Производные n-(1-фенил-3-алкилпиразол-5-ил)-n’-арилмочевины
Номер патента: 20641
Опубликовано: 30.12.2014
Авторы: Стронг Питер, Чаррон Кэтрин Элизабет, Уилльямс Джонатан Гарет, Ито Кадзухиро, Херст Саймон Кристофер, Кинг-Андервуд Джон, Рейппорт Уилльям Гарт, Мюррей Питер Джон, Таддеи Давид Мишель Адриен, Онионз Стюарт Томас







Формула / Реферат
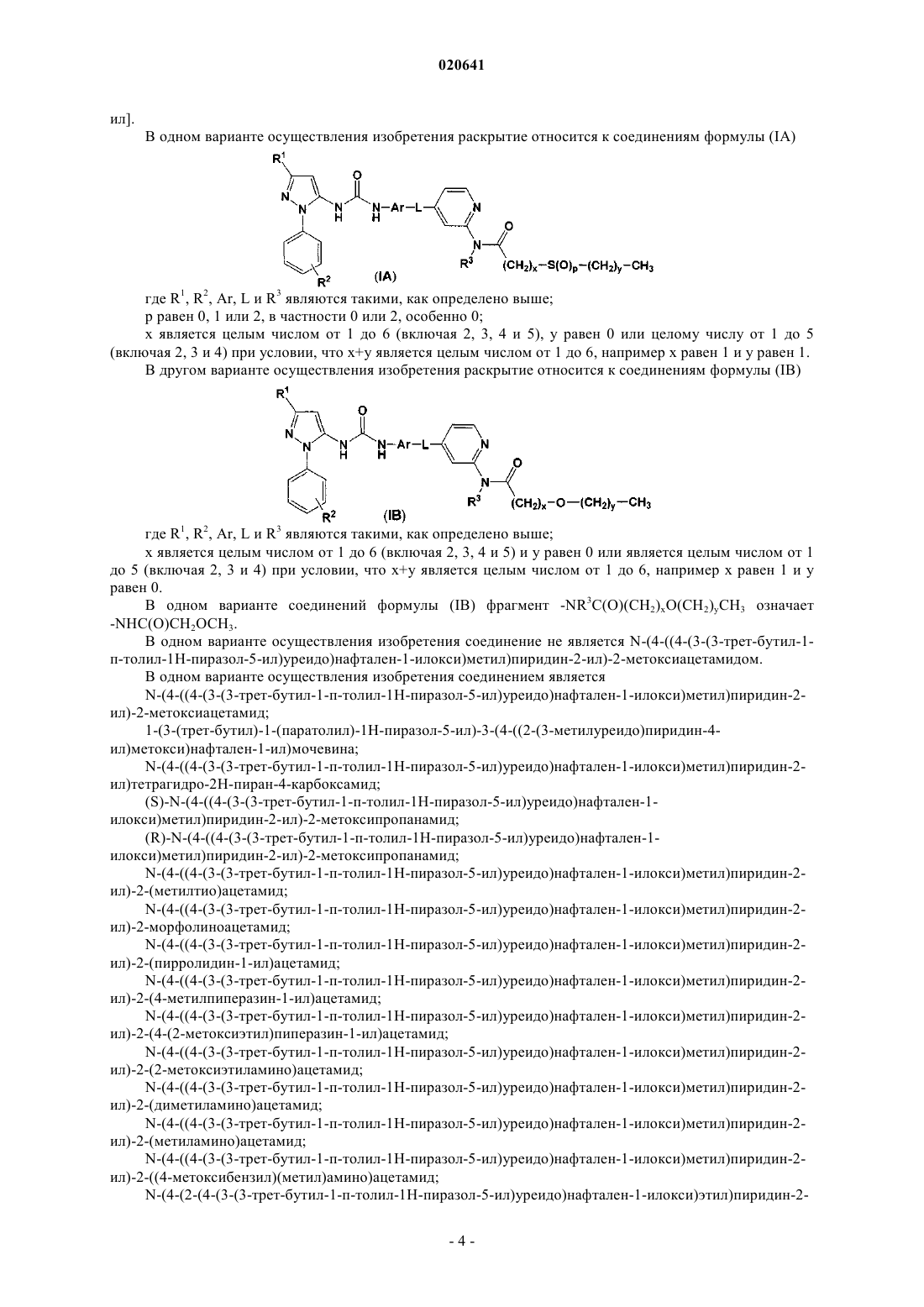
1. Соединение формулы (I)

где R1 означает C1-6алкил, необязательно замещенный гидроксильной группой;
R2 означает Н или C1-6алкил, необязательно замещенный гидроксильной группой;
R3 означает Н;
Ar означает нафтильное или фенильное кольцо, любое из которых может быть необязательно замещено одной или более группами, независимо выбранными из C1-6алкила, С1-6алкокси, амино и C1-4моно- или диалкиламино;
L представляет собой -O(CH2)m-, где m равен 0, 1 или 2;
-NR3C(O)Q выбирают из группы, содержащей
-NHC(O)CH2O(CH2)2OCH3;
-NHC(O)CH(CH3)OCH3;
-NHC(O)CH2NHCH3;
-NHC(О)CH2NHCH2CH2OCH3;
-NHC(O)NH2;
-NHC(O)NHC1-7алкил;
-NHC(O)N(C1-4алкил)C1-5алкил;
-NHC(О)CHN[(CH2)2OCH3]2;
-NHC(O)N(CH3)2;
-NHC(O)NHCH3;
-NHC(O)CH2NHCH2C6H4(OCH3);
-NHC(O)CH2N(CH3)CH2C6H4(OCH3);
-NHC(O)-(тетрагидропиранил);
-NHC(O)-(морфолинил);
-NHC(O)-(пирролидинил);
-NHC(O)-(пиперазинил);
-NHC(O)-(метилпиперазинил);
-NHC(О)-[(метоксиэтил)пиперазинил];
-NHC(O)CH2-(тетрагидропиранил);
-NHC(О)CH2-(морфолинил);
-NHC(O)CH2-(пирролидинил);
-NHC(О)CH2-(пиперазинил);
-NHC(O)CH2-(метилпиперазинил);
-NHC(О)CH2-[(метоксиэтил)пиперазинил];
-NHC(O)CH2N(CH3)2;
-NHC(O)-(CH2)x-S(O)p-(CH2)y-CH3 и
-NHC(O)(CH2)x-O-(CH2)y-CH3,
где х означает целое число от 1 до 6 и у означает 0 или целое число от 1 до 5 при условии, что x+у означает целое число от 1 до 6; и
p равен 0, 1 или 2,
или его фармацевтически приемлемая соль, включая все его стереоизомеры и таутомеры.
2. Соединение по п.1, где Ar означает нафтил.
3. Соединение по п.1 или 2, где R1 означает трет-бутил.
4. Соединение по любому одному из пп.1-3, где R2 означает метил.
5. Соединение по любому одному из пп.1-4, где R2 находится в пара-положении.
6. Соединение по любому из пп.1-5, где Q выбирают из -NHC(O)CH2OC1-6алкила,
-NHC(O)CH2O(CH2)2OCH3, -NHC(O)CH(CH3)OCH3, -NHC(O)CH2NHCH3, -NHC(O)CH2NHCH2CH2OCH3,
-NHC(O)CH2SCH3, -NHC(O)NH2, -NHC(O)CH2S(O)2CH3, -NHC(O)NHC1-7алкила, -NHC(O)N(C1-4алкил)C1-5алкила и -NHC(O)CHN[(CH2)2OCH3]2.
7. Соединение по п.6, где Q выбирают из -NHC(O)CH2OCH3, -NHC(O)CH2O(CH2)2OCH3,
-NHC(О)CH(CH3)OCH3, -NHC(O)CH2NHCH3, -NHC(О)CH2NH(CH2)2OCH3, -NHC(О)CH2SCH3, -NHC(O)NH2, -NHC(O)CH2S(O)2CH3, -NHC(O)NHCH3, -NHC(O)N(CH3)2 и -NHC(O)CHN[(CH2)2OCH3]2.
8. Соединение по п.1, представленное формулой (IC)

где x равен целому числу от 1 до 6 и у равен 0 или целому числу от 1 до 5 при условии, что х+у равно целому числу от 1 до 6, например x равен 1 и у равен 1,
или его фармацевтически приемлемая соль, включая все его стереоизомеры и таутомеры.
9. Соединение по п.1, представленное формулой (ID)

где x равен целому числу от 1 до 6 и у равен 0 или целому числу от 1 до 5 при условии, что х+у равно целому числу от 1 до 6, например x равен 1 и у равен 0,
или его фармацевтически приемлемая соль, включая все его стереоизомеры и таутомеры.
10. Соединение по п.1, выбранное из
N-(4-((4-(3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-метоксиацетамида;
1-(3-(трет-бутил)-1-(паратолил)-1H-пиразол-5-ил)-3-(4-((2-(3-метилуреидо)пиридин-4-ил)метокси)нафтален-1-ил)мочевины;
N-(4-((4-(3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)тетрагидро-2H-пиран-4-карбоксамида;
(S)-N-(4-((4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-метоксипропанамида;
(R)-N-(4-((4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-метоксипропанамида;
N-(4-((4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-(метилтио)ацетамида;
N-(4-((4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-морфолиноацетамида;
N-(4-((4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-(пирролидин-1-ил)ацетамида;
N-(4-((4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-(4-метилпиперазин-1-ил)ацетамида;
N-(4-((4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-(4-(2-метоксиэтил)пиперазин-1-ил)ацетамида;
N-(4-((4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-(2-метоксиэтиламино)ацетамида;
N-(4-((4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-(диметиламино)ацетамида;
N-(4-((4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-(метиламино)ацетамида;
N-(4-((4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-((4-метоксибензил)(метил)амино)ацетамида;
N-(4-(2-(4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)этил)пиридин-2-ил)-2-метоксиацетамида;
N-(4-(2-(4-(3-(3-трет-бутил-1-п-толил-1Н-пиразол-5-ил)уреидо)нафтален-1-илокси)этил)пиридин-2-ил)-2-(2-метоксиэтокси)ацетамида;
1-(3-(трет-бутил)-1-(паратолил)-1H-пиразол-5-ил)-3-(4-(2-(2-(3-метилуреидопиридин-4-ил)этокси)нафтален-1-ил)мочевины и
1-(3-(трет-бутил)-1-(паратолил)-1H-пиразол-5-ил)-3-(4-(2-(2-уреидопиридин-4-ил)этокси)нафталенил-1-ил)мочевины,
или его фармацевтически приемлемая соль, включая все его стереоизомеры и таутомеры.
11. Фармацевтическая композиция, содержащая соединение по любому одному из пп.1-10, в комбинации с одним или более фармацевтически приемлемыми разбавителями или носителями.
12. Применение соединения по любому одному из пп.1-10 в качестве лекарственного средства.
13. Применение соединения по любому одному из пп.1-10 для лечения или предотвращения состояния, выбранного из ХОБЛ (включая хронический бронхит и эмфизему), астмы, астмы у детей, кистозного фиброза, саркоидоза, идиопатического фиброза легких, аллергического ринита, ринита, синусита, аллергического конъюнктивита, конъюнктивита, аллергического дерматита, контактного дерматита, псориаза, язвенного колита, воспаления суставов, вторичного по отношению к ревматоидному артриту или остеоартриту, ревматоидного артрита, панкреатита, кахексии, ингибирования роста и метастазирования опухолей, включающих немелкоклеточную карциному легких, карциному молочной железы, карциному желудка, колоректальные карциномы и злокачественную меланому.
14. Применение соединения формулы (I) согласно пп.12 и 13 в сочетании с одним или более активным ингредиентом, представляющим собой лекарственное средство для лечения или предупреждения респираторных нарушений, где активный ингредиент представляет собой стероид, бета-агонист или ксантин.
15. Применение по п.14, где активный ингредиент представляет собой стероид, который выбран из группы, состоящей из будесонида, беклометазона дипропионата, флутиказона пропионата, мометазона фуроата и флутиказона фуроата, или активный ингредиент представляет собой бета-агонист, который выбран из группы, состоящей из тербуталина, сальбутамола, салметерола и формотерола, или активный ингредиент представляет собой ксантин, такой как теофиллин.
16. Применение соединения по любому одному из пп.1-10 для получения лекарственного средства для лечения или предотвращения состояния, выбранного из ХОБЛ (включая хронический бронхит и эмфизему), астмы, астмы у детей, кистозного фиброза, саркоидоза, идиопатического фиброза легких, аллергического ринита, ринита, синусита, аллергического конъюнктивита, конъюнктивита, аллергического дерматита, контактного дерматита, псориаза, язвенного колита, воспаления суставов, вторичного по отношению к ревматоидному артриту или остеоартриту, ревматоидного артрита, панкреатита, кахексии, ингибирования роста и метастазирования опухолей, включающих немелкоклеточную карциному легких, карциному молочной железы, карциному желудка, колоректальные карциномы и злокачественную меланому.
Текст
Изобретение предоставляет соединения формулы (I), где R1, Ar, L, X, R3 и Q являются такими, как определено в описании, а также фармацевтическую композицию на их основе и их применение в терапии, особенно в лечении воспалительных заболеваний. Ито Кадзухиро, Стронг Питер,Рейппорт Уилльям Гарт, Мюррей Питер Джон, Кинг-Андервуд Джон,Уилльямс Джонатан Гарет, Онионз Стюарт Томас, Херст Саймон Кристофер, Таддеи Давид Мишель Адриен, Чаррон Кэтрин Элизабет Область изобретения Изобретение относится к соединениям, которые являются ингибиторами митоген-активированных ферментов протеинкиназ p38 (названными в описании ингибиторами p38 МАР-киназ), в особенности ингибиторами подтипов киназ альфа и гамма, и к применению соединений в терапии, включая фармацевтические комбинации, особенно в лечении воспалительных заболеваний, включающих воспалительные заболевания легкого, таких как ХОБЛ (хроническая обструктивная болезнь легких). Предшествующий уровень техники Четыре изоформы MAPK р 38 (альфа, бета, гамма и дельта соответственно) идентифицированы, каждая имеющая тканеспецифическую картину экспрессии. Альфа- и бета-изоформы протеинкиназы p38MAPK экспрессируются по всему организму и обнаруживаются во многих различных типах клеток. Альфа- и бета-изоформы протеинкиназы p38 MAPK ингибируются некоторыми известными низкомолекулярными ингибиторами p38 MAPK. Полученные ранее соединения были высокотоксичными из-за повсеместного распределения экспрессии указанных выше изоформ и отсутствия направленного действия соединений. Более поздние ингибиторы являются улучшенными высокоизбирательными ингибиторами альфа- и бета-изоформ p38 MAPK и имеют более широкий предел безопасности. Менее известными являются гамма- и дельта-изоформы p38 MAPK. Указанные изоформы экспрессируются в специфических тканях/клетках (в отличие от альфа- и бета-изоформ p38). Дельта-изоформаp38 MAPK экспрессируется более всего в поджелудочной железе, яичках, легком, тонком кишечнике и почке. Указанная изоформа также имеется в избытке в макрофагах (Smith, S.J. (2006), Br. J. Pharmacol.,149:393-404) и определяется в нейтрофилах, CD4+ T-клетках и эндотелиальных клетках(www.genecard.org, Hale, K.K. (1999), J. Immunol., 62(7):4246-4252.). Весьма немного известно об экспрессии гамма-изоформы p38 MAPK, но более всего указанная изоформа экспрессируется в головном мозге и сердце, а также в лимфоцитах и макрофагах (www.genecard.org). В настоящее время селективные низкомолекулярные ингибиторы гамма- и дельта-изоформ p38MAPK недоступны, но одно имеющееся соединение, BIRB 796, как известно, проявляет общую для всех изоформ ингибиторную активность. Ингибирование гамма- и дельта-изоформ p38 наблюдается при более высоких концентрациях соединения, чем требуется для ингибирования альфа- и бета-изоформ p38(Kuma, Y. (2005), J. Biol. Chem. 280:19472-19479). Соединение BIRB 796 также уменьшает фосфорилирование протеинкиназ p38 MAPK или киназ JNK, осуществляемое посредством вышестоящей в каскаде реакций киназы МКК 6 или МКК 4. Исследователь Kuma высказал предположение, что конформационное изменение, вызванное связыванием ингибитора с MAPK, может нарушать структуру как участка фосфорилирования, так и участка связывания для вышестоящего активатора, тем самым ухудшая фосфорилирование протеинкиназ p38 MAPK или киназ JNK. Полагают, что p38 МАР-киназа играет ведущую роль во многих путях передачи сигнала, которые участвуют в инициации и поддержании хронического, стойкого воспаления при заболевании человека,например тяжелой астме и ХОБЛ (хронической обструктивной болезни легких). В настоящее время имеются многочисленные литературные данные, которые свидетельствуют о том, что p38 МАР-киназа активируется рядом провоспалительных цитокинов и что ее активация приводит к пополнению и высвобождению других провоспалительных цитокинов. Действительно, данные некоторых клинических исследований демонстрируют благоприятные изменения в активности заболевания у больных в процессе лечения ингибиторами p38 MAP-киназы. Например, Smith, S.J. (2006), Br. J. Pharmacol., 149:393-404 описывает влияние ингибирования ингибиторов p38 МАР-киназы на высвобождение цитокина из макрофагов человека. Предлагается применение ингибиторов p38 МАР-киназы в лечении хронической обструктивной болезни легких (ХОБЛ). Было показано, что низкомолекулярные ингибиторы, направленные на киназу p38 MAPK /, оказались эффективными ингибиторами, снижающими различные параметры воспаления в клетках и тканях больных ХОБЛ, которые в большинстве случаев не проявляли чувствительность к кортикостероиду (Smith, S.J. (2006), Br. J. Pharmacol., 149:393-404), и указанные ингибиторы были эффективными in vivo на животных моделях (Underwood, D.С. et al. (2000), 279:895-902; Nath, P. etal. (2006), Eur. J. Pharmacol., 544:160-167). Исследователь Irusen и коллеги также предположили возможность вовлечения киназы p38 MAPK/ в отсутствие чувствительности к кортикостероиду через снижение связывающего сродства глюкокортикоидного рецептора (GR) в ядрах (Irusen, E. et al. (2002), J. Allergy Clin. Immunol., 109:649-657). Клинический опыт с рядом ингибиторов p38 МАР-киназы, включающих AMG548, BIRB 796, VX702, SCIO469 и SCIO323, описан в публикации Lee et al. (2005), Current Med.Chem. 12:2979-2994. ХОБЛ является состоянием, при котором основное воспаление, как было показано, в значительной степени проявляет устойчивость к противовоспалительному действию ингалируемых кортикостероидов. Поэтому эффективная стратегия для лечения ХОБЛ может заключаться в достижении такого воздействия, которое проявляет как характерные противовоспалительные эффекты, так и способно повысить чувствительность тканей легкого больного ХОБЛ к ингалируемым кортикостероидам. Недавняя публикация может осуществляться "двойная" польза от использования ингибитора p38 МАР-киназы при лечении ХОБЛ. Однако основным препятствием, затрудняющим применение ингибиторов p38 МАР-киназы в лечении хронических воспалительных заболеваний человека, была токсичность, наблюдаемая у больных. Это обстоятельство было достаточно серьезной причиной, чтобы отказаться от клинической разработки многих улучшенных соединений, включающих все соединения, особо упомянутые выше. Международная патентная заявка WO 03/072569 раскрывает 1-фенил-1H-пиразол-5-илуреидо производные и их применение для лечения цитокин-опосредованных заболеваний. До настоящего времени сохраняется потребность в идентификации и разработке новых соединений,терапевтически используемых в качестве ингибиторов p38 МАР-киназы, которые имеют улучшенную терапевтическую способность, в частности, которые являются более эффективными, дольше действующими и/или менее токсичными при более приемлемой терапевтической дозе. Задачей настоящего изобретения является предоставить соединения, ингибирующие p38 МАР-киназу с определенной для подтипов специфичностью, которые проявляют хорошую противовоспалительную способность. Сущность изобретения Изобретение представляет соединение формулы (I) где R1 означает C1-6 алкил, необязательно замещенный гидроксильной группой;R2 означает Н или C1-6 алкил, необязательно замещенный гидроксильной группой;Ar означает нафтильное или фенильное кольцо, любое из которых может быть необязательно замещено одной или более группами, независимо выбранными из C1-6 алкила, C1-6 алкокси, амино и C1-4 моноили диалкиламино;-NHC(O)(CH2)x-O-(CH2)У-CH3,где х означает целое число от 1 до 6 и у означает 0 или целое число от 1 до 5 при условии, что х+у означает целое число от 1 до 6; р равен 0, 1 или 2,-2 020641 или его фармацевтически приемлемая соль, включая все его стереоизомеры и таутомеры. Краткое описание фигур На фиг. 1 представлен график зависимости числа нейтрофилов в БАЛ (BALF) от времени до введения препарата для соединения примера 1 в тесте накопления нейтрофилов, индуцированном LPS. На фиг. 2 представлен график зависимости % ингибирования нейтрофиллеза от времени до введения дозы для соединения примера 1 в тесте накопления нейтрофилов, индуцированном LPS. Подробное описание изобретения Алкил, как используют в описании, относится к прямой цепи или разветвленной алкильной цепи,такой как, без ограничения, метил, этил, пропил, изопропил, бутил и трет-бутил. В одном варианте осуществления изобретения алкил относится к прямой алкильной цепи. Алкокси, как используют в описании, относится к прямой или разветвленной алкоксицепи, например метокси, этокси, пропокси, бутокси. Алкокси, как используют в описании, также распространяется на варианты осуществления изобретения, в которых атом кислорода локализован в алкильной цепи, например -CH2CH2OCH3 или -CH2OCH3. В одном варианте осуществления изобретения алкокси связывается через кислород с остальной частью молекулы. В одном варианте осуществления изобретения раскрытием изобретения является прямая цепь алкокси. В одном варианте осуществления изобретения описание представляет соединения формулы (I), гдеR1 означает метил, этил, пропил, изопропил, бутил или трет-бутил, в частности трет-бутил. В одном варианте осуществления изобретения R1 означает -C(CH3)2CH2OH. В одном варианте осуществления изобретения R2 означает метил, этил, пропил, изопропил, бутил или трет-бутил, в частности метил. В одном варианте осуществления изобретения R2 является -CH2OH. В одном варианте осуществления изобретения R2 расположен во 2, 3 или 4 положении (т.е. орто-,мета- или пара-положении), в частности пара-положении (4). В одном варианте осуществления изобретения Ar является нафтилом. В одном варианте осуществления изобретения Ar не замещен необязательными заместителями. В одном варианте осуществления изобретения Ar замещен 1 или 2 группами. В одном варианте осуществления изобретения L представляет собой -O(CH2)m-, где m равен 1 или 2. Галоген включает фтор, хлор, бром или йод, в частности фтор, хлор или бром, особенно фтор или хлор. В одном варианте осуществления изобретения описанные соединения включают такие, в которых фрагмент -NR3C(O)Q в формуле (I) представляет собой:-NHC(O)CHN[(CH2)2OCH3]2. Таким образом, в одном варианте осуществления изобретения атом азота в алкильной цепи непосредственно связан с карбонилом фрагмента -NR3C(O) и, кроме того, может, например, входить в состав концевой аминогруппы, соответственно -NR3C(О)N(CH3)2 или -NR3C(O)NHCH3. В одном варианте осуществления изобретения примеры фрагмента -NR3C(O)Q представляют собой 3-NR C(O)CH2NHCH2C6H5(OCH3) и -NR3C(O)CH2N(CH3)CH2C6H5(OCH3). В одном варианте осуществления изобретения раскрытые соединения включают соединения формулы (I), у которых фрагмент -NR3C(O)Q представляет собой: ил]. В одном варианте осуществления изобретения раскрытие относится к соединениям формулы (IA)x является целым числом от 1 до 6 (включая 2, 3, 4 и 5), y равен 0 или целому числу от 1 до 5(включая 2, 3 и 4) при условии, что х+у является целым числом от 1 до 6, например x равен 1 и y равен 1. В другом варианте осуществления изобретения раскрытие относится к соединениям формулы (IB)x является целым числом от 1 до 6 (включая 2, 3, 4 и 5) и у равен 0 или является целым числом от 1 до 5 (включая 2, 3 и 4) при условии, что х+у является целым числом от 1 до 6, например x равен 1 и у равен 0. В одном варианте соединений формулы (IB) фрагмент -NR3C(O)(CH2)xO(CH2)yCH3 означает-NHC(O)CH2OCH3. В одном варианте осуществления изобретения соединение не является N-(4-4-(3-(3-трет-бутил-1 п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-метоксиацетамидом. В одном варианте осуществления изобретения соединением являетсяN-(4-(2-(4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)этил)пиридин-2 ил)-2-(2-метоксиэтокси)ацетамид; 1-(3-(трет-бутил)-1-(паратолил)-1H-пиразол-5-ил)-3-(4-(2-(2-(3-метилуреидопиридин-4 ил)этокси)нафтален-1-ил)мочевина или 1-(3-(трет-бутил)-1-(паратолил)-1H-пиразол-5-ил)-3-(4-(2-(2-уреидопиридин-4 ил)этокси)нафталенил-1-ил)мочевина,или его фармацевтически приемлемая соль, включая все его стереоизомеры и таутомеры. Примеры солей соединения формулы (I) включают все фармацевтически приемлемые соли, такие как, без ограничения, кислотно-аддитивные соли сильных минеральных кислот, такие как HCl- и HBrсоли, и кислотно-аддитивные соли сильных органических кислот, такие как соль метансульфокислоты. Примеры сольватов включают гидраты. Соединения, представленные в описании, могут включать один или более хиральных центров, и описание предназначено включить рацематы, энантиомеры и стереоизомеры указанных соединений. В одном варианте осуществления изобретения одна энантиомерная форма присутствует в значительно очищенном виде, которая практически не имеет соответствующей энантиомерной формы. Раскрытие также распространяется на все полиморфные формы соединений, представленных в описании. Соединения формулы (I) могут быть получены способом, включающим взаимодействие соединений формулы (II) где Ar, L, R1, R2 и R3 являются такими, как определено выше для соединений формулы (I),с соединением формулы (III) где Q является группой, определенной выше для соединений формулы (I); и LG1 является уходящей группой, например галогеном, таким как хлор. Реакцию подходящим образом осуществляют в присутствии основания (например, диизопропилэтиламина). Реакцию подходящим образом осуществляют в апротонном растворителе или смеси растворителей, например ДХМ и ДМФ. Соединения формулы (II) могут быть получены путем взаимодействия соединения формулы (IV) где R1 и R2 являются такими, как определено выше для соединений формулы (I),с соединением формулы (V) где Ar, L и R3 являются такими, как определено выше для соединений формулы (I),и соединением формулы (VI) где группы LG2 и LG3, каждая независимо, представляют собой уходящие группы (например, LG2 иLG3, обе, представляют собой имидазолил или галоген, такой как хлор). Реакцию подходящим образом осуществляют в апротонном растворителе (например, дихлормета-5 020641 не), используя соответствующие защитные группы для химически чувствительных групп. Соединение формулы (V) может быть получено восстановлением соединения формулы (VII) где Ar, L, X и R3 являются такими, как определено выше для соединений формулы (I),например, путем гидрирования в присутствии катализатора, такого как платина, нанесенная на углерод. Реакцию подходящим образом осуществляют в полярном протонном растворителе или в смеси растворителей (например, метанол и уксусная кислота). Соединение формулы (VII) может быть получено взаимодействием соединения формулы (VIIIa) или (VIIIb) где R3 и m являются такими, как определено выше для соединений формулы (I),с соединением формулы (IX) или (X) где соединения (IX) и (X) могут нести необязательные заместители, как определено выше для соединений формулы (I). Реакция может быть осуществлена в условиях реакции Мицунобу, таких как в присутствии трифенилфосфина и диизопропилазодикарбоксилата. Реакцию подходящим образом осуществляют в полярном апротонном растворителе (например, тетрагидрофуране, в частности безводном тетрагидрофуране). Соединения формул (III), (IV), (VI), (VIII), (IX) и (X) являются либо коммерчески доступными, либо известными, либо новыми и могут быть легко получены обычными способами. См., например, публикацию Regan, J. et al., J. Med. Chem., 2003, 46, 4676-4686, WO 00/043384, WO 2007/087448 иWO 2007/089512. Защитные группы могут потребоваться для защиты химически чувствительных групп в течение одной или более реакций, описанных выше, для уверенности в эффективности способа. Таким образом,если желательно или необходимо, промежуточные соединения могут быть защищены путем использования стандартных защитных групп. Защитные группы и средства для их удаления описаны в "ProtectiveGroups in Organic Synthesis", Theodora W. Greene and Peter G.M. Wuts, published by John WileySons Inc.; 4th Rev Ed., 2006, ISBN-10: 0471697540. Новые промежуточные соединения заявлены как аспект изобретения. В одном аспекте соединения пригодны в лечении, например для лечания ХОБЛ и/или астмы. Соединения, разработанные к настоящему времени, предназначены для перорального введения. Такая стратегия включает оптимизацию соединений, которые достигают продолжительности действия посредством соответствующего фармакокинетического профиля. Данный фармакокинетический профиль подтверждает, что существует достаточная концентрация, установленная и поддерживаемая после и между дозами для достижения клинического эффекта. Неизбежным результатом данного подхода является то, что все ткани организма, особенно печень и кишка, вероятно, подвергаются воздействию терапевтически активных концентраций лекарственного средства, оказывают ли они или нет отрицательное влияние на заболевание, подвергаемое лечению. Альтернативная стратегия заключается в разработке терапевтических подходов, в которых лекарственное средство непосредственно доставляют к воспаленному органу (местная терапия). Хотя данный подход не годится для всех хронических воспалительных заболеваний, он широко используется при легочных заболеваниях (астма, ХОБЛ), кожных заболеваниях (атопический дерматит и псориаз), назальных заболеваниях (аллергический ринит) и желудочно-кишечных заболеваниях (язвенный колит). При местной терапии эффективность может быть достигнута (i) при гарантии того, что лекарственное средство оказывает пролонгированное действие и сохраняется в данном органе для снижения риска системной токсичности, или (ii) путем приготовления препарата, который образует "депо" активного лекарственного средства, доступное для поддержания желаемых эффектов лекарственного средства. Примером подхода (i) является антихолинергическое лекарственное средство тиотропиум (Spiriva), которое вводят местно к легкому в качестве лечения ХОБЛ и которое имеет исключительно высокое сродство к рецептору-мишени, что приводит к замедлению скорости и получающемуся в результате пролонгированному действию. В одном аспекте раскрытия описанные соединения являются особенно подходящими для местной доставки, такой как местная доставка к легким, в частности для лечения ХОБЛ. В одном аспекте соединения оказывают более продолжительное действие, чем BIRB 796. В одном варианте осуществления изобретения соединения являются подходящими для больных,чувствительных к лечению кортикостероидом. Описанные соединения также могут быть пригодными для лечения ревматоидного артрита. Кроме того, настоящее изобретение представляет фармацевтическую композицию, содержащую соединение в соответствии с раскрытием сущности изобретения в комбинации с одним или более фармацевтически приемлемыми разбавителями или носителями. Разбавители и носители могут включать такие средства, которые являются подходящими для парентерального, перорального, местного, мукозального и ректального введения. Как описано выше, такие соединения могут быть получены, например, для парентерального, подкожного, внутримышечного, внутривенного, внутрисуставного или околосуставного введения, в частности, в виде жидких растворов или суспензий; для перорального введения, в частности, в виде таблеток или капсул; для местного, например легочного или интраназального, введения, в частности, в виде порошков, капель в нос или аэрозолей и трансдермального введения; для мукозального введения, например, к слизистой рта, под язык или слизистой влагалища и для ректального введения, например, в виде суппозитория. Композиции могут быть подходящим образом введены в стандартной лекарственной форме и могут быть приготовлены любым из способов, хорошо известных в фармацевтической области, например, как описано в публикации Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, PA.(1985). Препараты для парентерального введения могут содержать в качестве наполнителей стерилизованную воду или физиологический раствор, алкиленовые гликоли, такие как пропиленгликоль, полиалкиленгликоли, такие как полиэтиленгликоль, масла растительного происхождения, гидрогенизованные нафталены и т.п. Препараты для назального введения могут быть твердыми и могут содержать наполнители, например лактозу или декстран, или могут быть водными или масляными растворами для использования в виде капель в нос или дозированных распыляемых растворов. Для трансбуккального введения типичные наполнители включают сахара, стеарат кальция, стеарат магния, прежелатинизированный крахмал и т.п. Композиции, подходящие для перорального введения, могут содержать один или более физиологически совместимых носителей и/или наполнителей и могут быть в твердом или жидком виде. Таблетки и капсулы могут быть изготовлены с использованием связующих средств, например сиропа, камеди, желатина, сорбита, трагаканта или поливинилпирролидона; наполнителей, таких как лактоза, сахароза, кукурузный крахмал, фосфат кальция, сорбит или глицин; смазывающих средств, таких как стеарат магния,тальк, полиэтиленгликоль или диоксид кремния; и поверхностно-активных веществ, таких как лаурилсульфат натрия. Жидкие композиции могут содержать обычные добавки, такие как суспендирующие средства, например сорбитовый сироп, метилцеллюлоза, сахарный сироп, желатин, карбоксиметилцеллюлоза или пищевые жиры; эмульгирующие средства, такие как лецитин или аравийская камедь; растительные масла, такие как миндальное масло, кокосовое масло, рыбий жир или арахисовое масло; консерванты, такие как бутилированный гидроксианизол (ВНА) и бутилированный гидрокситолуол (ВНТ). Жидкие композиции могут быть капсулированы, например, в желатиновые капсулы для предоставления стандартной лекарственной формы. Твердые пероральные дозированные формы включают таблетки, состоящие из 2 частей, твердые капсулы и мягкие эластичные желатиновые (SEG) капсулы. В состав сухой оболочки обычно входит желатин с концентрацией приблизительно от 40 до 60%,пластификатор с концентрацией приблизительно от 20 до 30% (такой как глицерин, сорбит или пропиленгликоль) и вода с концентрацией приблизительно от 30 до 40%. Другие материалы, такие как консерванты, красители, замутнители и ароматизаторы, также могут присутствовать. Жидкий наполнитель включает твердое лекарственное средство, которое растворено, солюбилизировано или диспергировано(с суспендирующими средствами, такими как пчелиный воск, гидрогенизованное касторовое масло или полиэтиленгликоль 4000), или жидкое лекарственное средство в носителях или в комбинации носителей,таких как минеральное масло, растительные масла, триглицериды, гликоли, многоатомные спирты и поверхностно-активные вещества. Предпочтительно, когда соединение формулы (I) вводят местно к легкому. Поэтому авторы изобретения предоставляют согласно изобретению фармацевтическую композицию, содержащую описанное соединение необязательно в комбинации с одним или более местно допустимыми разбавителями или носителями. Местное введение к легкому может быть достигнуто при использовании аэрозольного препарата. Аэрозольные препараты обычно включают активный ингредиент, суспендированный или растворенный в более приемлемом аэрозольном пропелленте, таком как хлорфторуглерод (CFC) или гидрофторуглерод (HFC). Подходящие CFC-пропелленты включают трихлормонофторметан (пропеллент 11), дихлортетрафторметан (пропеллент 114) и дихлордифторметан (пропеллент 12). Подходящие HFCпропелленты включают тетрафторэтан (HFC-134a) и гептафторпропан (HFC-227). Пропеллент обычно содержит от 40 до 99,5%, например от 40 до 90 мас.% общей композиции для ингаляции. Препарат может содержать наполнители, включающие сорастворители (например, этанол) и поверхностно-активные вещества (например, лецитин, сорбитантриолеат и т.п.). Аэрозольные препараты упакованы в контейнеры, и подходящая доза доставляется посредством дозирующего клапана (например, как подается устройством Bespak, Valois или 3 М). Местное введение к легкому также может быть достигнуто при использовании препарата, не находящегося под давлением, такого как водный раствор или суспензия. Такой препарат может быть введен посредством небулайзера. Местное введение к легкому также может быть достигнуто при использовании препарата в виде сухого порошка. Сухой порошкообразный препарат будет содержать описанное соединение в тонкоизмельченном виде, обычно со средним диаметром частиц (MMAD) 1-10 мкм. Препарат обычно содержит допустимый для местного введения разбавитель, такой как лактоза, обычно большого размера частицы, например средний диаметр частицы (MMAD) 100 мкм или более. Примеры систем доставки сухого порошка включают SPINHALER, DISKHALER, TURBOHALER, DISKUS и CLICKHALER. Соединения согласно раскрытию сущности изобретения предназначены проявлять терапевтическую активность. В другом аспекте настоящее изобретение представляет соединение согласно раскрытию сущности изобретения для использования его в качестве лекарственного средства. Соединения согласно раскрытию сущности изобретения также могут быть пригодны в лечении респираторных нарушений, включающих ХОБЛ (включая хронический бронхит и эмфизему), астму, астму у детей, кистозный фиброз, саркоидоз, идиопатический фиброз легких, аллергический ринит, ринит, синусит, особенно астму, хронический бронхит и ХОБЛ. Соединения согласно раскрытию сущности изобретения также могут вновь сделать чувствительным состояние больного к лечению кортикостероидом, когда состояние больного не поддается лечению кортикостероидом. Соединения согласно раскрытию сущности изобретения также предполагается применять для лечения некоторых состояний, которые можно лечить посредством местной или локальной терапии, включающих аллергический конъюнктивит, конъюнктивит, аллергический дерматит, контактный дерматит,псориаз, язвенный колит, воспаление суставов, вторичное по отношению к ревматоидному артриту или остеоартриту. Соединения согласно раскрытию сущности изобретения также предполагается применять для лечения некоторых других состояний, включающих ревматоидный артрит, панкреатит, кахексию, ингибирование роста и метастазирования опухолей, включающих немелкоклеточную карциному легких, карциному молочной железы, карциному желудка, колоректальные карциномы и злокачественную меланому. Таким образом, в другом аспекте настоящее изобретение представляет соединение, представленное в описании, для применения в лечении указанных выше состояний. В другом аспекте настоящее изобретение относится к использованию соединения, представленного в описании, для получения лекарственного средства для лечения указанных выше состояний. В другом аспекте настоящее изобретение представляет способ лечения указанных выше состояний,включающий введение пациенту эффективного количества соединения согласно раскрытию сущности изобретения или его фармацевтической композиции. Слово "лечение" предназначено охватить профилактическое, а также терапевтическое лечение. Соединение согласно раскрытию сущности изобретения также может быть введено в комбинации с одним или более другими активными ингредиентами, например активными ингредиентами, подходящими для лечения указанных выше состояний. Например, возможные комбинации для лечения респираторных нарушений включают комбинации со стероидами (например, будесонид, беклометазона дипропионат, флутиказона пропионат, мометазона фуроат, флутиказона фуроат), бета-агонистами (например, тербуталин, сальбутамол, салметерол, формотерол) и/или ксантинами (например, теофиллин). Сокращения:BALF - жидкость бронхоальвеолярного лаважа (БАЛ);COPD - хроническая обструктивная болезнь легких (ХОБЛ);PBS - забуференный фосфатом физиологический раствор;RP HPLC - высокоэффективная жидкостная хроматография с обращенной фазой;TNF- - фактор альфа некроза опухоли. Общие методики. Все исходные материалы и растворители либо были получены из коммерческих источников, либо приготовлены согласно данным литературы. Гидрирование осуществляли в проточном реакторе Thales H-cube в установленных условиях. Органические растворы сушили, как принято, над сульфатом магния. Колонки КОХ приобретали в компании Supelco и сорбент обрабатывали 1 М водным растворомHCl до использования. Реакционную смесь, подвергаемую очистке, вначале разбавляли MeOH и подкисляли несколькими каплями AcOH. Полученный раствор прямо наносили на колонку КОХ и промывалиMeOH. Затем желаемый материал элюировали путем промывания раствором 1% NH3 в MeOH. Колоночную хроматографию осуществляли на предварительно укомплектованных картриджах с силикагелем компании Silicycle (230-400 меш, 40-63 мкМ), используя указанное количество. Препаративная высокоэффективная жидкостная хроматография с обращенной фазой. Колонка С 18 Agilent Scalar, 5 мкм (21,250 мм), скорость потока 28 мл/мин, элюирование градиентом смеси H2O-MeCN, содержащей 0,1% об./об. муравьиной кислоты, в течение 10 мин с использованием УФ-детекции при 215 и 254 нм. Условия градиента: 0,0-0,5 мин: 95% H2O-5% MeCN; 0,5-7,0 мин; линейное изменение от 95% H2O5% MeCN до 5% H2O-95% MeCN; 7,0-7,9 мин: поддерживание при 5% H2O-95% MeCN; 7,9-8,0 мин: возврат к 95% H2O-5% MeCN; 8,0-10,0 мин: поддерживание при 95% H2O-5% MeCN. Аналитические методы. Высокоэффективная жидкостная хроматография с обращенной фазой. Колонка С 18 Agilent Scalar, 5 мкм (4,650 мм) или колонка Waters XBridge C18, 5 мкм (4,650 мм),скорость потока 2,5 мл/мин, элюция градиентом смеси H2O-MeCN, содержащей 0,1% об./об. муравьиной кислоты, в течение 7 мин с использованием УФ-детекции при 215 и 254 нм. Условия градиента: 0,0-0,1 мин: 95% H2O-5% MeCN; 0,1-5,0 мин; линейное изменение от 95% H2O-5% MeCN до 5% H2O-95% MeCN; 5,0-5,5 мин: поддерживание при 5% H2O-95% MeCN; 5,5-5,6 мин: поддерживание при 5% H2O-95% MeCN, скорость потока увеличилась до 3,5 мл/мин; 5,6-6,6 мин: поддерживание при 5% H2O-95% MeCN, скорость потока 3,5 мл/мин; 6,6-6,75 мин: возврат к 95% H2O-5% MeCN, скорость потока 3,5 мл/мин; 6,75-6,9 мин: поддерживание при 95% H2O-5% MeCN,скорость потока 3,5 мл/мин; 6,9-7,0 мин: поддерживание при 95% H2O-5% MeCN, скорость потока снизилась до 2,5 мл/мин. 1 Н ЯМР спектроскопия. Спектрометр Bruker Avance III 400 МГц с использованием остаточного недейтерированного растворителя в качестве эталона. К раствору 4-нитронафтола (5,17 г, 27,3 ммоль), PPh3 (10,75 г, 41,0 ммоль) и 2-аминопиридин-4 метанола (1) (5,09 г, 41,0 ммоль) в ТГФ (50 мл) добавляли по каплям DIAD (8,07 мл, 41,0 ммоль) при-15 С. Смесь перемешивали в течение ночи при комнатной температуре и летучие компоненты удаляли в вакууме. Сырой продукт растирали в EtOAc (150 мл), отфильтровывали и промывали EtOAc (100 мл). Второе растирание в MeOH (100 мл) привело к 2-амино-4-4-нитронафтален-1-илокси)метил)пиридину(2) (4,54 г, 56%) в виде желтого твердого вещества.AcOH (200 мл) пропускали через реактор Thales H-cube (2,0 млмин-1, 40 С, 55 мм 10% Pt/C Cat-Cart, тип полной подачи водорода) и летучие компоненты удаляли в вакууме. Сырой продукт подвергали КОХ путем захвата и освобождения материала, используя элюцию 1% раствором NH3 в MeOH, и растворитель удаляли в вакууме с получением 2-амино-4-4-аминонафтален-1-илокси)метил)пиридина (3) (3,82 г,94%) в виде твердого вещества пурпурного цвета. К раствору CDI (4,18 г, 25,8 ммоль) в ДХМ (15 мл) добавляли по каплям в атмосфере азота раствор 3-трет-бутил-1-п-толил-1H-пиразол-5-амина (4) (WO 2000/043384) (5,91 г, 25,8 ммоль) в ДХМ (15 мл) в течение 40 мин. Образовавшийся раствор перемешивали при комнатной температуре в течение 1 ч, затем добавляли по каплям в атмосфере азота к раствору 2-амино-4-4-аминонафтален-1 илокси)метил)пиридина (3) (3,80 г, 12,9 ммоль). Смесь перемешивали в течение ночи и летучие компоненты удаляли в вакууме. Сырой материал очищали колоночной хроматографией (120 г); элюция от 0 до 6% MeOH в ДХМ с получением 1-(4-2-аминопиридин-4-ил)метокси)нафтален-1-ил)-3-(3-трет-бутил-1 п-толил-1 Н-пиразол-5-ил)мочевины (промежуточное соединение А) в виде не совсем белого твердого вещества (4,27 г, 63%). К смеси 1-(4-2-аминопиридин-4-ил)метокси)нафтален-1-ил)-3-(3-трет-бутил-1-п-толил-1Hпиразол-5-ил)мочевины (промежуточное соединение А) (526 мг, 0,96 ммоль) и DIPEA (184 мкл,1,06 ммоль) в смеси ДХМ/ДМФ (10:1, 11 мл) добавляли метоксиацетилхлорид (92 мкл, 1,01 ммоль). После перемешивания в течение 1 ч при комнатной температуре дополнительные количества DIPEA(184 мкл, 1,06 ммоль) и метоксиацетилхлорида (92 мкл, 1,01 ммоль) добавляли последовательно и перемешивание продолжали в течение 1 ч. После добавления 1% раствора NH3 в MeOH (40 мл) смесь перемешивали в течение 15 мин и упаривали в вакууме. Сырой продукт очищали колоночной хроматографией (40 г); элюция от 0 до 6% MeOH в ДХМ привела к получению N-(4-4-(3-(3-трет-бутил-1-п-толил-1 Нпиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-метоксиацетамида (пример 1) в виде белого твердого вещества (286 мг, 49%).(1,5 мл) добавляли метилизоцианат (14 мкл, 0,24 ммоль) и смесь оставляли перемешивать при комнатной температуре в течение 72 ч. Пиридин удаляли в вакууме и остаток растирали в ДХМ (3,0 мл). Фильтрование привело к получению не совсем белого порошка, 1-(3-(трет-бутил)-1-(паратолил)-1 Н-пиразол-5 ил)-3-(4-2-(3-метилуреидо)пиридин-4-ил)метокси)нафтален-1-ил)мочевина (пример 2) (36 мг, 45%). ДМФ (2 капли) добавляли к перемешиваемому раствору тетрагидропиран-2H-4-карбоновой кислоты и оксалилхлорида (21 мкл, 0,25 ммоль) в ДХМ (1,0 мл) и полученный раствор перемешивали при комнатной температуре в течение 1 ч. Раствор упаривали в вакууме с получением бесцветного масла,которое вновь растворяли в ДХМ (1,0 мл), и добавляли по каплям к перемешиваемой смеси 1-(4-2-аминопиридин-4-ил)метокси)нафтален-1-ил)-3-(3-трет-бутил-1-п-толил-1H-пиразол-5 ил)мочевины (промежуточное соединение А) (50 мг, 0,10 ммоль) и DIPEA (84 мкл, 0,50 ммоль) в ДХМ(1,0 мл). Перемешивание продолжали в течение 18 ч. Реакционную смесь перемешивали в 1% раствореNH3 в MeOH (20 мл) в течение 30 мин, упаривали в вакууме, предварительно адсорбировали на силикагеле и очищали колоночной хроматографией (12 г, 0-5% MeOH в ДХМ, градиентная элюция) с получением N-(4-4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2 ил)тетрагидро-2 Н-пиран-4-карбоксамида (пример 3) в виде светло-коричневого твердого вещества(S)-2-метоксипропионовой кислоты (50 мг, 0,48 ммоль) в ДХМ (1,0 мл) и полученный желтый раствор перемешивали при комнатной температуре в течение 1 ч. Раствор добавляли по каплям к перемешиваемой смеси 1-(4-2-аминопиридин-4-ил)метокси)нафтален-1-ил)-3-(3-трет-бутил-1-п-толил-1 Н-пиразол-5 ил)мочевины (промежуточное соединение А) (50 мг, 0,10 ммоль) и DIPEA (167 мкл, 0,96 ммоль) в ДХМ(1,0 мл). Перемешивание продолжали в течение ночи. Реакционную смесь перемешивали в 1% раствореNH3 в MeOH (20 мл), упаривали в вакууме, предварительно адсорбировали на диоксиде кремния и очищали колоночной хроматографией (12 г, 10-50% EtOAc в изогексане, градиентная элюция) с получением(S)-N-(4-4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2 ил)-2-метоксипропанамида (пример 4) в виде бесцветного твердого вещества (18 мг, 30%).(R)-2-метоксипропионовой кислоты (37 мг, 0,36 ммоль) в ДХМ (1,0 мл) и полученный раствор перемешивали при комнатной температуре в течение 1 ч. Раствор добавляли по каплям к перемешиваемой смеси 1-(4-2-аминопиридин-4-ил)метокси)нафтален-1-ил)-3-(3-трет-бутил-1-п-толил-1H-пиразол-5 ил)мочевины (промежуточное соединение А) (75 мг, 0,14 ммоль) и DIPEA (75 мкл, 0,43 ммоль) в ДХМ(2,0 мл) при 0 С. Перемешивание продолжали в течение дополнительных 48 ч. Смесь выливали в 1% раствор NH3 в MeOH (20 мл), перемешивали в течение 1 ч и упаривали в вакууме с получением желтого остатка. Колоночной хроматографией (12 г, 20-50% EtOAc в изогексане) получали (R)-N-(4-4-(3-(3 трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2 метоксипропанамид (пример 5) в виде твердого вещества светло-розового цвета (39 мг, 43%). К раствору DIPEA (1,37 мл, 7,68 ммоль) и 1-(4-2-аминопиридин-4-ил)метокси)нафтален-1-ил)-3(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)мочевины (промежуточное соединение А) (2,00 г, 3,84 ммоль) в ДХМ (40 мл) и ДМФ (8,0 мл) добавляли хлорацетилхлорид (0,61 мл, 7,68 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. ЖХ-МС обнаружено, что исходный материал израсходовался полностью. Дополнительную порцию хлорацетилхлорида (100 мкл, 1,25 ммоль) добавляли. После перемешивания в течение 1 ч при комнатной температуре реакционную смесь распределяли между ДХМ (40 мл) и насыщенным водным раствором NaHCO3 (40 мл). Органическую фазу концентрировали в вакууме и очищали колоночной хроматографией (80 г, 0-10% MeOH в ДХМ, градиентная элюция). Фракции продукта концентрировали в вакууме и остаток растирали в диэтиловом эфире (20 мл) и изогексане (20 мл). Твердое вещество собирали фильтрованием с получением N-(4-4-(3-(3-трет-бутил 1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-хлорацетамида (промежуточное соединение В) в виде твердого вещества светло-пурпурного цвета (1,07 г, 42%).N-(4-4-(3-(3-трет-Бутил-1-п-толил-1 Н-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2 ил)-2-хлорацетамид (промежуточное соединение В) (100 мг, 0,17 ммоль) добавляли порциями к перемешиваемой смеси тиометоксида натрия (35 мг, 0,50 ммоль) в MeOH (5,0 мл) и полученную смесь перемешивали в течение 1 ч при комнатной температуре. Смесь упаривали в вакууме и распределяли между насыщенным раствором соли (20 мл) и ДХМ (30 мл). Органический слой концентрировали в вакууме,остаток предварительно адсорбировали на диоксиде кремния и очищали колоночной хроматографией(12 г, 10-100% EtOAc в изогексане, градиентная элюция). Фракции продукта упаривали в вакууме с получениемN-(4-4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1 илокси)метил)пиридин-2-ил)-2-(метилтио)ацетамида (пример 6) в виде твердого вещества светложелтого цвета (28 мг, 26%).N-(4-4-(3-(3-трет-бутил-1-п-толил-1 Н-пиразол-5-ил)уреидо)нафтален-1 илокси)метил)пиридин-2-ил)-2-хлорацетамида (промежуточное соединение В) (50 мг, 0,08 ммоль) в ДХМ (1,0 мл), ДМФ (0,1 мл) и DIPEA (21,9 мкл, 0,13 ммоль) добавляли морфолин (11,0 мкл, 0,13 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. ЖХ-МС обнаружено, что 20% исходного материала превратилось в продукт. Реакционную смесь нагревали до 40C и перемешивали в течение 12 ч. ЖХ-МС обнаружено, что 87% исходного материала превратилось в продукт. Дополнительную порцию морфолина (11,0 мкл, 0,13 ммоль) добавляли и реакционную смесь перемешивали при 40C в течение 5 ч. ЖХ-МС обнаружено, что 94% исходного материала превратилось в продукт. Сырую реакционную смесь очищали колоночной хроматографией (12 г, 0-10% MeOH в ДХМ, градиентная элюция). Фракции продукта концентрировали в вакууме и остаток растирали в MeOH (5,0 мл). Твердое вещество собирали фильтрованием с получением N-(4-4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5 ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-морфолиноацетамида (пример 7) в виде твердого вещества светло-желтого цвета (11 мг, 20%).N-(4-4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1 илокси)метил)пиридин-2-ил)-2-хлорацетамида (промежуточное соединение В) (50 мг, 0,08 ммоль) в ДХМ (1,0 мл), ДМФ (0,1 мл) и DIPEA (22 мкл, 0,13 ммоль) добавляли пирролидин (7,0 мкл, 0,08 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. ЖХ-МС обнаружено, что 50% исходного материала превратилось в продукт. Реакционную смесь нагревали до 40C и перемешивали в течение 12 ч. ЖХ-МС обнаружено, что 95% исходного материала превратилось в продукт. Дополнительную порцию пирролидина (7,0 мкл, 0,08 ммоль) добавляли и реакционную смесь продолжали перемешивать при 40 С в течение 5 ч. Данные ЖХ-МС указали на полное превращение исходного материала в продукт. Сырую реакционную смесь очищали колоночной хроматографией (12 г, 0-10% MeOH в ДХМ, градиентная элюция) с получением N-(4-4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5 ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-(пирролидин-1-ил)ацетамида (пример 8) в виде твердого вещества светло-оранжевого цвета (17 мг, 32%).N-(4-4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1 илокси)метил)пиридин-2-ил)-2-хлорацетамида (промежуточное соединение В) (50 мг, 0,08 ммоль) в ДХМ (1,0 мл), ДМФ (0,1 мл) и DIPEA (22 мкл, 0,13 ммоль) добавляли N-метилпиперазин (9,3 мкл,0,08 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. ЖХ-МС об- 15020641 наружено, что 20% исходного материала превратилось в продукт. Реакционную смесь нагревали до 40C и перемешивали в течение 12 ч. ЖХ-МС обнаружено, что 91% исходного материала превратился в продукт. Дополнительную порцию N-метилпиперазина (9,0 мкл, 0,08 ммоль) добавляли и реакционную смесь продолжали перемешивать при 40C в течение 5 ч. ЖХ-МС обнаружено, что 98% исходного материала превратилось в продукт. Сырую реакционную смесь очищали колоночной хроматографией (12 г,0-10% MeOH в ДХМ, градиентная элюция). Фракции продукта концентрировали в вакууме и остаток растирали в смеси диэтилового эфира, ДХМ и изогексана (2:1:2, 5,0 мл) с получением N-(4-4-(3-(3-третбутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-(4 метилпиперазин-1-ил)ацетамида (пример 9) в виде твердого вещества светло-оранжевого цвета (26 мг,47%).(12,5 мкл, 0,08 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 ч. ЖХ-МС обнаружено, что 20% исходного материала превратилось в продукт. Реакционную смесь нагревали до 40C и перемешивали в течение 12 ч. ЖХ-МС обнаружено, что 78% исходного материала превратилось в продукт. Дополнительную порцию N-метоксиэтилпиперазина (12,5 мкл, 0,08 ммоль) добавляли и реакционную смесь продолжали перемешивать при 40C в течение 5 ч. ЖХ-МС обнаружено, что 89% исходного материала превратилось в продукт. Сырую реакционную смесь очищали колоночной хроматографией (12 г, 0-10% MeOH в ДХМ, градиентная элюция) с получением N-(4-4-(3-(3-третбутил-1-п-толил-1 Н-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-(4-(2 метоксиэтил)пиперазин-1-ил)ацетамида (пример 10) в виде твердого вещества светло-оранжевого цветаN-(4-4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1 илокси)метил)пиридин-2-ил)-2-хлорацетамида (промежуточное соединение В) (50 мг, 0,08 ммоль) в ДХМ (1,0 мл), ДМФ (0,1 мл) и DIPEA (17 мкл, 0,10 ммоль) добавляли 2-метоксиэтиламин (7,0 мкл,0,08 ммоль). Реакционную смесь нагревали до 40C и перемешивали в течение 12 ч. Сырую реакционную смесь очищали колоночной хроматографией (12 г, 0-10% MeOH в ДХМ, градиентная элюция). Фракции продукта концентрировали в вакууме и остаток растирали в смеси диэтилового эфира, ДХМ и изогексана (2:1:2, 5,0 мл) с получением N-(4-4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5 ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-(2-метоксиэтиламино)ацетамида (пример 11) в виде не совсем белого твердого вещества (6 мг, 11%).(41 мкл, 0,08 ммоль). Реакционную смесь нагревали до 40C и перемешивали в течение 12 ч. Сырую реакционную смесь очищали колоночной хроматографией (12 г диоксида кремния, 0-10% MeOH в ДХМ,градиентная элюция). Фракции продукта концентрировали в вакууме и остаток растирали в смеси диэтилового эфира, ДХМ и изогексана (2:1:2, 5,0 мл) с получением N-(4-4-(3-(3-трет-бутил-1-п-толил-1Hпиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-(диметиламино)ацетамида (пример 12) в виде оранжевого твердого вещества (18 мг, 35%).(41 мкл, 0,08 ммоль). Реакционную смесь нагревали до 40C и перемешивали в течение 12 ч. Сырую реакционную смесь очищали колоночной хроматографией (12 г, 0-10% MeOH в ДХМ, градиентная элюция). Фракции продукта были загрязнены примесью; сырой материал вновь подвергали очистке колоночной хроматографией (12 г, 0-10% MeOH в ДХМ, градиентная элюция) с получением N-(4-4-(3-(3 трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2(метиламино)ацетамида (пример 13) в виде твердого вещества светло-коричневого цвета (6 мг, 12%).N-(4-4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1 илокси)метил)пиридин-2-ил)-2-хлорацетамида (промежуточное соединение В) (50 мг, 0,08 ммоль) в ДХМ (1,0 мл), ДМФ (0,2 мл) и DIPEA (17,5 мкл, 0,1 ммоль) добавляли N-(4-метоксибензил)-Nметиламин (15,5 мкл, 0,09 ммоль). Реакционную смесь перемешивали при 55C в течение 12 ч. Сырую реакционную смесь очищали колоночной хроматографией (12 г, 0-10% MeOH в ДХМ, градиентная элюция). Фракции продукта концентрировали в вакууме и остаток растирали в смеси диэтилового эфира,ДХМ и изогексана (2:1:2, 5,0 мл) с получением N-(4-4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5 ил)уреидо)нафтален-1-илокси)метил)пиридин-2-ил)-2-4-метоксибензил)(метил)амино)ацетамида (пример 14) в виде белого твердого вещества (7 мг, 11%). К раствору 2-(2-(трет-бутоксикарбониламино)пиридин-4-ил)ацетата (5) (WO 2007/089512) (10,0 г,35,7 ммоль) в атмосфере азота в ТГФ (100 мл) при -78C добавляли DIBAL (1 M раствор в ТГФ, 71,3 мл,71,3 ммоль) в течение 1 ч. Реакционную смесь перемешивали при температуре от -78 до -60C в течение 40 мин и затем нагревали до -15C в течение 1 ч. Раствор вновь охлаждали до -78C и обрабатывали дополнительным количеством DIBAL (1 M раствор в ТГФ, 35 мл, 35,7 ммоль). Смесь оставляли нагреваться до -40C и перемешивали в течение 1 ч. Воду (10 мл) осторожно добавляли, чтобы погасить реакцию,с последующим добавлением MgSO4 (20 г) и твердые частицы удаляли фильтрацией. Фильтрат концентрировали досуха при пониженном давлении и остаток подвергали колоночной хроматографии (330 г),- 18020641(6,00 г, 64%) в виде твердого вещества желтого цвета.(70 мл) добавляли гидрид натрия (2,52 г, 63,0 ммоль, 60 мас.%) при 0C. Суспензию ярко-желтого цвета перемешивали в течение 20 мин при 0C до добавления 1-фтор-4-нитронафталена (4,81 г, 25,2 ммоль) в одну порцию. После перемешивания при комнатной температуре в течение 2 ч воду (100 мл) добавляли с последующим добавлением EtOAc (100 мл). Твердое вещество, образованное между слоями, собирали фильтрацией и органическую фазу промывали насыщенным водным раствором NaHCO3 (100 мл), насыщенным раствором соли (100 мл) и сушили. Летучие компоненты удаляли с получением твердого вещества оранжевого цвета. Твердые частицы объединяли и растирали в MeOH (50 мл) с получением третбутил 4-(2-(4-нитронафтален-1-илокси)этил)пиридин-2-илкарбамата (7) в виде твердого вещества желтого цвета (11,0 г, 98%). трет-Бутил 4-(2-(4-нитронафтален-1-илокси)этил)пиридин-2-илкарбамат (7) (5,20 г, 12,7 ммоль) и железную сетку (4,30 г, 76 ммоль) суспендировали в смеси AcOH и EtOH (1:2, 120 мл). Суспензию помещали в предварительно нагретую масляную баню при 60C и быстро перемешивали до завершения реакции, как оценивали посредством ЖХ-МС. Смесь охлаждали до комнатной температуры, осторожно выливали в насыщенный водный раствор (1000 мл) и экстрагировали EtOAc (2500 мл). Объединенные органические слои промывали также насыщенным водным раствором NaHCO3 (1000 мл), водой(1000 мл), насыщенным раствором соли (1000 мл) и сушили. Раствор фильтровали и упаривали с получением трет-бутил 4-(2-(4-аминонафтален-1-илокси)этил)пиридин-2-илкарбамата (8) в виде желтого масла К суспензии CDI (3,00 г, 18,18 ммоль) в ДХМ (15 мл) добавляли раствор 3-трет-бутил-1-п-толил 1 Н-пиразол-5-амина (4) (WO 2000/043384) (4,17 г, 18,18 ммоль) в ДХМ (40 мл) в течение 1,5 ч. После перемешивания при комнатной температуре в течение 2 ч раствор трет-бутил-4-(2-(4-аминонафтален-1 илокси)этил)пиридин-2-илкарбамата (8) (3,00 г, 7,91 ммоль) в ДХМ (15 мл) добавляли. После перемешивания в течение ночи раствор разбавляли MeOH (10 мл) и подвергали абсорбции на силикагеле (30 г) и колоночной хроматографии (330 г), элюируя раствором от 30 до 100% EtOAc в изогексане и затем от 0 до 6% MeOH в EtOAc с получением трет-бутил-4-(2-(4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5 ил)уреидо)нафтален-1-илокси)этил)пиридин-2-илкарбамата (9) в виде твердого вещества бежевого цвета К суспензии трет-бутил-4-(2-(4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5-ил)уреидо)нафтален-1 илокси)этил)пиридин-2-илкарбамата (9) (1,35 г, 2,20 ммоль) в ДХМ (10 мл) добавляли ТФУ (10 мл). После перемешивания при комнатной температуре в течение 2 ч летучие компоненты упаривали и остаток брали в EtOAc (50 мл) и экстрагировали насыщенным водным раствором NaHCO3 (50 мл). Слои разделяли, органический слой промывали насыщенным раствором соли (50 мл), сушили и упаривали с получением 1-(4-(2-(2-аминопиридин-4-ил)этокси)нафтален-1-ил)-3-(3-трет-бутил-1-п-толил-1H-пиразол-5 ил)мочевины (промежуточное соединение С) в виде твердого вещества бледно-розового цвета (1,20 г,100%).DIPEA (23 мкл, 0,131 ммоль) и метоксиацетилхлорид (7 мкл, 0,072 ммоль). Смесь перемешивали при комнатной температуре до тех пор, пока реакция не завершалась, как оценивали посредством ЖХ-МС; разбавляли насыщенным водным раствором NaHCO3 (1,5 мл) и слои разделяли посредством элемента разделения фаз. Органические слои собирали, упаривали при пониженном давлении и остаток подвергали КОХ путем захвата и освобождения материала. Полученный после КОХ остаток далее очищали препаративной ОФ-ВЭЖХ с получениемN-(4-(2-(4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5 ил)уреидо)нафтален-1-илокси)этил)пиридин-2-ил)-2-метоксиацетамида (пример 15) в виде белого твердого вещества (5 мг, 13%). вали при комнатной температуре до тех пор, пока реакция не завершалась, как оценивали ЖХ-МС; разбавляли насыщенным водным раствором NaHCO3 (1,5 мл) и слои разделяли посредством элемента разделения фаз. Органические слои собирали, упаривали при пониженном давлении и остаток подвергали КОХ путем захвата и освобождения материала. Полученный после КОХ остаток далее очищали препаративной ОФ-ВЭЖХ с получениемN-(4-(2-(4-(3-(3-трет-бутил-1-п-толил-1H-пиразол-5 ил)уреидо)нафтален-1-илокси)этил)пиридин-2-ил)-2-(2-метоксиэтокси)ацетамида (пример 16) в виде не совсем белого твердого вещества (13 мг, 31%). К раствору 1-(4-(2-(2-аминопиридин-4-ил)этокси)нафтален-1-ил)-3-(3-трет-бутил-1-п-толил-1 Нпиразол-5-ил)мочевины (7) (промежуточное соединение С) (50 мг, 0,094 ммоль) в пиридине (1,0 мл) добавляли метилизоцианат (5,34 мг, 0,094 ммоль). Смесь перемешивали при комнатной температуре в течение 72 ч и растворитель упаривали при пониженном давлении. Образовавшийся остаток растирали вMeOH (5,0 мл) с получением 1-(3-(трет-бутил)-1-(паратолил)-1 Н-пиразол-5-ил)-3-(4-(2-(2-(3 метилуреидопиридин-4-ил)этокси)нафтален-1-ил)мочевины (пример 17) в виде не совсем белого твердого вещества (7 мг, 13%). К раствору 1-(4-(2-(2-аминопиридин-4-ил)этокси)нафтален-1-ил)-3-(3-трет-бутил-1-п-толил-1Hпиразол-5-ил)мочевины (7) (промежуточное соединение С) (50 мг, 0,094 ммоль) в пиридине (1,0 мл) добавляли трихлорацетилизоцианат (12 мкл, 0,103 ммоль). Смесь перемешивали при комнатной температуре до тех пор, пока реакция не завершалась, как оценивали ЖХ-МС, и растворитель упаривали при пониженном давлении. Образовавшийся остаток подвергали КОХ путем захвата и освобождения остатка и растирали в ДХМ (10 мл) с получением 1-(3-(трет-бутил)-1-(паратолил)-1H-пиразол-5-ил)-3-(4-(2-(2 уреидопиридин-4-ил)этокси)нафталенил-1-ил)мочевины (пример 18) в виде не совсем белого твердого вещества (25 мг, 44%). Биологическое исследование. Все примеры соединений продемонстрировали величины ЕС 50 менее чем 1 мкМ в зависимости от высвобождения LPS-индуцированного фактора TNF- в дифференцированных клетках U937 (см. ниже относительно деталей исследования). Суммирование свойств соединения примера 1, установленных посредством исследования как in vitro, так и in vivo, представлено ниже.In vitro тестирование для соединения примера 1 50% эффективная концентрация по отношению к действию 10 мкг/мл BIRB 796 (как 100%). Никакого значительного токсического действия не наблюдали в МТТ-тесте. Описание указанных выше анализов следующее. Исследование ингибирования фермента. Ингибирующую фермент активность соединения определяли методом резонансного переноса энергии флуоресценции (FRET), используя синтетические пептиды, меченные как донорными, так и акцепторными флуорофорами (Z-LYTE, Invitrogen). Кратко, рекомбинантную, фосфорилированную гаммаизоформу p38 MAPK (MAPK12: Millipore) разбавляли в буфере HEPES, смешивали с соединением при желательных конечных концентрациях и инкубировали в течение 2 ч при комнатной температуре. ЗатемFRET-пептид (2 мкМ) и АТР (100 мкМ) добавляли к смеси фермент/соединение и инкубировали в течение 1 ч. Проявляющий реагент (протеаза) добавляли в течение 1 ч до количественного определения в считывающем устройстве для микропланшетов с флуоресцентным детектором. Сайт-специфическая протеаза только расщепляет нефосфорилированный пептид и выделяет FRET-сигнал. Уровни фосфорилирования каждой реакции рассчитывали, используя отношение испускания кумарина (донор) к испусканию флуоресцеина (акцептор), с высокими отношениями, указывающими на высокое фосфорилирование, и низкими отношениями, указывающими на низкие уровни фосфорилирования. Процент ингибирования каждой реакции рассчитывали относительно неингибированного контроля и затем рассчитывали концентрацию, вызывающую 50% ингибирования (величина IC50), исходя из кривой концентрация-ответ. Для альфа-изоформы p38 MAPK (MAPK14: Invitrogen) ферментативную активность оценивали непрямо путем определения активации/фосфорилирования нижестоящей в каскаде реакций молекулы,MAPKAP-K2. Белок p38 MAPKсмешивали с его неактивной мишенью MAPKAP-K2 (Invitrogen) и соединением в течение 2 ч при комнатной температуре. Затем FRET-пептид (2 мкМ), который является мишенью фосфорилирования для MAPKAP-K2, и АТР (10 мкМ) добавляли к смеси ферменты/соединение и инкубировали в течение 1 ч. Затем проявляющий реагент добавляли и смесь инкубировали в течение 1 ч до измерения флуоресценции, завершающего протокол исследования. Высвобождение LPS-индуцированного фактора TNF-альфа в клетках U937: активность. Клетки U937, линия моноцитов человека, были дифференцированы в клетки типа макрофагов путем инкубации с форболмиристатацетатом (РМА; 100 нг/мл) в течение времени от 48 до 72 ч. При необходимости клетки предварительно инкубировали с конечными концентрациями соединения в течение 2 ч. Затем клетки подвергали стимуляции 0,1 мкг/мл LPS (из E.coli: 0111:B4, Sigma) в течение 4 ч и супернатант собирали для определения концентрации TNF- методом сэндвич-ELISA (Duo-set, системыRD). Клетки ТНР-1, линию моноцитов человека, также использовали для данного исследования. Клетки ТНР-1 подвергали стимуляции 1 мкг/мл LPS (из E.coli: 0111:В 4, Sigma) в течение 4 ч и супернатант собирали для определения концентрации TNF-. Ингибирование продукции TNF- рассчитывали как процент ингибирования, достигаемого действием 10 мкг/мл BIRB 796 при каждой концентрации испытуемого соединения путем сравнения с носителем в качестве контроля. Исходя из выстроенной кривой концентрация-ответ, была определена 50% эффективная концентрация (ЕС 50). Высвобождение LPS-индуцированного фактора TNF-альфа в клетках ТНР-1: активность. Клетки ТНР-1, линия моноцитов человека, подвергали стимуляции 1 мкг/мл LPS (из E.coli; 0111:В 4,Sigma) в течение 4 ч и супернатант собирали для определения концентрации TNF- анализом сэндвичELISA (Duo-set, системы RD). Ингибирование продукции TNF- рассчитывали при каждой концентра- 22020641 ции путем сравнения с носителем в качестве контроля. Исходя из выстроенной кривой концентрацияответ, определяли 50% ингибиторную концентрацию (IC50). МТТ-тест. Дифференцированные клетки U937 предварительно инкубировали с соединением в течение 4 ч в 5% FCS или 10% FCS в течение 24 и 72 ч. Супернатант заменяли 200 мкл новых сред и 10 мкл исходного раствора МТТ (5 мг/мл) добавляли к каждой лунке. После 1 ч инкубации среды удаляли 200 мкл ДМСО добавляли к каждой лунке и планшеты слегка встряхивали в течение 1 ч до прочтения абсорбции при 550 нм. Потерю жизнеспособности клеток, выраженной в процентах, рассчитывали для каждой лунки относительно обработки носителем (0,5% ДМСО). В результате, кажущееся увеличение жизнеспособности клеток при обработке лекарственным средством относительно носителя представлено в таблице в виде отрицательного процента.In vivo исследование для примера 1.LPS-индуцированный нейтрофиллез у мыши: продолжительность действия. Мышам, которых не держали на голоде, вводили внутрь трахеи либо носитель, либо испытуемое вещество в моменты времени ("до введения препарата"), указанные в соответствии с началом обработкиLPS. В момент времени Т=0 мышей помещали в камеру для исследования и подвергали воздействиюLPS. Через 8 ч после стимуляции LPS животным давали наркоз, трахею канюлировали и БАЛ извлекали путем вливания и отбора 1 мл PBS в легкие через трахеальный катетер. Общий и дифференцированный подсчет лейкоцитов в образцах БАЛ осуществляли, используя счетную камеру Нейбауэра. Цитоспиновые мазки образцов БАЛ получали путем центрифугирования при 200 об/мин в течение 5 мин при комнатной температуре и окрашивали, используя систему окрашивания DiffQuik (Dade Behring). Клетки считали, используя микроскоп с масляной иммерсией. Результаты представлены на фиг. 1 и 2. Данные для числа нейтрофилов представлены как общий и дифференциальный счет (испытуемое вещество относительно носителя) клеток на 1 мл БАЛ, среднее значениеS.E.M. (n=8). Краткие выводы. Биологические исследования in vitro показали, что соединение примера 1 является мощным ингибитором подтипов альфа и гамма p38 МАР-киназы с хорошей эффективностью в in vitro модели противовоспалительной активности (LPS-индуцированное высвобождение TNF- из дифференцированных клеток U937 и клеток ТНР-1). Исходя из результатов МТТ-теста, можно заключить, что соединение не проявляет явной клеточной токсичности при используемых концентрациях. Биологические исследования in vivo показали, что соединение примера 1 проявляет эффективность в ингибировании LPS-индуцированном накоплении нейтрофилов в модели животных, с продолжительным эффектом действия, как показано посредством существенного ингибирования даже в течение 12 ч или более до введения препарата. Следует понимать, что по всему описанию и формуле изобретения, которая следует за описанием,если контекст не требует иного, слово "включать" и вариации, такие как "включения" и "включающий",подразумевает включение установленного числа, этапа, группы чисел или группы этапов, но не исключение любого другого числа, этапа, группы чисел или группы этапов. Все патенты и заявки на патенты, относящиеся к описанию, включены посредством ссылки во всей полноте. Заявка, у которой описание и формула изобретения образует часть, может быть использована как основа для приоритета в отношении любой последующей заявки. Пункты формулы изобретения такой последующей заявки могут быть направлены на любое свойство или комбинацию свойств, представленных в описании. Они могут охватывать форму продукта, композицию, способ или использовать пункты формулы изобретения и могут включать, путем примера и без ограничения, формулу изобретения. где R1 означает C1-6 алкил, необязательно замещенный гидроксильной группой;R2 означает Н или C1-6 алкил, необязательно замещенный гидроксильной группой;Ar означает нафтильное или фенильное кольцо, любое из которых может быть необязательно замещено одной или более группами, независимо выбранными из C1-6 алкила, С 1-6 алкокси, амино и C1-4 моноили диалкиламино;-NHC(O)(CH2)x-O-(CH2)y-CH3,где х означает целое число от 1 до 6 и у означает 0 или целое число от 1 до 5 при условии, что x+у означает целое число от 1 до 6; иp равен 0, 1 или 2,или его фармацевтически приемлемая соль, включая все его стереоизомеры и таутомеры. 2. Соединение по п.1, где Ar означает нафтил. 3. Соединение по п.1 или 2, где R1 означает трет-бутил. 4. Соединение по любому одному из пп.1-3, где R2 означает метил. 5. Соединение по любому одному из пп.1-4, где R2 находится в пара-положении. 6. Соединение по любому из пп.1-5,где где x равен целому числу от 1 до 6 и у равен 0 или целому числу от 1 до 5 при условии, что х+у равно целому числу от 1 до 6, например x равен 1 и у равен 1,или его фармацевтически приемлемая соль, включая все его стереоизомеры и таутомеры. 9. Соединение по п.1, представленное формулой (ID) где x равен целому числу от 1 до 6 и у равен 0 или целому числу от 1 до 5 при условии, что х+у равно целому числу от 1 до 6, например x равен 1 и у равен 0,или его фармацевтически приемлемая соль, включая все его стереоизомеры и таутомеры. 10. Соединение по п.1, выбранное изN-(4-(2-(4-(3-(3-трет-бутил-1-п-толил-1 Н-пиразол-5-ил)уреидо)нафтален-1-илокси)этил)пиридин-2 ил)-2-(2-метоксиэтокси)ацетамида; 1-(3-(трет-бутил)-1-(паратолил)-1H-пиразол-5-ил)-3-(4-(2-(2-(3-метилуреидопиридин-4 ил)этокси)нафтален-1-ил)мочевины и 1-(3-(трет-бутил)-1-(паратолил)-1H-пиразол-5-ил)-3-(4-(2-(2-уреидопиридин-4 ил)этокси)нафталенил-1-ил)мочевины,или его фармацевтически приемлемая соль, включая все его стереоизомеры и таутомеры. 11. Фармацевтическая композиция, содержащая соединение по любому одному из пп.1-10, в комбинации с одним или более фармацевтически приемлемыми разбавителями или носителями. 12. Применение соединения по любому одному из пп.1-10 в качестве лекарственного средства. 13. Применение соединения по любому одному из пп.1-10 для лечения или предотвращения состояния, выбранного из ХОБЛ (включая хронический бронхит и эмфизему), астмы, астмы у детей, кистозного фиброза, саркоидоза, идиопатического фиброза легких, аллергического ринита, ринита, синусита, аллергического конъюнктивита, конъюнктивита, аллергического дерматита, контактного дерматита, псориаза, язвенного колита, воспаления суставов, вторичного по отношению к ревматоидному артриту или остеоартриту, ревматоидного артрита, панкреатита, кахексии, ингибирования роста и метастазирования опухолей, включающих немелкоклеточную карциному легких, карциному молочной железы, карциному желудка, колоректальные карциномы и злокачественную меланому. 14. Применение соединения формулы (I) согласно пп.12 и 13 в сочетании с одним или более активным ингредиентом, представляющим собой лекарственное средство для лечения или предупреждения респираторных нарушений, где активный ингредиент представляет собой стероид, бета-агонист или ксантин. 15. Применение по п.14, где активный ингредиент представляет собой стероид, который выбран из группы, состоящей из будесонида, беклометазона дипропионата, флутиказона пропионата, мометазона фуроата и флутиказона фуроата, или активный ингредиент представляет собой бета-агонист, который выбран из группы, состоящей из тербуталина, сальбутамола, салметерола и формотерола, или активный ингредиент представляет собой ксантин, такой как теофиллин. 16. Применение соединения по любому одному из пп.1-10 для получения лекарственного средства для лечения или предотвращения состояния, выбранного из ХОБЛ (включая хронический бронхит и эмфизему), астмы, астмы у детей, кистозного фиброза, саркоидоза, идиопатического фиброза легких, аллергического ринита, ринита, синусита, аллергического конъюнктивита, конъюнктивита, аллергического дерматита, контактного дерматита, псориаза, язвенного колита, воспаления суставов, вторичного по отношению к ревматоидному артриту или остеоартриту, ревматоидного артрита, панкреатита, кахексии,ингибирования роста и метастазирования опухолей, включающих немелкоклеточную карциному легких,карциному молочной железы, карциному желудка, колоректальные карциномы и злокачественную меланому. Влияние соединения примера 1 на LPS-индуцированное накопление нейтрофилов в БАЛ Фиг. 1 Влияние соединения примера 1 на LPS-индуцированное накопление нейтрофилов в БАЛ
МПК / Метки
МПК: C07D 403/12, A61P 29/00, A61K 31/415, C07D 401/12, A61P 11/00
Метки: производные, n-(1-фенил-3-алкилпиразол-5-ил)-n'-арилмочевины
Код ссылки
<a href="https://eas.patents.su/27-20641-proizvodnye-n-1-fenil-3-alkilpirazol-5-il-n-arilmocheviny.html" rel="bookmark" title="База патентов Евразийского Союза">Производные n-(1-фенил-3-алкилпиразол-5-ил)-n’-арилмочевины</a>
Производные 5-[4-(азетидин-3-илокси)фенил]-2-фенил-5н-тиазоло[5,4-c]пиридин-4-она и их использование в качестве рецепторов mch

Номер патента: 15559
Опубликовано: 31.08.2011
Авторы: Секереш Хелен Джейн, Брунавс Майкл, Гардинир Кевин Мэттью, Хембр Эрик Джеймс, Гармен Дэвид Джозеф
МПК: C07D 513/04, A61K 31/437
Метки: 5-[4-(азетидин-3-илокси)фенил]-2-фенил-5н-тиазоло[5,4-c]пиридин-4-она, качестве, производные, рецепторов, использование
Формула / Реферат:
1. Соединение формулыгде ------ отсутствует или возможно представляет собой связь;q представляет собой 1 или 2;R1 независимо выбран из водорода, -C1-C2-алкила, галогена, гидрокси, -C1-C2-галогеналкила, -C1-C3-алкокси, циано, -O-C3-C4-циклоалкила и -OC1-C2-галогеналкила;R2 выбран из группы, состоящей из водорода, -C1-C3-алкила, гидрокси, -C1-C3-алкокси, циано, -C1-C2-галогеналкила, -OC1-C2-галогеналкила и галогена;R3 выбран из группы, состоящей...
Производные замещенных 3-фенил-1-(фенилтиенил)пропан-1-онов и 3-фенил-1-(фенилфуранил)пропан-1-онов, их получение и применение

Номер патента: 17449
Опубликовано: 28.12.2012
Автор: Дельомель Жан-Франсуа
МПК: C07D 333/22, A61P 3/00, A61K 31/381...
Метки: 3-фенил-1-(фенилтиенил)пропан-1-онов, 3-фенил-1-(фенилфуранил)пропан-1-онов, замещенных, получение, применение, производные
Формула / Реферат:
1. Соединение общей формулы (I)в которой X1 представляет собой галоген, R1, -SR1 или -OR1 группу;Х2 представляет собой атом серы или кислорода;Х3 представляет собой галоген, R3, -SR3 или -OR3 группу;Х4 представляет собой галоген, R4, -SR4 или -OR4 группу;Х5 представляет собой Y, -S-Y или -O-Y, где Y является группой R5-COOR12 или R5-CONR12R13;Х6 представляет собой галоген, R6, -SR6 или -OR6 группу;Х7 представляет собой галоген, R7, -SR7 или -OR7...
Производные фенил- и бензодиоксинилзамещенных индазолов

Номер патента: 18629
Опубликовано: 30.09.2013
Авторы: Дамен Ян, Лепистё Матти, Бергер Маркус, Нильссон Стинабритт, Хеммерлинг Мартин, Эрикссон Андерс, Ханссон Томас, Хоссаин Нафизал, Клингстенд Томас, Ревинкель Хартмут, Эдман Карл
МПК: A61K 31/416, A61K 31/4439, A61P 11/00...
Метки: производные, фенил, бензодиоксинилзамещенных, индазолов
Формула / Реферат:
1. Соединение формулы (Ib)где А представляет собой С1-2фторалкил;R3 представляет собой бензодиоксинил;W представляет собой фенил, замещенный -C(O)NR7R8;R7 представляет собой водород;R8 выбран из метила, этила, пропила и бутила (замещенных одной или двумя группами, выбранными из гидроксила, фенила или пиридинила) или R8 выбран из циклопентила, гидроксициклопентила, оксидотетрагидротиофенила, диоксидотетрагидротиофенила, тетрагидрофуранила и...
Производные n-фенил-2-пиримидинамина и способ их получения

Номер патента: 15103
Опубликовано: 30.06.2011
Авторы: Ким Ёнг-Сеок, Ким Хонг-Ёуб, Ким Донг-Ён, Чо Дае-Жин, Хан Бьенг-Чеол, Лии Сан-Эйх, Лии Гонг-Ял, Вуу Сеок-Хун
МПК: A61K 31/44, A61K 31/4965, A61K 31/425...
Метки: n-фенил-2-пиримидинамина, производные, получения, способ
Формула / Реферат:
1. Производное N-фенил-2-пиримидинамина формулы (1)или его соль, гдеR1 представляет селективно замещенный тиазол, имидазол или пиразин, где заместитель является аминогруппой или алкилом, содержащим от 1 до 6 атомов углерода;R2, R3и R6, каждый независимо, представляет водород, галоген, алкил, содержащий от 1 до 6 атомов углерода, или алкокси, содержащий от 1 до 6 атомов углерода;R4 и R5, каждый независимо, представляют водород, и R4и R5 не...
Производные 5-амино-6-фенил-7-галоген[1,2,4]триазолo[1,5-a]пиримидинa

Номер патента: 6710
Опубликовано: 24.02.2006
Авторы: Пес Клаус-Юрген, Аммерманн Эберхард, Альберт Гуидо, Термо И Бласко Хорди, Рениг Аннерозе, Серч Дебра
МПК: C07D 487/04
Метки: 5-амино-6-фенил-7-галоген[1,2,4]триазолo[1,5-a]пиримидинa, производные
Формула / Реферат:
1. Производные 5-амино-5-фенил-7-галоген[1,2,4]триазоло[1,5-a]пиримидина формулы I в которой R1 означает водород, фтор, C1-C10-алкил, C2-C10-алкенил, C2-C10-алкинил, C2-C10-алкадиенил, причем углеродные цепи этих остатков могут быть частично или полностью галогенированными или могут иметь от одной до трех групп Ra, Ra означает циано, нитро, гидроксил, C1-C6-алкил, C1-C6-галогеналкил, C1-C6-алкилкарбонил, C3-C6-циклоалкил, C1-C6-алкокси,...
Предыдущий патент: Способ изготовления слоистого оконного стекла
Следующий патент: Огнестойкая полимерная композиция с улучшенными механическими свойствами
Случайный патент: Бета - карболины, применяемые для лечения воспалительных заболеваний