ситуацией. Как указано выше, такое повышение уровня образования A42 является отличительным признаком развития или наличия болезни Альцгеймера. Одно из преимуществ соединений или части соединений настоящего изобретения может заключаться в усиленном проникновении в ЦНС. Кроме того, изобретение относится к фармацевтической композиции, включающей соединения формулы I в форме смеси с инертным носителем. Модуляторы -секретазы, полученные из соединений формулы I, могут входить в состав фармацевтических препаратов, включающих соединение формулы I в форме смеси с инертным носителем, являющимся носителем для фармацевтически активных веществ. Термин "носитель" относится к растворителям, адъювантам, вспомогательным веществам или жидкости, с которой вводится соединение. Такими носителями могут быть стерильные жидкости, например вода или масла, включая производные нефти, животные, растительные или синтетические, включающие,среди прочих, арахисовое масло, соевое масло, минеральное масло, кунжутное масло и т.п. Для внутренних форм предпочтительным носителем является вода. Для внутривенных форм предпочтительными носителями являются физиологический раствор и водный раствор декстрозы. Солевые физиологические растворы, а также водные растворы декстрозы и глицерина являются предпочтительными жидкими носителями для инъекционных форм. Подходящие вспомогательные вещества для фармацевтических препаратов включают крахмал, глюкозу, лактозу, сахарозу, желатин, солод, рис, муку, уголь, силикагель,натрия стеарат, глицерина моностеарат, тальк, натрия хлорид, сухое снятое молоко, глицерин, пропилен,гликоль, воду, этанол и т.п. В состав, если требуется, можно также включить незначительные количества смачивающих или эмульгирующих добавок или буферных веществ для регулировки рН. Эти составы могут принимать формы растворов, суспензий, эмульсий, таблеток, пилюль, капсул, порошков, форм замедленного высвобождения и др. Возможны также формы свечей, с традиционными связующими компонентами и носителями, такими как триглицериды. Внутренние формы могут включать стандартные носители для применения в фармацевтике, например маннит, лактоза, крахмал, стеарат магния, сахарин натрия, целлюлоза, карбонат магния и др. Примеры подходящих носителей описаны в работе Remington'sPharmaceutical Sciences, E.W. Martin. Такие составы будут содержать терапевтически эффективное количество соединения, предпочтительнее в очищенной форме, вместе с подходящим количеством носителя,для получения формы, удобной для введения больному. Состав должен соответствовать способу введения. Соединения, относящиеся к изобретению, а также их фармацевтически приемлемы соли, возможно,в сочетании с другими фармакологически активными компонентами подходят для лечения или профи- 11019710 лактики болезни Альцгеймера или ее симптомов. Такие дополнительные соединения включают препараты, улучшающие познавательную функцию, например ингибиторы ацетилхолинэстеразы (например, донепезил, такрин, галантамин, ривастигмин), антагонисты NMDA (например, мемантин), ингибиторыPDE4 (например, Арифло) или любые другие препараты, известные специалистам в данной области, для лечения или профилактики болезни Альцгеймера. Такие соединения также включают препараты, понижающие концентрацию холестерина, например статины (в частности, симвастатин). Эти соединения могут вводиться животным, предпочтительнее млекопитающим, в особенности людям, в качестве лекарств сами по себе, в смесях или в форме фармацевтических препаратов. Могут присутствовать консерванты и другие добавки, например антимикробные вещества, антиоксиданты, хелатирующие вещества, инертные газы и т.п. Все носители можно смешивать, по мере необходимости, с разрыхлителями, растворителями, гранулирующими добавками, смазочными веществами,связующими агентами и т.п. по традиционным технологиям. Далее, это изобретение дает способ лечения заболеваний, течение которых можно облегчить путем модуляции -секретазной активности, которая включает введение больному терапевтически эффективной дозы фармацевтического препарата немедленного действия. Использующийся в данном документе термин "субъект" включает, без ограничений, любое животное или экспериментально измененное животное с заболеванием, течение которого улучшается при модуляции активности -секретазы. Предпочтительной вариацией является такая, в которой субъектом является человек. Использующийся в настоящем документе термин "терапевтически эффективная доза" означает количество, достаточное для остановки, обращения или замедления прогресса заболевания. "Профилактическая эффективная доза" фармацевтического препарата соответствует количеству, достаточному для профилактики заболевания, т.е. исключения, облегчения или отсрочки его проявления. Существуют принятые способы определения терапевтических и профилактических эффективных доз для фармацевтических препаратов немедленного действия. Эффективную дозу фармацевтических препаратов для людей можно вычислить по результатам испытаний на животных. Известны различные системы доставки, которые можно использовать для введения компонента, относящегося к изобретению, при лечении болезни Альцгеймера или для модуляции активности секретазы, например инкапсуляция в липосомах, макрочастицах или микрокапсулах: если препарат не доставляется непосредственно в центральную нервную систему, предпочтительнее в головной мозг,лучше выбрать и/или изменить способы введения таким образом, чтобы компонент мог проникать через гематоэнцефалический барьер. Способы введения включают, среди прочих, внутрикожное, внутримышечное, внутрибрюшинное,подкожное, интраназальное, эпидуральное и оральное введение. Соединения могут вводиться любым удобным путем, например путем инфузии, болюсной инъекции, всасывания через эпителий или слизистую оболочку, а также вместе с другими биологически активными веществами. Введение может быть общим или местным. Кроме того, может оказаться желательным вводить лекарственные составы, относящиеся к изобретению, в центральную нервную систему любым подходящим путем, в том числе с помощью внутрижелудочковой или интратекальной инъекцией, возможно, через установленный в желудочек катетер, соединенный с резервуаром, например Оммайа. Возможно также введение через легкие, например, с помощью ингалятора плп аэрозольного аппарата, и создание лекарственной формы с аэрозолеобразующим компонентом. Модуляторы активности -секретазы из соединений формулы I могут доставляться в организм в пузырьках, в частности липосомах (Langer (1990), Science 249, 1527). Модуляторы активности -секретазы из соединений формулы I могут доставляться в организм с помощью системы контролируемого высвобождения. Можно также использовать насос (Sefton (1987),CRC Crit. Ref. Biomed. Eng. 14, 201; Buchwald et al. (1980), Surgery 88, 507; Saudek et al. (1989), N. Engl. J.Med. 321, 574). В другом осуществлении можно использовать полимерные материалы (Ranger and Peppas(1989), Ann. Neurol. 25, 351; Howard et al. (1989), J. Neurosurg. 71, 858). В другом осуществлении возможно использование системы контролируемого высвобождения в непосредственной близости от органамишени, в данном случае головного мозга, таким образом, требуется только небольшая часть системной дозы (Goodson, 1984, In: Medical Applications of Controlled Release, supra, Vol. 2, 115). Прочие системы контролируемого высвобождения обсуждаются в обзоре Langer (1990, Science 249, 1527). При выборе подходящего способа введения специалисты также учитывают способы, использующиеся для других известных препаратов против болезни Альцгеймера. Например, арисепт/донепезил и когнекс/такрин (все из которых относятся к ингибиторам ацетилхолинэстеразы) принимаются внутрь, а аксура/мемантин (антагонист рецептора NMDA) выпускаются как в форме таблеток/жидкости, так и в форме раствора для внутривенного введения. Кроме того, специалисты учитывают имеющиеся данные о способах введения препаратов семейст- 12019710 ва НСПВП во время клинических испытаний и других исследований влияния этих лекарств на течение болезни Альцгеймера. При выборе подходящей дозировки специалисты подбирают дозу, не проявлявшую токсических свойств при доклинических и клинических испытаниях и согласующуюся с приведенными ранее значениями, однако возможны отклонения. Точная доза будет также зависеть от пути введения и серьезности заболевания и должна подбираться лечащим врачом в зависимости от обстоятельств в каждом конкретном случае. Однако допустимый диапазон доз для внутривенного введения обычно составляет около 20-500 мкг активного соединения на 1 кг массы тела. Дозировка для интраназального введения обычно составляет около 0,01-1 мг/кг массы тела. Эффективные дозы можно вычислить экстраполяцией кривых доза-ответ, полученных in vitro или в экспериментах на животных. Примером животной модели является трансгенный штамм мыши Tg2576, содержащий разновидность белка АРР 695, подвергнутый двойной мутации (KM670/671NL). Для справки см. патентLehman et al. (2003), Neurobiol. Aging 24, 645. Для подбора дозировки, соответствующей выбранному курсу лечения, специалисты в этой области могут использовать существенные данные нескольких проведенных ранее исследований. Опубликованы результаты многочисленных исследований, описывающие влияние этих молекул на активность -секретазы. Это, в частности, следующие исследования: Lim et al. (2001), Neurobiol. Aging 22, 983; Lim et al. (2000), J. Neurosci. 20, 5709; Weggen et al. (2001), Nature 414, 212; Eriksen et al. (2003), J.Clin Invest. 112, 440; Yan et al. (2003), J. Neurosci. 23, 7504. Определения Термин "алкил" обозначает радикалы с линейной или разветвленной цепью длиной до 12 углеродных атомов, в основном до 6, если не указано иное, и включает, среди прочих, метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, пентил, изопентил, гексил, изогексил, гептил, октил,2,2,4-триметилпентил, нонил, децил, ундецил и додецил. Общее описание синтеза Следующее общее описание приведено только для ознакомления и никоим образом не ограничивает изобретение. Соединения формулы I, где X, A, R1, R3, R4, R5, R6, R7 и R8 определяются как входящие в формулу I,9a R представляет собой Н, могут быть получены гидролизом эфиров II в стандартной кислой или щелочной среде, включая реакцию с NaOH, при комнатной температуре, в течение нескольких часов, в соответствующей смеси растворителей, например вода, тетрагидрофуран (ТГФ) и метанол или этанол. Для иллюстративных целей эфиры II показаны с R9, являющимся алкилом, однако специалистам известно,что эфирный гидролиз действует для любых R9, указанных в формуле I Соединения IIa, где X является карбонильной группой, могут быть получены путем реакции соединений IIIa или IIIb с арилбороновой кислотой и моноксидом углерода в присутствии катализаторовPd(II), таких как Pd(dppf)2Cl2 и карбоната калия и иодида калия при повышенной температуре (60-150 С). Соединения IIb могут быть получены путем восстановления соединений IIa гидридными восстановителями, такими как борогидрид натрия. Альтернативно, соединения IIb могут быть получены путем добавления реактивов Гриньяра или реактивов лития (R9MgX или R9Li) к соединениям IIa при общих условиях алкилирования кетонов. Соединения IIc, IId и IIe могут быть получены путем реакции соединений IIIa или IIIb с арилацетилен-бороновыми кислотами в условиях Сузуки, например в водном растворе карбоната натрия вDME в присутствии Pd(PPh3)4 при повышенной температуре (60-180 С). Альтернативно, соединения IIc, IId и IId могут быть получены путем реакции соединений IIIa илиIIIb с хлоридами арил-винил-цинковой кислоты или арилметилен-цинковой кислоты в условиях Нигиши,например в ТГФ в присутствии каталитической концентрации Pd(dppf)2Cl2. Соединения IIf могут быть получены путем восстановления соединений IId и IIe в условиях каталитической гидрогенизации с использованием Pd-C, двуокиси платины или других катализаторов. Альтернативно, соединения IIc могут быть получены путем восстановления спирта IIb триметилсиланом во ТФУК-ТГФ или каталитической гидрогенизацией в кислой среде. Соединения IIIa могут быть получены путем реакции фенолов IV с трифторметансульфоновым ангидридом в DCM в присутствии основания, например пиридина, или триэтиламина при 0 С. Промежуточные продукты IIIb можно получить путем реакции фенолов IV с концентрированной HCl, HBr или HI при повышенной температуре (от 25 до 120 С). Альтернативно, соединения IIIb можно получить в мягких условиях путем обработки соответствующих трифталатов IIIa пинаколбораном в диоксане в присутствии триэтиламина и катализатора PdCl2 для получения эфиров пинаколбороната, на которые затем воздействуют галидами меди (II) в смеси метанола и воды; методика описана Несмеянов и др. (Chem Ber. 1960, 2729). Описанные выше эфиры пинаколбороната можно также получить с помощью Nal в водном ТГФ в присутствии хлораминов-Т для получения арилйодидов, как описано J.W. Huffman et al. Соединения IV можно получить дебензилированием соединений V путем гидрогенизации в спирте,например МеОН или EtOH в присутствии Pd-C. Возможно дебензилирование другими методами, например BBr3 в дихлорметане, NaCN в DMSO/120-200 С или LiCl в DMF/120-200 С. Соединения V могут быть получены алкилированием соединений VE путем взаимодействия с галогенопроизводными алкилов или алкенилов. Воздействие на соединения VI в ТГФ или другом апротонном растворителе основанием, например лития бис-(триметилсилил)амидом, натрия бис(триметилсилил)амидом или лития диизопропиламидом, при -78 С с последующим добавлением электрофильных компонентов, например галогенопроизводных алкилов или алкенилов, позволяет получить алкилированные соединения V. Альтернативно, соединения VI могут быть получены из соединений VII путем реакции сочетания с арилбороновыми кислотами при условиях Сузуки в водном растворе карбоната натрия в DME в присутствии Pd(PPh3)4. Альтернативно, трифлаты можно превратить в эфиры бороната в условиях, описанных выше, а затем провести реакцию сочетания с арилборомидами или арилхлоридами для получения соединений VI. Промежуточные трифлатные соединения VII можно получить из соединений VIII с помощью триф- 14019710 Промежуточное соединение VIII можно получить путем монодебензилирования соединения IX. Избирательное монодебензилирование соединения IX возможно путем селективного гидрогенолиза соединения IX в этаноле или метаноле с добавлением 1,1 экв. основания, например, гидроксида натрия или калия, в присутствии катализатора Pd-C в шейкере Парра. Реакцию проводят до расхода 1 экв. водорода. Промежуточный продукт IX можно легко получить путем реакции метилового эфира 3,5 дигидроксифенилуксусной кислоты, соединения X (имеющегося в продаже) с бензилбромидом или карбонатом калия в DMF при комнатной температуре. Соединения формулы I имеют хиральный центру карбоксильной группы и могут существовать в форме одного или двух энантиомеров (или их смеси, с избытком одного из них или без). Показаны энантиомеры Ia (R энантиомер) и Ib (S энантиомер). Чистые энантиомеры Ia и Ib можно получить разделением на хиральной колонке. Энантиомеры Ia и Ib также можно разделить путем перевода их в соли аминов с последующей фракционной перекристаллизацией. Кроме того, энантиомеры Ia и Ib можно получить благодаря кинетическому разрешению рацемата соответствующих эфиров с помощью ферментов липаз,например, Amano lipase Ak, Amano lipase PS, Araano lipase A, Amano lipase M, Amano lipase F-15 Amanolipase G (производства Biocatalytics Inc) в водных и органических растворителях, например, водных смесях с DMF, DMSO, t-бутиловым эфиром или тритоном Х-100. Альтернативно, соединения Ia и Ib можно получить путем хирального синтеза. Соединения Ia илиIb можно получить из хиральных фенольных соединений IVa и IVb, как описано выше. Хиральные соединения IVa и IVb можно получить путем удаления хиральных вспомогательных групп и последующей этерификации из соединений XIIIa и XIIIb соответственно с гидроксидом лития и пероксидом водорода в водном растворе ТГФ. Соединения XIIIa и XIIIb можно получить путем дебензилирования соединения XIVa и XIVb соответственно с помощью гидрогенизации в спиртовом растворителе, например МеОН или EtOH, в присут- 15019710 Соединения XIVa и XIVb можно получить алкилированием соединений XVa и XVb соответственно с помощью подходящего алкилбромида, включая втор-бутилбромид или втор-бутенилбромид для присоединения R1 группы к -углеродному атому карбоксильной группы. Воздействие на соединения XVa иXVb в ТГФ или в другом апротонном растворителе с основанием, например лития бис(триметилсилил)амидом, натрия бис-(триметилсилил)амидом или лития диизопропиламидом, при -78 С с последующим добавлением электрофильных компонентов, втор-бутилбромида или вторбутенилбромида позволяет получить алкилированные соединения XIVa и XIVb соответственно. Соединения XVa и XVb можно получить из промежуточных соединений XVI путем сочетания с Rизомером 4-бензил-оксазолидин-она (XVIIa) или S-изомером 4-бензил-оксазолидин-она (XVIIb) по методике Эванса. Промежуточные продукты XVI можно получить путем реакции с пивалоилхлоридом, оксалилхлоридом или изопропилхлорформиатом в ТГФ в присутствии основания, например триэтиламина или N-метилморфолина, для получения смешанных ангидридов или кислых хлоридов, которые затем реагируют с солями лития XVIIa или XVIIb в ТГФ. Альтернативно, можно использовать другие хиральные вспомогательные группы для хирального синтеза соединений IVa и IVb, например псевдоэфедрин в условиях Майерса (J. Am. Chem. Soc. 1994,116, 9361-9362). Например, взаимодействие хлоропроизводных или ангидридов карбоновых кислот с (+) или (-) псевдоэфедрином дает соединения XVIIIa и XVIIIb. Затем амиды обрабатываются сильным основанием, например, лития диизопропиламидом в присутствии лития хлорида с последующим добавлением алкилирующего агента для получения алкилированных продуктов XIXa и XIXb. Хиральные фенольные соединения IVa и IVb можно получить также из соединений XIXa и XIXb путем удаления хиральной вспомогательной группы псевдоэфедрина в водном растворе серной кислоты и последующей обработки BBr3/DCM для удаления защитной бензильной группы. Кроме того, хиральные фенольные соединения XIIIa, XIIIb, XXa и XXb могут служить как хиральные промежуточные продукты для получения хиральных соединений Ia и Ib. Хиральные вспомогательные группы удаляются на конечной стадии синтеза в условиях, описанных выше. Соединения XXIa и XXIb можно получить из хиральных фенольных соединений XIIIa и XIIIb в условиях, подобных вышеописанным. Например, трифлаты XXIIa и XXIIb, полученные из фенольных соединений XIIIa и XIIIb путем реакции с трифторметилсульфониловым ангидридом в растворе пиридинметиленхлорида, могут дать сочетающиеся соединения XXIa и XXIb в условиях Бухвальда-Хартвига, как описано выше. При аналогичных условиях соединения XXIIIa и XXIIIb могут быть получены из компонентовXXIIa и XXIIb с помощью реакции с бензофенонимином в присутствии трифенилфосфина и каталитической концентрации тетракис-трифенилфосфина палладия (0), как упоминалось ранее. Восстановительное аминирование соединений XXIIIa и XXIIIb арилкарбоксиальдегидами или гетерокарбоксиальдегидами с последующим алкилированием азота через восстановительное аминирование или алкилирование при взаимодействии с галогенопроизводными алкилов с последующим удалением хиральных вспомогательных групп с использованием гидроксида лития и пероксида водорода в водном растворе ТГФ дает хиральные соединения Ia и Ib. Альтернативно, соединения XXIVa и XXIVb могут быть получены из XXIIa и XXIIb описанным выше способом с использованием цианида цинка и тетракис-трифенилфосфина палладия с последующим восстановлением цианистых соединений двуокисью платины в кислотно-спиртовой среде. Хиральные амины XXIVa и XXIVb могут использоваться для получения окончательных целевых соединений Ia и Ib способами, аналогичными описанным выше. Методики синтеза. Все реакции проводили в инертной атмосфере, если не указано иное. ЯМР-спектры получали с помощью прибора Bruker dpx400. ЖХ-МС проводили с помощью системы Agilent 1100 с колонкой ZORBAX SB-C18, 4,675 мм, 3,5 мкм, для метода А. Скорость протекания через колонку была 1 мл/мин, в качестве растворителей использовали воду и ацетонитрил (0,1% трифторуксусной кислоты), объем ввода 10 мкл. Длины волн были 254 и 210 нм. Методы описаны ниже. Смесь (3,5-дигидроксифенил)уксусной кислоты метилового эфира (по Aldrich, 70 г, 0,385 моль),бензилбромида (137 мл, 1,16 моль), калия карбоната (160 г, 1,16 моль) и ДМФА (1,5 л) под слоем N2 механически перемешивали в течение ночи при комнатной температуре. Полученную реакционную смесь вливали в 1,5 л ледяной воды при перемешивании. Осадок отделяли фильтрованием и промывали гептаном для удаления бензилбромида и получения указанного в заголовке соединения (123,7 г) в форме коричневого твердого вещества, которое высушивали на воздухе для следующей реакции. 1 Н ЯМР (CDCl3):3,60 (с, 2 Н), 3,71 (с, 3 Н), 5,05 (с, 4 Н), 6,60 (с, 3 Н), 7,35-7,50 (м, 10 Н); вычисл. для С 23 Н 22 О 4 (М+Н) 363,15, обнаруж. 363. Раствор (3,5-бис-бензилоксифенил)уксусной кислоты метилового эфира (50 г, 1,38 моль) и NaOH(6,6 г, 1,65 моль) в 1 л EtOH в присутствии 10% Pd-C гидрогенизировали в шейкере Парра до потребления 1 экв. водорода. Смесь подкисляли добавлением концентрированной HCl, а затем удаляли катализатор и растворитель, оставляя масляный осадок. Неочищенный продукт очищали на колонке с силикагелем ISCO с использованием EtOAC-гептана в качестве элюента (градиент от 10 до 75% EtOAc) до получения 25 г (выход 65%) соединения, указанного в заголовке. 1 К раствору 3-(бензокси-5-гидроксифенил)уксусной кислоты этилового эфира (74,4 г, 0,26 моль) в дихлорметане (700 мл) добавляли пиридин (62,5 мл, 0,78 моль). Смесь охлаждали до 0 С. К охлажденному раствору добавляли трифторметансульфоновый ангидрид (65,6 мл, 0,39 моль) в течение 1,5 ч, поддерживая внутреннюю температуру ниже 5 С и перемешивали еще 0,5 ч при 0 С. Эту реакционную смесь приливали к смеси 1 N HCl (420 мл) с водой и льдом (105 г) и перемешивали 0,5 ч. Водный слой экстрагировали дихлорметаном (2100 мл). Фракции промывали водой (2100 мл), насыщенным водным раствором NaHCO3 (2100 мл) и насыщенным солевым раствором (2100 мл). Органические компоненты высушивали (MgSO4) и концентрировали под вакуумом до получения красноватой жидкости (108 г), которую использовали на следующей стадии без дальнейшей очистки. Вычисл. C18H17F3O6S (М+Н) 419,07, обнаруж. 419,1. Смесь (3-бензилокси-5-трифторметансульфонилоксифенил)уксусной кислоты этилового эфира (108 г, 0,26 моль), 4-(трифторметил)фенилбороновой кислоты (55,6 г, 0,29 моль), 1,2-диметоксиэтана (1,1 л) и водного раствора Na2CO3 (2 М, 129 мл, 0,26 моль) механически перемешивали при одновременной продувке N2 при комнатной температуре в течение 10 мин. К этой системе добавляли Pd(Ph3)4 (480 мг, 0,42 ммоль) и нагревали до кипения с обратным холодильником (95 С) в течение 2,5 ч. Красно-коричневую смесь разводили EtOAc (0,5 л) и промывали насыщенным водным раствором NaHCO3 (3200 мл) и насыщенным солевым раствором (2200 мл). Органическую фракцию высушивали (Na2SO4) и концентрировали под вакуумом. Неочищенную смесь очищали на колонке ISCO для получения (5-бензилокси-4'трифторметилбифенил-3-ил)уксусной кислоты этилового эфира (107 г, 100%). 1 К раствору соединения 1d (4,9 г, 11,8 ммоль) в ТГФ (50 мл) при -78 С добавляли Li[N(SiMe3)2] (1 N в ТГФ, 14,2 мл, 14,2 ммоль) по каплям. Реакционную смесь перемешивали в течение часа при -78 С, а затем добавляли по каплям 3-бром-2-метилпропен (1,25 мл, 12,4 ммоль). Раствор медленно нагревали до-35 С и перемешивали при -35 С в течение 0,5 ч. Реакцию останавливали добавлением насыщенного раствора NH4Cl и экстрагировали EtOAc. Органические экстракты высушивали (Na2SO4), концентрировали и очищали с помощью колоночной хроматографии для получения соединения 1 е (5,1 г, 92%) в форме прозрачного масла. 1 Н ЯМР (400 мГц, хлороформ-D)м.д. 1,19-1,29 (м, 3 Н), 1,74 (с, 3 Н), 2,47 (м, 1 Н), 2,85 (м, 1 Н), 3,83 Смесь соединения 1 е (5,1 г, 10,9 ммоль), 10% Pd/C (500 мг) в EtOH (50 мл) гидрогенизировали под слоем Н 2 (275,8 кПа (40 фунтов на кв.дюйм) в шейкере Парра в течение 20 ч. Полученную реакционную смесь фильтровали через целитовый фильтр и концентрировали фильтрат для получения соединения,указанного в заголовке (4,2 г, 100%) в форме прозрачного масла. 1 Н ЯМР (300 мГц, хлороформ-D)м.д. 0,92 (д, J=6,6 Гц, 6 Н), 1,25 (м, 3 Н), 1,49-1,61 (м, 1 Н), 1,651,70 (м, 1 Н), 1,95-2,05 (м, 1 Н), 3,67 (т, J=7,7 Гц, 1 Н), 4,10-4,29 (м, 2 Н), 6,91 (с, 1 Н), 6,97 (т, J=2,0 Гц, 1 Н),7,08 (с, 1 Н), 7,65 (с, 4 Н); вычисл. для C21H23F3O3 (М+Н) 381,16, обнаруж. 381. К раствору соединения 1f, 2-(5-гидрокси-4'-трифторметилбифенил-3-ил)-4-метилпентановой кислоты этилового эфира (2,8 г, 7,36 ммоль), и N-фенил-бис-(трифторметансульфонимида) (3,16 г, 8,83 ммоль) в ТГФ (30 мл) под слоем N2 добавляли Et3N (2,05 мл, 14,7 ммоль). Реакционную смесь нагревали с обратным холодильником в течение ночи. После охлаждения до комнатной температуры раствор концентрировали и очищали колоночной хроматографией для получения соединения, указанного в заголовке(3,7 г, 98%) в форме бесцветного густого масла. 1 Н ЯМР (400 мГц, хлороформ-D)м.д. 0,94 (дд, J=6,60, 1,47 Гц, 6 Н), 1,22-1,28 (м, 3 Н), 1,46-1,52 (м,1 Н), 1,69 (ддд, J=13,82, 7,09, 6,97 Гц, 1 Н), 1,98-2,06 (м, 1 Н), 3,75 (т, J=7,83 Гц, 1 Н), 4,10-4,21 (м, 2 Н), 7,31 Смесь соединения 1g (100 мг, 0,195 ммоль), 3,5-дихлорфенилбороновой кислоты (63 мг, 0,33 ммоль), Pd(dppf)2Cl2 (14,3 мг, 0,020 ммоль), K2CO3 (81 мг, 0,585 ммоль) и KI (97 мг, 0,585 ммоль) в анизоле (2 мл) при 85 С в атмосфере СО с использованием баллона, наполненного газом СО, нагревалась в течение 24 ч. После охлаждения до комнатной температуры раствор разделяли между EtOAc и Н 2 О. Органический слой высушивали (Na2SO4), концентрировали и очищали колоночной хроматографией для получения промежуточного этилового эфира.i) 2-[5-(3,5-Дихлоробензоил)-4'-трифторметилбифенил-3-ил]-4-метилпентановая кислота Смесь указанного выше промежуточного соединения и NaOH (2 N в H2O, 0,147 мл, 0,294 ммоль) в ТГФ-МеОН (0,6 мл-0,6 мл) перемешивали в течение 18 ч и концентрировали. Добавляли CH2Cl2 и воду,смесь подкисляли добавлением 1 N HCl. Органическую фазу отделяли, а водную фазу экстрагировалиCH2Cl2. Комбинированные органические слои высушивали, концентрировали и очищали колоночной хроматографией для получения 45 мг (45%, 2 стадии) соединения h в виде белого твердого вещества. 1 Н ЯМР (400 МГц, MeOD)0,96 (дд, J=6,60, 1,47 Гц, 6 Н), 1,53 (ддд, J=13,57, 6,72, 6,60 Гц, 1 Н), 1,77 Смесь соединения 1g (50 мг, 0,098 ммоль), 3,5-дихлорбензил цинка хлорида (0,5 М в ТГФ, 0,588 мл,0,294 ммоль) и Pd(dppf)2Cl2 (7,2 мг, 0,0098 ммоль) в ТГФ (1,5 мл) дегазировали в N2 в течение 6 мин и нагревали при 120 С и микроволновом излучении (300 Вт, 1723,7 кПа (250 фунтов на кв.дюйм в течение 20 мин. После охлаждения до комнатной температуры раствор разделяли между EtOAc и насыщенным раствором NH4Cl. Органический слой высушивали (Na2SO4), концентрировали и очищали колоноч- 21019710 ной хроматографией для получения промежуточного этилового эфира. Указанный промежуточный эфир гидролизовали с использованием процедуры гидролиза, описанной в примере 1, стадия (2), для получения соединения, указанного в заголовке. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,84 (дд, J=6,48, 3,30 Гц, 6 Н), 1,39 (ддд, J=13,57, 6,72, 6,60 Гц, 1 Н),1,58-1,65 (м, 1 Н), 1,83-1,91 (м, 1 Н), 3,65 (т, J=7,83 Гц, 1 Н), 3,97 (с, 2 Н), 7,12 (д, J=1,71 Гц, 2 Н), 7,16-7,19 Указанный в заголовке компонент получали путем реакции сочетания Нигиши 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты (промежуточное соединение 1g) с 2,4-дифторбензил цинка хлоридом при условиях, описанных в примере 2. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,75-0,85 (м, 6 Н), 1,37 (дт, J=13,39, 6,63 Гц, 1 Н), 1,54 (ддд, J=13,69,7,21, 6,97 Гц, 1 Н), 1,85 (ддд, J=13,51, 7,52, 7,34 Гц, 1 Н), 3,60 (т, J=7,83 Гц, 1 Н), 3,92 (с, 2 Н), 6,73-6,84 (м,2 Н), 7,12-7,19 (м, 2 Н), 7,29 (с, 1 Н), 7,37 (с, 1 Н), 7,58-7,66 (м, 4 Н); вычисл. для C26H23F5O2 (M+Na) 485,16, обнаруж. 485,1. Пример 4. 2-[5-(4-Хлоропиридин-3-илметил)-4'-трифторметилбифенил-3-ил]-4-метилпентановая кислота Указанное в заголовке соединение получали путем реакции сочетания Нигиши 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты (промежуточное соединение 1g) с 4-хлоропиридил-3 метил цинка хлоридом при условиях, описанных в примере 2. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,83 (тд, J=7,34, 1,96 Гц, 6 Н), 1,40 (ддд, J=13,33, 6,60, 6,48 Гц, 1 Н),1,49-1,57 (м, 1 Н), 1,87 (ддд, J=13,27, 8,50, 7,09 Гц, 1 Н), 3,55-3,61 (м, 1 Н), 3,99 (с, 2 Н), 7,19 (с, 1 Н), 7,267,33 (м, 2 Н), 7,44 (с, 1 Н), 7,58-7,70 (м, 5 Н) 8,20 (д, J=2,20 Гц, 1 Н); вычисл. для C25H23ClF3NO2 (М+Н) 462,14, обнаруж. 462,1. Пример 5. 4-Метил-2-[4'-трифторметил-5-(3-трифторметилбензил)бифенил-3-ил]пентановая кислота Указанное в заголовке соединение получали путем реакции сочетания Нигиши 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты(промежуточное соединение 1g) с 3 трифторметилбензил цинка хлоридом при условиях, описанных в примере 2. 1 Н ЯМР (400 МНц, MeOD)м.д. 0,76-0,86 (м, 6 Н), 1,36 (дт, J=13,51, 6,82 Гц, 1 Н), 1,54-1,62 (м, 1 Н),1,80-1,91 (м, 1 Н), 3,62 (т, J=7,83 Гц, 1 Н), 4,05 (с, 2 Н), 7,16 (с, 1 Н), 7,33-7,44 (м, 6 Н), 7,64 (кв., J=8,56 Гц,4 Н); вычисл. для C27H24F6O2 (M+Na) 517,17, обнаруж. 517,2. Указанное в заголовке соединение получали путем реакции сочетания Нигиши 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты (промежуточное соединение 1g) с 3,5-дифторбензил цинка хлоридом при условиях, описанных в примере 2. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,83 (дд, J=6,60, 3,18 Гц, 6 Н), 1,37-1,41 (м, 1 Н), 1,55-1,62 (м, 1 Н),1,86 (дд, J=7,70, 5,99 Гц, 1 Н), 3,63 (т, J=7,70 Гц, 1 Н), 3,97 (с, 2 Н), 6,64-6,67 (м, 1 Н), 6,75 (дд, J=8,56, 2,20 Гц, 2 Н), 7,17 (с, 1 Н), 7,34 (с, 1 Н), 7,41-7,42 (м, 1 Н), 7,62-7,65 (м, 2 Н), 7,67-7,70 (м, 2 Н); вычисл. для C26H23F5O2 (М+Н) 463,16, обнаруж. 463,3. Пример 7. 2-[5-(3,5-Дифторбензоил)-4'-трифторметилбифенил-3-ил]-4-метилпентановая кислота Указанное в заголовке соединение получали путем реакции сочетания Нигиши 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты(промежуточный компонент 1g) с 3,5 дифторфенилбороновой кислотой при условиях, описанных в примере 1. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,96 (дд, J=6,60, 1,71 Гц, 6 Н), 1,53 (дт, J=13,51, 6,82 Гц, 1 Н), 1,691,79 (м, 1 Н), 1,96-2,05 (м, 1 Н), 3,88 (т, J=7,83 Гц, 1 Н), 7,26-7,32 (м, 1 Н), 7,39 (ддд, J=12,35, 4,65, 2,32 Гц,2 Н), 7,76-7,86 (м, 5 Н), 7,92-8,00 (м, 2 Н); вычисл. для C26H21F5O3 (M+Na) 499,14, обнаруж. 499,0. Пример 8. 2-[5-(4-Цианобензоил)-4'-трифторметилбифенил-3-ил]-4-метилпентановая кислота Указанный в заголовке компонент получали путем реакции сочетания Нигиши 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты(промежуточное соединение 1g) с 4 цианофенилбороновой кислотой при условиях, описанных в примере 1. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,96 (дд, J=6, 60, 1,47 Гц, 6 Н), 1,50-1,57 (м, 1 Н), 1,74 (дд, J=13,82,6,97 Гц, 1 Н), 2,02 (дд, J=7,58, 5,87 Гц, 1 Н), 3,88 (т, J=7,83 Гц, 1 Н), 7,76-7,87 (м, 5 Н), 7,91-7,97 (м, 6 Н); вычисл. для C27H22F3NO3 (М+Н) 466,16, обнаруж. 466,2. Пример 9. 2-5-[(3,5-Дифторфенил)гидроксиметил]-4'-трифторметилбифенил-3-ил-4-метилпентановая кислота Замещение 3,5-дихлорфенилбороновой кислоты 3,5-дифторфенилбороновой кислотой, следующее за реакцией сочетания Сузуки, аналогичной получению соединения 1h, дает промежуточный эфир. К раствору указанного промежуточного эфира (10 мг, 0,02 ммоль) в ТГФ-EtOH (0,5 мл-0,5 мл) добавляли NaBH4 (1,1 мг, 0,03 ммоль). Реакционную смесь перемешивали в течение 4 ч при комнатной температуре и концентрировали. Остаток очищали препаративным TLC для получения соединения 9 а. 1 Н ЯМР (400 МГц, хлороформ-D)м.д. 0,77-0,87 (м, 6 Н), 1,14 (т, J=7,21 Гц, 3 Н), 1,36-1,46 (м, 1 Н),1,60 (дт, J=13,76, 6,94 Гц, 1 Н), 1,94 (дт, J=13,69, 7,70 Гц, 1 Н), 2,40 (дд, J=6,36, 3,42 Гц, 1 Н), 3,64 (т, J=7,83 Гц, 1H), 3,99-4,11 (м, 2H), 5,78 (д, J=2,20 Гц, 1 Н), 6,63 (тт, J=8,80, 2,32 Гц, 1 Н), 6,84-6,90 (м, 2 Н), 7,29 (с,1 Н), 7,38 (с, 1 Н), 7,42 (с, 1 Н), 7,56-7,65 (м, 4 Н); вычисл. для C28H27F5O3 (M+Na) 529,19, обнаруж. 529,2. Названный выше промежуточный компонент гидролизовали с использованием процедуры гидролизации, описанной в примере 1 для получения указанного в заголовке соединения. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,85-0,94 (м, 6 Н), 1,47 (дт, J=13,45, 6,72 Гц, 1 Н), 1,68 (ддд, J=13,69,7,21, 6,97 Гц, 1 Н), 1,92-2,02 (м, 1 Н), 3,75 (т, J=7,83 Гц, 1 Н), 5,84 (с, 1 Н), 6,79 (тт, J=9,05, 2,20 Гц, 1 Н),6,99-7,05 (м, 2 Н), 7,42 (с, 1 Н), 7,57 (д, J=17,12 Гц, 2 Н), 7,72-7,80 (м, 4 Н). Пример 10. 2-[5-(3,5-Дифторфенилэтинил)-4'-трифторметилбифенил-3-ил]-4-метилпентановая кислота Смесь соединения 1g (50 мг, 0,098 ммоль) и Pd(PPh3)2Cl2 (6,9 мг, 0,0098 ммоль) в Et2NH(2 мл) перемешивали под слоем N2 в течение 20 мин и добавляли CuI (1 мг, 0,0049 ммоль). После перемешивания в течение 20 мин. добавляли 1-этинил-3,5-дифторбензол (40 мг, 0,29 ммоль). Реакционную смесь нагревали при 85 С в течение 24 ч. После охлаждения до комнатной температуры раствор разделяли междуEtOAc и Н 2 О. Органический слой высушивали (Na2SO4), концентрировали и очищали препаративнымTLC для получения промежуточного этилового эфира. Названный выше промежуточный компонент гидролизовали с использованием процедуры гидролизации, описанной в примере 1 для получения указанного в заголовке соединения. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,77-0,88 (м, 6 Н), 1,43 (дт, J=13,45, 6,72 Гц, 1 Н), 1,62 (ддд, J=13,82,7,09, 6,97 Гц, 1 Н), 1,92 (дт, J=13,63, 7,61 Гц, 1 Н), 3,69 (т, J=7,83 Гц, 1 Н), 6,86-6,95 (м, 1 Н), 7,02-7,11 (м,2 Н), 7,48 (с, 1 Н), 7,58 (т, J=1,71 Гц, 1 Н), 7,65-7,74 (м, 5 Н). Указанное в заголовке соединение получали путем реакции сочетания Соногашира 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты (промежуточное соединение 1g) с 1-этинил-4 трифторметилбензолом при условиях, описанных в примере 10. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,86 (дд, J=6,60, 1,96 Гц, 6 Н), 1,44 (дт, J=13,45, 6,72 Гц, 1 Н), 1,62 Указанное в заголовке соединение получали путем реакции сочетания Соногашира 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты (промежуточное соединение 1g) с 1-этинил-4 хлорбензолом при условиях, описанных в примере 10. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,77-0,87 (м, 6 Н), 1,43 (дт, J=13,39, 6,63 Гц, 1 Н), 1,61 (ддд, J=13,63,7,09, 6,91 Гц, 1 Н), 1,91 (дт, J=13,51, 7,67 Гц, 1 Н), 3,68 (т, J=7,83 Гц, 1 Н), 7,23-7,30 (м, 2 Н), 7,37-7,46 (м,3 Н), 7,54 (т, J=1,71 Гц, 1 Н), 7,60 (т, J=1,47 Гц, 1 Н), 7,62-7,71 (м, 4 Н). Пример 13. 2-[5-(3,5-бис-Трифторметилфенилэтинил)-4'-трифторметилбифенил-3-ил]-4-метилпентановая кислота Указанное в заголовке соединение получали путем реакции сочетания Соногашира 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты (промежуточное соединение 1g) с 3,5-бис(трифторметил)бензолом при условиях, описанных в примере 10. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,89-0,99 (м, 6 Н), 1,53 (дт, J=13,51, 6,82 Гц, 1 Н), 1,73 (ддд, J=13,76,7,34, 7,03 Гц, 1 Н), 2,02 (ддд, J=13,57, 7,83, 7,70 Гц, 1 Н), 3,81 (т, J=7,70 Гц, 1 Н), 7,64 (с, 1 Н), 7,70-7,86 (м,6 Н), 7,94-8,02 (м, 1 Н), 8,15 (с, 2 Н). Смесь соединения 1g (60 мг, 0,117 ммоль), транс-2-(4-бифенил)винилбороновой кислоты (45 мг,0,199 ммоль), Pd(dppf)2Cl2 (10 мг, 0,0117 ммоль) и K2CO3 (32,3 мг, 0,234 ммоль) в 1,4-водно-диоксановой смеси (0,8-0,8 мл) нагревали до 85 С в течение 15 ч. После охлаждения до комнатной температуры раствор разделяли между EtOAc и Н 2 О. Органический слой высушивали (Na2SO4), концентрировали и очищали колоночной хроматографией для получения промежуточного этилового эфира. Названный выше промежуточный эфир гидролизовали с использованием процедуры гидролизации,описанной в примере 1, стадия (2), для получения указанного в заголовке соединения. 1 Н ЯМР (400 МГц, MeOD)0,88-0,99 (м, 6 Н), 1,57 (дт, J=13,39, 6,63 Гц, 1 Н), 1,74 (ддд, J=13,82,7,09, 6,97 Гц, 1 Н), 1,98-2,09 (м, 2 Н), 3,80 (т, J=7,70 Гц, 1 Н), 7,26-7,34 (м, 3 Н), 7,42 (т, J=7,58 Гц, 2 Н), 7,52 Указанное в заголовке соединение получали путем реакции сочетания Нигиши 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты (промежуточное соединение 1g) с транс-2-(4 трифтометилфенил)винилбороновой кислотой при условиях, описанных в примере 14. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,78-0,89 (м, 6H) 1,47 (дт, J=13,39, 6,63 Гц, 1 Н), 1, 62 (ддд, J=13,63,7,09, 6,91 Гц, 1 Н), 1,91-1,98 (м, 1 Н), 3,68 (т, J=7,83 Гц, 1 Н), 7,33 (д, J=16,4 Гц, 1 Н), 7,26 (д, J=16,5 Гц, 1 Н),7,48-7,58 (м, 4 Н), 7,64-7,70 (м, 5 Н), 7,75-7,78 (м, 2 Н). Пример 16. 4-Метил-2-[5-(2-р-толилвинил)-4'-трифторметилбифенил-3-ил]пентановая кислота Указанное в заголовке соединение получали путем реакции сочетания Нигиши 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты (промежуточное соединение 1g) с транс-2-(4 метилфенил)винилбороновой кислотой при условиях, описанных в примере 14. 1 Указанное в заголовке соединение получали путем реакции сочетания Нигиши 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты (промежуточное соединение 1g) с транс-2-(4 хлорфенил)винилбороновой кислотой при условиях, описанных в примере 14. 1 Н ЯМР (300 МГц, MeOD)м.д. 0,79 (дд, J=6,41, 2,64 Гц, 6 Н), 1,25-1,37 (м, 1 Н), 1,45 (ддд, J=13,47,7,16, 6,88 Гц, 1 Н), 1,67-1,77 (м, 1 Н), 3,51 (т, J=7,72 Гц, 1 Н), 6,56-6,65 (м, 2 Н), 7,09-7,18 (м, 5 Н), 7,34 (д,J=18,09 Гц, 2 Н), 7,54-7,63 (м, 4 Н); вычисл. для C27H24ClF3O2 (M+Na) 495,14, обнаруж. 495,3. Пример 18. 2-5-[2-(4-Фторфенил)винил]-4'-трифторметилбифенил-3-ил-4-метилпентановая кислота Указанное в заголовке соединение получали путем реакции сочетания Нигиши 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты (промежуточное соединение 1g) с транс-2-(4 хлорфенил)винилбороновой кислотой при условиях, описанных в примере 14. 1 Н ЯМР (400 МГц, хлороформ-D)м.д. 0,95 (д, J=6,60 Гц, 6 Н), 1,57 (ддд, J=13,33, 6,85, 6,72 Гц, 1 Н),1,76 (ддд, J=13,82, 7,09, 6,97 Гц, 1 Н), 2,00-2,10 (м, 1 Н), 3,78 (т, J=7,70 Гц, 1 Н), 7,03-7,13 (м, 4 Н), 7,43-7,54 Указанное в заголовке соединение получали путем реакции сочетания Нигиши 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты (промежуточное соединение 1g) с транс-2-(3,5 дифторфенил)винилбороновой кислотой при условиях, описанных в примере 14. 1 Указанное в заголовке соединение получали путем реакции сочетания Нигиши 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты (промежуточное соединение 1g) с транс-2-(4 метоксифенил)винилбороновой кислотой при условиях, описанных в примере 14. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,87 (дд, J=6,60, 3,18 Гц, 6 Н), 1,46 (дт, J=13,21, 6,60 Гц, 1 Н), 1,63 Смесь соединения 12 (20 мг, 0,042 ммоль), 5% Pd/CaCO3 (2 мг) и EtOAc (5 мл) гидрогенизировали под слоем Н 2 (137,9 кПа (20 фунтов на кв.дюйм в шейкере Парра в течение 9 ч. Полученную реакционную смесь фильтровали через целитовый фильтр, концентрировали и очищали ВЭЖХ для получения указанного в заголовке соединения (8 мг, 40%) в виде белого твердого вещества. 1 Н ЯМР (300 МГц, MeOD)м.д. 0,79 (дд, J=6,41, 2,64 Гц, 6 Н), 1,25-1,37 (м, 1 Н), 1,45 (ддд, J=13,47,7,16, 6,88 Гц, 1 Н), 1,67-1,77 (м, 1 Н), 3,51 (т, J=7,72 Гц, 1 Н), 6,56-6,65 (м, 2 Н), 7,09-7,18 (м, 5 Н), 7,31 (с,1 Н), 7,37 (с, 1 Н), 7,54-7,63 (м, 4 Н); вычисл. для C27H24ClF3O2 (M+Na) 495,14, обнаруж. 495,3. Пример 22. 4-Метил-2-4'-трифторметил-5-[2-(4-трифторметилфенил)винил]бифенил-3-илпентановая кислота Смесь соединения 11 (15 мг, 0,030 ммоль), 5% Pd/CaCO3 (1,5 мг) и МеОН (5 мл) гидрогенизировали под слоем Н 2 (137,9 кПа (20 фунтов на кв.дюйм) в шейкере Парра в течение 4 ч. Полученную реакционную смесь фильтровали через целитовый фильтр, концентрировали и очищали ВЭЖХ для получения указанного в заголовке соединения (6 мг, 39%) в виде белого твердого вещества. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,76-0,84 (м, 6 Н), 1,30-1,40 (м, 1 Н), 1,41-1,51 (м, 1 Н), 1,70-1,80 (м,1 Н), 3,50-3,60 (м, 1 Н), 6,88 (д, J=12,2 Гц, 1 Н), 6,81 (д, J=12,4 Гц, 1 Н), 7,11 (с, 1 Н), 7,27-7,40 (м, 4 Н), 7,48 Указанное в заголовке соединение путем реакции сочетания Нигиши 4-метил-2-(5 трифторметилбифенил-3-ил)пентановой кислоты (промежуточное соединение 1g) с транс-2-(3,5-бистрифторметилфенил)винилбороновой кислотой при условиях, описанных в примере 14. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,98 (дд, J=6,60, 3,18 Гц, 6 Н), 1,56 (дт, J=13,39, 6,63 Гц, 1 Н), 1,75 Смесь соединения 14 (20 мг, 0,039 ммоль), 10% Pd/C (10 мг) и МеОН (5 мл) гидрогенизировали под слоем Н 2 (275,8 кПа (40 фунтов на кв.дюйм в шейкере Парра в течение 20 ч. Полученную реакционную смесь фильтровали через целитовый фильтр и концентрировали для получения указанного в заголовке соединения (19 мг, 98%) в виде белого твердого вещества. 1 Н ЯМР (300 МГц, MeOD)0,83-0,92 (м, 6 Н), 1,46 (ддд, J=13,38, 6,59, 6,41 Гц, 1 Н), 1,63 (ддд,J=13,75, 7,16, 6,97 Гц, 1 Н), 1,87-1,98 (м, 1 Н), 2,94-3,07 (м, 4 Н), 3,68 (т, J=7,72 Гц, 1 Н), 7,15-7,23 (м, 3 Н),7,25-7,33 (м, 2 Н), 7,36-7,44 (м, 3 Н), 7,50 (д, J=8,29 Гц, 2 Н), 7,57 (д, J=7,54 Гц, 2 Н), 7,64-7,71 (м, 4 Н); вычисл. для C33H31F3O2 (M+Na) 539,23, обнаруж. 539,2. Пример 25. 4-Метил-2-4'-трифторметил-5-[2-(4-трифторметилфенил)этил]бифенил-3-илпентановая кислота Указанное в заголовке соединение получали гидрогенизацией соединения 15 при условиях, описанных в примере 24. 1 Н ЯМР (400 МГц, MeOD)м.д. 0,83 (д, J=6,60 Гц, 6 Н), 1,39-1,50 (м, 2 Н), 1,81-1,89 (м, 1 Н), 2,892,98 (м, 4 Н), 3,52 (т, J=7,58 Гц, 1 Н), 7,11-7,15 (м, 2 Н), 7,26 (д, J=8,07 Гц, 2 Н), 7,40-7,47 (м, 3 Н), 7,57-7,65
МПК / Метки
МПК: C07D 213/61, A61P 25/28, C07C 59/56, C07C 255/57, C07C 57/58, C07C 59/88, A61K 31/192, C07C 57/60, C07C 57/42
Метки: качестве, производные, кислоты, модуляторов, 2-арил-4-метилпентановой, gamma;-секретазы
Код ссылки
<a href="https://eas.patents.su/30-19710-proizvodnye-2-aril-4-metilpentanovojj-kisloty-v-kachestve-modulyatorov-gamma-sekretazy.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 2-арил-4-метилпентановой кислоты в качестве модуляторов γ-секретазы</a>
Замещенные бициклические производные имидазола в качестве модуляторов гамма-секретазы

Номер патента: 18625
Опубликовано: 30.09.2013
Авторы: Тресадерн Гэри Джон, Ван Брандт Свен Францискус Анна, Бертело Дидье Жан-Клод, Гейсен Хенрикус Якобус Мария, Трабанко-Суарес Андрес Авелино, Макдональд Грегор Джеймс, Бисхофф Франсуа Пол
МПК: C07D 471/04, A61P 25/28, A61K 31/4188...
Метки: бициклические, модуляторов, замещенные, производные, имидазола, качестве, gamma;-секретазы
Формула / Реферат:
1. Соединение формулы (I)или его стереоизомерные формы,где R0 представляет собой водород, галоген или С1-4алкил;R1 представляет собой водород, С1-4алкил или галоген;X представляет собой CR7 или N, где R7 является водородом или галогеном;А1 представляет собой CR2 или N;А2 представляет собой CR8 или N;каждый из А3 и А4 независимо представляет собой CH или N;при условии, что не более двух из А1, А2, А3 и А4 представляют собой N;R2 представляет...
Замещенные производные индазола и азаиндазола в качестве модуляторов гамма-секретазы

Номер патента: 19702
Опубликовано: 30.05.2014
Авторы: Бисхофф Франсуа Пол, Гейсен Хенрикус Якобус Мария, Питерс Серж Мария Алоисиус, Минн Гарретт Берлонд
МПК: C07D 403/12, A61K 31/416, A61P 25/28...
Метки: gamma;-секретазы, модуляторов, качестве, индазола, азаиндазола, замещенные, производные
Формула / Реферат:
1. Соединение формулы (I)или его стереоизомерная форма,где R1 представляет собой С1-6алкил, необязательно замещенный одним или более заместителями, каждый из которых независимо выбран из группы, состоящей из галогена, С1-4алкилокси, цикло-С3-7алкила, тетрагидропиранила, тетрагидрофуранила и фенила; цикло-С3-7алкил; тетрагидропиранил; тетрагидрофуранил; 1,3-бензодиоксолил или фенил, где каждый фенил независимо необязательно замещен одним или...
Производные фениламида 4-(бензил)пиперазин-1-карбоновой кислоты и родственные соединения в качестве модуляторов амида жирной кислоты гидролазы для лечения страхов, боли и других состояний

Номер патента: 12589
Опубликовано: 30.10.2009
Авторы: Аподака Ричард, Сейерстад Марк, Паттабираман Канака, Сяо Вэй, Брайтенбухер Дж.Гай
МПК: A61P 25/28, A61P 25/04, A61P 25/22...
Метки: фениламида, качестве, лечения, состояний, соединения, гидролазы, кислоты, родственные, страхов, модуляторов, других, 4-(бензил)пиперазин-1-карбоновой, жирной, боли, амида, производные
Формула / Реферат:
1. Соединение формулы (I) в которой Z означает -N- или >СН; R1 представляет собой -Н или -C1-4алкил; Ar1 означает 2-тиазолил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, каждый из которых является незамещенным или замещенным по атому углерода кольца одной или двумя Ra группами; где каждый остаток Ra независимо выбран из группы, включающей -C1-4алкил, -C2-4алкенил, -ОН, -OC1-4алкил, атом галогена, -CF3,...
Новые замещенные бициклические гетероциклические соединения в качестве модуляторов гамма-секретазы

Номер патента: 19685
Опубликовано: 30.05.2014
Авторы: Де Клейн Михел Анна Йозеф, Велтер Адриана Ингрид, Ву Тонгфей, Ван Брандт Свен Францискус Анна, Питерс Серж Мария Алоисиус, Бисхофф Франсуа Пол, Гейсен Хенрикус Якобус Мария, Макдональд Грегор Джеймс, Зая Мирко, Оельрих Дэниел, Бертело Дидье Жан-Клод, Суркин Михел
МПК: A61P 25/28, A61K 31/4353, C07D 403/10...
Метки: замещенные, качестве, модуляторов, гетероциклические, бициклические, соединения, gamma;-секретазы, новые
Формула / Реферат:
1. Соединение формулы (I)или его стереоизомерная форма, где Het1 представляет собой 5- или 6-членный ароматический гетероцикл, имеющий формулу (а-1), (а-2), (а-3) или (а-5)R0 представляет собой Н или С1-4алкил;R1 представляет собой Н, С1-4алкил или С1-4алкилоксиС1-4алкил;R2 представляет собой С1-4алкил;X представляет собой О или S;G1 представляет собой СН или N;G2 представляет собой СН, N или С, замещенный С1-4алкилом, при условии, что G1 и G2...
Способ разделения трехвалентных актинидов и, по меньшей мере, одного трехвалентного лантанида с помощью бис(арил)дитиофосфиновой кислоты в качестве экстрагента и органофосфата в качестве синергиста

Номер патента: 2721
Опубликовано: 29.08.2002
Авторы: Модоло Джузеппе, Одой Райнхард
МПК: G21F 9/12, C07F 9/16, C22B 3/34...
Метки: лантанида, бис(арил)дитиофосфиновой, одного, помощью, разделения, мере, трехвалентного, экстрагента, кислоты, актинидов, способ, трехвалентных, качестве, органофосфата, меньшей, синергиста
Формула / Реферат:
1. Способ разделения трехвалентных актинидов и, по меньшей мере, одного трехвалентного лантанида, и/или лантана, и/или иттрия, содержащихся в водном растворе при Н+-концентрации в пределах от 0,01 до 2 молей/л, в котором осуществляют экстракцию трехвалентного актинида с помощью бис(арил)дитиофосфиновой кислоты общей формулы (4) где R1 и R2 обозначают фенил или нафтил, необязательно замещенный, по меньшей мере, одним заместителем, выбранным из...
Предыдущий патент: Спирогетероциклы и их применение в качестве лекарственных средств
Случайный патент: Способ конверсии in situ с использованием нагревающей системы с замкнутым контуром