Агонисты рецептора меланокортина
Номер патента: 19146
Опубликовано: 30.01.2014
Авторы: Шим Донг Суп, Ли Коо, Ли Санг -Дае, Моон Санг Пил, Чои Сунг Пил, Ли Хиун Мин, Ахн Ин Ае, Чунг Соо Йонг, Ли Хиун Хо











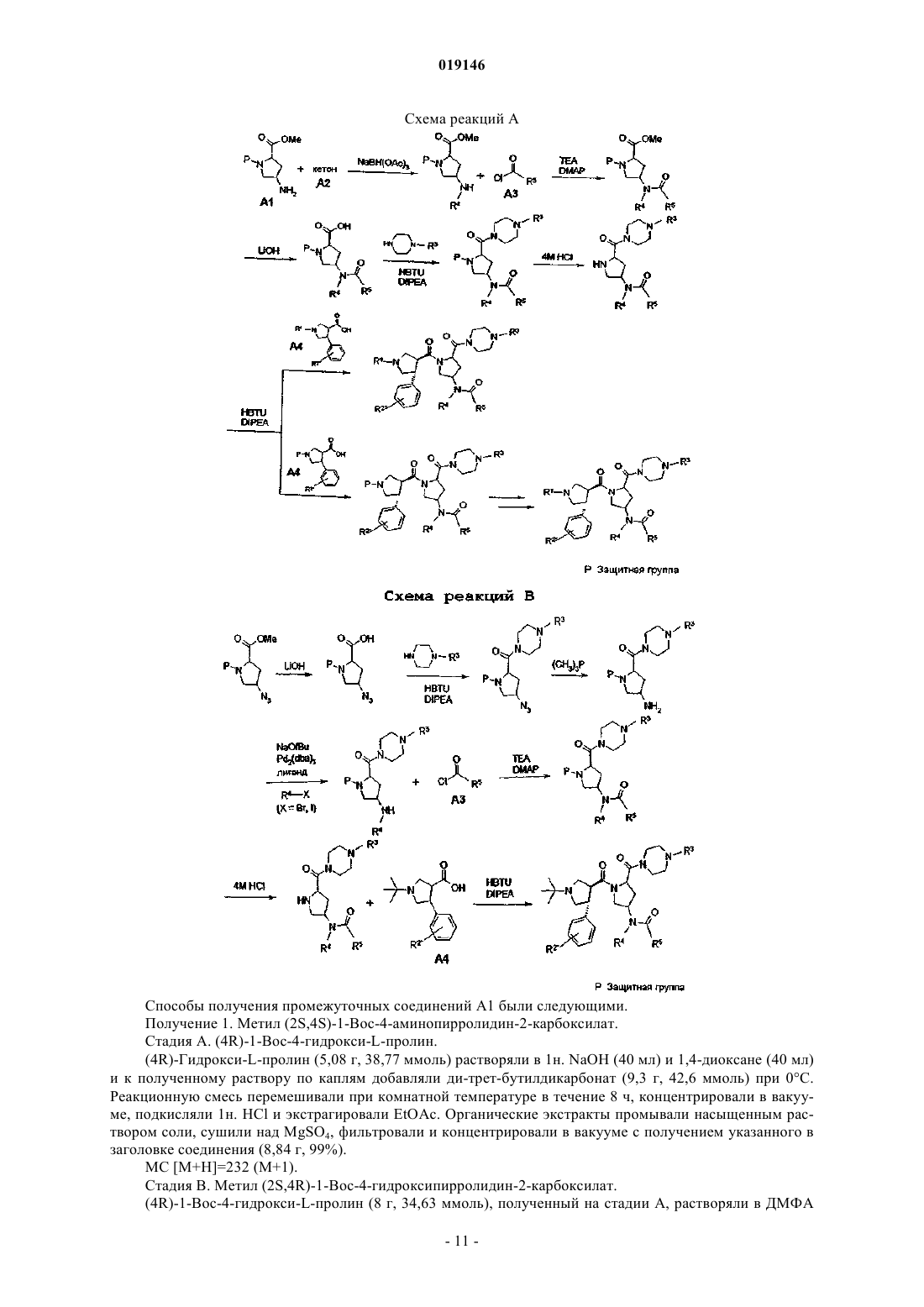

















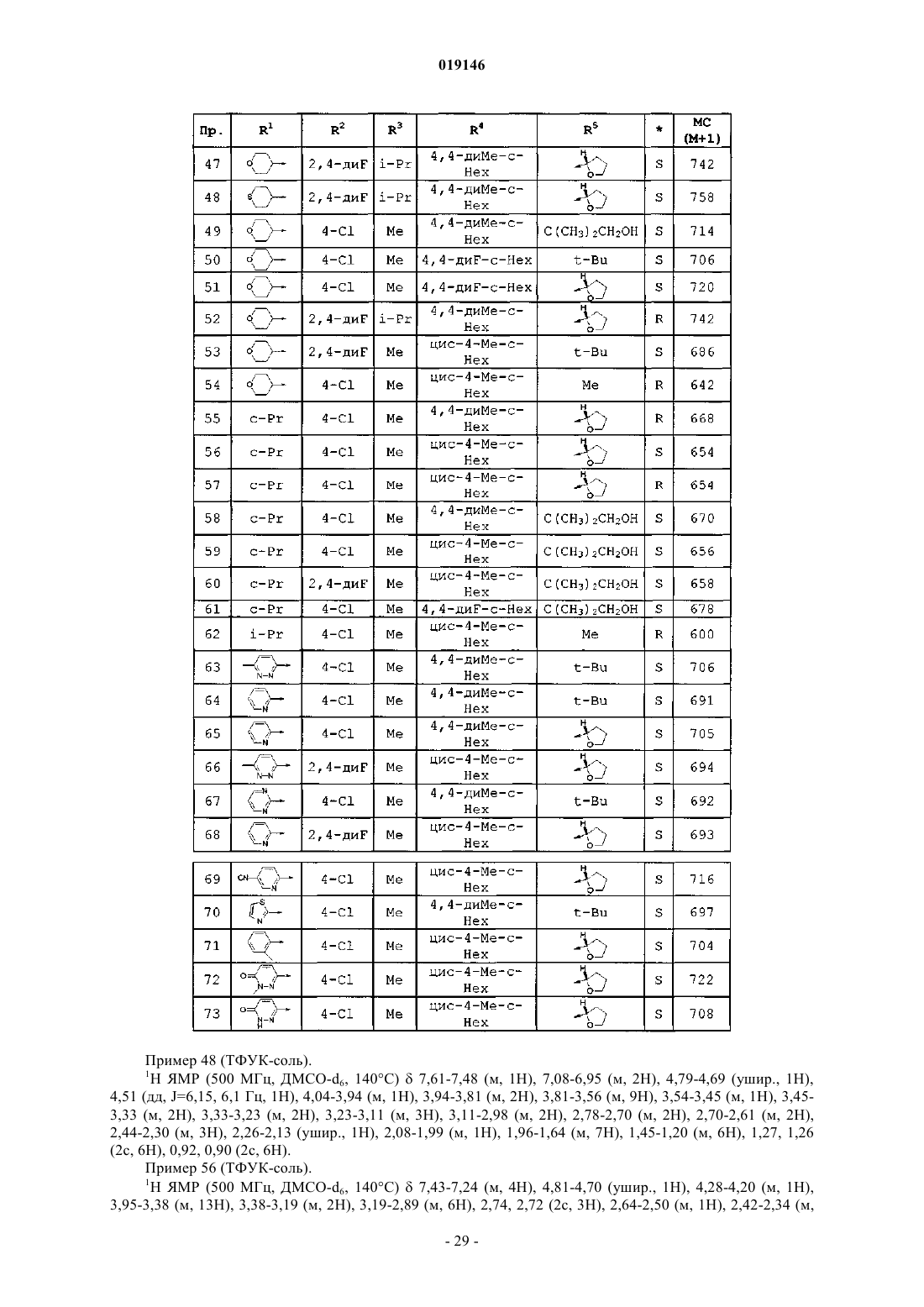
Формула / Реферат
1. Соединение следующей формулы 1

где R1 представляет собой водород или представляет собой C1-С10алкил, С3-С7циклоалкил, С6-С10арил, гетероцикл или гетероарил, каждый из которых является незамещенным или замещен по меньшей мере одним заместителем, выбранным из группы, состоящей из галогена, амино, С1-С4алкила, трифторметила, С1-С4алкокси, циано и оксо;
R2 представляет собой фенил или шестичленный гетероарил, каждый из которых является незамещенным или моно- или дизамещен заместителем(ями), выбранным(и) из группы, состоящей из галогена, гидрокси, С1-С4алкила, С1-С4алкокси, циано и амино;
R3 представляет собой водород или С1-С6алкил, каждый из которых является незамещенным или замещен заместителем(ями), выбранным(и) из группы, состоящей из галогена, метила, трифторметила, гидрокси и амино;
R4 представляет собой С4-С7циклоалкил или моноциклический гетероцикл, каждый из которых является незамещенным или моно- или полизамещен заместителем(ями), выбранным(и) из группы, состоящей из галогена, гидрокси, С1-С4алкила, трифторметила, C1-С4алкокси и оксо; или представляет собой фенил или шестичленный гетероарил, каждый из которых является незамещенным или моно- или дизамещен заместителем(ями), выбранным(и) из группы, состоящей из галогена, гидрокси, трифторметила и амино; и
R5 представляет собой С1-С6алкил, дифторметил, трифторметил, С3-С8циклоалкил, амино, С1-С4алкиламино, ди(С1-С4алкил)амино, моноциклический гетероарил или моноциклический гетероцикл, где алкил является незамещенным или замещен по меньшей мере одним заместителем, выбранным из группы, состоящей из фтора, гидрокси, меркапто, ацетокси, амино, ацетиламино, циано, карбамоила, диметилкарбамоила и оксо, и гетероарил является незамещенным или моно- или дизамещен заместителем(ями), выбранным(и) из группы, состоящей из галогена, гидрокси, метила, трифторметила, метокси и амино, где
"гетероцикл" представляет собой 4-8-членное кольцо, содержащее от 1 до 2 гетероатомов, выбранных из группы, состоящей из атома азота, атома кислорода и атома серы, и
"гетероарил" представляет собой ароматическое 5-6-членное кольцо, содержащее от 1 до 4 гетероатомов, выбранных из группы, состоящей из атома азота, атома кислорода и атома серы,
или его фармацевтически приемлемая соль или изомер.
2. Соединение формулы 1 по п.1, где R1 представляет собой водород, метил, этил, трифторэтил, пропил, изопропил, бутил, изобутил, трет-бутил, циклопропил, циклобутил, циклопентил или циклогексил или представляет собой фенил, оксазолинил, имидазолинил, тиазолинил, тетрагидропиранил, тетрагидротиопиранил, имидазолил, оксазолил, тиазолил, пиразолил, триазолил, пиридинил, пиримидинил, пиперидинил или пиридазинил, каждый из которых является незамещенным или замещен заместителем(ями), выбранным(и) из группы, состоящей из галогена, метила, циано и оксо или его фармацевтически приемлемая соль или изомер.
3. Соединение формулы 1 по п.2, где R1 представляет собой изопропил, трет-бутил или циклопропи; или представляет собой фенил, тетрагидропиранил, тиазолил, пиридинил, пиримидинил или пиридазинил, каждый из которых является незамещенным или замещен заместителем(ями), выбранным(и) из группы, состоящей из галогена, метила и циано, или его фармацевтически приемлемая соль или изомер.
4. Соединение формулы 1 по п.1, где R2 представляет собой фенил, который является незамещенным или моно- или дизамещен заместителем(ями), выбранным из группы, состоящей из фтора, хлора и брома, или его фармацевтически приемлемая соль или изомер.
5. Соединение формулы 1 по п.4, где R2 представляет собой 4-хлорфенил или 2,4-дифторфенил, или его фармацевтически приемлемая соль или изомер.
6. Соединение формулы 1 по п.1, где R3 представляет собой водород, метил, этил, пропил, изопропил, бутил, изобутил или трет-бутил, или его фармацевтически приемлемая соль или изомер.
7. Соединение формулы 1 по п.6, где R3 представляет собой водород, метил, этил или изопропил, или его фармацевтически приемлемая соль или изомер.
8. Соединение формулы 1 по п.1, где R4 представляет собой циклопентил, циклогексил, циклогептил, 4-метилциклогексил, 4,4-диметилциклогексил, 4-фторциклогексил, 4,4-дифторциклогексил или 4-трифторметилциклогексил или представляет собой фенил, который является незамещенным или моно- или дизамещен заместителем(ями), выбранным(и) из группы, состоящей из фтора, хлора, или его фармацевтически приемлемая соль или изомер.
9. Соединение формулы 1 по п.8, где R4 представляет собой циклогексил, 4-метилциклогексил, 4,4-диметилциклогексил, 4,4-дифторциклогексил или 2,4-дифторфенил, или его фармацевтически приемлемая соль или изомер.
10. Соединение формулы 1 по п.1, где R5 представляет собой метил, трифторметил, гидроксиметил, метоксиметил, этоксиметил, пропил, изопропил, изобутил, трет-бутил, -СН2СН2ОН, СН(СН3)СН2ОН,
-С(СН3)2СН2ОН, -С(СН3)(СН2ОН)2, оксазолинил, имидазолинил, тиазолинил, тетрагидропиранил, имидазолил, оксазолил, тиазолил, пиразолил, триазолил, фуранил, тетрагидрофуранил, дигидрофуранил, тетрагидропиранил, пиридинил или пиперидинил, или его фармацевтически приемлемая соль или изомер.
11. Соединение формулы 1 по п.10, где R5 представляет собой изопропил, трет-бутил, -С(СН3)2СН2ОН, фуранил или тетрагидрофуранил, или его фармацевтически приемлемая соль или изомер.
12. Соединение формулы 1 по п.1, где R1 представляет собой изопропил, трет-бутил или циклопропил или представляет собой фенил, тетрагидропиранил, тиазолил, пиридинил, пиримидинил или пиридазинил, каждый из которых является незамещенным или замещен заместителем(ями), выбранным(и) из группы, состоящей из галогена, метила и циано;
R2 представляет собой 4-хлорфенил или 2,4-дифторфенил;
R3 представляет собой водород, метил, этил или изопропил;
R4 представляет собой циклогексил, 4-метилциклогексил, 4,4-диметилциклогексил, 4,4-дифторциклогексил или 2,4-дифторфенил и
R5 представляет собой изопропил, трет-бутил, -С(СН3)2СН2ОН, фуранил или тетрагидрофуранил, или его фармацевтически приемлемая соль или изомер.
13. Соединение формулы 1 по п.12, которое выбирают из группы, включающей следующие соединения:
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)ацетамид;
N-[(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-(пиперазин-1-илкарбонил)пирролидин-3-ил]-N-(4,4-диметилциклогексил)ацетамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)-3-гидрокси-2,2-диметилпропанамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)ацетамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-этилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)ацетамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-изопропилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)ацетамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-этилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)ацетамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-изопропилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)ацетамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)-2,2-диметилпропанамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)-2,2-диметилпропанамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-этилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-изопропилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)-3-гидрокси-2,2-диметилпропанамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-2,2-диметил-N-(цис-4-метилциклогексил)пропанамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-2,2-диметил-N-(цис-4-метилциклогексил)пропанамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-3-гидрокси-2,2-диметил-N-(цис-4-метилциклогексил)пропанамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-3-гидрокси-2,2-диметил-N-(цис-4-метилциклогексил)пропанамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-этилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-изопропилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)-3-фурамид;
(2R)-N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-дифторциклогексил)-2,2-диметилпропанамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-дифторциклогексил)ацетамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-этилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-дифторциклогексил)ацетамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-изопропилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-дифторциклогексил)ацетамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-дифторциклогексил)-2,2-диметилпропанамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-дифторциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5R)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид;
N-{(3S,5R)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-2,2-диметил-N-(цис-4-метилциклогексил)пропанамид;
(2S)-N-{(3S,5R)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5R)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
N-{(3S,5R)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-3-гидрокси-2,2-диметил-N-(цис-4-метилциклогексил)пропанамид;
N-{(3S,5R)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)ацетамид;
N-{(3S,5R)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)ацетамид;
N-{(3S,5R)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-изопропилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)ацетамид;
(2R)-N-{(3S,5R)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-(тетрагидро-2Н-пиран-4-ил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-циклопропилпирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-(6-метилпиридазин-3-ил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-(6-хлорпиридазин-3-ил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1,6-дигидропиридазин-3-илпирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-фенилпирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-(тетрагидро-2Н-пиран-4-ил)пирролидин-3-ил]карбонил}-5-[(4-изопропилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-(тетрагидро-2Н-тиопиран-4-ил)пирролидин-3-ил]карбонил}-5-[(4-изопропилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид;
N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-(тетрагидро-2Н-пиран-4-ил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)-3-гидрокси-2,2-диметилпропанамид;
N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-(тетрагидро-2Н-пиран-4-ил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-дифторциклогексил)-2,2-диметилпропанамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-(тетрагидро-2Н-пиран-4-ил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-дифторциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5R)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-(тетрагидро-2Н-пиран-4-ил)пирролидин-3-ил]карбонил}-5-[(4-изопропилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид;
N-{(3S,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-(тетрагидро-2Н-пиран-4-ил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-2,2-диметил-N-(цис-4-метилциклогексил)пропанамид;
N-{(3S,5R)-1-{[(3S,4R)-4-(4-хлорфенил)-1-(тетрагидро-2Н-пиран-4-ил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4-метилциклогексил)ацетамид;
(2S)-N-{(3S,5R)-1-{[(3S,4R)-4-(4-хлорфенил)-1-циклопропилпирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-циклопропилпирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5R)-1-{[(3S,4R)-4-(4-хлорфенил)-1-циклопропилпирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-циклопропилпирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)-3-гидрокси-2,2-диметилпропанамид;
N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-циклопропилпирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)-3-гидрокси-2,2-диметилпропанамид;
N-{(3S,5S)-1-{[(3S,4R)-1-циклопропил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)-3-гидрокси-2,2-диметилпропанамид;
N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-циклопропилпирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-дифторциклогексил)-3-гидрокси-2,2-диметилпропанамид;
N-{(3S,5R)-1-{[(3S,4R)-4-(4-хлорфенил)-1-изопропилпирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)ацетамид;
N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-(6-метилпиридазин-3-ил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)-2,2-диметилпропанамид;
N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-пиридин-2-илпирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)-2,2-диметилпропанамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-пиридин-2-илпирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1,6-дигидропиридазин-3-илпирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-пиримидин-2-илпирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)-2,2-диметилпропанамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(2,4-дифторфенил)-1-пиридин-2-илпирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-(5-цианопиридин-2-ил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-(1,3-тиазол-2-ил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(4,4-диметилциклогексил)-2,2-диметилпропанамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-(2-метилфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-(1-метил-6-оксо-1,6-дигидропиридазин-3-ил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-4-(4-хлорфенил)-1-(6-оксо-1,6-дигидропиридазин-3-ил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3R,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4S)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3R,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид;
(2S)-N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(2,4-дифторфенил)тетрагидрофуран-2-карбоксамид;
N-[(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-(пиперазин-1-илкарбонил)пирролидин-3-ил]-N-(2,4-дифторфенил)-2-метилпропанамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(2,4-дифторфенил)-2-метилпропанамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(2,4-дифторфенил)-2,2-диметилпропанамид;
N-[(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-(пиперазин-1-илкарбонил)пирролидин-3-ил]-N-(2,4-дифторфенил)-2,2-диметилпропанамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(2,4-дифторфенил)-2,2-диметилпропанамид;
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(2,4-дифторфенил)-2-фурамид и
N-{(3S,5S)-1-{[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил}-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил}-N-(2,4-дифторфенил)-2-фурамид,
или его фармацевтически приемлемая соль или изомер.
14. Композиция, обладающая агонистической активностью к рецептору меланокортина, для лечения или профилактики заболеваний, опосредованных активностью указанного рецептора, содержащая соединение формулы 1, как определено в п.1, или его фармацевтически приемлемую соль или изомер в качестве активного ингредиента вместе с фармацевтически приемлемым носителем.
15. Композиция по п.14, где указанным заболеванием является ожирение.
16. Композиция по п.14, где указанным заболеванием является диабет.
17. Композиция по п.14, где указанным заболеванием является воспаление.
18. Композиция по п.14, где указанным заболеванием является эректильная дисфункция.
Текст
Настоящее изобретение относится к соединению, проявляющему хорошую агонистическую активность в отношении рецептора меланокортина, или к его фармацевтически приемлемой соли или изомеру и к агонистической композиции к рецептору меланокортина, содержащей указанное выше соединение в качестве активного ингредиента. Ли Коо, Ли Санг Дае, Моон Санг Пил,Ахн Ин Ае, Чои Сунг Пил, Ли Хиун Хо, Шим Донг Суп, Чунг Соо Йонг, Ли Хиун Мин (KR) Медведев В.Н. (RU)(71)(73) Заявитель и патентовладелец: ЭЛ ДЖИ ЛАЙФ САЙЕНСИЗ ЛТД. Область техники Настоящее изобретение относится к соединению следующей формулы 1, проявляющему хорошую агонистическую активность в отношении рецептора меланокортина, или к его фармацевтически приемлемой соли или изомеру где R1, R2, R3, R4 и R5 являются такими, как определено ниже. Настоящее изобретение также относится к способу получения соединения указанной выше формулы 1. Настоящее изобретение также относится к агонистической композиции для рецептора меланокортина, содержащей соединение указанной выше формулы 1 в качестве активного ингредиента, в частности композиции для предотвращения и лечения ожирения, диабета, воспаления и эректильной дисфункции. Предшествующий уровень техники В семействе меланокортинов было клонировано и охарактеризовано пять подтипов рецепторов. Gбелоксвязанные рецепторы (GPCR) стимулируют сАМР-зависимую сигнальную трансдукцию во многих различных тканях, осуществляя опосредованно широкий ряд физиологических функций. Рецептор 1 меланокортина (MC1R) экспрессируется, главным образом, в меланоцитах, моноцитах и тучных клетках для опосредования пигментации волос и кожи и торможения воспаления. Рецептор MC2R экспрессируется в адипоцитах и клетках надпочечников для опосредования стероидогенеза в надпочечной железе. Рецептор MC3R присутствует в головном мозге, гипоталамусе, сердце, кишке и плаценте и ассоциирован с энергетическим гомеостазом и воспалением. Рецептор MC4R экспрессируется единственно в головном мозге и контролирует пищевое поведение, энергетический гомеостаз и эректильную функцию. Нокаутированные по рецептору MC4R мыши обнаружили фенотип гиперфазии и ожирения. Рецептор MC5R обнаружен в широком ряде тканей и, как полагают, играет роль в функции экзокринных желез. Учитывая множество физиологических функций рецепторов меланокортинов, огромное число соединений было сконструировано и синтезировано в поисках потенциальных агонистов и антагонистов. Первыми примерами являются синтетические пептиды и пептидные аналоги, которые идентифицированы на основе эндогенного агониста, такого как MSH. Указанные пептидные агонисты были использованы для изучения функции данных рецепторов. NDP-MSH является высокоактивным и неселективным агонистом рецепторов MC1R, 3R, 4R и 5R и, как было показано, снижает потребность в пище и прирост массы тела на модели крыс. Циклический гептапептид MT-II является агонистом с подобным неселективным профилем, и его терапевтическое использование было апробировано в клинических испытаниях для лечения эректильной дисфункции. Было показано, что низкомолекулярные агонисты рецепторов меланокортинов проявляют значительную активность в испытаниях лекарственных средств, направленных на лечение ожирения, половой дисфункции или воспаления. Например, была идентифицирована серия эффективных и селективных агонистов рецептора MC4R, при этом один из агонистов продемонстрировал значительный эффект, осуществляя усиление эректильного ответа у мышей (J. Med. Chem. 2002, 45, 4849). Также был идентифицирован ряд агонистов рецептора MC4R, который проявлял гипофазическую активность и действие против ожирения на модели крыс (Bioorg. Med. Chem. Lett. 2005, 75, 171, Bioorg. Med. Chem. Lett. 2005, 75, 3430,Bioorg. Med. Chem. Lett. 2005, 75, 3501). Кроме того, MerckCo. Inc. были поданы патентные заявки на различные соединения в качестве агонистов MC4R (WO 01/55109, WO 01/70337, WO 01/70708, WO 02/081443, WO 02/15909, WO 02/067869, WO 02/068387, WO 02/068388, WO 2004/087159, WO 2004/078716, WO 2004/078717, WO 2006/019787, WO 2006/020277, WO 2007/041052, WO 2007/041061,WO 2007/047496). Другими фармацевтическими компаниями также были поданы патентные заявки на различные низкомолекулярные агонисты рецепторов MCR (WO 02/059095, WO 02/059107, WO 02/059117, WO 02/059108, WO 02/085925, WO 03/009847, WO 03/009850, WO 02/018327, WO 2005/040109, WO 2005/047251, WO 2005/077935, WO 2005/077935, WO 2006/072393, WO 2007/015157, WO 2007/015162, JP 2007131570, WO 2007/096186, WO 2007/096763, WO 2007/141343, WO 2008/039418, WO 2008/007930). Также сообщалось, что селективные низкомолекулярные агонисты рецептора MC1R проявляют противовоспалительную активность на модели острого воспаления у мыши (J. Med. Chem., 2003, 46,1123). Учитывая неразрешенные недостатки различных фармацевтических соединений, описанных выше,до сих пор существует необходимость в данной области в получении низкомолекулярных агонистов ре-1 019146 цепторов MCR и фармакологических композиций, имеющих улучшенные фармакологические профили. Поэтому целью настоящего изобретение является предоставление новых соединений, которые пригодны для лечения ожирения, диабета, эректильной дисфункции и воспаления. Раскрытие изобретения Указанные выше пептидные агонисты рецепторов MCR имеют значительные ограничения для использования в качестве перорально вводимого лекарственного средства из-за их молекулярных характеристик. Кроме того, большинство низкомолекулярных агонистов MCR непептидной природы, описанных до настоящего времени, должны быть улучшены в отношении пероральной поглощающей способности, проницаемости гематоэнцефалического барьера и эффективности с целью их использования в качестве лекарственного средства. Поэтому задачей настоящего изобретения является предоставление низкомолекулярных агонистовMCR непептидной природы с новой структурой, которые могут быть использованы для предотвращения и лечения ожирения, диабета, эректильной дисфункции и воспаления. Особенно задачей настоящего изобретения является предоставление непептидного соединения формулы 1, проявляющего превосходную агонистическую активность в отношении рецепторов MCR, в частности селективность относительно MC4R, или его фармацевтически приемлемой соли или изомера. Другой задачей настоящего изобретения является предоставление способа получения соединения формулы 1. Другой задачей настоящего изобретения является предоставление агонистической композиции для рецептора меланокортина, содержащей соединение формулы 1 или его фармацевтически приемлемую соль или изомер в качестве активного ингредиента вместе с фармацевтически приемлемым носителем. В частности, композиция согласно настоящему изобретению проявляет сильное действие по предотвращению и лечению ожирения, диабета, эректильной дисфункции и воспаления. Наилучший способ осуществления изобретения Настоящее изобретение предоставляет соединение следующей формулы 1 или его фармацевтически приемлемую соль или изомер где R1 представляет собой водород или представляет собой C1-С 10 алкил, С 3-С 7 циклоалкил, С 6-С 10 арил,гетероцикл или гетероарил, каждый из которых является незамещенным или замещен по меньшей мере одним заместителем, выбранным из группы, состоящей из галогена, амино, С 1-С 4 алкила, трифторметила,гидрокси, C1-С 4 алкокси, циано и оксо;R2 представляет собой фенил или шестичленный гетероарил, каждый из которых является незамещенным или моно- или дизамещен заместителями, выбранными из группы, состоящей из галогена, гидрокси, С 1-С 4 алкила, С 1-С 4 алкокси, циано и амино;R3 представляет собой водород или представляет собой C1-С 6 алкил или С 3-С 7 циклоалкил, каждый из которых является незамещенным или замещен заместителями, выбранными из группы, состоящей из галогена, метила, трифторметила, гидрокси и амино;R4 представляет собой С 4-С 7 циклоалкил или моноциклический гетероцикл, каждый из которых является незамещенным или моно- или полизамещен заместителями, выбранными из группы, состоящей из галогена, гидрокси, С 1-С 4 алкила, трифторметила, С 1-С 4 алкокси и оксо; или представляет собой фенил или шестичленный гетероарил, каждый из которых является незамещенным или моно- или дизамещен заместителями, выбранными из группы, состоящей из галогена, гидрокси, С 1-С 4 алкила, трифторметила,С 1-С 4 алкокси и амино; иR5 представляет собой С 1-С 6 алкил, дифторметил, трифторметил, С 3-С 8 циклоалкил, амино, С 1 С 4 алкиламино, ди(С 1-С 4 алкил)амино, фенил, моноциклический гетероарил или моноциклический гетероцикл, где алкил является незамещенным или замещен по меньшей мере одним заместителем, выбранным из группы, состоящей из фтора, гидрокси, меркапто, С 1-С 4 алкокси, ацетокси, амино, ацетиламино,циано, карбамоила, диметилкарбамоила и оксо, и фенил или гетероарил является незамещенным или моно- или дизамещен заместителями, выбранными из группы, состоящей из галогена, гидрокси, метила,трифторметила, метокси и амино. В определениях заместителей у соединения формулы (1) согласно настоящему изобретению, термин "алкил" при использовании его самого по себе или в комбинации как "алкилокси" означает углеводородный радикал с прямой или разветвленной цепью. Термин "циклоалкил" представляет собой насыщенное алифатическое кольцо, включающее циклогексил. Термин "арил" представляет собой 6-10-членную ароматическую группу, включающую фенил,нафтил и т.д. Термин "гетероарил" представляет собой ароматическое 3-6-членное кольцо, содержащее от 1 до 4 гетероатов, выбранных из группы, состоящей из атома азота, атома кислорода и атома серы, которое может быть необязательно конденсировано с бензо или С 3-С 8 циклоалкилом. Примерами моноциклического гетероарила являются, но не ограничиваются ими, тиазол, оксазол, тиофен, фуран, пиррол, имидазол,изоксазол, пиразол, триазол, тиадиазол, тетразол, оксадиазол, пиридин, пиридазин, пиримидин, пиразин и подобные им группы. Примерами бициклического гетероарила являются, но не ограничиваются ими,индол, бензотиофен, бензофуран, бензимидазол, бензоксазол, бензизоксазол, бензотиазол, бензотиадиазол, бензотриазол, хинолин, изохинолин, пурин, фуропиридин и подобные им группы. Термин "гетероцикл" представляет собой 4-8-членное кольцо, содержащее от 1 до 2 гетероатов, выбранных из группы, состоящей из атома азота, атома кислорода и атома серы, которое может быть необязательно конденсировано с бензо или С 3-С 8 циклоалкилом и является насыщенным или ненасыщенным с 1 или 2 двойными связями. Примерами их являются, но не ограничиваются ими, пиперидин, морфолин,тиаморфолин, пирролидин, имидазолидин, тетрагидрофуран, пиперазин и подобные им группы. Предпочтительными соединениями среди соединений формулы 1 согласно настоящему изобретению являются такие соединения, где(i) R1 представляет собой водород, метил, этил, трифторэтил, пропил, изопропил, бутил, изобутил,трет-бутил, циклопропил, циклобутил, циклопентил или циклогексил; или представляет собой фенил,оксазолинил, имидазолинил, тиазолинил, тетрагидропиранил, тетрагидротиопиранил, имидазолил, оксазолил, тиазолил, пиразолил, триазолил, пиридинил, пиримидинил, пиперидинил или пиридазинил, каждый из которых является незамещенным или замещен заместителем(ями), выбранным(и) из группы, состоящей из галогена, метила, циано, оксо и гидрокси, и более предпочтительно R1 представляет собой изопропил, трет-бутил или циклопропил; или представляет собой фенил, тетрагидропиранил, тиазолил,пиридинил, пиримидинил или пиридазинил, каждый из которых является незамещенным или замещен заместителем(ями), выбранным(и) из группы, состоящей из галогена, метила, циано и гидрокси,ii) R2 представляет собой фенил, который является незамещенным или моно- или дизамещен заместителем(ями), выбранным(и) из группы, состоящей из фтора, хлора, брома, метокси и метила, и более предпочтительно R2 представляет собой 4-хлорфенил или 2,4-дифторфенил,iii) R3 представляет собой водород, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил,циклопропил, циклобутил или циклопентил и более предпочтительно R3 представляет собой водород,метил, этил или изопропил,iv) R4 представляет собой циклопентил, циклогексил, циклогептил, 4-метилциклогексил, 4,4 диметилциклогексил, 4-фторциклогексил, 4,4-дифторциклогексил или 4-трифторметилциклогексил; или представляет собой фенил, который является незамещенным или моно- или дизамещен заместителями,выбранными из группы, состоящей из фтора, хлора, метила и метокси, и более предпочтительно R4 представляет собой циклогексил, 4-метилциклогексил, 4,4-диметилциклогексил, 4,4-дифторциклогексил или 2,4-дифторфенил,v) R5 представляет собой метил, трифторметил, гидроксиметил, метоксиметил, этоксиметил, пропил, изопропил, изобутил, трет-бутил, -СН 2 СН 2 ОН, -СН(СН 3)СН 2 ОН, -С(СН 3)2 СН 2 ОН, -С(СН 3)(СН 2 ОН)2,-С(СН 3)2 СН 2 ОМе, -С (СН 3) 2CH2OEt, фенил, оксазолинил, имидазолинил, тиазолинил, тетрагидропиранил, имидазолил, оксазолил, тиазолил, пиразолил, триазолил, фуранил, тетрагидрофуранил, дигидрофуранил, тетрагидропиранил, пиридинил или пиперидинил и более предпочтительно R5 представляет собой изопропил, трет-бутил, -С(СН 3)2 СН 2 ОН, фуранил или тетрагидрофуранил. Наиболее предпочтительными соединениями среди соединений формулы 1 согласно настоящему изобретению являются соединения, гдеR1 представляет собой изопропил, трет-бутил или циклопропил или представляет собой фенил, тетрагидропиранил, тиазолил, пиридинил, пиримидинил или пиридазинил, каждый из которых является незамещенным или замещен заместителем(ями), выбранным(и) из группы, состоящей из галогена, метила, циано и гидрокси;R5 представляет собой изопропил, трет-бутил, -С(СН 3)2 СН 2 ОН, фуранил или тетрагидрофуранил. Репрезентативные соединения формулы 1 согласно настоящему изобретению включают следующие перечисленные соединения:N-(3S,5S)-1-[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3-ил]карбонил-5-[(4 метилпиперазин-1-ил)карбонил]пирролидин-3-ил-N-(2,4-дифторфенил)-2-фурамид. Соединения согласно настоящему изобретению также могут образовывать фармацевтически приемлемые соли. Такие фармацевтически приемлемые соли включают кислотно-аддитивные соли, образованные кислотой, имеющей фармацевтически приемлемый анион для образования нетоксичной кислотно-аддитивной соли, включающей, например, неорганические кислоты, такие как хлористо-водородная кислота, серная кислота, азотная кислота, фосфорная кислота, бромисто-водородная кислота, йодистоводородная кислота и тому подобное; органическую карбоновую кислоту, такую как винная кислота,муравьиная кислота, лимонная кислота, уксусная кислота, трихлоруксусная кислота, трифторуксусная кислота, глюконовая кислота, бензойная кислота, молочная кислота, фумаровая кислота, малеиновая кислота и тому подобное; сульфоновую кислоту, такую как метансульфокислота, бензолсульфокислота, птолуолсульфокислота или нафталинсульфокислота и тому подобное; и более предпочтительно кислотноаддитивные соли, образованные серной кислотой, метансульфокислотой или галогеноводородными кислотами и тому подобное. Соединения формулы 1 согласно настоящему изобретению могут быть преобразованы в их соли обычными способами. Соединения согласно настоящему изобретению могут иметь асимметрический углеродный центр и,таким образом, могут быть представлены в виде R- или S-изомерных форм, рацематов, диастереомерных смесей и индивидуальных диастереомеров. Настоящее изобретение охватывает все из указанных изомерных форм и смесей. В другом аспекте настоящее изобретение предоставляет способ получения соединения формулы 1,включающий стадию образования амидной связи при взаимодействии соединения формулы 2 с соединением формулы 3-7 019146 где R1, R2, R3, R4 и R5 являются такими, как определено выше. Кроме того, настоящее изобретение предоставляет способ получения соединения формулы 1, включающий стадии образования амидной связи при взаимодействии соединения формулы 2 с соединением формулы 3 с образованием соединения формулы 1' и удаления защитной группы у соединения формулы 1' где R1 представляет собой водород,R2, R3, R4 и R5 являются такими, как определено выше, и Р представляет собой защитную группу, защищающую аминогруппу, предпочтительно третбутоксикарбонил (Boc), бензилоксикарбонил (Cbz) или фторенилметоксикарбонил (Fmoc). Кроме того, настоящее изобретение предоставляет способ получения соединения формулы 1, включающий стадии удаления защиты у соединения формулы 1' или 2' в указанном выше способе с последующим i) восстановительным аминированием с C1-С 10 алкилом, С 3-С 7 циклоалкилом или гетероциклом,содержащим оксозаместитель, или ii) сочетанием с арилгалогеном или гетероарилгалогеном где R1 представляет собой C1-С 10 алкил, С 3-С 7 циклоалкил, С 6-С 10 арил, гетероцикл или гетероарил, который является незамещенным или замещен по меньшей мере одним заместителем, выбранным из группы,состоящей из галогена, амино, С 1-С 4 алкила, трифторметила, гидрокси, С 1-С 4 алкокси, циано и оксо, иR2, R3, R4, R5 и Р являются такими, как определено выше. Предпочтительным является осуществление указанных выше способов согласно настоящему изобретению в обычном растворителе, который не оказывает неблагоприятного воздействия на реакцию, и особенно предпочтительно использовать один или более растворителей, выбранных из группы, состоящей из, но не ограничиваясь ими, диметилформамида, диметилацетамида, тетрагидрофурана, метиленхлорида и хлороформа. Реакцию удаления защиты с аминогруппы можно осуществлять в присутствии сильной кислоты,такой как хлористо-водородная кислота (HCl), трифторуксусная кислота (ТФУК) и т.д., в присутствии аминного основания, такого как триэтиламин, диизопропилэтиламин (DIPEA) и т.д., или путем гидрирования. Определенные условия реакции описаны в публикации T.W. GreenG.M. Wuts, Protective Groups ограничиваются ими, такие как дициклогексилкарбодиимид (DCC), 1-(3-диметиламинопропил)-3 этилкарбодиимид (EDC), 1,l'-дикарбонилдиимидазол (CDI) и т.д., в комбинации с 1-гидроксибензотриазолом (НОВТ) или 1-гидрокси-7-азабензотриазолом (НОАТ); или хлорангидрид бис-(2-оксо-3 оксазолидинил)фосфиновой кислоты (ВОР-Cl), дифенилфосфорилазид (DPPA), N-[диметиламино-1 Н 1,2,3-триазол[4,5-b]пиридин-1-илметилен]-N-метилметанаминий (HATU) и т.д. Соединения формулы 1, полученные способом настоящего изобретения, могут быть преобразованы в соли обычными способами. После завершения описанных выше реакций согласно способу настоящего изобретения продукты могут быть отделены и очищены общепринятыми последующими обработками, например, хроматографией, перекристаллизацией и т.д. Соединения согласно настоящему изобретению проявляют сильное агонистическое действие в отношении рецепторов меланокортина, и, таким образом, настоящее изобретение предоставляет агонистическую композицию для рецептора меланокортина, содержащую соединение формулы 1 или его фармацевтически приемлемую соль или изомер в качестве активного ингредиента вместе с фармацевтически приемлемым носителем. В частности, композиция согласно настоящему изобретению проявляет сильное действие по предотвращению и лечению, но не ограничиваясь ими, ожирения, эректильной дисфункции,диабета и воспаления. При введении соединения согласно настоящему изобретению для клинических целей, предпочтительная суточная доза должна быть в диапазоне 0,01-10 мг/кг массы тела в виде однократной дозировки или раздельной дозировки. Однако уровень дозирования, определенный для индивидуальных пациентов,может варьировать в зависимости от отдельного соединения, которое должно использоваться, массы тела, пола, состояния здоровья, питания, времени и способа введения лекарственного средства, скорости выведения, смеси лекарственных средств и тяжести заболевания. Соединения согласно настоящему изобретению могут быть введены любым путем, зависящим от цели. Инъекция и пероральное и назальное введение являются предпочтительными, однако введение может быть осуществлено подкожным, внутрибрюшинным, забрюшинным и ректальным путем. Инъекционный препарат, например водная или масляная суспензия для стерильной инъекции, может быть приготовлен известным способом при использовании подходящих диспергирующих средств,увлажняющих средств или суспендирующих средств. Растворителями, используемыми для этой цели,являются вода, раствор Рингера и изотоничный раствор NaCl. Стерилизованное нелетучее масло также используют обычно в качестве растворителя или суспендирующих сред. Любое нераздражающее нелетучее масло, включающее моно-, диглицерид, может быть использовано для указанной выше цели, и жирная кислота, такая как олеиновая кислота, может быть использована для инъекционного препарата. Твердыми дозированными формами для перорального введения являются капсулы, таблетки, пилюли, порошки и гранулы и, в частности, пригодными являются капсулы и таблетки. Таблетки и пилюли предпочтительно изготавливают с энтеросолюбильным покрытием. Твердые дозированные формы могут быть получены путем смешивания соединений формулы 1 согласно настоящему изобретению с одним или более инертными разбавителями, такими как сахароза, лактоза, крахмал и т.д., и носителями, например лубрикантами, подобно стеарату магния, дезинтегрирующими средствами, связующими веществами и т.д. Настоящее изобретение представлено более подробно следующими получениями и примерами, но не ограничивая каким-либо образом объема настоящего изобретения. Сокращения, используемые в последующих получениях и примерах, являются следующими: Ас - ацетил,АсОН - уксусная кислота,(Ас)2 О - уксусный ангидрид,Bn - бензил,n-Bu - н-бутил,t-Bu - трет-бутил,Bu - бутил,ВОС(Boc) - трет-бутоксикарбонил,с-Нех - циклогексил,c-Bu - циклобутил,с-Pen - циклопентил,с-Pr - циклопропил,CS2CO3 - карбонат цезия,CuSO45H2O - пентагидрат сульфата меди(II),DAST - трифторид диэтиламиносеры,DCE - дихлорэтан,DCM - дихлорметан,диМе - диметил,диF - дифтор,-9 019146DIPEA - диизопропилэтиламин,DMAP - 4-диметиламинопиридин,ДМФА - N,N-диметилформамид,ДМСО - диметилсульфоксид,EDC - гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида,Et - этил,EtOAc - этилацетат,Et2O - диэтиловый эфир,HClхлористо-водородная кислота,Н 2 О 2 - пероксид водорода,Hex - нормальный гексан,НОВТ - гидроксибензотриазол,HBTU - гексафторфосфат 2-(1 Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония,i-Pr - изопропил,i-Bu - изобутил,K2CO3 - карбонат калия,LHMDS - бис(триметилсилил)амид лития,LiBH4 - борогидрид лития,LiCl - хлорид лития,LiOH - гидроксид лития,Me - метил,МеОН - метанол,МТВЕ - метил-трет-бутиловый эфир,MgSO4 - сульфат магния,NaBH4 - борогидрид натрия,NaBH3CN - бороцианогидрид натрия,NaBH(OAc)3 - триацетоксиборогидрид натрия,NaIO4 - метапериодинат натрия,NaOtBu - трет-бутилат натрия,NaOH - гидроксид натрия,NaN3 - азид натрия,OsO4 - тетроксид осмия,Pyr - пиридин,Ph - фенил,Pr - пропил,t-Bu - трет-бутил,TEA - триэтиламин,ТФУК - трифторуксусная кислота,ТГФ - тетрагидрофуран. В частности, в следующих получениях и примерах соединения согласно настоящему изобретению были получены в соответствии со следующими методиками синтеза (схемы реакций А и В). Способы получения промежуточных соединений А 1 были следующими. Получение 1. Метил (2S,4S)-1-Вос-4-аминопирролидин-2-карбоксилат. Стадия А. (4R)-1-Вос-4-гидрокси-L-пролин.(4R)-Гидрокси-L-пролин (5,08 г, 38,77 ммоль) растворяли в 1 н. NaOH (40 мл) и 1,4-диоксане (40 мл) и к полученному раствору по каплям добавляли ди-трет-бутилдикарбонат (9,3 г, 42,6 ммоль) при 0 С. Реакционную смесь перемешивали при комнатной температуре в течение 8 ч, концентрировали в вакууме, подкисляли 1 н. HCl и экстрагировали EtOAc. Органические экстракты промывали насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения (8,84 г, 99%). МС [М+Н]=232 (М+1). Стадия В. Метил (2S,4R)-1-Вос-4-гидроксипирролидин-2-карбоксилат.(4R)-1-Вос-4-гидрокси-L-пролин (8 г, 34,63 ммоль), полученный на стадии А, растворяли в ДМФА(80 мл), к раствору добавляли K2CO3 (14 г, 101 ммоль) и по каплям добавляли метилйодид (2,6 мл, 51,9 ммоль) при 0 С. Реакционную смесь перемешивали при комнатной температуре в течение 5 ч, концентрировали в вакууме и экстрагировали EtOAc. Органические экстракты промывали водой и насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения (8,0 г, 95%). МС [М+Н]=246 (М+1). Стадия С. Метил (2S,4R)-1-Вос-4-[(метилсульфонил)окси]пирролидин-2-карбоксилат. Метил (2S,4R)-1-Вос-4-гидроксипирролидин-2-карбоксилат (8 г, 32,65 ммоль), полученный на стадии В, растворяли в DCM (80 мл), к раствору добавляли TEA (11,99 мл, 81,56 ммоль) и по каплям добавляли метансульфонилхлорид (3,77 мл, 48,9 ммоль) при 0 С. После перемешивания реакционной смеси при комнатной температуре в течение 3 ч органические экстракты промывали 1 н. HCl, насыщенным водным раствором NaHCO3 и насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения (9,4 г, 90%). МС [М+Н]=324 (М+1). Стадия D. Метил (2S,4S)-1-Вос-4-азидопирролидин-2-карбоксилат. Метил (2S,4R)-1-Вос-4-[(метилсульфонил)окси]пирролидин-2-карбоксилат (9 г, 27,86 ммоль), полученный на стадии С, растворяли в ДМФА (80 мл), к раствору добавляли NaN3 (2,7 г, 41,79 ммоль) и перемешивали при 90 С в течение 10 ч. Реакционную смесь концентрировали в вакууме, экстрагировалиEtOAc. Органические экстракты промывали насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали колоночной хроматографией (элюент,EtOAc/Hex=l/4) с получением указанного в заголовке соединения (6 г, 80%). МС [М+Н]=271 (М+1). Стадия Е. Метил (2S,4S)-1-Вос-4-аминопирролидин-2-карбоксилат. Метил (2S,4S)-1-Вос-4-азидопирролидин-2-карбоксилат (6 г, 22,22 ммоль), полученный на стадииD, растворяли в ТГФ (15 мл) и по каплям к раствору добавляли триметилфосфин (2,36 мл, 26,64 ммоль) при 0-5 С. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч, концентрировали в вакууме, подщелачивали насыщенным раствором NaHCO3 и экстрагировали EtOAc дважды. Органические экстракты концентрировали в вакууме с получением указанного в заголовке соединения в виде масла (5,34 г, 98,5%). МС [М+Н]=245 (М+1). Получение 2. Метил (2R,4S)-1-Вос-4-аминопирролидин-2-карбоксилат. Указанное в заголовке соединение получали, исходя из (4R)-гидрокси-D-пролина, согласно методике, аналогично описанной для получения 1. МС [М+Н]=245 (М+1). Получение 3. Метил (2S,4R)-1-Вос-4-аминопирролидин-2-карбоксилам. Указанное в заголовке соединение получали, исходя из (4S)-гидрокси-D-пролина, согласно методике, аналогично описанной для получения 1. МС [М+Н]=245 (М+1). Способы получения промежуточных соединений А 2 были следующими. Получение 4. 4,4-Диметилциклогексанон. 4,4-Диметилциклогексен-1-он (5 г, 40,3 ммоль) помещали в реакционный сосуд для гидрирования водородом, добавляли н-пентан (15 мл), а также Pd/C (500 мг). Сосуд для гидрирования водородом находился под давлением водорода (25 фунтов/кв.дюйм), и реакцию проводили в течение 30 мин. По окончании реакции твердые вещества отфильтровывали через целит и фильтрат концентрировали в вакууме с получением указанного в заголовке соединения (5 г, 98%). МС [М+Н]=127 (М+1). Получение 5. 4,4-Дифторциклогексанон. Стадия А. 8,8-Дифтор-1,4-диоксоспиро[4.5]декан. Коммерчески доступный 1,4-циклогександионмоноэтиленкеталь (25 г, 160 ммоль) растворяли вDCM (500 мл) и по каплям добавляли DAST (52 г, 2,0 ммоль) при 0 С. Реакционную смесь медленно нагревали до комнатной температуры и перемешивали до тех пор, пока не оканчивалась реакция. После подтверждения ТСХ того факта, что все реагирующие вещества израсходованы, реакционный раствор добавляли к насыщенному водному раствору NaHCO3 (700 мл) для остановки реакции и реакционную смесь экстрагировали DCM. Органические экстракты промывали насыщенным водным растворомNaHCO3 и насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме. Полученный остаток использовали для следующей реакции без дальнейшей очистки. Стадия В. 4,4-Дифторциклогексанон. 8,8-Дифтор-1,4-диоксоспиро[4.5]декан, полученный на стадии А, растворяли в ацетоне (90 мл) и 3 н.HCl (900 мл) и перемешивали до тех пор, пока реакция не завершалась. Затем реакционную смесь экстрагировали DCM, экстракт промывали насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме. Полученный остаток использовали для следующей реакции без дальнейшей очистки. МС [М+Н]=135 (М+1). Способы получения коммерчески недоступных промежуточных соединений A3 были следующими. Получение 6. (2S)-Тетрагидрофуран-2-карбонилхлорид.(2S)-Тетрагидрофуран-2-карбоновую кислоту (25 г, 0,215 моль) растворяли в DCM (25 мл), раствор охлаждали до 0 С и к раствору по каплям добавляли оксалилхлорид (43,7 г, 0,344 моль). К реакционной смеси добавляли ДМФА (50 мкл) и реакционную смесь перемешивали при комнатной температуре в течение 10 ч. Затем реакционную смесь концентрировали в вакууме при 20-30 С до тех пор, пока остаточный объем реакционной смеси не достигал приблизительно 30 мл. Реакционную смесь нагревали приблизительно при 150-160 С и перегоняли в вакууме при 80-100 С внутренней температуры, с получением указанного в заголовке соединения (24 г, 82,8%). Получение 7. (2R)-Тетрагидрофуран-2-карбонилхлорид. Указанное в заголовке соединение получали, исходя из (2R)-тетрагидрофуран-2-карбоновой кислоты, согласно методике, аналогично описанной для получения 6. Получение 8. Тетрагидрофуран-3-карбонилхлорид. Тетрагидрофуран-3-карбоновую кислоту (25 г, 0,215 моль) растворяли в DCM (25 мл), раствор охлаждали до 0 С и к раствору по каплям добавляли оксалилхлорид (43,7 г, 0,344 моль). К реакционной смеси добавляли ДМФА (50 мкл) и реакционную смесь перемешивали при комнатной температуре в течение 10 ч. Затем реакционную смесь концентрировали в вакууме при 20-30 С до тех пор, пока остаточный объем реакционной смеси не достигал приблизительно 30 мл. Полученный остаток использовали для следующей реакции без дальнейшей очистки. Получение 9. 3-Фуроилхлорид. 3-Фуранкарбоновую кислоту (25 г, 0,192 моль) растворяли в DCM (25 мл), раствор охлаждали до 0 С и по каплям добавляли оксалилхлорид (39,0 г, 0,307 моль). К реакционной смеси добавляли ДМФА(50 мкл) и реакционную смесь перемешивали при комнатной температуре в течение 10 ч. Затем реакционную смесь концентрировали в вакууме при 20-30 С до тех пор, пока остаточный объем реакционной смеси не достигал приблизительно 30 мл. Полученный остаток использовали для следующей реакции без дальнейшей очистки. Получение 10. 2,2-Диметил-3-ацетилоксипропионилхлорид. Стадия А. 2,2-Диметил-3-ацетилоксипропионовая кислота. 2,2-Диметил-3-гидроксипропионовую кислоту (11,8 г, 100 ммоль) растворяли в пиридине (30 мл) и реакционный раствор охлаждали до 0 С. Медленно по каплям добавляли ацетилхлорид (11,8 г, 15,0 ммоль), затем температуру повышали до комнатной температуры и перемешивали реакционный раствор при комнатной температуре в течение 3 ч. По окончании реакции, добавляли 1 н. HCl (30 мл) для доведения рН до 3-4 и затем реакционную смесь экстрагировали EtOAc. Органические экстракты промывали 1 н. HCl 4-5 раз, сушили над MgSO4 и концентрировали в вакууме с получением указанного в заголовке соединения (15,2 г, 95,0%). МС [М+Н]=161 (М+1). Стадия В. 2,2-Диметил-3-ацетилоксипропионилхлорид. Продукт со стадии А, 2,2-диметил-3-ацетилоксипропионовую кислоту (11,76 г, 80 ммоль) растворяли в бензоле (100 мл), реакционный раствор охлаждали до 0 С и затем медленно по каплям добавляли оксалилхлорид (15,0 г, 120 ммоль). Через 3 ч растворитель удаляли в вакууме, реакционную смесь перегоняли в вакууме с получением указанного в заголовке соединения. МС [М+Н]=179 (М+1). Способы получения промежуточных соединений А 4 были следующими. Получение 11. (3S,4R)-1-трет-Бутил-4-(2,4-дифторфенил)пирролидин-3-карбоновая кислота. Указанное в заголовке соединение получали согласно методике, описанной в международной публикации WO 2004/09126. Получение 12. (3S,4R)-1-трет-Бутил-4-(4-хлорфенил)пирролидин-3-карбоновая кислота. Указанное в заголовке соединение получали, исходя из коммерчески доступного 2-бром-1-(4 хлорфенил)этанона, согласно методике, аналогично описанной для получения 11. Получение 13. (3S,4R)-1-Boc-4-(2,4-дифторфенил)пирролидин-3-карбоновая кислота. Стадия A. (4R)-4-(2,4-Дифторфенил)пирролидин-3-карбонитрил.(4R)-1-трет-Бутил-4-(2,4-дифторфенил)пирролидин-3-карбонитрил (4 г, 15,15 ммоль), который получали согласно методике, описанной в международной публикации WO 2004/09126, растворяли в DCE(10 мл), и по каплям добавляли 1-хлорэтилхлорформиат (2,45 мл, 22,68 ммоль) при 0 С. Реакционный раствор нагревали до 70 С и при сохранении указанной температуры по каплям добавляли 1,8 бис(диметиламино)нафталин (4,87 г, 22,72 ммоль), растворенный в DCE (10 мл), в течение 2 ч. По окончании реакции добавляли метанол (10 мл), при сохранении указанной температуры реакционную смесь перемешивали в течение дополнительного 1 ч и концентрировали в вакууме. Полученный концентрат использовали для следующей реакции без дальнейшей очистки.(4R)-4-(2,4-Дифторфенил)пирролидин-3-карбонитрил, полученный на стадии A, DMAP (1,8 г, 15,15 ммоль) и TEA (5,56 мл, 15,15 ммоль) растворяли в DCM (10 мл) и по каплям добавляли ди-третбутилдикарбонат (4,9 г, 22,7 ммоль) при 0 С. Реакционную смесь перемешивали при комнатной температуре в течение 8 ч, концентрировали в вакууме и экстрагировали EtOAc. Органические экстракты промывали 1 н. раствором HCl и насыщенным раствором соли, сушили над MgSO4, концентрировали в вакууме и очищали колоночной хроматографией (элюент: EtOAc/Hex=l/6), с получением указанного в заголовке соединения (3,3 г, общий выход со стадий А и В: 72%). МС [М+Н]=309 (М+1). Стадия С. (3S,4R)-1-Вос-4-(2,4-Дифторфенил)пирролидин-3-карбоновая кислота.(4R)-1-Boc-4-(2,4-Дифторфенил)пирролидин-3-карбонитрил (3,3 г, 10,6 ммоль), полученный на стадии В, растворяли в этаноле (10 мл), добавляли 6 н. раствор NaOH (5 мл) и перемешивали при 70 С в течение 4 ч. По окончании реакции растворитель удаляли, реакционную смесь разбавляли эфиром, органический раствор достаточно подкисляли и промывали 6 н. HCl. Полученный органический раствор промывали насыщенным раствором соли, сушили над MgSO4 и концентрировали в вакууме с получением указанного в заголовке соединения (3,43 г, 99,0%). МС [М+1]=328 (М+1). Получение 14. (3S,4R)-1-Вос-4-(4-Хлорфенил)пирролидин-3-карбоновая кислота. Указанное в заголовке соединение получали, исходя из 4-хлорфенилпирролидин-3-карбонитрила,промежуточного соединения из получения 12, согласно методике, аналогично описанной для получения 13. МС [М+1]=326 (М+1). Получение 15. (3R,4R)-1-трет-Бутил-4-(4-хлорфенил)пирролидин-3-карбоновая кислота. Стадия A. (3R,4R)-1-трет-Бутил-4-(4-хлорфенил)пирролидин-3-карбонитрил. 3-трет-Бутил[(2S)-2-(4-хлорфенил)-2-гидроксиэтил]аминопропаннитрил (5 г, 17,80 ммоль), полученный способом, описанным в международной публикации WO 2004/09126, растворяли в ТГФ (27 мл). Реакционную смесь охлаждали до -20 С или ниже, к которой добавляли хлордиэтилфосфат (2,69 мл,18,70 ммоль). Температуру реакции поддерживали при 12-18 С, при указанной температуре по каплям добавляли 1 М LHMDS (37,4 мл, 37,38 ммоль) в течение 2 ч. По окончании реакции добавляли воду (45 мл), при этом температуру реакции поддерживали при 15 С или ниже. Образовавшуюся смесь перемешивали в течение 30 мин и экстрагировали EtOAc. Полученный таким образом органический экстракт концентрировали в вакууме и очищали колоночной хроматографией (элюент: EtOAc/Hex=1/3), с получением указанного в заголовке соединения (0,5 г, 10,69%). МС [М+1]=263 (М+1). Стадия В. (3R,4R)-1-трет-Бутил-4-(4-хлорфенил)пирролидин-3-карбоновая кислота. К (3R,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-карбонитрилу (0,4 г, 1,52 ммоль), полученному на стадии А, добавляли конц. HCl (10 мл) и смесь перемешивали в течение 5 ч при 110 С. По окончании реакции смесь охлаждали до 20-25 С и концентрировали в вакууме. И снова смесь концентрировали в вакууме 3 раза, используя EtOAc. EtOAc добавляли к остатку, смесь перемешивали в течение 3-4 ч и фильтровали с получением указанного в заголовке соединения (0,27 г, 62,95%). МС [М+1]=282 (М+1). Получение 16. (3S,4S)-1-трет-Бутил-4-(4-хлорфенил)пирролидин-3-карбоновая кислота. Стадия A. (3S,4S)-1-трет-Бутил-4-(4-хлорфенил)пирролидин-3-карбонитрил. 3- трет-Бутил[(2R)-2-(4-хлорфенил)-2-гидроксиэтил]аминопропаннитрил (5 г, 17,80 ммоль), который получали, исходя из (R)-1-метил-3,3-дифенилтетрагидропиррол[1,2-с][1,3,2]оксазаборола в три стадии способом, описанным в международной публикации WO 2004/09126, подвергали взаимодействию,согласно методике, аналогично стадии А получения 15 с получением указанного в заголовке соединения(0,6 г, 12,82%). МС [М+1]=263 (М+1). Стадия В. (3S,4S)-1-трет-Бутил-4-(4-хлорфенил)пирролидин-3-карбоновая кислота. К (3S,4S)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-карбонитрилу (0,4 г, 1,52 ммоль), полученному на стадии А, добавляли конц. раствор HCl (10 мл) и смесь перемешивали в течение 5 ч при 110 С. По окончании реакции смесь охлаждали до 20-25 С и концентрировали в вакууме. И снова смесь концентрировали в вакууме 2-3 раза, используя DCM. DCM добавляли к остатку, смесь перемешивали в течение 3-4 часов и фильтровали, с получением указанного в заголовке соединения (0,19 г, 44,30%). МС [М+1]=282 (М+1). Получение 17. (3S,4R)-4-(4-Хлорфенил)-1-(6-метилпиридазин-3-ил)пирролидин-3-карбоновая кислота. Стадия А. Метил (3S,4R)-4-(4-хлорфенил)пирролидин-3-карбоксилат. ванную согласно получению 14, растворяли в DCE (3 мл), по каплям добавляли 4 н. раствор HCl (3 мл) и смесь перемешивали при комнатной температуре в течение 2 ч. К реакционному раствору добавляли МеОН (5 мл). Затем реакционный раствор перемешивали при комнатной температуре в течение 3 ч и концентрировали в вакууме. К полученному остатку добавляли DCE (2 мл) и EtOAc (10 мл). Затем реакционную смесь перемешивали при комнатной температуре в течение 1 ч и фильтровали с получением указанного в заголовке соединения (0,46 г, 90,0%). МС [М+1]=240 (М+1). Стадия В. Метил (3S,4R)-4-(4-хлорфенил)-1-(6-метилпиридазин-3-ил)пирролидин-3-карбоксилат. Продукт со стадии А, метил (3S,4R)-4-(4-хлорфенил)пирролидин-3-карбоксилат (0,46 г, 1,92 ммоль), растворяли в диоксане (2 мл). Добавляли DIPEA (1,34 мл, 7,67 ммоль) и 3-хлор-6 метилпиридазин (0,74 г, 5,76 ммоль). Реакцию осуществляли при 180 С в течение 5 мин под воздействием микроволнового излучения. Затем реакционную смесь концентрировали в вакууме. К полученному остатку добавляли EtOAc. Смесь разбавляли EtOAc, промывали водой и очищали колоночной хроматографией (элюент: EtOAc/Hex=1/2) с получением указанного в заголовке соединения (0,48 г, 75%). МС [М+Н]=332 (М+1). Стадия С. (3S,4R)-4-(4-Хлорфенил)-1-(6-метилпиперазин-3-ил)пирролидин-3-карбоновая кислота. Продукт со стадии В, метил (3S,4R)-4-(4-хлорфенил)-1-(6-метилпиридазин-3-ил)пирролидин-3 карбоксилат (0,48 г, 1,45 ммоль), растворяли в МеОН (4 мл) и воде (0,4 мл) и добавляли LiOH (0,1 г, 4,18 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч и концентрировали в вакууме. Полученный остаток разбавляли водой. К разбавленной смеси добавляли 1 н. раствор HCl(4,18 мл). Затем реакционную смесь экстрагировали 3 раза смесью DCE/MeOH (10/1). Органические экстракты сушили над MgSO4 и концентрировали при пониженном давлении с получением указанного в заголовке соединения (0,32 г, 70%). МС [М+Н]=318 (М+1). Получение 18. (3S,4R)-4-(4-Хлорфенил)-1-(1,3-тиазол-2-ил)пирролидин-3-карбоновая кислота. Стадия А. Метил (3S,4R)-4-(4-хлорфенил)-1-(1,3-тиазол-2-ил)пирролидин-3-карбоксилат. Продукт со стадии А получения 17, метил (3S,4R)-4-(4-хлорфенил)пирролидин-3-карбоксилат (0,46 г, 1,92 ммоль), растворяли в 1,4-диоксане (2 мл). Добавляли DIPEA (1,34 мл, 7,67 ммоль) и 2-бром-1,3 тиазол (1,57 г, 9,6 ммоль). Взаимодействие осуществляли при 180 С в течение 20 мин при воздействии микроволнового излучения. Затем реакционную смесь концентрировали в вакууме. Полученный остаток разбавляли EtOAc, промывали водой и очищали колоночной хроматографией (элюент: EtOAc/Hex=1/3) с получением указанного в заголовке соединения (0,15 г, 25%). МС [М+Н]=325 (М+1). Стадия В. (3S,4R)-4-(4-Хлорфенил)-1-(1,3-тиазол-2-ил)пирролидин-3-карбоновая кислота. Указанное в заголовке соединение (0,10 г, 70%) получали, исходя из продукта со стадии А, метил(3S,4R)-4-(4-хлорфенил)-1-(1,3-тиазол-2-ил)пирролидин-3-карбоксилата (0,15 г, 0,46 ммоль), согласно методике, аналогично описанной на стадии А получения 17. МС [М+Н]=311 (М+1). Получение 19. (3S,4R)-4-(2,4-Дифторфенил)-1-(6-хлорпиридазин-3-ил)пирролидин-3-карбоновая кислота. Стадия А. Метил (3S,4R)-4-(2,4-дифторфенил)пирролидин-3-карбоксилат. Указанное в заголовке соединение (0,46 г, 90%) получали, исходя из продукта получения 13,(3S,4R)-1-Вос-4-(2,4-дифторфенил)пирролидин-3-карбоновой кислоты (0,70 г, 2,15 ммоль), согласно методике, аналогично описанной на стадии А получения 17. МС [М+1]=242 (М+1). Стадия В. Метил (3S,4R)-4-(2,4-дифторфенил)-1-(6-хлорпиридазин-3-ил)пирролидин-3-карбоксилат. Продукт со стадии А, метил (3S,4R)-4-(2,4-дифторфенил)пирролидин-3-карбоксилат (0,46 г, 1,91 ммоль), растворяли в диоксане (2 мл). Добавляли DIPEA (1,33 мл, 7,64 ммоль) и 3,6-дихлорпиридазин(1,42 г, 9,55 ммоль). Реакцию осуществляли при 120 С в течение 5 мин и при 150 С в течение 10 мин под воздействием микроволнового излучения. Затем реакционную смесь концентрировали в вакууме. Полученный остаток разбавляли EtOAc, промывали водой и очищали колоночной хроматографией (элюент:EtOAc/Hex=1/5), с получением указанного в заголовке соединения (0,44 г, 65%). МС [М+Н]=354 (М+1). Стадия С. (3S,4R)-4-(2,4-Дифторфенил)-1-(6-хлорпиридазин-3-ил)пирролидин-3-карбоновая кислота. Указанное в заголовке соединение (0,30 г, 70%) получали, исходя из продукта со стадии В, метил(3S,4R)-4-(2,4-дифторфенил)-1-(6-хлорпиридазин-3-ил)пирролидин-3-карбоксилата, согласно методике,аналогично описанной на стадии С получения 17. МС [М+Н]=340 (М+1). Получение 20. (3S,4R)-4-(4-Хлорфенил)-1-(6-хлорпиридазин-3-ил)пирролидин-3-карбоновая кислота. Указанное в заголовке соединение (0,30 г, 70%) получали, исходя из продукта получения 14,(3S,4R)-1-Вос-4-(4-хлорфенил)пирролидин-3-карбоновой кислоты, согласно методике, аналогично опи- 15019146 санной для получения 19. МС [М+1]=242 (М+1). Получение 21. (3S,4R)-4-(4-Хлорфенил)-1-(1,6-дигидропиридазин-3-ил)пирролидин-3-карбоновая кислота. Стадия А. Метил(3S,4R)-4-(4-хлорфенил)-1-(1,6-дигидропиридазин-3-ил)пирролидин-3 карбоксилат. Промежуточное соединение, полученное в получении 20, метил (3S,4R)-4-(4-хлорфенил)-1-(6 хлорпиридазин-3-ил)пирролидин-3-карбоксилат (1,0 г, 2,84 ммоль) растворяли в МеОН (5 мл). Добавляли 1,4-циклогексадиен (2,27 г, 28,4 ммоль) и 10% Pd/C (1,0 г). Реакционную смесь перемешивали при комнатной температуре в течение 10 ч и концентрировали в вакууме. Полученный остаток разбавлялиEtOAc и фильтровали через целит. Фильтрат концентрировали в вакууме и очищали колоночной хроматографией (элюент: EtOAc/Hex=1/2) с получением указанного в заголовке соединения (0,72 г, 80%). Стадия В. (3S,4R)-4-(4-Хлорфенил)-1-(1,6-дигидропиридазин-3-ил)пирролидин-3-карбоновая кислота. Указанное в заголовке соединение (0,48 г, 70%) получали, исходя из продукта со стадии A, (3S,4R)4-(4-хлорфенил)-1-(1,6-дигидропиридазин-3-ил)пирролидин-3-карбоксилата (0,72 г, 2,27 ммоль), согласно методике, аналогично описанной на стадии С получения 17. МС[М+Н]=304 (М+1). Получение 22.(4 мл). Взаимодействие осуществляли при 200 С в течение 5 мин под воздействием микроволнового излучения и реакционную смесь концентрировали в вакууме. Полученный остаток разбавляли EtOAc и промывали водой. Органический раствор концентрировали в вакууме и очищали колоночной хроматографией (элюент: EtOAc/Hex=1/l) с получением указанного в заголовке соединения (0,76 г, 80%). МС [М+Н]=334 (М+1). Стадия В. (3S,4R)-4-(4-Хлорфенил)-(6-оксо-1,6-дигидропиридазин-3-ил)пирролидин-3-карбоновая кислота. Указанное в заголовке соединение (0,51 г, 70%) получали, исходя из продукта со стадии А, метил(3S,4R)-4-(4-хлорфенил)-1-(6-оксо-1,6-дигидропиридазин-3-ил)пирролидин-3-карбоксилата (0,76 г, 2,28 ммоль), согласно методике, аналогично описанной на стадии С получения 17. МС [М+Н]=320 (М+1). Получение 23. (3S,4R)-4-(4-Хлорфенил)-1-(1-метил-6-оксо-1,6-дигидропиридазин-3-ил)пирролидин-3-карбоновая кислота. Стадия А. Метил (3S,4R)-4-(4-хлорфенил)-1-(1-метил-6-оксо-1,6-дигидропиридазин-3-ил)пирролидин-3-карбоксилат. Метил (3S,4R)-4-(4-хлорфенил)-1-(6-оксо-1,6-дигидропиридазин-3-ил)пирролидин-3-карбоксилат,синтезированный на стадии А получения 22, растворяли в ДМФА (5 мл). Добавляли CS2CO3 (1,11 г, 3,41 ммоль) и по каплям добавляли метилйодид (0,71 мл, 11,40 ммоль). Реакционную смесь перемешивали при 20-30 С в течение 2 ч и концентрировали в вакууме. Полученный остаток разбавляли EtOAc, промывали водой и очищали колоночной хроматографией (элюент: EtOAc/Hex=l/l), с получением указанного в заголовке соединения (0,55 г, 70%). МС [М+Н]=348 (М+1). Стадия В. (3S,4R)-4-(4-Хлорфенил)-1-(1-метил-6-оксо-1,6-дигидропиридазин-3-ил)пирролидин-3 карбоновая кислота. Указанное в заголовке соединение (0,37 г, 70%) получали, исходя из продукта со стадии А, метил(3S,4R)-4-(4-хлорфенил)-1-(1-метил-6-оксо-1,6-дигидропиридазин-3-ил)пирролидин-3-карбоксилата (0,55 г, 1,58 ммоль), согласно методике, аналогично описанной на стадии С получения 17. МС [М+Н]=334 (М+1). Соединения примеров, синтезированные способом, указанным на реакционной схеме А, были следующими. Пример 1. ТФУК-соль N-(3S,5S)-1-[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил-5-[(4-метилпилеразин-1-ил)карбонил]пирролидин-3-ил-N-(4,4-диметилциклогексил)ацетамида. Стадия А. Метил (2S,4S)-1-Boc-4-[(4,4-диметилциклогексил)амино]пирролидин-2-карбоксилат. Метил (2S,4S)-1-Вос-4-аминопирролидин-2-карбоксилат (1,07 г, 4,38 ммоль), синтезированный согласно получению 1, и 4,4-диметилциклогексанон (0,66 г, 5,25 ммоль) растворяли в DCE (10 мл) и к раствору добавляли NaBH(OAc)3 (1,39 г, 6,57 ммоль) при комнатной температуре. После перемешивания реакционной смеси при комнатной температуре в течение 4 ч, органические слои экстрагировали насыщенным водным раствором NaHCO3. Органические экстракты сушили над MgSO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали колоночной хроматографией (элюент: EtOAc/(2S,4S)-1-Boc-4-[ацетил-(4,4-диметилциклогексил)амино]пирролидин-2 карбоксилат. Метил (2S,4S)-1-Boc-4-[(4,4-диметилциклогексил)амино]пирролидин-2-карбоксилат (1,01 г, 2,84 ммоль), синтезированный на стадии А, растворяли в пиридине (5 мл) и к раствору добавляли Ас 2 О (1,34 мл, 14,20 ммоль) при комнатной температуре. Реакционный раствор нагревали до 90 С и перемешивали в течение 2 ч. По окончании реакции добавляли водный раствор CuSO4 5H2O и смесь экстрагировалиEtOAc. Органический экстракт сушили над MgSO4 и концентрировали в вакууме. Полученный остаток очищали колоночной хроматографией (элюент: EtOAc/Hex=l/l), с получением указанного в заголовке соединения (0,98 г, 87%). МС [М+Н]=397 (М+1). Стадия С. (4S)-1-Boc-4-[ацетил-(4,4-диметилциклогексил)амино]-L-пролин. Метил (2S,4S)-1-Boc-4-[ацетил-(4,4-диметилциклогексил)амино]пирролидин-2-карбоксилат (0,98 г,2,47 ммоль), синтезированный на стадии В, растворяли в МеОН (8 мл) и воде (1,6 мл). К раствору добавляли LiOH (0,18 г, 7,52 ммоль) при 0-5 С. После перемешивания реакционной смеси при комнатной температуре в течение 3 ч и концентрирования в вакууме, полученный остаток разбавляли водой и раствор экстрагировали EtOAc. Органический экстракт промывали насыщенным раствором соли, сушили над(4S)-1-Boc-4-[ацетил(4,4-диметилциклогексил)амино]-L-пролин (0,92 г, 2,41 ммоль), синтезированный на стадии С, растворяли в ДМФА (10 мл) и к раствору добавляли DIPEA (1,05 мл, 6,01 ммоль). Также последовательно добавляли 1-метилпиперазин (0,32 мл, 2,89 ммоль) и HBTU (0,91 г, 2,41 ммоль). После перемешивания реакционного раствора при комнатной температуре в течение 2 ч, раствор концентрировали в вакууме. Остаток разбавляли EtOAc и промывали насыщенным водным раствором NaHCO3. Органический экстракт сушили над MgSO4, концентрировали в вакууме и очищали колоночной хроматографией (элюент: МС/МеОН=10/1) с получением указанного в заголовке соединения (1,06 г, 95%). МС [М+Н]=465 (М+1). Стадия Е. N-(4,4-Диметилциклогексил)-N-(3S,5S)-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-илацетамид.(2S,4S)-1-Boc-4-[ацетил-(4,4-диметилциклогексил)амино]-2-[(4-метилпиперазин-1-ил)карбонил] пирролидин (1,06 г, 2,28 ммоль), синтезированный на стадии D, растворяли в DCM (1 мл) и по каплям добавляли 4 М раствор HCl (1 мл). После перемешивания реакционного раствора при комнатной температуре в течение 1 ч, раствор концентрировали в вакууме. Остаток концентрировали в вакууме с получением указанного в заголовке соединения (830 мг, 99,8%). МС [М+Н]=365 (М+1). Стадия F. ТФУК-соль N-(3S,5S)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-ил]карбонил)-5-[(4 метилпиперазин-1-ил)карбонил]пирролидин-3-ил)-N-(4,4-диметилциклогексил)ацетамида.N-(4,4-Диметилциклогексил)-N-(3S,5S)-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3 илацетамид (0,83 г, 2.2 8 ммоль), синтезированный на стадии Е, растворяли в ДМФА (5 мл) и к раствору добавляли DIPEA (0,99 мл, 5,69 ммоль). Последовательно также добавляли (3S,4R)-1-трет-бутил-4-(4 хлорфенил)пирролидин-3-карбоновую кислоту (0,64 г, 2,28 ммоль), синтезированную согласно получению 10, и HBTU (0,86 г, 2,28 ммоль). После перемешивания реакционного раствора при комнатной температуре в течение 2 ч раствор концентрировали в вакууме. Остаток разбавляли EtOAc и промывали насыщенным водным раствором NaHCO3. Органический экстракт сушили над MgSO4, концентрировали в(4S)-1-Вос-4-[ацетил-(4,4-диметилциклогексил)амино]-L-пролин (0,92 г, 2,41 ммоль), синтезированный на стадии С примера 1, растворяли в ДМФА (10 мл) и к раствору добавляли DIPEA (1,05 мл, 6,01 ммоль). Последовательно добавляли также пиперазин (0,25 г, 2,89 ммоль) и HBTU (0,91 г, 2,41 ммоль). После перемешивания реакционного раствора при комнатной температуре в течение 2 ч раствор концентрировали в вакууме. Остаток разбавляли EtOAc и промывали насыщенным водным раствором NaHCO3. Органический экстракт сушили над MgSO4, концентрировали в вакууме. Остаток очищали колоночной хроматографией (элюент: МС/МеОН=10/1) с получением указанного в заголовке соединения (1,01 г,93%). МС [М+Н]=451 (М+1). Стадия В.(1,01 г, 2,24 ммоль), синтезированный на стадии А, подвергали взаимодействию, согласно методике, аналогично описанной на стадии Е примера 1 с получением указанного в заголовке соединения (784 мг,99,8%). МС [М+Н]=351 (М+1). Стадия С. ТФУК-соль N-[(3S,5S)-1-[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3 ил]карбонил-5-(пиперазин-1-илкарбонил)пирролидин-3-ил]-N-(4,4-диметилциклогексил)ацетамида.(784 мг, 2,24 ммоль), синтезированный на стадии В, растворяли в ДМФА (5 мл) и к раствору добавлялиDIPEA (0,98 мл, 5,59 ммоль). Последовательно добавляли (3S,4R)-1-трет-бутил-4-(2,4 дифторфенил)пирролидин-3-карбоновую кислоту (0,63 г, 2,24 ммоль), синтезированную согласно получению 11, и HBTU (0,85 г, 2,24 ммоль). После перемешивания реакционного раствора при комнатной температуре в течение 2 ч раствор концентрировали в вакууме. Остаток разбавляли EtOAc и промывали насыщенным водным раствором NaHCO3. Органический экстракт сушили над MgSO4, концентрировали в вакууме. Остаток очищали ВЭЖХ с получением ТФУК-соли указанного в заголовке соединения (1,20 г, 87%). МС[М+Н]=616 (М+1). 1 Н ЯМР (500 МГц, ДМСО-d6, 140 С)7,64-7,50 (м, 1 Н), 7,11-6,96 (м, 2 Н), 4,77-4,64 (ушир., 1 Н),4,04-3,48 (м, 11 Н), 3,46-3,17 (м, 4 Н), 3,17-3,01 (м, 4 Н), 2,45-2,33 (м, 1 Н), 2,26-2,12 (ушир., 1 Н), 1,97 (с,3 Н), 1,88-1,72 (м, 1 Н), 1,68-1,50 (ушир., 1 Н), 1,49-1,20 (м, 6 Н), 1,38 (с, 9 Н), 0,91 (с, 6 Н). Пример 3. ТФУК-соль (2S)-N-(3S,5S)-1-[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3 ил]карбонил-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамида Стадия А. Метил (2S,4S)-1-Вос-4-(4,4-диметилциклогексил)[(2S)-тетрагидрофуран-2-илкарбонил] аминопирролидин-2-карбоксилат. Метил (2S,4S)-l-Boc-4-[(4,4-диметилциклогексил)амино]пирролидин-2-карбоксилат, синтезированный на стадии А примера 1 (1,01 г, 2,84 ммоль), растворяли в DCE (5 мл). К раствору добавляли ТЭА (5 мл) и DMAP (0,34 г, 2,84 ммоль). Добавляли (2S)-тетрагидрофуран-2-карбонилхлорид (1,14 г, 8,52 ммоль), синтезированный согласно получению 6. После перемешивания реакционного раствора при комнатной температуре в течение 2 ч и окончания реакции раствор концентрировали в вакууме. Насыщенный водный раствор NaHCO3 добавляли к остаточному раствору и экстрагировали EtOAc. Органический экстракт промывали 1 н. раствором HCl, сушили над MgSO4, концентрировали в вакууме и очищали колоночной хроматографией (элюент: EtOAc/Hex=l/4) с получением указанного в заголовке соединения(2S,4S)-1-Вос-4-(4,4-диметилциклогексил)[(2S)-тетрагидрофуран-2-илкарбонил]амино пирролидин-2-карбоксилат (1,05 г, 2,32 ммоль), синтезированный на стадии А, подвергали взаимодействию, согласно методике, аналогично описанной на стадии С примера 1, с получением указанного в заголовке соединения (0,99 г, 97%). МС [М+Н]=439 (М+1). Стадия С. (2S,4S)-1-Вос-4-(4,4-диметилциклогексил)[(2S)-тетрагидрофуран-2-илкарбонил]амино 2-[(4-метилпиперазин-1-ил)карбонил]пирролидин.(0,99 г, 2,2 6 ммоль), синтезированный на стадии В, подвергали взаимодействию, согласно методике,аналогично описанной на стадии D примера 1, с получением указанного в заголовке соединения (1,12 г,95%). МС[М+Н]=521 (М+1). Стадия D. (2S)-N-(4,4-Диметилциклогексил)-N-(3S,5S)-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-илтетрагидрофуран-2-карбоксамид.(2S,4S)-1-Вос-4-(4,4-диметилциклогексил)[(2S)-тетрагидрофуран-2-илкарбонил]амино-2-[(4 метилпиперазин-1-ил)карбонил]пирролидин (1,12 г, 2,15 ммоль), синтезированный на стадии С, подвергали взаимодействию, согласно методике, аналогично описанной на стадии Е примера 1, с получением указанного в заголовке соединения (903 мг, 99,8%). МС [М+Н]=421 (М+1). Стадия Е. ТФУК-соль (2S)-N-(3S,5S)-1-[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3 ил]карбонил-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамида.(2S)-N-(4,4-Диметилциклогексил)-N-(3S,5S)-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3 илтетрагидрофуран-2-карбоксамид (903 мг, 2,15 ммоль), синтезированный на стадии D, и (3S,4R)-1 трет-бутил-4-(4-хлорфенил)пирролидин-3-карбоновую кислоту, синтезированную согласно получению 12, подвергали взаимодействию по методике, аналогично описанной на стадии F примера 1, с получением ТФУК-соли указанного в заголовке соединения (1,25 г, 85%). МС [М+Н]=684 (М+1). 1 Стадия А. Метил (2S,4S)-1-Boc-4-([3-(ацетокси)-2,2-диметилпропаноил](4,4-диметилциклогексил) аминопирролидин-2-карбоксилат. Метил (2S,4S)-1-Boc-4-[(4,4-диметилциклогексил)амино]пирролидин-2-карбоксилат (1,01 г, 2,84 ммоль), синтезированный на стадии А примера 1, растворяли в DCE (5 мл). К раствору добавляли TEA (5 мл), а также добавляли DMAP (0,34 г, 2,84 ммоль) и 2,2-диметил-3-ацетилоксипропионилхлорид (1,01 г,5,68 ммоль), синтезированный согласно получению 10. Реакционный раствор нагревали до 90 С и перемешивали в течение 48 ч. По окончании реакции растворитель удаляли в вакууме. Насыщенный водный раствор NaHCO3 добавляли к остаточному раствору и экстрагировали EtOAc. Органический экстракт промывали 1 н. раствором HCl, сушили над MgSO4, концентрировали в вакууме и очищали колоночной хроматографией (элюент: EtOAc/Hex=l/4) с получением указанного в заголовке соединения (0,88 г, 63%). МС [М+Н]=497 (М+1). Стадия В. (4S)-1-Вос-4-[(4,4-диметилциклогексил)(3-гидрокси-2,2-диметилпропаноил)амино]-Lпролин. Метил (2S,4S)-l-Boc-4-[3-(ацетокси)-2,2-диметилпропаноил](4,4-диметилциклогексил)аминопирролидин-2-карбоксилат (0,88 г, 1,77 ммоль), синтезированный на стадии А, растворяли в МеОН (8 мл) и воде (1,6 мл). К раствору добавляли NaOH (213 мг, 5,31 ммоль). Реакционный раствор перемешивали в течение 12 ч. По окончании реакции реакционный раствор концентрировали в вакууме, подкисляли 1 н. раствором HCl и экстрагировали EtOAc. Органический экстракт промывали 1 н. раствором HCl, сушили над MgSO4 и концентрировали в вакууме с получением указанного в заголовке соединения (0,74 г, 95%). МС [М+Н]=441 (М+1). Стадия С. (2S,4S)-1-Вос-4-[(4,4-диметилциклогексил)(3-гидрокси-2,2-диметилпропаноил)амино]-2[(4-метилпиперазин-1-ил)карбонил]пирролидин.(4S)-1-Вос-4-[(4,4-диметилциклогексил)(3-гидрокси-2,2-диметилпропаноил)амино]-L-пролин (0,74 г, 1,68 ммоль), синтезированный на стадии В, подвергали взаимодействию согласно методике, аналогично описанной на стадии D примера 1, с получением указанного в заголовке соединения (0,83 г, 95%). МС [М+Н]=523 (М+1). Стадия D. N-(4,4-Диметилциклогексил)-3-гидрокси-2,2-диметил-N-(3S,5S)-5-[(4-метилпиперазин 1-ил)карбонил]пирролидин-3-илпропанамид.(2S,4S)-1-Вос-4-[(4,4-диметилциклогексил)(3-гидрокси-2,2-диметилпропаноил)амино]-2-[(4 метилпиперазин-1-ил)карбонил]пирролидин (0,83 г, 1,59 ммоль), синтезированный на стадии С, подвергали взаимодействию согласно методике, аналогично описанной на стадии Е примера 1, с получением указанного в заголовке соединения (0,67 г, 99,8%). МС [М+Н]=423 (М+1). Стадия Е. ТФУК-сольN-(4,4-Диметилциклогексил)-3-гидрокси-2,2-диметил-N-(3S,5S)-5-[(4-метилпиперазин-1 ил)карбонил]пирролидин-3-илпропанамид (0,67 г, 1,59 ммоль), синтезированный на стадии D, и(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3-карбоновую кислоту, синтезированную согласно получению 12, подвергали взаимодействию согласно методике, аналогично описанной на стадии F примера 1, с получением ТФУК-соли указанного в заголовке соединения (0,92 г, 85%). МС [М+Н]=686 (М+1). 1 Н ЯМР (500 МГц, ДМСО-d6, 140 С)7,43-7,31 (м, 4 Н), 4,78-4,68 (ушир., 1 Н), 3,94-3,55 (м, 10 Н),3,44-3,26 (м, 3 Н), 3,24-2,91 (м, 7 Н), 2,78 (с, 3 Н), 2,42-2,32 (м, 1 Н), 2,23-2,11 (ушир., 1 Н), 1,85-1,73 (м, 1 Н),1,68-1,56 (ушир., 1 Н), 1,47-1,18 (м, 6 Н), 1,39 (с, 9 Н), 1,14 (с, 6 Н), 0,92, 0,90 (2 с, 6 Н). Примеры 5-15. Соединения, синтезированные согласно получениям 1-23, подвергали взаимодействию по методике,аналогично описанной в примерах 1-4, с получением соединений следующих примеров. Стадия А. Метил (2S,4S)-1-Вос-4-[(цис-4-метилциклогексил)амино]пирролидин-2-карбоксилат. Метил (2S,4S)-1-Вос-4-аминопирролидин-2-карбоксилат (1,07 г, 4,38 ммоль), синтезированный согласно получению 1, и 4-метилциклогексанон растворяли в DCE (30 мл) и к раствору добавлялиNaBH(OAc)3 (1,39 г, 6,57 ммоль) при комнатной температуре. После перемешивания реакционного раствора в течение 4 ч при комнатной температуре, добавляли насыщенный водный раствор NaHCO3 и экстрагировали DCM (50 мл 2) и EtOAc. Органический экстракт промывали насыщенным раствором соли,сушили над MgSO4, фильтровали и концентрировали в вакууме. Цис- и транс-соединения разделяли из полученного остатка колоночной хроматографией (элюент: EtOAc/Hex=1/2), с получением указанного в заголовке соединения (0,84 г, 57%). Стадия В. Метил (2S,4S)-1-Вос-4-(цис-4-метилциклогексил)[(2S)-тетрагидрофуран-2-илкарбонил] аминопирролидин-2-карбоксилат. Метил (2S,4S)-l-Boc-4-[(цис-4-метилциклогексил)амино]пирролидин-2-карбоксилат (0,84 г, 2,49 ммоль), синтезированный на стадии А, подвергали взаимодействию согласно методике, аналогично описанной на стадии А примера 3, с получением указанного в заголовке соединения (0,92 г, 87%). МС [М+Н]=439 (М+1). Стадия С. (4S)-1-Вос-4-(цис-4-метилциклогексил)[(2S)-тетрагидрофуран-2-илкарбонил]амино-Lпролин. Метил(2S,4S)-1-Вос-4-(цис-4-метилциклогексил)[(2S-тетрагидрофуран-2-илкарбонил]амино пирролидин-2-карбоксилат (0,92 г, 2,10 ммоль), синтезированный на стадии В, подвергали взаимодействию согласно методике, аналогично описанной на стадии С примера 1, с получением указанного в заголовке соединения (0,85 г, 95%). МС [М+Н]=425 (М+1). Стадия D. (2S,4S)-1-Вос-4-(цис-4-метилциклогексил)[(2S)-тетрагидрофуран-2-илкарбонил]амино 2-[(4-метилпиперазин-1-ил)карбонил]пирролидин.(0,85 г, 2,00 ммоль), синтезированный на стадии С, подвергали взаимодействию согласно методике, аналогично описанной на стадии D примера 1, с получением указанного в заголовке соединения (0,95 г,94%). МС [М+Н]=507 (м+1). Стадия Е. (2S)-N-(цис-4-метилциклогексил)-N-(3S,5S)-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-илтетрагидрофуран-2-карбоксамид. метилпиперазин-1-ил)карбонил]пирролидин (0,95 г, 1,87 ммоль), синтезированный на стадии D, подвергали взаимодействию согласно методике, аналогично описанной на стадии Е примера 1, с получением указанного в заголовке соединения (0,76 г, 99,8%). МС [М+Н]=407 (М+1). Стадия F. ТФУК-соль (2S)-N-(3S,5S)-1-[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3 ил]карбонил-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил-N-(цис-4 метилциклогексил)тетрагидрофуран-2-карбоксамида.(2S)-N-(цис-4-метилциклогексил)-N-(3S,5S)-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3 илтетрагидрофуран-2-карбоксамид (0,76 г, 1,87 ммоль), синтезированный на стадии Е, и (3S,4R)-1-третбутил-4-(4-хлорфенил)пирролидин-3-карбоновую кислоту, синтезированную согласно получению 12,подвергали взаимодействию согласно методике, аналогично описанной на стадии F примера 1, с получением указанного в заголовке соединения (1,10 г, 88%). МС [М+Н]=670 (М+1). Стадия G. HCl-соль (2S)-N-(3S,5S)-1-[(3S,4R)-1-трет-бутил-4-(4-хлорфенил)пирролидин-3 ил]карбонил-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамида. ТФУК-соль соединения, синтезированного на стадии F, подщелачивали 1 н. раствором NaOH и экстрагировали EtOAc. Полученный органический раствор сушили над MgSO4, концентрировали в вакууме и к остатку добавляли 4 М HCl/диоксан. Реакционный раствор перемешивали в течение 1 ч при комнатной температуре и концентрировали в вакууме без дальнейшей очистки, с получением HCl-соли соединения. 1 Н ЯМР (500 МГц, ДМСО-d6, 140 С)7,54-7,22 (м, 4 Н), 4,79-4,65 (ушир., 1 Н), 4,53-4,43 (м, 1 Н),3,96-3,58 (м, 10 Н), 3,58-3,37(м, 2 Н), 3,37-3,22 (м, 2 Н), 3,22-2,94 (м, 5 Н), 2,73 (с, 3 Н), 2,64-2,50 (м, 1 Н),2,43-2,27 (м, 1 Н), 2,20-2,09 (м, 1 Н), 2,09-1,97 (м, 1 Н), 1,97-1,74 (м, 5 Н), 1,61-1,44 (м, 4 Н), 1,40 (с, 9 Н),1,37-1,22 (м, 3 Н), 0,95 (с, 3 Н). Примеры 17-25. Соединения, синтезированные согласно получениям 1-23, подвергали взаимодействию по методике,аналогично описанной в примерах 1-4, 16, с получением соединений следующих примеров. Стадия А. Метил (2S,4S)-1-Вос-4-[(4,4-дифторциклогексил)амино]пирролидин-2-карбоксилат. Метил (2S,4S)-1-Вос-4-аминопирролидин-2-карбоксилат (29 г, 120 ммоль), синтезированный согласно получению 1, и 4,4-дифторциклогексанон (19,31 г, 144 ммоль), синтезированный согласно получению 5, растворяли в DCE. К раствору добавляли NaBH(OAc)3 (37 г, 180 ммоль). Реакционный раствор перемешивали в течение 6 ч при комнатной температуре. По окончании реакции раствор концентрировали в вакууме и к остатку добавляли водный раствор NaHCO3. Раствор экстрагировали EtOAc, сушили надMgSO4, концентрировали в вакууме и очищали колоночной хроматографией (элюент: EtOAc/Hex=l/4) с получением указанного в заголовке соединения (23,66 г, 55%), отделенного от метил (2S,4S)-1-Вос-4-[(4'фторциклогекс-3-ен-1-ил)амино]пирролидин-2-карбоксилата. МС [М+Н]=363 (М+1). Стадия В. Метил (2S,4S)-1-Вос-4-[(4,4-дифторциклогексил)(2,2-диметилпропаноил)амино]пирролидин-2-карбоксилат. Метил (2S,4S)-1-Вос-4-[(4,4-дифторциклогексил)амино]пирролидин-2-карбоксилат (1,03 г, 2,84 ммоль), синтезированный на стадии А, растворяли в DCE (5 мл) и к раствору добавляли TEA (5 мл) иDMAP (0,36 г, 2,84 ммоль). Коммерчески доступный пивалоилхлорид (1,03 г, 8,52 ммоль) также добавляли. Реакционный раствор нагревали до 90 С и перемешивали в течение 24 ч. По окончании реакции растворитель удаляли в вакууме и добавляли насыщенный водный раствор NaHCO3. Раствор экстрагировали EtOAc. Органический экстракт промывали 1 н. раствором HCl, сушили над MgSO4, концентрировали в вакууме и очищали колоночной хроматографией (элюент: EtOAc/Hex=l/4) с получением указанного в заголовке соединения (1,04 г, 82%). МС [М+Н]=447 (М+1). Стадия С. (4S)-1-Вос-4-[(4,4-дифторциклогексил)(2,2-диметилпропаноил)амино]-L-пролин. Метил(2S,4S)-1-Вос-4-[(4,4-дифторциклогексил)(2,2-диметилпропаноил)амино]пирролидин-2 карбоксилат (1,02 г, 2,32 ммоль), синтезированный на стадии В, подвергали взаимодействию согласно методике, аналогично описанной на стадии С примера 1, с получением указанного в заголовке соединения (0,95 г, 95%). МС [М+Н]=433 (М+1). Стадия(4S)-1-Вос-4-[(4,4-дифторциклогексил)(2,2-диметилпропаноил)амино]-L-пролин (0,95 г, 2,2 ммоль),синтезированный на стадии С, подвергали взаимодействию согласно методике, аналогично описанной на стадии D примера 1, с получением указанного в заголовке соединения (1,05 г, 93%). МС [М+Н]=515 (М+1). Стадия Е. N-(4,4-Дифторциклогексил)-2,2-диметил-N-(3S,5S)-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-илпропанамид.(2S,4S)-1-Вос-4-[(4,4-дифторциклогексил)(2,2-диметилпропаноил)амино]-2-[(4-метилпиперазин-1 ил)карбонил]пирролидин (1,05 г, 2,04 ммоль, синтезированный на стадии D, подвергали взаимодействию согласно методике, аналогично описанной на стадии Е примера 1, с получением указанного в заголовке соединения (845 мг, 99,9%). МС [М+Н]=415 (М+1). СтадияN-(4,4-Дифторциклогексил)-2,2-диметил-N-(3S,5S)-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-илпропанамид (845 мг, 2,04 ммоль), синтезированный на стадии Е, и (3S,4R)-1-трет-бутил-4-(4 хлорфенил)пирролидин-3-карбоновую кислоту, синтезированную согласно получению 12, подвергали взаимодействию согласно методике, аналогично описанной на стадии F примера 1, с получением ТФУКсоли указанного в заголовке соединения (1,18 г, 85%). МС [М+Н]=678 (М+1). 1H ЯМР (500 МГц, ДМСО-d6, 140 С)7,42-7,30 (м, 4 Н), 4,80-4,70 (ушир., 1 Н), 3,91-3,82 (м, 1 Н),3,82-3,55 (м, 10 Н), 3,39-3,28 (м, 2 Н), 3,24-3,12 (м, 1 Н), 3,02-2,85 (м, 4 Н), 2,65 (с, 3 Н), 2,39-2,30 (м, 1 Н),2,22-2,13 (ушир., 1 Н), 2,13-1,81 (м, 6 Н), 1,63-1,56 (м, 1 Н), 1,56-1,48 (м, 1 Н), 1,38 (с, 9 Н), 1,17 (с, 9 Н). Примеры 27-31. Соединения, синтезированные согласно получениям 1-23, подвергали взаимодействию по методике,- 23019146 аналогично описанной в примерах 1-4, 26, с получением соединений следующих примеров. Стадия А. Метил (2R,4S)-1-Вос-4-[(4,4-диметилциклогексил)амино]пирролидин-2-карбоксилат. Метил (2R,4S)-1-Вос-4-аминопирролидин-2-карбоксилат (1,07 г, 4,38 ммоль), синтезированный согласно получению 2, и 4,4-диметилциклогексанон (0,66 г, 5,25 ммоль) растворяли в DCE (20 мл) и к раствору добавляли NaBH(OAc)3 (1,39 г, 6,57 ммоль) при комнатной температуре. Реакционный раствор перемешивали в течение 4 ч при комнатной температуре и экстрагировали DCM (50 мл 2) и EtOAc. Органический экстракт промывали насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали колоночной хроматографией (элюент,EtOAc/Hex=1/2) с получением указанного в заголовке соединения (1,16 г, 75%). МС [М+Н]=355 (М+1). Стадия В. Метил (2R,4S)-1-Вос-4-(4,4-диметилциклогексил)[(2S)-тетрагидрофуран-2-илкарбонил] аминопирролидин-2-карбоксилат. Метил (2R,4S)-l-Boc-4-[(4,4-диметилциклогексил)амино]пирролидин-2-карбоксилат (1,16 г, 3,27 ммоль), синтезированный на стадии А, подвергали взаимодействию согласно методике, аналогично описанной на стадии А примера 3, с получением указанного в заголовке соединения (1,21 г, 82%). МС [М+Н]=453 (М+1). Стадия С. ( 4S)-1-Вос-4-(4,4-диметилциклогексил)[(2S)-тетрагидрофуран-2-илкарбонил]амино-Dпролин. Метил(2R,4S)-1-Вос-4-(4,4-диметилциклогексил)[(2S)-тетрагидрофуран-2-илкарбонил]амино пирролидин-2-карбоксилат (1,21 г, 2,67 ммоль), синтезированный на стадии В, подвергали взаимодействию согласно методике, аналогично описанной на стадии С примера 1, с получением указанного в заголовке соединения (1,14 г, 97%). МС [М+Н]=439 (М+1). Стадия D. (2R,4S)-1-Вос-4-(4,4-диметилциклогексил)[(2S)-тетрагидрофуран-2-илкарбонил]амино 2-[(4-метилпиперазин-1-ил)карбонил]пирролидин.(1,14 г, 2,60 ммоль), синтезированный на стадии С, подвергали взаимодействию согласно методике, аналогично описанной на стадии D примера 1, с получением указанного в заголовке соединения (1,29 г,95%). МС [М+Н]=521 (М+1). Стадия Е. (2S)-N-(4,4-диметилциклогексил)-N-(3S,5R)-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-илтетрагидрофуран-2-карбоксамид.(2R,4S)-1-Вос-4-(4,4-диметилциклогексил)[(2S)-тетрагидрофуран-2-илкарбонил]амино-2-[(4 метилпиперазин-1-ил)карбонил]пирролидин (1,29 г, 2,48 ммоль), синтезированный на стадии D, подвер- 24019146 гали взаимодействию согласно методике, аналогично описанной на стадии Е примера 1, с получением указанного в заголовке соединения (1,04 г, 99,8%). МС [М+Н]=421 (М+1). Стадия F. ТФУК-соль (2S)-N-(3S,5R)-1-[(3S,4R)-1-трет-бутил-4-(2,4-дифторфенил)пирролидин-3 ил]карбонил-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамида.(2S)-N-(4,4-Диметилциклогексил)-N-(3S,5R)-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3 илтетрагидрофуран-2-карбоксамид (1,04 г, 2,47 ммоль), синтезированный на стадии Е, и (3S,4R)-1-третбутил-4-(2,4-дифторфенил)пирролидин-3-карбоновую кислоту, синтезированную согласно получению 11, подвергали взаимодействию согласно методике, аналогично описанной на стадии F примера 1, с получением указанного в заголовке соединения (1,44 г, 85%). МС [М+Н]=686 (М+1). 1 Н ЯМР (500 МГц, ДМСО-d6, 140 С)7,63-7,53 (м, 1 Н), 7,08-6,98 (м, 2 Н), 5,08-4,98 (ушир., 1 Н),4,51 (дд, J=6,7, 6,15 Гц, 1 Н), 4,19-4,08 (м, 1 Н), 3,96-3,86 (м, 1 Н), 3,82-3,66 (м, 9 Н), 3,66-3,53 (м, 2 Н), 3,533,42 (м, 1 Н), 3,34 (дд, J=ll,6, 11,0 Гц, 1 Н), 3,23-3,13 (м, 1 Н), 3,13-3,00 (м, 4 Н), 2,74 (с, 3 Н), 2,66-2,56 (м,1 Н), 2,08-1,98 (м, 1 Н), 1,98-1,88 (м, 1 Н), 1,88-1,78 (м, 2 Н), 1,78-1,63 (м, 1 Н), 1,63-1,49 (м, 1 Н), 1,45-1,24(м, 7 Н), 1,38 (с, 9 Н), 0,90 (с, 6 Н). Примеры 33-40. Соединения, синтезированные согласно получениям 1-23, подвергали взаимодействию, согласно методике, аналогичной описанной в примерах 1-4, с получением соединений следующих примеров.(2S)-N-(4,4-Диметилциклогексил)-N-(3S,5S)-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3 илтетрагидрофуран-2-карбоксамид (903 мг, 2,15 ммоль), синтезированный на стадии D примера 3, растворяли в ДМФА (5 мл). Затем к раствору добавляли DIPEA (0,94 мл, 5,37 ммоль), (3S,4R)-1-Вос-4-(4 хлорфенил)пирролидин-3-карбоновую кислоту (0,70 г, 2,15 ммоль), синтезированную согласно получению 14, и HBTU (0,81 г, 2,15 ммоль). Реакционный раствор перемешивали в течение 2 ч и концентрировали в вакууме. Остаток разбавляли EtOAc и промывали насыщенным водным раствором NaHCO3. Органический экстракт сушили над MgSO4 и концентрировали в вакууме. Полученный остаток очищали колоночной хроматографией (элюент, DCM/MeOH=15/l) с получением указанного в заголовке соедине- 25019146(2S)-N-(3S,5S)-1-[(3S,4R)-4-(4-хлорфенил)пирролидин-3-ил]карбонил-5-[(4 метилпиперазин-1-ил)карбонил]пирролидин-3-ил-N-(4,4-диметилциклогексил)тетрагидрофуран-2 карбоксамид. 1-Boc-(3R,4S)-3-(4-хлорфенил)-4-2S,4S)-4-(4,4-диметилциклогексил)[(2S)-тетрагидрофуран-2 илкарбонил]амино-2-[(4-метилпиперазин-1-ил)карбонил]пирролидин-1-илкарбонил)пирролидин (1,33 г, 1,83 ммоль), синтезированный на стадии А, растворяли в DCM (1 мл) и по каплям добавляли 4 М раствор HCl (1 мл). Реакционный раствор перемешивали в течение 1 ч при комнатной температуре и концентрировали в вакууме с получением указанного в заголовке соединения (1,14 г, 99,8%). МС [М+Н]=628 (М+1). Стадия С. ТФУК-соль (2S)-N-(3S,5S)-1-[(3S,4R)-4-(4-хлорфенил)-1-(тетрагидро-2 Н-пиран-4 ил)пирролидин-3-ил]карбонил-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил-N-(4,4 диметилциклогексил)тетрагидрофуран-2-карбоксамида.(2S)-N-(3S,5S)-1-[(3S,4R)-4-(4-хлорфенил)пирролидин-3-ил]карбонил-5-[(4-метилпиперазин-1 ил)карбонил]пирролидин-3-ил-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид (1,14 г,1,81 ммоль), синтезированный на стадии В, и тетрагидро-4 Н-пиран-4-он (0,34 мл, 3,62 ммоль) растворяли в DCE (10 мл) и добавляли NaBH(OAc)3 (0,58 г, 2,74 ммоль) при комнатной температуре. После перемешивания реакционного раствора в течение 2 ч при комнатной температуре, добавляли насыщенный водный раствор NaHCO3 и экстрагировали раствор EtOAc. Органический экстракт промывали насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме. Полученный остаток очищали ВЭЖХ с получением ТФУК-соли указанного в заголовке соединения (1,16 г, 90%). МС [М+Н]=712 (М+1). 1 Н ЯМР (500 МГц, ДМСО-d6, 140 С)7,42-7,29 (м, 4 Н), 4,81-4,70 (ушир., 1 Н), 4,50 (дд, J=6,75, 6,7 Гц, 1 Н), 3,99-3,89 (м, 2 Н), 3,89-3,54 (м, 12 Н), 3,54-3,20 (м, 7 Н), 3,16-2,94 (м, 4 Н), 2,73 (с, 3 Н), 2,43-2,30 (м,1 Н), 2,27-2,12 (ушир., 1 Н), 2,07-1,94 (м, 3 Н), 1,94-1,76 (м, 4 Н), 1,74-1,62 (м, 3 Н), 1,48-1,20 (м, 6 Н), 0,93,0,90 (2 с, 6 Н). Пример 42. ТФУК-соль (2S)-N-(3S,5S)-1-[(3S,4R)-4-(4-хлорфенил)-1-циклопропилпирролидин-3 ил]карбонил-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамида(2S)-N-(3S,5S)-l-[(33,4R-4-(4-хлорфенил)пирролидин-3-ил]карбонил-5-[(4-метилпиперазин-1 ил)карбонил]пирролидин-3-ил-N-(4,4-диметилциклогексил)тетрагидрофуран-2-карбоксамид (1,14 г,1,81 ммоль), синтезированный на стадии В примера 41, растворяли в DCE (20 мл). К раствору добавляли 1-этоксициклопропокситриметилсилан (0,47 г, 2,70 ммоль) и NaBH3CN (228 мг, 3,63 ммоль). После добавления каталитического количества уксусной кислоты, реакционный раствор перемешивали в течение 2 ч при 80 С. По окончании реакции раствор концентрировали в вакууме. Остаток разбавляли EtOAc и промывали насыщенным водным раствором NaHCO3. Органический экстракт сушили над MgSO4 и концентрировали в вакууме. Полученный остаток очищали ВЭЖХ, с получением ТФУК-соли указанного в заголовке соединения (1,03 г, 85%). МС [М+Н]=668 (М+1). 1 Н ЯМР (500 МГц, ДМСО-d6, 140 С)7,44-7,24 (м, 4 Н), 4,80-4,71 (ушир., 1 Н), 4,29-4,20 (м, 1 Н),3,95-3,38 (м, 13 Н), 3,38-3,20 (м, 2 Н), 3,20-2,89 (м, 6 Н), 2,74, 2,72 (2 с, 3 Н), 2,64-2,49 (м, 1 Н), 2,42-2,34 (м,1 Н), 2,25-2,10 (м, 1 Н), 2,10-1,96 (м, 1 Н), 1,96-1,76 (м, 3 Н), 1,76-1,62 (м, 2 Н), 1,62-1,46 (м, 3 Н), 1,46-1,20(2S)-N-(4,4-Диметилциклогексил)-N-(3S,5S)-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3 илтетрагидрофуран-2-карбоксамид (903 мг, 2,15 ммоль), синтезированный на стадии D примера 3, растворяли в ДМФА (5 мл). К раствору добавляли DIPEA (0,94 мл, 5,38 ммоль) и последовательно (3S,4R)4-(4-хлорфенил)-1-(6-метилпиридазин-3-ил)пирролидин-3-карбоновую кислоту (0,68 г, 2,15 ммоль), синтезированную согласно получению 17, и HBTU (0,82 г, 2,15 ммоль). Реакционный раствор перемешивали в течение 2 ч при комнатной температуре и концентрировали в вакууме. Остаток растворяли EtOAc и промывали раствор насыщенным водным раствором NaHCO3 и водой. Органический экстракт сушили над MgSO4 и концентрировали в вакууме. Остаток очищали ВЭЖХ, с получением ТФУК-соли указанного в заголовке соединения (1,31 г, 85%). МС [М+Н]=668 (М+1). 1 Н ЯМР (500 МГц, ДМСО-d6, 140 С)7,55-7,49 (м, 1 Н), 7,43-7,30 (м, 4 Н), 7,29-7,23 (м, 1 Н), 4,844,73 (ушир., 1 Н), 4,55 (дд, J=6,75, 6,1 Гц, 1 Н), 4,14-3,79 (м, 5 Н), 3,79-3,62 (м, 7 Н), 3,62-3,32 (м, 4 Н), 3,223,03 (м, 4 Н), 2,78 (с, 3 Н), 2,52-2,42 (м, 1 Н), 2,46 (с, 3 Н), 2,21-2,10 (ушир., 1 Н), 1,97-1,86 (м, 1 Н), 1,82-1,51(2S)-N-(цис-4-метилциклогексил)-N-(3S,5S)-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3 илтетрагидрофуран-2-карбоксамид (0,76 г, 1,87 ммоль), синтезированный на стадии Е примера 16, и(3S,4R)-4-(2,4-дифторфенил)-1-(6-хлорпиридазин-3-ил)пирролидин-3-карбоновую кислоту, синтезированную согласно получению 19, подвергали взаимодействию по методике, аналогично описанной на стадии F примера 1, с получением указанного в заголовке соединения (1,20 г, 88%). МС [М+Н]=728 (М+1). 1 Н ЯМР (500 МГц, ДМСО-d6, 140 С)7,51-7,42 (м, 1 Н), 7,37(д, 1 Н), 7,05-6,94 (м, 3 Н), 4,80-4,72(2S)-N-(4,4-Диметилциклогексил)-N-(3S,5S)-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3 илтетрагидрофуран-2-карбоксамид (903 мг, 2,15 ммоль), синтезированный на стадии D примера 3, и(3S,4R)-4-(4-хлорфенил)-1-(1,6-дигидропиридазин-3-ил)пирролидин-3-карбоновую кислоту, синтезированную согласно получению 21, подвергали взаимодействию по методике, аналогично описанной на стадии F примера 1, с получением указанного в заголовке соединения (1,34 г, 88%). МС [М+Н]=708 (М+1). 1 Н ЯМР (500 МГц, ДМСО-d6, 140 С)8,57-8,52 (д, 1 Н), 7,57-7,43 (м, 2 Н), 7,22-7,14 (м, 1 Н), 7,086,97 (м, 2 Н), 4,84-4,72 (ушир., 1 Н), 4,56-4,51 (м, 1 Н), 4,15-4,01 (м, 3 Н), 4,01-3,88 (м, 2 Н), 3,83-3,45 (м,11 Н), 3,25-3,07 (м, 4 Н), 2,81 (с, 3 Н), 2,46-2,35 (м, 1 Н), 2,27-2,14 (ушир., 1 Н), 2,11-2,01 (м, 1 Н), 1,99-1,74(2S)-N-(цис-4-метилциклогексил)-N-(3S,5S)-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3 илтетрагидрофуран-2-карбоксамид (0,76 г, 1,87 ммоль), синтезированный на стадии Е примера 16, и(3S,4R)-1-Вос-4-(4-хлорфенил)пирролидин-3-карбоновую кислоту, синтезированную согласно получению 14, подвергали взаимодействию по методике, аналогично описанной на стадии F примера 1, с получением указанного в заголовке соединения (1,18 г, 88%). МС [М+Н]=714 (М+1). Стадия В. (2S)-N-(3S,5S)-1-[(3S,4R)-4-(4-хлорфенил)пирролидин-3-ил]карбонил-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил-N-(цис-4-метилциклогексил)тетрагидрофуран-2 карбоксамид. 1-Вос-(3R,4S)-3-(4-хлорфенил)-4-2S,4S)-4-(цис-4-метилциклогексил)[(2S)-тетрагидрофуран-2 илкарбонил]амино-2-[(4-метилпиперазин-1-ил)карбонил]пирролидин-1-илкарбонил)пирролидин (1,18 г, 1,65 ммоль), синтезированный на стадии А, растворяли в DCM (1 мл). По каплям добавляли 4 М раствор HCl (1 мл). Реакционный раствор перемешивали в течение 1 ч при комнатной температуре, концентрировали в вакууме, с получением указанного в заголовке соединения (1,01 г, 99,8%). МС [М+Н]=614 (М+1). Стадия С. ТФУК-соль(2S)-N-(3S,5S)-1-[(3S,4R)-4-(4-хлорфенил)-1-фенилпирролидин-3 ил]карбонил-5-[(4-метилпиперазин-1-ил)карбонил]пирролидин-3-ил-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамида. 2S)-N-(3S,5S)-1-[(3S,4R)-4-(4-хлорфенил)пирролидин-3-ил]карбонил-5-[(4-метилпиперазин-1 ил)карбонил]пирролидин-3-ил-N-(цис-4-метилциклогексил)тетрагидрофуран-2-карбоксамид (1,01 г,1,64 ммоль), синтезированный на стадии В, растворяли в толуоле (10 мл). К раствору добавляли третбутилат натрия (0,18 г, 1,87 ммоль), 2-(ди-трет-бутилфосфино)бифенил (42 мг, 0,14 ммоль),трис(дибензилиденацетон)дипалладий(0) (80 мг, 0,09 ммоль) и бромбензол (29 мг, 1,87 ммоль) и перемешивали в течение 10 ч при 110 С. По окончании реакции, твердые вещества отфильтровывали, используя целит. Реакционный раствор разбавляли EtOAc и промывали насыщенным водным растворомNaHCO3. Органический слой сушили над MgSO4 и концентрировали в вакууме. Остаток очищали ВЭЖХ,с получением указанного в заголовке соединения (0,89 г, 78%). МС [М+Н]=690(М+1). 1 Н ЯМР (500 МГц, ДМСО-d6, 140 С)7,75-7,64 (м, 1 Н), 7,62-7,52 (м, 2 Н), 7,52-7,40 (м, 2 Н), 7,407,27 (м, 3 Н), 7,24-7,10 (м, 1 Н), 4,92-4,83 (ушир., 1 Н), 4,56-4,45 (м, 1 Н), 4,15-3,80 (м, 5 Н), 3,80-3,44 (м,11 Н), 3,25-3,07 (м, 4 Н), 2,77 (с, 3 Н), 2,46-2,35 (м, 1 Н), 2,27-2,14 (м, 1 Н), 2,11-2,01 (м, 1 Н), 1,99-1,74 (м,6 Н), 1,65-1,47(м, 4 Н), 1,41-1,29 (м, 2 Н), 0,98 (д, 3 Н). Примеры 47-73. Соединения, синтезированные согласно получениям 1-23, подвергали взаимодействию по методике,аналогично описанной в примерах 1-4, 41-46, с получением соединений следующих примеров.
МПК / Метки
МПК: C07D 403/14
Метки: меланокортина, рецептора, агонисты
Код ссылки
<a href="https://eas.patents.su/30-19146-agonisty-receptora-melanokortina.html" rel="bookmark" title="База патентов Евразийского Союза">Агонисты рецептора меланокортина</a>
Пиридоновые агонисты сопряженного с g-белком рецептора gpr119

Номер патента: 18709
Опубликовано: 30.10.2013
Авторы: Росси Карен А., Ван Инг, Уокер Дин А.
МПК: C07D 401/12, A61P 3/10, A61K 31/454...
Метки: пиридоновые, gpr119, g-белком, рецептора, агонисты, сопряженного
Формула / Реферат:
1. Соединение, выбранное из соединения формулы I или IAФормула I Формула IAили его энантиомера, диастереомера, сольвата или фармацевтически приемлемой соли, имеющих кольцо А и кольцо В, гдекольцо А необязательно замещено одним или большим количеством R, представленного как R20 и R21,G представляет собой СН или N,Q представляет собой С,X представляет собой СН или N,Y представляет собой О, OCR9R9 или S,n1 имеет значения 0-2,n2 имеет значения...
Агонисты в2-адренергического рецептора

Номер патента: 3453
Опубликовано: 26.06.2003
Авторы: Чои Сеок-Ки, Моран Эдмунд Дж.
МПК: A61K 38/00, A61K 39/00, A61K 39/395...
Метки: рецептора, в2-адренергического, агонисты
Формула / Реферат:
1. Соединение формулы (II) где Ar1 и Ar3 независимо выбирают из группы, включающей арил, гетероарил и гетероциклил; Ar2 выбирают из группы, включающей фенилен, где группы W и X присоединены в 1,2-, 1,3 и 1,4 положениях фениленового кольца; циклогексилен, необязательно замещенный метилом, где группы W и X присоединены в 1,3 и 1,4 положениях циклогексиленового кольца, и пиперазинилен, где группы W и X присоединены в 1,4 положениях...
Агонисты β2-адренергического рецептора

Номер патента: 4263
Опубликовано: 26.02.2004
Авторы: Чои Сеок-Ки, Моран Эдмунд Дж.
МПК: A61P 11/00, A61K 31/135, A61K 31/165...
Метки: beta;2-адренергического, рецептора, агонисты
Формула / Реферат:
1. Соединение формулы или его фармацевтически приемлемая соль. 2. Соединение формулы где стереохимия при *C и **C представляет (RS) и (RS); (R) и (R); (R) и (S); (S) и (R) или (S) и (S); или его фармацевтически приемлемая соль. 3. Соединение по п.2, где стереохимия при *C представляет (R) и стереохимия при **C представляет (R). 4. Соединение по п.2, где стереохимия при *C представляет (R) и стереохимия при **C представляет (S). 5....
Селективные пептидные агонисты рецептора vpac2

Номер патента: 12930
Опубликовано: 26.02.2010
Авторы: Чжан Ляньшань, Боквист Бенгт Кристер, Алсина-Фернандес Хорхе
МПК: A61K 38/00, C07K 14/575, A61K 47/48...
Метки: vpac2, пептидные, агонисты, селективные, рецептора
Формула / Реферат:
1. Пегилированный пептидный агонист рецептора VPAC2, содержащий последовательность, выбранную изи С-концевой удлиняющий сегмент, где N-конец С-концевого удлиняющего сегмента связан с С-концом пептидной последовательности и где С-концевой удлиняющий сегмент содержит последовательность аминокислот формулыгде Xaa1 представляет собой Gly, Cys или отсутствует;Хаа2 представляет собой Gly, Arg или отсутствует;Хаа3 представляет собой Pro, Thr или...
Селективные агонисты рецептора y2 для терапевтического воздействия

Номер патента: 11860
Опубликовано: 30.06.2009
Автор: Шварц Ту
МПК: A61P 3/04, A61K 8/00, A61K 38/17...
Метки: воздействия, агонисты, терапевтического, селективные, рецептора
Формула / Реферат:
1. Применение Y-рецепторного агониста, отличного от PYY3-36, селективного к Y2 рецептору по сравнению с Y1 и Y4 рецепторами, для получения композиции для лечения состояний, восприимчивых к активации Y2 рецепторов, причем: (а) указанный агонист является PP-складчатым пептидом или PP-складчатым пептидом-миметиком, выбранным из PYY, NPY, миметиков PYY и миметиков NPY, которые содержат С-концевую аминокислотную последовательность Y2-рецепторного...
Предыдущий патент: Активирующие подложки на основе перфторированных бороновых кислот
Следующий патент: Предварительно спрессованные быстро распадающиеся лекарственные формы соединений с низкой пероральной биодоступностью
Случайный патент: Способ утяжеления пластмассовых труб и утяжеленные пластмассовые трубы