Пиридинметиленазолидиноны и их применение
Номер патента: 12178
Опубликовано: 28.08.2009
Авторы: Бишофф Александер, Помель Винсэн, Рюкле Томас, Ковини Давид, Кваттропани Анна, Дорбэ Жером



























Формула / Реферат
1. Производное пиридинметиленазолидинона согласно формуле

в которой R1 выбран из Н;
R2 выбран из Н, необязательно замещенного арила, необязательно замещенного индолила, необязательно замещенного C3-C8-гетероциклоалкила, содержащего от 1 до 3 гетероатомов, выбранных из О, S, NR, где R представляет собой водород или метил, необязательно замещенного изоиндолила и необязательно замещенного бензотиенила;
причем эти группы необязательно замещены от 1 до 5 заместителями, выбранными из группы, состоящей из галогена, тригалогенметила, -O-C1-C6-алкила, CO-C1-C6-алкила, SO2-C1-C6-алкила, необязательно замещенного галогенами, SO2-C1-C6-алкилгетероциклоалкила, CO2-C1-C6-алкила, OSO2-C1-C6-алкила, необязательно замещенного галогенами, OSO2-C1-C6-алкилгетероциклоалкила, OCO-C1-C6-алкила, SO2-C1-C6-алкилгетероарила, CO2-C1-C6-алкилгетероарила и C1-C6-алкил-C1-C6-алкокси;
X выбран из S и О;
Y выбран из S и О;
А обозначает пиримидин, пиразин, фуран, имидазол;
n обозначает целое число, выбранное из 1 и 2;
а также его геометрические изомеры и их оптически активные формы, такие как энантиомеры, диастереомеры, и их рацемические формы, а также его фармацевтически приемлемые соли.
2. Производное пиридинметиленазолидинона по п.1, в котором R1 обозначает Н.
3. Производное пиридинметиленазолидинона по пп.1, 2, в котором R2 обозначает Н.
4. Производное пиридинметиленазолидинона по пп.1, 2, в котором R2 обозначает C3-C8-гетероциклоалкил.
5. Производное пиридинметиленазолидинона по пп.1, 2, в котором R2 выбран из арила и необязательно замещенного [2,3-дигидро]бензофурила, необязательно замещенного индолила, необязательно замещенного изоиндолила, необязательно замещенного бензотиенила, необязательно замещенного бензимидазолила, необязательно замещенного бензотриазолила, необязательно замещенного бензотиазолила, необязательно замещенного бензоксазолила, необязательно замещенного изобензотиенила, необязательно замещенного оксадиазолила,
причем эти группы необязательно замещены от 1 до 5 заместителями, выбранными из группы, состоящей из галогена, тригалогенметила, -O-C1-C6-алкила, CO-C1-C6-алкила, SO2-C1-C6-алкила, необязательно замещенного галогенами, SO2-C1-C6-алкилгетероциклоалкила, CO2-C1-C6-алкила, OSO2-C1-C6-алкила, необязательно замещенного галогенами, OSO2-C1-C6-алкилгетероциклоалкила, OCO-C1-C6-алкила, SO2-C1-C6-алкилгетероарила, CO2-C1-C6-алкилгетероарила и C1-C6-алкил-C1-C6-алкокси.
6. Производное пиридинметиленазолидинона по любому из пп.1-5, в котором X обозначает S.
7. Производное пиридинметиленазолидинона по любому из пп.1-6, в котором Y обозначает О.
8. Производное пиридинметиленазолидинона по любому из пп.1-7, в котором Y обозначает S.
9. Производное пиридинметиленазолидинона по любому из пп.1-8, в котором n=1.
10. Производное пиридинметиленазолидинона по любому из пп.1-9, в котором А вместе с пиридиновым кольцом образует следующую группу:

в которой R1 и R2 имеют значения, определенные в любом из пп.1-9.
11. Производное пиридинметиленазолидинона по любому из пп.1-9, в котором А вместе с пиридиновым кольцом образует следующую группу:

в которой R1 и R2 имеют значения, определенные в любом из пп.1-10.
12. Производное пиридинметиленазолидинона по любому из пп.1-9, в котором А вместе с пиридиновым кольцом образует следующую группу:

в которой R1 и R2 имеют значения, определенные в любом из пп.1-11.
13. Производное пиридинметиленазолидинона по любому из пп.1-9, в котором А вместе с пиридиновым кольцом образует следующую группу:

в которой R1 и R2 имеют значения, определенные в любом из пп.1-12.
14. Производное пиридинметиленазолидинона по любому из пп.1-10, в котором R1 обозначает Н; R2 обозначает C3-C8-гетероциклоалкил; X обозначает S; Y обозначает О или S и А вместе с пиридиновым кольцом образует группу формулы (Ia).
15. Производное пиридинметиленазолидинона по любому из пп.1-14, в котором R1 обозначает Н; X обозначает S; Y обозначает О и А вместе с пиридиновым кольцом образует группу формулы (Ib).
16. Производное пиридинметиленазолидинона по любому из пп.1-15, в котором R1 обозначает Н; X обозначает S; Y обозначает О и А вместе с пиридиновым кольцом образует группу формулы (Ic).
17. Производное пиридинметиленазолидинона по любому из пп.1-16, в котором R1 обозначает Н; X обозначает S; Y обозначает О и А вместе с пиридиновым кольцом образует группу формулы (Id).
18. Производное пиридинметиленазолидинона по любому из пп.1-17, выбранное из следующей группы:
(5Z)-5-{[4-(1-пиперидинил)пиридо[3,2-d]пиримидин-6-ил]метилен}-1,3-тиазолидин-2,4-дион;
(5Z)-5-{[4-(4-фтор-1-пиперидинил)пиридо[3,2-d]пиримидин-6-ил]метилен}-1,3-тиазолидин-2,4-дион;
(5Z)-5-({4-[4-(трифторметил)-1-пиперидинил]пиридо[3,2-d]пиримидин-6-ил}метилен)-1,3-тиазолидин-2,4-дион;
5-пиридо[2,3-b]пиразин-6-илметилентиазолидин-2,4-дион;
5-фуро[3,2-b]пиридин-5-илметилентиазолидин-2,4-дион;
5-[4-(4-фторпиперидин-1-ил)пиридо[3,2-d]пиримидин-6-илметилен]-2-тиоксотиазолидин-4-он;
5-(3-фенил-3H-имидазо[4,5-b]пиридин-5-илметилен)тиазолидин-2,4-дион;
5-[3-3,5-диметоксифенил)-3H-имидазо[4,5-b]пиридин-5-илметилен]тиазолидин-2,4-дион;
трет-бутиловый эфир 5-[5-(2,4-диоксотиазолидин-5-илиденметил)имидазо[4,5-b]пиридин-3-ил]-2,3-дигидроиндол-1-карбоновой кислоты;
5-[3-(2,3-дигидро-1Н-индол-5-ил)-3H-имидазо[4,5-b]пиридин-5-илметилен]тиазолидин-2,4-дион;
5-[3-(1-ацетил-2,3-дигидро-1Н-индол-5-ил)-3H-имидазо[4,5-b]пиридин-5-илметилен]тиазолидин-2,4-дион;
5-{3-[1-(4-диметиламинобутирил)-2,3-дигидро-1Н-индол-5-ил]-3H-имидазо[4,5-b]пиридин-5-илметилен}тиазолидин-2,4-дион;
5-[3-(1-метансульфонил-2,3-дигидро-1Н-индол-5-ил)-3H-имидазо[4,5-b]пиридин-5-илметилен]тиазолидин-2,4-дион;
5-[3-(1-хлорметансульфонил-2,3-дигидро-1Н-индол-5-ил)-3H-имидазо[4,5-b]пиридин-5-илметилен]тиазолидин-2,4-дион;
5-{3-[1-(3-морфолин-4-илпропан-1-сульфонил)-2,3-дигидро-1Н-индол-5-ил]-3H-имидазо[4,5-b]пиридин-5-илметилен}тиазолидин-2,4-дион;
трет-бутиловый эфир 6-[5-(2,4-диоксотиазолидин-5-илиденметил)имидазо[4,5-b]пиридин-3-ил]-2,3-дигидроиндол-1-карбоновой кислоты;
5-[3-(1-метансульфонил-2,3-дигидро-1Н-индол-6-ил)-3H-имидазо[4,5-b]пиридин-5-илметилен]тиазолидин-2,4-дион.
19. Применение соединений по пп.1-18 в качестве лекарственного средства.
20. Применение производного пиридинметиленазолидинона по пп.1-18, а также его изомеров и смесей для получения лекарственного средства для профилактики и/или лечения аутоиммунных нарушений и/или воспалительных заболеваний, сердечно-сосудистых заболеваний, нейродегенеративных заболеваний, бактериальных или вирусных инфекций, заболеваний почек, агрегации тромбоцитют, рака, трансплантации, отторжения трансплантата или повреждений легкого.
21. Применение по п.20, в котором указанные заболевания выбраны из группы, включающей рассеянный склероз, псориаз, ревматоидный артрит, системную красную волчанку, воспалительное заболевание кишечника, воспаление легких, тромбоз или инфекцию/воспаление мозга, такую как менингит или энцефалит.
22. Применение по п.20, в котором указанные заболевания выбраны из группы, включающей болезнь Альцгеймера, болезнь Гентингтона, травму ЦНС, инсульт или ишемические состояния.
23. Применение по п.20, в котором указанные заболевания выбраны из группы, включающей атеросклероз, гипертрофию сердечной мышцы, дисфункцию кардиомиоцитов, повышенное кровяное давление или вазоконстрикцию.
24. Применение по п.20, в котором указанные заболевания выбраны из группы, включающей хроническое обструктивное заболевание легких, фиброз анафилактического шока, псориаз, аллергические заболевания, астму, инсульт или ишемические состояния, ишемию-реперфузию, агрегацию/активацию тромбоцитов, атрофию/гипертрофию скелетных мышц, рекрутинг лейкоцитов в раковой ткани, ангиогенез, инвазию метастаз, меланому, саркому Капоши, острые и хронические бактериальные и вирусные инфекции, сепсис, отторжение трансплантата, гломерулосклероз, гломерулонефрит, прогрессивный фиброз почек, эндотелиальные и эпителиальные повреждения в легком или в общем воспаление дыхательных путей легкого.
25. Применение по любому из пп.20-24 для модуляции, в особенности для ингибирования, активности PI3 киназы.
26. Применение по п.25, в котором указанная PI3 киназа представляет собой PI3 киназу g.
27. Фармацевтическая композиция, содержащая по меньшей мере одно производное пиридинметиленазолидинона по любому из пп.1-18 и фармацевтически приемлемый носитель, разбавитель или эксципиент.
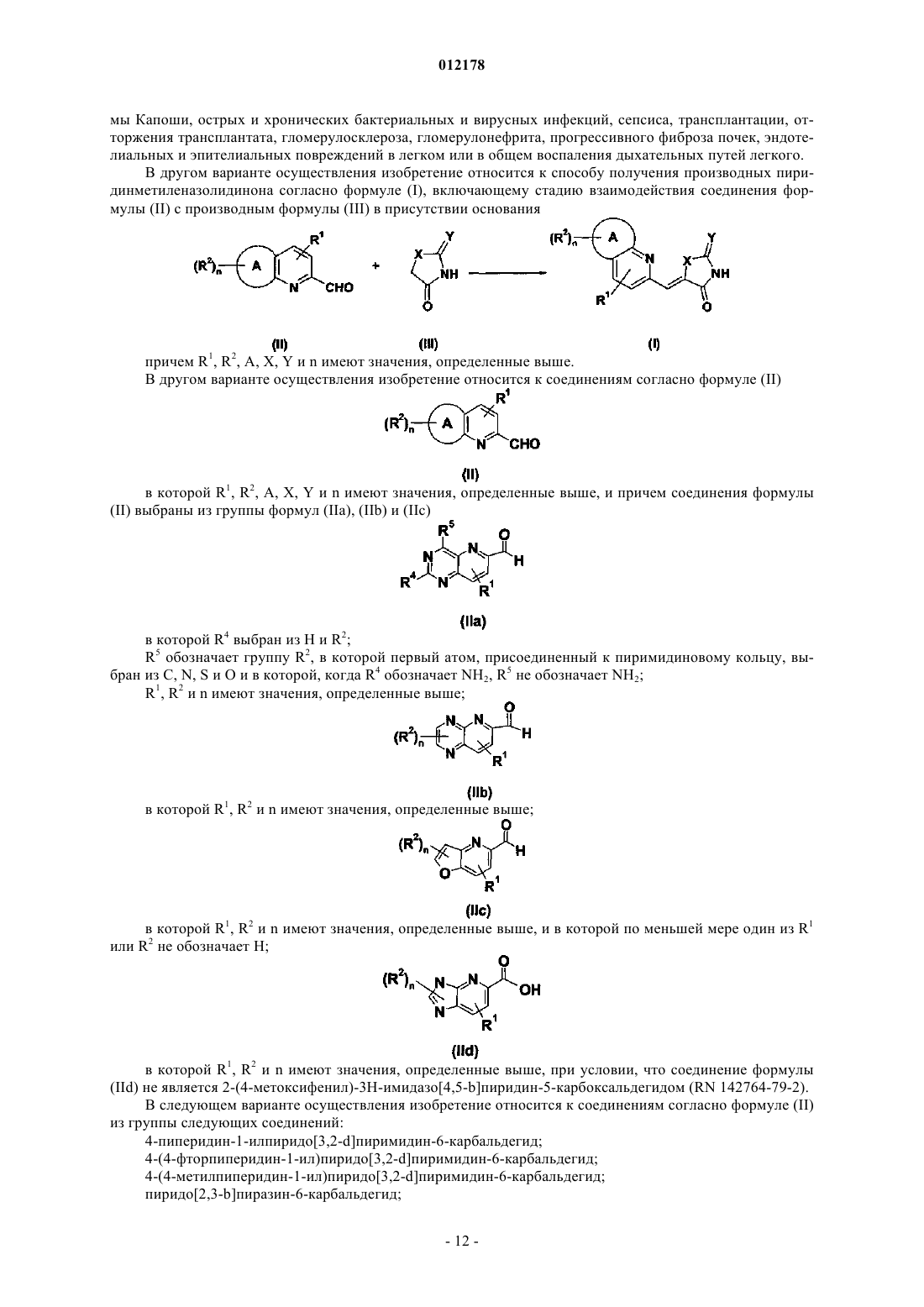
28. Способ получения производного пиридинметиленазолидинона по любому из пп.1-18, включающий стадию взаимодействия соединения формулы (II) с производным формулы (III) в присутствии основания

причем R1, R2, А, X, Y и n имеют значения, определенные в любом из пп.1-27.
29. Соединение согласно формуле (II)

в котором R1, R2, А, X, Y и n имеют значения, определенные в любом из пп.1-28, и причем соединение формулы (II) выбрано из группы формул (IIa), (IIb), (IIc) и (IId)

в которой R4 выбран из Н и R2; R5 обозначает группу R2, в которой первый атом, присоединенный к пиримидиновому кольцу, выбран из С, N, S и О, и в которой, когда R4 обозначает NH2, R5 не обозначает NH2; R1, R2 и n имеют значения, определенные в любом из пп.1-28;

в которой R1, R2 и n имеют значения, определенные в любом из пп.1-28;

в которой R1, R2 и n имеют значения, определенные в любом из пп.1-28, и в которой по меньшей мере один из R1 или R2 не обозначают Н;

в которой R1, R2 и n имеют значения, определенные в любом из пп.1-28, и при условии, что соединение формулы (IId) не является 2-(4-метоксифенил)-3H-имидазо[4,5-b]пиридин-5-карбоксальдегидом.
30. Соединение по п.29, выбранное из следующей группы:
4-пиперидин-1-илпиридо[3,2-d]пиримидин-6-карбальдегид;
4-(4-фторпиперидин-1-ил)пиридо[3,2-d]пиримидин-6-карбальдегид;
4-(4-метилпиперидин-1-ил)пиридо[3,2-d]пиримидин-6-карбальдегид;
пиридо[2,3-b]пиразин-6-карбальдегид;
2-триметилсиланилфуро[3,2-b]пиридин-5-карбальдегид;
3-фенил-1Н-имидазо[4,5-b]пиридин-5-карбальдегид;
3-(3,5-диметоксифенил)-3H-имидазо[4,5-b]пиридин-5-карбальдегид;
трет-бутил-5-(5-формил-3H-имидазо[4,5-b]пиридин-3-ил)индолин-1-карбоксилат;
3-(1-ацетил-2,3-дигидро-1Н-индол-5-ил)-3H-имидазо[4,5-b]пиридин-5-карбальдегид;
3-{1-[4-(диметиламино)бутаноил]-2,3-дигидро-1Н-индол-5-ил}-3H-имидазо[4,5-b]пиридин-5-карбальдегид;
3-[1-(метилсульфонил)-2,3-дигидро-1Н-индол-5-ил]-3H-имидазо[4,5-b]пиридин-5-карбальдегид;
3-{1-[(хлорметил)сульфонил]-2,3-дигидро-1Н-индол-5-ил}-3H-имидазо[4,5-b]пиридин-5-карбальдегид;
3-{1-[(3-морфолин-4-илпропил)сульфонил]-2,3-дигидро-1Н-индол-5-ил}-3H-имидазо[4,5-b]пиридин-5-карбальдегид;
трет-бутил-6-(5-формил-3H-имидазо[4,5-b]пиридин-3-ил)индолин-1-карбоксилат и
3-[1-(метилсульфонил)-2,3-дигидро-1Н-индол-6-ил]-3H-имидазо[4,5-b]пиридин-5-карбальдегид.
Текст
012178 Область изобретения Настоящее изобретение относится к применению производных пиридинметиленазолидинона формулы (I) для лечения и/или профилактики аутоиммунных нарушений и/или воспалительных заболеваний, сердечно-сосудистых заболеваний, нейродегенеративных заболеваний, бактериальных или вирусных инфекций, аллергии, астмы, панкреатита, мультиорганной недостаточности, заболеваний почек, агрегации тромбоцитов, рака, подвижности сперматозоидов, отторжения трансплантата или повреждения легкого. В частности, настоящее изобретение относится к производным пиридинметиленазолидинона для модуляции, особенно для ингибирования активности или функции фосфоинозитид-3-киназ (PI3K). Предпосылки создания изобретения Фосфоинозитид-3-киназы (PI3K) играют критическую сигнальную роль в пролиферации клеток,выживании клетки, васкуляризации, мембранном переносе, транспорте глюкозы, росте невритов, образовании складок мембраны, продукции супероксида, перестройке актина и хемотаксисе (Cantley, 2000, Science, 296, 1655-1657 and Vanhaesebroeck et al., 2001, Annu. Rev. Biochem., 70, 535-602). Термин РI3K относится к семейству липидкиназ, которое у млекопитающих состоит из восьми идентифицированных PI3K, которые делятся на три подсемейства согласно их структуре и их специфичности в отношении субстрата. Группа класса I PI3K состоит из двух подгрупп, класса IA и класса IB. Класс IA состоит из регуляторных единиц размером 85 кДа (ответственных за белок-белковые взаимодействия через взаимодействие домена гомологии Src 2 (SH2) с фосфотирозиновыми остатками других белков) и каталитической субъединицы размером 110 кДа. В этом классе существуют три каталитических формы (p100, p110 и р 110) и пять регуляторных изоформ (р 85, р 85, p55, p55 и р 50). Класс IB стимулируется субъединицамигетеродимерных белков G. Единственный охарактеризованный член класса IB PI3K (каталитическая субъединица р 110, скомплексованная с 101-кДа регуляторным белком, р 101). Класс II PI3K включает изоформы ,и , которые имеют размер приблизительно 170 кДа и характеризуются наличием С-концевого домена С 2. Класс III PI3K включает фосфатидилинозитолспецифические 3-киназы. Эволюционно консервативные изоформы p110 иэкспрессируются повсеместно, в то время какиболее специфично экспрессируются в клетках гематопоэтической системы, гладкомышечных клетках, миоцитах и эндотелиальных клетках (Vanhaesebroeck et al., 1997, Trends Biochem Sci., 22(7), 267-72). Их экспрессия может также регулироваться индуцируемым образом в зависимости от типа клетки, ткани и стимулов, а также контекста заболевания.PI3K представляют собой ферменты, участвующие в фосфолипидной передаче сигналов и активируются в ответ на множество внеклеточных сигналов, таких как факторы роста, митогены, интегрины(взаимодействия клетка-клетка), гормоны, цитокины, вирусы и нейромедиаторы, а также внутриклеточной перекрестной регуляцией другими сигнальными молекулами (перекрестная связь, где первоначальный сигнал может активизировать некоторые параллельные пути передачи, которые на второй стадии передают сигналы к PI3K за счет внутриклеточных сигнальных событий), такими как, например, малые ГТФ-азы, киназы или фосфатазы. Фосфатидилинозитол (PtdIns) представляет собой основное строительное звено для внутриклеточных инозитольных липидов в эукариотических клетках; он состоит из D-миоинозитол-1-фосфата (Ins1P),связанного через фосфатную группу с диацилглицерином. Головная инозитольная группа Ptdlns имеет пять свободных гидроксильных групп, и обнаружено, что три из них фосфорилируются в клетках в различных комбинациях. PtdIns и его фосфорилируемые производные все вместе называют инозитолфосфолипидами или фосфоинозитидами (PI). Восемь разновидностей PI были зарегистрированы в эукариотические клетки (Vanhaesebroeck et al., 2001, выше). Все PI находятся в мембранах и являются субстратами для киназ, фосфатаз и липаз.In vitro, PI3K фосфорилируют 3-гидроксильную группу инозитольного кольца на трех различных субстратах: фосфатидилинозитол (PtdIns), фосфатидилинозитол-4-фосфат (PI(4)P) и фосфатидилинозитол-4,5 бифосфат (PI(4,5)Р 2),соответственно приводя к трем липидным продуктам, а именно фосфатидилинозитол-3 монофосфату (PI(3)Р), фосфатидилинозитол-3,4-бифосфату (PI(3,4)P2) и фосфатидилинозитол-3,4,5 трифосфату (PI(,4,5)P3 (см. схему А ниже). Предпочтительным субстратом для класса I PI3K является PI(4,5)Р 2. Класс II PIK проявляет выраженное предпочтение в отношении PtdIns в качестве субстрата по сравнению с PI(4)Р и PI(4,5)P2. КлассIII PI3K может использовать как субстрат in vivo только PtdIns и, вероятно, является ответственным за генерацию большинства PI(3)Р в клетках (Vanhaesebroeck et al., 2001, выше). Фосфоинозитидный внутриклеточный сигнальный путь начинается со связывания сигнальной молекулы (внеклеточные лиганды, раздражители, димеризация рецептора, трансактивация гетерологичным рецептором (например, рецептором тирозинкиназы со связанным с белком G трансмембранным рецептором, интегрированным в плазматическую мембрану, что приводит к активации PI3K. После активации PI3K преобразовывают мембранный фосфолипид PI(4,5)P2 в PI(3,4,5)Р 3, который,в свою очередь, может быть далее преобразован в другую 3'-фосфорилированную форму фосфоинозитидов 5'-специфичными фосфоинозитидфосфатазами; таким образом, ферментативная активность PI3K приводит прямо или косвенно к генерации двух подтипов 3'-фосфоинозитида, которые функционируют как вторые мессенджеры во внутриклеточной передаче сигнала (Leslie et al., 2001, Chem. Rev. 101(8) 2365-80; Katso et al., 2001, Annu. Rev. Cell Dev. Biol. 1, 615-75 and Toker et al., 2002, Cell Mol. Life Sci. 59(5), 761-79). Роль фосфорилированных продуктов PtdIns в качестве вторых мессенджеров заключается в том, что они участвуют во множестве путей передачи сигнала, включая значимые для пролиферации клеток,дифференцировки клеток, роста клеток, размера клеток, выживания клеток, апоптоза, адгезии, подвижности клеток, миграции клеток, хемотаксиса, инвазии, перестройки цитоскелета, изменений формы клетки, переноса везикул и метаболического пути (Stein, 2000, Mol. Med. Today 6(9) 347-57). Хемотаксис направленное движение клеток по градиенту концентраций химических аттрактантов, также названных хемокинами, участвует во многих важных заболеваниях, таких как воспалительные/аутоиммунные заболевания, нейродегенерация, ангиогенез, инвазия/метастаз и заживление ран (Wyman et al., 2000, ImmunolToday, 21(6), 260-4; Hirsch et al., 2000, Science, 287(5455) 1049-53; Hirsch et al., 2001, FASEB J. 15(11) 2019-21 and Gerard et al., 2001, Nat Immunol. 2(2), 108-15). Поэтому считается, что активация PI3-киназы участвует в широком диапазоне клеточных реакций,включая рост клеток, дифференцировку и апоптоз (Parker et al., 1995, Current Biology, 5, 577-99; Yao etal., 1995, Science, 267, 2003-05). Недавние биохимические исследования показали, что класс I PI3K (например, изоформа PI3K класса IB) представляет собой киназные ферменты с двойной специфичностью, т.е. они демонстрируют как липидкиназную (фосфорилирование фосфоинозитидов), так и протеинкиназную активность, поскольку они могут вызывать фосфорилирование другого белка в качестве субстрата, включая автофосфорилирование как внутримолекулярный регуляторный механизм.PI3K, по-видимому, участвует во множестве аспектов активации лейкоцитов. Было показано, что р 85-связанная PI3-киназная активность физически ассоциирована с эндоплазматическим доменом CD28,который является важной костимулирующей молекулой для активации Т-клеток в ответ на антиген(Pages et al., 1994, Nature, 369, 321-29). Эти эффекты связаны с увеличенным уровнем транскрипции множества генов, включая интерлейкин-2 (IL-2), важный фактор роста Т-клеток (Fraser et al., 1991,Science, 251, 313-16). Мутация CD28, при которой он может дольше взаимодействовать с PI3-киназой,приводит к невозможности инициировать продукцию IL-2, что предполагает критическую роль PI3 киназы в активации Т-клеток. Клеточные процессы, в которых PI3K играют существенную роль, включают супрессию апоптоза,перестройку актинового скелета, рост кардиомиоцитов, стимуляцию гликогенсинтазы инсулином, TNFопосредованное примирование нейтрофила и генерацию супероксида, и миграцию лейкоцитов и адгезию к эндотелиальным клеткам.PI3K был идентифицирован как медиатор G бета-гамма-зависимой регуляции активности JNK, гдеG бета-гамма являются субъединицами гетеротримерных белков G (Lopez-Ilasaca et al., 1998, J. Biol.Chem. 273(5), 2505-8). Недавно было описано, что PI3K передает воспалительные сигналы через различныеG(i)-связанные рецепторы (Laffargue et al., 2002, Immunity, 16(3), 441-51) и играет центральную роль в функции тучных клеток, раздражителей в контексте лейкоцитов, иммунологии, включая, например, цитокины, хемокины, аденозины, антитела, интегрины, факторы агрегации, факторы роста, вирусы или гормоны (Lawlor et al., 2001, J. Cell. Sci., 114 (Pt 16), 2903-1 and Stephens et al., 2002, Curr. Opinion CellBiol. 14(2), 203-13). Специфические ингибиторы против отдельных членов семейства ферментов представляют собой ценные инструменты для расшифровки функции каждого фермента. Два соединения, LY294002 и вортманнин (см. ниже), широко использовались как ингибиторыPI3-киназы. Эти соединения представляют собой неспецифические ингибиторы PI3K, поскольку они не различают четыре члена класса I PI3-киназ Значения IC50 вортманнина против каждой из различных PI3-киназ класса I составляют 1-10 нМ, а значения IC50 для LY294002 против каждой из этих PI3-киназ составляют приблизительно 15-20 мкМ(Fruman et al., 1998, Ann. Rev. Biochem., 61, 481-507), также 5-10 мМ в отношении протеинкиназы CK2 и наблюдается некоторая ингибирующая активность в отношении фосфолипаз. Вортманнин представляет собой грибковый метаболит, который необратимо ингибирует активность PI3K за счет ковалентного связывания с каталитическим доменом этого фермента. Ингибирование активности PI3K вортманнином делает невозможной последующую клеточную реакцию на внеклеточный фактор (Thelen et al., 1994, Proc. Natl. Acad. Sci. USA, 91, 4960-64). Эксперименты с вортманнином показали, что активность PI3K в клетках гематопоэтической системы, особенно нейтрофилы, моноциты и другие типы лейкоцитов, участвует во многих видах иммунного ответа без запоминания, связанного с острым и хроническим воспалением. На основе исследований с использованием вортманнина было показано, что функция PI3-киназы также требуется для некоторых аспектов передачи сигнала лейкоцитами через G-белок-связанные рецепторы (Thelen et al., 1994). Более того, показано, что вортманнин и LY294002 блокируют миграцию нейтрофилов и высвобождение супероксида. Однако, поскольку эти соединения не различают различные изоформы PI3K, остается неясным, какая специфическая изоформа или изоформы PI3K участвуют в этих явлениях. Некоторые результаты показали, что ингибиторы PI3K, например, LY294002, могут увеличить invivo противоопухолевую активность некоторых цитотоксических средств (например, паклитаксела)(Grant, 2003, IDrugs, 6(10), 946-948). Недавно были получены производные тиазолидина в качестве ингибиторов PI3K (WO 2004/007491; В WO 2004/056820 описаны производные бензоксазина следующей структуры: Высокая релевантность пути PI3K в некоторых широко распространенных заболеваниях приводит к необходимости разработать ингибиторы, включая селективные ингибиторы, PIKs. Краткое описание изобретения Объектом изобретения являются вещества, которые пригодны для лечения и/или профилактики нарушений, связанных с фосфоинозитид-3-киназами, PI3K. Также объектом настоящего изобретения являются вещества, которые пригодны для лечения и/или профилактики аутоиммунных и/или воспалительных нарушений. Также объектом настоящего изобретения являются вещества, которые пригодны для лечения и/или профилактики сердечно-сосудистых заболеваний. Также объектом настоящего изобретения являются вещества, которые пригодны для лечения и/или профилактики нейродегенеративных нарушений. Также объектом настоящего изобретения являются вещества, которые пригодны для лечения и/или профилактики нарушения, выбранного из бактериальных и вирусных инфекций, заболеваний почек, агрегации тромбоцитов, рака, трансплантации, отторжения трансплантата, повреждений легкого, респираторных заболеваний и ишемических состояний. В частности, объектом настоящего изобретения являются химические соединения, которые способны модулировать, особенно ингибировать, активность или функцию фосфоинозитид-3-киназ, PI3K, при болезненных состояниях у млекопитающих, особенно у людей. Кроме того, объектом настоящего изобретения является новая категория фармацевтических составов для лечения и/или заболеваний, выбранных из аутоиммунных, воспалительных нарушений, сердечно-сосудистых заболеваний, нейродегенеративных нарушений, бактериальных и вирусных инфекций,заболеваний почек, агрегации тромбоцитов, рака, трансплантации, отторжения трансплантата, повреждений легкого, респираторных заболеваний и ишемических состояний. Кроме того, объектом настоящего изобретения является способ лечения и/или профилактики нарушений, выбранных из аутоиммунных, воспалительных нарушений, сердечно-сосудистых заболеваний,нейродегенеративных нарушений, бактериальных и вирусных инфекций, заболеваний почек, агрегации тромбоцитов, рака, трансплантации, отторжения трансплантата или повреждений легкого, респираторных заболеваний и ишемических состояний. В первом аспекте изобретение относится к производным пиридинметиленазолидинона формулы (I)-4 012178 Во втором аспекте изобретение относится к соединению согласно формуле (I) для применения в качестве лекарственного средства. В третьем аспекте изобретение относится к применению соединения согласно формуле (I) для получения фармацевтической композиции для лечения нарушения, выбранного из аутоиммунных, воспалительных нарушений, сердечно-сосудистых заболеваний, нейродегенеративных нарушений, бактериальных и вирусных инфекций, заболеваний почек, агрегации тромбоцитов, рака, трансплантации, отторжения трансплантата или повреждений легкого, респираторных заболеваний и ишемических состояний и других заболеваний и нарушений, связанных с фосфоинозитид-3-киназами, PI3K, включая PI3Kи . В четвертом аспекте изобретение относится к фармацевтической композиции, включающей по меньшей мере одно соединение согласно формуле (I) и фармацевтически приемлемый носитель, растворитель или эксципиент. В пятом аспекте изобретение относится к способу лечения пациента, у которого имеется нарушение, выбранное из аутоиммунных, воспалительных нарушений, сердечно-сосудистых заболеваний, нейродегенеративных нарушений, бактериальных и вирусных инфекций, заболеваний почек, агрегации тромбоцитов, рака, трансплантации, отторжения трансплантата или повреждений легкого, респираторных заболеваний и ишемических состояний и других заболеваний и нарушений, связанных с фосфоинозитид-3-киназами, PI3K. Способ включает введение соединения согласно формуле (I). В шестом аспекте изобретение относится к способу синтеза соединения согласно формуле (I). В седьмом аспекте изобретение относится к соединениям согласно формуле (II). Подробное описание изобретения В следующих параграфах представлены определения различных химических групп, которые составляют соединения согласно изобретению, которые должны однородно пониматься по всему описанию и формуле изобретения, если не дано более четкого определения.C1-C6-алкил относится к одновалентной алкильной группе, имеющей 1-6 атомов углерода. Этот термин иллюстрируется такими группами, как метил, этил, н-пропил, изопропил, н-бутил, изобутил,трет-бутил, н-гексил и т.п. По аналогии, C1-C12-алкил относится к одновалентной алкильной группе,имеющей 1-12 атомов углерода, включая метил, этил, н-пропил, изопропил, н-бутил, изобутил, третбутил, н-гексил, гептил, октил, нонил, децил, ундецил, додецил и т.п. Арил относится к ненасыщенной ароматической карбоциклической группе, содержащей от 6 до 14 атомов углерода, имеющей единственное кольцо (например, фенил) или множество конденсированных колец (например, нафтил). Арил включает фенил, нафтил, фенантренил и т.п.C1-C6-алкиларил относится к C1-C6-алкильным группам, имеющим арильный заместитель, включая бензил, фенетил и т.п. Гетероарил относится к моноциклической гетероароматической, или бициклической или трициклической конденсированной гетероароматической группе. Частные примеры гетероароматических групп включают необязательно замещенный пиридил, пирролил, пиримидинил, фурил, тиенил,имидазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, пиразолил, 1,2,3-триазолил, 1,2,4-триазолил,1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, 1,3,4-триазинил,1,2,3-триазинил, бензофурил, [2,3-дигидро]бензофурил, изобензофурил, бензотиенил, бензотриазолил,изобензотиенил, индолил, изоиндолил, 3H-индолил, бензимидазолил, имидазо[1,2-а]пиридил, бензотиазолил, бензоксазолил, хинолизинил, хиназолинил, фталазинил, хиноксалинил, циннолинил, нафтиридинил, пиридо[3,4-b]пиридил, пиридо[3,2-b]пиридил, пиридо[4,3-b]пиридил, хинолил, изохинолил, тетразолил, 5,6,7,8-тетрагидрохинолил, 5,6,7,8-тетрагидроизохинолил, пуринил, птеридинил, карбазолил,ксантенил или бензохинолил.C2-C6-алкенил относится к алкенильным группам, предпочтительно имеющим от 2 до 6 атомов углерода и имеющим по меньшей мере 1 или 2 участка ненасыщенности алкенила. Предпочтительные алкенильные включают группы винил (-СН=CH2), н-2-пропенил (аллил, -СН 2 СН=СН 2) и т.п.C2-C6-алкинил относится к алкинильным группам, предпочтительно имеющим от 2 до 6 атомов углерода и имеющим по меньшей мере 1-2 участка алкинильной ненасыщенности, предпочтительные алкинильные группы включают этинил (-CСН), пропаргил (-СН 2 ССН) и т.п.C3-C8-циклоалкил относится к насыщенной карбоциклической группе, содержащей от 3 до 8 атомов углерода, имеющей единственное кольцо (например, циклогексил) или множество конденсированных колец (например, норборнил). C3-C8-циклоалкил включает циклопентил, циклогексил, норборнил и т.п. Гетероциклоалкил относится к C3-C8-циклоалкильной группе согласно данному выше определению, у которой до 3 атомов углерода заменены гетероатомами, выбранными из группы, состоящей из О,S, NR, причем R представляет собой водород или метил. Гетероциклоалкил включает пирролидин, пиперидин, пиперазин, 1-метилпиперазин, морфолин, тетрагидрофуран и т.п.C3-C8-(гетеро)циклоалкилацил относится к 3-8-членным циклоалкильным или гетероциклоалкильным группам, имеющим ацильный заместитель. Ацилокси относится к группе -OC(O)R, где R включает Н, C1-C6-алкил, C2-C6-алкенил,C2-C6-алкинил, C3-C8-циклоалкил, гетероциклоалкил, арил, гетероарил, C1-C6-алкиларил илиC1-C6-алкилацилокси относится к C1-C6-алкильным группам, имеющим ацилоксизаместитель,включая этиловый эфир аминопропионовой кислоты и т.п. Алкокси относится к группе -O-R, где R включает C1-C6-алкил, или арил, или гетероарил илиC1-C6-алкилалкокси относится к C1-C6-алкильным группам, имеющим алкоксизаместитель, включая метокси, метоксиэтил и т.п. Алкоксикарбонил относится к группе -С(О)OR, где R включает Н, C1-C6-алкил, или арил, или гетероарил или C1-C6-алкиларил, или C1-C6-алкилгетероарил.C1-C6-алкилалкоксикарбонил относится к C1-C6-алкильным группам, имеющим алкоксикарбонильный заместитель, включая 2-(бензилоксикарбонил)этил и т.п. Аминокарбонил относится к группе -C(O)NRR', где каждый R, R' включает независимо водород или C1-C6-алкил или арил или гетероарил или C1-C6-алкиларил или C1-C6-алкилгетероарил.C1-C6-алкилгетероарил,C2-C6-алкениларил,C2-C6-алкенилгетероарил,C2-C6-алкиниларил, C2-C6-алкинилгетероарил, C1-C6-алкилциклоалкил, C1-C6-алкилгетероциклоалкил и где R' и R", вместе с атомом азота, к которому они присоединены, могут необязательно образовывать 3-8-членное гетероциклоалкильное кольцо.-6 012178 Амино относится к группе -NRR', где каждый R, R' обозначает независимо водород илиC1-C6-алкил, или арил, или гетероарил или C1-C6-алкиларил, или C1-C6-алкилгетероарил, или циклоалкил,или гетероциклоалкил и где R и R', вместе с атомом азота, к которому они присоединены, могут необязательно образовывать 3-8-членное гетероциклоалкильное кольцо.C1-C6-алкиламино относится к C1-C6-алкильным группам, имеющим аминозаместитель, включая 2-(1-пирролидинил)этил и т.п. Аммоний относится к положительно заряженной группе -N+RR'R", где каждый R, R', R" обозначает независимо C1-C6-алкил или C1-C6-алкиларил или C1-C6-алкилгетероарил, или циклоалкил, или гетероциклоалкил, и где R и R', вместе с атомом азота, к которому они присоединены, могут необязательно образовывать 3-8-членное гетероциклоалкильное кольцо.C1-C6-алкилсульфинил относится к C1-C6-алкильным группам, имеющим сульфинильный заместитель, включая 2-(метилсульфинил)этил и т.п. Сульфанил относится к группам -S-R, где R включает Н, C1-C6-алкил, C1-C6-алкил, замещенный галогенами, например к группе -SO-CF3, C2-C6-алкенил, C2-C6-алкинил, C3-C8-циклоалкил, гетероциклоалкил, арил, гетероарил, C1-C6-алкиларил или C1-C6-алкилгетероарил, C2-C6-алкениларил,C2-C6-алкенилгетероарил, C2-C6-алкиниларил, C2-C6-алкинилгетероарил, C1-C6-алкилциклоалкил,C1-C6-алкилгетероциклоалкил. Предпочтительные сульфанильные группы включают метилсульфанил,этилсульфанил и т.п.C1-C6-алкилсульфониламино относится к C1-C6-алкильным группам, имеющим сульфонаминозаместитель, включая 2-(этилсульфониламино)этил и т.п. Аминосульфонил относится к группе -SO2-NRR', где каждый R, R' включает независимо водород,С 1-С 6-алкил, С 2-С 6-алкенил, С 2-С 6-алкинил, С 3-С 8-циклоалкил, гетероциклоалкил, арил, гетероарил,С 1-С 6-алкиларил или С 1-С 6-алкилгетероарил,С 2-С 6-алкениларил,С 2-С 6-алкенилгетероарил,С 2-С 6-алкиниларил, С 2-С 6-алкинилгетероарил, С 1-С 6-алкилциклоалкил, С 1-С 6-алкилгетероциклоалкил. С 1-С 6-алкиламиносульфонил относится к С 1-С 6-алкильным группам, имеющим аминосульфонильный заместитель, включая 2-(циклогексиламиносульфонил)этил и т.п. Замещенный или незамещенный: если не оговорено в определении индивидуального заместителя, вышеуказанные группы, такие как алкенил, алкинил, арил, гетероарил, циклоалкил, гетероциклоалкил и т.д., могут быть необязательно замещены от 1 до 5 заместителями, выбранными из группы, состоящей из С 1-С 6-алкила, С 2-С 6-алкенила, С 2-С 6-алкинила, циклоалкила, гетероциклоалкила,С 1-С 6-алкиларила, С 1-С 6-алкилгетероарила, С 1-С 6-алкилциклоалкила, С 1-С 6-алкилгетероциклоалкила,амино, аммония, ацила, ацилокси, ациламино, аминокарбонила, алкоксикарбонила, уреидо, арила, карбамата, гетероарила, сульфинила, алкокси, сульфанила, галогена, карбокси, тригалогенметила, циано,гидрокси, меркапто, нитро и т.п.-7 012178 Замещенный относится к группам, замещенным от 1 до 5 заместителями, выбранными из группы,состоящей из С 1-С 6-алкила, С 2-С 6-алкенила, С 2-С 6-алкинила, циклоалкила, гетероциклоалкила,С 1-С 6-алкиларила, С 1-С 6-алкилгетероарила, С 1-С 6-алкилциклоалкила, С 1-С 6-алкилгетероциклоалкила,амино, аминосульфонила, аммония, ациламино, аминокарбонила, арила, гетероарила, сульфинила,сульфонила, алкокси, алкоксикарбонила, карбамата, сульфанила, галогена, тригалогенметила, циано,гидрокси, меркапто, нитро и т.п. Фармацевтически приемлемые соли или комплексы относятся к солям или комплексам определенных ниже соединений формулы (I), которые сохраняют желаемую биологическую активность. Примеры таких солей включают, но не ограничены ими, кислотно-аддитивные соли, образованные с неорганическими кислотами (например, соляной кислотой, бромисто-водородной кислотой, серной кислотой,фосфорной кислотой, азотной кислотой и т.п.), и соли, образованные с органическими кислотами, такими как уксусная кислота, щавелевая кислота, винная кислота, янтарная кислота, яблочная кислота, фумаровая кислота, малеиновая кислота, аскорбиновая кислота, бензойная кислота, дубильная кислота, памовая кислота, альгиновая кислота, полиглутаминовая кислота, нафталинсульфокислота, нафталиндисульфокислота и полигалактуроновая кислота. Указанные соединения могут также быть введены в форме известных специалисту фармацевтически приемлемых солей четвертичного основания, которые, в частности, включают четвертичную соль аммония формулы -NR, R', R"+Z-, где R, R', R" обозначают независимо водород, алкил или бензил, C1-C6-алкил, C2-C6-алкенил, C2-C6-алкинил, C1-C6-алкиларил,C1-C6-алкилгетероарил, циклоалкил, гетероциклоалкил, и Z обозначает противоион, включая хлорид,бромид, йодид, -О-алкил, толуолсульфонат, метилсульфонат, сульфонат, фосфат или карбоксилат (такой как бензоат, сукцинат, ацетат, гликолят, малеат, малат, фумарат, цитрат, тартрат, аскорбат, циннамоат,манделоат и дифенилацетат). Фармацевтически активное производное относится к любому соединению, которое при введении реципиенту является способным к обеспечению, прямо или косвенно, описанной здесь активности. Термин косвенно также охватывает пролекарства, которые могут быть преобразованы в активную форму лекарственного средства через эндогенные ферменты или метаболизм. В настоящее время обнаружено, что соединения согласно настоящему изобретению представляют собой модуляторы фосфатоинозитид-3-киназ (PI3K), включая PI3Kи . Когда фосфатоинозитид-3 киназный (PI3K) фермент ингибируется соединениями согласно настоящему изобретению, PI3K оказывается неспособным проявлять свои ферментативные, биологические и/или фармакологические эффекты. Соединения согласно настоящему изобретению поэтому могут быть использованы при лечении и профилактике аутоиммунных нарушений и/или воспалительных заболеваний, сердечно-сосудистых заболеваний, нейродегенеративных заболеваний, бактериальных или вирусных инфекций, заболеваний почек,агрегации тромбоцитов, рака, трансплантации, отторжения трансплантата или повреждений легкого. Общая формула (I) согласно настоящему изобретению также включает таутомеры, геометрические изомеры, оптически активные формы, такие как энантиомеры, диастереомеры и рацемические формы,так же как и фармацевтически приемлемые соли. Предпочтительными фармацевтически приемлемыми солями формулы (I) являются кислотно-аддитивные соли, образованные с фармацевтически приемлемыми кислотами, такие как гидрохлорид, гидробромид, сульфат или бисульфат, фосфат или гидрофосфат,ацетат, бензоат, сукцинат, фумарат, малеат, лактат, цитрат, тартрат, глюконат, метансульфонат, бензолсульфонат и п-толуолсульфонат. Соединения согласно формуле (I) являются пригодными для модуляции, особенно ингибирования активности фосфатоинозитид-3-киназ (PI3K). Поэтому предполагается, что соединения согласно настоящему изобретению также особенно пригодны для лечения и/или профилактики нарушений, опосредованных PI3K, особенно PI3Kи/или PI3K . Указанное лечение включает модуляцию - особенно ингибирование или даун-регуляцию - фосфатоинозитид-3-киназ. Соединения согласно формуле (I) являются пригодными для использования в качестве лекарственного средства. В одном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I)C1-C6-алкилалкокси, необязательно замещенного алкоксикарбонила, необязательно замещенного ацила,необязательно замещенного сульфонила, необязательно замещенного сульфанила, необязательно заме-8 012178 щенного сульфинила, необязательно замещенного алкокси и необязательно замещенного амино;C2-C6-алкенила, C2-C6-алкинила, необязательно замещенного арила, такого как фенил и 3,5-диметоксифенил, необязательно замещенного гетероарила, такого как необязательно замещенный 2,3-дигидроиндолил (например, трет-бутиловый эфир 2,3-дигидроиндол-1-карбоновой кислоты,2,3-дигидро-1 Н-индол-5-ил,ацетил-2,3-дигидро-1 Н-индол-5-ил,1-(4-диметиламинобутирил)-2,3 дигидро-1 Н-индол-5-ил, 1-метансульфонил-2,3-дигидро-1 Н-индол-5-ил, 1-хлорметансульфонил-2,3 дигидро-1 Н-индол-5-ил, 1-(3-морфолин-4-илпропан-1-сульфонил)-2,3-дигидро-1 Н-индол-5-ил), необязательно замещенного C3-C8-циклоалкила, необязательно замещенного C3-C8-гетероциклоалкила, включая необязательно замещенный пиперидинил, такой как 1-пиперидинил, 4-фтор-1-пиперидинил,4-(трифторметил)-1-пиперидинил, необязательно замещенного арил-C1-C6-алкила, необязательно замещенного гетероарил-C1-C6-алкила, необязательно замещенного C3-C8-циклоалкил-C1-C6-алкила и необязательно замещенного C3-C8-гетероциклоалкил-C1-C6-алкила, необязательно замещенногоC1-C6-алкилалкокси, необязательно замещенного алкоксикарбонила, необязательно замещенного ацила,необязательно замещенного сульфонила, необязательно замещенного сульфанила, необязательно замещенного сульфинила, необязательно замещенного алкокси и необязательно замещенного амино;Y выбран из О, S и NR3, где R3 выбран из Н, необязательно замещенного C1-C6-алкокси, необязательно замещенного C1-C6-алкила, необязательно замещенного C2-C6-алкенила, необязательно замещенного C2-C6-алкинила, необязательно замещенного C1-C6-алкиларила, циано и необязательно замещенного сульфонила; А обозначает необязательно замещенный гетероарил, включая необязательно замещенный пиримидинил, необязательно замещенный пиразинил, необязательно замещенный фурил и необязательно замещенный имидазолил;n обозначает целое число, выбранное из 1 и 2; а также их геометрические изомеры, их оптически активные формы, такие как энантиомеры, диастереомеры и их рацемические формы, а также их фармацевтически приемлемые соли. В частном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которой R1 обозначает Н. В другом частном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которой R2 обозначает Н. В другом частном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которой R2 обозначает необязательно замещенный C3-C8-гетероциклоалкил. В другом частном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которой R2 выбран из необязательно замещенного арила и необязательно замещенного гетероарила. В другом частном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которой R3 обозначает Н. В другом частном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которой X обозначает S. В другом частном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которой Y обозначает О. В другом частном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которой Y обозначает S. В другом частном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которой n=1. В другом частном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которой n=2. В предпочтительном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которой А является таким, что он вместе с пиридиновым кольцом образует следующую группу (Ia): в которой R1, R2 и n имеют значения, как определено выше. В другом предпочтительном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которой А является таким, что он вместе с пиридиновым кольцом образует следующую группу (Ib): в которой R1, R2 и n имеют значения, как определено выше. В другом предпочтительном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которой А является таким, что он вместе с пиридиновым кольцом образует следующую группу (Ic): в которой R1, R2 и n имеют значения, как определено выше. В другом предпочтительном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которой А является таким, что он вместе с пиридиновым кольцом образует следующую группу (Id): в которой R1, R2 и n имеют значения, как определено выше. В предпочтительном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которойR2 обозначает необязательно замещенный C3-C8-гетероциклоалкил;Y обозначает О или S; А вместе с пиридиновым кольцом образует группу формулы (Ia) иn=1. В предпочтительном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которойY обозначает О и А вместе с пиридиновым кольцом образует группу формулы (Ib). В предпочтительном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которойY обозначает О и А вместе с пиридиновым кольцом образует группу формулы (Iс). В предпочтительном варианте осуществления изобретение относится к производным пиридинметиленазолидинона формулы (I), в которойY обозначает О и А вместе с пиридиновым кольцом образует группу формулы (Id).- 10012178 Соединения согласно настоящему изобретению включают, в частности, соединения группы, состоящей из Соединения согласно настоящему изобретению пригодны как лекарственные средства. Они могут использоваться для получения лекарственного средства для профилактики и/или лечения аутоиммунных нарушений и/или воспалительных заболеваний, сердечно-сосудистых заболеваний, нейродегенеративных заболеваний, бактериальных или вирусных инфекций, заболеваний почек, агрегации тромбоцитов,рака, трансплантации, отторжения трансплантата или повреждений легкого. В одном варианте осуществления соединения формулы (I) пригодны для лечения и/или профилактики аутоиммунных заболеваний или воспалительных заболеваний, таких как рассеянный склероз, псориаз, ревматоидный артрит, системная красная волчанка, воспалительное заболевание кишечника, воспаление легких, тромбоз или мозговая инфекция/воспаление, такая как менингит или энцефалит. В другом варианте осуществления соединения формулы (I) пригодны для лечения и/или профилактики нейродегенеративных заболеваний, включая болезнь Альцгеймера, болезнь Гентингтона, травму ЦНС, инсульт или ишемические состояния. В следующем варианте осуществления изобретения соединения формулы (I) пригодны для лечения и/или профилактики сердечно-сосудистых заболеваний, таких как атеросклероз, гипертрофия сердечной мышцы, дисфункция кардиомиоцитов, повышенное кровяное давление или вазоконстрикция. В следующем варианте осуществления согласно изобретению соединения формулы (I) пригодны для лечения и/или профилактики хронического обструктивного заболевания легких, фиброза анафилактического шока, псориаза, аллергических заболеваний, астмы, инсульта или ишемических состояний,ишемии-реперфузии, агрегации/активации тромбоцитов, атрофии/гипертрофии скелетной мышцы, рекрутинга лейкоцитов в раковые ткани, ангиогенеза, инвазии/метастаза, в частности, при меланоме, сарко- 11012178 мы Капоши, острых и хронических бактериальных и вирусных инфекций, сепсиса, трансплантации, отторжения трансплантата, гломерулосклероза, гломерулонефрита, прогрессивного фиброза почек, эндотелиальных и эпителиальных повреждений в легком или в общем воспаления дыхательных путей легкого. В другом варианте осуществления изобретение относится к способу получения производных пиридинметиленазолидинона согласно формуле (I), включающему стадию взаимодействия соединения формулы (II) с производным формулы (III) в присутствии основания причем R1, R2, А, X, Y и n имеют значения, определенные выше. В другом варианте осуществления изобретение относится к соединениям согласно формуле (II)(II) выбраны из группы формул (IIa), (IIb) и (IIc) в которой R1, R2 и n имеют значения, определенные выше, и в которой по меньшей мере один из R1 или R2 не обозначает Н;(IId) не является 2-(4-метоксифенил)-3H-имидазо[4,5-b]пиридин-5-карбоксальдегидом (RN 142764-79-2). В следующем варианте осуществления изобретение относится к соединениям согласно формуле (II) из группы следующих соединений: 4-пиперидин-1-илпиридо[3,2-d]пиримидин-6-карбальдегид; 4-(4-фторпиперидин-1-ил)пиридо[3,2-d]пиримидин-6-карбальдегид; 4-(4-метилпиперидин-1-ил)пиридо[3,2-d]пиримидин-6-карбальдегид; пиридо[2,3-b]пиразин-6-карбальдегид;- 12012178 2-триметилсиланилфуро[3,2-b]пиридин-5-карбальдегид; 3-фенил-1 Н-имидазо[4,5-b]пиридин-5-карбальдегид; 3-(3,5-диметоксифенил)-3H-имидазо[4,5-b]пиридин-5-карбальдегид; трет-бутил-5-(5-формил-3H-имидазо[4,5-b]пиридин-3-ил) индолин-1-карбоксилат; 3-(1-ацетил-2,3-дигидро-1 Н-индол-5-ил)-3H-имидазо[4,5-b]пиридин-5-карбальдегид; 3-1-[4-(диметиламино)бутаноил]-2,3-дигидро-1H-индол-5-ил-3H-имидазо[4,5-b]пиридин-5 карбальдегид; 3-[1-(метилсульфонил)-2,3-дигидро-1H-индол-5-ил]-3H-имидазо[4,5-b]пиридин-5-карбальдегид; 3-1-[(хлорметил)сульфонил]-2,3-дигидро-1H-индол-5-ил-3H-имидазо[4,5-b]пиридин-5 карбальдегид; 3-1-[(3-морфолин-4-илпропил)сульфонил]-2,3-дигидро-1H-индол-5-ил-3H-имидазо[4,5-b]пиридин-5-карбальдегид; трет-бутил-6-(5-формил-3H-имидазо[4,5-b]пиридин-3-ил) индолин-1-карбоксилат; 3-[1-(метилсульфонил)-2,3-дигидро-1H-индол-6-ил]-3H-имидазо[4,5-b]пиридин-5-карбальдегид. Производные пиридинметиленазолидинона, иллюстрируемые этим изобретением, могут быть получены из легкодоступных исходных веществ, с использованием следующих общих способов и методик. Следует отметить, что, если приведены типичные или предпочтительные экспериментальные условия(т.е. температуры реакции, время, число молей реагентов, растворители и т.д.), другие экспериментальные условия могут также использоваться, если не указано иное. Оптимальные условия реакции могут варьировать в зависимости от конкретных используемых реагентов или растворителей, но такие условия могут быть определены специалистом с использованием обычных методик оптимизации. Когда они используются как фармацевтические средства, соединения согласно настоящему изобретению обычно вводятся в форме фармацевтической композиции. Следовательно, фармацевтические композиции, включающие соединение формулы (I) и фармацевтически приемлемый носитель, растворитель или эксципиент, также входят в объем настоящего изобретения. Специалисту известен весь спектр таких носителей, растворителей или эксципиентов, пригодных для составления фармацевтической композиции. Соединения по изобретению, вместе с традиционно используемым адъювантом, носителем, растворителем или эксципиентом, могут быть переведены в форму фармацевтических композиций и их разовых доз, и в такой форме могут использоваться как твердые препараты, такие как таблетки или наполненные капсулы, или жидкие препараты, такие как растворы, суспензии, эмульсии, эликсиры, или наполненные ими капсулы, для перорального использования, или в форме стерильных растворов для инъекций для перентерального (включая подкожное использование). Такие фармацевтические композиции и их разовые формы могут включать ингредиенты в обычных соотношениях, в присутствии или в отсутствие дополнительных активных соединений или действующих начал, и такие разовые формы могут содержать любое подходящее эффективное количество активного ингредиента, соразмерного с намеченным ежедневным диапазоном дозировки. Фармацевтические композиции, содержащие производные пиридинметиленазолидинона по изобретению, могут быть получены способом, известным в уровне техники в области фармации и могут включать по меньшей мере одно активное соединение. Вообще, соединения согласно настоящему изобретению применяются в фармацевтически эффективном количестве. Количество фактически используемого соединения будет определено врачом в свете обстоятельств, включая состояние, подлежащее лечению,выбранный путь введения, фактическое используемое соединение, возраст, массу тела и индивидуальную реакцию пациента, серьезность симптомов у пациента и т.п. Фармацевтические композиции согласно настоящему изобретению могут быть введены различными путями, включая пероральный, ректальный, чрескожный, подкожный, внутривенный, внутримышечный и внутриносовой. Композиции для перорального введения могут иметь форму жидких растворов или суспензий, или порошков в развес. Более обычно, однако, композиции представлены в формах разовых доз, чтобы облегчить точное дозирование. Термин формы разовых доз относится к физически дискретным единицам, пригодным в виде разовых доз для человека и других млекопитающих, причем каждая доза содержит предопределенное количество активного вещества, вычисленное таким образом, чтобы произвести желаемый терапевтический эффект, в комбинации с подходящим фармацевтическим эксципиентом. Типичные разовые формы включают предварительно наполненные, предварительно замеренные ампулы или шприцы с жидкими композициями или пилюли, таблетки, капсулы и т.п. в случае твердых композиций. В таких композициях производное пиридинметиленазолидинона обычно является минорным компонентом (от приблизительно 0,1 до приблизительно 50 вес.% или предпочтительно от приблизительно 1 до приблизительно 40 вес.%), где остаток образован различными основами или носителями и технологическими добавками, пригодными для получения желаемой лекарственной формы. Жидкие формы, подходящие для перорального введения, могут включать подходящий водный или неводный носитель с буферами, суспендирующими и диспергирующими средствами, красителями, ароматизаторами и т.п. Твердые формы могут включать, например, любой из следующих ингредиентов или соединений подобной природы: связующее, такое как микрокристаллическая целлюлоза, трагакант или желатин; эксципиент, такой как крахмал или лактоза, дезинтегратор, такой как альгиновая кислота,- 13012178Primogel, или кукурузный крахмал; лубрикант, такой как стеарат магния; глидант, такой как коллоидный диоксид кремния; подслащивающее средство, такое как сахароза или сахарин; или вкусовая добавка, такая как мята, метилсалицилат, или апельсиновая добавка. Инъецируемые композиции обычно основаны на стерильном солевом растворе для инъекций или забуференном фосфатом солевом растворе или других инъецируемых носителях, известных в уровне техники. Как указано выше, производные пиридинметиленазолидинона формулы (I) в таких композициях обычно составляют минорный компонент, часто составляющий от 0,05 до 10 вес.%, где остаток является инъецируемым носителем и т.п. Вышеописанные компоненты для перорально вводимых или инъецируемых композиций названы просто в качестве примера. Другие материалы, а также технологии производства и т.п. изложены в части 5 Remington's Pharmaceutical Sciences, 20th Edition, 2000, Marck Publishing Company, Easton, Pennsylvania,который включен в настоящее описание путем ссылки. Соединения согласно настоящему изобретению могут также вводиться в формах замедленного высвобождения или системах замедленного высвобождения для доставки лекарственных средств. Описание примеров материалов замедленного высвобождения может также быть найдено в Remington's Pharmaceutical Sciences. Синтез соединений по изобретению Новые производные пиридинметиленазолидинона согласно формуле (I) могут быть получены из легкодоступных исходных веществ несколькими синтетическими подходами с использованием протоколов как химии растворимой фазы, так и твердофазной (Brummond et al., 1999, J.O.С, 64, 1123-1126). Примеры путей синтеза будут описаны далее. Следующие сокращения относятся, соответственно, к определениям, приведенным ниже:- ангстрем,см - сантиметр,экв. - эквивалент,ч - час,г - грамм,М - молярный,МГц - мегагерц,мкл - микролитр,мин - минута,мг - миллиграмм,мл - миллилитр,мм - миллиметр,ммоль - миллимоль,мМ - миллимолярный,нм - нанометр,к.т. - комнатная температура,CN - ацетонитрил,АТФ - аденозид трифосфат,BSA - бычий сывороточный альбумин,DCM - дихлорметан,DIBAL - диизобутилалюминийгидрид,ДМФА - диметилформамид,ДМСО - диметилсульфоксид,ВЭЖХ - высокоэффективная жидкостная хроматография,Ins1P - D-миоинозитол-1-фосфат,ИК - инфракрасный,ЖХ - жидкостная хроматография,МС - масс-спектрометрия,ЯМР - ядерный магнитный резонанс,PBS - забуференный фосфатом солевой раствор,PI - фосфоинозитиды,PI3K - фосфоинозитид-3-киназы,PI(3)Р - фосфатидилинозитол-3-монофосфат,PI(3,4)Р 2 - фосфатидилинозитол-3,4-бифосфат,PI(3,4,5)P3 - фосфатидилинозитол-3,4,5-трифосфат,PI(4)Р - фосфатидилинозитол-4-фосфат,PI(4,5)P2 - фосфатидилинозитол-4,5-бифосфат,PtdIns - фосфатидилинозитол,PVT - поливинилтолуол,SPA - анализ близости сцинтилляции,- 14012178TEA - триэтиламин,ТФУК - трифторуксусная кислота,ТГФ - тетрагидрофуран,ТСХ - тонкослойная хроматография,TMS - триметилсилил,УФ - ультрафиолетовый. Производные пиридинметиленазолидинона, иллюстрируемые этим изобретением, могут быть получены из легкодоступных исходных веществ с использованием следующих общих способов и методик. Следует отметить, что, если приведены типичные или предпочтительные экспериментальные условия(т.е. температуры реакции, время, число молей реагентов, растворители и т.д.), другие экспериментальные условия могут также использоваться, если не указано иное. Оптимальные условия реакции могут варьировать в зависимости от конкретных используемых реагентов или растворителей, но такие условия могут быть определены специалистом с использованием обычных методик оптимизации. В способе, иллюстрируемом на следующих схемах, R1, R2, А, X, Y и n имеют значения, определенные выше в описании. Как правило, производные пиридинметиленазолидинона согласно общей формуле (I) могут быть получены несколькими синтетическими подходами с использованием протоколов как химии растворимой фазы, так и твердофазной (Brummond et al., 1999, выше) или обычными способами или методиками с дополнительным использованием микроволнового облучения. На первой стадии альдегидный реагент P1 (P1a, P1b, P1c, P1d) и 1-2 экв. реагента Р 2 (в частности,тиазолидиндиона или роданина) нагревают в присутствии предпочтительно слабого основания, чтобы получить соответствующий олефин формулы (I), как показано на схеме 1. На первой стадии P1 может быть заменен предшественниками P1a, P1b, P1c и P1d, чтобы получить конечные соединения (Ia), (Ib)(Ic) и (Id) соответственно, как описано выше в описании. Схема 1 Особенно предпочтительные способы согласно изобретению иллюстрируются в соответствии со следующими схемами 2, 3, 4 и 5, в которых соединения формулы (Ia), (Ib), (Ic) и (Id) соответственно могут быть получены с использованием тех же самых условий реакции, как указано выше. Схема 2 Хотя эта стадия может быть выполнена в отсутствие растворителя при температуре, достаточно высокой, чтобы вызвать, по меньшей мере частичное, плавление реакционной смеси, предпочтительно ее осуществляют в присутствии инертного растворителя. Предпочтительный диапазон температур составляет от приблизительно 70 до 250 С и особенно предпочтительный от приблизительно 80 до 120 С. Примеры таких растворителей для указанной реакции включают такие растворители, как диметоксиметан, ксилол, толуол, о-дихлорбензол и метанол. Примерами подходящих слабых оснований для указанной реакции являются соли щелочного и щелочно-земельного металла и слабых кислот, таких как(C1-C12)-алкилкарбоновые кислоты и бензойная кислота, карбонаты и бикарбонаты щелочного и щелочно-земельного металла, такие как карбонат кальция, карбонат магния, бикарбонат калия и вторичные амины, такие как пиперидин, морфолин или пирролидин, а также третичные амины, такие как пиридин,триэтиламин, диизопропилэтиламин, N-метилморфолин, N-этилпиперидин, N-метилпиперидин и т.п. Особенно предпочтительными слабыми основаниями являются ацетат натрия или пирролидин по причинам экономичности и эффективности. В такой типичной реакции (Tietze et al., in "The Knoevenagel reaction", p. 341 ff., Pergamon Press,Oxford 1991, Eds.: Trost B.M., Fleming I.) альдегид P1 и другое исходное вещество (например, тиазолидиндион) Р 2 комбинируют в приблизительно эквимолярных количествах с 0,5-1 экв. пирролидина в метанольном или подобном растворителе и нагревают до температуры от 70 до 200 С, при которой реакция в основном завершается приблизительно через 15 мин-3 ч. Желаемый олефин формулы (I) выделяют фильтрованием, в случае, если он осаждается из реакционной смеси после охлаждения, или, например,смешиванием с водой и последующим фильтрованием, чтобы получить сырой продукт. Сырой продукт очищают, если желательно, например, кристаллизацией или стандартными хроматографическими способами. Альтернативно, соединения формулы (I) можно получить обычным образом, смешивая эквимолярные количества тиазолидиндиона Р 2 с альдегидом P1 с молярным избытком безводного ацетата натрия и нагревая смесь при температуре, достаточно высокой, чтобы вызвать плавление, при которой реакция в основном завершается приблизительно через 5-60 мин. Предпочтительно указанную реакцию осуществляют в кислой среде, такой как уксусная кислота, в присутствии ацетата натрия или бета-аланина. Более предпочтительно указанную реакцию осуществляют в метаноле, используя от 1,1 до 2,0 экв. тиазолидиндиона Р 2, 1 экв. альдегида P1 и от 0,2 до 0,5 экв. пирролидина в метаноле. Реакции, описанные выше, могут быть выполнены альтернативно в условиях микроволнового облучения в качестве источника тепла. Как правило, исходное вещество альдегид P1 и тиазолидиндион Р 2 комбинируют в приблизительно эквимолярных количествах с 0,5-1 экв. пиперидина в диметоксиметане или подобном растворителе и нагревают до температуры от 140 до 240 С, при которой реакция в основном завершается приблизительно через 3-10 мин. Фармацевтически приемлемые катионные соли соединений согласно настоящему изобретению легко можно получить путем взаимодействия кислотных форм с подходящим основанием, обычно 1 экв., в сорастворителе. Типичными основаниями являются гидроксид натрия, метоксид натрия, этоксид натрия,гидрид натрия, гидроксид калия, метоксид калия, гидроксид магния, гидроксид кальция, бензатин, холин, диэтаноламин, этилендиамин, меглумин, бенетамин, диэтиламин, пиперазин и трометамин. Соль- 16012178 выделяют концентрацией досуха или добавлением вещества, не являющегося растворителем. В некоторых случаях соли можно получить, смешивая раствор кислоты с раствором катиона (этилгексаноата натрия, олеата магния), используя растворитель, в котором желаемая катионная соль осаждается, или она может быть выделена концентрацией и добавлением вещества, не являющегося растворителем. Производные 2,4-азолидинона Р 2 коммерчески доступны из различных источников. Способы получения промежуточных продуктов соединений формулы (I). Альдегиды формулы P1 получают различными известными способами, например окислениемвосстановлением, исходя из соответствующего алкилового эфира карбоновой кислоты или из карбоновой кислоты. В стандартных методиках восстановления алкилового эфира карбоновой кислоты, галогенидов карбоновой кислоты или карбоновой кислоты до бензиловых спиртов используют алюмогидрид лития, диизопропилалюминий, три-трет-бутоксигидрид лития-алюминия и т.д. В конечном счете, соответствующий бензиловый спирт повторно окисляют до соответствующего альдегида окислением в мягких условиях с использованием таких реагентов, как двуокись марганца,хромовая кислота, реагент Десса-Мартина, или окислением Сверна в других условиях, известных для получения альдегидов из первичных спиртов. Альтернативный путь может представлять собой прямое восстановление соответствующего алкилового эфира карбоновой кислоты или карбоновой кислоты до соответствующего альдегида, используя DIBAL при низкой температуре или любые другие методики,известные в данной области. Альтернативным способом получения подходящего альдегида P1 является селективное восстановление нитрильной группы до соответствующего альдегида с использованием известных способов, таких как, например, DIBAL. Другим способом получения альдегидов формулы P1 является селективное восстановление соответствующего ацилхлорида с использованием, например, три-трет-бутоксигидрида лития-алюминия (Chaet al., 1993, J.O.С, 58, p. 4732-34). Другой способ синтеза альдегидов P1 - исходя из соответствующих 2-пиридингалогенидов, которые подвергают реакции с металлорганическим ассистированием, чтобы получить соответствующие 2-винилпиридины, которые в конечном счете могут быть окислены до соответствующих альдегидов P1 с использованием стандартных окислителей для олефиновых связей, таких как тетроксид осмия, тетроксид рутения, озон, хлорид рутения (III), в присутствии перйодата натрия и других известных специалисту. Другой способ получения соответствующих альдегидов P1 представляет собой окисление 2-метилпиридина с использованием таких окислителей, как диоксид селена или бензолселениновый ангидрид. В соответствии с более предпочтительным способом по изобретению, как иллюстрируется ниже в соответствии со схемой 6, реагент P1 а можно получить, исходя из производного формулы Р 3a, где R выбран из метила, этила или любой другой известной специалисту группы, которая может быть восстановлена, необязательно используя последовательность восстановления/окисления с использованием предпочтительно алюмогидрида лития в тетрагидрофуране, с последующей стадией окисления с использованием предпочтительно диоксида марганца в дихлорметане. Схема 6 Промежуточным продуктом, который может использоваться для указанного синтеза, является 2,4,8-трихлорпиридо[3,2-d]пиримидин-6-метилкарбоксилат (промежуточный продукт 1.3), этот синтез описан в литературе (Srinivasan et al., 1919, J.O.С, 1979, 44, 3, p. 435), как показано на схеме 7. Схема 7- 17012178 Селективная замена 3 групп хлора может позволить введение групп R1 и R2, приводя к различным промежуточным продуктам формулы P3a (Р 3 а(1), Р 3 а(2), P3a(3), Р 3 а(4), Р 3 а(5), P3a(6), Р 3 а(7, как показано на схеме 8. Схема 8 Стадии восстановления на схеме 8 могут быть выполнены с использованием стандартных восстановителей, таких как водород или никель Ренея (Srinivasan et al., 1979, выше). Предпочтительно восстановление проводят в умеренных условиях, используя формиат аммония в присутствии палладия. Количество формиата аммония определяется числом атомов хлора, подлежащих удалению(2-12 экв.). Введение групп R2 и R1 осуществляют стандартными методиками реакции, известными специалисту. В соответствии с другим особенно предпочтительным способом согласно изобретению, как иллюстрируется схемой 9, альдегид P1b может быть получен, исходя из промежуточного продукта Р 3b, окислительным расщеплением олефиновой двойной связи. Схема 9 где R выбран из Н, необязательно замещенного C1-C6-алкила, необязательно замещенного арила. В такой реакции олефиновую двойную связь расщепляют, используя окислители для олефиновых связей, такие как тетроксид осмия, тетроксид рутения, озон, хлорид рутения (III), в присутствии перйодата натрия и других окислителей, известных специалисту. Промежуточный продукт P3b можно синтезировать, исходя из производных 2-галогенпиридина, с использованием реакций сочетания с металлорганическим ассистированием для введения виниловой группы стандартным способом, известным специалисту. Соответствующие 2-галогенпиридины легко доступны исходя, например, из 2-галоген-4-нитро-6-аминопиридина, как изображено на схеме 10, где В соответствии с другим особенно предпочтительным способом согласно изобретению, как иллюстрируется схемой 11, где R выбран из Н, необязательно замещенного C1-C6-алкила, необязательно замещенного арила, промежуточный продукт P1 с может быть получен, исходя из промежуточного продукта P3 с, окислением 2-метилпиридинов. Схема 11 Такое окисление можно осуществить, используя диоксид селена или бензолселениновый ангидрид в инертном растворителе при температурах от 150 до 250 С. Предпочтительно такую реакцию проводят,используя микроволновое облучение в качестве источника тепла. На второй стадии десилилирование выполняют в нормальных условиях, как описано в Kocienski, 1994 (выше) и Greene et al., 1999 (выше). Предпочтительно триметилсилильную группу расщепляют, используя 2-5 н. гидроксид натрия. Введение R2 может быть выполнено, как описано в WO2004/007491. Согласно другому более предпочтительному способу промежуточный продукт P1 с может быть получен из промежуточного продукта P3 с через перегруппировку пиколин-N-оксида: Обычно промежуточный продукт P3 с подвергают N-окислению, приводящему к промежуточному продукту P3', с использованием таких окислителей, как м-хлор-пербензойная кислота (m-СРВА) при комнатной температуре,или любого известного специалисту окислителя. Последующая обработка основанием и нагревание P3 с' в уксусном ангидриде при 100 С в течение 5-15 мин (Cava et al., 1958, JOC, 23, 1616) приводит к соответствующему защищенному ацетилом спирту, который, в свою очередь, может быть подвергнут снятию защиты и десилилированию одновременной обработкой гидроксидом натрия (2 н.) в метаноле при комнатной температуре. Наконец, первичный спирт P3 с" может быть окислен до соответствующего промежуточного альдегида P1 с с использованием таких окислителей, как диоксид марганца в дихлорметане,или любых окислителей, известных специалисту (схема 11 выше). В соответствии с другим особенно предпочтительным способом согласно изобретению, где представлены азабензимидазолы, промежуточный продукт P4d может быть получен из промежуточного продукта P5d, как изображено на схеме 12, где Hal обозначает галоген. Замещение 2-галогена группой R2NH2 в спиртах (например, этаноле) в присутствии основания сопровождается восстановлением нитрогруппы, катализируемым металлическим индием в присутствии источника водорода. P4d получают последующей циклизацией посредством конденсации с амидинами с последующим присоединением виниловой группы, используя реакции сочетания с металлорганическим ассистированием стандартным способом, известным специалисту. Когда R2 в промежуточном продукте P4d представляет собой химическую группу, которая должна быть подвергнута синтетическим превращениям, эти превращения осуществляют после завершения сочетания с винильной группой. Эти синтетические превращения включают, но не ограничены ими, снятия защиты, сочетания, окисления, восстановления. Схема 13 В соответствии с особенно предпочтительным способом по изобретению образованную винильную олефиновую связь промежуточного продукта P3d (схема 13 выше) расщепляют, используя окислители для олефиновых связей, такие как тетроксид осмия или хлорид рутения (III), в присутствии перйодата натрия, озона и других окислителей, известных специалисту. Согласно другому общему способу соединения формулы (I) могут быть преобразованы в альтернативные соединения формулы (I) с использованием подходящих методик взаимного превращения, известных специалисту. Если указанный набор общих синтетических способов не применим для получения соединений согласно формуле (I) и/или необходимых промежуточных продуктов для синтеза соединений формулы (I),должны использоваться подходящие способы получения, известные специалисту. Как правило, пути синтеза для любого индивидуального соединения формулы (I) будут зависеть от конкретных заместителей каждой молекулы и от доступности необходимых промежуточных продуктов; опять же, такие факторы определяются специалистом. В отношении всех способов установления и снятия защиты, см.Kocienski, 1994 (выше) и Green et al., 1999 (выше). Соединения согласно настоящему изобретению могут быть выделены в комбинации с молекулами растворителя кристаллизацией путем выпаривания соответствующего растворителя. Фармацевтически приемлемые кислотно-аддитивные соли соединений формулы (I), которые содержат основной центр, могут быть получены обычным образом. Например, раствор свободного основания может быть обработан подходящей кислотой, в чистом виде или в виде подходящего раствора, и полученную соль выделяют либо фильтрованием, либо выпариванием в вакууме растворителя реакции. Фармацевтически приемлемые основно-аддитивные соли могут быть получены аналогичным образом путем обработки раствора соединения формулы (I) подходящим основанием. Оба типа солей можно получить или преобразовать друг в друга методами с использованием ионообменной смолы. Далее настоящее изобретение иллюстрируется посредством некоторых примеров, которые не должны рассматриваться как ограничение объема изобретения. Примеры Использовали следующие коммерчески доступные исходные соединения: 5-аминоурацил, коммерчески доступный от Aldrich; диметилацетилендикарбоксилат, коммерчески доступный от Aldrich;N,N-диэтиланилин, коммерчески доступный от Aldrich; хлорангидрид фосфорной кислоты, коммерчески доступный от Aldrich;N-этилдиизопропиламин, коммерчески доступный от Aldrich; формиат аммония, коммерчески доступный от Aldrich; алюмогидрид лития, коммерчески доступный от Aldrich; оксид марганца, коммерчески доступный от Aldrich; 2,4-тиазолидиндион, коммерчески доступный от Aldrich; роданин, коммерчески доступный от Aldrich; бета-аланин, коммерчески доступный от Aldrich;- 20012178 4-фторпиперидин, коммерчески доступный от Fluorochem; 4-трифторметилпиперидин, коммерчески доступный от Lancaster; глиоксаль (оксальдегид), коммерчески доступный от Aldrich; тетракис-(трифенилфосфин)палладий, коммерчески доступный от Aldrich; винилтрибутилстаннан, коммерчески доступный от Aldrich; 6-йод-2-пиколин-5-ол, коммерчески доступный от Acros;(триметилсилил)ацетилен, коммерчески доступный от Aldrich; дихлор-бис-(трифенилфосфин)палладий(II), коммерчески доступный от Aldrich; 1,2-дихлорбензол, коммерчески доступный от Aldrich; 2-амино-3-нитро-6-хлорпиридин, коммерчески доступный от ACROS; порошок индия, коммерчески доступный от Aldrich; формамидинацетат, коммерчески доступный от Aldrich; трибутил(винил)олово, коммерчески доступное от Aldrich; тетроксид осмия, коммерчески доступный от Aldrich; перйодат натрия, коммерчески доступный от Aldrich; 2,6-дихлор-3-нитропиридин, коммерчески доступный от Aldrich; 3,5-диметоксианилин, коммерчески доступный от Aldrich; 5-нитроиндолин, коммерчески доступный от Aldrich; 6-нитроиндолин, коммерчески доступный от Aldrich; гидрохлорид 4-(диметиламино)масляной кислоты, коммерчески доступный от Aldrich; 3-хлорпропансульфонилхлорид, коммерчески доступный от Aldrich; хлорметансульфонилхлорид, коммерчески доступный от Alfa Aesar. Данные ВЭЖХ, ЯМР и МС, приведенные в примерах, описанных ниже, получены следующим образом: ВЭЖХ: колонка Waters Symmetry С 8 504,6 мм; условия: MeCN/H2O, 5-100% (8 мин), детекция при 230-400 нм; масс-спектры: PE-SCIEX API 150 EX (APCI и ESI); спектры ЖХ/МС: Waters ZMD (ES); 1H-ЯМР: Bruker DPX-300 МГц. Очистки препаративной ВЭЖХ осуществляли с использованием HPLC Waters Prep LC 4000 System,оборудованной колонками Prep Nova-PakHR C186 мкм 60, 4030 мм (до 100 мг) или с использованием XTerra Prep MS C8, 10 мкм, 50300 мм (до 1 г). Все очистки выполняли с градиентом MeCN/H2O 0,09% ТФУК. Полупрепаративную обращенно-фазовую ВЭЖХ выполняли с Biotage Parallex Flex System,оборудованной колонками Supelcosil ABZ+Plus (25 см 21,2 мм, 12 мкм); УФ-детекция при 254 нм и 220 нм; поток 20 мл/мин (до 50 мг). Анализ ТСХ выполняли на планшетах Merck Precoated 60 F254. Очистки флэш-хроматографией выполняли на подложке SiO2, используя смеси циклогексан/EtOAc или(2 Е)-2-[(2,4-диоксо-1,2,3,4-тетрагидро-5 пиримидинил)амино]-2-бутендиоат (схема 7). К суспензии 5-аминоурацила (В) (4,0 г; 31,5 ммоль; 1 экв.) в МеОН (120,00 мл) добавляли диметилацетилендикарбоксилат (С) (5,0 г; 35,2 ммоль; 1,1 экв.). Суспензию перемешивали при комнатной температуре в течение 46 ч. Реакцию отслеживали с помощью ЯМР. Твердое вещество отфильтровывали, получая диметил (2 Е)-2-[(2,4-диоксо-1,2,3,4-тетрагидро-5-пиримидинил)амино]-2-бутендиоат (8,0 г, 95%)(2 Е)-2-[(2,4-диоксо-1,2,3,4-тетрагидро-5-пиримидинил)амино]-2-бутендиоат (промежуточный продукт 1.1) (38,5 г; 0,14 моль; 1 экв.) даутерм(R) (1 л) (эвтектика фениловый эфир-дифенила). Суспензию перемешивали приводной мешалкой в атмосфере аргона и нагревали при 220 С. Реакцию отслеживали с использованием ВЭЖХ/ЖХ/МС. Через 3 ч реакцию останавливали охлаждением с последующим добавлением 300 мл петролейного эфира. Полученный осадок отфильтровывали и промывали ДМФА(2100 мл). Метил 2,4,8-триоксо-1,2,3,4,5,8-гексагидропиридо[3,2-d]пиримидин-6-карбоксилат (21,02 г; 62%) (промежуточный продукт 1.2) выделяли в виде порошка желтого цвета со 100% чистотой по ВЭЖХ. Количество: 21,0 г; выход: 62%; формула: C9H7O5N3; Н-ЯМР (ДМСО-d6):3,87 (с, 3H), 7,58 (с, 1 Н), 10,90 (с, 1 Н), 11,56 (с, 1 Н), 12,10 (уш., 1 Н). Промежуточный продукт 1.3. Метил 2,4,8-трихлорпиридо[3,2-d]пиримидин-6-карбоксилат (схема 7). Раствор метил 2,4,8-триоксо-1,2,3,4,5,8-гексагидропиридо[3,2-d]пиримидин-6-карбоксилата (промежуточный продукт 1.2) (9 г; 37,95 ммоль; 1 экв.) и N,N-диэтиланилина (10 мл) в хлорангидриде фосфорной кислоты (174 мл) нагревали с обратным холодильником в течение ночи. Раствор концентрировали в вакууме. Черное масло медленно выливали на лед. Добавляли этилацетат и органическую фазу промывали водой до рН 6. Органические слои высушивали над сульфатом магния, фильтровали и концентрировали. Метил 2,4,8-трихлорпиридо[3,2-d]пиримидин-6-карбоксилат (промежуточный продукт 1.3)(6,5 г, 59%) осаждали в циклогексане в виде твердого вещества розового цвета с 98% чистотой по ВЭЖХ. Количество: 6,5 г; выход: 59%; формула: C9H4O2Cl3N3; ВЭЖХ (H2O ТФУК 0,1%-ACN ТФУК 0,05%): Rt (мин); область %=3,07; 98; 1 Н-ЯМР (CDCl3)4,12 (с, 3H), 8,70 (с, 1 Н); ЖХ-МС: M/Z ESI: Rt (мин) 1,58; 293 (М+1). Промежуточный продукт 1.4. Метил 2,8-дихлор-4-(1-пиперидинил)пиридо[3,2-d]пиримидин-6 карбоксилат (схема 8). К раствору метил 2,4,8-трихлорпиридо[3,2-d]пиримидин-6-карбоксилата (4,65 г; 15,9 ммоль; 1 экв.)(промежуточный продукт 1.3) в ацетонитриле (14 0 мл) добавляли N-этилдиизопропиламин (4 мл; 23,8 ммоль; 1,5 экв.). Смесь охлаждали до 0 С. Добавляли по каплям раствор пиперидина (1,57 мл; 15,9 ммоль; 1 экв.) в ацетонитриле (20 мл). Смесь перемешивали 15 мин при 0 С. Смесь частично концентрировали и осадок отфильтровывали, промывали МеОН и высушивали в вакууме, получая метил 2,8-дихлор-4-(1-пиперидинил)пиридо[3,2-d]пиримидин-6-карбоксилат (промежуточный продукт 1.4) 3,98 г; 73%) в виде твердого вещества розового цвета с чистотой по ВЭЖХ 98,8%. Количество: 3,98 г; выход: 73%; формула: C14H14O2Cl2N4; ВЭЖХ (H2O ТФУК 0,1%-ACN ТФУК 0,05%): Rt (мин); область %=4,27; 98,84; 1 Н-ЯМР (ДМСО-d6):1,71 (шир.с, 6 Н), 3,92 (с, 3H), 4,01 (шир.с, 2 Н), 4,82 (шир.с, 2 Н), 8,42 (с, 1 Н); ЖХ-МС: M/Z ESI: Rt (мин) 2,02; 341,02, 342,89 (М+1). Промежуточный продукт 1.5. Метил 4-(1-пиперидинил)пиридо[3,2-d]пиримидин-6-карбоксилат(схема 8). В круглодонную колбу добавляли палладий (540 мг; 0,51 ммоль; 0,05 экв.) в изопропаноле (90 мл). В эту смесь барботировали Ar. Добавляли дегазированный раствор формиата аммония в воде (2,56 г,40,6 ммоль, 4 экв., в 4 мл воды), затем метил 2,8-дихлор-4-(1-пиперидинил)пиридо[3,2-d]пиримидин-6 карбоксилат (промежуточный продукт 1.4) (3,46 г; 10,5 ммоль; 1 экв.) и дегазированный изопропанол(10 мл). Через 30 мин добавляли вторую загрузку формиата аммония в виде раствора в воде (2,56 г,40,6 ммоль, 4 экв., в 4 мл воды). Наконец, через дополнительные 30 мин добавляли еще 8 экв. формиата аммония в воде (5,12 г, 81,2 ммоль, 8 экв., в 8 мл воды). Смесь перемешивали при комнатной температуре в течение ночи и фильтровали через целит. Фильтрат упаривали. Сырой продукт растворяли в DCM и промывали водой и рассолом. Органическую фазу высушивали над сульфатом магния, фильтровали и упаривали, получая метил 4-(1-пиперидинил)пиридо[3,2-d]пиримидин-6-карбоксилат (2,29 г; 83%) (промежуточный продукт 1.5) в виде твердого вещества желтого цвета с чистотой по ВЭЖХ 92,9%. Этот продукт использовали на следующей стадии без дальнейшей очистки. Количество: 2,29 г; выход: 82%; формула: C14H16O2N4; ВЭЖХ (H2O ТФУК 0,1%-ACN ТФУК 0,05%): Rt (мин); область %=1,83; 92,88; 1(4,4 г; 16,2 ммоль; 1 экв.) растворяли в ТГФ (176 мл) и раствор охлаждали до -35 С (внутренняя температура). Алюмогидрид лития (8,1 мл; 1,00 М.; 8,1 ммоль; 0,50 экв.) добавляли по каплям. Через 2 ч 30 мин при -35 С реакция завершалась. Добавляли воду (8,1 мл) и температуре давали подняться до комнатной. После добавления МеОН (8 мл) смесь фильтровали через целит и обильно промывали смесью DCM/MeOH 1:1. Растворители удаляли при пониженном давлении, получая[4-(1-пиперидинил)пиридо[3,2-d]пиримидин-6-ил] метанол (промежуточный продукт 1.6) (3.,9 г; количественный выход) с чистотой по ВЭЖХ 92,9%. Этот продукт использовали на следующей стадии без дальнейшей очистки. Количество: 3,99 г; выход: 100%; формула: C13H16ON4; ВЭЖХ (H2O ТФУК 0,1%-ACN ТФУК 0,05%): Rt (мин); область %=1,39; 92,88; 1[4-(1-Пиперидинил)пиридо[3,2-d]пиримидин-6-ил]метанол (промежуточный продукт 1.6) 3,95 г; 16,2 ммоль; 1,00 экв.) растворяли в DCM (160 мл). Раствор охлаждали до 0 С и добавляли оксид марганца (16,5 г; 0,162 моль; 10 экв.). Реакционную смесь перемешивали 5 мин при 0 С в течение ночи при комнатной температуре. Чтобы закончить преобразование, MnO2 добавляли через 12 и 20 ч (две загрузки 4,96 г; 48,48 ммоль; 3 экв.). Через 20 ч реакция завершалась. Добавляли МеОН (100 мл) и смесь фильтровали через целит и обильно промывали смесью DCM/MeOH 1:1. Растворители удаляли при пониженном давлении, получая 4-(1-пиперидинил)пиридо[3,2-d]пиримидин-6-карбальдегид (промежуточный продукт 1.7). Этот продукт использовали на следующей стадии без дальнейшей очистки. Количество: 4,1 г; формула: C13H14ON4; чистота по ВЭЖХ: 58,84%; ВЭЖХ (H2O ТФУК 0,1%-ACN ТФУК 0,05%): Rt (мин); область %=1,39; 58,84; 1(схемы 6 и 8). Указанное в заголовке соединение получали, используя 4-фторпиперидин, согласно общей методике, описанной для синтеза промежуточного продукта 1.7 (схемы 5 и 7). Количество: 4,15 г; формула: C13H13FON4; чистота по ВЭЖХ: 89,16%; ВЭЖХ (H2O ТФУК 0,1%-ACN ТФУК 0,05%): Rt (мин); область %=1,19; 89,16; 1H-ЯМР (ДМСО-d6):10,01 (с, 1 Н), 8,60 (с, 1 Н), 8,23 (м, 2 Н), 5,00 (м, 1 Н), 4,51 (уш., 4 Н), 1,91 (уш.,4 Н); ЖХ-МС: M/Z ESI: Rt (10 мин) 2,26; 261,08 (М+1). Промежуточный продукт 3.1. 4-(4-(Трифторметил)пиперидин-1-ил)пиридо[3,2-d]пиримидин-6 карбальдегид (схемы 6 и 8). Указанное в заголовке соединение получали, используя 4-трифторметилпиперидин, согласно общей методике, описанной для синтеза промежуточного продукта 1.7 (схемы 5 и 7). Количество: 4,8 г; формула: C14H13OF3N4; чистота по ВЭЖХ: 67,12%; ВЭЖХ (H2O ТФУК 0,1%-ACN ТФУК 0,05%): Rt (мин); область %=1,89; 67,12; 1H-ЯМР (ДМСО-d6):10,03 (с, 1 Н), 8,62 (с, 1 Н), 8,18 (с, 2 Н), 3,22 (т, 2 Н), 2,48 (м, 2 Н), 2,08 (д, 2 Н),1,80 (м, 3H); Промежуточный продукт 4.1. 6-Хлорпиридин-2,3-диамин (схема 10); ЖХ-МС: M/Z ESI: Rt (3 мин) 1,75; 311,04 (М+1). 2-Амино-3-нитро-6-хлорпиридин (3 г, 17,3 ммоль, 1 экв.) растворяли в ТГФ (50 мл) при комнатной температуре. Медленно добавляли дигидрат хлорида олова (15,6 г, 70 ммоль, 4 экв.), предварительно растворенный HClконц (5 мл), и реакционную смесь перемешивали при комнатной температуре в течение 4 ч. По завершении реакции реакционную смесь охлаждали до 0 С и обрабатывали гидроксидом натрия 5 М (12 мл) до рН 14 и соответствующее соединение экстрагировали этилацетатом. Органические фазы высушивали над сульфатом магния, выпаривали в вакууме и полученный сырой продукт очищали флэш-хроматографией, используя циклогексан/этилацетат (1/1), с получением 1,5 г масла красного цвета (промежуточный продукт 4.1). Количество: 1,5 г; выход: 60%; формула: C5H6N3Cl; чистота по ВЭЖХ: 98%; ВЭЖХ (H2O ТФУК 0,1%-ACN ТФУК 0,05%): Rt (мин); область %=0,5 мин; 98%; 1H-ЯМР (ДМСО-d6):6,67 (д, 1 Н, 5 Н, J=8 Гц), 6,36 (д, 1 Н, 4 Н, J=8 Гц), 5,78 (м, 2 Н, NH2), 4,75 (м,2 Н, NH2); ЖХ-МС: M/Z ESI: Rt (мин) 0,1 мин, 144,0 (М+1). Промежуточный продукт 4.2. 6-Хлорпиридо[2,3-b]пиразин (схема 10). 6-Хлор-2,3-пиридиндиамин (промежуточный продукт 4.1) (1 г, 6,96 ммоль, 1 экв.) растворяли в ТГФ (15 мл). Добавляли глиоксаль (0,84 мл, 18,1 ммоль, 2,5 экв.) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакцию отслеживали с использованием обращенно-фазовой ВЭЖХ. ТГФ выпаривали, остаток повторно растворяли в этилацетате (30 мл). Органические фазы промывали дважды насыщенным Na2 СO3, высушивали над сульфатом магния и упаривали в вакууме, получая 1,15 г ожидаемого соединения в виде твердого вещества белого цвета (промежуточный продукт 4.2). Количество: 1,15 г; выход: 100 %; формула: C7H4N3Cl; чистота по ВЭЖХ: 98%; ВЭЖХ (H2O ТФУК 0,1%-ACN ТФУК 0,05%): Rt (мин); область %=1,2 мин; 98%; 1H-ЯМР (CDCl3)9,0 (с, 1 Н), 8,88 (с, 1 Н), 8,36 (д, 1 Н, J=8 Гц), 7,67 (д, 1 Н, J=8 Гц); ЖХ-МС: M/Z ESI: Rt (мин) 0,68 мин, 167,0 (М+1). Промежуточный продукт 4.3. 6-Винилпиридо[2,3-b]пиразин (схема 10). 6-Хлорпиридо[2,3-b]пиразин (промежуточный продукт 4.2) (3 г, 18,12 ммоль, 1,00 зкв.) растворяли в ТГФ (150 мл) и дегазировали азотом при комнатной температуре в течение 10 мин. Добавляли тетракис-(трифенилфосфин)палладий(0) (1,46 г, 1,27 ммоль, 0,07 экв.) и винилтрибутилстаннан (7,47 мл,23,5 ммоль, 1,3 экв.) и реакционную смесь перемешивали при 65 С в течение 3 ч. ТГФ выпаривали и сы- 23012178 рой продукт очищали непосредственно флэш-хроматографией, используя циклогексан/этилацетат (8/2), с получением 2,3 г ожидаемого соединения (промежуточный продукт 4.3) в виде масла оранжевого цвета. Количество: 2,3 г; выход: 81%; формула: C9H7N3; чистота по ВЭЖХ: 98%; ВЭЖХ (H2O ТФУК 0,1%-ACN ТФУК 0,05%): Rt (мин); область %=1,32 мин; 98%; 1H-ЯМР (ДМСО-d6):9,10 (с, 1 Н), 8,98 (с, 1 Н), 8,46 (д, 1 Н, J=8 Гц), 7,95 (д, 1 Н, J=8 Гц), 6,90 (дд,1 Н, Jтранс=17 Гц, Jцис=10 Гц), 6,50 (дд, 1 Н, Jтранс=17 Гц, Jгем=1,5 Гц), 5,80 (дд, 1 Н, Jцис=10 Гц, Jгем=1,5 Гц); ЖХ-МС: M/Z ESI: Rt (мин) 0,78 мин, 158,13 (М+1). Промежуточный продукт 4.4. Пиридо[2,3-b]пиразин-6-карбальдегид (схема 9). 6-Винилпиридо[2,3-b]пиразин (промежуточный продукт 4.3) (1 г, 6,37 ммоль, 1 экв.) растворяли в метаноле (20 мл) и охлаждали до -70 С. Спокойный поток смеси кислорода/озона барботировали через раствор в течение 20 мин. Реакцию отслеживали с помощью ТСХ, используя смесь циклогексан/этилацетат (8/2). По завершении реакции добавляли диметилсульфид (0,1 мл) и реакционную смесь оставляли при комнатной температуре на 30 мин. Метанол выпаривали в вакууме и рекуперировали 600 мг пиридо[2,3-b]пиразин-6-карбальдегида. Сырой продукт анализировали без дальнейшей очистки(20,00 мл) добавляли (триметилсилил)ацетилен (1 г; 10,19 ммоль; 2,80 экв.), йодид меди (90,07 мг; 0,47 ммоль; 0,13 экв.) и дихлор-бис-(трифенилфосфин)палладий(II) (229,82 мг; 0,33 ммоль; 0,09 экв.). Раствор нагревали с обратным холодильником. Через 3 ч реакция завершалась, и реакционной смеси давали остыть до комнатной температуры. Раствор фильтровали через целит (промытый AcOEt и МеОН). Растворители удаляли. Добавляли AcOEt и воду и объединенные органические слои высушивали над сульфатом магния, фильтровали и концентрировали, получая ожидаемое соединение. Сырой продукт очищали короткой флэш-хроматографией, используя циклогексан, затем AcOEt/циклогексан 20/80 с получением 603 мг желательного соединения в виде твердого вещества (промежуточный продукт 5.1). Количество: 603 мг; выход: 81%; формула: C11H15NOSi; чистота по ВЭЖХ: 93,14%; ВЭЖХ (H2O ТФУК 0,1%-ACN ТФУК 0,05%): Rt (мин); область %=2,17 мин; 93,14%; 1H-ЯМР (CDCl3)7,65 (д, 1 Н, J=8,5 Гц), 7,10 (с, 1 Н), 7,06 (д, 1 Н, J=8,5 Гц), 2,67 (с, 3H), 0,36 (с, 9 Н); ЖХ-МС: M/Z ESI: Rt (мин) 1,89 мин, 206,06 (М+1). Промежуточный продукт 5.2. 2-Триметилсиланилфуро[3,2-b]пиридин-5-карбальдегид (схема 11). К раствору 5-метил-2-(триметилсилил)фуро[3,2-b]пиридина (промежуточный продукт 5.1) (600 мг; 2,92 ммоль; 1 экв.) в 1,2-дихлорбензоле (12 мл) добавляли диоксид селена (486 мг; 4,38 ммоль; 1,5 экв.). Реакционную смесь нагревали микроволновым облучением до 220 С в течение 6 ч. Раствор концентрировали в вакууме. Добавляли Et2O и отфильтровывали твердое вещество черного цвета. Фильтрат концентрировали и очищали флэш-хроматографией, используя циклогексан, затем циклогексан/AcOEt 90/10, с получением твердого вещества (промежуточный продукт 5.2). Количество: 130 мг; выход: 20%; формула: C11H13NO2Si; чистота по ВЭЖХ: 81,8%; ВЭЖХ (H2O ТФУК 0,1%-ACN ТФУК 0,05%): Rt (мин); область %=3,84 мин; 81,83%; 1H-ЯМР (CDCl3)10,19 (с, 1 Н), 7,99 (д, 1 Н, J=8,50 Гц), 7,89 (д, 1 Н, J=8,50 Гц), 7,27 (с, 1 Н), 0,40 (с,9 Н); ЖХ-МС: M/Z ESI: Rt (мин) 1,86 мин, 220 (М+1). Общие методики для синтеза промежуточных соединений 6-16.3. Общая методика I для замещения промежуточного продукта P5d группой R2NH2 (схема 12). Раствор 2,6-дибром-3-нитропиридина (промежуточный продукт 6 формулы P5d, в котором Hal обозначает Br и R1 обозначает Н) (1 экв.), ариламина (1,0-1,2 экв.) и триэтиламина (2 экв.) в этаноле(5 мл/ммоль) перемешивали в течение 48 ч при температуре окружающей среды. Фильтрованием полученного осадка получали соответствующий продукт замещения высокой чистоты. Общая методика II для восстановления (схема 12). Смесь бромпиридина (1 экв.), порошка индия (3-6 экв.), насыщенного водного хлорида аммония(8 мл/ммоль) и этанола (20 мл/ммоль) перемешивали при нагревании с обратным холодильником в течение 4 ч. Фильтрование через Celite и концентрация фильтрата в вакууме сопровождались основной экстракцией. Органический слой высушивали над сульфатом натрия и концентрировали в вакууме. Полученный соответствующий диаминопиридин использовали на следующей стадии без дальнейшей очистки.- 24012178 Общая методика для циклизации (схема 12). Смесь диаминопиридина (1 экв.), формамидинацетата (3-5 экв.) и 2-метоксиэтнола (30 мл/ммоль) перемешивали при нагревании с обратным холодильником в течение 15 ч. Смесь концентрировали в вакууме и очищали хроматографией (градиент EtOAc/гексан) с получением соответствующего бромимидазо[4,5-b]пиридина. Общая методика IV для реакции сочетания (схема 12). Раствор бромимидазо[4,5-b]пиридина (1 экв.), трибутил(винил)олова (1,5-3 экв.) и тетракис(трифенилфосфин)палладия(0) (0,1 экв.) в толуоле (дезоксигенированном с помощью N2, 20 мл/ммоль) перемешивали при кипении с обратным холодильником в течение 4 ч. Концентрация в вакууме и хроматографическая очистка (градиент EtOAc/гексан) дают соответствующий винилимидазо[4,5-b]пиридин. Общая методика V для окисления промежуточного продукта 4 (схема 13). Смесь винилимидазо[4,5-b]пиридина (1 экв.), тетроксида осмия (0,1 экв.), перйодата натрия(3-4 экв.), 1,4-диоксана (30 мл/ммоль) и воды (25 мл/ммоль) перемешивали в течение 15-30 мин при температуре окружающей среды. Полученную суспензию разбавляли равными количествами воды и этилацетата. После фильтрования через Celite органическую фазу высушивали над сульфатом натрия, концентрировали в вакууме и очищали посредством флэш-хроматографии с получением соответствующего формилимидазо[4,5-b]пиридина Промежуточный продукт 6. 2,6-Дибром-3-нитропиридин (схема 12). Смесь коммерчески доступного 2,6-дихлор-3-нитропиридина (10,0 г; 51,8 ммоль) и 33 вес.%HBr/АсОН (120 мл) нагревали при 80 С в течение 3 ч. Раствор концентрировали в вакууме, полученный остаток помещали в EtOAc и обрабатывали насыщенным водным бикарбонатом натрия. Органическую фазу высушивали над сульфатом натрия и концентрировали в вакууме. Полученный продукт 14,4 г (99%) использовали без дальнейшей очистки (промежуточный продукт 6). Промежуточный продукт 7.1. 6-Бром-3-нитро-2-фениламинопиридин (схема 12). Указанное в заголовке соединение получали из 2,6-дибром-3-нитропиридина (промежуточный продукт 6) и анилина с выходом 95% в соответствии с общей методикой I (промежуточный продукт 7.1). Промежуточный продукт 7.2. 6-Бром-N2-фенилпиридин-2,3-диамин (схема 12). Указанное в заголовке соединение получали из N-(5-бром-2-нитрофенил)-N-фениламина (промежуточный продукт 7.1) с выходом 97% в соответствии с общей методикой II. Промежуточный продукт 7.3. 5-Бром-3-фенил-3H-имидазо[4,5-b]пиридин (схема 12). Указанное в заголовке соединение получали из 6-бром-N2-фенилпиридин-2,3-диамина (промежуточный продукт 7.2) с выходом 71% в соответствии с общей методикой III. Промежуточный продукт 7.4. 3-Фенил-5-винил-3H-имидазо[4,5-b]пиридин (схема 12). Указанное в заголовке соединение получали из 5-бром-3-фенил-3H-имидазо[4,5-b]пиридина (промежуточный продукт 7.3) с выходом 92% в соответствии с общей методикой IV. Промежуточный продукт 7.5. 3-Фенил-3H-имидазо[4,5-b]пиридин-5-карбальдегид (схема 13). Указанное в заголовке соединение получали из 3-фенил-5-винил-3H-имидазо[4,5-b]пиридина (промежуточный продукт 7.4) с выходом 36% в соответствии с общей методикой V. Промежуточный продукт 8.1. 6-Бром-3-нитро-2-(3,5-диметоксифенил)аминопиридин (схема 12). Указанное в заголовке соединение получали из коммерчески доступных 2,6-дибром-3 нитропиридина и 3,5-диметоксианилина с выходом 85% в соответствии с общей методикой I. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 98% чистота, tR 10,12 мин (tR(SM):нитропиридин) 7,98 мин).H-ЯМР (400 МГц, ДМСО-d6):10,04 (с, 1 Н), 8,42 (д, 1 Н), 7,19 (д, 1 Н), 6,94 (с, 2 Н), 6,33 (с, 1 Н),3,76 (с, 6 Н) м.д. Промежуточный продукт 8.2. 6-Бром-N2-(3,5-диметоксифенил)пиридин-2,3-диамин (схема 12). Указанное в заголовке соединение получали изN-(5-бром-2-нитрофенил)-N-(3,5 диметоксифенил)амина (промежуточный продукт 8.1) с выходом 93%, используя общую методику II. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 96% чистота, tR 8,38 мин (tR(SM) 10,12 мин); Указанное в заголовке соединение получали из 6-бром-N2-(3,5-диметоксифенил)пиридин-2,3 диамина (промежуточный продукт 8.2) с выходом 43% в соответствии с общей методикой III. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 98% чистота, tR 8,51 мин (tR(SM) 8,40 мин); Промежуточный продукт 8.4. 3-(3,5-Диметоксифенил)-5-винил-3H-имидазо[4,5-b]пиридин (схема 12). Указанное в заголовке соединение получали из 5-бром-3-(3,5-диметоксифенил)-3H-имидазо[4,5b]пиридина (промежуточный продукт 8.3) с выходом 57% в соответствии с общей методикой IV. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 99% чистота, tR 8,39 мин (tR(SM) 8,51 мин);(схема 13). Указанное в заголовке соединение получали из 3-(3,5-диметоксифенил)-5-винил-3H-имидазо[4,5b]пиридина (промежуточный продукт 8.4) с выходом 56% в соответствии с общей методикой V. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 98% чистота, tR 7,26 мин (tR(SM) 8,39 мин); Промежуточный продукт 9.1. трет-Бутил-5-[(6-бром-3-нитропиридин-2-ил)амино]индолин-1 карбоксилат (схема 12). Указанное в заголовке соединение получали из 2,6-дибром-3-нитропиридина и трет-бутил-5 аминоиндолин-1-карбоксилата (полученного из коммерчески доступного 5-нитроиндолина присоединением защитной группы N-Boc и последующим восстановлением нитрогруппы при помощи H2/Pd/C в Промежуточный продукт 9.2. трет-Бутил-5-[(3-амино-6-бромпиридин-2-ил)амино]индолин-1 карбоксилат (схема 12). Указанное в заголовке соединение получали из трет-бутил-5-[(5-бром-2 нитрофенил)амино]индолин-1-карбоксилата (промежуточный продукт 9.1) с выходом 96% в соответствии с общей методикой II. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 98% чистота, tR 9,86 мин (tR(SM) 11,66 мин); МС (ESI) m/z (C18H21BrN4O2) 405,1/407,0 (M+1, 100), 349,1/351,1 (82) Finnegan LCQ Промежуточный продукт 9.3. трет-Бутил-5-(5-бром-3H-имидазо[4,5-b]пиридин-3-ил)индолин-1 карбоксилат (схема 12). Указанное в заголовке соединение получали из трет-бутил-5-[(3-амино-6-бромпиридин-2 ил)амино]индолин-1-карбоксилата (промежуточный продукт 9.2) с выходом 91% в соответствии с общей методикой III. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 97% чистота, tR 10,08 мин (tR(SM) 9,85 мин); МС (ESI) m/z (C19H19BrN4O2) 415,0/416,9 (M+1, 91), 359,1/361,0 (100), 315,1/317,2 (51) Finnegan Промежуточный продукт 9.4. трет-Бутил-5-(5-винил-3H-имидазо[4,5-b]пиридин-3-ил)индолин-1 карбоксилат (схема 12). Указанное в заголовке соединение получали из трет-бутил-5-(5-бром-3H-имидазо[4,5-b]пиридин-3 ил)индолин-1-карбоксилата (промежуточный продукт 9.3) с выходом 94% в соответствии с общей методикой IV. ВЭЖХ (за 10 мин 10-85% MeCN/0,1% ТФУК/H2O): 96% чистота, tR 7,21 мин (tR(SM) 8,51 мин); 1 Н-ЯМР (400 МГц, CDCl3):8,21 (с, 1 Н), 8,04 (д, 1 Н), 7,98 (уш.с, 0,5 Н), 7,56 (с, 1 Н), 7,55 (уш.с,0,5 Н), 7,48 (д, 1 Н), 7,35 (д, 1 Н), 6,89 (дд, 1 Н), 6,19 (д, 1 Н), 5,42 (д, 1 Н), 4,05 (т, 2 Н), 3,18 (т, 2 Н), 1,55 (с,9 Н) м.д. Промежуточный продукт 9.5. трет-Бутил 5-(5-формил-3H-имидазо[4,5-b]пиридин-3-ил)индолин-1 карбоксилат (схема 13). Указанное в заголовке соединение получали из трет-бутил-5-(5-винил-3H-имидазо[4,5-b]пиридин-3 ил)индолин-1-карбоксилата (промежуточный продукт 9.4) с выходом 69% в соответствии с общей методикой V. ВЭЖХ (за 10 мин 10-85% MeCN/0,1% ТФУК/H2O): 96% чистота, tR 7,31 мин (tR(SM) 7,21 мин)(200 мл), 2-пропанола (30 мл) и диоксана (50 мл) перемешивали в течение 1,5 ч при температуре окружающей среды. Смесь концентрировали досуха с получением 9,50 г (98% выход) тригидрохлоридной соли соответствующего свободного амина. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 99% чистота, tR 6,64 мин (tR(SM) 10,06 мин); 1 Промежуточный продукт 10.2. 3-(1-Ацетил-2,3-дигидро-1 Н-индол-5-ил)-5-винил-3H-имидазо[4,5b]пиридин (схема 12). Смесь 3-(2,3-дигидро-1 Н-индол-5-ил)-5-винил-3H-имидазо[4,5-b]пиридина (промежуточный продукт 10.1) (150,0 мг, 0,57 ммоль), ледяной уксусной кислоты (39,3 мкл, 0,69 ммоль), гидрохлорида(419,2 мг, 3,43 ммоль) и дихлорметана (10 мл) перемешивали в течение 24 ч при температуре окружаю- 28012178 щей среды. Смесь последовательно экстрагировали насыщенным водным хлоридом аммония и насыщенным водным бикарбонатом натрия. Органический слой высушивали над сульфатом натрия и концентрировали в вакууме с получением 139,1 мг (80%) соответствующего амида. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 99% чистота, tR 6,43 мин (tR(SM) 6,64 мин); Промежуточный продукт 10.3. 3-(1-Ацетил-2,3-дигидро-1 Н-индол-5-ил)-3H-имидазо[4,5b]пиридин-5-карбальдегид (схема 13). Указанное в заголовке соединение получали из 3-(1-ацетил-2,3-дигидро-1 Н-индол-5-ил)-5-винил 3H-имидазо[4,5-b]пиридина (промежуточный продукт 10.2) с выходом 44% в соответствии с общей методикой V. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 95% чистота, tR 5,38 мин (tR(SM) 6,43 мин);(промежуточный продукт 10.1), гидрохлорида 4-(диметиламино)масляной кислоты (36,3 мг, 0,21 ммоль),гидрохлорида N-этил-N'-(3-диметиламинопропил)карбодиимида (54,9 мг, 0,29 ммоль), 4-диметиламинопиридина (122,5 мг, 1,00 ммоль) и дихлорметана (8 мл) перемешивали в течение 24 ч при температуре окружающей среды. Смесь последовательно экстрагировали насыщенным водным хлоридом аммония и насыщенным водным бикарбонатом натрия. Органический слой высушивали над сульфатом натрия и концентрировали в вакууме с получением 50,4 мг (94%) соответствующего амида. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 92% чистота, tR 6,27 мин (tR(SM) 6,61 мин) Промежуточный продукт 11.2. 3-1-[4-(Диметиламино)бутаноил]-2,3-дигидро-1 Н-индол-5-ил-3Hимидазо[4,5-b]пиридин-5-карбальдегид (схема 13). Указанное в заголовке соединение получали из N,N-диметил-N-4-оксо-4-[5-(5-винил-3Hимидазо[4,5-b]пиридин-3-ил)-2,3-дигидро-1 Н-индол-1-ил]бутиламина (промежуточный продукт 11.1) с выходом 45% в соответствии с общей методикой V. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 94% чистота, tR 5,02 мин (tR(SM) 6,27 мин)(промежуточный продукт 10.1), метансульфонилхлорида (1,12 мл, 14,47 ммоль) и триэтиламина (2,94 мл,21,22 ммоль) в дихлорметане (50 мл) перемешивали в течение 30 мин при температуре окружающей среды. Смесь последовательно экстрагировали насыщенным водным хлоридом аммония и насыщенным- 29012178 водным бикарбонатом натрия. Органический слой высушивали над сульфатом натрия и концентрировали в вакууме с получением 3,24 г (99%) соответствующего сульфонамида. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 92% чистота, tR 7,16 мин (tR(SM) 6,64 мин); МС (ESI) m/z (C17H16N4O2S) 341,1 (M+1, 100) Finnegan LCQ Промежуточный продукт 12.2. 3-[1-(Метилсульфонил)-2,3-дигидро-1 Н-индол-5-ил]-3Hимидазо[4,5-b]пиридин-5-карбальдегид (схема 13). Указанное в заголовке соединение получали из 3-[1-(метилсульфонил)-2,3-дигидро-1 Н-индол-5-ил]5-винил-3H-имидазо[4,5-b]пиридина (промежуточный продукт 12.1) с выходом 60% в соответствии с общей методикой V. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 98% чистота, tR 6,13 мин (tR(SM) 7,16 мин)N,N-диизопропилэтиламина (11,98 мл, 68,58 ммоль) в дихлорметане (100 мл) перемешивали в течение 20 мин при температуре окружающей среды. Смесь последовательно экстрагировали насыщенным водным хлоридом аммония и насыщенным водным бикарбонатом натрия. Органический слой высушивали над сульфатом натрия и концентрировали в вакууме с получением 3,61 г (98%) соответствующего сульфонамида. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 92% чистота, tR 8,05 мин (tR(SM) 6,64 мин); 1 Промежуточный продукт 13.2. 3-1-[(Хлорметил)сульфонил]-2,3-дигидро-1 Н-индол-5-ил-3Hимидазо[4,5-b]пиридин-5-карбальдегид (схема 13). Указанное в заголовке соединение получали из 3-1-[(хлорметил)сульфонил]-2,3-дигидро-1 Ниндол-5-ил-5-винил-3H-имидазо[4,5-b]пиридина (промежуточный продукт 13.1) с выходом 86% в соответствии с общей методикой V. ВЭЖХ (за 10 мин 10-85% MeCN/100 мМ водн. NaOAc): 97% чистота, tR 7,03 мин (tR(SM) 8,05 мин); МС (ESI) m/z (C16H13ClN4O3S) 377,0 (M+1, 100) Finnegan LCQ
МПК / Метки
МПК: C07D 471/04, A61K 31/519, C07D 491/04, A61K 31/4355, C07D 487/04
Метки: применение, пиридинметиленазолидиноны
Код ссылки
<a href="https://eas.patents.su/30-12178-piridinmetilenazolidinony-i-ih-primenenie.html" rel="bookmark" title="База патентов Евразийского Союза">Пиридинметиленазолидиноны и их применение</a>
Производные диазепинкарбоксамида, способ их получения, их применение в качестве лекарственных средств, фармацевтические композиции и их применение

Номер патента: 5288
Опубликовано: 30.12.2004
Авторы: Бхатнагар Неерджа, Можер Жак
МПК: A61K 31/551, C07D 487/04, C07K 5/02...
Метки: лекарственных, качестве, применение, производные, композиции, способ, фармацевтические, диазепинкарбоксамида, получения, средств
Формула / Реферат:
1. Соединения формулы (I) в которой R1 означает радикал -C(O)-R5, -SO2-R5 или -C(O)-NR6R5, в которых R6 означает атом водорода или линейный или разветвленный алкил, содержащий не более 4 атомов углерода, и R5 означает линейный или разветвленный алкил, содержащий не более 6 атомов углерода, циклоалкил, содержащий не более 6 атомов углерода, фенил, нафтил или насыщенный или ненасыщенный 5- или 6-членный гетероциклический моноциклический радикал,...
Фармацевтическая композиция, способ ее получения и ее применение для лечения респираторных заболеваний, фармацевтический продукт и его применение, набор

Номер патента: 10886
Опубликовано: 30.12.2008
Авторы: Бойме Рольф, Ваймар Кристиан, Воллин Штефан-Лутц, Бундшух Даниела
МПК: A61K 31/4425, A61K 31/4375, A61K 31/4523...
Метки: фармацевтическая, способ, применение, продукт, набор, композиция, фармацевтический, лечения, респираторных, заболеваний, получения
Формула / Реферат:
1. Фармацевтическая композиция, содержащая первое действующее вещество, выбранное из группы, состоящей из 3-циклопропилметокси-4-дифторметокси-N-(3,5-дихлорпирид-4-ил)бензамида [MHH рофлумиласт] и его фармацевтически приемлемых производных, в смеси со вторым действующим веществом, выбранным из группы, состоящей из [2-[4-[(R)-n-хлор-a-фенилбензил]-1-пиперазинил]этокси]уксусной кислоты [МНН левоцетиризин] и ее фармацевтически приемлемых...
Термостабилизированная пресс-композиция, ее применение, формованные детали, состоящие из указанной композиции, способ получения формованных деталей и их применение

Номер патента: 11324
Опубликовано: 27.02.2009
Авторы: Янссен Роберт Хендрик Катарина, Гийсман Питер, Сур Вильхельмус Йозефус Мария, Рулкенс Руди
МПК: C08K 5/00, C08L 77/00, C08K 3/00...
Метки: термостабилизированная, способ, формованные, указанной, получения, детали, состоящие, деталей, пресс-композиция, применение, формованных, композиции
Формула / Реферат:
1. Термостабилизированная термопластичная пресс-композиция включающая: a) композицию термопластичного полиамида и b) систему стабилизации, включающую термостабилизатор, выбранный из группы, состоящей из фенольного термостабилизатора, органических фосфитов, ароматических аминов, солей металлов IB, IIB, III и IV групп Периодической таблицы и галидов щелочных и щёлочно-земельных металлов, и их комбинаций, характеризующаяся тем, что композиция...
Устройство для получения плазмы, способ ионизации, применение способа и применение устройства (варианты)

Номер патента: 6412
Опубликовано: 29.12.2005
Автор: Сеза Валентен
МПК: H05H 1/24, B01J 19/10
Метки: устройства, применение, устройство, способ, плазмы, получения, способа, варианты, ионизации
Формула / Реферат:
1. Устройство для получения плазмы путем реакции горения вещества или смеси веществ M, отличающееся тем, что содержит резонансную камеру (1), предназначенную для создания стационарной циркуляции потока вещества или смеси веществ (M), подаваемых в резонансную камеру (1) по меньшей мере через одно средство (2) питания и выходящих из резонансной камеры (1) по меньшей мере через один выход (3), выполненный в виде по меньшей мере одного вытянутого...
Фармацевтическая композиция на основе 3,6-ангидрогалактопиранозы и/или ее производных, и/или растворимого сахарида, содержащего данное соединение, применение композиции и входящих в ее состав ингредиентов, пищевой продукт, содержащий указанную композицию, и применение пищевого продукта

Номер патента: 4148
Опубликовано: 26.02.2004
Авторы: Еноки Татсудзи, Койама Нобуто, Сакаи Такеси, Томинага Таканари, Икаи Катсусиге, Йу Фу-Гонг, Като Икуносин, Сагава Хироаки, Нисийама Еидзи
МПК: A01N 1/00, A23L 1/30, A61K 31/70...
Метки: композиция, растворимого, входящих, пищевой, производных, ингредиентов, фармацевтическая, состав, пищевого, композицию, применение, продукта, данное, продукт, 3,6-ангидрогалактопиранозы, указанную, композиции, содержащий, основе, соединение, содержащего, сахарида
Формула / Реферат:
1. Фармацевтическая композиция, содержащая в качестве активного ингредиента по меньшей мере одно соединение, выбранное из группы, состоящей из 3,6-ангидрогалактопиранозы, представленной формулой I ее альдегида и гидрата и 2-O-метилированных производных 3,6-ангидрогалактопиранозы, ее альдегида и гидрата; и растворимого сахарида, содержащего данное соединение на восстанавливающем конце. 2. Фармацевтическая композиция по п.1, где сахарид...
Предыдущий патент: Новая композиция биотоплива
Следующий патент: Промышленный способ получения высокочистого диарилкарбоната, высокочистый дифенилкарбонат и установка для получения высокочистого диарилкарбоната
Случайный патент: Способ разделения газового потока (варианты)