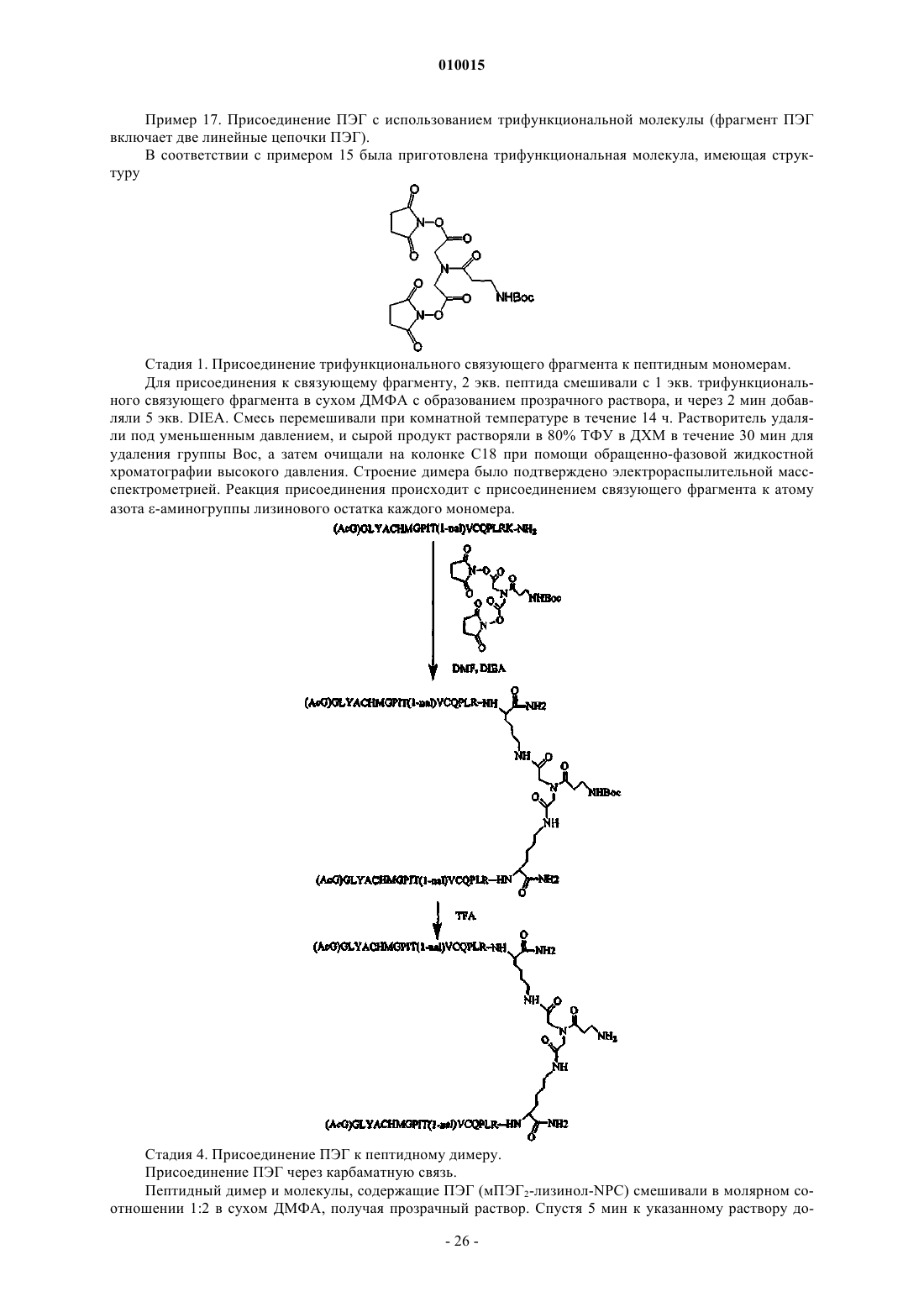
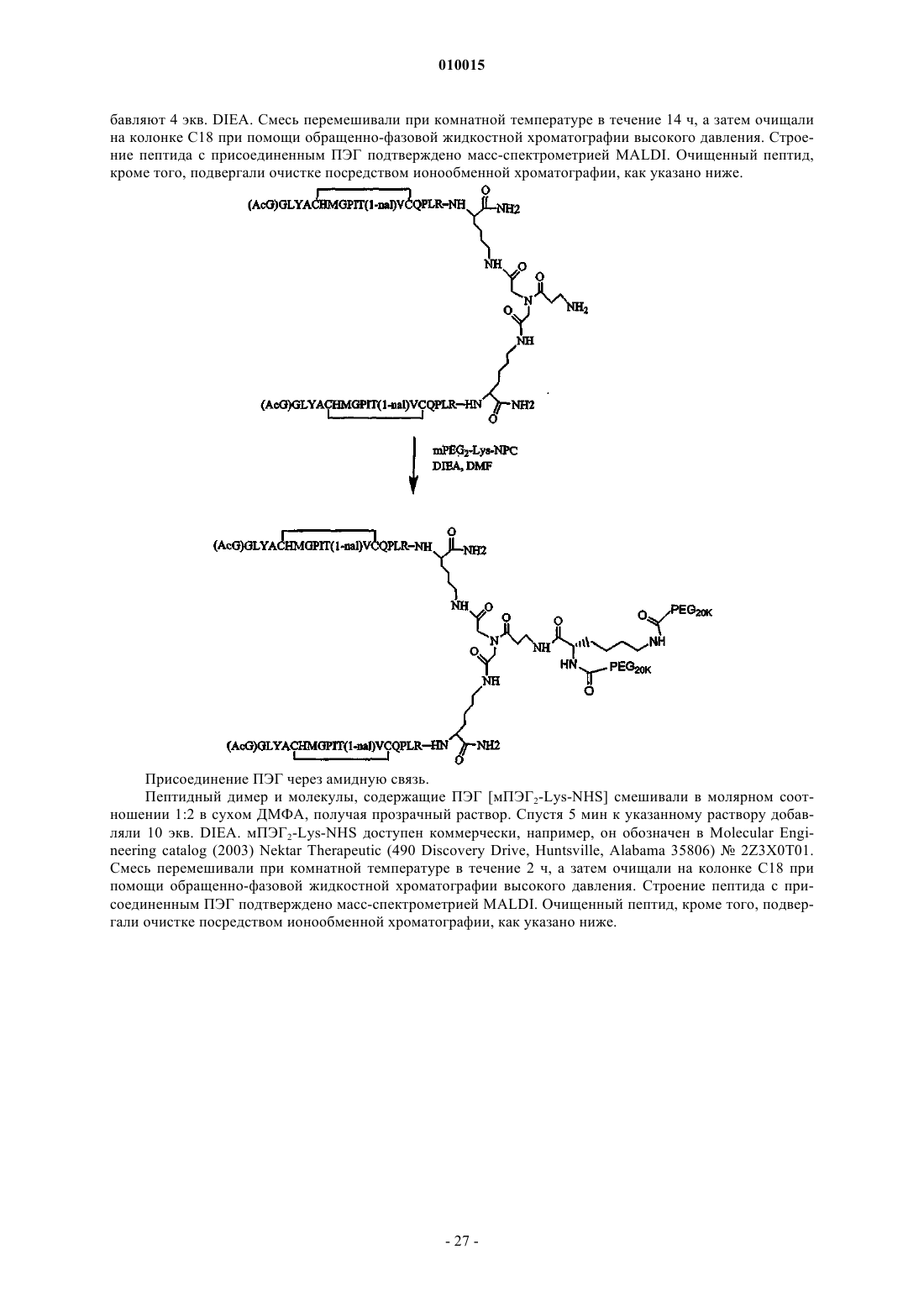
Новый разделительный фрагмент (спейсер) для модифицированных полиэтиленгликолем соединений на основе пептидов


















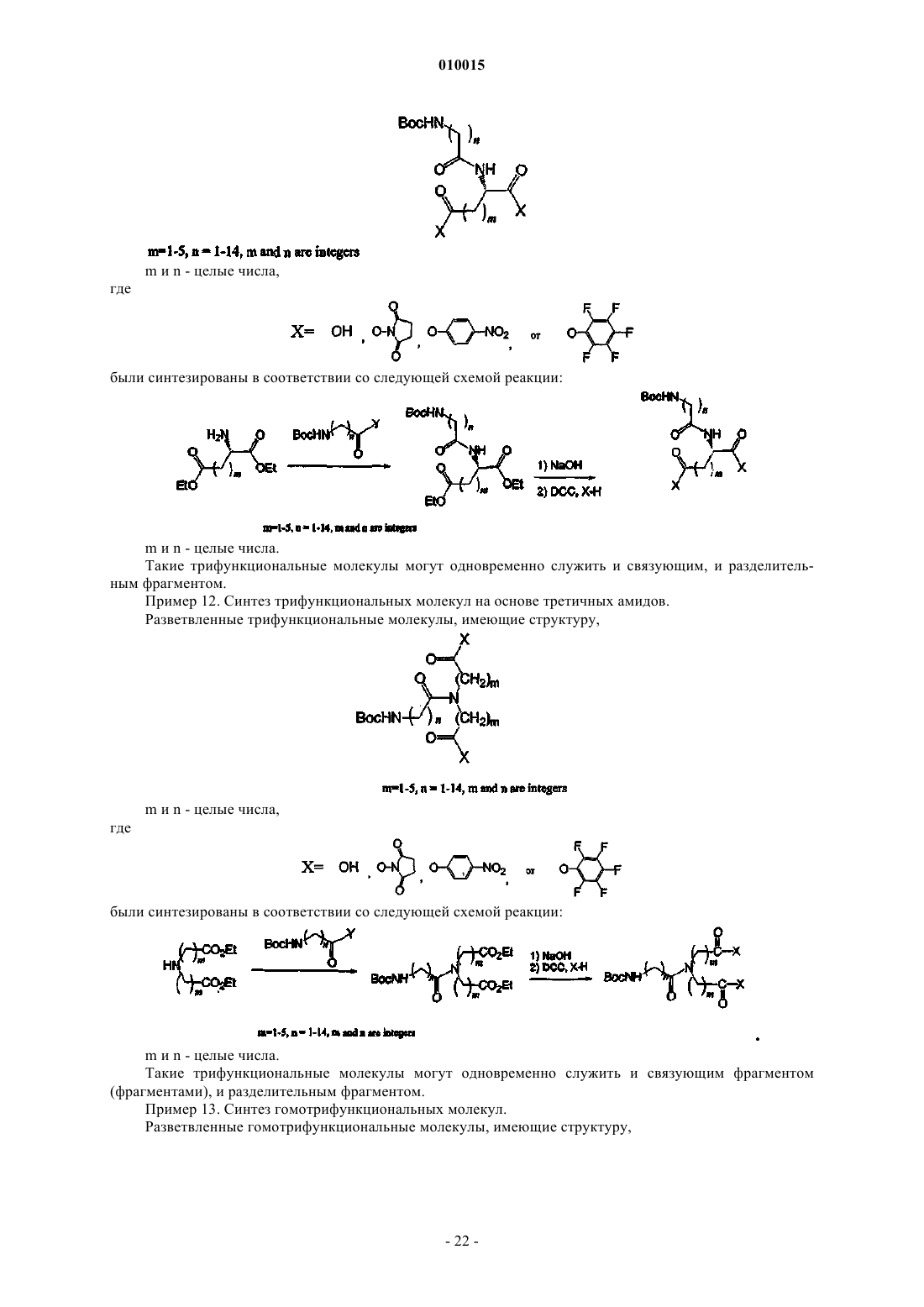
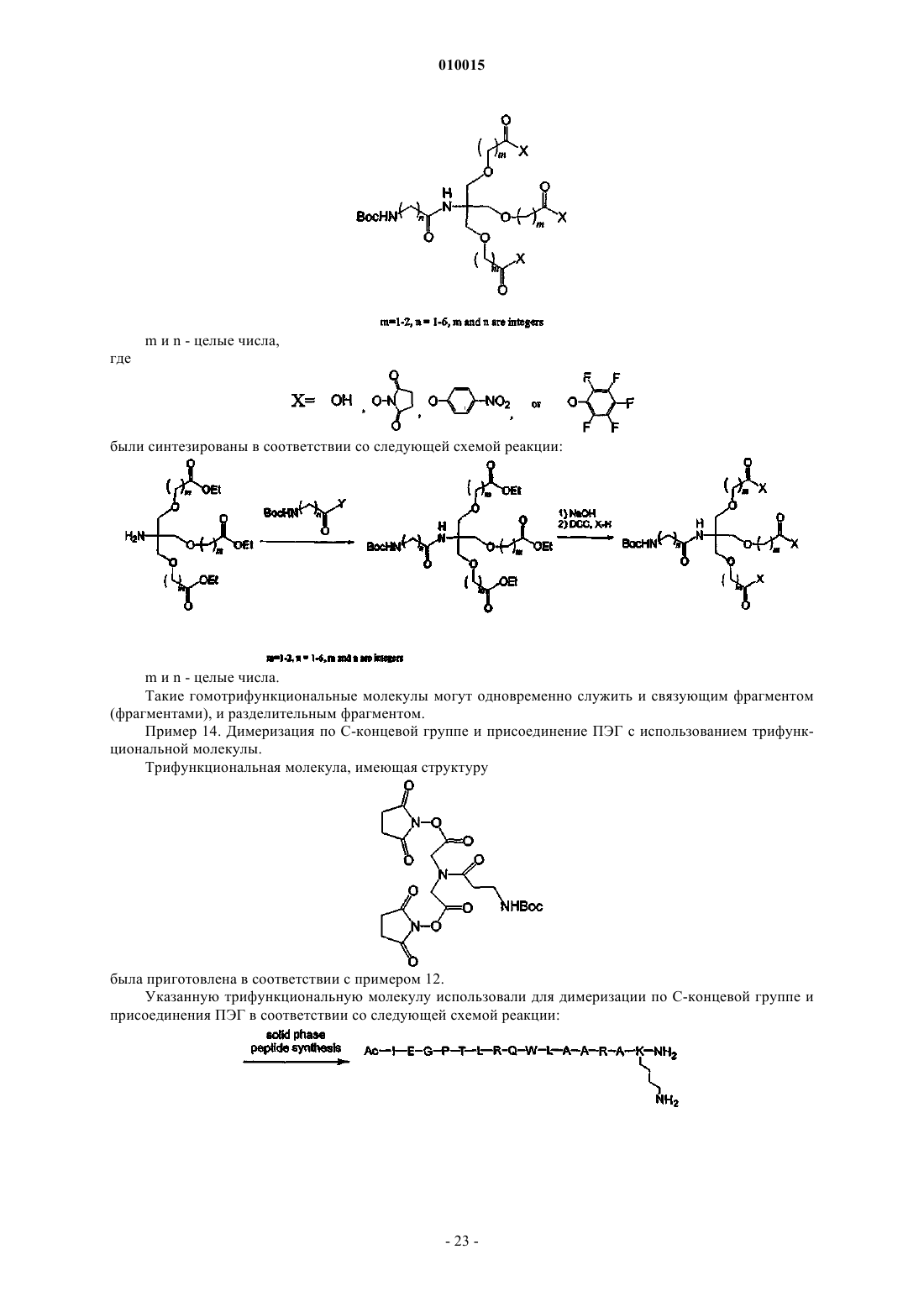
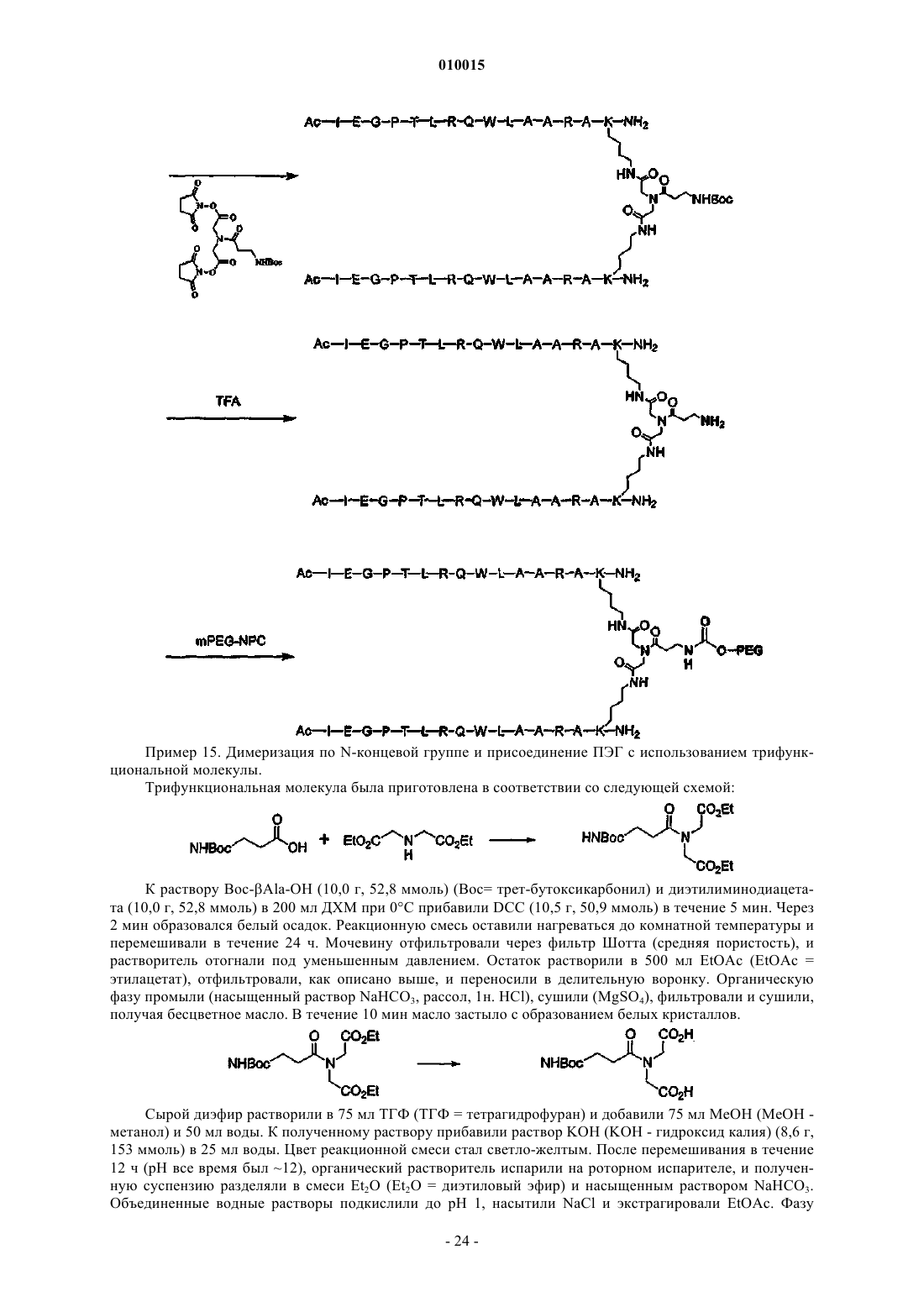
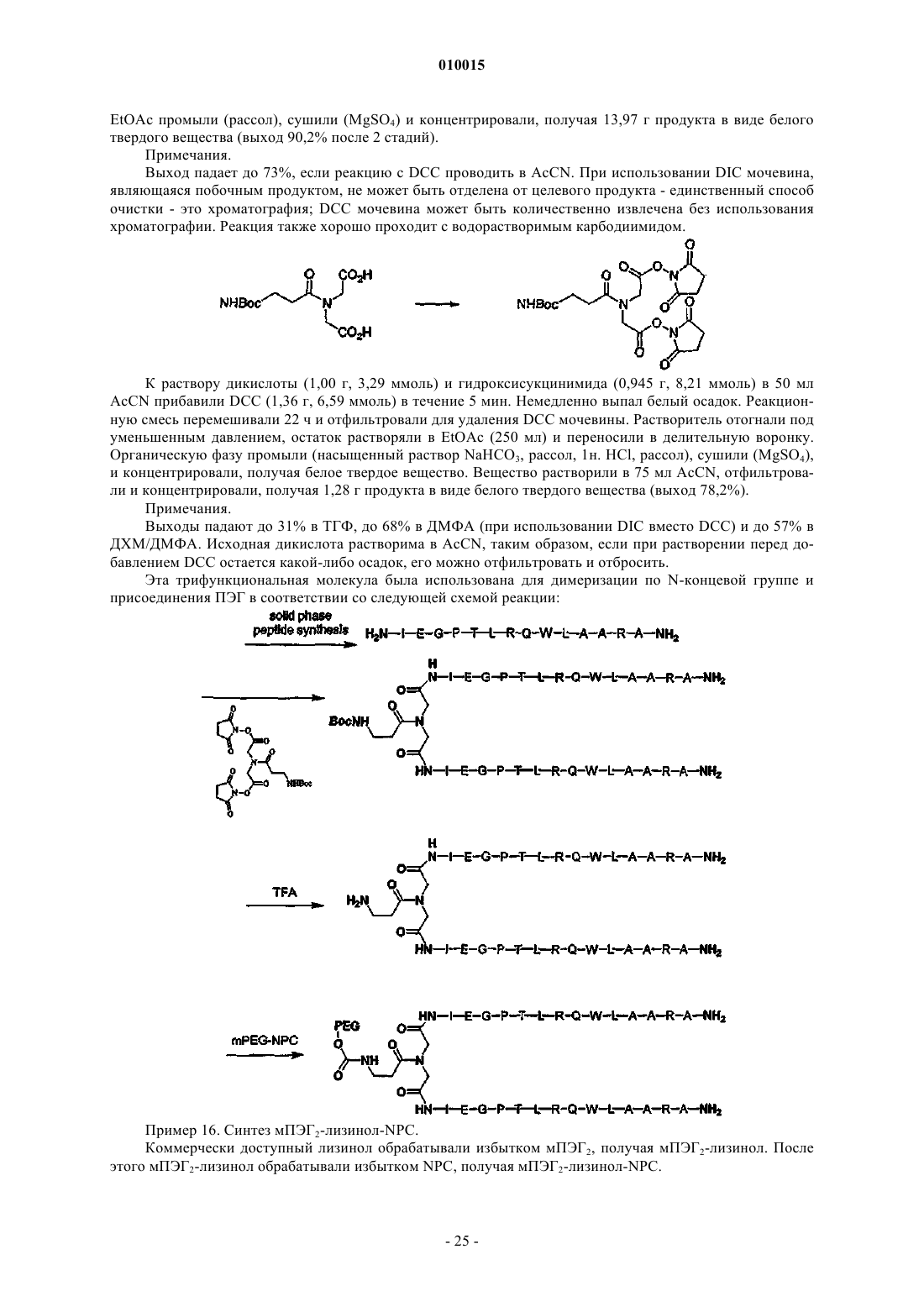


Формула / Реферат
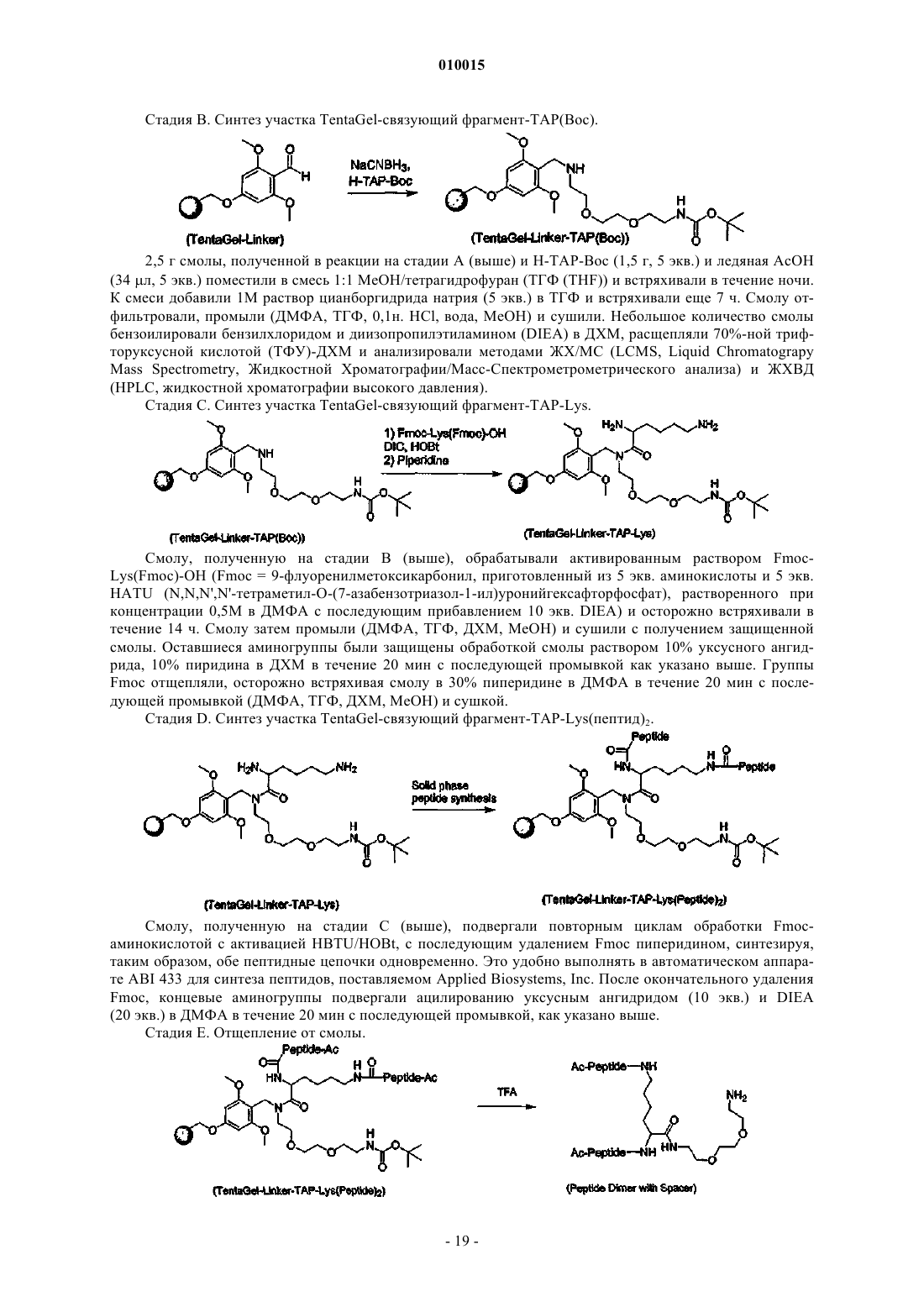
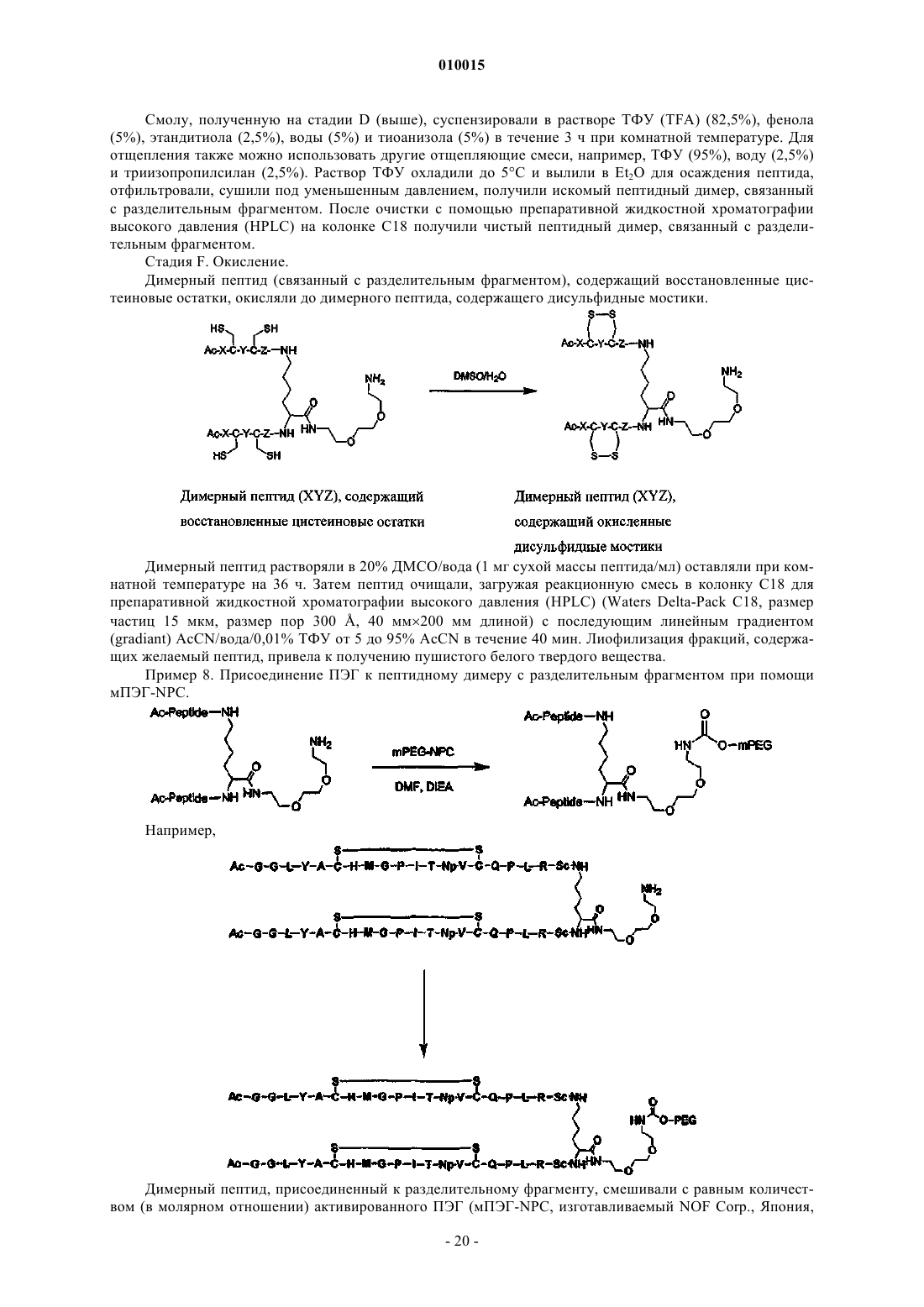
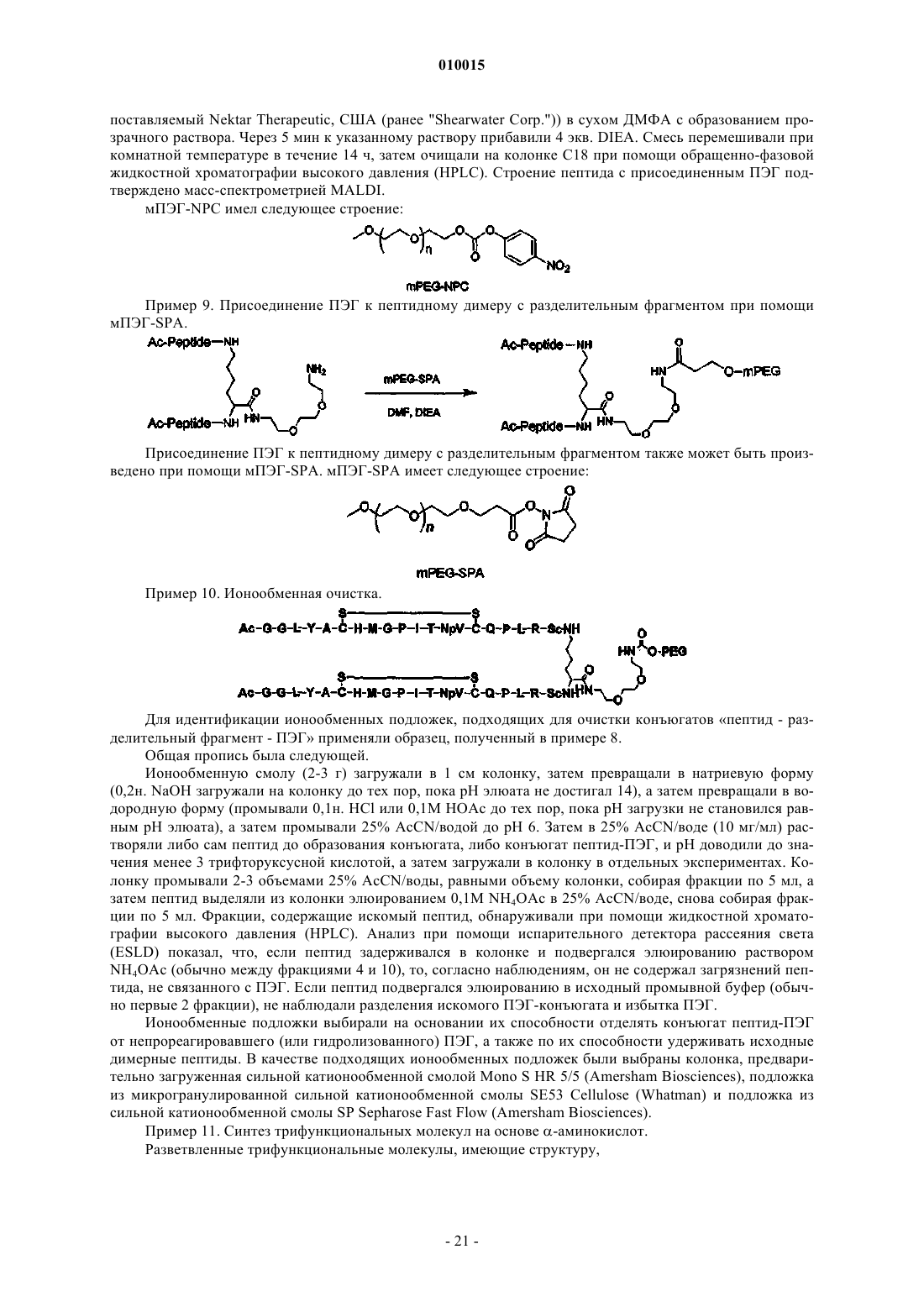
1. Соединение, включающее пептидный фрагмент, разделительный фрагмент (спейсер) и фрагмент полиэтиленгликоля, в котором разделительный фрагмент находится между пептидным фрагментом и фрагментом полиэтиленгликоля и имеет следующее строение:
![]()
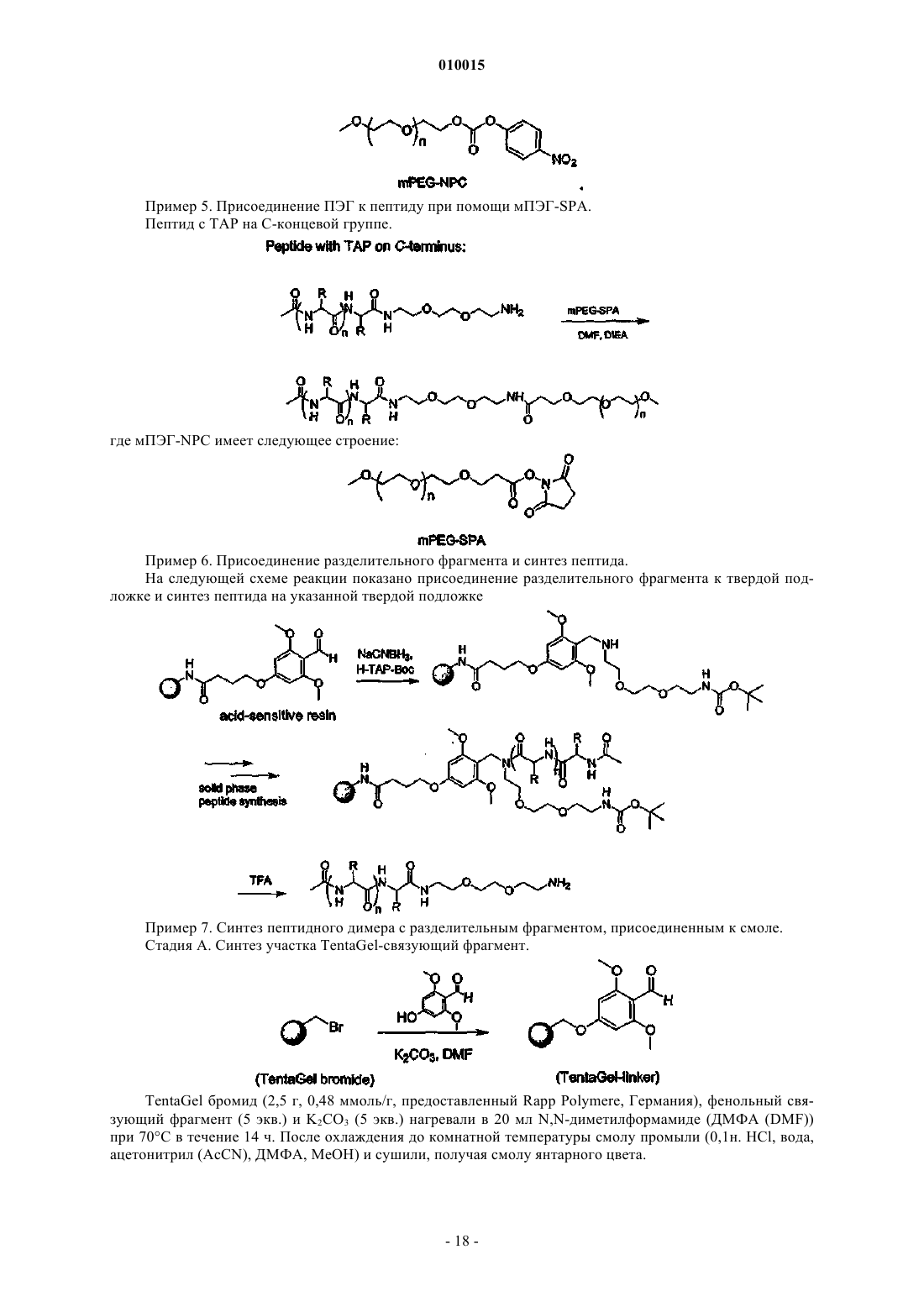
где каждый из коэффициентов a, b, g, d и e представляет собой целое число, значения которых выбирают независимо друг от друга, причем g или d не равен 0, а фрагмент полиэтиленгликоля ковалентно присоединен посредством карбаматной или амидной связи и имеет молекулярную массу более 20 кДа.
2. Соединение по п.1, в котором a - это целое число, 1_a_6; b - это целое число, 1_b_6; e - это целое число, 1_e_6; d равно 0 или 1; g - это целое число, 1_g_10 и Y представляет собой либо NH, либо CO.
3. Соединение по п.2, в котором g>1 и b=2.
4. Соединение по п.1, в котором a=b=e=2; g=d=1 и Y представляет собой NH.
5. Соединение по п.1, в котором фрагмент полиэтиленгликоля имеет линейное строение.
6. Соединение по п.1, в котором фрагмент полиэтиленгликоля имеет значение полидисперсности (Mw/Mn) менее 1,20.
7. Соединение по п.1, в котором пептидный фрагмент представляет собой пептидный мономер, включающий один пептид.
8. Соединение по п.1, в котором пептидный фрагмент представляет собой пептидный димер, включающий два пептида, соединенные связующим фрагментом.
9. Соединение по п.7 или 8, в котором каждый пептид включает не более 50 аминокислотных мономеров, предпочтительно каждый пептид включает приблизительно от 10 до 25 аминокислотных мономеров.
10. Соединение по п.1, в котором пептидный фрагмент включает один или более пептидов, связывающих рецепторы эритропоетина.
11. Соединение по п.1, в котором пептидный фрагмент включает один или более пептидов, связывающих рецепторы тромбопоетина.
12. Соединение по п.1, в котором a=2; g=d=b=e=0 и Y представляет собой CO.
13. Соединение по п.1, в котором фрагмент полиэтиленгликоля включает по меньшей мере одну мономерную цепочку полиэтиленгликоля.
14. Соединение по п.13, в котором молекулярная масса каждой цепочки полиэтиленгликоля составляет от 20 до 40 кДа.
15. Фармацевтическая композиция, которая содержит соединение по любому из пп.1-14 и один или более фармацевтически приемлемых разбавителей, консервантов, солюбилизаторов, эмульсификаторов, вспомогательных средств и/или носителей.
16. Соединение, включающее пептидный фрагмент, разделительный фрагмент (спейсер) и фрагмент водорастворимого полимера, в котором разделительный фрагмент находится между пептидным фрагментом и фрагментом водорастворимого полимера и имеет следующее строение:
![]()
где a=b=e=2; g=d=1 и Y представляет собой NH.
17. Фармацевтическая композиция, которая содержит:
а) соединение, включающее пептидный фрагмент, разделительный фрагмент (спейсер) и фрагмент водорастворимого полимера, в котором разделительный фрагмент находится между пептидным фрагментом и фрагментом водорастворимого полимера и имеет следующее строение:
![]()
где a=b=e=2; g=d=1 и Y представляет собой NH и
б) один или более фармацевтически приемлемый разбавитель, консервант, солюбилизатор, эмульсификатор, адьювант и/или носитель.
Текст
010015 В заявке на данное изобретение согласно 35 U.S.С.1199(e) испрашивается приоритет по находящейся одновременно с ней на рассмотрении предварительной патентной заявке США 60/469996 от 12 мая 2003 г. Содержание указанной предварительной заявки полностью включено в настоящее описание по ссылке. Область техники Настоящее изобретение относится к новому способу ковалентного присоединения водорастворимого полимера, например полиэтиленгликоля (ПЭГ), к пептидам и соединениям, полученным на основе пептидов. Кроме того, изобретение относится к новым терапевтическим композициям, включающим указанные соединения. Уровень техники В последние годы в связи с ростом интереса к исследованию белков было открыто большое количество пептидов с различными функциями. В связи с развитием методик генной рекомбинации и способов органического синтеза пептидов появилась возможность получать такие физиологически активные пептиды и их структурные аналоги в больших количествах. Многие из таких пептидов, обладающих специфической активностью, представляют собой чрезвычайно полезные фармацевтические препараты. Примеры таких пептидов включают пептиды, которые присоединяются к эритропоетиновым (ЕРО) рецепторам (EPO-R). Эритропоетин (ЕРО) представляет собой гормон гликопротеина, который включает 165 аминокислот и имеет 4 центра гликозилирования в 24, 38, 83 и 136 положениях аминокислот, а также имеет молекулярную массу приблизительно 34000 Дальтон (Да). Он стимулирует митотическое деление и дифференцирование клеток-предшественников эритроцитов и, таким образом, обеспечивает выработку эритроцитов. ЕРО необходим для процесса образования красных кровяных клеток; в перспективе, этот гормон может быть применен как в диагностике, так и в лечении заболеваний крови, характеризуемых низкой или нарушенной выработкой красных кровяных клеток. Был обнаружен ряд пептидов, взаимодействующих с EPO-R. (См., например, патент США 5773569, Wrighton et al.; патент США 5830851, Wrighton et al.; и WO 01/91780 Smith-Swintosky et al.) Однако клиренс (clearance) пептидов, особенно при введении в кровеносную систему, обычно происходит очень быстро. Таким образом, желательно увеличить стойкость указанных пептидов. Кроме того, при получении пептидов от животных различных видов, созданных при помощи пептиднопротеиновой инженерии (peptide protein engineering), и/или имеющих строение, отличное от пептидов пациента, всегда имеется риск возникновения нежелательных симптомов из-за выработки антител. Следовательно, желательно также улучшить антигенность таких пептидов. Для того чтобы указанные пептиды можно было применять в качестве фармацевтических препаратов, необходимо улучшить как их стойкость, так и их антигенность. Было показано, что химическая модификация пептидов макромолекулярными соединениями помогает улучшить антигенность и стойкость различных пептидов. Так, в качестве макромолекулярных реагентов для модификации пептидов широко используют полиэтиленгликоль и производные полиэтиленгликоля. Наиболее обычная форма полиэтиленгликоля имеет следующее строение: Вышеуказанный полимер, альфа-, омега-дигидроксилполиэтиленгликоль, может быть представлен при помощи укороченной формулы HO-PEG-OH, где символом -PEG- обозначена следующая структурная единица: Не прибегая к какой-либо частной теории механизма или действия, можно предположить, что в водной среде длинная, цепеобразная молекула или фрагмент PEG быстро движется и находится в сильно гидратированном состоянии. Полагают, что такое быстрое движение обуславливает охватывание молекулой ПЭГ большого объема и предотвращает приближение к ней и взаимодействие с ней других молекул. В результате, присоединившись к другому химическому образованию (такому, как пептид), полимерная цепь ПЭГ может защищать указанное образование от иммунного ответа или других механизмов клиренса. В результате, присоединение ПЭГ приводит к улучшенной эффективности и безопасности медикамента, полученной при помощи оптимальной фармакокинетики, повышенной биодоступности и пониженной иммуногенности и частоте приема. Например, некоторые активные производные ПЭГ удалось присоединить к пептидам, белкам и ферментам, что привело к хорошим результатам. ПЭГ растворим в органических растворителях. Присоединение ПЭГ к ферментам может приводить к образованию конъюгатов ПЭГ-фермент, которые растворимы и активны в органических растворителях. Присоединение ПЭГ к белку может снижать иммуногенность и скорость почечного клиренса конъюгата ПЭГ-белок по сравнению с немодифицированным белком, что может приводить к существенному увеличению продолжительности циркулирования конъюгата в кровеносной системе. Например, имеются сообщения о том, что ковалентное присоединение ПЭГ к терапевтическим белкам, таким как интерлейкины (Knauf, M. J. et al., J. Biol. Chem. 1988, 263, 15, 064; Tsutsumi, Y. et al., J.(Abuchowski, A. et al., J. Biol. Chem. 1977, 252, 3, 582), супероксиддисмутаза (Beauchamp, С. О. et al., Annal. Biochem. 1983, 131, 25) и аденозиндеаминаза (Chen, R. et al., Biochim. Biophy. Acta 1981, 660, 293) увеличивает их время полужизни (halflife) in vivo и/или снижает их иммуногенность и антигенность. Кроме того, присоединение ПЭГ к поверхности может снижать адсорбцию белков и клеток на поверхности и изменять электрические свойства поверхности. Аналогично, присоединение ПЭГ к липосомам может привести к значительному увеличению времени полужизни этих частиц в кровеносной системе и, таким образом, может способствовать их роли в доставки медикамента. (J. M. Harris, Ed., "Biomedical and Biotechnical Applications of Polyethylene Glycol Chemistry", Plenum, New York, 1992). Наличие аминокислотного или пептидного мостика между ПЭГ и присоединенной макромолекулой дает некоторые преимущества, поскольку позволяет придавать различные свойства, которые могут быть введены при помощи подходящей аминокислоты или пептида. Примерами таких аминокислотных или пептидных мостиков могут служить: норлейцин (Nle), применяемый для аналитических целей; Gly, меченый 14 С или тритием, применяемый для фармакокинетического исследования; Lys, применяемый для создания разветвленных структур; и Met-Nle или Met-Ala, применяемые для удаления ПЭГ при помощи обработки BrCN (Veronese, F. M. Biomaterials, 2001, 22, 405). Другой известный тип производного ПЭГ, содержащего аминокислотный мостик между ПЭГ и присоединенными макромолекулами, характеризуется наличием двух линейных цепочек ПЭГ, связанных друг с другом посредством двух функциональных групп трехфункционального разделительного фрагмента (спейсера), третья функциональная группа которого связывает белок. Лизин представляет собой трифункциональный аминокислотный спейсер, который связывает две цепочки ПЭГ при помощи своих альфа- и эпсилон-аминогрупп, в то время как карбоксильная группа лизина активирована с образованием гидроксисукцинимидного сложного эфира, предназначенного для связывания белка. Преимуществом этого производного ПЭГ является более низкая инактивация фермента во время связывания, а его зонтообразная структура эффективна при защите белков от протеолиза (расщепления белков) при приближении антител, а также при снижении иммуногенности (Veronese, F. M. Biomaterials, 2001, 22, 405). Если часть активирующей группы была захвачена конечным аддуктом вида ПЭГ-пептид или ПЭГлипосома, то иногда образуются нежелательные побочные продукты типа ПЭГ - связующий мостик(linker) - пептид или ПЭГ - связующий мостик - липосома. Frances et al. (Int. J. Hematol. 1998, 68, 1) сообщают, что такие связующие мостики могут иметь несколько видов нежелательных воздействий: (1) такие связующие мостики не обязательно иммунологически инертны, и существуют экспериментальные свидетельства, что такие группы отвечают за иммуногенность/антигенность протеинов ПЭГ; (2) некоторые связующие фрагменты содержат подвижные связи, которые могут расщепляться под действием химических агентов или ферментов; (3) связующие фрагменты, полученные из активированных ПЭГ, которые часто бывают относительно токсичными, могут создавать регуляторные проблемы; (4) определенные связующие группы, такие как триазиновые кольца, могут вызывать поперечную сшивку молекул. Несмотря на достижения в области модифицированных ПЭГ соединений, полученных на основе пептидов, все еще существует необходимость создания новых соединений, модифицированных ПЭГ,обладающих улучшенной антигенностью и стойкостью. Цитирование и/или обсуждение ссылок, использованных в настоящем разделе, а также во всем описании, не должно рассматриваться в качестве прототипа настоящего изобретения. Краткое описание изобретения Настоящее изобретение относится к соединению, полученному на основе пептида, включающему фрагмент пептида, разделительный фрагмент (спейсер) и фрагмент водорастворимого полимера, такого как полиэтиленгликоль. Разделительный фрагмент (спейсер) находится между фрагментом пептида и фрагментом водорастворимого полимера. Настоящее изобретение также относится к соединению, полученному на основе пептида, которое также включает связующий фрагмент (линкер). Разделительный фрагмент (спейсер) находится между связующим фрагментом (линкером) и фрагментом водорастворимого полимера, а связующий фрагмент(линкер) находится между разделительным фрагментом (спейсером) и пептидом. В альтернативном случае, связующий фрагмент (линкер) может включать разделительный фрагмент (спейсер). В одном из примеров реализации настоящего изобретения разделительный фрагмент (спейсер) имеет следующее строение: где каждый из коэффициентов , , ,ипредставляет собой целое число, значения которых выбирают независимо друг от друга. В предпочтительных примерах реализации- это целое число, 16;- это целое число, 16;- это целое число, 16;равно 0 или 1;- это целое число, 110; и Y представляет собой либо NH,либо CO. В некоторых предпочтительных примерах реализации =2, если 1.-2 010015 В одном особенно предпочтительном примере реализации ===2; ==1; и Y представляет собойNH. В других примерах реализации ==0; 2+5; и Y представляет собой CO. В некоторых других примерах реализации ==0; +=5; и Y представляет собой CO. В других примерах реализации =2; ====0; и Y представляет собой CO. В одном из примеров реализации настоящего изобретения связующий фрагмент (линкер) имеет следующее строение: где каждый из коэффициентовипредставляет собой целое число, значения которых выбирают независимо друг от друга, а N может быть ковалентно связан с Y разделительного фрагмента. В предпочтительных примерах реализации- это целое число, 16; и- это целое число, 16. В одном особенно предпочтительном примере реализации ==1. В другом примере реализации связующий фрагмент представляет собой остаток лизина или лизинамида (остаток лизина, в котором карбоксильная группа превращена в амидный фрагмент CONH2). Предпочтительно фрагмент водорастворимого полимера - это фрагмент полиэтиленгликоля. Более предпочтительно, фрагмент полиэтиленгликоля имеет линейное строение и имеет молекулярную массу,превышающую 10 кДа. Еще более предпочтительно фрагмент полиэтиленгликоля имеет молекулярную массу от 20 до 60 кДа. Наиболее предпочтительно фрагмент полиэтиленгликоля имеет молекулярную массу, равную 20 кДа. Предпочтительно фрагмент полиэтиленгликоля имеет значение полидисперсности(Mw/Mn) менее 1,20, более предпочтительно менее 1,1 и наиболее предпочтительно менее 1,05. Фрагмент водорастворимого полимера может быть двухмерным и включать два мономерных водорастворимых полимера, связанных посредством связующего или разделительного фрагмента. В одном из примеров реализации настоящего изобретения молекула пептида представляет собой димер и включает два мономерных пептида, связанных связующим фрагментом. В одном из примеров реализации пептидный фрагмент выбирают из пептидов, которые связывают эритропоетин-рецепторы. Неограничивающие примеры таких EPO-R связывающих пептидов включают пептиды, описанные в опубликованных международных заявках PCT/US00/32224 (номер публикацииWO 01/38342 А 2, с указанием U.S.), PCT/US96/09810 (номер публикации WO 96/40749, с указанием U.S.) и PCT/US01/16654 (номер публикации WO 01/91780 А 1); и патенты США 5767078, 5773569, 5830851,5986047 и 6221608. Дополнительные неограничивающие примеры таких EPO-R связывающих пептидов описаны в предварительных патентных заявках США, сер.60/470245; 60/469993 и 60/470244, каждая из которых была заявлена 12 мая 2004 г. В другом примере реализации пептидный фрагмент выбирают из пептидов, которые связывают тромбопоетин-рецепторы ("TRO-R"). Неограничивающие примеры таких TRO-R связывающих пептидов включают пептиды, описанные в патентах США 6552008, 6506362, 6498155, 6465430, 6333031, 6251864,6121238, 6083913, 5932546, 5869451, 5683983, 5677280, 5668110 и 5654276; и в опубликованных патентных заявках США 2003/0083361, 2003/0009018, 2002/0177166 и 2002/0160013. Настоящее изобретение также относится к фармацевтической композиции, включающей соединение (соединения), описанные выше. Подробное описание изобретения Определения. Аминокислотные остатки в пептидах обозначены следующим образом: фенилаланин обозначен Phe или E; лейцин - Leu или L; изолейцин - Ile или I; метионин - Met или M; Валин - Val или V; серин - Ser или S; пролин - Pro или P; треонин - Thr или Т; аланин - Ala или А; тирозин - Tyr или Y; гистидин - His или H; глутамин - Gln или Q; аспарагин - Asn или N; лизин - Lys или K; аспарагиновая кислота (asparaticacid) - Asp или D; глутаминовая кислота - Glu или Е; цистеин - Cys или C; триптофан - Trp или W; аргинин - Arg или R; глицин - Gly или G. Нетрадиционные аминокислоты в пептидах обозначены следующим образом: 1-нафтиламин обозначен 1-nal или Np; 2-нафтиламин - 2-nal; N-метилглицин (также известный как саркозин) - MeG или Sc; и ацетилированный глицин (N-ацетилглицин) - AcG. Термин пептид или полипептид относится к полимеру, в котором мономеры представляют собой альфа-аминокислоты, соединенные друг с другом амидными связями. Пептиды состоят из двух и часто большего количества мономеров. Пептиды, применяемые в настоящем изобретении, предпочтительно содержат в своей цепи менее 50 мономерных аминокислот. В настоящем описании словосочетание фармацевтически приемлемый относится к молекулярным структурам и композициям, которые обычно считают безопасными, т.е. которые физиологически переносимы организмом принимающего их человека и обычно не вызывают аллергических или подобных неблагоприятных реакций, таких как расстройство пищеварения, головокружения и подобных симптомов. Предпочтительно в настоящем описании термин фармацевтически приемлемый означает, что вещество одобрено органами государственного регулирования Федерального правительства или штата,или упомянуто Фармакопеей США или другой общеизвестной фармакопеей и разрешено для приема-3 010015 животными и, более конкретно, людьми. Термин носитель относится к разбавителю, вспомогательному веществу, наполнителю или транспортному средству, вместе с которым производят прием соединения. Такие фармацевтические носители могут представлять собой стерильные жидкости, такие как вода или масла, включая масла белкового, животного, растительного или синтетического происхождения, такие как арахисовое масло, соевое масло, минеральное масло, конопляное масло и подобные масла. В качестве носителей, в частности, в качестве растворов для инъекций предпочтительно применяют воду или водные растворы, солевые растворы и водные растворы декстрозы и глицерина. Подходящие фармацевтические носители описаны в публикации "Remington's Pharmaceutical Sciences" E. W. Martin. В настоящем описании термин агонист относится к биологически активному лиганду, который связывает комплементарный ему биологически активный рецептор и активирует последний, либо вызывая в указанном рецепторе биологический отклик, либо усиливая уже имеющуюся биологическую активность указанного рецептора. Разделительный фрагмент (спейсер). В соответствии с настоящим изобретением пептидный фрагмент, присоединенный к фрагменту ПЭГ через разделительный фрагмент, имеет следующее строение. В одном из примеров реализации настоящего изобретения разделительный фрагмент имеет следующее строение: где каждый из коэффициентов , , ,ипредставляет собой целые числа, значения которых выбраны независимо друг от друга. В предпочтительных примерах реализации- это целое число, 16;- это целое число, 16;- это целое число, 16;равно 0 или 1;- это целое число, 110; и Y представляет собой либо NH,либо CO. В некоторых предпочтительных примерах реализации =2, если 1. В одном особенно предпочтительном примере реализации ===2; ==1; и Y представляет собойNH. В других примерах реализации ==0; 2+5; и Y представляет собой CO. В некоторых других примерах реализации ==0; +=5; и Y представляет собой CO. В других примерах реализации =2; ====0; и Y представляет собой CO. В соответствии с настоящим изобретением фрагмент водорастворимого полимера (предпочтительно, ПЭГ) присоединен к концевой группе NH разделительного фрагмента. Водорастворимый фрагмент может быть присоединен непосредственно к разделительному фрагменту или он может быть присоединен опосредовано, например, при помощи амидного или карбаматного мостика (мостиков). В таких примерах реализации фрагмент ПЭГ включает по меньшей мере одну мономерную цепочку ПЭГ. В некоторых примерах реализации фрагмент ПЭГ включает две мономерных цепочки ПЭГ. Две мономерные цепочки ПЭГ, предпочтительно, связаны друг с другом через лизиновый остаток или лизинамид (остаток лизина, в котором карбоксильная группа превращена в амидный фрагмент CONH2). Более предпочтительно две цепочки ПЭГ связаны с альфа- и эпсилон-аминогруппами лизина, в то время как карбоксильная группа активирована с образованием гидроксисукцинимидных сложных эфиров для связывания разделительного фрагмента. Например, если лизинамид связывает две мономерные цепочки ПЭГ, то структура полученного димера может быть представлена формулой I, и в обобщенном виде - формулой II. В формуле I символом N2 обозначен атом азота -аминогруппы лизина, а символом N1 обозначен атом азота -аминогруппы лизина. В предпочтительных примерах реализации С-концевой лизин двух пептидных мономеров представляет собой L-лизин. В альтернативных примерах реализации, один или более остатков лизина может быть D-лизином. В соответствии с настоящим изобретением пептидный фрагмент присоединен к концевой группе Y разделительного фрагмента. Разделительный фрагмент может быть присоединен как к С-концевой группе, так и к N-концевой группе пептида. Таким образом, в тех примерах реализации, в которых разделительный фрагмент присоединен к С-концевой группе пептида, Y представляет собой NH. В тех же при-4 010015 мерах реализации, в которых разделительный фрагмент присоединен к N-концевой группе пептида, Y представляет собой CO. В альтернативных предпочтительных примерах реализации, разделительный фрагмент, предлагаемый в соответствии с настоящим изобретением, в котором Y представляет собойNH, присоединен при помощи амидной связи к -аминогруппе С-концевой группы лизинового остатка пептидного мономера. Связующий фрагмент (линкер). В другом предпочтительном примере реализации разделительный фрагмент, предлагаемый в соответствии с настоящим изобретением, присоединен к пептиду в виде части трифункционального связующего фрагмента (описанного ниже). В этом примере реализации единица Y разделительного фрагмента представляет собой CO, и Y разделительного фрагмента образует амидную связь с атомом N трифункционального связующего фрагмента. В одном особенно предпочтительном примере реализации настоящего изобретения связующий фрагмент представляет собой трифункциональный связующий фрагмент, имеющий следующее строение: где каждый из коэффициентовипредставляет собой целое число, значения которых выбирают независимо друг от друга, N ковалентно связан с Y связующего фрагмента, a Y связующего фрагмента представляет собой CO. В предпочтительных примерах реализации- это целое число, 16; и- это целое число, 16. В одном особенно предпочтительном примере реализации ==1. Разделительный фрагмент может быть введен в пептид во время синтеза указанного пептида. Например, если разделительный фрагмент содержит свободную аминогруппу и вторую функциональную группу (например, карбоксильную группу или аминогруппу), которая может быть связана с другим молекулярным фрагментом, то разделительный фрагмент может быть присоединен к твердой подложке. После этого, пептид может быть синтезирован непосредственно на свободной аминогруппе разделительного фрагмента при помощи стандартных методик твердофазного синтеза. В предпочтительном примере реализации разделительный фрагмент, содержащий две функциональные группы, сначала присоединяют к твердой подложке при помощи первой функциональной группы. Если необходимо синтезировать димерный пептид, то возможен следующий путь: связующий фрагмент Lk, содержащий две или более функциональные группы, которые могут служить исходными точками для синтеза пептида, и дополнительную функциональную группу (например, карбоксильную группу или аминогруппу), которая может быть связана с другим молекулярным фрагментом, присоединяют к разделительному фрагменту, образуя связь между второй функциональной группой разделительного фрагмента и третьей функциональной группой связующего фрагмента. После этого, непосредственно на двух реакционноспособных азотных группах связующего фрагмента Lk могут быть синтезированы два пептидных мономера при помощи любой методики твердофазного синтеза. Например, разделительный фрагмент, присоединенный к твердой подложке и имеющий свободную аминогруппу, может быть введен в реакцию со свободной карбоксильной группой лизинового связующего фрагмента. В другом примере реализации разделительный фрагмент может быть присоединен к пептиду после синтеза пептида. Такое присоединение может быть осуществлено при помощи способов, хорошо известных в настоящей области техники. В одном из примеров реализации связующий фрагмент содержит по меньшей мере одну функциональную группу, способную связываться с целевой функциональной группой синтезируемого пептида. Например, разделительный фрагмент, имеющий свободную аминогруппу,может быть введен в реакцию с С-концевой карбоксильной группой пептида. Фрагмент водорастворимого полимера/ПЭГ. Фрагмент водорастворимого полимера, предлагаемый в соответствии с настоящим изобретением,включает (а) полиалкиленгликоль и его производные, включающие ПЭГ, мПЭГ, гомополимеры ПЭГ,гомополимеры полипропиленгликоля, сополимеры этиленгликоля с пропиленгликолем, причем указанные гомополимеры и сополимеры могут быть как незамещенными, так и замещенными с одного конца алкильной группой; (b) целлюлозу и производные целлюлозы, включая метилцеллюлозу и карбоксиметилцеллюлозу; (с) крахмал и декстрины, а также их производные; (d) декстран и производные декстрана,включая сульфат декстрана, поперечно сшитый декстрин и карбоксиметилдекстрин; (е) гепарин и фрагменты гепарина; (f) поливиниловый спирт и этиловые эфиры поливинилового спирта; (g) поливинилпирролидон; (h) а,b-поли[(2-гидроксиэтил)-DL-аспартамид]; и (i) полиоксиэтилированные высокомолекулярные спирты (полиолы), но не ограничен указанными соединениями. Указанные полимеры могут быть линейными, разветвленными или звездообразными, а их молекулярные массы могут изменяться в широком диапазоне. Предпочтительно фрагмент водорастворимого полимера представляет собой ПЭГ. Для применения в соответствии с настоящим изобретением предпочтительным ПЭГ является линейный ПЭГ с молекулярной массой, превышающей 20 кДа. Предпочтительно указанный ПЭГ имеет молекулярную массу приблизительно от 20 до 60 кДа. Более предпочтительно указанный ПЭГ имеет молекулярную массу приблизительно от 20 до 40 кДа. Наиболее предпочтительно указанный ПЭГ имеет молекулярную массу,-5 010015 приблизительно равную 20 кДа. Фрагмент водорастворимого полимера ковалентно связан с разделительным или связующим фрагментом. В одном из примеров реализации фрагмент ПЭГ связан с N-концевой группой разделительного фрагмента. Соединения, предлагаемые в соответствии с настоящим изобретением, могут включать несколько фрагментов водорастворимого полимера (предпочтительно, ПЭГ) (например, 2, 3, 4 или более), и по меньшей мере один из нескольких указанных фрагментов водорастворимого полимера связан через разделительный фрагмент. Если соединение включает более одного фрагмента водорастворимого полимера,то эти несколько фрагментов водорастворимого полимера могут быть одинаковыми или химически отличными друг от друга фрагментами (например, фрагменты ПЭГ с различными молекулярными массами). В одном из примеров реализации изобретения фрагмент водорастворимого полимера представляет собой димер и включает два мономерных ПЭГ, связанных через разделительный фрагмент. В некоторых случаях на степень ПЭГилирования (количество фрагментов ПЭГ, присоединенных к пептиду и/или общее количество пептидов, к которым присоединен ПЭГ) может влиять отношение количества молекул ПЭГ к количеству молекул пептидов в реакции присоединения ПЭГ (ПЭГилирования), а также общая концентрация каждого из указанных веществ в реакционной смеси. В общем, оптимальное отношение количества молекул ПЭГ к количеству молекул пептидов (с точки зрения эффективности реакции, чтобы не оставалось непрореагировавших молекул пептидов и/или ПЭГ) определяется такими факторами, как желаемая степень ПЭГилирования (например, моно-, ди-, три- и т.д.), молекулярная масса выбранного полимера, строение полимера - разветвленное или неразветвленное, и условия реакции конкретного способа присоединения. Существует несколько способов присоединения ПЭГ, доступных специалистам в настоящей области техники [см., например, Goodson et al., (1990) Bio/Technology 8:343 (PEGylation of interleukin-2 at its(присоединение ПЭГ к ЕРО-пептидам); патент США 5672662 (Полиэтиленгликоль и родственные полимеры, монозамещенные пропионовой или бутановой кислотами и их функциональные производные для биотехнологического применения); патент США 6077939 (присоединение ПЭГ к N-концевому углероду в пептиде); Veronese et al., (1985) Appl. Biochem. Biotechnol. 11:141-142 (PEGylation of Nterminal -carbon of a peptide with PEG-nitrophenylcarbonate ("PEG-NPC") or PEG-trichlorophenylcarbonate); и Veronese (2001) Biomaterials 22:405-417 (Обзорная статья по присоединению ПЭГ к пептидам и белкам)]. Например, ПЭГ может быть ковалентно связан с остатками аминокислот через реакционноспособную группу. Реакционноспособные группы - это группы, к которым может быть присоединена активированная молекула ПЭГ (например, свободная аминогруппа или карбоксильная группа). Например, остаткиN-концевых аминокислот и остатки лизина (K) содержат свободную аминогруппу; а остатки С-концевых аминокислот содержат свободную карбоксильную группу. В качестве реакционноспособных групп для присоединения ПЭГ могут быть использованы сульфгидрильные группы (например, находящиеся в остатках цистеина). Кроме того, были описаны способы введения активированных групп (например, гидразидных, альдегидных и ароматических аминогрупп) по С-концевой группе полипептида при помощи ферментов [Schwarz et al. (1990) Methods Enzymol. 184:160; Rose et al. (1991) Bioconjugate Chem. 2:154;Gaertner et al. (1994) J. Biol. Chem. 269:7224]. Например, молекулы ПЭГ могут быть присоединены к аминогруппам при помощи метоксилированного ПЭГ (мПЭГ), имеющего иные реакционноспособные фрагменты. Неограничивающие примеры таких реакционноспособных фрагментов включают сукцинимидилсукцинат (SS), сукцинимидилкарбонат (SC), мПЭГ-имидат, пара-нитрофенилкарбонат (NPC), сукцинимидилпропионат (SPA) и циануридхлорид. Неограничивающие примеры таких мПЭГ включают мПЭГ-сукцинимидилсукцинат (mPEGSS), мПЭГ 2-сукцинимидилсукцинат (mPEG2-SS); мПЭГ-сукцинимидилкарбонат (mPEG-SC), мПЭГ 2 сукцинимидилкарбонат (mPEG2-SC); мПЭГ-имидат, мПЭГ-пара-нитрофенилкарбонат (mPEG-NPC),мПЭГ-имидат; мПЭГ 2-пара-нитрофенилкарбонат (mPEG2-NPC); мПЭГ-сукцинимидилпропионат (mPEGSPA); мПЭГ 2-сукцинимидилпропионат (mPEG2-SPA); мПЭГ-N-гидроксисукцинимид (mPEG-NHS); мПЭГ 2-N-гидроксисукцинимид (mPEG2-NHS); мПЭГ-циануридхлорид; мПЭГ 2-циануридхлорид; mPEG2Lysinol-NPC и mPEG2-Lys-NHS. Если присоединение ПЭГ неспецифично, а требуется пептид, содержащий специфично присоединенный ПЭГ, то требуемое соединение, к которому присоединен ПЭГ, выделяют очисткой из всей смеси соединений, к которым присоединен ПЭГ. Например, если необходимо получить пептид, в котором ПЭГ присоединен по концевому атому N, то форма с ПЭГ, присоединенным по концевому атому N, может быть выделена очисткой из множества пептидов со случайным присоединением ПЭГ (т.е. отделяют нужный фрагмент от других фрагментов с моноприсоединением ПЭГ).-6 010015 В некоторых примерах реализации ПЭГ присоединяют сайт-специфическим образом к пептиду или разделительному фрагменту. Ранее сайт-специфическое присоединение ПЭГ по N-концевой группе, боковой цепи и С-концевой группе потенциального аналога фактора высвобождения гормона роста осуществляли при помощи твердофазного синтеза [Felix et al. (1995) Int. J. Peptide Protein Res. 46:253]. Другой сайт-специфический способ включает присоединение пептида к крайним участкам цепочек ПЭГ, привитых на поверхность липосом сайт-специфическим способом при помощи реакционноспособной альдегидной группы на N-концевом участке, полученной при окислении N-концевого треонина периодатом натрия [Zalipsky et al. (1995) Bioconj. Chem. 6:705]. Однако этот способ ограничен полипептидами, содержащими N-концевые остатки серина или треонина. В одном из способов селективное N-концевое присоединение ПЭГ может быть осуществлено посредством восстановительного алкилирования, в основе которого лежит различная реакционная способность различных типов первичных аминогрупп (лизина относительно N-концевой группы), находящихся в конкретном фрагменте пептида или разделительном фрагменте и доступных для проведения реакций с образованием новых производных. В соответствующих реакционных условиях карбонильную группу,содержащую ПЭГ, селективно присоединяют к N-концевой группе пептида или разделительного фрагмента. Например, селективное присоединение ПЭГ к протеину по N-концевой группе осуществимо при помощи проведения реакции при таких значениях pH, которые выбирают в зависимости от разницы между pKa -аминогруппы лизинового остатка и -аминогруппы N-концевого остатка пептида или разделительного фрагмента. При таком селективном присоединении ПЭГ главным образом присоединяется поN-концевой группе протеина, практически не затрагивая других реакционноспособных групп (например,аминогруппы боковой цепочки лизина). Для того чтобы можно было применять восстановительное алкилирование, ПЭГ должен содержать только одну реакционноспособную альдегидную группу, способную присоединяться к протеину (например, может быть применен ПЭГ-пропиональдегид). Сайт-специфический мутагенез представляет собой еще один подход, который можно использовать для приготовления пептидов, применяемых для сайт-специфического присоединения полимеров. При помощи этого способа в пептиде создают последовательность аминокислот, пригодную для введения подходящей реакционноспособной группы в нужное положение внутри пептида. Например, в заявкеWO 90/12874 описано сайт-специфическое присоединение ПЭГ к белкам, модифицированным введением остатков цистеина или замещением других остатков остатками цистеина. В указанной публикации также описано приготовление мПЭГ-эритропоетина (мПЭГ-ЭПО) по реакции цистеин-специфического производного мПЭГ с остатком цистеина, введенным рекомбинантным способом и находящимся на ЭПО. Если фрагмент ПЭГ нужно присоединить к разделительному фрагменту или связующему фрагменту, то могут быть использованы аналогичные способы присоединения. В этом случае, разделительный или связующий фрагмент содержит реакционноспособную группу, а для осуществления ковалентного присоединения используют активированную молекулу ПЭГ, содержащую соответствующую комплементарную реакционноспособную группу. В предпочтительных примерах реализации реакционноспособная группа разделительного или связующего фрагмента - это концевая реакционноспособная группа (т.е. находящаяся на конечном (концевом) участке разделительного или связующего фрагмента). Пептиды, димеры пептидов и другие молекулы, полученные на основе пептидов, предлагаемые в соответствии с настоящим изобретением, могут быть присоединены к водорастворимым полимерам (например, ПЭГ) при помощи любой химической реакции, применяемой для присоединения водорастворимого полимера (полимеров) к участку молекулы, связывающему рецептор (receptor-binding portion) (например, пептид + разделительный фрагмент). В типичных примерах реализации для ковалентного присоединения водорастворимого полимера (полимеров) к участку молекулы, связывающему рецептор,применяют присоединение по одиночному центру (single attachment junction); однако в альтернативных примерах реализации может быть применено присоединение по множественным центрам (multiple attachment junctions), включающее дополнительные изменения, при которых различные типы водорастворимого полимера присоединяют к участку молекулы, связывающему рецептор, на определенных центрах для присоединения, которые могут включать центр (центры) для ковалентного присоединения разделительного фрагмента и/или к одной или обеим пептидным цепочкам. В некоторых примерах реализации димер или мультимер более высокого порядка включает различные виды пептидных цепочек (т.е. гетеродимер или иной гетеромультимер). В качестве неограничивающего примера можно назвать димер,который может включать первую пептидную цепочку, содержащую центр для присоединения ПЭГ, а вторая пептидная цепочка может либо вовсе не содержать центров для присоединения ПЭГ, или подвергаться другим реакциям присоединения, отличным от реакций, характерных для первой цепочки; а в некоторых альтернативных примерах разделительный фрагмент может содержать или не содержать центр для присоединения ПЭГ, причем указанный разделительный фрагмент, при проведении реакции присоединения ПЭГ, может подвергаться другим реакциям присоединения, отличным от реакций, характерных для первой и/или второй пептидной цепочки. В альтернативном примере реализации применяют ПЭГ,присоединенный к разделительному фрагменту участка молекулы, связывающего рецептор, и другой водорастворимый полимер (например, углевод), присоединенный к боковой цепи одной из аминокислот пептидной части молекулы.-7 010015 Для присоединения ПЭГ к участку молекулы, связывающему рецептор (пептиды + разделительный фрагмент), может быть использован ряд различных типов полиэтиленгликоля (ПЭГ). По существу, может быть применен любой подходящий реакционноспособный ПЭГ-реагент. В предпочтительных примерах реализации реакция реакционноспособного ПЭГ-реагента приводит к образованию карбаматной или амидной связи при присоединении реагента к участку молекулы, связывающего рецептор. Неограничивающие примеры подходящих реакционноспособных типов ПЭГ включают коммерчески доступные соединения, перечисленные в каталоге Drug Delivery Systems catalog (2003) Корпорации NOF (Yebisucatalog (2003), предоставляемом Nektar Therapeutics (490 Discovery Drive, Huntsville, Alabama 35806). В качестве неограничивающих примеров можно назвать следующие ПЭГ-реагенты, часто применяемые в различных примерах реализации: mPEG2-NHS, mPEG2-ALD, multi-Arm PEG, mPEG(MAL)2,mPEG2(MAL), mPEG-NH2, mPEG-SPA, mPEG-SBA, мПЭГ-тиоэфиры (mPEG-thioesters), мПЭГ-двойные сложные эфиры (mPEG-Double Esters), mPEG-BTC, mPEG-ButyrALD, mPEG-ACET, гетерофункциональные PEG (NH2-PEG-COOH, Boc-PEG-NHS, Fmoc-PEG-NHS, NHS-PEG-VS, NHS-PEG-MAL), ПЭГакрилаты (ACRL-PEG-NHS); ПЭГ-фосфолипиды (например, mPEG-DSPE), многолучевые (multiarmed)PEG серии SUNBRITE, включающие ПЭГ серии GL, полученные на основе глицерина и активированные при помощи химических реакций, выбираемых специалистами в данной области техники; любые из активированных ПЭГ серии SUNBRITE (неограничивающие примеры которых включают различные карбоксил-ПЭГ, p-NP-PEGs, Tresyl-PEGs, альдегидные ПЭГ, различные ацеталь-ПЭГ, различные аминоПЭГ, различные тиол-ПЭГ, различные малеимидо-ПЭГ, гидроксил-ПЭГ-амин, амино-ПЭГ-COOH, гидроксил-ПЭГ-альдегид, ПЭГ типа ангидрида карбоновой кислоты, функционализированные ПЭГфосфолипиды и другие подобные и/или подходящие реакционноспособные ПЭГ, которые могут быть выбраны специалистами в данной области техники с целью конкретного применения и использования). Пептидный фрагмент. В качестве пептидного фрагмента могут быть использованы любые пептиды, полученные из организмов различных животных, включая человека, микроорганизмов или растений, а также пептиды, полученные при помощи генной инженерии, или синтетические пептиды. Примеры таких пептидов включают пептиды, связывающие EPO-R; пептиды, связывающие TPO-R, цитокины, такие как различные интерфероны (например, интерферон-, интерферон-, интерферон-); интерлейкин-2 и интерлейкин-3; гормоны,такие как инсулин, фактор высвобождения гормона роста (GRF); кальцитонин; пептид, связанный с геном кальцитонина (calcitonin gene related peptide) (CGRP); натрийуретический пептид предсердия (ANP); вазопрессин; фактор высвобождения кортикотропина (CRF); вазоактивный пептид кишечника (VIP); секретин, гормон стимуляции -меланоцитов (-MSH); адренокортикотропный гормон (АСТН); холецитокинин (CCK); глюкагон; паратироидный гормон (РТН); соматостатин; эндотелин; вещество Р; динорфин; окситоцин и пептид высвобождения гормона роста [GHRP, см., например, Endocrinology, 114, 1537(1984)]; факторы роста, такие как гормон роста (GH), инсулиноподобный фактор роста (IGF-I, IGF-II),фактор роста нервов (-NGF), базовый (основной) фактор роста фибробластов (bFGF), трансформирующий фактор роста, эритропоетин, фактор, стимулирующий рост колонии гранулоцитов (G-CSF), фактор,стимулирующий рост гранулоцитарно-моноцитарно-макрофагальной колонии (granulocyte macrophagecolony-stimulating factor (GM-CSF, тромбоцитарный фактор роста (PDGF) и фактор роста эпидермиса(EGF); ферменты, такие как активатор плазминогена тканей (t-PA); эластаза, супероксиддисмутаза(ubiquitin); протеин активации островковых клеток поджелудочной железы (islet activating protein (IAP; тимический фактор сыворотки крови (serum thymic factor (STF; пептид-Т и ингибитор трипсина; а также производные указанных соединений. Предпочтительно пептидный фрагмент включает один или более пептидов, причем длина каждого пептида составляет менее 50 аминокислот, более предпочтительно приблизительно от 10 до 25 аминокислот и наиболее предпочтительно приблизительно от 12 до 18 аминокислот. В одном из предпочтительных примеров реализации пептидный фрагмент выбирают из пептидов,связывающих EPO-R, таких как, например, пептиды, описанные в патентах США 5773569, 5830851 и 5986047 Wrighton'ом, et al.; в публикации международной заявки WO 96/40749, Wrighton'ом, et al.; в патенте США 5767078 и публикации заявки WO 96/40772, Johnson'ом и Zivin'ым; заявке WO 01/38342, Balu; и в публикации заявки WO 01/91780, Smith-Swintosky, et al. Еще несколько примеров пептидов, связывающих EPO-R, которые могут быть использованы в качестве пептидного фрагмента, предлагаемого в соответствии с настоящим изобретением, описаны в предварительной патентной заявке CIIIA60/470245, поданной 12 мая 2003 г. Еще несколько примеров пептидов, связывающих EPO-R, которые могут быть использованы в качестве пептидного фрагмента, предлагаемого в соответствии с настоящим изобретением, описаны в предварительной патентной заявке США 60/469993, поданной 12 мая 2003 г. И еще несколько примеров пептидов, связывающих EPO-R, которые могут быть использованы в качестве пептидного фрагмента, предлагаемого в соответствии с настоящим изобретением, описаны в предварительной патентной заявке США 60/470244, поданной 12 мая 2003 г.-8 010015 В другом предпочтительном примере реализации пептидный фрагмент выбирают из пептидов, связывающих тромбопоетиновые рецепторы (thrombopoietin-receptors ("TPO-R". Неограничивающие примеры таких TPO-R связывающих пептидов включают примеры, описанные в патентах США 6552008,6506362, 6498155, 6465430, 6333031, 6251864, 6121238, 6083913, 5932546, 5869451, 5683983, 5677280,5668110 и 5654276; а также в опубликованных патентных заявках США 2003/0083361, 2003/0009018,2002/0177166 и 2002/0160013. В одном примере реализации пептидный фрагмент представляет собой мономерный пептид, содержащий в длину от 10 до 40 или более остатков аминокислот и имеющий последовательностьX3X4X5GPX6TWX7X8, в которой каждая аминокислота обозначена стандартным однобуквенным обозначением: Х 3 представляет собой С; Х 4 представляет собой R, H, L или W; X5 представляет собой M, F илиI; а Х 6 выбирают независимо от других аминокислот из 20 генетически закодированных (genetically encoded) L-аминокислот; Х 7 представляет собой D, Е, I, L или V; a X8 представляет собой С, который связывает и активирует эритропоетиновый рецептор (EPO-R), или, в другом случае, действует как агонист ЕРО. В другом примере реализации пептидный фрагмент представляет собой мономерный пептид, содержащий от 17 до приблизительно 40 аминокислот в длину, который включает следующую последовательность основной цепи аминокислот: LYACHMGPITX1CQPLR, в которой каждая аминокислота обозначена стандартным однобуквенным обозначением, a X1 представляет собой триптофан (W),1-нафтилаланин (1-nal) или 2-нафтилаланин (2-nal). В еще одном примере реализации пептидный фрагмент включает один или более TPO-R связывающих пептидов со следующей последовательностью аминокислот: Ac-Ile-Glu-Gly-Pro-Thr-Leu-ArgGln-Nal(l)-Leu-Ala-Ala-Arg-Sar или Ac-Ile-Glu-Gly-Pro-Thr-Leu-Arg-Gln-Trp-Leu-Ala-Ala-Arg-Sar. В одном из примеров реализации пептидный фрагмент непосредственно присоединен к разделительному фрагменту. В другом примере реализации пептидный фрагмент присоединен к разделительному фрагменту через связующий фрагмент. В соответствии с некоторыми примерами реализации настоящего изобретения два или более, предпочтительно приблизительно от двух до шести остатков аминокислот, независимо выбираемых из любых 20 генетически закодированных L-аминокислот или стереоизомерных им D-аминокислот, присоединяют к одному из концов или к обоим концам основной цепочки, описанной выше. Например, последовательность GG часто присоединяют к одному из концов или к обоим концам основной цепочки для облегчения синтеза пептида. В соответствии с настоящим изобретением также предложены конъюгаты указанных пептидов, а также их производные и пептидомиметики пептидов, сохраняющие способность связывать EPO-R. Стереоизомеры (например, D-аминокислоты) двадцати традиционно применяемых аминокислот,аминокислот неприродного происхождения, таких как ,-дизамещенные аминокислоты, Nалкиламинокислоты, молочная кислота и другие нетрадиционные аминокислоты, также могут быть использованы в качестве соединений, подходящих для настоящего изобретения. Неограничивающие примеры нетрадиционных аминокислот включают -аланин, 3-пиридил-аланин, 4-гидроксипролин, Офосфосерин, N-метилглицин, N-ацетилсерин, N-формилметионин, 3-метилгистидин, 5-гидроксилизин,норлейцин, 1- или 2-нафтилаланин, саркозин и другие подобные аминокислоты и иминокислоты. В предпочтительных примерах реализации пептидные фрагменты, предлагаемые в соответствии с настоящим изобретением, содержат внутримолекулярную дисульфидную связь между двумя остатками цистеина, находящимися в основной цепи аминокислот. Например или Димерные и олигомерные пептиды. В предпочтительном примере реализации мономерные пептидные фрагменты, предлагаемые в соответствии с настоящим изобретением, подвергают димеризации или олигомеризации с образованием димеров или олигомеров. Кроме того, такие димеры и другие мультимеры могут быть гетеродимерами или гетеромультимерами. В одном из примеров реализации, пептидные мономеры, предлагаемые в соответствии с настоящим изобретением, могут быть подвергнуты олигомеризации при помощи системы биотин/стрептавидин. Биотинилированные аналоги пептидных мономеров могут быть синтезированы обычными способами. Например, пептидные мономеры могут быть подвергнуты биотинилированию по С-концевому атому. Такие биотинилированные мономеры затем подвергают олигомеризации при помощи инкубации со стрептавидином [например, при молярном соотношении 4:1 при комнатной температуре в солевом растворе фосфатного буфера (PBS) или среде RPMI с буфером HEPES (Invitrogen) в течение 1 ч]. В модификации этого примере реализации биотинилированные пептидные мономеры затем могут быть подвергнуты олигомеризации при помощи инкубации с любым из коммерчески доступных антибиотиновых антител [например, антибиотин козы IgG, полученный в KirkegaardPerry Laboratories, Inc. (Washington,-9 010015DC)]. Связующие фрагменты (линкеры). В предпочтительных примерах реализации пептидные мономеры, предлагаемые в соответствии с настоящим изобретением, подвергают димеризации путем ковалентного присоединения по меньшей мере к одному связующему фрагменту. Связующий фрагмент (LK), предпочтительно, но не обязательно,представляет собой азотсодержащий связующий фрагмент. Наиболее предпочтительно связующий фрагмент представляет собой трифункциональный связующий фрагмент, возможно, имеющий в концевом положении одну или две связи -CO-. В предпочтительном примере реализации настоящего изобретения связующий фрагмент LK включает следующую структуру: где каждый из коэффициентовипредставляет собой целое число, значения которых выбирают независимо друг от друга, а N ковалентно связан с Y разделительного фрагмента, и Y разделительного фрагмента представляет собой CO. Наиболее предпочтительно связующий фрагмент связывает два пептидных мономера по С-концевым группам, путем одновременного присоединения к С-концевой аминокислоте каждого мономера. Например, если связующий фрагмент LK образует амидную связь с аминогруппой С-концевого остатка лизина первого и второго пептидного мономера, то димер может быть представлен структурной формулой III, и в обобщенном виде - формулой IV. В формуле IV символом N2 обозначен атом азота -аминогруппы лизина, и символом N1 обозначен атом азота -аминогруппы лизина. Для обозначения пептида, содержащего остаток лизина, в котором аминогруппа С-концевого остатка лизина связана со связующим фрагментом, может быть написана следующая димерная структура: [peptide-Lys]2-LK; для обозначения пептида, содержащего остаток лизина, в котором -аминогруппа С-концевого остатка лизина связана со связующим фрагментом, причем каждый пептид содержит внутримолекулярный дисульфидный мостик, может быть написана следующая димерная структура: [peptide-Lys, disulfide]2-LK; для обозначения пептида, содержащего остаток лизина, в котором -аминогруппа С-концевого остатка лизина связана со связующим фрагментом, причем каждый пептид содержит внутримолекулярный дисульфидный мостик, а молекула разделительного фрагмента образует ковалентную связь между С-концевой группой лизина и фрагментом ПЭГ, может быть написана следующая димерная структура: [peptide-Lys, disulfide]2-LK-Spacer-PEG, или для обозначения пептида,содержащего остаток лизина, в котором -аминогруппа С-концевого остатка лизина связана со связующим фрагментом, причем каждый пептид содержит внутримолекулярный дисульфидный мостик, а молекула разделительного фрагмента образует две ковалентные связи между С-концевой группой лизина и фрагментом ПЭГ, может быть написана следующая димерная структура: [peptide-Lys, disulfide]2-LKSpacer-PEG2. В другом предпочтительном примере реализации связующий фрагмент представляет собой остаток лизина или лизинамида (остаток лизина, в котором карбоксильная группа была превращена в амидный фрагмент CONH2). Более предпочтительно связующий фрагмент связывает С-концевые группы двух пептидных мономеров путем одновременного присоединения к С-концевым аминокислотам каждого мономера. Например, если С-концевый связующий фрагмент LK представляет собой лизинамид, то димер может быть представлен структурной формулой V, и в обобщенном виде - формулой VI. В формуле V символом N2 обозначен атом азота -аминогруппы лизина, а символом N1 обозначен атом азота -аминогруппы лизина. Для обозначения пептида, связанного как с -, так и с аминогруппами связующего фрагмента, включающего лизин, может быть написана следующая димерная структура: [peptide]2-LK; или для обозначения N-терминально ацетилированного пептида, связанного как с -, так и с -аминогруппами связующего фрагмента, включающего лизин, может быть написана следующая димерная структура: [Ac-peptide]2-LK; или для обозначения N-терминально ацетилированного пептида, связанного как с -, так и с -аминогруппами связующего фрагмента, включающего лизин, причем каждый из пептидов содержит внутримолекулярный дисульфидный мостик, может быть написана следующая димерная структура: [Ас-peptide, disulfide]2-LK; или для обозначения N-терминально ацетилированного пептида, связанного как с -, так и с -аминогруппами связующего фрагмента, включающего лизин, причем каждый из пептидов содержит внутримолекулярный дисульфидный мостик, а молекула разделительного фрагмента образует ковалентную связь между С-концевой группой лизина и фрагментом ПЭГ, может быть написана следующая димерная структура: [Ас-peptide, disulfide]2-LK-Spacer-PEG. Обычно, хотя и не обязательно, димеры пептидов, получаемые в соответствии с методиками, отличными от методик образования межмолекулярного дисульфидного мостика, также содержат одну или более дисульфидных связей между цистеиновыми остатками пептидных мономеров. Например, два таких мономера могут быть сшиты одним или более межмолекулярным дисульфидным мостиком. Предпочтительно два мономера содержат по меньшей мере одну внутримолекулярную дисульфидную связь. Наиболее предпочтительно оба мономера пептидного димера содержат внутримолекулярную дисульфидную связь, так что каждый мономер содержит циклический фрагмент. Модификация пептидов. Для получения других соединений, предлагаемых в соответствии с настоящим изобретением, может быть модифицирована концевая аминогруппа и/или карбоксильная группа пептидных соединений,предлагаемых в соответствии с настоящим изобретением. Модификация концевой аминогруппы включает метилирование (т.е. получение -NHCH3, -N(СН 3)2), ацетилирование (например, уксусной кислотой или ее галогенопроизводным, таким как -хлоруксусная кислота, -бромуксусная кислота или йодуксусная кислота), присоединение бензилоксикарбонильной группы (Cbz) или блокирование концевой аминогруппы при помощи любой блокирующей группы, содержащей карбоксилатную функциональную группу, определяемую как группа RCOO-, или сульфонильную функциональную группу, определяемую как группа R-SO2-, где R выбирают из группы, состоящей из алкила, арила, гетероарила, алкиларила и подобных им радикалов и групп. Для снижения восприимчивости к протеазам или для ограничения конформационных изменений пептидного соединения к N-концевой группе также может быть присоединена дезаминокислота (так что N-концевая аминогруппа будет при этом отсутствовать). В предпочтительных примерах реализации N-концевую группу ацетилируют. В наиболее предпочтительных примерах реализации N-концевую группу глицина ацетилируют, получая N-ацетилглицин (AcG). Модификация концевой карбоксильной группы включает замещение свободной кислоты карбоксамидной группой или образование циклического лактама на концевой карбоксильной группе с целью введения структурных ограничений. Пептиды, предлагаемые в соответствии с настоящим изобретением,также могут быть подвергнуты циклизации, или к концевым группам пепетидов могут быть присоединены дезаминирующие или декарбоксилирующие остатки, так что полученный пептид не содержит концевых амино- или карбоксильных групп, что снижает восприимчивость к протеазам или ограничивает конформационные изменения пептида. С-концевые функциональные группы соединений, предлагаемых в соответствии с настоящим изобретением, включают амидную группу, алкиламидную группу, образованную низшим алкилом, диалкиламидную группу, образованную низшим алкилом, алкоксильную группу,образованную низшим алкилом, гидроксигруппу, карбоксильную группу или ее сложноэфирную группу,образованную низшим алкилом, а также ее фармацевтически приемлемые соли. Боковые цепочки природного происхождения 20 генетически закодированных (genetically encoded) аминокислот (или стереоизомерных D-аминокислот) могут быть замещены другими боковыми цепочками, например, такими группами, как алкил, низший алкил, циклический алкил, содержащий 4, 5, 6 или 7 атомов в цикле, амидной группой, алкиламидной группой, образованной низшим алкилом, диалкила- 11010015 мидной группой, образованной низшим алкилом, алкоксильной группой, образованной низшим алкилом,гидроксигруппой, карбоксильной группой и ее сложноэфирной группой, образованной низшим алкилом,или 4-, 5-, 6- или 7-членным гетероциклом. В частности, могут быть использованы аналоги пролина, в которых 5-членный циклический фрагмент пролинового остатка замещен 4-, 6- или 7-членным циклом. Циклические группы могут быть как насыщенными, так и ненасыщенными, а ненасыщенные группы могут быть как ароматическими, так и неароматическими. Гетероциклические группы, предпочтительно,содержат один или более гетероатомов азота, кислорода и/или серы. Примеры таких групп включают следующие радикалы: фуразанил, фурил, имидазолидинил, имидазолил, имидазолинил, изотиазолил,изоксазолил, морфолинил (например, морфолино), оксазолил, пиперазинил (например, 1-пиперазинил),пиперидил (например, 1-пиперидил, пиперидино), пиранил, пиразинил, пиразолидинил, пиразолинил,пиразолил, пиридазинил, пиридил, пиримидинил, пирролидинил (например, 1-пирролидинил), пирролинил, пирролил, тиадиазолил, тиазолил, тиенил, тиоморфолинил (например, тиоморфолино) и тиазолил. Указанные гетероциклические группы могут быть как замещенными, так и незамещенными. Если группа имеет заместители, то такими заместителями могут быть следующие радикалы: алкил, алкоксильный радикал, галоген, кислород или замещенный или незамещенный фенил. Пептидные фрагменты также могут быть легко модифицированы посредством фосфорилирования и других способов [например, описанных Hruby et al., в (1990) Biochem. J. 268:249-262]. Пептидные фрагменты, предлагаемые в соответствии с настоящим изобретением, также могут представлять собой модели для непептидных соединений, имеющих аналогичную биологическую активность. Специалистам в данной области техники должно быть известно, что существует множество способов создания соединений с такой же, как и у начального (lead) пептидного соединения биологической активностью или аналогичной желаемой биологической активностью, но с улучшенными относительно этого соединения качествами, такими как растворимость, стабильность и подверженность гидролизу и протеолизу [см., Morgan and Gainor (1989) Ann. Rep. Med. Chem. 24:243-252]. Указанные способы включают замещение основной цепи пептида основной цепью, составленной из фосфонатов, амидатов, карбаматов,сульфонамидов, вторичных аминов и N-метиламинокислот. Фармацевтические композиции. В соответствии с еще одной целью настоящего изобретения предложены фармацевтические композиции на основе вышеописанных пептидов, модифицированных ПЭГ. Симптомы, которые могут быть смягчены или устранены при приеме таких композиций, включают симптомы, описанные выше. Такие фармацевтические композиции могут быть назначены для следующих типов приема: перорального, парентерального (внутримышечных, внутрибрюшных, внутривенных (ВВ) или подкожных инъекций),трансдермального (как пассивного, так и с использованием ионтофореза или электрофореза (electroporation, через слизистую оболочку (носовую, вагинальную, ректальную или подъязычную), или при помощи биоразрушаемых вставных элементов (bioerodible inserts), и могут быть приготовлены в виде лекарственных форм, приемлемых для соответствующего способа приема. В общем, изобретение охватывает фармацевтические композиции, включающие эффективные количества терапевтически активных пептидов (например, пептидов, способных связывать EPO-R), вместе с фармацевтически приемлемыми разбавителями, консервантами, солюбилизаторами, эмульсификаторами, вспомогательными средствами и/или носителями. Такие композиции включают разбавители с различными содержаниями буферного вещества(например, трис-HCl, ацетат, фосфат), с разными pH и ионной силой; добавки, такие как детергенты и солюбилизирующие агенты (например, Tween 80, Polysorbate 80); антиоксиданты (например, аскорбиновую кислоту, метабисульфит натрия); консерванты (например, Thimersol, бензиловый спирт) и наполнители (например, лактоза, маннит); также они включают введение материала в порошковые рецептуры полимерных соединений, таких как полимолочная кислота, полигликолевая кислота и т.д., или в липосомы. Также может быть использована гилауроновая кислота. Такие композиции могут влиять на физическое состояние, стабильность, скорость высвобождения in vivo и скорость клиренса in vivo указанных протеинов и их производных. См., например, издание Remington's Pharmaceutical Sciences, 18th Ed. (1990,Mack Publishing Co., Easton, PA 18042) с.1435-1712, включенное в настоящее описание по ссылке. Указанные композиции могут быть приготовлены в жидкой форме или могут находиться в виде сухого порошка (например, в лиофилизированной форме). Пероральный прием. Настоящее изобретение охватывает применение твердых лекарственных форм для перорального приема, которые, в основном, описаны в издании Remington's Pharmaceutical Sciences, 18th Ed. (1990,Mack Publishing Co., Easton, PA 18042), глава 89, включенном в настоящее описание по ссылке. Твердые лекарственные формы включают таблетки, капсулы, пилюли, драже или лепешки, облатки, подушечки,порошки или гранулы. Также для изготовления предлагаемых композиций может быть применено введение их в липосомную или протеноидную капсулу (как, например, введение в протеноидные микросферы, описанное в патенте США 4925673). Описание возможных твердых лекарственных форм, применяемых для терапевтических целей, имеется в публикации Marshall, K., находящейся в ModernPharmaceutics, изданной G.S. Banker и С.Т. Rhodes, Часть 10, 1979, включенной в настоящее описание по ссылке. В целом, рецептуры включают пептиды - агонисты EPO-R (или их химически модифицирован- 12010015 ные формы) и инертные ингредиенты, которые позволяют защищать активные ингредиенты от желудочных сред и высвобождать биологически активные материалы только в кишечнике. Настоящее изобретение также охватывает применение жидких лекарственных форм, пригодных для перорального приема, включающих фармацевтически приемлемые эмульсии, растворы, суспензии и сиропы, которые могут содержать и другие компоненты, включающие инертные разбавители; вспомогательные вещества, такие как смачивающие агенты, эмульгирующие и суспензирующие агенты; а также подсластители, вкусовые вещества и ароматизирующие вещества. Пептиды могут быть химически модифицированы таким образом, что пероральный прием производного производит необходимый эффект. В общем случае, химическую модификацию определяют как присоединение по меньшей мере одного фрагмента к самой молекуле компонента, причем указанный фрагмент позволяет достичь следующего действия: (а) замедления протеолиза; и (b) поступления в кровоток из желудка или кишечника. Желательно также обеспечить повышенную стабильность компонента или компонентов и увеличить время их циркуляции в организме. Как уже было сказано выше, предпочтительной химической модификацией для фармацевтического применения является присоединение ПЭГ(ПЭГилирование). Также могут быть введены и другие фрагменты, которые включают пропиленгликоль,сополимеры этиленгликоля и пропиленгликоля, карбоксиметилцеллюлоза, декстран, жирные кислоты[см., например, Abuchowski and Davis (1981) "Soluble Polymer-Enzyme Adducts", Enzymes and Drugs. Hocenberg and Roberts, eds. (Wiley-Interscience: New York, NY), и Newmark et al., (1982) J. Appl. Biochem. 4:185-189]. Для композиций, принимаемых перорально, областью высвобождения лекарственного средства может быть желудок, тонкий кишечник (двенадцатиперстная кишка, тощая кишка (jejunem) или подвздошная кишка) или толстый кишечник. Специалистам в данной области техники известны композиции, которые не растворяются в желудке, однако, высвобождают содержащийся в них материал в двенадцатиперстной кишке или в другой области тонкого кишечника. Предпочтительно высвобожденный материал не попадает в область разрушительного действия желудочных сред, что происходит либо благодаря защите пептида (или его производного), либо за счет высвобождения пептида (или его производного) за пределами желудка, например в кишечнике. Для обеспечения полной защиты от воздействия желудочных сред, на лекарственную форму необходимо нанести покрытие, непроницаемое, по меньшей мере, при значениях pH, достигающих 5,0. Примерами наиболее часто применяемых инертных ингредиентов, которые используют в качестве кишечнорастворимых покрытий, являются целлюлозы ацетата тримеллитат (CAT), гидроксипропилметилцеллюлозы фталат (НРМСР), НРМСР 50, НРМСР 55, поливинилацетата фталат (PVAP), Eudragit L30D, Aquateric, целлюлозы ацетата фталат (CAP), Eudragit L, Eudragit S и шеллак (Shellac). Указанные покрытия могут быть использованы в виде смешанных пленок. На таблетки также может быть нанесено покрытие или смесь покрытий, которые применяют с иной целью, нежели для защиты от желудочных сред. Такие покрытия могут включать покрытие сахаром или покрытия, облегчающие глотание таблетки. Капсулы могут состоять из твердой оболочки (такой, как желатин) для доставки сухого терапевтического средства (т.е. порошка), в то время как для жидких форм может быть использована оболочка из мягкого желатина. Материалом оболочки для облаток может быть плотный крахмал или другой пищевой бумажный материал. Для изготовления пилюль, лепешек, прессованных таблеток и таблеточных порошков подходят большинство методик обработки в массе. Пептид (или его производное) может быть введен в композицию в виде мелких отдельных частиц в виде гранул или подушечек с размером частицы, приблизительно равным 1 мм. Композиция материала,применяемого для приема в виде капсул, также может находиться в виде порошка, слегка уплотненных гранул или даже таблеток. Такие терапевтические средства могут быть изготовлены при помощи прессования. В композиции также могут быть включены красящие вещества и/или вкусовые вещества. Например, пептид (или его производное) могут быть включены в композицию (например, при помощи введения в липосомную капсулу или микросферу), а затем помещены в пищевой продукт, такой как охлажденный напиток, содержащий красящие вещества и вкусовые вещества. Содержание пептида (или его производного) может быть увеличено или снижено за счет содержания инертного материала. Такие разбавители могут включать углеводы, в особенности маннит, лактозу, безводную лактозу, целлюлозу, сахарозу, модифицированные декстраны и крахмал. В качестве наполнителей могут быть также использованы определенные неорганические соли, включающие трифосфат кальция, карбонат магния и хлорид натрия. Примерами некоторых коммерчески доступных разбавителей являются Fast-Flo, Emdex, STA-Rx 1500, Emcompress и Avicell. При изготовлении твердой лекарственной формы, содержащей терапевтическую композицию, в нее также могут быть включены дезинтегрирующие агенты. Неограничивающие примеры материалов, используемых в качестве дезинтегрирующих агентов, включают крахмал, включая коммерчески доступный дезинтегрирующий материал, изготовленный на основе крахмала, Explotab. Также могут быть использо- 13010015 ваны натрий-крахмала гликолят, Amberlite, натрий-карбоксиметилцеллюлоза, ультрамилопектин, альгинат натрия, желатин, апельсиновая корка, кислая карбоксиметилцеллюлоза, природная губка и бетонит. В качестве дезинтегрирующих агентов также могут быть использованы нерастворимые катионообменные смолы. В качестве дезинтегрирующих агентов и связывающих веществ могут быть использованы порошкообразные природные полимеры (гумми), которые могут включать такие порошкообразные природные полимеры (гумми), как агар-агар, смолу Кагауа или трагакантовую камедь. Альгиновая кислота и ее натриевая соль также могут быть использованы в качестве дезинтегрирующих агентов. Для связывания пептида (или его производного) с образованием твердой таблетки могут быть использованы связывающие вещества, включающие материалы, полученные из природных продуктов, таких как акация, трагакант, крахмал и желатин. Другие связывающие вещества включают метилцеллюлозу (МЦ (МС, этилцеллюлозу (ЭЦ (ЕС и карбоксиметилцеллюлозу (КМЦ (CMC. Для получения гранул, содержащих пептид (или его производное), могут быть использованы поливинилпирролидон (PVP) и гидроксипропилметилцеллюлоза (НРМС) в спиртовом растворе. Для предотвращения слипания во время изготовления композиции пептида (или его производного) в композицию может быть включен антифрикционный агент. Для создания слоя между пептидом (или его производным) и стенкой прессформы могут быть использованы смазочные вещества, неограничивающие примеры которых могут включать стеариновую кислоту, включая ее магниевую и кальциевую соль, политетрафторэтилен (PTFE),жидкий парафин, растительные масла и воски. Также могут быть использованы растворимые смазочные вещества, такие как лаурилсульфат натрия, лаурилсульфат магния, полиэтиленгликоль с различной молекулярной массой, Carbowax 4000 и 6000. В композицию могут быть добавлены скользящие вещества, которые могут улучшать характеристики текучести медикамента во время изготовления композиции и способствовать его перераспределению во время прессования. Скользящие вещества могут включать крахмал, тальк, пирогенный оксид кремния и гидратированный кремнийалюминат. Для улучшения растворимости пептида (или его производного) в водных средах может быть добавлено поверхностно-активное вещество, используемое в качестве смачивающего агента. Поверхностноактивные вещества могут включать анионные детергенты, такие как лаурилсульфат натрия, сульфосукцинат диоктил-натрия и сульфонат диоктил-натрия. Могут быть использованы катионные детергенты,которые могут включать бензальконийхлорид (benzalkonium chloride) или бензэтомийхлорид (benzethomium chloride). Список потенциальных неионных детергентов, которые могут быть включены в композицию в качестве поверхностно-активных веществ, включает лауромакрогол (lauromacrogol) 40,полиоксилстеарат-40 (polyoxyl 40 stearate), полиокиэтилен-гидрированное касторовое масло 10, 50 и 60,моностеарат глицерина, полисорбат 40, 60, 65 и 80, сложный эфир жирной кислоты и сахарозы, метилцеллюлозу и карбокисметилцеллюлозу. Указанные поверхностно-активные вещества могут присутствовать в композиции протеина или его производного либо самостоятельно, либо в смеси компонентов в различных соотношениях. Добавками, которые потенциально могут усиливать всасывание пептида (или его производного),являются, например, жирные кислоты: олеиновая кислота, линолевая кислота и линоленовая кислота. Может возникнуть необходимость приготовления композиций для перорального приема с контролируемым высвобождением лекарственного средства. Пептид (или его производное) может быть введен в инертную матрицу, способную высвобождать вещество либо посредством диффузии, либо выщелачивания, например, в матрицу природного полимера (гумми). В композицию также могут быть включены медленно разлагаемые матрицы. Некоторые кишечно-растворимые покрытия также обладают свойством замедленного высвобождения. Другим видом контролируемого высвобождения является способ, разработанный на основе терапевтической системы Oros (Alza Corp.), т.е. лекарственное средство заключают в полупроницаемую мембрану, в которую может поступать вода и выталкивать лекарственное средство через небольшое единичное отверстие в мембране за счет осмотического эффекта. Для приготовления композиции могут быть использованы и другие покрытия. Эти покрытия включают разнообразные сахара, которые могут быть нанесены в чане для нанесения покрытия. Пептид (или его производное) также может быть введен пациенту в таблетке с пленочным покрытием, и материалы,применяемые для этой цели, могут быть разделены на две группы. Первая группа состоит из кишечнонерастворимых (nonenteric) материалов и включает метилцеллюлозу, этилцеллюлозу, гидроксиэтилцеллюлозу, метилгидроксиэтилцеллюлозу, гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу,натрий-карбоксиметилцеллюлозу, провидон и полиэтиленгликоли. Вторая группа состоит из кишечнорастворимых (enteric) материалов, которые, в общем случае, представляют собой сложные эфиры фталевой кислоты. Для получения оптимального пленочного покрытия может быть использована смесь материалов. Нанесение пленочного покрытия может быть произведено в чане для нанесения покрытий, или в псевдоожиженном слое, или посредством прессовочного нанесения покрытия. Парентеральное введение. Препараты для парентерального введения, предлагаемые в соответствии с настоящим изобретением, включают стерильные водные и неводные растворы, суспензии или эмульсии. Примерами неводных- 14010015 растворителей или доставочных средств являются пропиленгликоль, полиэтиленгликоль, растительные масла, такие как оливковое масло и кукурузное масло, желатин и пригодные для введения органические сложные эфиры, такие как этилолеат. Указанные лекарственные формы также могут содержать вспомогательные вещества, такие как консерванты, смачивающие, эмульгирующие и диспергирующие агенты. Стерилизация таких форм может быть произведена, например, фильтрованием через фильтр, задерживающий бактерии, введением стерилизующих веществ в композиции, облучением композиций или их нагреванием. Они также могут быть изготовлены с использованием стерильной воды или другой стерильной и пригодной для введения жидкости непосредственно перед введением. Ректальное или вагинальное введение. Композиции для ректального или вагинального введения предпочтительно представляют собой суппозитории, которые, кроме активного вещества, могут содержать наполнители, такие как масло какао или суппозиторный воск. Композиции для введения через нос или для подъязычного введения также приготавливают, используя стандартные наполнители, хорошо известные в данной области техники. Внутрилегочное (пульмональное) введение. Настоящее изобретение также охватывает внутрилегочное введение пептидов - агонистов EPO-R(или их производных). Пептид (или его производное) вводят в легкие млекопитающего посредством вдоха, и далее он попадает в кровоток через эпителиальные ткани легких [см., например, Adjei et al. (1990)Debs et al. (1988) J. Immunol. 140:3482-3488 (интерферон- и фактор некроза опухоли ) и патент США 5284656, заявленный Platz et al. (фактор, стимулирующий рост гранулоцитарной колонии)]. Способ и композиция, применяемая для внутрилегочного введения лекарственных средств с целью достижения систематического эффекта, описаны в патенте США 5451569, заявленном Wong et al. Практическое применение настоящего изобретения охватывает широкий спектр механических устройств, предназначенных для внутрилегочного введения терапевтических продуктов; неограничивающие примеры таких устройств включают аэрозольные аппараты, ингаляторы, снабженные дозаторами, и ингаляторы порошкообразных средств; все вышеперечисленные устройства известны специалистам в данной области техники. Некоторые специфические примеры коммерчески доступных устройств, пригодных для применения настоящего изобретения, представляют собой аэрозольный аппарат Ultravent (Mallinckrodt Inc., St. Louis, МО); аэрозольный аппарат Acorn II (Marquest Medical Products, Englewood, CO); ингалятор Ventolin, снабженный дозатором (Glaxo Inc., Research Triangle Park, NC); и ингалятор порошкообразных средств Spinhaler (Fisons Corp., Bedford, MA). Для всех указанных устройств необходимо применение композиций, пригодных для распыления пептида (или его производного). Как правило, каждая композиция специфична для каждого применяемого устройства и может включать, наряду с обычными разбавителями, вспомогательным веществами и/или носителями, применяемыми для терапии, использование подходящего газа-вытеснителя. Настоящее изобретение также охватывает применение липосом, микрокапсул или микросфер, соединений включения или носителей других типов. Химически модифицированные пептиды также могут быть изготовлены в виде различных композиций, тип которых зависит от типа химической модификации или типа применяемого устройства. Композиции, пригодные для введения при помощи аэрозольного аппарата, как струйного, так и ультразвукового, обычно включают пептид (или его производное), растворенное в воде в концентрации приблизительно от 0,1 до 25 мг биологически активного протеина на 1 мл раствора. Композиция также может включать буферное вещество и простой сахар (например, для стабилизации протеина и регулировки осмотического давления). Композиции для аэрозольных аппаратов также могут содержать поверхностно-активное вещество для снижения или предотвращения поверхностной агрегации пептида (или его производного), вызываемой распылением раствора при образовании аэрозоля. Композиции, пригодные для применения в ингаляторах, снабженных дозатором, обычно включают мелко размолотый порошок, содержащий пептид (или его производное), суспензированный в газевытеснителе при помощи поверхностно-активного вещества. Газом-вытеснителем может служить любой обычно применяемый для этой цели материал, такой как хлорфторуглерод, хлороводородофторуглерод,фтороводородоуглерод или углеводород, включающий трихлорфторметан, дихлордифторметан, дихлортетрафторэтанол и 1,1,1,2-тетрафторэтан, или комбинация указанных соединений. Подходящие поверхностно-активные вещества включают сложный триэфир сорбита и олеиновой кислоты и соевый лецитин. В качестве поверхностно-активного вещества также может быть применена олеиновая кислота. Композиции, пригодные для введения через ингалятор для порошкообразного материала, включают мелко размолотый сухой порошок, содержащий пептид (или его производное), который также может включать наполнитель, такой как лактоза, сорбит, сахароза или маннит в количествах, которые способст- 15010015 вуют рассеиванию порошка из устройства, например от 50 до 90 мас.% от массы композиции. Для наиболее эффективного ввода в периферическую часть легкого, лучше всего, если пептид (или его производное) приготовлен в виде частиц со средним размером менее 10 мм (или микрон), более предпочтительно от 0,5 до 5 мм. Введение через носовые отверстия. Настоящее изобретение также охватывает введение пептидов - агонистов EPO-R (или их производных) через носовые отверстия. Введение через носовые отверстия позволяет пептиду проникать непосредственно в кровоток сразу после введения терапевтического продукта в нос, что позволяет избежать осаждения продукта в легких. Композиции, предназначенные для введения через носовые отверстия,включают композиции, содержащие декстран и циклодекстран. Дозировка. По мере проведения дальнейших исследований будет предоставлена информация относительно уровня дозы, подходящего для каждого пептида, в зависимости от различных симптомов у конкретного пациента; таким образом, обычный специалист в данной области техники после рассмотрения истории болезни (therapeutic context), возраста и общего состояния здоровья пациента сможет назначить нужный уровень дозы. Выбранная дозировка зависит от желаемого терапевтического эффекта, способа введения средства и желаемой продолжительности лечения. Обычно млекопитающим вводят суточные дозы, составляющие от 0,001 до 10 мг на кг массы тела. Обычно для внутривенного введения или вливания дозы могут быть меньше. График приема лекарственного средства может изменяться в зависимости от времени полужизни в кровеносной системе и типа применяемой композиции. Пептиды (или их производные), предлагаемые в соответствии с настоящим изобретением, могут быть введены в сочетании с одним или более дополнительных активных ингредиентов или фармацевтических композиций. Примеры. Ниже рассмотрены иллюстративные, не ограничивающие примеры реализации изобретения. Пример 1. Синтез молекулы H-TAP-Boc. Стадия А. Синтез Cbz-TAP.(ДХМ (DCM охладили до 0 С. К раствору ТАР медленно, в течение 6-7 ч, добавили через капельную воронку раствор бензилхлороформата (Cbz-Cl, Cbz = карбоксибензилокси) (4,82 мл, 33,7 ммоль) в безводном ДХМ (50 мл), поддерживая температуру реакционной смеси 0 С в течение всего прибавления. Полученной смеси затем дали нагреться до комнатной температуры (25 С). Спустя еще 16 ч ДХМ отогнали в вакууме, и остаток разделяли в смеси 3 н. HCl и эфира. Водные слои собирали и нейтрализовали 50% водным NaOH до pH 8-9 и экстрагировали этилацетатом. Слой этилацетата сушили безводнымNa2SO4 и затем концентрировали в вакууме, получая сырой моно-Cbz-ТАР (5 г, приблизительно 50% выход). Это соединение использовали далее для выполнения стадии В без очистки. Стадия В. Синтез Cbz-TAP-Boc.Boc2O (3,86 г, 17,7 ммоль, Boc = трет-бутоксикарбонил) добавили к сильно перемешиваемой суспензии Cbz-TAP (5 г, 17,7 ммоль) в гексане (25 мл). Перемешивание продолжали в течение ночи при комнатной температуре. Реакционную смесь разбавили ДХМ (25 мл) и промывали 10% водной лимонной кислотой (2 раза), водой (2 раза) и солевым раствором. Органический слой сушили безводным Na2SO4 и концентрировали в вакууме. Сырой продукт (выход 5 г) далее вводили непосредственно в реакцию на стадии С. Сырой Cbz-TAP-Boc, полученный на стадии В, растворили в метаноле (25 мл) и гидрировали в присутствии 5% Pd на угле (5% мас./мас.) в колбе под давлением в течение 16 ч. Смесь профильтровали,промыли метанолом и концентрировали фильтрат в вакууме, получая сырой продукт H-TAP-Boc (выход 3,7 г). Общий выход операций А-С составил приблизительно 44% (рассчитано исходя из количества использованного Cbz-Cl). Пример 2. Присоединение разделительного фрагмента к пептиду по С-концевой группе. На следующей схеме реакции показано присоединение разделительного фрагмента к пептиду по Сконцевой группе. Пептид со свободной С-концевой группой.H-TAP-Boc был приготовлен в соответствии с примером 1. DDC = N,N'-дициклогексилкарбодиимид. Пример 3. Присоединение разделительного фрагмента к пептиду со свободной кислотой в боковой цепи. На следующей схеме реакции показано присоединение разделительного фрагмента к пептиду со свободной кислотой в боковой цепи. Пептид со свободной кислотой в боковой цепи.TFA = трифторуксусная кислота (ТФУ). Пример 4. Присоединение ПЭГ к пептиду при помощи мПЭГ-NPC. Пептид с ТАР на С-концевой группе. Пример 5. Присоединение ПЭГ к пептиду при помощи мПЭГ-SPA. Пептид с ТАР на С-концевой группе. Пример 6. Присоединение разделительного фрагмента и синтез пептида. На следующей схеме реакции показано присоединение разделительного фрагмента к твердой подложке и синтез пептида на указанной твердой подложке Пример 7. Синтез пептидного димера с разделительным фрагментом, присоединенным к смоле. Стадия А. Синтез участка TentaGel-связующий фрагмент.TentaGel бромид (2,5 г, 0,48 ммоль/г, предоставленный Rapp Polymere, Германия), фенольный связующий фрагмент (5 экв.) и K2CO3 (5 экв.) нагревали в 20 мл N,N-диметилформамиде (ДМФА (DMF при 70 С в течение 14 ч. После охлаждения до комнатной температуры смолу промыли (0,1 н. HCl, вода,ацетонитрил (AcCN), ДМФА, MeOH) и сушили, получая смолу янтарного цвета. 2,5 г смолы, полученной в реакции на стадии А (выше) и H-TAP-Boc (1,5 г, 5 экв.) и ледяная AcOH(34 л, 5 экв.) поместили в смесь 1:1 MeOH/тетрагидрофуран (ТГФ (THF и встряхивали в течение ночи. К смеси добавили 1 М раствор цианборгидрида натрия (5 экв.) в ТГФ и встряхивали еще 7 ч. Смолу отфильтровали, промыли (ДМФА, ТГФ, 0,1 н. HCl, вода, MeOH) и сушили. Небольшое количество смолы бензоилировали бензилхлоридом и диизопропилэтиламином (DIEA) в ДХМ, расщепляли 70%-ной трифторуксусной кислотой (ТФУ)-ДХМ и анализировали методами ЖХ/МС (LCMS, Liquid Chromatograpy(HPLC, жидкостной хроматографии высокого давления). Стадия С. Синтез участка TentaGel-связующий фрагмент-TAP-Lys. Смолу, полученную на стадии В (выше), обрабатывали активированным раствором FmocLys(Fmoc)-OH (Fmoc = 9-флуоренилметоксикарбонил, приготовленный из 5 экв. аминокислоты и 5 экв.HATU (N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)уронийгексафторфосфат), растворенного при концентрации 0,5 М в ДМФА с последующим прибавлением 10 экв. DIEA) и осторожно встряхивали в течение 14 ч. Смолу затем промыли (ДМФА, ТГФ, ДХМ, MeOH) и сушили с получением защищенной смолы. Оставшиеся аминогруппы были защищены обработкой смолы раствором 10% уксусного ангидрида, 10% пиридина в ДХМ в течение 20 мин с последующей промывкой как указано выше. ГруппыFmoc отщепляли, осторожно встряхивая смолу в 30% пиперидине в ДМФА в течение 20 мин с последующей промывкой (ДМФА, ТГФ, ДХМ, MeOH) и сушкой. Стадия D. Синтез участка TentaGel-связующий фрагмент-ТАР-Lys(пептид)2. Смолу, полученную на стадии С (выше), подвергали повторным циклам обработки Fmocаминокислотой с активацией HBTU/HOBt, с последующим удалением Fmoc пиперидином, синтезируя,таким образом, обе пептидные цепочки одновременно. Это удобно выполнять в автоматическом аппарате ABI 433 для синтеза пептидов, поставляемом Applied Biosystems, Inc. После окончательного удаленияFmoc, концевые аминогруппы подвергали ацилированию уксусным ангидридом (10 экв.) и DIEA- 19010015 Смолу, полученную на стадии D (выше), суспензировали в растворе ТФУ (TFA) (82,5%), фенола(5%), этандитиола (2,5%), воды (5%) и тиоанизола (5%) в течение 3 ч при комнатной температуре. Для отщепления также можно использовать другие отщепляющие смеси, например, ТФУ (95%), воду (2,5%) и триизопропилсилан (2,5%). Раствор ТФУ охладили до 5 С и вылили в Et2O для осаждения пептида,отфильтровали, сушили под уменьшенным давлением, получили искомый пептидный димер, связанный с разделительным фрагментом. После очистки с помощью препаративной жидкостной хроматографии высокого давления (HPLC) на колонке С 18 получили чистый пептидный димер, связанный с разделительным фрагментом. Стадия F. Окисление. Димерный пептид (связанный с разделительным фрагментом), содержащий восстановленные цистеиновые остатки, окисляли до димерного пептида, содержащего дисульфидные мостики. Димерный пептид растворяли в 20% ДМСО/вода (1 мг сухой массы пептида/мл) оставляли при комнатной температуре на 36 ч. Затем пептид очищали, загружая реакционную смесь в колонку С 18 для препаративной жидкостной хроматографии высокого давления (HPLC) (Waters Delta-Pack С 18, размер частиц 15 мкм, размер пор 300 , 40 мм 200 мм длиной) с последующим линейным градиентом(gradiant) AcCN/вода/0,01% ТФУ от 5 до 95% AcCN в течение 40 мин. Лиофилизация фракций, содержащих желаемый пептид, привела к получению пушистого белого твердого вещества. Пример 8. Присоединение ПЭГ к пептидному димеру с разделительным фрагментом при помощи мПЭГ-NPC. Например, Димерный пептид, присоединенный к разделительному фрагменту, смешивали с равным количеством (в молярном отношении) активированного ПЭГ (мПЭГ-NPC, изготавливаемый NOF Corp., Япония,- 20010015 поставляемый Nektar Therapeutic, США (ранее "Shearwater Corp." в сухом ДМФА с образованием прозрачного раствора. Через 5 мин к указанному раствору прибавили 4 экв. DIEA. Смесь перемешивали при комнатной температуре в течение 14 ч, затем очищали на колонке С 18 при помощи обращенно-фазовой жидкостной хроматографии высокого давления (HPLC). Строение пептида с присоединенным ПЭГ подтверждено масс-спектрометрией MALDI. мПЭГ-NPC имел следующее строение: Пример 9. Присоединение ПЭГ к пептидному димеру с разделительным фрагментом при помощи мПЭГ-SPA. Присоединение ПЭГ к пептидному димеру с разделительным фрагментом также может быть произведено при помощи мПЭГ-SPA. мПЭГ-SPA имеет следующее строение: Для идентификации ионообменных подложек, подходящих для очистки конъюгатов пептид - разделительный фрагмент - ПЭГ применяли образец, полученный в примере 8. Общая пропись была следующей. Ионообменную смолу (2-3 г) загружали в 1 см колонку, затем превращали в натриевую форму(0,2 н. NaOH загружали на колонку до тех пор, пока pH элюата не достигал 14), а затем превращали в водородную форму (промывали 0,1 н. HCl или 0,1 М НОАс до тех пор, пока pH загрузки не становился равным pH элюата), а затем промывали 25% AcCN/водой до pH 6. Затем в 25% AcCN/воде (10 мг/мл) растворяли либо сам пептид до образования конъюгата, либо конъюгат пептид-ПЭГ, и pH доводили до значения менее 3 трифторуксусной кислотой, а затем загружали в колонку в отдельных экспериментах. Колонку промывали 2-3 объемами 25% AcCN/воды, равными объему колонки, собирая фракции по 5 мл, а затем пептид выделяли из колонки элюированием 0,1 М NH4OAc в 25% AcCN/воде, снова собирая фракции по 5 мл. Фракции, содержащие искомый пептид, обнаруживали при помощи жидкостной хроматографии высокого давления (HPLC). Анализ при помощи испарительного детектора рассеяния света(ESLD) показал, что, если пептид задерживался в колонке и подвергался элюированию растворомNH4OAc (обычно между фракциями 4 и 10), то, согласно наблюдениям, он не содержал загрязнений пептида, не связанного с ПЭГ. Если пептид подвергался элюированию в исходный промывной буфер (обычно первые 2 фракции), не наблюдали разделения искомого ПЭГ-конъюгата и избытка ПЭГ. Ионообменные подложки выбирали на основании их способности отделять конъюгат пептид-ПЭГ от непрореагировавшего (или гидролизованного) ПЭГ, а также по их способности удерживать исходные димерные пептиды. В качестве подходящих ионообменных подложек были выбраны колонка, предварительно загруженная сильной катионообменной смолой Mono S HR 5/5 (Amersham Biosciences), подложка из микрогранулированной сильной катионообменной смолы SE53 Cellulose (Whatman) и подложка из сильной катионообменной смолы SP Sepharose Fast Flow (Amersham Biosciences). Пример 11. Синтез трифункциональных молекул на основе -аминокислот. Разветвленные трифункциональные молекулы, имеющие структуру,были синтезированы в соответствии со следующей схемой реакции:m и n - целые числа. Такие трифункциональные молекулы могут одновременно служить и связующим, и разделительным фрагментом. Пример 12. Синтез трифункциональных молекул на основе третичных амидов. Разветвленные трифункциональные молекулы, имеющие структуру,были синтезированы в соответствии со следующей схемой реакции:m и n - целые числа. Такие трифункциональные молекулы могут одновременно служить и связующим фрагментом были синтезированы в соответствии со следующей схемой реакции:m и n - целые числа. Такие гомотрифункциональные молекулы могут одновременно служить и связующим фрагментом была приготовлена в соответствии с примером 12. Указанную трифункциональную молекулу использовали для димеризации по С-концевой группе и присоединения ПЭГ в соответствии со следующей схемой реакции: Пример 15. Димеризация по N-концевой группе и присоединение ПЭГ с использованием трифункциональной молекулы. Трифункциональная молекула была приготовлена в соответствии со следующей схемой: К раствору Вос-Ala-ОН (10,0 г, 52,8 ммоль) (Boc= трет-бутоксикарбонил) и диэтилиминодиацетата (10,0 г, 52,8 ммоль) в 200 мл ДХМ при 0 С прибавили DCC (10,5 г, 50,9 ммоль) в течение 5 мин. Через 2 мин образовался белый осадок. Реакционную смесь оставили нагреваться до комнатной температуры и перемешивали в течение 24 ч. Мочевину отфильтровали через фильтр Шотта (средняя пористость), и растворитель отогнали под уменьшенным давлением. Остаток растворили в 500 мл EtOAc (EtOAc = этилацетат), отфильтровали, как описано выше, и переносили в делительную воронку. Органическую фазу промыли (насыщенный раствор NaHCO3, рассол, 1 н. HCl), сушили (MgSO4), фильтровали и сушили,получая бесцветное масло. В течение 10 мин масло застыло с образованием белых кристаллов. Сырой диэфир растворили в 75 мл ТГФ (ТГФ = тетрагидрофуран) и добавили 75 мл MeOH (MeOH метанол) и 50 мл воды. К полученному раствору прибавили раствор KOH (KOH - гидроксид калия) (8,6 г,153 ммоль) в 25 мл воды. Цвет реакционной смеси стал светло-желтым. После перемешивания в течение 12 ч (pH все время был 12), органический растворитель испарили на роторном испарителе, и полученную суспензию разделяли в смеси Et2O (Et2O = диэтиловый эфир) и насыщенным раствором NaHCO3. Объединенные водные растворы подкислили до pH 1, насытили NaCl и экстрагировали EtOAc. ФазуEtOAc промыли (рассол), сушили (MgSO4) и концентрировали, получая 13,97 г продукта в виде белого твердого вещества (выход 90,2% после 2 стадий). Примечания. Выход падает до 73%, если реакцию с DCC проводить в AcCN. При использовании DIC мочевина,являющаяся побочным продуктом, не может быть отделена от целевого продукта - единственный способ очистки - это хроматография; DCC мочевина может быть количественно извлечена без использования хроматографии. Реакция также хорошо проходит с водорастворимым карбодиимидом.AcCN прибавили DCC (1,36 г, 6,59 ммоль) в течение 5 мин. Немедленно выпал белый осадок. Реакционную смесь перемешивали 22 ч и отфильтровали для удаления DCC мочевины. Растворитель отогнали под уменьшенным давлением, остаток растворяли в EtOAc (250 мл) и переносили в делительную воронку. Органическую фазу промыли (насыщенный раствор NaHCO3, рассол, 1 н. HCl, рассол), сушили (MgSO4),и концентрировали, получая белое твердое вещество. Вещество растворили в 75 мл AcCN, отфильтровали и концентрировали, получая 1,28 г продукта в виде белого твердого вещества (выход 78,2%). Примечания. Выходы падают до 31% в ТГФ, до 68% в ДМФА (при использовании DIC вместо DCC) и до 57% в ДХМ/ДМФА. Исходная дикислота растворима в AcCN, таким образом, если при растворении перед добавлением DCC остается какой-либо осадок, его можно отфильтровать и отбросить. Эта трифункциональная молекула была использована для димеризации по N-концевой группе и присоединения ПЭГ в соответствии со следующей схемой реакции: Пример 16. Синтез мПЭГ 2-лизинол-NPC. Коммерчески доступный лизинол обрабатывали избытком мПЭГ 2, получая мПЭГ 2-лизинол. После этого мПЭГ 2-лизинол обрабатывали избытком NPC, получая мПЭГ 2-лизинол-NPC.- 25010015 Пример 17. Присоединение ПЭГ с использованием трифункциональной молекулы (фрагмент ПЭГ включает две линейные цепочки ПЭГ). В соответствии с примером 15 была приготовлена трифункциональная молекула, имеющая структуру Стадия 1. Присоединение трифункционального связующего фрагмента к пептидным мономерам. Для присоединения к связующему фрагменту, 2 экв. пептида смешивали с 1 экв. трифункционального связующего фрагмента в сухом ДМФА с образованием прозрачного раствора, и через 2 мин добавляли 5 экв. DIEA. Смесь перемешивали при комнатной температуре в течение 14 ч. Растворитель удаляли под уменьшенным давлением, и сырой продукт растворяли в 80% ТФУ в ДХМ в течение 30 мин для удаления группы Boc, а затем очищали на колонке С 18 при помощи обращенно-фазовой жидкостной хроматографии высокого давления. Строение димера было подтверждено электрораспылительной массспектрометрией. Реакция присоединения происходит с присоединением связующего фрагмента к атому азота -аминогруппы лизинового остатка каждого мономера. Стадия 4. Присоединение ПЭГ к пептидному димеру. Присоединение ПЭГ через карбаматную связь. Пептидный димер и молекулы, содержащие ПЭГ (мПЭГ 2-лизинол-NPC) смешивали в молярном соотношении 1:2 в сухом ДМФА, получая прозрачный раствор. Спустя 5 мин к указанному раствору до- 26010015 бавляют 4 экв. DIEA. Смесь перемешивали при комнатной температуре в течение 14 ч, а затем очищали на колонке С 18 при помощи обращенно-фазовой жидкостной хроматографии высокого давления. Строение пептида с присоединенным ПЭГ подтверждено масс-спектрометрией MALDI. Очищенный пептид,кроме того, подвергали очистке посредством ионообменной хроматографии, как указано ниже. Присоединение ПЭГ через амидную связь. Пептидный димер и молекулы, содержащие ПЭГ [мПЭГ 2-Lys-NHS] смешивали в молярном соотношении 1:2 в сухом ДМФА, получая прозрачный раствор. Спустя 5 мин к указанному раствору добавляли 10 экв. DIEA. мПЭГ 2-Lys-NHS доступен коммерчески, например, он обозначен в Molecular Engineering catalog (2003) Nektar Therapeutic (490 Discovery Drive, Huntsville, Alabama 35806)2Z3X0T01. Смесь перемешивали при комнатной температуре в течение 2 ч, а затем очищали на колонке С 18 при помощи обращенно-фазовой жидкостной хроматографии высокого давления. Строение пептида с присоединенным ПЭГ подтверждено масс-спектрометрией MALDI. Очищенный пептид, кроме того, подвергали очистке посредством ионообменной хроматографии, как указано ниже. Настоящее изобретение не ограничено указанными в описании примерами реализации. Действительно, ознакомившись с вышеуказанным описанием и сопроводительными рисунками, специалисты в данной области техники могут понять, что, кроме указанных примеров, существуют многочисленные модификации настоящего изобретения. Такие модификации также защищены прилагаемой формулой изобретения. В описании настоящего изобретения упомянуты и обсуждены многочисленные публикации, включая патенты, патентные заявки, протоколы и различные публикации. Упоминание и/или обсуждение таких публикаций предназначено для разъяснения описания настоящего изобретения, но при этом ни одна из ссылок не является прототипом настоящего изобретения. Все публикации, упомянутые или обсужденные в настоящей спецификации, включены в описание в том объеме, который соответствует полному объему цитируемого источника. ФОРМУЛА ИЗОБРЕТЕНИЯ 1. Соединение, включающее пептидный фрагмент, разделительный фрагмент (спейсер) и фрагмент полиэтиленгликоля, в котором разделительный фрагмент находится между пептидным фрагментом и фрагментом полиэтиленгликоля и имеет следующее строение: где каждый из коэффициентов , , ,ипредставляет собой целое число, значения которых выбирают независимо друг от друга, причемилине равен 0, а фрагмент полиэтиленгликоля ковалентно присоединен посредством карбаматной или амидной связи и имеет молекулярную массу более 20 кДа. 2. Соединение по п.1, в котором- это целое число, 16;- это целое число, 16;- это целое число, 16;равно 0 или 1;- это целое число, 110 и Y представляет собой либо NH, либо CO. 3. Соединение по п.2, в котором 1 и =2. 4. Соединение по п.1, в котором ===2; ==1 и Y представляет собой NH. 5. Соединение по п.1, в котором фрагмент полиэтиленгликоля имеет линейное строение. 6. Соединение по п.1, в котором фрагмент полиэтиленгликоля имеет значение полидисперсности- 28010015 7. Соединение по п.1, в котором пептидный фрагмент представляет собой пептидный мономер,включающий один пептид. 8. Соединение по п.1, в котором пептидный фрагмент представляет собой пептидный димер, включающий два пептида, соединенные связующим фрагментом. 9. Соединение по п.7 или 8, в котором каждый пептид включает не более 50 аминокислотных мономеров, предпочтительно каждый пептид включает приблизительно от 10 до 25 аминокислотных мономеров. 10. Соединение по п.1, в котором пептидный фрагмент включает один или более пептидов, связывающих рецепторы эритропоетина. 11. Соединение по п.1, в котором пептидный фрагмент включает один или более пептидов, связывающих рецепторы тромбопоетина. 12. Соединение по п.1, в котором =2; ====0 и Y представляет собой CO. 13. Соединение по п.1, в котором фрагмент полиэтиленгликоля включает по меньшей мере одну мономерную цепочку полиэтиленгликоля. 14. Соединение по п.13, в котором молекулярная масса каждой цепочки полиэтиленгликоля составляет от 20 до 40 кДа. 15. Фармацевтическая композиция, которая содержит соединение по любому из пп.1-14 и один или более фармацевтически приемлемых разбавителей, консервантов, солюбилизаторов, эмульсификаторов,вспомогательных средств и/или носителей. 16. Соединение, включающее пептидный фрагмент, разделительный фрагмент (спейсер) и фрагмент водорастворимого полимера, в котором разделительный фрагмент находится между пептидным фрагментом и фрагментом водорастворимого полимера и имеет следующее строение: где ===2; ==1 и Y представляет собой NH. 17. Фармацевтическая композиция, которая содержит: а) соединение, включающее пептидный фрагмент, разделительный фрагмент (спейсер) и фрагмент водорастворимого полимера, в котором разделительный фрагмент находится между пептидным фрагментом и фрагментом водорастворимого полимера и имеет следующее строение: где ===2; ==1 и Y представляет собой NH и б) один или более фармацевтически приемлемый разбавитель, консервант, солюбилизатор, эмульсификатор, адьювант и/или носитель.
МПК / Метки
МПК: A61K 47/48, A61K 38/00
Метки: модифицированных, пептидов, разделительный, основе, соединений, новый, полиэтиленгликолем, спейсер, фрагмент
Код ссылки
<a href="https://eas.patents.su/30-10015-novyjj-razdelitelnyjj-fragment-spejjser-dlya-modificirovannyh-polietilenglikolem-soedinenijj-na-osnove-peptidov.html" rel="bookmark" title="База патентов Евразийского Союза">Новый разделительный фрагмент (спейсер) для модифицированных полиэтиленгликолем соединений на основе пептидов</a>
Дисперсионные покрытия на основе (со)полимеров винилиденфторида, модифицированных акриловыми полимерами

Номер патента: 1802
Опубликовано: 27.08.2001
Авторы: Дрюон Ксавьер Ф., Гэбоури Скотт Р.
МПК: C09D 127/16
Метки: основе, сополимеров, модифицированных, винилиденфторида, акриловыми, покрытия, полимерами, дисперсионные
Формула / Реферат:
1. Основа краски для дисперсионного покрытия, содержащая выделенный из дисперсии продукт эмульсионной полимеризации 100-200 вес.ч. акриловых мономеров, предпочтительно 20-80 вес. ч. акриловых мономеров, в присутствии 100 вес.ч. затравочных частиц (со)полимеров винилиденфторида и разбавитель. 2. Основа краски по п.1, отличающаяся тем, что разбавитель является практически неводным. 3. Основа краски по п.1, отличающаяся тем, что разбавитель имеет...
Новый способ синтеза соединений (2s, 3аs, 7аs)-1-[(s)-аланил]-октагидро -1h-индол-2-карбоновой кислоты и применение в синтезе периндоприла

Номер патента: 5490
Опубликовано: 24.02.2005
Авторы: Порч-Маккаи Марта, Мезей Тибор, Шимиг Дьюла
МПК: C07K 5/02
Метки: 7аs)-1-[(s)-аланил]-октагидро, синтеза, периндоприла, кислоты, новый, синтезе, способ, 1h-индол-2-карбоновой, 3аs, соединений, применение
Формула / Реферат:
1. Способ синтеза соединения формулы (I) в которой R1 представляет собой атом водорода, линейную или разветвлённую (C1-C6)алкильную группу или бензильную группу, и R2 представляет собой группу, которая защищает аминную функцию, отличающийся тем, что сложный эфир формулы (V) в которой R1 принимает значения, указанные для формулы (I), вводят в реакцию с соединением аланина формулы (VI) в которой R2 принимает значения, указанные для формулы...
Новый способ синтеза соединений ( 2s, 3аs, 7as )-1-[ (s)-аланил]октагидро -1н-индол-2-карбоновой кислоты и их применение для синтеза периндоприла

Номер патента: 9062
Опубликовано: 26.10.2007
Авторы: Ланглуа Паскаль, Дюбюффе Тьерри
МПК: C07K 5/02, C07K 5/06, C07D 209/42...
Метки: синтеза, 1, способ, s)-аланил]октагидро, периндоприла, 3аs, соединений, кислоты, 1н-индол-2-карбоновой, применение, новый
Формула / Реферат:
1. Способ синтеза соединений (I) в которой R1 представляет собой атом водорода или бензильную группу, a R2 - это защитная группа для группы амина, представляющая собой трет-бутоксикарбонильную группу, характеризующийся тем, что 1-(1-циклогексен-1-ил)пирролидин формулы (III) подвергают реакции с соединением серина формулы (IV) в которой R1 является таким, как определено для формулы (I), a R3 - это защитная группа для группы амина,...
Связанные с полиэтиленгликолем соединения гпп-1

Номер патента: 9366
Опубликовано: 28.12.2007
Авторы: Вик Эндрю Марк, Жанг Лианшан, Милликан Рон Ли Джуниор, Димарчи Ричард Дэнниз, Гласнер Вольфган
МПК: A61K 38/26, C07K 14/00, A61K 38/00...
Метки: гпп-1, полиэтиленгликолем, соединения, связанные
Формула / Реферат:
1. ПЭГилированное соединение ГПП-1, содержащее аминокислотную последовательность формулы IV (SEQ ID No.: 6) Формула IV (SEQ ID No.: 6), где Хаа7 обозначает L-гистидин, D-гистидин, дезаминогистидин, 2-аминогистидин, b-гидроксигистидин, гомогистидин, a-фторметилгистидин или a-метилгистидин и где 2 или 1 из Cys остатков ковалентно связан(ы) с молекулой ПЭГ, или 3, 2 или 1 из Lys остатков ковалентно связан(ы) с молекулой ПЭГ, и при условии, что,...
Диспергируемые композиции на основе макромолекулярных соединений

Номер патента: 956
Опубликовано: 28.08.2000
Авторы: Плэтз Роберт М., Бордмэн Теренс Д., Бревер Томас К.
МПК: A61K 9/12
Метки: макромолекулярных, композиции, диспергируемые, основе, соединений
Формула / Реферат:
1. Способ получения диспергируемых сухих порошков биологических макромолекулярных соединений, заключающийся в получении способной испаряться жидкой среды с заданной концентрацией макромолекулярного соединения и наполнителей; распылении жидкой среды в условиях, выбранных с целью образования капель со средним размером, меньшим заданного максимума; сушке капель в условиях, выбранных с целью образования диспергируемых частиц композиционного...
Предыдущий патент: Способ получения стабильной дисперсии полимеров, стабильная дисперсия полимеров и полиуретановая пена
Следующий патент: Новые модифицированные полиэтиленгликолем соединения и их применение
Случайный патент: Горловина загрузочная шприца вакуумного