Производные 5-(3-аминофенил)-5-алкил-5,6-дигидро-2h-[1,4]оксазин-3-амина
Номер патента: 24716
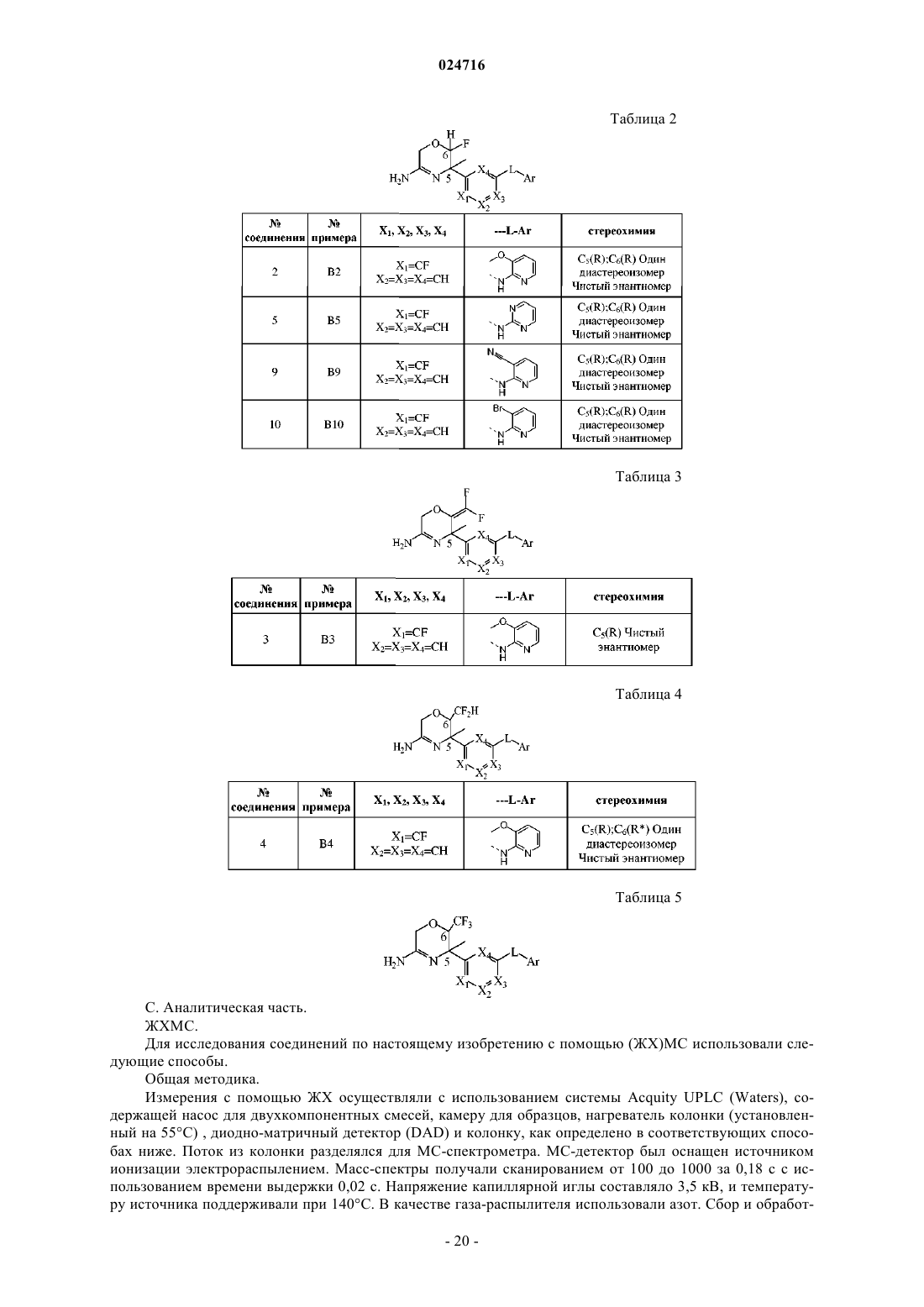
Опубликовано: 31.10.2016
Авторы: Ван Брандт Свен Францискус Анна, Гейсен Хенрикус Якобус Мария

























Формула / Реферат
1. Соединение формулы (I)

где R1 представляет собой фтор, дифторметил или трифторметил;
R2 представляет собой водород или трифторметил; или
R1 и R2 образуют двухвалентный радикал =CF2;
R3 представляет собой C1-3алкил, циклопропил, моно- или полигалоген-C1-3алкил;
R4 представляет собой водород или фтор;
Ar представляет собой гетероарил;
где гетероарил представляет собой пиридил или пиримидил, каждый из которых необязательно замещен одним, двумя или тремя заместителями, выбранными из галогена, циано, C1-3алкила, C2-3алкинила, С1-3алкилокси, моно- и полигалоген-C1-3алкила, моно- и полигалоген-С1-3алкилокси и С1-3алкилоксиС1-3алкилокси;
или его стереоизомерная форма или его фармацевтически приемлемая соль.
2. Соединение по п.1, где
R1 представляет собой фтор, дифторметил или трифторметил,
R2 представляет собой водород; или
R1 представляет собой фтор,
R2 представляет собой трифторметил; или
R1 и R2 образуют двухвалентный радикал =CF2;
R3 представляет собой метил или циклопропил;
R4 представляет собой водород или фтор;
Ar представляет собой гетероарил;
где гетероарил представляет собой пиридил, замещенный метокси или пиримидил;
или его фармацевтически приемлемая соль.
3. Соединение по п.1, где атом углерода, замещенный R3, характеризуется R-конфигурацией.
4. Соединение по п.1, выбранное из
(5R,6S)-5-циклопропил-6-фтор-5-{3-[(3-метоксипиридин-2-ил)амино]фенил}-6-(трифторметил)-5,6-дигидро-2H-1,4-оксазин-3-амина,
(5R,6R)-6-фтор-5-{2-фтор-5-[(3-метоксипиридин-2-ил)амино]фенил}-5-метил-5,6-дигидро-2H-1,4-оксазин-3-амина,
(5R)-6-(дифторметилиден)-5-{2-фтор-5-[(3-метоксипиридин-2-ил)амино]фенил}-5-метил-5,6-дигидро-2H-1,4-оксазин-3-амина,
(5R,6R*)-6-(дифторметил)-5-{2-фтор-5-[(3-метоксипиридин-2-ил)амино]фенил}-5-метил-5,6-дигидро-2H-1,4-оксазин-3-амина,
(5R,6R)-6-фтор-5-[2-фтор-5-(пиримидин-2-иламино)фенил]-5-метил-5,6-дигидро-2H-1,4-оксазин-3-амина,
(5R,6R)-5-{2-фтор-5-[(3-метоксипиридин-2-ил)амино]фенил]-5-метил-6-(трифторметил)-5,6-дигидро-2H-1,4-оксазин-3-амина,
(5R,6R)-5-[2-фтор-5-(пиримидин-2-иламино)фенил]-5-метил-6-(трифторметил)-5,6-дигидро-2H-1,4-оксазин-3-амина,
(5R,6R)-6-фтор-5-{2-фтор-5-[(3-метоксипиразин-2-ил)амино]фенил}-5-метил-5,6-дигидро-2H-1,4-оксазин-3-амина и
(5R,6R)-5-{2-фтор-5-[(3-метоксипиразин-2-ил)амино]фенил}-5-метил-6-(трифторметил)-5,6-дигидро-2H-1,4-оксазин-3-амина.
5. Фармацевтическая композиция для лечения расстройства, выбранного из болезни Альцгеймера, умеренного когнитивного нарушения, старения, деменции, деменции с тельцами Леви, церебральной амилоидной ангиопатии, слабоумия вследствие множественных инфарктов, синдрома Дауна, деменции, ассоциированной с инсультом, деменции, ассоциированной с болезнью Паркинсона, и деменции, ассоциированной с бета-амилоидом, содержащая терапевтически эффективное количество соединения по любому из пп.1-4 и фармацевтически приемлемый носитель.
6. Способ получения фармацевтической композиции по п.5, отличающийся тем, что фармацевтически приемлемый носитель тщательно смешивают с терапевтически эффективным количеством соединения по любому из пп.1-4.
7. Применение соединения по любому из пп.1-4 для лечения или профилактики болезни Альцгеймера (AD), умеренного когнитивного нарушения, старения, деменции, деменции с тельцами Леви, церебральной амилоидной ангиопатии, слабоумия вследствие множественных инфарктов, синдрома Дауна, деменции, ассоциированной с инсультом, деменции, ассоциированной с болезнью Паркинсона, или деменции, ассоциированной с бета-амилоидом.
8. Способ лечения расстройства, выбранного из болезни Альцгеймера, умеренного когнитивного нарушения, старения, деменции, деменции с тельцами Леви, церебральной амилоидной ангиопатии, слабоумия вследствие множественных инфарктов, синдрома Дауна, деменции, ассоциированной с инсультом, деменции, ассоциированной с болезнью Паркинсона, и деменции, ассоциированной с бета-амилоидом, у субъекта, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции по п.5.
Текст
Гейсен Хенрикус Якобус Мария, Ван Брандт Свен Францискус Анна (BE) Медведев В.Н. (RU) где определения заместителей R1-4 и Ar даны в формуле изобретения, в качестве ингибиторов бета-секретазы, также известной как фермент, расщепляющий предшественник амилоида по бета-сайту, BACE, BACE1, Asp2 или мемапсин 2. Настоящее изобретение также относится к фармацевтическим композициям, содержащим такие соединения, к способам получения таких соединений и композиций и к применению таких соединений и композиций для предупреждения и лечения нарушений, в которые вовлечена бета-секретаза, таких как болезнь Альцгеймера (AD),умеренное когнитивное нарушение, старение, деменция, деменция с тельцами Леви, церебральная амилоидная ангиопатия, слабоумие вследствие множественных инфарктов, синдром Дауна,деменция, ассоциированная с инсультом, деменция, ассоциированная с болезнью Паркинсона, и деменция, ассоциированная с бета-амилоидом.(71)(73) Заявитель и патентовладелец: ЯНССЕН ФАРМАЦЕВТИКА НВ (BE) Область изобретения Настоящее изобретение относится к новым производным 5-(3-аминофенил)-5-алкил-5,6-дигидро 2 Н-[1,4]оксазин-3-амина в качестве ингибиторов бета-секретазы, также известной как фермент, расщепляющий предшественник амилоида по бета-сайту, BACE, BACE1, Asp2 или мемапсин 2. Настоящее изобретение также относится к фармацевтическим композициям, содержащим такие соединения, к способам получения таких соединений и композиций и к применению таких соединений и композиций для предупреждения и лечения расстройств, в которые вовлечена бета-секретаза, таких как болезнь Альцгеймера(AD), умеренное когнитивное нарушение, старение, деменция, деменция с тельцами Леви, церебральная амилоидная ангиопатия, слабоумие вследствие множественных инфарктов, синдром Дауна, деменция,ассоциированная с инсультом, деменция, ассоциированная с болезнью Паркинсона, и деменция, ассоциированная с бета-амилоидом. Предпосылки изобретения Болезнь Альцгеймера (AD) представляет собой нейродегенеративное заболевание, ассоциированное со старением. Пациенты с AD страдают дефицитом когнитивных функций и потерей памяти, а также поведенческими проблемами, такими как тревожность. Более 90% пораженных AD имеют спорадическую форму расстройства, тогда как менее 10% случаев являются семейными или наследственными. В Соединенных Штатах приблизительно 1 из 10 людей в возрасте 65 лет имеет AD, тогда как в возрасте 85 лет 1 из каждых двух индивидов поражен AD. Средняя предполагаемая продолжительность жизни от первоначального диагноза составляет 7-10 лет, и пациенты с AD нуждаются во всестороннем уходе или в доме престарелых, что является очень дорогостоящим или осуществляется членами семьи. При возрастающем количестве пожилых людей в популяции AD представляет собой растущую медицинскую проблему. Доступные в настоящее время виды терапии AD обеспечивают лечение лишь симптомов заболевания и включают ингибиторы ацетилхолинэстеразы для улучшения когнитивных свойств, а также анксиолитики и нейролептики для контроля поведенческих проблем, ассоциированных с данной болезнью. Отличительными патологическими признаками в головном мозге пациентов с AD являются нейрофибриллярные клубки, которые возникают в результате гиперфосфорилирования тау-белка, и амилоидные бляшки, которые образуются в результате агрегации пептида бета-амилоида 1-42 (абета 1-42). Абета 1-42 образует олигомеры и затем фибриллы и, в конечном итоге, амилоидные бляшки. Олигомеры и фибриллы считаются особенно нейротоксичными и могут вызывать большинство нейрологических повреждений, ассоциированных с AD. Средства, которые предупреждают образование абета 1-42, имеют потенциал, чтобы стать средствами, модифицирующими течение заболевания, для лечения AD. Абета 142 образуется из белка-предшественника амилоида (APP), состоящего из 770 аминокислот. N-конец абета 1-42 расщепляется с помощью бета-секретазы (BACE), и затем гамма-секретаза расщепляет C-конец. В дополнение к абета 1-42 гамма-секретаза также высвобождает абета 1-40, который является преобладающим продуктом расщепления, так же как абета 1-38 и абета 1-43. Эти формы абета также могут агрегироваться с образованием олигомеров и фибрилл. Таким образом, предполагается, что ингибиторыBACE будут предупреждать образование абета 1-42, а также абета 1-40, абета 1-38 и абета 1-43 и будут являться потенциальными терапевтическими средствами для лечения AD. В WO-2011/009943 (Novartis) раскрывают незамещенные и 2-замещенные производные оксазина и их применение в качестве ингибиторов BACE для лечения нейрологических нарушений. В WO2011/020806 (Hoffmann-LaRoche) раскрывают производные 2,6-незамещенного 3-амино-5-фенил-5,6 дигидро-2H-[1,4]оксазина, характеризующиеся ингибиторными свойствами BACE1 и/или BACE2. Краткое описание изобретения Настоящее изобретение относится к производным 5,6-дигидро-2 Н-[1,4]оксазин-3-иламина формулы а также их стереоизомерным формам, гдеR2 представляет собой водород или трифторметил; илиR1 и R2 образуют двухвалентный радикал =CF2;R4 представляет собой водород или фтор;Ar представляет собой гетероарил; где гетероарил выбран из группы, состоящей из пиридила и пиримидила, каждый из которых необязательно замещен одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена, циано, C1-3 алкила, С 2-3 алкинила, С 1-3 алкилокси, моно-и полигалоген-C1-3 алкила, моно- и полигалоген-С 1-3 алкилокси и С 1-3 алкилоксиС 1-3 алкилокси, и их фармацевтически приемлемым солям присоединения. Примером настоящего изобретения является фармацевтическая композиция, содержащая фармацевтически приемлемый носитель и любое из соединений, описанных выше. Примером настоящего изобретения является фармацевтическая композиция, полученная путем смешивания любого из соединений,описанных выше, и фармацевтически приемлемого носителя. Примером настоящего изобретения является способ получения фармацевтической композиции, включающий смешивание любого из соединений,описанных выше, и фаомацевтически приемлемого носителя. Примером настоящего изобретения являются способы лечения расстройства, опосредованного ферментом бета-секретазой, включающие введение субъекту, нуждающемуся в этом, терапевтически эффективного количества любого из соединений или фармацевтических композиций, описанных выше. Дополнительным примером настоящего изобретения являются способы ингибирования фермента бета-секретазы, включающие введение субъекту, нуждающемуся в этом, терапевтически эффективного количества любого из соединений или фармацевтических композиций, описанных выше. Примером настоящего изобретения является способ лечения расстройства, выбранного из группы,состоящей из болезни Альцгеймера, умеренного когнитивного нарушения, старения, деменции, деменции с тельцами Леви, церебральной амилоидной ангиопатии, слабоумия вследствие множественных инфарктов, синдрома Дауна, деменции, ассоциированной с инсультом, деменции, ассоциированной с болезнью Паркинсона, и деменции, ассоциированной с бета-амилоидом, предпочтительно болезни Альцгеймера, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества любого из соединений или фармацевтических композиций, описанных в данном документе. Другим примером настоящего изобретения является любое из соединений, описанных выше, для применения в лечении: (a) болезни Альцгеймера, (b) умеренного когнитивного нарушения, (c) старения,(d) деменции, (e) деменции с тельцами Леви, (f) синдрома Дауна, (g) деменции, ассоциированной с инсультом, (h) деменции, ассоциированной с болезнью Паркинсона, и (i) деменции, ассоциированной с бета-амилоидом, у субъекта, нуждаюшегося в этом. Подробное описание изобретения Настоящее изобретение относится к соединениям формулы (I), определенным выше в данном документе, и их фармацевтически приемлемым солям и сольватам. Соединения формулы (I) являются ингибиторами фермента бета-секретазы (также известного как фермент, расщепляющий по бета-сайту,BACE, BACE1, Asp2 или мемапсин 2) и являются применимыми в лечении болезни Альцгеймера, умеренного когнитивного нарушения, старения, деменции, деменции, ассоциированной с инсультом, деменции с тельцами Леви, синдрома Дауна, деменции, ассоциированной с болезнью Паркинсона, и деменции,ассоциированной с бета-амилоидом, предпочтительно болезни Альцгеймера, умеренного когнитивного нарушения или деменции, более предпочтительно болезни Альцгеймера. В одном варианте осуществления настоящего изобретенияR2 представляет собой водород или трифторметил; илиR1 и R2 образуют двухвалентный радикал =CF2;R4 представляет собой водород или фтор;Ar представляет собой гетероарил; где гетероарил представляет собой пиридил или пиримидил, каждый из которых необязательно замещен одним, двумя или тремя заместителями, выбранными из группы, состоящей из галогена, циано,С 1-3 алкила, С 2-3 алкинила, С 1-3 алкилокси, моно- и полигалоген-С 1-3 алкила, моно- и полигалоген-С 13 алкилокси и С 1-3 алкилоксиС 1-3 алкилокси; и их фармацевтически приемлемых солей присоединения или сольватов. В другом варианте осуществленияR1 и R2 образуют двухвалентный радикал =CF2;R3 представляет собой метил или циклопропил;R4 представляет собой водород или фтор;Ar представляет собой гетероарил; где гетероарил представляет собой пиридил, замещенный метокси, или пиримидил; и их фармацевтически приемлемые соли присоединения или сольваты. В другом варианте осуществления атом углерода, замещенный R3, характеризуется Rконфигурацией. В другом варианте осуществления соединение формулы (I) выбрано из"Галоген" будет обозначать фтор, хлор или бром; "С 1-3 алкил" будет обозначать неразветвленную или с разветвленной цепью насыщенную алкильную группу с 1, 2 или 3 атомами углерода, например метил, этил, 1-пропил и 2-пропил; "C1-3 алкилокси" будет обозначать простой эфирный радикал, где С 13 алкил является таковым, как определено выше; "моно- и полигалогенС 1-3 алкил" будут обозначать определенный ранее С 1-3 алкил, замещенный 1, 2, 3 или, если возможно, большим количеством атомов галогенов, определенных выше; "моно- и полигалогенС 1-3 алкилокси" будут обозначать простой эфирный радикал, где моно- и полигалогенС 1-3 алкил является таковым, как определено выше; "C3-6 циклоалкил" будет обозначать циклопропил, циклобутил, циклопентил и циклогексил; "C3-6 циклоалкандиил" будет обозначать двухвалентный радикал, такой как циклопропандиил, циклобутандиил, циклопентандиил и циклогександиил. Используемое в данном документе выражение "субъект" относится к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, который является или являлся объектом лечения,наблюдения или эксперимента. Используемое в данном документе выражение "терапевтически эффективное количество" означает такое количество активного соединения или фармацевтического средства, которое вызывает биологический или медицинский эффект в системе тканей у животного или человека, желаемый исследователем,ветеринаром, врачом или другим клиницистом, что включает облегчение симптомов заболевания или расстройства, лечение которого осуществляют. Используемое в данном документе выражение "композиция" предназначена для охвата продукта,содержащего определенные ингредиенты в определенных количествах, а также любого продукта, который получают, прямо или опосредованно, в результате комбинаций определенных ингредиентов в определенных количествах. Выше и ниже в данном документе выражение "соединение формулы (I)" следует понимать как включающее его соли присоединения, сольваты и стереоизомеры. Выражения "стереоизомеры" или "стереохимически изомерные формы" ранее и далее в данном документе используют взаимозаменяемо. Настоящее изобретение включает все стереоизомеры соединения формулы (I) или в виде чистого стереоизомера, или в виде смеси из двух или более стереоизомеров. Энантиомеры являются стереоизомерами, которые представляют собой не совпадающие при наложении зеркальные отображения друг друга. Смесь 1:1 пары энантиомеров является рацематом или рацемической смесью. Диастереомеры (или диастереоизомеры) являются стереоизомерами, которые не являются энантиомерами, т.е они не соотносятся как зеркальные изображения. Если соединение содержит двойную связь, заместители могут находиться в E- или Z-конфигурации. Если соединение содержит двузамещенную циклоалкильную группу, заместители могут находиться в цис- или транс-конфигурации. Таким образом, настоящее изобретение включает энантиомеры, диастереомеры, рацематы, E-изомеры, Zизомеры, цис-изомеры, транс-изомеры и их смеси. Абсолютная конфигурация определяется согласно системе Кана-Ингольда-Прелога. Конфигурация при асимметричном атоме определяется или как R, или как S. Анализируемые соединения, абсолютная конфигурация которых неизвестна, могут быть обозначены как (+) или (-) в зависимости от направления,в котором они вращают плоскость поляризованного света. Если указан конкретный стереоизомер, это означает, что указанный стереоизомер практически чистый, т.е. связан с менее 50%, предпочтительно менее 20%, более предпочтительно менее 10%, еще более предпочтительно менее 5%, в частности менее 2% и наиболее предпочтительно менее 1% других изомеров. Таким образом, если соединение формулы (I), например, указано как (R), то это означает, что соединение практически не содержит S-изомер; если соединение формулы (I), указано, например, как E, то это означает, что соединение практически не содержит Z-изомер; если соединение формулы (I) указано, например, как цис, это означает, что соединение практически не содержит транс-изомер. Соединения формулы (I) сосуществуют в динамическом равновесии с таутомерами формулы (I-a) где L представляет собой -NH-. Что касается применения в медицине, соли соединений по настоящему изобретению относятся к нетоксичным "фармацевтически приемлемым солям". Однако, при получении соединений согласно настоящему изобретению или их фармацевтически приемлемых солей могут быть использованы другие соли. Подходящие фармацевтически приемлемые если соединений включают соли присоединения кислоты, которые можно получать, например, путем смешивания раствора соединения с раствором фармацевтически приемлемой кислоты, такой как соляная кислота, серная кислота, фумаровая кислота, малеиновая кислота, янтарная кислота, уксусная кислота, бензойная кислота, лимонная кислота, винная кислота, угольная кислота или фосфорная кислота. Кроме того, если соединения по настоящему изобретению несут кислотный фрагмент, их подходящие фармацевтически приемлемые соли могут включать соли щелочных металлов, например соли натрия или калия; соли щелочно-земельных металлов, например соли кальция или магния; и соли, образованные подходящими органическими лигандами, например соли четвертичного аммония. Типичные кислоты, которые можно использовать при получении фармацевтически приемлемых солей, включают, но без ограничений, следующие: уксусную кислоту, 2,2-дихлоруксусную кислоту, ацилированные аминокислоты, адипиновую кислоту, альгиновую кислоту, аскорбиновую кислоту, Lаспарагиновую кислоту, бензолсульфоновую кислоту, бензойную кислоту, 4-ацетамидобензойную кислоту, (+)-камфорную кислоту, камфорсульфоновую кислоту, каприновую кислоту, капроевую кислоту,каприловую кислоту, коричную кислоту, лимонную кислоту, цикламовую кислоту, этан-1,2 дисульфоновую кислоту, этансульфоновую кислоту, 2-гидрокси-этансульфоновую кислоту, муравьиную кислоту, фумаровую кислоту, галактаровую кислоту, гентизиновую кислоту, глюкогептоновую кислоту,D-глюконовую кислоту, D-глюкуроновую кислоту, L-глутаминовую кислоту, бета-оксо-глутаровую кислоту, гликолевую кислоту, гиппуровую кислоту, бромисто-водородную кислоту, хлористо-водородную кислоту, (+)-L-молочную кислоту, (+)-DL-молочную кислоту, лактобионовую кислоту, малеиновую кислоту, (-)-L-яблочную кислоту, малоновую кислоту, -DL-миндальную кислоту, метансульфоновую кислоту, нафталин-2-сульфоновую кислоту, нафталин-1,5-дисульфоновую кислоту, 1-гидрокси-2 нафтойную кислоту, никотиновую кислоту, азотную кислоту, олеиновую кислоту, оротовую кислоту,щавелевую кислоту, пальмитиновую кислоту, памоевую кислоту, фосфорную кислоту, Lпироглутаминовую кислоту, салициловую кислоту, 4-аминосалициловую кислоту, себациновую кислоту,стеариновую кислоту, янтарную кислоту, серную кислоту, дигалловую кислоту, (+)-L-винную кислоту,тиоциановую кислоту, п-толуолсульфоновую кислоту, трифторметилсульфоновую кислоту и ундециленовую кислоту. Типичные основания, которые можно использовать в получении фармацевтически приемлемых солей без ограничения включают следующие: аммиак, L-аргинин, бенетамин, бензатин, гидроксид кальция, холин, диметилэтаноламин, диэтаноламин, диэтиламин, 2-(диэтиламино)-этанол, этаноламин, этилендиамин, N-метилглюкамин, гидрабамин, 1H-имидазол, L-лизин, гидроксид магния, 4-(2 гидроксиэтил)-морфолин, пиперазин, гидроксид калия, 1-(2-гидроксиэтил)-пирролидин, вторичный амин, гидроксид натрия, триэтаноламин, трометамин и гидроксиц цинка. Названия соединений по настоящему изобретению составлены в соответствии с правилами номенклатуры, согласованными с Химической реферативной службой (CAS) с использованием программного обеспечения от Advanced Chemical Development, Inc. (ACD/название продукта, версия 10.01; сборка 15494, 1 декабря 2006 г.) или в соответствии с правилами номенклатуры, согласованными с Международным союзом теоретической и прикладной химии (IUPAC) с использованием программного обеспечения от Advanced Chemical Development, Inc. (ACD/название продукта, версия 10.01.0.14105, октябрь 2006 г.). В случае таутомерных форм составляли название представленных таутомерных форм структуры. Другие не представленные таутсмерные формы также включены в объем настоящего изобретения. Получение соединений. Экспериментальная методика 1. Конечные соединения в соответствии с формулой (I) можно получить путем взаимодействия промежуточного соединения формулы (II) и соединения формулы (III) в соответствии со схемой реакции (1),реакции, которую проводили в подходящем реакционно-инертном растворителе, таком как, например,изопропанол или 1,4-диоксан, в присутствие подходящей кислоты, такой как, например, H2SO4 или HCl,-4 024716 при температурных условиях, таких как, например, нагревание реакционной смеси при 100C, например,в течение 16 ч. Это превращение также можно проводить в присутствие Pd-комплексного катализатора,такого как, например, трис(дибензилиденацетон) дипалладий (0) [CAS 51364-51-3] в подходящем реакционно-инертном растворителе, таком как, например, 1,4-диоксан, этанол или смеси инертных растворителей, в присутствие подходящего основания, такого как, например, водный K3PO4, Na2CO3 или Cs2CO3,и подходящего лиганда, такого как, например, 1,1'-бис(дифенилфосфино)-ферроцен [CAS 12150-46-3],при температурных условиях, таких как, например, нагревание реакционной смеси при 160C в микроволновом излучении до завершения реакции, например, в течение 1 ч. В схеме реакции (1) все переменные величины определены в формуле (I) и W представляет собой галоген, L представляет собой -NH-,все заместители X1-X4 представляют собой CH, и R5 представляет собой водород. Схема реакции 1 Экспериментальная методика 2. Конечные соединения в соответствии с формулой (I-b), где R3 представляет собой дифторметил и 4R представляет собой водород, можно получить путем каталитической гидрогенизации промежуточного соединения формулы (I-a) в соответствии со схемой реакции (2). Указанное превращение можно проводить путем обработки промежуточного соединения формулы (I-a) водородом в присутствие подходящего катализатора, такого как, например, палладий или углерод, подходящего каталитического яда, такого как, например, тиофен, в подходящем реакционно-инертном растворителе, таком как, например, этилацетат. Смесь перемешивают в атмосфере водорода при подходящей температуре, как правило, комнатной температуре, при подходящем давлении, таком как, например, атмосферное давление, например, в течение 16 ч. На схеме реакции (2) все переменные величины определены как в формуле (I) , L представляет собой -NH-, все заместители X1-X4 представляют собой CH, и R5 представляет собой водород. Схема реакции 2 Экспериментальная методика 3. В общем, промежуточные соединения формулы (II-a) и (II-b) можно получить после стадий реакции, показанных на схеме реакции (3) ниже, где переменные величины определены как в формуле (I), L представляет собой -NH-, все заместители X1-X4 представляют собой CH, и R5 представляет собой водород. Схема реакции 3D - сочетание по типу Бухвальда-Хартвига (где W представляет собой галоген).E - восстановление нитросоединения до амина (где R6 представляет собой H).F - превращение бромсоединения в амин (где R6 представляет собой H). Производные амидина из вышеуказанной схемы реакции можно беспрепятственно получить из соответствующих производных тиоамида, действуя в соответствии с известным из уровня техники методикам превращения тиоамида в амидин (стадия реакции A). Указанное превращение можно беспрепятственно осуществлять путем обработки указанных тиоамидов источником аммиака, таким как, например,хлорид аммония или водный аммиак, в подходящем реакционно-инертном растворителе, таком как, например, вода или метанол и т. п., при температурных условиях, таких как, например, нагревание реакционной смеси при 60C, например, в течение 6 ч. Альтернативно, промежуточные соединения формулы (II-a), где R6 представляет собой водород, в приведенной выше схеме реакции (3) можно получить из соответствующих промежуточных соединений формулы (II-b) посредством типа методики сочетания, катализируемого медью (стадия реакции F). Указанное сочетание можно проводить путем обработки промежуточных соединений формулы (II-b) азидом натрия в подходящем реакционно-инертном растворителе, таком как, например, ДМСО, в присутствии смеси подходящих оснований, таких как, например, диметилэтилендиамин и Na2CO3, медного катализатора, такого как, например, CuI, при температурных условиях, таких как, например, нагревание реакционной смеси при 110C до завершения реакции, например, в течение 1 ч. Производные тиоамида из вышеуказанной схемы реакции (3) можно получить из производных амида, следуя известным из уровня техники методикам тионирования (стадия реакции B). Указанное превращение можно беспрепятственно осуществлять путем обработки указанных амидов средством для тионирования, таким как, например, пентасульфид фосфора или 2,4-бис-(4-метоксифенил)-1,3-дитиа-2,4 дифосфетан-2,4-дисульфид [реагент Лавессона, CAS 19172-47-5], в реакционно-инертном растворителе,таком как, например, тетрагидрофуран или 1,4-диоксан и т. п., при температурных условиях, таких как,например, нагревание реакционной смеси при 50C, например, в течение 50 ч. Производные амида в указанной выше схеме реакции (3) можно получить из производных бетааминоспирта формулы (VIII) и промежуточных соединений формулы (VII) в результате известных в данной области методик циклизации (стадия реакции C). Указанную циклизацию можно беспрепятственно проводить путем обработки указанных бета-аминоспиртов промежуточным соединением формулы (VII) в присутствии основания, такого как трет-бутоксид калия, или смеси оснований, такой как, третбутоксид калия/N,N-диизопропилэтиламин, реакционно-инертного растворителя, такого как, например,тетрагидрофуран и т. п., при -(80-100)C, предпочтительно -(15-25)C, в течение 30 мин-100 ч, предпочтительно 1-24 ч. Помимо этого, промежуточные соединения формулы (IV-a) и (VI-a) в описанной выше схеме реакции (3) можно получить из соответствующих промежуточных соединений формул (IV-b) и (VI-b), следуя известным в данной области методикам реакции сочетания типа Бухвальда-Хартвига (стадия реакции D). Указанное сочетание можно проводить путем обработки промежуточных соединений формул (IV-b) и(VI-b) с промежуточным соединением формулы (V) в подходящем реакционно-инертном растворителе,таком как, например, этанол, или смеси инертных растворителей, таких как, например, 1,2 диметоксиэтан/вода/этанола, в присутствие подходящего основания, такого как, например, водныйCs2CO3,Pd-комплексного катализатора,такого как,например,[1,1'бис(дифенилфосфино)ферроцен]-дихлорпалладий (II) [CAS 72287-26-4] или диацетат трансбис(дициклогексиламин)палладия [DAPCy, CAS 628339-96-8] при температурных условиях, таких как,например, нагревание реакционной смеси при 80C, например, в течение 20 ч, или, например, нагревание реакционной смеси при 130C, например, в течение 10 мин в микроволновом излучении. Кроме того, промежуточные соединения формул (IV-a) и (VI-а) в описанной выше схеме реакции(3), где R6 представляет собой Н, можно получить из соответствующих промежуточных соединений формул (IV-b) и (VI-b), следуя известным в данной области методикам реакции сочетания типа Бухвальда-Хартвига (стадия реакции E) . Указанное восстановление можно беспрепятственно провести, следуя известным в данной области методикам каталитической гидрогенизации. Например, указанное восстановление можно осуществлять путем перемешивания реагирующих веществ в атмосфере водорода и в присутствии соответствующего катализатора, такого как, например, палладий на угле, платина на угле,никель Ренея и им подобные катализаторы. Подходящими растворителями являются, например, вода,алканолы, например метанол, этанол и т. п., сложные эфиры, например этилацетат и т. п. Для повышения скорости указанной реакции восстановления может быть предпочтительным повышение температуры и/или давления в реакционной смеси. Нежелательное дополнительное гидрирование определенных функциональных групп в реагирующих веществах и продуктах реакции можно предотвратить добавлением каталитического яда, такого как, например, тиофен и т. п., к реакционной смеси. В общем, промежуточные соединения формул (VII), (VIII-a), (VIII-b) и (VIII-c) можно получить,-6 024716 следуя известным в данной области методикам по типу Штрекера, описанным в литературе, с последующими стандартными химическими трансформациями цианогруппы. Экспериментальная методика 4. В общем, промежуточные соединения формул (IX-a), (IX-b) и (IX-c) можно получить после стадий реакции, показанных на схеме реакции (4) ниже, где все заместители X1-X4 представляют собой CH, иK - циклизация. Промежуточные соединения формул (IX-a) и (IX-b) в указанной выше схеме реакции (4) можно получить из промежуточных соединений формул (X-a) и (X-b), следуя известным в данной области методикам фторирования (стадия реакции G). Такое превращение можно проводить путем обработки промежуточных соединений формул (X-a) и (X-b) в присутствие фторирующего средства, такого как, например, диэтиламиносеры трифторид (DAST), в подходящем реакционно-инертном растворителе, таком как,например, дихлорметан. Реакционную смесь перемешивают при подходящей температуре, например 0C, в течение необходимого времени для завершения реакции, например 20-40 мин. Промежуточные соединения формулы (IX-c) из вышеуказанной схемы реакции (4) можно получить из промежуточных соединений формулы (XI), следуя известными из уровня техники методиками хлорирования (стадия реакции H). Указанное превращение можно проводить путем обработки промежуточного соединения формулы (X-а) соответствующим хлорирующим средством, таким как, например, тионилхлорид, в присутствие основания, такого как, например, пиридин, в реакционно-инертном растворителе,таком как, например, дихлорметан. Реакционную смесь перемешивают при подходящей температуре,например 0C, в течение времени, необходимого для завершения реакции, например в течение 30-60 мин. Промежуточные соединения формулы (X-a) из вышеуказанной схемы реакции (4) можно получить из промежуточных соединений формулы (XI), следуя известным в данной области методикам трихлорметилирования (стадия реакции I). Указанное превращение можно осуществить путем обработки промежуточного соединения формулы (XI) в присутствие фторида тетрабутиламмония (TBAF), трифторметилирующим средством, таким как, например, (трифторметил)триметилсилан, в подходящем реакционноинертном растворителе, таком как, например, тетрагидрофуран. Реакционную смесь перемешивают при подходящей температуре, например при комнатной температуре, в течение времени, необходимого для завершения реакции, например в течение 2 ч. Промежуточные соединения формулы (X-b) из вышеуказанной схеме реакции (4) можно получить из промежуточных соединений формулы (XI), следуя известными из уровня техники методикам хлорирования (стадия реакции E). Указанное превращение можно проводить путем обработки промежуточного соединения формулы (XI) восстанавливающим средством, таким как, например, гидрид диизобутилалюминия, в подходящем реакционно-инертном растворителе, таком как, например, тетрагидрофуран. Реакционную смесь перемешивают при подходящей температуре, как правило, от -78C до комнатной температуры в течение необходимого времени для завершения реакции, например 2 ч. Промежуточные соединения формулы (XI) из вышеуказанной схемы реакции (4) можно получить из промежуточных соединений формулы (XII), следуя известным из уровня техники методикам циклизации (стадия реакции K). Указанное превращение можно проводить, во-первых, путем обработки промежуточных соединений формулы (XII) промежуточным соединением формулы (VII), таким как, например,хлорацетилхлорид, в присутствие основания, такого как, например, NaOH, в подходящей смеси инертных растворителей, таких как, например, вода и 1,4-диоксан или вода и ТГФ. pH реакционной смеси доводят до подходящего значения pH, например 10-11, путем добавления подходящего основания, такого как, например, NaOH. Реакционную смесь перемешивают при подходящей температуре, например от 0 до 25C, в течение времени, необходимого для завершения реакции, например в течение 1-4 ч. Полученный неочищенный остаток в дальнейшем можно циклизировать для получения промежуточного соединения (XI) путем добавления подходящего основания, такого как, например, K2CO3, Cs2CO3, N,Nдиизопропилэтиламин или NaHCO3, в подходящем реакционно-инертном растворителе, таком как, например, ацетонитрил или ДМФА. Реакционную смесь перемешивают при температурных условиях, таких как, например, нагревание реакционной смеси при от 25 до 80C в течение 2-24 ч или, например, нагревание реакционной смеси при 140C в течение 15-30 мин в микроволновом излучении. Это превращение также можно выполнить в отсутствие основания в подходящем реакционно-инертном растворителе,таком как, например, ацетонитрил или ДМФА, при подходящей температуре, как правило, от 40 до 110C, в течение периода, например 24-48 ч. Экспериментальная процедура 5. В общем, промежуточные соединения формул (VIII) и (IX) можно получить из промежуточных соединений формулы (X), следуя известным из уровня техники методикам (стадия реакции L), где все заместители X1-X4 представляют собой CH, и R5 представляет собой водород. Указанное превращение можно осуществить путем обработки промежуточного соединения формулы (IX-c) подходящим цинковым реагентом, таким как, например, цинковая пыль или цинк-медная пара, в подходящем растворителе,таком как уксусная кислота, при подходящей температуре, как правило, от комнатной температуры до 80C, в течение времени, необходимого для завершения реакции, например в течение 1-16 ч. Данное превращение обеспечивает смесь промежуточных соединений формулы (VIII) и (IX) в различном соотношении в зависимости от условий реакции и реактивов. Схема реакции 5 Фармацевтические композиции. В настоящем изобретении также представлены композиции для предупреждения или лечения заболеваний, при которых ингибирование бета-секретазы является благоприятным, таких как болезнь Альцгеймера (AD), умеренное когнитивное нарушение, старение, деменция, деменция с тельцами Леви, синдром Дауна, деменция, ассоциированная с инсультом, деменция, ассоциированная с болезнью Паркинсона, и деменция, ассоциированная с бета-амилоидом. Указанные композиции содержат терапевтически эффективное количество соединения в соответствии с формулой (I) и фармацевтически приемлемый носитель или разбавитель. Хотя активный ингредиент можно вводить отдельно, предпочтительно предоставлять его в виде фармацевтической композиции. Следовательно, в настоящем изобретении дополнительно представлена фармацевтическая композиция, содержащая соединение согласно настоящему изобретению вместе с фармацевтически приемлемым носителем или разбавителем. Носитель или разбавитель должны быть"приемлемыми" в том смысле, что они должны быть совместимыми с другими ингредиентами композиции и должны быть не вредны для получающих их пациентов. Фармацевтические композиции по настоящему изобретению могут быть получены любыми способами, хорошо известными в области фармацевтики. Терапевтически эффективное количество конкретного соединения в форме основания или в форме соли присоединения в качестве активного ингредиента комбинируют в однородную смесь с фармацевтически приемлемым носителем, который может иметь разнообразные формы в зависимости от формы препарата, который необходимо ввести. Желательно,чтобы данные фармацевтические композиции находились в единичной лекарственной форме, подходящей предпочтительно для системного введения, такого как пероральное, чрескожное или парентеральное введение; или для местного введения, такого как с помощью ингаляции, назального спрея, глазных капель или с помощью крема, геля, шампуня и т. п. Например, при получении композиций в виде пероральной лекарственной формы можно использовать любую общепринятую фармацевтическую среду,такую как, например, вода, гликоли, масла, спирты и т. п., в случае пероральных жидких препаратов,таких как суспензии, сиропы, эликсиры и растворы; или твердые носители, такие как крахмалы, сахара,каолин, смазывающие вещества, связующие вещества, средства для улучшения распадаемости и т. п., в случае порошков, пилюль, капсул и таблеток. Благодаря простоте их введения таблетки и капсулы представляют собой наиболее предпочтительные пероральные единичные лекарственные формы, в случае которых несомненно используют твердые фармацевтические носители. Для парентеральных композиций носитель будет, как правило, по меньшей мере, в значительной степени содержать стерильную воду, хотя может включать и другие ингредиенты, например, для улучшения растворимости. Например, могут быть получены растворы для инъекций, в которых носитель содержит физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Также могут быть получены суспензии для инъекций, в случае которых могут быть использованы подходящие жидкие носители, суспендирую-8 024716 щие средства и т. п. В композициях, подходящих для чрескожного введения, носитель необязательно содержит средство, повышающее проникновение, и/или подходящее смачиваемое средство, необязательно в комбинации с подходящими добавками любого происхождения в минимальных пропорциях,при этом добавки не оказывают никаких существенных вредных воздействий на кожу. Указанные добавки могут облегчать нанесение на кожу и/или могут быть полезными при получении необходимых композиций. Данные композиции могут наноситься различными путями, например, в виде трансдермального пластыря, точечно или в виде мази. Особенно предпочтительно для простоты введения и равномерности дозирования составлять вышеупомянутые фармацевтические композиции в единичные лекарственные формы. Единичная лекарственная форма, как используется в описании и формуле изобретения в данном документе, относится к физически дискретным единицам, подходящим в качестве единичных дозировок, причем каждая единица содержит предварительно определенное количество активного ингредиента, рассчитанное так, чтобы получить необходимый терапевтический эффект, совместно с требуемым фармацевтическим носителем. Примерами таких единичных лекарственных форм являются таблетки (в том числе делимые или покрытые таблетки), капсулы, пилюли, пакеты с порошком, облатки, растворы или суспензии для инъекций,чайные ложки, столовые ложки и т. п. и их отдельные множества. Точная дозировка и частота введения зависят от конкретного применяемого соединения формулы(I), конкретного состояния, лечение которого осуществляют, тяжести состояния, лечение которого осуществляют, возраста, веса, пола, степени тяжести расстройства и общего физического состояния конкретного пациента, а также от другой лекарственной терапии, которую может получать индивидуум, как хорошо известно специалисту в данной области. Более того, очевидно, что указанное эффективное суточное количество можно снижать или увеличивать в зависимости от ответа субъекта, подвергаемого лечению, и/или в зависимости от оценки врача, назначающего соединения по настоящему изобретению. В зависимости от способа введения фармацевтическая композиция будет содержать от 0,05 до 99 вес.%, предпочтительно от 0,1 до 70 вес.%, более предпочтительно от 0,1 до 50 вес.% активного ингредиента и от 1 до 99,95 вес.%, предпочтительно от 30 до 99,9 вес.%, более предпочтительно от 50 до 99,9 вес.% фармацевтически приемлемого носителя, при этом все процентные содержания основываются на общем весе композиции. Данные соединения можно применять для системного введения, такого как пероральное, чрескожное или парентеральное введение; или для местного введения, такого как с помощью ингаляции, назального спрея, глазных капель или с помощью крема, геля, шампуня или т. п. Соединения преимущественно являются перорально вводимыми. Точная дозировка и частота введения зависят от конкретного применяемого соединения в соответствии с формулой (I), конкретного состояния, лечение которого осуществляют, тяжести состояния, лечение которого осуществляют, возраста, веса, пола, степени тяжести расстройства и общего физического состояния конкретного пациента, а также другой лекарственной терапии, которую может получать индивидуум, что хорошо известно специалистам в данной области. Более того, очевидно, что указанное эффективное ежедневное количество можно снижать или увеличивать в зависимости от реакции субъекта, подвергаемого лечению, и/или в зависимости от оценки врача, назначающего соединения по настоящему изобретению. Количество соединения формулы (I), которое можно комбинировать с материалом носителя для получения разовой лекарственной формы, будет изменяться в зависимости от заболевания, лечение которого осуществляют, видов млекопитающих и конкретного способа введения. Однако, в качестве общего руководства подходящие стандартные дозы для соединений по настоящему изобретению могут, например, предпочтительно содержать от 0,1 до приблизительно 1000 мг активного соединения. Предпочтительная стандартная доза составляет от 1 до приблизительно 500 мг. Более предпочтительная стандартная доза составляет от 1 до приблизительно 300 мг. Еще более предпочтительная стандартная доза составляет от 1 до приблизительно 100 мг. Такие стандартные дозы можно вводить более одного раза в день, например 2, 3, 4, 5 или 6 раз в день, но предпочтительно 1 или 2 раза в день, чтобы общая дозировка для взрослого человека весом 70 кг была в диапазоне от 0,001 до приблизительно 15 мг на кг веса субъекта за введение. Предпочтительная доза составляет от 0,01 до приблизительно 1,5 мг на кг веса субъекта на введение, и такая терапия может длиться в течение нескольких недель или месяцев, а в некоторых случаях в течение нескольких лет. Однако, следует понимать, что определенный уровень дозы для любого конкретного пациента будет зависеть от ряда факторов, включающих активность определенного используемого соединения; возраст, вес тела, общее состояние здоровья, пол и режим питания индивидуума, подлежащего лечению; время и путь введения; скорость экскреции; другие лекарственные средства, которые были введены ранее; и тяжесть конкретного заболевания, подвергаемого терапии, что хорошо понятно специалистам в данной области. Типичной дозой может быть одна таблетка от 1 до приблизительно 100 мг или от 1 до приблизительно 300 мг, принимаемая один раз в день или несколько раз в день, или одна капсула или таблетка пролонгированного действия, принимаемая один раз в день и содержащая пропорционально более высокое содержание активного ингредиента. Эффекта пролонгированного действия можно добиться с помощью материалов капсулы, которые растворяются при различных значениях pH, с помощью капсул, кото-9 024716 рые медленно высвобождают лекарственное средство при осмотическом давлении, или с помощью любых иных известных средств с контролируемым высвобождением. В некоторых случаях может понадобиться применение доз вне этих диапазонов, что будет очевидно специалистам в данной области. Кроме того, следует отметить, что клиницист или лечащий врач будет знать, как и когда начинать, прерывать, корректировать или завершать терапию в соответствии с реакцией отдельного пациента. В отношении композиций, способов и наборов, приведенных выше, специалисту в данной области будет понятно, что предпочтительными соединениями для использования в каждом являются такие соединения, отмеченные как предпочтительные выше. Еще более предпочтительными соединениями для композиций, способов и наборов являются соединения, приведенные в неограничивающих примерах ниже. Экспериментальная часть. Далее в данном документе термин "т.п." означает точку плавления, "водн." означает водный, "р.с." означает реакционную смесь, "к.т." означает комнатную температуру, "DIPEA" означает N,Nдиизопропилэтиламин, "DIPE" означает диизопропилэфир, "ТГФ" означает тетрагидрофуран, "ДМФА" означает диметилформамид, "DCM" означает дихлорметан, "EtOH" означает этанол, "EtOAc" означает этилацетат, "AcOH" означает уксусную кислоту, "iPrOH" означает изопропанол, "iPrNH2" означает изопропиламин, "MeCN" означает ацетонитрил, "MeOH" означает метанол, "Pd(OAc)2" означает диацетат палладия (II), "рац." означает рацемический, "нас." означает насыщенный, "SFC" означает сверхкритическую жидкостную хроматографию, "SFC-MC" означает сверхкритическую жидкостную хроматографию/масс-спектрометрию, "ЖХ-MC" означает жидкостную хроматографию/масс-спектрометрию, "GCMC" означает газовую хроматографию/масс-спектрометрию, "ВЭЖХ" означает высокоэффективную жидкостную хроматографию, "RP" означает обращенно-фазовую, "UPLC" означает сверхпроизводительную жидкостную хроматографию, "Rt" означает время удерживания (в минутах), "[M+H]+" означает протонированную массу свободного основания соединения, "DAST" означает трифтори диэтиламиносеры,"DMTMM" означает 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолина хлорид, "HATU" означаетO-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат, "Xantphos" означает (9,9 диметил-9H-ксантен-4,5-диил)бис[дифенилфосфин], "TBAT" означает тетрабутиламмония трифенилдифторсиликат, "TFA" означает трифторуксусную кислоту, "Et2O" означает диэтиловый эфир, "ДМСО" означает диметилсульфоксид, "MeCN" означает ацетонитрил. Для ключевых промежуточных соединений, как и для некоторых конечных соединений, абсолютную конфигурацию хиральных центров (обозначена как R и/или S) устанавливали посредством сравнения с образцами с известной конфигурацией или путем использования аналитических методик, пригодных для определения абсолютной конфигурации, таких как VCD (вибрационный круговой дихроизм) или рентгеноструктурная кристаллография. Если абсолютная конфигурация хирального центра не известна, его произвольно обозначают R.A. Получение промежуточных соединений. Пример A1. Получение промежуточного соединения 1 Изопропоксид титана (IV) (202 мл, 658 ммоль) добавляли к перемешанной смеси этил-2-(3-бромфенил)-2-оксо-ацетата [(CAS 62123-80-2), 80 г, 329 ммоль] и (S)-2-метил-2-пропансульфинамида (47,9 г,395 ммоль) в н-гептане (740 мл). Смесь перемешивали при 80C в течение 4 ч. Смесь охлаждали при комнатной температуре и добавляли воду. Полученную в результате смесь фильтровали через прокладку из диатомитовой земли и промывали н-гептаном. Органический слой отделяли, высушивали (MgSO4),фильтровали и концентрировали в вакууме. Остаток очищали колоночной флэш-хроматографией (силикагель; элюент: н-гептан/EtOAc от 100/0 до 50/50). Требуемые фракции собирали и концентрировали в вакууме с получением промежуточного соединения 1 (91 г, выход 74%). Пример A2. Получение промежуточного соединения 2 Бромид циклопропилмагния (1 М, 300 мл, 300 ммоль) по каплям добавляли к перемешанному раствору промежуточного соединения 1 (91 г, 243 ммоль) в DCM (1500 мл) при -40C. Смесь перемешивали при данной температуре в течение 30 мин, а затем реакцию гасили путем добавления насыщенного водного раствора NH4Cl с последующим добавлением воды. Смесь экстрагировали с помощью DCM. Органический слой отделяли, высушивали (MgSO4), фильтровали и растворители выпаривали в вакууме с получением промежуточного соединения 2 (100 г, выход 99%), которое использовали без дополнитель- 10024716 ной очистки на следующей стадии. Пример A3. Получение промежуточного соединения 3 1 М водный раствор NaOH (750 мл, 750 ммоль) добавляли к раствору неочищенного промежуточного соединения 2 (100 г, 240 ммоль) в MeOH (400 мл). Полученную смесь перемешивали при дефлегмировании в течение 4 ч. Смесь охлаждали до комнатной температуры, а затем распределяли между водой иEtOAc. Водный слой отделяли и нейтрализовали путем добавления 1 М водного раствора HCl (750 мл), а затем экстрагировали с помощью DCM. Органический слой отделяли, высушивали (MgSO4), фильтровали и растворители выпаривали в вакууме. Остаток растирали в порошок с помощью DIPE/MeCH, и полученные твердые частицы отфильтровывали и высушивали в вакууме с получением промежуточного соединения 3 (37 г, выход 41%).D: +37,59 (589 нм, конц. 0,564 вес./об.%, MeOH, 20C). Абсолютную конфигурацию определяли путем рентгенодифракции. Пример A4. Получение промежуточного соединения 4 Соль хлористо-водородной кислоты Промежуточное соединение 3 (37 г, 99 ммоль) перемешивали в 4 М растворе HCl в диоксане (74 мл) и 1,4-диоксане (75 мл) при комнатной температуре в течение 30 мин. К полученной суспензии добавлялиDIPE, и осадок отфильтровывали и высушивали в вакууме с получением промежуточного соединения 4D: -68,89 (589 нм, с 0,646 вес./об.%, MeOH, 20C). Пример A5. Получение промежуточного соединения 5 1 М водный раствор NaOH (182,6 мл, 182,6 ммоль) добавляли к раствору промежуточного соединения 4 (28 г, 91,3 ммоль) и смесь охлаждали в ледяной бане. К этой смеси по каплям добавляли раствор хлорацетилхлорида (21,8 мл, 274 ммоль) в ТГФ (280 мл) при 15C в течение более одного часа, добавляя одновременно раствор 25%-ного водного раствора NaOH для поддержания рН приблизительно 10-11. После завершения реакции концентрированный водный раствор HCl осторожно добавляли к реакционной смеси до pH 2. Смесь частично концентрировали в вакууме и полученный в результате осадок отфильтровывали, промывали с помощью DIPE и высушивали в вакууме с получением промежуточного соединения 5 (2 6 г, выход 82%).D: -6,49 (589 нм, конц. 0,5855 вес./об.% MeOH, 20C) Пример A6. Получение промежуточного соединения 6(17 мл), и реакционную смесь перемешивали при 80C в течение 2 ч. Смесь частично концентрировали при пониженном давлении, охлаждали до комнатной температуры и затем фильтровали через диатомитовую землю. Фильтрат концентрировали в вакууме, и остаток очищали путем колончной флэшхроматографии (силикагель; элюент: н-гептан/EtOAc от 100/0 до 50/50). Требуемые фракции собирали и концентрировали в вакууме с получением промежуточного соединения 6 (0,54 г, выход 86%).D: -15,68 (589 нм, конц. 0,37 вес./об.%, MeOH, 20C). Пример A7. Получение промежуточного соединения 7 К раствору промежуточного соединения 6 (4,2 г, 13,54 ммоль) в ТГФ (55 мл) добавляли ТВАТ (0,73 г, 1,35 ммоль). Затем по каплям добавляли (трифторметил)триметилсилан (4,0 мл, 27 ммоль), и реакци- 11024716 онную смесь перемешивали при комнатной температуре в течение 2 ч. Смесь гасили водным NaCl, экстрагировали с помощью EtOAc, органическую фазу отделяли, высушивали (MgSO4) и концентрировали в вакууме. Полученное масло очищали колоночной хроматографией (силикагель; элюент: DCM/EtOAc от 100/0 до 0/100). Требуемые фракции собирали и концентрировали в вакууме с получением промежуточного соединения 7 (3 г, выход 58%) в виде смеси цис- и транс-изомеров, которую без дополнительной очистки использовали в следующей стадии. Пример A8. Получение промежуточного соединения 8DAST (1,16 мл, 9,5 ммоль) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 1 ч и затем реакционную смесь концентрировали при пониженном давлении. Остаток распределяли между DCM и водным насыщенным раствором NaHCO3. Органический слой отделяли и водный слой экстрагировали с помощью DCM. Объединенные органические слои высушивали(MgSO4), фильтровали, и растворитель выпаривали в вакууме. Остаток очищали колоночной флэшхроматографией на силикагеле (элюент: н-гептан/EtOAc от 100/0 до 0/100). Требуемые фракции собирали и концентрировали в вакууме с получением промежуточного соединения 8 (2 г, выход 6 6%) в виде смеси цис- и транс-изомеров, которую без дополнительной очистки использовали в следующей стадии. Пример A9. Получение промежуточного соединения 9P2S5 (1,16 г, 5,23 ммоль) добавляли к раствору промежуточного соединения 8 (2 г, 5,23 ммоль) в ТГФ (43 мл) при комнатной температуре. Смесь перемешивали при 70C в течение 3 ч. Затем смесь охлаждали до комнатной температуры, отфильтровывали, и органический растворитель выпаривали в вакууме. Неочищенный остаток очищали колоночной флэш-хроматографии (силикагель; элюент: нгептан/DCM от 80/100 до 0/100). Требуемые фракции собирали и выпаривали в вакууме с получением промежуточного соединения 9 (1,6 г, выход 77%) в виде смеси цис- и транс-изомеров. Пример A10. Получение промежуточных соединений 10 и 11 Промежуточное соединение 9 (4,2 г, 10,55 ммоль) добавляли к смеси 7 н. аммиака в MeOH (16 мл) и водного раствора NH4OH (40 мл) и реакционную смесь перемешивали при 14 0C в течение 1 ч в микроволновом излучении. Затем растворитель выпаривали и остаток растворяли в DCM, высушивали(Mg2SO4), фильтровали, и растворитель выпаривали в вакууме. Остаток очищали колоночной флэшхроматографией (силикагель; элюент: н-гептан/EtOAc от 100/0 до 50/50). Требуемые фракции собирали и концентрировали в вакууме с получением промежуточного соединения 10 (2,44 г, выход 61%) и промежуточного соединения 11 (0,7 г, выход 17%). Пример A11. Получение промежуточного соединения 12 Промежуточное соединение 10 (2,44 г, 6,4 ммоль) объединяли с NaN3 (1,04 г, 16 ммоль), CuI (1,52 г,8,0 ммоль) и Na2CO3 (1,357 г, 12,8 ммоль) в ДМСО (92 мл), и реакционную смесь дегазировали. После этого добавляли N,N'-диметилэтилендиамин (1,2 мл, 11,2 ммоль), и смесь нагревали при 110C до завершения реакции приблизительно 6 ч. Реакционную смесь вливали в DCM. Добавляли гидроксид аммония(28% в воде) и органический слой отделяли и промывали три раза раствором гидроксида аммония. Затем органический слой высушивали (MgSO4), фильтровали и концентрировали в вакууме с получением промежуточного соединения 12 (2 г, выход 98%). Пример A12. Получение промежуточного соединения 13 вали при комнатной температуре в течение 3 дней. Затем растворитель выпаривали в вакууме и остаток поглощали в EtOAc (80 мл). Твердую фазу фильтровали, и фильтрат выпаривали в вакууме с получением промежуточного соединения 13 (колич. выход 27,9 г), которое использовали на следующей стадии без дополнительной очистки. Пример A13. Получение промежуточного соединения 14 Промежуточное соединение 13 (27 г, 111 ммоль) растворяли в HCl (37% в H2O) (130 мл) и уксусной кислоте (130 мл), и смесь кипятили с обратным холодильником в течение 16 ч. После охлаждения до комнатной температуры смесь концентрировали в вакууме. Добавляли воду и водный слой экстрагировали с помощью EtOAc. Водный слой ощелачивали водным раствором NaOH (25%) до рН 7. Водный слой частично концентрировали в вакууме. Смесь охлаждали в ледяной бане, и осадок отфильтровывали,промывали водой и затем Et2O и высушивали в вакууме с получением промежуточного соединения 14(18 г, выход 62%) в виде белого твердого вещества. Пример A14. Получение промежуточного соединения 15 Смесь промежуточного соединения 14 (15 г, 57,2 ммоль) в растворе 10% H2SO4 в метаноле (330 мл) кипятили с обратным холодильником в течение 48 ч. Реакционную смесь концентрировали в вакууме. Добавляли воду, и раствор ощелачивали до рН 8 насыщенным водным раствором NHCO3. Затем водный слой экстрагировали с помощью EtOAc. Органический слой отделяли, высушивали (Mg4SO4), фильтровали и концентрировали в вакууме с получением промежуточного соединения 15 (15 г, выход 95%). Пример A15. Получение промежуточного соединения 16 Промежуточное соединение 15 (10 г) разделяли на соответствующие энантиомеры препаративнойSFC на (Chiralpak Daicel AD 30250 мм). Подвижная фаза (CO2, MeOH с 0,2% iPrNH2), при этом получали промежуточное соединение 16 (4,2 г, выход 42%) .D: -10,1 (365 нм, конц. 0,762 вес./об.% MeOH, 20C). Пример A16. Получение промежуточного соединения 17 ТГФ (150 мл) добавляли к раствору промежуточного соединения 16 (40 г, 145 ммоль) в NaOH (1M в Н 2 О, 360 мл). Смесь перемешивали в течение 4 ч при комнатной температуре. Смесь концентрировали в вакууме с получением промежуточного соединения 17 (42 г) в виде белого твердого вещества, которое использовали без дополнительной очистки на следующей стадии реакции. Пример A17. Получение промежуточного соединения 18 К охлажденному раствору промежуточного соединения 17 (41,3 г, 145 ммоль) в Н 2 О (150 мл) по каплям добавляли раствор хлорацетилхлорида (24 мл, 304,5 ммоль) в 1,4-диоксане (75 мл). Одновременно добавляли NaOH (5M в Н 2 О, 2 9 мл) для доведения рН до 10-11. Органический слой отделяли, и водный слой экстрагировали с помощью Et2O. Затем водный слой подкисляли с помощью HCl (6 М, в Н 2 О) до рН 2. Осажденное белое твердое вещество собирали фильтрацией, промывали с помощью Н 2 О и высушивали с получением промежуточного соединения 18 (42 г, выход 86%). Пример A18. Получение промежуточного соединения 19(1000 мл), и реакционную смесь перемешивали при 80C в течение 3 ч. Смесь частично концентрировали при пониженном давлении, охлаждали до комнатной температуры и затем фильтровали через диатомитовую землю. Фильтрат концентрировали в вакууме, и остаток очищали колоночной флэш- 13024716 хроматографией (силикагель; элюент: MeOH/DCM от 0/100 до 5/95). Требуемые фракции собирали и концентрировали в вакууме с получением промежуточного соединения 19 (36 г, выход 96%). Пример A19. Получение промежуточного соединения 20 Раствор промежуточного соединения 19 (10 г, 21,5 ммоль) в ТГФ (105 мл) охлаждали до -78C в атмосфере N2. Затем медленно добавляли гидрид диизобутилалюминия (43 мл, 4 3 ммоль). Реакционную смесь перемешивали в течение 2 ч, способствуя ее медленному подогреву до комнатной температуры. Реакционную смесь охлаждали до 0C и гасили путем медленного добавления водного раствора 1 н. HCl. Затем смесь экстрагировали с помощью EtOAc, органические слои отделяли, высушивали (Na2SO4),фильтровали и растворители выпаривали в вакууме с получением промежуточного соединения 20 (колич. выход 6,6 г, смесь диастереоизомеров 80/20), которое использовали без дополнительной очистки на следующей стадии реакции. Пример A20. Получение промежуточного соединения 21 Промежуточное соединение 20 (6,3 г, 20,7 ммоль) растворяли в DCM (84 мл), и реакционную смесь охлаждали до 0C. Затем по каплям добавляли DAST (3 мл, 24,9 ммоль). Через 20 мин при 0C реакционную смесь гасили насыщенным водным раствором NaHCO3 и экстрагировали с помощью DCM. Объединенные органические слои высушивали (MgSO4), фильтровали, и растворитель выпаривали в вакууме. Неочищенный продукт выделяли из DIPE, отфильтровывали и высушивали в вакууме при 60C с получением промежуточного соединения 21 (4,2 г, выход 66%, смесь диастереоизомеров 80/20). Пример A21. Получение промежуточного соединения 22 Промежуточное соединение 22 синтезировали, действуя в соответствии с тем же подходом, описанным в примере A9. Начиная с промежуточного соединения 21 (4,2 г, 13,7 ммоль) получали промежуточное соединение 22 (3 г, выход 68%, смесь диастереоизомеров 60/40). Пример A22. Получение промежуточных соединений 23 и 24 Промежуточное соединение 22 (6 г, 18,6 ммоль) растворяли в 7 н. аммиаке в MeOH (300 мл), и реакционную смесь перемешивали при 60C в течение 18 ч. Растворитель выпаривали и добавляли дополнительно 7 н. аммиак в MeOH (300 мл) и смесь дополнительно перемешивали при 60C в течение 18 ч. Затем растворитель выпаривали и неочищенный продукт очищали колоночной хроматографией (силикагель; элюент: 7 М раствор аммиака в метаноле/DCM от 0/100 до 10/90). Необходимые фракции собирали и концентрировали в вакууме с получением промежуточного соединения 23 (3,7 г, выход 65%) и промежуточного соединения 24 (0,6 г, выход 11%). Пример A23. Получение промежуточного соединения 25 Промежуточное соединение 23 (1,6 г, 5,24 ммоль) объединяли с NaN3 (0,85 г, 13 ммоль), CuI (1,25 г,6,5 ммоль) и Na2CO3 (1,1 г, 10,5 ммоль) в ДМСО (75 мл), и реакционную смесь дегазировали. После этого добавляли N,N'-диметилэтилендиамин (1 мл, 9,1 ммоль), и смесь нагревали при 110C до завершения реакции приблизительно 4 ч. Реакционную смесь вливали в DCM. Добавляли гидроксид аммония (28% в воде), и органический слой отделяли и промывали три раза гидроксидом аммония. Затем органический слой высушивали (Mg2SO4), фильтровали и концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией (силикагель; элюент: 7 М раствор аммиака в метаноле/DCM от 0/100 до 10/90). Необходимые фракции собирали и концентрировали в вакууме с получением промежуточного соединения 25 (0,3 г, выход 24%). Пример A24. Получение промежуточного соединения 26 К раствору промежуточного соединения 19 (11,6 г, 38,5 ммоль) в ТГФ (117 мл) добавляли ТВАТ(2,08 г, 3,85 ммоль). Затем по каплям добавляли (трифторметил)триметилсилан (12,5 мл, 84,6 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 20 мин. Смесь гасили воднымNaCl и экстрагировали с помощью EtOAc. Объединенные органические слои высушивали (MgSO4),фильтровали и концентрировали в вакууме с получением промежуточного соединения 26 (14 г, выход 98%) в виде смеси цис- и транс-изомеров, которую без дополнительной очистки использовали на следующей стадии. Пример A25. Получение промежуточного соединения 27 Промежуточное соединение 26 (14 г, 37,6 ммоль) растворяли в DCM (600 мл) и охлаждали до 0C и затем по каплям добавляли хлорид тионила (11,2 мл, 150 ммоль). Реакционную смесь перемешивали в течение 30 мин при 0C и затем добавляли пиридин (18,2 мл, 225,7 ммоль). Через 30 мин реакционную смесь гидролизировали водным раствором 1 н. HCl, а затем экстрагировали с помощью DCM. Органические слои отделяли, высушивали (MgSO4), фильтровали и выпаривали в вакууме. Неочищенный продукт очищали колоночной флэш-хроматографией (силикагель; элюент: 7 М раствор аммиака в метаноле/DCM от 0/100 до 2/98). Необходимые фракции собирали и концентрировали в вакууме с получением промежуточного соединения 27 (6 г, выход 41%, смесь диастереоизомеров). Пример A26. Получение промежуточного соединения 28 Промежуточное соединение 27 (7 г, 17,9 ммоль) и цинк-медную пару (8,55 г, 66,3 ммоль) перемешивали в уксусной кислоте (420 мл) при комнатной температуре в течение 16 ч. Реакционную смесь фильтровали, промывали с помощью DCM и концентрировали в вакууме. Добавляли раствор гидроксида аммония (28% в воде) и DCM, и смесь перемешивали при комнатной температуре в течение одного часа. Органический слой отделяли, и водный слой экстрагировали с помощью DCM. Объединенные органические слои высушивали (Mg4SO4), фильтровали и выпаривали в вакууме с получением промежуточного соединения 28 (6 г, выход 99%) . Пример A27. Получение промежуточного соединения 29P2S5 (5,95 г, 26,8 ммоль) добавляли к раствору промежуточного соединения 28 (6 г, 17,9 ммоль) в ТГФ (145 мл) при комнатной температуре. Смесь перемешивали при 70C в течение 90 мин. Затем смесь охлаждали до комнатной температуры, отфильтровывали и выпаривали органический растворитель в вакууме с получением промежуточного соединения 29 (5,9 г), которое использовали без дополнительной очистки на следующей стадии. Пример A28. Получение промежуточного соединения 30 Промежуточное соединение 30 синтезировали, действуя в соответствии с тем же подходом, описанным в примере A22. Начиная с промежуточного соединения 29 (5,9 г, 16,8 ммоль) получали промежуточное соединение 30 (4,04 г, выход 72%). Пример A29. Получение промежуточного соединения 31 Промежуточное соединение 31 синтезировали, действуя в соответствии с тем же подходом, описанным в примере A23. Начиная с промежуточного соединения 30 (3,6 г, 10,7 ммоль) получали промежуточное соединение 31 (1,52 г, выход 52%). Пример A30. Получение промежуточного соединения 32 К раствору промежуточного соединения 27 (3 г, 7,68 ммоль) в уксусной кислоте (136 мл) добавляли цинк (1,26 г, 19,2 ммоль). Затем реакционную смесь перемешивали при 80C в течение 3 ч, после чего реакционную смесь фильтровали горячей и концентрировали в вакууме. Остаток растворяли в DCM и промывали раствором гидроксида аммония. Органическую фазу отделяли, высушивали (MgSO4), и растворитель концентрировали в вакууме. Неочищенный продукт очищали колоночной хроматографией(силикагель; элюент: 7 М раствор аммиака в метаноле/DCM от 0/100 до 3/97). Необходимые фракции собирали и концентрировали в вакууме с получением промежуточного соединения 32 (2,7 г, выход 99%). Пример A31. Получение промежуточного соединения 33 Реагент Лавессона (6,82 г, 16,85 ммоль) добавляли к раствору промежуточного соединения 32 (6 г,16,85 ммоль), растворенного в ТГФ (68 мл), при комнатной температуре. Смесь перемешивали при 60C в течение 4 ч. Затем смесь охлаждали до комнатной температуры, отфильтровывали и органический растворитель выпаривали в вакууме. Неочищенный остаток очищали колоночной флэш-хроматографией(силикагель; элюент: н-гептан/DCM от 100/0 до 50/50). Требуемые фракции собирали и выпаривали в вакууме с получением промежуточного соединения 33 (6 г, выход 96%) в виде желтоватого масла. Пример A32. Получение промежуточных соединений 34 и 35 Промежуточное соединение 33 (6 г, 16,1 ммоль) растворяли в 7 н. аммиаке в MeOH (97 мл), и реакционную смесь перемешивали при 80C в течение 24 ч. Затем растворитель выпаривали и неочищенный продукт очищали колоночной хроматографией (силикагель; элюент: 7 М раствор аммиака в метаноле/DCM от 0/100 до 2/98). Требуемые фракции собирали и концентрировали в вакууме с получением промежуточного соединения 34 (3,4 г, выход 59%) и промежуточного соединения 35 (0,75 г, выход 13%). Пример A33. Получение промежуточного соединения 36 Промежуточное соединение 34 (3,4 г, 9,6 ммоль) объединяли с NaN3 (1,56 г, 24 ммоль), CuI (2,28 г,12 ммоль) и Na2CO3 (2,03 г, 19,1 ммоль) в ДМСО (137 мл), и реакционную смесь дегазировали. После этого добавляли N,N'-диметилэтилендиамин (1,8 мл, 16,8 ммоль), и смесь нагревали при 110C до завершения реакции приблизительно 1 ч. Реакционную смесь отфильтровывали и фильтровальный осадок промывали с помощью EtOAc. К фильтрату добавляли воду и EtOAc, и смесь подкисляли путем добавления HCl (1MB H2O). Затем органический слой отделяли и водный слой отмывали с помощью EtOAc. Затем водный слой ощелачивали с помощью водн. раствора аммиака и снова экстрагировали с помощьюEtOAc. Объединенные органические слои высушивали (Na2SO4), фильтровали и концентрировали в вакууме с получением промежуточного соединения 36 (2,5 г, выход 90%). Промежуточное соединение 12 (0,07 г, 0,221 ммоль) растворяли в изопропаноле (5 мл) и добавляли 2-бром-3-метоксипиридин (0,083 г, 0,441 ммоль) и серную кислоту (0,108 г, 1,1 ммоль). Смесь перемешивали в течение 40 ч при 80C. Смеси давали охладиться до комнатной температуры. Добавляли DCM и насыщенный водный раствор NaHCO3. Органические слои отделяли, высушивали (MgSO4), фильтровали, и растворители выпаривали в вакууме. Неочищенный продукт очищали колоночной флэшхроматографией (силикагель; элюент: 7 М раствор аммиака в метаноле/DCM от 0/100 до 10/90). Необходимые фракции собирали и концентрировали в вакууме. Затем остаток растворяли в DIPE и превращали в соль HCl путем добавления HCl в изопропаноле. Полученное твердое вещество фильтровали и высушивали в вакууме с получением соединения 1 (0,025 г, выход 25%) в виде хлористо-водородной соли. Пример B2. Получение соединения 2: (5R,6R)-6-фтор-5-2-фтор-5-[(3-метоксипиридин-2 ил)амино]фенил-5-метил-5,6-дигидро-2H-1,4-оксазин-3-амин Промежуточное соединение 25 (0,3 г, 1,24 ммоль) растворяли в изопропаноле (15 мл) и добавляли 2-бром-3-метоксипиридин (0,467 г, 2,49 ммоль) и серную кислоту (0,61 г, 6,22 ммоль). Смесь перемешивали в течение 40 ч при 80C. Смеси давали остыть до комнатной температуры. Добавляли DCM и насыщенный водный раствор NaHCO3. Органические слои отделяли, высушивали (MgSO4), фильтровали, и растворители испаряли в вакууме. Неочищенный продукт очищали препаративной ВЭЖХ на (RP Vydac Промежуточное соединение 31 (0,35 г, 1,29 ммоль) растворяли в изопропаноле (15 мл) и добавляли 2-бром-3-метоксипиридин (0,485 г, 2,58 ммоль) и серную кислоту (0,34 мл, 6,45 ммоль). Смесь перемешивали в течение 72 ч при 80C. Смеси давали остыть до комнатной температуры. Добавляли DCM и насыщенный водный раствор NaHCO3. Органические слои отделяли, высушивали (MgSO4), фильтровали,и растворители испаряли в вакууме. Неочищенный продукт очищали колоночной флэш-хроматографией(силикагель; элюент: 7 М раствор аммиака в метаноле/DCM от 0/100 до 2/98). Требуемые фракции собирали и концентрировали в вакууме. Остаток выделяли из DIPE/гептанов, фильтровали и высушивали при сильном разрежении с получениемсоединения 3 (0,279 г, выход 57%) в виде белого порошка. Пример B4. Получение соединения 4: (5R,6R)-6-(дифторметил)-5-2-фтор-5-[(3-метоксипиридин 2-ил)амино]фенил-5-метил-5,6-дигидро-2H-1,4-оксазин-3-амин Соединение 3 (0,228 г, 0,603 ммоль) растворяли в EtOAc (4 мл) и добавляли палладий на угле (10%)(0,064 г, 0,06 ммоль) и тиофен (0,4%-ный раствор в ТГФ, 0,8 мл, 0,041 ммоль). Смесь гидрогенизировали при комнатной температуре и атмосферном давлении в течение 16 ч. Катализатор отфильтровывали и растворители выпаривали в вакууме. Неочищенный продукт очищали колоночной флэш-хроматографией(силикагель; элюент: 7 М раствор аммиака в метаноле/DCM от 0/100 до 2/98). Необходимые фракции собирали и концентрировали в вакууме. Остаток выделяли из DIPE/гептанов, фильтровали и высушивали при сильном разрежении с получением соединения 4 (0,074 г, выход 32%). Промежуточное соединение 25 (0,048 г, 0,199 ммоль) растворяли в 1,4-диоксане (2 мл) и добавляли 2-бромпиримидин (0,032 г, 0,199 ммоль) и 4 М раствор HCl в диоксане (0,1 мл, 0,4 ммоль). Смесь перемешивали в течение 16 ч при 100C. Смеси давали охладиться до комнатной температуры. ДобавлялиDCM и насыщенный водный раствор NaHCO3. Органические слои отделяли, высушивали (MgSO4),фильтровали, и растворители испаряли в вакууме. Неочищенный продукт очищали колоночной флэшхроматографией (силикагель; элюент: 7 М раствор аммиака в метаноле/DCM от 0/100 до 5/95). Требуемые фракции собирали и концентрировали в вакууме с получением соединения 5 (0,013 г, выход 20%). Пример B6. Получение соединения 6: (5R,6R)-5-2-фтор-5-[(3-метоксипиридин-2-ил)амино]фенил 5-(трифторметил)-5,6-дигидро-2 Н-1,4-оксазин-3-амин Промежуточное соединение 36 (0,1 г, 0,343 ммоль) растворяли в изопропаноле (4 мл) и добавляли 2-бром-3-метоксипиридин (0,129 г, 0,687 ммоль) и серную кислоту (0,09 мл, 1,7 ммоль). Смесь перемешивали в течение 40 ч при 80C. Смеси давали охладиться до комнатной температуры. Добавляли DCM и насыщенный водный раствор NaHCO3. Органические слои отделяли, высушивали (MgSO4), фильтровали, и растворители выпаривали в вакууме. Неочищенный продукт очищали колоночной флэшхроматографией (силикагель; элюент: 7 М раствор аммиака в метаноле/DCM от 0/100 до 5/95) с получением соединения 6 (0,013 г, выход 20%). Пример B7. Получение соединения 7: (5R,6R)-5-[2-фтор-5-(пиримидин-2-иламино)фенил]-5-метил 6-(трифторметил)-5,6-дигидро-2H-1,4-оксазин-3-амин Промежуточное соединение 36 (0,1 г, 0,343 ммоль) растворяли в 1,4-диоксане (3,4 мл) и добавляли 2-бромпиримидин (0,055 г, 0,343 ммоль) и 4 М раствор HCl в диоксане (0,17 мл, 0,69 ммоль). Смесь перемешивали в течение 16 ч при 100C. Смеси давали охладиться до комнатной температуры. ДобавлялиDCM и насыщенный водный раствор NaHCO3. Органические слои отделяли, высушивали (MgSO4),фильтровали, и растворители выпаривали в вакууме. Неочищенный продукт очищали колоночной флэшхроматографией (силикагель; элюент: 7 М раствор аммиака в метаноле/DCM от 0/100 до 5/95). Требуемые фракции собирали и концентрировали. Неочищенный продукт очищали препаративной ВЭЖХ на(Chiralpal Diacel AS 20250 мм). Подвижная фаза (CO2, MeOH с 0,2% iPrNH2), при этом получали соединение 7 (0,036 г, выход 28%) . Пример B8. Получение соединения 8: (5R,6R)-6-фтор-5-2-фтор-5-[(3-метоксипиразин-2 ил)амино]фенил-5-метил-5,6-дигидро-2H-1,4-оксазин-3-амин Промежуточное соединение 25 (0,15 г, 0,622 ммоль) растворяли в 1,4-диоксане (6 мл). Добавляли 2 йод-3-метоксипиразин (0,12 г, 0,508 ммоль), карбонат цезия (0,405 г, 1,244 ммоль), 1,1'бис(дифенилфосфино)ферроцен (0,052 г, 0,093 ммоль) и трис(дибензилиденацетон)дипалладий(0) (0,028 г, 0,031 ммоль). Реакционную пробирку запаивали и смесь перемешивали при 160C в течение 1 ч в микроволновом излучении. После охлаждения реакционную смесь разбавляли с помощью DCM и фильтровали через дикалит. Фильтрат концентрировали в вакууме. Неочищенный продукт очищали колоночной флэш-хроматографией (силикагель; элюент: 7 М раствор аммиака в метаноле/DCM от 0/100 до 5/95). Требуемые фракции собирали и концентрировали. Неочищенный продукт дополнительно очищали препаративной ВЭЖХ на (Chiralpal Diacel AS 20250 мм). Подвижная фаза (CO2, MeOH с 0,2% iPrNH2), при этом получали соединение 8 (0,010 г, выход 5%). Пример B9. Получение соединения 9 Промежуточное соединение 25 (0,30 г, 1,244 ммоль) и 2-бромникотинонитрил (455 мг, 2,487 ммоль) растворяли в 1,4-диоксане (12,5 мл), затем по каплям добавляли 4 М раствор HCl в диоксане (0,933 мл,3,731 ммоль). Реакционную смесь перемешивали в течение 16 ч при 120C. Затем добавляли насыщенный водный раствор NaHCO3, после чего DCM. Фазы разделяли, и органический слой концентрировали при пониженном давлении. Полученное в результате масло очищали колоночной флэш-хроматографией(силикагель; элюент: 7 М раствор аммиака в метаноле/DCM от 0/100 до 5/95). Необходимые фракции собирали и далее очищали препаративной SFC на (Chiralcel Diacel OD 20250 мм). Подвижная фаза (CO2,iPrOH с 0,2% iPrNH2), при этом получали соединение 9 (0,006 г, 1%). Пример B10. Получение соединения 10 Промежуточное соединение 25 (0,482 г, 2 ммоль) растворяли в iPrOH (24 мл), затем добавляли 2,3 дибромопиридин (0,948 г, 4 ммоль) и серную кислоту (0,533 мл, 10 ммоль). Реакционную смесь перемешивали в течение 4 дней при 80C. Реакционной смеси давали охладиться, затем добавляли DCM и насыщенный водный раствор NaHCO3. Фазы разделяли и органический слой высушивали и концентрировали при пониженном давлении. Полученное масло очищали колоночной хроматографией (силикагель; элюент: 7 М раствор аммиака в метаноле/DCM от 0/100 до 10/90). Необходимые фракции собирали и далее очищали препаративной SFC (Chiralpak Daicel AS 20 микроом 500 гр). Подвижная фаза(CO2, iPrOH с 0,2% iPrNH2), при этом получали соединение 10 (0,135 г, 17%). Пример B11. Получение соединения 11 Промежуточное соединение 36 (0,358 г, 1,229 ммоль) растворяли в 1,4-диоксане (14,7 мл). Добавляли 2-бром-3-цианопиридин (0,191 г, 1,045 ммоль), карбонат цезия (0,800 г, 2,458 ммоль), 1,1'бис(дифенилфосфино)ферроцен (0,104 г, 0,184 ммоль) и смесь перемешивали в атмосфере аргона в течение нескольких минут. Затем добавляли трис(дибензилиден-ацетон)дипалладий(0) (0,056 г, 0,062 ммоль). Пробирку для реакций запаивали, и смесь перемешивали при 160C в течение 1 ч в микроволновом излучении. После охлаждения реакционную смесь разбавляли с помощью DCM и фильтровали через дикалит. Фильтрат концентрировали в вакууме. Неочищенный продукт очищали колоночной флэшхроматографией (силикагель; элюент: 7 М раствор аммиака в метаноле/DCM от 0/100 до 5/95). Требуемые фракции собирали и концентрировали. Неочищенный продукт дополнительно очищали препаративной SFC на (Chiralpal Diacel AS 20250 мм). Подвижная фаза (CO2, MeOH с 0,2% iPrNH2) с получением соединения 11 (0,052 г, выход 11%). Соединения 1-11 в табл. 1-5 представляют собой соединения, полученные согласно одному из приведенных выше примеров. В случае, если не указана форма соли, соединение получали в виде свободного основания. " примера" означает номер примера, в соответствии с протоколом которого соединение было синтезировано. " соединения" означает номер соединения. Таблица 1C. Аналитическая часть. ЖХМС. Для исследования соединений по настоящему изобретению с помощью (ЖХ)МС использовали следующие способы. Общая методика. Измерения с помощью ЖХ осуществляли с использованием системы Acquity UPLC (Waters), содержащей насос для двухкомпонентных смесей, камеру для образцов, нагреватель колонки (установленный на 55C) , диодно-матричный детектор (DAD) и колонку, как определено в соответствующих способах ниже. Поток из колонки разделялся для МС-спектрометра. МС-детектор был оснащен источником ионизации электрораспылением. Масс-спектры получали сканированием от 100 до 1000 за 0,18 с с использованием времени выдержки 0,02 с. Напряжение капиллярной иглы составляло 3,5 кВ, и температуру источника поддерживали при 140C. В качестве газа-распылителя использовали азот. Сбор и обработ- 20024716 ку данных проводили с помощью системы обработки данных Waters-Micromass MassLynx-Openlynx. Способ 1. Обращенно-фазовую UPLC (сверхпроизводительную жидкостную хроматографию) проводили на колонке C18 с мостиковым гибридом этилсилоксан/диоксид кремния (BEH) (1,7 мкм, 2,150 мм; WatersAcquity) со скоростью потока 0,8 мл/мин. Две подвижные фазы (подвижная фаза A: 10 мМ аммония ацетат в смеси 95/5 H2O/ацетонитрил; подвижная фаза B: ацетонитрил) использовали для соблюдения условия - градиент от 95% A и 5% B до 5% A и 95% B за 1,3 мин при удерживании в течение 0,2 мин. Использовали объем вводимой пробы 0,5 мкл. Напряжение на конусе составляло 10 B для режима положительной ионизации и 20 B для режима отрицательной ионизации. Способ 2. Обращенно-фазную UPLC (сверхпроизводительную жидкостную хроматографию) проводили на колонке С 18 с мостиковым гибридом этилсилоксан/диоксид кремния (BEH) (1,7 мкм, 2,150 мм; WatersAcquity) со скоростью потока 0,8 мл/мин. Две подвижные фазы (10 мМ ацетата аммония вH2O/ацетонитриле 95/5; подвижная фаза B: ацетонитрил) применяли для соблюдения условия градиента от 95% A и 5% B до 5% A и 95% B за 1,3 мин при удерживании в течение 0,3 мин. Использовали объем вводимой пробы 0,5 мкл. Напряжение на конусе составляло 10 В для режима положительной ионизации и 20 В для режима отрицательной ионизации. Способ 3. Обращенно-фазную UPLC (сверхпроизводительную жидкостную хроматографию) проводили на колонке C18 с мостиковым гибридом этилсилоксан/диоксид кремния (BEH) (1,7 мкм, 2,150 мм; WatersAcquity) со скоростью потока 0,8 мл/мин. Две подвижные фазы (10 мМ ацетата аммония вH2O/ацетонитриле 95/5; подвижная фаза B: ацетонитрил) использовали для соблюдения условия градиента от 95% A и 5% B до 5% A и 95% B за 1,3 мин при удерживании в течение 0,3 мин. Использовали объем вводимой пробы 0,5 мкл. Напряжение на конусе составляло 10 В для режима положительной ионизации и 20 В для режима отрицательной ионизации. Точки плавления. Значения представляют собой или пиковые значения, или диапазоны плавления, и их получают с экспериментальными погрешностями, которые обычно связаны с данным аналитическим способом.DSC823e (обозначено DSC в табл. 6). Для ряда соединений точки плавления определяли с помощью DSC823e (Mettler-Toledo). Точки плавления измеряли с градиентом температуры 30C/мин. Максимальная температура составляла 400C. Таблица 6 Аналитические данные - Rt означает время удерживания (мин), [M+H]+ означает протонированную массу соединения, способ относится к способу,используемому для (ЖХ)МС Оптические вращения. Оптические вращения измеряли на поляриметре Perkin-Elmer 341 с натриевой лампой и обозначали следующим образом: []tC (конц. г/100 мл, растворитель). Таблица 7 Аналитические данные - значения оптического вращения для энантиомерно чистых соединений ЯМР. Для ряда соединений спектры 1H-ЯМР регистрировали на спектрометре Bruker DPX-360, BrukerDPX-400 или Bruker Avance 600 со стандартными последовательностями импульсов, действующими при 360, 400 и 600 МГц соответственно, с использованием хлороформа-d (дейтерированного хлороформа,CDCl3) или ДМСО-d6 (дейтерированного ДМСО, диметил-d6 сульфоксида) в качестве растворителей. Химические сдвигирегистрировали в частях на миллион (м.д.) относительно тетраметилсиланаD. Фармакологические примеры. Соединения, предложенные в настоящем изобретении, являются ингибиторами APPрасщепляющего по бета-сайту фермента 1 (BACE1). Полагают, что ингибирование BACE1, аспарагиновой протеазы, является существенным для лечения болезни Альцгеймера (AD). Полагают, что продуцирование и накопление бета-амилоидных пептидов (Абета) из белка-предшественника бета-амилоида(APP) играет ключевую роль в возникновении и прогрессировании AD. Абета образуется из белкапредшественника амилоида (APP) в результате последовательного расщепления на N- и C-концах домена абета бета-секретазой и гамма-секретазой соответственно. Предполагается, что соединения формулы (I) действуют главным образом на BACE1 в силу своей способности ингибировать ферментативную активность. Поведение таких ингибиторов, которые тестировали с помощью биохимического анализа на основе резонансного переноса энергии флуоресценции(FRET) и клеточного анализа "Lisa" на клетках SKNBE2, которые описаны ниже и которые являются подходящими для выявления таких соединений и, в частности, соединений в соответствии с формулой(I), показано в табл. 9 и 10. Биохимический анализ на основе FRET. Данный анализ представляет собой анализ на основе резонансного переноса энергии флуоресценции (FRET). Субстратом для данного анализа является полученный из APP 13 аминокислотный пептид,который содержит "шведскую" мутацию Lys-Met/Asn-Leu сайта расщепления белка-предшественника амилоида (APP) бета-секретазой. Данный субстрат также содержит два флуорофора: (7-метоксикумарин 4-ил)уксусную кислоту (Mca), которая является донором флуоресценции с длиной волны возбуждения 320 нм и излучения 405 нм, и 2,4-динитрофенил (Dnp), который является соответствующим акцепторомгасителем. Расстояние между этими двумя группами выбирали таким образом, чтобы при световом возбуждении энергия флуоресценции донора в значительной степени гасилась акцептором посредством резонансного переноса энергии. При расщеплении с помощью BACE1 флуорофор Mca отделяется от гасящей группы Dnp, восстанавливая полный выход флуоресценции донора. Увеличение флуоресценции линейно зависит от степени протеолиза. Вкратце, в 384-луночном формате рекомбинантный белок BACE1 при конечной концентрации 1 мкг/мл инкубировали в течение 120 мин при комнатной температуре с 10 мкМ субстрата в инкубационном буфере (40 мМ цитратного буфера, рН 5,0, 0,04% PEG, 4% ДМСО) в отсутствии или в присутствии соединения. Далее величину протеолиза непосредственно измеряли путем измерения флуоресценции в момент T=0 и T=120 (возбуждение при 320 нм и излучение при 405 нм). Результаты выражали в RFU(относительных единицах флуоресценции) как разницу между T120 и T0. Кривую наилучшего приближения подбирали при помощи методики суммы наименьших квадратов для графика зависимости % мин. контроля от концентрации соединения. На его основании можно получить значение IC50 (ингибирующая концентрация, вызывающая 50% ингибирование активности).% мин контроля=(образец-LC)/(HC-LC)100. Следующие соединения, приведенные в качестве примера, тестировали главным образом, как описано выше, и они проявляли следующую активность: Таблица 9 Клеточный анализ Lisa на клетках SKNBE2. В двух анализах Lisa количественно определяли уровни общего абета и абета 1-42, продуцируемых и секретируемых в среду клеток SKNBE2 нейробластомы человека. Анализ основан на SKNBE2 нейробластомы человека, экспрессирующих белок-предшественник амилоида дикого типа (hAPP695). Соединения разбавляли и добавляли к этим клеткам, инкубировали в течение 18 ч, а затем осуществляли измерения количества абета 1-42 и общего абета. Общий абет и абет 1-42 измеряли с помощью сендвич Lisa.Lisa представляет собой сэндвич-анализ с использованием биотинилированного антитела AbN/25, связанного с покрытыми стрептавидином гранулами, или антитела Ab4G8 или cAb42/26, конъюгированного с груналами-акцепторами, для определения общего абета и абета 1-42 соответственно. В присутствии общего абета или абета 1-42 гранулы приближаются друг к другу. Возбуждение гранул-доноров вызывает высвобождение молекул синглетного кислорода, что запускает каскад переноса энергии на гранулыакцепторы, приводящий в результате к излучению света. Излучение света измеряли через 1 ч после инкубирования (возбуждение при 650 нм и излучение при 615 нм). Кривую наилучшего приближения подбирали при помощи методики суммы наименьших квадратов для графика зависимости % мин контроля от концентрации соединения. На его основании можно получить значение IC50 (ингибирующая концентрация, вызывающая 50% ингибирование активности).=Низкий контроль: клетки, предварительно инкубированные без соединения, без биотинилированного Ab в Lisa;=Высокий контроль: клетки, предварительно инкубированные без соединения;% мин контроля=(образец-LC)/(HC-LC)100. Следующие соединения, приведенные в качестве примера, тестировали главным образом, как описано выше, и они проявляли следующую активность: Таблица 10R2 представляет собой водород или трифторметил; илиR1 и R2 образуют двухвалентный радикал =CF2;R4 представляет собой водород или фтор;Ar представляет собой гетероарил; где гетероарил представляет собой пиридил или пиримидил, каждый из которых необязательно замещен одним, двумя или тремя заместителями, выбранными из галогена, циано, C1-3 алкила, C23 алкинила, С 1-3 алкилокси, моно- и полигалоген-C1-3 алкила, моно- и полигалоген-С 1-3 алкилокси и С 13 алкилоксиС 1-3 алкилокси; или его стереоизомерная форма или его фармацевтически приемлемая соль. 2. Соединение по п.1, гдеR1 и R2 образуют двухвалентный радикал =CF2;R3 представляет собой метил или циклопропил;R4 представляет собой водород или фтор;Ar представляет собой гетероарил; где гетероарил представляет собой пиридил, замещенный метокси или пиримидил; или его фармацевтически приемлемая соль. 3. Соединение по п.1, где атом углерода, замещенный R3, характеризуется R-конфигурацией. 4. Соединение по п.1, выбранное из(5R,6R)-5-2-фтор-5-[(3-метоксипиразин-2-ил)амино]фенил-5-метил-6-(трифторметил)-5,6 дигидро-2H-1,4-оксазин-3-амина. 5. Фармацевтическая композиция для лечения расстройства, выбранного из болезни Альцгеймера,умеренного когнитивного нарушения, старения, деменции, деменции с тельцами Леви, церебральной амилоидной ангиопатии, слабоумия вследствие множественных инфарктов, синдрома Дауна, деменции,ассоциированной с инсультом, деменции, ассоциированной с болезнью Паркинсона, и деменции, ассоциированной с бета-амилоидом, содержащая терапевтически эффективное количество соединения по любому из пп.1-4 и фармацевтически приемлемый носитель. 6. Способ получения фармацевтической композиции по п.5, отличающийся тем, что фармацевтически приемлемый носитель тщательно смешивают с терапевтически эффективным количеством соединения по любому из пп.1-4. 7. Применение соединения по любому из пп.1-4 для лечения или профилактики болезни Альцгеймера (AD), умеренного когнитивного нарушения, старения, деменции, деменции с тельцами Леви, церебральной амилоидной ангиопатии, слабоумия вследствие множественных инфарктов, синдрома Дауна,деменции, ассоциированной с инсультом, деменции, ассоциированной с болезнью Паркинсона, или деменции, ассоциированной с бета-амилоидом. 8. Способ лечения расстройства, выбранного из болезни Альцгеймера, умеренного когнитивного нарушения, старения, деменции, деменции с тельцами Леви, церебральной амилоидной ангиопатии, слабоумия вследствие множественных инфарктов, синдрома Дауна, деменции, ассоциированной с инсультом, деменции, ассоциированной с болезнью Паркинсона, и деменции, ассоциированной с бетаамилоидом, у субъекта, включающий введение субъекту, нуждающемуся в этом, терапевтически эффективного количества фармацевтической композиции по п.5.
МПК / Метки
МПК: A61P 25/00, A61K 31/5377, C07D 265/30, C07D 413/12
Метки: 5-(3-аминофенил)-5-алкил-5,6-дигидро-2h-[1,4]оксазин-3-амина, производные
Код ссылки
<a href="https://eas.patents.su/26-24716-proizvodnye-5-3-aminofenil-5-alkil-56-digidro-2h-14oksazin-3-amina.html" rel="bookmark" title="База патентов Евразийского Союза">Производные 5-(3-аминофенил)-5-алкил-5,6-дигидро-2h-[1,4]оксазин-3-амина</a>
Производные 6-дифторметил-5,6-дигидро-2h-[1,4]оксазин-3-амина

Номер патента: 23909
Опубликовано: 29.07.2016
Авторы: Суркин Михел, Трабанко-Суарес Андрес Авелино, Прокопкова Хана, Гейсен Хенрикус Якобус Мария
МПК: A61P 25/28, A61K 31/5377, A61P 25/16...
Метки: производные, 6-дифторметил-5,6-дигидро-2h-[1,4]оксазин-3-амина
Формула / Реферат:
1. Соединение формулы (I)или его таутомерная или стереоизомерная форма, гдеR1 представляет собой C1-3алкил;R2 представляет собой водород или фтор;L представляет собой связь или -NHCO-;Ar выбран из группы, состоящей из пиридинила, пиримидинила и пиразинила, каждый из которых необязательно замещен галогеном или С1-3алкокси;или его фармацевтически приемлемая соль присоединения.2. Соединение по п.1, где R1 представляет собой метил или этил.3....
Производные 5,6-дигидро-2h-[1,4]оксазин-3-иламина в качестве ингибиторов бета-секретазы (bace)

Номер патента: 21240
Опубликовано: 29.05.2015
Авторы: Мартинес Ламенка Каролина, Гейсен Хенрикус Якобус Мария, Трабанко-Суарес Андрес Авелино, Ромбаутс Фредерик Ян Рита, Макдональд Грегор Джеймс, Ван Гол Михиль Люк Мария, Тресадерн Гэри Джон
МПК: A61P 25/16, A61K 31/5355, A61K 31/535...
Метки: бета-секретазы, производные, качестве, 5,6-дигидро-2h-[1,4]оксазин-3-иламина, ингибиторов, bace
Формула / Реферат:
1. Соединение формулы (I)или его таутомерная или стереоизомерная форма, гдеR1, R2 и R3 независимо выбраны из группы, состоящей из водорода и фтора;R4 представляет собой фтор или трифторметил;R5 выбран из группы, состоящей из C1-3-алкила и циклопропила;X1, X2, X3, X4 независимо представляют собой C(R6);каждый R6 выбран из группы, состоящей из водорода и галогена;L представляет собой связь или -N(R7)CO-, где R7 представляет собой водород или...
Производные 1-[алкил], 1-[(гетероарил)алкил] и 1-[(арил)алкил]-7-пиридинил-имидазо[1, 2-a]пиримидин-5(1н)она

Номер патента: 4933
Опубликовано: 28.10.2004
Авторы: Маргери Северен, Локхид Алистер, Саади Мурад, Йеш Филипп
МПК: C07D 487/04, A61K 31/519, A61P 25/00...
Метки: 1-[(гетероарил)алкил, 1-[(арил)алкил]-7-пиридинил-имидазо[1, 1-[алкил, 2-a]пиримидин-5(1н)она, производные
Формула / Реферат:
1. Производное имидазо[1,2-a]пиримидона, представленное формулой (I), или его соль, или их сольват, либо их гидрат где X представляет собой связь, этениленовую группу, этиниленовую группу, метиленовую группу, возможно замещенную одной или двумя группами, выбранными из C1-6алкильной группы, гидроксигруппы и C1-4алкоксигруппы; карбонильную группу, атом кислорода, атом серы, сульфонильную группу, сульфинильную группу или атом азота, возможно...
Производные 1-[алкил]-, 1-[(гетероарил)алкил]- и 1-[(арил)алкил]-7-(пиримидин-4-ил)имидазо[1, 2-a]пиримидин-5(1н)-она

Номер патента: 7459
Опубликовано: 27.10.2006
Авторы: Локхид Алистер, Йеш Филипп, Саади Мурад
МПК: A61K 31/437, A61P 25/28, C07D 487/04...
Метки: 2-a]пиримидин-5(1н)-она, 1-[(гетероарил)алкил, 1-[(арил)алкил]-7-(пиримидин-4-ил)имидазо[1, 1-[алкил, производные
Формула / Реферат:
1. Производное имидазо[1,2-а]пиримидона, представленное формулой (I), или его соль, или сольват, или гидрат где X представляет собой связь, метиленовую группу, возможно замещенную одной или двумя группами, выбранными из С1-6алкильной группы, гидроксильной группы и С1-4алкоксильной группы; карбонильную группу; R1 представляет собой 2, 4 или 5-пиримидинил, возможно замещенный С1-4алкильной группой, С1-4 алкоксильной группой или атомом галогена;...
Производные 1,3-оксазин-4-она как гербициды.

Номер патента: 1102
Опубликовано: 30.10.2000
Авторы: Кудо Сатио, Крэмп Майкл Колин, Усуи Йосихиро
МПК: A01N 43/86, C07D 265/06
Метки: гербициды, производные, 1,3-оксазин-4-она
Формула / Реферат:
1. Производные 1,3-оксазин-4-она формулы I где R1 обозначает фенил или тиенил, необязательно замещенный одной или более группами, выбранными из: галогена, C1-4 алкила и C1-4 галогеналкила; R2 обозначает линейную или разветвленную группу алкила, содержащую от одного до шести атомов углерода; R3 обозначает: -(CH2)r - (фенильную группу, необязательно замещенную одной-двумя группами, выбранными из галогена или алкила); тиенила, фурила,...
Предыдущий патент: Пустотелая плита с межпустотными усилителями
Следующий патент: Способ получения оксида цинка из руды
Случайный патент: Способ и установка для разделения газов