Производные циклогексилпиразол-лактама в качестве ингибиторов 11-бета-гидроксистероиддегидрогеназы 1
Номер патента: 15675
Опубликовано: 31.10.2011
Авторы: Красутский Алексей Павлович, Сюй Яньпин, Анцевено Петер Бьяджо, Виннероски Леонард Лэрри, Снайдер Нэнси Джун, Уоллэйс Оуэн Брендан, Стефенсон Грегори Алан, Айкер Томас Дэниел, Мэбри Томас Эдвард, Тянь Хунци, Саеед Ашраф, Ли Жэньхуа





























Формула / Реферат
1. Соединение, структура которого представлена формулой

где R1представляет собой -H, -галоген;
R2 представляет собой -галоген;
R3 представляет собой -H;
R4 представляет собой -OH, -галоген, -(C1-C6)алкокси, -O-CH2-C(O)NH2, -C(O)N(R10)(R11),

при этом пунктирная линия указывает на точку присоединения к положению R4в формуле I;
где n равно 1;
R5 представляет собой -H, -галоген, -OH, -CN, -(C1-C4)алкил (необязательно замещенный 1-3 атомами галогена), -C(O)OH, -O-(C1-C4)алкил (необязательно замещенный 1-3 атомами галогена), -SO2-(C1-C4)алкил, -N(R8)(R8), -фенил (R21) (R21), -C(O)-NH-(C3-C6) циклоалкил,

при этом пунктирная линия указывает на точку присоединения к положению, обозначенному как R5;
где m равно 1;
где n равно 1;
R6 представляет собой -H, -галоген, -CN, -O-(C1-C4)алкил (необязательно замещенный 1-3 атомами галогена) или

R7 представляет собой -H, -галоген или -(C1-C4)алкил (необязательно замещенный 1-3 атомами галогена);
R8 в каждом случае независимо представляет собой -H, -(C1-C6) алкил или -S(O2)-(C1-C3)алкил;
R9 представляет собой -Н или -галоген;
R10 и R11, каждый независимо, представляют собой -H или -(C1-C4)алкил, или R10 и R11 вместе с азотом, к которому они присоединены, образуют пиперидинил;
R20 в каждом случае представляет собой -H;
R21 в каждом случае независимо представляет собой -H или -галоген; и
R22 в каждом случае представляет собой -H;
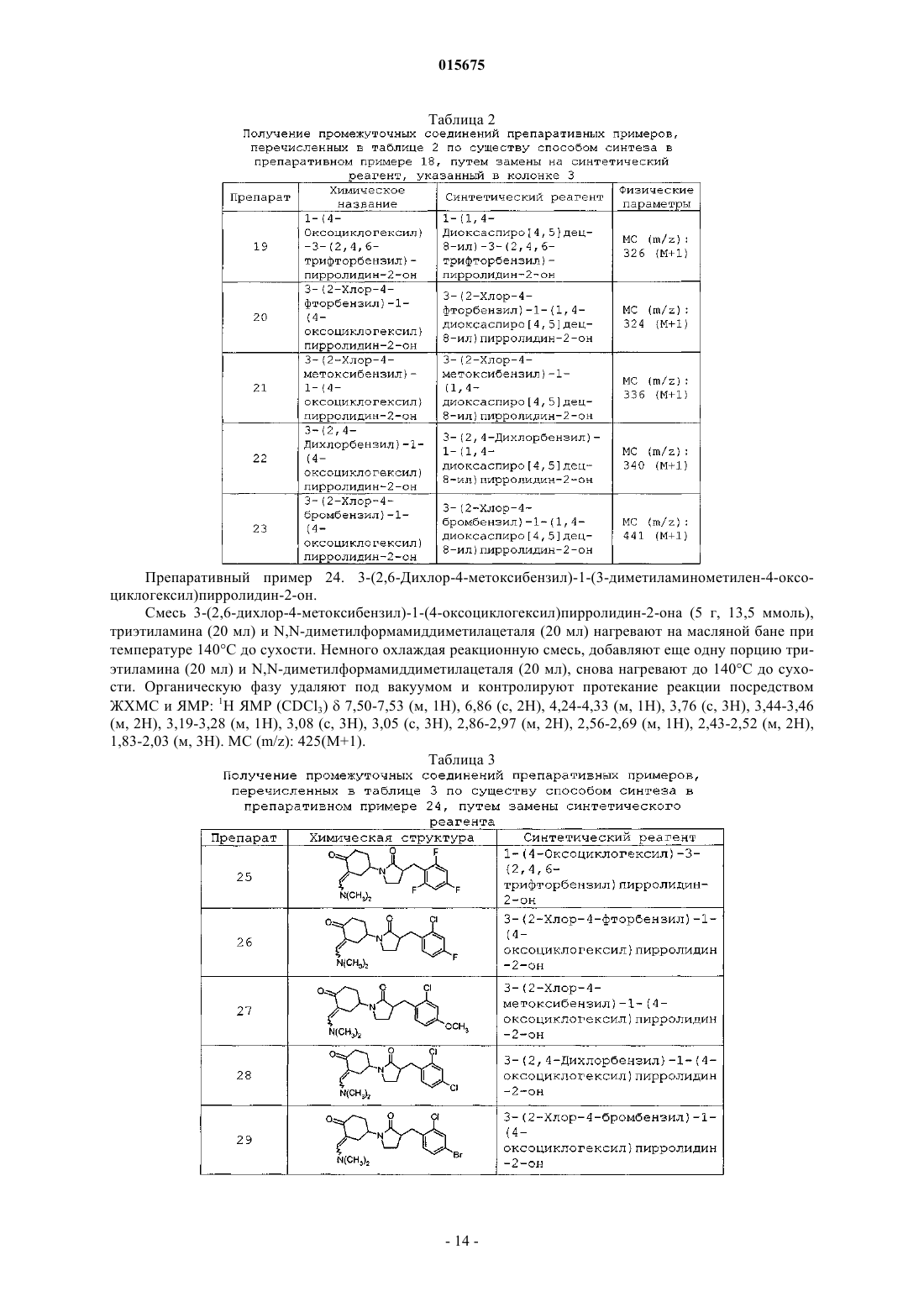
или его стереоизомер; или его фармацевтически приемлемая соль.
2. Соединение по п.1, структура которого представлена формулой

где R0 представляет собой

или его стереоизомер; или его фармацевтически приемлемая соль.
3. Соединение по п.1 или 2, где R1 и R2представляют собой хлор, или его стереоизомер; или его фармацевтически приемлемая соль.
4. Соединение по любому из пп.1-3, где R3 представляет собой водород, или его стереоизомер; или его фармацевтически приемлемая соль.
5. Соединение по любому из пп.2-4, где R0 представляет собой

или его фармацевтически приемлемая соль.
6. Соединение по любому из пп.2-4, где R0 представляет собой

или его фармацевтически приемлемая соль.
7. Соединение по любому из пп.1-6, где R4 представляет собой

и R7представляет собой водород,
или его стереоизомер; или его фармацевтически приемлемая соль.
8. Соединение по любому из пп.1-6, где R4 представляет собой

и R6представляет собой водород,
или его стереоизомер; или его фармацевтически приемлемая соль.
9. Соединение по любому из пп.1-8, где R5 представляет собой

или его стереоизомер; или его фармацевтически приемлемая соль.
10. Соединение по любому из пп.1-8, где R5 представляет собой хлор или фтор, или его стереоизомер; или его фармацевтически приемлемая соль.
11. Соединение по любому из пп.1-8, где R5 представляет собой фтор, или его стереоизомер; или его фармацевтически приемлемая соль.
12. Соединение по п.1, которое представляет собой (3R)-3-[(3,5-дихлор-4'-фтор[1,1'-бифенил]-4-ил)метил]-1-[(5S)-4,5,6,7-тетрагидро-1H-индазол-5-ил]-2-пирролидинон, или его фармацевтически приемлемая соль.
13. Соединение по п.1, которое представляет собой (3R)-3-(3,5-дихлор-4'-фторбифенил-4-илметил)-1-(4,5,6,7-тетрагидро-1H-индазол-4-ил)пирролидин-2-он, или его фармацевтически приемлемая соль.
14. Фармацевтическая композиция, которая содержит соединение по любому из пп.1-13 или его стереоизомер, или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель.
15. Применение соединения по любому из пп.1-13, или его стереоизомера, или его фармацевтически приемлемой соли для получения лекарственного средства.
16. Промежуточное соединение для получения соединения по п.12, представляющее собой

17. Промежуточное соединение для получения соединения по п.12, представляющее собой

18. Кристаллический (3R)-3-[(3,5-дихлор-4'-фтор[1,1'-бифенил]-4-ил)метил]-1-[(5S)-4,5,6,7-тетрагидро-1H-индазол-5-ил]-2-пирролидинон.
19. Кристаллический (3R)-3-[(3,5-дихлор-4'-фтор[1,1'-бифенил]-4-ил)метил]-1-[(5S)-4,5,6,7-тетрагидро-1H-индазол-5-ил]-2-пирролидинон по п.18, имеющий характерные пики на рентгенограмме при 2θ углах дифракции 6,0±0,1°, 12,0±0,1° и 18,1±0,1°.
Текст
Дата публикации и выдачи патента Номер заявки В изобретении описаны новые соединения формулы I обладающие антагонистической активностью по отношению к 11-HSD первого типа, а также способы получения указанных соединений. Согласно другому варианту реализации в изобретении описаны фармацевтические композиции, содержащие соединения формулы I, а также способы применения указанных соединений и композиций для лечения диабета, гипергликемии, ожирения,гипертензии, гиперлипидемии, метаболического синдрома и других состояний, связанных с активностью 11-HSD 1 типа.(71)(73) Заявитель и патентовладелец: ЭЛИ ЛИЛЛИ ЭНД КОМПАНИ (US) 015675 Настоящая заявка испрашивает приоритет предварительной заявки США 60/745320, поданной 21 апреля 2006 г. Изобретение относится к соединениям, представляющим собой ингибиторы 11 гидроксистероиддегидрогеназы 1 типа ("11HSD1"), к фармацевтическим композициям, содержащим такие соединения,и применению таких соединений и композиций для лечения человека или животного, а также к новым промежуточным соединениям, которые можно применять для получения ингибиторов. Настоящие соединения демонстрируют эффективное и селективное ингибирование 11HSD1, и в этом качестве их можно применять при лечении чувствительных к модуляции 11HSD1 нарушений, например диабета,метаболического синдрома, расстройства познавательной способности и т.п. Глюкокортикоиды, функционирующие в печени, жировой ткани и в мышцах, являются важными регуляторами метаболизма глюкозы, липидов и белков. Хронический избыток глюкокортикоидов связан с резистентностью к инсулину, висцеральным ожирением, гипертензией и дислипидемией, которые также представляют собой классические признаки метаболического синдрома. 11HSD1 катализирует преобразование неактивного кортизона в активный кортизол и, как полагают, вовлечен в развитие метаболического синдрома. Данные, полученные для грызунов и человека, свидетельствуют о связи 11-HSD1 с метаболическим синдромом. Данные позволяют предположить, что лекарственное средство,специфично ингибирующее 11HSD1 у пациентов с диабетом 2 типа, снижает уровень глюкозы в крови, замедляя глюконеогенез в печени, снижает центральное ожирение, улучшает атерогенный липопротеиновый фенотип, уменьшает кровяное давление и снижает резистентность к инсулину. Действие инсулина в мышцах должно усиливаться, и секреция инсулина -клетками островков также может возрастать. Данные, полученные при исследовании животных и человека, также свидетельствуют о том, что избыток глюкокортикоидов нарушает познавательную функцию. Недавние результаты показывают, что инактивация 11HSD1 усиливает функционирование памяти как у человека, так и у мышей. Было показано,что ингибитор 11HSD карбеноксолон улучшает когнитивную функцию у здоровых пожилых людей и у людей, страдающих диабетом 2 типа, а инактивация гена 11HSD1 предотвращает обусловленные старением повреждения у мышей. Недавно было показано, что селективное ингибирование 11HSD1 фармацевтическим агентом улучшает сохранение информации в памяти у мышей. В последние годы появился ряд публикаций, сообщающих об агентах, ингибирующих 11HSD1. См. международную публикацию заявки WO 2004/056744, описывающую адамантилацетамиды в качестве ингибиторов 11HSD1, международную публикацию WO 2005/108360, в которой в качестве ингибиторов 11HSD описаны производные пирролидин-2-она и пиперидин-2-она и международную публикацию WO 2005/108361, в которой в качестве ингибиторов 11HSD1 описаны производные адамантилпирролидин-2-она. Несмотря на существование ряда способов лечения заболеваний, в которые вовлечена 11HSD1, современные способы лечения имеют ряд недостатков, в том числе низкую или недостаточную эффективность, неприемлемые побочные эффекты и противопоказания для некоторых групп пациентов. Таким образом, сохраняется потребность в улучшении способов лечения с применением альтернативных или усовершенствованных фармацевтических агентов, которые ингибируют 11HSD1, и таким образом, могут оказаться полезными при лечении заболеваний, при которых ингибирование 11-HSD1 может оказывать благоприятный эффект. Настоящее изобретение обеспечивает такой вклад в данную область техники благодаря тому, что был обнаружен новый класс соединений, оказывающих сильное и селективное ингибирующее действие на 11HSD1. Отличительной особенностью настоящего изобретения являются представленные конкретные структуры и их активность. Сохраняется потребность в новых способах лечения диабета, метаболического синдрома и расстройств познавательной способности, и задачей настоящего обретения является удовлетворение данной и других потребностей. В настоящем изобретении предложено соединение, структура которого представлена формулой I: при этом пунктирная линия указывает на точку присоединения к положению R4 в формуле I; где n равно 1; при этом пунктирная линия указывает на точку присоединения к положению, обозначенному какR8 в каждом случае независимо представляет собой -H, -(C1-C6) алкил или -S(O2)-(C1-C3)алкил;R20 в каждом случае представляет собой -H;R21 в каждом случае независимо представляет собой -H или -галоген; иR22 в каждом случае представляет собой -H; или его стереоизомер; или его фармацевтически приемлемая соль. В настоящем изобретении предложены соединения формулы I, которые могут быть использованы в качестве мощных и селективных ингибиторов 11HSD1. В настоящем изобретении также предложена фармацевтическая композиция, которая содержит соединение формулы I или его стереоизомер, или его фармацевтическую соль, и фармацевтически приемлемый носитель, разбавитель или эксципиент. Кроме того, в настоящем изобретении предложено применение соединения формулы I или его стереоизомера,или его фармацевтически приемлемой соли для получения лекарственного средства. Предложен также способ лечения метаболического синдрома и связанных с ним нарушений, который включает введение пациенту, при необходимости такого лечения, эффективного количества соединения формулы I или его фармацевтически приемлемой соли. Согласно одному варианту реализации, в настоящем изобретении предложены соединения формулы I или их фармацевтически приемлемая соль, как подробно описано выше. Несмотря на то что для лечения пригодны все соединения согласно настоящему изобретению, некоторые из этих соединений представляют особый интерес и поэтому являются предпочтительными. Ниже перечислены некоторые груп-2 015675 пы предпочтительных соединений. Согласно другому варианту реализации, в изобретении предложено соединение, структура которого представлена формулой: или его стереоизомер; или его фармацевтически приемлемая соль. Согласно другому варианту реализации, в изобретении предложено соединение, структура которого представлена вышеуказанными формулами, где R1 и R2 представляют собой хлор, или его стереоизомер; или его фармацевтически приемлемая соль. Согласно другому варианту реализации, в изобретении предложено соединение, структура которого представлена вышеуказанными формулами, где R3 представляет собой водород, или его стереоизомер; или его фармацевтически приемлемая соль. Согласно другому варианту реализации, в изобретении предложено соединение, структура которого представлена вышеуказанными формулами, где R0 представляет собой или его фармацевтически приемлемая соль. Согласно другому варианту реализации, в изобретении предложено соединение, структура которого представлена вышеуказанными формулами, где R0 представляет собой или его фармацевтически приемлемая соль. Согласно другому варианту реализации, в изобретении предложено соединение, структура которого представлена вышеуказанными формулами, где R4 представляет собойR7 представляет собой водород, или его стереоизомер; или его фармацевтически приемлемая соль. Согласно другому варианту реализации, в изобретении предложено соединение, структура которого представлена вышеуказанными формулами, где R4 представляет собой и R6 представляет собой водород, или его стереоизомер; или его фармацевтически приемлемая соль. Согласно другому варианту реализации, в изобретении предложено соединение, структура которого представлена вышеуказанными формулами, где R5 представляет собой или его стереоизомер; или его фармацевтически приемлемая соль. Согласно другому варианту реализации, в изобретении предложено соединение, структура которого представлена вышеуказанными формулами, где R5 представляет собой хлор или фтор, или его стереоизомер; или его фармацевтически приемлемая соль.-3 015675 Согласно другому варианту реализации, в изобретении предложено соединение, структура которого представлена вышеуказанными формулами, где R5 представляет собой фтор, или его стереоизомер; или его фармацевтически приемлемая соль. Согласно другому варианту реализации, в изобретении предложено соединение, которое представляет собой (3R)-3-[3,5-дихлор-4'-фтор[1,1'-бифенил]-4-ил)метил]-1-[(5S)-4,5,6,7-тетрагидро-1H-индазол 5-ил]-2-пирролидинон, или его фармацевтически приемлемая соль. Согласно другому варианту реализации, в изобретении предложено соединение, которое представляет собой (3R)-3-(3,5-дихлор-4'-фторбифенил-4-илметил)-1-(4,5,6,7-тетрагидро-1H-индазол-4-ил)пирроидин-2-он, или его фармацевтически приемлемая соль. Существуют и другие примеры реализации настоящего изобретения, представляющие собой частные случаи описанных выше примеров реализации, ограниченные перечисленными ниже предпочтительными аспектами. В частности, каждый из предпочтительных аспектов может быть использован в сочетании с каждым из вышеперечисленных примеров реализации, и полученная конкретная комбинация будет представлять собой другой пример реализации изобретения, в котором предпочтительный изменяемый параметр имеет более узкое значение по сравнению с исходным предпочтительным параметром. Дополнительный вариант реализации изобретения представляет собой новые промежуточные соединения, описанные в настоящем документе, которые можно применять для получения 11HSD1 ингибиторов согласно формуле I и вариантов реализации изобретения, описанных в настоящем документе. Дополнительный вариант реализации изобретения представляет собой новые промежуточные соединения, описанные в настоящем документе, которые можно применять для получения (3R)-3-[3,5-дихлор-4'фтор[1,1'-бифенил]-4-ил)метил]-1-[(5S)-4,5,6,7-тетрагидро-1H-индазол-5-ил]-2-пирролидинона или его фармацевтически приемлемой соли. Так, одно новое промежуточное соединение для получения указанного выше целевого соединения представляет собой Другое новое промежуточное соединение для получения указанного выше целевого соединения представляет собой Еще один вариант реализации изобретения представляет собой кристаллический (3R)-3-[3,5 дихлор-4'-фтор[1,1'-бифенил]-4-ил)метил]-1-[(5S)-4,5,6,7-тетрагидро-1H-индазол-5-ил]-2-пирролидинон,в частности, имеющий характерные пики на рентгенограмме при 2 углах дифракции 6,00,1, 12,00,1 и 18,10,1. У пациентов, страдающих диабетом 2 типа, часто развивается "инсулинорезистентность", которая приводит к развитию аномального гомеостаза глюкозы и гипергликемии, которые, в свою очередь, приводят к повышенной заболеваемости и ранней смертности. Аномальный гомеостаз глюкозы связан с ожирением, гипертензией и изменениями в метаболизме липидов, липопротеинов и аполипопротеинов. Диабетики 2 типа характеризуются повышенным риском развития нарушений сердечно-сосудистой системы, например атеросклероза, коронарной болезни сердца, удара, заболеваний периферических сосудов,гипертензии, нефропатии, нейропатии и ретинопатии. Следовательно, терапевтический контроль гомеостаза глюкозы, липидного метаболизма, ожирения и гипертензии является важным при регулировании и лечении сахарного диабета. У многих пациентов с инсулинорезистентностью, но у которых еще не развивается диабет 2 типа, также высока вероятность развития "индрома X" или "метаболического синдрома". Метаболический синдром характеризуется инсулинорезистентностью, сопровождаемой брюшным ожирением, гиперинсулинемией, высоким кровяным давлением, низкими уровнями HDL (high-densitylipoproteins - липопротеинов очень низкой плотности), гипертензией, атеросклерозом, коронарной болезнью сердца и хронической почечной недостаточностью. У этих пациентов высок риск развития перечисленных выше сердечно-сосудистых нарушений, независимо от того, развивается у них сахарный диабет или нет. Поскольку предлагаемые соединения способны ингибировать 11HSD1, их можно применять для лечения разнообразных состояний и нарушений, при которых состояние может быть улучшено за счет-4 015675 ингибирования 11HSD1. В настоящем описании такие нарушения и состояния называются "диабетическими нарушениями" и "нарушениями метаболического синдрома". Специалист в данной области способен идентифицировать "диабетические нарушения" и "нарушения метаболического синдрома" в соответствии с активностью 11HSD1, проявляемой либо в патофизиологии нарушения, либо в гомеостатической реакции на нарушение. Таким образом, предлагаемые соединения могут быть использованы, например, для предотвращения, лечения или облегчения заболеваний, или состояний, или симптомов, или последствий, связанных с диабетическими нарушениями" и "нарушениями метаболического синдрома"."Диабетические нарушения" и "нарушения метаболического синдрома" включают, но не ограничиваются ими, диабет, диабет 1 типа, диабет 2 типа, гипергликемию, гиперинсулинемию, прекращение деятельности бета-клеток, усиленную функцию бета-клеток при восстановлении ответной реакции первой фазы, гипергликемию после приема пищи, предотвращение апоптоза, нарушенную гликемию натощак(impaired fasting glucose (IFG, метаболический синдром, гипогликемию, гипер-/гипокалиемию, нормализацию уровней глюкагона, улучшение отношения LDL/HDL (липопротеинов низкой/высокой плотности), уменьшение потребления пищи между регулярными приемами пищи, нарушение потребления пищи, снижение массы тела, синдром поликистозных яичников (olycystic ovarian syndrome (PCOS, ожирение как следствие заболевания диабетом, латентный аутоиммунный диабет взрослых (latent autoimmunediabetes in adults (LADA, инсулит (insulitis), пересадку островковой ткани, детский диабет, диабет, обусловленный беременностью, отсроченные осложнения диабета, микро-/макроальбуминурию, нефропатию, ретинопатию, нейропатию, изъязвление ступней при диабете, сниженную перистальтику кишечника из-за введения глюкагона, синдром укороченной тонкой кишки, антидиарейные эффекты, повышение желудочной секреции, снижение кровотока, эректильную дисфункцию, глаукому, послеоперационный стресс, уменьшение повреждения ткани органов, вызванного реперфузией тока крови вследствие ишемии, ишемическое повреждение сердца, сердечную недостаточность, застойную сердечную недостаточность, удар, инфаркт миокарда, аритмию, преждевременную смерть, антиапоптоз, заживление ран, нарушенную толерантность к глюкозе (impaired glucose tolerance (IGT, синдромы инсулинорезистентности, метаболический синдром, синдром X, гиперлипидемию, дислипидемию, гипертриглицеридемию,гиперлипопротеинемию, гиперхолестеринемию, артериосклероз, включая атеросклероз, глюкагоному,острый панкреатит, сердечно-сосудистые заболевания, гипертензию, гипертрофию сердца, желудочнокишечные нарушения, ожирение, диабет как следствие ожирения, диабетическую дислипидемию и т.д. Таким образом, настоящее изобретение также относится к способу лечения "диабетических нарушений" и "нарушений метаболического синдрома", который также включает снижение или устранение одного или нескольких нежелательных побочных эффектов, имеющихся у существующих способов лечения. Кроме того, в настоящем изобретении предложено соединение формулы I или его фармацевтически приемлемая соль, или фармацевтическая композиция, которая содержит соединение формулы I или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, разбавитель или эксципиент, применяемые для ингибирования активности 11HSD1; применяемые для ингибирования клеточной ответной реакции, медиатором которой является активность 11HSD1, у млекопитающего; применяемые для снижения гликемического уровня у млекопитающего; применяемые для лечения заболевания, вызываемого избыточной активностью 11HSD1; применяемые для лечения диабетических и других метаболических нарушений у млекопитающего; и применяемые для лечения диабета, метаболического синдрома, ожирения, гипергликемии, атеросклероза, ишемической болезни сердца, удара, нейропатии и для заживления ран. Таким образом, способы, предлагаемые в этом изобретении, охватывают профилактическое и терапевтическое введение соединения формулы I. В настоящем изобретении также предложено применение соединения формулы I или его фармацевтически приемлемой соли для производства лекарственного средства, пригодного для ингибирования активности 11HSD1; для изготовления лекарственного средства, пригодного для ингибирования клеточной ответной реакции, медиатором которой является активность 11HSD1, у млекопитающего; для изготовления лекарственного средства, пригодного для снижения гликемического уровня у млекопитающего; для изготовления лекарственного средства, пригодного для лечения заболевания, вызываемого избыточной активностью 11HSD1; для изготовления лекарственного средства, пригодного для лечения диабетических и других метаболических нарушений у млекопитающего; и для изготовления лекарственного средства, пригодного для предотвращения или лечения диабета, метаболического синдрома,ожирения, гипергликемии, атеросклероза, ишемической болезни сердца, удара, нейропатии и плохого заживления ран. В настоящем изобретении также предложены способ лечения состояний, вызываемых избыточной активностью 11HSD1 у млекопитающего; способ ингибирования активности 11HSD1 у млекопитающего; способ ингибирования клеточной ответной реакции, медиатором которой является активность 11HSD1, у млекопитающего; способ снижения гликемического уровня у млекопитающего; способ лечения диабетических и других метаболических нарушений у млекопитающего; способ предотвращения или лечения диабета, метаболического синдрома, ожирения, гипергликемии, атеросклероза, ишемической болезни сердца, удара, нейропатии и плохого заживления ран; причем указанные способы вклю-5 015675 чают введение млекопитающему, при необходимости такого лечения, такого количества соединения формулы I или его фармацевтически приемлемой соли, или фармацевтической композиции, которая содержит соединение формулы I или его фармацевтически приемлемую соль и фармацевтически приемлемый носитель, разбавитель или эксципиент, которое пригодно для ингибирования активности 11-HSD1. Кроме того, в настоящем изобретении предложена фармацевтическая композиция, которая содержит соединение формулы I или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель, разбавитель или эксципиент; адаптированная для применения при ингибировании активности 11HSD1; адаптированная для применения при ингибировании клеточных ответных реакций, медиатором которых является активность 11HSD1; адаптированная для применения при снижении гликемического уровня у млекопитающего; адаптированная для применения при лечении диабетических и других метаболических нарушений у млекопитающего; и адаптированная для применения при предотвращении или лечении диабета, метаболического синдрома, ожирения, гипергликемии, атеросклероза, ишемической болезни сердца, удара, невропатии и плохого заживления ран. Согласно дополнительному аспекту изобретения, настоящие соединения вводят в комбинации с одним или несколькими дополнительными активными веществами в любых подходящих соотношениях. Такие дополнительные активные вещества можно, например, выбрать из противодиабетических средств,средств, предназначенных для лечения ожирения, гипотензивных средств, средств, предназначенных для лечения осложнений, вызываемых или связанных с диабетом, и средств, предназначенных для лечения осложнений и нарушений, вызываемых или связанных с ожирением. Ниже представлен список нескольких групп таких комбинаций. Следует понимать, что каждое из указанных средств может быть использовано в комбинации с другими указанными средствами, что позволяет получать дополнительные комбинации. Таким образом, согласно дополнительному варианту реализации изобретения, настоящие соединения можно вводить в комбинации с одним или несколькими противодиабетическими средствами. Подходящие противодиабетические средства включают инсулин, аналоги и производные инсулина,например, описанные в патентной заявке EP 792290 (Novo Nordisk A/S), например, NB29-тетрадеканоилдез-(B30)-человеческий инсулин, в патентных заявках EP 214826 и EP 705275 (Novo Nordisk A/S), например, AspB28-человеческий инсулин, в патенте США 5504188 (Eli Lilly), например, LysB28 ProB29 человеческий инсулин, в патентной заявке EP 368187 (Aventis), например, производные Lantus, GLP-1 и GLP-1, подобные описанным в международной патентной заявке WO 98/08871 (Novo Nordisk A/S), а также гипогликемические средства, активные при пероральном приеме. Гипогликемические средства, активные при пероральном введении, предпочтительно включают имидазолины, сульфонилмочевины, бигуанидины, меглитиниды, оксадиазолидиндионы, тиазолидиндионы, активаторы инсулина, средства, усиливающие секрецию инсулина, например глимепирид, ингибиторы -глюкозидазы, средства, воздействующие на АТФ-зависимые калиевые каналы -клеток, например вещества, открывающие калиевые каналы, например, описанные в заявках WO 97/26265, WO 99/03861 иWO 00/37474 (Novo Nordisk A/S) или митиглинид, или блокиратор калиевых каналов, например, BTS67582, натеглинид, антагонисты глюкагона, например, описанные в заявках WO 99/01423 и WO 00/39088(Peroxisome proliferator-activated receptor (рецептор активатора пролиферации пероксисом, включающие подтипы PPAR-альфа, PPAR-гамма и PPAR-дельта, и агонисты RXR (ретиноидных X рецепторов), например, ALRT-268, LG-1268 или LG-1069. Согласно другому варианту реализации изобретения, предлагаемые соединения вводят в комбинации с инсулином или аналогом или производным инсулина, например, NB29-тетрадеканоил-дез-(B30)человеческим инсулином, AspB28-человеческим инсулином, LysB28 ProB29-человеческим инсулином, Lantus или смешанным препаратом, включающим одно или несколько указанных соединений. Согласно дополнительному варианту реализации изобретения, предлагаемые соединения вводят в комбинации с сульфонилмочевиной, например, глибенкламидом, глипизидом, толбутамидом, хлоропамидемом, толазамидом, глимепридом, гликазидом и глибуридом. Согласно другому варианту реализации изобретения, настоящие соединения вводят в комбинации с бигуанидом, например метформином. Согласно еще одному варианту реализации изобретения, настоящие соединения вводят в комбинации с меглитинидом, например репаглинидом или натеглинидом.-6 015675 Согласно еще другому варианту реализации изобретения, предлагаемые соединения вводят в комбинации с активатором инсулина на основе тиазолидиндиона, например с троглитазоном, циглитазоном,пиоглитазоном, розиглитазоном, изаглитазоном, дарглитазоном, энглитазоном, CS-011/CI-1037 или T 174 или соединениями, описанными в заявках WO 97/410 97, WO 97/41119, WO 97/41120, WO 00/41121 иWO 98/45292 (Dr. Reddy's Research Foundation). Согласно еще другому варианту реализации изобретения, предлагаемые соединения могут быть введены в комбинации с активатором инсулина, таким как, например, GI 262570, YM-440, MCC-555,JTT-501, AR-H039242, KRP-297, GW-409544, CRE-16336, AR-H049020, LY510929, MBX-102, CLX-0940,GW-501516 или соединениями, описанными в заявках WO 99/19313, WO 00/50414, WO 00/63191, WO 00/63192, WO 00/63193, например, рагаглитазаром (NN 622 или (-)DRF 2725) (Dr. Reddy's Research Foundation) и WO 00/23425, WO 00/23415, WO 00/23451, WO 00/23445, WO 00/23417, WO 00/23416, WO 00/63153, WO 63196, WO 00/63209, WO 00/63190 и WO 00/63189 (Novo Nordisk A/S). Согласно дополнительному варианту реализации изобретения, настоящие соединения вводят в комбинации с ингибитором -глюкозидазы, например воглибозой, эмиглитатом, миглитолом или акарбозой. Согласно другому варианту реализации изобретения, настоящие соединения вводят в комбинации с препаратом, воздействующим на АТФ-зависимые калиевые каналы -клеток, например толбутамидом,глибенкламидом, глипизидом, гликазидом, BTS-67582 или репаглинидом. Согласно еще одному варианту реализации изобретения, настоящие соединения можно вводить в комбинации с натеглинидом. Согласно еще другому варианту реализации изобретения, настоящие соединения вводят в комбинации с антилипидемическим средством или антигиперлипидемическим средством, например холестирамином, колестиполом, клофибратом, гемфиброзилом, ловастатином, правастатином, симвастатином, питавастатином, розувастатином, пробуколом, декстротироксином, фенофибратом или аторвастином. Согласно еще другому варианту реализации изобретения, настоящие соединения вводят в комбинации с соединениями, понижающими потребление пищи. Согласно другому варианту реализации изобретения, настоящие соединения вводят в комбинации с несколькими из вышеупомянутых соединений, например, в комбинации с метформином и сульфонилмочевиной, например, глибуридом; сульфонилмочевиной и акарбозом; натеглинидом и метформином; репаглинидом и метформином; акарбозом и метформином, сульфонилмочевиной метформином и троглитазоном; инсулином и сульфонилмочевиной; инсулином и метформином; инсулином, метформином и сульфонилмочевиной; инсулином и троглитазоном; инсулином и ловастатином, и так далее. Применяемые в настоящем документе при описании соединений общие термины имеют свои обычные значения. Применяемые в настоящем документе термины "(C1-C3)алкил", "(C1-C4)алкил" или "(C1-C6)алкил" относятся к насыщенным алифатическим группам с прямой или разветвленной цепью с указанным количеством углеродных атомов, таких как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил,трет-бутил, и тому подобное. Термин "(C1-C6)алкокси" означает C1-C6 алкильную группу, присоединенную через атом кислорода, и включает такие группы как, например, метокси, этокси, н-пропокси, изопропокси и тому подобное. Термин "галоген" относится к фтору, хлору, брому и йоду. Термин "(C3-C8) циклоалкил" относится к насыщенному или частично насыщенному карбоциклическому кольцу из 3-8 углеродных атомов, как правило, 3-7 углеродных атомов. Примеры (C3-C8) циклоалкила включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и тому подобное. Применяемый в настоящем документе термин "необязательно замещенный" или "необязательные заместители" означает, что данные группы являются или незамещенными или замещены одним или несколькими указанными заместителями. Если рассматриваемые группы замещены более чем одним заместителем, эти заместители могут быть как одинаковыми, так и различными. Кроме того, применение терминов "независимо", "независимо представляют собой" и "независимо выбирают из" означает, что рассматриваемые группы могут быть как одинаковыми, так и различными. Некоторые из определенных в данном документе терминов могут встречаться в структурных формулах более одного раза, и при таком появлении каждый термин должен быть определен независимо от другого. Следует понимать, что морские свинки, собаки, кошки, крысы, мыши, хомяки и приматы, в том числе люди, являются примерами пациентов в рамках значения термина "пациент". Предпочтительные пациенты включают людей. Термин "пациент" включает домашний скот. К домашнему скоту относятся животные, которых выращивают для производства продуктов питания. Жвачные животные, такие как коровы, быки, телки, волы, овцы, буйволы, бизоны, козы и антилопы, являются примерами жвачных животных. Другие примеры домашнего скота включают свиней и домашних птиц, таких как куры, утки,индейки и гуси. Предпочтительно, если пациент, подвергаемый лечению, является млекопитающим, в частности человеком. Применяемые в настоящем документе термины "лечение" и "лечить" включают их общепринятые-7 015675 значения, т.е. терапию и медицинский уход за пациентом с целью предотвращения, снижения риска возникновения или развития данного состояния или заболевания, предотвращения, ограничения, облегчения течения, улучшения состояния, замедления развития, остановки развития, отсрочки начала возникновения или обращения вспять прогрессирования или снижения тяжести заболевания или состояния у пациента, а также удержания на существующем уровне и/или лечения существующих симптомов заболевания, нарушения или патологического состояния, рассмотренного в настоящем описании, включая смягчение или облегчение симптомов, или осложнений, или вылечивание, или устранение заболевания, нарушения, или состояния. В зависимости от необходимости, предлагаемые способы включают медикаментозное лечение и/или профилактическую терапию. Применяемый в настоящем документе термин "терапевтически эффективное количество" означает количество соединения, предлагаемого в настоящем изобретении, которое способно облегчить симптомы различных патологических состояний, описанных в настоящей заявке. Конкретная доза соединения, вводимого согласно настоящему изобретению, конечно, должна быть определена с учетом конкретных обстоятельств, сопутствующих данному случаю, в том числе например, вводимого соединения, способа введения, состояния самого пациента и патологического состояния, подвергаемого лечению. Термин "композиция" означает фармацевтическую композицию и охватывает фармацевтический продукт, содержащий активный ингредиент (ингредиенты), в том числе, соединение (соединения) формулы I, и инертный ингредиент (ингредиенты), которые составляют носитель. Соответственно, фармацевтические композиции, предлагаемые в настоящем изобретении, включают любые композиции, составленные путем смешивания соединения, предлагаемого в настоящем изобретении, и фармацевтически приемлемого носителя. Термин "по существу чистый" относится к чистой кристаллической форме соединения, содержащей более примерно 90% требуемой кристаллической формы, и предпочтительно, более примерно 95% требуемой кристаллической формы. Термин "подходящий растворитель" относится к любому растворителю или смеси растворителей,инертному по отношению к протекаемой реакции, который в достаточной мере солюбилизирует реагирующие вещества, создавая среду для проведения требуемой реакции. Термин "единичная дозированная форма" означает физически дискретные единицы, применяемые в виде однократных дозировок, вводимых человеку и другим животным, при этом каждая единица содержит заранее установленное количество активного материала, которое по расчетам должно вызвать желаемый терапевтический эффект, вместе с подходящим фармацевтическим носителем. Соединения, предлагаемые в настоящем изобретении, могут иметь один или несколько хиральных центров и могут существовать в разнообразных стереоизомерных конфигурациях. Вследствие существования этих хиральных центров, соединения, предлагаемые в настоящем изобретении, могут встречаться в виде рацематов, в виде индивидуальных энантиомеров или смесей энантиомеров, а также в виде диастереомеров и смесей диастереомеров. Все указанные рацематы, энантиомеры, диастереомеры и их смеси,как разделенные, так и частично разделенные или неразделенные смеси, находятся в пределах объема настоящего изобретения. Для примеров, представленных в настоящем описании, если имеется молекула,которая содержит хиральный центр или центры известной конфигурации, то ее стереохимию указывают в названии и в структурном представлении молекулы. Если стереохимия молекулы неизвестна или не определена, то ее стереохимию не указывают в названии и в структурном представлении молекулы. Варианты реализации изобретения включают приведенные в настоящем документе примеры соединений, и хотя рассматриваемое соединение может отвечать хиральной или конформационной форме или представлять собой соль этой формы, другие примеры реализации настоящего изобретения включают все другие стереоизомерные или конформационные формы описываемых примеров, а также их фармацевтически приемлемые соли. Указанные примеры реализации включают любые выделенные энантиомеры,диастереомеры и/или конформеры рассматриваемых структур, а также любые смеси, содержащие более одной формы. Кроме того, когда в молекуле присутствует двойная связь или полностью, или частично насыщенная циклическая система, или более одного центра асимметрии, или связь с ограниченным вращением вокруг нее, могут образоваться диастереомеры. В объем настоящего изобретения включены любые диастереомеры, в виде разделенных, чистых или частично разделенных диастереомеров или их смесей. Кроме того, некоторые из соединений, предлагаемых в настоящем изобретении, могут существовать в различных таутомерных формах, и в объем настоящего изобретения включены любые таутомерные формы, которые могут образовать указанные соединения. Применяемый в настоящем документе термин "обогащение энантиомером" означает повышение доли одного энантиомера по сравнению с другим. Имеющийся удобный способ выражения достигаемого обогащения энантиомером представляет собой концепцию энантиомерного избытка или "ee", в которой,как показано, используют следующее уравнение:-8 015675 где E1 представляет собой количество первого энантиомера, а E2 представляет собой количество второго энантиомера. Таким образом, если исходное отношение двух энантиомеров равно 50:50, то есть равно отношению энантиомеров в рацемической смеси, и при этом может быть достигнуто энантиомерное обогащение, достаточное для получения конечного отношения, равного 70:30, то значение ee по отношению к первому энантиомеру будет составлять 40%. В то же время, если конечное отношение будет равно 90:10, то значение ee по отношению к первому энантиомеру будет составлять 80%. Предпочтительными являются значения ee, превышающие 90%, еще более предпочтительными являются значенияee, превышающие 95%, и наиболее предпочтительными являются значения ee, превышающие 99%. Специалист в данной области может легко определить значение энантиомерного обогащения при помощи стандартных методик и процедур, например, посредством газовой хроматографии или высокоэффективной жидкостной хроматографии с использованием хиральной колонки. Принцип выбора соответствующей хиральной колонки, элюента и условий, необходимых для разделения энантиомерной пары, хорошо известен специалистам в данной области. Кроме того, конкретные стереоизомеры и энантиомеры соединений формулы I, могут быть получены специалистом в данной области при помощи хорошо известных методик и способов, например, подобных описанным в публикации J. Jacques, et al., "Enantiomers, Racemates, and Resolutions", John Wiley and Sons, Inc., 1981, и E.L. Eliel, S.H. Wilen, "Stereochemistry of OrganicCompounds", (Wiley-Interscience 1994), и в Европейской патентной заявкеEP-A-838448 от 29 апреля 1998 г. Примеры способов разделения включают способы перекристаллизации или хиральной хроматографии. Соединения формулы I могут быть синтезированы специалистом в данной области в соответствии с рядом методик, некоторые из которых проиллюстрированы в процедурах и схемах, представленных ниже. Точная последовательность стадий, необходимых для получения соединений формулы I, зависит от конкретного синтезируемого соединения, исходного соединения и относительной неустойчивости замещенных групп. Необходимые реагенты или исходные материалы обычно доступны специалисту в данной области, а в тех случаях, когда они отсутствуют в продаже, обычный специалист в данной области легко синтезирует их по стандартным методикам, обычно применяемым в данной области техники, наряду с различными процедурами и схемами, представленными ниже. Нижеследующие схемы, препаративные примеры, примеры и процедуры приведены с целью лучшего понимания настоящего изобретения и в любом случае не должны рассматриваться как ограничение объема изобретения. Специалисты в данной области понимают, что могут быть сделаны различные модификации, которые не изменяют объема и сущности настоящего изобретения. Все публикации, упомянутые в описании изобретения, доступны по уровню специалистам в данной области, для которых предназначено это изобретение. Оптимальное время проведения реакций в указанных схемах, препаративных примерах, примерах и процедурах можно определить путем традиционных хроматографических способов отслеживания хода реакции. Более того, предпочтительно проводить реакции, предлагаемые в изобретении, в инертной атмосфере, такой как, например, аргон или азот. Выбор растворителя, как правило, не является решающим, поскольку применяемый растворитель является инертным по отношению к протекающей реакции и существенно повышает растворимость реагирующих веществ с целью осуществления требуемой реакции. Предпочтительно выделить и очистить соединения перед их применением в последующих реакциях. Некоторые соединения могут кристаллизоваться из реакционных растворов в процессе их образования и затем отделяться посредством фильтрации, или реакционный растворитель можно удалить путем экстракции, испарения или декантации. При необходимости, промежуточные и конечные продукты формулы I можно дополнительно очистить посредством таких традиционных способов, как перекристаллизация или хроматография на твердых носителях, таких как силикагель или оксид алюминия. Опытный специалист понимает, что не все заместители совместимы со всеми условиями реакции. В такие соединения на определенном этапе синтеза при помощи способов, известных в данной области техники, могут быть введены защитные группы. Термины и сокращения, применяемые в настоящих схемах, препаративных примерах, примерах и процедурах, имеют свои обычные значения, если не указано иное. Например, применяемые в настоящем документе следующие термины имеют указанные значения:"psi" относится к фунту на квадратный дюйм; "TCX (TLC)" относится к тонкослойной хроматографии;"ВЭЖХ (HPLC)" относится к высокоэффективной жидкостной хроматографии; "Rf" относится к коэффициенту удерживания; "Rt" относится к времени удерживания; относится к части на миллион (м.д.) в сторону уменьшения напряженности магнитного поля относительно сигнала тетраметилсилана; "MC" относится к масс-спектрометрии, если не указано особо, термин "наблюдаемая масса (Observed Mass)" означает [M+H]. "MS(APCi) означает хемоионизационную масс-спектрометрию при атмосферном давлении, "УФ" относится к ультрафиолетовой спектрометрии, "1H ЯМР" относится к спектрометрии протонного ядерного магнитного резонанса. "ЖХМС" относится к жидкостной хроматомасс-спектрометрии,"ГХ/МС" относится к газовой хроматографии/масс-спектрометрии. "ИК" относится к инфракрасной спектрометрии, при этом в ИК спектрах указаны только максимумы поглощения интересующих фраг-9 015675 ментов, а не все максимумы поглощения. "КТ" относится к комнатной температуре.N-этил-N'-(3-диметиламинопропил)карбодиимида,"DBU" относится к 1,8 диазабицикло[5.4.0]ундецену-7, "TBSCl" относится к трет-бутилдиметилсиланилоксиметилхлориду,"NBS" относится к N-бромсукцинимиду, "TsOH" относится к пара-толуолсульфоновой кислоте, "DCE" относится к дихлорэтану, "DAST" относится к (диэтиламино)сера трифториду, "EA/H" относится к смеси этилацетат/гексан, "Pd2(dba)3" относится к бис(дибензилиденацетон)палладию, "BINAP" относится к 2,2'бис(дифенилфосфино-1,1'-бинафталину, "HMP" относится к N-метилпирроллидину, "TMSCN" относится к триметилсилилцианиду, "TBAF" относится к фториду тетрабутиламмония, "Tf2O" относится к трифторметансульфоновому ангидриду, "TBSO" относится к трет-бутилдиметилсиланилокси, "OTf" относится к трифторметансульфонату, MeTi(Oi-Pr)3 относится к метилтитантриизопропоксиду, "BBr3" относится к трибромиду бора, "PBr3" относится к трибромиду фосфора, "Pd(PPh3)4" относится к тетракис(трифенилфосфин)палладию(0), "OAc" относится к ацетату, "ДМЭ" относится к диметилэтану, "Et2O" относится к диэтиловому эфиру, "(Ph3P)4Pd" относится к тетракис(трифенилфосфин)палладию(0),"DMFDMA" относится к N,N-диметилформамиддиметилацеталю, "Et3N" относится к триэтиламину,"tBu" относится к трет-бутилу, "DIPEA" относится к диизопропилэтиламину, "EDC" относится к гидрохлориду -(3-диметиламинопропил)-3-этилкарбодиимида, "НОАс" относится к уксусной кислоте, "boc" относится к трет-бутоксикарбонилу. В структурной формуле, "Ph" относится к фенилу, "Me" относится к метилу, "Et" относится к этилу, "Bn" относится к бензилу, "MeOH" относится к метанолу, "OTf" относится к трифторметансульфонату, "TIPSO" относится к триизопропилсиланилокси, "TBSO" относится к трет-бутилдиметилсиланилокси, "NaBH(OAc)3" относится к триацетоксиборгидриду натрия,"[Ir(cod)Cl]2" относится к ди-хлор-бис 1,2,5,6-эта)-1,5-циклооктадиен)дииридию. Примеры, представленные в настоящем документе, иллюстрируют изобретение и, никоим образом не ограничивают его объем. Названия соединений, указанных в примерах и синтезах, получены с использованием программ AutoNcm 2.2 в ChemDraw Ultra или AutoNom 2000 в MDL ISIS/Draw, версия 2.5SP1, предоставляемых MDL Information Systems, Inc. или предоставлены Chemical Abstracts Services. Спектрометр Varian INOVA 400 МГц применяют для получения 1H ЯМР спектров в указанном растворителе. Прибор Agilent HP1100, оборудованный масс-спектрометром (Agilent MSD SL), применяют для получения ЖХМС. В качестве неподвижной фазы применяют Waters Xterra C18 (2,150 мм, 3,5 микрон), и стандартный способ включал использование градиента 5-100% ацетонитрил/метанол (50:50) с добавлением 0,2% формиата аммония в течение 3,5 мин, а затем выдерживание при 100% В в течение 0,5 мин при температуре колонки 50C и скорости потока 1,0 мл/мин. Другой стандартный способ включал использование градиента 5-100% ацетонитрил/метанол (50:50) с добавлением 0,2% формиата аммония в течение 7,0 мин, а затем выдерживание при 100% В в течение 1,0 мин при температуре колонки 50C и скорости потока 1,0 мл/мин. Дополнительный MC-анализ, проводимый на Agilent MSD (циклический прибор), представляет собой стандартный проточно-инжекционный анализ (FIA), в отсутствие колонки при скорости потока 80% MeOH с добавлением 6,5 мМ ацетата аммония, равной 0,5 мл/мин выдерживаемой в течение 30 с. Схема А Согласно схеме A, в необязательно замещенный фенол (1) вводят защитную группу (например, с помощью TBSCl) и затем превращают в альдегид (2). Соединение 2 реагирует с соединением, содержащим защитную группу (Pg) и уходящую группу (Lg) с образованием эфирного соединения 3. Pg может представлять собой -CH3 или -CH2-фенил и Lg может представлять собой мезилат или галоген. Предпоч- 10015675 тительно, если соединение Lg-Pg представляет собой I-CH3 или Br-CH2-фенил. Альдегид восстанавливают с образованием спирта (4) и затем превращают в соединение 5. Предпочтительно, если соединение 4 галогенируют посредством PBr3 с получением 2-бромметильного соединения. Защита и снятие защиты указанных соединений с целью синтеза соединений формулы I и других веществ хорошо известны специалистам в данной области техники и описаны в литературе. (Например,см.: Greene and Wuts, Protective Groups in Organic Synthesis, Third Edition, John Wiley and Sons Inc., 1999). Препаративный пример 1. Трет-бутил-(3,5-дихлорфенокси)диметилсилан. 3,5-Дихлорфенол (1 кг, 6,13 моль) растворяют в 3 л диметилформамида и охлаждают до 0C. Добавляют имидазол (918,74 г, 6,75 моль), а затем трет-бутилдиметилсилилхлорид (1017,13 г, 6,75 моль). Смесь нагревают до комнатной температуры и перемешивают в течение 15 мин. Смесь выливают в воду(6 л) и экстрагируют эфиром (4 л). Перед высушиванием над сульфатом натрия органическую фазу промывают 2 раза водой, 10% водным раствором хлорида лития, затем соляным раствором. Фильтруют и концентрируют под вакуумом до получения 135 г масла. Препаративный пример 2. 2,6-Дихлор-4-гидроксибензальдегид. Трет-бутил-(3,5-дихлорфенокси)диметилсилан (425 г, 1,5 моль) растворяют в 4 л сухого тетрагидрофурана и охлаждают до -68C. Медленно добавляют 1,1 эквивалентов втор-бутиллития (103,1 г, 1,61 моль) при -68C (1,75 ч). После того как добавление завершено, реакционную смесь перемешивают при-70C в течение 30 мин. Добавляют диметилформамид (168,5 г, 2,3 моль) и реакционную смесь перемешивают при -70C в течение 1 ч. Добавляют 1 М соляной кислоты в воде (3,5 л) и оставляют реакцию нагреваться до комнатной температуры. Реакционную смесь выливают в эфир (5 л), промывают водой, затем соляным раствором. Высушивают над сульфатом натрия и концентрируют под вакуумом с образованием оранжевого твердого вещества. Растирают в порошок, применяя холодный дихлорметан, и фильтруют, выделяя 250 г (80%) бледножелтого твердого вещества. Препаративный пример 3. 2,6-Дихлор-4-метоксибензальдегид. Смешивают 2,6-дихлор-4-гидроксибензальдегид (120 г, 628,24 ммоль) и карбонат калия (173,65 г,1256,5 ммоль) в 900 мл диметилформамида и обрабатывают йодметаном (107 г, 753,9 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 3 ч. Твердое вещество отфильтровывают и выливают в 6 л воды. Твердое вещество фильтруют, промывают несколько раз водой, высушивают на воздухе и растворяют в этилацетате. Промывают водой, затем соляным раствором, а затем высушивают над сульфатом натрия. Фильтруют и концентрируют под вакуумом до объема 100 мл, в этот момент твердое вещество начинает крошиться. Фильтруют, затем фильтрат концентрируют с получением второй партии. Промывают гексаном, объединяют все твердое вещество и высушивают под вакуумом с получением 112,3 г грязно-белого твердого вещества: 1H ЯМР (400 МГц, CDCl3)10,41 (с, 1H), 6,90 (с,2H), 3,87 (с, 3H). Препаративный пример 4. 2,6-Дихлор-4-бензилоксибензальдегид. Смесь 2,6-дихлор-4-гидроксибензальдегида (250 г, 1,3 моль) и карбоната калия (361,8 г, 2,62 моль) в 2 л диметилформамида обрабатывают бензилбромидом (268,64 г, 1,57 моль). Реакционную смесь перемешивают при комнатной температуре в течение 1 ч. Твердое вещество отфильтровывают и выливают в 12 л воды. Твердое вещество фильтруют, промывают несколько раз водой, высушивают на воздухе и растворяют в этилацетате. Высушивают над сульфатом магния, фильтруют и концентрируют под вакуумом до 1,5 л. Оставляют на ночь, затем фильтруют. Твердое вещество промывают минимальным количеством гексана и высушивают под вакуумом. Фильтрат концентрируют под вакуумом и растирают в порошок, применяя гексан, с получением второй партии продукта, что, при смешивании с первой партией, позволяет получить 245 г белых кристаллов. Процедуру повторяют и получают третью партию 80 г в виде светло-коричневого порошка (88% суммарный выход): 1H ЯМР (400 МГц, ДМСО-d6)10,26 (с, 1H),7,43 (м, 5H), 7,28 (с, 2H), 5,25 (с, 2H). Препаративный пример 5. (2,6-Дихлор-4-метоксифенил)метанол. 2,6-Дихлор-4-метоксибензальдегид (112 г, 546 ммоль) суспендируют в 1500 мл этанола и охлаждают на ледяной бане до 7C. Добавляют порциями боргидрид натрия (20,67, 546 ммоль) и получают раствор. Удаляют ледяную баню и перемешивают в течение 2 ч. Осторожно добавляют реакционную смесь к насыщенному раствору хлорида аммония (4 л) и перемешивают до полного гашения. Экстрагируют дихлорметаном (31 л) и высушивают объединенные органические экстракты над сульфатом натрия. Фильтруют и концентрируют под вакуумом с получением 113 г светло-коричневого твердого вещества: 1H ЯМР (400 МГц, CDCl3)6,86 (с, 2H), 4,86 (с, 2H), 3,78 (с, 3H), 2,07 (с, 1H). Препаративный пример 6. (2,6-Дихлор-4-бензилоксифенил)метанол. Вышеназванное соединение, по существу, получают способом синтеза в препаративном примере 5,применяя в качестве исходного вещества 2,6-дихлор-4-бензилоксибензальдегид: 1 Н ЯМР (400 МГц,ДМСО-d6)7,38 (м, 4H), 7,33 (м, 1H), 7,12 (с, 2H), 5,14 (с, 2H), 5,05 (т, 1H), 4,59 (д, 2H). Препаративный пример 7. 2-Бромметил-1,3-дихлор-5-метоксибензол.(2,6-Дихлор-4-метоксифенил)метанол ( 113 г, 545,76 ммоль) растворяют в 1200 мл сухого тетрагид- 11015675 рофурана и охлаждают до 0C в атмосфере азота. В атмосфере азота добавляют PBr3 (59,1 г, 218,3 ммоль) и перемешивают при 0C в течение 30 мин. Выливают в насыщенный водный бикарбонат натрия и экстрагируют этилацетатом. Высушивают и концентрируют под вакуумом с получением 129,4 г продукта в виде грязно-белого твердого вещества: 1H ЯМР (400 МГц, CDCl3)6,88 (с, 2H), 4,73 (с, 2H), 3,79 (с, 3H). Препаративный пример 8. 2-Бромметил-1,3-дихлор-5-бензилоксибензол. Вышеназванное соединение по существу получают способом синтеза в препаративном примере 6 при 89% выходе, применяя в качестве исходного вещества 2,6-дихлор-4-бензилоксифенил)метанол: ES Согласно схеме B, обработка замещенного метилбензола 6 посредством NXS (X=Cl, Nхлорсукцинамид; X=Br, N-бромсукцинамид; X=I, N-йодсукцинамид) в присутствии бензоилпероксида вCCl4 при температуре кипения с обратным холодильником позволяет получить соответствующий бензилгалогенид 7. Препаративный пример 9. 2-Хлор-4-метокси-1-метилбензол. Раствор 3-хлор-4-метилфенола (15 г, 0,11 моль), йодметана (9,8 мл, 0,16 моль), и карбоната калия(22 г, 0,16 моль) в ДМФ (200 мл) нагревают до 50C и перемешивают в течение 2 ч. Охлаждают реакцию до комнатной температуры и гасят посредством 1 н водного HCl. Водную фазу экстрагируют диэтиловым эфиром (Et2O). Органическую фазу промывают соляным раствором, высушивают над MgSO4 и фильтруют. Удаляют растворитель и получают 16,4 г (100%) требуемого продукта. Препаративный пример 10. 1-Бромметил-2-хлор-4-метоксибензол. Раствор 2-хлор-4-метокси-1-метилбензола (2,0 г, 13 ммоль), N-бромсукцинимида (2,7 г, 15 ммоль),и бензоилпероксида (50 мг) в CCl4 (50 мл) нагревают при кипении с обратным холодильником и перемешивают в течение 3 ч. Реакцию охлаждают до комнатной температуры и гасят водой. Водную фазу экстрагируют CH2Cl2, высушивают над MgSO4 и фильтруют. Растворитель удаляют с получением 3,0 г (98%) требуемого продукта. Схема C Согласно схеме C обработка 1,4-диоксаспиро[4,5]декан-8-она хлористо-водородной солью этилового эфира 4-аминомасляной кислоты в присутствии NaBH(OAc)3 приводит к образованию 1-(1,4 диоксаспиро[4,5]дец-8-ил)пирролидин-2-она. Алкилирование достигают путем применения LDA и бензилбромида 5 с получением требуемого лактама 6. Удаление кетальной защиты в кислотных условиях приводит к образованию кетона 7. Соединение 8 получают путем обработки кетона диметоксиметилдиметиламином или реагентом Бредерика. Обработка 8 гидразином дает пиразол Ia. Препаративный пример 11. 1-(1,4-Диоксаспиро[4,5]дец-8-ил)пирролидин-2-он. 1,4-Диоксаспиро 4,5 декан-8-он (100 г, 640,3 ммоль), гидрохлорид метил-4-аминобутирата (98,5 г,640,3 ммоль), триэтиламин (90 мл, 640,3 ммоль) и дихлорметан (2 л) растворяют и перемешивают при комнатной температуре. Добавляют триацетоксиборогидрид натрия (135,7 г, 640,3 ммоль), перемешивают 17 ч при комнатной температуре. Гасят водой (1 л), разделяют, промывают водный слой дихлормета- 12015675 ном (3500 мл), объединяют органические фазы и высушивают над безводным сульфатом натрия, фильтруют и концентрируют. Материал очищают на 1,5 кг кварцевой колонке, 6 дюймов в диаметре, при элюировании смесью 8:2 гексан/этилацетат - 95:5 этилацетат/метанол с получением 73 г вышеназванного соединения в виде воскообразного коричневого твердого вещества. 1H ЯМР (CDCl3)3,99-4,10 (м, 1H),3,93 (с, 4H), 3,32-3,36 (м, 2H), 2,36-2,40 (м, 2H), 1,94-2,03 (м, 2H), 1,65-1,83 (м, 8H). Препаративный пример 12. 3-(2,6-Дихлор-4-метоксибензил)-1-(1,4-диоксаспиро[4,5]дец-8-ил) пирролидин-2-он. Раствор 1-(1,4-диоксаспиро[4,5]дец-8-ил)пирролидин-2-она (5 г, 22,2 ммоль) в тетрагидрофуране(100 мл) охлаждают до -78C, продувая раствор азотом. Добавляют LDA (2,0 М, 15 мл, 30 ммоль) с такой скоростью, чтобы внутренняя температура реакции не превышала -67C. Перемешивают 30 мин при-78C, добавляют раствор 2-бромметил-1,3-дихлор-5-метоксибензола (6,6 г, 24,4 ммоль) в ТГФ (20 мл) на протяжении 1-2 мин, удаляют холодную баню и оставляют реакцию нагреваться в течение 3 ч. Реакцию гасят насыщенным водным хлоридом аммония (100 мл), экстрагируют этилацетатом (3100 мл), экстракты объединяют и высушивают над безводным сульфат натрия. Очищают на кварцевой колонке, элюируя смесями 8:2 гексан:этилацетат - 1:1 гексан:этилацетат с получением продукта в виде твердого вещества цвета слоновой кости, 6,5 г, 71%. 1H ЯМР (CDCl3)6,86 (с, 2H), 4,06-4,12 (м, 1H), 3,94 (с, 4H), 3,77 (с,3H), 3,32-3,41 (м, 2H), 3,15-3,21 (м, 1H), 2,83-2,97 (м, 2H), 1,68-2,04 (м, 8H). ЖХМС m+1 414. Таблица 1 Препаративный пример 18. 3-(2,6-Дихлор-4-метоксибензил)-1-(4-оксоциклогексил)пирролидин-2 он. 3-(2,6-Дихлор-4-метоксибензил)-1-(1,4-диоксаспиро[4,5]дец-8-ил)пирролидин-2-он (6,5 г, 15,7 ммоль) растворяют в ацетоне (100 мл), добавляют гидрат паратолуолсульфоновой кислоты (3 г, 15,7 ммоль) и перемешивают в течение 24 ч при комнатной температуре. Добавляют 5 н. HCl (10 мл) и нагревают до 45C в течение 1 ч. Протекание реакции можно контролировать посредством TCX. Концентрируют реакционную смесь, разбавляют насыщенным водным бикарбонатом натрия (500 мл) и экстрагируют этилацетатом (3150 мл). Объединенные экстракты промывают водой (100 мл) и соляным раствором (100 мл), высушивают над безводным сульфатом натрия, фильтруют и концентрируют до объема примерно 50 мл, разбавляют гексаном (50 мл) и фильтруют с получением 5,5 г продукта, 95%, в виде твердого белого вещества. 1H ЯМР (CDCl3)6,78 (с, 2H), 4,44-4,52 (м, 1H), 3,78 (с, 3H), 3,37-3,47 (м, 1H),3,29-3,36 (м, 1H), 3,15-3,23 (м, 2H), 2,39-2,62 (м, 4H), 1,80-2,11 (м, 6H). ЖХМС m+1 370. Препаративный пример 24. 3-(2,6-Дихлор-4-метоксибензил)-1-(3-диметиламинометилен-4-оксоциклогексил)пирролидин-2-он. Смесь 3-(2,6-дихлор-4-метоксибензил)-1-(4-оксоциклогексил)пирролидин-2-она (5 г, 13,5 ммоль),триэтиламина (20 мл) и N,N-диметилформамиддиметилацеталя (20 мл) нагревают на масляной бане при температуре 140C до сухости. Немного охлаждая реакционную смесь, добавляют еще одну порцию триэтиламина (20 мл) и N,N-диметилформамиддиметилацеталя (20 мл), снова нагревают до 140C до сухости. Органическую фазу удаляют под вакуумом и контролируют протекание реакции посредством ЖХМС и ЯМР: 1H ЯМР (CDCl3)7,50-7,53 (м, 1H), 6,86 (с, 2H), 4,24-4,33 (м, 1H), 3,76 (с, 3H), 3,44-3,46 Согласно схеме D, соединение 10 получают путем обработки соединения 9 различными арилбороновыми кислотами или эфирами при стандартных условиях реакции сочетания Сузуки, т.е. в присутствии Pd(PPh)4, Na2CO3 в ДМЭ. Обработка кетона 10 реагентом Бредерика позволяет получить соединение 11. Пиразол Ib получают при обработке 11 гидразином. Препаративный пример 30. 3-[2-Хлор-4-(1-метил-1H-пиразол-4-ил)бензил]-1-(4-оксоциклогексил) пирролидин-2-он. Смешивают 3-(4-бром-2-хлорбензил)-1-(4-оксоциклогексил)пирролидин-2-он (0,5, 1,3 ммоль), 1 метил-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1H-пиразол (0,814 г, 3,9 ммоль), карбонат натрия(0,689 г, 6,5 ммоль) в ДМЭ (8 мл)/H2O (3 мл) и дегазируют потоком азота. Добавляют (Ph3P)4Pd (0,150 г,0,13 ммоль), и перемешивают при 80C в течение 17 ч в атмосфере азота. Охлаждают до температуры окружающей среды и добавляют этилацетат (20 мл) и воду (10 мл). Водную фазу экстрагируют этилацетатом (220 мл), высушивают (сульфат натрия) и конденсируют при пониженном давлении. Применение хроматографии (диоксид кремния, EtOAc) позволяет получить 0,287 г продукта (42%) в виде твердого белого вещества МС (m/z): 386 (M+1). Препаративный пример 31. 3-[2-Хлор-4-(1-метил-1H-пиразол-4-ил)бензил]-1-(3-диметиламинометилен-4-оксоциклогексил)пирролидин-2-он. Смесь 3-[2-хлор-4-(1-метил-1H-пиразол-4-ил)бензил]-1-(4-оксоциклогексил)пирролидин-2-она(0,287 г, 0,745 ммоль) и реагента Бредерика (0,17 мл) в толуоле (3 мл) нагревают до 90C в течение 30 мин. Органическую фазу удаляют под вакуумом с получением 0,327 г масла, которое применяют без дополнительной очистки. МС (m/z): 441 (M+1). Схема E Согласно схеме E, при обработке соединения 12 с помощью BBr3 получают фенол 13, который затем можно трифлатировать, применяя Tf2O, в присутствии пиридина, с получением соединения 14. Ic можно получить путем применения различных бороновых кислот или эфиров при стандартных условиях реакции сочетания Сузуки, т.е. в присутствии Pd(PPh)4 и Na2CO3 в ДМЭ. Препаративный пример 32. 3-(2,6-Дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-2H-индазол-5 ил)пирролидин-2-он. 3-(2,6-Дихлор-4-метоксибензил)-1-(4,5,6,7-тетрагидро-2H-индазол-5-ил)пирролидин-2-он (пример 1) (15,0 г, 38 ммоль) растворяют в метиленхлориде (1,5 л) и охлаждают до 0C. По каплям обрабатывают трибромидом бора (19,9 мл, 190 ммоль) в течение 30 мин и оставляют реакционную смесь нагреваться до комнатной температуры. Перемешивают в течение 28 ч при комнатной температуре, повторно охлаждают на ледяной бане, осторожно в течение 15 мин при перемешивании к реакционной смеси добавляют метанол (38 мл). Органические растворители удаляют с помощью роторного испарителя и остаток рас- 15015675 творяют в смеси 4:1 хлороформ/изопропиловый спирт (600 мл) и воде (100 мл). Нейтрализуют до pH 7,применяя 5 н. NaOH, разделяют, сохраняют органическую фазу, а водную фазу экстрагируют двумя порциями смеси хлороформ/изопропиловый спирт. Объединенные органические фазы высушивают над сульфатом натрия, фильтруют и выпаривают до образования твердого вещества, которое высушивают под вакуумом при 90C с получением 10,7 г (75%) вышеназванного соединения. МС (m/z): 380 (m+1). Препаративный пример 33. 3,5-Дихлор-4-[2-оксо-1-(2-трифторметансульфонил-4,5,6,7-тетрагидро 2H-индазол-5-ил)пирролидин-3-илметил]фениловый эфир трифторметансульфоновой кислоты 3-(2,6-Дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-2H-индазол-5-ил)пирролидин-2-он (5,0 г,13,2 ммоль) растворяют в пиридине (32 мл) и охлаждают до 0C. По каплям в течение 5 мин обрабатывают трифталатным ангидридом (7,1 мл, 42 ммоль) и оставляют реакционную смесь нагреваться до комнатной температуры. Перемешивают в течение 4 ч при комнатной температуре в атмосфере азота, добавляют воду, разбавляют смесью этилацетат/диэтиловый эфир и разделяют. Органическую фазу промывают водой, соляным раствором, и высушивают над сульфатом натрия. Применение хроматографии (диоксид кремния, 75:25 гексан/этилацетат, краситель САМ для визуализации фракций) позволяет получить 4,97 г продукта (58%) в виде смеси N1/N2 трифлатных изомеров. МС (m/z): 645 (m+1). Схема F Согласно схеме F, Id получают из соединения 15, которое сначала гидролизуют до кислоты, а затем сочетают с различными аминами в присутствии EDC. Препаративный пример 34. Трифторметансульфоновая кислота 3-(3,5-дихлор-4'-карбоксиметилбифенил-4-илметил)-1-(4,5,6,7-тетрагидро-2H-индазол-5-ил)пирролидин-2-он. Смешивают 3,5-дихлор-4-[2-оксо-1-(2-трифторметансульфонил-4,5,6,7-тетрагидро-2H-индазол-5 ил)пирролидин-3-илметил]фениловый эфир трифторметансульфоновой кислоты (1,0 г, 1,55 ммоль), 4 карбоксиметилфенилбороновую кислоту (0,416 г, 2,3 ммоль), карбонат натрия (2,3 мл 2,0 М, 5,4 ммоль) в ДМЭ (12 мл) и дегазируют потоком азота. Добавляют (Ph3P)4Pd (0,078 г, 0,15 ммоль) и перемешивают при 80C в течение 17 ч в атмосфере азота. Охлаждают до температуры окружающей среды и добавляют этилацетат (20 мл) и воду (10 мл). Водную фазу экстрагируют этилацетатом (220 мл), высушивают(сульфат натрия) и конденсируют при пониженном давлении. Применение хроматографии (диоксид кремния, 95:5 CH2Cl2/метанол) позволяет получить 0,750 г продукта в виде твердого белого вещества МС(m/z): 632 (M+2). Препаративный пример 35. 3-(2,6-Дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-2H-индазол-5 ил)пирролидин-2-он. Смешивают соединение примера 7 (изомер 1, см. примеры 7-10) 3-(2,6-дихлор-4-метоксибензил)-1(4,5,6,7-тетрагидро-2H-индазол-5-ил)пирролидин-2-он (0,38 г, 0,96 ммоль), BBr3 (5,3 мл 1,0 М, 5,3 ммоль) в дихлорэтане (20 мл) при 0C, оставляют реакцию нагреваться до комнатной температуры и перемешивают в течение 7 ч. Охлаждают до 0C, гасят метанолом, нейтрализуют с помощью 5,0 н. NaOH, экстрагируют продукт смесью 3:1 CHCl3/изопропиловый спирт и высушивают над сульфатом натрия. Применение хроматографии (диоксид кремния, 95:5: CHCl3/EtOH/NH3) позволяет получить 0,290 г продукта в виде твердого белого вещества. МС (m/z): 380 (M+1). Согласно схеме G, соединение 16 получают из 4-бензилоксазолидин-2-она, который сначала ацилируют пентеноилхлоридом, а затем, после предварительной обработки LiHMDS, алкилируют бензилбромидом. Окисление винильной группы в соединении 16 можно достичь обработкой озоном и PPh3 илиOsO4 в присутствии NaIO4 и получить альдегид 17. Препаративный пример 36. (R)-4-Бензил-3-пент-4-еноилоксазолидин-2-он. Струей азота в течение 20 мин промывают 12-л 3-горлую круглодонную колбу, оборудованную механической мешалкой, зондом для измерения внутренней температуры/N2 входным отверстием и 1 л воронкой, затем добавляют (R)-4-бензил-2-оксазолидинон (250 г, 1,41 моль). Разбавляют ТГФ (1,8 л) и охлаждают на бане с сухим льдом/ацетоном до тех пор, пока внутренняя температура не составит -74C. 1,6 М гексановый раствор н-бутиллития (970 мл, 1,552 моль) переносят в воронку через канюлю и добавляют к раствору оксазолидинона с такой скоростью, чтобы внутренняя температура не превышала -65C. После завершения добавления оставляют реакционную смесь перемешиваться на охлаждающей бане на 30 мин. 4-Пентеноилхлорид (175 мл, 1,585 моль) переносят в воронку и добавляют по каплям в анионный раствор в течение 25 мин. Реакционную смесь перемешивают в течение 45 мин на охлаждающей бане. Охлаждающую баню удаляют и реакционную смесь перемешивают 18 ч, по мере того как она медленно достигает комнатной температуры. Смесь разбавляют 1 н. водной соляной кислотой (1,5 л) и диэтиловым эфиром (1,0 л). Фазы разделяют и органическую фазу промывают водой (21 л), затем соляным раствором (1 л). Экстрагируют объединенные водные промывки эфиром (1 л). Высушивают объединенные органические фазы над безводным сульфатом магния, фильтруют и концентрируют до образования 390 г желто-коричневого масла. Этот материал очищают способом хроматографии на силикагеле, применяя смесь гексан:этилацетат с получением 345 г (94,5%) прозрачного желтого масла. Препаративный пример 37. (R)-4-Бензил-3-[(S)-2-(4-бензилокси-2,6-дихлорбензил)пент-4-еноил] оксазолидин-2-он. Смесь (R)-4-бензил-3-пент-4-еноилоксазолидин-2-она (345 г, 1,33 моль) и ТГФ (1,8 л) перемешивают в 12-л 3-горлой круглодонной колбе, оборудованной зондом для измерения внутренней температуры/выходным отверстием для азота и воронкой, в атмосфере азота и охлаждают до -75C. 1 М LiHMDS(1,6 л) переносят в воронку и добавляют с такой скоростью, чтобы внутренняя температура не превышала -60C. После завершения добавления оставляют реакционную смесь перемешиваться при -25C в течение 30 мин, затем охлаждают до примерно -60C. В этот момент добавляют порциями твердый 2 бромметил-1,3-дихлор-5-бензилоксибензол в течение 5 мин. Когда добавление завершено, переносят реакционный сосуд в -10C ацетоновую баню и поддерживают внутреннюю температуру реакции ниже 10C в течение 1 ч. Смесь охлаждают до 0C, затем гасят 2 л водной 1 н соляной кислоты. Смесь переносят в 22-л делительную воронку и разбавляют 2,5 л воды и 2 л эфира. Фазы разделяют и водную фазу экстрагируют эфиром. Объединенную органическую фазу высушивают над безводным сульфатом магния, фильтруют и концентрируют до получения 800 г вязкого масла. Очищают способом хроматографии на силикагеле, применяя смесь гексан:этилацетат с получением 597 г, (86%) бесцветного масла. Препаративный пример 38. (R)-4-R)-4-Бензил-2-оксооксазолидин-3-ил)-3-(4-бензилокси-2,6 дихлорбензил)-4-оксобутиральдегид. Смесь (R)-4-бензил-3-[(S)-2-(4-бензилокси-2,6-дихлорбензил)пент-4-еноил]оксазолидин-2-она (100 г, 190,68 ммоль) и дихлорметана (800 мл) охлаждают до -74C. Барботируют озон, полученный в озоновом генераторе A-113 со скоростью 75% через реакционную смесь с помощью транспортирующего воздуха со скоростью 5 CFM до тех пор, пока раствор не приобретет голубой цвет (примерно 3 ч). Трифенилфосфин (60 г, 228,8 ммоль) добавляют в виде раствора в 200 мл дихлорметана и оставляют реакционную смесь перемешиваться на ночь, пока температура достигает комнатной. Раствор концентрируют под вакуумом и очищают способом хроматографии на силикагеле, применяя градиент 20-50% этилацетата в гексане с получением 82,1 г (82%) продукта в виде белой пены: МС (m/z): 526 (M+). Альтернативным образом, смесь (R)-4-бензил-3-[(S)-2-(4-бензилокси-2,6-дихлорбензил)пент-4 еноил]оксазолидин-2-она (0,96 г, 1,8 ммоль), ТГФ (21 мл) и воды (7 мл) обрабатывают посредством 2,5% тетроксида осмия в трет-бутаноле (46 мг, 0,18 ммоль). Добавляют периодат натрия (1,17 г, 5,5 ммоль) и реакционную смесь перемешивают 4 ч при комнатной температуре. Реакцию гасят водой и экстрагируют этилацетатом. Органическую фазу промывают водным 1 н. тиосульфатом натрия, затем соляным раство- 17015675 ром. Органическую фазу высушивают над сульфатом магния, фильтруют и концентрируют под вакуумом. Неочищенный материал очищают способом хроматографии на силикагеле, применяя смесь гексан:этилацетат для элюирования чистого продукта. Фракции, содержащие продукт, концентрируют под вакуумом с получением 0,46 г (48%) требуемого продукта. МС (m/z): 526 (M+). Схема H Согласно схеме H кетон 18 превращают в пиразол, применяя реагент Бредерика, с последующей обработкой N2H4 в EtOH. Энантиомерно чистый амин 19 получают после разделения энантиомеров способом хиральной хроматографии и затем удаления Вос-группы в кислотных условиях, т.е. в присутствииHCl или ТФК. Препаративный пример 39. Трет-бутиловый эфир (+/-)-(4,5,6,7-тетрагидро-2H-индазол-5 ил)карбаминовой кислоты. Смесь трет-бутилового эфир (4-оксоциклогексил)карбаминовой кислоты (100 г, 0,469 моль), N,Nдиметилформамиддиметилацеталя (100 мл) и Et3N (100 мл) кипятят до сухости на масляной бане (140C) в течение 30 мин. Добавляют еще по 100 мл каждого реагента и оставляют выкипать. Повторяют эту операцию три раза, используя в сумме по 300 мл DMFDMA и Et3N. Концентрируют смесь, содержащую трет-бутиловый эфир (3-диметиламинометилен-4-оксоциклогексил)карбаминовой кислоты до образования стекла, повторно растворяют в 250 мл этанола и затем добавляют 65 мл гидразингидрата и реакционную смесь перемешивают в течение ночи при комнатной температуре. Смесь концентрируют, помещают в 500 мл EtOAc и промывают 2500 мл водой. Водный слой экстрагируют 150 мл EtOAc, и объединенные органические фазы промывают 250 мл соляного раствора, высушивают на безводном MgSO4,фильтруют и концентрируют. Применение хроматографии (диоксид кремния, этилацетат) позволяет получить 87 г продукта в виде светло-желтого твердого вещества. МС (m/z): 237. Отдельные энантиомеры получают способом хиральной хроматографии (Chiralpak AD-H, 4,6150 мм, 80:10:10, C7 (гексан)/3A/MeOH мас./0,2% ДМЭА, 0,6 мл/мин, при 235 нм), выделяя: Изомер 1 (время удерживания = 6,8 мин, % е.е.99, []25D -41,7 (с 1, MeOH). Изомер 2 (время удерживания =9,5 мин, % е.е.95, []25D 40,7 (с 1, MeOH). Препаративный пример 40. 4,5,6,7-Тетрагидро-2H-индазол-5-иламин (свободное основание). Смешивают изомер 1 (из препаративного примера 39), трет-бутиловый эфир (-)-(4,5,6,7-тетрагидро 2H-индазол-5-ил) карбаминовой кислоты, (5,3 г, 22,3 ммоль) в CH2Cl2 (200 мл), обрабатывают ТФК (16,5 мл, 223 ммоль) и перемешивают при комнатной температуре в течение 3 ч. Растворитель удаляют при пониженном давлении, получая вязкое масло. При выделении свободного основания способом твердофазной экстракции (50 г, Mega-bond Elut, Varian, 0,79 миллиэквивалент/г смолы) путем применения от метанола до влажной смолы, с последующим элюированием раствором 95:5 CH2Cl2/метанол и в заключении раствором 95:5 CH2Cl2/7 M NH3 метанол получают 2,66 г (86%) желто-коричневого масла, которое затвердевает при стоянии. Препаративный пример 41. 4,5,6,7-Тетрагидро-2H-индазол-5-иламин (свободное основание). Смешивают изомер 2 (из препаративного примера 39), трет-бутиловый эфир (+)-(4,5,6,7 тетрагидро-2H-индазол-5-ил) карбаминовой кислоты (5,01 г, 2,11 ммоль) в CH2Cl2 (200 мл), обрабатывают ТФК (16,0 мл, 211 ммоль) и перемешивают при комнатной температуре в течение 3 ч. Растворитель удаляют при пониженном давлении, получая вязкое масло. При выделении свободного основания способом твердофазной экстракции (50 г, Mega-bond Elut, Varian, 0,79 миллиэквивалент/г смолы) путем применения от метанола до влажной смолы, с последующим элюированием раствором 95:5 CH2Cl2/метанол и в заключении раствором 95:5 CH2Cl2/7 M NH3 метанол получают 2,1 г (71%) желто-коричневого масла,которое затвердевает при стоянии. Согласно схеме I, при обработке альдегида 17 амином 19 в присутствии NaBH(OAc)3 получают пиразол 20. Удаление бензильной защитной группы при гидрировании в присутствии Pd(OH)2 позволяет получить фенол 21, который затем переводят в трифлат 22. Ie получают путем обработки трифлата 22 различными бороновыми кислотами или сложными эфирами при стандартных условиях реакции сочетания Сузуки, т.е. в присутствии Pd(PPh3)4 и Na2CO3. Согласно схеме I, соединение 19 может представлять собой любой энантиомер. Реакция соединения 17 с 19 позволяет получить при C-3 стереоконфигурацию "R", обозначенную стрелкой. В соединении Ie стереоконфигурацию при C-3 в пирролидиноне обозначают "R", а обозначение стереоконфигурации приC-5 в тетрагидроиндазоле определяют по стереоконфигурации в соединении 19. Препаративный пример 42. (3R,5S)-3-(4-Гидрокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-2Hиндазол-5-ил)пирролидин-2-он.Pd(OH)2 (6,0 г) и перемешивают при давлении водорода 30 psi. После 6 ч результаты ESMS (массспектрометрии с электрораспылением) указывают на потребление исходного материала и образование продукта. Катализатор удаляют путем фильтрации, промывают этанолом и выпаривают до образования темной пены. Фильтрация через 50 г SCX mega-bond elut с применением 95:5 CH2Cl2/MeOH, а затем 95:5CH2Cl2/7,0 М NH3/MeOH позволяет получить 5,6 г продукта (79%) в виде аморфного твердого вещества МС (m/z): 380 (M+1). Препаративный пример 43. 3,5-Дихлор-4-[2-оксо-1-(2-трифторметансульфонил-4,5,6,7-тетрагидро 2H-индазол-5-ил)пирролидин-3-илметил]фениловый эфир(3R,5S)-трифторметансульфоновой кислоты.(препаративный пример 42) (5,56 г, 14,6 ммоль) растворяют в сухом пиридине (35 мл), охлаждают до 0C и добавляют по каплям трифталатный ангидрид (7,4 мл, 4 4 ммоль) в течение 60 с. Реакционную смесь оставляют нагреваться до комнатной температуры и при непрерывном перемешивании в течение 3,0 ч в атмосфере азота. Реакцию гасят водой (20 мл) и разбавляют этилацетатом (75 мл). Слои разделяют, органическую фазу промывают 0,1 н. HCl (2), соляным раствором и высушивают над сульфатом натрия. С помощью фильтрации и выпаривания получают 7,71 г (80%) аморфного оранжевого твердого вещества. МС (m/z): 645 (M+1). Препаративный пример 44. 1-Хлор-3,4-дифтор-2-йодметил-5-трифторметилбензол. Раствор 1-хлор-2-хлорметил-3,4-дифтор-5-трифторметилбензола (1,0 г, 3,8 ммоль) в ацетоне (10 мл) обрабатывают иодидом натрия (2,8 г, 19 ммоль) и реакционную смесь перемешивают 1 ч при комнатной температуре. Реакцию разбавляют в EtOAc (50 мл) и фильтруют. Фильтрат концентрируют с получением- 19015675 1,35 г (100%) продукта. 1H ЯМР (d6-CDCl3)7,39 (д, 1 Н, J=5,7 Гц), 4,46 (д, 2 Н, J=2,2 Гц). Препаративный пример 45. (R)-4-Бензил-3-[(S)-2-(6-хлор-2,3-дифтор-4-трифторметилбензил)пент 4-еноил]оксазолидин-2-он. Раствор (R)-4-бензил-3-пент-4-еноилоксазолидин-2-она (0,94 г, 3,6 ммоль) в ТГФ (10 мл) охлаждают до -78C. Указанный раствор обрабатывают по каплям 1,0 М LiHMDS в ТГФ (4 мл, 4,0 ммоль) и перемешивают при -78C в течение 30 мин. Раствор обрабатывают 1-хлор-3,4-дифтор-2-йодметил-5 трифторметилбензолом (1,35 г, 3,8 ммоль) и оставляют медленно нагреваться до комнатной температуры. Реакционную смесь перемешивают 4 ч при комнатной температуре. Реакцию гасят 1 н. HCl (водным) и экстрагируют Et2O. Органическую фазу промывают соляным раствором, высушивают над MgSO4,фильтруют и удаляют растворитель. Неочищенный остаток очищают способом хроматографии на силикагеле, применяя смесь гексан:EtOAc для элюирования чистого продукта. Растворитель удаляют с получением 0,725 г (41%) требуемого продукта. МС (m/e): 488 (M+1). Препаративный пример 46. (R)-4-R)-4-Бензил-2-оксооксазолидин-3-ил)-3-(6-хлор-2,3-дифтор-4 трифторметилбензил)-4-оксобутиральдегид. Раствор(R)-4-бензил-3-[(S)-2-(6-хлор-2,3-дифтор-4-трифторметилбензил)пент-4-еноил]оксазолидин-2-она (препаративный пример 45) (0,72 г, 1,5 ммоль) в ТГФ (9 мл) и воде (3 мл) обрабатывают посредством 2,5% OsO4 в tBuOH (1,5 г, 0,15 ммоль). Затем к раствору добавляют периодат натрия (0,96 г,4,5 ммоль) и реакционную смесь перемешивают в течение 4 ч при комнатной температуре. Реакцию гасят водой и экстрагируют EtOAc. Органическую фазу промывают 1 н. тиосульфатом натрия и соляным раствором. Органическую фазу отделяют, высушивают над MgSO4, фильтруют и удаляют растворитель. Неочищенный остаток очищают способом хроматографии на силикагеле, применяя смесь гексан:EtOAc для элюирования чистого продукта. Растворитель удаляют с получением 0,60 г (82%) требуемого продукта. МС (m/e): 490 (M+1). Препаративный пример 47. (3R,5R)-3-(2,6-Дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-2Hиндазол-5-ил)пирролидин-2-он.(3R,5R)-3-(4-Бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-2H-индазол-5-ил)пирролидин-2 он (пример 57) (0,977 г, 2,1 ммоль) растворяют в ТГФ (90 мл), обрабатывают 20% Pd(OH)2 (0,977 г) и перемешивают при давлении водорода 30 psi. После перемешивания в течение ночи, согласно ESMS соотношение продукта к исходному материалу составляет 2:1. Катализатор удаляют путем фильтрации,промывают этанолом и выпаривают до образования темной пены. Применение хроматографии (диоксид кремния, 95:5 CH2Cl2/EtOH/NH3) позволяет получить 0,26 г (33%) вышеназванного соединения в виде аморфного твердого вещества МС (m/z): 380 (M+1). Препаративный пример 48. 3,5-Дихлор-4-[2-оксо-1-(2-трифторметансульфонил-4,5,6,7-тетрагидро 2H-индазол-5-ил)пирролидин-3-илметил]фениловый эфир (3R,5R)-трифторметансульфоновой кислоты.(препаративный пример 47) (0,261 г, 0,86 ммоль) растворяют в сухом пиридине (2,0 мл), охлаждают до 0C и добавляют по каплям трифталатный ангидрид (0,359 мл, 2,1 ммоль) в течение 60 с. Реакционную смесь оставляют нагреваться до комнатной температуры и продолжают перемешивание в течение 4,0 ч в атмосфере азота. Реакцию гасят водой (20 мл) и разбавляют этилацетатом (75 мл). Слои разделяют, органическую фазу промывают 0,1 н. HCl (2), соляным раствором и высушивают над сульфатом натрия. Применение фильтрации и хроматографии (диоксид кремния, 95:5 CH2Cl2/MeOH) позволяет получить 0,34 г (77%) аморфного оранжевого твердого вещества. МС (m/z): 645 (M+1) Препаративный пример 49. Трет-бутиловый эфир (3R,5S)-5-[3-(4-бензилокси-2,6-дихлорбензил)-2 оксопирролидин-1-ил]-4,5,6,7-тетрагидроиндазол-2-карбоновой кислоты и трет-бутиловый эфир. Раствор (3R,5S)-3-(4-бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-2H-индазол-5-ил)пирролидин-2-она (пример 35) (0,429 г, 0,91 ммоль) и пиридина (0,144 г, 1,82 ммоль) в CH2Cl2 (15 мл) обрабатывают ди-трет-бутилдикарбонатом (0,24 г, 1,10 ммоль) и перемешивают в течение 5 ч при комнатной температуре в атмосфере N2. Реакцию экстрагируют 1 н. HCl и водой. Органическую фазу высушивают,применяя Na2SO4, растворитель удаляют в вакууме и получают неочищенный продукт, который очища- 20015675 ют на диоксиде кремния, применяя градиент от 0 до 100% этилацетата в гексане с получением 0,443 г Смесь трет-бутилового эфира (3R,5S)-5-[3-(4-бензилокси-2,6-дихлорбензил)-2-оксопирролидин-1 ил]-4,5,6,7-тетрагидроиндазол-2-карбоновой кислоты и трет-бутилового эфира (3R,5S)-5-[3-(4-бензилокси-2,6-дихлорбензил)-2-оксопирролидин-1-ил]-4,5,6,7-тетрагидроиндазол-1-карбоновой кислоты (препаративный пример 49) (0,44 г, 0,77 ммоль) и 20% Pd(OH)2 на углероде (0,44 г) в этилацетате (50 мл) барботируют с применением N2, а затем H2. Смесь перемешивают под баллоном H2 в течение 16 ч при комнатной температуре. Смесь обрабатывают посредством Na2SO4 и затем фильтруют через слой Hyflow для удаления катализатора и удаляют растворитель в вакууме с получением 0,388 г (100%) вышеназванных продуктов в виде смеси. Rf=0,42 (9/1 CH2Cl2/метанол). Препаративный пример 51. 1-Бром-3-бромметил-2,4-дихлорбензол. Смесь 2,6-дихлортолуола (50,0 г, 0,31 моль), йода (0,10 г, 0,39 ммоль) и 325 порошка железа (0,70 г,12,5 ммоль) в CCl4 (60 мл) в течение 20 мин по каплям обрабатывают бромом (52,8 г, 0,33 моль) и перемешивают в течение 3 ч при комнатной температуре. Смесь выливают в ледяную воду и экстрагируют 1,2-дихлорэтаном. Органическую фазу промывают насыщенным бисульфитом натрия и высушивают посредством Na2SO4. Растворитель удаляют в вакууме с получением 76,01 г (100%) 1-бром-2,4-дихлор-3 метилбензола. Смесь 1-бром-2,4-дихлор-3-метилбензола (76,01 г, 0,316 моль) и N-бромсукцинимида (59,2 г, 0,332 моль) в CCl4 (500 мл) обрабатывают бензоилпероксидом (0,77 г, 3,18 ммоль) и нагревают при кипении с обратным холодильником в течение 6 ч в атмосфере N2. Реакционную смесь охлаждают до 0C и фильтруют, применяя гексан для промывания твердого вещество. Фильтрат экстрагируют водой и насыщенным NaHCO3. Органическую фазу высушивают (Na2SO4) и растворитель удаляют в вакууме с получением 97,89 г (97%) вышеназванного продукта. Rf=0,34 (100% гексан). Препаративный пример 56. Этил-2-(4-бром-2-хлорбензил)пент-4-еноат.(125 мл) при -78C и перемешивают в течение 15 мин. Добавляют 4-бром-2-хлорбензилбромид (3,3 г,10,5 ммоль) и нагревают реакцию до комнатной температуры. Гасят раствором хлорида аммония, реакционную смесь экстрагируют метиленхлоридом и органическую фазу промывают соляным раствором. Высушивают над сульфатом натрия, фильтруют и концентрируют. Очищают остаток на силикагелевой колонке (гексан) с получением вышеназванного соединения (1,65 г, 73%) в виде бесцветного масла. Препаративный пример 57. Этил-2-(4-бром-2-хлорбензил)-4-оксобутаноат. Периодат натрия (41 г, 190 ммоль) добавляют в раствор этил-2-(4-бром-2-хлорбензил)пент-4-еноата(21 г, 63 ммоль), 2,5% вес. OsO4 (64 г, 6,3 ммоль) в ТГФ (400 мл) и воде (160 мл) и перемешивают в течение 2 ч. Реакционную смесь экстрагируют этилацетатом, органический слой промывают раствором тиосульфата натрия и соляным раствором. Высушивают над сульфатом натрия, фильтруют и концентрируют. Остаток очищают на силикагелевой колонке с получением вышеназванного соединения (15,9 г, 75%) в виде бесцветного масла: 1H ЯМР (CDCl3) 9,73 (с, 1H), 7,53 (д, J=2,0 Гц, 1H), 7,32 (дд, J=8,20, 2,0 Гц,1H), 7,08 (д, J=8,2 Гц, 1H), 4,05-4,15 (м, 2H), 3,20-3,28 (м, 1H), 3,07-3,15 (м, 1H), 2,84-2,92 (м, 2H), 2,532,61 (м, 1H), 1,51-1,21 (м, 3H). Препаративный пример 58. (4,4-Дифторпиперидин-1-ил)-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]метанон. Стадия 1. 4-Бромбензойную кислоту (4,98 г, 24 ммоль), гидрохлорид 4,4,-дифторпиперидин (4,3 г,27 ммоль), HOBt (4,09 г, 29 ммоль) и DIPEA (15,5 мл, 8 9 ммоль) растворяют в ТГФ (50 мл). ДобавляютEDC (5,7 г, 29 ммоль) и перемешивают при комнатной температуре в течение 24 ч. Реакционную смесь разбавляют этилацетатом, промывают 0,1 н. HCl, бикарбонатом натрия (насыщенным), соляным раствором, высушивают над сульфатом натрия, фильтруют и выпаривают до образования масла, получая (4 бромфенил)-(4,4-дифторпиперидин-1-ил)метанон в виде вышеназванного продукта: МС (m/z): 305(M+1). Стадия 2. (4-Бромфенил)-(4,4-дифторпиперидин-1-ил)метанон (3,0 г, 9,86 ммоль), бис(пинаколато) диборан (2,74 г, 10,7 ммоль), ацетат калия (2,9 г, 29 ммоль), хлорид [1,1'-бис(дифенилфосфино) ферроцен]палладия(II) (0,804 г, 0,9 ммоль) добавляют в ДМСО (20 мл) и перемешивают при 80C в течение 17 ч. Охлаждают, разбавляют этилацетатом, промывают соляным раствором (5), высушивают над сульфа- 21015675 том натрия, фильтруют и выпаривают. Применение хроматографии (диоксид кремния, CH2Cl2/MeOH,98:2) позволяет получить 1,7 г (47%) (4,4-дифторпиперидин-1-ил)-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]метанона. Препаративный пример 59. 4-Бензилокси-2,6-диметилбензальдегид. Раствор 2,6-диметил-4-гидроксибензальдегида (4,0 г, 27 ммоль) и бензилбромида (3,3 мл, 28 ммоль) в ДМФ (50 мл) обрабатывают карбонатом калия (4,5 г, 32 ммоль). Нагревают реакционную смесь до 60C и перемешивают в течение 1 ч. Реакционную смесь охлаждают и гасят 1 н водного HCl. Водную фазу экстрагируют Et2O. Органическую фазу промывают соляным раствором, высушивают над MgSO4 и фильтруют. Растворитель удаляют с получением 6,4 г (100%) продукта. 1 Н ЯМР (d6-CDCl3)10,46 (с,1H), 7,31-7,43 (м, 5H), 6,65 (с, 2H), 5,08 (с, 2H), 2,58 (с, 6H). Препаративный пример 60. (4-Бензилокси-2,6-диметилфенил)метанол. Раствор 4-бензилокси-2,6-диметилбензальдегида (6,4 г, 27 ммоль) в метаноле (60 мл) обрабатывают борогидридом натрия (0,82 г, 22 ммоль). Реакционную смесь перемешивают в течение 1 ч при комнатной температуре. Реакцию гасят насыщенным раствором бикарбоната натрия в воде и экстрагируют Et2O. Органическую фазу промывают соляным раствором, высушивают над MgSO4 и фильтруют. Растворитель удаляют с получением 6,3 г (97%) продукта. 1H ЯМР (d6-CDCl3)7,31-7,43 (м, 5H), 6,65 (с, 2H), 5,01(с, 2H), 4,66 (с, 2H), 2,38 (с, 6H). Препаративный пример 61. 5-Бензилокси-2-бромметил-1,3-диметилбензол. Раствор (4-бензилокси-2,6-диметилфенил)метанола (5,70 г, 24 ммоль) в ТГФ (100 мл) охлаждают до 0C. Раствор обрабатывают фосфористым трибромидом (0,9 мл, 9,4 ммоль) и реакционную смесь перемешивают в течение 2 ч при 0C. Реакцию гасят водой и экстрагируют Et2O. Органическую фазу промывают соляным раствором, высушивают над MgSO4 и фильтруют. Растворитель удаляют с получением 7,1 г (99%) продукта. 1H ЯМР (d6-CDCl3)7,31-7,43 (м, 5H), 6,65 (с, 2H), 5,01 (с, 2H), 4,56 (с, 2H), 2, 38 (с,6H). Препаративный пример 62. (R)-4-Бензил-3-[(S)-2-(4-бензилокси-2,6-диметилбензил)пент-4-еноил] оксазолидин-2-он. Раствор (R)-4-бензил-3-пент-4-еноилоксазолидин-2-она (4,8 г, 19 ммоль) в ТГФ (100 мл) охлаждают до -78 C. Раствор по каплям обрабатывают 1,0 М LiHMDS в ТГФ (20 мл, 20 ммоль) и перемешивают при-78C в течение 30 мин. Раствор обрабатывают 5-бензилокси-2-бромметил-1,3-диметилбензолом (6,8 г,22 ммоль) и оставляют медленно нагреваться до комнатной температуры. Реакционную смесь перемешивают 3 ч при комнатной температуре. Реакцию гасят 1 н. HCl (водн.) и экстрагируют Et2O. Органическую фазу промывают соляным раствором, высушивают над MgSO4, фильтруют и удаляют растворитель. Неочищенный остаток очищают способом колоночной хроматографии на силикагеле, применяя смесь гексан:EtOAc для элюирования чистого продукта. Растворитель удаляют с получением 7,0 г (78%) требуемого продукта. МС (m/e): 484 (M+1). Препаративный пример 63. (R)-4-R)-4-Бензил-2-оксооксазолидин-3-ил)-3-(4-бензилокси-2,6-диметилбензил)-4-оксобутиральдегид. Раствор (R)-4-бензил-3-[2-(4-бензилокси-2,6-диметилбензил)пент-4-еноил]оксазолидин-2-она (7,5 г,16 ммоль) в ТГФ (120 мл) и воде (40 мл) обрабатывают 2,5% OsO4 в tBuOH (16 г, 1,6 ммоль). К раствору добавляют периодат натрия (10 г, 47 ммоль) и реакционную смесь перемешивают в течение 3 ч при комнатной температуре. Реакцию гасят водой и экстрагируют EtOAc. Органическую фазу промывают 1 н тиосульфатом натрия и соляным раствором. Органическую фазу отделяют, высушивают над MgSO4,фильтруют и растворитель удаляют. Неочищенный остаток очищают способом колоночной хроматографии на силикагеле, применяя смесь гексан:EtOAc для элюирования чистого продукта. Растворитель удаляют с получением 3,25 г (43%) требуемого продукта. МС (m/e): 486 (M+1). Препаративный пример 64. (R)-3-(4-Бензилокси-2,6-диметилбензил)-1-(4,5,6,7-тетрагидро-2Hиндазол-5-ил)пирролидин-2-он. Смешивают раствор (R)-4-R)-4-бензил-2-оксооксазолидин-3-ил)-3-(4-бензилокси-2,6-диметилбензил)-4-оксобутиральдегида (3,0 г, 6,2 ммоль) и рацемического 4,5,6,7-тетрагидро-2H-индазол-5-иламина(0,85 г, 6,2 ммоль) в дихлорэтане (50 мл) и ацетонитриле (50 мл). Раствор обрабатывают триацетоксиборогидридом натрия (6,6 г, 31 ммоль) и перемешивают 1 ч при комнатной температуре. Реакционную смесь обрабатывают N,N-диизопропилэтиламином (5,6 мл, 31 ммоль) и перемешивают в течение ночи при комнатной температуре. Реакционную смесь концентрируют до получения остатка. Остаток гасят насыщенным карбонатом натрия и экстрагируют EtOAc. Органическую фазу промывают соляным раствором, высушивают над MgSO4, фильтруют и растворитель удаляют. Неочищенный остаток очищают способом колоночной хроматографии на силикагеле, применяя CH2Cl2 и 2 М аммиак в MeOH для элюирования чистого продукта. Растворитель удаляют с получением 1,66 г (62%) требуемого продукта. МС(m/e): 430 (M+1). Препаративный пример 65. (R)-3-(4-Гидрокси-2,6-диметилбензил)-1-(4,5,6,7-тетрагидро-2H-индазол-5-ил)пирролидин-2-он. Раствор (R)-3-(4-бензилокси-2,6-диметилбензил)-1-(4,5,6,7-тетрагидро-2H-индазол-5-ил)пирроли- 22015675 дин-2-она (препаративный пример 64) (1,65 г, 3,8 ммоль) в EtOH (20 мл) обрабатывают гидроксидом палладия-на-углероде (1,7 г). Раствор продувают водородом и повышают давление до 35 psi. Реакционную смесь перемешивают 2 дня при давлении водорода 35 psi. Реакционную смесь фильтруют через слой целита для удаления катализатора. Неочищенный остаток очищают способом колоночной хроматографии на силикагеле, применяя CH2Cl2 и 2 М аммиак в MeOH для элюирования чистого продукта. Растворитель удаляют с получением 0,85 г (65%) требуемого продукта. МС (m/e): 340 (M+1). Препаративный пример 66. 3,5-Диметил-4-[(R)-2-оксо-1-(2-трифторметансульфонил-4,5,6,7-тетрагидро-2H-индазол-5-ил)-пирролидин-3-илметил]фениловый эфир трифторметансульфоновой кислоты. Раствор (R)-3-(4-гидрокси-2,6-диметилбензил)-1-(4,5,6,7-тетрагидро-2H-индазол-5-ил)пирролидин 2-она (препаративный пример 65) (0,85 г, 2,5 ммоль) в пиридине (20 мл) охлаждают до 0C и обрабатывают трифторметансульфоновым ангидридом (1,3 мл, 7,5 ммоль). Оставляют реакционную смесь нагреваться до комнатной температуры. После перемешивания в течение 2 ч при комнатной температуре реакцию гасят посредством 1 н HCl и экстрагируют EtOAc. Органическую фазу промывают соляным раствором, высушивают над MgSO4 и фильтруют. Растворитель удаляют с получением 1,05 г (70%) требуемого продукта. МС (m/e): 604 (M+1). Схема J Согласно схеме J, 4- или 6-амино-4,5,6,7-тетрагидро-2H-индазол можно получить из 3 аминоциклогексанола, аминогруппа которого первоначально защищена Cbz, с последующим окислением спирта до кетона посредством PCC. Тетрагидроиндазольное кольцо получают сначала путем обработки кетона реагентом Бредерика, а затем N2H4. После снятия защиты Cbz группы при гидрировании получают требуемые 4- и 6-амино 4,5,6,7-тетрагидро-2H-индазолы в виде смеси. Препаративный пример 67. Бензиловый эфир (3-гидроксициклогексил)карбаминовой кислоты. Смесь 3-гидроксициклогексиламина (10 г, 86,96 ммоль), карбоната калия (18 г, 130 ммоль), этилацетата (150 мл) и воды (70 мл) обрабатывают бензилхлорформиатом (22,17 г, 130 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 12 ч. Органический слой отделяют и высушивают над сульфатом натрия, фильтруют и концентрируют. К остатку добавляют диэтиловый эфир. Образовавшийся белый осадок фильтруют и высушивают на воздухе с получением 19,1 г вышеназванного соединения (88%). MC (m/z): 250 (M+). Препаративный пример 68. Бензиловый эфир (3-оксоциклогексил)карбаминовой кислоты. Раствор бензилового эфира (3-гидроксициклогексил)карбаминовой кислоты (15,5 г, 62,24 ммоль) в дихлорметане (300 мл) обрабатывают PCC (16,73 г, 77,81 ммоль) и смесь перемешивают при комнатной температуре в течение 12 ч. Реакционную смесь фильтруют через слой целита. Растворитель удаляют в вакууме. Остаток очищают на силикагелевой колонке, применяя градиент от 25% до 50% этилацетата в гексане с получением 12,6 г (82%) вышеназванного соединения. MC (m/z): 248 (M+). Препаративный пример 69. Бензиловый эфир (4,5,6,7-тетрагидро-1H-индазол-4-ил)карбаминовой кислоты. Смесь бензилового эфира (3-оксоциклогексил)карбаминовой кислоты (12,3 г, 49,8 ммоль), толуола(60 мл) и трет-бутоксибис(диметиламино)метана (9,53 г, 54,77 ммоль) перемешивают при 90C в течение 1,5 ч. Реакцию охлаждают и растворитель удаляют в вакууме. К остатку добавляют метанол (60 мл) и гидразингидрат (2,74 г, 54,77 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 3 ч. Растворитель удаляют в вакууме. К остатку добавляют этилацетат и промывают водой. После высушивания органического слоя над сульфатом натрия фильтруют и концентрируют с получением 9,8 г вышеназванного соединения с 80% чистотой. Препаративный пример 70. 4,5,6,7-Тетрагидро-1H-индазол-4-иламин. Смесь бензилового эфира (4,5,6,7-тетрагидро-1H-индазол-4-ил)карбаминовой кислоты (3,5 г), метанола (50 мл) и палладия-на-углероде (10%, 0,7 г) перемешивают на гидрогенизационном вибраторе Парра при 50 psi в течение 3 ч. Реакционную смесь удаляют из вибратора Парра и фильтруют смесь через слой целита. Фильтрат концентрируют с получением 1,7 г вышеназванного соединения. Согласно схеме K, при обработке альдегида 4-амино-4,5,6,7-тетрагидро-2H-индазолом в присутствии NaBH(OAc)3 получают пиразол. После удаления бензильной защитной группы при гидрировании в присутствии Pd(OH)2 получают фенол, который затем переводят в трифлат. Конечный продукт получают путем обработки трифлата различными бороновыми кислотами или эфирами при стандартных условиях реакции сочетания Сузуки, т.е. в присутствии Pd(PPh3)4 и Na2CO3. Препаративный пример 71. (R)-3-(4-Бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-1H-индазол-4-ил)пирролидин-2-он. К раствору (R)-4-R)-4-бензил-2-оксооксазолидин-3-ил)-3-(4-бензилокси-2,6-дихлорбензил)-4-оксобутиральдегида, препаративный пример 38, (2 г, 3,81 ммоль) в DCE (75 мл) при комнатной температуре,добавляют 4,5,6,7-тетрагидро-1H-индазол-4-иламин (0,66 г, 4,77 ммоль) и триацетоксиборгидрид натрия(2,42 г, 11,45 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 12 ч и при 50C в течение 1 ч. Реакционную смесь охлаждают, разбавляют дихлорметаном и промывают водой. После высушивания органического слоя над сульфатом натрия фильтруют и концентрируют под вакуумом. Остаток очищают способом хроматографии на силикагеле, применяя смеси от 50% этилацетата в гексане до 100% этилацетата с получением 1,32 г вышеназванного соединения. МС (m/z): 470 (M+). Препаративный пример 72. (R)-3-(2,6-Дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-1 Н-индазол 4-ил)пирролидин-2-он. К раствору (R)-3-(4-бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-1H-индазол-4-ил)пирролидин-2-она (1,32 г) в этаноле (50 мл), добавляют гидроксид палладия-на-углероде (10%, 0,65 г). Смесь перемешивают при гидрировании (50 psi) при комнатной температуре в течение 3 ч. Смесь фильтруют через слой целита. Растворитель удаляют в вакууме с получением 0,93 г вышеназванного соединения. MC(m/z): 380 (M+). Препаративный пример 73. 3,5-Дихлор-4-[(R)-2-оксо-1-(1-трифторметансульфонил-4,5,6,7-тетрагидро-1H-индазол-4-ил)пирролидин-3-илметил]фениловый эфир трифторметансульфоновой кислоты и 3,5-дихлор-4-[(R)-2-оксо-1-(2-трифторметансульфонил-4,5,6,7-тетрагидро-2H-индазол-4-ил)пирролидин 3-илметил]фениловый эфир трифторметансульфоновой кислоты При 0C раствор (R)-3-(2,6-дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-1H-индазол-4-ил)пирролидин-2-она (0,93 г, 2,45 ммоль) и пиридина (0,97 г, 12,27 ммоль) в CH2Cl2 (20 мл) обрабатывают трифторметансульфоновым ангидридом (2,07 г, 7,36 ммоль) в течение 20 мин. Реакцию нагревают до комнатной температуры и перемешивают в течение 30 мин. Реакционную смесь разбавляют CH2Cl2 и промывают 1 н. HCl и водой. Органический слой высушивают (Na2SO4) и растворитель удаляют. Остаток очищают способом хроматографии на силикагеле с получением 1,45 г смеси вышеназванных продуктов. МС (m/z): 644 (M+). Препаративный пример 74. Трет-бутиловый эфир 5-[3-(4'-карбокси-3,5-дихлорбифенил-4-илметил)2-оксопирролидин-1-ил]-4,5,6,7-тетрагидроиндазол-2-карбоновой кислоты.- 24015675 При 0C раствор (3R,5S)-3-[2,6-дихлор-4-(морфолин-4-карбонил)бензил]-1-(4,5,6,7-тетрагидро-2Hиндазол-5-ил)пирролидин-2-она, пример 73, (0,066 г, 0,14 ммоль) в CH2Cl2 (5 мл) и пиридине (2,0 мл) обрабатывают Вос-ангидридом (0,036 г, 0,16 ммоль) в течение 5 ч. Реакционную смесь нагревают до комнатной температуры, разбавляют CH2Cl2 и промывают 1 н. HCl и водой. Органический слой высушивают (Na2SO4) и растворитель удаляют с получением 0,074 г смеси вышеназванных продуктов. Препаративный пример 75. (4-Трифторметилпиперидин-1-ил)-[4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]метанон. Стадия 1. 4-Бромбензойную кислоту (4,98 г, 24 ммоль), 4-трифторметилпиперидин гидрохлорид(4,3 г, 27 ммоль), HOBt (4,09 г, 29 ммоль) и DIPEA (15,5 мл, 89 ммоль) растворяют в ТГФ (50 мл). Добавляют EDC (5,7 г, 29 ммоль) и перемешивают при комнатной температуре в течение 24 ч. Реакционную смесь разбавляют этилацетатом, промывают 0,1 н. HCl, бикарбонатом натрия (насыщенным), соляным раствором, высушивают над сульфатом натрия фильтруют и выпаривают до образования масла, получая(4-бромфенил)-(4-трифторметилпиперидин-1-ил)метанон в виде вышеназванного продукта. Стадия 2. (4-Бромфенил)-(4-трифторметилпиперидин-1-ил)метанон (3,13 г, 9,86 ммоль),бис(пинаколато)диборан (2,74 г, 10,7 ммоль), ацетат калия (2,9 г, 2 9 ммоль), хлорид [1,1'-бис (дифенилфосфино)ферроцен]палладия(II) (0,804 г, 0,9 ммоль) добавляют в ДМСО (20 мл) и перемешивают при 80C в течение 17 ч. Охлаждают, разбавляют этилацетатом, промывают соляным раствором (5), высушивают над сульфатом натрия фильтруют и выпаривают. Применение хроматографии (диоксид кремния,CH2Cl2/MeOH, 97:3) позволяет получить 1,04 г (27%) (4-трифторметилпиперидин-1-ил)-[4-(4,4,5,5 тетраметил-[1,3,2]диоксаборолан-2-ил)фенил]метанона. Схема M Согласно схеме M, 4,5,6,7-тетрагидро-1H-индазол-7-иламин получают из 1H-индазол-7-иламина при гидрировании, применяя 5% Rh/C в качестве катализатора. При обработке 4,5,6,7-тетрагидро-1Hиндазол-7-иламина альдегидом в присутствии NaBH(OAc)3 получают пиразол. После удаления бензильной защитной группы с помощью CH3SO3H в присутствии диметилсульфоксида получают фенол, который затем переводят в трифлат. Конечный продукт получают путем обработки трифлата различными бороновыми кислотами или эфирами при стандартных условиях реакции сочетания Сузуки, т.е. в присутствии Pd(PPh3)4 и Na2CO3. Препаративный пример 76. 4,5,6,7-Тетрагидро-1H-индазол-7-иламин. Смешивают 1H-индазол-7-иламин (5,0 г, 37,6 ммоль) и 5% Rh/C (2,45 г) в этаноле (120 мл) и нагревают при 120C в течение 48 ч при давлении H2 1000 psi. Реакционную смесь охлаждают и фильтруют через слой Hyflo. Растворитель удаляют в вакууме и очищают неочищенный продукт с помощью 5% 2 М(R)-4-R)-4-бензил-2-оксооксазолидин-3-ил)-3-(4-бензилокси-2,6-дихлорбензил)-4-оксо- 25015675 бутиральдегида, препаративный пример 38, (9,95 г, 18,9 ммоль) в CH2Cl2 (300 мл) добавляют к раствору 4,5,6,7-тетрагидро-1H-индазол-7-иламина (2,60 г, 19,00 ммоль) в ацетонитриле (300 мл) и перемешивают в течение 30 мин при комнатной температуре в атмосфере N2. К реакционной смеси добавляют триацетоксиборгидрид натрия (12,02 г, 56,9 ммоль) и перемешивают в течение 72 ч. Растворитель удаляют в вакууме и твердое вещество экстрагируют этилацетатом, водой и насыщенным NaHCO3. Органическую фазу высушивают (Na2SO4) фильтруют и концентрируют под вакуумом. Остаток очищают способом хроматографии на силикагеле с градиентом 0-10% метанола в CH2Cl2 с получением 2,53 г (28%) вышеназванного соединения. МС (m/z): 470 (M+). Препаративный пример 78. (3R)-3-(2,6-Дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-1Hиндазол-7-ил)пирролидин-2-он. К раствору (R)-3-(4-бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-1H-индазол-7-ил)пирролидин-2-она (2,46 г, 5,23 ммоль) в диметилсульфоксиде (28 мл), добавляют метансульфоновую кислоту(8,23 г, 85,7 ммоль) и энергично перемешивают смесь при комнатной температуре в течение 18 ч. Растворитель удаляют в вакууме и осадок разбавляют водой, устанавливают pH=7 с помощью 5 н. NaOH, и несколько раз экстрагируют смесь этилацетатом и ТГФ. После высушивания объединенной органической фазы (Na2SO4) фильтруют и концентрируют под вакуумом с получением 2,20 г (100%) вышеназванного соединения. МС (m/z): 381 (M+). Препаративный пример 79. 3,5-Дихлор-4-[(R)-2-оксо-1-(1-трифторметансульфонил-4,5,6,7 тетрагидро-1H-индазол-7-ил)пирролидин-3-илметил]фениловый эфир трифторметансульфоновой кислоты и 3,5-дихлор-4-[(R)-2-оксо-1-(2-трифторметансульфонил-4,5,6,7-тетрагидро-2H-индазол-7-ил)пирролидин-3-илметил]фениловый эфир трифторметансульфоновой кислоты(R)-3-(2,6-дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-1H-индазол-7 ил)пирролидин-2-она (2,10 г, 5,52 ммоль) в пиридине (50 мл) по каплям обрабатывают трифталатным ангидридом (4,99 г, 17,7 ммоль) и перемешивают при 0C в течение 15 мин. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 90 мин. Реакционную смесь разбавляютCH2Cl2 и промывают 1 н. HCl (3 300 мл). Органический слой высушивают (Na2SO4) и растворитель удаляют с получением 2,93 г (82%) смеси вышеназванных продуктов. МС (m/z): 645 (M+). Схема N- 26015675 Согласно схеме N, 4,5,6,7-тетрагидро-1H-индазол-6-иламин получают из 1H-индазол-6-иламина при гидрировании, применяя 5% Rh/C в качестве катализатора. При обработке 4,5,6,7-тетрагидро-1Hиндазол-6-иламина альдегидом в присутствии NaBH(OAc)3 получают пиразол. Удаление бензильной защитной группы с помощью CH3SO3H в присутствии диметилсульфоксида позволяет получить фенол,который затем переводят в трифлат. Конечный продукт получают путем обработки трифлата различными бороновыми кислотами или эфирами при стандартных условиях реакции сочетания Сузуки, т.е. в присутствии Pd(PPh3)4 и Na2CO3. Препаративный пример 80. 4,5,6,7-Тетрагидро-1H-индазол-6-иламин. Смешивают 1H-индазол-6-иламин (12,45 г, 93,6 ммоль) и 5% Rh/C (6,13 г) в этаноле (300 мл) и нагревают при 120C в течение 72 ч при давлении H2 1000 psi. Реакционную смесь охлаждают и фильтруют через слой Hyflo. Растворитель удаляют в вакууме и неочищенный продукт очищают 10% 2 М NH3 вMeOH в CH2Cl2 и смесь фракций повторно очищают 15% 2 М NH3 в MeOH в CH2Cl2 с получением 4,80 г(R)-4-R)-4-бензил-2-оксооксазолидин-3-ил)-3-(4-бензилокси-2,6-дихлорбензил)-4-оксобутиральдегида, препаративный пример 38, (8,17 г, 15,5 ммоль) в CH2Cl2 (100 мл) добавляют к раствору 4,5,6,7-тетрагидро-1H-индазол-6-иламина (2,13 г, 15,5 ммоль) в ацетонитриле (200 мл) и перемешивают в течение 30 мин при комнатной температуре в атмосфере N2. К реакционной смеси добавляют триацетоксиборгидрид натрия (9,89 г, 46,7 ммоль) и перемешивают в течение 18 ч. Растворитель удаляют в вакууме и твердое вещество экстрагируют этилацетатом, водой и насыщенным NaHCO3. Органическую фазу высушивают (Na2SO4), фильтруют и концентрируют под вакуумом. Остаток очищают способом хроматографии на силикагеле с градиентом от 0 до 10% метанола в CH2Cl2 с получением 3,93 г (54%) вышеназванного соединения. МС (m/z): 470 (M+). Препаративный пример 82. (3R)-3-(2,6-Дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-1Hиндазол-6-ил)пирролидин-2-он. К раствору (3R)-3-(4-бензилокси-2,6-дихлорбензил)-1-(4,5,6,7-тетрагидро-1H-индазол-6-ил)пирролидин-2-она (3,93 г, 8,36 ммоль) в диметилсульфоксиде (45 мл) добавляют метансульфоновую кислоту(13,23 г, 138 ммоль) и энергично перемешивают смесь в атмосфере азота при комнатной температуре в течение 18 ч. Растворитель удаляют в вакууме и осадок разбавляют водой, устанавливают pH=7 с помощью 5 н. NaOH и несколько раз экстрагируют смесь этилацетатом и ТГФ. После высушивания объединенной органической фазы (Na2SO4) фильтруют и концентрируют под вакуумом с получением 3,60 г(100%) вышеназванного соединения. МС (m/z): 381 (M+). Препаративный пример 83. 3,5-Дихлор-4-[(R)-2-оксо-1-(1-трифторметансульфонил-4,5,6,7 тетрагидро-1H-индазол-6-ил)пирролидин-3-илметил]фениловый эфир трифторметансульфоновой кислоты и 3,5-дихлор-4-[(R)-2-оксо-1-(2-трифторметансульфонил-4,5,6,7-тетрагидро-2H-индазол-6-ил)пирролидин-3-илметил]фениловый эфир трифторметансульфоновой кислоты При 0C раствор (R)-3-(2,6-дихлор-4-гидроксибензил)-1-(4,5,6,7-тетрагидро-1H-индазол-6-ил)пирролидин-2-она (3,58 г, 9,42 ммоль) в пиридине (100 мл) по каплям обрабатывают трифталатным ангидридом (8,52 г, 30,2 ммоль) и перемешивают при 0C в течение 15 мин. Реакционную смесь нагревают до комнатной температуры и перемешивают в течение 90 мин. Реакционную смесь разбавляют CH2Cl2 и промывают 1 н. HCl (3600 мл). Органический слой высушивают (Na2SO4) и удаляют растворитель с получением 5,12 г (84%) смеси вышеназванных продуктов. МС (m/z): 645 (M+). Препаративный пример 84. 7-(4,4,5,5-Тетраметил-[1,3,2]диоксаборолан-2-ил)имидазо[1,2a]пиридин. Смешивают 7-хлоримидазо[1,2-a]пиридин (500,4 г, 3,28 моль), бис(пинаколато)дибор (999 г, 3,93 моль), трициклогексилфосфин (92 г; 328,06 ммоль) и калия ацетат (483 г; 4,92 моль) в диглиме (простом диметиловом эфире диэтиленгликоля) (4 л) и воде (4,83 мл) и перемешивают в течение 5 мин. Добавляют ацетат палладия(II) (36,81 г, 163,96 ммоль) и дополнительное количество диглима (1 л) и нагревают до 100C в течение 17 ч. Реакцию охлаждают, добавляют карбонат калия (340 г, 2,46 моль) и перемешивают 18 ч. Суспензию отфильтровывают и твердое вещество промывают диглимом (21 л). Твердое вещество суспендируют в воде (5 л) и затем фильтруют и промывают водой (21 л) и гептаном (1 л). Твердое вещество высушивают в вакуумной печи при 60C с получением 695,1 г (90%) вышеназванного продукта. МС (m/z): 245 (M+1).Ir. Боронат присоединяется к пара-бромфторбензолу в реакции сочетания Сузуки с получением биарилтолуола. Биарилтолуол бромируют в положении бензила, применяя NBS в условиях радикального бромирования. Препаративный пример 85. 4-Бромметил-3,5-дихлор-4'-фторбифенил. Стадия 1). Смесь 1,3-дихлортолуола (250 мл, 1,95 моль), гептана (625 мл), бис(пинаколато)дибора(237,9 г, 937 ммоль), 2,2'-бипиридина (3,08 г, 19,47 ммоль), и ди-хлорбис 1,2,5,6-эта)-1,5 циклооктадиен)дииридия (0,662 г; 0,98 ммоль) нагревают при 100C в течение 4 ч. Смесь охлаждают до 55C и добавляют 940 мл трет-бутилметилового эфира. Раствор пропускают через 133 г флэш-силикагеля и силикагель промывают 200 мл трет-бутилметилового эфира. Растворитель заменяют изопропанолом(приблизительно 1 л) и перемешивают суспензию при 5C в течение 1 ч. Твердое вещество собирают путем фильтрации и промывают 200 мл холодного изопропанола. Твердое вещество высушивают под вакуумом с получением 410 г (73% выход) 2-(3,5-дихлор-4-метилфенил)-4,4,5,5-тетраметил[1,3,2]диоксаборолана в виде грязно-белого твердого вещества. 1H ЯМР (CDCl3, 500 МГц)1,34 (12H, с),2,48 (3H, с), 7,68 (2H, с). Стадия 2). К суспензии 2-(3,5-дихлор-4-метилфенил)-4,4,5,5-тетраметил-[1,3,2]диоксаборолана (119 г; 414,64 ммоль) в 1,2-диметоксиэтане (240 мл) в атмосфере азота, добавляют пара-бромфторбензол (57 мл; 518,54 ммоль), воду (120 мл), карбонат калия (115,7 г; 828,79 ммоль), трифенилфосфин (2,72 г; 10,37 ммоль) и Pd(OAc)2 (0,47 г; 2,09 ммоль). Смесь нагревают при 80C в течение 12 ч и затем оставляют охлаждаться до комнатной температуры. К смеси добавляют 240 мл трет-бутилметилового эфира и 480 мл воды, и затем слои разделяют. Органический слой промывают водным раствором TMT (тритиоциануровая кислота, гидрат тринатриевой соли, 5 г) в 120 мл, а затем насыщенным водным NaCl (120 мл). Органический слой заменяют на приблизительно 600 мл изопропанола с целью получения суспензии. Добавляют воду (120 мл) и суспензию охлаждают до 3C. Твердое вещество собирают путем фильтрации и промывают 120 мл холодного метанола. Твердое вещество высушивают под вакуумом с получением 92,8 г 3,5-дихлор-4'-фтор-4-метилбифенила в виде светло-желтого твердого вещества. 1H ЯМР (CDCl3, 500 МГц)2,49 (3H, с), 7,13 (2H, т, J=9 Гц), 7,46 (2H, с), 7,49 (2H, дд, J=9, 5 Гц). Альтернативная стадия 2). Суспензию 2-(3,5-дихлор-4-метилфенил)-4,4,5,5-тетраметил-[1,3,2]диоксаборолана (300 г, 1,05 моль) в 2 л изопропанола перемешивают под защитным слоем азота. Добавляют парабромфторбензол 61619 (150 мл; 1,36 моль), а затем водный раствор карбоната калия (160,5 г; 1,15 моль) в 300 мл воды. Воронку промывают 400 мл изопропанола. Добавляют трифенилфосфин (12,3 г; 46,89 ммоль) и Pd(OAc)2 (2,35 г; 10,47 ммоль) и смесь нагревают при 75C в течение 8 ч. Добавляют воду(300 мл) и оставляют смесь охлаждаться до комнатной температуры с получением суспензии. Суспензию охлаждают на ледяной бане, твердое вещество собирают путем фильтрации и промывают 350 мл смеси 6:1 IPA-вода. Твердое вещество высушивают в вакуумной печи при комнатной температуре в течение ночи с получением 345 г неочищенного продукта. Неочищенный продукт суспендируют в 2,2 л этанола и нагревают при 70C с получением раствора. Добавляют Darco (35 г) и перемешивают в течение 30 мин. Смесь фильтруют через слой Hyflo (целит) и оставляют раствор охлаждаться до комнатной температуры для получения суспензии. Суспензию охлаждают до 5C, твердое вещество собирают путем фильтрации и промывают холодным этанолом (530 мл). Твердое вещество высушивают под вакуумом с получением 230 г 3,5-дихлор-4'-фтор-4-метилбифенила в виде грязно-белого твердого вещества (86% выход). Стадия 3). К суспензии 3,5-дихлор-4'-фтор-4-метилбифенила (138 г; 541 ммоль) в 965 мл ацетонитрила, добавляют N-бромсукцинимид (109 г; 603 ммоль), и затем добавляют дополнительное количество 140 мл ацетонитрила (промывание). Добавляют бензоилпероксид (1,37 г; 5,5 ммоль) и смесь нагревают при 80C в течение 2,5 ч. Добавляют тиосульфат натрия (1,75 г) в 276 мл воды и оставляют смесь постепенно охлаждаться. Добавляют к смеси еще 138 мл воды при 55C и вводят затравку кристаллов 4 бромметил-3,5-дихлор-4'-фторбифенила. После того как температура суспензии достигнет комнатной,- 28015675 добавляют еще 280 мл воды и суспензию охлаждают на ледяной бане. Твердое вещество собирают путем фильтрации, промывают 275 мл холодного метанола и высушивают с получением 170 г (94%) 4 бромметил-3,5-дихлор-4'-фторбифенила в виде твердого вещества. Оптимальная очистка - уменьшение содержания основной примеси от 2 до 1%. 100 г порцию вышеуказанного материала суспендируют в 200 мл ACN и нагревают до 75C. Добавляют метанол (200 мл),позволяя температуре уменьшиться до 54C. Суспензию охлаждают на ледяной бане и твердое вещество собирают путем фильтрации. Твердое вещество высушивают под вакуумом с получением 79,5 г 4 бромметил-3,5-дихлор-4'-фторбифенила в виде твердого вещества. 1H ЯМР (ДМСО-d6, 500 МГц)4,81NBS (6457 г) и продолжают перемешивание. Добавляют бензоилпероксид (73 г), продолжают перемешивание и медленно нагревают реакционную смесь до 85C. После снижения экзотермы продолжают нагревать реакционную смесь до 120C и поддерживают эту температуру реакции до тех пор, пока реакция согласно данным ГХ не будет завершена (1-2 ч). Охлаждают реакционную смесь до 50-55C и гасят тиосульфат пентагидратом натрия (224 г) в воде (25,4 л), затем перемешивают смесь в течение как минимум 30 мин. Слои разделяют. Органический слой промывают водой (120 л). Водные промывки еще раз экстрагируют (стадии 8 и 10) хлорбензолом (16 л). Органические фазы объединяют, высушивают MgSO4,добавляют древесный уголь и хорошо перемешивают. Смесь фильтруют через воронку Бюхнера, применяя стекловолоконный фильтр, промывают метиленхлоридом (3500 мл). Фильтрат концентрируют до 3 л суммарного объема; красное масло выпаривают вместе с изопропанолом (37 л) для удаления всего остаточного хлорбензола (изопропанол можно заменить метанолом). Круглодонную колбу с широким горлом помещают в охлаждающую баню, масло разбавляют метанолом (5 л) и энергично перемешивают до осаждения продукта. Смесь разбавляют дополнительным количеством метанола (10 л) и перемешивают при комнатной температуре в течение ночи. Охлаждают смесь до 0-5C (баня со льдом/водой) в течение как минимум 2 ч. Твердое вещество фильтруют на воронке Бюхнера, применяя полипропиленовую фильтровальную ткань. Твердое вещество промывают метанолом (34 л, -20C). Твердое вещество помещают на поддон и высушивают в вакуумной печи при 25C до 30C в течение как минимум 12 ч. Схема P Согласно схеме P, 5-нитроиндазол защищают в виде N-ацетилпроизводного и затем нитрогруппу и бензольное кольцо восстанавливают в условиях гидрирования в присутствии аммиака. Аммиак приводит к деацилированию при восстановительных условиях. Образовавшийся рацемический амин разделяют с помощью ди-п-толуоилвинной кислоты. Образовавшуюся разделенную соль переводят в свободное основание путем перемешивания суспензии соли с карбонатом калия в ACN/воде. В качестве альтернативы, можно избежать образования защищенного ацетила и сразу восстановить 5-нитроиндазол. Альтернативный препаративный пример амина без ацетильной защитной группы. Смесь 25 г 5-нитроиндазола, 500 мл 8,3 М гидроксида аммония и 25 г 10% палладия-на-углероде с влажностью 50% нагревают при 100C при давлении водорода 200 psi в течение 24 ч. 10 г порцию этой смеси очищают способом хроматографии, применяя смесь NH3/MeOH/CH2Cl2 с получением 235 мг (58% расчетный выход) рацемического амина (R,S)-(4,5,6,7-тетрагидро-1H-индазол-5-ил)амин. Рацемический амин разделяют, как описано в способе получения препаративного примера 86. Препаративный пример 86. (S)-(4,5,6,7-Тетрагидро-1H-индазол-5-ил)амин 1/2 DTTA. Стадия 1). Коммерчески доступный 5-нитроиндазол (100 г, 613 ммоль) растворяют в 2 л ТГФ и затем добавляют триэтиламин (260 мл; 1,87 моль). Раствор перемешивают при комнатной температуре до тех пор, пока все твердое вещество не перейдет в раствор, и затем добавляют ангидрид уксусной кислоты(94 г; 927 ммоль). После 2 ч при комнатной температуре добавляют 2 л EtOAc и экстрагируют смесь 1 н.HCl (2800 мл). Органическую фазу промывают водным бикарбонатом натрия, а затем насыщенным водным NaCl. Органическую фазу высушивают сульфатом магния и концентрируют до образования густой суспензии. Добавляют трет-бутилметиловый эфир (300 мл) и фильтруют суспензию. Собирают твер- 29
МПК / Метки
МПК: C07D 403/04, A61P 9/10, A61P 3/10, A61K 31/416
Метки: ингибиторов, циклогексилпиразол-лактама, 11-бета-гидроксистероиддегидрогеназы, качестве, производные
Код ссылки
<a href="https://eas.patents.su/30-15675-proizvodnye-ciklogeksilpirazol-laktama-v-kachestve-ingibitorov-11-beta-gidroksisteroiddegidrogenazy-1.html" rel="bookmark" title="База патентов Евразийского Союза">Производные циклогексилпиразол-лактама в качестве ингибиторов 11-бета-гидроксистероиддегидрогеназы 1</a>
Производные бифениламидлактама в качестве ингибиторов 11-бета-гидроксистероиддегидрогеназы 1

Номер патента: 14718
Опубликовано: 28.02.2011
Авторы: Айкер Томас Дэниел, Хайт Гари Алан, Хинклин Рональд Джей, Тот Джеймс Ли, Сюй Яньпин, Уоллэйс Оуэн Брендан, Чэнь Чжаогэнь, Виннероски Джуниор Леонард Ларри, Маккауан Джефферсон Рэй, Саеед Ашраф, Снайдер Нэнси Джун, Йорк Джереми Шуленбург, Ли Жэньхуа, Красутский Алексей Павлович
МПК: C07D 401/10, C07D 401/12, C07D 207/12...
Метки: 11-бета-гидроксистероиддегидрогеназы, качестве, ингибиторов, бифениламидлактама, производные
Формула / Реферат:
1. Соединение, структура которого представлена формулойгде R1представляет собой или где пунктирной линией обозначено место присоединения в положение R1;R2 представляет собой -Н, -галоген, -СН3(необязательно замещенный 1-3 атомами галогенов) или -О-СН3(необязательно замещенный 1-3 атомами галогенов);R3 представляет собой -галоген, -СН3(необязательно замещенный 1-3 атомами галогенов) или -O-СН3 (необязательно замещенный 1-3 атомами галогенов);R4...
Производные лактама циклогексилимидазола в качестве ингибиторов 11-бета-гидроксистероид-дегидрогеназы 1

Номер патента: 14717
Опубликовано: 28.02.2011
Авторы: Снайдер Нэнси Джун, Саеед Ашраф, Уоллэйс Оуэн Брендан, Сюй Яньпин, Мэбри Томас Эдвард, Гюнтер Ребекка Линн
МПК: A61P 3/00, C07D 401/14, A61K 31/4015...
Метки: циклогексилимидазола, ингибиторов, 11-бета-гидроксистероид-дегидрогеназы, качестве, производные, лактама
Формула / Реферат:
1. Соединение, структура которого представлена формулойв которой R1 представляет собой -Н, -галоген, -О-СН3(необязательно, имеющий в качестве заместителей от одного до трех атомов галогена) или -СН3 (необязательно, имеющий в качестве заместителей от одного до трех атомов галогена);R2 представляет собой -Н, -галоген, -О-СН3(необязательно, имеющий в качестве заместителей от одного до трех атомов галогена) или -СН3 (необязательно, имеющий в...
Триазолопиридины в качестве ингибиторов 11-бета-гидроксистероиддегидрогеназы типа i

Номер патента: 13017
Опубликовано: 26.02.2010
Авторы: Робл Джеффри А., Ли Джеймс Дж., Руан Жеминг, Ванг Хайксиа, Хейманн Лоуренс Дж., Ли Юнь, Купер Кристофер Б.
МПК: A61K 31/435, A61P 3/00, C07D 471/04...
Метки: качестве, триазолопиридины, 11-бета-гидроксистероиддегидрогеназы, типа, ингибиторов
Формула / Реферат:
1. Соединение формулы (I)его энантиомеры, диастереоизомеры, сольваты или соли, гдеW представляет собой арил, циклоалкил, гетероарил или гетероциклил, каждый из которых может быть необязательно замещен R1, R1a, R1b, R1c и R1d;R1, R1a, R1b, R1c и R1dнезависимо представляют собой водород, галоген, -ОН, -CN, -NO2, -CO2R2a, -CONR2R2a, -SO2NR2R2a, -SOR2a, -SO2R2a, -NR2SO2R6, -NR2CO2R6, алкил, галогеналкил, циклоалкил, алкокси, арилокси, алкенил,...
Производные адамантилпирролидин-2-она в качестве ингибиторов 11-бета-гидроксистероид-дегидрогеназы

Номер патента: 11021
Опубликовано: 30.12.2008
Авторы: Бюйк Кристоф Франсис Роберт Нестор, Линдерс Йоаннес Теодорус Мария, Яроскова Либуше, Ван Дер Векен Луи Йозеф Элизабет
МПК: A61K 31/402, C07D 207/26, C07D 211/76...
Метки: 11-бета-гидроксистероид-дегидрогеназы, адамантилпирролидин-2-она, ингибиторов, качестве, производные
Формула / Реферат:
1. Соединение, имеющее формулу его N-оксидные формы, фармацевтически приемлемые аддитивные соли и стереохимически изомерные формы, где n равно 1 или 2; М представляет собой прямую связь или C1-3алкильный линкер, необязательно замещенный одним или двумя заместителями, выбранными из С1-4алкила, C1-3алкилокси-С1-4алкила, гидрокси-С1-4алкила, гидрокси, C1-3алкилокси- или фенил-С1-4алкила; каждый из R1 и R2 независимо представляет собой водород,...
Производные n-2-адамантанил-2-феноксиацетамида в качестве ингибиторов 11-бета- гидроксистероид-дегидрогеназы

Номер патента: 12263
Опубликовано: 28.08.2009
Авторы: Линдерс Йоаннес Теодорус Мария, Ван Дер Векен Луи Йозеф Элизабет, Бисхофф Франсуа Пол, Виллемсенс Густаф Хенри Мария, Яроскова Либуше
МПК: A61K 31/165, A61P 19/10, A61P 25/28...
Метки: производные, n-2-адамантанил-2-феноксиацетамида, ингибиторов, качестве, гидроксистероид-дегидрогеназы, 11-бета
Формула / Реферат:
1. Применение соединения формулы (I) его N-оксидных форм, фармацевтически приемлемых аддитивных солей и стереохимически изомерных форм, где n=1, 2, 3 или 4; Z представляет О, S, NR6, SO или SO2; R1 представляет водород, циано, гидрокси или C1-4алкил, необязательно замещенный галогеном; R2 представляет водород, C1-4алкил или C1-4алкилокси-; R3 представляет водород, С1-4алкил, C1-4алкилокси- или R3 в комбинации с R2 вместе образуют двухвалентный...
Предыдущий патент: Жидкие составы, содержащие диалкилсульфосукцинат и ингибиторы гидроксифенилпируват-диоксигеназы
Следующий патент: Композиции и способы ингибирования экспрессии гена pcsk9