Способы и интермедиаты для получения ингибиторов интегразы




















Формула / Реферат
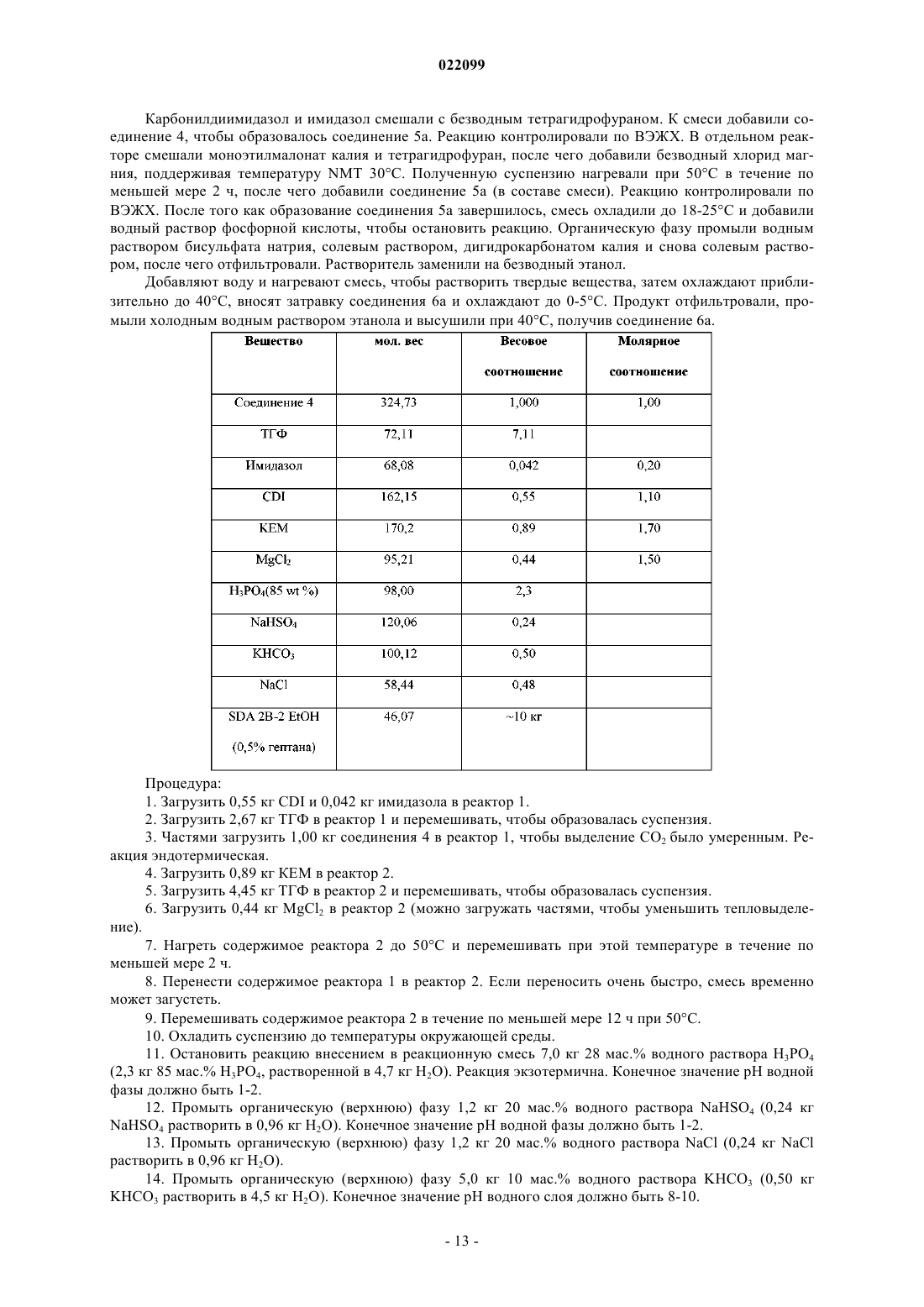
1. Соединение формулы 3

или его соль.
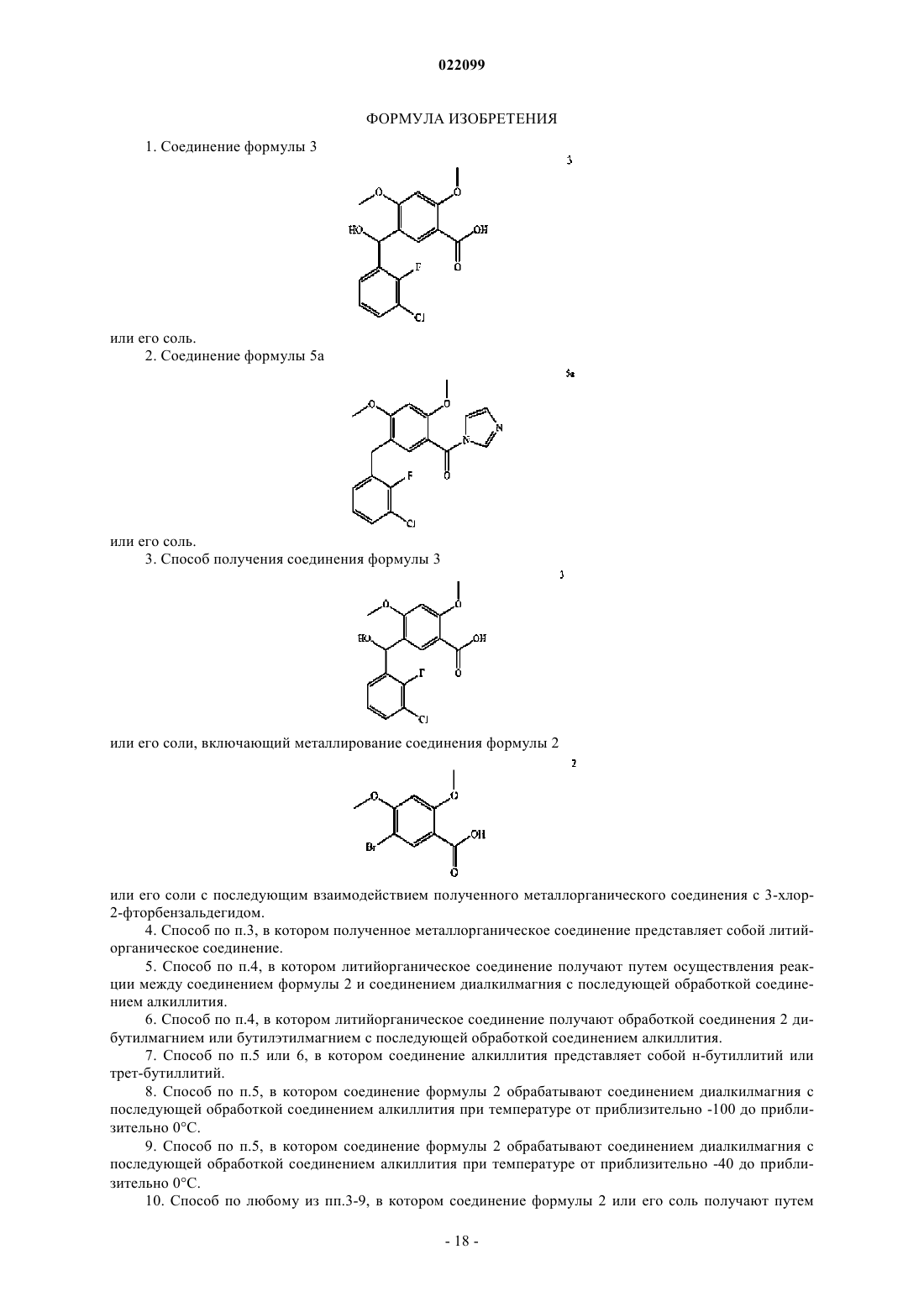
2. Соединение формулы 5а

или его соль.
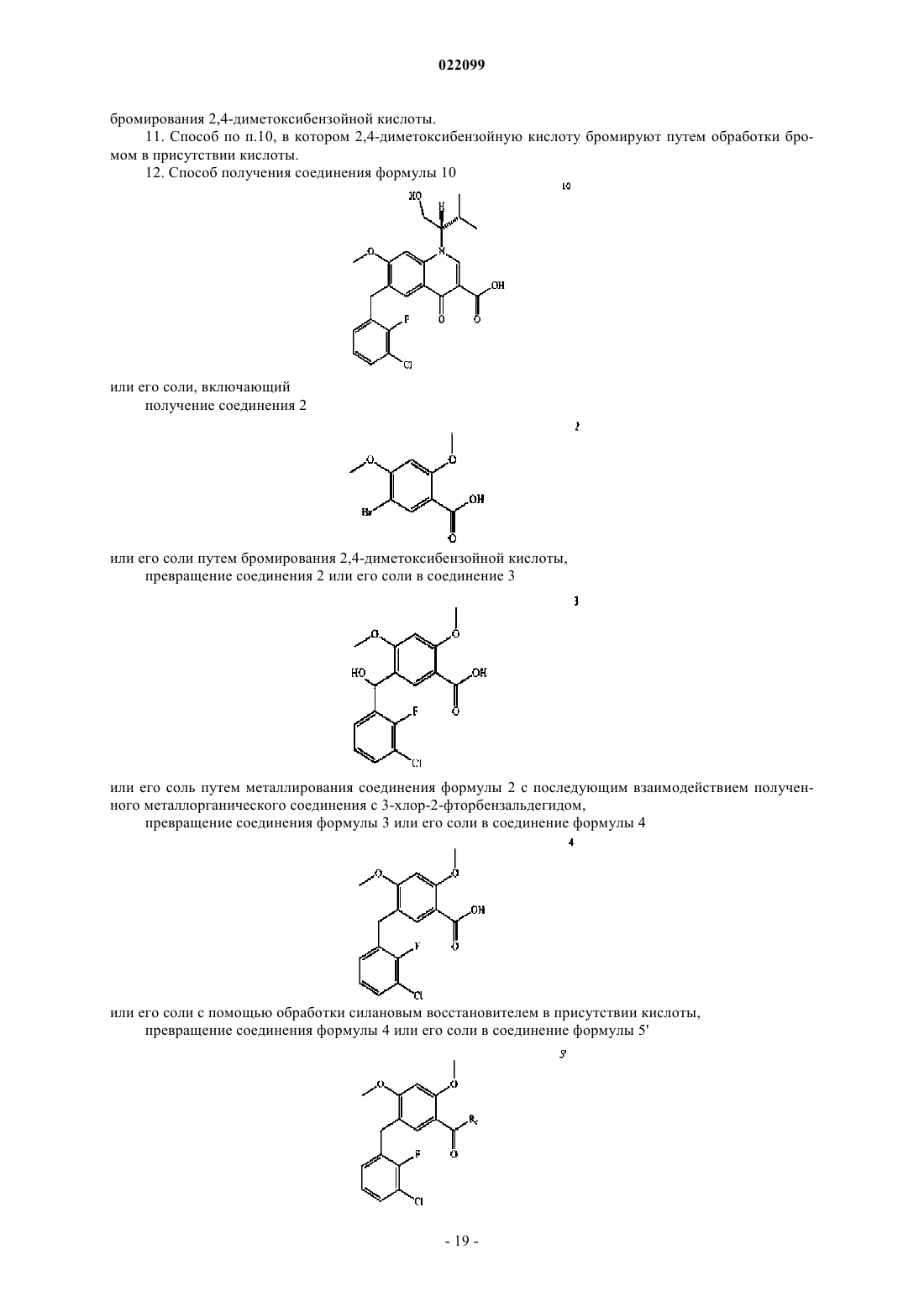
3. Способ получения соединения формулы 3

или его соли, включающий металлирование соединения формулы 2

или его соли с последующим взаимодействием полученного металлорганического соединения с 3-хлор-2-фторбензальдегидом.
4. Способ по п.3, в котором полученное металлорганическое соединение представляет собой литий-органическое соединение.
5. Способ по п.4, в котором литийорганическое соединение получают путем осуществления реакции между соединением формулы 2 и соединением диалкилмагния с последующей обработкой соединением алкиллития.
6. Способ по п.4, в котором литийорганическое соединение получают обработкой соединения 2 дибутилмагнием или бутилэтилмагнием с последующей обработкой соединением алкиллития.
7. Способ по п.5 или 6, в котором соединение алкиллития представляет собой н-бутиллитий или трет-бутиллитий.
8. Способ по п.5, в котором соединение формулы 2 обрабатывают соединением диалкилмагния с последующей обработкой соединением алкиллития при температуре от приблизительно -100 до приблизительно 0°С.
9. Способ по п.5, в котором соединение формулы 2 обрабатывают соединением диалкилмагния с последующей обработкой соединением алкиллития при температуре от приблизительно -40 до приблизительно 0°С.
10. Способ по любому из пп.3-9, в котором соединение формулы 2 или его соль получают путем бромирования 2,4-диметоксибензойной кислоты.
11. Способ по п.10, в котором 2,4-диметоксибензойную кислоту бромируют путем обработки бромом в присутствии кислоты.
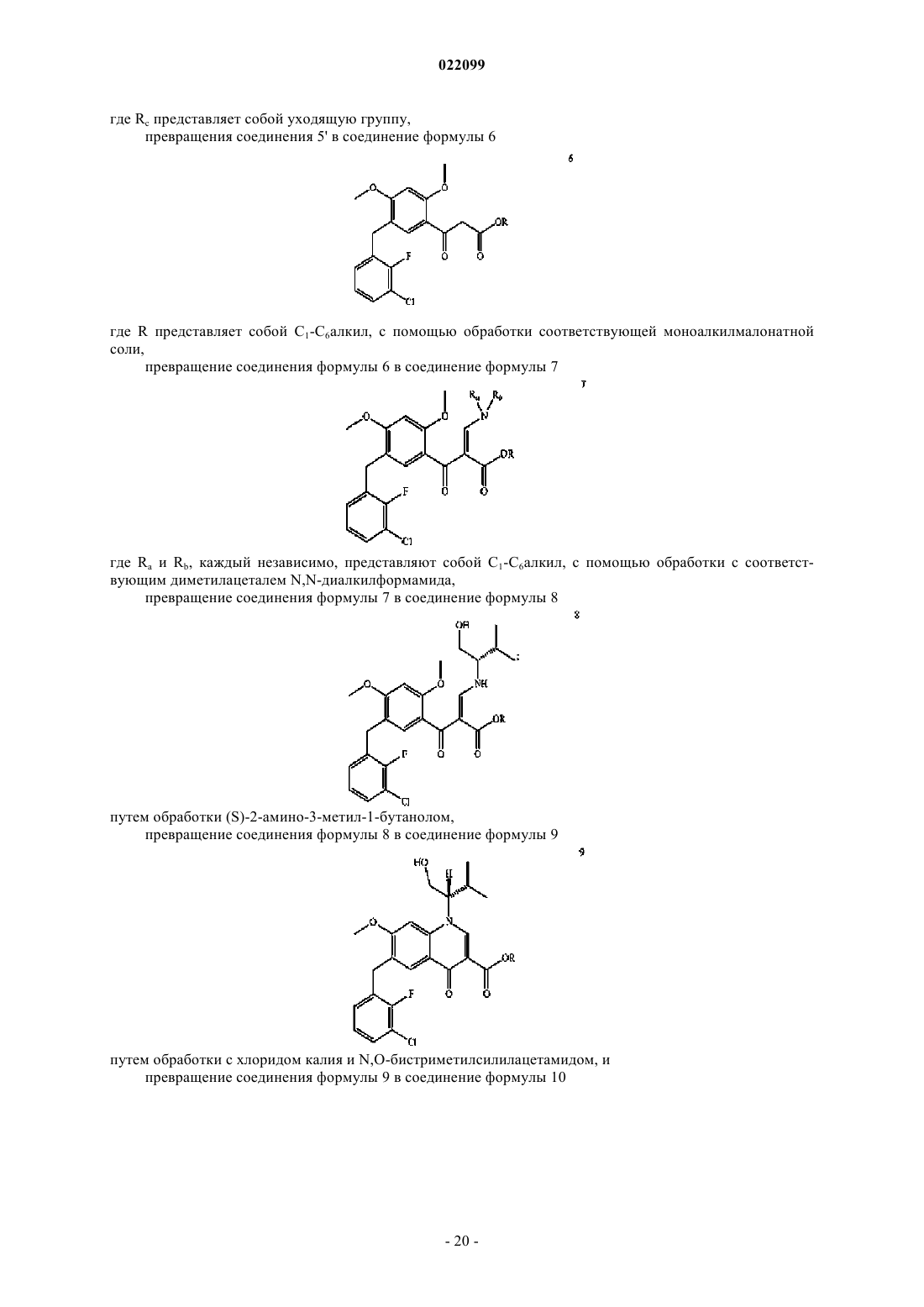
12. Способ получения соединения формулы 10

или его соли, включающий
получение соединения 2

или его соли путем бромирования 2,4-диметоксибензойной кислоты,
превращение соединения 2 или его соли в соединение 3

или его соль путем металлирования соединения формулы 2 с последующим взаимодействием полученного металлорганического соединения с 3-хлор-2-фторбензальдегидом,
превращение соединения формулы 3 или его соли в соединение формулы 4

или его соли с помощью обработки силановым восстановителем в присутствии кислоты,
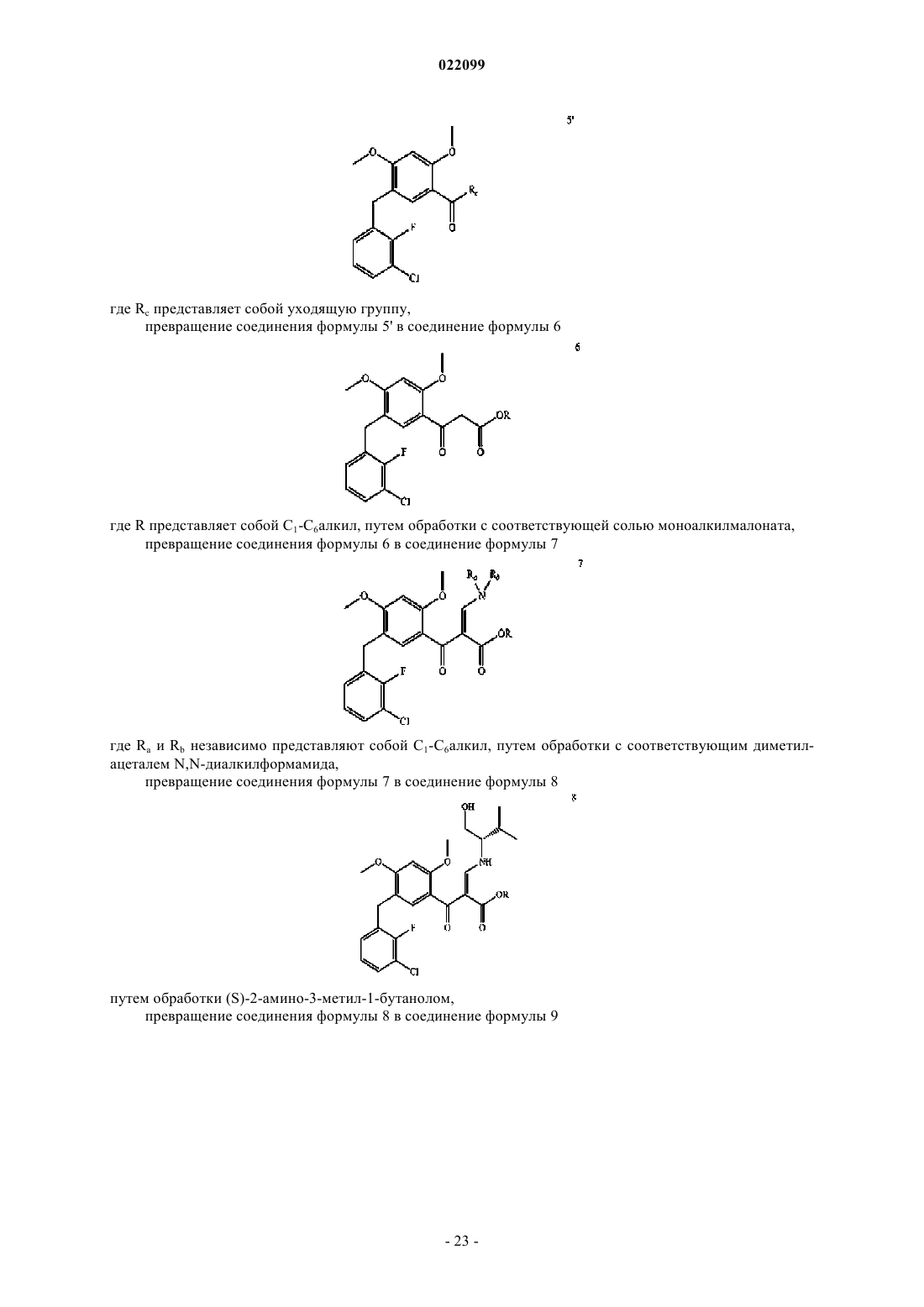
превращение соединения формулы 4 или его соли в соединение формулы 5'

где Rc представляет собой уходящую группу,
превращения соединения 5' в соединение формулы 6

где R представляет собой C1-C6алкил, с помощью обработки соответствующей моноалкилмалонатной соли,
превращение соединения формулы 6 в соединение формулы 7

где Ra и Rb, каждый независимо, представляют собой C1-C6алкил, с помощью обработки с соответствующим диметилацеталем N,N-диалкилформамида,
превращение соединения формулы 7 в соединение формулы 8

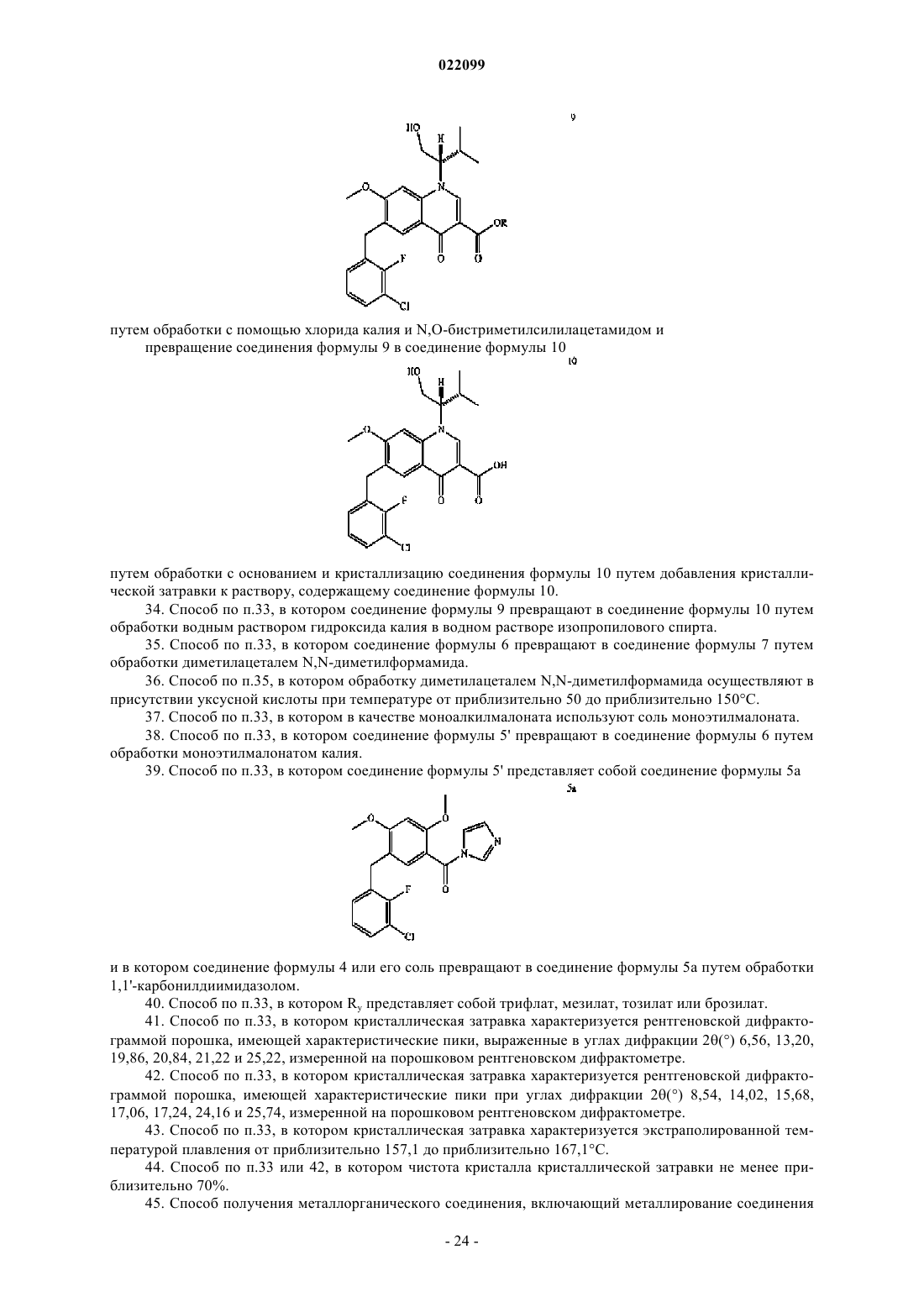
путем обработки (S)-2-амино-3-метил-1-бутанолом,
превращение соединения формулы 8 в соединение формулы 9

путем обработки с хлоридом калия и N,О-бистриметилсилилацетамидом, и
превращение соединения формулы 9 в соединение формулы 10

путем обработки основанием и кристаллизацию соединения формулы 10 путем добавления кристаллической затравки к раствору, содержащему соединение формулы 10.
13. Способ по п.12, отличающийся тем, что полученное металлорганическое соединение представляет собой литийорганическое соединение.
14. Способ по п.13, отличающийся тем, что литийорганическое соединение получают путем приведения во взаимодействие соединения формулы 2 с соединением диалкилмагния с последующей обработкой соединением алкиллития.
15. Способ по п.13, отличающийся тем, что литийорганическое соединение получают путем обработки соединения формулы 2 с дибутилмагнием или бутилэтилмагнием с последующей обработкой соединением алкиллития.
16. Способ по п.14 или 15, отличающийся тем, что соединение алкиллития представляет собой н-бутиллитий или трет-бутиллитий.
17. Способ по п.14, отличающийся тем, что соединение формулы 2 обрабатывают соединением диалкилмагния с последующей обработкой соединением алкиллития при температуре приблизительно от
-100 до 0°С.
18. Способ по п.14, отличающийся тем, что соединение формулы 2 обрабатывают с помощью соединения диалкилмагния с последующей обработкой соединением алкиллития при температуре приблизительно от
-40 до 0°С.
19. Способ по п.12, отличающийся тем, что 2,4-диметоксибензойную кислоту бромируют путем обработки бромом в присутствии кислоты.
20. Способ по п.12, в котором силановый восстановитель представляет собой триэтилсилан, а кислота представляет собой трифторуксусную кислоту.
21. Способ по п.12, в котором указанная уходящая группа представляет собой галоген или 1-имидазолил.
22. Способ по п.12, в котором соединение формулы 5' представляет собой соединение формулы 5а

23. Способ по п.22, в котором соединение формулы 5а получают обработкой соединения формулы 4 1,1'-карбонилдиимидазолом.
24. Способ по п.12, в котором R в соединении формулы 6 представляет собой этил.
25. Способ по п.24, в котором соединение формулы 5' превращают в соединение формулы 6 путем обработки моноэтилмалонатом калия.
26. Способ по п.12, в котором соединение формулы 6 превращают в соединение формулы 7 путем обработки диметилацеталем N,N-диметилформамида.
27. Способ по п.26, в котором обработку диметилацеталем N,N-диметилформамида проводят в присутствии уксусной кислоты при температуре от приблизительно 50 до приблизительно 150°С.
28. Способ по п.12, в котором соединение формулы 9 превращают в соединение формулы 10 путем обработки водным раствором гидроксида калия в водном растворе изопропилового спирта.
29. Способ по п.12, отличающийся тем, что кристаллическая затравка характеризуется рентгеновской дифрактограммой порошка, имеющей характеристические пики, выраженные в углах 2θ(°) при 6,56, 13,20, 19,86, 20,84, 21,22 и 25,22, измеренной на порошковом рентгеновском дифрактометре.
30. Способ по п.12, отличающийся тем, что кристаллическая затравка характеризуется рентгеновской дифрактограммой порошка, имеющей характеристические пики, выраженные в углах 2θ(°) при 8,54, 14,02, 15,68, 17,06, 17,24, 24,16 и 25,74, измеренной на порошковом рентгеновском дифрактометре.
31. Способ по п.12, отличающийся тем, что кристаллическая затравка имеет экстраполированную температуру плавления от приблизительно 157,1 до приблизительно 167,1°С.
32. Способ по любому из пп.12 или 30, отличающийся тем, что кристаллическая затравка имеет чистоту по меньшей мере приблизительно 70%.
33. Способ получения соединения формулы 10

или его соли, включающий
получение соединения формулы 4

или его соли путем металлирования соединения формулы 2

или его соли с последующим взаимодействием полученного металлорганического соединения с соединением формулы 11

где Ry представляет собой уходящую группу,
превращение соединения формулы 4 или его соли в соединение формулы 5'

где Rc представляет собой уходящую группу,
превращение соединения формулы 5' в соединение формулы 6

где R представляет собой C1-С6алкил, путем обработки с соответствующей солью моноалкилмалоната,
превращение соединения формулы 6 в соединение формулы 7

где Ra и Rb независимо представляют собой C1-С6алкил, путем обработки с соответствующим диметилацеталем N,N-диалкилформамида,
превращение соединения формулы 7 в соединение формулы 8

путем обработки (S)-2-амино-3-метил-1-бутанолом,
превращение соединения формулы 8 в соединение формулы 9

путем обработки с помощью хлорида калия и N,О-бистриметилсилилацетамидом и
превращение соединения формулы 9 в соединение формулы 10

путем обработки с основанием и кристаллизацию соединения формулы 10 путем добавления кристаллической затравки к раствору, содержащему соединение формулы 10.
34. Способ по п.33, в котором соединение формулы 9 превращают в соединение формулы 10 путем обработки водным раствором гидроксида калия в водном растворе изопропилового спирта.
35. Способ по п.33, в котором соединение формулы 6 превращают в соединение формулы 7 путем обработки диметилацеталем N,N-диметилформамида.
36. Способ по п.35, в котором обработку диметилацеталем N,N-диметилформамида осуществляют в присутствии уксусной кислоты при температуре от приблизительно 50 до приблизительно 150°С.
37. Способ по п.33, в котором в качестве моноалкилмалоната используют соль моноэтилмалоната.
38. Способ по п.33, в котором соединение формулы 5' превращают в соединение формулы 6 путем обработки моноэтилмалонатом калия.
39. Способ по п.33, в котором соединение формулы 5' представляет собой соединение формулы 5а

и в котором соединение формулы 4 или его соль превращают в соединение формулы 5а путем обработки 1,1'-карбонилдиимидазолом.
40. Способ по п.33, в котором Ry представляет собой трифлат, мезилат, тозилат или брозилат.
41. Способ по п.33, в котором кристаллическая затравка характеризуется рентгеновской дифрактограммой порошка, имеющей характеристические пики, выраженные в углах дифракции 2θ(°) 6,56, 13,20, 19,86, 20,84, 21,22 и 25,22, измеренной на порошковом рентгеновском дифрактометре.
42. Способ по п.33, в котором кристаллическая затравка характеризуется рентгеновской дифрактограммой порошка, имеющей характеристические пики при углах дифракции 2θ(°) 8,54, 14,02, 15,68, 17,06, 17,24, 24,16 и 25,74, измеренной на порошковом рентгеновском дифрактометре.
43. Способ по п.33, в котором кристаллическая затравка характеризуется экстраполированной температурой плавления от приблизительно 157,1 до приблизительно 167,1°С.
44. Способ по п.33 или 42, в котором чистота кристалла кристаллической затравки не менее приблизительно 70%.
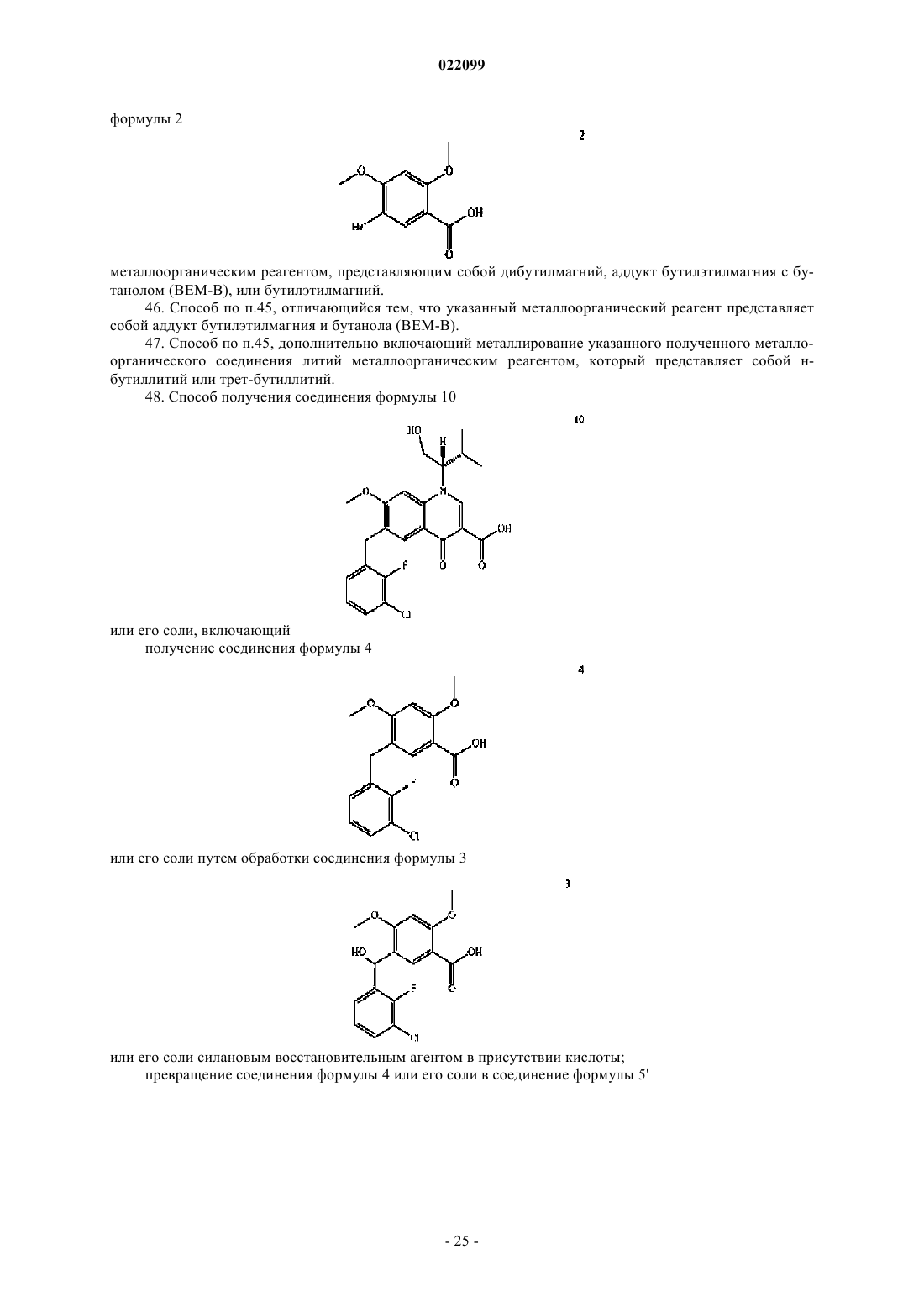
45. Способ получения металлорганического соединения, включающий металлирование соединения формулы 2

металлоорганическим реагентом, представляющим собой дибутилмагний, аддукт бутилэтилмагния с бутанолом (ВЕМ-В), или бутилэтилмагний.
46. Способ по п.45, отличающийся тем, что указанный металлоорганический реагент представляет собой аддукт бутилэтилмагния и бутанола (BEM-B).
47. Способ по п.45, дополнительно включающий металлирование указанного полученного металлоорганического соединения литий металлоорганическим реагентом, который представляет собой н-бутиллитий или трет-бутиллитий.
48. Способ получения соединения формулы 10

или его соли, включающий
получение соединения формулы 4

или его соли путем обработки соединения формулы 3

или его соли силановым восстановительным агентом в присутствии кислоты;
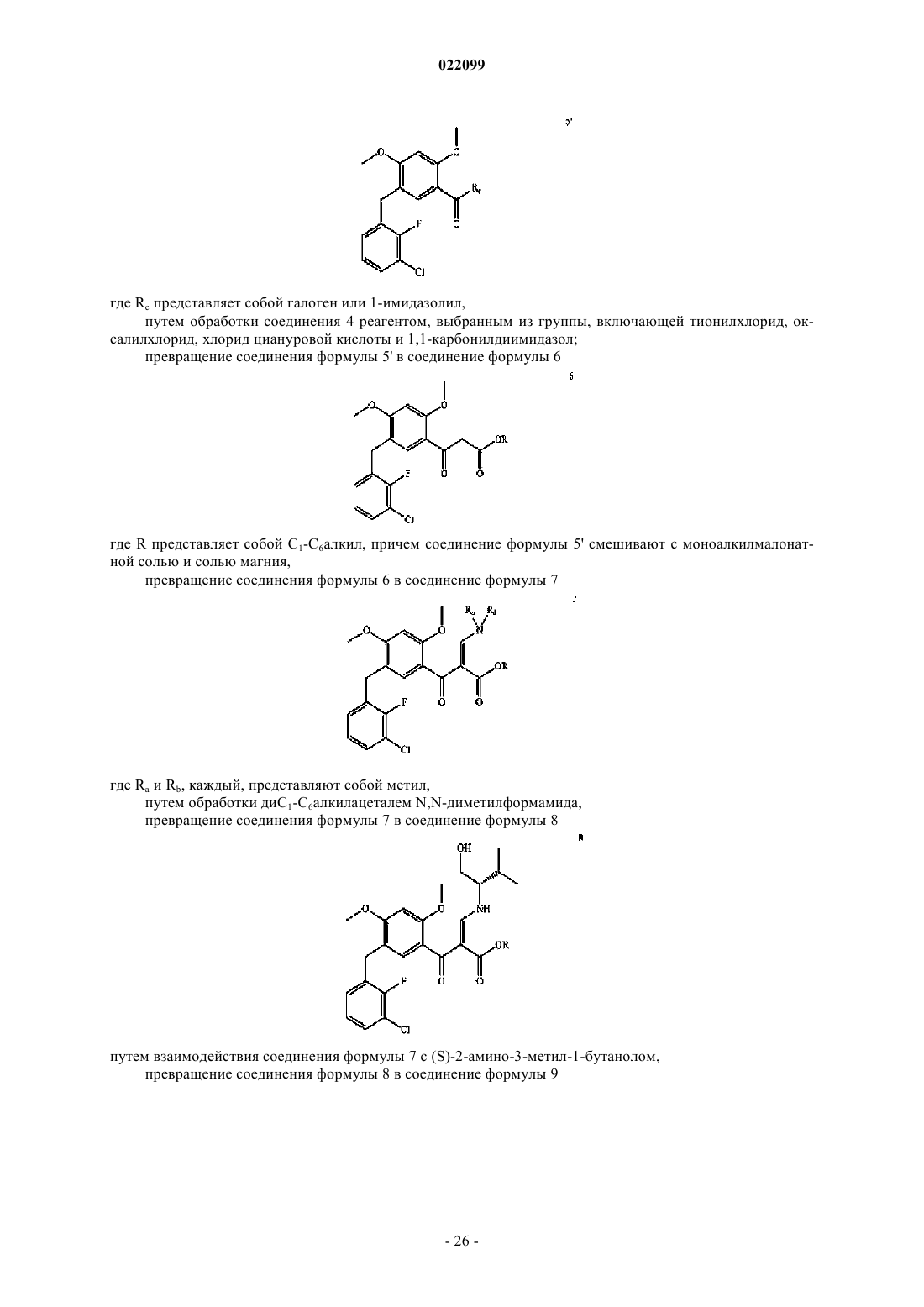
превращение соединения формулы 4 или его соли в соединение формулы 5'

где Rc представляет собой галоген или 1-имидазолил,
путем обработки соединения 4 реагентом, выбранным из группы, включающей тионилхлорид, оксалилхлорид, хлорид циануровой кислоты и 1,1-карбонилдиимидазол;
превращение соединения формулы 5' в соединение формулы 6

где R представляет собой C1-С6алкил, причем соединение формулы 5' смешивают с моноалкилмалонатной солью и солью магния,
превращение соединения формулы 6 в соединение формулы 7

где Ra и Rb, каждый, представляют собой метил,
путем обработки диС1-С6алкилацеталем N,N-диметилформамида,
превращение соединения формулы 7 в соединение формулы 8

путем взаимодействия соединения формулы 7 с (S)-2-амино-3-метил-1-бутанолом,
превращение соединения формулы 8 в соединение формулы 9

путем обработки соединения формулы 8 силилирующим агентом, выбранным из группы, включающей N,О-бистриметилсилилацетамид, N,О-бис(триметилсилил)трифторацетамид и гексаметилдисилазан, при температуре 100±20°С, и
превращение соединения формулы 9 в соединение формулы 10 путем обработки соединения формулы 9 основанием.
49. Способ по п.48, отличающийся тем, что силановый восстановитель представляет собой триэтилсилан и кислота представляет собой трифторуксусную кислоту.
50. Способ по п.48, отличающийся тем, что указанный диС1-С6алкилацеталь N,N-диметилформамида представляет собой диметилацеталь N,N-диметилформамида.
51. Способ по п.48 или 50, отличающийся тем, что обработку диметилацеталем N,N-диметилформамида проводят в присутствии уксусной кислоты при температуре 100±50°С.
52. Способ по п.48, отличающийся тем, что указанная обработка соединения формулы 8 силилирующим агентом выполняется в присутствии соли, выбранной из группы, включающей хлорид калия, хлорид лития, хлорид натрия и хлорид магния, в полярном апротонном растворителе, выбранном из группы, включающей диметилформамид, диметилацетамид, N-метилпирролидинон и ацетонитрил.
53. Способ по п.48, отличающийся тем, что соединение формулы 8 превращают в соединение формулы 9 путем обработки соединения формулы 8 хлоридом калия и N,O-бис(триметилсилил)ацетамидом.
Текст
СПОСОБЫ И ИНТЕРМЕДИАТЫ ДЛЯ ПОЛУЧЕНИЯ ИНГИБИТОРОВ ИНТЕГРАЗЫ Изобретение относится к способам синтеза и синтетическим интермедиатам, которые можно использовать для получения 4-оксохинолоновых соединений с полезными свойствами ингибиторов интегразы. Область техники, к которой относится изобретение Изобретение относится к области органической химии, в частности к способам получения 4 оксохинолоновых соединений и новым интермедиатам для получения ингибиторов интегразы. Сведения о предшествующем уровне техники Публикация международной патентной заявки номер WO 2004/046115 относится к некоторым 4 оксохинолоновым соединениям, полезным в качестве ингибиторов ВИЧ-интегразы. Сообщается, что соединения могут быть полезны в качестве агентов против вируса иммунодефицита человека (ВИЧ). Публикация международной патентной заявки номер WO 2005/113508 относится к некоторым особым кристаллическим формам одного из этих 4-оксохинолоновых соединений, а именно к 6-(3-хлоро-2 фторобензил)-1-[(S)-1-гидроксиметил-2-метилпропил]-7-метокси-4-оксо-1,4-дигидрохинолон-3-карбоновой кислоте. Сообщается, что особые кристаллические формы характеризуются улучшенной физической и химической стабильностью по сравнению с другими физическими формами соединения. В настоящее время существует потребность в улучшенных способах получения 4-оксохинолоновых соединений, о которых сообщается в публикации международной патентной заявки WO 2004/046115 и в публикации международной патентной заявки WO 2005/113508. В частности, имеется потребность в новых упрощенных или в более дешевых синтетических методах, обеспечивающих повышенный выход или избавляющих от необходимости использовать токсичные или дорогие реагенты. Сущность изобретения Настоящее изобретение относится к новым способам синтеза и синтетическим интермедиатам, полезным для получения 4-оксохинолоновых соединений, о которых сообщается в публикации международной патентной заявки WO 2004/046115 и в публикации международной патентной заявки WO 2005/113508. Таким образом, в соответствии с одним своим аспектом изобретение относится к соединению формулы 3 или его соли. В соответствии с другим аспектом изобретение относится к соединению формулы 5 а или его соли. В соответствии с другим аспектом изобретение относится к способу получения соединения формулы 3 или его соли, включающему превращение соответствующего соединения формулы 2 или его соли в соединение формулы 3 или его соль. Изобретение относится также к другим описанным здесь способам синтеза и синтетическим интермедиатам, полезным для приготовления 4-оксохиноновых соединений. Если не указано иное, перечисленные ниже термины означают следующее: галоген - это фтор, хлор,бром или йод. Термин "алкил" включает в себя нормальную и разветвленную углеродную цепь. Однако названия индивидуальных радикалов, таких как пропил, предполагают только прямую цепь, изомеры с разветвленной цепью, такие как изопропил, называют отдельно. Специалист понимает, что соединения с хиральным центром могут существовать в оптически активной и рацемической формах и в таких формах их можно выделить. Некоторые соединения могут демонстрировать полиморфизм. Следует понимать, что настоящее изобретение включает в себя описание способов получения любых рацемических, оптически активных,полиморфных, таутомерных или стереоизомерных форм описанного здесь соединенияи и их смесей,причем в уровне техники хорошо известно, как можно получать оптически активные формы (например,разделение рацемической формы методами рекристаллизации, синтез из оптически активных исходных материалов, хиральный синтез или хроматографическое разделение с использованием хиральной неподвижной фазы). Конкретные и предпочтительные значения, перечисленные ниже для радикалов, заместителей и интервалов, предназначены только для иллюстрации; они не исключают других заданных значений или других значений в заданных интервалах для радикалов и заместителей. Конкретно,C1-С 6 алкил может представлять собой метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил,пентил, 3-пентил или гексил. Специфическое значение для Ra - метил. Специфическое значение для Rb - метил. Специфическое значение для Rc - 1-имидазолил. Специфическое значение для R - этил. В соответствии с одним аспектом изобретение относится к способу получения соединения формулы 3 или его соли, включающему превращение соответствующего соединения формулы 2 или его соли в соединение формулы 3 или его соль. Как показано ниже, реакцию можно легко осуществить путем смешивания соединения 2 с полярным апротонным растворителем (например, тетрагидрофураном) и охлаждения смеси до температуры ниже комнатной (например, приблизительно до -20 С). Сначала смесь обрабатывают органометаллическим реагентом (например, диалкилмагнием, диалкилцинком, алкилмагний галидом, триалкилалюминием или гидридом металла) с образованием соли карбоксилата. Например, смесь можно обработать приблизительно 0,5 экв. дибутилмагния или бутилэтилмагния либо приблизительно 1 экв. аддукта бутилэтилмагния-бутанола с получением соединения А. Полученную смесь можно смешать со вторым органометаллическим реагентом (например, алкиллитий или алкилмагний галидом) с получением органометаллического соединения (соединения В 1 или В 2). Как правило, это делается при пониженной температуре, чтобы повлиять на обмен металл-галоген. Например, полученную смесь можно смешать приблизительно с 1,2-2,2 экв. алкиллития (например, приблизи-2 022099 тельно с 1,8 экв. н-бутиллития или трет-бутиллития) при температуре -5050 С с получением литийорганического соединения (соединение В 1). В соответствии с одним аспектом изобретения реакцию обмена металл-галоген можно проводить при температуре -2020 С. Ход такой реакции можно отслеживать любым удобным способом (например, ВЭЖХ). По завершении реакции в систему можно добавить 3 хлоро-2-фторобензальдегид (около 1,3 экв.). За ходом реакции присоединения можно следить любым подходящим способом (например, методом ВЭЖХ). Соединение 3 выделяют также любым подходящим способом (например, хроматографией или кристаллизацией). Такой подход позволяет избежать загрязнений и снизить стоимость, связанную с использованием других реагентов (например, переходных металлов, таких как реагенты палладия). В соответствии с одним аспектом изобретения соединение формулы 2 или его соль получают бромированием 2,4-диметоксибензойной кислоты. Реакцию проводят в стандартных условиях бромирования. В соответствии с одним аспектом изобретения соединение формулы 3 или его соль затем превращают в соединение формулы 4 или его соль. Около 1-5 гидридных экв. силанового восстановителя (например, фенилдиметилсилана, полиметилгидросилоксана или хлородиметилсилана или триалкилсилана, такого как триэтилсилан) смешивают с подходящей кислотой (например, трифторуксусной кислотой, трифторометансульфоновой кислотой или уксусной кислотой). Удобно проводить реакцию приблизительно с 1,2-2,0 гидридными экв. триэтилсилана и 5-10 экв. трифтороуксусной кислоты. К этой смеси добавляют соединение 3 или его соль. Это можно удобно сделать при пониженной температуре, например 010 С. За ходом реакции можно следить любым подходящим способом (например, ВЭЖХ). По завершении реакции соединение 4 или его соль выделяют также любым подходящим способом (например, хроматографией или кристаллизацией). Соединение 4 или его соль можно также получать, добавляя трифторуксусную кислоту к соединению 3 в подходящем растворителе, а затем введением силанового восстановителя получая соединение 4. Альтернативно, соединение 4 или его соль можно приготовить, получив соответствующее металлоорганическое соединение из соединения 2 и проведя реакцию между ним и соединением 11 где Ry представляет собой подходящую уходящую группу (например, трифлат, мезилат, тозилат,брозилат и т.д.). В соответствии с одним аспектом изобретения соединение формулы 4 или его соль затем превращают в соединение формулы 5' или его соль, где Rc представляет собой подходящую уходящую группу. Функциональную группу карбоновой кислоты соединения 4 можно активировать, например, превратив в ацилхлорид или ацилимидазолид (соединение 5'), обработав подходящим реагентом, например тионилхлоридом, оксалилхлоридом, цианурхлоридом или 1,1'-карбонилдиимидазолом в подходящем растворителе (например, толуоле или тетрагидрофуране). В молекулу можно ввести любую подходящую уходящую группу Rc, главное, чтобы соединение формулы 5' можно было затем преобразовать в соединение формулы 6. Например, реакцию можно легко провести с использованием 1 экв. 1,1'карбонилдиимидазола в тетрагидрофуране. В соответствии с другим аспектом изобретения соединение формулы 5' или его соль затем превращают в соединение формулы 6 или его соль, где R представляет собой C1-С 6 алкил. Например, соединение формулы 5' можно смешать с 1-5 экв. соли моноалкилмалоната и 1-5 экв. соли магния в подходящем растворителе. Удобно бывает смешать соединение формулы 5' приблизительно с 1,7 экв. моноэтилмалоната калия и приблизительно с 1,5 экв. хлорида магния. В реакцию затем добавляют подходящее основание, например триэтиламин или имидазол. После этого реакцию проводят при повышенной температуре (например, около 10050 С), а за ее ходом следят любым подходящим способом (например, ВЭЖХ). По завершении реакции соединение 6 можно выделить также любым подходящим способом (например, хроматографией или кристаллизацией). В соответствии с другим аспектом изобретения соединение формулы 6 или его соль можно преобразовать в соответствующее соединение формулы 7 где Ra и Rb независимо представляют собой C1-С 6 алкил и R представляет собой C1-С 6 алкил. Соединение 6 можно превратить в активированный алкилиденовый аналог, такой как соединение 7, обработав донором формиатной группы, таким как диметилформамида диалкилацеталь (например, диметилформамида диметилацеталь) или триалкилортоформиат. Реакцию проводят при повышенной температуре (например,около 10050 С). Реакцию можно ускорить введением кислого катализатора, такого как, например, алкановая кислота, бензойная кислота, сульфоновая кислота или минеральная кислота. Удобно использовать приблизительно 500 ppm-1% уксусной кислоты. За ходом реакции можно следить любым подходящим методом (например, ВЭЖХ). Соединение 7 можно выделять или непосредственно использовать для получения соединения формулы 8, как описано ниже. В соответствии с другим аспектом изобретения соединение формулы 7 можно преобразовать в соответствующее соединение формулы 8 где R представляет собой C1-С 6 алкил. Соединение 7 можно смешать с (S)-2-амино-3-метил-1-бутанолом (S-валинолом, около 1,1 экв.) с получением соединения 8. За ходом реакции можно следить любым подходящим методом (например,ВЭЖХ). Соединение формулы 8 можно выделять или непосредственно использовать для получения соединения формулы 9, как описано ниже. В соответствии с другим аспектом изобретение относится к способу получения соединения формулы 9 где R представляет собой C1-С 6 алкил, включающему циклизацию соответствующего соединения формулы 8(например, N,О-бис(триметилсилил)ацетамидом, N,O-бис(триметилсилил)трифтороацетамидом или гексаметилдисилазаном). Реакцию можно проводить в полярном апротонном растворителе (например, в диметилформамиде, диметилацетамиде, N-метилпирролидиноне или ацетонитриле). Можно добавить соль (например, хлорид калия, хлорид лития, хлорид натрия или хлорид магния) для ускорения реакции. Как правило, добавляют около 0,5 экв. соли, такой как хлорид калия. Реакцию можно проводить при повышенной температуре (например, около 10020), так можно добиться приемлемого времени завершения способа. За ходом реакции можно следить любым подходящим методом (например, ВЭЖХ). Введение кислоты в ходе работы дает возможность гидролизовать любой силиловый эфир, образующийся при реакции силилирующего реагента со спиртовой группой соединения 8. Примеры типичных кислот включают минеральные кислоты, сульфоновые кислоты и алкановые кислоты. Конкретным примером кислоты, использовать которую допустимо, является водный раствор хлороводородной кислоты. По завершении гидролиза соединение 9 можно выделить любым подходящим методом (например, хроматографией или кристаллизацией). В ходе описанного выше превращения силилирующий агент временно защищает спиртовую группу, а затем удаляется. Это избавляет от необходимости вводить отдельные этапы введения и снятия защиты, что повышает эффективность превращения. В соответствии с другим аспектом изобретения соединение формулы 9 превращают в соединение формулы 10 Соединение 9 конвертируют в соединение 10 обработкой подходящим основанием (например, гидроксидом калия, натрия или лития). Например, удобно использовать около 1,3 экв. гидроксида калия. Реакцию можно проводить в любом подходящем растворителе, таком как, например, тетрагидрофуран,метанол, этанол, изопропанол или их смесь. В качестве растворителя можно использовать также воду. Удобно применять смесь изопропанола и воды. За ходом реакции можно следить любым подходящим способом (например, ВЭЖХ). Образующуюся сначала соль карбоксилата можно нейтрализовать действием кислоты (например, соляной или уксусной). Например, удобно использовать приблизительно 1,5 экв. уксусной кислоты. После нейтрализации соединение 10 выделяют любым подходящим методом (например, хроматографией или кристаллизацией). В соответствии с другим аспектом изобретения соединение формулы 10 можно кристаллизовать,добавляя кристалл "затравки" к раствору, содержащему соединение формулы 10. Публикация международной патентной заявки WO 2005/113508 раскрывает некоторые особые кристаллические формы 6-(3-хлоро-2-фторобензил)-1-[(S)-1-гидроксиметил-2-метилпропил]-7-метокси-4 оксо-1,4-дигидрохинолон-3-карбоновой кислоты. Публикация международной патентной заявки WO 2005/113508 целиком и полностью инкорпорирована здесь посредством ссылки (в особенности, см. стр. 12-62 этой заявки). Особые кристаллические формы идентифицируются там как кристаллическая формаII и кристаллическая форма III. Картина рентгеновского рассеяния на порошке для кристаллической формы II характеризуется пиками дифракции при углах дифракции 2 , равных 6,56, 13,20, 19,86,20,84, 21,22 и 25,22, что было измерено на рентгеновском порошковом дифрактометре. Структура рентгеновского рассеяния на порошке для кристаллической формы III характеризуется пиками дифракции при углах дифракции 2 , равных 8,54, 14,02, 15,68, 17,06, 17,24, 24,16 и 25,74, что было измерено на рентгеновском порошковом дифрактометре. В публикации международной патентной заявки WO 2005/113508 также описаны способы получения кристаллической формы 6-(3-хлоро-2-фторобензил)-1-[(S)-1-гидроксиметил-2-метилпропил]-7 метокси-4-оксо-1,4-дигидрохинолон-3-карбоновой кислоты с полученной экстраполяцией температурой плавления около 162,1 С, а также способы получения кристаллической "затравки" с чистотой кристалла не менее 70%. Соответственно, кристаллы затравки 6-(3-хлоро-2-фторобензил)-1-[(S)-1-гидроксиметил 2-метилпропил]-7-метокси-4-оксо-1,4-дигидрохинолон-3-карбоновой кислоты можно получить, как описано в публикации международной патентной заявки WO 2005/113508. Представленный на приводимой далее схеме 1 способ позволяет благоприятно получить неочищенную ("сырую") смесь соединения 10,которое можно непосредственно закристаллизовать, получив кристаллическую форму III без дополнительной очистки (например, без предварительного получения другого полиморфа, такого как кристаллическая форма II, или без некоторых других форм предварительной очистки), см. пример 6 ниже. В случаях, если идентифицированные здесь соединения являются достаточно основными или кислыми для образования стабильных основных или кислых солей, изобретение относится также к солям таких соединений. Такие соли могут использоваться в качестве интермедиатов, например, для очистки этих соединений. Примеры полезных солей включают соли присоединения органической кислоты, образуемые с кислотами, например тозилат, метансульфонат, ацетат, цитрат, малонат, тартрат, сукцинат, бензоат, аскорбат, -кетоглутарат и -глицерофосфат. Можно получать также подходящие неорганические соли, включая гидрохлориды, сульфаты, нитраты, бикарбонаты и карбонаты. Соли можно получать обычными способами, хорошо известными в уровне техники, например, по реакции между достаточно основным соединением, таким как амин, с подходящий кислотой, обеспечивающей анион. Могут также использоваться соли щелочных металлов (например, калия, натрия или лития), щелочно-земельных металлов (например, кальция или магния) и карбоновых кислот. Сведения, подтверждающие возможность осуществления изобретения Ниже изобретение иллюстрируется следующими неограничивающими его область примерами. Ингибитор интегразы формулы 10 можно получить, как показано на следующей схеме 1. Схема 1 Пример 1. Получение соединения 3. Соединение 2 (10 г) добавили к 192 мл ТГФ и охладили смесь до -20 С. Затем смесь последователь-6 022099 но обработали 21 мл 1 М раствора дибутилмагния в гептане и 19,2 мл 2,5 М раствора н-бутиллития в гексане, поддерживая температуру -20 С. Добавили 3-хлор-2-фторбензальдегид (7,3 г) и дали смеси нагреться до 0 С. Через 2 ч при этой температуре реакция была остановлена добавлением 55 мл 2 М хлороводородной кислоты. Органическую фазу отделили и экстрагировали 92 мл этилацетата. Объединенные органические фазы промыли 92 мл насыщенного водного раствора хлорида натрия. Органическую фазу сконцентрировали и осадили продукт добавлением 200 мл гептана. Суспензию отфильтровали и полученный продукт высушили на воздухе, получив соединение 3. 1H ЯМР (ДМСО-d6, 400 МГц)12,15 (br s, 1H), 7,81 (s, 1H), 7,42 (t, J=7,2 Гц, 1H), 7,26 (t, J=6,8 Гц,1H), 7,15 (t, J=7,8 Гц, 1H), 6,77 (s, 1H), 6,09 (d, J=4,7 Гц, 1H), 5.90 (d, J=4,9 Гц, 1H), 3,84 (s, 3 Н), 3,80 (s,3 Н). Альтернативно, соединение 3 может быть получено следующим способом. Соединение 2 (20 г) добавили к 300 мл ТГФ и охладили до -20 С. Смесь последовательно обработали 75,93 г мл раствора ВЕМ-В (аддукт бутилэтилмагния и бутанола) в гептане и 35,08 г 28 мас.% раствора трет-бутиллития в гептане, поддерживая температуру -20 С. Затем добавили 3-хлор-2 фторбензальдегид (15,80 г) и дали смеси нагреться до 0 С. Через 2 ч при этой температуре реакция была остановлена добавлением 2 М хлороводородной кислоты. Органическую фазу отделили и экстрагировали этилацетатом, после чего высушили над сульфатом натрия и осадили продукт добавлением МТБЭ. Суспензию отфильтровали и высушили продукт на воздухе, получив соединение 3 (18,00 г, выход 69,1%). 1H ЯМР (ДМСО-d6, 400 МГц)12,15 (br s, 1H), 7,81 (s, 1H), 7,42 (t, J=7,2 Гц, 1H), 7,26 (t, J=6,8 Гц,1H), 7,15 (t, J=7,8 Гц, 1H), 6,77 (s, 1H), 6,09 (d, J=4,7 Гц, 1H), 5,90 (d, J=4,9 Гц, 1H), 3,84 (s, 3H), 3,80 (s,3H). Соединение 3 может быть также получено способом, приведенным на следующей схеме. Соединение 14 (10 г) добавили к 28 мл ТГФ и 9 мл бисдиметиламиноэтилового простого эфира,предварительно охлажденного до 0 С. Добавили изопропилмагний хлорид (22,9 мл 2,07 М раствора в ТГФ) и дали смеси нагреться до комнатной температуры в течение ночи. Перед добавлением 3-хлор-2 фторбензальдегида, чтобы улучшить степень конверсии, в реакционную смесь добавили дополнительную порцию изопропилмагний хлорида (5 мл). Затем добавили 3-хлор-2-фторбензальдегид (4,4 мл). После перемешивания при температуре окружающей среды в течение 2 ч в смесь добавили 38,6 г 14 мас.% раствора, содержащего комплекс изопропилмагний хлорида и хлорида лития в ТГФ. После перемешивания в течение ночи при температуре окружающей среды через реакционную смесь барботировали газообразный СО 2. Реакция была остановлена добавлением 2 М хлороводородной кислоты до рН 3. Органическую фазу отделили и экстрагировали этилацетатом. Объединенные органические фазы промыли насыщенным водным раствором хлорида натрия. Органическую фазу сконцентрировали, и осадили продукт добавлением МТБЭ. Суспензию отфильтровали и высушили продукт на воздухе, получив соединение 3. 1 Н ЯМР (ДМСО-d6, 400 МГц)12,15 (br s, 1H), 7,81 (s, 1H), 7,42 (t, J=7,2 Гц, 1H), 7,26 (t, J=6,8 Гц,1H), 7,15 (t, J=7,8 Гц, 1H), 6,77 (s, 1H), 6,09 (d, J=4,7 Гц, 1H), 5,90 (d, J=4,9 Гц, 1H), 3,84 (s, 3 Н), 3,80 (s,3 Н). Соединение 3 может быть также получено способом, приведенным на следующей схеме. Пример 2. Получение соединения 4. К трифторуксусной кислоте (33,13 г), предварительно охлажденной в бане со льдом, добавили триэтилсилан (6,83 г). Затем к смеси добавили соединение 3 (10 г), поддерживая температуру ниже 15 С. После перемешивания в течение 2 ч добавили МТБЭ, чтобы осадить продукт. Суспензию отфильтровали и продукт промыли дополнительным количеством МТБЭ. После высушивания получили 9,12 г соединения 4. 1 Н ЯМР (ДМСО-d6, 400 МГц)12,11 (br s, 1H), 7,47 (s, 1H), 7,42-7,38 (m, 1H), 7,14-7,08 (m, 2H), 6,67(s, 1H), 3,87-3,84 (m, 8H). В качестве альтернативы соединение 4 может быть получено следующим образом. К трифторуксусной кислоте (49,02 г), предварительно охлажденной в бане со льдом, добавили триэтилсилан (7,50 г). К смеси добавили соединение 3 (14,65 г), поддерживая температуру ниже 15 С. После перемешивания в течение 1 ч добавили раствор 17,63 г ацетата натрия в 147 мл метанола. Смесь кипятили в течение 3 ч, затем охладили до 0 С. Суспензию отфильтровали и промыли продукт дополнительным количеством метанола. После высушивания получили 12,3 г соединения 4 (выход 89,7%). 1(s, 1H), 3,87-3,84 (m, 8H). Пример 3. Получение соединения 5 а. Имидазол (0,42 г) и 1,1'-карбонилдиимидазол (5,49 г) суспендировали в 30 мл ТГФ при температуре окружающей среды. Затем за один прием добавили соединение 4 (10 г) и перемешивали смесь при температуре окружающей среды до окончания реакции (контроль по ВЭЖХ). Полученную суспензию отфильтровали и промыли осадок МТБЭ. Затем осадок высушили, получив соединение 5 а. 1H ЯМР (ДМСО-d6, 400 МГц)7,99 (s, 1H), 7,52 (s, 1H), 7,41-7,38 (m, 1H), 7,30 (s, 1H), 7,12-7,08 (m,2 Н), 7,04 (s, 1H), 6,81 (s, 1H), 3,91 (s, 2H), 3,90 (s, 3 Н), 3,79 (s, 3 Н). Пример 4. Получение соединения 6 а. Имидазол (0,42 г) и 1,1'-карбонилдиимидазол (5,49 г) суспендировали в 30 мл ТГФ при температуре окружающей среды. Затем за один прием добавили соединение 5 а (10 г) и смесь перемешивали при температуре окружающей среды в течение 4 ч, чтобы образовалась суспензия соединения 5 а. В отдельной колбе суспендировали 8,91 г моноэтил малоната калия в 40 мл ТГФ. Добавили хлорид магния (4,40 г),после чего выдерживали полученную суспензию при 55 С в течение 90 мин. Суспензию соединения 5 а добавили к смеси хлорида магния/моноэтил малоната калия и перемешивали при 55 С в течение ночи. Затем смесь охладили до комнатной температуры и остановили реакцию добавлением по каплям 80 мл 28 мас.% водного раствора Н 3 РО 4. Органическую фазу отделили и последовательно промыли водными растворами NaHSO4, KHCO3 и NaCl. Затем ее сконцентрировали, получив масло, которое заново упарили с этанолом. Полученное твердое вещество растворили в 30 мл этанола и 6 мл воды. Соединение 6 а кристаллизовали при охлаждении. Полученные кристаллы отделили фильтрацией и промыли продукт водным раствором этанола. После высушивания получили соединение 6 а. 1 Н ЯМР (ДМСО-d6, 400 МГц)7,51 (s, 1H), 7,42-7,38 (m, 1H), 7,12-7,10 (m, 2 Н), 6,70 (s, 1H), 4,06 (q,J=7,0 Гц, 2 Н), 3,89 (s, 8H), 3,81 (s, 2H), 1,15 (t, J=7,0 Гц, 3 Н). Альтернативно, соединение 6 а может быть получено следующим образом. Карбонилдиимидазол (10,99 г) суспендировали в 60 мл ТГФ при температуре окружающей среды. Затем за один прием добавили соединение 4 (20 г) и перемешивали смесь при температуре окружающей среды в течение 30 мин, чтобы образовалась суспензия соединения 5 а. В отдельной колбе суспендировали 15,72 г моноэтил малоната калия в 100 мл ТГФ. Добавили хлорид магния (6,45 г) и нагревали полученную суспензию при 55 С в течение 5 ч. Суспензию соединения 5 а добавили к смеси хлорида маг-8 022099 ния/моноэтил малоната калия и перемешивали при 55 С в течение ночи. Затем смесь охладили до комнатной температуры и остановили реакцию добавлением 120 мл 28 мас.% водного раствора Н 3 РО 4. Органическую фазу отделили и последовательно промыли водными растворами KHCO3 и NaCl. Затем ее сконцентрировали, получив масло, и переупарили с этанолом. Полученное твердое вещество растворили в 100 мл этанола и 12 мл воды. Соединение 6 а кристаллизовали при охлаждении. Полученные кристаллы отделили фильтрацией и промыли продукт водным раствором этанола. После высушивания получили 21,74 г соединения 6 а (выход 89%). 1H ЯМР (ДМСО-d6, 400 МГц)7,51 (s, 1H), 7,42-7,38 (m, 1H), 7,12-7,10 (m, 2 Н), 6,70 (s, 1H), 4,06 (q,J=7,0 Гц, 2 Н), 3,89 (s, 8H), 3,81 (s, 2H), 1,15 (t, J=7,0 Гц, 3 Н). Пример 5. Получение соединения 9 а. Соединение 6 а (20 г) перемешивали с 6,6 г диметилформамид диметилацеталя, 66 г толуола и 0,08 г ледяной уксусной кислоты. Смесь нагревали при 90 С в течение 4 ч. Затем смесь охладили до комнатной температуры и добавили 5,8 г (S)-2-амино-3-метил-1-бутанола. Смесь перемешивали при температуре окружающей среды в течение 1 ч, а затем сконцентрировали, получив густое масло. После этого добавили диметилформамид (36 г), хлорид калия (1,8 г) и бис(триметилсилил)ацетамид (29,6 г) и нагревали смесь при 90 С в течение 1 ч. Смесь охладили до комнатной температуры и разбавили 200 г дихлорметана. Добавили разбавленную хлороводородную кислоту (44 г, примерно 1H) и перемешивали смесь при температуре окружающей среды 20 мин. Органическую фазу отделили, последовательно промыли водой,водным раствором гидрокарбоната натрия и снова водой. Растворитель заменили на ацетонитрил и довели объем смеси до 160 мл. Смесь нагревали, пока она не стала прозрачной, слегка охладили, ввели затравку и охладили, чтобы выделить кристаллическое соединение 9 а. Продукт отделили фильтрацией и дополнительно промыли холодным ацетонитрилом. После вакуумной сушки получили соединение 9 а. 1 Н ЯМР (ДМСО-d6, 400 МГц)8,61 (s, 1H), 7,86 (s, 1H), 7,45 (t, J=7,4 Гц, 1H), 7,26 (s, 1H), 7,23-7,14(m, 2 Н), 5,10 (br s, 1H), 4,62 (br s, 1H), 4,18 (q, J=7,0 Гц, 2 Н), 4,03 (s, 2H), 3,96 (s, 3H), 3,92-3,84 (m, 1H),3,78-3,75 (m, 1H), 2,28 (br s, 1H), 1,24 (t, J=7,0 Гц, 3 Н), 1,12 (d, J=6,4 Гц, 3 Н), 0,72 (d, J=6,4 Гц, 3 Н). В качестве альтернативы соединение 9 а может быть получено следующим образом. Соединение 6 а (50 г) перемешивали с 17,5 г диметилформамид диметил ацеталя, 90 г ДМФ и 0,2 г ледяной уксусной кислоты. Смесь нагревали до 65 С в течение 3 ч. Затем смесь охладили до комнатной температуры и добавили 14,5 г (S)-2-амино-3-метил-1-бутанола и 25 г толуола. Смесь перемешивали при комнатной температуре в течение ночи, после чего сконцентрировали дистилляцией. После этого добавили хлорид калия (4,5 г) и бис(триметилсилил)ацетамид (80,2 г) и смесь нагревали до 90 С в течение 2 ч. Смесь охладили до комнатной температуры и разбавили 250 г дихлорметана. Добавили разбавленную хлороводородную кислоту (110 г, 1N) и перемешивали смесь при температуре окружающей среды 30 мин. Органическую фазу отделили, последовательно промыли водой, водным раствором гидрокарбоната натрия и снова водой. Растворитель заменили на ацетонитрил путем дистилляции. Смесь нагревали, пока она не стала прозрачной, слегка охладили, ввели затравку и охладили, чтобы полностью выделить кристаллическое соединение 9 а. Продукт отделили фильтрацией и дополнительно промыли холодным ацетонитрилом. После вакуумной сушки получили 48,7 г (выход 81%) соединения 9 а. 1(m, 2H), 5,10 (br s, 1H), 4,62 (br s, 1H), 4,18 (q, J=7,0 Гц, 2H), 4,03 (s, 2 Н), 3,96 (s, 3 Н), 3,92-3,84 (m, 1H),3,78-3,75 (m, 1H), 2,28 (br s, 1H), 1,24 (t, J=7,0 Гц, 3 Н), 1,12 (d, J=6,4 Гц, 3 Н), 0,72 (d, J=6,4 Гц, 3 Н). Пример 6. Получение соединения 10. Соединение 9 а (6,02 г) суспендировали в 36 мл изопропанола и 24 мл воды. Добавили водный раствор хлорида калия (2,04 г 45 мас.% раствор) и нагрели смесь до 40 С. Через 3 ч добавили 1,13 г ледяной уксусной кислоты и внесли в смесь затравку соединения 10 (10 мг). Смесь охлаждали на ледяной бане 2 ч и полученное твердое вещество отделяли фильтрацией. Осадок промывали водным изопропанолом и высушивали, получив соединение 10. 1 Н ЯМР (ДМСО-d6, 400 МГц)15,42 (s, 1H), 8,87 (s, 1H), 8,02 (s, 1H), 7,48-7,45 (m, 2H), 7,23 (t, J= 6,8 Гц, 1H), 7,17 (t, J=7,8 Гц, 1H), 5,18 (br s, 1H), 4,86 (br s, 1H), 4,10 (s, 2H), 4,02 (s, 3 Н), 3,97-3,96 (m, 1H),3,79-3,76 (m, 1H), 2,36 (br s, 1H), 1,14 (d, J=6,3 Гц, 3 Н), 0,71 (d, J=6,3 Гц, 3 Н). Пример 7. Получение соединения 13. Синтез соединения 9 а из соединения 7 а, описанный в приведенном выше примере 5, приводит к образованию еще одного продукта, который, вероятнее всего, получается из-за наличия примеси (S)-2 амино-4-метил-1-пентанола в (S)-2-амино-3-метил-1-бутаноле. Чтобы подтвердить идентичность второго продукта, проведен независимый синтез соединения 13, как показано ниже. Соединение 13 получено из соединения 12 по способу, аналогичному приведенному выше для соединения 10 в примере 6. После описанной процедуры продукт экстрагировали анизолом. Описанный продукт выделили в виде пены после удаления растворителя. 1(t, J=7,6 Гц, 1H), 5,19 (br s, 1H), 4,09 (s, 2H), 4,00 (s, 3 Н), 3,77 (br s, 2H), 1,94-1,87 (m, 1H), 1,82-1,75 (m,1H), 1,43 (hept, J=6.4 Гц, 1H), 0,89 (d, J=6,4 Гц, 3 Н), 0,85 (d, J=6,8 Гц, 3 Н). Промежуточное соединение 12 получали следующим образом. а) Соединение 12 получали из соединения 6 а по способу, аналогичному приведенному выше для получения соединения 9 а, за исключением того, что вместо (S)-2-амино-3-метил-1-бутанола был использован (S)-(+)-2-амино-4-метил-1-пентанол. Целевой продукт выделили в виде пены после того, как досуха сконцентрировали конечный раствор ацетонитрила. 1 Н ЯМР (ДМСО-d6, 400 МГц)8,54 (s, 1H), 7,86 (s, 1H), 7,46-7,43 (m, 1H), 7,25 (s, 1H), 7,22-7,14 (m,2H), 4,97 (br s, 1H), 4,20-4,16 (m, 2H), 4,03 (s, 2H), 3,95 (s, 3 Н), 3,73 (br s, 2H), 1,83-1,82 (m, 1H), 1,72-1,69(m, 1H), 1,43 (hept, J=6,4 Гц, 1H), 1,24 (t, J=7,2 Гц, 3 Н), 0,88 (d, J=6,4 Гц, 3 Н), 0,84 (d, J=6,4 Гц, 3 Н). Соединение 13 может использоваться в качестве ингибитора ВИЧ интегразы, как описано в публикации международной патентной заявки WO 2004/046115. Соответственно, изобретение также относится к соединению 13 или его соли, а также и к способам получения соединения 13 или его соли. Изобретение относится также к композициям, содержащим соединение 10 или его соль и соединение 13 или его соль,а также к композициям, содержащим соединение 9 а или его соль и соединение 12 или его соль. Такие композиции полезны для приготовления ингибиторов интегразы, описанных в публикации международной патентной заявке WO 2004/046115. В качестве альтернативы соединение 10 может быть получено из соединения 2, как описано в следующих иллюстративных примерах 8-12, исходя из 1 кг стартового соединения. Пример 8. Получение соединения с формулой 3. Соединение 2 смешали с безводным тетрагидрофураном и нагрели, чтобы образовался раствор или тонкая суспензия. Смесь охладили до температуры от -20 до -30 С и добавили бутилэтилмагний в гептане. В отдельном реакторе бутиллитий в гексане смешали с охлажденным (до температуры от -20 до-30 С) тетрагидрофураном. Суспензию соединения 2 с бутилэтилмагнием добавили к раствору нбутиллития, поддерживая температуру смеси в интервале от -20 до -30 С. Реакцию обмена литий/галоген контролировали по ВЭЖХ. После завершения реакции добавили раствор 3-хлор-2-фторбензальдегида в тетрагидрофуране. Через 1 ч смесь нагрели до 0 С. Окончание реакции контролировали по ВЭЖХ. После завершения реакцию остановили водным раствором хлороводородной кислоты, доведя рН до 1-3. После разделения фаз водную фазу дважды экстрагировали этилацетатом. Объединенные органические фазы высушили сульфатом натрия при 18-25 С. После того как сульфат натрия отфильтровали, растворитель заменили на МТБЭ и полученную суспензию охладили до 0 С. Продукт отделили фильтрацией, промыли холодным МТБЭ и высушили при 40 С, получив соединение 3. 1. Загрузить 1,00 кг соединения 2 и 8,7 кг ТГФ в реактор (1). 2. Нагреть смесь до 45-50 С так, чтобы растворились все твердые вещества или образовалась тонкая однородная суспензия и на дне реактора не оставалось твердых частиц. 3. Охладить содержимое реактора (1) до температуры от -20 до -30 С. 4. Загрузить в реактор BuEtMg (15% мас./мас., в гептане) (-1,8 кг, 0,6 экв.) (1), поддерживая температуру реакционной смеси ниже -20 С в течение всего времени добавления. 5. В отдельный реактор (2) загрузить 2,6 кг ТГФ и охладить до температуры от -20 до -30 С. 6. Загрузить в реактор (2) н-BuLi (в гексане) (1,9 кг, 1,8 экв.), поддерживая температуру ниже -20 С в течение всего времени добавления. 7. Перенести содержимое реактора (1) в реактор (2), поддерживая температуру ниже -20 С в течение всего времени добавления. 8. Загрузить в реактор (3) 0,5 кг ТГФ и охладить до (-20) - (-30)С. 9. Перенести содержимое реактора (3) в реактор (1) затем в реактор (2) для прямой промывки. 10. Примерно через 15 мин после начала синтеза отобрать из реакционной смеси пробу и проанализировать на ВЭЖХ, чтобы определить, как идет обмен литий/галоген. (Обычно остаток соединения 2 составляет 1-8%. Если количество соединения 2 больше 8%, отобрать пробу еще через 30 мин и до внесения дополнительного количества н-BuLi). 11. Смешать в подходящем контейнере 0,79 кг альдегида и 0,79 кг ТГФ. 12. Добавить содержимое контейнера в реактор. При добавлении поддерживать температуру реакционной смеси ниже -20 С. 13. Перемешивать реакционную смесь при -20 С в течение 1 ч, затем нагреть до 0 С. 14. Остановить реакцию добавлением 2 М HCl (-3,8 кг) до рН 1-3. 15. Разделить фазы. 16. Проэкстрагировать водную фазу 2,3 кг EtOAc. 17. Проэкстрагировать водную фазу 2,3 кг EtOAc. 18. Отбросить водную фазу. 19. Объединить органические фазы и высушить над 2 кг Na2SO4 в течение по меньшей мере 1 ч. Температура органической фазы перед добавлением Na2SO4 должна быть 20-25 С. 20. Профильтровать суспензию, чтобы удалить Na2SO4. 21. Сконцентрировать объединенные органические фазы путем вакуумной дистилляции до 1,5 л(должна образоваться густая суспензия). 22. Добавить к суспензии 2,8 кг МТБЭ (метил трет-бутиловый простой эфир). 23. Сконцентрировать смесь до 1,5 л. 24. Добавить к суспензии 2,8 кг МТБЭ. 25. Сконцентрировать смесь до 1,5 л. 26. Добавить к суспензии 1,9 кг МТБЭ. 27. Охладить суспензию до -0 С и отделить соединение 3 фильтрацией. 28. Промыть посуду для дистилляции 1,9 кг МТБЭ, предварительно охлажденного до 0 С. 29. Осушать осадок до тех пор, пока не будут получены твердые гранулы. Чистота соединения 3 может быть повышена, если его ресуспендировать в 6 объемах смеси толуол:НОАс (85:15). 30. Сушить влажный осадок под вакуумом при температуре менее 40 С. Пример 9. Получение соединения с формулой 4. Соединение 3 смешали с трифторуксусной кислотой и перемешивали пока не образовался раствор. Раствор охладили до температуры от -3 до 3 С и добавили триэтилсилан, поддерживая температуру 15 С. Ход реакции контролировали по ВЭЖХ. После завершения реакции добавили МТБЭ, чтобы осадить соединение 4, и охладили смесь до 0 С. Продукт отделили фильтрацией, промыли МТБЭ и высушили при 60 С, получив соединение 4. 1. Растворить 1,00 кг соединения 3 в 1,7 кг ТФУК. 2. Охладить реакционную смесь до (-3)-(+3)С. 3. Загрузить 0,4 кг триэтилсилана в реакционную смесь. При добавлении поддерживать температуру реакционной смеси ниже 15 С. 4. Через 30 мин после добавления триэтилсилана отобрать из реакционной смеси пробу и проанализировать на ВЭЖХ, чтобы определить, завершилось ли превращение соединения 3 в соединение 4. 5. Загрузить в реакционную смесь 4,0 кг МТБЭ, поддерживая при этом температуру смеси ниже 15 С. 6. Охладить смесь до 0 С и перемешивать в течение по меньшей мере 30 мин. 7. Отделить соединение 4 фильтрацией и промыть реакционный сосуд 1,6 кг МТБЭ. 8. Высушить полученное соединение 4 под вакуумом при температуре менее 60 С. Примечание: чистоту соединения 4 можно увеличить путем ресуспендирования в 4 объемах ацетона. Суспензию нагревали до 40 С в течение 2 ч, перед фильтрацией охлаждали 12 ч до 18-25 С, затем дважды промыли 1 объемом ацетона. Пример 10. Получение соединения с формулой 6 а. Карбонилдиимидазол и имидазол смешали с безводным тетрагидрофураном. К смеси добавили соединение 4, чтобы образовалось соединение 5 а. Реакцию контролировали по ВЭЖХ. В отдельном реакторе смешали моноэтилмалонат калия и тетрагидрофуран, после чего добавили безводный хлорид магния, поддерживая температуру NMT 30 С. Полученную суспензию нагревали при 50 С в течение по меньшей мере 2 ч, после чего добавили соединение 5 а (в составе смеси). Реакцию контролировали по ВЭЖХ. После того как образование соединения 5 а завершилось, смесь охладили до 18-25 С и добавили водный раствор фосфорной кислоты, чтобы остановить реакцию. Органическую фазу промыли водным раствором бисульфата натрия, солевым раствором, дигидрокарбонатом калия и снова солевым раствором, после чего отфильтровали. Растворитель заменили на безводный этанол. Добавляют воду и нагревают смесь, чтобы растворить твердые вещества, затем охлаждают приблизительно до 40 С, вносят затравку соединения 6 а и охлаждают до 0-5 С. Продукт отфильтровали, промыли холодным водным раствором этанола и высушили при 40 С, получив соединение 6 а. Процедура: 1. Загрузить 0,55 кг CDI и 0,042 кг имидазола в реактор 1. 2. Загрузить 2,67 кг ТГФ в реактор 1 и перемешивать, чтобы образовалась суспензия. 3. Частями загрузить 1,00 кг соединения 4 в реактор 1, чтобы выделение СО 2 было умеренным. Реакция эндотермическая. 4. Загрузить 0,89 кг КЕМ в реактор 2. 5. Загрузить 4,45 кг ТГФ в реактор 2 и перемешивать, чтобы образовалась суспензия. 6. Загрузить 0,44 кг MgCl2 в реактор 2 (можно загружать частями, чтобы уменьшить тепловыделение). 7. Нагреть содержимое реактора 2 до 50 С и перемешивать при этой температуре в течение по меньшей мере 2 ч. 8. Перенести содержимое реактора 1 в реактор 2. Если переносить очень быстро, смесь временно может загустеть. 9. Перемешивать содержимое реактора 2 в течение по меньшей мере 12 ч при 50 С. 10. Охладить суспензию до температуры окружающей среды. 11. Остановить реакцию внесением в реакционную смесь 7,0 кг 28 мас.% водного раствора Н 3 РО 4(2,3 кг 85 мас.% Н 3 РО 4, растворенной в 4,7 кг Н 2 О). Реакция экзотермична. Конечное значение рН водной фазы должно быть 1-2. 12. Промыть органическую (верхнюю) фазу 1,2 кг 20 мас.% водного раствора NaHSO4 (0,24 кгNaHSO4 растворить в 0,96 кг Н 2 О). Конечное значение рН водной фазы должно быть 1-2. 13. Промыть органическую (верхнюю) фазу 1,2 кг 20 мас.% водного раствора NaCl (0,24 кг NaCl растворить в 0,96 кг Н 2 О). 14. Промыть органическую (верхнюю) фазу 5,0 кг 10 мас.% водного раствора KHCO3 (0,50 кгKHCO3 растворить в 4,5 кг Н 2 О). Конечное значение рН водного слоя должно быть 8-10. 15. Промыть органическую (водную) фазу 1,2 кг 20 мас.% водного раствора NaCl (0,24 кг NaCl, 0,96 кг Н 2 О). Конечное значение рН водной фазы должно быть 7-9. 16. Сконцентрировать органическую фазу и заменить растворитель на EtOH. 17. Довести концентрацию до 3,5 л/кг. 18. Загрузить 0,6 объема воды. 19. Нагревать 70-80 С, чтобы образовался прозрачный раствор. 20. Охладить до 40 С и внести затравку 0,1 мас.% соединения 6. 21. Медленно охладить до 5 С. 22. Выдерживать в течение по меньшей мере 2 ч. 23. Отфильтровать и дважды промыть осадок смесью EtOH:H2O 50:50 (1,2 кг EtOH на 1,5 кг Н 2 О),по 1,35 кг на каждую промывку. 24. Высушить осадок при температуре ниже 50 С. Пример 11. Получение соединения формулы 9 а. Соединение 6 а смешали с толуолом, диметилацеталем N,N-диметилформамида и ледяной уксусной кислотой, после чего нагрели до 100 С. Реакцию контролировали по ВЭЖХ. После того как образование соединения 7 а завершилось, смесь охладили до 18-25 С, после чего добавили (S)-(+)-валинол. Реакцию контролировали по ВЭЖХ. После того как образование соединения 8 а завершилось, смесь сконцентрировали. Остаток смешали с диметилформамидом, хлоридом калия и N,O-бистриметилсилилацетамидом и нагрели до 100 С. Реакцию контролировали по ВЭЖХ. После завершения реакции смесь охладили и добавили дихлорметан. После этого добавили водный раствор хлороводородной кислоты, чтобы десилилировать соединение 9 а. Реакцию контролировали по ТСХ. После завершения реакции органическую фазу промыли водой, водным раствором гидрокарбоната натрия и опять водой. Растворитель заменили на ацетонитрил и смесь нагрели. Внесли в смесь затравку и охладили, закристаллизовав соединение 9 а. Продукт отфильтровали, промыли холодным ацетонитрилом и высушили при 40 С, получив соединение 9 а. 1. Загрузить 1,00 кг соединения 6 а в реактор 1. 2. Загрузить 0,33 кг N,N-диметилформамид диметил ацеталя (1,1 экв.), 0,001 кг ледяной уксусной кислоты и 3,3 кг толуола в реактор 1. 3. Нагреть смесь до 100 С (нужно иметь ввиду, что МеОН при этом может отгоняться). 4. Через 1 ч реакция должна завершиться (контроль по ВЭЖХ) (может остаться 2% соединения 6 а)(пик ВЭЖХ оставшегося соединения 6 а растягивается артефактом базовой линии. ВЭЖХ на этом этапе показывает только 2% соединения 6 а относительно соединения 8 а. Эксперименты показали, что введение дополнительного количества реагентов и увеличение времени реакции, как правило, не позволяет снизить наблюдаемый уровень соединения 6 а). 5. Охладить смесь в реакторе 1 до 18-25 С. 6. Загрузить 0,29 кг (S)-(+)-валинола (1,1 экв.), растворенного в 1,0 кг толуола в реактор 1 и продолжать перемешивание при температуре окружающей среды. 7. Через 1 ч реакция должна завершиться (контроль по ВЭЖХ) (менее 1% соединения 6 а). 8. Сконцентрировать содержимое реактора 1 до 2 л/кг. 9. В реактор 1 загрузить 1,8 кг ДМФ, 0,09 кг хлорида калия (0,5 экв.) и 1,13 кг N,Обистриметилсилил ацетамида (2,2 экв.). 10. Нагреть смесь в реакторе 1 до 100 С. 11. Через 1 ч реакция должна завершиться (остается 5% соединения 8 а). 12. Охладить содержимое реактора 1 до 18-25 С. 13. Загрузить 10 кг ДХМ в реактор 1. 14. Загрузить в реактор 1 в течение 15 мин 2,0 кг 1 Н водного раствора HCl, поддерживая температуру смеси ниже 35 С. 15. Перемешивать смесь в течение по меньшей мере 10 мин, чтобы десилилировать соединение 8 а(контроль по ТСХ) (метод ТСХ: элюирующий растворитель: этилацетат 100%. Силилированное соединение 9 а Rf: 0,85. Соединение 9 а Rf: 0,50). 16. Разделить фазы. 17. Промыть органическую фазу 4,0 кг воды. 18. Промыть органическую фазу 4,0 кг 5% водного раствора гидрокарбоната натрия. 19. Промыть органическую фазу 4,0 кг воды. 20. Сконцентрировать органическую фазу дистилляцией до 1,5 л/кг соединения 6 а. 21. Заменить растворитель на ацетонитрил дистилляцией пока не образуется суспензия. Довести конечный объем до 8 л/кг соединения 6 а. 22. Нагреть смесь до кипения, чтобы твердое вещество растворилось. 23. Охладить раствор до 75 С и загрузить затравку соединения 9 а. 24. Охладить смесь до 0 С в течение по меньшей мере 2 ч и держать при этой температуре в течение по меньшей мере 1 ч. 25. Отделить соединение 9 а фильтрацией и промыть влажный осадок 1,6 кг холодного ацетонитрила. 26. Высушить влажный осадок при температуре ниже 40 С под вакуумом. Пример 12. Получение соединения формулы 10. Соединение 9 а смешали с водным раствором изопропилового спирта и нагрели до 30-40 С. Добавили водный раствор гидроксида калия, при этом реакцию контролировали по ВЭЖХ. После завершения реакции добавили ледяную уксусную кислоту и нагрели смесь до 60-70 С. Раствор профильтровали на горячем фильтре и охладили до 55-65 С. Внесли в раствор затравку (см. публикацию международной патентной заявки WO 2005/113508) и охладили до 0 С. Продукт отделили фильтрацией, промыли холодным водным раствором изопропилового спирта и высушили при 50 С, получив соединение 10. 1. Загрузить 1,00 кг соединения 9 а в реактор 1. 2. Загрузить 4,7 кг изопропилового спирта и 4,0 кг воды в реактор 1. 3. Загрузить 0,34 кг 45% водного раствора KOH в реактор 1. 4. Нагреть смесь в реакторе 1 до 30-40 С. 5. После окончания гидролиза добавить 0,19 кг ледяной уксусной кислоты. 6. Нагреть смесь до 60-70 С и пропустить раствор через барьерный фильтр в реактор 2. 7. Охладить смесь в реакторе 2 до 55-65 С. 8. Внести затравку соединения 10 (см. публикацию международную патентную заявку WO 2005/113508) в виде суспензии в 0,28 объема смеси изопропиловый спирт/вода 6:4. 9. Охладить смесь до 18-25 С в течение по меньшей мере 2 ч и перемешать, чтобы образовалась суспензия. 10. Охладить смесь до 0 С и перемешивать в течение по меньшей мере 2 ч. 11. Отделить соединение 10 фильтрацией и промыть осадок 31S холодной смеси изопропиловый спирт/вода (6:4). 12. Высушить выделенное твердое вещество при температуре менее 50 С под вакуумом. Пример 13. Получение соединения 15. Бисдиметиламиноэтиловый простой эфир (2,84 г) растворили в 42 мл ТГФ и охладили на ледяной бане. Затем последовательно медленно добавили изопропилмагний хлорид (8,9 мл 2 М раствора в ТГФ), а затем соединение 14 (5 г растворили в 5 мл ТГФ). Смеси дали нагреться до температуры окружающей среды и перемешивали в течение ночи. Добавили 2,1 мл 3-хлор-2-фторбензальдегида. После перемешивания в течение 1 ч реакцию остановили добавлением 2 Н HCl до рН 7. Продукт экстрагировали этилацетатом и высушили органическую фазу над сульфатом натрия. Растворитель заменили на гептан, чтобы осадить продукт, и затем добавили смесь гептан:МТБЭ (4:1), чтобы образовалась суспензия. После фильтрации твердое вещество суспендировали в толуоле, отфильтровали и высушили под вакуумом, получив соединение 15. 1 Соединение 14 (5 г), изопропилмагний хлорид (8,9 мл 2 М раствора в ТГФ) и ТГФ (56 мл) смешали при температуре окружающей среды и затем нагревали при 50 С в течение 5 ч. После охлаждения до температуры окружающей среды и перемешивания в течение ночи добавили по каплям 2,1 мл 3-хлор-2 фторбензальдегида, чтобы образовалась суспензия. После перемешивания в течение ночи твердое вещество отделили фильтрацией и промыли МТБЭ, получив соединение 15 а. Пример 15. Получение соединения 16. К трифторуксусной кислоте (2,3 мл), охлажденной на ледяной бане, добавили триэтилсилан (1,2 мл). К смеси прибавили соединение 15 (1,466 г), поддерживая температуру ниже 5 С. После перемешивания в течение 2 ч добавили лед, чтобы остановить реакцию. Продукт экстрагировали дихлорметаном и промыли органическую фазу водным раствором NaHCO3. Органическую фазу высушили над Na2SO4 и сконцентрировали досуха. Продукт очистили колоночной хроматографией на силикагеле, получив 1,341 г соединения 16. 1H ЯМР (CDCl3, 400 МГц)7,20 (t, J=7,0 Гц, 1H), 6,99-6,91 (m, 3H), 6,46 (s, 1H), 3,91 (s, 3H), 3,81 (s,5H). Все публикации, патенты и патентная документация включены сюда по ссылке так, как будто они были бы включены по ссылке индивидуально. Изобретение было описано со ссылками на различные специфические и предпочтительные аспекты и методы. Однако следует понимать, что сохраняя дух изобретения и не выходя за пределы его области, в него можно внести множество различных вариаций и модификаций. или его соль. 3. Способ получения соединения формулы 3 или его соли, включающий металлирование соединения формулы 2 или его соли с последующим взаимодействием полученного металлорганического соединения с 3-хлор 2-фторбензальдегидом. 4. Способ по п.3, в котором полученное металлорганическое соединение представляет собой литийорганическое соединение. 5. Способ по п.4, в котором литийорганическое соединение получают путем осуществления реакции между соединением формулы 2 и соединением диалкилмагния с последующей обработкой соединением алкиллития. 6. Способ по п.4, в котором литийорганическое соединение получают обработкой соединения 2 дибутилмагнием или бутилэтилмагнием с последующей обработкой соединением алкиллития. 7. Способ по п.5 или 6, в котором соединение алкиллития представляет собой н-бутиллитий или трет-бутиллитий. 8. Способ по п.5, в котором соединение формулы 2 обрабатывают соединением диалкилмагния с последующей обработкой соединением алкиллития при температуре от приблизительно -100 до приблизительно 0 С. 9. Способ по п.5, в котором соединение формулы 2 обрабатывают соединением диалкилмагния с последующей обработкой соединением алкиллития при температуре от приблизительно -40 до приблизительно 0 С. 10. Способ по любому из пп.3-9, в котором соединение формулы 2 или его соль получают путем бромирования 2,4-диметоксибензойной кислоты. 11. Способ по п.10, в котором 2,4-диметоксибензойную кислоту бромируют путем обработки бромом в присутствии кислоты. 12. Способ получения соединения формулы 10 или его соли, включающий получение соединения 2 или его соли путем бромирования 2,4-диметоксибензойной кислоты,превращение соединения 2 или его соли в соединение 3 или его соль путем металлирования соединения формулы 2 с последующим взаимодействием полученного металлорганического соединения с 3-хлор-2-фторбензальдегидом,превращение соединения формулы 3 или его соли в соединение формулы 4 или его соли с помощью обработки силановым восстановителем в присутствии кислоты,превращение соединения формулы 4 или его соли в соединение формулы 5' где Rc представляет собой уходящую группу,превращения соединения 5' в соединение формулы 6 где R представляет собой С 1-С 6 алкил, с помощью обработки соответствующей моноалкилмалонатной соли,превращение соединения формулы 6 в соединение формулы 7 путем обработки с хлоридом калия и N,О-бистриметилсилилацетамидом, и превращение соединения формулы 9 в соединение формулы 10 путем обработки основанием и кристаллизацию соединения формулы 10 путем добавления кристаллической затравки к раствору, содержащему соединение формулы 10. 13. Способ по п.12, отличающийся тем, что полученное металлорганическое соединение представляет собой литийорганическое соединение. 14. Способ по п.13, отличающийся тем, что литийорганическое соединение получают путем приведения во взаимодействие соединения формулы 2 с соединением диалкилмагния с последующей обработкой соединением алкиллития. 15. Способ по п.13, отличающийся тем, что литийорганическое соединение получают путем обработки соединения формулы 2 с дибутилмагнием или бутилэтилмагнием с последующей обработкой соединением алкиллития. 16. Способ по п.14 или 15, отличающийся тем, что соединение алкиллития представляет собой нбутиллитий или трет-бутиллитий. 17. Способ по п.14, отличающийся тем, что соединение формулы 2 обрабатывают соединением диалкилмагния с последующей обработкой соединением алкиллития при температуре приблизительно от-100 до 0 С. 18. Способ по п.14, отличающийся тем, что соединение формулы 2 обрабатывают с помощью соединения диалкилмагния с последующей обработкой соединением алкиллития при температуре приблизительно от -40 до 0 С. 19. Способ по п.12, отличающийся тем, что 2,4-диметоксибензойную кислоту бромируют путем обработки бромом в присутствии кислоты. 20. Способ по п.12, в котором силановый восстановитель представляет собой триэтилсилан, а кислота представляет собой трифторуксусную кислоту. 21. Способ по п.12, в котором указанная уходящая группа представляет собой галоген или 1 имидазолил. 22. Способ по п.12, в котором соединение формулы 5' представляет собой соединение формулы 5 а 23. Способ по п.22, в котором соединение формулы 5 а получают обработкой соединения формулы 4 1,1'-карбонилдиимидазолом. 24. Способ по п.12, в котором R в соединении формулы 6 представляет собой этил. 25. Способ по п.24, в котором соединение формулы 5' превращают в соединение формулы 6 путем обработки моноэтилмалонатом калия. 26. Способ по п.12, в котором соединение формулы 6 превращают в соединение формулы 7 путем обработки диметилацеталем N,N-диметилформамида. 27. Способ по п.26, в котором обработку диметилацеталем N,N-диметилформамида проводят в присутствии уксусной кислоты при температуре от приблизительно 50 до приблизительно 150 С. 28. Способ по п.12, в котором соединение формулы 9 превращают в соединение формулы 10 путем обработки водным раствором гидроксида калия в водном растворе изопропилового спирта. 29. Способ по п.12, отличающийся тем, что кристаллическая затравка характеризуется рентгеновской дифрактограммой порошка, имеющей характеристические пики, выраженные в углах 2 при 6,56,13,20, 19,86, 20,84, 21,22 и 25,22, измеренной на порошковом рентгеновском дифрактометре. 30. Способ по п.12, отличающийся тем, что кристаллическая затравка характеризуется рентгенов- 21022099 ской дифрактограммой порошка, имеющей характеристические пики, выраженные в углах 2 при 8,54,14,02, 15,68, 17,06, 17,24, 24,16 и 25,74, измеренной на порошковом рентгеновском дифрактометре. 31. Способ по п.12, отличающийся тем, что кристаллическая затравка имеет экстраполированную температуру плавления от приблизительно 157,1 до приблизительно 167,1 С. 32. Способ по любому из пп.12 или 30, отличающийся тем, что кристаллическая затравка имеет чистоту по меньшей мере приблизительно 70%. 33. Способ получения соединения формулы 10 или его соли, включающий получение соединения формулы 4 или его соли путем металлирования соединения формулы 2 или его соли с последующим взаимодействием полученного металлорганического соединения с соединением формулы 11 где Ry представляет собой уходящую группу,превращение соединения формулы 4 или его соли в соединение формулы 5' где Rc представляет собой уходящую группу,превращение соединения формулы 5' в соединение формулы 6 где Ra и Rb независимо представляют собой C1-С 6 алкил, путем обработки с соответствующим диметилацеталем N,N-диалкилформамида,превращение соединения формулы 7 в соединение формулы 8 путем обработки с помощью хлорида калия и N,О-бистриметилсилилацетамидом и превращение соединения формулы 9 в соединение формулы 10 путем обработки с основанием и кристаллизацию соединения формулы 10 путем добавления кристаллической затравки к раствору, содержащему соединение формулы 10. 34. Способ по п.33, в котором соединение формулы 9 превращают в соединение формулы 10 путем обработки водным раствором гидроксида калия в водном растворе изопропилового спирта. 35. Способ по п.33, в котором соединение формулы 6 превращают в соединение формулы 7 путем обработки диметилацеталем N,N-диметилформамида. 36. Способ по п.35, в котором обработку диметилацеталем N,N-диметилформамида осуществляют в присутствии уксусной кислоты при температуре от приблизительно 50 до приблизительно 150 С. 37. Способ по п.33, в котором в качестве моноалкилмалоната используют соль моноэтилмалоната. 38. Способ по п.33, в котором соединение формулы 5' превращают в соединение формулы 6 путем обработки моноэтилмалонатом калия. 39. Способ по п.33, в котором соединение формулы 5' представляет собой соединение формулы 5 а и в котором соединение формулы 4 или его соль превращают в соединение формулы 5 а путем обработки 1,1'-карбонилдиимидазолом. 40. Способ по п.33, в котором Ry представляет собой трифлат, мезилат, тозилат или брозилат. 41. Способ по п.33, в котором кристаллическая затравка характеризуется рентгеновской дифрактограммой порошка, имеющей характеристические пики, выраженные в углах дифракции 2 6,56, 13,20,19,86, 20,84, 21,22 и 25,22, измеренной на порошковом рентгеновском дифрактометре. 42. Способ по п.33, в котором кристаллическая затравка характеризуется рентгеновской дифрактограммой порошка, имеющей характеристические пики при углах дифракции 2 8,54, 14,02, 15,68,17,06, 17,24, 24,16 и 25,74, измеренной на порошковом рентгеновском дифрактометре. 43. Способ по п.33, в котором кристаллическая затравка характеризуется экстраполированной температурой плавления от приблизительно 157,1 до приблизительно 167,1 С. 44. Способ по п.33 или 42, в котором чистота кристалла кристаллической затравки не менее приблизительно 70%. 45. Способ получения металлорганического соединения, включающий металлирование соединения металлоорганическим реагентом, представляющим собой дибутилмагний, аддукт бутилэтилмагния с бутанолом (ВЕМ-В), или бутилэтилмагний. 46. Способ по п.45, отличающийся тем, что указанный металлоорганический реагент представляет собой аддукт бутилэтилмагния и бутанола (BEM-B). 47. Способ по п.45, дополнительно включающий металлирование указанного полученного металлоорганического соединения литий металлоорганическим реагентом, который представляет собой нбутиллитий или трет-бутиллитий. 48. Способ получения соединения формулы 10 или его соли, включающий получение соединения формулы 4 или его соли путем обработки соединения формулы 3 или его соли силановым восстановительным агентом в присутствии кислоты; превращение соединения формулы 4 или его соли в соединение формулы 5' где Rc представляет собой галоген или 1-имидазолил,путем обработки соединения 4 реагентом, выбранным из группы, включающей тионилхлорид, оксалилхлорид, хлорид циануровой кислоты и 1,1-карбонилдиимидазол; превращение соединения формулы 5' в соединение формулы 6 где R представляет собой C1-С 6 алкил, причем соединение формулы 5' смешивают с моноалкилмалонатной солью и солью магния,превращение соединения формулы 6 в соединение формулы 7 путем взаимодействия соединения формулы 7 с (S)-2-амино-3-метил-1-бутанолом,превращение соединения формулы 8 в соединение формулы 9 путем обработки соединения формулы 8 силилирующим агентом, выбранным из группы, включающейN,О-бистриметилсилилацетамид, N,О-бис(триметилсилил)трифторацетамид и гексаметилдисилазан, при температуре 10020 С, и превращение соединения формулы 9 в соединение формулы 10 путем обработки соединения формулы 9 основанием. 49. Способ по п.48, отличающийся тем, что силановый восстановитель представляет собой триэтилсилан и кислота представляет собой трифторуксусную кислоту. 50. Способ по п.48, отличающийся тем, что указанный диС 1-С 6 алкилацеталь N,N-диметилформамида представляет собой диметилацеталь N,N-диметилформамида. 51. Способ по п.48 или 50, отличающийся тем, что обработку диметилацеталем N,N-диметилформамида проводят в присутствии уксусной кислоты при температуре 10050 С. 52. Способ по п.48, отличающийся тем, что указанная обработка соединения формулы 8 силилирующим агентом выполняется в присутствии соли, выбранной из группы, включающей хлорид калия,хлорид лития, хлорид натрия и хлорид магния, в полярном апротонном растворителе, выбранном из группы, включающей диметилформамид, диметилацетамид, N-метилпирролидинон и ацетонитрил. 53. Способ по п.48, отличающийся тем, что соединение формулы 8 превращают в соединение формулы 9 путем обработки соединения формулы 8 хлоридом калия и N,O-бис(триметилсилил)ацетамидом.
МПК / Метки
МПК: C07C 51/367, C07C 63/68, C07D 233/54, C07D 215/56, C07C 43/205, C07B 49/00
Метки: получения, интегразы, способы, ингибиторов, интермедиаты
Код ссылки
<a href="https://eas.patents.su/28-22099-sposoby-i-intermediaty-dlya-polucheniya-ingibitorov-integrazy.html" rel="bookmark" title="База патентов Евразийского Союза">Способы и интермедиаты для получения ингибиторов интегразы</a>
Способ и промежуточные соединения для получения ингибиторов интегразы

Номер патента: 19431
Опубликовано: 31.03.2014
Авторы: Дауди Эрик, Пфайффер Стивен
МПК: C07C 43/23, C07C 41/30, C07C 43/225...
Метки: получения, промежуточные, интегразы, ингибиторов, способ, соединения
Формула / Реферат:
1. Способ получения соединения формулы 4или соли указанного соединения из соединения формулы 15или соли указанного соединения путем проведения стадий замещения атома брома карбоксильной группой и замещения гидроксильной группы атомом водорода.2. Способ по п.1, отличающийся тем, что соединение формулы 4 или его соль получают путем металлирования соединения формулы 15 или его соли и обработки диоксидом углерода с получением соединения формулы 3или...
Трициклические соединения – ингибиторы интегразы вич, способ их получения (варианты), фармацевтическая композиция на их основе, способ ее получения и способы их использования в лечении болезней

Номер патента: 11399
Опубликовано: 27.02.2009
Авторы: Джин Хаолун, Фардис Мерайя, Чен Сяову, Ким Чанг Ю., Шахерер Лаура Н., Чен Джеймс М.
МПК: C07D 471/04, C07D 498/20, C07D 487/04...
Метки: основе, фармацевтическая, интегразы, ингибиторы, лечении, болезней, варианты, трициклические, композиция, вич, способы, использования, способ, получения, соединения
Формула / Реферат:
1. Соединение формулы где А1 независимо выбирают из ряда: C(R2)2, CR2OR, CR2OC(=O)R, C(=O), C(=S), CR2SR, C(=NR); А2 независимо выбирают из ряда: О, S, NR, C(R2)2-C(R3)2, C(R2)=C(R3), C(=O)C(R3)2, NR-C(R3)2, N=C(R3), N=N или C(R2)=N; Q означает N или CR4; L выбирают из ряда: связь, О, S, S-S, S(=O), S(=O)2, S(=O)2NR, NR, N-OR, C1-С12алкилен, замещенный C1-C12алкилен, С2-С12алкенилен, замещенный С2-С12алкенилен, С2-С12алкинилен, замещенный...
Способ получения дулоксетина и новые ключевые интермедиаты для его применения

Номер патента: 15828
Опубликовано: 30.12.2011
Авторы: Шиндлер Йожеф, Вукич Кристина, Боди Йожеф, Фараго Янош, Елеш Янош, Гати Тамаш, Темешвари Кристина, Цоке Каталин, Фогашши Эелемер
МПК: C07D 333/20
Метки: применения, ключевые, дулоксетина, получения, новые, интермедиаты, способ
Формула / Реферат:
1. Способ получения дулоксетина((S)-N-метил-3-(1-нафталинилокси)-3-(2-тиенил)пропанамина) формулы I и его фармацевтически приемлемых солей, который включает следующие стадии:расщепление рацемического 3-(N-метил-N-бензиламино)-1-(2-тиенил)-1-пропанола формулы IIпроизводным D-фенилглицина общей формулы IIIгде R1, R2 и R3представляют собой независимо друг от друга водород, галоген, циано, нитро или аминогруппу, необязательно замещенную одной или...
Способы получения ингибиторов jak киназ и родственных промежуточных соединений

Номер патента: 20643
Опубликовано: 30.12.2014
Авторы: Пань Юнчунь, Лин Циянь, Чжоу Цзячэн, Ван Хайшэн, Лю Пинли, Меткаф Брайан В., Мелони Дэвид, Ксиа Майкл, Ли Мей, Юэ Тай-Юйэнь, Роджерс Джеймс Д.
МПК: C07D 487/04, C07F 5/04, C07D 231/16...
Метки: родственных, соединений, получения, промежуточных, способы, киназ, ингибиторов
Формула / Реферат:
1. Способ получения композиции, содержащей рацемат соединения формулы Iaкоторый включает:(а) обработку композиции, содержащей энантиомерный избыток (R)- или (S)-энантиомера соединения формулы Ia, соединением формулы D-1в присутствии первого основания в условиях, достаточных для получения соединения формулы IV(b) реакцию соединения формулы IV с соединением формулы D-1 в присутствии второго основания, где * указывает на хиральный углерод; P1...
Способы и соединения, предназначенные для получения ингибиторов котранспортера натрий-глюкозы 2 типа

Номер патента: 17411
Опубликовано: 28.12.2012
Авторы: Гудвин Николь Кэтлин, Харрисон Брайс Олден, Чжан Хаймин, У Вэньсюэ, Янь Цзе, Сонг Цюлин, Мейбон Росс, Иимура Синия, Чжао Мэттью Манчжу
МПК: C07H 5/04, C07H 15/04, C07H 7/04...
Метки: соединения, типа, котранспортера, предназначенные, натрий-глюкозы, способы, ингибиторов, получения
Формула / Реферат:
1. Способ получения соединения формулы Iили его соли, который включает взаимодействие соединения формулы IIс основанием с получением соединения формулы I,где Y представляет собой О, S, NR4 или C(R4)2;Z1 представляет собой О, S, SO или SO2;каждый Р1 независимо представляет собой гидроксизащитную группу, устойчивую в кислых условиях;каждый R1 независимо представляет собой водород, галоген, циано, OR1A SR1A или C1-10алкил;каждый R1A независимо...
Следующий патент: Способ получения лекарственного средства, содержащего тадалафил
Случайный патент: Модуляторы сск-1 рецепторов