Кристаллические формы производных фениламинопиримидина
Номер патента: 19223
Опубликовано: 28.02.2014
Авторы: Адибхатла Калисатия Бхуджанга Рао, Компелла Амала Кишан, Венкайах Човдари Наннапанени, Рачаконда Сринивас



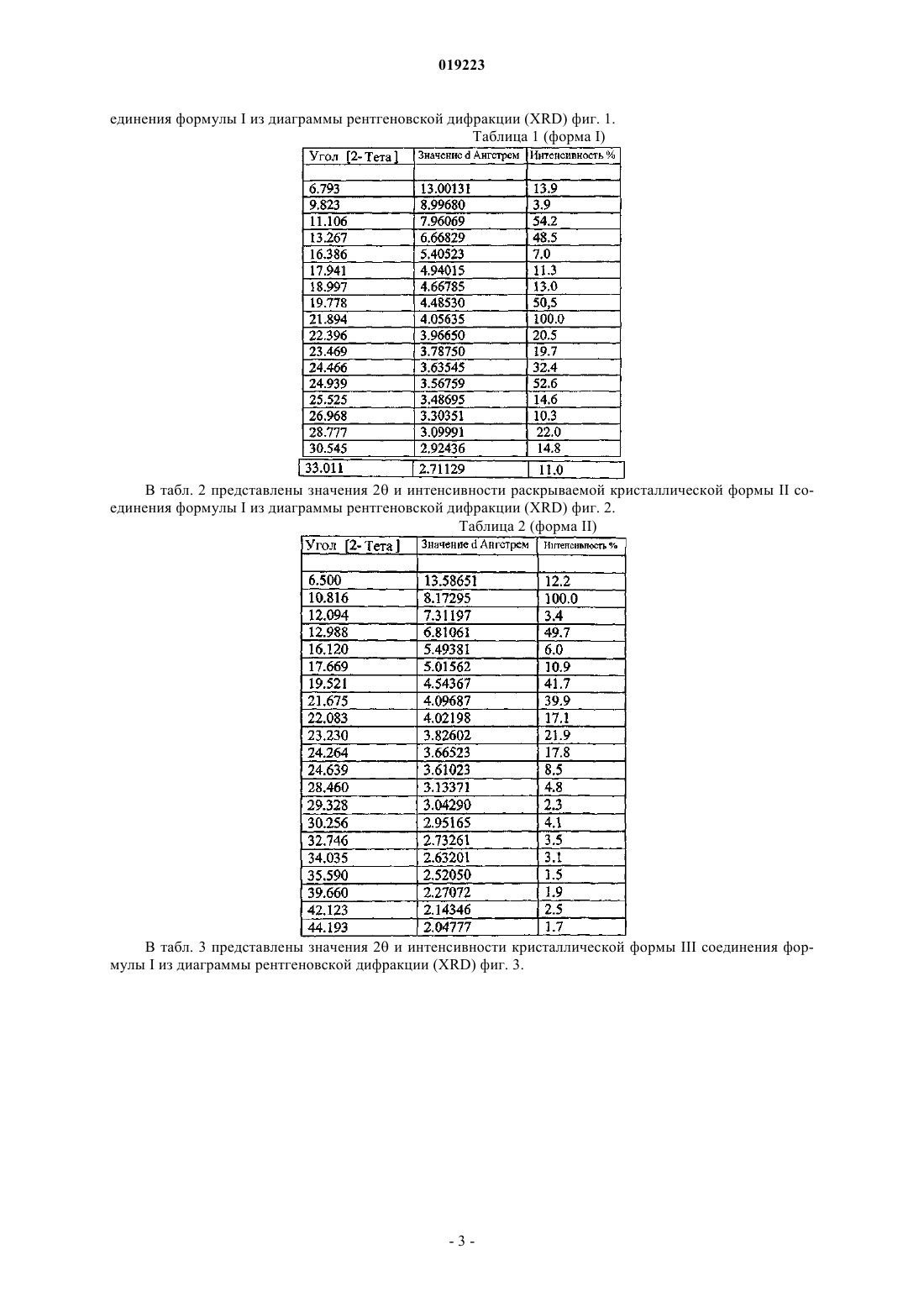
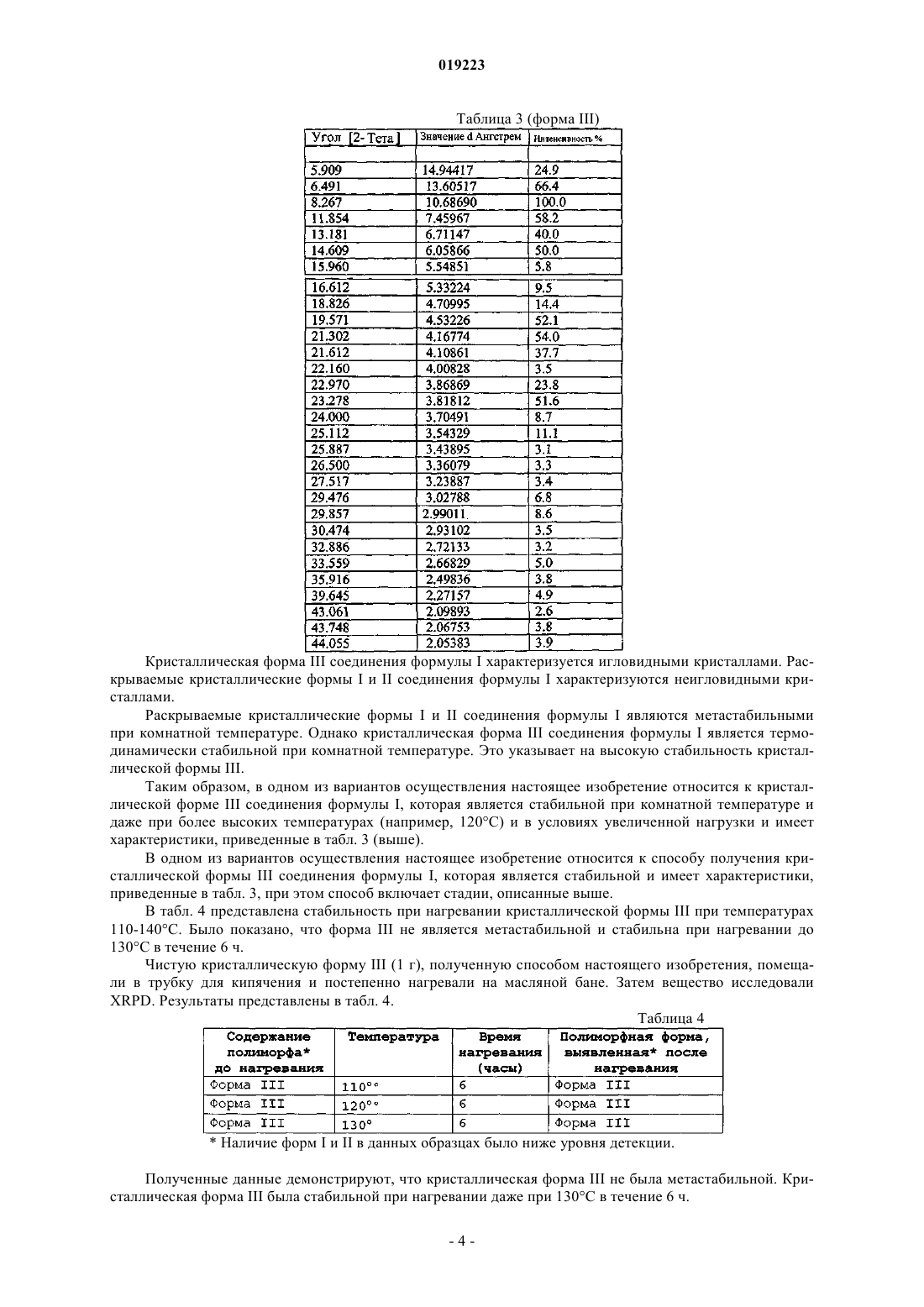
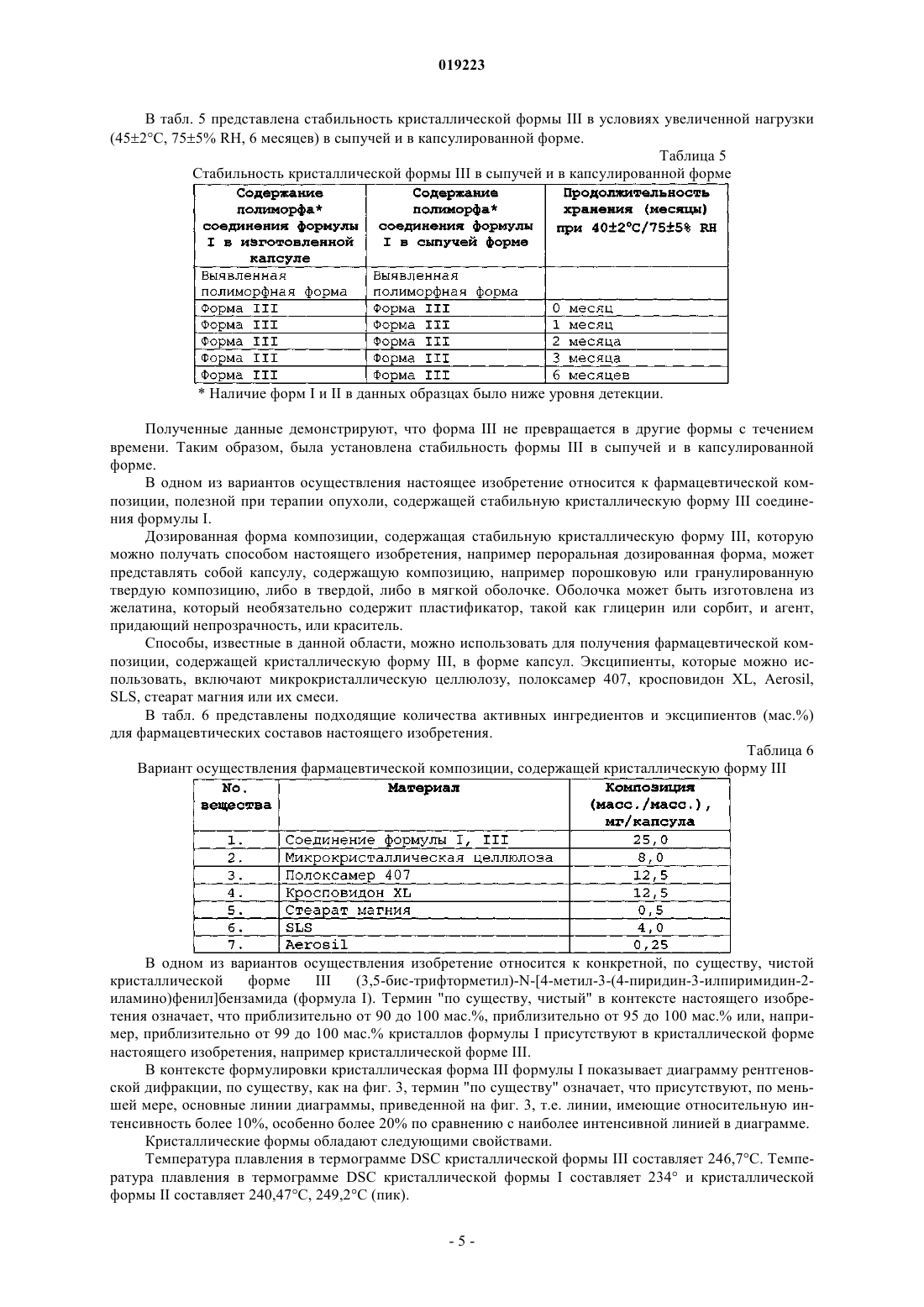


Формула / Реферат
1. Кристаллическая форма III (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамида (формула I), которая имеет характеристики XRPD
2. Кристаллическая форма III по п.1, которая имеет температуру плавления 240°С или выше.
3. Кристаллическая форма III по п.1, которая является, по существу, чистой.
4. Способ получения кристаллической формы III (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамида (формула I), включающий обработку соединения формулы I в кристаллической форме I или II, имеющих следующие характеристики XRPD:
Форма I
Форма II
уксусной кислотой или смесью диметилформамида и ацетона, гексана или толуола и последующую обработку однократно обработанного соединения уксусной кислотой, ацетоном, гексаном или толуолом.
5. Способ по п.4, включающий
обработку соединения формулы I в кристаллической форме I или II уксусной кислотой и
последующую обработку однократно обработанного соединения уксусной кислотой.
6. Способ по п.4, включающий
обработку соединения формулы I в кристаллической форме I или II смесью диметилформамида и ацетона и
последующую обработку однократно обработанного соединения ацетоном.
7. Способ по п.4, включающий
обработку соединения формулы I в кристаллической форме I или II смесью диметилформамида и гексана и
последующую обработку однократно обработанного соединения гексаном.
8. Способ по п.4, включающий
обработку соединения формулы I в кристаллической форме I или II смесью диметилформамида и толуола и
последующую обработку однократно обработанного соединения толуолом.
9. Фармацевтическая композиция, содержащая фармацевтически приемлемый эксципиент и кристаллическую форму III (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамида (формула I), как определено в п.1.
10. Применение кристаллической формы III (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамида (формула I), где кристаллическая форма III является такой, как определено в п.1, для изготовления антипролиферативного лекарственного средства для лечения опухолевого заболевания.
11. Способ лечения субъекта, нуждающегося в антипролиферативной терапии, включающий введение субъекту кристаллической формы III (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамида (формула I), где кристаллическая форма III является такой, как определено в п.1.
Текст
КРИСТАЛЛИЧЕСКИЕ ФОРМЫ ПРОИЗВОДНЫХ ФЕНИЛАМИНОПИРИМИДИНА Настоящее изобретение относится к кристаллической форме III (3,5-бис-трифторметил)-N[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамида (формула I), способам ее получения, фармацевтическим композициям, содержащим данную кристаллическую форму, и применению указанной кристаллической формы III в качестве противоопухолевого средства у человека. Соединение формулы I, также известное как AN-019, представляет собой Компелла Амала Кишан, Рачаконда Сринивас, Адибхатла Калисатия Бхуджанга Рао, Венкайах Човдари Наннапанени (IN) Медведев В.Н. (RU)(71)(73) Заявитель и патентовладелец: НАТКО ФАРМА ЛИМИТЕД (IN) Область техники Настоящее изобретение относится к кристаллической форме III (3,5-бис-трифторметил)-N-[4 метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамида (формула I), к способам ее получения,фармацевтическим композициям, содержащим такую кристаллическую форму, и применению указанной кристаллической формы III в качестве противоопухолевого средства у человека. Соединение формулы I,также известное как AN019, представляет собой Уровень техники, к которой относится изобретение Получение(3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил] бензамида формулы I и его применение, особенно в качестве противоопухолевого средства, описано в примерах 3 и 4 WO 2006/027795 (PCT/IN 05/00243, поданной 19 июля 2005 г.), которая опубликована 16 марта 2006 г., и в соответствующих заявках многих других стран, включая США (Pub. No. US 2007/0232633). В этих публикациях полиморфизм не рассмотрен. В настоящее время неожиданно обнаружено, что в определенных условиях образуется новая полиморфная форма соединения формулы I, которая в дальнейшем в данном описании описана как кристаллическая форма III, и она обладает преимущественными свойствами. Сущность изобретения Настоящее изобретение относится к кристаллической форме III (3,5-бис-трифторметил)-N-[4 метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамида (формула I), способам ее получения,фармацевтическим композициям, содержащим такую кристаллическую форму, и к применению указанной кристаллической формы III в качестве противоопухолевого средства у человека. Соединение формулы I, также известное как AN019, представляет собой Указанная кристаллическая форма III (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3 илпиримидин-2-иламино)фенил]бензамида (формула I) обладает характеристиками XRPD, приведенными в табл. 3 ниже. Данная кристаллическая форма может иметь температуру плавления 240 С или выше. Кристаллическая форма может быть, по существу, чистой. Указанная кристаллическая форма III может использоваться в способе лечения субъекта, нуждающегося в антипролиферативной терапии. Этот способ включает введение субъекту кристаллической формы III (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамида (формула I), которая имеет характеристики XRPD, приведенные в табл. 3 ниже. Настоящее изобретение также относится к фармацевтической композиции. Фармацевтическая композиция содержит фармацевтически приемлемый эксципиент и кристаллическую форму III (3,5-бистрифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамида (формула I), которая имеет характеристики XRPD, приведенные в табл. 3 ниже. Настоящее изобретение также относится к применению кристаллической формы III (3,5-бистрифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамида (формула I), которая имеет характеристики XRPD, приведенные в табл. 1 ниже, для получения антипролиферативного лекарственного средства для лечения опухолевого заболевания. Настоящее изобретение относится к способу получения кристаллической формы III (3,5-бистрифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамида (формула I), которая имеет характеристики XRPD, приведенные в табл. 3 ниже. Данный способ включает обработку соединения формулы I в кристаллической форме I или II уксусной кислотой или смесью диметилформамида и ацетона, гексана или толуола. Данный способ также включает последующую обработку однократно обработанного соединения уксусной кислотой, ацетоном, гексаном или толуолом. Также раскрывается кристаллическая форма I (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3 илпиримидин-2-иламино)фенил]бензамида (формула I), которая имеет характеристики XRPD, приведенные в табл. 1 ниже. Также раскрывается кристаллическая форма II (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3 илпиримидин-2-иламино)фенил]бензамида (формула I), которая имеет характеристики XRPD, приведенные в табл. 2 ниже. Краткое описание чертежей На фиг. 1 представлена диаграмма рентгеновской дифракции (XRD) кристаллической формы I соединения формулы I. Значения 2 и интенсивности приведены в табл. 1. На фиг. 2 представлена диаграмма рентгеновской дифракции (XRD) кристаллической формы II соединения формулы I. Значения 2 и интенсивности приведены в табл. 2. На фиг. 3 представлена диаграмма рентгеновской дифракции (XRD) кристаллической формы III соединения формулы I. Значения 2 и интенсивности приведены в табл. 3. На фиг. 4 представлена термограмма дифференциальной сканирующей калориметрии (DSC) кристаллической формы I соединения формулы I. На фиг. 5 представлена термограмма дифференциальной сканирующей калориметрии (DSC) кристаллической формы II соединения формулы I. На фиг. 6 представлена термограмма дифференциальной сканирующей калориметрии (DSC) кристаллической формы III соединения формулы I. На фиг. 7 представлена морфология кристаллической формы I соединения формулы I. На фиг. 8 представлена морфология кристаллической формы II соединения формулы I. На фиг. 9 представлена морфология кристаллической формы III соединения формулы I. На фиг. 10 А и 10 В проиллюстрировано, что внутрибрюшинные инъекции соединения формулы I(пример 1) вызывали регрессию лейкоза у голых мышей. Мазки крови, взятые из хвостовой вены, показали повышение LGI у контролей и прогрессирующее снижение LGI у мышей, обработанных иматинибом и AN019 (фиг. 10 А). На 48 сутки было определено, что LGI у мышей, обработанных иматинибом,составил 205, и LGI у мышей, обработанных AN019, составил 102 с практически нормальным числом клеток (фиг. 10 В). На фиг. 11 проиллюстрировано, что клетки, обработанные AN019, не показывали какого-либо увеличения селезенки, в то время как у мышей, обработанных иматинибом, наблюдалось увеличение селезенки (пример 2). На фиг. 12 проиллюстрировано, что AN019 в концентрации 1 мг/кг не индуцировал снижение экспрессии люциферазы, 5 мг/кг вызывали фиксацию лейкозных клеток на постоянных уровнях экспрессии,и концентрации 10 и 20 мг/кг вызывали снижение экспрессии люциферазы. На фиг. 13 проиллюстрировано, что пероральное введение иматиниба (1, 5, 10 и 20 мг/кг) голым мышам, которым имплантировали клетки лейкоза человека K562luc, приводило к регрессии лейкоза при более высоких концентрациях. Введение иматиниба в концентрации 1 мг/кг не индуцировало снижения экспрессии люциферазы, в то время как 10 и 20 мг/кг показали замедление экспрессии люциферазы. На фиг. 14 А, 14 В и 14C проиллюстрированы результаты исследований, аналогичных исследованиям, проиллюстрированным на фиг. 12 и 13, но с использованием контрольного лекарственного средства дасатиниба (пример 16). Фиг. 15 и 16 - анализ инвазии в матригель in vitro клеток глиомы 4910, 5310 и U87 и клеток рака молочной железы MDAMB231 и ZR71 в присутствии указанных концентраций AN019, AN024 при облучении и без него. Фиг. 17 - анализ ангиогенеза in vitro клеток глиомы 4910, 5310 и U87 и клеток рака молочной железыMDAMB231 и ZR71 в присутствии указанных концентраций AN019, AN024 при облучении и без него. Фиг. 18 - вестерн-блот анализ клеток глиомы 4910, 5310 и U87 и клеток рака молочной железыMDAMB231 и ZR71 в присутствии указанных концентраций AN019, AN024 при облучении и без него. Фиг. 19 - экспрессия люциферазы у мышей, которым имплантировали K562luc, после обработкиAN024, AN019 или иматинибом. Фиг. 20 - количество животных, вылеченных после обработки AN024 или AN019 на 58 сутки. Обработку лекарственным средством прекращали на 42 сутки, лечебное воздействие AN024 и AN019 у животных продолжалось после прекращения обработки лекарственным средством (данные, основанные на люминесценции). Фиг. 21 - число бластных клеток в мазках крови, взятых у животных на указанные сутки. Обработку лекарственным средством прекращали на 42 сутки. AN024 и AN019 показали эффективность после прекращения обработки лекарственным средством. Было обнаружено, что иматиниб является неэффективным. Фиг. 22 - полуколичественный анализ внутричерепных опухолей у голых мышей после введенияTMZ, AN024 или AN019 с облучением или без него (5Gly). Фиг. 23 - графическое представление голых мышей, показывающих отсутствие опухолей после обработки лекарственным средством AN019, AN024 с облучением или без него. Подробное описание изобретения В табл. 1 представлены значения 2 и интенсивности раскрываемой кристаллической формы I со-2 019223 единения формулы I из диаграммы рентгеновской дифракции (XRD) фиг. 1. Таблица 1 (форма I) В табл. 2 представлены значения 2 и интенсивности раскрываемой кристаллической формы II соединения формулы I из диаграммы рентгеновской дифракции (XRD) фиг. 2. Таблица 2 (форма II) В табл. 3 представлены значения 2 и интенсивности кристаллической формы III соединения формулы I из диаграммы рентгеновской дифракции (XRD) фиг. 3. Кристаллическая форма III соединения формулы I характеризуется игловидными кристаллами. Раскрываемые кристаллические формы I и II соединения формулы I характеризуются неигловидными кристаллами. Раскрываемые кристаллические формы I и II соединения формулы I являются метастабильными при комнатной температуре. Однако кристаллическая форма III соединения формулы I является термодинамически стабильной при комнатной температуре. Это указывает на высокую стабильность кристаллической формы III. Таким образом, в одном из вариантов осуществления настоящее изобретение относится к кристаллической форме III соединения формулы I, которая является стабильной при комнатной температуре и даже при более высоких температурах (например, 120 С) и в условиях увеличенной нагрузки и имеет характеристики, приведенные в табл. 3 (выше). В одном из вариантов осуществления настоящее изобретение относится к способу получения кристаллической формы III соединения формулы I, которая является стабильной и имеет характеристики,приведенные в табл. 3, при этом способ включает стадии, описанные выше. В табл. 4 представлена стабильность при нагревании кристаллической формы III при температурах 110-140 С. Было показано, что форма III не является метастабильной и стабильна при нагревании до 130 С в течение 6 ч. Чистую кристаллическую форму III (1 г), полученную способом настоящего изобретения, помещали в трубку для кипячения и постепенно нагревали на масляной бане. Затем вещество исследовали Наличие форм I и II в данных образцах было ниже уровня детекции. Полученные данные демонстрируют, что кристаллическая форма III не была метастабильной. Кристаллическая форма III была стабильной при нагревании даже при 130 С в течение 6 ч. В табл. 5 представлена стабильность кристаллической формы III в условиях увеличенной нагрузки(452 С, 755% RH, 6 месяцев) в сыпучей и в капсулированной форме. Таблица 5 Стабильность кристаллической формы III в сыпучей и в капсулированной форме Наличие форм I и II в данных образцах было ниже уровня детекции. Полученные данные демонстрируют, что форма III не превращается в другие формы с течением времени. Таким образом, была установлена стабильность формы III в сыпучей и в капсулированной форме. В одном из вариантов осуществления настоящее изобретение относится к фармацевтической композиции, полезной при терапии опухоли, содержащей стабильную кристаллическую форму III соединения формулы I. Дозированная форма композиции, содержащая стабильную кристаллическую форму III, которую можно получать способом настоящего изобретения, например пероральная дозированная форма, может представлять собой капсулу, содержащую композицию, например порошковую или гранулированную твердую композицию, либо в твердой, либо в мягкой оболочке. Оболочка может быть изготовлена из желатина, который необязательно содержит пластификатор, такой как глицерин или сорбит, и агент,придающий непрозрачность, или краситель. Способы, известные в данной области, можно использовать для получения фармацевтической композиции, содержащей кристаллическую форму III, в форме капсул. Эксципиенты, которые можно использовать, включают микрокристаллическую целлюлозу, полоксамер 407, кросповидон XL, Aerosil,SLS, стеарат магния или их смеси. В табл. 6 представлены подходящие количества активных ингредиентов и эксципиентов (мас.%) для фармацевтических составов настоящего изобретения. Таблица 6 Вариант осуществления фармацевтической композиции, содержащей кристаллическую форму III В одном из вариантов осуществления изобретение относится к конкретной, по существу, чистой кристаллической форме(3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2 иламино)фенил]бензамида (формула I). Термин "по существу, чистый" в контексте настоящего изобретения означает, что приблизительно от 90 до 100 мас.%, приблизительно от 95 до 100 мас.% или, например, приблизительно от 99 до 100 мас.% кристаллов формулы I присутствуют в кристаллической форме настоящего изобретения, например кристаллической форме III. В контексте формулировки кристаллическая форма III формулы I показывает диаграмму рентгеновской дифракции, по существу, как на фиг. 3, термин "по существу" означает, что присутствуют, по меньшей мере, основные линии диаграммы, приведенной на фиг. 3, т.е. линии, имеющие относительную интенсивность более 10%, особенно более 20% по сравнению с наиболее интенсивной линией в диаграмме. Кристаллические формы обладают следующими свойствами. Температура плавления в термограмме DSC кристаллической формы III составляет 246,7 С. Температура плавления в термограмме DSC кристаллической формы I составляет 234 и кристаллической формы II составляет 240,47 С, 249,2 С (пик). Диаграммы рентгеновской дифракции также демонстрируют другие выраженные отличия. В одном из вариантов осуществления, по существу, чистая кристаллическая форма III соединения формулы I имеет диаграмму рентгеновской дифракции, указанную на фиг. 3. В одном из вариантов осуществления изобретение относится к кристаллической форме III соединения формулы I, которая характеризуется наличием кристаллов, имеющих форму, представленную на фиг. 9, например кристаллическую форму III, по существу, в чистой форме. В одном из вариантов осуществления кристаллическая форма III соединения формулы I имеет температуру плавления более 240 С, например от 245 до 250 С. Кристаллическую форму III (например, по существу, чистую) можно получить следующим способом: обработкой другой кристаллической формы, например кристаллической формы I или II соединения формулы I, подходящим полярным растворителем, например N,N-ди-низший алкил-низшим алканкарбоксамидом, таким как N,N-диметилформамид или N,N-диметилацетамид, или, например, алифатической карбоновой кислотой, такой как уксусная кислота, или смесью указанных выше соединений с кетоном, таким как ацетон, или гидрофобным углеводородом, например толуолом или гексаном, или их смесью при подходящей температуре. В одном из вариантов осуществления соединение формулы I обрабатывают смесью диметилформамида и ацетона с последующей обработкой ацетоном. В одном из вариантов осуществления соединение формулы I обрабатывают смесью диметилформамида и гексана с последующей обработкой гексаном. В одном из вариантов осуществления соединение формулы I обрабатывают смесью диметилформамида и толуола с последующей обработкой толуолом. В одном из вариантов осуществления соединение формулы I обрабатывают один или несколько раз уксусной кислотой. В этом контексте "проведение обработки" или "обработка" относятся к нагреванию, охлаждению, кипячению с обратным холодильником, промыванию, суспендированию или подобному в растворителе или смеси растворителей. Каждый из указанных вариантов осуществления приводит к кристаллической форме III соединения формулы I. В определенных вариантах осуществления способ настоящего изобретения получения кристаллической формы III не включает обработку другой кристаллической формы соединения формулы I хлороформом, метанолом, дихлорметаном, простым эфиром (например, диэтиловым эфиром), водой, этилацетатом или их смесью. В определенных вариантах осуществления кристаллическую форму III не получают обработкой другой кристаллической формы соединения формулы I хлороформом, метанолом, дихлорметаном, простым эфиром (например, диэтиловым эфиром), водой, этилацетатом или их смесью. Кристаллическая форма III соединения формулы I, а также другие формы обладают подходящими фармакологическими свойствами, и их можно использовать, например, в качестве противоопухолевых средств. Также раскрывается способ получения соединения формулы I. Данный способ может включать предоставление соединения формулы VI, 3,5-бис-трифторметилбензоилхлорида; или получение данного соединения известными способами; конденсацию 4-метил-3-нитроанилина с соединением формулы IV при приблизительно от 0 до-10 С в хлоруглеводородном растворителе с добавлением основного соединения, с получением соединения формулы III, 3,5-бис-трифторметил)-N-(4-метил-3-нитрофенилбензамида; восстановление соединения формулы III хлоридом олова(II)/концентрированной HCl при температуре кипения с обратным холодильником в течение приблизительно от 0,5 до приблизительно 1 ч, с получением соединения формулы IV, (3,5-бис-трифторметил)-N-(3-амино-4-метилфенил)бензамида; конденсацию соединения формулы IV с водным раствором цианамида при температуре кипения с обратным холодильником в н-бутанольном растворителе, с получением соединения формулы V, (3,5 бис-трифторметил)-N-(3-гуанидино-4-метилфенил)бензамида; конденсацию соединения формулы (V) с 3-диметиламино-1-пиридин-3-ил-пропеноном в присутствии основания при температуре кипения с обратным холодильником, с получением соединения формулы I. Способ может быть осуществлен, как описано в примере 10. Варианты осуществления настоящего изобретения описаны в примерах, приведенных ниже, которые предоставлены только для иллюстрации изобретения и, таким образом, их не следует истолковывать,как ограничивающие объем изобретения. Примеры Общий комментарий Обнаружено, что основное соединение настоящего изобретения формулы I (указанное с помощью кода разработки AN019), обладает полезной противоопухолевой активностью, превосходящей некоторые из существующих одобренных лекарственных средств этого класса. Обнаружено, что данное соединение обладает тремя различными полиморфными формами I, II и III, как обсуждалось выше. Несмотря на то,что все формы обладают ценными фармакологическими свойствами, и их можно использовать в качестве противоопухолевых средств, форма III была выбрана для оценки биологической активности, исходя из ее термодинамической стабильности. Код разработки AN024 относится к другому соединению того же класса, описанному авторами настоящего изобретения в другом месте. Биологическую эффективность и активность соединений настоящего изобретения сравнивали с одобренными лекарственными средства-6 019223 ми, такими как иматиниба мезилат и дасатиниб, чтобы они служили в качестве положительных контролей в данном исследовании. "Иматиниба мезилат" сокращают и обозначают в данном исследовании как"иматиниб". Пояснительный пример 1. Установление активности AN019 против CML у голых мышей, которым имплантировали клетки k562 (фиг. 10 А и 10 В). Для определения активности соединений настоящего изобретения против CML клетки K562 получали от АТСС и эти клетки имплантировали голым мышам. В качестве контроля для сравнения использовали иматиниб в той же концентрации на 1 кг массы тела. Обработку лекарственным средством начинали через 15 суток после имплантации и продолжали до 48 суток с помощью внутрибрюшинных инъекций 10 мг/кг раз в сутки. Кровь отбирали из хвостовой вены каждые 6 сутки, определяли процент клетокK562 и вычисляли процентный индекс роста лейкоза (LGI). У мышей, обработанных AN019, наблюдалось устойчивое снижение LGI после начала обработки лекарственным средством. LGI для мышей, обработанных иматинибом, также показал снижение, но значительно отстающее от мышей, обработанных AN019. Через 48 суток после постоянных внутрибрюшинных инъекций мыши, обработанные AN019, были сравнимы с нормальными контрольными мышами, и у мышей, обработанных иматинибом, LGI составлял 205 по сравнению с контролями. Иммуногистохимия селезенки на белок Crk выявила локализацию клеток K562 в селезенке контрольных мышей. У мышей,обработанных AN019, наблюдались базальные уровни экспрессии Crk, в то время как у мышей, обработанных иматинибом, наблюдалось меньшее количество колоний от низких до не поддающихся детекции уровней экспрессии Crk. Способы. Имплантация K562. Голым мышам (nu/nu) имплантировали клетки K562 (1106) через хвостовую вену. Использовали четыре мыши на группу и их разделяли на 4 группы. В трех группах имплантировали клетки K562 и одну группу использовали в качестве нормальных контролей. Группу 2 инокулировали клетками K562, и она служила в качестве положительных контролей без введения. Две другие группы, которым имплантировали клетки K562, обрабатывали либо иматинибом, либо AN019. Внутрибрюшинная инъекция AN019 и иматиниба. Мышей, которым имплантировали K562, обрабатывали либо иматинибом, либо AN019 (10 мг/кг массы тела) посредством внутрибрюшинных инъекций. Исходные растворы иматиниба и AN019 получали в концентрации 100 мкг/мкл в ДМСО. Мышам делали инъекции указанных выше дозировок внутрибрюшинным путем. Было определено, что средняя масса голых мышей составляет 303 г, и было вычислено, что дозировка на мышь составляет 30030 мкг/мышь. 3 мкл исходных растворов разводили до 100 мкл стерильной водой непосредственно перед внутрибрюшинными инъекциями. Внутрибрюшинные инъекции проводили раз в сутки, начиная с 15 суток после имплантации до 48 суток, всего проводя 33 инъекции. Определение индекса роста лейкоза (LGI). Индекс роста лейкоза определяли с использованием формулы где Vc означает среднее количество бластных клеток (K562) на 1 мл крови у контрольных животных в определенное время измерения и Vt означает среднее количество бластных клеток на 1 мл крови у тестируемых животных в определенное время измерения. Отбор крови у мышей (обработанных и необработанных) проводили на шестые сутки после имплантации клеток K562 и продолжали с интервалом каждые 6 суток в течение 48 суток. Отбор крови проводили через хвостовую вену. Количественный анализLGI представляли графически. Иммуноцитохимия. Контрольных мышей умерщвляли на 42 сутки, когда наблюдали вторичные симптомы лейкоза, такие как увеличение живота и отсутствие кровотока в периферических сосудах, приводящее к потере пальцев и красным пятнам под поверхностью кожи. Селезенку и любые вторичные опухоли извлекали,фиксировали в формальдегиде и погружали в парафин в соответствии со стандартными протоколами. Получали парафиновый срез и обрабатывали его для иммуноцитохимии. Регидратированные срезы обрабатывали 0,3% Н 2 О 2 для инактивации каких-либо нативных пероксидаз перед иммунологическим исследованием. Срезы блокировали стерилизованным фильтрацией (0,22-мкм фильтр) 1% BSA-PBS при комнатной температуре в течение 1 ч. После блокирования срезы подвергали иммунозондированию со специфичным против Crk человека первичным моноклональным антителом, индуцированного у кролика,против синтетического пептида, соответствующего остаткам в SH2 домене около N-конца белка CrkL человека (Abeam, Cambridge, MA, USA) в 1% BSA-PBS в течение ночи. Вторичное антитело (антитело против антител кролика), конъюгированное с HRP, использовали для детекции наличия первичного антитела. Срезы быстро промывали три раза в растворе PBS после иммунозондирования вторичным антителом и добавляли субстрат HRP DAB согласно инструкциям изготовителя (Sigma St Louis MO USA). Реакции давали возможность протекать до тех пор, пока не наблюдался четкий контраст между положи-7 019223 тельными и отрицательными контролями. Для отрицательных контролей первичное антитело удаляли. Селезенка мышей с лейкозом служила в качестве положительных контролей. Результаты. Внутрибрюшинные инъекции AN019 вызывали регрессию лейкоза у голых мышей. Мазки крови,которые получали каждые шесть суток после имплантации, показали повышение LGI у контрольных мышей по сравнению с мышами без введения. Мазки крови, взятые из хвостовой вены, показали повышение LGI у контролей и прогрессирующее снижение LGI у мышей, обработанных иматинибом и AN019(фиг. 10 А). Начиная с пятнадцатых суток после имплантации, мышам проводили внутрибрюшинные инъекции AN019 или иматиниба в дозировке 10 мг/кг массы тела. Мыши, обработанные иматинибом,отставали от мышей, обработанных AN019, в каждый момент времени. На 48 сутки было определено,что LGI мышей, обработанных иматинибом, составляет 205, в то время как было обнаружено, что LGI мышей, обработанных AN019, составляет 102 с практически нормальным количеством клеток (фиг. 10 В). Пояснительный пример 2. Инъекции AN019 голым мышам, которым имплантировали клетки K562,не показали увеличения селезенки и экспрессии Crk (фиг. 11). Голым мышам, которым имплантировали клетки K562, вводили иматиниб или AN019 внутрибрюшинными инъекциями. После того как наблюдали снижение LGI, мышей умерщвляли и селезенки извлекали. Увеличение селезенок наблюдали при макроскопическом исследовании, и было определено, что у контрольных мышей селезенки были увеличенными, что указывает на локализацию клеток K562. Клетки, обработанные AN019, не показали какого-либо увеличения селезенки, в то время как у мышей, обработанных иматинибом, селезенка была немного увеличена (фиг. 11). Парафиновые срезы селезенки, подвергнутые иммунозондированию в отношении наличия белка Crk, показали выраженную локализацию экспрессии Crk, сопровождающуюся повышенной клеточной плотностью, указывающей на локализациюK562 у контрольных мышей. У мышей, обработанных AN019, наблюдался только базальный уровень экспрессии Crk, сравнимый с отрицательным контролем. У мышей, обработанных иматинибом, наблюдались локализованные области экспрессии Crk, указывающие на клетки K562 в селезенке. У контрольных мышей также развивались случайным образом расположенные подкожные опухоли, демонстрирующие экспрессию Crk, что указывало на присутствие клеток K562. Из полученных результатов, очевидно, что введение AN019 вызывало регрессию LGI у голых мышей. У мышей, обработанных иматинибом, также наблюдалось значимое снижение LGI, но оно отставало от снижения у мышей, обработанных AN019. Мыши, обработанные как AN019, так и иматинибом, не имели физиологических, фенотипических или поведенческих отклонений. У контрольных мышей наблюдалось наличие случайным образом расположенных подкожных опухолей с потерей пальцев, сопровождающейся красноватыми пятнами под кожей и небольшим увеличением живота. Из полученных результатов очевидно, что AN019 представляет собой перспективное лекарственное средство для лечения лейкоза. Пояснительный пример 3. Исследования in vivo с использованием клеточных линий CML мышиBaf3, устойчивых к иматинибу. Для определения in vivo противолейкозной активности AN019 и AN024 голым мышам внутрибрюшинно имплантировали клетки лейкоза мыши Baf3 (Wt, T315I, М 351 Т и E225K) и через 15 суток после имплантации мышей обрабатывали иматинибом (10 мг/кг), AN019 (20 мг/кг) и AN024 (20 мг/кг) путем перорального принудительного кормления или внутрибрюшинных инъекций. Мазки крови получали через хвостовую вену или через бедренную вену каждые 6 суток, проводили подсчет бластных клеток и получали его графическое представление. Мазки крови мышей, которым имплантировали Baf3Wt и обрабатывали иматинибом (10 мг/кг),AN019 (20 мг/кг) или AN024 (20 мг/кг), были сходными с нормальными контролями через 42 суток. Мазки крови мышей, которым имплантировали Baf3T315I и вводили иматиниб (10 мг/кг), AN019(20 мг/кг) или AN024 (20 мг/кг), показали значимое снижение количества бластных клеток в случае мышей, обработанных AN024 и AN019. У мышей, обработанных пероральной дозировкой иматиниба, не наблюдалось снижения количества бластных клеток, и оно было сходным с контролями без введения и мышами, которым имплантировали Baf3M351T. Мыши, которым имплантировали Baf3E255K, также имели сходное поведение с мышами, которым имплантировали Baf3M351T и Baf3T315I. Голых мышей, которым имплантировали мутантные клетки Baf3Wt, E255K, T315I и М 351 Т, обрабатывали иматинибом, AN019 и AN024 (перорально и внутрибрюшинно). Кратко, мышам вводили иматиниб (10 мг/кг), AN019 (20 мг/кг) и AN024 (20 мг/кг) принудительным пероральным кормлением или внутрибрюшинными инъекциями через 15 суток после имплантации. Мониторинг увеличения живота и снижения активности проводили каждые сутки, каждые 6 суток получали мазки крови из хвостовой вены или бедренной вены и окрашивали их НЕ согласно стандартным протоколам. Через 42 суток после имплантации мышей умерщвляли и селезенки извлекали. Определяли увеличение селезенки и сопоставляли его с количеством бластных клеток в мазках крови. Результаты. Микроскопическое определение количества бластных клеток. У мышей, которым имплантировали клетки Baf3, каждые 6 суток до 42 суток отбирали кровь из хвостовой вены или бедренной вены. На 15 сутки после имплантации мышей обрабатывали иматинибом,AN019 и AN024 (перорально и внутрибрюшинно), как описано выше. Было выявлено, что у мышей, которым имплантировали Baf3Wt, при всех условиях введения наблюдалось прогрессирующее снижение количества бластных клеток. Baf3M351T, T315I и Е 225K плохо отвечали на обработку иматинибом. Пероральное введение иматиниба не имело существенного воздействия на мышей, которым имплантировали Baf3M351T, T315I и E225K. В целом, внутрибрюшинное введение AN024 и AN019 было значительно лучше в отношении снижения количества бластных клеток у всех мышей, которым имплантировалиBaf3. Обработка AN019 мышей, которым имплантировали Baf3Wt, индуцировала полную регрессию лейкозных бластных клеток, и они были сравнимы с контролями без введения. Внутрибрюшинное введение превосходило пероральное введение. Обработка AN019 мышей, которым имплантировали Baf3M351T,T315I и E225K, показала значимое снижение количества бластных клеток, и на 42 сутки они были сравнимы с мышами, которых на 12 сутки после имплантации подвергали обработке; мыши, которым проводили внутрибрюшинное введение, продемонстрировали большую регрессию бластных клеток, чем мыши, которым проводили пероральное введение. Обработка AN024 мышей, которым имплантировалиBaf3 М 351 Т, T315I и Е 225K, продемонстрировала значимое снижение количества бластных клеток, и она превосходила обработку AN019 на 42 сутки; у мышей, которым проводили внутрибрюшинное введение,наблюдалась большая регрессия бластных клеток, чем у мышей, которым проводили пероральное введение. Пояснительный пример 4. Ответ голых мышей, которым имплантировали лейкозные клетки человека k562 normal/luc, с низкой дозой AN024, AN019 и иматиниба (фиг. 12 и 13). Голым мышам (nu/nu) имплантировали клетки K562 (1106) normal/luc через хвостовую вену. Использовали по пять мышей на группу и их подразделяли на 24 группы+2 контроля. Всем группам имплантировали клетки K562 normal/luc. Из 24 групп 12 использовали для исследований люциферазы, в то время как другие 12 использовали для исследований с подсчетом в мазках крови. Пероральное введение AN019, AN024 и иматиниба. Лекарственные средства вводили с помощью принудительного перорального кормления (2% гуммиарабик и 2% SLS в водной суспензии). Результаты. Пероральное введение AN019 (1, 5, 10 и 20 мг/кг) голым мышам, которым имплантировали лейкозные клетки человека K562luc, привело к регрессии лейкоза при более высоких концентрациях. Концентрация AN019 1 мг/кг не индуцировала снижения экспрессии люциферазы, в то время как концентрация 5 мг/кг вызывала фиксацию клеток лейкоза на постоянных уровнях экспрессии, и концентрация 10 и 20 мг/кг вызывала снижение экспрессии люциферазы (фиг. 12). Пероральное введение иматиниба (1, 5, 10 и 20 мг/кг) голым мышам, которым имплантировали лейкозные клетки человека K562luc, приводило к регрессии лейкоза при более высоких концентрациях. Введение иматиниба в концентрации 1 мг/кг не индуцировало снижения экспрессии люциферазы, в то время как 10 и 20 мг/кг показали замедление экспрессии люциферазы (фиг. 13). Данное исследование демонстрирует превосходство AN019 над иматинибом в отношении регрессии лейкоза, в частности, через 24 суток после введения. Пояснительный пример 5. Определение эффективности лекарственного средства (De) и определение проницаемости для лекарственного средства с течением времени. Определение эффективности лекарственного средства (De). Эффективность лекарственного средства определяли с использованием уравнения гдеживых означает общее количество живых мышей при данной концентрации в конце эксперимента,умноженное на количество фотонов, luc означает общее количество живых мышей, демонстрирующих экспрессию люциферазы при данной концентрации в конце эксперимента,с означает количество контрольных животных без введения, умноженное на количество фотонов,с-исходное означает исходное количество животных в контрольной группе в начале эксперимента,умноженное на количество фотонов. Результаты были представлены графически как процент эффективности лекарственного средства Результаты. В табл. 7 представлена эффективность лекарственного средства при различных концентрациях иматиниба, AN019 и AN024 при определении в исследованиях in vivo. Из табл. 7 очевидно, что AN019 является дозозависимым, в то время как иматиниб и AN024 не являются дозозависимыми, и они были эффективными в низких концентрациях. Таблица 7 Определение проницаемости для лекарственного средства с течением времени (Tp). Проницаемость с течением времени вычисляли с использованием следующего уравнения: где Р означает количество фотонов на сутки "а", сутки "ter" или на сутки "b", где "а" означает сутки, когда обработку лекарственным средством прекращали, "ter" означает сутки, когда эксперимент завершали,и "b" означает сутки, когда Р являлось минимальным после суток "а", но до суток "ter",n равен числу живых животных во время измерения Р. Большее значение указывает на высокую эффективность лекарственного средства после прекращения обработки лекарственным средством, т.е. проницаемость для лекарственного средства с течением времени. Результаты. Для определения проницаемости с течением времени для AN019, AN024 и иматиниба голым мышам имплантировали клетки K562luc. Животных после трансплантации фотографировали с интервалами 6 суток. Обработку лекарственным средством (AN019 20 мг/кг, AN024 20 мг/кг и иматинибом 10 мг/кг) начинали через 15 суток после имплантации посредством внутрибрюшинных введений раз в сутки. Обработку лекарственным средством прекращали на 35 сутки, животных фотографировали до 45 суток и проводили вычисления, как описано в способах. С использованием уравнения для проницаемости с течением времени были определены следующие значения Tp:AN019=2,0,AN024=2,4,Иматиниб=0,8. Значения Tp указывают на то, что AN024 имел активность выше контроля без обработки и был значительно лучше, чем AN019, в отношении активности с течением времени после прекращения обработки лекарственным средством. Пояснительный пример 6. Эффект дасатиниба у голых мышей, которым имплантировали Baf3, по сравнению с AN024, AN019 и иматинибом (фиг. 14 А, 14 В и 14 С). В примере 3 показана эффективность AN024 и AN019 для лечения лейкоза по сравнению с иматинибом с использованием мутантных клеточных линий Baf3 (wt, T315I, М 351 Т и E255K). В данном случае авторы настоящего изобретения использовали дасатиниб в качестве контрольного лекарственного средства для определения ответа мутантных клеток Baf3 на введение по сравнению с AN019, AN024 и иматинибом. Способ. Схема эксперимента приведена ниже в форме таблицы Голым мышам внутрибрюшинно имплантировали мутантные клетки Baf3 (wt, T315I, М 351 Т илиE255K). Через 15 суток после имплантации мышам вводили либо перорально, либо внутрибрюшинно 10 мг/кг дасатиниба в течение 27 суток. Каждые 6 суток отбирали кровь из бедренной вены или хвостовой вены, определяли количество бластных клеток и его графически представляли. Результаты. На 6 сутки мазки крови голых мышей, которым имплантировали мутантные клетки Baf3 (wt, T315I,М 351 Т или E255K), продемонстрировали нормальное количество бластных клеток, и количество бластных клеток постепенно возрастало, как наблюдали на 12 сутки. Введение дасатиниба начинали на 15 сутки после имплантации. Введение дасатиниба давало результаты не лучше, чем введение иматиниба. На 42 сутки при завершении эксперимента мыши, которым имплантировали клетки wt, продемонстрировали выраженный ответ на дасатиниб, что указывает на то, что внутрибрюшинное введение превосходило пероральное. Мыши, которым имплантировали T315I и М 351 Т, были сходны с контролями без значимого снижения количества бластных клеток. У мышей, которым имплантировали E255K, результаты были немного лучше, чем в случае имплантированных клеток T315I, в ответ на введение дасатиниба. В общем,введение дасатиниба приводило только к замедлению прогрессирования лейкоза без значимого лечебного эффекта. Количество бластных клеток было значительно более низким в группе, которой вводилиAN019. Полученные результаты проиллюстрированы на фиг. 14 А, 14 В и 14 С. Пояснительный пример 7. Исследования in vitro на клеточных линиях глиомы и рака молочной железы (фиг. 15-18). Материалы и способы. Для определения эффекта AN019, AN024 и темозоломида с облучением или без него на клетки глиомы и рака молочной железы, клетки обрабатывали конкретными дозами и определяли инвазию, ангиогенез и изменения определенных молекул передачи сигнала. Анализ инвазии в матригеле. Инвазивность in vitro клеток глиомы 4910, 5310 и U87 и клеток рака молочной железы MDAMB231 и ZR71 в присутствии определенных концентраций соединений оценивали с использованием модифицированного анализа в камере Бойдена. Клетки обрабатывали данными соединениями в течение 48 ч. 1106 клеток суспендировали в 600 мкл бессывороточной среды, дополненной 0,2% BSA и помещенной в верхнее отделение камер transwell (Corning Costar Fischer Scientific, каталожный 07-200-158, Pittsburgh PA),покрытых матригелем (0,7 мг/мл). Нижнее отделение камеры заполняли 200 мкл сывороточной среды и клеткам давали возможность мигрировать в течение 24 ч. После инкубации клетки фиксировали, окрашивали Hema-3 и количественно определяли, как описано ранее (Mohanam et al. 1993). Мигрировавшие клетки визуализировали под микроскопом для определения снижения инвазивности, индуцированной соединениями настоящего изобретения. Анализ ангиогенеза. Ангиогенез in vitro клеток глиомы 4910, 5310 и U87 и клеток рака молочной железы MDAMB231 иZR71 в присутствии определенных концентраций соединений определяли следующим образом. Клетки(2104/лунка) высевали на предметные стекла с 8-луночными камерами и обрабатывали различными концентрациями тестируемых соединений. После инкубации в течение 24 ч кондиционированные среды удаляли и добавляли к 4104 эндотелиальных клеток дермы человека (монослой в предметных стеклах с 8-луночными камерами), эндотелиальным клеткам дермы человека давали возможность расти в течение 72 ч. Затем клетки фиксировали в 3,7% формальдегиде, окрашивали НЕ и фотографировали. Вестерн-блот анализ. Вестерн-блот анализ клеток глиомы 4910, 5310 и U87 и клеток рака молочной железы MDAMB231 и ZR71 в присутствии определенных концентраций соединений оценивали в соответствии со стандартными протоколами. Клетки обрабатывали AN019, AN024 или темозоломидом в определенных концентрациях. Через 24 ч после обработки клетки собирали и клеточные лизаты экстрагировали. Равные количества белков фракционировали с помощью SDS-PAGE. Фракционированные белки подвергали блоттингу на нейлоновые мембраны и иммунозондировали на AKT, ERK и Pi3k. Выделенные белки клеток рака молочной железы, кроме того, иммунозондировали на EGFR, ErbB1, ErbB2 и ErbB3. Результаты. Анализ инвазии в матригеле. Инвазивность in vitro клеток глиомы 4910, 5310 и U87 и клеток рака молочной железы MDAMB231 и ZR71 в присутствии определенных концентраций соединений оценивали с использованием модифицированного анализа в камере Бойдена. Клетки обрабатывали данными соединениями в течение 48 ч. В табл. 2 представлены результаты исследований in vitro в анализе инвазии в матригеле клеток глиомы 4910, 5310 и U87 и клеток рака молочной железы MDAMB231 и ZR71 в присутствии определенных концентраций соединений, при облучении и без него. Изменение инвазивности различных клеточных линий приведено в табл. 8. Из анализа инвазии очевидно, что AN019 и AN024 были наиболее эффективными в отношении ингибирования инвазии большинства клеток как при облучении, так и без него. На фиг. 15 и 16 проиллюстрированы результаты анализа инвазии в матригеле in vitro клеток глиомы 4910, 5310 и U87 и клеток рака молочной железы MDAMB231 и ZR71 в присутствии определенных концентраций AN019 при облучении и без него. Анализ ангиогенеза. Из экспериментов с анализом ангиогенеза было выявлено, что AN019 является наиболее эффективным в отношении ингибирования ангиогенеза. Обработка темозоломидом вызывала полное ингибирование ангиогенеза в клетках ZR-71, в то время как в клетках MDA-MB-231 наблюдали только небольшое ингибирование в контрольных условиях с возрастанием ингибирования после облучения. Клетки ксенотрансплантата глиомы 4910 показали выраженное ингибирование ангиогенеза как при облучении, так и без него. В случае клеток 5310 ингибирование ангиогенеза наблюдали в контрольных условиях, в то время как после обработки облучением происходила стимуляция ангиогенеза. Клетки глиомы U87 продемонстрировали сходный характер ингибирования как при облучении, так и без него. Обработка AN024 вызывала полное ингибирование ангиогенеза в клетках ZR-71, в то время как в клетках MDA-MB-231 наблюдали только небольшое ингибирование в контрольных условиях и при обработке облучением. Клетки ксенотрансплантата глиомы 4910 продемонстрировали значимое ингибирование ангиогенеза как при облучении, так и без него. В случае клеток 5310 ингибирование ангиогенеза наблюдали в контрольных условиях, в то время как ангиогенез далее ингибировался после обработки облучением. Клетки глиомы U87 продемонстрировали значимое замедление ангиогенеза с увеличением ингибирования после облучения. Обработка AN019 вызвала полное ингибирование ангиогенеза в клетках ZR-71, в то время как в клетках MDA-MB-231 наблюдали небольшое ингибирование как в контрольных условиях, так и при обработке облучением. Клетки ксенотрансплантата глиомы 4910 продемонстрировали ингибирование ангиогенеза, сходное с клетками MDA-MB-231, с повышением ингибирования ангиогенеза после облучения. В случае клеток 5310 ингибирование ангиогенеза было большим в контрольных условиях, чем после обработки облучением. Клетки глиомы U87 продемонстрировали сходное замедление ангиогенеза как при облучении, так и без него. На фиг. 17 проиллюстрированы результаты, полученные в анализе ангиогенеза in vitro клеток глиомы 4910, 5310 и U87 и клеток рака молочной железы MDAMB231 и ZR71 в присутствии определенных концентраций AN019 при облучении и без него. Вестерн-блот анализ. Вестерн-блот анализ клеток глиомы 4910, 5310 и U87 и клеток рака молочной железы MDAMB231 и ZR71 в присутствии определенных концентраций соединений настоящего изобретения показал, что клетки U87 не показывают значимого изменения уровней AKT или PI3k как при облучении, так и без него, в то время как в клетках, обработанных AN024, наблюдали небольшое снижение уровней ERK, и после облучения снижение усиливалось. Клетки 4910 были сходны с клетками U87, со снижением уровней AKT после обработки AN024, и снижение уровней AKT усиливалось после облучения. В случае клеток 5310 не наблюдали значимых отличий в экспрессии ERK, в то время как обработка AN019 вызывала снижение уровней экспрессии AKT. В клетках, обработанных AN019 без облучения, уровни PBk практически не поддавались детекции, но повторно появлялись после обработки облучением. В случае клеток рака молочной железы MDAMB231 не наблюдали значимого изменения в AKT, ERK или PI3k, в то время как в случае ZR71 обработка AN019 вызывала снижение уровней AKT, которое усиливалось после облучения. Обработка AN024 не показала какого-либо значимого изменения в условиях без облучения, в то время как после облучения клетки, обработанные AN024, показали снижение экспрессии AKT. Уровни PDk отсутствовали при обработке AN019 как при облучении, так и без него. Обработка AN024 приводила к снижению уровней PBk после облучения. Уровни pAKT значительно не изменялись при любой из обработок, с облучением или без него, в то время как в клетках, обработанных AN019, уровни pERK значительно снижались как при облучении, так и без него. AN024 также показал снижение уровней pERK,но в меньшей степени, чем AN019. Обработка темозоломидом как при облучении, так и без него, не показала каких-либо значимых изменений в уровнях pAKT или ERK. На фиг. 18 проиллюстрированы результаты вестерн-блот анализа клеток глиомы 4910, 5310 и U87 и клеток рака молочной железы MDAMB231 и ZR71 в присутствии определенных концентраций AN024 при облучении и без него. Пояснительный пример 8. Анализ выживания при лейкозе (фиг. 19-21). Клетки K562, экспрессирующие люциферазу, имплантировали внутрибрюшинно голым мышам; мышей сканировали с использованием устройства для визуализации xenogeny IVIS после внутрибрюшинных инъекций люциферина для определения имплантации. Обработку лекарственным средством начинали, как и в предыдущих исследованиях, через 15 суток после имплантации. Животных обрабатывали до 42 суток, затем обработку лекарственным средством прекращали и определяли выживаемость животных в соответствии с правилами ухода за животными. Было выявлено, что у контрольных животных развивался лейкоз, и смерть происходила на 34 и 35 сутки, согласно правилам, авторы настоящего изобретения приняли решение умертвить оставшихся 8 животных на 35 сутки. Обработку лекарственным средством прекращали на 42 сутки после имплантации и определяли выживание животных. У животных, обработанных AN024, наблюдалась смертность на 38 сутки, у погибших животных при дальнейшем исследовании не было выявлено увеличения селезенки и было определено, что причина смерти отлична от лейкоза, при этом мазки крови от погибших животных взять было нельзя. Среди 10 использованных животных у 8 животных на 55 сутки не было признаков лейкоза. Среди животных, обработанных AN019, не было какой-либо смертности, и у 7 из 10 животных наблюдалось полное отсутствие симптомов лейкоза. У животных, обработанных иматинибом, наблюдалось повторное возникновение симптомов лейкоза после прекращения введения, и у них смертность наступала на 55, 56, 57 и 58 сутки. У выживших животных было выявлено наличие симптомов лейкоза. На фиг. 19 проиллюстрирована величина экспрессии люциферазы на K562luc, полученная у имплантированных мышей после обработки AN024, AN019 или иматинибом. На фиг. 20 проиллюстрировано количество животных, вылеченных после обработки AN024 илиAN019, на 58 сутки. Обработку лекарственным средством прекращали на 42 сутки, у животных после обработки AN024 и AN019 наблюдалось продолжение эффекта излечения после прекращения обработки лекарственным средством. На фиг. 21 проиллюстрировано количество бластных клеток из мазков крови, взятых у животных на указанные сутки. Обработку лекарственным средством прекращали на 42 сутки. AN024 и AN019 продемонстрировали эффективность после прекращения обработки лекарственным средством. Было выявлено,что иматиниб является неэффективным. Пояснительный пример 8 А. Исследования ED50, LD50, MTD и терапевтического индекса. В представленной ниже таблице обобщены ED50, LD50, цитированная выше MTD (максимальная переносимая доза) и терапевтический индекс соединений настоящего изобретения по сравнению с иматинибом. Использовали способы согласно J. Pharmacol. Exp. Ther., (1949), 96: 96-113. мыши с лейкозом (K562). Пояснительный пример 9. Исследования глиомы с облучением (фиг. 22 и 23). Голым мышам интракраниально имплантировали клетки ксенотрансплантата глиомы человека 4910(1106 клеток). Через десять суток после имплантации мышам вводили AN019, AN024 или темозоломид с облучением или без него (5 Gy/неделя). Эксперимент завершали на 40 сутки после имплантации. Из результатов было выявлено, что у 100% контрольных животных развились внутричерепные опухоли и облучение отдельно имело очень небольшой эффект на снижение размера опухоли. У животных,обработанных TMZ отдельно, было выявлено снижение внутричерепных опухолей, причем у 3 из 10 животных наблюдалось полное отсутствие опухолей. Обработка облучением, комбинированная с введениемTMZ, вызывала дальнейшее уменьшение размера опухолей, причем у животных в меньшей степени наблюдались симптомы внутричерепного давления (дугообразная спина), в этом случае у 2 из 10 животных наблюдалось отсутствие видимых внутричерепных опухолей. У животных, обработанных AN024 без облучения, наблюдалось наличие внутричерепных опухолей, однако опухоли были четко ограниченными и не демонстрировали диффузных краев, как наблюдали у контролей или при введении TMZ, 3 из 10 животных излечились. После обработки облучением 7 из 10 животных излечились, животные, у которых наблюдалось наличие опухолей, продемонстрировали четко ограниченные приемлемые для хирургической операции опухоли. У животных, обработанных AN019 отдельно, наблюдались опухоли, сходные с опухолями животных, обработанных AN024, и в этом случае как при облучении, так и без него, 6 из 10 животных излечились. Наблюдали, что после облучения размер опухоли значительно снижался. На фиг. 22 проиллюстрированы результаты, полученные в полуколичественном анализе внутричерепных опухолей у голых мышей после введения TMZ, AN024 или AN019 с облучением или без него (5 Гр). На фиг. 23 показано графическое представление голых мышей, показавших отсутствие внутричерепных опухолей после обработки лекарственным средством AN019, AN024 и без обработки облучением. В следующих примерах проиллюстрированы препаративный синтез и другие аспекты соединений настоящего изобретения. Иллюстративный пример 1. Получение формы I.(3,5-бис-Трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамид формулы I получали согласно четырехстадийному способу примера 3 из WO 2006/027795 (US 2007/0232633) следующим образом. Получение(3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил] бензамида (I). Стадия (I). Получение нового (3,5-бис-трифторметил)-N-(4-метил-3-нитрофенил)бензамида. Сначала получали 3,5-бис-трифторметилбензоилхлорид, который использовали в качестве одного из исходных веществ, следующим образом. К раствору 3,5-бис-трифторметилбензойной кислоты (Lancaster) (250,0 г, 0,97 моль) в хлороформе(2,5 л) добавляли тионилхлорид (576,0 г, 4,8 моль) в течение 15 мин при комнатной температуре. Реакционную смесь кипятили с обратным холодильником в течение 1 ч. К раствору 3,5-бис-трифторметилбензойной кислоты (Lancaster) (250,0 г, 0,97 моль) в хлороформе(2,5 л) добавляли тионилхлорид (576,0 г, 4,8 моль) в течение 15 мин при комнатной температуре. Реакционную смесь кипятили с обратным холодильником в течение 1 ч. Избыток тионилхлорида удаляли совместной дистилляцией с хлороформом при пониженном давлении. После завершения дистилляции полученный 3,5-бис-трифторметилбензоилхлорид охлаждали до комнатной температуры и растворяли в 400 мл хлороформа. Раствор 4-метил-3-нитроанилина (92,0 г, 0,60 моль) в хлороформе (1,2 л) охлаждали до -5 С и добавляли триэтиламин (304,8 г, 3,0 моль). По каплям добавляли раствор 3,5-бистрифторметилбензоилхлорида в хлороформе при -5 С в течение 60-75 мин. Полученную суспензию перемешивали в течение 1 ч при -5 С. Суспензию подвергали дистилляции до конечного объема 800 мл и фильтровали, промывали охлажденным хлороформом (200 мл) и сушили в вакууме с получением 160,0 г нового (3,5-бис-трифторметил)-N-(4-метил-3-нитрофенил)бензамида (68%) в виде кремовых кристаллов(чистота по ВЭЖХ 98,2%) MR-123-130C. Стадия (II). Получение (3,5-бис-трифторметил)-N-(3-амино-4-метилфенил)бензамида. Суспензию нового (3,5-бис-трифторметил)-N-(4-метил-3-нитрофенил)бензамида (160 г, 0,41 моль) и хлорида олова(II) (460,8 г, 2,0 моль) в абсолютном этаноле (850 мл) кипятили с обратным холодильником в течение 40 мин. Затем полученную суспензию охлаждали до комнатной температуры и гасили 5 л ледяной воды, pH реакционной смеси доводили до 8,0 с помощью 4,3 л 5% раствора гидроксида натрия и экстрагировали 22 л этилацетатом. Слой этилацетата промывали последовательно водой и насыщенным раствором соли и сушили над сульфатом натрия. Этилацетат полностью отгоняли, к осадку добавляли 500 мл гексана и фильтровали. Осадок на фильтре сушили при высоком вакууме при 60 С с получением 96,0 г нового (3,5-бис-трифторметил)-N-(3-амино-4-метилфенил)бензамида (65%) в виде желтых кристаллов. (Чистота по ВЭЖХ 98,5%) MR-153-156C. Стадия (III). Получение (3,5-бис-трифторметил)-N-(3-гуанидино-4-метилфенил)бензамида. Суспензию (3,5-бис-трифторметил)-N-(3-амино-4-метилфенил)бензамида (90 г, 0,20 моль) в нбутаноле (500 мл) обрабатывали последовательно концентрированной азотной кислотой до тех пор, пока значение рН не достигало 2,5 (15,9 г), и раствором цианамида (15,7 г, 0,37 моль) в воде (15 мл) в течение 30 мин. Полученную реакционную смесь перемешивали при кипячении с обратным холодильником в течение 6 ч. Затем реакционную смесь полностью отгоняли в вакууме и осадку давали возможность ох- 14019223 ладиться до комнатной температуры. К реакционной массе добавляли смесь 180 мл метанола и 180 млIPE и ее перемешивали при комнатной температуре в течение 1 ч. Продукт фильтровали при отсасывании, промывали смесью метанола и IPE (350 мл) и сушили в вакууме при 60 С с получением 72 г нитратной соли нового (3,5-бис-трифторметил)-N-(3-гуанидино-4-метилфенил)бензамида с выходом 62% от теоретического (чистота по ВЭЖХ 99,2%), MR-285-287C. Стадия (IV). Получение (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2 иламино)фенил]бензамида (I). Суспензию нитрата (3,5-бис-трифторметил)-N-(3-гуанидино-4-метилфенил)бензамида (70 г, 0,15 моль) в н-бутаноле (470 мл) в атмосфере азота обрабатывали последовательно хлопьями гидроксида натрия (7,0 г, 0,18 моль) и 3-диметиламино-1-пиридин-3-илпропеноном (28,0 г, 0,16 моль). Полученную суспензию кипятили с обратным холодильником в течение 2 ч. Реакционные смеси превращались в гомогенный темно-оранжевый раствор и диметиламин удаляли отгонкой с н-бутанолом. Реакционную массу охлаждали до кт, добавляли смесь воды и хлороформа (300 мл+300 мл) и слой хлороформа отделяли. Слой хлороформа промывали водой и подвергали дистилляции до конечного объема 70 мл. К реакционной массе добавляли этилацетат (350 мл) и фильтровали при отсасывании, выделенное твердое вещество промывали этилацетатом (250 мл) и водой (250 мл) и сушили в вакууме при 60 С. Выход: 48,0 г (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил] бензамида формулы I. Соединение формулы I, полученное четырехстадийным способом, суспендировали в 480 мл хлороформа и нагревали до 50-55 С. Реакционную массу охлаждали до комнатной температуры и затем охлаждали до (-5) - 0 С. Продукт фильтровали и промывали хлороформом (250 мл). Влажный осадок сушили в течение 6 ч при 60 С. Выход составил 40 г. Температура плавления 230-237 С (DSC). На фиг. 1, прилагаемой к данному описанию, представлена порошковая рентгенограмма (XRPD), на которой показан, главным образом, по существу, чистый образец формы I, полученной согласно способу,описанному в данном примере. Значения 2 и интенсивности приведены в табл. 1. На фиг. 4 представлена термограмма DSC кристаллической модификации I, полученной способом,описанным в данном примере (в интервале от 40,0 до 325 С, 10,00 С/мин; N2 80 мл/мин). На фиг. 7 показаны кристаллы кристаллической формы I соединения формулы (I). Иллюстративный пример 2. Получение формы II.(3,5-бис-Трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамид формулы I получали способом примера 4 в WO 2006/027795 (US 2007/0232633) следующим образом. Альтернативный способ получения(3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3 илпиримидин-2-иламино)фенил]бензамида. Сначала получали 3,5-бис-трифторметилбензоилхлорид, который использовали в качестве одного из исходных веществ, следующим образом. Тионилхлорид (2,04 кг, 17,2 моль) добавляли в течение 15 мин к раствору 3,5-бистрифторметилбензойной кислоты (855,0 г, 3,3 моль) и ДМФА (9 мл) в хлороформе (9 л) при комнатной температуре. Реакционную смесь кипятили с обратным холодильником в течение 1 ч. Избыток тионилхлорида удаляли совместной дистилляцией с хлороформом при пониженном давлении при 40 С. После окончания дистилляции полученный 3,5-бис-трифторметилбензоилхлорид охлаждали до комнатной температуры и растворяли в 700 мл хлороформа. Раствор N-(5-амино-2-метилфенил)-(3-пиридил)-2-пиримидинамина (0,73 кг, 2,64 моль) в хлороформе (9 л) охлаждали до -5 С и добавляли триэтиламин (1,03 кг, 10,2 моль). По каплям добавляли 3,5 бис-трифторметилбензоилхлорид в хлороформе при -5 С в течение 60-75 мин. Полученную суспензию перемешивали в течение 1 ч при -5 С. Суспензию фильтровали, промывали D.M. водой и метанолом с получением 1,3 кг неочищенного влажного указанного в заголовке соединения, которое при перекристаллизации из метанола давало 0,82 кг (60%) (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3 илпиримидин-2-иламино)фенил]бензамида (I). Соединение формулы I, полученное указанным выше способом, суспендировали в 5 л метанола и кипятили с обратным холодильником. Реакционную массу выдерживали при той же температуре в течение 30-40 мин, медленно охлаждали до 40-45 С и выдерживали при этой температуре в течение 60 мин. Продукт фильтровали и промывали 0,5 л метанола при 40-45 С. Влажный осадок сушили в течение 6 ч при 60 С. Выход: 650 г. На фиг. 2, прилагаемой к данному описанию, представлена порошковая рентгенограмма (XRPD), на которой показан, главным образом, по существу, чистый образец формы II, полученной способом, описанным в данном примере. Значения 2 и интенсивности приведены в табл. 2. На фиг. 5 представлена термограмма DSC кристаллической формы II, полученной способом, описанным в данном примере (интервал от 30,0 до 350 С, 10,00 С/мин; N2 80 мл/мин). На фиг. 8 показаны кристаллы кристаллической формы II соединения формулы I. Пример 1. Получение формы III. мулы I получали способом примера 4 в WO 2006/027795, как указано в представленном выше примере 11. Соединение формулы I, полученное указанным выше способом, суспендировали в смеси 2,5 л диметилформамида и 4,1 л ацетона и нагревали до 50 С. Реакционную массу выдерживали при той же температуре в течение 30-40 мин, затем медленно охлаждали до 25-30 С и выдерживали при этой температуре в течение 60 мин. Суспензию продукта затем охлаждали до -5 С, фильтровали и промывали 0,8 л ацетона. Влажный осадок суспендировали в 0,4 л ацетона и кипятили с обратным холодильником. Охлажденную суспензию фильтровали и промывали 0,4 л ацетона. Влажный осадок сушили в течение 6 ч при 60 С. Выход: 500 г. На фиг. 3, прилагаемой к данному описанию, представлена порошковая рентгенограмма (XRPD), на которой показан, главным образом, по существу, чистый образец формы III, полученной способом, описанным в данном примере. Значения 2 и интенсивности приведены в табл. 3. На фиг. 6 представлена термограмма DSC кристаллической модификации формы III, полученной способом, описанным в данном примере (в интервале от 40,0 до 325 С, 10,00 С/мин; N2 80 мл/мин). На фиг. 9 показаны кристаллы кристаллической формы III соединения формулы I. Пример 2. Получение формы III.(3,5-бис-Трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамид формулы I получали согласно способу примера 4 в WO 2006/027795, как указано в представленном выше примере 11. Соединение формулы I, полученное указанным выше способом, суспендировали в смеси 2,5 л диметилформамида и 4,1 л гексана и нагревали до 50 С. Реакционную массу выдерживали при той же температуре в течение 30-40 мин, медленно охлаждали до 25-30 С и выдерживали при этой температуре в течение 12 ч. Суспензию продукта затем охлаждали до -5 С, фильтровали и промывали 0,8 л гексана. Влажный осадок сушили в течение 6 ч при 60 С. Выход: 400 г; DSC: 248,2 (пик). Пример 3. Получение формы III.(3,5-бис-Трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамид формулы I, получали согласно способу примера 4 в WO 2006/027795, как указано в представленном выше примере 11. Соединение формулы I, полученное указанным выше способом, суспендировали в смеси 2,5 л диметилформамида и 4,1 л толуола и нагревали до 50 С. Реакционную массу выдерживали при той же температуре в течение 30-40 мин, медленно охлаждали до 25-30 С и выдерживали при этой температуре в течение 12 ч. Суспензию далее охлаждали до -5 С, фильтровали и промывали 0,8 л толуола. Влажный осадок сушили в течение 6 ч при 60 С. Выход: 450 г; DSC: 246,7 (пик). Пример 4. Получение формы III.(3,5-бис-Трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2-иламино)фенил]бензамид формулы I получали согласно способу примера 4 в WO 2006/027795, как указано в представленном выше примере 11. Соединение формулы I, полученное указанным выше способом, суспендировали в смеси 2,5 л уксусной кислоты и нагревали до 50 С. Реакционную массу выдерживали при той же температуре в течение 30-40 мин, медленно охлаждали до 25-30 С и выдерживали при этой температуре в течение 96 ч. Продукт фильтровали и промывали 0,4 л охлажденной уксусной кислоты. Влажный осадок сушили в течение 6 ч при 60 С. Выход: 650 г; DSC: 246,3 (пик). Пример 5. Получение формы III. Форму I, полученную по примеру 10 (40 г), суспендировали в смеси 120 мл диметилформамида и 200 мл ацетона и нагревали до 50 С. Реакционную массу выдерживали при той же температуре в течение 30-40 мин, медленно охлаждали до 25-30 С и выдерживали при этой температуре в течение 60 мин. Суспензию охлаждали до -5 С, фильтровали и промывали 40 мл ацетона. Влажный осадок суспендировали в 200 мл ацетона, нагревали до температуры кипения с обратным холодильником и выдерживали в течение 60 мин при температуре кипячения с обратным холодильником. Суспензию охлаждали до 25 С, фильтровали и промывали 40 мл ацетона. Влажный осадок сушили в течение 6 ч при 60 С. Выход: 25 г; DSC: 247,0 С (пик). Пример 6. Получение формы III. Форму II, полученную по примеру 11 (650 г), суспендировали в смеси 1,95 л диметилформамида и 3,25 л ацетона и нагревали до 50 С. Реакционную массу выдерживали при той же температуре в течение 30-40 мин. Реакционную массу медленно охлаждали до 25-30 С и выдерживали при этой температуре в течение 60 мин. Продукт фильтровали и промывали 0,7 л ацетона. Влажный осадок суспендировали в 3,5 л ацетона и нагревали до температуры кипячения с обратным холодильником. Суспензию охлаждали до 25 С, фильтровали и промывали 0,7 л ацетона. Влажный осадок сушили в течение 6 ч при 60 С. Выход: 395 г; DSC: 246,5 С (пик). Как использовано в данном описании, термин "приблизительно" относится к отклонению в количестве или диапазоне, которое является общепринятым в области органической химии, например к конкретному отклонению в температурах или времени при измерении в реальных ситуациях в лаборатории органической химии, при увеличении масштаба или в производственном помещении, или в оценке анти- 16019223 пролиферативных средств. Любой диапазон или количество, используемое в описании настоящего изобретения, которые модифицированы термином "приблизительно", также являются частью изобретения,если они не дополнены термином "приблизительно". Например, указание на "приблизительно от 10 до приблизительно 20" также включает указание на "от 10 до 20". Следует отметить, как использовано в данном описании и прилагаемой формуле изобретения, что форма единственного числа включает множественное число, если содержание явно не указывает на иное. Таким образом, например, указание на композицию, содержащую "соединение", включает смесь двух или более соединений. Также следует отметить, что термин "или" обычно используют в его значении"и/или", если содержание явно не указывает на иное. Все публикации и патентные заявки в данном описании соответствуют уровню среднего специалиста в области, к которой относится настоящее изобретение. Изобретение описано с помощью различных и предпочтительных вариантов осуществления и способов. Однако следует понимать, что могут быть проведены изменения и модификации, сохраняя сущность и объем изобретения. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Кристаллическая форма III (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3-илпиримидин-2 иламино)фенил]бензамида (формула I), которая имеет характеристики XRPD 2. Кристаллическая форма III по п.1, которая имеет температуру плавления 240 С или выше. 3. Кристаллическая форма III по п.1, которая является, по существу, чистой. 4. Способ получения кристаллической формы III (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин 3-илпиримидин-2-иламино)фенил]бензамида (формула I), включающий обработку соединения формулыI в кристаллической форме I или II, имеющих следующие характеристики XRPD: Форма II уксусной кислотой или смесью диметилформамида и ацетона, гексана или толуола и последующую обработку однократно обработанного соединения уксусной кислотой, ацетоном, гексаном или толуолом. 5. Способ по п.4, включающий обработку соединения формулы I в кристаллической форме I или II уксусной кислотой и последующую обработку однократно обработанного соединения уксусной кислотой. 6. Способ по п.4, включающий обработку соединения формулы I в кристаллической форме I или II смесью диметилформамида и ацетона и последующую обработку однократно обработанного соединения ацетоном. 7. Способ по п.4, включающий обработку соединения формулы I в кристаллической форме I или II смесью диметилформамида и гексана и последующую обработку однократно обработанного соединения гексаном. 8. Способ по п.4, включающий обработку соединения формулы I в кристаллической форме I или II смесью диметилформамида и толуола и последующую обработку однократно обработанного соединения толуолом. 9. Фармацевтическая композиция, содержащая фармацевтически приемлемый эксципиент и кристаллическую форму иламино)фенил]бензамида (формула I), как определено в п.1. 10. Применение кристаллической формы III (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3 илпиримидин-2-иламино)фенил]бензамида (формула I), где кристаллическая форма III является такой,как определено в п.1, для изготовления антипролиферативного лекарственного средства для лечения опухолевого заболевания. 11. Способ лечения субъекта, нуждающегося в антипролиферативной терапии, включающий введение субъекту кристаллической формы III (3,5-бис-трифторметил)-N-[4-метил-3-(4-пиридин-3 илпиримидин-2-иламино)фенил]бензамида (формула I), где кристаллическая форма III является такой,как определено в п.1.
МПК / Метки
МПК: A61K 31/506, C07D 401/04, A61P 35/02
Метки: фениламинопиримидина, формы, кристаллические, производных
Код ссылки
<a href="https://eas.patents.su/30-19223-kristallicheskie-formy-proizvodnyh-fenilaminopirimidina.html" rel="bookmark" title="База патентов Евразийского Союза">Кристаллические формы производных фениламинопиримидина</a>
Новые кристаллические формы периндоприл эрбумина

Номер патента: 8603
Опубликовано: 29.06.2007
Авторы: Штресслер Кристоф, Леллек Вит, Фесслер Роже
МПК: C07D 209/42
Метки: кристаллические, периндоприл, новые, формы, эрбумина
Формула / Реферат:
1. Кристаллическая форма d периндоприл эрбумина, обладающая следующими параметрами дифракции при порошковой рентгенографии с использованием облучения СuKa: 2. Кристаллическая форма e периндоприл эрбумина, обладающая следующими параметрами дифракции при порошковой рентгенографии с использованием облучения СuKa: 3. Лекарственное средство, содержащее кристаллическую форму периндоприл эрбумина по п.1 или 2. 4. Применение кристаллических форм...
Новые кристаллические формы гидробромида клопидогреля и способы их получения

Номер патента: 8972
Опубликовано: 26.10.2007
Авторы: Пигера Павел, Степанкова Гана, Гайичек Йосеф
МПК: C07D 221/00, C07D 333/00, C07D 495/04...
Метки: получения, клопидогреля, кристаллические, гидробромида, формы, способы, новые
Формула / Реферат:
1. Гидробромид клопидогреля в кристаллической форме I, отличающийся рентгенограммой с характеристическими межплоскостными расстояниями d, равными 4,01; 4,39 и 3,17 Е. 2. Гидробромид клопидогреля в кристаллической форме I по п.1, отличающийся межплоскостными расстояниями d, равными 3,12; 6,99; 5,5; 4,29 и 3,65 Е. 3. Гидробромид клопидогреля в кристаллической форме I по п.1 или 2, отличающийся наличием в инфракрасном спектре полос поглощения при...
Кристаллические формы макролидных соединений, обладающих противовоспалительной активностью

Номер патента: 13082
Опубликовано: 26.02.2010
Авторы: Брешелло Роберто, Мораззони Габриеле, Вердзини Массимо, Ди Мария Алессандро, Мелотто Элиза, Браджа Дарио, Рестелли Анджело, Пеллачини Франко, Наполетано Мауро, Массаччеси Франко, Мараньи Паоло, Микьелетто Иван, Котарка Ливиус
МПК: C07H 17/08, A61P 29/00, A61K 31/7048...
Метки: соединений, обладающих, формы, кристаллические, макролидных, противовоспалительной, активностью
Формула / Реферат:
1. Кристаллическая форма I соединения формулы (I)характеризующаяся порошковой рентгенограммой, содержащей значения угла 2q, составляющие приблизительно 4,9; приблизительно 8,5; приблизительно 9,1; приблизительно 9,6; приблизительно 10,3; приблизительно 11,1; приблизительно 14,5; приблизительно 17,0; приблизительно 18,2; приблизительно 19,3.2. Кристаллическая форма по п.1, характеризующаяся порошковой рентгенограммой, в основном соответствующей...
Кристаллические формы eto2c-ch2-(r)cgl-aze-pab-oh

Номер патента: 3264
Опубликовано: 27.02.2003
Авторы: Эдвардссон Даниель, Хедстрём Лена, Петтерссон Урсула, Лундблад Анита
МПК: C07K 5/062, A61K 38/55, A61P 7/02...
Метки: eto2c-ch2-(r)cgl-aze-pab-oh, формы, кристаллические
Формула / Реферат:
1. По существу, кристаллическая форма EtO2C-CH2-(R)Cgl-Aze-Pab-OH или его фармацевтически приемлемой соли. 2. Соединение по п.1, которое находится в безводной форме. 3. Соединение по п.2, которое находится не в форме соли. 4. Соединение по любому из пп.1-3, которое содержит не более 2 маc.% воды. 5. Соединение по любому из пп.1-4, характеризуемое кривой дифференциальной сканирующей калориметрии, на которой при скорости нагрева 5шC/мин в...
Новые кристаллические формы макролидного антибиотика

Номер патента: 3417
Опубликовано: 24.04.2003
Авторы: Мортон Барри Джеймс, Рейган Коулман Брендан, Рафка Роберт Джон, Аллен Даглас Джон Мелдрам
МПК: A61K 31/70, C07H 17/08
Метки: формы, кристаллические, новые, макролидного, антибиотика
Формула / Реферат:
1. Кристаллическая форма соединения формулы 1 2. Кристаллическая форма по п.1, представляющая собой одну из форм: безводную форму, моногидрат и сесквигидрат. 3. Кристаллическая форма по п.1, которая имеет картину дифракции рентгеновских лучей на порошке, показывающую характеристические пики, выраженные в 2q , при примерно 6,0: 8,6; 9,7; 15,4; 15,9; 17,5; 18,2; 18,7 и 21. 4. Кристаллическая форма по п.3, которая имеет картину дифракции...
Предыдущий патент: Способ получения сложных диэфиров из динитрильных соединений
Следующий патент: Композиции и методы для лечения рака молочной железы
Случайный патент: Сухая грануляция металлургического шлака