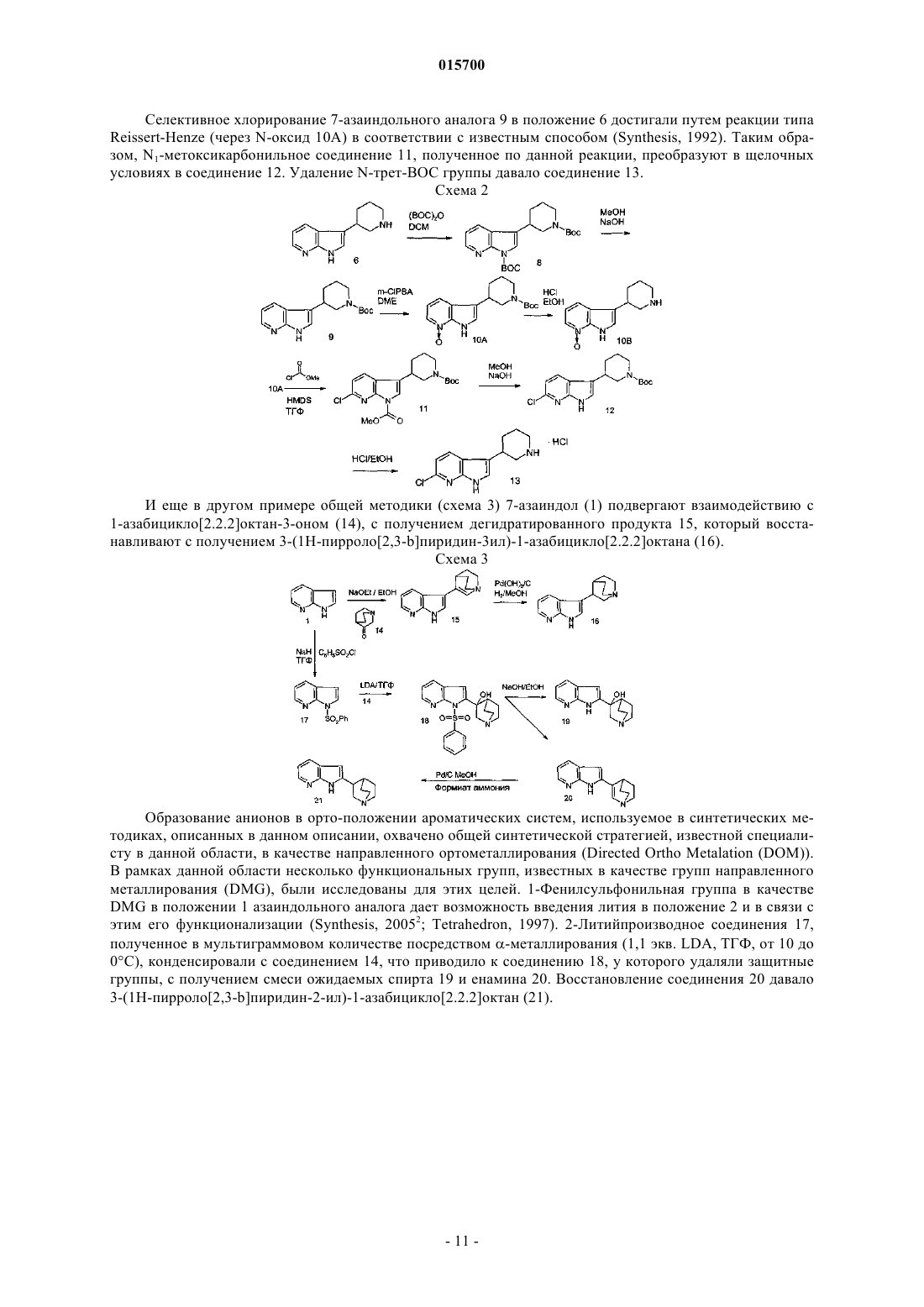
Азаиндольные производные с сочетанием частичного агонизма к никотиновому ацетилхолиновому рецептору и ингибирования обратного захвата дофамина
Номер патента: 15700
Опубликовано: 31.10.2011
Авторы: Стойт Аксел, Крусе Корнелис Г., Ван Дер Нэт Мартина А.В., Колен Хейн К.А.С.




























Формула / Реферат
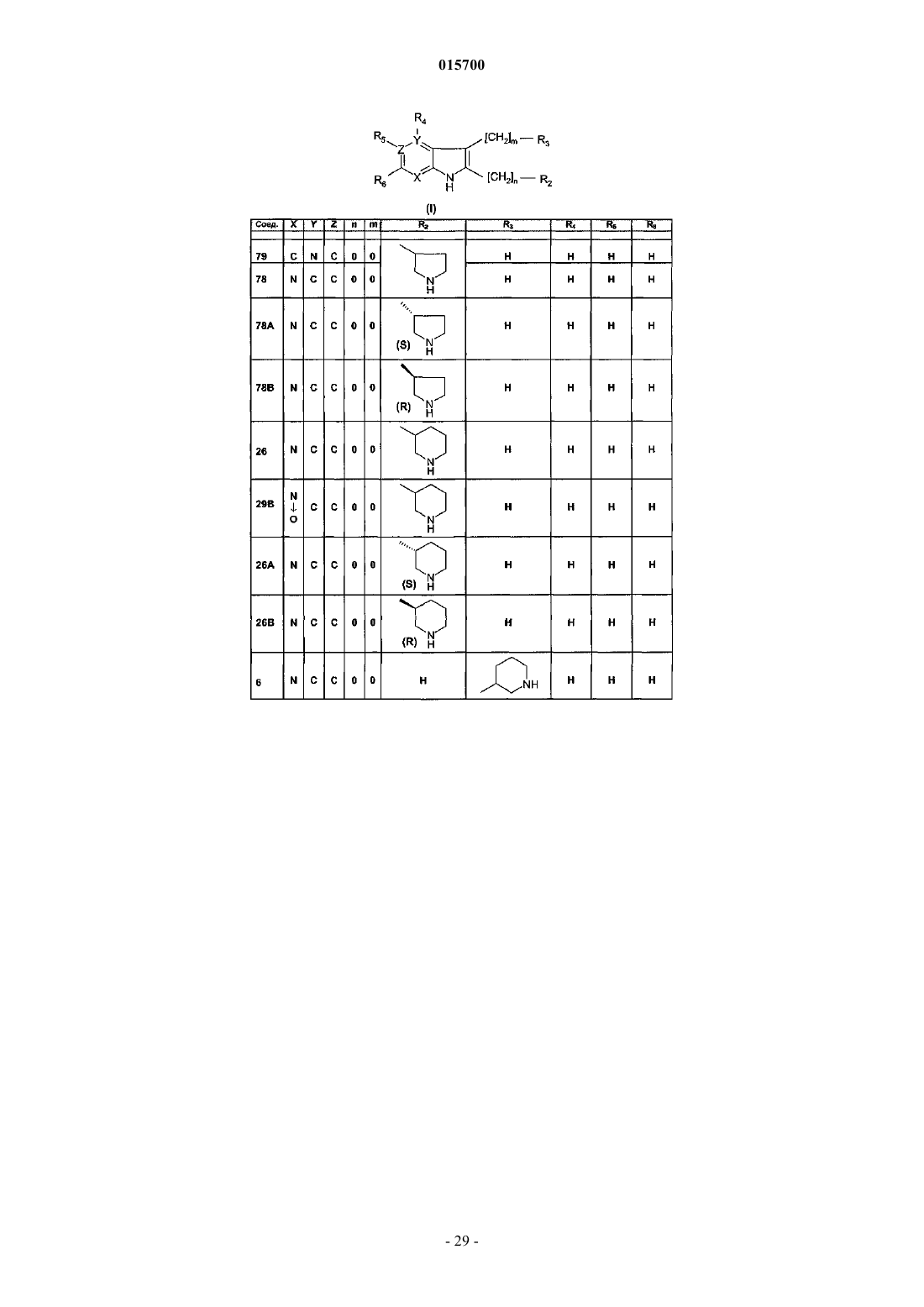
1. Соединения формулы (I)

или таутомер, стереоизомер, N-оксид или фармакологически приемлемая соль, гидрат или сольват любого из вышеуказанного,
где X, Y и Z независимо представляют собой N или С с учетом того, что кольцо содержит по меньшей мере один N-атом и не более 2;
m и n независимо равны или 0, или 1 при условии, что когда Y и Z представляют собой углерод и X представляет собой азот, m равен 0;
R2 и R3независимо представляют собой водород, галоген, (C1-3)алкил, (C1-3)алкинил, NH(C1-3)алкил, CF3, гидроксил, (C1-3)алкилокси или пиперидинильную, пирролидинильную, тетрагидропиридинильную, морфолинильную, азепанильную, 1-азабицикло[2.2.2]октанильную или 1-азабицикло[2.2.2]окт-2-енильную группу, причем любая группа является или незамещенной, или замещена одним или несколькими заместителями, выбранными из галогена, (C1-3)алкила, фенила или бензила;
R4, R5и R6 независимо представляют собой водород, галоген, (C1-3)алкил, (С2-3)алкинил, CF3, NH(C1-3)алкил, гидроксил или (C1-3)алкилокси с учетом того, что R4присутствует только тогда, когда Y представляет собой С, и R5присутствует только тогда, когда Z представляет собой С,
при условии, что когда X и Z представляют собой N, Y представляет собой С, R4 представляет собой Cl, R5и R6 представляют собой Н, m и n равны 0, R2представляет собой Н, R3не является иодом или 1,2,3,6-тетрагидропиридин-4-илом;
при условии, что когда X представляет собой N, Y и Z представляют собой С, R4 представляет собой Cl, R5представляет собой Br, R6 представляет собой Н, m и n равны 0, R3 представляет собой Н, R2не является иодом или Н;
при условии, что когда X и Z представляют собой С, Y представляет собой N, R5 представляет собой гидрокси, R2, R4и R6 представляют собой Н, m и n равны 0, R3не является 1,2,3,6-тетрагидропиридин-4-илом;
при условии, что когда X представляет собой N, Y и Z представляют собой С, m и n равны 0, R2, R3, R4 и R5 представляют собой Н, R6не является хлором или фтором;
при условии, что когда X представляет собой N, Y и Z представляют собой С, m и n равны 0, R2, R3, R4 и R6 представляют собой Н, R5не является бромом, хлором или фтором;
при условии, что когда X представляет собой N, Y и Z представляют собой С, m равен 1, n равен 0, R2, R4, R5 и R6представляют собой Н, R3не является этилом или морфолин-4-илом;
при условии, что когда X представляет собой N, Y и Z представляют собой С, m и n равны 0, R2, R4, R5 и R6 представляют собой Н, R3не является бромом;
при условии, что когда X представляет собой N, Y и Z представляют собой С, m и n равны 0, R2, R3, R5 и R6 представляют собой Н, R4не является хлором;
при условии, что когда X представляет собой N, Y и Z представляют собой С, m равен 0, n равен 1, R3, R4, R5 и R6представляют собой Н, R2не является водородом или пиперидин-1-илом;
при условии, что когда X представляет собой N, Y и Z представляют собой С, m равен 0, n равен 1, R4, R5и R6 представляют собой Н, R3представляет собой бром, R2не является пиперидин-1-илом, 2-метилпиперидинил-1-илом, 2-этилпиперидинил-1-илом или морфолин-4-илом.
2. Соединения по п.1 формулы (I), где R2 и R3независимо представляют собой водород или пиперидинильную, пирролидинильную, тетрагидропиридинильную, морфолинильную, азепанильную, 1-азабицикло[2.2.2]октанильную или 1-азабицикло[2.2.2]окт-2-енильную группу, причем любая группа является или незамещенной, или замещена одним или несколькими заместителями, выбранными из галогена, (C1-3)алкила, фенила или бензила, R4, R5 и R6 независимо представляют собой водород, галоген, (C1-3)алкил или алкил(C1-3)окси с учетом того, что R4 присутствует только тогда, когда Y представляет собой С, и что R5 присутствует только тогда, когда Z представляет собой С, и X, Y, Z, m и n имеют значения, как указано в п.1.
3. Соединение по п.1, которое представляет собой
2-пирролидин-3-ил-1Н-пирроло[3,2-b]пиридин,
2-пирролидин-3-ил-1Н-пирроло[2,3-b]пиридин,
(S)-2-пирролидин-3-ил-1Н-пирроло[2,3-b]пиридин,
(R)-2-пирролидин-3-ил-1Н-пирроло[2,3-b]пиридин,
2-пиперидин-3-ил-1Н-пирроло[2,3-b]пиридин,
2-пиперидин-3-ил-1Н-пирроло[2,3-b]пиридин 7-оксид,
(S)-2-пиперидин-3-ил-1Н-пирроло[2,3-b]пиридин,
(R)-2-пиперидин-3-ил-1Н-пирроло[2,3-b]пиридин,
3-пиперидин-3-ил-1Н-пирроло[2,3-b]пиридин,
3-пиперидин-3-ил-1Н-пирроло[2,3-b]пиридин 7-оксид,
4-хлор-2-пиперидин-3-ил-1Н-пирроло[2,3-b]пиридин,
6-хлор-2-пиперидин-3-ил-1Н-пирроло[2,3-b]пиридин,
(S)-6-хлор-2-пиперидин-3-ил-1Н-пирроло[2,3-b]пиридин,
(R)-6-хлор-2-пиперидин-3-ил-1Н-пирроло[2,3-b]пиридин,
6-хлор-3-пиперидин-3-ил-1Н-пирроло[2,3-b]пиридин,
2-(1-метилпиперидин-3-ил-1Н-пирроло[2,3-b]пиридин,
2-(1,2,5,6-тетрагидропиридин-3-ил)-1Н-пирроло[2,3-b]пиридин,
3-(1Н-пирроло[2,3-b]пиридин-2-ил)пиперидин-3-ол,
3-(1Н-пирроло[2,3-b]пиридин-2-ил)-1-азабицикло[2.2.2]октан-3-ол,
3-(1Н-пирроло[2,3-b]пиридин-2-ил)-1-азабицикло[2.2.2]октан,
3-(1Н-пирроло[2,3-b]пиридин-3-ил)пиперидин-3-ол,
3-(1Н-пирроло[2,3-b]пиридин-3-ил)-1-азабицикло[2.2.2]октан,
3-(1Н-пирроло[2,3-b]пиридин-3-ил)-1-азабицикло[2.2.2]окт-2-ен,
2-пирролидин-2-илметил-1Н-пирроло[2,3-b]пиридин,
(R)-2-пирролидин-2-илметил-1Н-пирроло[2,3-b]пиридин,
(R)-6-пирролидин-2-илметил-7Н-пирроло[2,3-d]пиримидин,
(R)-6-фтор-2-пирролидин-2-илметил-1Н-пирроло[2,3-b]пиридин,
(R)-6-хлор-2-пирролидин-2-илметил-1Н-пирроло[2,3-b]пиридин,
(R)-6-бром-2-пирролидин-2-илметил-1Н-пирроло[2,3-b]пиридин,
(R)-6-метокси-2-пирролидин-2-илметил-1Н-пирроло[2,3-b]пиридин,
(R)-5-бром-2-пирролидин-2-илметил-1Н-пирроло[2,3-b]пиридин,
(R)-5-метил-2-пирролидин-2-илметил-1Н-пирроло[2,3-b]пиридин,
(R)-5-метокси-2-пирролидин-2-илметил-1Н-пирроло[3,2-b]пиридин,
(R)-2-пирролидин-2-илметил-1Н-пирроло[3,2-b]пиридин,
(R)-3-пирролидин-2-илметил-1Н-пирроло[3,2-b]пиридин,
(S)-2-пирролидин-2-илметил-1Н-пирроло[3,2-b]пиридин,
(S)-2-пирролидин-2-илметил-1Н-пирроло[2,3-b]пиридин,
(S)-6-фтор-2-пирролидин-2-илметил-1Н-пирроло[2,3-b]пиридин,
(S)-6-хлор-2-пирролидин-2-илметил-1Н-пирроло[2,3-b]пиридин,
(S)-6-бром-2-пирролидин-2-илметил-1Н-пирроло[2,3-b]пиридин,
(S)-5-бром-2-пирролидин-2-илметил-1Н-пирроло[2,3-b]пиридин,
(S)-2-(1-метилпирролидин-2-илметил)-1Н-пирроло[2,3-b]пиридин,
(R)-2-(1-метилпирролидин-2-илметил)-1Н-пирроло[2,3-b]пиридин,
2-пирролидин-3-илметил-1Н-пирроло[2,3-b]пиридин,
2-(1-метилпирролидин-3-илметил)-1Н-пирроло[2,3-b]пиридин,
2-(1-бензилпирролидин-3-илметил)-1Н-пирроло[2,3-b]пиридин.
4. (R)-Энантиомеры соединений по п.1, где R2 или R3 независимо представляют собой пиперидинильную, пирролидинильную, тетрагидропиридинильную, морфолинильную, азепанильную, 1-азабицикло[2.2.2]октанильную или 1-азабицикло[2.2.2]окт-2-енильную группу, причем любая группа является или незамещенной, или замещена одним или несколькими заместителями, выбранными из галогена, (C1-3)алкила, фенила или бензила, и кольцо которого содержит асимметричный атом углерода или непосредственно связанный с азаиндольным ядром (когда m и n равны 0), или связанный через метиленовый мостик (когда m или n равны 1), и где все другие символы имеют значения, как указано в п.1.
5. Фармацевтическая композиция, содержащая в дополнение к фармацевтически приемлемому носителю и/или по меньшей мере одному фармацевтически приемлемому вспомогательному веществу фармакологически активное количество по меньшей мере одного соединения по одному из пп.1-4 или его соли в качестве активного ингредиента.
6. Способ получения фармацевтических композиций по п.5, заключающийся в том, что соединение по одному из пп.1-4 приводят в форму, подходящую для введения.
7. Применение соединения по любому из пп.1-4 или его соли в качестве лекарственного средства.
8. Соединение формулы (I*)

где Q представляет собой защитную группу

X, Y и Z независимо представляют собой N или С с учетом того, что кольцо содержит по меньшей мере один N-атом и не более 2;
m и n независимо равны или 0, или 1, при условии, что когда Y и Z представляют собой углерод и X представляет собой азот, m равен 0,
R2 и R3независимо представляют собой пиперидинильную, пирролидинильную, тетрагидропиридинильную, морфолинильную, азепанильную, 1-азабицикло[2.2.2]октанильную или 1-азабицикло[2.2.2]окт-2-енильную группу, причем любая группа является или незамещенной, или замещена одним или несколькими заместителями, выбранными из галогена, (C1-3)алкила, фенила или бензила, в котором кольцо, когда оно содержит один атом азота, указанный атом азота замещен атомом водорода, бензильной группой, трет-ВОС-группой или SO2-OH группой;
R4, R5и R6 независимо представляют собой водород, галоген, (C1-3)алкил, (C1-3)алкинил, CF3, NH(C1-3)алкил, гидроксил или алкилоксигруппу с учетом того, что R4 присутствует только тогда, когда Y представляет собой С, и R5 присутствует только тогда, когда Z представляет собой С,
в качестве промежуточного соединения для получения некоторых соединений формулы (I).
9. Применение соединения по любому из пп.1-4 для получения фармацевтической композиции для лечения заболеваний ЦНС, таких как нейроэндокринные, неврологические и невропсихиатрические заболевания, шизофрения, затруднения с памятью и при обучении, расстройство гиперактивности с дефицитом внимания, тревожные расстройства, депрессивные заболевания, нейродегенеративные заболевания, болезнь Альцгеймера, аддикция, боль при расстройстве пищевого поведения, воспалительные процессы, судорожные синдромы, офтальмологические заболевания, глаукома, дегенерация желтого пятна, диабетическая ретинопатия, сердечно-сосудистые и желудочно-кишечные заболевания и рак.
10. Применение по п.9, где указанная аддикция выбрана из группы, состоящей из никотиновой зависимости, кокаиновой зависимости и амфетаминовой зависимости.
Текст
Дата публикации и выдачи патента Номер заявки АЗАИНДОЛЬНЫЕ ПРОИЗВОДНЫЕ С СОЧЕТАНИЕМ ЧАСТИЧНОГО АГОНИЗМА К НИКОТИНОВОМУ АЦЕТИЛХОЛИНОВОМУ РЕЦЕПТОРУ И ИНГИБИРОВАНИЯ ОБРАТНОГО ЗАХВАТА ДОФАМИНА Изобретение относится к азаиндольным производным, имеющим общую формулу (I), где символы имеют значения, приведенные в описании. Такие соединения обладают сочетанием частичного агонизма в отношении никотинового ацетилхолинового рецептора и ингибирования обратного захвата дофамина. Изобретение также относится к фармацевтическим композициям, содержащим такие соединения, способам их получения, способам получения новых промежуточных соединений, пригодных для их синтеза, способам получения композиций и применению таких соединений и композиций, в частности их применению при введении их пациентам для достижения терапевтического эффекта при заболеваниях, в которых затронуты никотиновые рецепторы и/ или дофаминовые переносчики, или которые можно лечить посредством воздействия на данные рецепторы. Стойт Аксел, Колен Хейн К.А.С.,Ван Дер Нэт Мартина А.В., Крусе Корнелис Г. (NL) Медведев В.Н. (RU) 015700 Область техники, к которой относится изобретение Данное изобретение относится к азаиндольным производным, имеющим общую формулу (I) где символы имеют значения, приведенные в описании. Данные соединения обладают сочетанием частичного агонизма в отношении никотинового ацетилхолинового рецептора и ингибирования обратного захвата дофамина. Изобретение также относится к фармацевтическим композициям, содержащим указанные соединения, к способам их получения, способам получения новых промежуточных соединений,пригодных для их синтеза, способам получения композиций и применению таких соединений и композиций, в частности их применению при введении пациентам для достижения терапевтического эффекта при заболеваниях, в которых затронуты никотиновые рецепторы и/или дофаминовые переносчики или которые можно лечить посредством воздействия на эти рецепторы. Предшествующий уровень техники Как предполагается, никотин обладает рядом фармакологических действий (Pullan, 1994). Некоторые из этих действий связаны с воздействиями в результате высвобождения нейромедиатора. Высвобождение ацетилхолина, дофамина, норэпинефрина, серотонина и глутамата после введения никотина было описано Toth, 1992. Подтверждающие публикации и дополнительные последние исследования включают модуляцию ЦНС глутаматом, оксидом азота, ГАМК, тахикининами, цитокинами и пептидами (Brioni,1997). Кроме того, нейропротекторные и различные другие положительные фармакологические действия никотина предполагаются (Sjak-shie, 1993; Onaivi, 1994). Различные соединения, для которых мишенью являются рецепторы nAChR, как сообщалось, являются полезными при лечении широкого спектра состояний и заболеваний (Damaj, 1999; Bannon, 1998;Bencherif, 2002; Levin, 2002; O'Neill, 2002; Breining, 2005). Терапевтические показания, обсуждаемые в приведенной выше литературе, включают заболевания ЦНС, такие как нейроэндокринные, неврологические и невропсихиатрические заболевания, шизофрения, затруднения с памятью и при обучении, расстройство гиперактивности с дефицитом внимания, тревожные расстройства, депрессивные заболевания,нейродегенеративные заболевания, болезнь Альцгеймера, аддикция (болезненное пристрастие), никотиновая зависимость, кокаиновая зависимость, амфетаминовая зависимость, боль при расстройстве пищевого поведения, воспалительные процессы, судорожные синдромы, офтальмологические заболевания,глаукома, дегенерация желтого пятна, диабетическая ретинопатия, сердечно-сосудистые и желудочнокишечные заболевания и рак. Антагонисты никотиновых рецепторов обладают хорошим потенциалом в качестве терапевтических агентов, поскольку они обеспечивают другие механизмы модуляции функционирования никотинового рецептора. Никотиновые агонисты быстро десенсибилизируют эти рецепторы, существенно ингибируя их функционирование. Таким образом, ингибирование функционирования никотинового рецептора может быть тем действием, которое дает полезное клиническое свойство, указывающее на то, что антагонисты никотиновых рецепторов могут также быть полезными в лечении заболеваний, для которых в настоящее время разрабатываются никотиновые агонисты. Например, шизофрения и лекарственная зависимость, и то, и другое, тесно связаны с гиперактивностью дофаминергических систем ЦНС, и ингибирование никотиновых рецепторов может быть эффективным при снижении такой гиперактивности. Заболевания ЦНС, тип неврологических заболеваний, могут быть вызваны действием лекарственного средства; могут объясняться генетической предрасположенностью, инфекцией или травмой; или могут быть неизвестной этиологии. Они включают невропсихиатрические заболевания, неврологические заболевания и расстройства умственной деятельности и включают нейродегенеративные заболевания,поведенческие нарушения, нарушения познавательных способностей и когнитивные заболевания, вызывающие расстройство психики. Существует несколько заболеваний ЦНС, клинические проявления которых объясняются нарушением функции ЦНС (т.е. заболевания, возникающие в результате неадекватных уровней высвобождения нейромедиатора, неадекватных свойств нейромедиаторных рецепторов и/или неадекватного взаимодействия между нейромедиаторами и нейромедиаторными рецепторами). Некоторые заболевания ЦНС объясняются недостатком ацетилхолина, дофамина, норэпинефрина и/или серотонина. В общем и целом, обычные заболевания ЦНС включают предстарческое слабоумие (болезнь Альцгеймера с ранним началом), старческое слабоумие (слабоумие по типу болезни Альцгеймера), микроинфарктное слабоумие, связанное со СПИДом слабоумие, мультиинфарктное слабоумие, болезнь Крейцфельда-Якоба, болезнь Пика, паркинсонизм, включая болезнь Паркинсона, слабоумие с тельцами Леви,прогрессирующий супрануклеарный паралич, хорею Гентингтона, позднюю дискинезию, гиперкинезию,эпилепсию, манию, синдром дефицита внимания, состояние тревоги, дислексию, шизофрению, депрессию, обсессивно-компульсивные расстройства и синдром Туретта. Действие многих нейрофармакологических терапевтических агентов включает модуляцию высво-1 015700 бождения дофамина, норэпинефрина и серотонина, поглощение и сохранение в пределах их соответствующих границ в ЦНС. Большая часть нейромедиаторов сохраняется в синаптических везикулах, которые являются выступающими фрагментами нервных окончаний. Концентрация в везикулах, повидимому, ответственна за поддержание быстрого обеспечения нейромедиаторами, доступными для нейронного экзоцитотического выброса в синаптическую щель. Везикулы также выполняют задачу защиты нейромедиатора от метаболического распада. Один транспортный участок везикулярной мембраны представляет собой везикулярный моноаминный переносчик-2 (VMAT2), задача которого заключается в транспорте переносчика из цитозоля в синаптическую везикулу. Дигидротетрабеназин, структурно родственный метокситетрабеназину, использовали в качестве радиоактивной метки для исследования взаимодействия лекарственных средств с VMAT2. Оба соединения действовали на том же самом участке наVMAT2. После того как нейромедиатор высвобождается из нервного окончания в синаптическое пространство, он взаимодействует с постсинаптическими рецепторами и далее возвращается в нервное окончание с помощью переносчика на цитоплазматической мембране (например, дофаминового переносчика и/или серотонинового переносчика). Таким образом, транспортные белки изменяют концентрацию нейромедиатора в цитозольном и везикулярном пулах, благодаря чему имеют способность изменять последующую нейротрансмиссию. Дофамин представляет собой моноаминный нейромедиатор, который играет крайне необходимую роль в функционировании гипоталамической-гипофизарно-адреналовой системы и в интеграции информации в сенсорной, лимбической и двигательной системах. Основной механизм прекращения нейротрансмиссии дофамина заключается в обратном захвате высвобожденного дофамина с помощью Na+/Cl- зависимых переносчиков на цитоплазматической мембране. В зависимости от ионного состояния окружающей среды дофаминовый переносчик может функционировать в качестве медиатора дофаминового транспорта как во внутреннее пространство (т.е. "обратного захвата"), так и направленного дофаминового транспорта во внешнее пространство (т.е. "высвобождения"). Функциональная значимость дофаминового переносчика заключается в его регулировании дофаминовой нейротрансмиссии путем прекращения действия дофамина в синапсе посредством обратного захвата. Дофаминергические предпочтительные пути метаболизма вовлечены в заболевания, происходящие в результате пагубных привычек. Модификации гена дофаминового D2 рецептора связаны с алкоголизмом, ожирением, патологическим пристрастием к риску, расстройством гиперактивности с дефицитом внимания, синдромом Туретта, зависимостью от кокаина, зависимостью от никотина, злоупотреблением многими веществами и другой лекарственной зависимостью. В связи с тем что пониженные дофаминергические функции были обнаружены у индивидуумов с минорной А 1 аллелью дофаминового D2 рецептора, предположили, что дофаминовый D2 рецептор может быть генетически усилен или подкреплен. Кроме того, несколько исследований позволили предположить, что ассоциированные полиморфизмы гена дофаминового D2 рецептора связаны с импульсивным-вызывающим привыкание-компульсивным поведением, т.е. "синдромом отсутствия компенсации" (Blum, 1995). Дофаминовый переносчик представляет собой пресинаптически локализованную макромолекулу,которая играет важную роль в патофизиологических процессах ЦНС. Дофаминовый переносчик прекращает дофаминергическую нейротрансмиссию путем реаккумуляции высвобожденного дофамина в пресинаптические нейроны. В случае кокаиновой зависимости связывание кокаина с дофаминовым переносчиком и являющаяся результатом этого блокировка поглощения дофамина, по-видимому, имеет отношение к усиливающим свойствам лекарственного средства. Кроме того, связанной с функцией переносчика является концентрация нейротоксических химических веществ в дофаминергических нейронах,которые вовлечены в болезнь Паркинсона. Макромолекула-переносчик может быть маркером болезни Паркинсона, что подтверждается ее отсутствием в срезах ткани оболочек мозга у болеющих болезнью Паркинсона. Дофаминовый переносчик,кроме того, играет значимую роль в нейротоксическом действии 1-метил-4-фенил-1,2,3,6-тетрагидропиридина (МРТР), который вызывает идиопатический синдром Паркинсона у людей. Вследствие этого,сильнодействующие и еще селективные лиганды для дофаминового переносчика имеют потенциальную возможность применения для in vivo мониторинга основных мишеней кокаина в мозге, для характеристики участков связывания кокаина, для фармакотерапевтических агентов для лечения пополнения кокаина и для мониторинга болезни Паркинсона. Многие лекарственные средства могут вызвать физическое и/или психологическое привыкание. Такие наиболее известные лекарственные средства включают опиаты, такие как героин, опиум и морфин; симпатомиметики, включающие кокаин и амфетамины; седативно-снотворные средства, включающие спирт, бензодиазепины и барбитураты; и никотин, который имеет воздействия, подобные опиоидам и симпатомиметикам. Привыкание к лекарственному средству характеризуется пристрастием или иррациональной навязчивой тягой к приему лекарственного средства и неспособностью ограничить его употребление. Кроме того, зависимость от лекарственного средства тесно связана с толерантностью к лекарственному средству, с утратой эффекта лекарственного средства, наступающей при повторном введении,и с синдромом отмены, с появлением соматических и поведенческих симптомов в случае, когда лекарственное средство не употребляется. Повышение чувствительности происходит, если повторное введение лекарственного средства приводит к возросшей ответной реакции на каждую дозу. Толерантность, по-2 015700 вышенная чувствительность и синдром отмены представляют собой феномены, подтверждающие изменение в центральной нервной системе, наступившие в результате продолжительного применения лекарственного средства. Это изменение обуславливает привыкание пациента к продолжению применения лекарственного средства, несмотря на серьезные социальные, правовые, физические и/или профессиональные последствия. Зависимость от кокаина остается одной из важнейших проблем охраны здоровья в Соединенных Штатах. Фундаментальные исследования многих лабораторий показали, что кокаин блокирует поглощение дофамина из синаптической щели дофаминового переносчика. Номифензин и бупропион представляют собой два соединения, применяемые в качестве фармакологических стандартов для ингибиторов дофаминового переносчика. Оба соединения клинически используются в качестве антидепрессантов, и бупропион также представляет собой одно из немногочисленных соединений, используемых в терапии зависимости от никотина. Класс соединений "GBR" известен его чрезвычайно высокой селективностью и активностью в отношении дофаминового переносчика. Два из этих соединений обладают аффинностью в низком наномолярном диапазоне (DeVries, 1997). Введение радиоактивной метки в данные соединения облегчало выявление нейрофармакологической активности. GBR 12909 (ваноксерин) диссоциирует очень медленно из DAT и ослабляет возрастание уровней содержания внеклеточного дофамина, вызванное кокаином, что определено посредством микродиализа. Это соединение проявляло себя как нестимулирующее для испытуемых людейдобровольцев, и было показано, что оно блокировало действие кокаина при бесконтрольном его приеме у макак-резус (Dutta, 1993). Такие исследования подтвердили возможность того, что подходящие соединения могут служить в качестве антагонистов кокаина без привыкания к ним. Одно из воздействий никотина заключается в высвобождении дофамина. Ингибиторы дофаминового переносчика, по существу, оказывают такое же воздействие, хотя и путем совершенно другого механизма действия. Таким образом, в условиях, в которых повышение эндогенных уровней содержания дофамина необходимо или желательно, вероятность того, что соединение, которое имеет двойной механизм действия, скорее всего, будет эффективным, выше, чем вероятность этого для соединений, имеющих только один способ воздействия. Соединения с таким двойным механизмом действия известны. Первым таким идентифицированным соединением был, как это часто упоминается в истории современной медицины, продукт природного происхождения.-Лобелин (лобелин), липофильный, не содержащий пиридин, алкалоидный компонент Indian tobacco, представляет собой преобладающий алкалоид в семействе структурно-родственных соединений,обнаруженных в Lobelia inflata (лобелия одутлая). Лобелин, как было опубликовано, оказывает много никотиноподобных воздействий, включая тахикардию и гипертензию, гиперальгезию и улучшение обучаемости и памяти. Лобелин обладает высокой аффинностью в отношении никотиновых рецепторов, но не выявлено заметного структурного сходства лобелина с никотином, и структурно-функциональные взаимосвязи между S(-)-никотином и лобелином не дают возможность предполагать обычный фармакофор. Кроме того, характерные воздействия лобелина и никотина говорят о том, что данные лекарственные средства могут не проявлять активности путем обычного ЦНС механизма несмотря на то, что лобелин рассматривается как смешанный никотиновый агонист/антагонист. Лобелин вызывает высвобождение дофамина из стриарных срезов у крыс. При этом вызванное лобелином высвобождение дофамина также не зависит от внеклеточного содержания кальция и не чувствительно к мекамиламину, неконкурентному антагонисту никотинового рецептора. Таким образом, вызванное лобелином высвобождение дофамина происходит по другому механизму, нежели высвобождение дофамина, которое вызывает никотин. В связи с этим лобелин также ингибирует поглощение дофамина в стриарных синаптических везикулах крысы путем взаимодействия с дигидротетрабеназиновым участком VMAT2, таким образом повышая доступность цитозольного дофамина для обратного транспорта цитоплазматическим переносчиком (DAT) (Teng, 1997, 1998). Таким образом, лобелин взаимодействует с никотиновыми рецепторами и блокирует вызванное никотином высвобождение дофамина, но также взаимодействует с дофаминпереносящими белками (DAT и VMAT2), изменяя концентрацию до-3 015700 фамина в цитозольном и везикулярном накопительных пулах, посредством этого изменяя последующую дофаминергическую нейротрансмиссию. В US 20030100547 и US 20040266824 раскрыты серии 2,6-дизамещенных пиперидиновых и пиперазиновых производных, структурных аналогов лобелина Данные соединения были синтезированы и протестированы на активность в биологических испытаниях на никотиновом рецепторе и дофаминовом переносчике, и в анализе высвобождения для оценки взаимодействия таких соединений с такими специфическими белками на пресинаптическом окончании моноаминергических нейронов ЦНС. Некоторые из таких соединений обладают большой селективностью при взаимодействии с DAT, чем при взаимодействии с никотиновыми рецепторами, в то время как другие соединения взаимодействуют как с никотиновыми рецепторами, так и с DAT, более похожи на лобелин. Другие соединения были более селективны в отношении никотинового рецептора, чем в отношении DAT. Такие сочетания фармакологической активности рассматриваются как полезные для лечения злоупотребления психостимуляторами и синдрома отмены, расстройств пищевого поведения и заболеваний и патологий центральной нервной системы. Азаиндолэтиламиновые производные в качестве агентов, связывающихся с никотиновым ацетилхолиновым рецептором, полезны при лечении состояний, связанных с истощением никотиновых рецепторов у млекопитающих, а именно с пристрастием к никотину, были раскрыты в ЕР 0870768 А 1 и ЕР 1178045 А 1. Некоторые из раскрытых соединений структурно родственны соединениям настоящего изобретения. Они представляют собой сильнодействующие заменители [3 Н]-никотина со значениямиIC50 менее чем 2 мкМ, таким образом со значениями pIC50 5,7 или выше. Аффинности индивидуальных специфических соединений не были раскрыты. Области применения, приведенные выше, умалчивают об ингибировании обратного захвата дофамина. Следовательно, правомерно, поскольку синтез и тестирование ряда соединений заявлены, еще раз скрыть, что они лишены активности в качестве ингибиторов в процессе ингибирования обратного захвата дофамина. Целью изобретения было предоставить дополнительные соединения с двойным механизмом действия: (частичный) агонизм никотиновых ацетилхолиновых рецепторов и ингибирование обратного захвата дофамина. Подробное описание изобретения Неожиданно было обнаружено, что соединения общей формулы (I) где X, Y и Z независимо представляют собой N или С, с учетом того, что кольцо содержит по меньшей мере один N-атом и не более чем 2;m и n независимо равны или 0, или 1, при условии, что когда Y и Z представляют собой углерод и X представляет собой азот, m равен 0;R2 и R3 независимо представляют собой водород, галоген, (C1-3)алкил, (C1-3)алкинил, NH(C1-3)алкил,CF3, гидроксил, (C1-3)алкилокси или пиперидинильную, пирролидинильную, тетрагидропиридинильную,морфолинильную, азепанильную, 1-азабицикло[2.2.2]октанильную или 1-азабицикло[2.2.2]окт-2-енильную группу, причем любая группа является или незамещенной, или замещена одним или несколькими заместителями, выбранными из галогена, (C1-3)алкила, фенила или бензила;R4, R5 и R6 независимо представляют собой водород, галоген, (C1-3)алкил, (С 2-3)алкинил, CF3,NH(C1-3)алкил, гидроксил или (С 1-3)алкилокси с учетом того, что R4 присутствует только тогда, когда Y представляет собой С, и R5 присутствует только тогда, когда Z представляет собой С, и их таутомеры,стереоизомеры и N-оксиды, а также фармакологически приемлемые соли, гидраты и сольваты указанных соединений формулы (I), и их таутомеры, стереоизомеры и N-оксиды представляют собой новые соединения и обладают сочетанием частичного агонизма к никотиновому ацетилхолиновому рецептору и ингибирования обратного захвата дофамина. Данное изобретение, в частности, относится к соединениям общей формулы (I), где R2 и R3 независимо представляют собой водород или пиперидинильную, пирролидинильную, тетрагидропиридинильную, морфолинильную, азепанильную, 1-азабицикло[2.2.2]октанильную или 1-азабицикло[2.2.2]окт-2 енильную группу, причем любая группа является или незамещенной, или замещена одним или несколькими заместителями, выбранными из галогена, (C1-3)алкила, фенила или бензила, R4, R5 и R6 независимо представляют собой водород, галоген, (C1-3)алкил или алкил (C1-3)окси с учетом того, что R4 присутству-4 015700 ет только тогда, когда Y представляет собой С, и что R5 присутствует только тогда, когда Z представляет собой С, и X, Y, Z, m и n имеют значения, как указано выше. Соединения изобретения общей формулы (I), а также их фармакологически приемлемые соли обладают (частичной) агонистической активностью в отношении никотиновых ацетилхолиновых рецепторов и ингибируют обратный захват дофамина. Они полезны при лечении заболеваний, затрагивающих указанные выше рецепторы, или для терапии посредством воздействия на такие рецепторы. Например, нейроэндокринных, неврологических и невропсихиатрических заболеваний, шизофрении, затруднения с памятью и при обучении, расстройства гиперактивности с дефицитом внимания, тревожных расстройств,депрессивных заболеваний, нейродегенеративных заболеваний, болезни Альцгеймера, аддикции (болезненное пристрастие), никотиновой зависимости, кокаиновой зависимости, амфетаминовой зависимости,боли при расстройстве пищевого поведения, воспалительных процессов, судорожных синдромов, офтальмологических заболеваний, глаукомы, дегенерации желтого пятна, диабетической ретинопатии, сердечно-сосудистых и желудочно-кишечных заболеваний и рака. Изобретение также охватывает фармацевтические композиции для лечения, например, заболевания или состояния, поддающегося лечению посредством активации и/или блокирования указанных выше рецепторов, причем композиция содержит соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель; способы лечения заболевания или состояния, поддающегося лечению посредством активации и/или блокирования указанных выше рецепторов, причем способ включает введение млекопитающему, нуждающемуся в таком лечении, соединения формулы (I) или его фармацевтически приемлемой соли; фармацевтические композиции для лечения, например, заболевания или состояния, выбранного из группы, состоящей из нейроэндокринных, неврологических и невропсихиатрических заболеваний, шизофрении, затруднений с памятью и при обучении, расстройства гиперактивности с дефицитом внимания, тревожных расстройств, депрессивных заболеваний, нейродегенеративных заболеваний, болезни Альцгеймера, аддикции, никотиновой зависимости, кокаиновой зависимости, амфетаминовой зависимости, боли при расстройстве пищевого поведения, воспалительных процессов, судорожных синдромов,офтальмологических заболеваний, глаукомы, дегенерации желтого пятна, диабетической ретинопатии,сердечно-сосудистых и желудочно-кишечных заболеваний и рака; способы лечения заболевания или состояния, выбранного из группы, состоящей из перечисленных выше заболеваний, причем способы включают введение млекопитающему, нуждающемуся в таком лечении, соединения формулы (I) или его фармацевтически приемлемой соли; фармацевтические композиции для лечения заболевания или состояния, выбранного из группы, состоящей из перечисленных выше заболеваний, причем композиции содержат соединение формулы (I) или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель; способы лечения заболевания или состояния, выбранного из группы, состоящей из перечисленных выше заболеваний, причем способы включают введение пациенту, нуждающемуся в таком лечении, соединения формулы (I) или его фармацевтически приемлемой соли; способы активации никотинового рецептора и/или ингибирования накопления дофамина, которые включают введение субъекту, нуждающемуся в этом, эффективного количества соединения формулы (I). Изобретение также представляет применение соединения или соли, соответствующего формуле (I),для производства лекарственного средства. Изобретение, кроме того, относится к комбинированной терапии, при которой соединение изобретения или его фармацевтически приемлемую соль, или фармацевтическую композицию, или препарат, содержащие соединение изобретения, вводят одновременно или последовательно, или в качестве комбинированного препарата с другим терапевтическим агентом или агентами, для лечения одного или нескольких перечисленных состояний. Такого рода другой(ие) терапевтический(ие) агент(ы) может(могут) вводиться перед, одновременно с или после введения соединений изобретения. Изобретение также представляет соединения, фармацевтические композиции, наборы и способы лечения заболеваний или состояний, выбранных из группы, состоящей из перечисленных выше заболеваний, причем способ включает введение пациенту, нуждающемуся в таком лечении, соединения формулы (I) или его фармацевтически приемлемой соли. Соединения изобретения обладают (частичной) агонистической активностью в отношении никотиновых ацетилхолиновых рецепторов и ингибируют обратный захват дофамина. Такие активности полностью демонстрируют, например, используя биологические испытания, описанные в данном описании или известные в данной области. Изобретение также представляет способы получения соединений изобретения и промежуточные соединения, используемые в указанных способах. Соединения настоящего изобретения могут содержать один или несколько асимметрических центров и могут, таким образом, существовать в виде рацематов и рацемических смесей, отдельных энантиомеров, диастериомерных смесей и индивидуальных диастереомеров. В зависимости от природы различных заместителей молекула может иметь дополнительные асимметрические центры. Каждый такой асимметрический центр будет независимо от других давать два оптических изомера. Все возможные оп-5 015700 тические изомеры и диастереомеры, в виде смесей и в качестве чистых или частично очищенных соединений относятся к настоящему изобретению. Настоящее изобретение охватывает все такие изомерные формы указанных соединений. Формула (I) показывает структуру класса соединений без предоставления стереохимии. Отдельные синтезы таких диастереомеров или их хроматографическое разделение могут быть осуществлены в соответствии с известным уровнем техники путем соответствующей модификации раскрываемой в данном описании методики. Их действительная стереохимия может быть определена с помощью рентгеноструктурного анализа кристаллических продуктов или кристаллических промежуточных продуктов, из которых получают производные, если это необходимо, путем взаимодействия с реагентом, содержащим асимметрический центр действительно известной конфигурации. Рацемические смеси соединений могут быть разделены на индивидуальные энантиомеры способами, хорошо известными в данной области, такими как сочетание в рацемической смеси соединений энантиомерно чистого соединения с образованием диастереомерной смеси, с последующей очисткой индивидуальных диастереомеров стандартными методами, такими как фракционированная кристаллизация или хроматография. Сочетание часто заключается в образовании солей с использованием энантиомерно чистых кислоты или основания, например (-)-ди-п-толуоил-D-виноградной кислоты и/или (+)-ди-п-толуоил-L-виноградной кислоты. Диастереомерные производные затем могут быть преобразованы в чистые энантиомеры путем отщепления добавленного хирального остатка. Рацемическая смесь соединений также может быть непосредственно разделена хроматографическими методами, использующими хиральные неподвижные фазы,которые хорошо известны в данной области. Альтернативно, любой энантиомер соединения может быть получен путем стереоселективного синтеза с использованием оптически чистых исходных веществ или реагентов известной конфигурации способами, хорошо известными в данной области. Таутомеры соединения формулы (I) или его фармацевтически приемлемой соли также относятся к изобретению. Некоторые кристаллические формы соединений могут существовать в виде полиморфных форм,при этом подразумевается, что такие формы относятся к изобретению. Кроме того, некоторые соединения могут образовывать сольваты с водой (т.е. гидраты) или с обычными органическими растворителями. Такие сольваты также входят в объем данного изобретения. Изотопно-меченное соединение формулы (I) или его фармацевтически приемлемая соль, включая соединения формулы (I), изотопно-меченные для их определения путем PET или SPECT, также входят в объем данного изобретения. То же касается соединений формулы (I), меченных с помощью [13 С]-, [14 С]-,[3 Н]-, [18F]-, [125I]- или других изотопно-обогащенных атомов, пригодных для исследований рецепторного связывания или метаболизма. Определения химических и других терминов Термин "алкил" относится к прямым или разветвленным насыщенным углеводородным радикалам."Алкил(C1-3)", например, означает метил, этил, н-пропил или изопропил и "алкил(С 1-4)" означает "метил,этил, н-пропил, изопропил, н-бутил, 2-бутил, изобутил или 2-метил-н-пропил". Термин "алкенил" указывает на прямые или разветвленные углеводородные радикалы, имеющие одну или несколько углеродуглеродных двойных связей, такие как винил, аллил, бутенил и т.д., и предпочтительно относится к(С 2-4)алкенилу. В "алкинильных" группах прямые или разветвленные углеводородные радикалы имеют одну или несколько углерод-углеродных тройных связей, таких как этинил, пропаргил, 1-бутинил,2-бутинил и т.д., и предпочтительно они представляют собой (С 2-4)алкинил. Термин "ацил" означает алкил (C1-3)карбонил, арилкарбонил или арилалкил (C1-3)карбонил."Гало" или "галоген" означают хлор, фтор, бром или иод; "гетеро", как в "гетероалкиле, гетероароматическом" и т.д., означает содержание одного или нескольких N, О или S атомов, "гетероалкил" включает алкильные группы с гетероатомами в каком-либо положении, таким образом, включающиеN-связанные, О-связанные или S-связанные алкильные группы. Термины "окси", "тио" и "карбо", как они использованы в качестве составной части другой группы, соответственно относятся к кислородному атому, к атому серы и к карбонильной (С=O) группе, служащей в качестве линкера между двумя группами, такой как, например, гидроксил, оксиалкил, тиоалкил, карбоксиалкил и т.д. Термин "амино", как он использован сам по себе или в качестве составной части другой группы, относится к атому азота, который может быть или концевым, или может быть линкером между двумя другими группами, где группа может быть первичным, вторичным или третичным (два водородных атома связаны с атомом азота, один водородный атом связан с атомом азота и ни одного водородного атома не связано с атомом азота соответственно) амином. Термины "сульфинил" и "сульфонил", как они использованы в данном описании в качестве составной части другой группы, соответственно относятся к -SO- группе или к -SO2- группе. Как он использован в данном описании, если не указано иное, термин "уходящая группа" должен означать заряженный или незаряженный атом или группу, которые уходят в процессе замещения или реакции вытеснения. Подходящие примеры включают, но не ограничиваются ими, Br, Cl, I, мезилат, тозилат и подобные.N-оксиды соединений, которые указаны выше, относятся к изобретению. Третичные амины могут быть или не быть источником метаболитов N-оксида. Степень, до которой N-окисление осуществляется,изменяется от следовых количеств до преобразования, близкого к количественному выходу. N-оксиды-6 015700 могут быть более активны, чем соответствующие им третичные амины, или менее активны. ХотяN-оксиды и могут быть легко восстановлены до соответствующих третичных аминов с помощью химических средств, в человеческом организме это происходит в различной степени. Некоторые N-оксиды претерпевают почти количественное восстановительное преобразование до соответствующих третичных аминов, в других случаях преобразование представляет собой всего лишь реакцию в следовых количествах или даже полностью отсутствует (Bickel, 1969). Со ссылкой на заместители термин "независимо" означает, что в случае, когда более одного таких заместителей возможно, они могут быть одинаковыми или различными по отношению друг к другу. Чтобы предоставить более четкое описание, некоторые из приведенных в данном описании количественных выражений не сопровождаются термином "приблизительно". Понятно, что если термин "приблизительно" применяется явным образом или нет, каждое приведенное в данном описании количество предназначено как относящееся к фактически существующей данной величине и оно также предназначено как относящееся к приближенному значению для такой данной величины, которое с достаточным основанием получено специалистом в данной области, включая приближенные значения, соответствующие экспериментальным и/или измерительным условиям для такой данной величины. На всем протяжении описания и формулы изобретения в данном описании изобретения слово "включать" и вариации слова,такие как "включая" и "включающий", не предназначены для того, чтобы исключать другие добавки,компоненты, числа или стадии. Любое соединение, метаболизирующееся in vivo с образованием биологически активного агента(т.е. соединения формулы (I, представляет собой пролекарство в рамках объема и по сущности настоящего изобретения. Пролекарства представляют собой терапевтические агенты, сами по себе неактивные,но преобразуемые в один или несколько активных метаболитов. Таким образом, в способах лечения настоящего изобретения термин "введение" должен охватывать лечение различных заболеваний, описанное с конкретно раскрываемым соединением или с соединением, которое конкретно не раскрыто, но которое превращается в конкретное соединение in vivo после введения пациенту. Пролекарства представляют собой биореверсируемые производные молекул лекарственного средства, используемые для преодоления некоторых ограничений полезности исходной молекулы лекарственного средства. Эти ограничения включают, но не ограничиваются ими, растворимость, проницаемость, стабильность, пресистемный метаболизм и ограничения направленности действия (Bundgaard, 1985; King, 1994; Stella, 2004; Ettmayer,2004; Jarvinen, 2005). Пролекарства, т.е. соединения, которые при введении людям посредством какоголибо известного способа введения метаболизируют в соединения, имеющие формулу (I), относятся к изобретению. А именно это относится к соединениям с первичными или вторичными амино или гидроксигруппами. Такие соединения могут быть подвергнуты реакции с органическими кислотами, давая соединения, имеющие формулу (I), где присутствует дополнительная группа, которая легко удаляется после введения, например, но не ограничиваясь ими, амидин, енамин, основание Манниха, гидроксиметиленовое производное, производное О-(ацилоксиметиленкарбамата), карбамат, сложный эфир, амид или енаминон. Термин "композиция", как он используется в данном описании, охватывает продукт, содержащий определенные требуемые ингредиенты в заранее установленных количествах или пропорциях, а также какой-либо продукт, который получается в результате, прямо или косвенно, путем объединения определенных требуемых ингредиентов в определенных требуемых количествах. В отношении фармацевтических композиций данный термин охватывает продукт, содержащий один или несколько активных ингредиентов и необязательный носитель, включающий инертные ингредиенты, а также какой-либо продукт,который получается в результате, прямо или косвенно, путем объединения, комплексообразования или агрегации каких-либо двух или нескольких ингредиентов, или путем диссоциации одного или нескольких ингредиентов, или путем других типов реакций или взаимодействий одного или нескольких ингредиентов. Как правило, фармацевтические композиции получают путем равномерного и непосредственного введения активного ингредиента в ассоциацию с жидким носителем или тонкоизмельченным твердым носителем либо с ними обоими и с последующим, если это необходимо, формованием продукта в желаемый препарат. Фармацевтическая композиция содержит достаточное количество активного целевого соединения для получения желаемого эффекта при прогрессировании или определенном состоянии заболеваний. В соответствии с этим фармацевтические композиции настоящего изобретения охватывают любую композицию, полученную посредством смешивания соединения настоящего изобретения и фармацевтически приемлемого носителя. Под "фармацевтически приемлемым" подразумевается носитель, разбавитель или эксципиент, который должен быть совместимым с другими ингредиентами препарата и сам по себе безвреден для реципиента. Доза. Аффинность соединений изобретения в отношении никотиновых рецепторов и участков накопления дофамина определяли, как описано ниже. Из аффинности связывания, измеренной для данного соединения формулы (I), можно установить самую низшую эффективную теоретическую дозу. При концентрации соединения, равной удвоенной измеренной Kj-величине, почти 100% возможных рецепторов будут заняты соединением. Путем пересчета этой концентрации в мг соединения на кг массы пациента получают самую низшую теоретическую эффективную дозу, предполагая идеальную биодоступность.-7 015700 Фармакокинетические, фармакодинамические и другие факторы могут вносить изменения фактически вводимой дозы в сторону повышения или понижения ее величины. Доза вводимого соединения будет зависеть от учитываемых показаний, возраста, массы тела и пола пациента и может быть установлена врачом. Дозировка предпочтительно должна быть в диапазоне от 0,01 до 10 мг/кг. Обычная дневная доза активных ингредиентов изменяется в широком диапазоне и будет зависеть от различных факторов, таких как учитываемые показания, способ введения, возраст, масса тела и пол пациента, и может быть установлена врачом. Как правило, пероральные и парентеральные дозировки должны быть в диапазоне от 0,1 до 1000 мг в день общего количества активных ингредиентов. Термин "терапевтически эффективное количество", как он использован в данном описании, относится к количеству терапевтического агента для лечения состояния, подвергаемого лечению, путем введения композиции изобретения. Это количество представляет собой количество, достаточное для проявления заметного терапевтического или благоприятного ответа в тканях организма животного или человека. Эффект может охватывать, например, лечение перечисленных выше состояний. Точное эффективное количество для субъекта будет зависеть от размера и здоровья субъекта, от природы и объема подвергаемого лечению состояния, рекомендаций лечащего врача (исследователя, ветеринара, лечащего врача или другого клинициста) и лекарственных средств или комбинации лекарственных средств, выбранных для введения. Таким образом, нет какой-либо пользы в том, чтобы точно указывать требуемое эффективное количество заблаговременно. Термин "фармацевтически приемлемая соль" относится к таким солям, которые представляют собой, в рамках обоснованных медицинских взглядов, подходящие для применения в контакте с тканями человека и низших животных без чрезмерной токсичности, болезненной чувствительности, аллергической реакции и им подобных, и которые соизмеримы с приемлемым соотношением польза/риск. Фармацевтически приемлемые соли хорошо известны в данной области. Они могут быть получены in situ при окончательном выделении и очистке соединений изобретения или отдельно путем взаимодействия с фармацевтически приемлемыми нетоксичными основаниями или кислотами, включая неорганические или органические основания и неорганические или органические кислоты. Термин "лечение", как он использован в данном описании, относится к любому лечению состояния или заболевания млекопитающих, предпочтительно человека, и включает: (1) ингибирование заболевания или состояния, т.е. прекращение его развития, (2) облегчение заболевания или состояния, т.е. осуществление регресса состояния, или (3) прекращение симптомов заболевания. Как он используется в данном описании, термин "лекарственная терапия" подразумевает включение профилактических, диагностических и терапевтических схем лечения, осуществляемых in vivo или exvivo для людей или других млекопитающих. Термин "субъект", как он использован в данном описании, относится к животному, предпочтительно к млекопитающему, более предпочтительно к человеку, который являлся объектом лечения, наблюдения или экспериментальной работы. СокращенияAPT - тест на присоединенные протоны 9-BBN - 9-борбицикло[3.3.1]нонан ВОС - трет-бутоксикарбонил н-BuLi - н-бутиллитий ЦНС - центральная нервная системаDOM - направленное ортометилирование ЕР - входной потенциалMel - метилиодид МеОН - метанол мг - миллиграмм(ы) мин - минута(ы) мл - миллилитр(ы) Т.пл. - точка плавления, интервал плавления МС - масс-спектер, масс-спектрометрияSPECT - однофотонная эмиссионная компьютерная томографияVMAT2 - везикулярный моноаминовый преносчик-2 Примеры Пример 1. Аналитические методы. Спектры ядерного магнитного резонанса (1 Н-ЯМР и 13 С-ЯМР, APT) регистрировали в указанном растворителе, используя прибор Bruker ARX 400 (1 Н: 400 МГц, 13 С: 100 МГц) при 300 K, за исключением иначе указанных случаев. 19F-HMP и 13 С-ЯМР-спектры получали на спектрофотометре Varian Inova 500,работающим при 11,74 Т (499,9 МГц для 1H; 125,7 МГц для 13 С; 50,7 МГц, 470,4 МГц для 19F), использующим 5 мм SW датчик. Спектры регистрировали в дейтерированных хлороформе или дихлорметане,полученных от Cambridge Isotope Laboratories Ltd. Химические сдвиги (5) приведены в м.д. относительно тетраметилсилана (1 Н, 13 С) или CCl3F (19F). Константы спин-спинового взаимодействия J приведены в Гц. Формы пиков в ЯМР-спектре указаны с помощью символов "кв" (квартет), "дкв" (двойной квартет), "т"(триплет), "дт" (двойной триплет), "д" (дублет), "дд" (двойной дублет), "с" (синглет), "ушир.с" (уширенный синглет) и "м" (мультиплет). Сигналы для NH и ОН определяли после смешивания образца с каплейD2O. Флэш-хроматография относится к очистке с использованием указанного элюента и силикагеля (либо Acros: 0,030-0,075 мм или Merck silica gel 60: 0,040-0,063 мм). Колоночную хроматографию осуществляли, используя силикагель 60 (0,063-0,200 мм, Merck). Точки плавления регистрировали на приборе для определения точки плавления Bchi B-545. Масс-спектры снимали на приборе Micromass QTOF-2 с программным обеспечением MassLynx для сбора и преобразования данных. Четкое измерение массы было сделано для квазимолекулярного иона[М+Н]+. Точные измерения массы осуществляли, используя масс-спектрометр JEOL JMS-SX/SX 102 АTandem Mass Spectrometer, применяющий бомбардировку быстрыми атомами. Разрешающую способность 10000 (10% минимум зоны четкого определения) применяли для FAB масс-спектрометрии высокого разрешения. Все реакции, включающие влагочувствительные соединения или условия, осуществляли в безводной атмосфере азота. Реакции контролировали с использованием тонкослойной хроматографии на покрытых слоем силикагеля пластинах (Merck precoated silica gel 60 F254) с указанным элюентом. Пятна детектировали в УФ-свете (254 нм) или с помощью I2. Коэффициенты экстинкции определяли на HP 8453 UV-Vis спектрофотометре. Аналитическую ВЭЖХ осуществляли на С 18 колонке (Inertsil ODS-3, размер частиц 3; 4,6, 50 мм),-9 015700 используя следующий элюирующий градиент: линейный градиент от 5 до 95% водный CH3CN, содержащий 0,04% НСО 2 Н .в течение 5 мин, затем 95% водный CH3CN, содержащий 0,04% НСО 2 Н в течение 2 мин при 2,0 мл мин-1. Продукты детектировали при =254 нм. Жидкостная хроматография-масс-спектрометрия (ЖХ-МС). Система для ЖХ-МС состоит из 2 Perkin elmer series 200 микронасосов. Микронасосы соединены друг с другом посредством 50 мкл Т-образного смесителя, связанного с Gilson 215 автоматическим пробоотборником. Способ представляет собой следующее: Автоматический пробоотборник имеет 2-мкл инъекционную петлю. Автоматический пробоотборник связан с Waters Atlantis С 18 304,6 мм колонкой, заполненной частицами размером 3 мкм. Колонка термостатирована в Perkin Elmer series 200 колоночном термостате при 40 С. Колонка присоединена кPerkin Elmer series 200 UV измерительному прибору с 2,7-мкл проточной ячейкой. Длина волны установлена 254 нм. УФ-измерительный прибор соединен с Sciex API 150EX масс-спектрометром. Массспектрометр имеет следующие параметры: диапазон измерения: 150-900 е.а.м.; полярная корреляция: положительная; способ изображения результатов измерения: линия графика; разрешение Q1: UNIT; шаговый размер: 0,10 е.а.м.; время на измерение: 0,500 с; NEB: 10; CUR: 10 IS: 5200; ТЕМ: 325; DF: 30; FP: 225 и ЕР: 10. Детектор светорассеяния присоединен к Sciex API 150. Детектор светорассеяния представляет собой Sedere Sedex 55, работающий при 50 С и 3 бар N2. Укомплектованная система контролируется посредством G3 powermac. Пример 2. Общие аспекты синтезов. Соединения общей формулы (I) получали из легкодоступных исходных веществ. Замещенные 1 Нпирроло[2,3-b]пиридины доступны из коммерческих источников или известны в химической литературе(Synthesis, 1992; Heterocycles, 1999; US 2002/0061892; Current Organic Chemistry, 2001). В примере общей методики (схема 1) 1 Н-пирроло[2,3-b]пиридин (7-азаиндол (1 подвергают взаимодействию с 1-бензилпиперидин-3-оном (2) в присутствии основания с образованием пиперидин-3-ола соединения 3 и небензильного аналога соединения 5 (BioorganicMedicinal Chemistry Letters, 2002). Удаление бензильной группы может быть осуществлено с применением хорошо известных методов. Специальные условия представляют собой формиат аммония и гидроксид палладия в метаноле с получением соединения 4, которое дегидратировали (5) и восстанавливали с образованием желаемого 3-пиперидин-3-ил-1 Н-пирроло[2,3-b]пиридина (6). Некоторые конкретные соединения структуры 7 могут быть получены с применением методики,проиллюстрированной на схеме 1, исходя из (4, 5 или 6) замещенного 1 Н-пирроло[2,3-b]пиридина. В конкретных примерах R представляет собой низший алкил, алкилокси и фтор. Схема 1 Схема 2 иллюстрирует альтернативный способ получения соединений структуры 7. Более конкретно синтез 6-хлор-3-пиперидин-3-ил-1 Н-пирроло[2,3-b]пиридина (13). Таким образом, соединение 6 подвергают взаимодействию с ди-трет-бутилдикарбонатом, с образованием соединения 9 за две последовательные стадии. В Greene (1999) представлена еще информация по присоединению и последующему удалению защитных групп в органическом синтезе.- 10015700 Селективное хлорирование 7-азаиндольного аналога 9 в положение 6 достигали путем реакции типаReissert-Henze (через N-оксид 10 А) в соответствии с известным способом (Synthesis, 1992). Таким образом, N1-метоксикарбонильное соединение 11, полученное по данной реакции, преобразуют в щелочных условиях в соединение 12. Удаление N-трет-ВОС группы давало соединение 13. Схема 2 И еще в другом примере общей методики (схема 3) 7-азаиндол (1) подвергают взаимодействию с 1-азабицикло[2.2.2]октан-3-оном (14), с получением дегидратированного продукта 15, который восстанавливают с получением 3-(1 Н-пирроло[2,3-b]пиридин-3 ил)-1-азабицикло[2.2.2]октана (16). Схема 3 Образование анионов в орто-положении ароматических систем, используемое в синтетических методиках, описанных в данном описании, охвачено общей синтетической стратегией, известной специалисту в данной области, в качестве направленного ортометаллирования (Directed Ortho Metalation (DOM. В рамках данной области несколько функциональных групп, известных в качестве групп направленного металлирования (DMG), были исследованы для этих целей. 1-Фенилсульфонильная группа в качествеDMG в положении 1 азаиндольного аналога дает возможность введения лития в положение 2 и в связи с этим его функционализации (Synthesis, 20052; Tetrahedron, 1997). 2-Литийпроизводное соединения 17,полученное в мультиграммовом количестве посредством -металлирования (1,1 экв. LDA, ТГФ, от 10 до 0 С), конденсировали с соединением 14, что приводило к соединению 18, у которого удаляли защитные группы, с получением смеси ожидаемых спирта 19 и енамина 20. Восстановление соединения 20 давало 3-(1 Н-пирроло[2,3-b]пиридин-2-ил)-1-азабицикло[2.2.2]октан (21). Ссылаясь на схему 3, исходное вещество 17 преобразуют в соединение 22 (схема 4). После щелочного гидролиза N1-фенилсульфонильной группы (23) предпочтительной последовательностью превращений было удаление бензильной группы (24) с последующей дегидратацией (25). Восстановление давало желаемое соединение 26 (2-пиперидин-3-ил-1 Н-пирроло[2,3-b]пиридин). Ссылаясь на схему 2, восстановление соединения 27 с помощью сильного восстанавливающего агента, например LiAlH4, представляет собой предпочтительный способ получения N-CH3 соединения (28). Кроме того, путем последовательных превращений, описанных на схеме 2, исходное вещество 27 преобразовывали в разделяемую смесь соединений 30 и 31 (схема 4). Неселективность этой специфической реакции типа Reissert-Henze (через N-оксид 29 А) понятна специалисту в данной области. Щелочной гидролиз, последующее удаление N-трет-ВОС защитной группы приводит к соответствующим 6-(или 4)хлор-2-пиперидин-3-ил-1 Н-пирроло[2,3-b]пиридинам (32 и 33). Схема 5 В другом аспекте 5-(R)-[3,3,0]-1-аза-2-тиа-3-оксабициклооктан-2,2-диоксид (соединение 35, схема 5) или 5-(S)-аналог (соединение 36, Tetrahedron Asymmetry, 1990) используют в качестве исходных веществ для соединений настоящего изобретения, проиллюстрированных формулой (I). 2-Литийпроизводное соединения 17 подвергали взаимодействию с (R)-сульфамидатом 35, с получением литийсульфоната 37, который впоследствии гидролизовали до образования соединения 38 А.- 12015700 Удаление N1-фенилсульфонильной группы может быть осуществлено с помощью хорошо известных способов, например необязательно взаимодействием соединения 38 А с гидроксидом калия в диэтиленгликоле в присутствии гидразина, что дает (R)-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин(39 А). (S)-производное 39 В получали исходя из (S)-сульфамидата 36. Восстановительное алкилирование соединения 39 А до соединения 41 А (схема 5) может быть осуществлено с использованием различных способов. Как альтернатива преобразованию соединения 26 в соединение 28 (схема 4), восстановительное метилирование соединения 38 А и последующее удаление N1-фенилсульфонильной группы в соединении 40 А дает желаемый (R)-2-(1-метилпирролидин-2 илметил)-1 Н-пирроло[2,3-b]пиридин (41 А). Схема 6 Схема 6 иллюстрирует получение соединений формулы (I), где R5 и R6 представляют собой галоген,(C1-3)алкил или алкилокси. Следующая методика (применение хиральных сульфамидатов 35 и 36) используется для преобразования соединения 42 и 43 (Synthesis, 1992), 44 (схема 7), 50 (Heterocycles, 1999) и 51 (Current Organic Chemistry, 2001) в соответствующие соединения 45 А/В, 46 А/В, 47 А/В, 52 А/В и 53 А/В. Примером функционализации 7-азаиндольной системы в положении 6 является проиллюстрированное преобразование соединения 48, катализируемое Cu(I)бромидом, (и последующее удаление третВОС защитной группы), дающее (R)-6-метокси-2-пирролидин-2-илметил-1-Н-пирроло[2,3-b]пиридин(49). Следует подчеркнуть, что синтез 6-фтор-7-азаиндола (44) еще не был опубликован. Таким образом,настоящее изобретение представляет способ получения соединения 44, хотя и основывается на синтетических стратегиях, известных специалисту в данной области и проиллюстрированных на схеме 7. Схема 7 Коммерчески доступный 2,6-дифторпиридин (54) преобразовывали в 2-амино-6-фторпиридин (55,Tetrahedron, 2002). Различные DMG группы проверяли (например, 2,2-диметилпропанамид, Heterocycles,1999; WO 2003/053970). Однако было обнаружено, что карбамат в качестве DMG (Chem. Pharm. Bull.,1987) представляется наиболее важным для селективного иодирования соединения 56 с образованием соединения 57, этилового эфира (6-фтор-3-иодпиридин-2-ил)карбаминовой кислоты. Последующие химические преобразования по Соногашира и следующее за этим замыкание кольца в присутствииCu(I)иодида (Synthesis, 20051) давало соединение 59. Отщепление Ni-карбамата приводило к получению 6-фтор-1 Н-пирроло[2,3-b]пиридина (44). Синтез соединений 47 А/В проиллюстрирован на схеме 6 в соответствии с методиками, показанными на схеме 5. Другая иллюстрация соединений настоящего изобретения формулы (I) показана на схеме 8. Мягкость и селективность 9-BBN продемонстрирована его способностью к селективному восстановлению лактамной группы в коммерчески доступном третичном лактаме 61 (Tetrahedron Letters, 1999) до циклического амина 62, который преобразовывали в амид Вайнреба 63 (Tetrahedron Letters, 1997). Ссылаясь на схему 5, 2-литийпроизводное 17 подвергали взаимодействию с амидом Вайнреба 63, с образованием соединения 64. В общем способе осуществления реакции с 2-литийпроизводным 17 и подходящим электрофилом (см. приведенные ниже схемы) оптимальными условиями являются температура от приблизительно -50 до -10 С, предпочтительно от -30 до -20 С в течение приблизительно 2 ч. Соединение 64 подвергают восстановлению по Huang-Minion с сопутствующим отщеплениемN1-фенилсульфонильной группы с получением соединения 65. Последующее восстановительное отщепление бензильной группы может быть осуществлено в условиях, которые описаны выше (схема 4), с получением 2-пирролидин-3-илметил-1 Н-пирроло[2,3-b]пиридина (66). Преобразование соединения 66 в соединение 67 с использованием методики, описанной на схеме 4,и последующее восстановление приводит к получению соединения 68. Схема 9 В еще другом примере общей методики (схема 9) 2-литийпроизводное 17 (или 70) подвергают взаимодействию с Се(III)хлоридом (J. Med. Chem., 2002) с последующим конденсированием с третбутиловым эфиром 3-оксопирролидин-1-карбоновой кислоты (71), с образованием соответствующих третичных спиртов (72, 73). Ссылаясь на схему 5, N1-отщепление фенилсульфонильной группы приводит к получению соединений 74, 75. Кислотная среда (предпочтительно 6 М HCl) катализирует удалениеN-трет-ВОС защитной группы, и последующая дегидратация приводит к получению соединений 76, 77,которые восстанавливали до желаемых 2-пирролидин-3-ил-1 Н-пирроло[2,3-b]пиридина (78) и 2-пирролидин-3-ил-1 Н-пирроло[3,2-b]пиридина (79). Разделение изомеров может быть достигнуто при использовании хиральной колонки. Для получения соединения 83 (схема 10, European Journal of Medicinal Chemistry, 2004). 2-Литийпроизводное 70 и/или 83 (полученное предпочтительно с t-BuLi/TMEDA в ТГФ) подвергали взаимодействию с соединением 35 и/или 36, получая соответствующие соединения 80 А/В и 84. Преобразование соединений 80 А/В и 84 в соединения 81 А/В и 85 может быть выполнено с применением методики, описанной на схеме 5. Бромирование положения 3 в различных изомерных азаиндолах может быть осуществлено различными способами (Heterocycles, 1999, 2000; Tetrahedron Letters, 1969; WO 2004/078 757). Неожиданно было обнаружено, что предпочтительные условия, представляющие собойNBS/ДМФА, позволяют проводить бромирование (от соединения 69 до соединения 86) почти с количественным выходом. Правильный выбор защитной группы, а именно триизопропилсилильной группы(TIPS), дает возможность заменить 3-бром- на 3-литийпроизводное 87, которое впоследствии конденсировали с (R)-сульфамидатом (соединение 35). Кислотное удаление TIPS с последующим гидролизом сульфонатной группы (ссылаясь на схему 5) приводит к получению желаемого (R)-3-пирролидин-2 илметил-1 Н-пирроло[3,2-b]пиридина (88). Другая иллюстрация соединений настоящего изобретения формулы (I) показана на схеме 11. Коммерчески доступный 4-хлор-7 Н-пирроло[2,3-d]пиримидин (89) путем восстановления преобразовывают в 7 Н-пирроло[2,3-d]пиримидин (90). 2-(Триметилсилил)этоксиметильная группа в качестве DMG в положении 1 индольных производных дает возможность введения лития в положение 2 (Helvetica Chimica Используя данную хорошо известную методику, 2-литий-производное 91 конденсировали с (например) соединением 35. Во избежание проблем, связанных с применением литийалкилов или литийдиалкиламидов, правильным выбором был литийтетраметилпиперидин (92). Таким образом, 2-литийпроизводное 91 подвергали взаимодействию с (R)-сульфамидатом (35) с образованием соединения 93, с последующим удалением SEM защитной группы (94). Гидролиз сульфонатной группы в стандартных условиях, как описано выше, приводит к получению желаемого (R)-6-пирролидин-2-илметил-7 Н-пирроло[2,3-d]пиримидина (95). Фармацевтически приемлемые соли могут быть получены с использованием стандартных методик,хорошо известных в данной области, например путем смешивания соединения настоящего изобретения с подходящей кислотой, например неорганической кислотой или органической кислотой. Пример 3. Синтезы конкретных соединений. Конкретные соединения, для которых синтез описан ниже, предназначены для дополнительной более подробной иллюстрации изобретения и поэтому не предполагают каким-либо образом ограничить объем изобретения. Другие примеры осуществления изобретения будут очевидны специалисту в данной области из рассмотрения раскрываемых описания и практического осуществления изобретения. Предполагается, таким образом, чтобы описание и примеры рассматривались в качестве иллюстраций. 1-Бензил-3-(1 Н-пирроло[2,3-b]пиридин-3-ил)пиперидин-3-ол (соединение 3). Дисперсию 60%-ную NaH в минеральном масле (9,5 г, 179 ммоль) медленно добавляли к 150 млEtOH (0C). Полученный раствор добавляли к 7-азаиндолу (5,3 г, 44,9 ммоль) и 11,25 г (44,9 ммоль) 1-бензилпиперидин-3-она (в виде HCl соли). Полученную в результате смесь перемешивали в течение 72 ч при комнатной температуре. Добавляли этилацетат к смеси и органической слой промывали три раза насыщенным раствором NaHCO3, сушили (Na2SO4), фильтровали и концентрировали. Полученный в- 15015700 результате остаток очищали флэш-хроматографией (диэтиловый эфир/этилацетат градиент (от 1:1 до чистого этилацетата, получая соединение 3 в виде масла (10,3 г, 74,7%). 1 Н-ЯМР (400 МГц, CDCl3):10,0 (ушир.с, 1 Н), 8,27 (дд, J=5 Гц, 2 Гц, 1 Н), 8,14 (дд, J=8 Гц, 2 Гц, 1 Н), 7,35-7,24 (м, 6 Н), 7,03 (дд,J=5 Гц, 8 Гц, 1 Н), 3,96-3,88 (ушир.с, 1 Н), 3,60 (дд, Jгем=13 Гц, 2 Н), 3,07-3,01 (м, 1 Н), 2,95-2,89 (м, 1 Н),2,39 (д, J=10 Гц, 1 Н), 2,16-1,96 (м, 2 Н), 1,92-1,78 (м, 2 Н), 1,72-1,65 (м, 1 Н) (ТСХ EtOAc Rf 0,09). 3-(1 Н-Пирроло[2,3-b]пиридин-3-ил)пиперидин-3-ол (соединение 4). Соединение 3 (2,4 г, 7,8 ммоль), 1,5 г формиата аммония (23,8 ммоль) и 20% Pd(OH)2/C (240 мг) смешивали в МеОН (50 мл) и нагревали до температуры кипения с обратным холодильником в течение 2 ч. Смесь охлаждали, фильтровали, концентрировали и повторно растворяли в МеОН. Фильтрование через 25 г SCX-2 (МеОН, затем 1 н. NH3/MeOH) приводило к получению указанного в заголовке соединения (1,5 г, 6,9 ммоль, 88%) в виде аморфного вещества. 1 Н-ЯМР (400 МГц, D6DMCO):11,3 (ушир.с,1 Н), 8,17-8,13 (м, 2 Н), 7,24 (с, 1 Н), 7,0-6,95 (м, 1 Н), 3,0-2,89 (м, 2 Н), 2,85 (д, J=13 Гц, 1 Н), 2,6-2,51 (м, 1 Н),2,06-1,8 (м, 3 Н), 1,5-1,42 (м, 1 Н) (ТСХ МеОН/триэтиламин (97/3 Rf 0,16. 3-(1,2,5,6-Тетрагидропиридин-3-ил)-1 Н-пирроло[2,3-b]пиридин (соединение 5). 10 мл ацетилхлорида медленно добавляли к 200 мл EtOH (-10 С). Через 15 мин полученный раствор добавляли к соединению 4 (5 г, 16,3 ммоль) и нагревали до температуры кипения с обратным холодильником в течение 1 ч. Смесь охлаждали, концентрировали и повторно растворяли в МеОН. Фильтрование через 25 г SCX-2 (МеОН, затем 1 н. NH3/MeOH) приводило к получению указанного в заголовке соединения (1,43 г, 7,18 ммоль, 44,1%) в виде аморфного вещества. 1 Н-ЯМР (400 МГц, CDCl3):11,4-10,8(м, 1 Н), 3,76-3,72 (м, 2 Н), 3,10-3,04 (м, 2 Н), 2,36-2,29 (м, 2 Н) (ТСХ МеОН/триэтиламин (97/3 Rf 0,25. 3-Пиперидин-3-ил-1 Н-пирроло[2,3-b]пиридин (соединение 6). 10 мл ацетилхлорида медленно добавляли к 200 мл МеОН (-10 С). Через 15 мин полученный раствор добавляли к соединению 5 (1,43 г, 7,18 ммоль) и 20% Pd(OH)2/C (130 мг). Смесь гидрировали при 50 psi в течение 1 ч. Смесь фильтровали и концентрировали. Последующее фильтрование через SCX-2 с последующей флэш-хроматографией (МеОН/триэтиламин (97/3) приводило к получению соединения 6(0,81 г, 4,02 ммоль, 55%), которое подвергали взаимодействию с 1 экв. фумаровой кислоты в EtOH и концентрировали. Перекристаллизация из смеси EtOH/этилацетат давала твердое вещество (свободное основание/фумаровая кислота (2:1. Т.пл 225 С (с разложением). 1 Н-ЯМР (400 МГц, D2O):8,04(ушир.д, J=5 Гц, 1 Н), 7,93 (ушир.д, J=8 Гц, 1 Н), 7,13 (ушир.с, 1 Н), 7,01 (дд, J=8 Гц, 5 Гц, 1 Н), 6,36 (с, 1 Н),3,46-3,40 (м, 1 Н), 3,37-3,31 (м, 1 Н), 3,14-3,04 (м, 1 Н), 2,92-2,82 (м, 2 Н), 2,0-1,87 (м, 2 Н), 1,8-1,54 (м, 2 Н). трет-Бутиловый эфир 3-(1 Н-пирроло[2,3-b]пиридин-3-ил)пиперидин-1-карбоновой кислоты (соединение 9). Соединение 6 (0,3 г, 1,5 ммоль), 1,0 г ди-трет-бутилдикарбоната (4,58 ммоль) и 0,5 мл триэтиламина смешивали в дихлорметане (20 мл) и нагревали до температуры кипения с обратным холодильником в течение 15 мин. Смесь охлаждали и концентрировали в вакууме. Полученный в результате остаток помещали в этилацетат, промывали насыщенным раствором соли, сушили (Na2SO4), фильтровали и концентрировали в вакууме, с получением соединения 8 в виде маслянистого остатка, который использовали на следующей стадии (дополнительной очистки не требовалось). 1H-ЯМР (400 МГц, CDCl3):8,5 (дд,J=5 Гц, 2 Гц, 1 Н), 7,98-7,91 (ушир.д, J=8 Гц, 1 Н), 7,39 (с, 1 Н), 7,18 (дд, J=8 Гц, 5 Гц, 1 Н), 4,48-4,04 (м,2 Н), 2,97-2,75 (м, 3 Н), 2,2-2,14 (м, 1 Н), 1,84-1,63 (м, 3 Н), 1,67 (с, 9 Н), 1,49 (с, 9 Н) (ТСХ диэтиловый эфирRf 0,39). Полученное вещество (8) растворяли в 10 мл МеОН и 3 мл 2 н. NaOH. Реакционную смесь перемешивали в течение 0,5 ч при комнатной температуре. Добавляли этилацетат к смеси и органический слой промывали три раза 2 н. NaOH, сушили (Na2SO4), фильтровали и концентрировали. Полученный в результате остаток очищали флэш-хроматографией (диэтиловый эфир) с получением соединения 9 в виде масла (0,25 г, 0,83 ммоль, 55% (суммарный. 1H-ЯМР (400 МГц, CDCl3):9,8 (ушир.с, 1 Н), 8,3 (ушир.д,J=5 Гц, 1 Н), 8,04-7,98 (ушир.д, J=8 Гц, 1 Н), 7,13 (ушир.с, 1 Н), 7,07 (дд, J=8 Гц, 5 Гц, 1 Н), 4,45-4,02 (м,2 Н), 3,04-2,76 (м, 3 Н), 2,2-2,14 (м, 1 Н), 1,83-1,60 (м, 3 Н), 1,49 (с, 9 Н) (ТСХ диэтиловый эфир Rf 0,13). трет-Бутиловый эфир 3-(7-окси-1 Н-пирроло[2,3-b]пиридин-3 ил)пиперидин-1-карбоновой кислоты(соединение 10 А). Соединение 9 (0,23 г, 0,76 ммоль) и 0,23 г метахлорпербензойной кислоты (1,4 экв., 1,07 ммоль) смешивали в диметоксиэтане (10 мл) и перемешивали в течение 10 мин при комнатной температуре. Смесь концентрировали на SiO2 и очищали флэш-хроматографией (этилацетат, затем этилацетат/МеОН/триэтиламин (90/10/1) с получением соединения 10 А (аморфное вещество, 0,23 г, 0,72 ммоль,95%). 1 Н-ЯМР (400 МГц, CDCl3):8,2 (ушир.д, J=5 Гц, 1 Н), 7,8-7,5 (ушир.с, 1 Н), 7,22 (с, 1 Н), 7,04 (дд,J=8 Гц, 5 Гц, 1 Н), 4,44-4,02 (м, 2 Н), 2,99-2,78 (м, 3 Н), 2,2-2,12 (м, 1 Н), 1,81-1,60 (м, 3 Н), 1,48 (с, 9 Н). 3-Пиперидин-3-ил-1 Н-пирроло[2,3-b]пиридин-7-оксид (соединение 10 В). 0,4 мл ацетилхлорида медленно добавляли к 10 мл EtOH (-10 С). Через 15 мин полученныйHCl/EtOH раствор добавляли к соединению 10 А (0,27 г, 0,85 ммоль) и нагревали до температуры кипения с обратным холодильником в течение 1 ч. Смесь охлаждали и частично концентрировали, добавляли- 16015700 этилацетат и полученный в результате осадок отделяли фильтрованием и промывали диизопропиловым эфиром. Указанное в заголовке соединение (10 В, в виде HCl соли) получали в виде твердого вещества(м, 1 Н), 3,04-2,90 (м, 2 Н), 2,14-2,06 (м, 1 Н), 2,05-1,94 (м, 1 Н), 1,86-1,66 (м, 2 Н). 6-Хлор-3-пиперидин-3-ил-1 Н-пирроло[2,3-b]пиридин (соединение 13). Соединение 10 А (0,22 г, 0,69 ммоль) и 0,15 мл 1,1,1,3,3,3-гексаметилдисилазана (0,72 ммоль) смешивали в 10 мл ТГФ. К полученному раствору добавляли 0,14 мл (1,8 ммоль) метилхлорформиата и реакционную смесь перемешивали в течение 30 мин при комнатной температуре. Реакционную смесь концентрировали в вакууме. Полученный в результате остаток помещали в этилацетат, промывали 5% раствором NaHCO3, затем насыщенным раствором соли, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Очистка флэш-хроматографией (диэтиловый эфир/РА: 1/1) приводила к получению соединения 11 в виде масла (0,16 г, 58,6%). 1H-ЯМР (400 МГц, CDCl3)7,95-7,84 (ушир.д, J=8 Гц, 1 Н), 7,53 (с,1 Н), 7,24 (д, J=8, 1H), 4,42-4,20 (м, 1 Н), 4,10-4,01 (м, 1 Н), 4,09 (с, 3 Н), 3,14-2,84 (м, 2 Н), 2,84-2,68 (м, 1 Н),2,19-2,11 (м, 1 Н), 1,82-1,60 (м, 3 Н), 1,49 (с, 9 Н) (ТСХ диэтиловый эфир/РЕ (1/1) Rf 0,19). Соединение 11 (0,16 г) растворяли в 25 мл МеОН, содержащего 3 мл 2 н. NaOH, и перемешивали в течение 18 ч при комнатной температуре. Реакционную смесь концентрировали в вакууме. Полученный в результате остаток помещали в дихлорметан, промывали 5% раствором NaHCO3, затем насыщенным раствором соли, сушили (Na2SO4), фильтровали и концентрировали в вакууме с получением соединения 12 (0,13 г, 95%). 1 Н-ЯМР (400 МГц, CDCl3):7,99-7,91 (ушир.д, J=8 Гц, 1 Н), 7,12-7,07 (м, 2 Н), 4,42-4,20(м, 1 Н), 4,16-4,02 (м, 1 Н), 3,02-2,75 (м, 3 Н), 2,20-2,12 (м, 1 Н), 1,82-1,60 (м, 3 Н), 1,49 (с, 9 Н) (ТСХ диэтиловый эфир/РЕ (1/1) Rf 0,17). 0,2 мл ацетилхлорида медленно добавляли к 10 мл EtOH (-10 С). Через 15 мин полученный раствор добавляли к соединению 12 (0,13 г, 0,39 ммоль) и нагревали до температуры кипения с обратным холодильником в течение 1,5 ч. Смесь охлаждали и частично концентрировали, добавляли этилацетат, полученный в результате осадок отделяли фильтрованием и промывали диизопропиловым эфиром. Указанное в заголовке соединение 6-хлор-3-пиперидин-3-ил-1 Н-пирроло[2,3-b]пиридин 13 (в виде HCl соли) получали в виде твердого вещества (80 мг, 76%). Т.пл. 279 С (с разложением). 1 Н-ЯМР (400 МГц, D2O):7,60 (д, J=8 Гц, 1 Н), 7,14 (с, 1 Н), 6,99 (д, J=8 Гц, 1 Н), 3,52-3,44 (м, 1 Н), 3,40-3,32 (м, 1 Н), 3,19-3,09 (м, 1 Н),2,97-2,87 (м, 2 Н), 2,08-1,91 (м, 2 Н), 1,84-1,60 (м, 2 Н). 3-(1 Н-Пирроло[2,3-b]пиридин-3-ил)-1-азабицикло[2.2.2]октан (соединение 16). Дисперсию 60%-ную NaH в минеральном масле (4 г, 75 ммоль) медленно добавляли к 75 мл EtOH(0C). Полученный раствор добавляли к 7-азаиндолу (2 г, 16,9 ммоль) и 3,4 г (21 ммоль) 1-азабицикло[2.2.2]октан-3-ону (14) (в виде HCl соли). Полученную в результате смесь перемешивали в течение 24 ч при 60 С. Раствору давали возможность достичь комнатной температуры. К реакционной смеси добавляли 0,5 мл H2O и 25 г SiO2 и затем концентрировали в вакууме. Полученный в результате остаток очищали флэш-хроматографией (дихлорметан/МеОН/7 н. NH3/МеОН (от 960/35/5 до 910/100/10, получая соединение 15 в виде твердого вещества (2,5 г, 69%). Т.пл. 180 С. 1 Н-ЯМР (400 МГц, D6DMCO):11,8 (ушир.с, 1 Н), 8,24 (дд, J=5 Гц, 2 Гц, 1 Н), 8,12 (дд, J=8 Гц, 2 Гц, 1 Н), 7,61 (с, 1 Н), 7,08 (дд, J=5 Гц,8 Гц, 1 Н), 6,87 (д, J=2 Гц, 1 Н), 3,14-3,09 (м, 1 Н), 3,01-2,91 (м, 2 Н), 2,64-2,54 (м, 2 Н), 1,78-1,69 (м, 2 Н),1,59-1,49 (м, 2 Н). Соединение 15 (1,9 г, 7,11,95 ммоль) и 20% Pd(OH)2/C (190 мг) смешивали в МеОН (100 мл). Смесь гидрировали при 50 psi в течение 72 ч. Смесь фильтровали и концентрировали на 25 г SiO2. Полученный в результате остаток очищали флэш-хроматографией (дихлорметан/МеОН/7 н. NH3/MeOH (от 960/35/5 до 910/100/10, получая указанное в заголовке соединение 16 в виде твердого вещества (1,55 г, 57%). Т.пл. 185 С. 1H-ЯМР (400 МГц, D6DMCO):11,35 (ушир.с, 1 Н), 8,17 (дд, J=5 Гц, 2 Гц, 1 Н), 7,85 (дд, J=8 Гц,2 Гц, 1 Н), 7,31 (д, J=2 Гц, 1 Н), 6,98 (дд, J=5 Гц, 8 Гц, 1 Н), 3,32-3,24 (м, 1 Н), 3,22-2,95 (м, 1 Н), 3,0-2,83 (м,4 Н), 2,78-2,68 (м, 1 Н), 1,95-1,91 (м, 1 Н), 1,84-1,54 (м, 3 Н), 1,35-1,24 (м, 1 Н). 3-(1-Бензолсульфонил-1 Н-пирроло[2,3-b]пиридин-2-ил)-1-азабицикло[2.2.2]октан-3-ол (соединение 18).LDA (2,0 М в ТГФ/гептан) (5,5 мл, 11 ммоль) добавляли к 100 мл безводного ТГФ при -10 С в атмосфере N2. Раствор безводного ТГФ (10 мл), содержащий соединение 17 (Tetrahedron, 1997) (2,58 г,10 ммоль), добавляли по каплям. После добавления полученный в результате раствор перемешивали в течение 30 мин при -10 С-0 С. Затем температуру понижали до -70 С. При этой температуре раствор соединения 14 (1,25 г, 10 ммоль) в 10 мл ТГФ добавляли по каплям. Свободное основание из HCl соли коммерчески доступного 1-азабицикло[2.2.2]октан-3-она (14) получали после фильтрования через SCX-2(МеОН, затем 1 н. NH3/MeOH) и последующего упаривания. После добавления соединения 14 температуру повышали до -30 С-25 С и полученный в результате раствор перемешивали в течение 60 мин. Раствору давали возможность достичь комнатной температуры. К реакционной смеси добавляли 5 мл Н 2 О и 50 г SiO2 и затем концентрировали в вакууме. Полученный в результате остаток очищали флэшхроматографией (дихлорметан/МеОН/7 н. NH3/MeOH (960/35/5, получая соединение 18 в виде масла(960/35/5 Rf 0,33). 3-(1 Н-Пирроло[2,3-b]пиридин-2-ил)-1-азабицикло[2.2.2]октан (соединение 21). Соединение 18 (2,4 г, 6,26 ммоль) и 31 мл 2 н. NaOH смешивали в EtOH (310 мл) и нагревали до температуры кипения с обратным холодильником в течение 2 ч. Смесь охлаждали и концентрировали. К остатку добавляли 50 г SiO2 и 100 мл МеОН. Полученную смесь концентрировали в вакууме и затем очищали флэш-хроматографией (дихлорметан/МеОН/7 н. NH3/МеОН (960/35/5, получая соединение 20 в виде масла (0,54 г, 38,3%). 1 Н-ЯМР (200 МГц, CDCl3):11,3 (ушир.с, 1 Н), 8,33 (дд, J=5 Гц, 2 Гц, 1 Н),7,87 (дд, J=8 Гц, 2 Гц, 1 Н), 7,14-7,02 (м, 2 Н), 6,52 (ушир.с, 1 Н), 3,25-2,65 (м, 5 Н), 1,95-1,40 (м, 4 Н) (ТСХ дихлорметан/МеОН/7 н. NH3/MeOH (960/35/5) Rf 0,20). Второе соединение (19), полученное в процессе хроматографии, получали в виде твердого вещества(дд, J=8 Гц, 2 Гц, 1 Н), 7,02 (дд, J=8 Гц, 5 Гц, 1 Н), 6,43 (д, J=2 Гц, 1 Н), 5,32-5,28 (ушир.с, 1 Н), 3,5-3,43 (м,1 Н), 2,97-2,88 (м, 2 Н), 2,84-2,64 (м, 3 Н), 2,24-2,12 (м, 2 Н), 1,48-1,37 (м, 2 Н), 1,33-1,24 (м, 1 Н) (ТСХ дихлорметан/МеОН/7 н. NH3/MeOH (960/35/5) Rf 0,09). Соединение 20 (0,54 г, 2,4 ммоль), 0,76 г формиата аммония (12 ммоль) и 20% Pd(OH)2/C (54 мг) смешивали в МеОН (50 мл) и нагревали до температуры кипения с обратным холодильником в течение 2 ч. Смесь охлаждали, фильтровали, концентрировали и повторно растворяли в МеОН, затем осуществляли дальнейшее фильтрование через 5 г SCX-2 (МеОН, затем 1 н. NH3/MeOH). Очистка флэшхроматографией (дихлорметан/МеОН/7 н. NH3/MeOH (960/35/5 приводила к получению соединения 21 в виде твердого вещества (0,25 г, 46%). Т.пл. 190 С. 1 Н-ЯМР (400 МГц, CDCl3):11,65 (ушир.с, 1 Н), 8,21LDA (2,0 М в ТГФ/гептан) (28 мл, 56 ммоль) добавляли к 200 мл безводного ТГФ при -10 С в атмосфере N2. Раствор безводного ТГФ (20 мл), содержащий соединение 17 (14,54 г, 56 ммоль), добавляли по каплям. После добавления полученный в результате раствор перемешивали в течение 30 мин при-10 С-0 С. Затем температуру понижали до -70 С. При этой температуре раствор соединения 2 (11,5 г,60,8 ммоль) в 25 мл ТГФ добавляли по каплям (10 мин). Свободное основание из HCl соли коммерчески доступного 1-бензилпиперидин-3-она (2) получали после фильтрования через SCX-2 (МеОН, затем 1 н.NH3/MeOH) и последующего упаривания. После добавления соединения 2 температуру повышали до-35 С-30 С и полученный в результате раствор перемешивали в течение 2 ч. Смеси давали возможность нагреться до температуры окружающей среды и выливали в раствор NH4Cl (10 г/50 мл H2O). Добавляли этилацетат и органический слой промывали 5% раствором NaHCO3, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный в результате остаток очищали флэш-хроматографией (диэтиловый эфир/РА градиент от 1:2 до чистого диэтилового эфира), получая соединение 22 в виде масла(5,8 г, 23%). 1H-ЯМР (400 МГц, CDCl3):8,31 (дд, J=5 Гц, 2 Гц, 1 Н), 8,10-8,05 (м, 2 Н), 7,55 (дд, J=8 Гц,2 Гц, 1 Н), 7,51-7,45 (м, 1 Н), 7,41-7,24 (м, 7 Н), 7,09 (дд, J=5 Гц, 8 Гц, 1 Н), 6,80 (с, 1 Н), 4,97-4,86 (ушир.с,1 Н), 3,69 и 3,59 (дд, Jгем=13 Гц, 2 Н), 3,12-2,87 (м, 2 Н), 2,58-2,38 (м, 2 Н), 2,14-2,04 (м, 1 Н), 1,96-1,83 (м,1 Н), 1,68-1,44 (м, 2 Н) (ТСХ диэтиловый эфир Rf 0,2). Соединение 22 (1,49 г, 3,3 ммоль) и 3,1 мл 2 н. NaOH смешивали в МеОН (31 мл) и нагревали до температуры кипения с обратным холодильником в течение 2 ч. Смесь охлаждали и концентрировали. Добавляли этилацетат и органический слой промывали 5% раствором NaHCO3, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный в результате остаток очищали флэш-хроматографией(этилацетат), получая указанное в заголовке соединение (23) в виде твердого вещества. Перекристаллизация из смеси этилацетат/диизопропиловый эфир. Выход 0,68 г, 66,5%. Т.пл. 122-126 С. 1 Н-ЯМР(400 МГц, CDCl3):10,9 (ушир.с, 1 Н), 8,26 (дд, J=5 Гц, 2 Гц, 1 Н), 7,84 (дд, J=8 Гц, 2 Гц, 1 Н), 7,34-7,22 (м,5 Н) 7,02 (дд, J=5 Гц, 8 Гц, 1 Н), 6,28 (д, J=2 Гц, 1 Н), 3,98-3,86 (ушир.с, 1 Н), 3,59 (дд, Jгем=13 Гц, 2 Н),2,93-2,83 (м, 2 Н), 2,45-2,39 (м, 1 Н), 2,25-1,95 (м, 1 Н), 2,04-1,80 (м, 3 Н), 1,75-1,66 (м, 1 Н) (ТСХ диэтиловый эфир Rf 0,2). 3-(1 Н-Пирроло [2,3-b]пиридин-2-ил)пиперидин-3-ол (соединение 24). Соединение 23 (3,07 г, 10 ммоль), 3,5 г формиата аммония (58,3 ммоль) и 20% Pd(OH)2/C (340 мг) смешивали в МеОН (100 мл) и нагревали до температуры кипения с обратным холодильником в течение 1 ч. Смесь охлаждали, фильтровали, концентрировали и повторно растворяли в МеОН. Фильтрование через 40 г SCX-2 (МеОН, затем 1 н. NH3/MeOH) и последующая очистка флэш-хроматографией(97/3 Rf 0,26). 2-Пиперидин-3-ил-1 Н-пирроло[2,3-b]пиридин (соединение 26). Соединение 24 (1,73 г, 7,97 ммоль), растворенное в 75 мл 6 н. HCl, нагревали до температуры кипения с обратным холодильником в течение 18 ч. Смесь охлаждали и концентрировали. Кристаллизация из смеси этилацетат/EtOH приводила к получению соединения 25 (1,99 г, 92% (в виде HCl соли по двум положениям. Т.пл. 275 С (с разложением). 1 Н-ЯМР (400 МГц, D6DMCO):12,75 (ушир.с, 1 Н), 8,33(ушир.с, 1 Н), 4,10-4,04 (м, 2 Н), 3,34-3,27 (м, 2 Н), 2,65-2,58 (м, 2 Н) (ТСХ МеОН/триэтиламин (97/3) Rf 0,25). Соединение 25 (1,71 г, 6,3 ммоль) и 20% Pd(OH)2/C (210 мг) смешивали в 100 мл МеОН и гидрировали при 50 psi в течение 1 ч. Смесь фильтровали и концентрировали. Последующее фильтрование через 30 г SCX-2 (МеОН, затем 1 н. NH3/MeOH) приводило к получению указанного в заголовке соединения 26(1,06 г, 84%), которое подвергали взаимодействию с 1 экв. фумаровой кислоты в EtOH и концентрировали. Перекристаллизация из смеси EtOH/этилацетат давала твердое вещество (свободное основание/фумаровая кислота (1:1. Т.пл. 211 С (с разложением). 1 Н-ЯМР (400 МГц, D6DMCO):12,75(ушир.с, 1 Н), 8,33 (дд, J=5 Гц, 2 Гц, 1 Н), 8,24 (дд, J=8 Гц, 2 Гц, 1 Н), 7,30 (дд, J=8 Гц, J=5 Гц, 1 Н), 6,896,85 (м, 1 Н), 6,42 (ушир.с, 1 Н), 4,10-4,04 (м, 2 Н), 3,34-3,27 (м, 2 Н), 2,65-2,58 (м, 2 Н) (ТСХ МеОН/триэтиламин (97/3) Rf 0,14). Разделения энантиомерно чистых изомеров достигали, используя хиральную колонку (Chiralpak AD 20 мкм, 2504,6, MeOH/EtOH 1/1, 2 мл/мин, =220 нм, Rt: 5,6 мин (26 А), []D25 +4 (с 1, толуол) и Rt: 8,3 мин (26 В), []D25 -4 (с 1, толуол). трет-Бутиловый эфир 2-(1 Н-пирроло[2,3-b]пиридин-3-ил)пиперидин-1-карбоновой кислоты (соединение 27). Соединение 26 (8,3 г, 41,29 ммоль) преобразовывали в указанное в заголовке соединение (27) способом, аналогично описанным для соединения 9. Выход 10,9 г (36,21 ммоль, 87,7%). Т.пл. 144-145 С;(ТСХ диэтиловый эфир Rf 0,20). 2-(1-Метилпиперидин-3-ил)-1 Н-пирроло[2,3-b]пиридин (соединение 28). Соединение 27 (0,54 г, 1,8 ммоль) растворяли в 5 мл безводного ТГФ. Полученный в результате раствор медленно добавляли к перемешиваемому раствору LiAlH4 (0,2 г, 5,2 ммоль) в 25 мл безводного ТГФ (60 С в атмосфере N2). После перемешивания в течение 1,5 ч смесь охлаждали. К полученной в результате смеси добавляли последовательно 0,2 мл Н 2 О, 0,4 мл 2 н. NaOH и 0,2 млH2O и нагревали при 60 С в атмосфере N2. Смесь охлаждали, фильтровали и промывали МеОН, затем осуществляли дальнейшее фильтрование через 5 г SCX-2 (МеОН, затем 1 н. NH3/MeOH). Очистка флэшхроматографией (МеОН/триэтиламин (97/3 давала указанное в заголовке соединение 28 в виде твердого вещества (0,32 г, 83%), которое подвергали взаимодействию с 1 экв. фумаровой кислоты в EtOH и концентрировали. Перекристаллизация из смеси EtOH/этилацетат приводила к получению твердого вещества (0,47 г, свободное основание/фумаровая кислота (1:1. Т.пл.218 С (с разложением). 1H-ЯМР(дд, J=8 Гц, 5 Гц, 1 Н), 6,56 (с, 2 Н), 6,20 (ушир.с, 1 Н), 3,36-3,30 (м, 1 Н), 3,18-3,04 (м, 2 Н), 2,56-2,50 (м,1 Н), 2,47 (с, 3 Н), 2,41-2,32 (м, 1 Н), 2,11-2,03 (м, 1 Н), 1,86-1,67 (м, 2 Н), 1,58-1,45 (м, 1 Н). 2-Пиперидин-3-ил-1 Н-пирроло[2,3-b]пиридин 7-оксид (соединение 29 В). Соединение 27 (5,95 г, 19,7 ммоль) и 5 г метахлорпербензойной кислоты (23,2 ммоль) смешивали в диметоксиэтане (60 мл) и перемешивали в течение 10 мин при комнатной температуре. Реакционную смесь концентрировали в вакууме. Полученный в результате остаток помещали в этилацетат, промывали 2 н. NaOH, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Смесь концентрировали на SiO2 и очищали флэш-хроматографией (этилацетат/МеОН/триэтиламин (90/10/1), с получением соединения 29 А (аморфное вещество, 5,48 г, 87,5%). ЖХ/МС; Rt: 1,50 мин, ([М+Н]+=318), (ТСХ МеОН/этилацетат/триэтиламин (10/90/1) Rf 0,46). Соединение 29 А (0,44 г, 1,38 ммоль) преобразовывали в указанное в заголовке соединение (29 В) способом, как описано для соединения 10 В. Указанное в заголовке соединение (29 В, в виде HCl соли) получали в виде твердого вещества (0,33 г, 95%). Т.пл. 245 С (с разложением). ЖХ/МС; Rt: 0,63 мин.(м, 1 Н), 6,53 (ушир.с, 1 Н), 3,64-3,57 (м, 1 Н), 3,40-3,33 (м, 1 Н), 3,31-3,22 (м, 1 Н), 3,10 (ушир.т, J=10 Гц,1 Н), 2,99-2,90 (м, 1 Н), 2,19-2,12 (м, 1 Н), 2,01-1,94 (м, 1 Н), 1,85-1,66 (м, 2 Н). 6-Хлор-2-пиперидин-3-ил-1 Н-пирроло[2,3-b]пиридин (соединение 32) и 4-хлор-2-пиперидин-3-ил 1 Н-пирроло[2,3-b]пиридин (соединение 33). Соединение 29 А (4,87 г, 15,36 ммоль) и 3,4 мл 1,1,1,3,3,3-гексаметилдисилазана (16,30 ммоль) смешивали в 100 мл ТГФ. К полученному раствору добавляли 3,2 мл (41,4 ммоль) метилхлорформиата и реакционную смесь перемешивали в течение 1,5 ч при температуре кипения с обратным холодильником. Реакционную массу охлаждали и концентрировали в вакууме. Полученный в результате остаток поме- 19015700 щали в этилацетат, промывали 5% раствором NaHCO3, затем насыщенным раствором соли, сушили(Na2SO4), фильтровали и концентрировали в вакууме. Очистка флэш-хроматографией (диэтиловый эфир/РА:1/2, затем диэтиловый эфир) приводила к получению соединения 30 А (аморфное вещество,2,08 г, 34,4%) (ТСХ диэтиловый эфир/РЕ (1/1) Rf 0,17). Второе соединение, полученное в процессе хроматографии, (31 А) получали в виде масла (2,7 г, 44,6%) (ТСХ диэтиловый эфир/РЕ (1/1) Rf 0,08). Щелочное отщепление N1-карбаматной группы осуществляли в соответствии со способом, описанным для соединения 12. Таким образом, соединение 30 А (2,08 г, 5,29 ммоль) растворяли в 75 мл МеОН,содержащего 20 мл 2 н. NaOH, и перемешивали в течение 18 ч при комнатной температуре. Реакционную смесь концентрировали в вакууме. Полученный в результате остаток помещали в дихлорметан, промывали раствором 5% NaHCO3, затем насыщенным раствором соли, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Очистка флэш-хроматографией (диэтиловый эфир/РЕ: 1/1) приводила к получению соединения 30 В (аморфное вещество, 1,74 г, 98%) (ТСХ диэтиловый эфир/РЕ (1/1) Rf 0,21). ЖХ/МС; Rt: 2,33 мин, ([М+Н]+=336). Соединение 31 А (0,35 г, 0,89 ммоль) растворяли в 8 мл МеОН, содержащего 2 мл 2 н. NaOH, и перемешивали в течение 18 ч при комнатной температуре. Реакционную массу концентрировали в вакууме. Полученный в результате остаток помещали в дихлорметан, промывали 5% раствором NaHCO3, затем насыщенным раствором соли, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Повторная очистка флэш-хроматографией (диэтиловый эфир/РЕ: 1/1) приводила к получению соединения 31 В(аморфное вещество, 0,1 г, 33%) (ТСХ диэтиловый эфир Rf 0,3). ЖХ/МС; Rt: 2,27 мин, ([М+Н]+=336). 2 мл ацетилхлорида медленно добавляли к 40 мл EtOH (-10 С). Через 15 мин полученный раствор добавляли к соединению 30 В (1,67 г, 5 ммоль) и нагревали до температуры кипения с обратным холодильником в течение 1,5 ч. Смесь охлаждали и частично концентрировали, добавляли этилацетат, полученный в результате осадок отделяли фильтрованием и промывали диизопропиловым эфиром. Указанное в заголовке соединение, 6-хлор-2-пиперидин-3-ил-1 Н-пирроло [2,3-b] пиридин 32 (в видеHCl соли), получали в виде твердого вещества (1,29 г, 100%). Т.пл 290 С (с разложением). 1 Н-ЯМР(400 МГц, D6DMCO):12,0 (ушир.с, 1 Н), 7,93 (д, J=8 Гц, 1 Н), 7,08 (д, J=8 Гц, 1 Н), 6,31 (д, J=2 Гц, 1 Н),3,58-3,49 (м, 1 Н), 3,34-3,24 (м, 2 Н), 3,15-3,02 (м, 1 Н), 2,92-2,79 (м, 1 Н), 2,18-2,10 (м, 1 Н), 1,94-1,80 (м,2 Н), 1,78-1,64 (м, 1 Н). Разделения энантиомерно чистых изомеров достигали, используя хиральную колонку (Chiralpak(с 1, толуол) и Rt: 25,2 мин (32 В), []D25 +10 (с 1, толуол). Оба изомера подвергали взаимодействию с 1 экв. фумаровой кислоты в EtOH и концентрировали. Перекристаллизация из смеси EtOH/этилацетат приводила к получению твердого вещества (свободное основание/фумаровая кислота (1:1. Т.пл. 206 С (с разложением). 0,22 мл ацетилхлорида медленно добавляли к 5 мл EtOH (-10 С). Через 15 мин полученный раствор добавляли к соединению 31 В (0,1 г, 0,3 ммоль) и нагревали до температуры кипения с обратным холодильником в течение 1,5 ч. Смесь охлаждали и частично концентрировали, добавляли этилацетат, полученный в результате осадок отделяли фильтрованием и промывали диизопропиловым эфиром с получением соединения 33,4-хлор-2-пиперидин-3-ил-1 Н-пирроло[2,3-b]пиридина (в виде HCl соли), (52 мг, 64%). Т.пл. 250 С (с разложением). 1 Н-ЯМР (400 МГц, D2O):N1-H не наблюдали, 8,30 (ушир.д, J=8 Гц, 1 Н), 7,39 (ушир.д,J=8 Гц, 1 Н), 6,63 (ушир.с, 1 Н), 3,66-3,58 (м, 1 Н), 3,42-3,23 (м, 2 Н), 3,16-3,06 (м, 1 Н), 3,0-2,90 (м, 1 Н), 2,232,14 (м, 1 Н), 2,04-1,92 (м, 1 Н), 1,86-1,67 (м, 2 Н).(R)-2-Пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 39 А) и (S)-2-пирролидин-2 илметил-1 Н-пирроло[2,3-b]пиридин (соединение 39 В). К раствору безводного ТГФ (75 мл), содержащему соединение 17 (6,2 г, 24 ммоль), добавляли 12 мл(24 ммоль) LDA (2,0 М в ТГФ/гептан) по каплям при -10 С в атмосфере N2. После добавления полученный в результате раствор перемешивали в течение 30 мин при -10 С-0 С. Затем температуру понижали до -70 С. При этой температуре раствор соединения 35 (4 г, 24,5 ммоль) в 25 мл ТГФ добавляли по каплям (10 мин). После добавления соединения 35 температуру повышали до -20 С и полученный в результате раствор перемешивали в течение 2 ч. Смеси давали возможность нагреться до температуры окружающей среды и перемешивали в течение еще 2 ч. Реакционную смесь концентрировали и остаток растворяли в 40 мл 1 н. HCl, 40 мл EtOH и 40 мл ТГФ. Смесь перемешивали при 80 С в течение 18 ч. Реакционную смесь концентрировали в вакууме. Полученный в результате остаток помещали в этилацетат, промывали 2 н. NaOH, сушили (Na2SO4),фильтровали и концентрировали в вакууме. Очистка флэш-хроматографией (МеОН/триэтиламин 97/3) приводила к получению соединения 38 А (аморфное вещество, 3,63 г, 44%). 1H-ЯМР (400 МГц, CDCl3):8,35 (ушир.д, J=5 Гц, 1 Н), 8,09 (ушир.д, J=8 Гц, 2 Н), 7,67 (ушир.д, J=8 Гц, 1 Н), 7,57-7,41 (м, 3 Н), 7,12 (дд,J=8 Гц, 5 Гц, 1 Н), 6,46 (с, 1 Н), 3,70-3,58 (м, 1 Н), 3,39-3,31 (м, 1 Н), 3,23-3,15 (м, 1 Н), 3,13-3,03 (м, 1 Н),2,98-2,86 (м, 1 Н), 2,10-1,70 (м, 3 Н), 1,57-1,44 (м, 1 Н) (ТСХ МеОН/триэтиламин 97/3 Rf 0,18). ЖХ/МС; Rt:- 20015700 1,27 мин, ([М+Н]+=342). Соединение 38 А (3,63 г, 10,6 ммоль), 11 г KOH и 22 мл гидразинмоногидрата смешивали в 2-(2 гидроксиэтокси)этаноле (100 мл) и перемешивали 1 ч при 100 С. К охлажденной реакционной смеси добавляли этилацетат и полученный в результате органический слой промывали 2 н. NaOH, сушили(Na2SO4), фильтровали и концентрировали в вакууме. Смесь растворяли в МеОН и фильтровали через 40 г SCX-2 (МеОН, затем 1 н. NH3/MeOH). Последующая очистка флэш-хроматографией (МеОН/триэтиламин (98/2) приводила к получению указанного в заголовке соединения 39 А (ЖХ/МС; Rt: 0,91 мин, ([М+Н]+=202), (аморфное вещество, 1,42 г, 66%), ([]D25 -50 (с 1, толуол, которое подвергали взаимодействию с 1 экв. фумаровой кислоты в EtOH и концентрировали. Перекристаллизация из смесиEtOH/этилацетат давала твердое вещество (свободное основание/фумаровая кислота (1:1. Т.пл. 163 С(S)-2-Пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 39 В) получали, применяя методику, описанную для соединения 39 А, при использовании сульфамидата 36. []D25 +50 (с 1, толуол).(R)-2-(1-Метилпирролидин-2-илметил)-1 Н-пирроло[2,3-b]пиридин (соединение 41 А) и (S)-2-(1 метилпирролидин-2-илметил)-1 Н-пирроло[2,3-b]пиридин (соединение 41 В). Соединение 38 А (0,39 г, 1,14 ммоль), 0,5 г NaBH(OAc)3 и 0,2 мл формальдегида (37%) смешивали в дихлорэтане (15 мл) и перемешивали 1 ч при комнатной температуре. Реакционную массу концентрировали в вакууме. Полученный в результате остаток помещали в этилацетат, промывали 2 н. NaOH, сушили(Na2SO4), фильтровали и концентрировали в вакууме с получением соединения 40 А (аморфное вещество,0,40 г, 98%). 1 Н-ЯМР (400 МГц, CDCl3):8,35 (дд, J=5 Гц, 2 Гц, 1 Н), 8,12-8,07 (м, 2 Н), 7,69 (дд, J=8 Гц,2 Гц, 1 Н), 7,56-7,41 (м, 3 Н), 7,12 (дд, J=8 Гц, 5 Гц, 1 Н), 6,40 (с, 1 Н), 3,73-3,67 (м, 1 Н), 3,16-3,10 (м, 1 Н),2,87-2,73 (м, 2 Н), 2,47 (с, 3 Н), 2,33-2,25 (м, 1 Н), 1,95-1,50 (м, 4 Н). Соединение 40 А (0,4 г, 11,2 ммоль), 1,1 г KOH и 2,2 мл гидразинмоногидрата смешивали в 2-(2 гидроксиэтокси)этаноле (10 мл) и перемешивали в течение 1 ч при 100 С. К охлажденной реакционной смеси добавляли этилацетат и полученный в результате органический слой промывали 2 н. NaOH, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Смесь растворяли в МеОН и фильтровали через 40 г SCX-2 (МеОН, затем 1 н. NH3/MeOH). Последующая очистка флэш-хроматографией (МеОН/триэтиламин (99/1 приводила к получению указанного в заголовке соединения 41 А (твердое вещество, 0,19 г,76%). ([]D25 +44 (с 1, толуол. 1 Н-ЯМР (400 МГц, CDCl3):10,4 (ушир.с, 1 Н), 8,18 (дд, J=5 Гц, 2 Гц,1 Н), 7,79 (дд, J=8 Гц, 2 Гц, 1 Н), 6,99 (дд, J=8 Гц, 5 Гц, 1 Н), 6,18 (ушир.с, 1 Н), 3,18-3,07 (м, 2 Н), 2,90-2,83(S)-2-(1-Метилпирролидин-2-илметил)-1 Н-пирроло[2,3-b]пиридин (соединение 41 В) получали, используя методику, описанную для соединения 41 А. ([]D25 -42 (с 1, толуол. Т.пл. 130-131 С.(R)-6-Хлор-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 45 А) и (S)-6-хлор-2 пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 45 В). 6-Хлор-7-азаиндол (9,3 г, 61,1 ммоль), полученное как описано в литературе (Synthesis, 1992), растворяли в 100 мл безводного ТГФ в атмосфере N2. При 0 С добавляли 60%-ную дисперсию NaH 3,5 г(65,9 ммоль) в минеральном масле. После перемешивания в течение 1 ч при комнатной температуре смесь охлаждали (0 С) и 8,7 мл (67,2 ммоль) бензолсульфонилхлорида, растворенного в 20 мл безводного ТГФ, добавляли. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Добавляли этилацетат к смеси и органический слой промывали три раза насыщенным раствором NaHCO3, сушили (Na2SO4), фильтровали и концентрировали. Полученный в результате остаток очищали флэшхроматографией (диэтиловый эфир/РЕ градиент (от 1:4 до чистого диэтилового эфира, получая 1 бензолсульфонил-6-хлор-1 Н-пирроло[2,3-b]пиридин (аморфное вещество, 14,4 г, 80,7%) (ТСХ диэтиловый эфир/РЕ (1/1) Rf 0,37). ЖХ/МС; Rt: 1,98 мин, ([М+Н]+=293). К раствору безводного ТГФ (100 мл), содержащего соединение 1-бензолсульфонил-6-хлор-1 Нпирроло[2,3-b]пиридин (2,52 г, 8,6 ммоль), добавляли 4,3 мл (24 ммоль) LDA (2,0 М в ТГФ/гептан) по каплям при -78 С в атмосфере N2. После добавления полученный в результате раствор перемешивали в течение 60 мин при -78 С. При этой температуре раствор соединения 35 (1,4 г, 8,6 ммоль) в 10 мл ТГФ добавляли по каплям (5 мин). После добавления соединения 35 температуру повышали до -20 С и полученный в результате раствор перемешивали в течение 2 ч. Смеси давали возможность нагреться до температуры окружающей среды и перемешивали в течение еще 2 ч. Реакционную смесь концентрировали и остаток растворяли в 40 мл 1 н. HCl, 40 мл EtOH и 40 мл ТГФ. Смесь перемешивали при 80 С в течение 18 ч. Реакционную смесь концентрировали в вакууме. Полученный в результате остаток помещали в этилацетат, промывали 2 н. NaOH, сушили (Na2SO4),фильтровали и концентрировали в вакууме. Очистка флэш-хроматографией (МеОН/триэтиламин 98/2) давала (R)-1-бензолсульфонил-6-хлор-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (аморфное вещество, 1,01 г, 31%) (ТСХ МеОН/триэтиламин 98/2 Rf 0,32). (R)-1-Бензолсульфонил-6-хлор-2 пирролидин-2-илметил-1 Н-пирроло [2,3-b]пиридин (1,01 г, 2,69 ммоль), 20 мл 2 н. NaOH и 30 мл изопро- 21015700 панола смешивали и перемешивали 3 ч при 100 С. К охлажденной реакционной смеси добавляли этилацетат и полученный в результате органический слой промывали 2 н. NaOH, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Смесь растворяли в МеОН и фильтровали через 10 г SCX-2 (МеОН,затем 1 н. NH3/МеОН). Последующая очистка флэш-хроматографией (МеОН/триэтиламин (98/2) приводила к получению указанного в заголовке соединения 45 А (твердое вещество, 366 мг, 58%). ([]D25 -48 (с 1, толуол. 1 Н-ЯМР (400 МГц, CDCl3):11,0-10,0 (ушир.с, 1 Н), 7,52 (д, J=8 Гц, 1 Н), 7,01 (д, J=8 Гц, 1 Н),6,16 (с, 1 Н), 3,50-3,42 (м, 1 Н), 3,02-2,90 (м, 3 Н), 2,81-2,73 (м, 1 Н), 1,95-1,85 (м, 1 Н), 1,82-1,64 (м, 2 Н),1,43-1,33 (м, 1 Н). Соединение 45 А подвергали взаимодействию с 1 экв. фумаровой кислоты в EtOH и концентрировали. Перекристаллизация из смеси EtOH/этилацетат давала твердое вещество (свободное основание/фумаровая кислота (2:1. Т.пл. 222 С (с разложением).(S)-6-Хлор-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 45 В) получали (из соединения 36), применяя методику, описанную для соединения 45 А. ([]D25 +44 (с 1, толуол. Соединение 45 В подвергали взаимодействию с 1 экв. фумаровой кислоты в EtOH и концентрировали. Перекристаллизация из смеси EtOH/этилацетат позволяла получить твердое вещество (свободное основание/фумаровая кислота (1:1. Т.пл. 189-190 С.(R)-6-Бром-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 46 А) и (S)-6-бром-2 пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 46 В). 6-Бром-7-азаиндол (4,65 г, 23,6 ммоль), полученный как описано в литературе (Synthesis, 1992),преобразовывали в 1-бензолсульфонил-6-бром-1 Н-пирроло[2,3-b]пиридин, используя методику, описанную выше (твердое вещество, т.пл. 121-124 С), (7,57 г, 95%). 1 Н-ЯМР (400 МГц, CDCl3):8,28-8,22 (м,2 Н), 7,70-7,50 (м, 5 Н), 7,32 (д, J=8 Гц, 1 Н), 6,56 (д, J=4 Гц, 1 Н) (ТСХ диэтиловый эфир Rf 0,55). 1-Бензолсульфонил-6-бром-1 Н-пирроло[2,3-b]пиридин (2,07 г, 6,1 ммоль) преобразовывали в (R)-1 бензолсульфонил-6-бром-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин, используя методику,описанную выше (аморфное вещество, 1,24 г, 48%). 1 Н-ЯМР (400 МГц, CDCl3):8,21-7,97 (м, 2 Н), 7,617,47 (м, 5 Н), 7,25 (д, J=8 Гц, 1 Н), 6,46 (с, 1 Н), 3,65-3,57 (м, 1 Н), 3,37-3,31 (м, 1 Н), 3,09-3,03 (м, 1 Н), 2,962,89 (м, 1 Н), 2,06-1,73 (м, 3 Н), 1,54-1,45 (м, 1 Н) (ТСХ МеОН/триэтиламин 97/3 Rf 0,22).(R)-1-Бензолсульфонилил-6-бром-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (1,24 г,2,95 ммоль), 30 мл 2 н. NaOH и 50 мл МеОН смешивали и перемешивали в течение 30 мин при 60 С. К охлажденной реакционной смеси добавляли этилацетат и полученный в результате органический слой промывали 2 н. NaOH, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Смесь растворяли в МеОН и фильтровали через 10 г SCX-2 (МеОН, затем 1 н. NH3/MeOH). Последующая очистка флэшхроматографией (МеОН/триэтиламин (97/3) приводила к получению указанного в заголовке соединения 46 А (аморфное вещество, 630 мг, 76%), ([]D25 -50 (с 1, толуол, которое преобразовывали в его соль(S)-6-Бром-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 46 В) получали (из соединения 36), используя методику, описанную для соединения 46 А. ([]D25 +50 (с 1, толуол.(R)-6-Фтор-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 47 А) и (S)-6-фтор-2 пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 47 В). Соединение 55 (18 г, 161 ммоль), 70 г K2CO3 (506 ммоль) и 15,4 мл (161 ммоль) этилхлорформиата смешивали в CH3CN (400 мл) и перемешивали в течение 4 дней при 40 С. ТСХ показала небольшое превращение. Дополнительные 15,4 мл этилхлорформиата добавляли и перемешивание продолжали в течение 2 дней. Смесь охлаждали. Добавляли этилацетат к смеси и органический слой промывали насыщенным раствором NaHCO3, сушили (Na2SO4), фильтровали и концентрировали. Полученный в результате остаток очищали флэш-хроматографией (диэтиловый эфир/РЕ (1:3, что приводило к получению этилового эфира (6-фторпиридин-2-ил)карбаминовой кислоты (56) (аморфное вещество, 19,68 г, 66,5%). 1 НЯМР (400 МГц, CDCl3):7,83 (дд, J=8 Гц, 2 Гц, 1 Н), 7,80-7,73 (м, 1 Н), 7,50-7,40 (ушир.с, 1 Н), 6,61-6,57(м, 1 Н), 4,25 (кв, J=8 Гц, 2 Н), 1,32 (т, J=8 Гц, 3 Н) (ТСХ диэтиловый эфир/РЕ 1/1 Rf 0,5). 28,6 мл (0,18 моль) TMEDA добавляли к раствору соединения 56 (13,41 г, 72,9 ммоль), растворенного в 300 мл безводного ТГФ. Смесь охлаждали до -78 С (в атмосфере N2). К перемешиваемой реакционной смеси добавляли 76 мл (2,5 М н-BuLi) и смесь перемешивали в течение 2 ч при -78 С. После добавления I2 (48 г, 0,17 моль) смесь перемешивали в течение 1 ч при -78 С. Реакционную смесь далее гасили насыщенным раствором Na2S2O3 и оставляли нагреваться до температуры окружающей среды. Добавляли этилацетат к смеси и органический слой промывали насыщенным раствором NaHCO3, сушили(Na2SO4), фильтровали и концентрировали. Полученный в результате остаток очищали флэшхроматографией (диэтиловый эфир/РЕ (от 1/3) до 1/1), что приводило к получению этилового эфира(1,29 ммоль) Cu(I)иодида, 454 мг (0,65 ммоль) PdCl2 (PPh3)2 (454 мг, 0,64 ммоль) и триэтиламина (6,3 мл) перемешивали и дегазировали (N2). Полученную в результате реакционную смесь перемешивали в течение 10 ч при 100 С (закрытый сосуд), охлаждали и выливали в этилацетат и Н 2 О. Органический слой промывали Н 2 О, сушили (Na2SO4), фильтровали и концентрировали. Остаток очищали флэшхроматографией (диэтиловый эфир/РЕ (1/3, что приводило к получению этилового эфира (6-фтор-3 триметилсиланилэтинилпиридин-2-ил)карбаминовой кислоты (58) (маслянистое вещество, 1,5 г, 42%). 1 Н-ЯМР (200 МГц, CDCl3):7,76 (ушир.т, J=8 Гц, 1 Н), 7,68-7,60 (ушир.с, 1 Н), 6,56 (дд, J=8 Гц, 3 Гц, 1 Н),4,29 (кв, J=8 Гц, 2 Н), 1,34 (т, J=8 Гц, 3 Н), 0,3 (с, 9 Н) (ТСХ диэтиловый эфир/РЕ 1/1 Rf 0,39). Смесь соединения 58 (6 г, 21,4 ммоль) и 8,2 г (43 ммоль) Cu(I)иодида растворяли в 100 мл ДМФА и дегазировали в течение 0,5 ч. Реакционную смесь перемешивали при 150 С (предварительно нагретая масляная баня) в течение 30 мин. Смесь охлаждали, разбавляли этилацетатом и фильтровали. Остаток промывали Н 2 О, сушили (Na2SO4), фильтровали и концентрировали. Последующая очистка флэшхроматографией (диэтиловый эфир/РЕ (1/1 приводила к получению соединения 59 (аморфное вещество,2,58 г, 12,4 ммоль, 57,9%). 1 Н-ЯМР (200 МГц, CDCl3):7,95 (ушир.т, J=8 Гц, 1 Н), 7,69 (д, J=4 Гц, 1 Н),6,87 (дд, J=8 Гц, 2 Гц, 1 Н), 6,57 (д, J=4 Гц, 1 Н), 4,55 (кв, J=8 Гц, 2 Н), 1,49 (т, J=8 Гц, 3 Н) (ТСХ диэтиловый эфир/РЕ 1/1 Rf 0,42). Соединение 59 (2,58 г, 12,4 ммоль) растворяли в 50 мл МеОН и 20 мл 2 н. NaOH. Реакционную смесь перемешивали в течение 30 мин при комнатной температуре. Добавляли этилацетат к смеси и органический слой промывали 5% водным раствором NaHCO3, сушили (Na2SO4), фильтровали и концентрировали, что приводило к получению 6-фтор-1 Н-пирроло[2,3-b]пиридина (6-фтор-7-азаиндол, соединение 44) в виде полутвердого вещества (1,68 г, 12,3 ммоль, 99%). 1H-ЯМР (400 МГц, CDCl3):9,6(ТСХ диэтиловый эфир/РЕ (1/1) Rf 0,34). ЖХ/МС; Rt: 1,39 мин, ([М+Н]+=137). 6-Фтор-7-азаиндол (соединение 44, 1,72 г, 12,6 ммоль) подвергали взаимодействию с бензолсульфонилхлоридом, как описано для синтеза соединения 45 А/В, получая 1-бензолсульфонил-6-фтор-1 Нпирроло[2,3-b]пиридин (соединение 60), (твердое вещество, 3,03 г, 86,6%). Т.пл. 130-132 С; (ТСХ диэтиловый эфир/РЕ (1/1) Rf 0,29). ЖХ/МС; Rt: 1,79 мин, ([М+Н]+=277). 1-Бензолсульфонил-6-фтор-1 Н-пирроло[2,3-b]пиридин (0,8 г, 2,9 ммоль) преобразовывали в (R)-1 бензолсульфонил-6-фтор-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин, используя методику, аналогично описанной для соединения 45 А/В (аморфное вещество, 0,31 г, 30%) (ТСХ МеОН/триэтиламин(R)-1-Бензолсульфонил-6-фтор-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (0,26 г, 0,72 ммоль) преобразовывали в (R)-6-фтор-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 47 А, 90 мг, 57%), используя условия, описанные для соединения 45 А/В. Указанное в заголовке соединение 47 А (аморфное вещество), ([]D -38 (с 1, толуол, преобразовывали в его (аморфное вещество) соль (свободное основание/фумаровая кислота (1:1. 1 Н-ЯМР (400 МГц,D6DMCO):12,2-11,7 (ушир.с, 1 Н), 8,0 (ушир.т, J=8 Гц, 1 Н), 6,76 (ушир.д, J=8 Гц, 1 Н), 6,51 (с, 2 Н), 6,37(S)-6-Фтор-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 47 В) получали (из соединения 36), используя методику, описанную для соединения 47 А. ([]D +38 (с 1, толуол.(R)-6-Бром-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (46 А, 0,67 г, 2,39 ммоль) преобразовывали в пирролидин (Boc защищенный) аналог 48 (аморфное вещество, 0,72 г, 79% суммарный), используя методику, описанную для соединения 9. 1H-ЯМР (400 МГц, CDCl3):9,4 (ушир.с, 1 Н), 7,66 (ушир.д, J=8 Гц, 1 Н), 7,18 (ушир.д, J=8 Гц, 1 Н),6,19 (ушир.с, 1 Н), 4,10-4,03 (м, 1 Н), 3,42-3,25 (м, 2 Н), 3,16-3,07 (м, 1 Н), 3,04-2,88 (м, 1 Н), 2,0-1,89 (м, 1 Н),1,8-1,63 (м, 3 Н). Соединение 48 (0,34 г, 0,89 ммоль) растворяли в 4 мл ДМФА и 2,5 мл МеОН (в атмосфере N2). К полученной смеси добавляли 1,6 г (29,6 ммоль) NaOMe и 0,25 г (1,74 ммоль) Cu(I)бромида и смесь перемешивали в течение 1 ч при комнатной температуре. Добавляли этилацетат и органический слой промывали 5% раствором NaHCO3, сушили (Na2SO4),фильтровали и концентрировали в вакууме. Полученный в результате остаток очищали флэшхроматографией (диэтиловый эфир), получая пирролидиновый (Boc защищенный) предшественник соединения 49 в виде масла (0,32 г, 56%) (ТСХ диэтиловый эфир Rf 0,8). 1 Н-ЯМР (400 МГц, CDCl3):8,7(с, 3 Н), 3,46-3,23 (м, 2 Н), 3,18-2,96 (м, 1 Н), 2,93-2,78 (м, 1 Н), 1,97-1,84 (м, 1 Н), 1,83-1,71 (м, 3 Н), 1,51 (с,9 Н), для которого удаляли защитные группы (HCl/EtOH, как описано выше), получая указанное в заголовке соединение (49) в виде HCl соли (аморфное вещество, гигроскопичное). Выход 0,180 мг (80%),([]D25 -12 (с 1, МеОН. ЖХ/МС; R1: 1,17 мин, ([М+Н]+=232).- 23015700 Соединение 49 (свободное основание, полученное после фильтрования HCl соли через SCX-2), преобразовывали в его соль (используя методику, описанную выше) (свободное основание/фумаровая кислота (1:1), аморфное вещество). 1 Н-ЯМР (400 МГц, D6DMCO):11,61 (ушир.с, 1 Н), 7,75 (д, J=8 Гц, 1 Н),6,49 (с, 2 Н), 6,47 (д, J=8 Гц, 1 Н), 6,21 (с, 1 Н), 3,84 (с, 3 Н), 3,78-3,71 (м, 1 Н), 3,23-3,18 (м, 1 Н), 3,14-3,08 (м,1 Н), 3,01-2,95 (м, 1 Н), 2,05-1,80 (м, 3 Н), 1,67-1,59 (м, 1 Н).(R)-5-Бром-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 52 А) и (S)-5-бром-2 пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 52 В). 5-Бром-7-азаиндол (5,19 г, 26,3 ммоль), коммерчески доступный, преобразовывали в 1 бензолсульфонил-5-бром-1 Н-пирроло[2,3-b]пиридин, используя методику, описанную для соединения 45 А/В, твердое вещество, Т.пл. 141-142 С. (6,9 г, 78%). 1H-ЯМР (400 МГц, CDCl3):8,45 (д, J=2 Гц, 1 Н),8,19-8,15 (м, 2 Н), 7,98 (д, J=2 Гц, 1 Н), 7,74 (д, J=4 Гц, 1 Н), 7,62-7,47 (м, 3 Н), 6,55 (д, J=4 Гц, 1 Н). ЖХ/МС;Rt: 1,92 мин, ([М+Н]+=337). 1-Бензолсульфонил-5-бром-1 Н-пирроло[2,3-b]пиридин (5,52 г, 16,3 ммоль) преобразовывали в (R)1-бензолсульфонил-5-бром-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин, используя методику,описанную для соединения 45 А/В (аморфное вещество, 3,02 г, смесь соединений, содержащая приблизительно 70% ожидаемого С 2-региоизомера) (ТСХ МеОН/триэтиламин (97/3) Rf 0,3). ЖХ/МС; Rt: 1,44 мин,([М+Н]+=420, 422). Вышеуказанную смесь, содержащую (R)-1-бензолсульфонил-5-бром-2-пирролидин-2-илметил-1 Нпирроло[2,3-b]пиридин (3,02 г, приблизительно 7,1 ммоль), преобразовывали в указанное в заголовке соединение 52 А, используя методику, описанную для соединения 45 А/В (многократная флэшхроматография), (аморфное вещество, 0,5 г, приблизительно 25%), которое преобразовывали в его соль(S)-5-Бром-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 52 В) получали (из соединения 36), используя методику, описанную для соединения 52 А. ЖХ/МС; Rt: 1,27 мин, ([М+Н]+=280).(R)-5-Метил-2-пирролидин-2-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 53 А). 5-Метил-7-азаиндол (4,15 г, 31,4 ммоль) преобразовывали в 1-бензолсульфонил-5-метил-1 Нпирроло[2,3-b]пиридин, используя методику, описанную для соединения 45 А/В (аморфное вещество,7,07 г, 82%). 1 Н-ЯМР (400 МГц, CDCl3):8,25 (ушир.д, J=2 Гц, 1 Н), 8,19-8,15 (м, 2 Н), 7,66 (д, J=4 Гц,1 Н), 7,62 (ушир.д, J=2 Гц, 1 Н), 7,58-7,43 (м, 3 Н), 6,51 (д, J=2 Гц, 1 Н), 2,38 (с, 3 Н).(R)-1-Бензолсульфонил-5-метил-2-пирролидин-2-илметил-1 Н-пирроло [2,3-b] пиридин (1,5 г,4,22 ммоль), 3,5 г KOH и 1 мл гидразинмоногидрата смешивали в 2-(2-гидроксиэтокси)этаноле (25 мл) и перемешивали 1 ч при 100 С. К охлажденной реакционной смеси добавляли МеОН и реакционную смесь фильтровали через 60 г SCX-2 (МеОН, затем 1 н. NH3/МеОН). Последующая очистка флэшхроматографией (МеОН/триэтиламин (90/2) приводила к получению указанного в заголовке соединения 53 А (аморфное вещество, 0,46 г, 50%); ([]D25 -10 (с 1, диоксан, которое подвергали взаимодействию с 1 экв. фумаровой кислоты в МеОН и концентрировали, что приводило к получению указанного в заголовке соединения 53 А (аморфное вещество) (свободное основание/фумаровая кислота (1:1. 1H-ЯМР(400 МГц, D6DMCO):11,6 (ушир.с, 1 Н), 7,97 (ушир.с, 1 Н), 7,63 (ушир.с, 1 Н), 6,51 (с, 2 Н), 6,22 (с, 1 Н),3,84-3,77 (м, 1 Н), 3,26-3,03 (м, 4 Н), 2,33 (с, 3 Н), 2,07-1,81 (м, 3 Н), 1,69-1,61 (м, 1 Н). 2-Пирролидин-3-илметил-1 Н-пирроло[2,3-b]пиридин (соединение 66). 8,83 г (91 ммоль) метоксиметиламина (HCl соль) перемешивали в 200 мл безводного бензола (0 С, в атмосфере N2). Добавляли 46,2 мл смеси триметилалюминий/толуол (2,5 М) и смесь перемешивали в течение 2,5 ч при 0 С. Соединение 62 (6,73 г, 30,7 ммоль), растворенное в 100 мл бензола, добавляли и полученную в результате смесь перемешивали в течение 1 ч при комнатной температуре. К смеси последовательно добавляли насыщенный раствор NaHCO3 (0C) и этилацетат. Органический слой промывали три раза 5% раствором NaHCO3, сушили (Na2SO4), фильтровали и концентрировали. Полученный в результате остаток очищали флэш-хроматографией (МеОН/этилацетат (1/9, получая метоксиметиламид 1 бензилпирролидин-3-карбоновой кислоты (соединение 63, 6,76 г, 88,7%). 1H-ЯМР (400 МГц, CDCl3):7,38-7,22 (м, 5 Н), 3,69-3,62 (м, 5 Н), 3,45-3,35 (м, 1 Н), 3,19-3,18 (2 с, 3 Н), 3,06-2,99 (м, 1 Н), 2,88-2,81 (м,1 Н), 2,56-2,41 (м, 2 Н), 2,14-2,03 (м, 2 Н). К раствору безводного ТГФ (75 мл), содержащему соединение 17 (7,2 г, 27,1 ммоль), добавляли 13,6 мл LDA (2,0 М в ТГФ/гептан) по каплям при -10 С в атмосфере N2. После добавления полученный в результате раствор перемешивали в течение 30 мин при -10-0 С. Затем температуру понижали до -70 С.- 24015700 При этой температуре раствор соединения 63 (6,72 г, 27 ммоль) в 25 мл ТГФ добавляли по каплям (10 мин). После добавления соединения 63 температуру повышали до -30 С (0,5 ч) и полученный в результате раствор перемешивали в течение 1 ч при -30 С. Затем смесь гасили добавлением насыщенного раствора NH4Cl при -30 С и оставляли нагреваться до температуры окружающей среды. Добавляли этилацетат и органический слой промывали 5% раствором NaHCO3, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Очистка флэш-хроматографией (диэтиловый эфир, затем этилацетат) приводила к получению соединения 64 (аморфное вещество, 5,38 г, 44,6%). ЖХ/МС; Rt: 1,27 мин, ([М+Н]+=342). Соединение 64 (1,05 г, 2,35 ммоль), 2 г KOH и 3,56 мл гидразинмоногидрата смешивали в 2-(2 гидроксиэтокси)этаноле (20 мл) и перемешивали 30 мин при 100 С (в атмосфере N2), затем дополнительно 45 мин при 200 С. К охлажденной реакционной смеси добавляли этилацетат и полученный в результате органический слой промывали 2 н. NaOH, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Смесь растворяли в МеОН и фильтровали через 80 г SCX-2 (МеОН, затем 1 н. NH3/MeOH). Последующая очистка флэш-хроматографией (МеОН/этилацетат) приводила к получению соединения 65(аморфное вещество, 0,6 г, 83%). 1 Н-ЯМР (400 МГц, CDCl3):10,65 (ушир.с, 1 Н), 8,20 (дд, J=5 Гц, 2 Гц,1 Н), 7,80 (дд, J=8 Гц, 2 Гц, 1 Н), 7,37-7,20 (м, 5 Н), 7,02 (дд, J=8 Гц, 5 Гц, 1 Н), 6,17 (с, 2 Н), 3,63 (дд,Jгем=13 Гц, 2 Н), 2,95-2,90 (м, 2 Н), 2,77-2,54 (м, 4 Н), 2,43-2,38 (м, 1 Н), 2,12-2,02 (м, 1 Н), 1,65-1,55 (м, 1 Н). ЖХ/МС; Rt: 1,46 мин, ([М+Н]+=292). Соединение 65 (0,52 г, 1,8 ммоль), 0,3 г формиата аммония (4,7 ммоль) и 20% Pd(OH)2/C (50 мг) смешивали в МеОН (10 мл) и нагревали до температуры кипения с обратным холодильником в течение 1 ч. Смесь охлаждали, фильтровали, концентрировали и растворяли в МеОН. Последующее фильтрование через 25 г SCX-2 (МеОН, затем 1 н. NH3/МеОН) приводило к получению указанного в заголовке соединения, 2-пирролидин-3-илметил-1 Н-пирроло[2,3-b]пиридина (соединение 66, 0,34 г, 94%), которое подвергали взаимодействию с 1 экв. фумаровой кислоты в МеОН и концентрировали, что приводило к получению указанного в заголовке соединения 66 (аморфное вещество) (свободное основание/фумаровая кислота (1:1. 1 Н-ЯМР (400 МГц, D6DMCO):11,6 (ушир.с, 1 Н), 8,11 (дд, J=5 Гц, 2 Гц, 1 Н), 7,81 (дд, J=8 Гц, 2 Гц, 1 Н), 6,99 (дд, J=8 Гц, 5 Гц, 1 Н), 6,5 (с, 2 Н), 6,21 (ушир.с, 1 Н), 3,29-3,22 (м, 2 Н), 3,17-3,09 (м, 1 Н),2,90-2,76 (м, 3 Н), 2,72-2,61 (м, 1 Н), 2,06-1,97 (м, 1 Н), 1,69-1,59 (м, 1 Н) (ТСХ МеОН/триэтиламин (97/3)Rf 0,08). 2-(1-Метилпирролидин-3-илметил)-1 Н-пирроло[2,3-b]пиридин (соединение 68). Соединение 66 (0,3 г, 1,49 ммоль) преобразовывали в соединение 67, используя методику, описанную для соединения 9 (выход 0,362 г (80,7%); (ТСХ диэтиловый эфир Rf 0,11, которое как таковое использовали для получения указанного в заголовке соединения (68), используя методику, аналогично описанной для соединения 28. (Выход 0,17 г, 65%). Т.пл. 96-97 С. 1 Н-ЯМР (400 МГц, CDCl3):N1-H не наблюдали, 8,16 (дд, J=5 Гц, 2 Гц, 1 Н), 7,77 (дд, J=8 Гц, 2 Гц, 1 Н), 6,98 (дд, J=8 Гц, 5 Гц, 1 Н), 6,16 (ушир.с,1 Н), 2,92-2,84 (м, 2 Н), 2,76-2,64 (м, 2 Н), 2,62-2,50 (м, 2 Н), 2,34 (с, 3 Н), 2,40-2,31 (м, 1 Н), 2,11-2,01 (м, 1 Н),1,63-1,53 (м, 1 Н) (ТСХ МеОН/триэтиламин (97/3) Rf 0,2). 2-Пирролидин-3-ил-1 Н-пирроло[2,3-b]пиридин (соединение 78). К раствору безводного ТГФ (75 мл), содержащему соединение 17 (6,0 г, 23,2 ммоль), добавляли 12,5 мл (24 ммоль) LDA (2,0 М в ТГФ/гептан) по каплям при -10 С в атмосфере N2. После добавления полученный в результате раствор перемешивали в течение 30 мин при -10 С-0 С и затем температуру понижали до -70 С. Безводный CeCl3 (6 г, 24,3 ммоль) добавляли к 50 мл безводного ТГФ, полученную в результате смесь перемешивали в течение 0,5 ч при 30 С (в атмосфере N2). Полученную смесь добавляли к 2-литийпроизводному 17, поддерживая при этом температуру -70 С. Температуру повышали до -10 С и перемешивали в течение 10 мин. Далее температуру понижали до -50 С. Соединение 71 (5 г, 28,5 ммоль), растворенное в 50 мл ТГФ, добавляли к реакционной смеси, полученную в результате смесь перемешивали в течение 2 ч при -30 С. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение еще 2 ч. Смесь концентрировали в вакууме, добавляли этилацетат и органический слой промывали 5% раствором NaHCO3, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Полученный в результате остаток очищали флэш-хроматографией (диэтиловый эфир/РЕ градиент от 1:1 до чистого диэтилового эфира), получая соединение 72, трет-бутиловый эфир 3-(1-бензолсульфонил-1 Нпирроло[2,3-b]пиридин-2-ил)-3-гидроксипирролидин-1-карбоновой кислоты (аморфное вещество, 6,0 г,58%). 1 Н-ЯМР (400 МГц, CDCl3):8,38-8,32 (м, 1 Н), 8,17-8,12 (м, 2 Н), 7,80-7,50 (м, 1 Н), 7,60-7,43 (м,3 Н), 7,17-7,12 (м, 1 Н), 6,55 (с, 1 Н), 4,90 и 4,85 (2 ушир.с, 1 Н), 4,24-4,13 (м, 1 Н), 3,92-3,79 (м, 1 Н), 3,753,48 (м, 2 Н), 2,68-2,47 (м, 2 Н), 1,49 (с, 9 Н). ЖХ/МС; Rt: 1,83 мин, ([М+Н]+=444). Соединение 72 (8,6 г, 19,4 ммоль), 12 г КОН и 20 мл гидразинмоногидрата смешивали в 2-(2 гидроксиэтокси)этаноле (150 мл) и перемешивали 30 мин при 100 С (в атмосфере N2). К охлажденной реакционной смеси добавляли этилацетат и полученный в результате органический слой промывали несколько раз 2 н. NaOH, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Последующая очистка флэш-хроматографией (диэтиловый эфир, затем этилацетат) приводила к получению соединения 74(маслянистое вещество, 4,97 г, 84%). 1 Н-ЯМР (400 МГц, D6DMCO):11,6 (ушир.с, 1 Н), 8,15 (дд, J=5 Гц,- 25015700 2 Гц, 1 Н), 7,87 (дд, J=8 Гц, 2 Гц, 1 Н), 7,01 (дд, J=8 Гц, 5 Гц, 1 Н), 6,38 (д, J=2 Гц, 1 Н), 5,67 (ушир.с, 1 Н),3,61-3,41 (м, 4 Н), 2,37-2,28 (м, 1 Н), 2,16-2,07 (м, 1 Н), 1,43 и 1,41 (2 с, 9 Н). ЖХ/МС; Rt: 1,48 мин,([М+Н]+=304). Соединение 74 (4,92 г, 16,23 ммоль), 100 мл Н 2 О и 100 мл 38% раствора HCl смешивали и нагревали до температуры кипения с обратным холодильником в течение 12 ч. Смесь охлаждали, концентрировали и растворяли в МеОН. Фильтрование через 50 г SCX-2 (МеОН, затем 1 н. NH3/МеОН) с последующей флэш-хроматографией (МеОН/триэтиламин (99/1 приводили к получению соединения 76 (маслянистое вещество, 1,75 г, 58%). 1 Н-ЯМР (400 МГц, D6DMCO):12,1 (ушир.с, 1 Н), 8,22 (дд, J=5 Гц, 2 Гц,1 Н), 7,92 (дд, J=8 Гц, 2 Гц, 1 Н), 7,05 (дд, J=8 Гц, 5 Гц, 1 Н), 6,55-6,50 (м, 1 Н), 6,49 (с, 1 Н), 4,23-4,17 (м,2 Н), 4,07-4,02 (м, 2 Н). ЖХ/МС; Rt: 0,91 мин, ([М+Н]+=186). Соединение 76 (1,8 г, 9,73 ммоль) и 20% Pd(OH)2/C (180 мг) смешивали в МеОН (100 мл). Смесь гидрировали при 50 psi в течение 2 ч. Смесь фильтровали, концентрировали, повторно растворяли в МеОН и фильтровали через 50 г SCX-2 (МеОН, затем 1 н. NH3/МеОН), затем проводили флэшхроматографию (МеОН/триэтиламин (98/2, получая указанное в заголовке соединение, 2-пирролидин 3-ил-1 Н-пирроло[2,3-b]пиридин (соединение 78), (аморфное вещество, 1,09 г, 59%). ЖХ/МС; Rt: 0,75 мин, ([М+Н]+=188). Разделения энантиомерно чистых изомеров достигали, используя хиральную колонку (Chiralpak AD 20 мкм, 2504,6, 20% МеОН, 20% EtOH, 60% гептана, 2 мл/мин, =220 нм, Rt: 6,0 мин (78 А), ([]D25 -10 (с 1, МеОН и Rt: 7,9 мин (78 В), ([]D25 +12 (с 1, МеОН. Оба изомера подвергали взаимодействию с 1 экв. фумаровой кислоты в МеОН и концентрировали, что приводило к получению соли указанного в заголовке соединения, Т.пл. 130-133 С (свободное основание/фумаровая кислота (1:1,5. 1 Н-ЯМР (400 МГц,D6DMCO):11,7 (ушир.с, 1 Н), 8,08 (дд, J=5 Гц, 2 Гц, 1 Н), 7,78 (дд, J=8 Гц, 2 Гц, 1 Н), 6,95 (дд, J=8 Гц,5 Гц, 1 Н), 6,4 (с, 3 Н), 6,27 (ушир.с, 1 Н), 3,63-3,53 (м, 2 Н), 3,33-3,15 (м, 3 Н), 2,36-2,25 (м, 1 Н), 2,08-1,97 (м,1 Н). 2-Пирролидин-3-ил-1 Н-пирроло[3,2-b]пиридин (соединение 79). Соединение 69 (коммерчески доступное) подвергали взаимодействию с бензолсульфонилхлоридом,как описано для соединения 45 А/В, или описано в Eur. J. of Med. Chem (2004). Выход 80-90%. Соединение 70 получали в виде масла, которое кристаллизовалось при стоянии. 1 Н-ЯМР (400 МГц, CDCl3):8,54(дд, J=5 Гц, 2 Гц, 1 Н), 8,27 (ушир.д, J=8 Гц, 1 Н), 8,0-7,85 (м, 2 Н), 7,81 (д, J=4 Гц, 1 Н), 7,60-7,54 (м, 1 Н),7,49-7,43 (м, 2 Н), 7,24 (дд, J=8 Гц, 5 Гц, 1 Н), 6,88 (ушир.д, J=4 Гц, 1 Н). Соединение 70 (2,58 г, 10 ммоль) подвергали взаимодействию с соединением 71, используя методику, описанную для синтеза указанного выше соединения 72, с получением соединения 73. Соединение 73. трет-Бутиловый эфир 3-(1-бензолсульфонил-1 Н-пирроло[3,2-b]пиридин-2-ил)-3 гидроксипирролидин-1-карбоновой кислоты (аморфное вещество, 1,23 г, 28%). 1 Н-ЯМР (400 МГц,CDCl3):8,52-8,48 (м, 1 Н), 8,28-8,21 (м, 2 Н), 7,82-7,78 (м, 2 Н), 7,58-7,51 (м, 1 Н), 7,45-7,39 (м, 2 Н), 7,237,17 (м, 1 Н), 6,93 (ушир.с, 1 Н), 4,58 и 4,53 (2 ушир.с, 1 Н), 4,14-4,08 (м, 1 Н), 3,81-3,50 (м, 3 Н), 2,64-2,45(м, 2 Н), 1,49 (с, 9 Н). Соединение 75. трет-Бутиловый эфир 3-гидрокси-3-(1 Н-пирроло[3,2-b]пиридин-2-ил)-3 гидроксипирролидин-1-карбоновой кислоты получали из соединения 73 (1,2 г, 2,7 ммоль), используя методику, описанную для синтеза указанного выше соединения 74. Соединение 75 (аморфное вещество, 0,42 г, 51%). 1 Н-ЯМР (400 МГц, CDCl3) выявлены поворотные изомеры (важные изомеры описаны):9,02 и 9,0 (2 с, 1 Н), 8,40-8,32 (м, 1 Н), 7,66-7,59 (м, 1 Н), 7,08-7,02(ущир.дд, J=8 Гц, 5 Гц, 1 Н), 6,41 и 6,33 (2 ушир.с, 1 Н). Соединение 77. 2-(2,5-Дигидро-1 Н-пиррол-3-ил)-1 Н-пирроло[3,2]пиридин получали из соединения 75, (0,42 г, 1,38 ммоль), используя методику, описанную для синтеза указанного выше соединения 76. Соединение 77 (аморфное вещество, 0,2 г, 78%). 1 Н-ЯМР (400 МГц, D6DMCO):11,4 (ушир.с, 1 Н),8,19 (ушир.д, J=5 Гц, 1 Н), 7,61 (ушир.д, J=8 Гц, 1 Н), 7,0 (дд, J=8 Гц, 5 Гц, 1 Н), 6,43-6,38 (м, 2 Н), 3,93-3,88(м, 2 Н), 3,77-3,72 (м, 2 Н). Указанное в заголовке соединение: 2-пирролидин-3-ил-1 Н-пирроло[3,2-b]пиридин (соединение 79),получали из соединения 77 (0,19 г, 1,02 ммоль), используя методику, описанную для синтеза указанного выше соединения 78. Соединение 79 (аморфное вещество, 0,16 г, 83%) подвергали взаимодействию с 1 экв. фумаровой кислоты в МеОН и концентрировали, что приводило к получению соли указанного в заголовке соединения (аморфное вещество, свободное основание/фумаровая кислота (1:1,5. 1 Н-ЯМР (400 МГц,D6DMCO):11,6 (ушир.с, 1 Н), 8,26 (ушир.д, J=5 Гц, 1 Н), 7,69 (ушир.д, J=8 Гц, 1 Н), 7,05 (ушир.дд, J=8 Гц, 5 Гц, 1 Н), 6,54 (с, 3 Н), 6,48 (ушир.с, 1 Н), 3,74-3,62 (м, 2 Н), 3,40-3,25 (м, 2 Н), 2,44-2,37 (м, 1 Н), 2,091,97 (м, 1 Н).(R)-2-Пирролидин-2-илметил-1 Н-пирроло[3,2-b]пиридин (соединение 81 А) и (S)-2-пирролидин-2 илметил-1 Н-пирроло[3,2-b]пиридин (соединение 81 В). Получение литийпроизводного (LDA) соединения 70 (1,42 г, 5,05 ммоль) и последующее взаимодействие с соединением 35 осуществляли, используя методику, описанную для синтеза (первая стадия)(S)-2-Пирролидин-2-илметил-1 Н-пирроло[3,2-b]пиридин (соединение 81 В) получали, применяя методику, описанную для соединения 81 А, при использовании сульфамидата 36. ([]D25 + 74 (с 1, толуол.(1,5 М в пентане) по каплям при -70 С в атмосфере N2. После добавления полученный в результате раствор перемешивали в течение 15 мин при -70 С. При этой температуре раствор соединения 83 (3,7 г, 14,3 ммоль) в 25 мл ТГФ добавляли по каплям (10 мин). Смесь перемешивали в течение 60 мин при -70 С. При этой температуре раствор соединения 35 (2,33 г, 14,3 ммоль) в 30 мл безводного ТГФ добавляли,смесь перемешивали в течение 30 мин при -70 С и затем повышали температуру до -20 С. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение еще 20 ч. Реакционную смесь концентрировали и остаток растворяли в 100 мл 1 н. HCl и 100 мл ТГФ. Смесь перемешивали при 70 С в течение 20 ч. Реакционную смесь концентрировали в вакууме. Полученный в результате остаток помещали в этилацетат, промывали 2 н. NaOH, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Очистка флэш-хроматографией (МеОН/триэтиламин (97/3 приводила к получению соединения 84 (твердое вещество, 2,83 г, 53%). Т.пл. 101-103 С. 1 Н-ЯМР (400 МГц, CDCl3):8,30 (д, J=9 Гц, 1 Н),7,71-7,65 (м, 2 Н), 7,57-7,50 (м, 1 Н), 7,45-7,35 (м, 2 Н), 6,65 (д, J=9 Гц,1 Н), 6,54 (с, 1 Н), 3,94 (с, 3 Н), 3,563,47 (м, 1 Н), 3,15-2,98 (м, 4 Н), 2,05-1,65 (м, 3 Н), 1,48-1,37 (м, 1 Н). Соединение 84 преобразовывали в указанное в заголовке соединение (85), применяя методику, использованную для синтеза соединения 39 А. Указанное в заголовке соединение: (R)-5-метокси-2 пирролидин-2-илметил-1 Н-пирроло[3,2-b]пиридин, получали в виде сиропа (1,18 г, 68%), []D25 -42 (с 1,CHCl3), которое подвергали взаимодействию с 1 эквивалентом фумаровой кислоты в МеОН и концентрировали, что приводило к получению указанного в заголовке соединения 95 (аморфное вещество)(R)-3-Пирролидин-2-илметил-1 Н-пирроло[3.2-b]пиридин (соединение 88). 4,6 г (25,8 ммоль) NBS добавляли к 3,05 г (25,8 ммоль) 4-азаиндола в 40 мл ДМФА (0 С). Смесь перемешивали в течение 30 мин при 0 С. МеОН добавляли и смесь фильтровали через SCX-2, с последующей флэш-хроматографией (этилацетат, затем МеОН), с получением 3-бром-1 Н-[3,2-b]пиридина (86) в виде твердого вещества. Т.пл. 241 С (4,52 г, 89%). 1H-ЯМР (400 МГц, D6DMCO):11,7 (ушир.с, 1 Н),8,39 (дд, J=5 Гц, 2 Гц, 1 Н), 7,85 (с, 1 Н), 7,82 (дд, J=8 Гц, 2 Гц, 1 Н), 7,19 (дд, J=8 Гц, 5 Гц, 1 Н). К раствору безводного ТГФ (75 мл), содержащему соединение 86 (2,28 г, 11,6 ммоль), добавляли 4,6 мл н-BuLi (2,5 М в гексане) по каплям при -78 С в атмосфере N2. После добавления полученный в результате раствор перемешивали в течение 45 мин при -78 С. При этой температуре раствор TIPS-C1 (2,73 мл) в 10 мл ТГФ добавляли по каплям. После добавления полученный в результате раствор перемешивали в течение 1 ч при -78 С. Затем смесь оставляли нагреваться до комнатной температуры. Реакционную смесь концентрировали и полученный в результате остаток помещали в этилацетат, промывали 5% раствором NaHCO3, сушили (Na2SO4), фильтровали и концентрировали в вакууме. Очистка флэшхроматографией (диэтиловый эфир/РЕ (1/1 приводила к получению 3-бром-1-три-трет-бутилсиланил 1 Н-пирроло[3,2-b]пиридина (87) в виде твердого вещества. Т.пл. 79-80 С (3,52 г, 86%). 1H-ЯМР (400 МГц, CDCl3):8,55 (дд, J=5 Гц, 2 Гц, 1 Н), 7,76 (дд, J=8 Гц, 2 Гц, 1 Н), 7,49 (с, 1 Н), 7,13 (дд, J=8 Гц, 5 Гц,1 Н), 1,73-1,60 (м, 3 Н), 1,15 и 1,13 (2 с, 18 Н). К раствору безводного ТГФ (100 мл), содержащему соединение 87 (3,42 г, 9,69 ммоль), добавляли 3,9 мл н-BuLi (2,5 М в гексане) по каплям (-78 С в атмосфере N2). После добавления полученный в результате раствор перемешивали в течение 60 мин при -78 С. При этой температуре добавляли по каплям(5 мин) раствор соединения 35 (1,58 г, 9,69 ммоль) в 10 мл ТГФ. После добавления соединения 35 температуру повышали до -20 С и полученный в результате раствор перемешивали в течение 2 ч. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение еще 2 ч. Реакционную смесь концентрировали и остаток растворяли в 40 мл 1 н. HCl, 40 мл EtOH и 40 мл ТГФ. Смесь перемешивали при 80 С в течение 18 ч. Реакционную смесь концентрировали в вакууме.- 27015700 МеОН (25 мл) добавляли и смесь концентрировали на 25 г SiO2. Последующая флэш-хроматография(51,6 ммоль) и 20% Pd(OH)2/C (140 мг) смешивали в МеОН (50 мл) и нагревали до температуры кипения с обратным холодильником в течение 2 ч. Смесь охлаждали, фильтровали, концентрировали и повторно растворяли в МеОН. Фильтрование через 25 г SCX-2 (МеОН, затем 1 н. NH3/MeOH) с последующей флэш-хроматографией (этилацетат/МеОН (9/1 приводило к получению соединения 90 (1,2 г,4,02 ммоль, 97%) аморфное вещество. 1 Н-ЯМР (400 МГц, CDCl3):11,1 (ушир.с, 1 Н), 9,1 (ушир.с, 1 Н),9,0 (ушир.с, 1 Н), 7,45-7,40 (м, 1 Н), 6,68-6,62 (м, 1 Н). 7 Н-Пирроло[2,3-d]пиримидин (1,16 г, 9,74 ммоль) растворяли в 100 мл безводного ТГФ в атмосфереN2. При 0 С добавляли 60%-ную дисперсию NaH 0,51 г в минеральном масле. Смесь перемешивали при температуре окружающей среды и добавляли 2,93 мл (9,8 ммоль) (2-хлорметоксиэтил)триметилсилана,растворенного в 15 мл безводного ТГФ. Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и затем концентрировали в вакууме. Добавляли этилацетат к смеси и органический слой промывали три раза насыщенным раствором NaHCO3, сушили (Na2SO4), фильтровали и концентрировали. Полученный в результате остаток очищали флэш-хроматографией (диэтиловый эфир) с получением 7-(2-триметилсиланилэтоксиметил-7 Н-пирроло[2,3-d]пиримидина (соединение 91), (аморфное вещество,0,84 г, 35%). 1 Н-ЯМР (400 МГц, CDCl3):9,1 (ушир.с, 1 Н), 9,0 (ушир.с, 1 Н), 7,43 (ушир.д, J=4 Гц, 1 Н),6,68 (ушир.д, J=4 Гц, 1 Н), 5,73 (с, 1 Н), 3,6 (т, J=8 Гц, 2 Н), 0,97 (т, J=8 Гц, 2 Н), 0,1 (с, 9 Н). В атмосфере азота 0,62 мл (3,7 ммоль) 2,2,6,6-тетраметилпиперидина добавляли к 20 мл безводного ТГФ (-78 С). н-BuLi 1,3 мл (2,5 М, 3,33 ммоль) добавляли и реакционную смесь перемешивали в течение 30 мин. Соединение 91 (0,83 г, 2,16 ммоль), растворенное в 10 мл ТГФ, добавляли и смесь перемешивали в течение 30 мин. Добавляли соединение 35 (557 мг, 2,16 ммоль), растворенное в 5 мл ТГФ. После добавления соединения 35 температуру повышали до -30 С и полученный в результате раствор перемешивали в течение 2 ч. Смесь оставляли нагреваться до температуры окружающей среды и перемешивали в течение еще 2 ч. Насыщенный раствор NH4Cl (5 мл) добавляли к реакционной смеси, затем 25 г SiO2 и смесь впоследствии концентрировали в вакууме. Полученный в результате остаток очищали флэшхроматографией (дихлорметан/МеОН/NH4OH) (90/10/1), получая соединение 93 (0,4 г, 44,9%). ЖХ/МС;Rt: 1,38 мин, ([М+Н]+=413). Соединение 93 (0,63 г, 1,53 ммоль), 11,4 мл тетрабутиламмонийфторида (1 М в ТГФ) и 25 мл ТГФ смешивали и смесь нагревали до температуры кипения с обратным холодильником (36 ч). Смесь охлаждали, добавляли 25 г SiO2 и затем концентрировали в вакууме. Полученный в результате остаток очищали флэш-хроматографией (дихлорметан/МеОН/NH4OH) (90/10/0,5), получая соединение 94 (0,35 г, 81%). 1H-ЯМР (400 МГц, CDCl3):11,3 (ушир.с, 1 Н), 8,80 (ушир.с, 1 Н), 8,65 (ушир.с, 1 Н), 6,2(ушир.с, 1 Н), 3,95-3,88 (м, 1 Н), 3,65-3,58 (м, 1 Н), 3,24-3,17 (м, 2 Н), 2,89-2,82 (м, 1 Н), 2,05-1,96 (м, 2 Н),1,74-1,6 (м, 2 Н). Соединение 94 (0,35 г, 1,24 ммоль), 10 мл 1 н. HCl и 10 мл EtOH смешивали и нагревали до температуры кипения с обратным холодильником в течение 18 ч. Реакционную смесь охлаждали и концентрировали в вакууме. Полученный в результате остаток помещали в этилацетат, промывали 2 н. раствором(МеОН/триэтиламин 97/3) приводила к получению указанного в заголовке соединения: (R)-6 пирролидин-2-илметил-7 Н-пирроло[2,3-d]пиримидина (95), аморфное вещество, 70 мг, 27,9%); ЖХ/МС; Rt: 0,68 мин, ([М+Н]+=203, которое подвергали взаимодействию с 1 экв. фумаровой кислоты в МеОН и концентрировали с получением указанного в заголовке соединения 95 (аморфное вещество)(свободное основание/фумаровая кислота (1:2. 1 Н-ЯМР (400 МГц, D6DMCO):12,8-11,8 (ушир.с, 1 Н),8,86 (с, 1 Н), 8,68 (с, 1 Н), 6,54 (с, 4 Н), 6,45 (с, 1 Н), 3,85-3,79 (м, 1 Н), 3,25-3,19 (м, 2 Н), 3,17-3,09 (м, 2 Н),2,10-2,03 (м, 1 Н), 2,00-1,92 (м, 1 Н, 1,90-1,82 (м, 1 Н), 1,69-1,62 (м, 1 Н). Структуры соединений изобретения, для которых синтезы описаны выше, приведены в таблице ниже.
МПК / Метки
МПК: C07D 487/04, A61P 25/16, C07D 519/00, A61K 31/40, A61P 25/28, A61P 25/30, C07D 471/04, A61K 31/445
Метки: ингибирования, никотиновому, производные, ацетилхолиновому, сочетанием, частичного, обратного, дофамина, захвата, азаиндольные, рецептору, агонизма
Код ссылки
<a href="https://eas.patents.su/30-15700-azaindolnye-proizvodnye-s-sochetaniem-chastichnogo-agonizma-k-nikotinovomu-acetilholinovomu-receptoru-i-ingibirovaniya-obratnogo-zahvata-dofamina.html" rel="bookmark" title="База патентов Евразийского Союза">Азаиндольные производные с сочетанием частичного агонизма к никотиновому ацетилхолиновому рецептору и ингибирования обратного захвата дофамина</a>
Производные аминоиндана в качестве ингибиторов обратного захвата серотонина и захвата норэпинефрина

Номер патента: 7655
Опубликовано: 29.12.2006
Авторы: Келер Ян, Бегесе Клаус Петер, Пюшль Аск, Брегнедаль Петер
МПК: A61K 31/135, A61K 31/357, A61P 25/22...
Метки: норэпинефрина, аминоиндана, обратного, качестве, серотонина, захвата, ингибиторов, производные
Формула / Реферат:
1. Соединение аминоиндана, имеющее формулу I где X означает -O-, -S- или -CR4R5-; Y означает -CR6R7-, -CR6R7-CR8R9- или -CR6=CR7-; или X и Y вместе образуют группу -CR4=CR5- или -CR4=CR5-CR6R7-; и U означает -O-, -S- или -CR10R11 или X означает -O-, -S- или -CR4R5-; и Y и U вместе образуют группу -CR6=CR7-, -CR6=CR7-CR10R11- или -CR6R7-CR10=CR11-; или X, и Y, и U вместе образуют -CR4=CR5-CR6=CR7-; R1 и R2 независимо выбраны из водорода,...
Производные биариловых эфиров, полезные в качестве ингибиторов обратного захвата моноамина

Номер патента: 5671
Опубликовано: 28.04.2005
Авторы: Хауард Гарри Ралф Младший, Адам Мейвис Дайан
МПК: A61K 31/13, C07C 217/58, A61P 25/24...
Метки: производные, биариловых, полезные, захвата, ингибиторов, моноамина, обратного, качестве, эфиров
Формула / Реферат:
1. Соединение формулы где n и m независимо выбраны из единицы и двух; R1 и R2 независимо выбраны из водорода и (C1-C4)алкила; R3 и R4 независимо выбраны из водорода и (C1-C4)алкила; каждый X независимо выбран из фенила и 5- или 6-членного гетероцикла, содержащего от 1 до 3 гетероатомов, выбранных из N, O и S, где каждый X может быть дополнительно замещен водородом, галогено, (C1-C4)алкилом, оксо, амино и группировкой NR5(C=O)(C1-C4)алкил, где...
Производные n-пирролидин-3-ил-амида в качестве ингибиторов обратного захвата серотонина и норадреналина

Номер патента: 9881
Опубликовано: 28.04.2008
Авторы: Фрэй Майкл Джонатан, Уитлок Гэвин Алистер, Браун Алан Дэниел, Лэнсделл Марк Айан, Эндрюс Марк Дейвид, Уэйкенхат Флориан, Стоби Алан, Фиш Пол Винсент
МПК: A61P 25/24, A61P 13/00, A61K 31/40...
Метки: n-пирролидин-3-ил-амида, захвата, обратного, производные, серотонина, ингибиторов, норадреналина, качестве
Формула / Реферат:
1. Соединение формулы (I) и его фармацевтически и/или ветеринарно приемлемые соль, сольват, эфир или амид либо соль или сольват такого эфира или амида; где R2 представляет собой арил или гетероарил, каждый из которых возможно замещен по меньшей мере одним заместителем, независимо выбранным из С1-8алкила, С1-8алкокси, ОН, галогено, CF3, OCF3, SCF3, гидрокси-С1-6алкила, С1-4алкокси-С1-6алкила и С1-4алкил-S-С1-4алкила; R3 представляет собой...
N-[(3s)пирролидин-3-ил]бензамидные производные в качестве ингибиторов обратного захвата моноаминов

Номер патента: 11217
Опубликовано: 27.02.2009
Авторы: Стоби Алан, Фиш Пол Винсент, Райкманс Томас, Уитлок Гэвин Алистер, Уэйкенхат Флориан
МПК: C07D 207/14, A61K 31/40
Метки: захвата, качестве, ингибиторов, производные, моноаминов, n-[(3s)пирролидин-3-ил]бензамидные, обратного
Формула / Реферат:
1. Соединение формулы (I) и его фармацевтически и/или ветеринарно приемлемые производные, где каждый из R1, R2, R3 и R20 независимо представляет собой Н, Cl, Br, F, I, CF3, OCF3, Me или Et; R4 представляет собой het или C3-7циклоалкил, возможно замещенный С1-4алкилом, C1-4алкокси, алкоксиалкилом, содержащим 2-4 атома углерода, или группой -S-(C1-4алкил); а равен 0 или 1; и het представляет собой неароматический 4-, 5- или 6-членный гетероцикл,...
Производные фенил-гетероциклильных эфиров в качестве ингибиторов обратного захвата серотонина

Номер патента: 8936
Опубликовано: 26.10.2007
Авторы: Хепворт Дейвид, Адам Мейвис Дайан, Хауард Гарри Ральф Мл., Эндрюс Марк Дейвид, Джаймер Джеффри Эдвард, Стоуби Алан, Миддлтон Доналд Стюарт
МПК: A61K 31/44, A61P 25/24, C07D 213/65...
Метки: обратного, производные, серотонина, эфиров, фенил-гетероциклильных, ингибиторов, качестве, захвата
Формула / Реферат:
1. Соединение общей формулы (I), его фармацевтически приемлемые соли, сольваты или полиморфы где L и U, которые могут быть одинаковыми или разными, представляют собой -N- или -С(Н)-; M представляет собой -N-, -N+(-O-)- или -C(R4)-; Q представляет собой -N- или -C(R4)-; кольцо А содержит 1 или 2 атома азота, в том случае, когда M представляет собой -N+(O-)-, тогда кольцо А не содержит других атомов азота; R1 и R2, которые могут быть одинаковыми...