N-[(3s)пирролидин-3-ил]бензамидные производные в качестве ингибиторов обратного захвата моноаминов
Номер патента: 11217
Опубликовано: 27.02.2009
Авторы: Райкманс Томас, Уэйкенхат Флориан, Уитлок Гэвин Алистер, Фиш Пол Винсент, Стоби Алан







Формула / Реферат
1. Соединение формулы (I)

и его фармацевтически и/или ветеринарно приемлемые производные, где
каждый из R1, R2, R3 и R20 независимо представляет собой Н, Cl, Br, F, I, CF3, OCF3, Me или Et;
R4 представляет собой het или C3-7циклоалкил, возможно замещенный С1-4алкилом, C1-4алкокси, алкоксиалкилом, содержащим 2-4 атома углерода, или группой -S-(C1-4алкил);
а равен 0 или 1; и
het представляет собой неароматический 4-, 5- или 6-членный гетероцикл, содержащий по меньшей мере один гетероатом N, О или S, возможно конденсированный с 5- или 6-членной карбоциклической группой либо вторым 4-, 5- или 6-членным гетероциклом, содержащим по меньшей мере один гетероатом N, О или S, при этом группа het возможно замещена по меньшей мере одним заместителем, независимо выбранным из С1-8алкила, С1-8алкокси, ОН, галогено, CF3, OCF3, SCF3, гидрокси-C1-6алкила, C1-4алкокси-C1-6алкила и C1-4алкил-S-C1-4алкила;
при условии, что по меньшей мере один из R1, R2 и R3 отличен от Н.
2. Соединение по п.1, где R1 представляет собой Cl, Br, F, I, CF3, Me или Et; и каждый из R2 и R3 независимо представляет собой Н, Cl, Br, F, I, CF3, Me или Et.
3. Соединение по п.2, где каждый из R1 и R2 независимо представляет собой Cl, Br, F, I, CF3, Me или Et; и R3 представляет собой Н, Cl, Br, F, I, CF3, Me или Et.
4. Соединение по п.1 или 2, где R1 представляет собой Cl, Me или CF3; R2 представляет собой Н, Cl или F; и R3 представляет собой Н, Cl или F.
5. Соединение по любому из пп.1-4, где R4 представляет собой C3-7циклоалкил.
6. Соединение по любому из пп.1-5, где а равен 0.
7. Соединение по п.1, представляющее собой
2,3-дихлор-N-циклопентил-N-[(3S)пирролидин-3-ил]бензамид;
2,3-дихлор-N-циклопентил-4-фтор-N-[(3S)пирролидин-3-ил]бензамид;
3-хлор-N-циклопентил-2-метил-N-[(3S)пирролидин-3-ил]бензамид;
N-циклопентил-3-фтор-2-метил-N-[(3S)пирролидин-3-ил]бензамид;
2-хлор-N-циклопентил-3-фтор-N-[(3S)пирролидин-3-ил]бензамид;
2,3-дихлор-N-циклогексил-N-[(3S)пирролидин-3-ил]бензамид;
2-хлор-N-циклогексил-3-фтор-N-[(3S)пирролидин-3-ил]бензамид;
N-циклогексил-3-фтор-2-метил-N-[(3S)пирролидин-3-ил]бензамид;
2,3-дихлор-N-циклобутил-N-[(3S)пирролидин-3-ил]бензамид;
N-циклобутилметил-2,3-дихлор-N-[(3S)пирролидин-3-ил]бензамид;
2,3-дихлор-N-(циклопропилметил)-N-[(3S)пирролидин-3-ил]бензамид;
2,3-дихлор-N-[(3S)пирролидин-3-ил]-N-тетрагидро-2H-пиран-4-илбензамид;
2-хлор-N-циклопентил-N-[(3S)пирролидин-3-ил]бензамид;
2-хлор-N-циклогексил-N-[(3S)пирролидин-3-ил]бензамид;
2-хлор-N-циклогептил-N-[(3S)пирролидин-3-ил]бензамид;
N-циклогептил-N-[(3S)пирролидин-3-ил]-2-(трифторметил)бензамид;
N-циклогексил-N-[(3S)пирролидин-3-ил]-2-(трифторметил)бензамид;
N-циклопентил-N-(3S)пирролидин-3-ил]-2-(трифторметил)бензамид;
2,3-дихлор-N-[(1-метилциклопропил)метил]-N-[(3S)пирролидин-3-ил]бензамид;
3-хлор-2-метил-N-[(1-метилциклопропил)метил]-N-[(3S)пирролидин-3-ил]бензамид;
N-(циклобутилметил)-N-[(3S)пирролидин-3-ил]-2-(трифторметил)бензамид; или
их фармацевтически и/или ветеринарно приемлемые производные.
8. Соединение по п.7, представляющее собой 2,3-дихлор-N-циклопентил-N-[(3S)пирролидин-3-ил]бензамид или его фармацевтически и/или ветеринарно приемлемые производные.
9. Фармацевтическая композиция, содержащая соединение по любому из пп.1-8 и фармацевтически приемлемый адъювант, разбавитель или носитель.
10. Применение соединения по любому из пп.1-8 в качестве лекарственного средства.
11. Применение соединения по любому из пп.1-8 в изготовлении лекарственного средства для лечения расстройства, в которое вовлечено регулирование функции транспортера моноаминов у млекопитающих.
12. Применение соединения по любому из пп.1-8 в изготовлении лекарственного средства для лечения расстройства, в которое вовлечено регулирование серотонина или норадреналина у млекопитающих.
13. Применение по п.12, где вовлечено регулирование серотонина и норадреналина.
14. Применение соединения по любому из пп.1-8 в изготовлении лекарственного средства для лечения уринарных расстройств, депрессии, боли, преждевременной эякуляции, синдрома дефицита внимания с гиперактивностью (ADHD) или фибромиалгии у млекопитающих.
15. Применение соединения по п.14 для лечения недержания мочи, такого как истинное недержание при напряжении (GSI) или недержание мочи при напряжении (SUI), у млекопитающих.
16. Способ лечения расстройства, в которое вовлечено регулирование функции транспортера моноаминов, включающий введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по любому из пп.1-8.
17. Способ лечения расстройства, в которое вовлечено регулирование серотонина или норадреналина, включающий введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по любому из пп.1-8.
18. Способ по п.17, где вовлечено регулирование серотонина и норадреналина.
19. Способ лечения уринарных расстройств, депрессии, боли, преждевременной эякуляции, ADHD или фибромиалгии, включающий введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по любому из пп.1-8.
20. Способ по п.19, где уринарное расстройство представляет собой недержание мочи, такое как GSI или SUI.
21. Способ получения соединения по любому из пп.1-8, включающий взаимодействие соединения формулы (IX)

где R4 и а являются такими, как определено в любом из пп.1-8, и PG представляет собой защитную группу, с кислотой или ацилгалогенидом формулы (II)

где X представляет собой ОН или галогено, и где R1, R2, R3 и R20 являются такими, как определено в любом из пп.1-8, и удаление защиты.
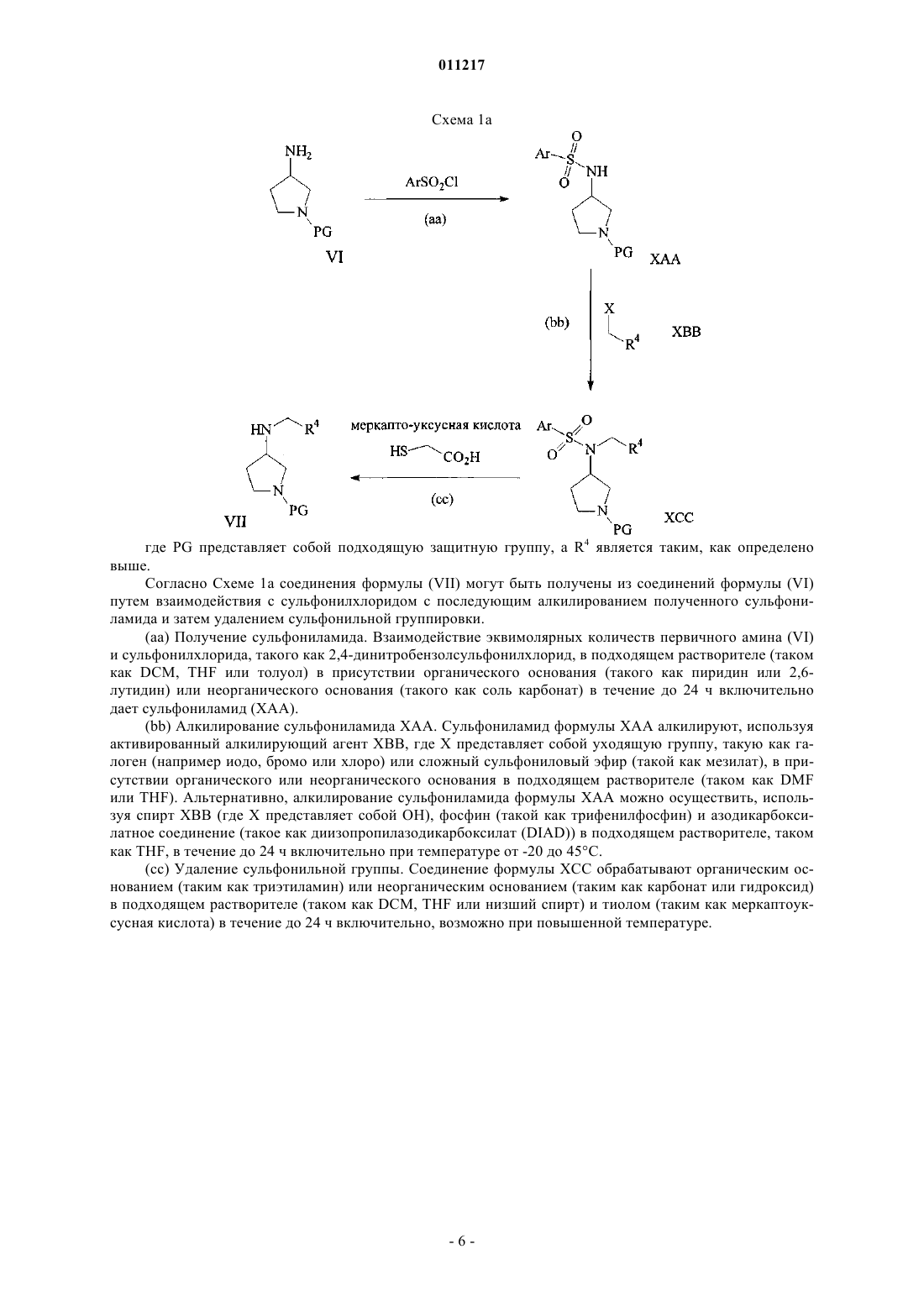
Текст
011217 Данное изобретение относится к новым амидным соединениям, которые ингибируют обратный захват моноаминов, к способам их получения, к фармацевтическим композициям, содержащим их, и к их применению в медицине. Соединения по изобретению проявляют активность ингибиторов обратного захвата как серотонина,так и норадреналина и, следовательно, находят применение в различных терапевтических областях. Например, соединения по изобретению находят применение в лечении расстройств, в которые вовлечено регулирование функции транспортеров моноаминов; более конкретно, расстройств, в которые вовлечено ингибирование обратного захвата серотонина или норадреналина. Кроме того, соединения по изобретению находят применение в лечении расстройств, в которые вовлечено ингибирование как серотонина,так и норадреналина, таких как недержание мочи. В дополнение, соединения по изобретению находят применение в лечении расстройств, в которых может быть желательно предпочтительное ингибирование обратного захвата одного из норадреналина или серотонина по отношению к другому, таких как боль,фибромиалгия, синдром дефицита внимания с гиперактивностью (ADHD) и депрессия. Согласно первому аспекту изобретения предложено соединение формулы (I) и его фармацевтически и/или ветеринарно приемлемые производные, где: каждый из R1, R2, R3 и R20 независимо представляет собой Н, Cl, Br, F, I, CF3, OCF3, Me или Et;het представляет собой неароматический 4-, 5- или 6-членный гетероцикл, содержащий по меньшей мере один гетероатом N, О или S, возможно конденсированный с 5- или 6-членной карбоциклической группой либо вторым 4-, 5- или 6-членным гетероциклом, содержащим по меньшей мере один гетероатом N, О или S, при этом группа het возможно замещена по меньшей мере одним заместителем, независимо выбранным из С 1-8 алкила, С 1-8 алкокси, ОН, галогено, CF3, OCF3, SCF3, гидрокси-C1-6 алкила, C14 алкокси-C1-6 алкила и C1-4 алкил-S-С 1-4 алкила; при условии, что по меньшей мере один из R1, R2 и R3 отличен от Н. В одном воплощении изобретения R1 представляет собой Cl, Br, F, I, CF3, Me или Et; и R2 и R3 каждый независимо представляет собой Н, Cl, Br, F, I, CF3, Me или Et. В дополнительном воплощении R1 иR2 каждый независимо представляет собой Cl, Br, F, I, CF3, Me или Et, и R3 представляет собой Н, Cl, Br,F, I, CF3, Me или Et. В еще одном дополнительном воплощении R1 представляет собой Cl, Me или CF3; R2 представляет собой Н, Сl или F; и R3 представляет собой Н, Cl или F. Согласно дополнительному воплощению изобретения R2 и R20 отличны от Н. В таком воплощении 2R и R20 каждый может независимо представлять собой Cl, F, CF3, Me или Et. Согласно еще одному дополнительному воплощению изобретения R1, R2 и R20 отличны от Н. В таком воплощении R1, R2 и R20 каждый может независимо представлять собой Cl, F, CF3, Me или Et. Согласно еще одному дополнительному воплощению изобретения R1, R3 и R20 отличны от Н. В таком воплощении R1, R3 и R20 каждый может независимо представлять собой Cl, F, CF3, Me или Et. Согласно любому описанному выше воплощению изобретения R4 может представлять собой C3-7 циклоалкил, возможно замещенный С 1-4 алкилом, C1-4 алкокси, алкоксиалкилом, содержащим 2-4 атома углерода, или группой -S-(C1-4 алкил). Согласно дополнительному воплощению изобретения R4 может представлять собой C3-7 циклоалкил,возможно замещенный Me или Et. Согласно любому описанному выше воплощению изобретения а может быть равен 0. Согласно любому описанному выше воплощению изобретения группа het может быть замещена одним, двумя или тремя заместителями, независимо выбранным из галогено, ОН, С 1-4 алкила и CF3. В дополнительном воплощении группа het соединения, как оно определено выше со ссылкой на первый аспект изобретения и любое из конкретных воплощений, может быть не замещена. В дополнительном воплощении изобретения предложено соединение, выбранное из: 2,3-дихлор-N-циклопентил-N- [(3S)-пирролидин-3-ил]бензамида; 2,3-дихлор-N-циклопентил-4-фтор-N-[(3S)-пирролидин-3-ил]бензамида; 3 -хлор-N-циклопентил-2-метил-N-[(3S)-пирролидин-3-ил]бензамида;N-(циклобутилметил)-N-[(3S)-пирролидин-3-ил]-2-(трифторметил)бензамида; или их фармацевтически и/или ветеринарно приемлемых производных. Под фармацевтически и/или ветеринарно приемлемым производным подразумеваются любые фармацевтически или ветеринарно приемлемые соль, сольват, эфир или амид соединения формулы (I), либо соль или сольват такого эфира или амида, либо любое другое соединение, которое при введении реципиенту способно давать (прямо или опосредованно) соединение формулы (I) либо его активный метаболит или радикал. В случае фармацевтического или ветеринарного применения соли, упомянутые выше, будут представлять собой фармацевтически или ветеринарно приемлемые соли, однако другие соли могут найти применение, например, в получении соединений формулы (I) и их фармацевтически или ветеринарно приемлемых солей. Вышеупомянутые фармацевтически или ветеринарно приемлемые соли включают соли присоединения кислоты и соли оснований. Подходящие соли присоединения кислоты получают из кислот, образующих нетоксичные соли. Примеры включают соли: ацетат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат,камзилат, цитрат, гемицитрат, эдизилат, гемиэдизилат, эзилат, фумарат, глюцептат, глюконат, глюкуронат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидроиодид/иодид, изетионат, лактат, малат,малеат, малонат, мезилат, метилсульфат, 2-напсилат, никотинат, нитрат, оротат, памоат, фосфат/гидрофосфат/дигидрофосфат, сахарат, стеарат, сукцинат, тартрат и тозилат. Подходящие соли оснований получают из оснований, образующих нетоксичные соли. Примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка. Обзор подходящих солей дан в Stahl and Wermuth, "Handbook of Pharmaceutical Salts: Properties, Selection, and Use" (Wiley-VCH, Weinheim, Germany, 2002). Фармацевтически приемлемую соль соединения формулы (I) можно легко получить, смешивая вместе растворы соединения и желаемой кислоты или основания, как это целесообразно. Соль можно осадить из раствора и собрать фильтрацией или ее можно получить выпариванием растворителя. Степень ионизации в соли может меняться от полностью ионизированной до почти неионизированной. Фармацевтически приемлемые сольваты по изобретению включают гидраты и сольваты соединения формулы (I). Также в объем изобретения входят комплексы, такие как клатраты, комплексы включения лекарственное средство - молекула-хозяин, где, в противоположность вышеупомянутым сольватам, лекарственное средство и молекула-хозяин присутствуют в стехиометрических или нестехиометрических количествах. Также в данное изобретение включены комплексы фармацевтического лекарственного средства,содержащие два или более органических и/или неорганических компонента, которые могут присутствовать в стехиометрических или нестехиометрических количествах. Полученные комплексы могут быть ионизированы, частично ионизированы или неионизированы. Обзор таких комплексов дан в Haleblian, J. Pharm. Sci., 64(8), 1269-1288 (August 1975). Соединения формулы (I) могут быть модифицированы с получением их фармацевтически или ветеринарно приемлемых производных по любым функциональным группам в соединениях. Примеры таких производных описаны в Drugs of Today, Volume 19, Number 9, 1983, pp. 499-538; Topics in Chemistry,Chapter 31, pp. 306-316; и в Н. Bundgaard, "Design of Prodrugs", Elsevier, 1985, Chapter 1 (сущность этих документов включена в данное описание посредством ссылки) и включают сложные эфиры, карбонатные эфиры, полуэфиры, фосфатные эфиры, нитроэфиры, сульфатные эфиры, сульфоксиды, амиды, сульфонамиды, карбаматы, азо-соединения, фосфамиды, гликозиды, простые эфиры, ацетали и кетали. Специалисту в данной области очевидно, что некоторые группировки, известные в данной области техники как про-группировки, например, как описано Н. Bundgaard в "Design of Prodrugs" (см. выше),могут быть заменены соответствующими функциональными группами, когда такие функциональные группы присутствуют в соединениях по изобретению.-2 011217 Соединения формулы (I) содержат один или более чем один хиральный центр благодаря асимметрическому атому углерода пирролидин-3-ильной группировки и дополнительным асимметрическим атомам углерода, определяемым конкретными значениями R4. Несмотря на то, что стереохимия по положению 3 является фиксированной, могут существовать любые другие хиральные центры в любой возможной стереоизомерной форме. Следует понимать, что настоящее изобретение охватывает все изомеры соединений по изобретению, включая все геометрические, таутомерные и оптические формы (за исключением хирального центра по положению 3 пирролидинильной группировки) и их смеси (например таутомерные или рацемические смеси). Соединения по изобретению могут существовать в одной или более таутомерных формах. Все таутомеры и их смеси включены в объем настоящего изобретения. Например, пункт формулы изобретения,относящийся к 2-гидроксипиридинилу, также будет охватывать его таутомерную форму -пиридонил. Следует понимать, что настоящее изобретение включает соединения формулы (I), меченые радиоактивным изотопом. Соединения формулы (I) и их фармацевтически и ветеринарно приемлемые производные также могут быть способны существовать в более чем одной кристаллической форме; свойство, известное как полиморфизм. Все такие полиморфные формы (полиморфы) входят в объем данного изобретения. В общем случае полиморфизм может иметь место как ответ на изменения температуры или давления, либо их обоих, а также может быть результатом отклонений в процессе кристаллизации. Полиморфы можно различить по различным физическим характеристикам, и обычно для различения полиморфов используют рентгеновские дифрактограммы, поведение при растворении и точку плавления соединения. Если не указано иное, любая алкильная группа может быть прямой или разветвленной и состоять из 1-8 атомов углерода, например 1-6 атомов углерода или 1-4 атомов углерода, как например группа метил,этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил. Когда алкильная группа содержит более чем один атом углерода, она может быть ненасыщенной. Таким образом, термин С 1-6 алкил включает С 1-6 алкенил и C2-6 алкинил. Аналогично, термин C1-8 алкил включает C2-8 алкенил и C2-8 алкинил,и термин C1-4 алкил включает C2-4 алкенил и С 2-4 алкинил. Термин галоген используют для обозначения фтора, хлора, брома или иода. Если не указано иное, термин het включает любой неароматический, насыщенный или ненасыщенный 4-, 5- или 6-членный гетероцикл, который содержит гетероатомы, выбранные из N, О и S, в количестве до 4 включительно. Примеры таких гетероциклических групп включают фурил, тиенил, пирролил, пирролинил, пирролидинил, имидазолил, диоксоланил, оксазолил, тиазолил, имидазолил, имидазолинил, имидазолидинил, пиразолил, пиразолинил, пиразолидинил, изоксазолил, изотиазолил, оксадиазолил, триазолил, тиадиазолил, пиранил, тетрагидропиранил, пиридил, пиперидинил, диоксанил, морфолино, дитианил, тиоморфолино, пиридазинил, пиримидинил, пиразинил, пиперазинил, сульфоланил, тетразолил, триазинил, азепинил, оксазепинил, тиазепинил, диазепинил и тиазолинил. В дополнение, термин гетероцикл включает конденсированные гетероциклические группы, например бензимидазолил, бензоксазолил, имидазопиридинил, бензоксазинил, бензтиазинил, оксазолопиридинил, бензофуранил, хинолинил, хиназолинил, хиноксалинил, дигидрохиназолинил, бензтиазолил, фталимидо, бенздиазепинил,индолил и изоиндолил. Термины het, гетероциклил и гетероциклический следует истолковывать одинаково. Во избежание неясности, если не указано иное, термин замещенный означает замещенный одной или более чем одной определенной группой. В том случае, когда группы могут быть выбраны из ряда альтернативных групп, эти выбранные группы могут быть одинаковыми или разными. Кроме того, термин независимо означает, что когда из ряда возможных заместителей выбран более чем один заместитель, эти заместители могут быть одинаковыми или разными. Далее соединения формулы (I) и их фармацевтически и ветеринарно приемлемые производные, меченые радиоактивным изотопом аналоги указанного выше, изомеры указанного выше и полиморфы указанного выше упоминаются как соединения по изобретению. В одном воплощении изобретения соединения по изобретению представляют собой фармацевтически и ветеринарно приемлемые производные соединений формулы (I), такие как фармацевтически или ветеринарно приемлемые соли или сольваты соединений формулы (I) (например фармацевтически или ветеринарно приемлемые соли соединений формулы (I. В еще одном воплощении изобретения предложено соединение по изобретению, представляющее собой ингибитор обратного захвата моноаминов серотонина и/или норадреналина, имеющее значенияSRI или NRI Ki (константы ингибирования обратного захвата серотонина или норадреналина, соответственно) 200 нМ или менее. В дополнительном воплощении соединение имеет значения SRI и/или NRI Ki 100 нМ или менее. В еще одном воплощении соединение имеет значения SRI или NRI Ki 50 нМ или менее. В еще одном воплощении соединение имеет значения SRI и NRI Ki 50 нМ или менее. В еще одном воплощении соединение имеет значения SRI и NRI Ki 25 нМ или менее. Согласно схеме 1 соединения формулы (V) (то есть соединение формулы (I), где а равен 1, могут быть получены из соединений формулы (VI) путем взаимодействия с альдегидом R4CHO с последующим-3 011217 взаимодействием с кислотой или галогенангидридом, как показано (то есть, где X представляет собой ОН или галогено), и удалением защиты. Схема 1 На приведенной выше схеме R1, R2, R3, R4 и R20 являются такими, как определено выше, а равен 1, иPG представляет собой подходящую защитную группу.(а) Восстановительное аминирование Взаимодействие 1 (первичного) амина (VI) с альдегидом с образованием 2 (вторичного) амина(VII) представляет собой реакцию восстановительного аминирования, в которой после дегидрирования амина и альдегида следует восстановление образовавшегося имина металлогидридным реагентом или гидрированием в подходящем растворителе при комнатной температуре. В данной реакции эквимолярные количества амина и альдегида обычно обрабатывают либо триацетоксиборгидридом натрия (STAB), NaCN(BH)3, либо NaBH4 в подходящем растворителе (например вDCM, THF) при комнатной температуре в течение 1-24 ч. Альтернативно, избыток восстанавливающего агента (например NaBH4, LiAlH4, STAB) в подходящем растворителе (например THF, MeOH, EtOH) добавляют после того, как амин и альдегид перемешивали в течение 1-18 ч, возможно в присутствии осушителя (например молекулярного сита) или с удалением воды, используя аппарат Дина-Старка, с подходящим растворителем (например толуолом, ксилолом). Дополнительная альтернатива включает каталитическое гидрирование в присутствии палладиевого или никелевого катализатора (например Pd/C,Raney Ni) в атмосфере H2, возможно при повышенных температуре и давлении, в подходящем растворителе (например EtOH). Более конкретный пример восстановительного аминирования включает обработку альдегида амином в присутствии либо 10% Pd/C, возможно в присутствии триэтиламина, в этаноле при давлении водорода приблизительно 415 кПа (приблизительно 60 фунт/кв.дюйм) при комнатной температуре в течение 18 ч, либо избытком боргидрида натрия в метаноле при комнатной температуре в течение 6 ч. Специалистам в данной области техники очевидно, что вместо альдегида на стадии восстановительного аминирования можно использовать в подходящих условиях кетон или другой карбонилсодержащий реагент.(b) Образование амида Образование пептидной связи между кислотой или галогенангидридом и амином (VII) может быть осуществлено путем использования либо:(1) ацилгалогенида и амина (VII) с избытком акцептора кислоты в подходящем растворителе, либо(2) кислоты, возможно с традиционным агентом сочетания, и амина (VII), возможно в присутствии катализатора с избытком акцептора кислоты в подходящем растворителе. Примерами такого взаимодействия являются следующие:(VII), возможно с избытком 3 (третичного) амина, такого как Et3N, основание Хюнига или NMM, вDCM, толуоле или диоксане, возможно при повышенной температуре в течение 1-24 ч;(VII) и избытком NMM, Et3N, основания Хюнига в THF, DCM или EtOAc при комнатной температуре в течение 4-48 ч; или(3) кислоту и РУВОР/РуВгОР/реагент Мукаямы подвергают взаимодействию с избытком амина(VII) и избытком NMM, Et3N, основания Хюнига в THF, толуоле, DCM или EtOAc при комнатной температуре в течение 4-24 ч. Когда галогенангидрид является хлорангидридом (то есть Х=Cl), он может быть получен in situ согласно стандартной методологии и затем приведен во взаимодействие с амином (VII) и триэтиламином в дихлорметане при 70 С в течение 90 мин.(с) Удаление защиты Когда PG является подходящей защитной группой амина, предпочтительно ВОС, трифторацетатом или бензилом, удаление PG из (VIII) с образованием незащищенного амина (V) осуществляют способом,избирательным по отношению к данной защитной группе, как подробно изложено в книге TW Greeneand PGM Wuts, "Protective Groups in Organic Synthesis", 3rd edition, John Wiley and Sons, Inc., 1999, которая включена в данное описание посредством ссылки. Примерами таких реакций удаления защиты являются следующие: Когда PG представляет собой ВОС, удаление защиты включает обработку (VIII) избытком: сильной кислоты (например HCl, TFA) при комнатной температуре в подходящем растворителе (например DCM,EtOAc, диоксане). Когда PG представляет собой трифторацетат, удаление защиты включает обработку (VIII) основанием (например K2CO3, Na2CO3, NH3, Ва(ОН)2) в спиртовом растворителе (например МеОН, EtOH) возможно с водой и возможно при повышенной температуре. Когда PG представляет собой Bz, удаление защиты включает либо гидрирование с переносом в присутствии катализатора гидрирования на основе переходного металла или соли переходного металла(например Pd/C, Pd(OH)2) в присутствии донора водорода (например NH4+HCO2-) в полярном растворителе (например тетрагидрофуране, этаноле, метаноле), возможно при повышенных температуре и/или давлении, либо каталитическое гидрирование в присутствии палладиевого или никелевого катализатора(например Pd/C, Raney Ni) в атмосфере H2, возможно при повышенных температуре и давлении, в подходящем растворителе. Более конкретно: когда PG представляет собой ВОС, удаление защиты включает обработку либо избытком 4 М соляной кислоты в диоксане в течение 18 ч при комнатной температуре, либо TFA в DCM в течение 4,5 ч при комнатной температуре (КТ); когда PG представляет собой трифторацетат, удаление защиты включает обработку K2CO3 в смеси метанол:вода (от 5:1 до 10:1) при комнатной температуре в течение 18 ч; когда PG представляет собой Bz, удаление защиты включает обработку с использованиемNH4+HCO2- И 10% Pd/C в этаноле в условиях спокойного кипячения с обратным холодильником в течение 6-20 ч. Альтернативные способы получения соединения (VII), являющегося вторичным амином, из соединения (VI), являющегося первичным амином, описаны ниже на схемах 1 а и 1b. где PG представляет собой подходящую защитную группу, a R4 является таким, как определено выше. Согласно Схеме 1 а соединения формулы (VII) могут быть получены из соединений формулы (VI) путем взаимодействия с сульфонилхлоридом с последующим алкилированием полученного сульфониламида и затем удалением сульфонильной группировки.(аа) Получение сульфониламида. Взаимодействие эквимолярных количеств первичного амина (VI) и сульфонилхлорида, такого как 2,4-динитробензолсульфонилхлорид, в подходящем растворителе (таком как DCM, THF или толуол) в присутствии органического основания (такого как пиридин или 2,6 лутидин) или неорганического основания (такого как соль карбонат) в течение до 24 ч включительно дает сульфониламид (ХАА).(bb) Алкилирование сульфониламида ХАА. Сульфониламид формулы ХАА алкилируют, используя активированный алкилирующий агент ХВВ, где X представляет собой уходящую группу, такую как галоген (например иодо, бромо или хлоро) или сложный сульфониловый эфир (такой как мезилат), в присутствии органического или неорганического основания в подходящем растворителе (таком как DMF или THF). Альтернативно, алкилирование сульфониламида формулы ХАА можно осуществить, используя спирт ХВВ (где X представляет собой ОН), фосфин (такой как трифенилфосфин) и азодикарбоксилатное соединение (такое как диизопропилазодикарбоксилат (DIAD в подходящем растворителе, таком как THF, в течение до 24 ч включительно при температуре от -20 до 45 С.(cc) Удаление сульфонильной группы. Соединение формулы ХСС обрабатывают органическим основанием (таким как триэтиламин) или неорганическим основанием (таким как карбонат или гидроксид) в подходящем растворителе (таком как DCM, THF или низший спирт) и тиолом (таким как меркаптоуксусная кислота) в течение до 24 ч включительно, возможно при повышенной температуре. На приведенной выше схеме R4 является таким, как определено выше, и PG представляет собой защитную группу. Ацилирование-восстановление Согласно схеме 1b соединения формулы (VII) могут быть получены из 1 (первичного) амина формулы (VI) путем взаимодействия с карбоновой кислотой или галогенангидридом AAA (возможно полученного in situ), R4COX (где X представляет собой ОН или галогено) с последующим взаимодействием с восстанавливающим агентом, таким как боран.(х) Образование амида Образование пептидной связи между кислотой или галогенангидридом и 1 (первичным) амином(VI) может быть осуществлено путем использования либо:(1) ацилгалогенида и амина (VI) с избытком акцептора кислоты в подходящем растворителе, либо(2) кислоты, возможно с традиционным агентом сочетания, и амина (VI), возможно в присутствии катализатора с избытком акцептора кислоты в подходящем растворителе. Примерами такого взаимодействия являются следующие:(VI), возможно с избытком 3 (третичного) амина, такого как Et3N, основание Хюнига или NMM, в DCM или диоксане, возможно при повышенной температуре в течение 1-24 ч;(VI) и избытком NMM, Et3N, основания Хюнига в THF, DCM или EtOAc при комнатной температуре в течение 4-48 ч; или(3) кислоту и циклический ангидрид сложного 1-пропил-фосфониевого эфира/PYBOP/PyBrOP/реагент Мукаямы подвергают взаимодействию с избытком амина (VI) и избыткомNMM, Et3N, основания Хюнига в THF, DCM или EtOAc при комнатной температуре в течение 4-24 ч. Более конкретный пример образования амида включает обработку кислоты амином в присутствии циклического ангидрида сложного 1-пропил-фосфониевого эфира и в присутствии триэтиламина в DCM при комнатной температуре в течение 1 ч. Когда галогенангидрид является хлорангидридом (то есть Х=Cl), он может быть получен in situ согласно стандартной методологии и затем приведен во взаимодействие с амином (VI) и триэтиламином в дихлорметане при 70 С в течение 90 мин.(у) Восстановление Взаимодействие (у) представляет собой восстановление амида, например с использованием в подходящих условиях гидридного восстанавливающего агента. Удобно проводить восстановление амида в присутствии борана в THF при температуре дефлегмации в течение 2 ч с последующим добавлением метанола и водного хлорида аммония при температуре дефлегмации в течение 4 ч. Согласно схеме 2 соединения формулы (IX) могут быть получены из соединений формулы (VI) путем взаимодействия в подходящих условиях с R4-(CH2)a-L, где а является таким, как определено выше, иL является уходящей группой. Полученное соединение формулы (IX) затем может быть превращено в соединение формулы (I) путем образования амида и удаления защиты способом, аналогичным описанному выше в отношении схемы 1. На приведенной выше схеме R1, R2, R3, R4, R20 и а являются такими, как определено выше, PG представляет собой подходящую защитную группу, и L является уходящей группой, значение которой будет зависеть, среди прочего, от характера взаимодействия и конкретных используемых условий реакции. Подходящие уходящие группы очевидны специалисту и описаны во многих общепринятых руководствах по органической химии, например "Advanced Organic Chemistry", Jerry March, Third Edition, Wiley (1985),стр. 587, включенной в данное описание посредством ссылки; они включают галоген (например Br) и сульфонатные эфиры (например метансульфонат или трифторметансульфонат). Согласно схеме 3 соединения формулы (IX) могут быть получены из кетона формулы (XII) путем взаимодействия с первичным амином R4-(CH2)a-NH2 в подходящих условиях. Полученное соединение формулы (IX) затем может быть превращено в соединение формулы (I) путем образования амида и удаления защиты способом, аналогичным описанному выше в отношении схемы 1. Схема 3-8 011217 На приведенной выше схеме R1, R2, R3, R4, R20 и а являются такими, как определено выше, и PG представляет собой подходящую защитную группу. Взаимодействие (е) первичного амина R4-(CH2)a-NH2 с кетоном (XII) для удобства может представлять собой реакцию восстановительного аминирования, в которой после дегидратации амина и кетона следует восстановление полученного имина, например, металлгидридным реагентом или гидрированием,в подходящих условиях. Удобно осуществлять взаимодействие амина и кетона в присутствии тетраизопропилата титана (IV) в THF при комнатной температуре в течение 18 ч с последующим восстановлением избытком боргидрида натрия в метаноле при комнатной температуре в течение 5 ч. Специалист способен выбрать наиболее приемлемый путь синтеза желаемого соединения формулы(I). Приведенные выше схемы, несомненно, можно модифицировать, если это целесообразно, используя общие знания специалистов в данной области. Специалистам в данной области техники очевидно, что в ходе синтеза соединения формулы (I) может требоваться защита и удаление защиты одной или более реакционноспособных функциональных групп. Это может быть достигнуто с использованием традиционных методик, например описанных в книге TW Greene and PGM Wilts, "Protective Groups in Organic Synthesis", 3rd edition, John Wiley and Sons,Inc., 1999, включенной в данное описание посредством ссылки, где также описаны способы удаления таких групп. Специалистам в данной области техники очевидно, что некоторые защищенные производные соединений по изобретению, которые могут быть получены до конечной стадии удаления защиты, могут не обладать фармакологической активностью как таковые, но в некоторых случаях могут быть введены перорально или парентерально и после этого подвергаться метаболизму в организме с образованием соединений по изобретению, которые фармакологически активны. Следовательно, такие производные могут быть определены как пролекарства. Кроме того, некоторые соединения по изобретению могут выступать в качестве пролекарств для других соединений по изобретению. Таким образом, согласно следующему аспекту изобретения предложен способ получения соединений формулы (I), включающий взаимодействие соединения формулы (IX) где R4 и а являются такими, как определено выше, и PG представляет собой защитную группу, с кислотой или ацилгалогенидом формулы (II) где X представляет собой ОН или галогено, и удаление защиты. Когда а равен 1, соединение формулы (IX) может быть получено путем взаимодействия соединения формулы (VI) с альдегидом R4CHO. Альтернативно, соединение формулы (IX) может быть получено путем взаимодействия соединения формулы (VI) с соединением R4-(CH2)a-L, где L представляет собой уходящую группу, возможно выбранную из галогенида, метансульфоната и трифторметансульфоната. Кроме того, соединение формулы (IX) может быть получено путем взаимодействия соединения формулы (XII) с соединением R4-(CH2)a-NH2.-9 011217 Некоторые промежуточные соединения, описанные выше, являются новыми соединениями, и следует понимать, что все новые промежуточные соединения в данном описании составляют дополнительные аспекты настоящего изобретения. Рацемические соединения могут быть разделены либо с использованием препаративной ВЭЖХ и колонки с хиральной стационарной фазой, либо разделены с получением индивидуальных энантиомеров с использованием способов, известных специалистам в данной области техники. Кроме того, хиральные промежуточные соединения могут быть разделены и использованы для получения хиральных соединений по изобретению. Согласно следующему аспекту изобретения предложен один или более чем один метаболит соединений по изобретению, образованный in vivo. Преимуществом соединений по изобретению может быть то, что они являются более сильнодействующими, обладают большей продолжительностью действия, имеют более широкий интервал активности, более стабильны, имеют меньше побочных эффектов, более селективны, обладают лучшими характеристиками в отношении таблетирования или имеют другие более полезные свойства по сравнению с соединениями предшествующего уровня техники. Соединения по изобретению полезны, так как обладают фармакологической активностью у млекопитающих, включая людей. Так, они полезны в лечении или предупреждении расстройств, в которые вовлечено регулирование функции транспортера моноаминов, более конкретно, расстройств, в которые вовлечено ингибирование обратного захвата серотонина или норадреналина, и особенно таких, в которые вовлечено ингибирование обратного захвата серотонина и норадреналина. Соответственно, соединения по изобретению полезны в лечении недержания мочи, такого как истинное недержание при напряжении (GSI), недержание мочи при напряжении (SUI) или недержание мочи в пожилом возрасте; гиперактивного мочевого пузыря (ОАВ), включая идиопатическую детрузорную нестабильность, детрузорную гиперактивность, вторичную по отношению к неврологическим расстройствам (например болезни Паркинсона, рассеянному склерозу, поражению спинного мозга и инсульту), и детрузорную гиперактивность, вторичную по отношению к закупорке оттока из мочевого пузыря (например доброкачественной гиперплазии предстательной железы (ВРН), уретральной стриктуре или стенозу); ночного энуреза; недержания мочи в результате сочетания вышеуказанных состояний (например истинного недержания при напряжении, ассоциированного с гиперактивным мочевым пузырем); и таких симптомов нижних мочевыводящих путей, как частота и неотложные позывы к мочеиспусканию. Предполагается, что термин ОАВ охватывает как влажный ОАВ, так и сухой ОАВ. Ввиду вышеуказанной фармакологической активности соединения по изобретению также полезны в лечении депрессии, такой как большая депрессия, рекуррентная депрессия, одиночный депрессивный эпизод, субсиндромальная симптоматическая депрессия, депрессия у раковых пациентов, депрессия у пациентов с болезнью Паркинсона, депрессия после инфаркта миокарда, депрессия у детей, депрессия,вызванная жестоким обращением с ребенком, депрессия у бесплодных женщин, послеродовая депрессия,предменструальная дисфория и синдром старческой ворчливости. Ввиду вышеуказанной фармакологической активности соединения по изобретению также полезны в лечении когнитивных расстройств, таких как деменция, в частности дегенеративная деменция (включая сенильную деменцию, болезнь Альцгеймера, болезнь Пика, хорею Гентингтона, болезнь Паркинсона и болезнь Крейцфельдта-Якоба) и сосудистая деменция (включая мультиинфарктную деменцию), а также деменция, ассоциированная с поражениями внутричерепного пространства, травмой, инфекциями и родственными состояниями (включая ВИЧ-инфекцию), метаболизмом, токсинами, аноксией и витаминной недостаточностью; легкой когнитивной недостаточности, ассоциированной со старением, в частности возрастного ослабления памяти (AAMI), амнестического расстройства и возрастного снижения когнитивных функций (ARCD); психотических расстройств, таких как шизофрения и маниакальный синдром; тревожных расстройств, таких как генерализованное тревожное расстройство, фобии (например агорафобия, социальная фобия и простые фобии), паническое расстройство, обсессивно-компульсивное расстройство, посттравматическое стрессовое расстройство, смешанное тревожное и депрессивное расстройство; расстройств личности, таких как уклоняющееся расстройство личности и синдром дефицита внимания с гиперактивностью (ADHD); сексуальной дисфункции, такой как преждевременная эякуляция, мужская эректильная дисфункция (MED) и женская сексуальная дисфункция (FSD) (например расстройство полового возбуждения у женщин (FSAD; предменструального синдрома; сезонного аффективного расстройства (SAD); расстройств приема пищи, таких как нервная анорексия и нервная булимия; ожирения; снижения аппетита; зависимости от химических веществ вследствие привыкания к лекарственным средствам или пристрастия к веществам, например пристрастия к никотину, алкоголю, кокаину,героину, фенобарбиталу и бензодиазепинам; абстинентных синдромов, таких как появляющиеся в результате вышеупомянутой зависимости от химических веществ; головной боли, такой как мигрень, кластерная головная боль, хроническая пароксизмальная гемикрания, головная боль, ассоциированная с сосудистыми расстройствами, головная боль, ассоциированная с зависимостью от химических веществ или абстинентными синдромами, являющимися следствиями зависимости от химических веществ, и головная боль напряжения; боли; болезней Паркинсона, таких как деменция при болезни Паркинсона, индуци- 10011217 рованный приемом нейролептиков паркинсонизм и поздняя дискинезия; эндокринных расстройств, таких как гиперпролактинемия; вазоспазма, например в сосудистой сети головного мозга; мозжечковой атаксии; синдрома Туретта; трихотилломании; клептомании; эмоциональной лабильности; патологической плаксивости; расстройства сна (катаплексии); и шока. Из вышеприведенных состояний особенный интерес вызывает ADHD. Диагностика ADHD основана на клинической оценке (М. Dulcan, et al. J. Am. Acad. Child Adolesc. Psychiatry, Oct. 1997, 36(10 Suppl),85S-121S; National Institutes of Health, 1998). Основным признаком ADHD является устойчивая картина невнимания и/или гиперактивности/импульсивности, которая наблюдается более часто и в более тяжелой форме, чем обычно у индивидуумов сопоставимого уровня развития (4-е изд. Руководства по диагностике и статистике психических расстройств (DSM-IV) Американской ассоциации психологов, Вашингтон, федеральный округ Колумбия, 1994) (Diagnostic and Statistical Manual of Mental Disorders (DSM-IV),American Psychiatric Association, Washington, D.C., 1994). Для того, чтобы пациентам поставить диагнозADHD, они должны демонстрировать симптомы ADHD, причиняющие ущерб здоровью, до возраста семи лет, и симптомы должны иметь место в течение более шести месяцев по меньшей мере в двух испытаниях (например в школе [или на работе] и дома) (см. DSM-IV). Ввиду вышеуказанной фармакологической активности соединения по изобретению также полезны в лечении ряда других состояний или расстройств, включая гипотензию; расстройства желудочнокишечного тракта (включая изменения моторики и секреции), такие как синдром раздраженного кишечника (IBS), непроходимость кишечника (например послеоперационная непроходимость кишечника и непроходимость кишечника при сепсисе), гастропарез (например диабетический гастропарез), пептическая язва, гастроэзофагеальное рефлюксное заболевание (GORD или его синоним GERD), метеоризм и другие функциональные расстройства кишечника, такие как диспепсия (например неязвенная диспепсия(NUD и некардиальная боль в грудной клетке (NCCP); и фибромиалгический синдром. Соединения по изобретению, являющиеся ингибиторами обратного захвата серотонина и/или норадреналина, являются потенциально полезными в лечении ряда расстройств, включая боль. Физиологическая боль представляет собой важный защитный механизм, предназначенный для предупреждения об опасности от потенциального поражающего действия раздражителей внешней окружающей среды. Эта система действует через специфическую группу первичных сенсорных нейронов и активируется исключительно вредными раздражителями посредством механизмов периферической трансдукции (см. Millan, 1999, Prog. Neurobiol. 57: 1-164, для обзора). Эти сенсорные волокна известны как ноцицепторы и характеризуются как аксоны небольшого диаметра с малыми скоростями проводимости. Ноцицепторы кодируют интенсивность, продолжительность и качество вредного раздражителя и,посредством своей топографически организованной проекции в направлении спинного мозга, локализацию раздражителя. Ноцицепторы обнаружены на ноцицептивных нервных волокнах, которых имеется два основных типа, А-дельта-волокна (миелинированные) и С-волокна (немиелинированные). Активность, генерированная началом действия ноцицепторов, передается после сложного процессинга в дорсальный роговидный отросток либо напрямую, либо посредством передающих ядер ствола мозга в вентробазальный таламус и затем в кору головного мозга, где создается ощущение боли. В общем случае боль можно классифицировать на острую и хроническую. Острая боль начинается внезапно и продолжается в течение короткого периода (обычно двенадцать недель или меньше). Обычно она ассоциирована с конкретной причиной, такой как конкретное повреждение, и зачастую является резкой и тяжелой. Существует вид боли, которая может иметь место после конкретных повреждений, вызванных хирургическим вмешательством, стоматологическими работами, напряжением или растяжением. Как правило, острая боль не приводит к устойчивому физиологическому ответу. В противоположность этому хроническая боль является продолжительной болью, в типичном случае являясь устойчивой в течение более трех месяцев и приводя к значительным физиологическим и эмоциональным проблемам. Типичными примерами хронической боли являются невропатическая боль (например болезненная диабетическая невропатия, постгерпетическая невралгия), запястный синдром, боль в спине, головная боль,боль при раке, боль при артрите и хроническая послеоперационная боль. Когда имеет место существенное поражение ткани организма из-за заболевания или травмы, особенности ноцицепторной активации меняются и появляется чувствительность на периферии, локально вокруг поражения и с центром в месте окончания ноцицепторов. Эти эффекты приводят к повышению восприятия боли. При острой боли эти механизмы могут быть полезными в стимулировании защитных реакций, которые могут дать большую возможность осуществления репарационных процессов. При обычном выжидательном методе лечения как только такое поражение излечивается, повышенная чувствительность возвращается к нормальной. Тем не менее, при многих хронических болевых состояниях повышенная чувствительность продолжает сохраняться дольше процесса излечивания и часто обусловлена поражением нервной системы. Это поражение зачастую приводит к нарушениям в сенсорных нервных волокнах, ассоциированных с маладаптацией и аберрантной активностью (WoolfSalter, 2000, Science, 288: 1765-1768). Клиническая боль имеет место, когда характерным признаком среди симптомов пациента являются дискомфорт и аномальная чувствительность. Пациенты имеют тенденцию к крайней гетерогенности и у- 11011217 них могут иметь место различные болевые симптомы. Такие симптомы включают: 1) спонтанную боль,которая может быть тупой, жгучей или внезапной острой; 2) преувеличенные болевые ответы на вредные раздражители (гипералгезия); и 3) боль, вызванную нормальными безвредными раздражителями (аллодиния - Meyer et al., 1994, Textbook of Pain, 13-44). Несмотря на то, что пациенты, страдающие от различных форм острой и хронической боли, могут иметь похожие симптомы, лежащие в основе механизмы могут отличаться и, следовательно, могут потребовать разных стратегий лечения. Следовательно, боль также можно разделить на ряд разных подтипов вследствие различной патофизиологии, включая ноцицептивную, воспалительную и невропатическую боль. Ноцицептивная боль индуцируется тканевым поражением или сильными раздражителями с возможностью поражения. Болевые афференты активируются посредством передачи раздражителей ноцицепторами от места поражения и активируют нейроны в спинном мозге на уровне их окончания. Этот сигнал далее передается вверх по спинномозговым нервным путям к головному мозгу, где боль воспринимается (Meyer et al., 1994, Textbook of Pain, 13-44). В результате активации ноцицепторов активируются два типа афферентных нервных волокон. Миелинированные А-дельта-волокна, быстро передающие сигнал, ответственны за резкие и внезапные острые болевые ощущения, в то время как немиелинированные С-волокна передают сигнал с более низкой скоростью и сообщают тупую или продолжительную боль. Острая ноцицептивная боль со степенью от средней до тяжелой представляет собой известную характерную особенность боли в результате травмы центральной нервной системы, напряжения/растяжения, ожогов, инфаркта миокарда и острого панкреатита, боли после хирургического вмешательства (боль после хирургической процедуры любого типа), посттравматической боли, почечной колики, боли при раке и боли в спине. Боль при раке может быть хронической болью, такой как опухолеассоциированная боль (например костная боль, головная боль, лицевая боль или висцеральная боль) или боль, ассоциированная с терапией рака (например постхимиотерапевтический синдром, хронический болевой синдром после хирургического вмешательства и синдром после облучения). Боль при раке также может иметь место в ответ на химиотерапию, иммунотерапию, гормональную терапию или лучевую терапию. Боль в спине может иметь место вследствие наличия грыжи или разрыва межпозвоночных дисков или аномальностей небольшой суставной поверхности синовиальных соединений в области поясницы, крестцово-подвздошных синовиальных соединений, параспинальных мышц или задних продольных связок. Боль в спине может быть устранена естественным образом, но у некоторых пациентов, у которых она продолжается более 12 недель, она становится хроническим состоянием, которое может быть особенно изнуряющим. Невропатическую боль в настоящее время определяют как боль, инициированную или вызванную первичным поражением или дисфункцией в нервной системе. Повреждение нервов может быть вызвано травмой и болезнью и поэтому термин невропатическая боль охватывает многие расстройства с разнообразной этиологией. Они включают периферическую невропатию, диабетическую невропатию, постгерпетическую невралгию, невралгию тройничного нерва, боль с спине, раковую невропатию, ВИЧневропатию, фантомную боль, запястный синдром, центральную боль после инсульта и боль, ассоциированную с хроническим алкоголизмом, гипотиреоидией, уремией, рассеянным склерозом, поражением спинного мозга, болезнью Паркинсона, эпилепсией или витаминной недостаточностью, но этим не ограничены. Невропатическая боль является патологической, поскольку не играет никакой защитной роли. Зачастую она явно проявляется после того, как ее первоначальная причина устраняется, обычно продолжается в течение многих лет, значительно снижая качество жизни пациентов (Woolf and Mannion, 1999,Lancet, 353: 1959-1964). Симптомы невропатической боли трудно поддаются лечению, поскольку зачастую они являются гетерогенными даже среди пациентов с одним и тем же заболеванием (WoolfDecosterd, 1999, Pain Supp. 6: S141-S147; Woolf and Mannion, 1999, Lancet, 353: 1959-1964). Они включают спонтанную боль, которая может быть непрерывной, и пароксизмальную или аномальную индуцированную боль, такую как гипералгезия (повышенная чувствительность к вредному раздражителю) и аллодиния (чувствительность к обычно безвредному раздражителю). Воспалительный процесс представляет собой сложный набор биохимических и клеточных событий,активируемых в ответ на тканевое поражение или присутствие чужеродных веществ, которые приводят к образованию припухлости и боли (Levine and Taiwo, 1994: Textbook of Pain, 45-56). Боль при артритах является самой распространенной воспалительной болью. Ревматоидное заболевание является одним из самых распространенных хронических воспалительных состояний в развитых странах, и ревматоидный артрит является обычной причиной потери трудоспособности. Точная этиология ревматоидного артрита неизвестна, однако согласно существующим в настоящее время гипотезам полагают, что могут быть важны как генетические, так и микробиологические факторы(GrennanJayson, 1994, Textbook of Pain, 397-407). Было установлено, что почти 16 миллионов американцев, большинству из которых более 60 лет, имеют диагноз симптоматический остеоартрит (ОА) или дегенеративное заболевание сустава, и ожидается увеличение до 40 миллионов, поскольку возраст населения увеличивается, делая это общенародной проблемой здоровья огромного значения (HougeMersfelder, 2002, Ann. Pharmacother. 36: 679-686; McCarthy et al., 1994, Textbook of Pain, 387-395). Большинство пациентов с диагнозом остеоартрит обращаются за медицинской помощью из-за боли. Артрит оказы- 12011217 вает значительное воздействие на психосоциальную и физическую функцию и известен как ведущая причина потери трудоспособности в более поздний период жизни. Анкилозирующий спондилит также представляет собой ревматическое заболевание, которое приводит к артриту в позвоночнике и крестцово-подвздошныъх суставах. Он изменяется от перемежающихся эпизодов боли в спине, которые имеют место в течение всей жизни, до тяжелого хронического заболевания, которое воздействует на позвоночник, периферические суставы и другие органы тела. Другим типом воспалительной боли является висцеральная боль, которая включает боль, ассоциированную с воспалительным заболеванием кишечника (IBD). Висцеральная боль представляет собой боль, ассоциированную с внутренними органами, которые охватывают органы брюшной полости. Эти органы включают половые органы, селезенку и часть пищеварительной системы. Боль, ассоциированную с внутренними органами, можно подразделить на висцеральную боль пищеварительной системы и висцеральную боль, не связанную с пищеварительной системой. Обычно встречаемые желудочно-кишечные(GI) расстройства, вызывающие боль, включают функциональное расстройство кишечника (FBD) и воспалительное заболевание кишечника (IBD). Эти GI расстройства включают большое разнообразие болезненных состояний, которые в настоящее время являются регулируемыми только в умеренной степени,включая, что касается FBD, гастроэзофагеальный рефлюкс, диспепсию, синдром раздраженного кишечника (IBS) и функциональный абдоминальный болевой синдром (FAPS), и что касается IBD, болезнь Крона, илеит и язвенный колит; все из которых регулярно дают висцеральную боль. Другие типы висцеральной боли включают боль, ассоциированную с дисменореей, циститом и панкреатитом и болью в области таза. Следует отметить, что некоторые типы боли имеют множественные этиологии и, таким образом,могут быть классифицированы более чем в одной области, например, боль в спине и боль при раке имеют как ноцицептивную, так и невропатическую составляющую. Другие виды боли включают боль в результате мышечно-скелетных расстройств, включая миалгию, фибромиалгию, спондилит,серонегативные (неревматоидные) артропатии, несуставной ревматизм, дистрофинопатию, гликогенолиз,полимиозит и пиомиозит; сердечную и сосудистую боль, включая боль, вызванную стенокардией, инфарктом миокарда, стенозом митрального клапана, перикардитом, феноменом Рейно, склеродермией и ишемией скелетных мышц; головную боль, такую как мигрень (включая мигрень с аурой и мигрень без ауры), кластерная головная боль, головная боль по типу напряжения, смешанная головная боль и головная боль, ассоциированная с сосудистыми заболеваниями; и орофасциальную боль, включая зубную боль, боль в ушах, синдром жжения во рту и височнонижнечелюстную миофасциальную боль. Представляющие особый интерес расстройства включают недержание мочи, например смешанное недержание, GSI и SUI; боль; депрессию; тревожные расстройства, такие как обсессивно-компульсивное расстройство и посттравматическое стрессовое расстройство; расстройства личности, такие как ADHD; сексуальную дисфункцию; и зависимость от химических веществ и абстинентные синдромы вследствие зависимости от химических веществ. Таким образом, согласно дополнительным аспектам изобретения предложено: 1) соединение по изобретению для применения в терапии человека или ветеринарии; 2) соединение по изобретению для применения в лечении расстройства, в которое вовлечено регулирование функции транспортера моноаминов, такого как недержание мочи; 3) применение соединения по изобретению в изготовлении лекарственного средства для лечения расстройства, в которое вовлечено регулирование функции транспортера моноаминов; 4) соединение по изобретению для применения в лечении расстройства, в которое вовлечено регулирование серотонина или норадреналина; 5) применение соединения по изобретению в изготовлении лекарственного средства для лечения расстройства, в которое вовлечено регулирование серотонина или норадреналина; 6) соединение по изобретению для применения в лечении расстройства, в которое вовлечено регулирование серотонина и норадреналина; 7) применение соединения по изобретению в изготовлении лекарственного средства для лечения расстройства, в которое вовлечено регулирование серотонина и норадреналина; 8) соединение по изобретению для применения в лечении недержания мочи, такого как GSI илиSUI; 9) применение соединения по изобретению в изготовлении лекарственного средства для лечения недержания мочи, такого как GSI или SUI; 10) способ лечения расстройства, в которое вовлечено регулирование функции транспортера моноаминов, включающий введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по изобретению; 11) способ лечения расстройства, в которое вовлечено регулирование серотонина или норадренали- 13011217 на, включающий введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по изобретению; 12) способ лечения расстройства, в которое вовлечено регулирование серотонина и норадреналина,включающий введение пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по изобретению; и 13) способ лечения недержания мочи, такого как GSI или SUI, включающий введение пациенту,нуждающемуся в таком лечении, терапевтически эффективного количества соединения по изобретению. Следует понимать, что в данном описании все ссылки на лечение включают куративное, паллиативное и профилактическое лечение, если ясно не указано иное. Соединения по изобретению могут быть введены сами по себе или как часть комбинированной терапии. Когда вводят комбинацию терапевтических агентов, тогда активные ингредиенты можно вводить либо последовательно, либо одновременно в отдельных или комбинированных фармацевтических композициях. Примеры подходящих агентов для дополнительной терапии включают опиоидный анальгетик, например морфин, героин, гидроморфон, оксиморфон, леворфанол, левалорфан, метадон, меперидин, фентанил, кокаин, кодеин, дигидрокодеин, оксикодон, гидрокодон, пропоксифен, налмефен, налорфин, налоксон, налтрексон, бупренорфин, буторфанол, налбуфин или пентазоцин; нестероидное противовоспалительное лекарственное средство (NSAID), например аспирин, диклофенак, дифлусинал, этодолак, фенбуфен, фенопрофен, флуфенизал, флурбипрофен, ибупрофен, индометацин, кетопрофен, кеторолак, меклофенамовая кислота, мефенамовая кислота, мелоксикам, набуметон,напроксен, нимесулид, нитрофлурбипрофен, оксалазин, оксапрозин, фенилбутазон, пироксикам, сульфасалазин, сулиндак, толметин или зомепирак; седативное средство на основе барбитуратов, например амобарбитал, апробарбитал, бутабарбитал,бутабитал, мефобарбитал, метарбитал, метогекситал, пентобарбитал, фенобарбитал, секобарбитал, талбутал, теамилал или тиопентал; бензодиазепин, обладающий седативным действием, например хлордиазепоксид, клоразепат, диазепам, флуразепам, лоразепам, оксазепам, темазепам или триазолам; Н 1-антагонист, обладающий седативным действием, например дифенгидрамин, пириламин, прометазин, хлорфенирамин или хлорциклизин; седативное средство, такое как глутетимид, мепробамат, метаквалон или дихлоралфеназон; миорелаксант скелетных мышц, например баклофен, каризопродол, хлорзоксазон, циклобензаприн,метокарбамол или орфренадин; антагонист NMDA-рецептора (N-метил-D-аспартатного рецептора), например декстрометорфан+)-3-гидрокси-N-метилморфинан) или его метаболит декстрорфан +)-3-гидрокси-N-метилморфинан),кетамин, мемантин, пирролохинолин хинин, цис-4-(фосфонометил)-2-пиперидинкарбоновая кислота,будипин, EN-3231 (MorphiDex, комбинированный препарат морфина и декстрометорфана), топирамат,нерамексан или перзинфотел, включая NR2B-антагонист, например ифенпродил, траксопродил или (-)(R)-6-2-[4-(3-фторфенил)-4-гидрокси-1-пиперидинил]-1-гидроксиэтил-3,4-дигидро-2(1 Н)-хинолинон; альфа-адренергическое средство, например доксазозин, тамсулозин, клонидин, кванфацин, дексметатомидин, модафинил, фентоламин, теразазин, празазин или 4-амино-6,7-диметокси-2-(5-метансульфонамидо-1,2,3,4-тетрагидроизохинол-2-ил)-5-(2-пиридил)хиназолин; трициклический антидепрессант, например дезипрамин, имипрамин, амитриптилин или нортриптилин; противосудорожное средство, например карбамазепин, ламотриджин, топиратмат или вальпроат; тахикининовый (NK) антагонист, в частности NK-3, NK-2 или NK-1 антагонист, например (R,9R)7-[3,5-бис(трифторметил)бензил]-8,9,10,11-тетрагидро-9-метил-5-(4-метилфенил)-7 Н-[1,4]диазоцино[2,1g][1,7]нафтиридин-6-13-дион (TAK-637), 5-(2R,3S)-2-[(1R)-1-[3,5-бис(трифторметил)фенил]этокси-3-(4 фторфенил)-4-морфолинил]-метил]-1,2-дигидро-3H-1,2,4-триазол-3-он (MK-869), апрепитант, ланепитант, дапитант или (2S,3S)-3-2-метокси-5-(трифторметокси)фенил]-метиламино]-2-фенилпиперидин; мускариновый антагонист, например оксибутинин, толтеродин, пропиверин, тропсия хлорид, дарифенацин, солифенацин, темиверин и ипратропий; селективный ингибитор циклооксигеназы-2 (СОХ-2), например целекоксиб, рофекоксиб, парекоксиб, валдекоксиб, деракоксиб, эторикоксиб или люмиракоксиб; анальгетик на основе каменноугольной смолы, в частности, парацетамол; нейролептическое средство, такое как дроперидол, хлорпромазин, галоперидол, перфеназин, тиоридазин, мезоридазин, трифлуоперазин, флуфеназин, клозапин, оланзапин, рисперидон, зипразидон, кветиапин, сертиндол, арипипразол, сонепипразол, блонансерин, илоперидон, пероспирон, раклоприд, зотепин, бифепрунокс, азенапин, луразидон, амисульприд, балаперидон, палиндор, эпливансерин, осанетант,римонабант, меклинертант, Miraxion или саризотан; агонист (например резинфератоксин) или антагонист (например капсазепин) ванилоидных рецепто- 14011217 ров; бета-адренергическое средство, такое как пропранолол; местный анестетик, такой как мексилетин; кортикостероид, такой как дексаметазон; агонист или антагонист 5-НТ-рецепторов, в частности 5-HT1B/1D-агонист, такой как элетриптан, суматриптан, наратриптан, золмитриптан или ризатриптан; агонист 5-НТ 2 А-рецепторов, такой как R(+)-альфа-(2,3-диметокси-фенил)-1-[2-(4-фторфенилэтил)]4-пиперидинметанол (MDL-100907); холинергический (никотиновый) анальгетик, такой как испрониклин (ТС-1734), (Е)-N-метил-4-(3 пиридинил)-3-бутен-1-амин (RJR-2403), (R)-5-(2-азетидинилметокси)-2-хлорпиридин (АВТ-594) или никотин;(варденафил),5-(5-ацетил-2-бутокси-3 пиридинил)-3-этил-2-(1-этил-3-азетидинил)-2,6-дигидро-7 Н-пиразоло[4,3-d]пиримидин-7-он,5-(5 ацетил-2-пропокси-3-пиридинил)-3-этил-2-(1-изопропил-3-азетидинил)-2,6-дигидро-7 Н-пиразоло[4,3d]пиримидин-7-он,5-[2-этокси-5-(4-этилпиперазин-1-илсульфонил)пиридин-3-ил]-3-этил-2-[2 метоксиэтил]-2,6-дигидро-7 Н-пиразоло[4,3-d]пиримидин-7-он,4-[(3-хлор-4-метоксибензил)амино]-2[(2S)-2-(гидроксиметил)пирролидин-1-ил]-N-(пиримидин-2-илметил)пиримидин-5-карбоксамид,3-(1 метил-7-оксо-3-пропил-6,7-дигидро-1 Н-пиразоло[4,3-d]пиримидин-5-ил)-N-[2-(1-метилпирролидин-2 ил)этил]-4-пропоксибензолсульфонамид; альфа-2-дельта-лиганд, такой как габапентин, прегабалин, 3-метилгабапентин, (1,3,5)(3-аминометилбицикло[3.2.0]гепт-3-ил)уксусная кислота, (3S,5R)-3-аминометил-5-метилгептановая кислота,(3S,5R)-3-амино-5-метилгептановая кислота, (3S,5R)-3-амино-5-метилоктановая кислота, (2S,4S)-4-(3 хлорфенокси)пролин, (2S,4S)-4-(3-фторбензил)пролин, [(1R,5R,6S)-6-(аминометил)бицикло[3.2.0]гепт-6 ил]уксусная кислота, 3-(1-аминометил-циклогексилметил)-4 Н-[1,2,4]оксадиазол-5-он, С-[1-(1 Н-тетразол 5-илметил)циклогептил]метиламин, (3S,4S)-(1-аминометил-3,4-диметилциклопентил)уксусная кислота,(3S,5R)-3-аминометил-5-метилоктановая кислота, (3S,5R)-3-амино-5-метилнонановая кислота, (3S,5R)-3 амино-5-метилоктановая кислота, (3R,4R,5R)-3-амино-4,5-диметилгептановая кислота и (3R,4R,5R)-3 амино-4,5-диметилоктановая кислота; каннабиноид; антагонист метаботропных глутаматных рецепторов 1 подтипа (mGluR1); ингибитор обратного захвата серотонина, такой как сертралин, метаболит сертралина деметилсертралин, флуоксетин, норфлуоксетин (дезметилированный метаболит флуоксетина), флувоксамин, пароксетин, циталопрам, метаболит циталопрама дезметилциталопрам, эсциталопрам, d,l-фенфлурамин, фемоксетин, ифоксетин, цианодотиепин, литоксетин, дапоксетин, нефазодон, церикламин и тразодон; ингибитор обратного захвата норадреналина (норэпинефрина), такой как мапротилин, лофепрамин,миртазепин, оксапротилин, фезоламин, томоксетин, миансерин, бупроприон, метаболит бупроприона гидроксибупроприон, номифензин и вилоксазин (Vivalan), в особенности селективный ингибитор обратного захвата норадреналина, такой как ребоксетин, в частности (S,S)-ребоксетин; смешанный ингибитор обратного захвата серотонина-норадреналина, такой как венлафаксин, метаболит венлафаксина О-дезметилвенлафаксин, кломипрамин, метаболит кломипрамина дезметилкломипрамин, дулоксетин, милнаципран и имипрамин; ингибитор индуцибельной синтазы оксида азота (iNOS), такой как S-[2-[(l-иминоэтил)амино]этил]L-гомоцистеин, S-[2-[(1-иминоэтил)амино]этил]-4,4-диоксо-L-цистеин, S-[2-[(l-иминоэтил)амино]этил]2-метил-L-цистеин, (2S,5Z)-2-амино-2-метил-7-[(1-иминоэтил)амино]-5-гептеновая кислота, 2-(1R,3S)3-амино-4-гидрокси-1-(5-тиазолил)бутил]тио]-5-хлор-3-пиридинкарбонитрил,2-(1R,3S)-3-амино-4 гидрокси-1-(5-тиазолил)бутил]тио]-4-хлорбензнитрил, (2S,4R)-2-амино-4-2-хлор-5-(трифторметил)фенил]тио]-5-тиазолбутанол, 2-(1R,3S)-3-амино-4-гидрокси-1-(5-тиазолил)бутил]тио]-6-(трифторметил)3-пиридинкарбонитрил, 2-(1R,3S)-3-амино-4-гидрокси-1-(5-тиазолил)бутил]тио]-5-хлорбензнитрил, N[4-[2-(3-хлорбензиламино)этил]фенил]тиофен-2-карбоксамидин или гуанидиноэтилдисульфид; ингибитор ацетилхолинэстеразы, такой как донепезил; антагонист простагландина E2 подтипа 4 (ЕР 4), такой как N-[(2-[4-(2-этил-4,6-диметил-1 Нимидазо[4,5-с]пиридин-1-ил)фенил]этиламино)карбонил]-4-метилбензолсульфонамид или 4-[(1S)-1([5-хлор-2-(3-фторфенокси)пиридин-3-ил]карбониламино)этил]бензойная кислота; антагонист лейкотриена В 4, такой как 1-(3-бифенил-4-илметил-4-гидроксихроман-7-ил)циклопентанкарбоновая кислота- 15011217 ингибитор 5-липоксигеназы, такой как зилейтон, 6-[(3-фтор-5-[4-метокси-3,4,5,6-тетрагидро-2 Нпиран-4-ил])феноксиметил]-1-метил-2-хинолон (ZD-2138) или 2,3,5-триметил-6-(3-пиридилметил)-1,4 бензохинон (CV-6504); блокатор натриевых каналов, такой как лидокаин; 5-НТ 3-антагонист, такой как ондансетрон, гранисетрон, трописетрон, азасетрон, доласетрон или алосетрон; агонист эстрогена или селективный модулятор рецепторов эстрогенов (например гормонозамещающие терапевтические средства (HRT) или лазофоксифен); агонист альфа-адренергических рецепторов, такой как фенилпропаноламин или R-450; агонист дофаминовых рецепторов (например апоморфин, инструкцию по применению которого в качестве фармацевтического средства можно найти в US-A-5945117), включая агонист дофаминовых D2 рецепторов (например премиприксал, соединение номер PNU95666 от Pharmacia Upjohn; или ропинирол); агонист рецепторов простагландина E1 (PGE1) (например алпростадил); и их фармацевтически приемлемые соли и сольваты. Таким образом, в следующем аспекте изобретения предложена комбинация, содержащая соединение по изобретению вместе с дополнительным терапевтическим агентом. Для применения у людей соединения по изобретению можно вводить сами по себе, но в общем случае при лечении людей их вводят в смеси с подходящим фармацевтическим эксципиентом, разбавителем или носителем, выбранным с учетом предполагаемого пути введения и общепринятой фармацевтической практики. Например, соединения по изобретению можно вводить перорально, трансбуккально или сублингвально в форме таблеток, капсул (в том числе мягких желатиновых капсул), овулей, эликсиров, растворов или суспензий, которые могут содержать корригенты или красители, для немедленного, замедленного, модифицированного, непрерывного, двойного, контролируемого или пульсирующего высвобождения. Соединения по изобретению также можно вводить посредством внутрикавернозной инъекции. Соединения по изобретению также можно вводить посредством быстродиспергируемых или быстрорастворимых лекарственных форм. Такие таблетки могут содержать эксципиенты, такие как микрокристаллическая целлюлоза, лактоза, цитрат натрия, карбонат кальция, двухосновный фосфат кальция, глицин и крахмал (предпочтительно кукурузный, картофельный или крахмал тапиоки), разрыхлители, такие как крахмалгликолят натрия,кроскармелоза натрия и некоторые комплексные силикаты, и связующие агенты для гранулирования,такие как поливинилпирролидон, гидроксипропилметилцеллюлоза (НРМС), гидроксипропилцеллюлоза(НРС), сахароза, желатин и аравийская камедь. Кроме того, могут быть включены такие смазывающие агенты, как стеарат магния, стеариновая кислота, глицерилбегенат и тальк. Твердые композиции аналогичного типа также можно использовать в качестве наполнителей в желатиновых капсулах. Предпочтительные эксципиенты в этом отношении включают лактозу, крахмал,целлюлозу, молочный сахар или высокомолекулярные полиэтиленгликоли. Для водных суспензий и/или эликсиров соединения по изобретению и их фармацевтически приемлемые соли можно объединять с различными подсластителями или корригентами, подкрашивающим веществом или красителями, эмульгирующими и/или суспендирующими агентами и с разбавителями, такими как вода, этанол, пропиленгликоль и глицерин, и их комбинациями. Лекарственные формы с модифицированным высвобождением и пульсирующим высвобождением могут содержать такие эксципиенты, как, например, подробно описанные для лекарственных форм с немедленным высвобождением, вместе с дополнительными эксципиентами, которые действуют как модификаторы скорости высвобождения, причем их наносят на поверхность и/или включают в ядро композиции. Модификаторы скорости высвобождения включают гидроксипропилметилцеллюлозу, метилцеллюлозу, натриевую соль карбоксиметилцеллюлозы, этилцеллюлозу, ацетат целлюлозы, полиэтиленоксид,ксантановую камедь, карбомер, аммоний-метакрилатный сополимер, гидрогенизированное касторовое масло, карнаубский воск, парафин, фталатацетат целлюлозы, фталат гидроксипропилметилцеллюлозы,сополимер метакриловой кислоты и их смеси, но исключительно ими не ограничены. Лекарственные формы с модифицированным высвобождением и пульсирующим высвобождением могут содержать один эксципиент, модифицирующий скорость высвобождения, или их комбинацию. Модифицирующие скорость высвобождения эксципиенты могут присутствовать как внутри лекарственной формы, то есть внутри матрицы, так и/или на лекарственной форме, то есть на поверхности или оболочке. Быстродиспергируемые или быстрорастворимые лекарственные препараты (FDDF) могут содержать следующие ингредиенты: аспартам, ацесульфам калия, лимонную кислоту, кроскармелозу натрия,кросповидон, диаскорбиновую кислоту, этилакрилат, этилцеллюлозу, желатин, гидроксипропилметилцеллюлозу, стеарат магния, маннит, метилметакрилат, мятный корригент, полиэтиленгликоль, белую сажу, диоксид кремния, крахмалгликолят натрия, стеарилфумарат натрия, сорбит, ксилит. Термины"диспергируемый" или "растворимый", при использовании в данном описании для описания FDDF, зависят от растворимости используемого лекарственного средства, то есть когда лекарственное средство не- 16011217 растворимо, может быть приготовлена быстродиспергируемая лекарственная форма, а когда лекарственное средство растворимо, может быть приготовлена быстрорастворимая лекарственная форма. Также соединения по изобретению можно вводить парентерально, например внутривенно, внутриартериально, внутрибрюшинно, интратекально, интравентрикулярно, интрауретрально, внутригрудинно,интракраниально, внутримышечно или подкожно, либо их можно вводить посредством инфузий. Для такого парентерального введения их лучше всего применять в форме стерильного водного раствора, который может содержать другие вещества, например достаточное количество солей или глюкозы, чтобы сделать данный раствор изотоничным крови. При необходимости водные растворы следует подходящим образом забуферить (предпочтительно до рН 3-9). Изготовление подходящих парентеральных композиций в стерильных условиях легко осуществить стандартными фармацевтическими методами, хорошо известными специалистам в данной области техники. Для перорального и парентерального введения пациентам людям уровень суточных дозировок соединений по изобретению или их солей либо сольватов обычно будет составлять от 10 до 500 мг (в одной или разделенных дозах). Так, например, таблетки или капсулы соединений по изобретению или их солей либо сольватов могут содержать от 5 до 250 мг активного соединения для введения их по одной или по две или более одновременно, как это приемлемо. Врач в любом случае определит реальную дозировку, которая будет наиболее подходящей для любого отдельного пациента и которая будет варьировать в зависимости от возраста, массы и реакции конкретного пациента. Вышеуказанные дозировки являются примерами усредненного случая. Несомненно, могут иметь место отдельные случаи, когда полезны более высокие или более низкие интервалы дозировок, которые также входят в объем данного изобретения. Специалисту также очевидно, что при лечении некоторых состояний (включая РЕ) соединения по изобретению можно принимать в виде разовой дозы "по мере необходимости" (то есть как необходимо или желательно). Пример композиции в виде таблетки В общем случае композиция в виде таблетки обычно может содержать от приблизительно 0,01 до 500 мг соединения по настоящему изобретению (или его соли), в то время как массы наполнителей в таблетках могут меняться от 50 до 1000 мг. Приведен пример композиции в виде таблетки 10 мг: Данное количество обычно уточняют в соответствии с активностью лекарственного средства, и оно основано на массе свободного основания. Соединения по изобретению также можно вводить интраназально или посредством ингаляции и удобно доставлять в форме сухого порошкового ингаляционного средства или аэрозольного спрея из находящегося под давлением контейнера, насоса, распылителя или небулайзера с использованием подходящего пропеллента, например дихлордифторметана, трихлорфторметана, дихлортетрафторэтана, гидрофторалкана, такого как 1,1,1,2-тетрафторэтан (HFA 134A [товарный знак]) или 1,1,1,2,3,3,3 гептафторпропан (HFA 227EA [товарный знак]), двуокиси углерода или другого подходящего газа. В случае находящегося под давлением аэрозоля единица дозировки может быть установлена путем обеспечения клапана, выпускающего отмеренное количество. Находящийся под давлением контейнер, насос,распылитель или небулайзер может содержать раствор или суспензию активного соединения, например с использованием смеси этанола и пропеллента в качестве растворителя, которые могут дополнительно содержать смазывающее вещество, например сорбитантриолеат. Капсулы и картриджи (изготовленные,например, из желатина) для применения в ингаляторе или в инсуффляторе могут быть изготовлены содержащими порошковую смесь соединения по изобретению и подходящей порошковой основы, такой как лактоза или крахмал. Аэрозольные или сухие порошковые композиции предпочтительно отрегулированы таким образом,чтобы каждая отмеренная доза или впрыск содержали от 1 до 50 мг соединения по изобретению для доставки пациенту. Общая суточная доза в случае аэрозоля будет находиться в диапазоне от 1 до 50 мг, и ее можно вводить в разовой дозе или, более часто, в разделенных дозах на протяжении суток. Кроме того, соединения по изобретению могут быть изготовлены для доставки при помощи пульверизатора. Композиции для пульверизаторных устройств могут содержать в качестве солюбилизаторов,эмульгаторов или суспендирующих агентов следующие ингредиенты: воду, этанол, глицерин, пропиленгликоль, низкомолекулярные полиэтиленгликоли, хлорид натрия, фторуглероды, простые эфиры полиэтиленгликоля, сорбитантриолеат, олеиновую кислоту. Альтернативно, соединения по изобретению могут быть введены в форме суппозитория или песса- 17011217 рия, либо они могут быть использованы местно в форме геля, гидрогеля, лосьона, раствора, крема, мази или присыпки. Кроме того, соединения по изобретению можно вводить дермально или трансдермально,например посредством использования кожных пластырей. Их также можно вводить глазным, легочным или ректальным путем. Для офтальмического применения соединения можно изготовить в виде микронизированных суспензий в изотоническом, с подведенным значением рН, стерильном физиологическом растворе или,предпочтительно, в виде растворов в изотоническом, с подведенным значением рН, стерильном физиологическом растворе, возможно в комбинации с консервантом, таким как бензалкония хлорид. Альтернативно, они могут быть изготовлены в виде мази, такой как вазелин. Для местного применения на коже соединения по изобретению могут быть изготовлены в виде подходящей мази, содержащей активное соединение, суспендированное или растворенное, например, в смеси одного или более из следующих: минеральное масло, жидкий вазелин, белый вазелин, пропиленгликоль, полиоксиэтилен-полиоксипропиленовое соединение, эмульгирующий воск и вода. Альтернативно,они могут быть изготовлены в виде подходящего лосьона или крема, суспендированными или растворенными, например, в смеси одного или более из следующих: минеральное масло, сорбитанмоностеарат,полиэтиленгликоль, вазелиновое масло, полисорбат 60, цетиловые эфиры, воск, цетеариловый спирт, 2 октилдодеканол, бензиловый спирт и вода. Кроме того, соединения по изобретению можно использовать в комбинации с циклодекстрином. Известно, что циклодекстрины образуют с молекулами лекарственного средства комплексы включения и комплексы без включения. Образование комплекса лекарственное средство-циклодекстрин может изменять растворимость, скорость растворения, биодоступность и/или стабильность молекулы лекарственного средства. Комплексы лекарственное средство-циклодекстрин, как правило, пригодны для большинства лекарственных форм и путей введения. В качестве альтернативы непосредственному образованию комплекса с лекарственным средством, циклодекстрин можно использовать в качестве вспомогательной добавки, например в качестве носителя, разбавителя или солюбилизатора. Наиболее часто используемыми являются альфа-, бета- и гамма-циклодекстрины, и подходящие примеры описаны в WO-A-91/11172,WO-A-94/02518 и WO-A-98/55148. Для перорального или парентерального введения пациентам людям уровни суточных доз соединений формулы (I) и их фармацевтически приемлемых солей будут составлять от 0,01 до 30 мг/кг (в разовой или разделенных дозах) и предпочтительно будут находиться в диапазоне от 0,01 до 5 мг/кг. Так,таблетки будут содержать от 1 мг до 0,4 г соединения для введения по одной или по две или более одновременно, как это приемлемо. Врач в любом случае определит фактическую дозировку, которая будет наиболее подходящей для любого конкретного пациента и которая будет варьировать в зависимости от возраста, массы и реакции конкретного пациента. Вышеуказанные дозировки несомненно являются лишь примерами усредненного случая, и могут быть случаи, когда будут полезны более высокие или более низкие дозы, которые входят в объем данного изобретения. Пероральное введение является предпочтительным. Для применения в ветеринарии соединение по изобретению вводят в виде подходящей приемлемой композиции в соответствии с обычной ветеринарной практикой, и ветеринар определяет схему дозирования и путь введения, наиболее подходящий конкретному животному. Таким образом, согласно следующему аспекту изобретения предложена фармацевтическая композиция, содержащая соединение по изобретению и фармацевтически приемлемый адъювант, разбавитель или носитель. Упоминаемые выше комбинации также могут быть удобно представлены для применения в форме фармацевтической композиции, и таким образом фармацевтические композиции, содержащие комбинацию, как она определена выше, вместе с фармацевтически приемлемым адъювантом, разбавителем или носителем, составляют еще один аспект изобретения. Отдельные компоненты таких комбинаций можно вводить либо последовательно, либо одновременно в отдельных или комбинированных фармацевтических композициях. Когда соединение по изобретению применяют в комбинации со вторым терапевтическим средством, доза каждого соединения может отличаться от дозы при применении одного соединения. Соответствующие дозы будут легко определены специалистами в данной области. Изобретение проиллюстрировано следующими ниже не ограничивающими примерами, в которых могут быть использованы следующие сокращения и определения: Подготовительные примеры и примеры, приведенные ниже, иллюстрируют, но никоим образом не ограничивают, данное изобретение. Все температуры приведены в С. Колоночную флэшхроматографию проводили, используя силикагель 60 Merck (9385). Хроматографию с применением твердофазной экстракции (SPE) проводили, используя картриджи Varian Mega Bond Elut (Si) (Anachem) в условиях вакуума 2 кПа (15 мм рт. ст Тонкослойную хроматографию (ТСХ) проводили на пластинах- 19011217 силикагеля 60 Merck (5729). Точки плавления определяли, используя аппарат Gallenkamp MPD350, и не корректировали. ЯМР-анализ осуществляли, используя ЯМР-спектрометр Varian-Unity Inova (400 МГц) или Varian Mercury (400 МГц). Масс-спектроскопию осуществляли, используя одноквадрупольный массспектрометр Finnigan Navigator с ионизацией электрораспылением или масс-спектрометр Finnigan aQaAPCI. Соединения по изобретению после обработки удобно выделять в форме свободного основания, однако, используя традиционные способы, можно получить фармацевтически приемлемые соли присоединения кислот соединений по изобретению. Сольваты (например гидраты) соединения по изобретению могут быть образованы в процессе обработки на одной из вышеуказанных стадий способа. Когда соединения получали способом, описанным для ранее приведенного Примера, специалисту очевидно, что для каждой конкретной реакции продолжительности реакций, количество эквивалентов реагентов и температуры реакций можно модифицировать и что, тем не менее, может быть необходимым или желательным использование других условий обработки или очистки. Подготовительный пример 1. трет-Бутил-(3S)-3-(циклопентиламино)пирролидин-1-карбоксилат Циклопентанон (12,7 мл; 143 ммоль) добавляли к трет-бутил-(3S)-3-аминопирролидин-1 карбоксилату (26,6 г; 143 ммоль) в смеси метанол:толуол 3:1 (600 мл:200 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч в атмосфере азота. Реакционную смесь затем упаривали до 50 мл, три раза подвергали азеотропной перегонке со смесью метанол:толуол 3:1 (600 мл:200 мл) и концентрировали в вакууме. Реакционную смесь помещали в метанол (250 мл), охлаждали до 0 С и порциями добавляли боргидрид натрия (7,5 г; 200,2 ммоль). После завершения реакции добавляли воду(50 мл) и растворитель выпаривали. Остаток разбавляли дополнительным количеством воды (150 мл) и три раза экстрагировали дихлорметаном (250 мл). Органические фазы объединяли, сушили над сульфатом магния и концентрировали в вакууме, получая указанное в заголовке соединение в виде смолы (36,1 г; 99,4%). 1 Триэтиламин (24 мл; 170 ммоль) добавляли к раствору амина из подготовительного примера 1 (36,1 г; 142 ммоль) в дихлорметане (350 мл) в атмосфере азота. Реакционную смесь охлаждали до 0 С и по каплям добавляли 2,3-дихлор-бензоилхлорид (29,8 г; 142 ммоль) в дихлорметане, поддерживая температуру ниже 5 С. Реакционную смесь затем перемешивали в течение 6 ч. Добавляли воду (200 мл) и собирали органическую фазу. Водный слой экстрагировали дихлорметаном (250 мл). Объединенные органические фазы промывали 2 М водным раствором гидроксида натрия и 10%-ным раствором лимонной кислоты, сушили над сульфатом магния и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией на силикагеле, элюируя смесью этилацетат:циклогексан (от 1:6 до 1:4, до 1:2, до 1:1 по объему) с получением указанного в заголовке продукта (50 г; 82,4%). 1 Н ЯМР (CDCl3, 400 МГц, ротамеры) : 1,43-1,47 (d, 9H), 1,56-1,66 (m, 5H), 1,79 (m, 0,5 Н), 1,98 (m,3H), 2,37 (m, 1 Н), 2,92 (m, 0,5 Н), 3,15 (m, 0,5 Н), 3,40 (m, 1 Н), 3,58 (m, 1,5 Н), 3,74 (m, 2 Н), 3,97 (m, 1 Н),7,10 (m, 1 Н), 7,24 (m, 1 Н), 7,46 (d, 1H).(4,25 г; 20,33 ммоль) (см. ЕР 0600317, пример 15) в безводном дихлорметане (41 мл) при комнатной температуре в атмосфере азота. Добавляли N,N-диметилформамид (80 мкл; 1 ммоль) и реакционную смесь перемешивали в течение 1 ч. Растворитель удаляли выпариванием при пониженном давлении, получая желтое твердое вещество, которое растворяли в дихлорметане (20 мл) и по каплям добавляли к раствору триэтиламина (4,72 мл; 33,9 ммоль) и амина из Подготовительного примера 1 (4,31 г; 16,95 ммоль) в дихлорметане (36 мл) в атмосфере азота. После перемешивания в течение 18 ч при комнатной температуре полученную смесь разбавляли дихлорметаном (100 мл) и 1 М водным карбонатом калия (90 мл). Органическую фазу сушили над сульфатом магния, фильтровали и растворитель удаляли выпариванием при пониженном давлении. Остаток растворяли в минимальном количестве дихлорметана и очищали хроматографией на силикагеле с элюированием градиентом растворителей от пентана до смеси этилацетат:пентан (20:80 по объему), получая указанное в заголовке соединение в виде белой пены (6,6 г; 73%). 1 Н ЯМР (CD3OD, 400 МГц, ротамеры) : 1,43-1,47 (d, 9H), 1,62 (m, 1,5H), 1,72 (m, 3H), 1,88 (m,1,5 Н), 1,97 (m, 0,5 Н), 2,13 (m, 1,5 Н), 2,32 (m, 0,5 Н), 2,74 (m, 1H), 3,40 (m, 1 Н), 3,51-3,59 (m, 1.5 Н), 3,76 (m,2 Н), 3,88 (m, 1 Н), 4,05 (m, 1H), 7,33 (m, 2 Н). Подготовительный пример 4. трет-Бутил-(3S)-3-(циклогексиламино)пирролидин-1-карбоксилат трет-Бутил-(3S)-3-(циклогексиламино)пирролидин-1-карбоксилат получали способом, аналогичным описанному для подготовительного примера 1, используя трет-бутил-(3S)-3-аминопирролидин-1 карбоксилат и циклогексанон с получением желаемого продукта (5,9 г; 82%). 1 Н ЯМР (CDCl3, 400 МГц) : 1,09 (m, 2 Н), 1,24 (m, 3H), 1,45 (s, 9 Н), 1,62 (m, 2 Н), 1,72 (m, 2 Н), 1,88 трет-Бутил-(3S)-3-[циклогексил(2,3-дихлорбензоил)амино]пирролидин-1-карбоксилат получали способом, аналогичным описанному для Подготовительного примера 2, используя амин из Подготовительного примера 4 и 2,3-дихлорбензоилхлорид с получением желаемого продукта (5,14 г; 83%). 1 Н ЯМР (CDCl3, 400 МГц, ротамеры) : 1,00-1,09 (m, 2 Н), 1,26 (m, 1 Н), 1,43-1,47 (d, 9 Н), 1,58 (m,3H), 1,73 (m, 2 Н), 1,89 (m, 2 Н), 2,68 (m, 1 Н), 2,89 (m, 1 Н), 3,10 (m, 1H), 3,39 (m, 1 Н), 3,52 (m, 1 Н), 3,69 (m,1 Н), 3,92 (m, 2 Н), 7,10 (m, 1 Н), 7,23 (m, 1 Н), 7,47 (d, 1 Н). трет-Бутил-(3S)-3-[циклогексил(2-хлор-3-фторбензоил)амино]пирролидин-1-карбоксилат получали способом, аналогичным описанному для подготовительного примера 3, используя амин из подготовительного примера 4 и 2-хлор-3-фторбензойную кислоту с получением желаемого продукта (158 мг; 29%). 1 Н ЯМР (CDCl3, 400 МГц, ротамеры) : 0,98-1,06 (m, 2,5H), 1,29 (m, 1,5 Н), 1,47 (d, 9 Н), 1,57 (m, 3H),1,73 (m, 2 Н), 1,87 (m, 2 Н), 2,66 (m, 0,5 Н), 2,89 (m, 1 Н), 3,11 (m, 1H), 3,38 (m, 1H), 3,53 (m, 1H), 3,69 (m,0,5H), 3,91 (m, 2H), 6,99 (m, 1H), 7,15 (t, 1H), 7,28 (m, 1H). трет-Бутил-(3S)-3-[циклогексил(3-фтор-2-метилбензоил)амино]пирролидин-1-карбоксилат получали способом, аналогичным описанному для подготовительного примера 3, используя амин из подготовительного примера 4 и 3-фтор-2-метилбензойную кислоту с получением желаемого продукта (63 мг; 12%). 1 Н ЯМР (CDCl3, 400 МГц, ротамеры) : 1,02 (m, 2,5H), 1,30 (m, 2,5 Н), 1,47 (d, 9 Н), 1,57 (m, 1 Н), 1,62 трет-Бутил-(3S)-3-(циклобутиламино)пирролидин-1-карбоксилат получали способом, аналогичным описанному для подготовительного примера 1, используя трет-бутил-(3S)-3-аминопирролидин-1 карбоксилат и циклобутанон (15 эквивалентов: сначала добавляли 10 экв., 5 экв. добавляли перед вторым азеотропным удалением воды), за исключением того, что неочищенный продукт очищали колоночной хроматографией на силикагеле с получением указанного в заголовке соединения (542 мг; 28%),1 Н ЯМР (CD3OD, 400 МГц) : 1,45 (s, 9H), 1,70 (m, 3H), 1,81 (m, 3H), 2,05 (m, 1H), 2,21 (m, 2 Н), 3,03 трет-Бутил-(3S)-3-[циклобутил(2,3-дихлорбензоил)амино]пирролидин-1-карбоксилат получали способом, аналогичным описанному для подготовительного примера 2, используя амин из подготовительного примера 8 и 2,3-дихлорбензоилхлорид с получением желаемого продукта (370 мг; 79%). 1 Н ЯМР (CD3OD, 400 МГц) : 1,43 (m, 1H), 1,48 (s, 9H), 1,66 (m, 1H), 1,95 (m, 1 Н), 2,12-2,27 (m, 4 Н),2,72 (m, 1 Н), 3,42 (m, 1 Н), 3,61 (m, 1 Н), 3,69 (m, 1 Н), 3,86 (m, 1 Н), 3,98 (m, 1 Н), 4,45 (m, 1H), 7,24 (dd, 1H),7,40 (t, 1H), 7,62 (d, 1H). трет-Бутил-(3S)-3-аминопирролидин-1-карбоксилат (5 г; 27 ммоль) добавляли к раствору 2,6 лутидина (6,2 мл; 54 ммоль) в дихлорметане (150 мл) в атмосфере азота. Реакционную смесь охлаждали до 0 С и в течение 15 мин при 0 С медленно добавляли раствор 2,4-динитробензолсульфонилхлорида(7,15 г; 27 ммоль) в дихлорметане (100 мл). Затем реакционную смесь перемешивали при комнатной температуре в течение 48 ч в атмосфере азота. Добавляли воду (100 мл) с последующим добавлением 2 н. водного хлористого водорода до тех пор, пока рН водного слоя не достигал 2. Затем слои разделяли и водный слой экстрагировали дополнительным количеством дихлорметана (100 мл). Органические фазы объединяли, дважды промывали водой (100 мл), сушили над сульфатом магния и концентрировали в вакууме с получением указанного в заголовке соединения в виде смолы (10 г; 89%). 1 Н ЯМР (CDCl3, 400 МГц) : 1,42 (s, 9 Н), 1,88 (m, 1 Н), 2,15 (m, 1H), 3,18 (m, 1 Н), 3,37-3,44 (m, 2 Н),4,07 (m, 1 Н), 5,58 (d, 1H), 8,40 (d, 1H), 8,57 (d, 1H), 8,68 (s, 1H). Подготовительный пример 11. трет-Бутил-(3S)-3-(циклобутилметиламино)пирролидин-1 карбоксилат Циклобутанметанол (0,2 мл; 2,11 ммоль), затем трифенилфосфин (465 мг; 2,3 ммоль) добавляли к раствору соединения из подготовительного примера 10 (0,8 г; 1,92 ммоль) в тетрагидрофуране (40 мл) в атмосфере азота. Реакционную смесь охлаждали до 0 С и по каплям, поддерживая температуру ниже 3 С, добавляли раствор диизопропилазодикарбоксилата (0,45 мл; 2,3 ммоль) в тетрагидрофуране (15 мл). Реакционную смесь перемешивали при 0 С в течение 0,5 ч и затем при комнатной температуре в течение 18 ч. Тетрагидрофуран выпаривали и остаток помещали в дихлорметан (20 мл). Добавляли триэтиламин(0,53 мл; 3,84 ммоль) и меркаптоуксусную кислоту (0,16 мл; 2,3 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Затем ее промывали 2 н. водным хлористым водородом. Водный слой подщелачивали до рН 11 с использованием 2 М гидроксида натрия и повторно трижды экстрагировали этилацетатом. Органические фазы затем концентрировали в вакууме с получением указанного в заголовке соединения (151 мг; 31%). 1 Н ЯМР (CDCl3, 400 МГц) : 1,44 (s, 9 Н), 1,65 (m, 3H), 1,87 (m, 2 Н), 2,05 (m, 3H), 2,45 (m, 1 Н), 2,62(d, 1 Н), 3,05 (m, 1 Н), 3,28-3,51 (m, 5 Н). Альтернативный способ Борантетрагидрофурановый комплекс (1M в тетрагидрофуране; 100 мл; 100 ммоль) добавляли к раствору соединения из подготовительного примера 27 (9 г; 33,54 ммоль) в безводном тетрагидрофуране(100 мл) в атмосфере азота. Реакционную смесь кипятили с обратным холодильником в течение 2 ч. Реакционную смесь охлаждали до комнатной температуры, гасили метанолом и концентрировали в вакууме. Остаток подвергали азеотропной перегонке с метанолом, затем перерастворяли в метаноле (200 мл),кипятили с обратным холодильником в течение 18 ч, далее концентрировали в вакууме. Очистка остатка хроматографией на силикагеле с элюированием смесью дихлорметан:метанол:0,88 аммиак (95:5:0,5 по объему) дала указанное в заголовке соединение в виде смолы (7,67 г; 90%). 1 Н ЯМР (400 МГц, CD3OD) : 1,44 (s, 9H), 1,70 (m, 3H), 1,90 (m, 2H), 2,08 (m, 3H), 2,47 (m, 1 Н), 2,62 трет-Бутил-(3S)-3-[циклобутилметил(2,3-дихлорбензоил)амино]пирролидин-1-карбоксилат получали способом, аналогичным описанному для подготовительного примера 2, используя амин из подготовительного примера 11 и 2,3-дихлорбензоилхлорид с получением желаемого продукта (94 мг; 37%). трет-Бутил-(3S)-3-(циклопропилметиламино)пирролидин-1-карбоксилат получали способом, аналогичным описанному для подготовительного примера 1, используя трет-бутил-(3S)-3-аминопирролидин 1-карбоксилат и циклопропанкарбоксальдегид, за исключением того, что неочищенный продукт очищали хроматографией на силикагеле с элюированием градиентом растворителей от дихлорметана до смеси дихлорметан:метанол:0,88 аммиак (90:10:1 по объему), получая указанное в заголовке соединение (5,2 г; 81%). 1 Н ЯМР (CDCl3, 400 МГц) : 0,15 (m, 2 Н), 0,48 (m, 2 Н), 0,97 (m, 1 Н), 1,43 (s, 9H), 1,75 (m, 1 Н), 2,05 трет-Бутил-(3S)-3-аминопирролидин-1-карбоксилат добавляли к раствору тетрагидро-4 Н-пиран-4 она и 10% Pd/C (300 мг) в этаноле и реакционную смесь оставляли под давлением газообразного водорода приблизительно 415 кПа (приблизительно 60 фунт/кв.дюйм) на 18 ч. Реакционную смесь фильтровали- 24011217 через Arbocel, тщательно промывая этилацетатом. Фильтрат концентрировали в вакууме и неочищенный продукт очищали колоночной хроматографией на силикагеле с получением желаемого продукта (6,7 г; 61%). 1 трет-Бутил-(3S)-3-[(2,3-дихлорбензоил)(тетрагидро-2H-пиран-4-ил)амино]пирролидин-1 карбоксилат получали способом, аналогичным описанному для подготовительного примера 2 (используяN-метилморфолин в качестве основания), из амина из подготовительного примера 14 и 2,3 дихлорбензоилхлорида с получением желаемого продукта (200 мг; 31%). 1 Н ЯМР (CD3OD, 400 МГц) : 1,43-1,47 (d, 9 Н), 1,58 (m, 1,5 Н), 1,76 (m, 0,5 Н), 1,94 (m, 2 Н), 2,11 (m,1H), 2,76 (m, 0,5 Н), 2,98 (m, 0,5 Н), 3,13 (m, 2 Н), 3,4-3,58 (m, 4 Н), 3,69 (m, 0,5 Н), 3,9 (m, 2,5 Н), 4,04 (m,0,5 Н), 4,2 (m, 0,5 Н), 7,29 (d, 1H), 7,43 (t, 1H), 7,62 (d, 1 Н) ротамеры. трет-Бутил-(3S)-3-[(2-хлорбензоил)(циклопентил)амино]пирролидин-1-карбоксилат получали способом, аналогичным описанному для подготовительного примера 2 (используя толуол в качестве растворителя, N-метилморфолин в качестве основания и нагревая реакционную смесь при 60 С в течение 18 ч),используя амин из подготовительного примера 1 и 2-хлорбензоилхлорид с получением желаемого продукта (1,16 г; 75%). 1 трет-Бутил-(3S)-3-[(2-хлорбензоил)(циклогексил)амино]пирролидин-1-карбоксилат получали способом, аналогичным описанному для подготовительного примера 2 (используя толуол в качестве растворителя, IV-метилморфолин в качестве основания и нагревая реакционную смесь при 60 С в течение 18 ч), используя амин из подготовительного примера 4 и 2-хлорбензоилхлорид с получением желаемого трет-Бутил-(3S)-3-(циклогептиламино)пирролидин-1-карбоксилат получали способом, аналогичным описанному для подготовительного примера 14, используя трет-бутил-(3S)-3-аминопирролидин-1 карбоксилат и циклогептанон с получением желаемого продукта (4,54 г; 100%). 1 Н ЯМР (CD3OD, 400 МГц) : 1,45 (s, 12H), 1,57 (m, 5H), 1,69 (m, 3H), 1,89 (m, 2 Н), 2,12 (m, 1 Н),2,71 (m, 1H), 3,02 (m, 1H), 3,26 (m, 1H), 3,45 (m, 2H), 3,59 (m, 1H). трет-Бутил-(3S)-3-[(2-хлорбензоил)(циклогептил)амино]пирролидин-1-карбоксилат получали способом, аналогичным описанному для подготовительного примера 2 (используя толуол в качестве растворителя, N-метилморфолин в качестве основания и нагревая реакционную смесь при 60 С в течение 18 ч),используя амин из подготовительного примера 18 и 2-хлорбензоилхлорид с получением желаемого продукта (1,27 г; 90%). трет-Бутил-(3S)-3-циклогептил[2-(трифторметил)бензоил]аминопирролидин-1-карбоксилат получали способом, аналогичным описанному для подготовительного примера 2 (используя толуол в качестве растворителя, N-метилморфолин в качестве основания и нагревая реакционную смесь при 60 С в течение 18 ч), используя амин из подготовительного примера 18 и 2-(трифторметил)бензоилхлорид с получением желаемого продукта (910 мг; 57%). трет-Бутил-(3S)-3-циклогексил[2-(трифторметил)бензоил]аминопирролидин-1-карбоксилат получали способом, аналогичным описанному для подготовительного примера 2 (используя толуол в качестве растворителя, N-метилморфолин в качестве основания и нагревая реакционную смесь при 60 С в течение 18 ч), используя амин из подготовительного примера 4 и 2-(трифторметил)бензоилхлорид с получением желаемого продукта (860 мг; 52%). трет-Бутил-(3S)-3-циклопентил[2-(трифторметил)бензоил]амино-пирролидин-1-карбоксилат получали способом, аналогичным описанному в подготовительном примере 2 (используя толуол в качестве растворителя, N-метилморфолин в качестве основания и нагревая реакционную смесь при 60 С в течение 18 ч), используя амин из подготовительного примера 1 и 2-(трифторметил)бензоилхлорид с получением желаемого продукта (910 мг; 54%). 1 Н ЯМР (CDCl3, 400 МГц, ротамеры) : 1,43-1,47 (br d, 11H), 1,55 (m, 2H), 1,65 (m, 1 Н), 1,76 (m, 2 Н),1,99 (m, 2 Н), 2,35 (m, 0,5 Н), 2,89 (m, 0,5 Н), 3,05 (m, 0,5 Н), 3,19 (m, 0,5 Н), 3,39 (m, 1 Н), 3,57 (m, 1 Н), 3,69 1-Метилциклопропанкарбоновую кислоту (2,96 г; 29,54 ммоль) добавляли к раствору трет-бутил(3S)-3-аминопирролидин-1-карбоксилата (5 г; 26,85 ммоль) в дихлорметане (135 мл) при комнатной температуре. Затем добавляли триэтиламин (9,4 мл; 67,13 ммоль). Реакционную смесь охлаждали до 0 С и добавляли 2,4,6-триоксид 2,4,6-трипропил-1,3,5,2,4,6-триоксатрифосфинана (50%-ного в этилацетате; 17,4 мл; 29,54 ммоль). Смесь перемешивали при комнатной температуре в течение 1 ч и затем концентрировали при пониженном давлении. Остаток разбавляли в дихлорметане (100 мл) и добавляли 20%ный водный раствор карбоната калия (80 мл). Смесь перемешивали при комнатной температуре в течение 18 ч и слои разделяли. Органический слой промывали 20%-ным раствором карбоната калия (50 мл),соляным раствором (80 мл), сушили над сульфатом магния, концентрировали в вакууме и подвергали азеотропной перегонке с диэтиловым эфиром, получая указанное в заголовке соединение в виде белого твердого вещества (6,815 г; 95%). Боран (1M в тетрагидрофуране; 75 мл; 75 ммоль) медленно добавляли к раствору соединения из Подготовительного примера 23 (6,815 г; 25,39 ммоль) в тетрагидрофуране (75 мл) в атмосфере азота. Смесь нагревали при температуре дефлегмации в течение 2 ч, затем перемешивали при комнатной температуре в течение 18 ч. Для гашения в реакционную смесь осторожно добавляли метанол и затем смесь концентрировали в вакууме. Остаток помещали в метанол (120 мл) и нагревали при температуре дефлегмации в течение 3 ч. Затем добавляли водный хлорид аммония и реакционную смесь нагревали при температуре дефлегмации в течение 4 ч. Реакционную смесь затем концентрировали в вакууме, остаток распределяли между этилацетатом и 1 н. водным NaOH, водную фазу один раз экстрагировали этилацетатом и объединенные вместе органические фазы сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле с элюированием смесью этилацетат:пентан (от 20:80 до 30:70), получая указанный в заголовке продукт в виде белого твердого вещества (3,85 г; 60%). 1 Н ЯМР (CD3OD, 400 МГц) : 0,55-0,58 (m, 2 Н), 0,62-0,64 (m, 2 Н), 1,22 (s, 3H), 1,46 (s, 9 Н), 2,08 (m,1 Н), 2,36 (m, 1 Н), 2,94 (q, 2 Н), 3,36-3,41 (m, 2 Н), 3,56 (m, 1 Н), 3,74-3,86 (m, 2 Н). трет-Бутил-(3S)-3-(2,3-дихлорбензоил)[(1-метилциклопропил)метил]амино-пирролидин-1-карбоксилат получали способом, аналогичным описанному в подготовительном примере 2, используя амин из подготовительного примера 24 и 2,3-дихлорбензоилхлорид с получением желаемого продукта (448 мг; 85%). 1 Н ЯМР (DMSO-D6, 400 МГц, ротамеры) : 0,15-0,18 (m, 1,5H), 0,26-0,31 (m, 1,5 Н), 0,42 (m, 0,5 Н),0,53 (m, 1 Н), 0,88 (d, 2H), 1,07 (s, 1,5H), 1,33 (s, 4H), 1,39 (s, 5H), 2,05 (m, 1 Н), 2,55 (m, 0,5 Н), 2,82 (m,0,5 Н), 2,96 (m, 0,5 Н), 3,10-3,22 (m, 2 Н), 3,31 (m, 1 Н), 3,49-3,56 (m, 1,5 Н), 3,65 (m, 0,5 Н), 4,23 (m, 0,5 Н),7,35 (m, 1H), 7,42 (m, 1H), 7,66 (m, 1 Н). трет-Бутил-(3S)-3-(3-хлор-2-метилбензоил)[(1-метилциклопропил)метил]аминопирролидин-1- 28011217 карбоксилат получали способом, аналогичным описанному в подготовительном примере 3, используя амин из подготовительного примера 24 и 3-хлор-2-метилбензойную кислоту с получением желаемого продукта (300 мг; 60%). 1 Н ЯМР (DMSO-D6, 400 МГц, ротамеры) : 0,18 (m, 1 Н), 0,24-0,32 (m, 2 Н), 0,51 (m, 1H), 0,83 (s, 2 Н),1,06 (s, 1 Н), 1,32 (s, 4 Н), 1,39 (s, 5 Н), 1,85 (m, 0,5 Н), 2,04 (m, 1 Н), 2,18 (s, 2 Н), 2,22 (s, 1H), 2,56 (m, 1 Н),2,95 (m, 1 Н), 3,09 (m, 1 Н), 3,18-3,22 (m, 2 Н), 3,48-3,55 (m, 1,5 Н), 3,66 (m, 0,5 Н), 4,22 (m, 0,5 Н), 7,15 (m,1H), 7,26 (t, 1 Н), 7,45 (d, 1H). MS APCI+ m/z 407 [МН]+. Подготовительный пример 27. трет-Бутил-(3S)-3-[(циклобутилкарбонил)амино]пирролидин-1 карбоксилат Циклобутанкарбонилхлорид (9 г; 76 ммоль) добавляли к раствору триэтиламина (12,5 мл; 89,7 ммоль) и трет-Бутил-(3S)-3-аминопирролидин-1-карбоксилата (12,87 г; 69 ммоль) в дихлорметане (385 мл) при комнатной температуре в атмосфере азота. После перемешивания в течение 18 ч при комнатной температуре реакционную смесь промывали водой, сушили над сульфатом магния и концентрировали в вакууме, получая указанный в заголовке продукт в виде светло-коричневого стекловидного материала(0,88 мл; 6,3 ммоль). Раствор перемешивали в течение 18 ч при комнатной температуре и растворитель удаляли при пониженном давлении. Неочищенный продукт очищали колоночной хроматографией на силикагеле, используя градиент пентан:этилацетат (4:1 по объему) с изменением до пентанэтилацетат(1:1 по объему) с получением указанного в заголовке соединения в виде бесцветного масла (1,40 г; неочищенное), которое используют непосредственно на следующей стадии. Вос-защищенный продукт из подготовительного примера 2 (46 г; 107 ммоль) растворяли в дихлорметане (85 мл) в атмосфере азота и реакционную смесь обрабатывали трифторуксусной кислотой (85 мл; 1 моль), добавляя по каплям при 0 С. Затем реакционную смесь перемешивали при комнатной температуре в течение 4 ч, упаривали при пониженном давлении, дважды подвергали азеотропной перегонке с толуолом и концентрировали в вакууме. Остаток помещали в дихлорметан (400 мл) и промывали 1 М водным раствором гидроксида натрия (200 мл). Органическую фазу отделяли, сушили над сульфатом магния и концентрировали в вакууме. Оста- 29011217 ток подвергали азеотропной перегонке с этилацетатом (10) и затем сушили под вакуумом с получением свободного основания указанного в заголовке продукта в виде смолы (34 г; 97%). Часть этого продукта(24 г; 70 ммоль) в изопропаноле (400 мл) обрабатывали раствором гидрата этан-дисульфоновой кислоты(6,65 г; 35 ммоль) в изопропаноле (70 мл). Растворитель удаляли в вакууме и остаток подвергали азеотропной перегонке с этилацетатом с получением бежевой пены, которую кристаллизовали из смеси изопропанол/диизопропиловый эфир, получая беловатое твердое вещество (23,3 г). Это твердое вещество перекристаллизовывали из смеси изопропанол/метанол (700 мл изопропанола и минимальное количество метанола, необходимого для достижения растворимости) и сушили под высоким вакуумом, получая указанное в заголовке соединение в виде белого твердого вещества (13,94 г). 1 Н ЯМР (CD3OD, 400 МГц) : 1,40-1,74 (m, 7 Н), 1,92 (m, 1 Н), 2,52 (m, 2 Н), 3,24 (s, 2 Н), 3,30 (m, 1H),3,56 (m, 1H), 3,78 (m, 3H), 4,33 (m, 1 Н), 7,35 (dd, 1H), 7,42 (t, 1H), 7,63 (d, 1H). Хлористый водород (4M в 1,4-диоксане, 37 мл, 148 ммоль) добавляли к раствору продукта из подготовительного примера 3 (6,65 г; 14,93 ммоль) в дихлорметане (40 мл). После перемешивания при комнатной температуре в течение 18 ч растворитель удаляли выпариванием при пониженном давлении с получением твердого вещества, которое растирали с эфиром с получением указанного в заголовке продукта (5,5 г) в виде белого твердого вещества. 1 Н ЯМР (CD3OD, 400 МГц) : 1,54 (m, 4H), 1,73 (m, 3H), 1,92 (m, 1H), 2,52 (m, 2H), 3,25 (m, 1H),3,52 (m, 1H), 3,78 (m, 3H), 4,32 (m, 1H), 7,37 (m, 2H). MS APCI+ m/z 345 [M]+. Пример 3. Гидрохлорид 3-хлор-N-циклопентил-2-метил-N-[(3S)-пирролидин-3-ил]бензамида трет-Бутил-(3S)-3-[циклопентил(3-хлор-2-метилбензоил)амино]пирролидин-1-карбоксилат получали способом, аналогичным описанному в подготовительном примере 3, используя амин из подготовительного примера 1 и 3-хлор-2-метилбензойную кислоту с получением желаемого продукта (606 мг; неочищенный).MS APCI+ m/z 407 [МН]+. Гидрохлорид 3-хлор-N-циклопентил-2-метил-N-[(3S)-пирролидин-3-ил]бензамида получали из упомянутого выше соединения способом, аналогичным описанному в примере 2, с получением указанного в заголовке продукта в виде твердого вещества (199 мг; 47%). 1 Н ЯМР (CD3OD, 400 МГц) : 1,44-1,60 (m, 4H), 1,79 (m, 4H), 2,33 (d, 3H), 2,44-2,54 (m, 2 Н), 3,24 (m,1 Н), 3,54 (m, 1 Н), 3,72 (m, 1 Н), 3,80 (m, 2 Н), 4,31 (m, 1 Н), 7,16 (dd, 1 Н), 7,28 (t, 1H), 7,47 (d, 1H).
МПК / Метки
МПК: C07D 207/14, A61K 31/40
Метки: захвата, ингибиторов, качестве, моноаминов, обратного, n-[(3s)пирролидин-3-ил]бензамидные, производные
Код ссылки
<a href="https://eas.patents.su/30-11217-n-3spirrolidin-3-ilbenzamidnye-proizvodnye-v-kachestve-ingibitorov-obratnogo-zahvata-monoaminov.html" rel="bookmark" title="База патентов Евразийского Союза">N-[(3s)пирролидин-3-ил]бензамидные производные в качестве ингибиторов обратного захвата моноаминов</a>
Производные n-пирролидин-3-ил-амида в качестве ингибиторов обратного захвата серотонина и норадреналина

Номер патента: 9881
Опубликовано: 28.04.2008
Авторы: Браун Алан Дэниел, Фиш Пол Винсент, Фрэй Майкл Джонатан, Лэнсделл Марк Айан, Уитлок Гэвин Алистер, Эндрюс Марк Дейвид, Уэйкенхат Флориан, Стоби Алан
МПК: A61K 31/40, A61P 25/24, A61P 13/00...
Метки: ингибиторов, обратного, норадреналина, производные, захвата, серотонина, качестве, n-пирролидин-3-ил-амида
Формула / Реферат:
1. Соединение формулы (I) и его фармацевтически и/или ветеринарно приемлемые соль, сольват, эфир или амид либо соль или сольват такого эфира или амида; где R2 представляет собой арил или гетероарил, каждый из которых возможно замещен по меньшей мере одним заместителем, независимо выбранным из С1-8алкила, С1-8алкокси, ОН, галогено, CF3, OCF3, SCF3, гидрокси-С1-6алкила, С1-4алкокси-С1-6алкила и С1-4алкил-S-С1-4алкила; R3 представляет собой...
Производные аминоиндана в качестве ингибиторов обратного захвата серотонина и захвата норэпинефрина

Номер патента: 7655
Опубликовано: 29.12.2006
Авторы: Келер Ян, Пюшль Аск, Брегнедаль Петер, Бегесе Клаус Петер
МПК: A61K 31/135, A61P 25/22, A61K 31/357...
Метки: обратного, аминоиндана, ингибиторов, захвата, норэпинефрина, производные, серотонина, качестве
Формула / Реферат:
1. Соединение аминоиндана, имеющее формулу I где X означает -O-, -S- или -CR4R5-; Y означает -CR6R7-, -CR6R7-CR8R9- или -CR6=CR7-; или X и Y вместе образуют группу -CR4=CR5- или -CR4=CR5-CR6R7-; и U означает -O-, -S- или -CR10R11 или X означает -O-, -S- или -CR4R5-; и Y и U вместе образуют группу -CR6=CR7-, -CR6=CR7-CR10R11- или -CR6R7-CR10=CR11-; или X, и Y, и U вместе образуют -CR4=CR5-CR6=CR7-; R1 и R2 независимо выбраны из водорода,...
Производные биариловых эфиров, полезные в качестве ингибиторов обратного захвата моноамина

Номер патента: 5671
Опубликовано: 28.04.2005
Авторы: Адам Мейвис Дайан, Хауард Гарри Ралф Младший
МПК: A61K 31/13, A61P 25/24, C07C 217/58...
Метки: качестве, производные, обратного, полезные, моноамина, захвата, ингибиторов, биариловых, эфиров
Формула / Реферат:
1. Соединение формулы где n и m независимо выбраны из единицы и двух; R1 и R2 независимо выбраны из водорода и (C1-C4)алкила; R3 и R4 независимо выбраны из водорода и (C1-C4)алкила; каждый X независимо выбран из фенила и 5- или 6-членного гетероцикла, содержащего от 1 до 3 гетероатомов, выбранных из N, O и S, где каждый X может быть дополнительно замещен водородом, галогено, (C1-C4)алкилом, оксо, амино и группировкой NR5(C=O)(C1-C4)алкил, где...
Производные фенил-гетероциклильных эфиров в качестве ингибиторов обратного захвата серотонина

Номер патента: 8936
Опубликовано: 26.10.2007
Авторы: Хепворт Дейвид, Хауард Гарри Ральф Мл., Адам Мейвис Дайан, Миддлтон Доналд Стюарт, Стоуби Алан, Эндрюс Марк Дейвид, Джаймер Джеффри Эдвард
МПК: C07D 213/65, A61P 25/24, A61K 31/44...
Метки: производные, серотонина, ингибиторов, захвата, обратного, эфиров, качестве, фенил-гетероциклильных
Формула / Реферат:
1. Соединение общей формулы (I), его фармацевтически приемлемые соли, сольваты или полиморфы где L и U, которые могут быть одинаковыми или разными, представляют собой -N- или -С(Н)-; M представляет собой -N-, -N+(-O-)- или -C(R4)-; Q представляет собой -N- или -C(R4)-; кольцо А содержит 1 или 2 атома азота, в том случае, когда M представляет собой -N+(O-)-, тогда кольцо А не содержит других атомов азота; R1 и R2, которые могут быть одинаковыми...
Производные 4-(2-фенилоксифенил)пиперидина или -1,2,3,6- тетрагидропиридина в качестве ингибиторов обратного захвата серотонина

Номер патента: 9417
Опубликовано: 28.12.2007
Авторы: Банг-Андерсен Бенни, Кролл Фридрих, Келер Ян
МПК: A61K 31/451, A61K 31/4418, A61P 25/00...
Метки: производные, обратного, качестве, 1,2,3,6, тетрагидропиридина, ингибиторов, захвата, серотонина, 4-(2-фенилоксифенил)пиперидина
Формула / Реферат:
1. Производное 4-(2-фенилоксифенил)пиперидина или 1,2,3,6-тетрагидропиридина общей формулы I где пунктирная линия ---- означает простую связь или двойную связь; R1, R2, R3, R4, R5 независимо выбраны из водорода, галогена, C1-6-алкила, C1-6-алкилокси, галоген-С1-6-алкила; или R2 и R3 вместе образуют гетероцикл, конденсированный с фенильным кольцом, выбранный из a R1, R4, R5 имеют определенные выше значения; R6, R7, R8, R9 независимо...